WO2009037705A2 - Esters of n-phenylanthranilic acid for use in the treatment of cancer and inflammation - Google Patents
Esters of n-phenylanthranilic acid for use in the treatment of cancer and inflammation Download PDFInfo
- Publication number
- WO2009037705A2 WO2009037705A2 PCT/IL2008/001255 IL2008001255W WO2009037705A2 WO 2009037705 A2 WO2009037705 A2 WO 2009037705A2 IL 2008001255 W IL2008001255 W IL 2008001255W WO 2009037705 A2 WO2009037705 A2 WO 2009037705A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- unit dosage
- group
- dosage form
- pharmaceutical composition
- substituted
- Prior art date
Links
- 0 *C1C(*)=C(*)C(*)=C(*)C1N Chemical compound *C1C(*)=C(*)C(*)=C(*)C1N 0.000 description 2
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/216—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acids having aromatic rings, e.g. benactizyne, clofibrate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention in some embodiments thereof, relates to methods, compositions and unit dosage forms for the treatment of cancer and inflammation, and, more particularly, but not exclusively, to methods, unit dosage forms and compositions for treating cancer and inflammation which utilize compounds that exhibit tissue-selective activity.
- Cancer is a leading cause of death in developed countries. Despite continuing advances in diagnosis and treatment regimens, most existing treatments have undesirable side effects and limited efficacy. Progress in this field has been hindered because a number of different cellular events contribute to the formation and metastasis of tumors, and many of these events are still not well understood.
- Cyclooxygenase (COX) enzymes catalyze the conversion of arachidonic acid into prostaglandin H2 (PGH2), the early step in the biosynthesis of prostanoids.
- the COX enzymes are functional homodimers, in which each subunit contains both a cyclooxygenase and a peroxidase active site.
- COX-1 is a ubiquitous constitutive form of the enzyme that is involved in the regulation of various physiological processes such as platelet aggregation or homeostasis of the gastrointestinal tract and kidneys
- COX-2 isozyme expression is mainly observed during inflammatory processes.
- COX-2 is constitutively expressed in some tissues, but can be induced by different stimuli such as growth factors or interleukin-1.
- Prostanoids are naturally produced in numerous cells and tissues in mammalian systems as players in a multi-layer cell stress signaling systems designed to regulate biological processes. These processes include recruitment of blood-born inflammatory leukocytes to sites of infection, inflammation or edema (mostly via the activation of inducible COX-2 enzyme).
- prostanoids produced by both COX-1 and COX-2 participate in the homeostatic regulation of blood cells and blood vessels metabolic functions throughout the vascular system.
- Prostanoids have been found to regulate anti-platelets aggregation as well as anti-acid secretion in the stomach (COX-1).
- Prostanoids have also been found to be involved in the regulation of the adhesion activity of vascular endothelium cells (COX-2), and regulation of renal blood flow, urine production and salts & water retention in the kidney (COX-1 and COX-2).
- Non-steroidal anti-inflammatory drugs are organic compounds clinically used as medicines to alleviate fever, pain and inflammation associated with acute and chronic inflammatory diseases, like OA, RA and others. All of the presently known NSAIDs inhibit the COX isozymes to different extents, an activity which accounts for their anti-inflammatory and analgesic properties as well as their gastrointestinal side effects, the latter being more related to COX-1 inhibition, mg.
- NSAIDs have been synthesized and marketed by drug companies.
- the major indication for general occasional use of NSAIDs is in acute pain (headache, muscle pain, etc.) whereas the indications for chronic treatment with NSAIDs are in RA and OA patients.
- the major drawbacks (adverse side effects) associated with the use of NSAIDs include initiation or exacerbation of peptic ulcer bleeding.
- An additional major drawback in the elderly population (which is usually the one who suffers most from OA & RA) is undesired changes in renal function that promote renal failure.
- NSAIDs such as Diclofenac
- the anti-tumor activity of NSAIDs has been attributed to their anti-COX activity and inhibition of prostaglandin synthesis [see, for example, Brown and DuBois Clinical Cancer Research 2004; 10: 4266s-4269s].
- the anti- carcinogenic effect of NSAIDs in a rodent cancer model was shown to be directly related to the dose of the drug.
- Celecoxib (Celebrex) is a highly selective COX-2 inhibitor which has been shown to be chemopreventive, in a dose dependent manner, in azoxymethane- induced colon cancer model in rats, in spontaneous adenomas of the small intestine in mice and in bladder cancer models in mice [Steele et al, Mutation Res. 2003; 523-524: 137-144].
- COX-2 selective NSAIDs e.g. Celebrex
- a daily basis administration of these drugs may be of therapeutic benefit, as a prophylactic treatment, against the development of cancer and inflammation-based degenerative diseases like Alzheimer and RA.
- the recent accumulating negative data on this class of compounds regarding their adverse cardiovascular side effects have limited their use in ameliorating existing disease conditions as well as in preventive, prophylactic treatments.
- Daily intake of a low dose of aspirin is used in the general population with age over 60 to reduce the risk of a thrombotic event leading to a heart attack. Yet still some patients are stomach-sensitive even to this low dose.
- the recommended daily dose of NSAIDs is limited by the adverse side effects induced thereby.
- the recommended daily dose of NSAIDs such as Diclofenac or Indomethacin
- the recommended daily dose of NSAIDs is 100-150 mg.
- the highest recommended Diclofenac and Indomethacin daily dose is 200 mg.
- Diclofenac is administered at an oral dose of 1-3 mg/kg body weight/day in divided doses.
- the recommended dosage is 100-150 mg/day.
- Indomethacin is administered at a total daily dose of 150-200 mg.
- U.S. Patent Application No. 11/110,669 having Publication No. 20050250833, by some of the present inventors, teaches a family of N-phenylanthranilic acid derivatives for use as potassium channel modulators.
- Some of the compounds taught in U.S. Patent Application No. 11/110,669 are ester derivatives of phenylanthranilic acid such as Diclofenac and Meclofenamic acid, in which the acid functional group of these compounds has been esterified. These ester derivatives have shown to exhibit COX inhibition activity.
- 20040024057 teaches nitrosated NSAIDs for treating gastrointestinal disorders, for facilitating wound healing, for treating and/or preventing gastrointestinal, renal and/or respiratory toxicity resulting from the use of NSAIDs, for treating inflammatory disease states and/or preventing ophthalmic diseases.
- the compounds taught in U.S. Patent Application No. 10/612,014 comprise a nitroso-containing alkyl moiety which is linked to the NSAID via an amide or ester bond.
- the compounds taught in this patent application are aimed at releasing NO.
- Esterified NSAIDs have also been described in U.S. Patent No. 4,851 ,426 and in U.S. Patent Application Nos. 10/767,581 and 10/678,430.
- U.S. Patent No. 4,851 ,426 teaches 1-ethoxycarbonyloxyethyl esters of NSAIDs possessing increased bioavailability and reduced ulceroginicity when administered orally as compared to the NSAIDs with a free carboxylic function from which they are derived.
- 4,851 ,426, include esters of aspirin, indomethacin, naproxen, ibuprofen, sulindac, diflusinal, ketoprofen, mefenamic acid, tolmetin, Diclofenac and flufenamic acid.
- 10/767,581 further teaches a method for reducing gastrointestinal side effects associated with NSAIDs by masking the carboxyl function of the drug to prevent localization of the drug in the gastric mucosa.
- the NSAID ester derivatives taught therein are prodrugs which are less toxic to the gastrointestinal system than the native drug and, when administered orally, are absorbed from the gut into the blood stream where they liberate their corresponding parent drugs or exhibit independent pharmacological activity.
- U.S. Patent Application No. 10/678,430 having Publication No. 20040067914 teaches esters of an R-enantiomer of NSAIDs, which is substantially free from the S-enantiomer.
- the compounds described therein may be used in treating a disease or illness in a mammal such as inflammation, cystic fibrosis, dementia, Alzheimer's disease, Parkinson's disease and neoplastic disease, whereby administration of compositions including R-isomers of NSAIDs, which are substantially free of the S-enantiomer of the selected NSAIDs, is accompanied by a significant reduction in NSAID associated adverse effects.
- esterifying agents to be linked to the NSAID include glycerol, propylene glycol, hydroxysuccinic acid, hydroxyglutamic acid, glyceric acid, tartaric acid, xylaric acid, maleic acid, lactic acid, hydroxybutyric acid and ascorbic acid.
- NSAIDs are widely used drugs, there are no "safe NSAIDS" on the market today, and the use of NSAIDs, chronic use in particular, is limited by the non-selective COX inhibition activity of these drugs, which is often associated with adverse side effects.
- NSAIDs do not reach their full potential in becoming disease-modifying drugs because their dose is limited by their side effects.
- esters of N-phenylanthranilic acid, and of derivatives thereof exhibit tissue-selective COX inhibition activity and hence can be used as NSAIDs for the treatment of neoplastic and inflammation related diseases yet their associated gastrointestinal and renal side effects are significantly reduced.
- tissue-selective COX inhibition activity of these compounds allows using dosage forms of these drugs, which are higher than those used for non-modified NSAIDs (having nonselective COX inhibition activity that leads to adverse side effects), thus rendering these compounds highly suitable for use in the treatment of acute conditions, as well as chronic conditions, associated with neoplasia and/or inflammation.
- A is alkyl or absent
- K is selected from the group consisting of O and S;
- Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
- Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino, -NR 12 R 13 , and a positively charged group, whereas R 12 and R 13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R 12 and R 13 form a five- or six-member heteroalicyclic ring;
- R 1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 and R 10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino and -NR 14 R 15 , or, alternatively, at least two of R , R , R , R 5 and R 6 , of R 7 , R 8 , R 9 and R 10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or heteroalicycl
- R 14 and R 15 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R 14 and R 15 form a five- or six- member heteroalicyclic ring, and a pharmaceutically acceptable carrier, wherein an amount of the ester derivative is at least 1 rng/kg body weight, the unit dosage form being identified for use, from 1 to 4 times daily, in the treatment of a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
- Y is selected from the group consisting of a substituted or unsubstituted alkyl and a substituted or unsubstituted polyalkylene glycol moiety.
- the polyalkylene glycol moiety has a general formula II:
- Ra is hydrogen. According to some embodiments of the invention, Ra is alkyl. According to some embodiments of the invention, Ra is a positively charged group. According to some embodiments of the invention, the positively charged group is an ammonium group.
- G is C; K is O; each of R 2 , R 3 , R 4 , R 5 and R 6 is independently selected from the group consisting of hydrogen, alkyl, halo and trihaloalkyl; and each of R 7 , R 8 , R 9 and R 10 is hydrogen.
- the N-phenylanthranilic acid is Diclofenac.
- ester derivative is selected from the group consisting of:
- a pharmaceutical composition in a unit dosage form comprising an ester derivative of N-phenylanthranilic acid having the general Formula I, as described hereinabove, wherein an amount of the ester derivative is at least 50 mg, the unit dosage form being identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
- a pharmaceutical composition unit dosage form comprising an ester derivative of N- phenylanthranilic acid having the general Formula I, as described hereinabove, wherein an amount of the ester derivative is at least 1 mg/kg body weight, the unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to 4 times daily, in the treatment of a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
- a pharmaceutical composition unit dosage form comprising an ester derivative of N- phenylanthranilic acid having the general Formula I, as described hereinabove, wherein an amount of the ester derivative is higher than 50 mg, the unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
- the amount of the ester derivative is at least 2 mg/kg body weight.
- the amount of the ester derivative is at least 3 mg/kg body weight.
- the amount of the ester derivative is at least 6 mg/kg body weight.
- the amount of the ester derivative ranges from 1 mg/kg body weight to 10 mg/kg body weight.
- the amount of the ester derivative is higher than 70 mg.
- the composition is used 2 to 4 times a day.
- the composition is identified for use such that a daily dose of the ester derivative is at least 2 mg/kg body weight.
- the composition is identified for use such that a daily dose of the ester derivative is at least 3 mg/kg body weight.
- the composition is identified for use such that a daily dose of the ester derivative is higher than 150 mg.
- the composition is identified for use such that a daily dose of the ester derivative is higher than 200 mg.
- the composition is formulated for intraperitoneal administration.
- the composition is identified for use such that a daily dose of the ester derivative is higher than 1 mg/kg body weight.
- the composition is identified for use such that a daily dose of the ester derivative is higher than 50 mg.
- the treatment is a chronic treatment.
- the composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of the medical condition. According to some embodiments of the invention, the composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of cancer and an inflammatory disease or disorder.
- a method of treating a medical condition selected from the group consisting of cancer and an inflammatory disease or disorder in a subject in need thereof comprising administering to the subject a therapeutically effective amount of an ester derivative of N- phenylanthranilic acid having a general Formula I, as described hereinabove,
- the therapeutically effective amount is at least 1 mg/kg body, and the administering is performed from 1 to 4 times a day.
- the therapeutically effective amount is higher than 50 mg, and the administering is performed from 1 to 4 times a day.
- the administering is effected intraperitoneal ⁇ , the therapeutically effective amount is higher than 1 mg/kg body, and the administering is performed from 1 to 4 times a day.
- the administering is effected intraperitoneally, the therapeutically effective amount being higher than 50 mg, and the administering is performed from 1 to 4 times a day. According to some embodiments of the invention, therapeutically effective amount is at least 2 mg/kg body weight.
- the therapeutically effective amount is at least 3 mg/kg body weight.
- the therapeutically effective amount is at least 6 mg/kg body weight.
- the therapeutically effective amount is ranges from 1 mg/kg body weight to 10 mg/kg body weight.
- the therapeutically effective amount is higher than 70 mg.
- the administering is performed from 2 to 4 times a day.
- a daily dose of the ester derivative is higher than 2 mg/kg body weight.
- a daily dose of the ester derivative is higher than 3 mg/kg body weight.
- a daily dose of the ester derivative is higher than 150 mg.
- a daily dose of the ester derivative is higher than 200 mg.
- the administering is performed intraperitoneally.
- an ester derivative of N-phenylanthranilic acid having a general Formula I as described hereinabove, in the manufacture of a medicament for treating a medical condition selected from the group consisting of cancer and an inflammatory disease or disorder.
- the treating of the medical condition comprises chronic treatment.
- the chronic treatment comprises administering the ester derivative for a time period that ranges from 20 to 40 days.
- the medical condition is cancer
- the inflammatory disease or disorder is selected from a group consisting of Alzheimer's disease, cortical dementia, vascular dementia, muli-infract dementia, pre-senile dementia, alcoholic dementia, senile dementia, memory loss or central nervous damage resulting from stroke, ischemia or trauma, multiple sclerosis, Parkinson's disease, Huntington's disease, epilepsy, cystic fibrosis, arthritis diseases such as osteoarthritis, rheumatoid arthritis, spondyloarthopathies, gouty arthritis, systemic lupus erythematosus, and juvenile arthritis fever, periarteritis; gastrointestinal disorders such as inflammatory bowel disease, Chron's disease, gastritis, irritable bowel syndrome, ulcerative colitis, cardiovascular disorders such as myocardial ischemia, reperfusion injury to an ischemic organ; angiogenesis, asthma, bronchitis, menstrual cramps, premature labor, tendinitis, bursit
- FIG. 1 presents the 2-D chemical structures of exemplary compounds according to some embodiments of the present invention.
- FIGs. 2A-C present plots showing the viability level of CT26 cells, incubated with Diclofenac ( Figure 2A), NH6 ( Figure2B) and NH17 ( Figure 2C), as a function of the compound's concentration.
- Mouse colon adenocarcinoma (CT26) cells were incubated for an hour at 37 0 C, with the tested compound, at various inhibitor concentrations, and the number of viable cells was quantified using an XTT reagent test.
- the fluorescence intensity, measured using a spectrophotometer, in each cell culture was proportional to the number of metabolic active cells.
- the viability of the cells was significantly decreased only when the cells were exposed to Diclofenac at a concentration above 1 mM; to NH6 at a concentration of above 300 ⁇ M; and to NH17 at a concentration of above 100 ⁇ M.
- FIGs. 3A-B present plots showing the spontaneous decomposition of NH6 ( Figure 3A) and NH17 ( Figure 3B) to Diclofenac as a function of time.
- NH6 or NH17 were diluted to a concentration of 500 ⁇ M in RPMI medium and subjected to HPLC analysis after 0, 5, 10, 15, 20, 25 and 30 hours.
- the amount of acid (Diclofenac) was quantified for every time point.
- FIGs. 4A-B present bar graphs depicting the ability of Diclofenac, NH6 and NH17 to inhibit the COX-1 dependent ( Figure 4A) or COX-2 dependent ( Figure 4B), prostaglandin PGE 2 synthesis from Arachidonic acid.
- 20 enzyme units of Bovine COX-1 or human recombinant COX-2 purified enzymes were added to a reaction vial containing either Diclofenac (1 ⁇ M), NH6 (1 ⁇ M or 3 ⁇ M in the experiments with COX-1 and COX-2, respectively) or NH17 (1 ⁇ M or 3 ⁇ M in the experiments with COX-1 and COX-2, respectively). 15 ⁇ l of 10 ⁇ M Arachidonic acid was added to each vial.
- FIGs. 5A-C present bar graphs showing the anti-COX activity of NH6 and NH17, as compared to Diclofenac, in tumor, stomach and kidney tissues of mice, upon administration at a single dose of 1 , 3 or 10 mg per kg body weight.
- Tumor growth was induced by CT26 cells injection into the right hind foot of a BALB/c mouse. Once the tumor has reached the volume of approximately 400 mm 3 (21-24 days post injection) 1 , 3 or 10 mg per kg (mpk) of Diclofenac, NH6 or NH17 was injected intraperitoneal ⁇ (I. P.) to each mouse in a single dose. 3 hours after the injection, the tumor was removed, and the stomach and kidneys were collected.
- I. P. intraperitoneal ⁇
- the extent of PGE2 synthesis as a measure of COX activity was assessed by quantifying PGE 2 levels by radioimmunoassay, in each organ, in the presence of the tested compound, as compared to PGE2 levels in tumor, stomach and kidney tissue from control, non-treated mice.
- Diclofenac, NH6 and NH17 each inhibited COX activity in tumor tissue to a similar extent (see, Figure 5A).
- NH6 and NH17 inhibited COX activity significantly less than Diclofenac (see, Figure 5B).
- the kidney at a concentration of 1 mg per kilogram, NH6 and NH17 inhibited COX activity significantly less than Diclofenac (see, Figure 5C).
- 6A-C present bar graphs showing the COX inhibition (anti-COX) activity of NH6 and NH17 when administered at a chronic dose of 3 mg per kg/day, as compared to lndomethacin administered at a chronic dose of 1 mg per kg/day and Diclofenac administered at a chronic dose of 2 mg per kg/day in tumor, stomach and kidney tissue in mice.
- Tumor growth was induced by CT26 cells injection into the right hind foot of a BALB/c mouse. Starting from the day of cells injection, the mice were injected intraperitoneally with 100 ⁇ l of 1 mg per kg lndomethacin or 2 mg per kg Diclofenac or 3 mg per kg NH6 or NH17 every 24 hours for 27-33 days.
- FIG. 7 presents comparative plots showing the tumor size as a function of days post intraperitoneal injection of lndomethacin (filled squares), Diclofenac (filled circles), NH6 (filled triangles) or NH17 (horizontal line) to tumor baring mice and compared to tumor size in control, not-treated tumor baring mice (filled diamonds).
- CT26 cells suspended in HBSS buffer were injected to the right hind foot of BALB/c mice. Starting from the day of cells injection, the mice were injected I. P. with 100 ⁇ l of 1 mg per kg lndomethacin or 2 mg per kg Diclofenac or 3 mg per kg NH6 or 3 mg NH17 every 24 hours for 27-33 days.
- the volume of the tumor was measured every day starting from the day of cells injection.
- the rate of tumor growth inhibition in mice injected with 3 mg per kg/day NH6 and NH17 was comparable to that of mice injected with 2 mg per kg/day lndomethacin and significantly higher as compared to mice injected with 1 mg per kg /day Diclofenac.
- the present invention in some embodiments thereof, relates to methods, compositions and unit dosage forms for the treatment of cancer and inflammation, and, more particularly, but not exclusively, to methods, compositions and unit dosage forms for treating cancer and inflammation which utilize compounds that exhibit tissue-selective activity.
- Non-steroidal anti-inflammatory drugs are organic compounds clinically used as medicines to alleviate fever, pain and inflammation associated with acute and chronic inflammatory conditions.
- NSAIDs such as Diclofenac
- NSAIDs are widely used drugs, their administration, and particularly their chronic administration, is limited by the adverse side effects induced thereby, gastrointestinal and renal side effects in particular.
- NSAID-related gastrointestinal side effects include ulceration, haemmorrhage, perforation and death.
- NSAID-related gastrointestinal adverse effects are mainly due to a reduction in the levels of cytoprotective, prostaglandins PGE 2 and PGI 2 in the stomach lining, which further leads to a reduced mucous secretion and impaired protection of the stomach against acidic insults.
- Renal NSAID-related adverse effects are also related to inhibition of the COX-dependent prostaglandin synthesis.
- Prostaglandins normally cause vasodilatation of the afferent arterioles of the glomeruli, which is crucial for the normal glomerular perfusion and glomerular filtration rate (GFR).
- the present inventors have now further tested the anti-COX activity of ester derivatives of N-phenylanthranilic acid and have surprisingly uncovered that these compounds exhibit a therapeutic activity that is superior to the parent, non-esterified, N-phenylanthranilic acid.
- ester derivatives exhibit tissue-selective anti-COX activity, namely, they exhibit a profound anti-COX activity in tumor tissues and inflammatory sites yet reduced anti-COX activity in stomach and kidney tissues.
- these ester derivatives cause an elevation rather than inhibition of COX activity in, for example, stomach tissue.
- Diclofenac exhibit a potent anti-tumor effect due to their anti-COX activity.
- Diclofenac in contrast to the parent, non-esterified compound Diclofenac, NH6 and NH17, at a concentration of 1-10 mg per kilogram, did not inhibit activity of COX enzymes in the stomach but rather induce COX activity (see, Example 5).
- ester derivatives of N-phenylanthranilic acid can be safely used at a dosage regimen higher than that of their parent compounds (e.g., Diclofenac), a dosage regimen which can result in a pronounced therapeutic effect without the risk of increased adverse side effects.
- parent compounds e.g., Diclofenac
- NH6 and NH17 were found to be therapeutically superior to the known NSAIDs lndomethacin and Diclofenac, administered at a similar dosage range (1 mpk/day and 2 mpk/day, respectively; see, Example 6), in the treatment of inflammation.
- Diclofenac and lndomethacin at the tested dosage regimen leads to renal and stomach adverse effects caused by the COX inhibition in these organs.
- NH17 also inhibits prostaglandin synthesis but, the extent of inhibition of prostaglandin synthesis in stomach and kidney tissue is significantly reduced as compared to lndomethacin and Diclofenac, thus rendering a chronic administration of these compounds, at relatively high dosages, applicable.
- NH6 and NH17 were further shown that the anti-tumor activity of NH6 and NH17 (as determined by observing the reduction in tumor size) was superior to that of Diclofenac and comparable to that of lndomethacin (see, Example 7).
- NH6 and NH17 in assays conducted in a rat model of inflammation, exhibit anti-inflammatory activity which is comparable to that of Diclofenac and lndomethacin administered at a same dosage (see, for example, Table 5 and Example 8.).
- the equivalence of the NH6, NH17, Diclofenac and lndomethacin dosages administered in the current experiments, in a human subject are dosages of 3 mg per kg and 9 mg per kg. Taking the average human weight as being about 70 kg, such dosages are equivalent to 210 mg per day and 630 mg per day.
- the recommended daily dose of NSAIDs, such as Diclofenac or lndomethacin is 100-150 mg and the highest recommended Diclofenac and lndomethacin daily dose is 200 mg.
- the dosages ,of Diclofenac and lndomethacin, administered to rats in the current experiments were well above the recommended daily dosage for humans and indicate that the tested compounds can be safely utilized at such doses since no adverse side effects are iduced thereby.
- NH6 and NH17 enhance rather than reduce COX activity in the stomach.
- NH6 and NH17 are superior to Diclofenac in the treatment of inflammation.
- a method of treating a medical condition such as cancer and an inflammatory disease or disorder.
- the method is effected by administering to a subject in need thereof a therapeutically effective amount of an ester derivative of N-phenylanthranilic acid, as described herein.
- an ester derivative of N-phenylanthranilic acid as described herein, in the manufacture of a medicament for treating a medical condition such as an inflammatory disease or disorder and cancer.
- ester derivatives of N-phenylanthranilic acid are also referred to interchangeably as “ester derivatives” or simply as “compounds”. As discussed hereinabove and is further demonstrated in the Examples section that follows, the ester derivatives described herein were found highly effective in the treatment of both cancer and inflammation.
- ester derivatives described herein were found to exhibit a tissue-selective anti-COX activity, such that they exert COX inhibition activity at the tumor tissue or inflammation site but, they do not affect COX activity, or, induce COX activity, at stomach and kidney tissues.
- the anti-cancer and anti-inflammatory activities of the ester derivatives described herein were shown to be at least comparable if not superior to common NSAIDs such as Diclofenac and lndomethacin, whereby the lack of COX inhibition activity in the stomach and kidney was observed only for the ester derivatives described herein and was not observed with the NSAIDs.
- COX inhibition activity describes the ability of a compound to inhibit cyclooxygenase, a family of isoenzymes responsible for the biosynthesis of protaglandins.
- the inhibition may be either of one of the three cyclooxygenase isoenzymes, namely cyclooxygenase-1 (COX-1 ), cyclooxygenase-2 (COX-2) and cyclooxygenase-3 (COX-3), or a combined inhibition of two or all three cyclooxygenase isoenzymes.
- the inhibition can be by any available pathway including for example, direct or allosteric inhibition, competitive or noncompetitive inhibition.
- COX inhibition activity is also referred to herein interchangeably as "anti-COX activity”.
- a therapeutically effective amount which is also referred to herein interchangeably as “pharmaceutically effective amount”, as used herein, describes an amount of an active ingredient needed to achieve the desired outcome, which is generally to prevent, alleviate or ameliorate a condition or symptoms of the condition. Determination of a therapeutically effective amount is within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- a “therapeutically effective amount” is referred to herein as a single dose, which can be administered once daily or a few times (e.g., 2, 3 or 4 times) daily.
- a “single dose” refers to a single event of administration at a certain time point.
- the single dose can be a single unit dosage form, a portion of a unit dosage form, or a number of unit dosage forms, depending on the unit dosage form available. If two or more unit dosage forms (e.g., two tablets or capsules) are taken together, at the same time point within a day, they are referred to herein as a single dose.
- a single dose also encompasses a continuous administration, such as, for example, a continuous intravenous administration.
- a therapeutically effective amount of the compounds described and utilized according to these aspects of the present embodiments indudes a single dose that is higher than 1 mg/kg body weight, which is utilized in a regimen of 1 to 4 times a day.
- the therapeutically effective amount of the compounds utilized in these aspects of the present embodiments can therefore be, for example, 2 mg/kg body weight, 3 mg/kg body weight, 4 mg/kg body weight, 5 mg/kg body weight, 6 mg/kg body weight, 7 mg/kg body weight, 8 mg/kg body weight, 9 mg/kg body weight or 10 mg/kg body weight, 11 mg/kg body weight, 12 mg/kg body weight, 13 mg/kg body weight, 14 mg/kg body weight, 15 mg/kg body weight, and even higher.
- the therapeutically effective amount is 2 mg/kg body weight. In another embodiment, the therapeutically effective amount is 3 mg/kg body weight. In yet another embodiment, the therapeutically effective amount is 6 mg/kg body weight.
- the therapeutically effective amount ranges from 1 mg/kg body weight to 10 mg/kg body weight.
- the above-described amounts correspond to dosages of 70 mg , 140 mg, 210 mg, 280 mg, 350 mg, 420 mg, 490 mg, 560 mg, 630 mg, 700 mg, 770 mg, 840 mg, 910 mg, 980 mg, 1050 mg, respectively, and higher.
- these amounts correspond to dosage administration of 20 mg , 40 mg, 60 mg, 80 mg, 100 mg, 120 mg, 140 mg, 160 mg, 180 mg, 200 mg, 220 mg, 240 mg, 260mg, 280 mg, 300 mg, respectively, and higher.
- the compounds described herein are administered in a therapeutically effective amount higher than 20 mg, administered from 1 to 4 times a day.
- the therapeutically effective amount is higher than 50 mg.
- the therapeutically effective amount is higher than 70 mg.
- the therapeutically effective dose may be administered once daily, or can be divided and administered from 2 to 6 times daily. In some embodiments, the therapeutically effective amount is divided and is administered, for example, twice daily, 3 times daily or 4 times daily.
- a therapeutically effective amount of 2 mg/kg body weight may be administered once daily as a 140 mg single dose or as a dose of 70 mg administered twice daily or a dose of 47 mg administered three times per day or a dose of 35 mg administered four times per day.
- a therapeutically effective amount of 3 mg/kg body weight may be administered once daily as a 60 mg single dose or a dose of 30 mg administered twice daily or a dose of 20 mg administered three times per day or a dose of 15 mg administered four times per day.
- the compounds described herein may be formulated for intravenous infusion and be administered in a continuous fashion, for a time period of a few minutes to a few hours, a day.
- the administration protocol of the compounds described herein can include a single administration or a few subsequent administrations, as part of a treatment protocol for treating an acute condition (e.g., pain).
- the administration protocol can include a chronic treatment protocol, for treating chronic conditions (e.g., RA).
- a chronic treatment includes, for example, a daily administration of an ester derivative as described herein, for a time period of 20 (consecutive) days or more.
- the ester derivative as described herein may be administered during a time period of 20 days, 21 days, 22 days, 23 days, 24 days, 25 days, 26 days, 27 days, 28 days, 29 days, 30 days, 31 days, 32 days, 33 days, 34 days, 35 days, 36 days, 37 days, 38 days, 39 days, 40 days and even more.
- the time period for utilizing the ester derivatives described herein in chronic treatment ranges from 27 to 33 days.
- an ester derivative as described herein may be administered at a therapeutically effective amount of 3 mg/kg body weight a day, for 27 days.
- the ester derivatives described herein are administered at a daily dose higher than 2 mg/kg body weight. In some embodiments, the ester derivatives described herein are administered at a daily dose higher than 3 mg/kg body weight.
- the ester derivatives described herein are administered, to a human patient (weighing 70 grams) at a daily dose higher than 150 mg, and even higher than 200 mg.
- the ester derivatives described herein may be administered via various routes of administration. These include, as non-limiting examples, oral, transdermal, intraperitoneal, sublingual, intravenous and rectal routes or by inhalation.
- the ester derivatives are administered intraperitoneally.
- Intraperitoneal administration refers to the injection of the compounds described herein through the peritoneum into the peritoneal cavity.
- the peritoneum is a thin, transparent membrane that lines the walls of the abdominal (peritoneal) cavity and contains/encloses the abdominal organs such as the stomach and intestines.
- the administration of drugs via injection to peritoneal cavity is a well known administration route.
- clinical investigators have confirmed the safety and pharmacokinetic advantage associated with the intraperitoneal delivery of a number of antineoplastic agents with known activity in ovarian cancer [Maurine Markman. J CHn Oncol 2003; 21 :145s-148s].
- the ester derivatives described herein are administered intraperitoneally in a therapeutically effective amount of at least 1 mg/kg body weight, wherein the administering is performed from 1 to 4 times a day.
- the ester derivatives are administered to a human patient intraperitoneally in a therapeutically effective amount of at least 50 mg, wherein the administering is performed from 1 to 4 times a day.
- intraperitoneal bioavailability is higher than oral bioavailability and hence doses that are considered safe, in terms of adverse side effects, for oral administration, may cause significant side effects upon intraperitoneal administration of such doses. Accordingly, doses that are therapeutically effective upon intraperitoneal administration may be less effective when administered orally.
- ester derivatives can be utilized either per se or as a part of a pharmaceutical composition, which further comprises a pharmaceutically acceptable carrier.
- such a pharmaceutical composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment or prevention of a medical condition such as cancer and an inflammatory disease or disorder,
- a "pharmaceutical composition” refers to a preparation of one or more of the compounds described herein (as active ingredient), or physiologically acceptable salts or prodrugs thereof, with other chemical components, including, but not limited to, physiologically suitable carriers, excipients, lubricants, buffering agents, antibacterial agents, bulking agents (e.g. mannitol), antioxidants (e.g., ascorbic acid or sodium bisulfite), and the like.
- physiologically suitable carriers including, but not limited to, physiologically suitable carriers, excipients, lubricants, buffering agents, antibacterial agents, bulking agents (e.g. mannitol), antioxidants (e.g., ascorbic acid or sodium bisulfite), and the like.
- the purpose of a pharmaceutical composition is to facilitate administration of a compound to a subject.
- active ingredient refers to a compound, which is accountable for a biological effect.
- physiologically acceptable carrier and “pharmaceutically acceptable carrier”, which are used interchangeably, describe a carrier or a diluent that does not cause significant irritation to the subject and does not abrogate the biological activity and properties of the administered compound.
- carrier refers to a diluent, adjuvant, excipient, or vehicle with which the therapeutic is administered.
- excipient refers to an inert substance added to a pharmaceutical composition to further facilitate administration of an active ingredient.
- excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
- compositions for use in accordance with the present invention thus may be formulated in conventional manner using one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the compounds into preparations which can be used pharmaceutically.
- Proper formulation is dependent upon the route of administration chosen.
- the dosage may vary depending upon the dosage form employed and the route of administration utilized. The exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition (see e.g., Fingl et al, 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p.1).
- the pharmaceutical composition may be formulated for administration in either one or more of routes depending on whether local or systemic treatment or administration is of choice, and on the area to be treated. Administration may be done orally, by inhalation, or parenterally, for example by intravenous drip or intraperitoneal, subcutaneous, intramuscular or intravenous injection, or topically (including ophthalmically, vaginally, rectally, intranasally).
- Formulations for topical administration may include but are not limited to lotions, ointments, gels, creams, suppositories, drops, liquids, sprays and powders.
- Conventional pharmaceutical carriers, aqueous, powder or oily bases, thickeners and the like may be necessary or desirable.
- compositions for oral administration include powders or granules, suspensions or solutions in water or non-aqueous media, sachets, pills, caplets, capsules or tablets. Thickeners, diluents, flavorings, dispersing aids, emulsifiers or binders may be desirable.
- Formulations for parenteral administration may include, but are not limited to, sterile solutions which may also contain buffers, diluents and other suitable additives. Slow release compositions are envisaged for treatment.
- compositions to be administered will, of course, be dependent on the subject being treated, the severity of the affliction, the manner of administration, the judgment of the prescribing physician, etc.
- compositions according to the present embodiments may, if desired, be presented in a pack or dispenser device, such as an FDA (the U.S. Food and Drug Administration) approved kit, which may contain one or more unit dosage forms containing the active ingredient.
- the pack may, for example, comprise metal or plastic foil, such as, but not limited to a blister pack or a pressurized container (for inhalation).
- the pack or dispenser device may be accompanied by instructions for administration.
- the pack or dispenser may also be accompanied by a notice associated with the container in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the compositions for human or veterinary administration. Such notice, for example, may be of labeling approved by the U.S.
- compositions comprising a compound of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of a medical condition, as defined herein.
- the compounds described herein have unique features that enable to use these compounds as therapeutic agents as dosages that are higher than the dosages commonly practiced with common NSAIDs.
- a pharmaceutical composition unit dosage form which comprises an ester derivative as described herein, wherein an amount of the ester derivative is at least 1 mg/kg body weight.
- the unit dosage form is identified for use, from 1 to 4 times daily, in the treatment of a medical condition such as an inflammatory disease or disorder and cancer.
- the term "unit dosage form”, as used herein, describes physically discrete units, each unit containing a predetermined quantity of one or more active ingredient(s) calculated to produce the desired therapeutic effect, in association with at least one pharmaceutically acceptable carrier, diluent, excipient, or combination thereof.
- the single unit dosage forms described herein can be formulated for oral, mucosal (e.g., nasal, sublingual, vaginal, buccal, or rectal), parenteral (e.g., intraperitoneal, subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial), or transdermal administration to a patient.
- mucosal e.g., nasal, sublingual, vaginal, buccal, or rectal
- parenteral e.g., intraperitoneal, subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial
- transdermal administration e.g., transdermal administration to a patient.
- unit dosage forms include, but are not limited to: tablets including orally dissolving tablets; thin films; gelcaps; caplets; granules, capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; enemas; pessary; vaginal tablets; ointments; cataplasms (poultices); pastes; powders; dressings; creams; plasters; solutions; patches; liquid sprays; metered and unmetered aerosols (e.g., nasal sprays or inhalers); drops; lyophilized compositions; transdermal patches; gels; liquid dosage forms suitable for oral or mucosal administration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, tinctures and elixirs; syrups, liquid dosage forms suitable for
- the pharmaceutical composition unit dosage form described herein comprises an amount of the ester derivative which is higher than 1 mg/kg body weight. In some embodiments, the unit dosage form comprises an amount of the ester derivative that is higher than 2 mg/kg body weight, and even higher than 3 mg/kg body weight. In some embodiments, the amount of the ester derivative in the unit dosage form is 6 mg/kg body weight or higher. In some embodiments, the pharmaceutical composition unit dosage form comprises an amount of the ester derivative which ranges from 1 mg/kg body weight to 10 mg/kg body weight.
- the amount of the ester derivative in the pharmaceutical composition unit dosage form described herein can be, for example, about 1 mg/kg body weight, about 2 mg/kg body weight, about 3 mg/kg body weight, about 4 mg/kg body weight, about 5 mg/kg body weight, about 6 mg/kg body weight, about 7 mg/kg body weight, about 8 mg/kg body weight, about 9 mg/kg body weight, about 10 mg/kg body weight, about 11 mg/kg body weight, about 12 mg/kg body weight, about 13 mg/kg body weight, about 14 mg/kg body weight, about 15 mg/kg body weight, about 16 mg/kg body weight, about 17 mg/kg body weight, about 18 mg/kg body weight, about 19 mg/kg body weight, about 20 mg/kg body weight, about 21 mg/kg body weight, about 22 mg/kg body weight, about 23 mg/kg body weight, about 24 mg/kg body weight, about 25 mg/kg body weight and even higher.
- a unit dosage form as described herein comprises an amount of the ester derivative which is higher than 50 mg.
- a pharmaceutical composition unit dosage form wherein an amount of the ester derivative is higher than 50 mg and the unit dosage form is identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition such as an inflammatory disease or disorder and cancer.
- an amount of the ester derivative in the pharmaceutical composition unit dosage form described herein can be, for example, about 50 mg, about 60 mg, about 70 mg, about 80 mg, about 90 mg, about 100 mg, about 150 mg, about 200 mg, about 250 mg, about 300 mg, about 350 mg, about 400 mg, about 450 mg, about 500 mg, about 550 mg, about 600 mg, about 650 mg, about 700 mg, about 750 mg, about 800 mg, about 850 mg, about 900 mg, about 950 mg, about 1000 mg and even higher.
- the optimal dosage of the ester derivatives in the unit dosage form depends upon factors such as the selected regime, the medical condition being treated and the carrier used, as is detailed herein.
- composition unit dosage form described herein can be formulated for various administration routes, such as, for example, orally, transdermal ⁇ , intreaperitoneally, sublingually, intravenously, rectally or by inhalation.
- the pharmaceutical composition unit dosage form is formulated for intraperitoneal administration. In other embodiments, the unit dosage form is formulated for oral administration.
- a pharmaceutical composition unit dosage form which comprises the ester derivative described herein, wherein an amount of the ester derivative is at least 1 mg/kg body weight, the unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to
- a pharmaceutical composition unit dosage form which comprises an ester derivative as described herein, wherein the amount of the ester derivative is higher than 50 mg and the unit dosage form is formulated for intraperitoneal administration to a human subject, 1 to 4 times daily.
- the pharmaceutical composition unit dosage form described herein may be formulated for use in divided doses.
- the unit dosage form may be administered 1 time per day, 2 times per day, 3 times per day, 4 times per day, 5 times per day, 6 times per day and so on.
- the composition is administered 2 to 4 times per day.
- the pharmaceutical composition unit dosage form is formulated such that the daily dose of the ester derivative is higher than 2 mg/kg body weight, and even higher than 3 mg/kg body weight. Accordingly, the pharmaceutical composition unit dosage form is formulated such that the daily dose of the ester derivative is higher than 150 mg, and even higher than 200 mg, when utilized by a human patient.
- the pharmaceutical composition unit dosage form described herein can be identified for use in acute and chronic treatment protocols, as detailed hereinabove. According to a feature of the present invention, the pharmaceutical composition unit dosage form described herein is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of a medical condition such as an inflammatory disease or disorder and cancer.
- cancer is used herein to describe the pathological process that results in the formation and growth of a neoplasm, i.e., an abnormal tissue that grows by cellular proliferation more rapidly than normal tissue and continues to grow after the stimuli that initiated the new growth ceases.
- a neoplasm i.e., an abnormal tissue that grows by cellular proliferation more rapidly than normal tissue and continues to grow after the stimuli that initiated the new growth ceases.
- Cancerous tissue exhibits partial or complete lack of structural organization and functional coordination with the normal tissue, and usually forms a distinct mass of tissue which may be benign (benign tumor) or malignant (carcinoma).
- cancer is used as a general term to describe any of various types of malignant neoplasms, most of which invade surrounding tissues, may metastasize to several sites and are likely to recur after attempted removal and to cause death of the patient unless adequately treated.
- tumor and/or cancer is used to describe all types of neoplasia, including benign and malignant.
- Exemplary cancers which may be effectively treated by the methods and compositions described herein, include, but are not limited to, brain cancer, a bone cancer, an epithelial cell- derived neoplasia (epithelial carcinoma), a basal cell carcinoma, an adenocarcinoma, a gastrointestinal cancer, a lip cancer, a mouth cancer, an esophageal cancer, a small bowel cancer, a stomach cancer, a colon cancer, a liver cancer, a bladder cancer, a pancreas cancer, an ovary cancer, a cervical cancer, a lung cancer, a breast cancer, a skin cancer, a squamus cell cancer, a prostate cancer, a renal cell carcinoma, a cancerous tumor, a growth, a polyp, an adenomatous polyp, a familial adenomatous polyposis or a fibros
- Inflammation is the complex biological response of vascular tissue to harmful stimuli, such as pathogens, damaged cells, or irritants. Inflammation can be classified as either acute or chronic. Acute inflammation is the initial response of the body to harmful stimuli and is achieved by the increased movement of plasma and leukocytes from the blood into the injured tissues. A cascade of biochemical events propagates and matures the inflammatory response, involving the local vascular system, immune system and various cells within the injured tissue. Inflammation is activated by PLA 2 , which liberates arachidonic acid, the substrate for COX enzymes. COX converts arachidonic acid to the prostaglandin PGE 2 , the major prostaglandin detected in inflammatory conditions ranging from acute edema to chronic arthritis.
- inflammatory diseases or disorders is used to describe disease or disorders which are characterized by inflammation.
- the term "inflammatory diseases or disorders” is used as a general term to describe acute inflammation as well as prolonged, chronic inflammation. Prolonged inflammation, known as chronic inflammation, leads to a progressive shift in the type of cells which are present at the site of inflammation and is characterized by simultaneous destruction and healing of the tissue from the inflammatory process. Chronic inflammation can lead to many diseases and disorders, including degenerative inflammation disorders.
- inflammatory diseases or disorders therefore encompasses also degenerative inflammation disorders.
- exemplary inflammatory diseases or disorders which are treatable by the ester derivatives described herein include, but are not limited to, Alzheimer's disease, cortical dementia, vascular dementia, muli-infract dementia, pre-senile dementia, alcoholic dementia, senile dementia, memory loss or central nervous damage resulting from stroke, ischemia or trauma, multiple sclerosis, Parkinson's disease, Huntington's disease, epilepsy, cystic fibrosis, arthritis diseases such as osteoarthritis, rheumatoid arthritis, spondyloarthopathies, gouty arthritis, systemic lupus erythematosus, and juvenile arthritis fever, periarteritis; gastrointestinal disorders such as inflammatory bowel disease, Chron's disease, gastritis, irritable bowel syndrome, ulcerative colitis, cardiovascular disorders such as myocardial ischemia, reperfusion injury to an ischemic organ; angio
- K is selected from the group consisting of O and S;
- Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
- Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino, -NR 12 R 13 , and a positively charged group, whereas R 12 and R 13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R 12 and R 13 form a five- or six-member heteroalicyclic ring;
- R 1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 and R 10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino and -NR 14 R 15 , or, alternatively, at least two of R 2 , R 3 , R 4 , R 5 and R 6 , of R 7 , R 8 , R 9 and R 10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or
- alkyl refers to a saturated aliphatic hydrocarbon including straight chain and branched chain groups.
- the alkyl group has 1 to 20 carbon atoms. Whenever a numerical range; e.g., "1-20", is stated herein, it means that the group, in this case the alkyl group, may contain 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 20 carbon atoms. More preferably, it is a medium size alkyl having 1 to 10 carbon atoms. Most preferably, it is a lower alkyl having 1 to 4 carbon atoms.
- the alkyl group may be substituted or unsubstituted.
- the substituent group can be, for example, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, halo, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N- thiocarbamyl, C-amido, N-amido, C-carboxy, O-carboxy, nitro, sulfonamido, trihalomethanesulfonamido, silyl, guanyl, guanidino, ureido or amino.
- alkenyl describes an unsaturated alkyl, as defined herein, having at least two carbon atoms and at least one carbon-carbon double bond.
- the alkenyl may be substituted or unsubstituted by one or more substituents, as described hereinabove.
- alkynyl as defined herein, is an unsaturated alkyl having at least two carbon atoms and at least one carbon-carbon triple bond.
- the alkynyl may be substituted or unsubstituted by one or more substituents, as described hereinabove.
- hydroxyalkyl describes an alkyl, as defined herein, substituted (e.g., terminated) by a hydroxy group.
- hydroxyalkenyl describes an alkenyl, as defined herein, substituted (e.g., terminated) by a hydroxy group.
- cycloalkyl describes an all-carbon monocyclic or fused ring (i.e., rings which share an adjacent pair of carbon atoms) group wherein one of more of the rings does not have a completely conjugated pi-electron system.
- examples, without limitation, of cycloalkyl groups are cyclopropane, cyclobutane, cyclopentane, cyclopentene, cyclohexane, cyclohexadiene, cycloheptane, cycloheptatriene, and adamantane.
- a cycloalkyl group may be substituted or unsubstituted.
- the substituent group can be, for example, alkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, halo, carbonyl, thiocarbonyl, C-carboxy, O-carboxy, O-carbamyl, N-carbamyl, C-amido, N- amido, nitro and amino, as defined herein.
- aryl describes an all-carbon monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups having a completely conjugated pi-electron system.
- aryl groups are phenyl, naphthalenyl and anthracenyl.
- the aryl group may be substituted or unsubstituted.
- the substituent group can be, for example, halo, trihalomethyl, alkyl, hydroxy, alkoxy, aryloxy, thiohydroxy, thiocarbonyl, C-carboxy, O-carboxy, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, sulfinyl, sulfonyl and amino, as defined herein.
- heteroalicyclic refers to a monocyclic or fused ring group having in the ring(s) one or more atoms such as nitrogen, oxygen and sulfur.
- the rings may also have one or more double bonds. However, the rings do not have a completely conjugated pi-electron system.
- the heteroalicyclic may be substituted or unsubstituted.
- the substituted group can be, for example, alkyl, cycloalkyl, aryl, heteroaryl, halo, trihalomethyl, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, carbonyl, thiocarbonyl, C-carboxy, O-carboxy, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, sulfinyl, sulfonyl, C-amido, N-amido and amino, as defined herein.
- the term "hydroxy" describes an -OH group.
- heteroaryl describes a monocyclic or fused ring (Ae., rings which share an adjacent pair of atoms) group having in the ring(s) one or more atoms, such as, for example, nitrogen, oxygen and sulfur and, in addition, having a completely conjugated pi-electron system.
- heteroaryl groups examples include pyrrole, furane, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrimidine, quinoline, isoquinoline and purine.
- the heteroaryl group may be substituted or unsubstituted.
- the substituent group can be, for example, alkyl, cycloalkyl, halo, trihalomethyl, hydroxy, alkoxy, aryloxy, thiohydroxy, thiocarbonyl, sulfonamido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, O-carbamyl, N-carbamyl, O- thiocarbamyl, N-thiocarbamyl, C-amido, N-amido and amino, as defined herein.
- halo describes fluorine, chlorine, bromine or iodine.
- alkoxy describes both an -O-alkyl and an -O-cycloalkyl group, as defined herein.
- aryloxy describes both an -O-aryl and an -O-heteroaryl group, as defined herein.
- thiohydroxy describes a -SH group.
- thioalkoxy describes both an -S-alkyl group, and an -S-cycloalkyl group, as defined herein.
- thioaryloxy describes both an -S-aryl and an -S-heteroaryl group, as defined herein.
- cyano describes a -C ⁇ N group.
- nitro describes an -NO 2 group.
- amino describes an -NH 2 or NR'R" group, with R' as defined herein and R" as defined for R'. This term further encompasses cyclic amino groups, in which R' and R" are linked therebetween to form a five- or six-membered cyclic group.
- positively charged group describes an atom or a group of atoms which forms a part of an organic molecule, and which is characterized by a positive electrostatic charge.
- Compounds which include one or more positively charged groups are molecular ions oftentimes referred to as molecular cations.
- a positively charged group of atoms has at least one electron less than the number of protons in these atoms.
- Positively charged groups include, for a non-limiting example, ammonium and sulfonium groups, as defined herein.
- ammonium group describes N + (R') 4 , wherein R' groups maybe the same or different and are as defined herein.
- sulfonium group describes S + (R') 3 , wherein R' groups maybe the same or different and are as defined herein.
- trihaloalkyl describes a -(alkyl)X3, wherein X is a halo group.
- Y is a substituted or unsubstituted alkyl.
- Ra is other than hydrogen.
- Ra is hydroxy.
- Y is a substituted or unsubstituted polyalkylene glycol moiety.
- the polyalkylene glycol moiety has a general formula II:
- each of m and n is independently an integer of 1-10.
- the Ra attached to the polyalkylene glycol moiety is, for example, hydrogen or alkyl.
- the Ra attached to the polyalkylene glycol moiety is a positively charged group, as defined herein.
- the positively charged group is an ammonium group.
- the polyalkylene glycol moiety does not include a nitroso (- ONO 2 ) group, or any other NO-releasing group, or an anti-oxidant moiety.
- G is C; K is O; each of R 2 , R 3 , R 4 , R 5 and R 6 is independently selected from the group consisting of hydrogen, alkyl, halo and trihaloalkyl; and each of R 7 , R 8 , R 9 and R 10 is hydrogen.
- N-phenylanthranilic acid is Diclofenac and the compounds described herein are ester derivatives of Diclofenac.
- R 2 and R 6 are chloro, and each of R 3 -R 5 and R 7 -R 10 is hydrogen.
- Diclofenac is a non-steroidal anti-inflammatory drug (NSAID) commonly used for reducing inflammation and as an analgesic for reducing pain in conditions such as arthritis or acute injury. The recommended dose for most conditions is 100-200 mg daily, administered orally. Dosing intervals depend on the diclofenac formulation used and the condition being treated. The most common side effects of diclofenac involve the gastrointestinal system. It can cause ulcerations, abdominal burning, pain, cramping, nausea, gastritis, and even serious gastrointestinal bleeding and liver toxicity. Rash, kidney impairment, ringing in the ears, and lightheadedness are also seen.
- Diclofenac ester derivatives include, but are not limited to:
- the present embodiments further encompass any enantiomers, diastereomers, prodrugs, solvates, hydrates and/or pharmaceutically acceptable salts of the compounds described herein.
- enantiomer refers to a stereoisomer of a compound that is superposable with respect to its counterpart only by a complete inversion/reflection (mirror image) of each other. Enantiomers are said to have "handedness” since they refer to each other like the right and left hand. Enantiomers have identical chemical and physical properties except when present in an environment which by itself has handedness, such as all living systems.
- diastereomers or “diastereoisomers”, as used herein, refer to stereoisomers that are not enantiomers with respect to one another. Diastereomers have more than one chiral center, and can have different physical properties and different reactivity. Diastereoisomers are not mirror images of each other but rather have one or more chiral centers inverted between the two stereoisomers. If a molecule exhibits two chiral centers (two asymmetric carbons), there are up to four possible stereo-configurations and hence up to four possible diastereomers.
- prodrug refers to an agent, which is converted into the active compound (the active parent drug) in vivo.
- Prodrugs are typically useful for facilitating the administration of the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent drug is not. A prodrug may also have improved solubility as compared with the parent drug in pharmaceutical compositions. Prodrugs are also often used to achieve a sustained release of the active compound in vivo.
- solvate refers to a complex of variable stoichiometry (e.g., di-, tri-, tetra-, penta-, hexa-, and so on), which is formed by a solute (the compound of the present invention) and a solvent, whereby the solvent does not interfere with the biological activity of the solute.
- Suitable solvents include, for example, ethanol, acetic acid and the like.
- hydrate refers to a solvate, as defined hereinabove, where the solvent is water.
- pharmaceutically acceptable salt refers to a charged species of the parent compound and its counter ion, which is typically used to modify the solubility characteristics of the parent compound and/or to reduce any significant irritation to an organism by the parent compound, while not abrogating the biological activity and properties of the administered compound. It is appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the invention, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable subcombination or as suitable in any other described embodiment of the invention. Certain features described in the context of various embodiments are not to be considered essential features of those embodiments, unless the embodiment is inoperative without those elements.
- Chemicals and solvents were either A.R. grade or purified by standard techniques.
- Diclofenac was purchased from Sigma-Aldrich (St. Louis, MO, USA).
- XTT reagent kit was obtained from Biological Industries, Kibbutz Bet Ha'emek.
- COX-1 purified from Bovine was obtained from Cayman Chemicals, USA.
- Arachidonic acid, Diclofenac, Indomethacin, Tris Buffer, Phenol, Trp and Hematin were obtained from Sigma.
- RPMI Rosewell Park Memorial Institute
- HBSS Hank Balanced Salt Solution
- PBS Phosphate Buffer Solution
- mice C57Black or BALB/c, weighing 20 gram on average, were obtained from the Animal facility of TeI Aviv University Medical School
- F344 female rats were obtained from the Animal facility of the Department of Psychology, TeI Aviv University
- TLC Thin layer chromatography
- Flash chromatography was performed using silica gel Merck 60 (particle size 0.040-0.063 mm) by Merck, and eluent given in parentheses.
- Spectrophotometric measurements were carried out using Spectramax 190 instrument.
- HPLC was performed on an Elite Lachrom Merck-Hitachi instrument, using a 45 % H2O (0.1 % TFA): 55% Acetaminophen as the stationary phase and a L-2450 detector, operated at 25 degrees, in a flow rate of Iml/min. Protein content assessment:
- Protein concentrations were measured by the Bradford method (1976) [Bradford M. M. 1976. Anal. Biochem. 72:248-254], using BSA as a standard.
- Diclofenac (100 mg, 0.338 mmol) was dissolved in DCM. Thereafter N-boc- diglycolamine (70 mg, 0.34 mmol) [Kim, Y. S.; Kim, K. M.; Song, R.; Jun, M. J.; Sohn, Y. S., J. Inorg. Biochem. 2001 , 87(3), 157-163] and DMAP (4 mg, 0.034 mmol) were added. To this mixture DCC (139 mg, 0.68 mmol) DCM was added dropwise. The reaction mixture was stirred at room temperature for 1 hour while monitoring by TLC (EtOAc:He, 1 :1). Upon completion of the reaction, the mixture was filtered and the solvent was removed under reduced pressure.
- CT26 cells Viability test: Mouse colon adenocarcinoma (CT26) cells were plated in 96-wells culture plates supplemented with rich medium [PLEASE INDICATE WHICH MEDIUM]. When the cells reached 70 % confluence, either Diclofenac, Compound 1 (NH6) or Compound 2 (NH17), was added at the appropriate concentration. The viability of the cultured cells was quantified using the XTT reagent test. The XTT assay is based on the ability of metabolic active cells to reduce the Tetrazolium salt XTT to orange colored compounds of formazan. The greater the number of active cells in the well, the greater the activity of mitochondria enzymes, and the higher the concentration of the dye formed.
- the plates were incubated for an hour at 37 0 C, the medium was thereafter washed and XTT reagent and an activation reagent were added. The plates were then put in incubation for additional 8 hours at 37 0 C. Thereafter, the fluorescence intensity of the wells was quantified using a spectrophotometer at a wave length of 450 nm and 690 nm. The fluorescence intensity in each well was proportional to the number of metabolic active cells.
- Cyclooxygenase inhibition assay in cell culture Mouse colon adenocarcinoma CT26 and mouse lewis lung carcinoma D122 cells were grown in DMEM (D122) or RPMI (CT26) supplemented with 10 % fetal calf serum, 4 % glutamine, 1 % penicillin-sterptomycin-nystatin and 3 % non essential amino acids (D122) and were kept in a humidified 37 0 C incubator with 5 % CO 2 . For the COX activity assays, cells were grown onto 24 multi-well plates.
- mice were sacrificed, the tumor was removed, and the stomach and kidneys were collected. All organs were homogenized in 1 ml PBS. 50 ⁇ l of the specific organ-cell suspension were kept for protein concentration determination, and the residual suspension centrifuged for 1 minute in 10,000 rpm. The supernatant, which did not contain any cells, was removed to a separate vial and the PGE 2 concentration in the supernatant was assessed by radioimmunoassay, as described hereinabove. In order to determine the overall protein concentration in the tissue, all the vials to which samples were removed were centrifuged in 12,000 rpm for 3 minutes. The supernatant was removed and 100 ⁇ l of NaOH 1 N were added to the pellet, to Iyse the cells. All samples were analyzed for protein content using the Bradford technique.
- the volume of the tumor was measured by a caliper every day starting from the day of cells injection Once the tumor has reached a volume of approximately 400 mm 3 , the mouse was sacrificed, the tumor removed, and the stomach and kidneys were collected. The last dose of the tested compound was injected 8 hours prior to organs collection. All organs were homogenized in 1 ml PBS. 50 ⁇ l of the organ-containing suspension were kept for protein concentration determination in the tissue, while the residual suspension was centrifuged for 1 minute in 10,000 rpm. The supernatant, which did not contain any cells, was removed to a separate vial and the PGE 2 concentration in the supernatant was assessed by radioimmunoassay, as described hereinabove.
- Acute footpad inflammation in F344 female rats was induced by injecting 100 ⁇ l of 1.5 % carrageenen (CR) solution (in PBS) into the right footpad.
- Control PBS solution i.e. not containing any compound
- the dimension of each footpad was measured with a caliber and the volume (in mm 3 ) was calculated according to the formula: thinckness 2 x width x 0.52.
- the rats were pre-treated with the tested compound at the indicated dose 30 minutes prior to the injection of CR.
- the tested compounds were prepared as 50 mM stock solutions in DMSO, diluted with PBS and injected intraperitoneally (100 ⁇ l per rat). The calculated results are means of 4 rats per treatment. SEM values for each mean were ⁇ 10 % of that mean. Percent inhibition of footpad inflammation was calculated as:
- NH6 and NH17 is via a mechanism of toxicity to cells In-vitro. CT26 cells were incubated with
- the observed COX inhibition activity by NH6 and NH17 may be attributed to (1) direct inhibition of COX-1 and COX-2 by NH6 and NH17 or (2) enzymatic cleavage of the ester bond in NH6 and NH17 to yield the parent Diclofenac, exhibits the anti-COX activity.
- the anti-COX activity of Compounds 1-3 was examined in Mouse colon adenocarcinoma CT26 and mouse lewis lung carcinoma D122 cell lines. Cells were incubated with each of the tested compounds and the level of the COX-dependent synthesis of PGE 2 was quantified using radioimmunoassay techniques. The results are presented in Table 3 and show that all compounds exhibited anti-COX activity comparable to or better than that of Diclofenac.
- Anti-COX activity of NH6 and NH17, at a single dose of 1-10 mpk in tumor tissue and in stomach and kidney tissues In vivo Studies Treatment of cancer with NSAIDs is problematic due to gastrointestinal and renal side effects related to the NSAID anti-COX activity in the stomach and kidney organs.
- NH6 and NH17 induced rather than inhibited COX activity, as manifested by promoting PGE 2 synthesis.
- a similar COX activity induction was not observed upon administration of Diclofenac (see, Figure 5B).
- NH6 and NH17 exhibited anti-COX activity comparable to that of Diclofenac (see Figure 5C).
- mice (10 per group) were treated as described in Example 6 hereinabove.
- the tumor volume was measured at different time points post daily injections of either Diclofenac, Indomethacin, NH6 or NH17, during a time period of 27-33 days.
- the volume of footpad without tumor cells was subtracted from all measurements.
- the results are presented in Figure 7 and show that the rate of tumor growth inhibition in mice injected with 3 mpk/day NH6 and NH17 was comparable to that of mice injected with 2mpk/day Indomethacin and significantly higher as compared to mice injected with 1 mpk/day Diclofenac.
- Table 4 below presents the extent of tumor volume reduction by the various tested compounds, 27 days post injection.
Landscapes
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Methods, unit dosage forms and compositions for treating cancer and inflammation, and which utilize ester derivatives of N-phenylanthranilic acid that exhibit tissue-selective anti-COX activity are disclosed.
Description
METHODS AND COMPOSITIONS FOR THE TREATMENT OF CANCER AND
INFLAMMATION
FIELD AND BACKGROUND OF THE INVENTION The present invention, in some embodiments thereof, relates to methods, compositions and unit dosage forms for the treatment of cancer and inflammation, and, more particularly, but not exclusively, to methods, unit dosage forms and compositions for treating cancer and inflammation which utilize compounds that exhibit tissue-selective activity.
Cancer is a leading cause of death in developed countries. Despite continuing advances in diagnosis and treatment regimens, most existing treatments have undesirable side effects and limited efficacy. Progress in this field has been hindered because a number of different cellular events contribute to the formation and metastasis of tumors, and many of these events are still not well understood.
Cyclooxygenase (COX) enzymes catalyze the conversion of arachidonic acid into prostaglandin H2 (PGH2), the early step in the biosynthesis of prostanoids. The COX enzymes are functional homodimers, in which each subunit contains both a cyclooxygenase and a peroxidase active site.
Three COX isozymes have been characterized so far; COX-1 , COX-2, and the recently discovered COX-3. While COX-1 is a ubiquitous constitutive form of the enzyme that is involved in the regulation of various physiological processes such as platelet aggregation or homeostasis of the gastrointestinal tract and kidneys, the COX-2 isozyme expression is mainly observed during inflammatory processes. COX-2 is constitutively expressed in some tissues, but can be induced by different stimuli such as growth factors or interleukin-1.
Prostanoids are naturally produced in numerous cells and tissues in mammalian systems as players in a multi-layer cell stress signaling systems designed to regulate biological processes. These processes include recruitment of blood-born inflammatory leukocytes to sites of infection, inflammation or edema (mostly via the activation of inducible COX-2 enzyme). In addition, prostanoids produced by both COX-1 and COX-2 participate in the homeostatic regulation of blood cells and blood vessels metabolic functions throughout the vascular system. Prostanoids have been found to regulate anti-platelets aggregation as well as anti-acid secretion in the stomach (COX-1). Prostanoids have also been found to be involved in the regulation of the adhesion activity of vascular endothelium cells (COX-2), and regulation of renal blood flow, urine production and salts & water retention in the kidney (COX-1 and COX-2).
Several chronic diseases, mostly associated with aging, are characterized by chronic excessive local production and secretion of prostanoids, which in turn promote the production and release of pro-inflammatory cytokines that promote angiogenesis (the generation of new blood vessels) and further exacerbate the inflammation and pain as seen in Osteoarthritis (OA) and Rheumatoid arthritis (RA). Such excessive production of prostanoids may therefore also
contribute to an accelerated progression of these diseases.
Non-steroidal anti-inflammatory drugs (NSAIDs) are organic compounds clinically used as medicines to alleviate fever, pain and inflammation associated with acute and chronic inflammatory diseases, like OA, RA and others. All of the presently known NSAIDs inhibit the COX isozymes to different extents, an activity which accounts for their anti-inflammatory and analgesic properties as well as their gastrointestinal side effects, the latter being more related to COX-1 inhibition, mg.
Since aspirin was introduced into clinical use over a century ago, numerous novel NSAIDs have been synthesized and marketed by drug companies. The major indication for general occasional use of NSAIDs is in acute pain (headache, muscle pain, etc.) whereas the indications for chronic treatment with NSAIDs are in RA and OA patients. For both patient populations the major drawbacks (adverse side effects) associated with the use of NSAIDs include initiation or exacerbation of peptic ulcer bleeding. An additional major drawback in the elderly population (which is usually the one who suffers most from OA & RA) is undesired changes in renal function that promote renal failure. In both the stomach (for peptide ulcer bleeding) and the kidney (for renal failure), the undesired side effects induced by NSAIDs have been linked to inhibition of constitutive COX activities important for these tissues homeostatic function, which result from the non-selective COX inhibition activity of these drugs. These adverse side effects have put a very heavy limitation on the chronic use of these drugs by OA & RA patients, with some having treatment halted.
The side effects induced by NSAIDs further limit the permitted dosing of these drugs. The inability to administer high doses of the NSAIDs often leads to incomplete inhibition of COX enzymes at the inflammatory sites to obtain a possible disease-modifying effect. In the past 7-8 years a group of NSAIDs selective for COX-2 (Celebrex, Vioxx) have appeared on the market with the aim of inhibiting only COX-2 (associated with most of the inflammation and pain processes) and not COX-1 (which controls stomach acid secretion). However, the recent withdrawal of diarylheterocyclic selective COX-2 inhibitors due to adverse cardiovascular side effects toned down the initial enthusiasm surrounding the launch of selective anti-COX-2 drugs. in addition to the pronounced anti-inflammatory effect of NSAIDs, recent studies on cancer and Alzheimer animal models in rodents have indicated that treatment with NSAIDs can attenuate the progression of these diseases too. Epidemiological data in human have supported these findings. The anti-tumor activity of NSAIDs (such as Diclofenac) has been attributed to their anti-COX activity and inhibition of prostaglandin synthesis [see, for example, Brown and DuBois Clinical Cancer Research 2004; 10: 4266s-4269s]. Furthermore, the anti- carcinogenic effect of NSAIDs in a rodent cancer model was shown to be directly related to the dose of the drug. For example, Celecoxib (Celebrex) is a highly selective COX-2 inhibitor which
has been shown to be chemopreventive, in a dose dependent manner, in azoxymethane- induced colon cancer model in rats, in spontaneous adenomas of the small intestine in mice and in bladder cancer models in mice [Steele et al, Mutation Res. 2003; 523-524: 137-144].
The scientific data on COX-2 selective NSAIDs (e.g. Celebrex) suggest that a daily basis administration of these drugs may be of therapeutic benefit, as a prophylactic treatment, against the development of cancer and inflammation-based degenerative diseases like Alzheimer and RA. However, the recent accumulating negative data on this class of compounds regarding their adverse cardiovascular side effects, have limited their use in ameliorating existing disease conditions as well as in preventive, prophylactic treatments. Daily intake of a low dose of aspirin is used in the general population with age over 60 to reduce the risk of a thrombotic event leading to a heart attack. Yet still some patients are stomach-sensitive even to this low dose.
In addition, it has been recognized that inhibition of COX enzymes in the heart muscle may be of therapeutic benefit. However, the use of NSAIDs to this effect is limited sue to their side effects.
Presently, the recommended daily dose of NSAIDs is limited by the adverse side effects induced thereby. Thus, for example, for the treatment of inflammatory related disease conditions, the recommended daily dose of NSAIDs, such as Diclofenac or Indomethacin, is 100-150 mg. The highest recommended Diclofenac and Indomethacin daily dose is 200 mg. For example, for the treatment of juvenile chronic arthritis, Diclofenac is administered at an oral dose of 1-3 mg/kg body weight/day in divided doses. For the treatment of osteoarthritis, the recommended dosage is 100-150 mg/day. For the treatment of rheumatic arthritis Indomethacin is administered at a total daily dose of 150-200 mg.
U.S. Patent Application No. 11/110,669, having Publication No. 20050250833, by some of the present inventors, teaches a family of N-phenylanthranilic acid derivatives for use as potassium channel modulators. Some of the compounds taught in U.S. Patent Application No. 11/110,669 are ester derivatives of phenylanthranilic acid such as Diclofenac and Meclofenamic acid, in which the acid functional group of these compounds has been esterified. These ester derivatives have shown to exhibit COX inhibition activity. U.S. Patent Application No. 10/612,014 having Publication No. 20040024057, teaches nitrosated NSAIDs for treating gastrointestinal disorders, for facilitating wound healing, for treating and/or preventing gastrointestinal, renal and/or respiratory toxicity resulting from the use of NSAIDs, for treating inflammatory disease states and/or preventing ophthalmic diseases. The compounds taught in U.S. Patent Application No. 10/612,014 comprise a nitroso-containing alkyl moiety which is linked to the NSAID via an amide or ester bond. The compounds taught in this patent application are aimed at releasing NO.
Esterified NSAIDs have also been described in U.S. Patent No. 4,851 ,426 and in U.S. Patent Application Nos. 10/767,581 and 10/678,430.
U.S. Patent No. 4,851 ,426 teaches 1-ethoxycarbonyloxyethyl esters of NSAIDs possessing increased bioavailability and reduced ulceroginicity when administered orally as compared to the NSAIDs with a free carboxylic function from which they are derived. The NSAID esters taught by U.S. Patent No. 4,851 ,426, include esters of aspirin, indomethacin, naproxen, ibuprofen, sulindac, diflusinal, ketoprofen, mefenamic acid, tolmetin, Diclofenac and flufenamic acid.
U.S. Patent Application No. 10/767,581 having Publication No. 20050004118, teaches ester derivatives of NSAID of the formula RC(O)O-spacer-OC(O)R' wherein RC(O)- is the acyl residue of an NSAID bearing a carboxylic acid function and R' is a substituted or unsubstituted heteroaryl or heterocyclic. The NSAID ester derivatives described in this patent application are for use in the treatment of inflammation and inflammatory disorders and other disease states. U.S. Patent Application No. 10/767,581 further teaches a method for reducing gastrointestinal side effects associated with NSAIDs by masking the carboxyl function of the drug to prevent localization of the drug in the gastric mucosa. According to the teachings of U.S. Patent Application No. 10/767,581 , the NSAID ester derivatives taught therein are prodrugs which are less toxic to the gastrointestinal system than the native drug and, when administered orally, are absorbed from the gut into the blood stream where they liberate their corresponding parent drugs or exhibit independent pharmacological activity.
U.S. Patent Application No. 10/678,430 having Publication No. 20040067914 teaches esters of an R-enantiomer of NSAIDs, which is substantially free from the S-enantiomer. According to the teachings of this patent application, the compounds described therein may be used in treating a disease or illness in a mammal such as inflammation, cystic fibrosis, dementia, Alzheimer's disease, Parkinson's disease and neoplastic disease, whereby administration of compositions including R-isomers of NSAIDs, which are substantially free of the S-enantiomer of the selected NSAIDs, is accompanied by a significant reduction in NSAID associated adverse effects. The esterifying agents to be linked to the NSAID, taught by this patent application, include glycerol, propylene glycol, hydroxysuccinic acid, hydroxyglutamic acid, glyceric acid, tartaric acid, xylaric acid, maleic acid, lactic acid, hydroxybutyric acid and ascorbic acid.
SUMMARY OF THE INVENTION
Although NSAIDs are widely used drugs, there are no "safe NSAIDS" on the market today, and the use of NSAIDs, chronic use in particular, is limited by the non-selective COX inhibition activity of these drugs, which is often associated with adverse side effects. Currently marketed NSAIDs do not reach their full potential in becoming disease-modifying drugs because their dose is limited by their side effects.
The present inventors have surprisingly uncovered that esters of N-phenylanthranilic acid, and of derivatives thereof, exhibit tissue-selective COX inhibition activity and hence can be
used as NSAIDs for the treatment of neoplastic and inflammation related diseases yet their associated gastrointestinal and renal side effects are significantly reduced.
The tissue-selective COX inhibition activity of these compounds allows using dosage forms of these drugs, which are higher than those used for non-modified NSAIDs (having nonselective COX inhibition activity that leads to adverse side effects), thus rendering these compounds highly suitable for use in the treatment of acute conditions, as well as chronic conditions, associated with neoplasia and/or inflammation.
According to an aspect of some embodiments of the present invention there is provided a pharmaceutical composition in unit dosage form comprising an ester derivative of N- phenylanthranilic acid having the general Formula I:

Formula I wherein:
Z is an A-C(=K)-O-Y-Ra group;
A is alkyl or absent;
K is selected from the group consisting of O and S;
Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino, -NR12R13, and a positively charged group, whereas R12 and R13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R12 and R13 form a five- or six-member heteroalicyclic ring;
R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R2, R3, R4, R5, R6, R7, R8, R9 and R10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy,
sulfinyl, sulfonyl, cyano, nitro, amino and -NR14R15, or, alternatively, at least two of R , R , R , R5 and R6, of R7, R8, R9 and R10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or heteroalicyclic ring; and
R14 and R15 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R14 and R15 form a five- or six- member heteroalicyclic ring, and a pharmaceutically acceptable carrier, wherein an amount of the ester derivative is at least 1 rng/kg body weight, the unit dosage form being identified for use, from 1 to 4 times daily, in the treatment of a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
According to some embodiments of the invention, Y is selected from the group consisting of a substituted or unsubstituted alkyl and a substituted or unsubstituted polyalkylene glycol moiety.
According to some embodiments of the invention, the polyalkylene glycol moiety has a general formula II:
[(CH2)m-0]n-
Formula Il wherein: each of m and n is independently an integer of 1-10.
According to some embodiments of the invention, Ra is hydrogen. According to some embodiments of the invention, Ra is alkyl. According to some embodiments of the invention, Ra is a positively charged group. According to some embodiments of the invention, the positively charged group is an ammonium group.
According to some embodiments of the invention, G is C; K is O; each of R2, R3, R4, R5 and R6 is independently selected from the group consisting of hydrogen, alkyl, halo and trihaloalkyl; and each of R7, R8, R9 and R10 is hydrogen.
According to some embodiments of the invention, the N-phenylanthranilic acid is Diclofenac.
According to some embodiments of the invention, the ester derivative is selected from the group consisting of:

and
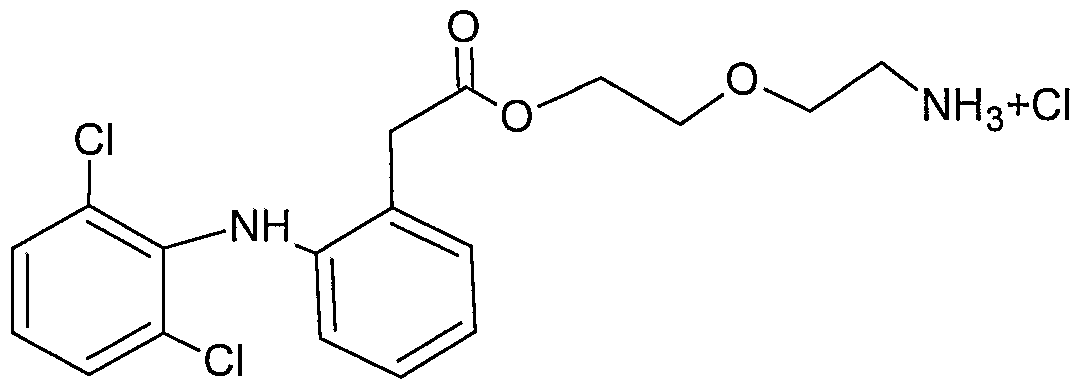
According to another aspect of some embodiments of the present invention there is provided a pharmaceutical composition in a unit dosage form comprising an ester derivative of N-phenylanthranilic acid having the general Formula I, as described hereinabove, wherein an amount of the ester derivative is at least 50 mg, the unit dosage form being identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
According to another aspect of some embodiments of the present invention there is provided a pharmaceutical composition unit dosage form comprising an ester derivative of N- phenylanthranilic acid having the general Formula I, as described hereinabove, wherein an amount of the ester derivative is at least 1 mg/kg body weight, the unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to 4 times daily, in the treatment of a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
According to another aspect of some embodiments of the present invention there is provided a pharmaceutical composition unit dosage form comprising an ester derivative of N- phenylanthranilic acid having the general Formula I, as described hereinabove, wherein an amount of the ester derivative is higher than 50 mg, the unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
According to some embodiments of the invention, in the unit dosage form the amount of
the ester derivative is at least 2 mg/kg body weight.
According to some embodiments of the invention, in the unit dosage form the amount of the ester derivative is at least 3 mg/kg body weight.
According to some embodiments of the invention, in the unit dosage form the amount of the ester derivative is at least 6 mg/kg body weight.
According to some embodiments of the invention, in the unit dosage form the amount of the ester derivative ranges from 1 mg/kg body weight to 10 mg/kg body weight.
According to some embodiments of the invention, in the unit dosage form the amount of the ester derivative is higher than 70 mg. According to some embodiments of the invention, the composition is used 2 to 4 times a day.
According to some embodiments of the invention, the composition is identified for use such that a daily dose of the ester derivative is at least 2 mg/kg body weight.
According to some embodiments of the invention, the composition is identified for use such that a daily dose of the ester derivative is at least 3 mg/kg body weight.
According to some embodiments of the invention, the composition is identified for use such that a daily dose of the ester derivative is higher than 150 mg.
According to some embodiments of the invention, the composition is identified for use such that a daily dose of the ester derivative is higher than 200 mg. According to some embodiments of the invention, the composition is formulated for intraperitoneal administration.
According to some embodiments of the invention, the composition is identified for use such that a daily dose of the ester derivative is higher than 1 mg/kg body weight.
According to some embodiments of the invention, the composition is identified for use such that a daily dose of the ester derivative is higher than 50 mg.
According to some embodiments of the invention, the treatment is a chronic treatment.
According to some embodiments of the invention, the composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of the medical condition. According to some embodiments of the invention, the composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of cancer and an inflammatory disease or disorder.
According to another aspect of some embodiments of the present invention there is provided a method of treating a medical condition selected from the group consisting of cancer and an inflammatory disease or disorder in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of an ester derivative of N- phenylanthranilic acid having a general Formula I, as described hereinabove,
According to some embodiments of the invention, the therapeutically effective amount is
at least 1 mg/kg body, and the administering is performed from 1 to 4 times a day.
According to some embodiments of the invention, the therapeutically effective amount is higher than 50 mg, and the administering is performed from 1 to 4 times a day.
According to some embodiments of the invention, the administering is effected intraperitoneal^, the therapeutically effective amount is higher than 1 mg/kg body, and the administering is performed from 1 to 4 times a day.
According to some embodiments of the invention, the administering is effected intraperitoneally, the therapeutically effective amount being higher than 50 mg, and the administering is performed from 1 to 4 times a day. According to some embodiments of the invention, therapeutically effective amount is at least 2 mg/kg body weight.
According to some embodiments of the invention, the therapeutically effective amount is at least 3 mg/kg body weight.
According to some embodiments of the invention, the therapeutically effective amount is at least 6 mg/kg body weight.
According to some embodiments of the invention, the therapeutically effective amount is ranges from 1 mg/kg body weight to 10 mg/kg body weight.
According to some embodiments of the invention, the therapeutically effective amount is higher than 70 mg. According to some embodiments of the invention, the administering is performed from 2 to 4 times a day.
According to some embodiments of the invention, a daily dose of the ester derivative is higher than 2 mg/kg body weight.
According to some embodiments of the invention, a daily dose of the ester derivative is higher than 3 mg/kg body weight.
According to some embodiments of the invention, a daily dose of the ester derivative is higher than 150 mg.
According to some embodiments of the invention, a daily dose of the ester derivative is higher than 200 mg. According to some embodiments of the invention, the administering is performed intraperitoneally.
According to another aspect of some embodiments of the present invention there is provided a use of an ester derivative of N-phenylanthranilic acid having a general Formula I, as described hereinabove, in the manufacture of a medicament for treating a medical condition selected from the group consisting of cancer and an inflammatory disease or disorder.
According to some embodiments of the invention, the treating of the medical condition comprises chronic treatment.
According to some embodiments of the invention, the chronic treatment comprises administering the ester derivative for a time period that ranges from 20 to 40 days.
According to some embodiments of the invention, the medical condition is cancer.
According to some embodiments of the invention, the inflammatory disease or disorder is selected from a group consisting of Alzheimer's disease, cortical dementia, vascular dementia, muli-infract dementia, pre-senile dementia, alcoholic dementia, senile dementia, memory loss or central nervous damage resulting from stroke, ischemia or trauma, multiple sclerosis, Parkinson's disease, Huntington's disease, epilepsy, cystic fibrosis, arthritis diseases such as osteoarthritis, rheumatoid arthritis, spondyloarthopathies, gouty arthritis, systemic lupus erythematosus, and juvenile arthritis fever, periarteritis; gastrointestinal disorders such as inflammatory bowel disease, Chron's disease, gastritis, irritable bowel syndrome, ulcerative colitis, cardiovascular disorders such as myocardial ischemia, reperfusion injury to an ischemic organ; angiogenesis, asthma, bronchitis, menstrual cramps, premature labor, tendinitis, bursitis, an autoimmune disease, an immunological disorder, systemic lupus erythematosus, inflammatory disorders of the skin such as psoriasis, eczema, burns and dermatitis; neoplasia, an inflammatory process in a disease, pulmonary inflammation, a central nervous system disorder, migraine headaches, allergic rhinitis, respiratory distress syndrome, endotoxin shock syndrome, a microbial infection, a bacterial-induced inflammation, a viral induced inflammation, a urinary disorder, a urological disorder, endothelial dysfunction, organ deterioration, tissue deterioration, adhesion and infiltration of neutrophils at the site of inflammation, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, rheumatic fever, myasthenia gravis, sarcoidosis, nephrotic syndrome, Behcet's syndrome, polymyositis, hypersensitivity, conjunctivitis, gingivitis and swelling occurring after injury.
Unless otherwise defined, all technical and/or scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which the invention pertains. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of embodiments of the invention, exemplary methods and/or materials are described below. In case of conflict, the patent specification, including definitions, will control. In addition, the materials, methods, and examples are illustrative only and are not intended to be necessarily limiting.
BRIEF DESCRIPTION OF THE DRAWINGS
Some embodiments of the invention are herein described, by way of example only, with reference to the accompanying drawings. With specific reference now to the drawings in detail, it is stressed that the particulars shown are by way of example and for purposes of illustrative discussion of embodiments of the invention. In this regard, the description taken with the drawings makes apparent to those skilled in the art how embodiments of the invention may be practiced.
In the drawings:
FIG. 1 presents the 2-D chemical structures of exemplary compounds according to some embodiments of the present invention.
FIGs. 2A-C present plots showing the viability level of CT26 cells, incubated with Diclofenac (Figure 2A), NH6 (Figure2B) and NH17 (Figure 2C), as a function of the compound's concentration. Mouse colon adenocarcinoma (CT26) cells were incubated for an hour at 37 0C, with the tested compound, at various inhibitor concentrations, and the number of viable cells was quantified using an XTT reagent test. The fluorescence intensity, measured using a spectrophotometer, in each cell culture was proportional to the number of metabolic active cells. The viability of the cells was significantly decreased only when the cells were exposed to Diclofenac at a concentration above 1 mM; to NH6 at a concentration of above 300 μM; and to NH17 at a concentration of above 100 μM.
FIGs. 3A-B present plots showing the spontaneous decomposition of NH6 (Figure 3A) and NH17 (Figure 3B) to Diclofenac as a function of time. NH6 or NH17 were diluted to a concentration of 500μM in RPMI medium and subjected to HPLC analysis after 0, 5, 10, 15, 20, 25 and 30 hours. The amount of acid (Diclofenac) was quantified for every time point.
FIGs. 4A-B present bar graphs depicting the ability of Diclofenac, NH6 and NH17 to inhibit the COX-1 dependent (Figure 4A) or COX-2 dependent (Figure 4B), prostaglandin PGE2 synthesis from Arachidonic acid. 20 enzyme units of Bovine COX-1 or human recombinant COX-2 purified enzymes were added to a reaction vial containing either Diclofenac (1 μM), NH6 (1μM or 3 μM in the experiments with COX-1 and COX-2, respectively) or NH17 (1μM or 3 μM in the experiments with COX-1 and COX-2, respectively). 15μl of 10μM Arachidonic acid was added to each vial. After 20 minutes, 5μM of lndomethacin was added to the reaction mixture in order to stop prostaglandin synthesis. PGE2 levels, as a measure of COX activity, were quantified using radioimmunoassay techniques. A vial to which no COX inhibitor was added (marked control) and a vial to which no Arachidonic acid was added (marked AA) served as a control. The results show that in the presence of NH6 and NH17 the synthesis of PGE2 by COX-1 and COX-2 was significantly inhibited.
FIGs. 5A-C present bar graphs showing the anti-COX activity of NH6 and NH17, as compared to Diclofenac, in tumor, stomach and kidney tissues of mice, upon administration at a single dose of 1 , 3 or 10 mg per kg body weight. Tumor growth was induced by CT26 cells injection into the right hind foot of a BALB/c mouse. Once the tumor has reached the volume of approximately 400 mm3 (21-24 days post injection) 1 , 3 or 10 mg per kg (mpk) of Diclofenac, NH6 or NH17 was injected intraperitoneal^ (I. P.) to each mouse in a single dose. 3 hours after the injection, the tumor was removed, and the stomach and kidneys were collected. The extent of PGE2 synthesis as a measure of COX activity was assessed by quantifying PGE2 levels by radioimmunoassay, in each organ, in the presence of the tested compound, as compared to PGE2 levels in tumor, stomach and kidney tissue from control, non-treated mice. Diclofenac,
NH6 and NH17 each inhibited COX activity in tumor tissue to a similar extent (see, Figure 5A). In the stomach, NH6 and NH17 inhibited COX activity significantly less than Diclofenac (see, Figure 5B). In the kidney, at a concentration of 1 mg per kilogram, NH6 and NH17 inhibited COX activity significantly less than Diclofenac (see, Figure 5C). FIGs. 6A-C present bar graphs showing the COX inhibition (anti-COX) activity of NH6 and NH17 when administered at a chronic dose of 3 mg per kg/day, as compared to lndomethacin administered at a chronic dose of 1 mg per kg/day and Diclofenac administered at a chronic dose of 2 mg per kg/day in tumor, stomach and kidney tissue in mice. Tumor growth was induced by CT26 cells injection into the right hind foot of a BALB/c mouse. Starting from the day of cells injection, the mice were injected intraperitoneally with 100μl of 1 mg per kg lndomethacin or 2 mg per kg Diclofenac or 3 mg per kg NH6 or NH17 every 24 hours for 27-33 days. When the tumor reached a volume of approximately 400 mm3, the mouse was sacrificed, the tumor was removed, and the stomach and kidneys were collected. The extent of PGE2 synthesis as a measure of COX activity was assessed by quantifying PGE2 levels by radioimmunoassay, in each organ, in the presence of the tested compound, as compared to PGE2 levels in tumor, stomach and kidney tissue from control non-treated mice. The results show that NH6 and NH17 inhibit COX activity in tumor tissue to a greater extent as compared to Diclofenac and to a similar extent as compared to lndomethacin (see Figure 6A). In the stomach (Figure 6B) and kidney (Figure 6C) NH6 and NH17 inhibited COX activity to a much lesser extent as compared to Diclofenac and lndomethacin.
FIG. 7 presents comparative plots showing the tumor size as a function of days post intraperitoneal injection of lndomethacin (filled squares), Diclofenac (filled circles), NH6 (filled triangles) or NH17 (horizontal line) to tumor baring mice and compared to tumor size in control, not-treated tumor baring mice (filled diamonds). CT26 cells suspended in HBSS buffer were injected to the right hind foot of BALB/c mice. Starting from the day of cells injection, the mice were injected I. P. with 100 μl of 1 mg per kg lndomethacin or 2 mg per kg Diclofenac or 3 mg per kg NH6 or 3 mg NH17 every 24 hours for 27-33 days. The volume of the tumor was measured every day starting from the day of cells injection. The rate of tumor growth inhibition in mice injected with 3 mg per kg/day NH6 and NH17 was comparable to that of mice injected with 2 mg per kg/day lndomethacin and significantly higher as compared to mice injected with 1 mg per kg /day Diclofenac.
DETAILED DESCRIPTION OF EMBODIMENTS OF THE INVENTION
The present invention, in some embodiments thereof, relates to methods, compositions and unit dosage forms for the treatment of cancer and inflammation, and, more particularly, but not exclusively, to methods, compositions and unit dosage forms for treating cancer and inflammation which utilize compounds that exhibit tissue-selective activity.
Before explaining at least one embodiment of the invention in detail, it is to be
understood that the invention is not necessarily limited in its application to the details set forth in the following description or exemplified by the Examples. The invention is capable of other embodiments or of being practiced or carried out in various ways.
Non-steroidal anti-inflammatory drugs (NSAIDs) are organic compounds clinically used as medicines to alleviate fever, pain and inflammation associated with acute and chronic inflammatory conditions.
As discussed hereinabove, scientific data from studies on cancer and Alzheimer animal models in rodents indicate that treatment with NSAIDs can attenuate the progression of these diseases. Epidemiological data in human further support these findings. The anti-carcinogenic effect of NSAIDs in a rodent cancer model was shown to be directly related to the dose of the drug.
The anti-tumor activity of NSAIDs (such as Diclofenac) has been attributed to their anti- COX activity and their activity in inhibiting prostaglandin synthesis [see, for example, Brown and DuBois Clinical Cancer Research 2004; 10: 4266s-4269s]. Although NSAIDs are widely used drugs, their administration, and particularly their chronic administration, is limited by the adverse side effects induced thereby, gastrointestinal and renal side effects in particular.
These adverse side effects are attributed, at least in part, to the non-selective COX inhibition activity of NSAIDs. Commonly observed NSAID-related gastrointestinal side effects include ulceration, haemmorrhage, perforation and death. NSAID-related gastrointestinal adverse effects are mainly due to a reduction in the levels of cytoprotective, prostaglandins PGE2 and PGI2 in the stomach lining, which further leads to a reduced mucous secretion and impaired protection of the stomach against acidic insults.
Renal NSAID-related adverse effects are also related to inhibition of the COX- dependent prostaglandin synthesis.
Prostaglandins normally cause vasodilatation of the afferent arterioles of the glomeruli, which is crucial for the normal glomerular perfusion and glomerular filtration rate (GFR).
Reduction in prostaglandin levels, due to NSAID administration, alters renal function, and leads to complications such as hypertension, salt and fluid retention and in some cases interstitial nephritis, nephritic syndrome acute renal failure and acute tubular necrosis.
In U.S. Patent Application No. 11/110,669, having Publication No. 20050250833, ester derivatives of N-phenylanthranilic acid, and of derivatives thereof, have been taught. Some of these ester derivatives were tested and found to exhibit COX inhibition activity.
The present inventors have now further tested the anti-COX activity of ester derivatives of N-phenylanthranilic acid and have surprisingly uncovered that these compounds exhibit a therapeutic activity that is superior to the parent, non-esterified, N-phenylanthranilic acid.
Specifically, the present inventors have now uncovered that these ester derivatives exhibit tissue-selective anti-COX activity, namely, they exhibit a profound anti-COX activity in
tumor tissues and inflammatory sites yet reduced anti-COX activity in stomach and kidney tissues. In fact, it was found that at some dose regimens, these ester derivatives cause an elevation rather than inhibition of COX activity in, for example, stomach tissue.
As detailed in the Example section that follows, the present inventors have shown that at a single dosage administration of 1-10 mg per kilogram body weight, Compound 1 (also referred to herein interchangeably as NH6) and Compound 2 (also referred to herein interchangeably as NH17), which are ester derivatives of the N-phenylanthraniiic acid
Diclofenac, exhibit a potent anti-tumor effect due to their anti-COX activity. Surprisingly, in contrast to the parent, non-esterified compound Diclofenac, NH6 and NH17, at a concentration of 1-10 mg per kilogram, did not inhibit activity of COX enzymes in the stomach but rather induce COX activity (see, Example 5).
Thus, since the gastrointestinal adverse effects of NSAIDs are related to the inhibition of prostaglandin synthesis, it is deduced that an administration of NH6 and NH17, at a dosage of 1-10 mg per kilogram body weight, to a subject suffering from cancer, results in amelioration of the disease with significantly reduced stomach-related adverse effects as compared to administration of a similar dosage of the parent NSAID Diclofenac.
Accordingly, these findings show that ester derivatives of N-phenylanthranilic acid can be safely used at a dosage regimen higher than that of their parent compounds (e.g., Diclofenac), a dosage regimen which can result in a pronounced therapeutic effect without the risk of increased adverse side effects.
As further detailed in the Examples section that follows, at a chronic administration of 3 mg per kilogram/day, for a time period of 27-33 days, NH6 and NH17 were found to be therapeutically superior to the known NSAIDs lndomethacin and Diclofenac, administered at a similar dosage range (1 mpk/day and 2 mpk/day, respectively; see, Example 6), in the treatment of inflammation.
In the case of lndomethacin and Diclofenac, the inhibition of prostaglandin synthesis in the tumor tissue, as a manifestation of the inhibition of COX activity, is accompanied by a similar inhibition of prostaglandin synthesis in the stomach and kidney. Thus, chronic administration of
Diclofenac and lndomethacin at the tested dosage regimen leads to renal and stomach adverse effects caused by the COX inhibition in these organs.
In contrast, it was found that a daily dosage of 3 mg per kilogram body weight of NH6 or
NH17 also inhibits prostaglandin synthesis but, the extent of inhibition of prostaglandin synthesis in stomach and kidney tissue is significantly reduced as compared to lndomethacin and Diclofenac, thus rendering a chronic administration of these compounds, at relatively high dosages, applicable.
The present inventors have further shown that the anti-tumor activity of NH6 and NH17 (as determined by observing the reduction in tumor size) was superior to that of Diclofenac and comparable to that of lndomethacin (see, Example 7).
As further detailed in the Examples section that follows, in assays conducted in a rat model of inflammation, NH6 and NH17, at a single dosage of 2 mg per kilogram body weight and of 6 mg per kilogram body weight, exhibit anti-inflammatory activity which is comparable to that of Diclofenac and lndomethacin administered at a same dosage (see, for example, Table 5 and Example 8.).
The conversion ratio between rat NSAID dosage and human NSAID dosage is known to be [rat dosage x 3 / 2 = human dosage]. Thus, the equivalence of the NH6, NH17, Diclofenac and lndomethacin dosages administered in the current experiments, in a human subject are dosages of 3 mg per kg and 9 mg per kg. Taking the average human weight as being about 70 kg, such dosages are equivalent to 210 mg per day and 630 mg per day. For the treatment of inflammatory related disease conditions, the recommended daily dose of NSAIDs, such as Diclofenac or lndomethacin, is 100-150 mg and the highest recommended Diclofenac and lndomethacin daily dose is 200 mg. Thus, the dosages ,of Diclofenac and lndomethacin, administered to rats in the current experiments were well above the recommended daily dosage for humans and indicate that the tested compounds can be safely utilized at such doses since no adverse side effects are iduced thereby.
As detailed hereinabove, at a single administration of 1-10 mg per kilogram weight, NH6 and NH17 enhance rather than reduce COX activity in the stomach. Thus, at therapeutically relevant dosages, NH6 and NH17 are superior to Diclofenac in the treatment of inflammation. Accordingly, according to an aspect of embodiments of the present invention there is provided a method of treating a medical condition such as cancer and an inflammatory disease or disorder. The method, according to this aspect of the present invention, is effected by administering to a subject in need thereof a therapeutically effective amount of an ester derivative of N-phenylanthranilic acid, as described herein. Further according to the present embodiments, there is provided a use of an ester derivative of N-phenylanthranilic acid as described herein, in the manufacture of a medicament for treating a medical condition such as an inflammatory disease or disorder and cancer.
Herein throughout the ester derivatives of N-phenylanthranilic acid are also referred to interchangeably as "ester derivatives" or simply as "compounds". As discussed hereinabove and is further demonstrated in the Examples section that follows, the ester derivatives described herein were found highly effective in the treatment of both cancer and inflammation.
Moreover, the ester derivatives described herein were found to exhibit a tissue-selective anti-COX activity, such that they exert COX inhibition activity at the tumor tissue or inflammation site but, they do not affect COX activity, or, induce COX activity, at stomach and kidney tissues. The anti-cancer and anti-inflammatory activities of the ester derivatives described herein were shown to be at least comparable if not superior to common NSAIDs such as Diclofenac and lndomethacin, whereby the lack of COX inhibition activity in the stomach and kidney was
observed only for the ester derivatives described herein and was not observed with the NSAIDs. These unique features of the compounds described herein enable to use these compounds as therapeutic agents as dosages that are higher than the dosages commonly practiced with common NSAIDs. The phrase "COX inhibition activity" describes the ability of a compound to inhibit cyclooxygenase, a family of isoenzymes responsible for the biosynthesis of protaglandins. The inhibition may be either of one of the three cyclooxygenase isoenzymes, namely cyclooxygenase-1 (COX-1 ), cyclooxygenase-2 (COX-2) and cyclooxygenase-3 (COX-3), or a combined inhibition of two or all three cyclooxygenase isoenzymes. The inhibition can be by any available pathway including for example, direct or allosteric inhibition, competitive or noncompetitive inhibition. The phrase "COX inhibition activity" is also referred to herein interchangeably as "anti-COX activity".
The phrase "a therapeutically effective amount", which is also referred to herein interchangeably as "pharmaceutically effective amount", as used herein, describes an amount of an active ingredient needed to achieve the desired outcome, which is generally to prevent, alleviate or ameliorate a condition or symptoms of the condition. Determination of a therapeutically effective amount is within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
A "therapeutically effective amount" is referred to herein as a single dose, which can be administered once daily or a few times (e.g., 2, 3 or 4 times) daily. A "single dose" refers to a single event of administration at a certain time point.
The single dose can be a single unit dosage form, a portion of a unit dosage form, or a number of unit dosage forms, depending on the unit dosage form available. If two or more unit dosage forms (e.g., two tablets or capsules) are taken together, at the same time point within a day, they are referred to herein as a single dose. A single dose also encompasses a continuous administration, such as, for example, a continuous intravenous administration.
Thus, in some embodiments, a therapeutically effective amount of the compounds described and utilized according to these aspects of the present embodiments, indudes a single dose that is higher than 1 mg/kg body weight, which is utilized in a regimen of 1 to 4 times a day.
The therapeutically effective amount of the compounds utilized in these aspects of the present embodiments can therefore be, for example, 2 mg/kg body weight, 3 mg/kg body weight, 4 mg/kg body weight, 5 mg/kg body weight, 6 mg/kg body weight, 7 mg/kg body weight, 8 mg/kg body weight, 9 mg/kg body weight or 10 mg/kg body weight, 11 mg/kg body weight, 12 mg/kg body weight, 13 mg/kg body weight, 14 mg/kg body weight, 15 mg/kg body weight, and even higher.
In one embodiment, the therapeutically effective amount is 2 mg/kg body weight. In another embodiment, the therapeutically effective amount is 3 mg/kg body weight. In yet
another embodiment, the therapeutically effective amount is 6 mg/kg body weight.
In some embodiments, the therapeutically effective amount ranges from 1 mg/kg body weight to 10 mg/kg body weight.
In a patient weighing 70 kg (an average human body weight), the above-described amounts correspond to dosages of 70 mg , 140 mg, 210 mg, 280 mg, 350 mg, 420 mg, 490 mg, 560 mg, 630 mg, 700 mg, 770 mg, 840 mg, 910 mg, 980 mg, 1050 mg, respectively, and higher.
In a child weighing 20 kg, these amounts correspond to dosage administration of 20 mg , 40 mg, 60 mg, 80 mg, 100 mg, 120 mg, 140 mg, 160 mg, 180 mg, 200 mg, 220 mg, 240 mg, 260mg, 280 mg, 300 mg, respectively, and higher.
Thus, in some embodiments, the compounds described herein are administered in a therapeutically effective amount higher than 20 mg, administered from 1 to 4 times a day. In some embodiments, the therapeutically effective amount is higher than 50 mg. In some embodiments, the therapeutically effective amount is higher than 70 mg. The therapeutically effective dose may be administered once daily, or can be divided and administered from 2 to 6 times daily. In some embodiments, the therapeutically effective amount is divided and is administered, for example, twice daily, 3 times daily or 4 times daily.
For example, to a patient weighing 70 mg, a therapeutically effective amount of 2 mg/kg body weight, may be administered once daily as a 140 mg single dose or as a dose of 70 mg administered twice daily or a dose of 47 mg administered three times per day or a dose of 35 mg administered four times per day. To a child weighing 20 mg, a therapeutically effective amount of 3 mg/kg body weight, may be administered once daily as a 60 mg single dose or a dose of 30 mg administered twice daily or a dose of 20 mg administered three times per day or a dose of 15 mg administered four times per day. Alternatively, the compounds described herein may be formulated for intravenous infusion and be administered in a continuous fashion, for a time period of a few minutes to a few hours, a day.
The administration protocol of the compounds described herein can include a single administration or a few subsequent administrations, as part of a treatment protocol for treating an acute condition (e.g., pain). Alternatively, the administration protocol can include a chronic treatment protocol, for treating chronic conditions (e.g., RA).
A chronic treatment includes, for example, a daily administration of an ester derivative as described herein, for a time period of 20 (consecutive) days or more. For example, the ester derivative as described herein may be administered during a time period of 20 days, 21 days, 22 days, 23 days, 24 days, 25 days, 26 days, 27 days, 28 days, 29 days, 30 days, 31 days, 32 days, 33 days, 34 days, 35 days, 36 days, 37 days, 38 days, 39 days, 40 days and even more.
In one embodiment, the time period for utilizing the ester derivatives described herein in chronic treatment ranges from 27 to 33 days. In one example, an ester derivative as described
herein may be administered at a therapeutically effective amount of 3 mg/kg body weight a day, for 27 days.
In some embodiments, the ester derivatives described herein are administered at a daily dose higher than 2 mg/kg body weight. In some embodiments, the ester derivatives described herein are administered at a daily dose higher than 3 mg/kg body weight.
Accordingly, in some embodiments, the ester derivatives described herein are administered, to a human patient (weighing 70 grams) at a daily dose higher than 150 mg, and even higher than 200 mg. The ester derivatives described herein may be administered via various routes of administration. These include, as non-limiting examples, oral, transdermal, intraperitoneal, sublingual, intravenous and rectal routes or by inhalation.
In one embodiment, the ester derivatives are administered intraperitoneally. Intraperitoneal administration refers to the injection of the compounds described herein through the peritoneum into the peritoneal cavity. The peritoneum is a thin, transparent membrane that lines the walls of the abdominal (peritoneal) cavity and contains/encloses the abdominal organs such as the stomach and intestines. The administration of drugs via injection to peritoneal cavity is a well known administration route. For example, clinical investigators have confirmed the safety and pharmacokinetic advantage associated with the intraperitoneal delivery of a number of antineoplastic agents with known activity in ovarian cancer [Maurine Markman. J CHn Oncol 2003; 21 :145s-148s].
Thus, according to some embodiments of the present invention, the ester derivatives described herein are administered intraperitoneally in a therapeutically effective amount of at least 1 mg/kg body weight, wherein the administering is performed from 1 to 4 times a day. In yet another preferred embodiment, the ester derivatives are administered to a human patient intraperitoneally in a therapeutically effective amount of at least 50 mg, wherein the administering is performed from 1 to 4 times a day.
It is noted herein that, as would be recognized by any person skilled in the art, intraperitoneal bioavailability is higher than oral bioavailability and hence doses that are considered safe, in terms of adverse side effects, for oral administration, may cause significant side effects upon intraperitoneal administration of such doses. Accordingly, doses that are therapeutically effective upon intraperitoneal administration may be less effective when administered orally.
The results presented in the Examples section there follows, which show a therapeutic effect at doses of 1-10 mg/kg body weight, with no adverse side effects in stomach and kidney tissues, upon intraperitoneal administration, therefore indicate that (i) the ester derivatives described herein can be used intreaperitoneally at doses similar or higher than those used orally with non-esterified compounds (e.g., Diclofenac), a fact which is surprising considering
the side effects associated with non-esterified compounds, the recommended daily dosage for oral administration of non-estereified compounds, and the higher bioavailability of intreaperitoneal administration versus oral administration; and (ii) the ester derivatives described herein can be used effectively even at doses lower than those recommended for oral administration, when given intraperitoneal^.
In any of the method and uses described herein, the ester derivatives can be utilized either per se or as a part of a pharmaceutical composition, which further comprises a pharmaceutically acceptable carrier.
According to embodiments of the present invention, such a pharmaceutical composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment or prevention of a medical condition such as cancer and an inflammatory disease or disorder,
As used herein, a "pharmaceutical composition" refers to a preparation of one or more of the compounds described herein (as active ingredient), or physiologically acceptable salts or prodrugs thereof, with other chemical components, including, but not limited to, physiologically suitable carriers, excipients, lubricants, buffering agents, antibacterial agents, bulking agents (e.g. mannitol), antioxidants (e.g., ascorbic acid or sodium bisulfite), and the like. The purpose of a pharmaceutical composition is to facilitate administration of a compound to a subject.
The phrase "active ingredient" refers to a compound, which is accountable for a biological effect.
Herein, the phrases "physiologically acceptable carrier" and "pharmaceutically acceptable carrier", which are used interchangeably, describe a carrier or a diluent that does not cause significant irritation to the subject and does not abrogate the biological activity and properties of the administered compound. As used herein, the term "carrier" refers to a diluent, adjuvant, excipient, or vehicle with which the therapeutic is administered.
Herein the term "excipient" refers to an inert substance added to a pharmaceutical composition to further facilitate administration of an active ingredient. Examples, without limitation, of excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
Techniques for formulation and administration of drugs may be found in "Remington's Pharmaceutical Sciences" Mack Publishing Co., Easton, PA, latest edition, which is incorporated herein by reference.
Pharmaceutical compositions for use in accordance with the present invention thus may be formulated in conventional manner using one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the compounds into preparations which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. The dosage may vary depending upon the dosage form
employed and the route of administration utilized. The exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patient's condition (see e.g., Fingl et al, 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p.1).
The pharmaceutical composition may be formulated for administration in either one or more of routes depending on whether local or systemic treatment or administration is of choice, and on the area to be treated. Administration may be done orally, by inhalation, or parenterally, for example by intravenous drip or intraperitoneal, subcutaneous, intramuscular or intravenous injection, or topically (including ophthalmically, vaginally, rectally, intranasally).
Formulations for topical administration may include but are not limited to lotions, ointments, gels, creams, suppositories, drops, liquids, sprays and powders. Conventional pharmaceutical carriers, aqueous, powder or oily bases, thickeners and the like may be necessary or desirable.
Compositions for oral administration include powders or granules, suspensions or solutions in water or non-aqueous media, sachets, pills, caplets, capsules or tablets. Thickeners, diluents, flavorings, dispersing aids, emulsifiers or binders may be desirable.
Formulations for parenteral administration may include, but are not limited to, sterile solutions which may also contain buffers, diluents and other suitable additives. Slow release compositions are envisaged for treatment.
The amount of a composition to be administered will, of course, be dependent on the subject being treated, the severity of the affliction, the manner of administration, the judgment of the prescribing physician, etc.
Compositions according to the present embodiments may, if desired, be presented in a pack or dispenser device, such as an FDA (the U.S. Food and Drug Administration) approved kit, which may contain one or more unit dosage forms containing the active ingredient. The pack may, for example, comprise metal or plastic foil, such as, but not limited to a blister pack or a pressurized container (for inhalation). The pack or dispenser device may be accompanied by instructions for administration. The pack or dispenser may also be accompanied by a notice associated with the container in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the compositions for human or veterinary administration. Such notice, for example, may be of labeling approved by the U.S. Food and Drug Administration for prescription drugs or of an approved product insert. Compositions comprising a compound of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of a medical condition, as defined herein. As described hereinabove, the compounds described herein have unique features that enable to use these compounds as therapeutic agents as dosages that are higher than the dosages commonly practiced with common NSAIDs.
Hence, according to an aspect of embodiments of the present invention there is
provided a pharmaceutical composition unit dosage form which comprises an ester derivative as described herein, wherein an amount of the ester derivative is at least 1 mg/kg body weight. The unit dosage form is identified for use, from 1 to 4 times daily, in the treatment of a medical condition such as an inflammatory disease or disorder and cancer. The term "unit dosage form", as used herein, describes physically discrete units, each unit containing a predetermined quantity of one or more active ingredient(s) calculated to produce the desired therapeutic effect, in association with at least one pharmaceutically acceptable carrier, diluent, excipient, or combination thereof.
The single unit dosage forms described herein can be formulated for oral, mucosal (e.g., nasal, sublingual, vaginal, buccal, or rectal), parenteral (e.g., intraperitoneal, subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial), or transdermal administration to a patient. Examples of unit dosage forms include, but are not limited to: tablets including orally dissolving tablets; thin films; gelcaps; caplets; granules, capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; enemas; pessary; vaginal tablets; ointments; cataplasms (poultices); pastes; powders; dressings; creams; plasters; solutions; patches; liquid sprays; metered and unmetered aerosols (e.g., nasal sprays or inhalers); drops; lyophilized compositions; transdermal patches; gels; liquid dosage forms suitable for oral or mucosal administration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, tinctures and elixirs; syrups, liquid dosage forms suitable for parenteral administration to a patient (e.g., ampoules, sterile bags); sterile solids (e.g., crystalline or amorphous solids) that can be reconstituted to provide liquid dosage forms suitable for parenteral administration to a patient; and as components of autoinjector devices.
In some embodiments, the pharmaceutical composition unit dosage form described herein comprises an amount of the ester derivative which is higher than 1 mg/kg body weight. In some embodiments, the unit dosage form comprises an amount of the ester derivative that is higher than 2 mg/kg body weight, and even higher than 3 mg/kg body weight. In some embodiments, the amount of the ester derivative in the unit dosage form is 6 mg/kg body weight or higher. In some embodiments, the pharmaceutical composition unit dosage form comprises an amount of the ester derivative which ranges from 1 mg/kg body weight to 10 mg/kg body weight.
Thus, the amount of the ester derivative in the pharmaceutical composition unit dosage form described herein can be, for example, about 1 mg/kg body weight, about 2 mg/kg body weight, about 3 mg/kg body weight, about 4 mg/kg body weight, about 5 mg/kg body weight, about 6 mg/kg body weight, about 7 mg/kg body weight, about 8 mg/kg body weight, about 9 mg/kg body weight, about 10 mg/kg body weight, about 11 mg/kg body weight, about 12 mg/kg body weight, about 13 mg/kg body weight, about 14 mg/kg body weight, about 15 mg/kg body weight, about 16 mg/kg body weight, about 17 mg/kg body weight, about 18 mg/kg body weight,
about 19 mg/kg body weight, about 20 mg/kg body weight, about 21 mg/kg body weight, about 22 mg/kg body weight, about 23 mg/kg body weight, about 24 mg/kg body weight, about 25 mg/kg body weight and even higher.
Taking the average human weight as being about 70 kg, when formulated for use in humans, a unit dosage form as described herein comprises an amount of the ester derivative which is higher than 50 mg.
Accordingly, according to another aspect of the present invention there is provided a pharmaceutical composition unit dosage form wherein an amount of the ester derivative is higher than 50 mg and the unit dosage form is identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition such as an inflammatory disease or disorder and cancer.
Thus, an amount of the ester derivative in the pharmaceutical composition unit dosage form described herein can be, for example, about 50 mg, about 60 mg, about 70 mg, about 80 mg, about 90 mg, about 100 mg, about 150 mg, about 200 mg, about 250 mg, about 300 mg, about 350 mg, about 400 mg, about 450 mg, about 500 mg, about 550 mg, about 600 mg, about 650 mg, about 700 mg, about 750 mg, about 800 mg, about 850 mg, about 900 mg, about 950 mg, about 1000 mg and even higher.
The optimal dosage of the ester derivatives in the unit dosage form depends upon factors such as the selected regime, the medical condition being treated and the carrier used, as is detailed herein.
The pharmaceutical composition unit dosage form described herein can be formulated for various administration routes, such as, for example, orally, transdermal^, intreaperitoneally, sublingually, intravenously, rectally or by inhalation.
In some embodiments, the pharmaceutical composition unit dosage form is formulated for intraperitoneal administration. In other embodiments, the unit dosage form is formulated for oral administration.
Thus, according to yet another aspect of the present invention there is provided a pharmaceutical composition unit dosage form which comprises the ester derivative described herein, wherein an amount of the ester derivative is at least 1 mg/kg body weight, the unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to
4 times daily.
According to yet another aspect of the present invention there is provided a pharmaceutical composition unit dosage form which comprises an ester derivative as described herein, wherein the amount of the ester derivative is higher than 50 mg and the unit dosage form is formulated for intraperitoneal administration to a human subject, 1 to 4 times daily.
The pharmaceutical composition unit dosage form described herein may be formulated for use in divided doses. For example, the unit dosage form may be administered 1 time per day, 2 times per day, 3 times per day, 4 times per day, 5 times per day, 6 times per day and so
on. Preferably, the composition is administered 2 to 4 times per day.
According to some embodiment of the present invention the pharmaceutical composition unit dosage form is formulated such that the daily dose of the ester derivative is higher than 2 mg/kg body weight, and even higher than 3 mg/kg body weight. Accordingly, the pharmaceutical composition unit dosage form is formulated such that the daily dose of the ester derivative is higher than 150 mg, and even higher than 200 mg, when utilized by a human patient.
The pharmaceutical composition unit dosage form described herein can be identified for use in acute and chronic treatment protocols, as detailed hereinabove. According to a feature of the present invention, the pharmaceutical composition unit dosage form described herein is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of a medical condition such as an inflammatory disease or disorder and cancer.
The term "cancer" is used herein to describe the pathological process that results in the formation and growth of a neoplasm, i.e., an abnormal tissue that grows by cellular proliferation more rapidly than normal tissue and continues to grow after the stimuli that initiated the new growth ceases. Cancerous tissue exhibits partial or complete lack of structural organization and functional coordination with the normal tissue, and usually forms a distinct mass of tissue which may be benign (benign tumor) or malignant (carcinoma). The term "cancer" is used as a general term to describe any of various types of malignant neoplasms, most of which invade surrounding tissues, may metastasize to several sites and are likely to recur after attempted removal and to cause death of the patient unless adequately treated. The term "tumor and/or cancer" is used to describe all types of neoplasia, including benign and malignant. Exemplary cancers which may be effectively treated by the methods and compositions described herein, include, but are not limited to, brain cancer, a bone cancer, an epithelial cell- derived neoplasia (epithelial carcinoma), a basal cell carcinoma, an adenocarcinoma, a gastrointestinal cancer, a lip cancer, a mouth cancer, an esophageal cancer, a small bowel cancer, a stomach cancer, a colon cancer, a liver cancer, a bladder cancer, a pancreas cancer, an ovary cancer, a cervical cancer, a lung cancer, a breast cancer, a skin cancer, a squamus cell cancer, a prostate cancer, a renal cell carcinoma, a cancerous tumor, a growth, a polyp, an adenomatous polyp, a familial adenomatous polyposis or a fibrosis resulting from radiation therapy.
Inflammation is the complex biological response of vascular tissue to harmful stimuli, such as pathogens, damaged cells, or irritants. Inflammation can be classified as either acute or chronic. Acute inflammation is the initial response of the body to harmful stimuli and is achieved by the increased movement of plasma and leukocytes from the blood into the injured tissues. A cascade of biochemical events propagates and matures the inflammatory response,
involving the local vascular system, immune system and various cells within the injured tissue. Inflammation is activated by PLA2, which liberates arachidonic acid, the substrate for COX enzymes. COX converts arachidonic acid to the prostaglandin PGE2, the major prostaglandin detected in inflammatory conditions ranging from acute edema to chronic arthritis. The phrase "inflammatory diseases or disorders" is used to describe disease or disorders which are characterized by inflammation. The term "inflammatory diseases or disorders" is used as a general term to describe acute inflammation as well as prolonged, chronic inflammation. Prolonged inflammation, known as chronic inflammation, leads to a progressive shift in the type of cells which are present at the site of inflammation and is characterized by simultaneous destruction and healing of the tissue from the inflammatory process. Chronic inflammation can lead to many diseases and disorders, including degenerative inflammation disorders.
The phrase "inflammatory diseases or disorders", as used herein, therefore encompasses also degenerative inflammation disorders. Exemplary inflammatory diseases or disorders which are treatable by the ester derivatives described herein include, but are not limited to, Alzheimer's disease, cortical dementia, vascular dementia, muli-infract dementia, pre-senile dementia, alcoholic dementia, senile dementia, memory loss or central nervous damage resulting from stroke, ischemia or trauma, multiple sclerosis, Parkinson's disease, Huntington's disease, epilepsy, cystic fibrosis, arthritis diseases such as osteoarthritis, rheumatoid arthritis, spondyloarthopathies, gouty arthritis, systemic lupus erythematosus, and juvenile arthritis fever, periarteritis; gastrointestinal disorders such as inflammatory bowel disease, Chron's disease, gastritis, irritable bowel syndrome, ulcerative colitis, cardiovascular disorders such as myocardial ischemia, reperfusion injury to an ischemic organ; angiogenesis, asthma, bronchitis, menstrual cramps, premature labor, tendinitis, bursitis, an autoimmune disease, an immunological disorder, systemic lupus erythematosus, inflammatory disorders of the skin such as psoriasis, eczema, burns and dermatitis; neoplasia, an inflammatory process in a disease, pulmonary inflammation, a central nervous system disorder, migraine headaches, allergic rhinitis, respiratory distress syndrome, endotoxin shock syndrome, a microbial infection, a bacterial-induced inflammation, a viral induced inflammation, a urinary disorder, a urological disorder, endothelial dysfunction, organ deterioration, tissue deterioration, adhesion and infiltration of neutrophils at the site of inflammation, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, rheumatic fever, myasthenia gravis, sarcoidosis, nephrotic syndrome, Behcet's syndrome, polymyositis, hypersensitivity, conjunctivitis, gingivitis and swelling occurring after injury. The ester derivatives utilized in any of the aspects described herein has the general
Formula I:

Formula I wherein:
Z is an A-C(= K)-O-Y-Ra group; A is alkyl or absent;
K is selected from the group consisting of O and S;
Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino, -NR12R13, and a positively charged group, whereas R12 and R13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R12 and R13 form a five- or six-member heteroalicyclic ring;
R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R2, R3, R4, R5, R6, R7, R8, R9 and R10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino and -NR14R15, or, alternatively, at least two of R2, R3, R4, R5 and R6, of R7, R8, R9 and R10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or heteroalicyclic ring; and R14 and R15 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R14 and R15 form a five- or six- member heteroalicyclic ring.
As used herein, the term "alkyl" refers to a saturated aliphatic hydrocarbon including straight chain and branched chain groups. Preferably, the alkyl group has 1 to 20 carbon atoms. Whenever a numerical range; e.g., "1-20", is stated herein, it means that the group, in this case the alkyl group, may contain 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 20 carbon atoms. More preferably, it is a medium size alkyl having 1 to 10 carbon
atoms. Most preferably, it is a lower alkyl having 1 to 4 carbon atoms. The alkyl group may be substituted or unsubstituted. When substituted, the substituent group can be, for example, cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, halo, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N- thiocarbamyl, C-amido, N-amido, C-carboxy, O-carboxy, nitro, sulfonamido, trihalomethanesulfonamido, silyl, guanyl, guanidino, ureido or amino.
The term "alkenyl" describes an unsaturated alkyl, as defined herein, having at least two carbon atoms and at least one carbon-carbon double bond. The alkenyl may be substituted or unsubstituted by one or more substituents, as described hereinabove. The term "alkynyl", as defined herein, is an unsaturated alkyl having at least two carbon atoms and at least one carbon-carbon triple bond. The alkynyl may be substituted or unsubstituted by one or more substituents, as described hereinabove.
The term "hydroxyalkyl" describes an alkyl, as defined herein, substituted (e.g., terminated) by a hydroxy group. The term "hydroxyalkenyl" describes an alkenyl, as defined herein, substituted (e.g., terminated) by a hydroxy group.
The term "cycloalkyl" describes an all-carbon monocyclic or fused ring (i.e., rings which share an adjacent pair of carbon atoms) group wherein one of more of the rings does not have a completely conjugated pi-electron system. Examples, without limitation, of cycloalkyl groups are cyclopropane, cyclobutane, cyclopentane, cyclopentene, cyclohexane, cyclohexadiene, cycloheptane, cycloheptatriene, and adamantane. A cycloalkyl group may be substituted or unsubstituted. When substituted, the substituent group can be, for example, alkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, halo, carbonyl, thiocarbonyl, C-carboxy, O-carboxy, O-carbamyl, N-carbamyl, C-amido, N- amido, nitro and amino, as defined herein.
The term "aryl" describes an all-carbon monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups having a completely conjugated pi-electron system. Examples, without limitation, of aryl groups are phenyl, naphthalenyl and anthracenyl. The aryl group may be substituted or unsubstituted. When substituted, the substituent group can be, for example, halo, trihalomethyl, alkyl, hydroxy, alkoxy, aryloxy, thiohydroxy, thiocarbonyl, C-carboxy, O-carboxy, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, sulfinyl, sulfonyl and amino, as defined herein.
The term "heteroalicyclic" group refers to a monocyclic or fused ring group having in the ring(s) one or more atoms such as nitrogen, oxygen and sulfur. The rings may also have one or more double bonds. However, the rings do not have a completely conjugated pi-electron system. The heteroalicyclic may be substituted or unsubstituted. When substituted, the substituted group can be, for example, alkyl, cycloalkyl, aryl, heteroaryl, halo, trihalomethyl, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, carbonyl,
thiocarbonyl, C-carboxy, O-carboxy, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, sulfinyl, sulfonyl, C-amido, N-amido and amino, as defined herein. The term "hydroxy" describes an -OH group.
The term "heteroaryl" describes a monocyclic or fused ring (Ae., rings which share an adjacent pair of atoms) group having in the ring(s) one or more atoms, such as, for example, nitrogen, oxygen and sulfur and, in addition, having a completely conjugated pi-electron system.
Examples, without limitation, of heteroaryl groups include pyrrole, furane, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrimidine, quinoline, isoquinoline and purine. The heteroaryl group may be substituted or unsubstituted. When substituted, the substituent group can be, for example, alkyl, cycloalkyl, halo, trihalomethyl, hydroxy, alkoxy, aryloxy, thiohydroxy, thiocarbonyl, sulfonamido, C-carboxy, O-carboxy, sulfinyl, sulfonyl, O-carbamyl, N-carbamyl, O- thiocarbamyl, N-thiocarbamyl, C-amido, N-amido and amino, as defined herein.
The term "halo" describes fluorine, chlorine, bromine or iodine.
The term "alkoxy" describes both an -O-alkyl and an -O-cycloalkyl group, as defined herein.
The term "aryloxy" describes both an -O-aryl and an -O-heteroaryl group, as defined herein.
The term "thiohydroxy" describes a -SH group.
The term "thioalkoxy" describes both an -S-alkyl group, and an -S-cycloalkyl group, as defined herein.
The term "thioaryloxy" describes both an -S-aryl and an -S-heteroaryl group, as defined herein.
The term "sulfinyl" describes an -S(=O)-R' group, where R' is hydrogen, alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring carbon) or heteroalicyclic (bonded through a ring carbon) as defined herein.
The term "sulfonyl" describes an -S(=O)2-R' group, where R' is as defined herein.
The term "cyano" describes a -C≡N group.
The term "nitro" describes an -NO2 group.
The term "amino" describes an -NH2 or NR'R" group, with R' as defined herein and R" as defined for R'. This term further encompasses cyclic amino groups, in which R' and R" are linked therebetween to form a five- or six-membered cyclic group.
The phrase "positively charged group" describes an atom or a group of atoms which forms a part of an organic molecule, and which is characterized by a positive electrostatic charge. Compounds which include one or more positively charged groups are molecular ions oftentimes referred to as molecular cations. A positively charged group of atoms has at least one electron less than the number of protons in these atoms. Positively charged groups include, for a non-limiting example, ammonium and sulfonium groups, as defined herein.
The term "ammonium group" describes N+(R')4, wherein R' groups maybe the same or
different and are as defined herein.
The term "sulfonium group" describes S+(R')3, wherein R' groups maybe the same or different and are as defined herein.
The term "carbonyl" describes a -C(=O)-R' group, wherein R1 is as defined herein. The term "trihaloalkyl" describes a -(alkyl)X3, wherein X is a halo group.
In some embodiments, Y is a substituted or unsubstituted alkyl.
According to these embodiments, Ra is other than hydrogen. In one example, Ra is hydroxy.
In some embodiments, Y is a substituted or unsubstituted polyalkylene glycol moiety. In some embodiments, the polyalkylene glycol moiety has a general formula II:
[(CH2)m-0]n-
Formula I!
wherein: each of m and n is independently an integer of 1-10.
In these embodiments, the Ra attached to the polyalkylene glycol moiety is, for example, hydrogen or alkyl.
Alternatively, the Ra attached to the polyalkylene glycol moiety is a positively charged group, as defined herein. In some embodiments, the positively charged group is an ammonium group.
In some embodiments, the polyalkylene glycol moiety does not include a nitroso (- ONO2) group, or any other NO-releasing group, or an anti-oxidant moiety.
In some embodiments, G is C; K is O; each of R2, R3, R4, R5 and R6 is independently selected from the group consisting of hydrogen, alkyl, halo and trihaloalkyl; and each of R7, R8, R9 and R10 is hydrogen.
According to some embodiments the N-phenylanthranilic acid is Diclofenac and the compounds described herein are ester derivatives of Diclofenac.
In these embodiments, R2 and R6 are chloro, and each of R3-R5 and R7-R10 is hydrogen. Diclofenac is a non-steroidal anti-inflammatory drug (NSAID) commonly used for reducing inflammation and as an analgesic for reducing pain in conditions such as arthritis or acute injury. The recommended dose for most conditions is 100-200 mg daily, administered orally. Dosing intervals depend on the diclofenac formulation used and the condition being treated. The most common side effects of diclofenac involve the gastrointestinal system. It can cause ulcerations, abdominal burning, pain, cramping, nausea, gastritis, and even serious gastrointestinal bleeding and liver toxicity. Rash, kidney impairment, ringing in the ears, and lightheadedness are also seen.
Exemplary Diclofenac ester derivatives according to the present embodiments include,
but are not limited to:

1 (NH6) and

2 (NH17).
The present embodiments further encompass any enantiomers, diastereomers, prodrugs, solvates, hydrates and/or pharmaceutically acceptable salts of the compounds described herein.
As used herein, the term "enantiomer" refers to a stereoisomer of a compound that is superposable with respect to its counterpart only by a complete inversion/reflection (mirror image) of each other. Enantiomers are said to have "handedness" since they refer to each other like the right and left hand. Enantiomers have identical chemical and physical properties except when present in an environment which by itself has handedness, such as all living systems.
The terms "diastereomers" or "diastereoisomers", as used herein, refer to stereoisomers that are not enantiomers with respect to one another. Diastereomers have more than one chiral center, and can have different physical properties and different reactivity. Diastereoisomers are not mirror images of each other but rather have one or more chiral centers inverted between the two stereoisomers. If a molecule exhibits two chiral centers (two asymmetric carbons), there are up to four possible stereo-configurations and hence up to four possible diastereomers.
The term "prodrug" refers to an agent, which is converted into the active compound (the active parent drug) in vivo. Prodrugs are typically useful for facilitating the administration of the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent drug is not. A prodrug may also have improved solubility as compared with the parent drug in pharmaceutical compositions. Prodrugs are also often used to achieve a sustained release of the active compound in vivo. The term "solvate" refers to a complex of variable stoichiometry (e.g., di-, tri-, tetra-, penta-, hexa-, and so on), which is formed by a solute (the compound of the present invention)
and a solvent, whereby the solvent does not interfere with the biological activity of the solute. Suitable solvents include, for example, ethanol, acetic acid and the like.
The term "hydrate" refers to a solvate, as defined hereinabove, where the solvent is water. The phrase "pharmaceutically acceptable salt" refers to a charged species of the parent compound and its counter ion, which is typically used to modify the solubility characteristics of the parent compound and/or to reduce any significant irritation to an organism by the parent compound, while not abrogating the biological activity and properties of the administered compound. It is appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the invention, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable subcombination or as suitable in any other described embodiment of the invention. Certain features described in the context of various embodiments are not to be considered essential features of those embodiments, unless the embodiment is inoperative without those elements.
Various embodiments and aspects of the present invention as delineated hereinabove and as claimed in the claims section below find experimental support in the following examples.
EXAMPLES
Reference is now made to the following examples, which together with the above descriptions illustrate some embodiments of the invention in a non limiting fashion.
MATERIALS AND EXPERIMENTAL METHODS Chemicals:
Abbreviations - NHS: N-hydroxysuccinimide; t-Boc: t-butoxycarbonyl; DCM: dichloromethane; DMAP: dimethylaminopyridine; DMF: dimethyl formamide; DCC: dicyclohexylcarbodiimide; EtOAc: ethylacetate; He: hexanes; and TFA: trifluoroacetic acid.
All reactions requiring anhydrous conditions were performed in oven-dried glassware under an Argon or N2 atmosphere.
Chemicals and solvents were either A.R. grade or purified by standard techniques.
Diclofenac was purchased from Sigma-Aldrich (St. Louis, MO, USA).
XTT reagent kit was obtained from Biological Industries, Kibbutz Bet Ha'emek.
COX-1 purified from Bovine was obtained from Cayman Chemicals, USA. Arachidonic acid, Diclofenac, Indomethacin, Tris Buffer, Phenol, Trp and Hematin were obtained from Sigma.
Rosewell Park Memorial Institute (RPMI) medium and Hank Balanced Salt Solution (HBSS) medium and Phosphate Buffer Solution (PBS) were obtained from Biological Industries, Bet Ha'emek. Unless noted otherwise, all other reagents, including salts and solvents, were purchased from Sigma-Aldrich (Milwaukee, Wl). human recombinant COX-2 was obtained from Cayman Chemicals, USA.
Animals:
Mice: C57Black or BALB/c, weighing 20 gram on average, were obtained from the Animal facility of TeI Aviv University Medical School
F344 female rats, aged 3-6 months, weighing 200-250 grams, were obtained from the Animal facility of the Department of Psychology, TeI Aviv University
Instrumental measurement:
Thin layer chromatography (TLC) was performed using silicagel 60 F254 plates or silicagel preparative plates Analtech 1000 microns by Merck, and the compounds were visualized by irradiation with UV light and/or by treatment with a solution of 25 grams phosphomolybdic acid, 10 grams Ce(SO4)2-H2O, 60 ml concentrated H2SO4 and 940 ml H2O, followed by heating and/or by staining with a solution of 12 grams 2,4-dinitrophenylhydrazine in
60 ml concentrated H2SO4; 80 ml H2O and 200 ml 95 % EtOH, followed by heating and/or immersing into an iodine bath (30 grams I2, 2 grams Kl, in 400 ml EtOH/H2O 1 :1) and warming.
Flash chromatography (FC) was performed using silica gel Merck 60 (particle size 0.040-0.063 mm) by Merck, and eluent given in parentheses.
1H NMR was performed using Bruker AMX 200 or 400. The chemical shifts are expressed in δ relative to TMS (δ = 0 ppm) and the coupling constants J in Hz. The spectra were recorded in CDCI3 or CD3OD as a solvent at room temperature unless stated otherwise.
HR-MS liquid secondary ionization (LSI-MS) was performed using VG ZAB-ZSE with 3- nitrobenzyl-alcohol matrix.
Spectrophotometric measurements were carried out using Spectramax 190 instrument.
HPLC was performed on an Elite Lachrom Merck-Hitachi instrument, using a 45 % H2O (0.1 % TFA): 55% Acetaminophen as the stationary phase and a L-2450 detector, operated at 25 degrees, in a flow rate of Iml/min. Protein content assessment:
Protein concentrations were measured by the Bradford method (1976) [Bradford M. M. 1976. Anal. Biochem. 72:248-254], using BSA as a standard.
CHEMICAL SYNTHESES Preparation of NH6:
NH6, an exemplary ester derivative according to embodiments of the present invention, was prepared as described in U.S. Patent Application No. 20050250833.
Diclofenac (50 mg, 0.17 mmol) was dissolved in dry DCM. Catalytic amount of DMAP and diethylene glycol (0.08 ml, 0.85mmol) were added, and the stirred mixture was cooled to 0 0C. Thereafter DCC (52.6 mg, 0.255 mmol) was dissolved in DCM and added dropwise to the reaction mixture. The suspension was then stirred at 0 0C for 30 minutes and monitored by TLC (EtOAσHe, 1 :1). The precipitating solid was removed by filtration and washed with DCM. The filtrate was concentrated under reduced pressure and purified by flash chromatography over silica gel to afford the pure ester NH6 (43 mg, 66 % yield). 1H-NMR (200 MHz, CDCI3): δ=7.35 ppm (2H, d, J=8); 7.19-7.06 (2H, m); 6.99 (1 H, d,
J=8); 6.96 (1 H, d, J=8.); 6.56 (1 H, d, J=8); 4.35 (2H, m); 3.8 (2H, s); 3.65-3.75 (4H, m); 3.53- 3.57 (2H, m). MS (FAB): [C18H19CI2NO4] 383.0. Preparation of NH17:
NH17, an exemplary ester derivative according to embodiments of the present invention, was prepared as illustrated in Scheme 1 below.
Scheme 1

NH17
Diclofenac (100 mg, 0.338 mmol) was dissolved in DCM. Thereafter N-boc- diglycolamine (70 mg, 0.34 mmol) [Kim, Y. S.; Kim, K. M.; Song, R.; Jun, M. J.; Sohn, Y. S., J. Inorg. Biochem. 2001 , 87(3), 157-163] and DMAP (4 mg, 0.034 mmol) were added. To this mixture DCC (139 mg, 0.68 mmol) DCM was added dropwise. The reaction mixture was stirred at room temperature for 1 hour while monitoring by TLC (EtOAc:He, 1 :1). Upon completion of the reaction, the mixture was filtered and the solvent was removed under reduced pressure.
The crude product was purified by column chromatography on silica gel (EtOAc: He, 1 :3) to afford the t-BOC intermediate as depicted in Scheme 1 (148 mg, 90 %) in the form of a color less oil.
1H-NMR (200 MHz, CDCI3): δ = 7.34ppm (d, J = 8 Hz, 2H); 7.28 (dd, J = 7.4, 1.5 Hz, 1 H); 7.14 (dt, J = 7.4, 1.5 Hz, 1 H); 7.02-6.93 (m, 2H); 6.87 (s, 1 H); 6.57 (d, J = 7.9 Hz, 1 H); 4.31-4.26 (m, 2H); 3.84 (s, 2H) 3.68-3.63 (m, 2H); 3.47 (t, J = 5.0 Hz, 2H); 3.27 (q, J = 5.0 Hz, 2H); 1.43 (s, 9H).
The t-BOC intermediate (127 mg, 0.26 mmol) was dissolved in 10 ml DCM, and 3 ml of a solution of HCI (1 M) in ether was added thereto. The mixture was stirred at room temperature overnight and monitored by TLC (EtOAc:MeOH, 9:1 ). Upon completion of the reaction, the solvents were removed under reduced pressure to afford NH17 (29 mg, 27 % yield) in the form of a white powder which was used without further purification.
NH17: 1H-NMR (200 MHz, CDCI3): δ= 7.32 ppm (d, J = 8.0 Hz, 2H); 7.26-7.22 (m, 1 H); 7.12 (t, J = 8.0 Hz, 1 H); 7.0-6.92 (m, 2H); 6.81 (s, 1 H); 6.51 (d, J = 7.6 Hz, 1 H); 4.30 (bs, 2H); 3.84 (s, 2H); 3.76 (bs, 4H); 3.25 (bs, 2H).
CT26 cells Viability test: Mouse colon adenocarcinoma (CT26) cells were plated in 96-wells culture plates supplemented with rich medium [PLEASE INDICATE WHICH MEDIUM]. When the cells reached 70 % confluence, either Diclofenac, Compound 1 (NH6) or Compound 2 (NH17), was added at the appropriate concentration. The viability of the cultured cells was quantified using the XTT reagent test. The XTT assay is based on the ability of metabolic active cells to reduce the Tetrazolium salt XTT to orange colored compounds of formazan. The greater the number of
active cells in the well, the greater the activity of mitochondria enzymes, and the higher the concentration of the dye formed. Briefly, the plates were incubated for an hour at 37 0C, the medium was thereafter washed and XTT reagent and an activation reagent were added. The plates were then put in incubation for additional 8 hours at 37 0C. Thereafter, the fluorescence intensity of the wells was quantified using a spectrophotometer at a wave length of 450 nm and 690 nm. The fluorescence intensity in each well was proportional to the number of metabolic active cells.
Assessment of Spontaneous decomposition of Compound 1 (NH6) and Compound 2 (NH17) into Diclofenac: NH6 or NH17 was diluted to a concentration of 500 μM in RPMI medium without serum, and the solution was subjected to a HPLC analysis under the following conditions: Mobile phase - 45 % H2O (0.1 % TFA):55 % Acetaminophen (CAN), flow rate - 1 ml/min, temperature - 25 0C, overall running time - 15 minutes. For assessment of the time-dependent decomposition the NH6 and NH17, solutions of these compounds were subjected to HPLC analyses after incubation at room temperature for 0, 5, 10, 15, 20, 25 and 30 hours and the amount of the acid form (i.e. Diclofenac) was quantified for each time point.
Cyclooxygenase inhibition assay using purified ovine COX-1 and human recombinant COX-2:
The effect of Compound 1 (NH6) and Compound 2 (NH17) on COX-1- or COX-2- dependent PGE2 synthesis from Arachidonic acid was tested. A reaction mixture containing 25 nM Tris Buffer, 0.5 mM Phenol, 1 mM Trp and 1 μM Hematin was added to vials. Then, 10 μl Diclofenac, NH6 or NH17, at the tested concentration, were added to each reaction vial and the mixture was stirred. A reaction vial to which no compound was added served as control. Bovine COX-1 or human recombinant COX-2 purified enzymes were diluted with Tris buffer containing 20 % TWEEN®. An enzyme solution corresponding to 20 enzyme units was added to each reaction vial and the vials were incubated for 5 minutes at room temperature. Following incubation, 15 μl of 10 μM aqueous solution of Arachidonic acid were added to each vial, and the vials were incubated in room temperature for additional 20 minutes. A reaction vial to which no Arachidonic acid was added served as an negative control. Finally, 10μl of a 5 μM aqueous solution of lndomethacin were added to the reaction mixture in order to stop prostaglandin synthesis. 400 μl of Tris Buffer were then added to the vial and the PGE2 concentration was quantified using radioimmunoassay (DuPont-NEN; Boston MA) according to manufacturer protocol.
Cyclooxygenase inhibition assay in cell culture: Mouse colon adenocarcinoma CT26 and mouse lewis lung carcinoma D122 cells were grown in DMEM (D122) or RPMI (CT26) supplemented with 10 % fetal calf serum, 4 % glutamine, 1 % penicillin-sterptomycin-nystatin and 3 % non essential amino acids (D122) and were kept in a humidified 37 0C incubator with 5 % CO2. For the COX activity assays, cells
were grown onto 24 multi-well plates.
Before the assay, cells were washed with serum-free medium; then the tested compounds were added to the medium and incubated for 30 minutes at 37 0C after which 30 μM arachidonic acid were added and incubated for additional 20 minutes. At the end of the incubation period, 10 μM lndomethacin were added to stop the reaction and medium was transferred to another 24 multi-well plate for measuring PGE2 production by radioimmunoassay (DuPont-NEN; Boston MA) according to manufacturer protocol. Protein determination was assayed by the Bradford method (as described hereinabove) on scraped cells and was used to normalize the amount of PGE2 production to indicate ngPGE2 produced /mg cell protein. Assessing anti-COX activity of a single dose administration of Compound 1 (NH6) and Compound 2 (NH17) in-vivo:
30 μl (total volume) of 5 x 104 CT26 cells suspended in 30 μl HBSS buffer were injected to the right hind foot of BALB/c mice (10 mice per experimental group) Once the tumor has reached a volume of approximately 400 mm3, as measured by caliper, (usually 21-24 days post injection), 100 μl of a solution of the tested compound (dissolved in 1 % DMSO in PBS), at the tested concentration, were injected to each mouse intraperitoneally (I. P.) in a single dose. The concentrations were calculated on a mg per kg (mpk) of body weight, assuming that each mouse weighs 20 gram on average.
Three hours post-injection, mice were sacrificed, the tumor was removed, and the stomach and kidneys were collected. All organs were homogenized in 1 ml PBS. 50 μl of the specific organ-cell suspension were kept for protein concentration determination, and the residual suspension centrifuged for 1 minute in 10,000 rpm. The supernatant, which did not contain any cells, was removed to a separate vial and the PGE2 concentration in the supernatant was assessed by radioimmunoassay, as described hereinabove. In order to determine the overall protein concentration in the tissue, all the vials to which samples were removed were centrifuged in 12,000 rpm for 3 minutes. The supernatant was removed and 100 μl of NaOH 1 N were added to the pellet, to Iyse the cells. All samples were analyzed for protein content using the Bradford technique.
Assessing anti-COX activity of a chronic administration of NHβ and NH17 in-vivo: 5 x 104 CT26 cells suspended in HBSS buffer in a volume of 30μl were injected to the right hind foot of the mouse (10 mice per group). Starting from the day of cells injection, 100 μl of a solution of the tested compound in 1% DMSO in PBS, at the indicated concentrations, were injected to each mouse intraperitoneally (I. P.) every 24 hours. The injected amount of compound were calculated on a "mg per kg of body weight basis", assuming that each mouse weighs 20 gram on average.
The volume of the tumor was measured by a caliper every day starting from the day of cells injection Once the tumor has reached a volume of approximately 400 mm3,, the mouse was sacrificed, the tumor removed, and the stomach and kidneys were collected. The last dose
of the tested compound was injected 8 hours prior to organs collection. All organs were homogenized in 1 ml PBS. 50 μl of the organ-containing suspension were kept for protein concentration determination in the tissue, while the residual suspension was centrifuged for 1 minute in 10,000 rpm. The supernatant, which did not contain any cells, was removed to a separate vial and the PGE2 concentration in the supernatant was assessed by radioimmunoassay, as described hereinabove.
In order to determine the overall protein concentration in the tissue, all the vials for which 50 μl samples were removed, were centrifuged in 12,000 rpm for 3 minutes. The supernatant was removed and 100 μl of NaOH 1 N were added to the pellet, to lyse the cells. All samples were analyzed for protein content using the Bradford technique. Measurements of extent of acute local inflammation in rats:
Acute footpad inflammation in F344 female rats (weighing 200-250 grams) was induced by injecting 100 μl of 1.5 % carrageenen (CR) solution (in PBS) into the right footpad. Control PBS solution (i.e. not containing any compound ) was injected into the left footpad. After 3 hours, the dimension of each footpad was measured with a caliber and the volume (in mm3) was calculated according to the formula: thinckness2 x width x 0.52. The rats were pre-treated with the tested compound at the indicated dose 30 minutes prior to the injection of CR. The tested compounds were prepared as 50 mM stock solutions in DMSO, diluted with PBS and injected intraperitoneally (100 μl per rat). The calculated results are means of 4 rats per treatment. SEM values for each mean were < 10 % of that mean. Percent inhibition of footpad inflammation was calculated as:
net paw volume in specific group
I [ i I . ™ i J Λv i I n UnU maximum net paw volume in control rats
EXPERIMENTAL RESULTS
EXAMPLE 1 Effect of Compound 1(NH6) and compound 2 (NH17) on CT26 cells viability
Viability tests were conducted in order to examine whether the therapeutic activity of
NH6 and NH17 is via a mechanism of toxicity to cells In-vitro. CT26 cells were incubated with
Diclofenac, NH6 or NH17 for 8 hours and the level of cells viability was measured using the XTT reagent test. The fluorescence intensity measured was proportional to the number of metabolic active cells.
The results are presented in Figure 2 and show that the viability of the cells was significantly decreased only when the cells were exposed to Diclofenac at a concentration above 1 mM (Figure 2A) or to NH6 at a concentration of above 300 μM (Figure 2B) or to NH17
at a concentration of above 100 μM (Figure 2C).
These results indicate that NH6 and NH17 are not toxic to the tested cells up to a concentration of 100 μM. This concentration is much higher than the concentrations likely obtained when Diclofenac or the tested ester derivatives reach the tumor site or other bodily organs (estimated maximal plasma concentration in the mouse is 10 μM). Therefore, cell toxicity is not responsible for the therapeutic activity of the derivatives, as observed in cell culture in vitro.
EXAMPLE 2 Determination of the rate of spontaneous decomposition of NH6 and NH17 to Diclofenac
In order to assess whether the anti-COX and anti-tumorogenic effects of NH6 and NH17 are due to a spontaneous decomposition of NH6 and NH17 into Diclofenac, the rate of decomposition was determined using HPLC measurements. The compounds were solubilized in an in RPMI medium without serum, and incubated at 37 degrees for various time lengths and the time dependent conversion rate of NH6 and NH17 to Diclofenac was measured The results are presented in Figures 3A and 3B, respectively. The percentages of conversion of NH6 and NH17 to Diclofenac, at various time points are also presented in Tables 1 and 2 below.
Table 1


The results show that both derivatives undergo a slow, time-dependent cleavage of the ester bond, so as to produce the parent compound, Diclofenac, which a substantial decomposition (e.g., of 10 % and more) observed only following 10-15 hours. Such a decomposition rate indicates that the activity of both derivatives in inhibiting prostaglandin synthesis (see, Example 3 below, in which inhibition of COX was observed within an hour) is not a result of their decomposition to Diclofenac), but is rather exhibited by these compounds in their ester form.
EXAMPLE 3 Anti-COX activities of Compound 1 ( NH6) and Compound 2 (NH17)
The observed COX inhibition activity by NH6 and NH17 may be attributed to (1) direct inhibition of COX-1 and COX-2 by NH6 and NH17 or (2) enzymatic cleavage of the ester bond in NH6 and NH17 to yield the parent Diclofenac, exhibits the anti-COX activity.
In order to determine if NH6 and NH17 have an inherent anti-COX activity, which is not attributed to the decomposition thereof to produce Diclofenac, the level of COX-dependent PGE2 synthesis from Arachidonic acid was evaluated in the presence of NH6 or NH17, with the level of PGE2 synthesis being indicative of the anti-COX activity of the tested compound. The experiments were conducted using purified COX-1 and COX-2, in the absence of enzymes capable of converting NH6 and NH17 to Diclofenac (such as esterase), thus isolating the observed effect on PGE2 synthesis to a NH6/NH17-dependent effect.
The obtained results are presented in Figures 4A and 4B, and show that in the presence of NH6 and NH17, the synthesis of PGE2 by COX-1 (Figure 4A) and COX-2 (Figure
4B) was significantly inhibited. These results clearly indicate that NH6 and NH17 both possess
an independent and direct anti-COX activity, which is unrelated to the enzymatic conversion of NH6 or NH17 into Diclofenac. Furthermore, in the case of COX-2, the results clearly indicate that both derivatives are superior to the parent molecule, Diclofenac, in inhibiting the enzyme and consequently inhibiting the extent of PGE2 synthesis. Also shown, as control, are the synthesis of PGE2 in the absence of any COX inhibitor (marked as control) as well as lack of PGE2 synthesis in the absence of the substrate Arachdonic acid (marked as no AA).
EXAMPLE 4 Anti-COX activities of compound in cell culture
The anti-COX activity of Compounds 1-3 was examined in Mouse colon adenocarcinoma CT26 and mouse lewis lung carcinoma D122 cell lines. Cells were incubated with each of the tested compounds and the level of the COX-dependent synthesis of PGE2 was quantified using radioimmunoassay techniques. The results are presented in Table 3 and show that all compounds exhibited anti-COX activity comparable to or better than that of Diclofenac.
Table 3

EXAMPLE 5
Anti-COX activity of NH6 and NH17, at a single dose of 1-10 mpk in tumor tissue and in stomach and kidney tissues In vivo Studies Treatment of cancer with NSAIDs is problematic due to gastrointestinal and renal side effects related to the NSAID anti-COX activity in the stomach and kidney organs.
The ability of NH6 and NH17, administered as a single dose, to exhibit the desired anti- COX activity it tumor tissue but not in stomach and kidney tissue was examined.
As shown in Figure 5A, Diclofenac, NH6 and NH17 at a single i.p. dose of 1 , 3 and 10 mg per kilogram, inhibit COX activity in tumor tissue to a similar extent. Surprisingly, as shown in Figure 5B, in the stomach, NH6 and NH17 induced rather than inhibited COX activity, as manifested by promoting PGE2 synthesis. In contrast, a similar COX activity induction was not observed upon administration of Diclofenac (see, Figure 5B). In the kidney, at a dosage of 3 mpk and 10 mpk, NH6 and NH17 exhibited anti-COX activity comparable to that of Diclofenac (see Figure 5C). Interestingly, at a dose of 1 mpk, NH6 and NH17 exhibited reduced anti-COX activity as compared to Diclofenac. In fact, in the case of 1 mpk of NH6, the levels of PGE2 were higher than in control, indicating that NH6 induced rather than reduced COX activity.
These results show that at a single dose administration of 1-10 mg per kilogram, NH6 and NH17 are potential anti-tumor agents due to their anti-COX activity. In contrast to Diclofenac, NH6 and NH17 at a concentration of 1-10 mg per kilogram, do not inhibit COX enzymes in the stomach and kidney but rather induce COX activity.
EXAMPLE 6 anti-COX activity of NH6 and NH17, administered at a chronic dosage of 3 mpk/day in tumor tissue and in stomach and kidney tissues
In vivo Studies
Assessment of the anti-COX activity of NH6 and NH17 in tumor, kidney and stomach tissue following chronic administratiom of the drugs, was evaluated. Tumor bearing BALB/c mice (10 mice per group) were injected intraperitoneally, daily, with either 2 mpk Indomethacin, 1 mpk Diclofenac or 3mpk NH6 or NH17 and the level of COX-inhibition was evaluated after 27- 33 days.
The results, presented in Figures 6A-C, show that NH6 and NH17, at a concentration of 3 mpk inhibited COX activity in the tumor tissue to a greater extent as compared to 1 mpk of Diclofenac and to a similar extent as compared to 2 mpk Indomethacin (see, Figure 6A). Surprisingly, in the stomach, NH6 and NH17 inhibited COX activity to a much lesser extent as compared to Diclofenac and Indomethacin (see, Figure 6B). Similar results were obtained in kidney tissue where, once again NH6 and NH17, exhibited reduced anti-COX activity as compared to Diclofenac and Indomethacin (see, Figure 6C).
EXAMPLE 7
Inhibition of tumor growth In vivo Studies
Mice (10 per group) were treated as described in Example 6 hereinabove. The tumor volume was measured at different time points post daily injections of either Diclofenac, Indomethacin, NH6 or NH17, during a time period of 27-33 days. The volume of footpad without tumor cells was subtracted from all measurements. The results are presented in Figure 7 and show that the rate of tumor growth inhibition in mice injected with 3 mpk/day NH6 and NH17 was comparable to that of mice injected with 2mpk/day Indomethacin and significantly higher as compared to mice injected with 1 mpk/day Diclofenac. Table 4 below presents the extent of tumor volume reduction by the various tested compounds, 27 days post injection.
Table 4

The results show that at a dosing of 3 mpk/day, NH6 and NH17 both exhibit anti-tumor activity which is comparable to that of 2 mpk/day Indomethacin and superior to that of 1 mpk/day Diclofenac.
EXAMPLE 8
Inhibition of acute local inflammation in rats
The ability of NH6 and NH17 to inhibit inflammation at a single dosage of 2 mpk or 6 mpk was examined and compared to a similar dosage of Diclofenac and Indomethacin. The results are presented in Table 5 below and show that NH6 and NH17 inhibit inflammation to a similar extent as compared to Diclofenac and Indomethacin.
Table 5

Although the invention has been described in conjunction with specific embodiments thereof, it is evident that many alternatives, modifications and variations will be apparent to those skilled in the art. Accordingly, it is intended to embrace all such alternatives, modifications and variations that fall within the spirit and broad scope of the appended claims.
All publications, patents and patent applications mentioned in this specification are herein incorporated in their entirety by reference into the specification, to the same extent as if each individual publication, patent or patent application was specifically and individually indicated to be incorporated herein by reference. In addition, citation or identification of any reference in this application shall not be construed as an admission that such reference is available as prior art to the present invention. To the extent that section headings are used, they should not be construed as necessarily limiting.
Claims
1. A pharmaceutical composition unit dosage form comprising an ester derivative of N-phenylanthranilic acid having the general Formula I:

Formula I wherein:
Z is an A-C(=K)-O-Y-Ra group;
A is alkyl or absent;
K is selected from the group consisting of O and S;
Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino, -NR12R13, and a positively charged group, whereas R12 and R13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R12 and R13 form a five- or six-member heteroalicyclic ring;
R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R2, R3, R4, R5, R6, R7, R8, R9 and R10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino and -NR14R15, or, alternatively, at least two of R2, R3, R4, R5 and R6, of R7, R8, R9 and R10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or heteroalicyclic ring; and
R14 and R15 are each independently selected from the group consisting of hydrogen,
14 alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R and R form a five- or six- 001255
44 member heteroalicyclic ring, and a pharmaceutically acceptable carrier, wherein an amount of said ester derivative is at least 1 mg/kg body weight, said unit dosage form being identified for use, from 1 to 4 times daily, in the treatment of a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
2. A pharmaceutical composition unit dosage form comprising an ester derivative of a N-phenylanthranilic acid having the general Formula I of claim 1 , and a pharmaceutically acceptable carrier, wherein an amount of said ester derivative is at least 50 mg, said unit dosage form being identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
3. A pharmaceutical composition unit dosage form comprising an ester derivative of a N-phenylanthranilic acid having the general Formula I of claim 1 and a pharmaceutically acceptable carrier, wherein an amount of said ester derivative is at least 1 mg/kg body weight, said unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to 4 times daily, in the treatment of a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
4. A pharmaceutical composition unit dosage form comprising an ester derivative of a N-phenylanthraniiic acid having the general Formula I of claim 1 , and a pharmaceutically acceptable carrier, wherein an amount of said ester derivative is higher than 50 mg, said unit dosage form being formulated for intraperitoneal administration and identified for use, from 1 to 4 times daily, in the treatment of a human subject having a medical condition selected from the group consisting of an inflammatory disease or disorder and cancer.
5. The pharmaceutical composition unit dosage form of claim 1, wherein said amount of said ester derivative is at least 2 mg/kg body weight.
6. The pharmaceutical composition unit dosage form of claim 1 , wherein said amount of said ester derivative is at least 3 mg/kg body weight.
7. The pharmaceutical composition unit dosage form of claim 1, wherein said amount of said ester derivative is at least 6 mg/kg body weight. IL2008/001255
45
8. The pharmaceutical composition unit dosage form of claim 1, wherein said amount of said ester derivative ranges from 1 mg/kg body weight to 10 mg/kg body weight.
9. The pharmaceutical composition unit dosage form of claim 2, wherein said amount of said ester derivative is higher than 70 mg.
10. The pharmaceutical composition unit dosage form of any of claims 1 to 9, wherein said unit dosage form is used 2 to 4 times a day.
11. The pharmaceutical composition unit dosage form of any of claims 1 , 5, 6 and 10, wherein said unit dosage form is identified for use such that a daily dose of said ester derivative is at least 2 mg/kg body weight.
12. The pharmaceutical composition unit dosage form of claim 11, wherein said daily dose of said ester derivative is at least 3 mg/kg body weight.
13. The pharmaceutical composition unit dosage form of any of claims 2, 9 and 10, wherein said unit dosage form is identified for use such that a daily dose of said ester derivative is higher than 150 mg.
14. The pharmaceutical composition unit dosage form of claim 13, wherein said daily dose of said ester derivative is higher than 200 mg.
15. The pharmaceutical composition unit dosage form of any of claims 1 , 2 and 5- 14, being formulated for intraperitoneal administration.
16. The pharmaceutical composition unit dosage form of any of claims 3 and 10, wherein said unit dosage form is identified for use such that a daily dose of said ester derivative is higher than 1 mg/kg body weight.
17. The pharmaceutical composition unit dosage form of any of claims 4 and 10, wherein said unit dosage form is identified for use such that a daily dose of said ester derivative is higher than 50 mg.
18. The pharmaceutical composition unit dosage form of any of claims 1-17, wherein said treatment is a chronic treatment.
19. The pharmaceutical composition unit dosage form of any of claims 1-18, being packaged in a packaging material and identified in print, in or on said packaging material, for use in the treatment of said medical condition.
20. A method of treating a medical condition selected from the group consisting of cancer and an inflammatory disease or disorder in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of an ester derivative of N-phenylanthranilic acid having a general Formula I:

Formula I wherein:
Z is an A-C(= K)-O-Y-Ra group;
A is alkyl or absent;
K is selected from the group consisting of O and S;
Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino, -NR12R13, and a positively charged group, whereas R12 and R13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R12 and R13 form a five- or six-member heteroalicyclic ring;
R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R2, R3, R4, R5, R6, R7, R8, R9 and R10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino and -NR14R15, or, alternatively, at least two of R2, R3, R4, R5 and R6, of R7, R8, R9 and R10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or heteroalicyclic ring; and R14 and R15 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and suifonyl, or, alternatively R14 and R15 form a five- or six- member heteroalicyclic ring.
21. The method of claim 20, wherein said therapeutically effective amount is at least 1 mg/kg body, and said administering is performed from 1 to 4 times a day.
22. The method of claim 20, wherein said therapeutically effective amount is higher than 50 mg, and said administering is performed from 1 to 4 times a day.
23. The method of claim 20, wherein said administering is effected intraperitoneal^, said therapeutically effective amount is higher than 1 mg/kg body, and said administering is performed from 1 to 4 times a day.
24. The method of claim 20, wherein said administering is effected intraperitoneally, said therapeutically effective amount being higher than 50 mg, and said administering is performed from 1 to 4 times a day.
25. The method of claim 20, wherein said therapeutically effective amount is at least 2 mg/kg body weight.
26. The method of claim 20, wherein said therapeutically effective amount is at least 3 mg/kg body weight.
27. The method of claim 20, wherein said therapeutically effective amount is at least 6 mg/kg body weight.
28. The method of claim 20, wherein said therapeutically effective amount is ranges from 1 mg/kg body weight to 10 mg/kg body weight.
29. The method of claim 22, wherein said therapeutically effective amount is higher than 70 mg.
30. The method of any of claims 20 to 29, wherein said administering is performed from 2 to 4 times a day.
31. The method of any of claims 20, 25, 26 and 30, wherein a daily dose of said ester derivative is higher than 2 mg/kg body weight.
32. The method of claim 31, wherein said daily dose of said ester derivative is higher than 3 mg/kg body weight.
33. The method of any of claims 22 and 30, wherein a daily dose of said ester derivative is higher than 150 mg.
34. The method of claim 33, wherein said daily dose of said ester derivative is higher than 200 mg.
35. The method of any of claims 20, 22 and 25-34, wherein said administering is performed intraperitoneally.
36. Use of an ester derivative of N-phenylanthranilic acid having a general Formula

Formula I wherein:
Z is an A-C(= K)-O-Y-Ra group;
A is alkyl or absent;
K is selected from the group consisting of O and S;
Y is selected from the group consisting of a substituted or unsubstituted alkyl, a substituted or unsubstituted alkenyl, a substituted or unsubstituted hydroxyalkyl, a substituted or unsubstituted hydroxyalkenyl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted heteroalicyclic, a substituted or unsubstituted heretroaryl and a substituted or unsubstituted polyalkylene glycol moiety;
Ra is independently selected from the group consisting of hydrogen, halo, hydroxy, alkoxy, aryloxy, heteroalicyclic, heteroaryl, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyi, sulfonyl, cyano, nitro, amino, -NR12R13, and a positively charged group, whereas R12 and R13 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R12 and R13 form a five- or six-member heteroalicyclic ring; R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl or aryl; each of R2, R3, R4, R5, R6, R7, R8, R9 and R10 is independently selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, halo, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, sulfinyl, sulfonyl, cyano, nitro, amino and -NR14R15, or, alternatively, at least two of R2, R3, R4, R5 and R6, of R7, R8, R9 and R10 form a five- or six-membered aromatic, heteroaromatic, alicyclic or heteroalicyclic ring; and
R14 and R15 are each independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, aryl, carbonyl and sulfonyl, or, alternatively R14 and R15 form a five- or six-member heteroalicyclic ring, in the manufacture of a medicament for treating a medical condition selected from the group consisting of cancer and an inflammatory disease or disorder.
37. The pharmaceutical composition unit dosage form, use or method of any of claims 1 to 36, wherein Y is selected from the group consisting of a substituted or unsubstituted alkyl and a substituted or unsubstituted polyalkylene glycol moiety.
38. The pharmaceutical composition unit dosage form, use or method of claim 37, wherein said polyalkylene glycol moiety has a general formula II:
[(CH2)m-0]n-
Formula Il wherein: each of m and n is independently an integer of 1-10.
39. The pharmaceutical composition unit dosage form, use or method of claim 38, wherein Ra is hydrogen.
40. The pharmaceutical composition unit dosage form, use or method of claim 38, wherein Ra is alkyl.
41. The pharmaceutical composition unit dosage form, use or method of claim 38, wherein Ra is a positively charged group.
42. The pharmaceutical composition unit dosage form, use or method of claim 41 , wherein said positively charged group is an ammonium group.
43. The pharmaceutical composition unit dosage form, use or method of any of claims 1-42, wherein:
G is C;
K is O; each of R2, R3, R4, R5 and R6 is independently selected from the group consisting of hydrogen, alkyl, halo and trihaloalkyl; and each of R7, R8, R9 and R10 is hydrogen.
44. The pharmaceutical composition unit dosage form, use or method of any of claims 1-43, wherein said N-phenylanthranilic acid is Diclofenac.
45. The pharmaceutical composition unit dosage form, use or method of any of claims 1-44, wherein said ester derivative is selected from the group consisting of:

and

46. The pharmaceutical composition unit dosage form, use or method of any of claims 1-45, wherein treating said medical condition comprises chronic treatment.
47. The pharmaceutical composition unit dosage form, use or method of claim 46, wherein said chronic treatment comprises administering said ester derivative for a time period that ranges from 20 to 40 days.
48. The pharmaceutical composition unit dosage form, use or method of any of claims 1-47, wherein said medical condition is cancer.
49. The pharmaceutical composition unit dosage form, use or method of any of claims 1-47 , wherein said inflammatory disease or disorder is selected from a group consisting of Alzheimer's disease, cortical dementia, vascular dementia, muli-infract dementia, pre-senile dementia, alcoholic dementia, senile dementia, memory loss or central nervous damage resulting from stroke, ischemia or trauma, multiple sclerosis, Parkinson's disease, Huntington's disease, epilepsy, cystic fibrosis, arthritis diseases such as osteoarthritis, rheumatoid arthritis, spondyloarthopathies, gouty arthritis, systemic lupus erythematosus, and juvenile arthritis fever, periarteritis; gastrointestinal disorders such as inflammatory bowel disease, Chron's disease, gastritis, irritable bowel syndrome, ulcerative colitis, cardiovascular disorders such as myocardial ischemia, reperfusion injury to an ischemic organ; angiogenesis, asthma, bronchitis, menstrual cramps, premature labor, tendinitis, bursitis, an autoimmune disease, an immunological disorder, systemic lupus erythematosus, inflammatory disorders of the skin such as psoriasis, eczema, burns and dermatitis; neoplasia, an inflammatory process in a disease, pulmonary inflammation, a central nervous system disorder, migraine headaches, allergic rhinitis, respiratory distress syndrome, endotoxin shock syndrome, a microbial infection, a bacterial-induced inflammation, a viral induced inflammation, a urinary disorder, a urological disorder, endothelial dysfunction, organ deterioration, tissue deterioration, adhesion and infiltration of neutrophils at the site of inflammation, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, rheumatic fever, myasthenia gravis, sarcoidosis, nephrotic syndrome, Behcet's syndrome, polymyositis, hypersensitivity, conjunctivitis, gingivitis and swelling occurring after injury.
50. The pharmaceutical composition unit dosage form of any of claims 1-18 packaged in a packaging material and identified in print, in or on said packaging material, for use in the treatment of cancer and an inflammatory disease or disorder.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US96021607P | 2007-09-20 | 2007-09-20 | |
| US60/960,216 | 2007-09-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009037705A2 true WO2009037705A2 (en) | 2009-03-26 |
| WO2009037705A3 WO2009037705A3 (en) | 2009-07-09 |
Family
ID=40468560
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IL2008/001255 WO2009037705A2 (en) | 2007-09-20 | 2008-09-18 | Esters of n-phenylanthranilic acid for use in the treatment of cancer and inflammation |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009037705A2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8278357B2 (en) | 2002-10-21 | 2012-10-02 | Ramot At Tel-Aviv University Ltd. | Derivatives of N-phenylanthranilic acid and 2-benzimidazolone as potassium channel and/or neuron activity modulators |
| WO2012012278A3 (en) * | 2010-07-20 | 2013-02-14 | Corning Incorporated | Compositions and methods for the treatment of pathological condition(s) related to gpr35 and/or gpr35-herg complex |
| WO2013135147A1 (en) * | 2012-03-14 | 2013-09-19 | 中国中化股份有限公司 | Use of substituted diphenylamine compounds in preparing anti-tumour drugs |
| US8765815B2 (en) | 2007-09-20 | 2014-07-01 | Ramot At Tel-Aviv University Ltd. | N-phenyl anthranilic acid derivatives and uses thereof |
| CN105267214A (en) * | 2014-07-21 | 2016-01-27 | 沈阳化工研究院有限公司 | Application of N-heteroaryl phenylamine compounds for preparation of antitumor drugs |
Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3652762A (en) * | 1970-04-14 | 1972-03-28 | Ciba Geigy Corp | Pharmaceutical compositions and methods employing substituted derivatives of 2-anilinophenylacetic acids and esters |
| FR2411825A1 (en) * | 1977-02-26 | 1979-07-13 | Hokuriku Pharmaceutical | N- (TRIFLUOROMETHYL-3-PHENYL) ALKYL ANTHRANILATE, USED AS ANTI-INFLAMMATORY AGENT |
| FR2462420A1 (en) * | 1979-07-31 | 1981-02-13 | Simes | O-ALCOXYCARBONYL-PHENYL ESTERS OF ANTHRANILIC ACID, THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP0131790A2 (en) * | 1983-07-01 | 1985-01-23 | Troponwerke GmbH & Co. KG | Etofenamate preparation |
| US4734434A (en) * | 1986-03-11 | 1988-03-29 | Rorer Pharmaceutical Corporation | Method for the treatment of pruritus and composition for the use therein |
| EP0307599A1 (en) * | 1987-08-12 | 1989-03-22 | Bayer Ag | Medicaments for the region of the buccal cavity |
| JPH0196158A (en) * | 1987-10-09 | 1989-04-14 | Santen Pharmaceut Co Ltd | Anti-inflammatory agent |
| EP0335164A2 (en) * | 1988-03-31 | 1989-10-04 | Merckle GmbH | Ester derivatives of neopentanol, process for their preparation and their use as medicines |
| EP0336147A2 (en) * | 1988-03-31 | 1989-10-11 | Merckle GmbH | Diethylene glycol monoesters, process for their preparation and their use as medicines |
| US5017604A (en) * | 1990-04-11 | 1991-05-21 | Warner-Lambert Company | Novel fenamic acid methyl hydroxamate derivatives having cyclooxygenase and 5-lipoxygenase inhibition |
| WO1999001421A1 (en) * | 1997-07-01 | 1999-01-14 | Warner-Lambert Company | 2-(4-bromo or 4-iodo phenylamino) benzoic acid derivatives and their use as mek inhibitors |
| WO2001093680A1 (en) * | 2000-06-02 | 2001-12-13 | Medinox, Inc. | Protected forms of a combination of pharmacologically active agents and uses therefor |
| US6355666B1 (en) * | 2000-06-23 | 2002-03-12 | Medinox, Inc. | Protected forms of pharmacologically active agents and uses therefor |
| WO2002065977A2 (en) * | 2001-02-20 | 2002-08-29 | Israel Institute For Biological Research | Compounds co-inducing cholinergic up-regulation and inflammation down-regulation and uses thereof |
| WO2003103602A2 (en) * | 2002-06-11 | 2003-12-18 | Nitromed, Inc. | Nitrosated and/or nitrosylated cyclooxygenase-2 selective inhibitors, compositions and methods of use |
| WO2004000300A1 (en) * | 2002-06-25 | 2003-12-31 | Nicox S.A. | Cyclooxygenase-2 inhibitors |
| WO2004026808A1 (en) * | 2002-09-20 | 2004-04-01 | Nicox Sa. | Manufacturing process for no-donating compounds such as no-donating diclofenac |
| WO2004052840A1 (en) * | 2002-12-12 | 2004-06-24 | Galderma Research & Development, S.N.C. | Compounds which modulate pparϝ type receptors, and use thereof in cosmetic or pharmaceutical compositions |
| WO2005009975A2 (en) * | 2003-07-24 | 2005-02-03 | Warner-Lambert Company Llc | Benzimidazole derivatives as mek inhibitors |
| US20050250833A1 (en) * | 2002-10-21 | 2005-11-10 | Ramot At Tel Aviv University Ltd. | Derivatives of N-phenylanthranilic acid and 2-benzimidazolone as potassium channel and/or neuron activity modulators |
| WO2006042387A1 (en) * | 2004-10-21 | 2006-04-27 | Cmax Otimizacão De Resultados S C Ltda | Drugs derived from diclofenac containing no-donor heterocycles, composition and method of inflammation treatment |
| WO2006117316A1 (en) * | 2005-04-29 | 2006-11-09 | Nycomed Gmbh | Mutual prodrug compounds for use us antiinflammatory agents with gastrointestinal protective activity |
| US20080004245A1 (en) * | 2005-05-27 | 2008-01-03 | Antibe Therapeutics Inc. | Hydrogen sulfide derivatives of non-steroidal anti-inflammatory drugs |
| WO2008012602A1 (en) * | 2006-07-25 | 2008-01-31 | Techfields Biochem Co. Ltd | Positively charged water-soluble prodrugs of diclofenac with very fast skin penetration rate |
| WO2008029199A1 (en) * | 2006-09-03 | 2008-03-13 | Techfields Biochem Co. Ltd | Positively charged water-soluble prodrugs of n-arylanthranilic acids with very fast skin penetration rate |
| US7378442B1 (en) * | 2007-03-11 | 2008-05-27 | Sami Labs Limited | Chemical entities with multiple modes of anti-inflammatory action |
| WO2009007827A2 (en) * | 2007-07-10 | 2009-01-15 | Chemisches Institut Schaefer Ag | Novel diclofenac esters and uses thereof |
| WO2009037707A2 (en) * | 2007-09-20 | 2009-03-26 | Ramot At Tel Aviv University Ltd. | N-phenyl anthranilic acid derivatives and uses thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63275549A (en) * | 1987-05-07 | 1988-11-14 | ファブリカ、エスパニョ−ラ、デ、プロドゥクトス、キミコス、イ、ファルマセウティコス、ソシエダ、アノニマ (エ−フェア−エ−エ−セ) | Diethyleneglycol ester and medicine containing same |
| JPS63275593A (en) * | 1987-05-08 | 1988-11-14 | Fujirebio Inc | Beta-aminoethylphosphoric acid ester derivative |
-
2008
- 2008-09-18 WO PCT/IL2008/001255 patent/WO2009037705A2/en active Application Filing
Patent Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3652762A (en) * | 1970-04-14 | 1972-03-28 | Ciba Geigy Corp | Pharmaceutical compositions and methods employing substituted derivatives of 2-anilinophenylacetic acids and esters |
| FR2411825A1 (en) * | 1977-02-26 | 1979-07-13 | Hokuriku Pharmaceutical | N- (TRIFLUOROMETHYL-3-PHENYL) ALKYL ANTHRANILATE, USED AS ANTI-INFLAMMATORY AGENT |
| FR2462420A1 (en) * | 1979-07-31 | 1981-02-13 | Simes | O-ALCOXYCARBONYL-PHENYL ESTERS OF ANTHRANILIC ACID, THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP0131790A2 (en) * | 1983-07-01 | 1985-01-23 | Troponwerke GmbH & Co. KG | Etofenamate preparation |
| US4734434A (en) * | 1986-03-11 | 1988-03-29 | Rorer Pharmaceutical Corporation | Method for the treatment of pruritus and composition for the use therein |
| EP0307599A1 (en) * | 1987-08-12 | 1989-03-22 | Bayer Ag | Medicaments for the region of the buccal cavity |
| JPH0196158A (en) * | 1987-10-09 | 1989-04-14 | Santen Pharmaceut Co Ltd | Anti-inflammatory agent |
| EP0335164A2 (en) * | 1988-03-31 | 1989-10-04 | Merckle GmbH | Ester derivatives of neopentanol, process for their preparation and their use as medicines |
| EP0336147A2 (en) * | 1988-03-31 | 1989-10-11 | Merckle GmbH | Diethylene glycol monoesters, process for their preparation and their use as medicines |
| US5017604A (en) * | 1990-04-11 | 1991-05-21 | Warner-Lambert Company | Novel fenamic acid methyl hydroxamate derivatives having cyclooxygenase and 5-lipoxygenase inhibition |
| WO1999001421A1 (en) * | 1997-07-01 | 1999-01-14 | Warner-Lambert Company | 2-(4-bromo or 4-iodo phenylamino) benzoic acid derivatives and their use as mek inhibitors |
| WO2001093680A1 (en) * | 2000-06-02 | 2001-12-13 | Medinox, Inc. | Protected forms of a combination of pharmacologically active agents and uses therefor |
| US6355666B1 (en) * | 2000-06-23 | 2002-03-12 | Medinox, Inc. | Protected forms of pharmacologically active agents and uses therefor |
| WO2002065977A2 (en) * | 2001-02-20 | 2002-08-29 | Israel Institute For Biological Research | Compounds co-inducing cholinergic up-regulation and inflammation down-regulation and uses thereof |
| WO2003103602A2 (en) * | 2002-06-11 | 2003-12-18 | Nitromed, Inc. | Nitrosated and/or nitrosylated cyclooxygenase-2 selective inhibitors, compositions and methods of use |
| WO2004000300A1 (en) * | 2002-06-25 | 2003-12-31 | Nicox S.A. | Cyclooxygenase-2 inhibitors |
| WO2004026808A1 (en) * | 2002-09-20 | 2004-04-01 | Nicox Sa. | Manufacturing process for no-donating compounds such as no-donating diclofenac |
| US20050250833A1 (en) * | 2002-10-21 | 2005-11-10 | Ramot At Tel Aviv University Ltd. | Derivatives of N-phenylanthranilic acid and 2-benzimidazolone as potassium channel and/or neuron activity modulators |
| WO2004052840A1 (en) * | 2002-12-12 | 2004-06-24 | Galderma Research & Development, S.N.C. | Compounds which modulate pparϝ type receptors, and use thereof in cosmetic or pharmaceutical compositions |
| WO2005009975A2 (en) * | 2003-07-24 | 2005-02-03 | Warner-Lambert Company Llc | Benzimidazole derivatives as mek inhibitors |
| WO2006042387A1 (en) * | 2004-10-21 | 2006-04-27 | Cmax Otimizacão De Resultados S C Ltda | Drugs derived from diclofenac containing no-donor heterocycles, composition and method of inflammation treatment |
| WO2006117316A1 (en) * | 2005-04-29 | 2006-11-09 | Nycomed Gmbh | Mutual prodrug compounds for use us antiinflammatory agents with gastrointestinal protective activity |
| US20080004245A1 (en) * | 2005-05-27 | 2008-01-03 | Antibe Therapeutics Inc. | Hydrogen sulfide derivatives of non-steroidal anti-inflammatory drugs |
| WO2008012602A1 (en) * | 2006-07-25 | 2008-01-31 | Techfields Biochem Co. Ltd | Positively charged water-soluble prodrugs of diclofenac with very fast skin penetration rate |
| WO2008029199A1 (en) * | 2006-09-03 | 2008-03-13 | Techfields Biochem Co. Ltd | Positively charged water-soluble prodrugs of n-arylanthranilic acids with very fast skin penetration rate |
| US7378442B1 (en) * | 2007-03-11 | 2008-05-27 | Sami Labs Limited | Chemical entities with multiple modes of anti-inflammatory action |
| WO2009007827A2 (en) * | 2007-07-10 | 2009-01-15 | Chemisches Institut Schaefer Ag | Novel diclofenac esters and uses thereof |
| WO2009037707A2 (en) * | 2007-09-20 | 2009-03-26 | Ramot At Tel Aviv University Ltd. | N-phenyl anthranilic acid derivatives and uses thereof |
Non-Patent Citations (8)
| Title |
|---|
| "Supporting Text S1. Synthesis of Compounds" 20071226, 26 December 2007 (2007-12-26), pages 1-13, XP007908469 * |
| BONINA F ET AL: "Oligoethylene ester derivatives of ketoprofen, naproxen and diclofenac as oral prodrugs: a pharmacological evaluation" PHARMAZIE, DIE, GOVI VERLAG, ESCHBORN, DE, vol. 57, no. 8, 1 August 2002 (2002-08-01), pages 552-555, XP001539451 ISSN: 0031-7144 * |
| CENZO CONGIU ET AL: "New Potential Anticancer Agents Based on the Anthranilic Acid Scaffold. Synthesis and Evaluation of Biological Activity" JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON, US, vol. 48, no. 26, 1 January 2005 (2005-01-01), pages 8245-8252, XP007908449 ISSN: 0022-2623 [retrieved on 2005-11-15] * |
| DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; KUTSUMA, TERUO ET AL: "Preparation of .beta.-aminoethyl phosphates containing indomethacin- and diclofenac residues as analgesic inflammation inhibitors" XP002526999 retrieved from STN Database accession no. 1989:193120 & JP 63 275593 A (FUJIREBIO, INC., JAPAN) 14 November 1988 (1988-11-14) * |
| DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; ORJALES VENERO, AURELIO ET AL: "Preparation of hydroxyethoxyethyl 2-(2,6-dichlorophenylamino)phenylacetate as an analgesic and antiinflammatory" XP002526998 retrieved from STN Database accession no. 1989:172889 & JP 63 275549 A (FABRICA ESPANOLA DE PRODUCTOS QUIMICOS Y FARMACEUTICOS S. A. , SPAIN) 14 November 1988 (1988-11-14) * |
| FREGNAN G B ET AL: "Local anti-inflammatory properties of etoclofene. A new fenamate derivative" PHARMACOLOGY, S. KARGER AG, vol. 11, no. 4, 1 January 1974 (1974-01-01), pages 213-219, XP009116638 ISSN: 0031-7012 * |
| KALGUTKAR A S ET AL: "Amide Derivatives of Meclofenamic Acid as Selective Cyclooxygenase-2 Inhibitors" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 12, 1 January 2002 (2002-01-01), pages 521-524, XP002319950 ISSN: 0960-894X * |
| PERETZ ASHER ET AL: "A tale of switched functions: from cyclooxygenase inhibition to M-channel modulation in new diphenylamine derivatives" PLOS ONE, PUBLIC LIBRARY OF SCIENCE, SAN FRANCISCO, CA, US, vol. 2, no. 12, 26 December 2007 (2007-12-26), pages e1332-1, XP009116544 ISSN: 1932-6203 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8278357B2 (en) | 2002-10-21 | 2012-10-02 | Ramot At Tel-Aviv University Ltd. | Derivatives of N-phenylanthranilic acid and 2-benzimidazolone as potassium channel and/or neuron activity modulators |
| US8618169B2 (en) | 2002-10-21 | 2013-12-31 | Ramot At Tel-Aviv University Ltd. | Derivatives of N-phenylanthranilic acid and 2-benzimidazolone as potassium channel and/or neuron activity modulators |
| US9403756B2 (en) | 2007-09-20 | 2016-08-02 | Ramot At Tel-Aviv University Ltd. | N-phenyl anthranilic acid derivatives and uses thereof |
| US8765815B2 (en) | 2007-09-20 | 2014-07-01 | Ramot At Tel-Aviv University Ltd. | N-phenyl anthranilic acid derivatives and uses thereof |
| WO2012012278A3 (en) * | 2010-07-20 | 2013-02-14 | Corning Incorporated | Compositions and methods for the treatment of pathological condition(s) related to gpr35 and/or gpr35-herg complex |
| CN103237791A (en) * | 2010-07-20 | 2013-08-07 | 康宁股份有限公司 | Compositions and methods for the treatment of pathological condition(s) related to GPR35 and/or GPR35-HERG complex |
| KR20140097444A (en) * | 2012-03-14 | 2014-08-06 | 시노켐 코포레이션 | Substitute diphenylamine compounds use thereof as antitumor agents |
| CN104136021A (en) * | 2012-03-14 | 2014-11-05 | 中国中化股份有限公司 | Use of substituted diphenylamine compounds in preparing anti-tumour drugs |
| EP2826474A1 (en) * | 2012-03-14 | 2015-01-21 | Sinochem Corporation | Use of substituted diphenylamine compounds in preparing anti-tumour drugs |
| EP2826474A4 (en) * | 2012-03-14 | 2015-04-08 | Sinochem Corp | Use of substituted diphenylamine compounds in preparing anti-tumour drugs |
| KR101599300B1 (en) | 2012-03-14 | 2016-03-03 | 시노켐 코포레이션 | Substitute diphenylamine compounds use thereof as antitumor agents |
| CN104136021B (en) * | 2012-03-14 | 2016-04-20 | 中国中化股份有限公司 | Substituted diphenylamine aminated compounds is as the application preparing antitumor drug |
| US9376376B2 (en) | 2012-03-14 | 2016-06-28 | Sinochem Corporation | Substitute diphenylamine compounds use thereof as antitumor agents |
| WO2013135147A1 (en) * | 2012-03-14 | 2013-09-19 | 中国中化股份有限公司 | Use of substituted diphenylamine compounds in preparing anti-tumour drugs |
| CN105267214A (en) * | 2014-07-21 | 2016-01-27 | 沈阳化工研究院有限公司 | Application of N-heteroaryl phenylamine compounds for preparation of antitumor drugs |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009037705A3 (en) | 2009-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5758395B2 (en) | Sepiapterin reductase inhibitors for the treatment of pain | |
| JP5424880B2 (en) | Positively charged water-soluble prodrugs of aryl- and heteroarylpropionic acids with very fast skin permeability | |
| CN106866571B (en) | Heterocyclic urea compound and its pharmaceutical composition and application | |
| ES2741439T3 (en) | Aromatic Compounds Substituted | |
| ES2922425T3 (en) | NSAIA prodrugs with very high skin and membrane penetration rates and their new medicinal uses | |
| TWI630194B (en) | Bicyclic analgesic compounds | |
| US8048926B2 (en) | L-DOPA amide derivatives and uses thereof | |
| IL229246A (en) | Partially saturated tricyclic compounds and methods of making and using same | |
| JPH11509519A (en) | Compositions and methods for preventing non-steroidal anti-inflammatory drug-induced toxicity | |
| JPH11302173A (en) | Histone deacetylase inhibitor | |
| CA2879456A1 (en) | Substituted aminoindane- and aminotetralincarboxylic acids and use thereof | |
| CN102260190B (en) | N-benzyl-N'-(terminal carboxylic acid substituted acyloxy)octanediamide compounds with anti-tumor effect and pharmaceutical salts thereof | |
| WO2007149782A2 (en) | Selective inhibitors for transferases | |
| WO2009037705A2 (en) | Esters of n-phenylanthranilic acid for use in the treatment of cancer and inflammation | |
| JPH11509540A (en) | Salt of amidine derivative and cyclooxygenase inhibitor, method for producing the same, application as a medicine, and pharmaceutical composition containing the same | |
| EP2124930A1 (en) | Isosorbide mononitrate derivatives for the treatment of intestinal disorders | |
| KR101497113B1 (en) | Aryloxyphenoxyacryl-based compounds having HIF-1 inhibition activity, preparation method thereof and pharmaceutical composition containing the same as an active ingredient | |
| WO2012078519A2 (en) | 3-acylidene-2-oxoindole derivatives for inhibition of transglutaminase 2 | |
| CN113004356B (en) | Novel genipin derivative, and preparation method and application thereof | |
| Qin et al. | Design and synthesis of pseudo-rutaecarpines as potent anti-inflammatory agents via regulating MAPK/NF-κB pathways to relieve inflammation-induced acute liver injury in mice | |
| WO2011125930A1 (en) | Prophylactic or therapeutic agent for inflammatory bowel diseases or autoimmune peripheral neuropathy | |
| WO2021023016A1 (en) | Thiazolidone derivative of lovatinib acid and application thereof | |
| JP2018505184A (en) | Compounds containing indoleacetic acid core structure and their applications | |
| JP2023535692A (en) | Entero-degradable co-drugs, preparation and use thereof | |
| JP5940036B2 (en) | Positively charged water-soluble prodrugs of aryl- and heteroarylpropionic acids with very fast skin permeability |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08808056 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08808056 Country of ref document: EP Kind code of ref document: A2 |