WO2010015653A1 - Pyrimidine derivatives as activators of soluble guanylate cyclase - Google Patents
Pyrimidine derivatives as activators of soluble guanylate cyclase Download PDFInfo
- Publication number
- WO2010015653A1 WO2010015653A1 PCT/EP2009/060146 EP2009060146W WO2010015653A1 WO 2010015653 A1 WO2010015653 A1 WO 2010015653A1 EP 2009060146 W EP2009060146 W EP 2009060146W WO 2010015653 A1 WO2010015653 A1 WO 2010015653A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- phenyl
- pyrimidin
- phenylmethyloxy
- carboxylic acid
- Prior art date
Links
- 0 C*1/C=C(\c2ccnc(Cl)n2)/C(/O*)=C\C(C)(*)/C=C1 Chemical compound C*1/C=C(\c2ccnc(Cl)n2)/C(/O*)=C\C(C)(*)/C=C1 0.000 description 11
- XNGGOXOLHQANRB-WAYWQWQTSA-N CCO/C=C(/C(C(F)(F)F)=O)\C(OCC)=O Chemical compound CCO/C=C(/C(C(F)(F)F)=O)\C(OCC)=O XNGGOXOLHQANRB-WAYWQWQTSA-N 0.000 description 2
- RUJPPJYDHHAEEK-UHFFFAOYSA-N CCOC(C1CCNCC1)=O Chemical compound CCOC(C1CCNCC1)=O RUJPPJYDHHAEEK-UHFFFAOYSA-N 0.000 description 2
- UZYDWJLYGWIVCE-UHFFFAOYSA-N CCCOc1c(C=O)ccc(Br)c1 Chemical compound CCCOc1c(C=O)ccc(Br)c1 UZYDWJLYGWIVCE-UHFFFAOYSA-N 0.000 description 1
- UIVYBQPWEFTPAI-UHFFFAOYSA-N CCOC(C(CC1)CCN1c1nc(-c(cc(C)cc2)c2OCc(cc2)c(C)cc2-c2ccc(C(F)(F)F)cc2C)ccn1)=O Chemical compound CCOC(C(CC1)CCN1c1nc(-c(cc(C)cc2)c2OCc(cc2)c(C)cc2-c2ccc(C(F)(F)F)cc2C)ccn1)=O UIVYBQPWEFTPAI-UHFFFAOYSA-N 0.000 description 1
- QAJXSXCLJDFEGP-UHFFFAOYSA-N CCOC(C(CC1)CCN1c1nc(-c(cc(C)cc2)c2OCc2ccc(B3OC(C)(C)C(C)(C)O3)cc2C)ccn1)=O Chemical compound CCOC(C(CC1)CCN1c1nc(-c(cc(C)cc2)c2OCc2ccc(B3OC(C)(C)C(C)(C)O3)cc2C)ccn1)=O QAJXSXCLJDFEGP-UHFFFAOYSA-N 0.000 description 1
- PBJTZLUVYOOMOF-UHFFFAOYSA-N CCOC(C(CC1)CCN1c1nc(-c2cc(C(F)(F)F)ccc2O)ccn1)=O Chemical compound CCOC(C(CC1)CCN1c1nc(-c2cc(C(F)(F)F)ccc2O)ccn1)=O PBJTZLUVYOOMOF-UHFFFAOYSA-N 0.000 description 1
- JMLHMCMDRLWLQM-UHFFFAOYSA-N CCOC(c1c(C(F)(F)F)[n](-c2nc(-c3cc(Cl)ccc3O)ccn2)nc1)=O Chemical compound CCOC(c1c(C(F)(F)F)[n](-c2nc(-c3cc(Cl)ccc3O)ccn2)nc1)=O JMLHMCMDRLWLQM-UHFFFAOYSA-N 0.000 description 1
- KPPGOLQXXYVSDK-UHFFFAOYSA-N CCOC(c1c(C(F)(F)F)[n](-c2nc(Cl)ncc2)nc1)=O Chemical compound CCOC(c1c(C(F)(F)F)[n](-c2nc(Cl)ncc2)nc1)=O KPPGOLQXXYVSDK-UHFFFAOYSA-N 0.000 description 1
- SRDPHYMKBGIQMT-UHFFFAOYSA-N Cc(cc1)cc(-c2ccnc(N(CC3)CCC3C(O)=O)n2)c1OCc(cc1)c(C)cc1-c(c(C)c1)ccc1C#N Chemical compound Cc(cc1)cc(-c2ccnc(N(CC3)CCC3C(O)=O)n2)c1OCc(cc1)c(C)cc1-c(c(C)c1)ccc1C#N SRDPHYMKBGIQMT-UHFFFAOYSA-N 0.000 description 1
- BTTNYQZNBZNDOR-UHFFFAOYSA-N Clc1ccnc(Cl)n1 Chemical compound Clc1ccnc(Cl)n1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 1
- MSKJYRJEURENQP-UHFFFAOYSA-N Oc(c(-c1ccnc(Cl)n1)c1)ccc1F Chemical compound Oc(c(-c1ccnc(Cl)n1)c1)ccc1F MSKJYRJEURENQP-UHFFFAOYSA-N 0.000 description 1
- GBJJCODOZGPTBC-UHFFFAOYSA-N Oc1c(C=O)ccc(F)c1 Chemical compound Oc1c(C=O)ccc(F)c1 GBJJCODOZGPTBC-UHFFFAOYSA-N 0.000 description 1
- HLVBEUWJSOBVEI-UHFFFAOYSA-N Oc1ccc(C(F)(F)F)cc1-c1ccnc(Cl)n1 Chemical compound Oc1ccc(C(F)(F)F)cc1-c1ccnc(Cl)n1 HLVBEUWJSOBVEI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to novel compounds, pharmaceutical compositions containing them, to their use in medicine, and to processes for their preparation.
- the present invention relates to compounds which, when administered to a patient, activate soluble guanylate cyclase (sGC) and to the use of such compounds for the activation of sGC in patients for a therapeutic effect.
- sGC soluble guanylate cyclase
- sGC is a member of a family of related enzymes which share homologous catalytic domains but are activated in different ways.
- This family includes the adenylate cyclases, a class of membrane bound enzymes that convert ATP to cAMP, which are regulated by G proteins, and the membrane-bound guanylate cyclases that make cyclic guanosine monophosphate (cGMP) in response to hormone signals via an extracellular ligand binding domain.
- cGMP cyclic guanosine monophosphate
- the active enzyme contains one heme unit in a heterodimer arrangement, composed of one alpha and one beta-subunit.
- Several subtypes of subunits have been described, which differ from each other with respect to sequence and tissue-specific distribution.
- the subtypes alpha-1 and beta-1 are thought to be mainly expressed in the brain and the lung but have also been shown to be expressed in heart, kidney, liver, skeletal muscle, placenta, colon, uterus, prostate, spleen, pancreas, platelets and isolated blood vessels.
- Alpha-2 subunits have been detected in the brain, placenta, uterus and pancreas, while beta-2 subunits seem to be expressed in the liver and kidney.
- the enzyme is thought to be a principal receptor for the ubiquitous signalling molecule, nitric oxide (NO), forming a NO-sGC-cGMP signal transduction axis. It is believed that soluble guanylate cyclase is a heme sensor protein that selectively binds NO at the heme iron, which activates the enzyme to convert guanosine triphosphate (GTP) to cGMP. It is thought that cGMP subsequently mediates a number of important physiological processes, including smooth muscle relaxation and neurotransmission.
- NO ubiquitous signalling molecule
- soluble guanylate cyclase is a heme sensor protein that selectively binds NO at the heme iron, which activates the enzyme to convert guanosine triphosphate (GTP) to cGMP. It is thought that cGMP subsequently mediates a number of important physiological processes, including smooth muscle relaxation and neurotransmission.
- cGMP is a critical component involved in the regulation of various (patho)physiological processes, for example in cardiovascular, respiratory, gastrointestinal, urogenital, nervous and immune systems including, neuronal excitability and particularly smooth muscle tone, thereby controlling, among other things, blood pressure, gastro-intestinal motility and genital erection.
- novel compounds of the invention are activators of sGC and consequently may have application in the treatment of one or more diseases or conditions, which include: cardiovascular diseases and conditions, such as angina (including stable and unstable angina pectoris), low cardiac output, cerebral ischemia, cardiac ischemia, myocardial infarction, coronary reperfusion injury, arterial hypertension (including pulmonary arterial hypertension), congestive heart failure (for example due to systolic and/or diastolic cardiac dysfunction, low cardiac output or high systemic vascular resistance), heart failure with preserved ejection fraction, acute heart failure syndromes (AHFS), cardiac hypertrophy, acute coronary syndrome, thromboses (including arterial or venous thrombosis), atherosclerosis, peripheral vascular disease, glomerulonephritis, restenosis (for example following percutaneous vascular intervention, vascular angioplasty or stent placement), Raynaud's disease, vascular complications of diabetes or of obesity, stroke, hereditary cerebral haemorrhage, endothelial
- cardiovascular hypertension including pulmonary arterial hypertension
- cardiac ischemia myocardial infarction
- congestive heart failure for example due to systolic and/or diastolic cardiac dysfunction, low cardiac output or high systemic vascular resistance
- cardiac hypertrophy acute coronary syndrome, atherosclerosis, peripheral vascular disease, cardiorenal syndrome, hepatorenal syndrome and restenosis (for example following percutaneous vascular intervention, vascular angioplasty or stent placement).
- a particular disease or condition for which the compounds of the invention may be useful is congestive heart failure. Another particular disease or condition for which the compounds of the invention may be useful is peripheral vascular disease. Another particular disease or condition for which the compounds of the invention may be useful is arterial hypertension (also known as systemic hypertension). Another particular disease or condition for which the compounds of the invention may be useful is pulmonary arterial hypertension. Another particular disease or condition for which the compounds of the invention may be useful is angina.
- the present invention provides a compound of formula (I)
- R 1 and R 2 are independently selected from hydrogen, halo, CF 3 and Ci -4 alkyl;
- one of U and V represents N and the other represents CH;
- R 3 represents CF 3 or C 1-4 alkyl
- Z is absent or represents O
- A represents CH or N; when A represents CH, R 5 is selected from hydrogen, methyl, C 1-4 alkoxy, methoxyC 2 - 3 alkoxy, chloro and fluoro and R 6 represents hydrogen;
- R 5 and R 6 each represent hydrogen or one of R 5 and R 6 represents hydrogen and the other represents methyl
- J and L both represent CH, or one represents N and the other represents CH, provided that only one of A, J and L represents N;
- R 8 represents hydrogen or chloro, fluoro, CF 3 , C 1-4 alkyl or C 1-4 alkoxy in a meta or ortho position relative to the R 9 substituent;
- R 8 represents hydrogen or halo in a meta or ortho position relative to the R 9 substituent
- R a represents hydrogen, halo, CF 3 , OCF 3 , C 1-4 -alkyl, C 1-4 -alkoxy, CN, CONR 10 R , CO 2 H or N 3 , wherein R ,10 and R are independently selected from hydrogen and C 1-4 -alkyl;
- R 1 and R 2 are independently selected from hydrogen, halo, CF 3 or C 1-4 alkyl; one of U and V represents N and the other represents CH;
- R 3 represents CF 3 or C 1-4 alkyl
- Z is absent or represents O
- A, J and L each represent CH; or one of A, J and L represents N and the other two each represents CH; and when A represents CH, R 5 is selected from hydrogen, methyl, C 1-4 alkoxy, methoxyC 2- 3 alkoxy, chloro and fluoro and R 6 represents hydrogen; or when A represents N, R 5 and R 6 each represent hydrogen or one of R 5 and R 6 represents hydrogen and the other represents methyl;
- R 8 represents hydrogen or chloro, fluoro, CF 3 , C 1- 4 alkyl or C 1-4 alkoxy in a meta or ortho position relative to the R 9 substituent; or when one of J and L represents N, R 8 represents hydrogen or halo in a meta or ortho position relative to the R 9 substituent; and R 9 represents hydrogen, halo, CF 3 , OCF 3 , C 1-4 -alkyl, C 1-4 -alkoxy, CN, CONR 10 R 11 , CO 2 H or N 3 , wherein R 10 and R 11 are independently selected from hydrogen and C 1-4 -alkyl;
- alkyl refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms.
- C 1-4 alkyl means a straight or branched alkyl containing at least 1 , and at most 4, carbon atoms.
- alkyl as used herein include, but are not limited to, methyl, ethyl, n-propyl, n-butyl, isobutyl, isopropyl, and t-butyl.
- alkoxy refers to a straight or branched alkoxy group containing the specified number of carbon atoms.
- C 1-4 alkoxy means a straight or branched alkoxy group containing at least 1 , and at most 4, carbon atoms.
- alkoxy as used herein include, but are not limited to, methoxy, ethoxy, propoxy, prop-2-oxy, butoxy, but-2-oxy, 2-methylprop-1-oxy, or 2-methylprop-2-oxy.
- halo refers to the elements fluorine, chlorine, bromine and iodine. In an embodiment halo represents bromine, fluorine and chlorine. In a further embodiment halo represents fluorine and chlorine.
- pharmaceutically acceptable means a compound which is suitable for pharmaceutical use.
- Y— represents
- R 1 is in a para position relative to the -OCH 2 - linker.
- R 2 is in an ortho position relative to the -OCH 2 - linker.
- R 1 is in a para position relative to the -OCH 2 - linker and R 2 is in an ortho position relative to the -OCH 2 - linker.
- R 1 is in an ortho position relative to the bond linking to the pyridine ring.
- R 2 is in a meta position relative to the -OCH 2 - linker.
- R 1 and R 2 do not both represent C 2-4 alkyl.
- R 1 and R 2 represents the other represents hydrogen
- R 1 represents C 1-4 alkyl, CF 3 or halo, in a further embodiment with R 1 in a para position relative to the -OCH 2 - linker.
- R 1 represents methyl, CF 3 , fluoro or chloro, in a further embodiment with R 1 in a para position relative to the -OCH 2 - linker.
- R 2 represents hydrogen
- R 1 represents chloro or fluoro and R 2 represents hydrogen, in a further embodiment with R 1 in an ortho position relative to the bond linking to the pyridine ring.
- R 2 represents chloro or fluoro and R 1 represents hydrogen, in a further embodiment with R 2 in a meta position relative to the -OCH 2 - linker.
- U represents CH and V represents N.
- R 1 and R 2 each represent hydrogen. In an embodiment R 1 represents chloro or fluoro and R 2 represents hydrogen. In an embodiment R 1 represents C 1-4 alkyl and R 2 represents hydrogen. In an embodiment R 1 represents methyl and R 2 represents hydrogen. In an embodiment R 1 represents CF 3 and R 2 represents hydrogen.
- R 3 represents methyl or CF 3 . In a further embodiment R 3 represents CF 3 .
- Z is absent. In an embodiment Z represents O.
- A represents CH.
- R 6 represents hydrogen
- R 5 represents hydrogen, methyl, methoxy, propyloxy, isopropyloxy, isobutyloxy, methoxyethoxy, fluoro or chloro. In an embodiment, R 5 represents hydrogen, methyl, methoxy or propyloxy.
- J and L both represent CH. In a further embodiment J represents N and L represents CH. In a further embodiment J represents CH and L represents N.
- R 8 represents hydrogen or chloro, CF 3 , methyl or methoxy in a meta or ortho position relative to the R 9 substituent. In an embodiment R 8 represents hydrogen or chloro, CF 3 , methyl or methoxy in a meta position relative to the R 9 substituent.
- J and L both represent CH and R 8 represents hydrogen or chloro, CF 3 , methyl or methoxy in a meta position relative to the R 9 substituent.
- J represents N and L represents CH and R 8 represents hydrogen, or chloro in a meta position relative to the R 9 substituent.
- J represents CH and L represents N and R 8 represents hydrogen.
- R 9 represents hydrogen, halo, CF 3 , OCF 3 , C 1-4 alkyl, C 1-4 alkoxy or CN. In a further embodiment R 9 represents halo, CF 3 , OCF 3 , C 1-4 alkoxy or CN. In a further embodiment R 9 represents chloro, fluoro, CF 3 , OCF 3 , methoxy or CN.
- R 8 represents hydrogen and R 9 represents methoxy, CN, CF 3 , OCF 3 or fluoro.
- R 8 represents methyl and R 9 represents CN, methoxy or CF 3 , with R 8 being in a meta position relative to the R 9 substituent.
- R 8 represents CF 3
- R 9 represents CN, methoxy or chloro, with R 8 being in a meta position relative to the R 9 substituent.
- R 8 represents chloro
- R 9 represents CF 3 , CN or methoxy, with R 8 being in a meta position relative to the R 9 substituent.
- Separation of the individual enantiomers of the relevant compounds may be carried out by standard methods well-known to the person skilled in the art, for example by chiral chromatography.
- Salts of compounds of formula (I) which are suitable for use in medicine are those wherein the counterion is pharmaceutically acceptable.
- salts having non-pharmaceutically acceptable counterions are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
- Solvates of the compounds of formula (I) and solvates of the salts of the compounds of formula (I) are included within the scope of the present invention.
- the term "solvate” refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I) or a salt thereof) and a solvent.
- solute in this invention, a compound of formula (I) or a salt thereof
- solvents for the purpose of the invention may not interfere with the biological activity of the solute.
- suitable solvents include, but are not limited to, water, methanol, ethanol and acetic acid.
- the solvent used is a pharmaceutically acceptable solvent.
- suitable pharmaceutically acceptable solvents include, without limitation, water, ethanol and acetic acid.
- the solvent used is water. Where the solvent used is water such a solvate may then also be referred to as a hydrate.
- the salts of the compounds of formula (I) will be pharmaceutically acceptable.
- the invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the salts of the compounds of formula (I).
- a salt may be readily prepared by using a desired acid or base as appropriate.
- the salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent.
- Suitable pharmaceutically acceptable salts can include acid addition salts or base addition salts and will be apparent to those skilled in the art.
- a pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic acid such as hydrochloric, hydrobromic, hydroiodic, sulfuric, nitric or phosphoric acid; or with a suitable organic acid such as succinic, maleic, malic, mandelic, formic, acetic, propionic, hexanoic, fumaric, glutamic, lactic, citric, tartaric, benzoic, salicylic, aspartic, benzenesulfonic, p-toluenesulfonic, methanesulfonic, ethanesulfonic or naphthalenesulfonic acid.
- a suitable inorganic acid such as hydrochloric, hydrobromic, hydroiodic, sulfuric, nitric or phosphoric acid
- a pharmaceutically acceptable base addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic or organic base, including salts of primary, secondary and tertiary amines, such as isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexyl amine, N-methyl-D-glucamine triethylamine, triethanolamine, choline, arginine, lysine or histidine.
- a suitable inorganic or organic base including salts of primary, secondary and tertiary amines, such as isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexyl amine, N-methyl-D-glucamine triethylamine, triethanolamine, choline, arginine, lysine or histidine.
- suitable pharmaceutically acceptable salts include pharmaceutically acceptable metal salts, for example pharmaceutically acceptable alkali- metal or alkaline-earth-metal salts such as sodium, potassium, calcium or magnesium salts; in particular pharmaceutically acceptable metal salts of the carboxylic acid moiety that is present in the compound of formula (I). Since the compounds of formula (I) include a carboxylic acid moiety together with one or more basic nitrogen atom(s) they have the possibility to also form internal salts, which salts are also included within the scope of the present invention.
- pharmaceutically acceptable metal salts for example pharmaceutically acceptable alkali- metal or alkaline-earth-metal salts such as sodium, potassium, calcium or magnesium salts
- pharmaceutically acceptable metal salts of the carboxylic acid moiety that is present in the compound of formula (I). Since the compounds of formula (I) include a carboxylic acid moiety together with one or more basic nitrogen atom(s) they have the possibility to also form internal salts, which salts are also included within the scope of the present invention.
- polymorphs of a compound of the invention are also included within the scope of the invention.
- the invention also includes all suitable isotopic variations of a compound of the invention.
- An isotopic variation of a compound of the invention is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2 H, 3 H, 13 C, 14 C, 15 N, 17 O, 18 O, 31 P, 32 P, 35 S, 18 F and 36 CI, respectively.
- isotopic variations of the invention for example, those in which a radioactive isotope such as 3 H or 14 C is incorporated, are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability.
- lsotopic variations of the compounds of the invention can generally be prepared by conventional procedures such as by the illustrative methods or by the preparations described in the Examples hereafter using appropriate isotopic variations of suitable reagents.
- compounds of the invention as activators of sGC, may be useful in the treatment of a disease or condition which is mediated by sGC activity.
- the invention provides a compound of the invention as defined above for use in therapy; in an embodiment the therapy is human therapy.
- the invention provides the use of a compound of the invention for the manufacture of a medicament for treating a disease or condition mediated by sGC activity.
- the invention provides a compound of the invention for use in the treatment of a disease or condition mediated by sGC activity.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention, in association with one or more pharmaceutically acceptable carrier(s), diluents(s) and/or excipient(s).
- the carrier, diluent and/or excipient must be "acceptable” in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
- the invention provides a method of treatment of a disease or condition which is mediated by the activity of sGC such as one or more of the diseases described above, for example arterial hypertension, pulmonary arterial hypertension, angina, cardiac ischemia, myocardial infarction, congestive heart failure, acute coronary syndrome, atherosclerosis, peripheral vascular disease or restenosis, comprising administration to a human subject in need of such treatment of a therapeutically effective amount of a compound of the invention, or of a pharmaceutical composition comprising a compound of the invention.
- sGC such as one or more of the diseases described above, for example arterial hypertension, pulmonary arterial hypertension, angina, cardiac ischemia, myocardial infarction, congestive heart failure, acute coronary syndrome, atherosclerosis, peripheral vascular disease or restenosis
- the invention provides a method of treatment of cardiorenal syndrome or hepatorenal syndrome comprising administration to a human subject in need of such treatment of a therapeutically effective amount of a compound of the invention, or of a pharmaceutical composition comprising a compound of the invention.
- the compounds of the invention may also be used in combination with other therapeutic agents.
- the invention thus provides, in a further aspect, a combination comprising a compound of the invention or a pharmaceutically acceptable derivative thereof together with a further therapeutic agent.
- a compound of the invention or a pharmaceutically acceptable derivative thereof is used in combination with a second therapeutic agent active against the same disease state the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art. It will be appreciated that the amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian.
- the compounds of the present invention may for example be used in combination with antihypertensive drugs such as diuretics (for example epitizide, bendroflumethiazide, chlortalidone, chlorthiazide, hydrochlorthiazide, indapamide, metolazone), ACE inhibitors (such as benzapril, captopril, enalapril, fosinopril, lisinopril, preindopril, quinapril, ramipril, trandopril), angiotensin receptor blockers (such as candesartan, irbesartan, losartan, telmisartan, valsartan), calcium channel inhibitors (such as amlodipine, felodipine, isradapine, nifedipine, niimodipine, nitrendipine, diltiazem, verapamil), ⁇ -adrenergic receptor antagonists (such
- compositions comprising a combination as defined above together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention.
- the individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations by any convenient route.
- either the compound of the invention or the second therapeutic agent may be administered first.
- the combination may be administered either in the same or different pharmaceutical composition.
- references herein to "treatment” extend to prophylaxis, prevention of recurrence and suppression or amelioration of symptoms (whether mild, moderate or severe) as well as the treatment of established conditions.
- the compound of the invention may be administered as the raw chemical but the active ingredient is preferably presented as a pharmaceutical formulation.
- the compound of the invention may be administered in conventional dosage forms prepared by combining a compound of the invention with one or more standard pharmaceutical excipients, carriers or diluents, according to conventional procedures well known in the art. These procedures may involve mixing, granulating and compressing or dissolving the ingredients as appropriate for the desired preparation.
- compositions of the invention may be formulated for administration by any route, and include those in a form adapted for oral, topical or parenteral administration to mammals including humans.
- compositions may be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
- topical formulations of the present invention may be presented as, for instance, ointments, creams or lotions, eye ointments and eye or ear drops, impregnated dressings and aerosols, and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration and emollients in ointments and creams.
- the formulations may also contain compatible conventional carriers, such as cream or ointment bases and ethanol or oleyl alcohol for lotions.
- suitable conventional carriers such as cream or ointment bases and ethanol or oleyl alcohol for lotions.
- Such carriers may be present as from about 1% up to about 98% of the formulation. More usually they will form up to about 80% of the formulation.
- Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatine, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants, for example potato starch; or acceptable wetting agents such as sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives, such as suspending agents, for example sorbitol, methyl cellulose, glucose syrup, gelatine, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and, if desired, conventional flavouring or colouring agents.
- suspending agents for example sorbitol, methyl cellulose, glucose syrup, gelatine, hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate
- Suppositories typically contain conventional suppository bases, e.g. cocoa-butter or other glyceride.
- fluid unit dosage forms are prepared utilising the compound and a sterile vehicle, water being preferred.
- the compound depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle.
- the compound can be dissolved in water for injection and filter-sterilised before filling into a suitable vial or ampoule and sealing.
- agents such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum.
- the dry lyophilised powder is then sealed in the vial and an accompanying vial of water for injection may be supplied to reconstitute the liquid prior to use.
- Parenteral suspensions are prepared in substantially the same manner except that the compound is suspended in the vehicle instead of being dissolved and sterilisation cannot be accomplished by filtration.
- the compound can be sterilised by exposure to ethylene oxide before suspending in the sterile vehicle.
- a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
- compositions of the invention may be formulated, for administration to mammals including humans, by any route, and include those in a form adapted for oral, topical or parenteral administration.
- the compositions may, for example, be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
- the invention provides a pharmaceutical composition for oral administration such as an oral suspension or liquid, for example an aqueous based fluid formulation, or a solid dosage formulation such as a tablet or capsule.
- a pharmaceutical composition for oral administration such as an oral suspension or liquid, for example an aqueous based fluid formulation, or a solid dosage formulation such as a tablet or capsule.
- compositions may contain from 0.1% by weight, preferably from 10-60% by weight, of the active material, depending on the method of administration. Where the compositions comprise dosage units, each unit will typically contain from 5-1000 mg of the active ingredient. It will be recognised by one of skill in the art that the optimal quantity and spacing of individual doses of a compound of the invention will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular mammal being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e. the number of doses of a compound of the invention given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
- the compounds of formula (I) are intended for use in pharmaceutical compositions it will readily be understood that they are each suitably provided in substantially pure form, for example at least 60% pure, for example at least 75% pure, for example at least 85%, for example at least 98% pure (% are on a weight for weight basis). Impure preparations of the compounds may be used for preparing the more pure forms used in the pharmaceutical compositions; these less pure preparations of the compounds typically contain at least 1 %, for example at least 5%, for example from 10 to 59% of a compound of the invention.
- step (i) the intermediate compounds of formula (II) can be prepared according to the processes set out in Schemes 2 a anndd 33.
- 2,4-dichloropyrimidine is commercially available from Aldrich.
- the compounds of formula (V) wherein R 3 represents CF 3 or CH 3 are commercially available (from Aldrich and Orphachem respectively).
- the compounds of formula (V) wherein R 3 represents C 2-4 alkyl can be prepared according to the process described in US2005096353 (see Scheme 6 at page 17 and the Examples).
- Ethyl isonipecotate is commercially available (from Aldrich).
- the compound of formula (lib) can also be prepared in a solvent such as acetone, CH 3 CN or THF in the presence of a base such as Na 2 CO 3 , K 2 CO 3 or Cs 2 CO 3 , under reflux.
- step (ii) the intermediate compounds of formula (III) can be prepared according to the processes set out in Schemes 4, 5 and 6.
- 2-hydroxyphenyl boronic acid is commercially available (from Aldrich) as is 5-fluoro-2- hydroxyphenyl boronic acid (from Apollo or Combi Blocks).
- the compounds of formula (NIb) can be prepared from the boronic acids of formula (VIb); these are either commercially available or can be prepared by standard methods well-known to the person skilled in the art.
- the compound of formula (VIb) where R 1 is ethyl and R 2 is hydrogen may be prepared as described in WO2005019151.
- Compounds of formula (VIb) where R 1 is propyl in a para position relative to the -OCH 2 - linker and R 2 is hydrogen may be prepared from the corresponding 4-propyl anisole by (i) bromination in the position ortho to the methoxy group and (ii) conversion of the bromine to a boronic acid group by standard methods.
- 2-hydroxy-phenylboronic acid and 5-methyl-2-methoxy-phenylboronic acid are commercially available from Aldrich.
- 5-chloro-2-hydroxy-phenylboronic acid, 5-fluoro-2-hydroxy- phenylboronic acid and 5-trifluoromethyl-2-methoxy-phenylboronic acid are commercially available from Combi-blocks.
- step (iii) the following general synthetic schemes can be used (Schemes 8, 9 and 10).
- Scheme 9 The synthesis described in Scheme 9 relates to compounds wherein Z is absent and both J and L represent CH or J represents CH and L represents N and is particularly suitable for compounds wherein R 5 represents F or Cl.
- the subsequent step (iv) may be carried out according to Scheme 11 a.
- the temperature used in this ester hydrolysis reaction will depend on the nature of the compound and the length of time for which the reaction is performed; this will be well appreciated by the person skilled in the art.
- ester hydrolysis reaction is instead suitably carried out using LiOH at room temperature to avoid hydrolysis of the cyano group to an amide group.
- Scheme 13 is particularly suitable to prepare compounds wherein R 9 represents CN, Z is absent or Z represents O.
- hal' is chloro or bromo if such compounds are commercially available. If not, then the corresponding compound wherein hal' is bromo may be used and may be prepared by standard methods. 2-chloro-5-chloromethylpyridine and 4-bromo-2-fluoro-benzyl bromide are both commercially available (from Aldrich). Other compounds of formula (VIII) may generally be prepared by the following methods (Schemes 14, 15 and 16) or by other standard methods well-known to the person skilled in the art.
- Scheme 17 is particularly suitable for compounds wherein one of A, J or L represents N.
- Pathway A is thus typically used where A represents N or L represents N; and pathway B is typically used where J represents N.
- 2- methoxypyridine-5-boronic acid is available from Aldrich and 2-cyanopyridine-5-boronic acid and 2-trifluoromethylpyridine-5-boronic acid are available from Frontier; other compounds of formula (IX) not commercially available may be prepared by standard methods well-known to the person skilled in the art.
- Certain compounds of formula (XV) are commercially available, for example: wherein J represents N, R 8 represents H and R 9 represents CN, CF 3 , COOR, Cl or OMe (Aldrich, Fluka or Acros); wherein J represents N, R 8 represents Cl and R 9 represents CF 3 (Aldrich).
- Other compounds of formula (XV) may be prepared by standard methods well-known to the person skilled in the art.
- Pathway B is also typically used where J and L both represent CH where hal represents Br.
- Scheme 18b is particularly suitable.
- Scheme 18c is particularly suitable.
- R 35 represents H, Me, OMe.
- Pathway B is thus typically used for A represents N or for A represents CH and hal represents F and R 5 represents H, Me, or OMe.
- Phenol derivatives of formula (XIX) are commercially available or may be prepared by standard methods well-known to the person skilled in the art.
- R 5 represents Cl or F:
- Certain compounds of formula (XVII) are commercially available, for example: wherein hal represents Br or Cl, J represents N, R 8 represents H, and R 9 represents CN, CF 3 , COOR, Cl or OMe (Aldrich, Fluka or Acros); wherein hal represents Cl, J represents N, R 8 represents Cl, and R 9 represents CF 3 (Aldrich); wherein J represents CH, hal represents F, R 8 represents H, F, Cl, OMe, Me, or CF 3 , and R 9 represents CN or COOR (Aldrich, Acros, Apin).
- Other compounds of formula (XVII) may be prepared by standard methods well-known to the person skilled in the art.
- R 5 represents OC 1-4 alkyl or O(CH 2 ) 2 OMe:
- MS mass spectra
- MS mass spectra
- Example 1 1 -(4-(5-methyl-2-(2-methyl-4-(4-cvano-2-methylphenyl)benzyloxy)- phenyl)pyrimidin-2-yl)-piperidine-4-carboxylic acid
- soluble guanylate cyclase can be tested in an assay based on measuring the fluorescent polarisation (FP) signal of fluorescently labelled cGMP.
- FP fluorescent polarisation
- cGMP displaces the interaction giving rise to a decrease in polarisation and FP signal which can be equated to enzyme activity.
- Compounds are incubated with human sGC, anti-cGMP antibody, the GTP substrate and fluorescently labelled cGMP. After a period of one hour the assay is stopped with the addition of EDTA and after a further hour the assay is read.
- Human sGC is thawed and resuspended in assay buffer (10OmM TRIS, 1 OmM MgCI 2 , 0.2mM Tween 20, pH7.4, containing 1 :100 dilution of sheep anti-cGMP) to give final concentration of 1 nM in the well.
- assay buffer (10OmM TRIS, 1 OmM MgCI 2 , 0.2mM Tween 20, pH7.4, containing 1 :100 dilution of sheep anti-cGMP) to give final concentration of 1 nM in the well.
- a substrate solution is prepared containing GTP and 8- fluo-cGMP in de-ionized water to a final concentration of 25 ⁇ M and 5OnM respectively.
- Assay plates containing 5 ⁇ l_ of various test compounds and of a standard agonist (50 ⁇ M - 5OnM) in 1 % DMSO as 6 point, four fold dilutions across a 96 well plate are used in the assay.
- the plate also contains 6 wells of DMSO (1 %) to produce high control and a cGMP standard curve (14nM to 10 ⁇ M) to convert FP data to cGMP concentration.
- 25 ⁇ l_ of enzyme mix and 20 ⁇ l of substrate mix described above are added to each well of the plate.
- Samples are mixed on an orbital shaker and then incubated at room temperature for 1 hour. After this incubation period 5 ⁇ l of 0.5M EDTA is added to all wells and the plates are incubated for a further hour at room temperature prior to reading the FP signal in an appropriate reader.
- FP data are converted to cGMP concentrations and then fitted using ActivityBase software.
- the activity of a test compound is determined as the pEC500 value which is the concentration able to increase by 5-fold basal cGMP.
- the compounds of Examples 1 to 47 were tested in the assay described above and gave pEC500 values of greater than 5.0. In an embodiment the compounds of the invention give a pEC500 value of ⁇ 6.0 when tested in this assay. In a further embodiment the compounds of the invention give a pEC500 value of ⁇ 7.0 when tested in this assay.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Cardiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Disclosed are compounds of formula (I) and or salts thereof which activate soluble guanylate cyclase (sGC), pharmaceutical compositions containing them, their use in the manufacture of a medicament for teating cardiovascular diseases, and processes for their preparation.
Description
PYRIMIDINE DERIVATIVES AS ACTIVATORS OF SOLUBLE GUANYLATE CYCLASE
The present invention relates to novel compounds, pharmaceutical compositions containing them, to their use in medicine, and to processes for their preparation. In particular the present invention relates to compounds which, when administered to a patient, activate soluble guanylate cyclase (sGC) and to the use of such compounds for the activation of sGC in patients for a therapeutic effect.
sGC is a member of a family of related enzymes which share homologous catalytic domains but are activated in different ways. This family includes the adenylate cyclases, a class of membrane bound enzymes that convert ATP to cAMP, which are regulated by G proteins, and the membrane-bound guanylate cyclases that make cyclic guanosine monophosphate (cGMP) in response to hormone signals via an extracellular ligand binding domain.
Whilst not wishing to be bound by theory, it is considered that the active enzyme contains one heme unit in a heterodimer arrangement, composed of one alpha and one beta-subunit. Several subtypes of subunits have been described, which differ from each other with respect to sequence and tissue-specific distribution. The subtypes alpha-1 and beta-1 are thought to be mainly expressed in the brain and the lung but have also been shown to be expressed in heart, kidney, liver, skeletal muscle, placenta, colon, uterus, prostate, spleen, pancreas, platelets and isolated blood vessels. Alpha-2 subunits have been detected in the brain, placenta, uterus and pancreas, while beta-2 subunits seem to be expressed in the liver and kidney.
The enzyme is thought to be a principal receptor for the ubiquitous signalling molecule, nitric oxide (NO), forming a NO-sGC-cGMP signal transduction axis. It is believed that soluble guanylate cyclase is a heme sensor protein that selectively binds NO at the heme iron, which activates the enzyme to convert guanosine triphosphate (GTP) to cGMP. It is thought that cGMP subsequently mediates a number of important physiological processes, including smooth muscle relaxation and neurotransmission. It has been suggested that cGMP is a critical component involved in the regulation of various (patho)physiological processes, for example in cardiovascular, respiratory, gastrointestinal, urogenital, nervous and immune systems including, neuronal excitability and particularly smooth muscle tone, thereby controlling, among other things, blood pressure, gastro-intestinal motility and genital erection.
Due to its ubiquitous nature, activation of this enzyme is likely to have significant pathological implications. This is particularly true of the cardiovascular system in which dysfunction of NO-sGC-cGMP signalling is thought to be involved in diseases and conditions such as atherosclerosis, stroke and sepsis. Thus, novel drugs based on selective activation of the enzyme have the potential for therapeutic benefit.
For a review of NO-independent activation of sGC see Oleg V. Evgenov et al.; Nature Reviews, Vol. 5, September 2006, pp755-768. Reference is made therein to the compounds BAY 58-2667 (see also WO01/19780) and HMR-1766 (see also WO00/02851 ) as sGC activators. The following more recent article discusses BAY 58-2667 in the context of treatment of congestive heart failure: Hypertension, 2007, 49, 1128-1133.
The novel compounds of the invention are activators of sGC and consequently may have application in the treatment of one or more diseases or conditions, which include: cardiovascular diseases and conditions, such as angina (including stable and unstable angina pectoris), low cardiac output, cerebral ischemia, cardiac ischemia, myocardial infarction, coronary reperfusion injury, arterial hypertension (including pulmonary arterial hypertension), congestive heart failure (for example due to systolic and/or diastolic cardiac dysfunction, low cardiac output or high systemic vascular resistance), heart failure with preserved ejection fraction, acute heart failure syndromes (AHFS), cardiac hypertrophy, acute coronary syndrome, thromboses (including arterial or venous thrombosis), atherosclerosis, peripheral vascular disease, glomerulonephritis, restenosis (for example following percutaneous vascular intervention, vascular angioplasty or stent placement), Raynaud's disease, vascular complications of diabetes or of obesity, stroke, hereditary cerebral haemorrhage, endothelial dysfunction, and other inflammatory cardiovascular disorders; erectile dysfunction; female sexual arousal disorder, respiratory failure, acute respiratory distress syndrome, gall bladder dysfunction, sickle cell disease, osteoporosis, inflammation, wound healing, chronic kidney insufficiency, renal fibrosis, renal failure, glomerulonephritis, chronic renal disease, cardiorenal syndrome, hepatorenal syndrome, liver cirrhosis, diabetes, metabolic syndrome, male pattern baldness; neuro-function disorders (including diseases or conditions displaying neuroinflammation pathology and neurodegenerative diseases, particularly chronic neurodegenerative conditions) such as Alzheimer's disease, dementia, age-related memory dysfunction, mild cognitive impairment, cognitive deficit, corticobasal degeneration, frontotemporal dementia, diffuse Lewis body type of Alzheimer's disease, and apoptotic insults caused by beta-amyloid treatment, epilepsy; pain of neuropathic origin including neuralgias, Parkinson's disease, subacute sclerosing panencephalitic Parkinsonism, postencephalitic Parkinsonism, guam Parkinsonism-dementia complex, progressive supranuclear palsy, pugilistic encephalitis, Pick's disease, Huntingdon's disease, AIDS-associated dementia; multiple sclerosis, amyotrophic lateral sclerosis; sleep disorders (including narcolepsy and sleep deficits associated with Parkinson's disease), and ALS (motor neuron disease).
Thus representative diseases and conditions for which the compounds of the invention may be useful include arterial hypertension (including pulmonary arterial hypertension), angina, cardiac ischemia, myocardial infarction, congestive heart failure (for example due to systolic and/or diastolic cardiac dysfunction, low cardiac output or high systemic vascular resistance), cardiac hypertrophy, acute coronary syndrome, atherosclerosis, peripheral vascular disease,
cardiorenal syndrome, hepatorenal syndrome and restenosis (for example following percutaneous vascular intervention, vascular angioplasty or stent placement).
A particular disease or condition for which the compounds of the invention may be useful is congestive heart failure. Another particular disease or condition for which the compounds of the invention may be useful is peripheral vascular disease. Another particular disease or condition for which the compounds of the invention may be useful is arterial hypertension (also known as systemic hypertension). Another particular disease or condition for which the compounds of the invention may be useful is pulmonary arterial hypertension. Another particular disease or condition for which the compounds of the invention may be useful is angina.
According to one aspect the present invention provides a compound of formula (I)

R1 and R2 are independently selected from hydrogen, halo, CF3 and Ci-4alkyl;
one of U and V represents N and the other represents CH;
— Y— represents

Z is absent or represents O;
A represents CH or N;
when A represents CH, R5 is selected from hydrogen, methyl, C1-4alkoxy, methoxyC2-3alkoxy, chloro and fluoro and R6 represents hydrogen;
when A represents N, R5 and R6 each represent hydrogen or one of R5 and R6 represents hydrogen and the other represents methyl; and
X represents

wherein: J and L both represent CH, or one represents N and the other represents CH, provided that only one of A, J and L represents N;
when both J and L represent CH, R8 represents hydrogen or chloro, fluoro, CF3, C1-4alkyl or C1-4alkoxy in a meta or ortho position relative to the R9 substituent;
when one of J and L represents N, R8 represents hydrogen or halo in a meta or ortho position relative to the R9 substituent; and
Ra represents hydrogen, halo, CF3, OCF3, C1-4-alkyl, C1-4-alkoxy, CN, CONR10R , CO2H or N3, wherein R ,10 and R are independently selected from hydrogen and C1-4-alkyl;
with the exception of 1-[4-(2-(4-(4-chlorophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid and 1-[4-(2-((4-phenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid.
According to a further aspect the present invention provides a compound of formula (I)

R1 and R2 are independently selected from hydrogen, halo, CF3 or C1-4alkyl;
one of U and V represents N and the other represents CH;
— Y— represents

Z is absent or represents O;
A, J and L each represent CH; or one of A, J and L represents N and the other two each represents CH; and when A represents CH, R5 is selected from hydrogen, methyl, C1-4alkoxy, methoxyC2- 3alkoxy, chloro and fluoro and R6 represents hydrogen; or when A represents N, R5 and R6 each represent hydrogen or one of R5 and R6 represents hydrogen and the other represents methyl;
X represents

with the exception of 1-[4-(2-(4-(4-chlorophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid and 1-[4-(2-((4-phenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid. As used herein, the term "alkyl" refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms. For example, C1-4alkyl means a straight or branched alkyl containing at least 1 , and at most 4, carbon atoms. Examples of "alkyl" as used herein include, but are not limited to, methyl, ethyl, n-propyl, n-butyl, isobutyl, isopropyl, and t-butyl.
As used herein, the term "alkoxy" refers to a straight or branched alkoxy group containing the specified number of carbon atoms. For example, C1-4alkoxy means a straight or branched
alkoxy group containing at least 1 , and at most 4, carbon atoms. Examples of "alkoxy" as used herein include, but are not limited to, methoxy, ethoxy, propoxy, prop-2-oxy, butoxy, but-2-oxy, 2-methylprop-1-oxy, or 2-methylprop-2-oxy.
As used herein, the term "halo" refers to the elements fluorine, chlorine, bromine and iodine. In an embodiment halo represents bromine, fluorine and chlorine. In a further embodiment halo represents fluorine and chlorine.
In an embodiment there is provided a compound of formula (I) as defined above or a pharmaceutically acceptable salt thereof.
As used herein, the term "pharmaceutically acceptable" means a compound which is suitable for pharmaceutical use.
In an embodiment — Y— represents

In an embodiment R1 is in a para position relative to the -OCH2- linker. In a further embodiment R2 is in an ortho position relative to the -OCH2- linker. In a further embodiment R1 is in a para position relative to the -OCH2- linker and R2 is in an ortho position relative to the -OCH2- linker.
In a further embodiment R1 is in an ortho position relative to the bond linking to the pyridine ring. In a further embodiment R2 is in a meta position relative to the -OCH2- linker.
In an embodiment R1 and R2 do not both represent C2-4alkyl.
In an embodiment, where one of R1 and R2 represents
 the other represents hydrogen.
the other represents hydrogen.
In an embodiment R1 represents C1-4alkyl, CF3 or halo, in a further embodiment with R1 in a para position relative to the -OCH2- linker. In an embodiment R1 represents methyl, CF3, fluoro or chloro, in a further embodiment with R1 in a para position relative to the -OCH2- linker.
In an embodiment R2 represents hydrogen.
In an embodiment R1 represents chloro or fluoro and R2 represents hydrogen, in a further embodiment with R1 in an ortho position relative to the bond linking to the pyridine ring.
In an embodiment, R2 represents chloro or fluoro and R1 represents hydrogen, in a further embodiment with R2 in a meta position relative to the -OCH2- linker.
In an embodiment U represents CH and V represents N.
In an embodiment R1 and R2 each represent hydrogen. In an embodiment R1 represents chloro or fluoro and R2 represents hydrogen. In an embodiment R1 represents C1-4alkyl and R2 represents hydrogen. In an embodiment R1 represents methyl and R2 represents hydrogen. In an embodiment R1 represents CF3 and R2 represents hydrogen.
In an embodiment R3 represents methyl or CF3. In a further embodiment R3 represents CF3.
In an embodiment Z is absent. In an embodiment Z represents O.
In an embodiment A represents CH.
In an embodiment R6 represents hydrogen.
In an embodiment R5 represents hydrogen, methyl, methoxy, propyloxy, isopropyloxy, isobutyloxy, methoxyethoxy, fluoro or chloro. In an embodiment, R5 represents hydrogen, methyl, methoxy or propyloxy.
In an embodiment J and L both represent CH. In a further embodiment J represents N and L represents CH. In a further embodiment J represents CH and L represents N.
In an embodiment R8 represents hydrogen or chloro, CF3, methyl or methoxy in a meta or ortho position relative to the R9 substituent. In an embodiment R8 represents hydrogen or chloro, CF3, methyl or methoxy in a meta position relative to the R9 substituent.
In an embodiment J and L both represent CH and R8 represents hydrogen or chloro, CF3, methyl or methoxy in a meta position relative to the R9 substituent. In an embodiment J represents N and L represents CH and R8 represents hydrogen, or chloro in a meta position relative to the R9 substituent. In an embodiment J represents CH and L represents N and R8 represents hydrogen.
In an embodiment R9 represents hydrogen, halo, CF3, OCF3, C1-4alkyl, C1-4alkoxy or CN. In a further embodiment R9 represents halo, CF3, OCF3, C1-4alkoxy or CN. In a further embodiment R9 represents chloro, fluoro, CF3, OCF3, methoxy or CN.
In an embodiment R8 represents hydrogen and R9 represents methoxy, CN, CF3, OCF3 or fluoro. In an embodiment R8 represents methyl and R9 represents CN, methoxy or CF3, with R8 being in a meta position relative to the R9 substituent. In an embodiment R8 represents
CF3, and R9 represents CN, methoxy or chloro, with R8 being in a meta position relative to the R9 substituent. In an embodiment R8 represents chloro, and R9 represents CF3, CN or methoxy, with R8 being in a meta position relative to the R9 substituent.
For the avoidance of doubt, the term "independently" means that where more than one substituent is selected from a number of possible substituents, those substituents may be the same or different.
In an embodiment there is provided a compound of formula (I) as defined above selected from:
1-[4-(2-(4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid;
1-[4-(5-chloro-2-(4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid 1 -[4-(2-(4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid;
1-[4-(2-(4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid; 1-[4-(5-chloro-2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid;
1-[4-(2-(4-(4-cyanophenyloxy)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid;
1-[4-(5-chloro-2-(4-(4-cyanophenyloxy)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5- trifluoromethyl-pyrazole-4-carboxylic acid;
1-[4-(5-methyl-2-(4-(3-chloro-5-trifluoromethylpyridin-2- yloxy)phenylmethyloxy)phenyl)pyrimidin-2-yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methoxy-4-(3-chloro-5-trifluoromethylpyridin-2- yloxy)phenylmethyloxy)phenyl)pyrimidin-2-yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid; 1-[4-(5-methyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2- yloxy)phenylmethyloxy)phenyl)pyrimidin-2-yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid;
1-[2-(2-(4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-4-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid;
1-[2-(2-(4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-4-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid; and
1-[2-(2-(4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-4-yl]-5-trifluoromethyl- pyrazole-4-carboxylic acid; or a salt thereof, in an embodiment a pharmaceutically acceptable salt thereof.
In an embodiment there is provided a compound of formula (I) as defined above selected from:
1-[4-(5-methyl-2-(2-methyl-4-(4-cyano-2-methylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-methoxy-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-piperidine-4- carboxylic acid; 1-[4-(2-(2-methyl-4-(2,4-dimethoxy-phenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-methoxy-2-methyl-phenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methoxy-4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-piperidine-4- carboxylic acid; 1-[4-(5-fluoro-2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(2-methyl-4-(4-fluorophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(4-(4-cyanophenyloxy)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-piperidine-4- carboxylic acid;
1-[4-(5-fluoro-2-(4-(5-trifluoromethylpyridin-2-yloxy)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid; 1-[4-(5-methyl-2-(2-methyl-4-(4-trifluoromethoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(2-methyl-4-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(2-chloro-4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-
2-yl]-piperidine-4-carboxylic acid; 1-[4-(5-methyl-2-(2-propyloxy-4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-
2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(2-chloro-4-methoxyphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(4-methoxy-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; 1-[4-(5-methyl-2-(2-propyloxy-4-(4-chloro-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(6-methoxypyridin-3-yl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(5-trifluoromethylpyridin-2-yl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2-yloxy)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-trifluoromethyl-2-(2-methyl-4-(4-chloro-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; 1-[4-(5-methyl-2-(2-methyl-4-(2-chloro-4-cyanophenoxy)phenylmethyloxy)-phenyl)pyrimidin-
2-yl]-piperidine-4-carboxylic acid; and
1-[4-(5-trifluoromethyl-2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid; or a salt thereof, in an embodiment a pharmaceutically acceptable salt thereof.
In an embodiment there is provided a compound of formula (I) as defined above selected from:
1-[4-(5-methyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2-yl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; and 1-[4-(5-trifluoromethyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2-yl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; or a salt thereof, in an embodiment a pharmaceutically acceptable salt thereof.
To the extent that certain compounds of formula (I) may exist in stereoisomeric forms (e.g. they may contain one or more asymmetric carbon atoms), the individual stereoisomers (enantiomers and diastereomers) and mixtures of these are included within the scope of the present invention. Similarly the invention also extends to conformational isomers of compounds of formula (I) and any geometric (cis and/or trans) isomers of said compounds. Likewise, it is understood that if the compounds of formula (I) may exist in tautomeric forms other than that shown above, then these tautomers are also included within the scope of the present invention.
Separation of the individual enantiomers of the relevant compounds (e.g. from racemic mixtures produced) may be carried out by standard methods well-known to the person skilled in the art, for example by chiral chromatography.
Salts of compounds of formula (I) which are suitable for use in medicine are those wherein the counterion is pharmaceutically acceptable. However, salts having non-pharmaceutically
acceptable counterions are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
Solvates of the compounds of formula (I) and solvates of the salts of the compounds of formula (I) are included within the scope of the present invention. As used herein, the term "solvate" refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I) or a salt thereof) and a solvent. Those skilled in the art of organic chemistry will appreciate that many organic compounds can form such complexes with solvents in which they are reacted or from which they are precipitated or crystallized. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute. Examples of suitable solvents include, but are not limited to, water, methanol, ethanol and acetic acid. Preferably the solvent used is a pharmaceutically acceptable solvent. Examples of suitable pharmaceutically acceptable solvents include, without limitation, water, ethanol and acetic acid. Most preferably the solvent used is water. Where the solvent used is water such a solvate may then also be referred to as a hydrate.
Because of their potential use in medicine, in one embodiment the salts of the compounds of formula (I) will be pharmaceutically acceptable. Reference is made to Berge et al. J. Pharm. ScL, 1977, 66, 1-19, which is incorporated herein by reference. The invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the salts of the compounds of formula (I).
Typically, a salt may be readily prepared by using a desired acid or base as appropriate. The salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent.
Suitable pharmaceutically acceptable salts can include acid addition salts or base addition salts and will be apparent to those skilled in the art. A pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic acid such as hydrochloric, hydrobromic, hydroiodic, sulfuric, nitric or phosphoric acid; or with a suitable organic acid such as succinic, maleic, malic, mandelic, formic, acetic, propionic, hexanoic, fumaric, glutamic, lactic, citric, tartaric, benzoic, salicylic, aspartic, benzenesulfonic, p-toluenesulfonic, methanesulfonic, ethanesulfonic or naphthalenesulfonic acid. Other non-pharmaceutically acceptable salts such as oxalates, may be used, for example in the isolation of compounds of formula (I) and are included within the scope of this invention. A pharmaceutically acceptable base addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic or organic base, including salts of primary, secondary and tertiary amines, such as isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexyl amine, N-methyl-D-glucamine triethylamine, triethanolamine, choline, arginine, lysine or histidine. Other suitable pharmaceutically acceptable salts include pharmaceutically acceptable metal salts, for example pharmaceutically acceptable alkali-
metal or alkaline-earth-metal salts such as sodium, potassium, calcium or magnesium salts; in particular pharmaceutically acceptable metal salts of the carboxylic acid moiety that is present in the compound of formula (I). Since the compounds of formula (I) include a carboxylic acid moiety together with one or more basic nitrogen atom(s) they have the possibility to also form internal salts, which salts are also included within the scope of the present invention.
It will be appreciated by those skilled in the art that certain protected derivatives of compounds of formula (I), which may be made prior to a final deprotection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or parenterally and thereafter metabolised in the body to form compounds of the invention which are pharmacologically active. Such derivatives may therefore be described as "prodrugs". All such prodrugs of compounds of the invention are included within the scope of the invention. Examples of pro-drug functionality suitable for the compounds of the present invention are described in Drugs of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31 , pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as "pro-moieties", for example as described by H. Bundgaard in "Design of Prodrugs" (the disclosure in which document is incorporated herein by reference) may be placed on appropriate functionalities when such functionalities are present within compounds of the invention.
Hereinafter, compounds of formula (I) (whether in solvated or unsolvated form) or their pharmaceutically acceptable salts (whether in solvated or unsolvated form) are referred to as "compounds of the invention".
Also included within the scope of the invention are polymorphs of a compound of the invention.
The invention also includes all suitable isotopic variations of a compound of the invention. An isotopic variation of a compound of the invention is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F and 36CI, respectively. Certain isotopic variations of the invention, for example, those in which a radioactive isotope such as 3H or 14C is incorporated, are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3H, and carbon-14, i.e., 14C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with isotopes such as deuterium, i.e., 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life
or reduced dosage requirements and hence may be preferred in some circumstances, lsotopic variations of the compounds of the invention can generally be prepared by conventional procedures such as by the illustrative methods or by the preparations described in the Examples hereafter using appropriate isotopic variations of suitable reagents.
As discussed above, it is believed that compounds of the invention, as activators of sGC, may be useful in the treatment of a disease or condition which is mediated by sGC activity.
According to a further aspect the invention provides a compound of the invention as defined above for use in therapy; in an embodiment the therapy is human therapy.
According to a further aspect the invention provides the use of a compound of the invention for the manufacture of a medicament for treating a disease or condition mediated by sGC activity.
According to a further aspect the invention provides a compound of the invention for use in the treatment of a disease or condition mediated by sGC activity.
According to a further aspect, the invention provides a pharmaceutical composition comprising a compound of the invention, in association with one or more pharmaceutically acceptable carrier(s), diluents(s) and/or excipient(s). The carrier, diluent and/or excipient must be "acceptable" in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
According to a further aspect the invention provides a method of treatment of a disease or condition which is mediated by the activity of sGC such as one or more of the diseases described above, for example arterial hypertension, pulmonary arterial hypertension, angina, cardiac ischemia, myocardial infarction, congestive heart failure, acute coronary syndrome, atherosclerosis, peripheral vascular disease or restenosis, comprising administration to a human subject in need of such treatment of a therapeutically effective amount of a compound of the invention, or of a pharmaceutical composition comprising a compound of the invention.
According to a further aspect the invention provides a method of treatment of cardiorenal syndrome or hepatorenal syndrome comprising administration to a human subject in need of such treatment of a therapeutically effective amount of a compound of the invention, or of a pharmaceutical composition comprising a compound of the invention.
The compounds of the invention may also be used in combination with other therapeutic agents. The invention thus provides, in a further aspect, a combination comprising a compound of the invention or a pharmaceutically acceptable derivative thereof together with a further therapeutic agent.
When a compound of the invention or a pharmaceutically acceptable derivative thereof is used in combination with a second therapeutic agent active against the same disease state the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art. It will be appreciated that the amount of a compound of the invention required for use in treatment will vary with the nature of the condition being treated and the age and the condition of the patient and will be ultimately at the discretion of the attendant physician or veterinarian.
The compounds of the present invention may for example be used in combination with antihypertensive drugs such as diuretics (for example epitizide, bendroflumethiazide, chlortalidone, chlorthiazide, hydrochlorthiazide, indapamide, metolazone), ACE inhibitors (such as benzapril, captopril, enalapril, fosinopril, lisinopril, preindopril, quinapril, ramipril, trandopril), angiotensin receptor blockers (such as candesartan, irbesartan, losartan, telmisartan, valsartan), calcium channel inhibitors (such as amlodipine, felodipine, isradapine, nifedipine, niimodipine, nitrendipine, diltiazem, verapamil), α-adrenergic receptor antagonists (such as doxazosin, prazosin, terazosin, phentolamine, indoramin, phenoxybenzamine, tolazoline), β-adrenergic receptor antagonists (such as atenolol, metoprolol, nadolol, oxprenolol, pindolol, propanolol, timolol), mixed α/β-adrenergic receptor antagonists (such as bucindalol, carvedilol, labetolol) or may be used in combination with PDE5 inhibitors (such as sildenafil, tadalafil, vardenafil), or may be used in combination with cholesterol-lowering or lipid-lowering drugs, for example statins (such as atorvastatin, cerivastatin, fluvastatin, lovastatin, mevastatin, pitavastatin, pravastatin, rosuvastatin, simvastatin), fibrates (such as bezafibrate, ciprofibrate, gemfibrozil, fenofibrate), or nicotinic acid.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical formulation and thus pharmaceutical formulations comprising a combination as defined above together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention. The individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations by any convenient route.
When administration is sequential, either the compound of the invention or the second therapeutic agent may be administered first. When administration is simultaneous, the combination may be administered either in the same or different pharmaceutical composition.
When combined in the same formulation it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the formulation. When formulated separately they may be provided in any convenient formulation, conveniently in such manner as are known for such compounds in the art.
It will be appreciated that references herein to "treatment" extend to prophylaxis, prevention of recurrence and suppression or amelioration of symptoms (whether mild, moderate or severe) as well as the treatment of established conditions.
The compound of the invention may be administered as the raw chemical but the active ingredient is preferably presented as a pharmaceutical formulation.
The compound of the invention may be administered in conventional dosage forms prepared by combining a compound of the invention with one or more standard pharmaceutical excipients, carriers or diluents, according to conventional procedures well known in the art. These procedures may involve mixing, granulating and compressing or dissolving the ingredients as appropriate for the desired preparation.
The pharmaceutical compositions of the invention may be formulated for administration by any route, and include those in a form adapted for oral, topical or parenteral administration to mammals including humans.
The compositions may be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
The topical formulations of the present invention may be presented as, for instance, ointments, creams or lotions, eye ointments and eye or ear drops, impregnated dressings and aerosols, and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration and emollients in ointments and creams.
The formulations may also contain compatible conventional carriers, such as cream or ointment bases and ethanol or oleyl alcohol for lotions. Such carriers may be present as from about 1% up to about 98% of the formulation. More usually they will form up to about 80% of the formulation.
Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatine, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants, for example potato starch; or acceptable wetting agents such as sodium lauryl sulphate. The tablets may be coated according to methods well known in normal pharmaceutical practice. Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives, such as suspending agents, for example sorbitol, methyl cellulose, glucose syrup, gelatine,
hydroxyethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and, if desired, conventional flavouring or colouring agents.
Suppositories typically contain conventional suppository bases, e.g. cocoa-butter or other glyceride.
For parenteral administration, fluid unit dosage forms are prepared utilising the compound and a sterile vehicle, water being preferred. The compound, depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle. In preparing solutions the compound can be dissolved in water for injection and filter-sterilised before filling into a suitable vial or ampoule and sealing.
Advantageously, agents such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle. To enhance the stability, the composition can be frozen after filling into the vial and the water removed under vacuum. The dry lyophilised powder is then sealed in the vial and an accompanying vial of water for injection may be supplied to reconstitute the liquid prior to use. Parenteral suspensions are prepared in substantially the same manner except that the compound is suspended in the vehicle instead of being dissolved and sterilisation cannot be accomplished by filtration. The compound can be sterilised by exposure to ethylene oxide before suspending in the sterile vehicle. Advantageously, a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
The pharmaceutical compositions of the invention may be formulated, for administration to mammals including humans, by any route, and include those in a form adapted for oral, topical or parenteral administration. The compositions may, for example, be in the form of tablets, capsules, powders, granules, lozenges, creams or liquid preparations, such as oral or sterile parenteral solutions or suspensions.
Thus in one aspect the invention provides a pharmaceutical composition for oral administration such as an oral suspension or liquid, for example an aqueous based fluid formulation, or a solid dosage formulation such as a tablet or capsule.
The compositions may contain from 0.1% by weight, preferably from 10-60% by weight, of the active material, depending on the method of administration. Where the compositions comprise dosage units, each unit will typically contain from 5-1000 mg of the active ingredient.
It will be recognised by one of skill in the art that the optimal quantity and spacing of individual doses of a compound of the invention will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular mammal being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e. the number of doses of a compound of the invention given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
Since the compounds of formula (I) are intended for use in pharmaceutical compositions it will readily be understood that they are each suitably provided in substantially pure form, for example at least 60% pure, for example at least 75% pure, for example at least 85%, for example at least 98% pure (% are on a weight for weight basis). Impure preparations of the compounds may be used for preparing the more pure forms used in the pharmaceutical compositions; these less pure preparations of the compounds typically contain at least 1 %, for example at least 5%, for example from 10 to 59% of a compound of the invention.
Compounds of the invention may be prepared in a variety of ways. These processes form further aspects of the present invention.
Throughout the specification, general formulae are designated by Roman numerals (I), (II), (III), (IV) etc.
Compounds of formula (I) can be prepared according to the following general synthetic process (Scheme 1 ):
Scheme 1

Scheme 2: pyrazole compounds of formula (Na)

(Ma)
2,4-dichloropyrimidine is commercially available from Aldrich. The compounds of formula (V) wherein R3 represents CF3 or CH3 are commercially available (from Aldrich and Orphachem respectively). The compounds of formula (V) wherein R3 represents C2-4 alkyl can be prepared according to the process described in US2005096353 (see Scheme 6 at page 17 and the Examples).
Scheme 3: piperidine compound of formula (lib)


Ethyl isonipecotate is commercially available (from Aldrich). The compound of formula (lib) can also be prepared in a solvent such as acetone, CH3CN or THF in the presence of a base such as Na2CO3, K2CO3 or Cs2CO3, under reflux.
Where U represents N and V represents CH, in respect of step (ii) the intermediate compounds of formula (III) can be prepared according to the processes set out in Schemes 4, 5 and 6.
Scheme 4

2-hydroxyphenyl boronic acid is commercially available (from Aldrich) as is 5-fluoro-2- hydroxyphenyl boronic acid (from Apollo or Combi Blocks).
Scheme 5
The compounds of formula (NIb) can be prepared from the boronic acids of formula (VIb); these are either commercially available or can be prepared by standard methods well-known to the person skilled in the art.

(N)
Suitable, for example, for
R1 = Cl and R2 =H
R1 = R2 = F
R1 = CH3 and R2 = H
R1 = CF, and R2 = H
The compound of formula (VIb) where R1 is ethyl and R2 is hydrogen may be prepared as described in WO2005019151.
Certain compounds of formula (VIb) are commercially available, for example where R1 is in a para position relative to the -OCH2- linker and R2 is in an ortho position relative to the - OCH2- linker:


Compounds of formula (VIb) where R1 is propyl in a para position relative to the -OCH2- linker and R2 is hydrogen may be prepared from the corresponding 4-propyl anisole by (i) bromination in the position ortho to the methoxy group and (ii) conversion of the bromine to a boronic acid group by standard methods.
Analogous processes may be used to prepare other compounds of formula (VIb) where R1 is C1-4alkyl and R2 is as defined above. For example, 2-bromo-4,6-dimethyl phenol is commercially available from Bionet.
Compounds of formula (VIb) where R1 is H and R2 is ethyl, isopropyl or t-butyl in an ortho position relative to the -OCH2- linker may be prepared from the corresponding 2-ethyl- anisole, 2-isopropyl-anisole or 2-t-butyl-anisole, by
(i) bromination in the position ortho to the methoxy group using NBS and diisopropylethylamine (by a method as described in US2004235852, page 29, compound (0231 )), and (ii) conversion of the bromine to a boronic acid group by standard methods.
Analogous processes may be used to prepare other compounds of formula (VIb) where R2 is and R1 is as defined above.
Where U represents CH and V represents N, in respect of steps (i) and (ii) the intermediate compounds of formula (II) and (III) can be prepared according to the processes set out in Scheme 7.
Scheme 7

2-hydroxy-phenylboronic acid and 5-methyl-2-methoxy-phenylboronic acid are commercially available from Aldrich. 5-chloro-2-hydroxy-phenylboronic acid, 5-fluoro-2-hydroxy- phenylboronic acid and 5-trifluoromethyl-2-methoxy-phenylboronic acid are commercially available from Combi-blocks.
In respect of step (iii), the following general synthetic schemes can be used (Schemes 8, 9 and 10).
The synthesis described in Scheme 8 is particularly suitable for compounds wherein either A, J or L represents N or Z represents O (although it can also be used wherein Z is absent).
Scheme 8

Scheme 9
The synthesis described in Scheme 9 relates to compounds wherein Z is absent and both J and L represent CH or J represents CH and L represents N and is particularly suitable for compounds wherein R5 represents F or Cl.

Compounds of formula (IX) are either commercially available or can be prepared by standard methods well-known to the person skilled in the art.
Compounds of formula (IVc) wherein A represents CH and Z is absent and/or J or L represent N may be prepared by the following method (Scheme 10) - as an alternative to using Scheme 8 or 9 above.
Scheme 10

(IVc)
The subsequent step (iv) may be carried out according to Scheme 11 a.
Scheme 11a
NaOH or LiOH MeOH / H2O rt or 60-70°C
(IVa) or (IVb) or (IVc) - (Ia) or (Ib) or (Ic)
The temperature used in this ester hydrolysis reaction will depend on the nature of the compound and the length of time for which the reaction is performed; this will be well appreciated by the person skilled in the art.
Where R9 represents CN, the ester hydrolysis reaction is instead suitably carried out using LiOH at room temperature to avoid hydrolysis of the cyano group to an amide group.
As an alternative, where R9 represents CN or CO2R12 (R12 represents C1-4-alkyl) Schemes 11 b and 1 1c may suitably be used to prepare compounds of formula (I):
Scheme 11b
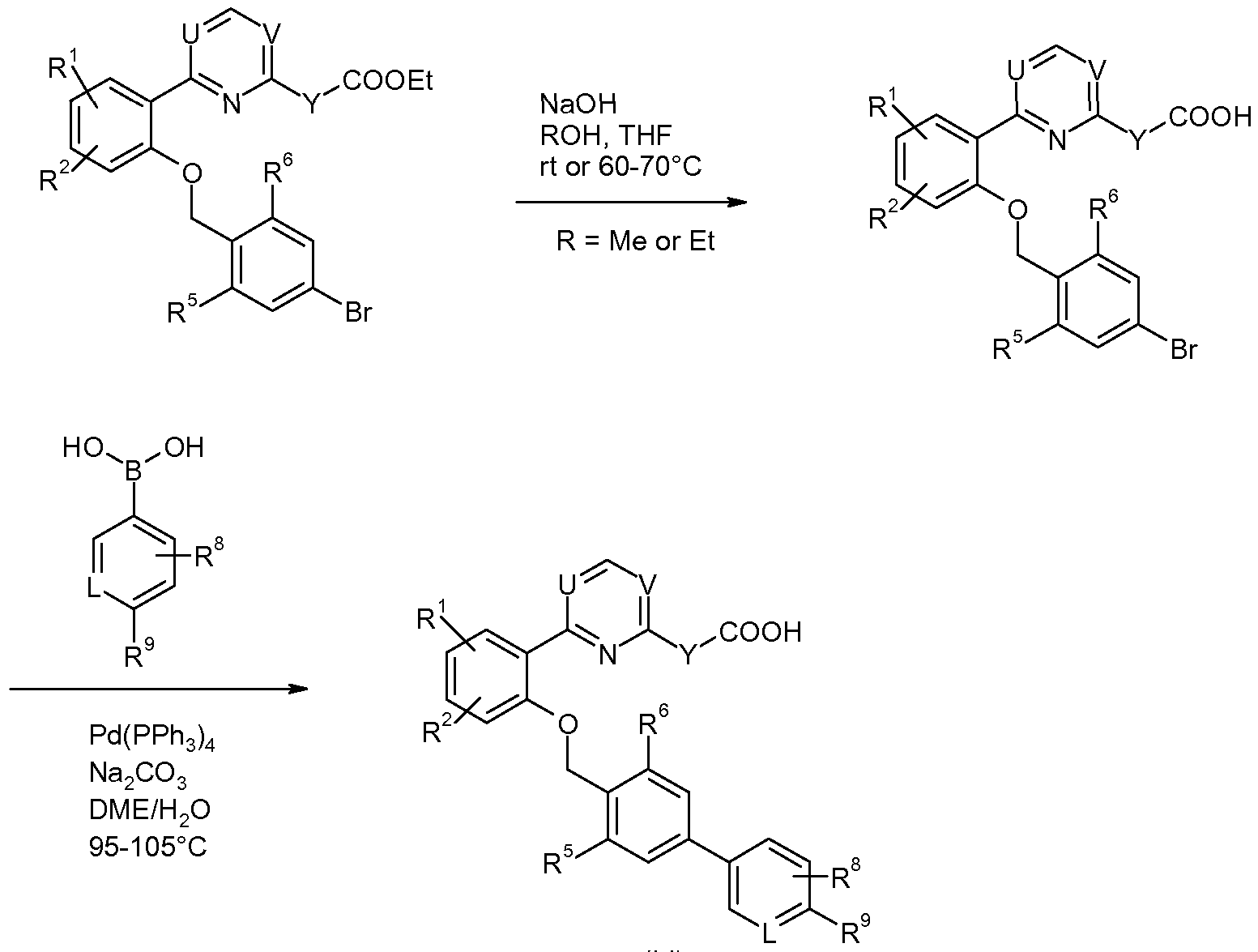
(Id)
Scheme 11c

Scheme 12

Scheme 13 Scheme 13 is particularly suitable to prepare compounds wherein R9 represents CN, Z is absent or Z represents O.

In respect of the compounds of formula (VIII):

Scheme 14
When R5 represents Me, OMe or Cl:

(XIIa) (VIIIb)

(XIIb)
Scheme 15
rt rt

(VIIIc)

R' =

Compounds of formula (XIIa), (XIIb), (XIIc) and (XIId) are commercially available: 4-bromo-2- methylbenzoic acid, 4-bromo-2-chlorobenzoic acid, methyl 4-bromo-2-methoxybenzoate (from Aldrich) or 4-bromo-2-hydroxybenzoic acid (from Apin) and 4-bromo-2- hydroxybenzaldehyde (from Apin); or may be prepared by standard methods well-known to the person skilled in the art.
Scheme 16a
rt rt


(XIII) (VIIId)
Certain compounds of formula (XIII) are commercially available, for example wherein R5 represents H and R6 represents Me or R5 represents Me and R6 represents H (from Asymchem). Other compounds of formula (XIII) may be prepared by standard methods well- known to the person skilled in the art.
Scheme 16b


Synthesis of biphenyl compounds of formula (VII) wherein Z is absent can be carried out according to Schemes 17 or 18.
Scheme 17
Scheme 17 is particularly suitable for compounds wherein one of A, J or L represents N.

(Vila)
Pathway A is thus typically used where A represents N or L represents N; and pathway B is typically used where J represents N. In respect of the compounds of formula (IX), 2- methoxypyridine-5-boronic acid is available from Aldrich and 2-cyanopyridine-5-boronic acid and 2-trifluoromethylpyridine-5-boronic acid are available from Frontier; other compounds of formula (IX) not commercially available may be prepared by standard methods well-known to the person skilled in the art.
Certain compounds of formula (XIV) are commercially available, for example: where A represents CH and R5 represents Me (Betapharma); A represents CH and R5 represents OMe (Aldrich); A represents CH and R5 represents F (Aldrich). Where A represents N, note the earlier reference to compounds of formula (XIII) in Scheme 16. Other compounds of formula (XIV) may be prepared by standard methods well-known to the person skilled in the art.
Certain compounds of formula (XV) are commercially available, for example: wherein J represents N, R8 represents H and R9 represents CN, CF3, COOR, Cl or OMe (Aldrich, Fluka or Acros); wherein J represents N, R8 represents Cl and R9 represents CF3 (Aldrich). Other compounds of formula (XV) may be prepared by standard methods well-known to the person skilled in the art.
Pathway B is also typically used where J and L both represent CH where hal represents Br.
Scheme 18a
When R iiss oother than CN or CO2R (R represents C1-4alkyl) and A and J each represent CH and R5/R6 is other than halogen, Scheme 18a is particularly suitable.

Scheme 18b
When R9 is CN or CO2R12 (R12 represents C1-4alkyl) and A and J each represent CH and/or R5/R6 is halogen, Scheme 18b is particularly suitable.
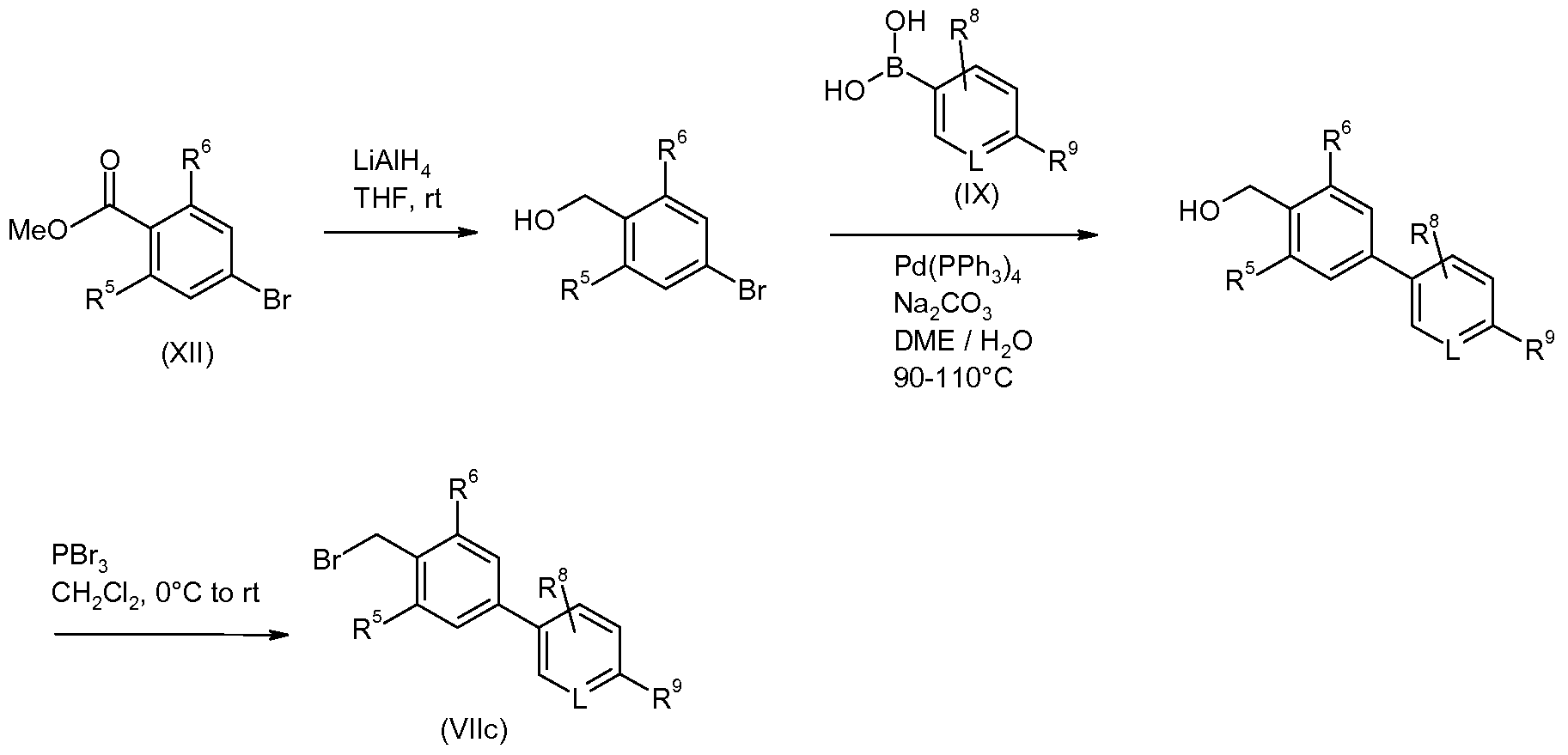
Scheme 18c
When R9 is CN or CO2R12 (R12 represents
 and A represents CH, and J represents N and/or R5/R6 is halogen, Scheme 18c is particularly suitable.
and A represents CH, and J represents N and/or R5/R6 is halogen, Scheme 18c is particularly suitable.

Synthesis of compounds of formula (VII) with Z representing O, can be carried out according to Schemes 19, 20 or 21.
Scheme 19
rt

:hway A is thus typically used for J represents N and/or R9 represents CN, CO<
R 35 represents H, Me, OMe. Pathway B is thus typically used for A represents N or for A represents CH and hal represents F and R5 represents H, Me, or OMe.
Compounds of formula (XVIa) are commercially available, for example: 4-hydroxy-2- methylbenzaldehyde (Interchim), 4-hydroxy-2-methoxybenzaldehyde (Fluka or Acros), and 4-hydroxybenzaldehyde (Aldrich).
In respect of the compounds of formula (XVIII), note the earlier reference to compounds of formula (XIII) in Scheme 16a. Certain compounds of formula (XVIII) are commercially
available, for example: wherein A represents CH and R5 represents H, Me or OMe (Aldrich, Apollo or Apin); or wherein A represents N and R5 and R6 each represents H (Aldrich). Other compounds of formula (XVIII) may be prepared by standard methods well-known to the person skilled in the art.
Phenol derivatives of formula (XIX) are commercially available or may be prepared by standard methods well-known to the person skilled in the art.
Scheme 20
Where R5 represents Cl or F:


Compounds of formula (XVIb) are commercially available, for example from Aldrich for R5 represents Cl and from Apin for R5 represents F.
Certain compounds of formula (XVII) are commercially available, for example: wherein hal represents Br or Cl, J represents N, R8 represents H, and R9 represents CN, CF3, COOR, Cl or OMe (Aldrich, Fluka or Acros); wherein hal represents Cl, J represents N, R8 represents Cl, and R9 represents CF3 (Aldrich); wherein J represents CH, hal represents F, R8 represents H, F, Cl, OMe, Me, or CF3, and R9 represents CN or COOR (Aldrich, Acros, Apin). Other compounds of formula (XVII) may be prepared by standard methods well-known to the person skilled in the art.
Scheme 21
Where R5 represents OC1-4alkyl or O(CH2)2OMe:
 R = C1 4alkyl or (CH2JnOMe, n= 2 or 3
R = C1 4alkyl or (CH2JnOMe, n= 2 or 3
Supporting Examples and Descriptions
The invention is illustrated by the Examples described below.
In the procedures that follow, after each starting material, reference to a Description by number is typically provided. This is provided merely for assistance to the skilled chemist. The starting material may not necessarily have been prepared from the batch referred to.
Where reference is made to the use of a "similar" procedure, as will be appreciated by those skilled in the art, such a procedure may involve minor variation, for example reaction temperature, reagent/solvent amount, reaction time, work-up conditions or chromatographic purification conditions.
Compounds are named using ACD/Name PRO 6.02 chemical naming software (Advanced Chemistry Development Inc., Toronto, Ontario, M5H2L3, Canada).
In the reporting of Proton Magnetic Resonance (1H NMR) spectral data, chemical shifts are reported in ppm (δ) using tetramethylsilane as the internal standard. Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet.
The following table lists the used abbreviations:


Analytical method LC-MS
Analytical HPLC was conducted on a X-terra MS C18 column (2,5 μm 3*30 mm id) eluting with 0.01 M ammonium acetate in water (solvent A) and 100% acetonitrile (solvent B) using the following elution gradient: 0 * 4 minutes, 5%B * 100%B; 4 * 5 minutes, 100%B at a flowrate of 1.1 mL/min with a temperature of 400C. The mass spectra (MS) were recorded on a Micromass ZQ-LC mass spectrometer using electrospray positive ionisation [ES+ve to give MH+ molecular ion] or electrospray negative ionisation [ES-ve to give (M-H)" molecular ion] modes.
Analytical LC-HRMS Methods:
(a) Analytical HPLC was conducted on a LUNA 3u C18 column (2,5 μm 30*3 mm id) eluting with 0.01 M ammonium acetate in water (solvent A) and 100% acetonitrile (solvent B) using the following elution gradient: 0 ■* 0.5 minutes, 5%B; 0.5 ■* 3.5 minutes, 5%B ■* 100%B; 3.5 ■* 4 minutes, 100%B; 4 ■* 4.5 minutes, 100%B ■* 5%B; 4.5 ■* 5.5 minutes, 5%B at a flowrate of 1.3 mL/min with a temperature of 400C. The mass spectra (MS) were recorded on a Micromass LCT mass spectrometer using electrospray positive ionisation [ES+ve to give MH+ molecular ion] or electrospray negative ionisation [ES-ve to give (M-H)" molecular ion] modes.
(b) Analytical HPLC was conducted on a X-Bridge C18 column (2,5 μm 30*3 mm id) eluting with 0.01 M ammonium acetate in water (solvent A) and 100% acetonitrile (solvent B) using the following elution gradient: 0 ■> 0.5 minutes, 5%B; 0.5 ■> 3.5 minutes, 5%B -> 100%B; 3.5 ■* 4 minutes, 100%B; 4 ■* 4.5 minutes, 100%B ■* 5%B; 4.5 ■* 5.5 minutes, 5%B at a flowrate of 1.3 mL/min with a temperature of 400C. The mass spectra (MS) were recorded on a Micromass LCT mass spectrometer using electrospray positive ionisation [ES+ve to give MH+ molecular ion] or electrospray negative ionisation [ES-ve to give (M-H)" molecular ion] modes.
Description 1 : 4-bromo-2-propyloxy-benzaldehvde (DD

To a solution of 4-bromo-2-hydroxybenzaldehyde (Apin, 5g, 24.87mmol) in DMF (80ml) was added portionwise NaH (60% in mineral oil, 1.19g, 29.85mmol) and the mixture was stirred at room temperature for 30 minutes. Then 1-bromopropane (2.71 ml, 29.85mmol) was added and the mixture was heated at 50°C for 4 hours and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was triturated with cHex, and the resulting precipitate was filtered and dried . The title compound was obtained as a cream powder (3.7g, yield= 61.2%). LC/MS: 245 (M+H), Rt= 3.43min. 1H NMR (300MHz, CDCI3, ppm): 7.7 (d, 1 H), 7.2 (d + s, 2H), 4.05 (t, 2H), 1.9 (m, 2H), 1.1 (t, 3H).
Description 2a: 4-(4-cvanophenyl)-benzaldehvde (D2a)

To a solution of 4-bromobenzaldehyde (Aldrich, 3g, 16.2mmol) in DME (80ml) and H2O (10ml) were added Pd(PhP3)4 (0.937g, 0.81 mmol), 4-cyanophenylboronic acid (Aldrich, 2.86g, 19.46mmol) and Na2CO3 (4.29g, 40.53mmol). The mixture was heated at 900C for 3 hours, cooled and poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. After purification by chromatography on silicagel (CH2CI2 / cHex, 4/1 ), the title compound was obtained as a cream solide (3g, yield: 89.4%). H1 NMR (300MHz, CDCI3, ppm): 10 (s, 1 H), 8.05 (d, 2H), 7.8 ( m, 6H).
Prepared by a similar method as described for D2a:


Description 3a: 4-(4-cyanophenoxy)-benzaldehyde (D3a)

To a solution of 4-hydroxybenzaldehyde (Aldrich, 5.04g, 41.28mmol), in DMF(50ml) was added portionwise Cs2CO3 (14.79g, 45.41 mmol) and the mixture was stirred at room temperature for 10 minutes. Then 4-fluorobenzonitrile (Aldrich, 5g, 41.28mmol) was added and the mixture was heated at 1 100C for 2 hours, then cooled and poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. After purification by chromatography on silicagel (CH2CI2 / cHex, 3/2, then 4/1 ), the title compound was obtained as a white solid(5.25g, yield: 57.0%). LC/MS: 224.1 (M+H) , Rt= 3.07min.
Prepared by a similar method as described for D3a:


Description 4a: 4-(4-cvanophenyl)-benzyl alcohol (D4a)

To a solution of 4-(4-cyanophenyl)-benzaldehyde (D2a, 1.5g, 7.25mmol) in MeOH (50ml) was added portionwise NaBH4 (0.303g, 7.97mmol). The mixture was stirred at room temperature for 30 minutes and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The title compound was obtained as a cream solid (1.49g, yield: 98.4%). H1 NMR (300MHz, CDCI3, ppm): 7.65 (q, 4H), 7.55 (d, 2H), 7.45 (d, 2H), 4.7 (s, 2H).
Prepared by a similar method as described for D4a:


Description 5a: 4-(4-trifluoromethylphenyl)-benzyl alcohol (D5a)

To a solution of 4-(4-trifluoromethylphenyl)-benzoic acid (Apollo, 3g, 11.27mmol) in THF (100ml) was added dropwise a solution of LiAIH4 1 M in THF (11.3ml, 11.27mmol) and the mixture was stirred at room temperature for 30 minutes. Water (50ml) was then added dropwise. The insoluble material was filtered on a Celite pad and washed with CH2CI2. The filtrate was washed with CH2CI2 and the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The title compound was obtained as a white solid (1.9g, yield= 66.8%). H1 NMR (300MHz, DMSO d6, ppm): 7.9 (d, 2H), 7.85 (d, 2H), 7.7 (d, 2H), 7.45 (d, 2H), 5.3 (t, 1 H), 4.55 (d, 2H).
Prepared by a similar method as described for D5a:
Description 5b: 2-methyl-4-(4-trifluoromethylphenyl)-benzyl alcohol

From ethyl 2-methyl-4-(4-trifluoromethylphenyl)-benzoate (D2c) as a grey powder (yield = 78%). 1H NMR (300MHz, CDCI3, ppm): 7.71 (bs, 4H), 7.49 (m, 2H), 7.44 (bs, 1 H), 4.79 (s, 2H), 2.46 (s, 3H).
Description 5c: 2-methyl-4-(2-methyl-4-trifluoromethylphenyl)-benzyl alcohol (D5c)

From ethyl 2-methyl-4-(2-methyl-4-trifluoromethylphenyl)-benzoate (D2d).
Description 5d: 4-bromo-2-methyl-benzyl alcohol (D5d)

Description 5ej r2-methyl-4-(4A5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyllmethanol (D5e)

4-bromo-2-methyl-benzyl alcohol (D5d, 30 g, 150 mmol), bis(pinacolato)diboron (40 g, 156 mmol), Pd(dppf)CI2(ll) (8 g, 10 mmol), and potassium acetate (44 g, 448 mmol) were dissolved in 1 ,4-dioxane (600 ml_), and the reaction mixture was heated to reflux under N2 for 4 h. After cooling, the reaction mixture was filtered, and the filtrate was concentrated. Purification by column chromatography (silica gel; petroleum ether: ethyl acetate = 5:1 ) afforded the title compound as a red oil (34 g, yield: 90%). 1H NMR (400 MHz, DMSO-c/6) δ 7.50-7.38 (m, 3H), 5.14 (t, 1 H), 4.51 (d, 2H), 2.22 (s, 3H), 1.28 (s, 12H).
Description 5f : 2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2-yl)-benzyl alcool (D5f)

To a solution of [2-methyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl]methanol (D5e, 5g, 20.2mmol) in dioxane (50ml) and H2O (50ml) were added 2,3-dichloro-5- trifluoromethylpyridine (5.2g, 24mmol), Pd(PPh3)4 (0.69g, O.θmmol) and K2CO3 (8.3g, 60.5mmol) and the mixture was heated at 1000C overnight. Dioxane was removed under reduced pressure and AcOEt and water were added. The organic phase was washed with brine, then water, dried (Na2SO4) and concentrated under reduced pressure. Purification by
chromatography on Silicagel (petroleum ether/AcOEt, 5/1 ) afforded the title compound (4 g, yield: 65.9%). LC/MS: 301.7 (M+H), Rt= 1.7min
Description 6a: 4-(4-cvanophenyl)-benzyl bromide (D6a)

To a solution of 4-(4-cyanophenyl)-benzyl alcohol (D4a, 1.49g, 7.13mmol) in CH2Cb (40ml) cooled in a ice bath, was added dropwise PBr3 (solution 11WCH2CI2, 3.56ml, 3.56mmol). The mixture was stirred at 00C for 30 minutes, then at room temperature for 1 hour and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The title compound was obtained as a cream solid(1.8g, yield: 92.8%). LC/MS: Rt= 3.53min.
Prepared by a similar method as described for D6a:



Description 7: ethyl 1 -(2-chloro-pyrimidin-4-yl)-5-(trifluoromethyl)-1 H-pyrazole-4- carboxylate (D7)

A solution of 2,4-dichloropyrimidine (2g, 13.42mmol) in THF (50ml) was added dropwise to a solution of hydrazine in THF (1 M/THF, 100ml, lOOmmol). The mixture was stirred at room temperature for 1 hour and then poured into water. After extraction with AcOEt, the organic phase was dried (Na2SO4), and concentrated under reduced pressure. The solid residue was dissolved in EtOH (50ml) and ethyl 2-(ethoxymethylene)-4,4,4-trifluoro-3-oxobutyrate (Aldrich, 3.22g, 13.4mmol) was added. The reaction mixture was stirred at room temperature for 18 hours and then concentrated under reduced pressure. The residue was purified by chromatography on silicagel (cHex/AcOEt, 95/5 to 8/2). The title compound was obtained as solid (1.5g, yield= 34.9%). LC/MS: 320.9 (M+H), Rt= 3.31 min. 1H NMR (DMSO d6, ppm): 9.06 (d, 1 H), 8.48 (s, 1 H), 8.03 (d, 1 H), 4.34 (q, 2H), 1.30 (t, 3H).
Description 8: ethyl 1 -r2-(2-hvdroxyphenyl)pyrimidin-4-yll-5-(trifluoromethyl)-1H- pyrazole-4-carboxylate (D8)

To a solution of ethyl 1-(2-chloro-pyrimidin-4-yl)-5-(trifluoromethyl)-1 H-pyrazole-4- carboxylate (D7, 1.5g, 4.68mmol) in DME (100ml) were added Pd(PPh3)4 (0.541 g, 0.468mmol), 2-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenol (Aldrich, 1.08g, 4.91 mmol) and Na2CO3 (0.744g, 7.02mmol) in water (15ml). The mixture was heated at 1100C for 4 hours and poured into water. After extraction with AcOEt, the organic phase was dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by chromatography on silicagel (CH2CI2). The title compound was obtained as a pale yellow
solid (1.4g, yield= 79%). 1H NMR (DMSO d6, ppm): 12.87 (s, OH), 9.16 (d, 1 H), 8.49 (s, 1 H), 8.29 (dd, 1 H), 7.96 (d, 1 H), 7.47 (t, 1 H), 7.01 (d, 1 H), 7 (t, 1 H), 4.35 (q, 2H), 1.33 (t, 3H).
Description 9: 2-chloro-4-(2-hvdroxyphenyl)pyrimidine (D9)

To a solution of 2,4-dichloropyrimidine (Aldrich, 5g, 33.56mmol) in DME (80ml) and H2O (8ml), were added 2-hydroxyphenylboronic acid (4.86g, 35.23mmol), Na2CO3 (5.34g, 50.34mmol), Pd(OAc)2 (0.188g, 0.84mmol) and triphenylphosphine (0.44g, 1.68mmol), and then the mixture was heated at 900C overnight, then cooled and poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. After crystallisation from CH3CN, the title compound was obtained as yellow crystals (4.5g, yield= 64.9%). LC/MS: 207.1 (M+H), Rt= 2.93min. 1H NMR (CDCI3, ppm) : 8.7 (d, 1 H), 7.8 (d, 1 H), 7.75 (d, 1 H), 7.45 (t, 1 H), 7.1 (d, 1 H), 7 (t, 1 H).
Description 10a: 2-chloro-4-(2-methoxy-5-methylphenyl)pyrimidine (D10a)



Description 11a: 2-chloro-4-(2-hvdroxy-5-chlorophenyl)pyrimidine (D11a)

To a solution of 2-chloro-4-(2-methoxy-5-chlorophenyl)pyrimidine (D10c) (0.2g, 0.784mmol) in anhydrous CH2CI2 (ml) cooled in an ice bath was added dropwise BBr3 (1.56ml of a solution 1 M in CH2CI2, 1.56mmol)and the mixture was stirred at 00C for 30 minutes then at room temperature overnight. The reaction mixture was neutralised with a saturated solution of NaHCO3 in water, then extracted with CH2CI2; The organic phase was washed with water, dried (Na2SO4) and concentrated under reduced pressure. The title compound was obtained as (0.185g, yield= 100%)
1H NMR (CDCI3, ppm) 12.41 (s, OH), 8.73 (d, 1 H), 7.75 (sd, 1 H), 7.71 (d, 1 H), 7.4 (dd, 1 H), 7.05 (d, 1 H)
Prepared by a similar method as described for D11 a:
Description 11b: 2-chloro-4-(2-hvdroxy-5-fluorophenyl)pyrimidine (D11 b)

From 2-chloro-4-(2-methoxy-5-fluorophenyl)pyrimidine (D10b). 1H NMR (CDCI3, ppm): 12.21 (s, OH), 8.73 (d, 1 H), 7.66 (d, 1 H), 7.46 (m, 1 H), 7.2 (m, 1 H), 7.06 (m, 1 H).
Description 12: 2-chloro-4-(2-hvdroxy-5-methylphenyl)pyrimidine (D12)

Description 13: 2-chloro-4-(2-hvdroxy-5-trifluoromethylphenyl)pyrimidine (D13)

From 2-chloro-4-(2-methoxy-5-trifluoromethylphenyl)pyrimidine (D10d) as a yellow solid (yield= ). LC/MS: 275.0 (M+H), Rt= 3.32min.
Description 14a: Ethyl 1 -(4-(5-chloro-2-hvdroxyphenyl)pyrimidin-2-yl)-5- trifluoromethyl-pyrazole-4-carboxylate (D14a)

To a solution of 2-chloro-4-(2-hydroxy-5-chlorophenyl)pyrimidine (D1 1a, 0.185g, 0.784mmol) in EtOH (ml) was added hydrazine hydrate (0.38ml, 7.84mmol) and the mixture was heated at 600C for 2 hours and then cooled. After dilution with water (10ml), the precipitate was filtered, washed with water, then CH2CI2 and then with iPr2O. The resulting yellow solid was
dissolved in EtOH (20ml), and ethyl 2-(ethoxymethylene)-4,4,4-trifluoro-3-oxobutyrate (Aldrich, 0.22g, 0.913mmol) was added. The reaction mixture was heated at 600C for 2 hours and then concentrated under reduced pressure. The residue was triturated with iPr2O and the resulting precipitate was filtered, washed with iPr2O and dried. The title compound was obtained as a cream powder (0.25g, yield= 80%). 1H NMR (CDCI3, ppm) : 12.39 (s, OH), 8.93 (d, 1 H), 8.22 (s, 1 H), 7.86 (d, 1 H), 7.83 (sd, 1 H), 7.42 (dd, 1 H), 7.06 (d, 1 H), 4.41 (q, 2H), 1.42 (t, 3H).
Prepared by a similar method as described for D14a:


Description 15: Ethyl 1-(4-(2-hydroxyphenyl)pyrimidin-2-yl)piperidine-4-carboxylate (D15)

To a solution of 2-chloro-4-(2-hydroxyphenyl)pyrimidine (D9, 3.6g, 17.43mmol) in EtOH (50ml), was added ethyl isonipecotate (Aldrich, 6.72ml, 43.58mmol) and the mixture was heated under reflux for 3hours and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography on silicagel (CH2CI2 then CH2CI2/Me0H, 99/1 ). The title compound was obtained as a yellow oil which crystallised (5.6g, yield= 98.23%) LC/MS: 328.1 (M+H), Rt= 3.55min. 1H NMR (CDCI3, ppm) : 8.45 (d, 1 H), 7.75 (d, 1 H), 7.4 (t, 1 H), 7.05 (2d, 2H), 6.95 (t, 1 H), 4.55 (Id, 2H), 4.2 (q, 2H), 3.2 (t, 2H), 2.65 (m, 1 H), 2.05 (m, 2H), 1.8 (m, 2H), 1.3 (t, 3H).
Prepared by a similar method as described for D15:
Description 16: Ethyl 1-(4-(2-hvdroxy-5-methylphenyl)pyrimidin-2-yl)piperidine-4- carboxylate (D16)

From 2-chloro-4-(2-hydroxy-5-methylphenyl)pyrimidine (D12) as a pale yellow oil (2.7g, yield= 63.4%). LC/MS: 342.1 (M+H), Rt= 3.72min. 1H NMR (CDCI3, ppm) : 13.75 (s, OH), 8.41 (d, 1 H), 7.54 (sd, 1 H), 7.18 (dd, 1 H), 7.01 (d, 1 H), 6.90 (d, 1 H), 4.51 (Id, 2H), 4.17 (q, 2H), 3.18 (m, 2H), 2.62 (m, 1 H), 2.34 (s, 3H), 2.05 (m, 2H), 1.80 (m, 2H), 1.28 (t, 3H).
Description 17: Ethyl 1-(4-(2-hvdroxy-5-trifluoromethylphenyl)pyrimidin-2- yl)piperidine-4-carboxylate (D17)

From 2-chloro-4-(2-hydroxy-5-trifluoromethylphenyl)pyrimidine (D13) as a pale yellow oil which crystallised (yield= 82%). LC/MS: 396.0 (M+H), Rt= 3.93min. 1H NMR (CDCI3, ppm) :8.39 (d, 1 H), 7.92 (s, 1 H), 7.5 (d, 1 H), 6.98 (d, 1 H), 6.95 (d, 1 H), 4.41 (Id, 2H), 4.09 (q, 2H), 3.13 (m, 2H), 2.55 (m, 1 H), 1.98 (m, 2H), 1.73 (m, 2H), 1.2 (t, 3H).
Description 18: Ethyl 1 -(4-(5-fluoro-2-hvdroxyphenyl)pyrimidin-2-yl)piperidine-4- carboxylate (D18)


Description 20: Ethyl 1-(4-( 2-(4-bromo-2-methylbenzyloxy)-phenyl)pyrimidin-2-yl)-5- trifluoromethyl-pyrazole-4-carboxylate (D20)

To a solution of 2-chloro-4-(2-hydroxyphenyl)pyrimidine (D9, 2g, 5.29mmol) in acetone (80ml), was added Cs2CO3 (2.59g, 7.94mmol) and the mixture was stirred at room temperature for 15 minutes. 4-bromo-2-methyl-benzyl bromide (D6n, 1.54g, 5.82mmol) was then added and the mixture was heated under reflux overnight and poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography on silicagel (CH2CI2 then CH2CI2/Me0H, 99/1 ). The title compound was obtained as a yellow oil (2.9g, yield= 97.7%). LC/MS: 562.8 (M+H), Rt= 4.26min. 1H NMR (CDCI3, ppm) : 8.8 (d, 1 H), 8.25 (s+d, 2H), 8.1 (d, 1 H), 7.55 (t, 1 H), 7.4 (m, 2H), 7.25 (d, 1 H), 7.2 (t, 1 H), 7.15 (d, 1 H), 5.15 (s, 2H), 4.4 (q, 2H), 2.35 (s, 3H), 1.4 (t, 3H).
Prepared by a similar method as described for D20:



Description 28: 1 -(4-( 5-trifluoromethyl-2-(2-methyl-4-bromo-benzyloxy)- phenyl)pyrimidin-2-yl)-piperidine-4-carboxylic acid (D28)

To a solution of ethyl 1-(4-(5-trifluoromethyl-2-(2-methyl-4-bromo-benzyloxy)- phenyl)pyrimidin-2-yl)-piperidine-4-carboxylate (D26, 0.7g, 1.210 mmol) in MeOH (10 ml)
and THF (10ml) was added sodium hydroxyde (6.05 ml of a solution 1 N, 6.05 mmol), and the mixture was heated at 600C for 1 hour and then neutralised with a solution 1 N of HCI. After evaporation under reduced pressure of MeOH and THF, the resulting precipitate was filtered, washed with water, then pentane and dried. The product was obtained as pale yellow solid powder (0.65g, yield= 98%). LC/MS: 551.9 (M+H), Rt= 3.36min
Prepared by a similar method as described for D28:
Description 29j 1 -(4-(5-trifluoromethyl-2-(2-propyloxy-4-bromo-benzyloxy)- phenyl)pyrimidin-2-yl)-piperidine-4-carboxylic acid (D29)

From ethyl 1-(4-(5-trifluoromethyl-2-(2-propyloxy-4-bromo-benzyloxy)-phenyl)pyrimidin-2-yl)- piperidine-4-carboxylate (D27) as pale yellow oil (yield = 99%). LC/MS : 595.9 (M+H), Rt= 3.64min.
Description 30: ethyl 1-f4-r5-methyl-2-αr2-methyl-4-(4.4.5.5-tetramethyl-1.3.2- dioxaborolan-2-yl)phenyllmethyl)oxy)phenyll-2-pyrimidinyl)-4-piperidinecarboxylate


To a solution of ethyl 1-(4-( 5-methyl-2-(2-methyl-4-bromo-benzyloxy)-phenyl)pyrimidin-2-yl)- piperidine-4-carboxylate (D24, 0.4g, 0.763 mmol) in DME (50ml) and water (5ml) were added Pd(PPh3)4 (0.044 g, 0.038 mmol), (4-cyano-2-methylphenyl)boronic acid (0.184 g, 1.144 mmol) and Na2CO3 (0.202 g, 1.907 mmol). The reaction was heated at 1050C overnight and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography on silicagel (CH2CI2 / MeOH, 99/1 ). The title compound was obtained as a colorless oil (0.37g, yield= 87%). LC/MS: 561.1 (M+H), Rt= 4.55min. 1H NMR (CDCI3, ppm): 8.27 (d, 1 H), 7.75 (s, 1 H), 7.58 (s, 1 H), 7.54 (d, 1 H), 7.49 (d, 1 H), 7.33 (d, 1 H), 7.24 (d, 1 H), 7.14 (m, 3H), 7.02 (d, 1 H), 5.14 (s, 2H), 4.79 (Id, 2H), 4.17 (q, 2H), 3.09 (t, 2H), 2.60 (m, 1 H), 2.4 (s, 3H), 2.38 (s, 3H), 2.32 (s, 3H), 2.02 (m, 2H), 1.76 (m, 2H), 1.28 (t, 3H).
Description 32: Ethyl 1 -(4-(5-methyl-2-(2-methyl-4-(4-methoxy-2- trifluoromethylphenyl)benzyloxy)-phenyl)pyrimidin-2-yl)-piperidine-4-carboxylate (D32)

To a solution of ethyl 1-(4-( 5-methyl-2-(2-methyl-4-bromo-benzyloxy)-phenyl)pyrimidin-2-yl)- piperidine-4-carboxylate (D24, 0.42 g, 0.801 mmol) in 1 ,2-Dimethoxyethane (DME) (50 ml.) and water (5 ml.) were added Pd(PPh3)4 (0.046 g, 0.040 mmol), [4-(methyloxy)-2- (trifluoromethyl)phenyl]boronic acid (0.264 g, 1.201 mmol) and sodium carbonate (0.212 g, 2.002 mmol) and the reaction mixture was heated at 95°C overnight and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated
under reduced pressure. The residue was purified by chromatography on silicagel (CH2CI2, 99/1 ). The product was obtained as a colorless oil ( 0.44g, yield= 89%). LC/MS: 620.1 (M+H), Rt= 4.67min. 1H NMR (CDCI3, ppm): 8.27 (d, 1 H), 7.76 (bs, 1 H), 7.42 (d, 1 H), 7.25 (m, 3H), 7.15 (m, 3H), 7.09 (dd, 1 H), 7.02 (d, 1 H), 5.13 (s, 2H), 4.79 (Id, 2H), 4.17 (q, 2H), 3.91 (s, 3H), 3.09 (m, 2H), 2.59 (m, 1 H), 2.4 (s, 3H), 2.36 (s, 3H), 2.02 (m, 2H), 1.78 (m, 2H), 1.28 (t, 3H).
Prepared by a similar method as described for D31 :







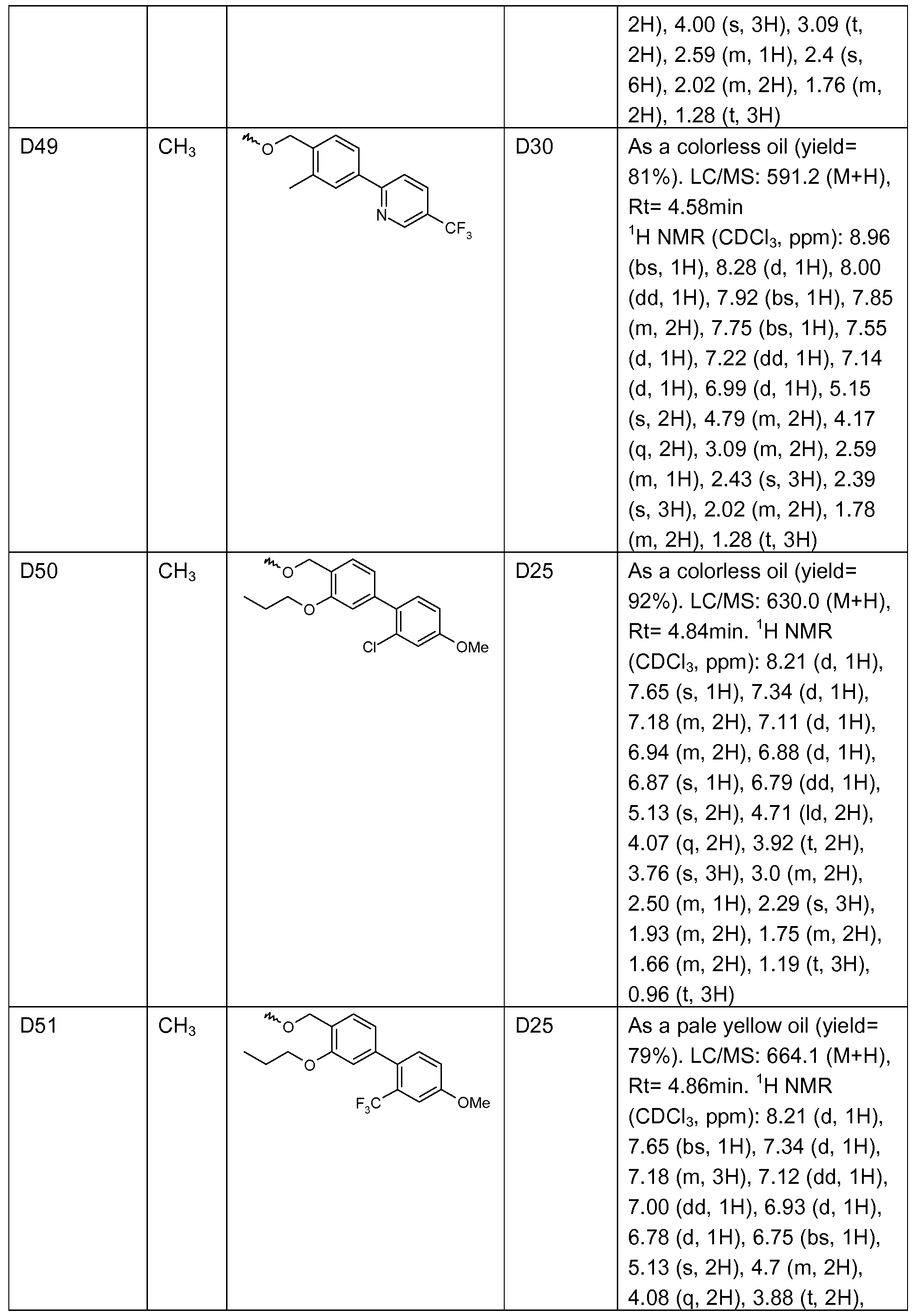


To a solution of ethyl 1-(4-(2-hydroxy-5-methylphenyl)pyrimidin-2-yl)piperidine-4-carboxylate (D16, 0.25g, 0.732 mmol) in acetone (30 ml) was added Cs2CO3 (0.358 g, 1.098 mmol), and the mixture was stirred at room temperature for 10 minutes. 2-methyl-4-(2-methyl-4- trifluoromethylphenyl)-benzyl bromide (D6e, 0.264 g, 0.769 mmol) was then added and the mixture was heated at 700C overnight and then poured into water. After extraction with CH2CI2, the organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography on silicagel (CH2CI2/Me0H, 98/2). The title compound was obtained as a colorless oil (0.4g, yield= 90%). LC/MS: 604.2 (M+H), Rt= 4.82min. 1H NMR (CDCI3, ppm): 8.28 (d, 1 H), 7.75 (bs, 1 H), 7.5 (m, 3H), 7.34 (d, 1 H), 7.24 (dd, 1 H), 7.15 (m, 3H), 7.02 (d, 1 H), 5.14 (s, 2H), 4.79 (Id, 2H), 4.17 (q, 2H), 3.10 (t, 2H), 2.59 (m, 1 H), 2.4 (s, 3H), 2.38 (s, 3H), 2.34 (s, 3H), 2.02 (m, 2H), 1.78 (m, 2H), 1.28 (t, 3H).
Prepared by a similar method as described for D54:





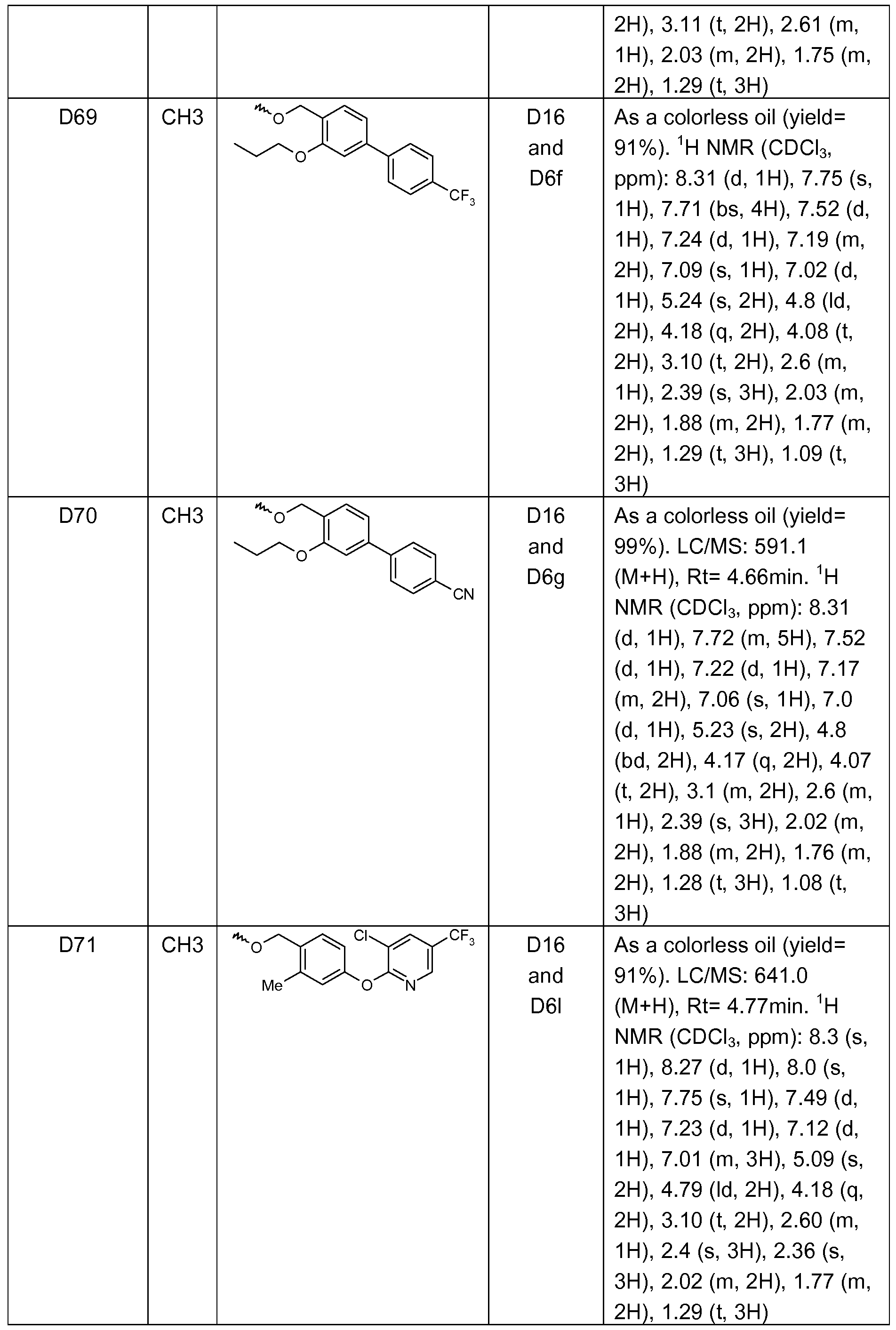



Example 1 : 1 -(4-(5-methyl-2-(2-methyl-4-(4-cvano-2-methylphenyl)benzyloxy)- phenyl)pyrimidin-2-yl)-piperidine-4-carboxylic acid

Example 2: 1-(4-(5-methyl-2-(2-methyl-4-(4-methoxy-2- trifluoromethylphenyl)benzyloxy)-phenyl)pyrimidin-2-yl)-piperidine-4-carboxylic acid

Prepared by a similar method as described for Example 1 for R9 = CN and by a similar method as described for Example 2 for R9≠ CN:








Prepared by a similar method as described for Example 1 for R = CN and by a similar method as described for Example 2 for R9≠ CN:












Prepared by a similar method as described for Example 1 for R = CN and by a similar method as described for Example 2 for R9≠ CN:



Example 47: 1-(4-(5-trifluoromethyl-2-(2-methyl-4-(4-cvanophenyl)benzyloxy)- phenyl)pyrimidin-2-yl)-piperidine-4-carboxylic acid

Biological Assay The activity of soluble guanylate cyclase (sGC) can be tested in an assay based on measuring the fluorescent polarisation (FP) signal of fluorescently labelled cGMP. FP increases on interaction with an anti-cGMP antibody as the motility of the molecule is reduced. Newly produced cGMP displaces the interaction giving rise to a decrease in polarisation and FP signal which can be equated to enzyme activity. Compounds are incubated with human sGC, anti-cGMP antibody, the GTP substrate and fluorescently labelled cGMP. After a period of one hour the assay is stopped with the addition of EDTA and after a further hour the assay is read.
Human sGC is thawed and resuspended in assay buffer (10OmM TRIS, 1 OmM MgCI2, 0.2mM Tween 20, pH7.4, containing 1 :100 dilution of sheep anti-cGMP) to give final concentration of 1 nM in the well. A substrate solution is prepared containing GTP and 8- fluo-cGMP in de-ionized water to a final concentration of 25μM and 5OnM respectively. Assay plates containing 5μl_ of various test compounds and of a standard agonist (50μM - 5OnM) in 1 % DMSO as 6 point, four fold dilutions across a 96 well plate are used in the assay. The plate also contains 6 wells of DMSO (1 %) to produce high control and a cGMP standard curve (14nM to 10μM) to convert FP data to cGMP concentration. 25μl_ of enzyme mix and 20μl of substrate mix described above are added to each well of the plate. Samples are mixed on an orbital shaker and then incubated at room temperature for 1 hour. After this incubation period 5μl of 0.5M EDTA is added to all wells and the plates are incubated for a further hour at room temperature prior to reading the FP signal in an appropriate reader. For data handling FP data are converted to cGMP concentrations and then fitted using ActivityBase software. The activity of a test compound is determined as the pEC500 value which is the concentration able to increase by 5-fold basal cGMP.
The compounds of Examples 1 to 47 were tested in the assay described above and gave pEC500 values of greater than 5.0. In an embodiment the compounds of the invention give
a pEC500 value of ≥6.0 when tested in this assay. In a further embodiment the compounds of the invention give a pEC500 value of ≥7.0 when tested in this assay.
The application of which this description and claims forms part may be used as a basis for priority in respect of any subsequent application. The claims of such subsequent application may be directed to any feature or combination of features described herein. They may take the form of product, composition, process, or use claims and may include, by way of example and without limitation, the following claims.
Claims
1. A compound of formula (I)

or a salt thereof; wherein
R1 and R2 are independently selected from hydrogen, halo, CF3 and  one of U and V represents N and the other represents CH;
one of U and V represents N and the other represents CH;
— Y— represents

Z is absent or represents O;
A represents CH or N;
when A represents CH, R5 is selected from hydrogen, methyl, d^alkoxy, methoxyC2- 3alkoxy, chloro and fluoro and R6 represents hydrogen;
when A represents N, R5 and R6 each represent hydrogen or one of R5 and R6 represents hydrogen and the other represents methyl; and
X represents 
wherein: J and L both represent CH, or one represents N and the other represents CH, provided that only one of A, J and L represents N;
when both J and L represent CH, R8 represents hydrogen or chloro, fluoro, CF3, C1- 4alkyl or C1-4alkoxy in a meta or ortho position relative to the R9 substituent;
when one of J and L represents N, R8 represents hydrogen or halo in a meta or ortho position relative to the R9 substituent; and
R9 represents hydrogen, halo, CF3, OCF3, C1-4-alkyl, C1-4-alkoxy, CN, CONR10R11, CO2H or N3, wherein R10 and R11 are independently selected from hydrogen and C1-4- alkyl;
with the exception of 1-[4-(2-(4-(4-chlorophenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid and 1-[4-(2-((4- phenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-5-trifluoromethyl-pyrazole-4-carboxylic acid.
2. A compound of formula (I) or a salt thereof as claimed in claim 1 wherein — Y—
represents 
3. A compound of formula (I) or a salt thereof as claimed in claim 2 wherein R1 represents C1-4alkyl, CF3 or halo in a para position relative to the -OCH2- linker and R2 represents hydrogen.
4. A compound of formula (I) or a salt thereof as claimed in claim 3 wherein R1 represents methyl, CF3, fluoro or chloro.
5. A compound of formula (I) or a salt thereof as claimed in claim 4 wherein R1 represents methyl or CF3.
6. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 5 wherein U represents CH and V represents N.
7. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 6 wherein Z is absent.
8. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 7 wherein A represents CH.
9. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 8 wherein R6 represents hydrogen.
10. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 9 wherein R5 represents hydrogen, methyl, methoxy or propyloxy.
1 1. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 10 wherein J and L both represent CH; or wherein J represents N and L represents CH.
12. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 11 wherein R8 represents hydrogen or R8 represents chloro, CF3, methyl or methoxy in a meta position relative to the R9 substituent.
13. A compound of formula (I) or a salt thereof as claimed in claim 11 or claim 12 wherein: J and L both represent CH and R8 represents hydrogen or R8 represents chloro, CF3, methyl or methoxy in a meta position relative to the R9 substituent; or J represents N and L represents CH, R8 represents hydrogen, or R8 represents chloro in a meta position relative to the R9 substituent.
14. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 13 wherein R9 represents chloro, fluoro, CF3, OCF3, methoxy or CN.
15. A compound of formula (I) or a salt thereof as claimed in any one of claims 2 to 14 wherein: R8 represents hydrogen and R9 represents methoxy, CN, CF3, OCF3 or fluoro; or R8 represents CF3 and R9 represents CN, methoxy or chloro, with R8 being in a meta position relative to the R9 substituent; or R8 represents chloro and R9 represents CF3, CN or methoxy, with R8 being in a meta position relative to the R9 substituent.
16. A compound of formula (I) as claimed in claim 2 selected from:
1-[4-(5-methyl-2-(2-methyl-4-(4-cyano-2-methylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-methoxy-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid; 1-[4-(2-(2-methyl-4-(2,4-dimethoxy-phenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-methoxy-2-methyl-phenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methoxy-4-(4-trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(2-methyl-4-(4-fluorophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(4-(4-cyanophenyloxy)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-fluoro-2-(4-(5-trifluoromethylpyridin-2-yloxy)phenylmethyloxy)-phenyl)pyrimidin- 2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-trifluoromethoxyphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-methoxyphenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]- piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(2-methyl-4-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(2-chloro-4-methoxyphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(4-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(4-cyanophenyl)phenylmethyloxy)-phenyl)pyrimidin-2- yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(2-chloro-4-methoxyphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(4-methoxy-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-propyloxy-4-(4-chloro-2-trifluoromethylphenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; 1-[4-(5-methyl-2-(2-methyl-4-(6-methoxypyridin-3-yl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; 1-[4-(5-methyl-2-(2-methyl-4-(5-trifluoromethylpyridin-2-yl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2- yloxy)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(2-chloro-4-cyanophenoxy)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-trifluoromethyl-2-(2-methyl-4-(4-chloro-2- trifluoromethylphenyl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-trifluoromethyl-2-(2-methyl-4-(4-cyanophenyl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid;
1-[4-(5-methyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2-yl)phenylmethyloxy)- phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; and
1-[4-(5-trifluoromethyl-2-(2-methyl-4-(3-chloro-5-trifluoromethylpyridin-2- yl)phenylmethyloxy)-phenyl)pyrimidin-2-yl]-piperidine-4-carboxylic acid; or a salt thereof.
17. A pharmaceutical composition comprising a compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, together with one or more pharmaceutically acceptable carrier(s), diluents(s) and/or excipient(s).
18. A compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as claimed in claim 17, for use in therapy.
19. A compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as claimed in claim 17, for use in the treatment of a disease or condition mediated by the activity of sGC.
20. A compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as claimed in claim 17, for use in the treatment of arterial hypertension, pulmonary arterial hypertension, angina, cardiac ischemia, myocardial infarction, congestive heart failure, acute coronary syndrome, atherosclerosis, peripheral vascular disease, cardiorenal syndrome, hepatorenal syndrome or restenosis.
21. Use of a compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, for the preparation of a medicament for the treatment of a disease or condition mediated by the activity of sGC.
22. Use of a compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, for the preparation of a medicament for the treatment of arterial hypertension, pulmonary arterial hypertension, angina, cardiac ischemia, myocardial infarction, congestive heart failure, acute coronary syndrome, atherosclerosis, peripheral vascular disease, cardiorenal syndrome, hepatorenal syndrome or restenosis.
23. A method of treatment of a disease or condition mediated by the activity of sGC comprising administration to a human subject in need of such treatment of a therapeutically effective amount of a compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, or of a pharmaceutical composition as claimed in claim 17.
24. A method of treatment of arterial hypertension, pulmonary arterial hypertension, angina, cardiac ischemia, myocardial infarction, congestive heart failure, acute coronary syndrome, atherosclerosis, peripheral vascular disease, cardiorenal syndrome, hepatorenal syndrome or restenosis comprising administration to a human subject in need of such treatment of a therapeutically effective amount of a compound of formula (I) as claimed in any one of claims 1 to 16, or a pharmaceutically acceptable salt thereof, or of a pharmaceutical composition as claimed in claim 17.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0814492A GB0814492D0 (en) | 2008-08-07 | 2008-08-07 | Compounds |
| GB0814492.5 | 2008-08-07 | ||
| GB0817052.4 | 2008-09-17 | ||
| GB0817052A GB0817052D0 (en) | 2008-09-17 | 2008-09-17 | Compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010015653A1 true WO2010015653A1 (en) | 2010-02-11 |
Family
ID=41010020
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/060146 WO2010015653A1 (en) | 2008-08-07 | 2009-08-05 | Pyrimidine derivatives as activators of soluble guanylate cyclase |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010015653A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2594270A2 (en) | 2011-11-18 | 2013-05-22 | BIP Patents | The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc) |
| US8455638B2 (en) | 2007-09-06 | 2013-06-04 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US8461348B2 (en) | 2008-04-04 | 2013-06-11 | Takeda Pharmaceutical Company Limited | Heterocyclic derivative and use thereof |
| US8507512B2 (en) | 2009-02-26 | 2013-08-13 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US8569339B2 (en) | 2011-03-10 | 2013-10-29 | Boehringer Ingelheim International Gmbh | Soluble guanylate cyclase activators |
| WO2014039434A1 (en) * | 2012-09-07 | 2014-03-13 | Boehringer Ingelheim International Gmbh | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| US8815857B2 (en) | 2011-08-12 | 2014-08-26 | Boehringer Ingelheim International Gmbh | Soluble guanylate cyclase activators |
| WO2016042536A1 (en) * | 2014-09-19 | 2016-03-24 | Glaxosmithkline Intellectual Property Development Limited | Novel soluble guanylate cyclase activators and their use |
| US9353090B2 (en) | 2014-07-22 | 2016-05-31 | Boehringer Ingelheim International Gmbh | Heterocyclic carboxylic acids as activators of soluble guanylate cyclase |
| WO2018069148A1 (en) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Combination containing sgc activators and mineralocorticoid receptor antagonists |
| US10208018B2 (en) | 2014-07-02 | 2019-02-19 | Novartis Ag | Indane and indoline derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2019081456A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | Use of activators and stimulators of sgc comprising a beta2 subunit |
| EP3498298A1 (en) | 2017-12-15 | 2019-06-19 | Bayer AG | The use of sgc stimulators and sgc activators alone or in combination with pde5 inhibitors for the treatment of bone disorders including osteogenesis imperfecta (oi) |
| WO2019211081A1 (en) | 2018-04-30 | 2019-11-07 | Bayer Aktiengesellschaft | The use of sgc activators and sgc stimulators for the treatment of cognitive impairment |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| EP3574905A1 (en) | 2018-05-30 | 2019-12-04 | Adverio Pharma GmbH | Method of identifying a subgroup of patients suffering from dcssc which benefits from a treatment with sgc stimulators and sgc activators in a higher degree than a control group |
| WO2020148379A1 (en) | 2019-01-17 | 2020-07-23 | Bayer Aktiengesellschaft | Methods to determine whether a subject is suitable of being treated with an agonist of soluble guanylyl cyclase (sgc) |
| WO2023237577A1 (en) | 2022-06-09 | 2023-12-14 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000027394A1 (en) * | 1998-11-05 | 2000-05-18 | University College London | Activators of soluble guanylate cyclase |
| WO2009032249A1 (en) * | 2007-09-06 | 2009-03-12 | Merck & Co., Inc. | Soluble guanylate cyclase activators |
| WO2009068652A1 (en) * | 2007-11-30 | 2009-06-04 | Smithkline Beecham Corporation | 2, 6-disubstituted pyridines and 2, 4-disubstituted pyrimidines as soluble guanylate cyclase activators |
| WO2009071504A1 (en) * | 2007-12-03 | 2009-06-11 | Smithkline Beecham Corporation | 2,6-disubstituted pyridines as soluble guanylate cyclase activators |
-
2009
- 2009-08-05 WO PCT/EP2009/060146 patent/WO2010015653A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000027394A1 (en) * | 1998-11-05 | 2000-05-18 | University College London | Activators of soluble guanylate cyclase |
| WO2009032249A1 (en) * | 2007-09-06 | 2009-03-12 | Merck & Co., Inc. | Soluble guanylate cyclase activators |
| WO2009068652A1 (en) * | 2007-11-30 | 2009-06-04 | Smithkline Beecham Corporation | 2, 6-disubstituted pyridines and 2, 4-disubstituted pyrimidines as soluble guanylate cyclase activators |
| WO2009071504A1 (en) * | 2007-12-03 | 2009-06-11 | Smithkline Beecham Corporation | 2,6-disubstituted pyridines as soluble guanylate cyclase activators |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8455638B2 (en) | 2007-09-06 | 2013-06-04 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US8461348B2 (en) | 2008-04-04 | 2013-06-11 | Takeda Pharmaceutical Company Limited | Heterocyclic derivative and use thereof |
| US8507512B2 (en) | 2009-02-26 | 2013-08-13 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US8569339B2 (en) | 2011-03-10 | 2013-10-29 | Boehringer Ingelheim International Gmbh | Soluble guanylate cyclase activators |
| US8815857B2 (en) | 2011-08-12 | 2014-08-26 | Boehringer Ingelheim International Gmbh | Soluble guanylate cyclase activators |
| EP2594270A2 (en) | 2011-11-18 | 2013-05-22 | BIP Patents | The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc) |
| KR20150048765A (en) * | 2012-09-07 | 2015-05-07 | 베링거 인겔하임 인터내셔날 게엠베하 | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| US8906904B2 (en) | 2012-09-07 | 2014-12-09 | Boehringer Ingelheim International Gmbh | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| USRE46886E1 (en) | 2012-09-07 | 2018-06-05 | Boehringer Ingelheim International Gmbh | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| CN104619695A (en) * | 2012-09-07 | 2015-05-13 | 勃林格殷格翰国际有限公司 | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| JP2015527394A (en) * | 2012-09-07 | 2015-09-17 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Alkoxypyrazoles as soluble guanylate cyclase activators |
| KR101692707B1 (en) | 2012-09-07 | 2017-01-04 | 베링거 인겔하임 인터내셔날 게엠베하 | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| EA027244B1 (en) * | 2012-09-07 | 2017-07-31 | Бёрингер Ингельхайм Интернациональ Гмбх | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| WO2014039434A1 (en) * | 2012-09-07 | 2014-03-13 | Boehringer Ingelheim International Gmbh | Alkoxy pyrazoles as soluble guanylate cyclase activators |
| US10550102B2 (en) | 2014-07-02 | 2020-02-04 | Novartis Ag | Indane and indoline derivatives and the use thereof as soluble guanylate cyclase activators |
| US10208018B2 (en) | 2014-07-02 | 2019-02-19 | Novartis Ag | Indane and indoline derivatives and the use thereof as soluble guanylate cyclase activators |
| US9353090B2 (en) | 2014-07-22 | 2016-05-31 | Boehringer Ingelheim International Gmbh | Heterocyclic carboxylic acids as activators of soluble guanylate cyclase |
| JP2017527602A (en) * | 2014-09-19 | 2017-09-21 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Novel soluble guanylate cyclase activators and their use |
| JP2020105189A (en) * | 2014-09-19 | 2020-07-09 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Novel soluble guanylate cyclase activators and their use |
| AU2015319724B2 (en) * | 2014-09-19 | 2018-05-10 | Glaxosmithkline Intellectual Property Development Limited | Novel soluble guanylate cyclase activators and their use |
| US9938260B2 (en) | 2014-09-19 | 2018-04-10 | Glaxosmithkline Intellectual Property Development Limited | Soluble guanylate cyclase activators and their use |
| CN106687456A (en) * | 2014-09-19 | 2017-05-17 | 葛兰素史密斯克莱知识产权发展有限公司 | Novel soluble guanylate cyclase activators and their use |
| JP2021155450A (en) * | 2014-09-19 | 2021-10-07 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Novel soluble guanylate cyclase activators and their use |
| WO2016042536A1 (en) * | 2014-09-19 | 2016-03-24 | Glaxosmithkline Intellectual Property Development Limited | Novel soluble guanylate cyclase activators and their use |
| EA033697B1 (en) * | 2014-09-19 | 2019-11-18 | Glaxosmithkline Ip Dev Ltd | Soluble guanylate cyclase activators and their use |
| US10472350B2 (en) | 2014-09-19 | 2019-11-12 | Glaxosmithkline Intellectual Property Development Limited | Soluble guanylate cyclase activators and their use |
| WO2018069148A1 (en) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Combination containing sgc activators and mineralocorticoid receptor antagonists |
| WO2019081456A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | Use of activators and stimulators of sgc comprising a beta2 subunit |
| EP3498298A1 (en) | 2017-12-15 | 2019-06-19 | Bayer AG | The use of sgc stimulators and sgc activators alone or in combination with pde5 inhibitors for the treatment of bone disorders including osteogenesis imperfecta (oi) |
| WO2019211081A1 (en) | 2018-04-30 | 2019-11-07 | Bayer Aktiengesellschaft | The use of sgc activators and sgc stimulators for the treatment of cognitive impairment |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| EP3574905A1 (en) | 2018-05-30 | 2019-12-04 | Adverio Pharma GmbH | Method of identifying a subgroup of patients suffering from dcssc which benefits from a treatment with sgc stimulators and sgc activators in a higher degree than a control group |
| WO2020148379A1 (en) | 2019-01-17 | 2020-07-23 | Bayer Aktiengesellschaft | Methods to determine whether a subject is suitable of being treated with an agonist of soluble guanylyl cyclase (sgc) |
| WO2023237577A1 (en) | 2022-06-09 | 2023-12-14 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010015653A1 (en) | Pyrimidine derivatives as activators of soluble guanylate cyclase | |
| WO2009071504A1 (en) | 2,6-disubstituted pyridines as soluble guanylate cyclase activators | |
| WO2009068652A1 (en) | 2, 6-disubstituted pyridines and 2, 4-disubstituted pyrimidines as soluble guanylate cyclase activators | |
| EP2401265B1 (en) | Derivatives of 1-pyri(mid)in-2-yl-pyrazole-4-carboxylic acid which are useful for therapy or prophylaxis of cardiovascular diseases | |
| WO2010015652A2 (en) | Thiazole compounds as activators of soluble guanylate cyclase | |
| TWI585088B (en) | Imidazo[1,2-b]pyridazine analogues as kinase inhibitors | |
| EP2509955B1 (en) | Pyrazole derivatives as modulators of calcium release-activated calcium channel | |
| EP2197551B1 (en) | Soluble guanylate cyclase activators | |
| EP1142890B1 (en) | Aminopyrazole derivatives | |
| TWI426910B (en) | Cept inhibitors, pharmaceutical composition and use thereof | |
| WO2012138648A1 (en) | Compositions and methods for modulating lpa receptors | |
| TW201838983A (en) | Ask1 inhibitor compounds and uses thereof | |
| WO1992018504A1 (en) | PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVE AND ANTI-INFLAMMATORY CONTAINING THE SAME | |
| EA020106B1 (en) | L-(piperidin-4-yl)pyrazole derivatives as gpr119 modulators | |
| EP2417121A1 (en) | 4, 5-dihydro-1h-pyrazole compounds and their pharmaceutical uses | |
| AU2006207300A1 (en) | Substituted triazole derivatives as oxytocin antagonists | |
| JP2018514535A (en) | Pyrazole derivatives useful as 5-lipoxygenase activating protein (FLAP) inhibitors | |
| CN110872285A (en) | Heterocyclic compounds as receptor interacting protein 1(RIP1) kinase inhibitors | |
| EP3218363B1 (en) | Sulfonyl piperidine derivatives and their use for treating prokineticin mediated gastrointestinal disorders | |
| KR20230074722A (en) | Pyrazole boronic acid compounds, pharmaceutical compositions containing them, and uses thereof | |
| KR20170102360A (en) | Pyrazole compound | |
| CN118420559A (en) | Deuterated oxadiazinone compound and application thereof | |
| KR20220044211A (en) | Bicyclic Heterocyclic Compounds Useful as IRAK4 Inhibitors | |
| EA040632B1 (en) | COMPOUNDS INHIBITING ASK1 AND THEIR USE |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09781511 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09781511 Country of ref document: EP Kind code of ref document: A1 |