WO2007089149A2 - Water-soluble cc-1065 analogs and their conjugates - Google Patents
Water-soluble cc-1065 analogs and their conjugates Download PDFInfo
- Publication number
- WO2007089149A2 WO2007089149A2 PCT/NL2007/050043 NL2007050043W WO2007089149A2 WO 2007089149 A2 WO2007089149 A2 WO 2007089149A2 NL 2007050043 W NL2007050043 W NL 2007050043W WO 2007089149 A2 WO2007089149 A2 WO 2007089149A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- isomer
- compound according
- compound
- moiety
- optionally substituted
- Prior art date
Links
- 0 CCC(C)(CC[n]1nnc(CNC(OC(C)CCOC(C)*)=O)c1)OCC*(C)O*=O Chemical compound CCC(C)(CC[n]1nnc(CNC(OC(C)CCOC(C)*)=O)c1)OCC*(C)O*=O 0.000 description 6
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/58—[b]- or [c]-condensed
- C07D209/60—Naphtho [b] pyrroles; Hydrogenated naphtho [b] pyrroles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
Definitions
- This invention relates to novel analogs of the DNA-binding alkylating agent CC-1065 and to their conjugates. Furthermore this invention concerns intermediates for the preparation of said agents and their conjugates.
- the conjugates are designed to release their (multiple) payload after one or more activation steps and/or at a rate and time span controlled by the conjugate in order to selectively deliver and/or controllably release one or more of said DNA alkylating agents.
- the agents, conjugates, and intermediates can be used to treat an illness that is characterized by undesired (cell) proliferation.
- the agents and the conjugates of this invention may be used to treat a tumor.
- duoca ⁇ nycins first isolated from a culture broth of Streptomyces species, are parent members of a family of antitumor antibiotics that also includes CC-1065. These extremely potent agents allegedly derive their biological activity from an ability to sequence-selectively alkylate DNA at the N3 of adenine in the minor groove, which initiates a cascade of events that terminates in an apoptotic cell death mechanism. 1 Although CC-1065 has shown very potent cytotoxicity, it could not be used in the clinic because of serious delayed hepatotoxicity. 2 This observation led to the development of synthetic analogs of CC-1065 (see for CC-1065 derivatives for example Aristoff et al, J. Org. Chem.
- these derivatives lack sufficient selectivity for tumor cells as the selectivity of these agents - and cytotoxic agents in general - is for a considerable part based on the difference in the rate of proliferation of tumor cells and normal cells, and therefore they also affect healthy cells that show a high proliferation rate. This typically leads to severe side effects. Drug concentrations that would completely eradicate the tumor cannot be reached because of dose-limiting side effects such as gastrointestinal tract and bone marrow toxicity. In addition, tumors can develop resistance against anticancer agents after prolonged treatment. In modem drug development, targeting of cytotoxic drugs to the tumor site can be considered one of the primary goals.
- tumor-associated antigens, receptors, and other receptive moieties which can serve as a target.
- a target may be upregulated or to some degree be specifically present in tumor tissue or in closely associated tissue, such as neovascular tissue, with respect to other tissues in order to achieve efficient targeting.
- Many targets have been identified and validated and several methods to identify and validate targets have been developed. 3
- a ligand e.g. an antibody or antibody fragment
- tumor-associated enzymes Another promising approach to obtain selectivity for tumor cells or tumor tissue is to exploit the existence of tumor-associated enzymes.
- a relatively high level of tumor-specific enzyme can convert a pharmacologically inactive prodrug, which consists of an enzyme substrate directly or indirectly linked to the toxic drug, to the corresponding drug in the vicinity of or inside the tumor.
- a high concentration of toxic anticancer agent can be selectively generated at the tumor site. All tumor cells may be killed if the dose is sufficiently high, which may decrease development of drug-resistant tumor cells.
- Enzymes have also been transported to the vicinity of or inside target cells or target tissue via for example antibody-directed enzyme prodrug therapy (ADEPT) 4 , polymer-directed enzyme prodrug therapy (PDEPT) or macromolecular-directed enzyme prodrug therapy (MDEPT) 5 , virus-directed enzyme prodrug therapy (VDEPT) 6 , or gene-directed enzyme prodrug therapy (GDEPT) 7 .
- ADEPT antibody-directed enzyme prodrug therapy
- PDEPT polymer-directed enzyme prodrug therapy
- MDEPT macromolecular-directed enzyme prodrug therapy
- VDEPT virus-directed enzyme prodrug therapy
- GDEPT gene-directed enzyme prodrug therapy
- EPR enhanced permeability and retention
- a therapeutic agent By coupling a therapeutic agent directly or indirectly to a macromolecule, said agent can be selectively targeted to tumor tissue.
- another criterion for the successful application of targeted conjugates of cytotoxic agents in tumor therapy is that the one or more agents are released efficiently from the conjugate.
- a further important criterion is that the conjugate is non-cytotoxic or only very weakly cytotoxic, whereas the cytotoxic agent itself exhibits highly potent cytotoxicity.
- the conjugate must have suitable pha ⁇ naco logical properties, such as sufficient stability in the circulation, low aggregation tendency, and good water solubility.
- glycoside conjugates of seco CC- 1065 analogs (analogs in which the cyclopropyl ring as present in CC- 1065 is "opened") have been described that can be activated at the lesion site via an ADEPT approach.
- the difference in cytotoxicity between the conjugates and the corresponding drugs (the cytotoxicity quotient, herein defined as IC 50 , c o n jugate/ICso, pa r ent dr ug ) was however relatively low, and the seco CC- 1065 analogs themselves did not show extremely potent cytotoxicity.
- the present invention fulfils the above-mentioned need with a compound of formula (I) or (II):
- R 1 is selected from halogen and OSO 2 R U ; wherein R u is selected from optionally substituted Ci -6 alkyl, Ci -6 perhaloalkyl, benzyl, and phenyl;
- R 2 is selected from H and optionally substituted Ci-s alkyl
- R 3 , R 3 , R 4 , and R 4 are independently selected from H and optionally substituted Ci-8 alkyl, wherein two or more of R 2 , R 3 , R 3 , R 4 , and R 4 are optionally joined to form one or more optionally substituted carbocycles or heterocycles;
- X 2 is selected from O, C(R 14 )(R 14' ), and NR 14' , wherein R 14 is selected from H and optionally substituted Ci -S alkyl or acyl and wherein R 14 may be absent or be selected from H and optionally substituted Ci -8 alkyl or acyl;
- Each R 5 , R 5' , R 6 , R 6' , R 7 , and R 7' is independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO,
- OS(O)R k OS(O) 2 R k , OS(O)OR k , OS(O) 2 OR k , OR k , NHR k , N(R k )R L , "N(R k )(R L )R m ,
- R L , and R m are independently selected from H and optionally substituted C 1-4 alkyl,
- X 1 is selected from O, S, and NR 13 , wherein R 13 is selected from H and optionally substituted
- X 3 is selected from O, S, and NR !5 , wherein R 15 is selected from H and optionally substituted
- X 4 is selected from N and CR 16 , wherein R 16 is selected from H and optionally substituted C 1 -8 alkyl or acyl; X s is selected from O, S, and NR 17 , wherein R 17 is selected from H and optionally substituted C i-8 aEcyl or acyl;
- R 8 , R 9 , R 10 , and R 11 are each independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO, CF 3 , CN, C(O)NH 2 , C(O)H, C(O)OH, halogen, R x , SR X , S(O)R X , S(O) 2 R X , S(O)OR X , S(O) 2 OR X , OS(O)R X , OS(O) 2 R*, OS(O)OR X , OS(O) 2 OR X , OR X , NHR X , N(R x )R y , + N(R ⁇ )R 2 , P(O)(OR x )(OR y ), OP(O)(OR x )(OR y ), SiR x R y R z , C(O)R X , C(O)OR X
- the invention relates to a compound of formula (III):
- V 2 is either absent or a functional moiety
- Each L 2 is independently absent or a linking group linking V 2 to L or to V 1 or Y when L is absent;
- Each L is independently absent or a linking group linking L 2 or V 2 when L 2 is absent to one or more V 1 and/or Y;
- Each V 1 is independently H or a conditionally-cleavable or conditionally-transformable moiety, which can be cleaved or transformed by a chemical, photochemical, physical, biological, or enzymatic process;
- Each Y is independently absent or a self-eliminating spacer system which is comprised of 1 or more self-elimination spacers and is linked to V 1 , optionally L, and one or more Z;
- Each p and q are numbers representing a degree of branching and are each independently a positive integer;
- z is a positive integer equal to or smaller than the total number of attachment sites for Z in the one or more V 1 -Y moieties;
- Each Z is independently a compound of formula (I) or (II) as defined hereinabove wherein one or more of X 1 , R 6 , R 7 , R 8 , R 9 , R 10 , and R 11 may optionally in addition be substituted by a substituent of formula (V):
- each V 2 ', L 2 ', L', V 1 ', Y', Z', p 1 , q', and z' have the same meaning as defined for V 2 , L 2 , L, V 1 , Y, Z, p, q, and z, the one or more substituents of formula (V) being independently connected to one or more of X 1 , R 6 , R 7 , R 8 , R 9 , R 10 , and R 11 via Y' or V 1 ' when Y' is absent, each Z being connected to Y or V 1 when Y is absent through either X 1 or an atom in R 6 , R 7 , R 8 , R 9 , R 10 , or R u ; with the proviso that at least one of the one or more V 1 and the one or more V 1 is not H.
- z does not represent a degree of polymerization; hence z does not indicate that a number of moieties Z are connected to one another.
- the present invention also relates to a compound of formula (IV):
- RM is a reactive moiety and L, V 1 , Y, Z, p, and z are as defined above, except that L is now linking RM to one or more V 1 and/or Y, and V 1 , Y, and Z may contain protecting groups and the one or more V 2 '-L 2 ' moieties optionally present in Z as defined hereinabove may optionally and independently be replaced by RM', which is a reactive moiety, and wherein, if there is more than 1 reactive moiety in (IV), the reactive moieties are the same or different.
- These linker-agent conjugates may or may not be considered intermediates for compounds of formula (III).
- this invention relates to the cyclopropyl ring-containing analogs of compounds of formulae (I) and (II) which result from rearrangement of and concomitant elimination of H-R 1 from the corresponding seco compounds of formulae (I) and (II) ( Figure 1).
- Said cyclopropyl ring- containing analogs are believed to be the active species, allegedly being formed from compounds of formulae (I) and (II) in vivo via said rearrangement.
- This invention relates to enantiomerically pure and/or diastereomerically pure compounds of formulae (I) - (IV) as well as to enantiomeric and/or diastereomeric mixtures of compounds of formulae (I) - (IV).
- Figure 1 illustrates the rearrangement of a seco compound to a cyclopropyl-containing compound.
- Figure 2 illustrates the preparation of some agents by coupling a DNA-alkylating unit to a DNA- binding unit.
- Figure 3 illustrates the preparation of some ⁇ -galactopyranose conjugates of this invention.
- FIG. 4 depicts the preparation of DNA binder 13.
- FIG. 5 illustrates the preparation of DNA binder 19.
- Figure 6 describes the preparation of DNA binder 27.
- Figure 7 illustrates the preparation of DNA binder 30.
- Figure 8 illustrates the preparation of compounds 37 - 39 from DNA binder 41.
- Figure 9 illustrates the synthesis of agent 45.
- Figure 10 describes the synthesis of agent 33.
- Figure 11 illustrates the preparation of conjugate 36.
- Figure 12 describes the synthesis of linker-agent conjugates 47a-f.
- Figure 13 depicts the synthesis of linker-agent conjugates 48c-d.
- Figure 14 illustrates the preparation of linker-agent conjugates 50a-c.
- Figure 15 describes the synthesis of activated linkers 57-60.
- Figure 16 depicts the in vitro cytotoxicity of conjugates 8a and 8a' against the human lung carcinoma cell line A549.
- Figure 17 depicts the treatment schedule for an ADEPT in vivo experiment.
- Figure 18 depicts the tumor volume of treated and control mice in an ADEPT in vivo experiment.
- antibody refers to a full length immunoglobulin molecule, an immunologically active portion of a full-length immunoglobulin molecule, or a derivative of a full length immunoglobulin molecule or an active portion thereof, i.e., a molecule that contains an antigen-binding site that immunospecifically binds an antigen of a target of interest or part thereof, such targets including, but not limited to, tumor cells.
- the immunoglobulin can be of any type (e.g., IgG 5 IgE, IgM, IgD, IgA, and IgY), class (e.g., IgGl, IgG2, IgG3, IgG4, IgAl, and IgA2), or subclass of immunoglobulin molecule.
- the immunoglobulin can be derived from any species, e.g., human, rodent (e.g., mouse, rat, or hamster), donkey, sheep, rabbit, goat, guinea pig, camelid, horse, cow, or chicken, but preferably, it is of human, murine, or rabbit origin.
- Antibodies useful in the invention include, but are not limited to, monoclonal, polyclonal, bispecific, human, humanized, or chimeric antibodies, single chain antibodies, Fv fragments, Fab fragments, F(ab') fragments, F(ab') 2 fragments, fragments produced by a Fab expression library, anti-idiotypic antibodies, CDRs, and epitope-binding fragments of any of the above which immunospecifically bind to an antigen-of- interest.
- the term "leaving group” refers to a group that can be substituted by another group.
- Such leaving groups are well-known in the art, and examples include, but are not limited to, a halide (fluoride, chloride, bromide, iodide), a sulfonate (e.g., methanesulfonate, p-toluenesulfonate, and trifluoromethanesulfonate), succinimide-N-oxide, p-nitrophenoxide, pentafluorophenoxide, tetrafluorophenoxide, a carboxylate, and an alkoxycarboxylate.
- a halide fluoride, chloride, bromide, iodide
- a sulfonate e.g., methanesulfonate, p-toluenesulfonate, and trifluoromethanesulfonate
- succinimide-N-oxide p-nitrophenoxide
- pentafluorophenoxide t
- water-soluble group refers to a functional group that is well solvated in aqueous environments and that imparts improved water solubility to the compound to which it is attached.
- water-soluble groups include, but are not limited to, alcohols and polyalcohols, straight chain or cyclic saccharides, primary, secondary, tertiary, or quaternary amines and polyamines, sulfate groups, carboxylate groups, phosphate groups, phosphonate groups, ascorbate groups, glycols, including polyethylene glycols, and polyethers.
- R h , R 1 , and R j are independently selected from H and optionally substituted C M5 alkyl, Ci-15 heteroalkyl, C3- 1 5 cycloalkyl, Ca-I 5 heterocycloalkyl, C 4- I 5 aryl, or C 4 -I 5 heteroaryl or a combination thereof, two or more of R h , R 1 , and R J optionally being joined to form one or more carbocycles or heterocycles.
- aryl refers to a carbocyclic aromatic substituent, which may consist of one ring or two or more rings fused together.
- aryl groups include, but are not limited to, phenyl, naphthyl, and anthracenyl.
- heteroaryl refers to a carbocyclic aromatic substituent, which may consist of one ring or two or more rings fused together, wherein at least one carbon in one of the rings is replaced by a heteroatom.
- heteroaryl groups include, but are not limited to, pyridinyl, furanyl, pyrrolyl, triazolyl, pyrazolyl, imidazolyl, thiophenyl, indolyl, benzofuranyl, benzimidazolyl, indazolyl, benzotriazolyl, benzisoxazolyl, and quinolinyl.
- alkyl refers to a straight chain or branched, saturated or unsaturated hydrocarbon substituent.
- alkyl groups include, but are not limited to, methyl, ethyl, propyl, butyl, pentyl, hexyl, octyl, decyl, isopropyl, sec-butyl, isobutyl, tert-butyl, isopentyl, 2-methylbutyl, vinyl, allyl, 1-butenyl, 2-butenyl, isobutylenyl, 1-pentenyl, and 2-pentenyl.
- heteroalkyl refers to a straight chain or branched, saturated or unsaturated hydrocarbon substituent in which at least one carbon is replaced by a heteroatom. Examples include, but are not limited to, methyloxymethyl, ethyloxymethyl, methyloxyethyl, ethyloxyethyl, methylaminomethyl, dimethylaminomethyl, methylaminoethyl, dimethylar ⁇ inoethyl, methylthiomethyl, ethylthiomethyL ethylthioethyl, and methylthioethyl.
- cycloalkyl refers to a saturated or unsaturated non-aromatic carbocycle substituent, which may consist of one ring or two or more rings fused together. Examples include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclopentadienyl, cyclohexyl, cyclohexenyl, 1,3-cyclohexadienyl, decalinyl, and 1,4-cyclohexadienyl.
- heterocycloalkyl refers to a saturated or unsaturated non-aromatic cyclic hydrocarbon substituent, which may consist of one ring or two or more rings fused together, wherein at least one carbon in one of the rings is replaced by a heteroatom. Examples include, but are not limited to, tetrahydrofuranyl, pyrrolidinyl, piperidinyl, 1,4-dioxanyl, decahydroquinolinyl, piperazinyl, oxazolidinyl, and morpholinyl.
- extension "-ylene” as opposed to “-yl” in for example "alkylene” as opposed to “alkyl” indicates that said for example “alkylene” is a divalent moiety connected to one or two other moieties via two covalent single bonds or one double bond as opposed to being a monovalent group connected to one moiety via one covalent single bond in said for example "alkyl”.
- alkylene therefore refers to a straight chain or branched, saturated or unsaturated hydrocarbon moiety;
- heteroalkylene refers to a straight chain or branched, saturated or unsaturated hydrocarbon moiety in which at least one carbon is replaced by a heteroatom;
- arylene refers to a carbocyclic aromatic moiety, which may consist of one ring or two or more rings fused together;
- heteroarylene refers to a carbocyclic aromatic moiety, which may consist of one ring or two or more rings fused together, wherein at least one carbon in one of the rings is replaced by a heteroatom;
- cycloalkylene refers to a saturated or unsaturated non-aromatic carbocycle moiety, which may consist of one ring or two or more rings fused together;
- heterocycloalkylene refers to a saturated or unsaturated
- poly indicates that two or more of such "-ylene” moieties, e.g., alkylene moieties, are joined together to form a branched or unbranched multivalent moiety containing one or more attachment sites for adjacent moieties.
- Certain compounds of the invention possess chiral centers or double bonds; the enantiomeric, diastereomeric, and geometric mixtures of two or more isomers, in any composition, as well as the individual isomers are encompassed within the scope of the present invention.
- the compounds of the invention may also contain unnatural proportions of atomic isotopes at one or more atoms that constitute such compounds. All isotopic variations of the compounds of this invention, whether radioactive or not, are intended to be encompassed within the scope of this invention.
- pharmaceutically active salt refers to a pharmaceutically acceptable organic or inorganic salt of a compound of the invention.
- acid addition salts can be formed.
- acidic groups e.g., a carboxylic acid group
- base addition salts can be formed.
- zwitterions may in addition be obtained as salts. Instances where multiple charged atoms are part of the pharmaceutically acceptable salt can have multiple counterions.
- pharmaceutically acceptable solvate refers to an association of one or more solvent molecules and a compound of the invention.
- solvents that form pharmaceutically acceptable solvates include, but are not limited to, water, isopropyl alcohol, ethanol, methanol, DMSO, ethyl acetate, and acetic acid.
- conjugate hereinbelow refers to a compound of formula (III).
- linker-agent conjugate hereinbelow refers to a compound of formula (IV).
- agent hereinbelow refers to a compound of formula (I), (II), (VII), (VIII), (I'), or (H').
- targeting moiety refers to any molecule that specifically binds or reactively associates or complexes with a moiety specifically or in relative excess present at or near the target site, on, in, or near the target cell, or in (the proximity of) the target tissue or organ, e.g., a receptor, a receptor complex, substrate, antigenic determinant, or other receptive moiety, or that can target the conjugate to the target site via other mechanisms by virtue of its nature, e.g., through the EPR effect.
- Examples of a targeting moiety include, but are not limited to, an aptamer, an antibody or antibody fragment, a polymer, a dendrimer, a lectin, a biologic response modifier, an enzyme, a vitamin, a growth factor, a steroid, a sugar residue, an oligosaccharide residue, a carrier protein, and a hormone, or any combination thereof.
- moiety that improves the pharmacokinetic properties of the compound refers to a moiety that changes the pharmacokinetic properties of a compound of this invention in such a way that a better therapeutic effect can be obtained.
- the moiety can for example increase the water solubility, increase the circulation time, or reduce iramunogenicity.
- linking group refers to a structural element of a compound that links one structural element of said compound to one or more other structural elements of said same compound.
- a number representing degree of branching is used to denote that the subscript number next to a closing bracket represents how many units of the moiety within, the brackets are each directly attached to the moiety immediately to the left of the corresponding opening bracket.
- A-(B) b with b being a number representing a degree of branching means that b units B are all directly attached to A. This means that when b is 2, the formula reduces to B-A-B.
- A-(B) b with b being a number representing a degree of polymerization means that when b is 2, the formula reduces to A-B-B.
- single-release spacer refers to a self-elimination spacer that can release one moiety upon self-immolation.
- multiple-release spacer refers to a self-elimination spacer that can release two or more moieties upon repetitive self-immolation.
- electro cascade spacer refers to a self-elimination spacer, either branched or unbranched, which may self-eliminate through one or more 1,2+2/7 electronic cascade eliminations
- ⁇ -amino aminocarbonyl cyclization spacer refers to a self-elimination spacer that may eliminate through a cyclization process under formation of a cyclic ureum derivative.
- spacer system refers to a single spacer moiety or to two or more of the same or different spacer moieties coupled together.
- a spacer system may be branched or unbranched and contain one or more attachment sites for Z as well as V 1 and optionally L.
- adjectives and/or adjective phrases to a noun that is a) the first in a list of nouns or b) that is anywhere in the middle of a list of nouns and said noun and adjectives together are preceded by the word "and", the adjectives do not only bear on said noun, but on all following nouns separately, unless the context dictates otherwise.
- a line ending in space (a "loose" end), i.e., at one end not having another line or specific atom connected to it, represents a CH 3 group.
- a line ending in space may indicate the position of attachment of another structural element of the compound. This has been indicated with a wavy line perpendicular to and crossing the "loose" line in most drawings.
- the present invention provides novel agents that can be classified as belonging to the class of DNA- binding alkylating agents CC-1065 and the duocarmycins. Furthermore, the invention relates to novel conjugates of these agents and to linker-agent conjugates, which may or may not serve as intermediates for said conjugates.
- the agents of the present invention are deemed to be used to treat an illness that is characterized by undesired (cell) proliferation.
- an agent of this invention can be used to treat a tumor, cancer, an autoimmune disease, or an infectious disease.
- the conjugates of the present invention are in one aspect deemed to be applicable to target agents of formulae (I) and (II) to a specific target site where the conjugate can be converted into one or more agents or be induced to be converted into one or more of said agents.
- This invention can furthermore find application in (non-specific) controlled release of one or more of said agents from a conjugate, with the aim of for example enhancing pharmacokinetic properties.
- the increased water-solubility of the agent may cause the compound to better be able to reach its (intracellular) target and thus increase its cytotoxicity after release from the conjugate, while it may also reduce aggregation of the conjugate and reduce side effects of the conjugate due to premature release of the agent from the conjugate as the liberated agent may be less effective in entering (non-targeted) cells due to its increased polarity.
- Steric hindrance may reduce direct alleviation of biomolecules by the conjugate and non-DNA alkylation by the agent, thereby reducing aspecific (cyto)toxicity.
- the inventive concept of combining water solubility and steric shielding in compounds of formulae (I) and (II) may lead to agents that are more water-soluble and more selective and to an increased cytotoxicity quotient for conjugates of this invention.
- the invention provides a compound of formula (I) or (II):
- R 1 is selected from halogen and OSO 2 R 11 , wherein R u is selected from optionally substituted Ci-6 alkyl, C 1 ⁇ perhaloalkyl, benzyl, and phenyl;
- R 2 is selected from H and optionally substituted Ci -8 alkyl
- R 3 , R 3 ', R 4 , and R 4' are independently selected from H and optionally substituted C 1 -S alkyl, wherein two or more of R 2 , R 3 , R 3 , R 4 , and R 4 are optionally joined to form one or more optionally substituted carbocycles or heterocycles;
- X 2 is selected from O, C(R l4 )(R 14' ), and NR 14' , wherein R 14 is selected from H and optionally substituted C 1 ⁇ alkyl or acyl and wherein R 14 may be absent or be selected from H and optionally substituted Ci. ⁇ alkyl or acyl;
- Each R 5 , R 5' , R 6 , R 6' , R 7 , and R 7' is independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO,
- X 1 is selected from O, S, and NR 13 , wherein R 13 is selected from H and optionally substituted Ci -8 alkyl;
- X 3 is selected from O, S, and NR 15 , wherein R 15 is selected from H and optionally substituted Ci-8 alkyl or acyl, or -X 3 - represents -X 3a and X 3b - wherein X 3a is connected to the carbon to which X 4 is attached and X 3b is connected to the phenyl ring ortho to R 10 , wherein X 3a is independently selected from H and optionally substituted Ci -8 alkyl or acyl and X 3b is selected from the same group of substituents as R 8 ;
- X 4 is selected from N and CR 16 , wherein R 16 is selected from H and optionally substituted Ci -8 alkyl or acyl;
- X 5 is selected from O, S, and NR 17 , wherein R 17 is selected from H and optionally substituted Ci-g alkyl or acyl;
- R 8 , R 9 , R 10 , and R 1 * are each independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO, CF 3 , CN, C(O)NH 2 , C(O)H, C(O)OH, halogen, R x , SR X , S(O)R X , S(O) 2 R X , S(O)OR X , S(O) 2 OR", OS(O)R X , OS(O) 2 R X , OS(O)OR X , OS(O) 2 OR X , OR X , NHR X , N(R x )R y , 4 N(R x )(R y )R 2 , P(O)(OR x )(OR
- this invention relates to enantiomerically pure and/or diastereomerically pure compounds of formulae (I) and (II) as well as to enantiomeric and/or diastereomeric mixtures of compounds of formulae (I) and (II).
- R 1 in a compound of formula (I) or (II) is a leaving group.
- the leaving group R 1 in a compound of formula (I) or (II) is a halide.
- R 1 is selected from chloride (Cl), bromide (Br), and iodide (I).
- R 1 is chloride (Cl).
- R 1 is bromide (Br).
- R 1 may cause the seco agent to become an alkylating agent as well, which may decrease the cytotoxicity quotient of conjugates of compounds of formulae (I) and (II) as the agent may be able to alley late while still being bound in the conjugate.
- R 1 is too bad a leaving group, the seco agent may not close to form a cyclopropyl-containing agent, believed to be the active species, which may reduce its cytotoxicity and, most likely, reduce the cytotoxicity quotient.
- Another means to tune the alkylating activity of the seco agents and their cyclopropyl-containing derivatives may be to somewhat shield the carbon to which the leaving group is attached or on which nucleophilic attack can occur by choosing at least one of R 2 , R 3 , R 3' , R 4 , R 4 ', R 5 , and R 5' present to be other than hydrogen. Shielding of said carbon may reduce aspecific alkylation by compounds of formulae (I) and (II) and their cyclopropyl-containing analogs and by their conjugates as well.
- R 2 in a compound of formula (I) is optionally substituted Ci-S allcyl.
- R 2 is optionally substituted linear Ci -8 alkyl.
- R 2 is unsubstituted linear Q -8 allcyl.
- R 2 is methyl.
- steric shielding of the carbon bearing R 1 may be introduced by choosing one or more of R 3 , R 3 , R 4 , and R 4 to be other than hydrogen.
- R 3 , R 3 , R 4 , and R 4 are each H.
- R 3 and R 3 are both H.
- R 4 and R 4' are both H.
- one of R 3 and R 3 is Ci -S alkyl while the other is H.
- one of R 4 and R 4 ' is Ci -8 alkyl while the other is H.
- one of R 3 and R 3 is Ci -8 alkyl and one of R 4 and R 4 is Ci-S allcyl while the others are H.
- both R 3 and R 3 are independently selected from Ci -8 alkyl.
- both R 4 and R 4 are independently selected from Ci -8 alkyl.
- R 2 is H and one of R 3 , R 3 ', R 4 , and R 4' is selected from Ci -8 alkyl.
- R 2 is H and one of R 3 , R 3' , R 4 , and R 4' is selected from methyl. In another embodiment, R 2 is H and two of R 3 , R 3' , R 4 , and R 4 ' are independently selected from Ci -8 allcyl. In yet another embodiment, R 2 is H and two of R 3 , R 3 , R 4 , and R 4' are methyl.
- the alkylating activity of a compound of formula (I) or (II) or its cyclopropyl-containing analog may also be affected by the nature of X 1 .
- the nature of X 1 may affect the rate at which and the conditions under which the seco agents ring close to the cyclopropyl analogs and/or the rate at which the cyclopropyl ring is opened by nucleophilic attack by DNA, and thus affect the alkylation behavior.
- X 1 is O.
- R 5 , R 5' , R 6 , R 6' , R 7 , R 7' , and X 2 as well as the size of the ring(s) connected to the left-hand side of the ring bearing X may for example, each independently or two or more taken together, affect the pharmacokinetic properties of the agent, affect the water solubility, affect the aggregation behavior, affect the DNA alkylation process, or affect the DNA binding strength. Furthermore, especially R 5 and R 5' may also affect the degree of shielding of the carbon on which nucleophilic attack can occur.
- R 5 and R 5 are both H.
- at least one of R 5 and R 5' is not hydrogen.
- R 5 is not hydrogen.
- R 2 is hydrogen and at least one of R 5 or R 5 is not hydrogen.
- R 5 is selected from nitro, halogen, amino, hydroxy, and optionally substituted alkylamino, alkyicarbonylamino, alkoxycarbonylamino, alkyloxy, alkylcarbonyloxy, aU ⁇ ylaminocarbonyloxy, or Ci -4 alkyl.
- R 5 is optionally substituted linear CM alkyl.
- R 5 is unsubstituted linear C M alkyl.
- R 5 is methyl.
- alkylation rate and efficiency of compounds like the ones of formulae (I) and (II) may be tuned in several ways, in one aspect of this invention, this is achieved by introducing steric shielding choosing for a compound of formula (I) at least one of R 2 , R 3 , R 3 , R 4 , R 4 , R 5 , and R 5 present to be other than hydrogen and for a compound of formula (II) optionally one or more of R 2 , R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present to be other than hydrogen.
- compounds of formulae (I) and (II) may be represented by compounds of formulae (Ia) and (Ha), respectively:
- R 6 and R 7 in (Ia) or (Ha) are both H.
- compounds of formulae (I) and (II) may be represented by compounds of formulae (Ib) and (lib), respectively:
- X in (Ib) or (lib) is N.
- X in (Ib) or (lib) is CH.
- R 5 , R 6 , and R 7 in (Ib) or (lib) are each H.
- R 5 , R 6 , and R 7 in (Ib) or (lib) are each H and X 2 is CH.
- R 5 and R 7 in (Ib) or (lib) are each H and R 6 is CO 2 Me.
- R 5 and R 7 in (Ib) or (lib) are each H and R 6 is OMe.
- R 5 and R 7 in (Ib) or (lib) are each H and R 6 is CN.
- R 5 in (Ib) or (Hb) is selected from nitro, halogen, amino, hydroxy, and optionally substituted alkylamino, alkylcarbonylamino, alkoxycarbonylamino, alkyloxy, alkylcarbonyloxy, alkylaminocarbonyloxy, or d- 4 alkyl.
- R 5 in (Ib) or (lib) is optionally substituted linear C M alkyl.
- R 5 in (Ib) or (lib) is unsubstituted linear Cj -4 alkyl.
- R 5 in (Ib) or (lib) is methyl.
- compounds of formulae (I) and (II) may be represented by compounds of formulae (Ic) and (lie), respectively:
- X 2 in (Ic) or (lie) is NH.
- R 6 and R 7 in (Ic) or (He) are H and CO 2 CH3, respectively, and X 2 is NH.
- R 7 and R 6 in (Ic) or (He) are H and CO2CH3, respectively, and X 2 is NH.
- R 6 in (Ic) or (lie) is CH3 and X 2 is NH.
- compounds of formulae (I) and (II) may be represented by compounds of formulae (Id) and (Hd), respectively:
- R 7 and R 7' in (Id) or (lid) are CO2CH3 and CH3, respectively.
- X 2 is NH, R 6 and R 6 together are
- R 7 and R 7 ' are CO 2 CH3 and CH 3 , respectively.
- X 3 is NH. In another embodiment, X 3 is O. In one embodiment, X 4 is CH. In one embodiment, X 5 is O.
- the water-soluble group is a group that imparts an increased solubility on the compounds of formulae (I) and (II) with respect to similar compounds from the prior art. It may also contribute to prevent or reduce aggregation of compounds of this invention or to reduce side effects.
- water-soluble groups include, but are not limited to, -NH 2 , -NH-, -NHR a , -NR a -, -N(R a )(R b ), - + N(R a )(R b )-, - "1 ⁇ (R 3 XR 1 O(R 0 ), -COOH, -COOR a , -OP(O)(OH) 2 , -OP(O)(OH)O-, -OP(O)(OR a )O-, -OP(O)(OH)OR 3 , -0P(0)(0R b )0R a , -P(O)(OH) 2 , -P(O)(OH)O-, -P(O)(OR a )OH, -P(0)(0R a )0-, -P(0)(0R a )(0R b ), -OS(O) 2 OH,
- the water-soluble group may be at any position within R 8 , R 9 , R 10 , and/or R 11 or may constitute the whole R 8 , R 9 , R 10 , or R 11 moiety.
- the water-soluble group may for example be located at any interior position, be part of the main chain, be part of a ring structure, be a functional group pending to the main chain or a ring, or be placed at the position at which the R 8 , R 9 , R 10 , or R ⁇ substituent is attached to the remainder of the molecule.
- one of R 8 , R 9 , R 10 , and R 11 contains a water-soluble group.
- one of R 8 , R 9 , and R 10 contains a water-soluble group.
- R 8 contains a water-soluble group.
- R 9 contains a water-soluble group.
- R 10 contains a water-soluble group.
- the water-soluble group is a carboxylic acid group.
- the water-soluble group is an amino group.
- the water-soluble group is a primary amino group.
- the water-soluble group is a secondary amino group.
- the water-soluble group is a tertiary amino group, hi another embodiment, the water-soluble group is a quaternary amino group.
- the water-soluble group is a dimethylamino group, hi another embodiment, the water-soluble group is a morpholino group.
- the water-soluble group is a l-methylpi ⁇ eridin-4-yl group.
- the water-soluble group is a methylamino group.
- the water-soluble group is a piperidin-4-yl group.
- the water-soluble group is an amino (NH 2 ) group.
- the water-soluble group is an N-methyl-N-(carboxymethyl)amino group.
- the water-soluble group is an N-methyl-N-(2-methoxy-2- oxoethyl)amino group.
- the water-soluble group is not an ether group (-O-, -OR a ), not being an oligoether or polyether group. In yet another embodiment, the water-soluble group is not an amide group, not being an oligopeptide or polypeptide group.
- the water-soluble group is not a primary, secondary, or tertiary amino group of which the nitrogen is directly connected (and thus conjugated) to an aromatic moiety or a secondary or tertiary amino group being part of an aromatic moiety, the nitrogen atom being an atom in the aromatic ring system.
- the water-soluble group is not a hydroxyl group connected to an aromatic ring system.
- R 8 , R 9 , R 10 , or R 11 is selected from -0-C 1-6 alkylene- ⁇ (R 100 ) 2 , -NC(O)-C 1-5 alkylene-N(R 100 ) 2 , (l-(R 100 )piperidin-4-yl)-Ci. 5 alkylene-O-, and (morpholin-4-yl)-Ci_ 8 alkylene-O, wherein each R 100 is independently selected from H and Ci .3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being CM alkyl.
- R 8 , R 9 , or R 10 is selected from -O-d -6 alkylene-N(R 100 ) 2 , -NC(O)-C 1-5 alkylene-N(R 100 ) 2 , (l-(R 100 )piperidin-4-yl)-Ci -5 alkylene-O-, and (morpholin-4-yl)-C 1-8 alkylene-O, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being CM alkyl.
- R 8 is selected from -0-Ci -6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-
- R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being CM alkyl.
- R 9 is selected from -0-Cu alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-
- R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C 14 alkyl.
- R 10 is selected from -O-Ci-6 alkylene-N(R 100 )2, -NC(O)-Ci -5 alkylene- N(R 100 ) 2 , (l-(R 100 )pi ⁇ eridin-4-yl)-Ci, 5 alkylene-O-, and (morpholin-4-yl)-Ci. 8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C M alkyl.
- R 8 , R 9 , R 10 , or R 11 is selected from 2-(mo ⁇ holin-4-yl)ethoxy 3 (l-methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, and 2-(N,N-dimethylamino)- acetylamino.
- R 8 , R 9 , or R 10 is selected from 2-(morpholin-4-yl)ethoxy
- R 8 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4- yl)methoxy, 2-(N,N-dimethylamino)ethoxy, and 2-(N,N-dimethylamino)acetylamino.
- R 8 is 2-(morpholin-4-yl)ethoxy.
- R 8 is (l-methylpiperidin-4-yl)methoxy.
- R 8 is 2-(N,N-dimethylamino)ethoxy.
- R is 2-(N,N-dimethylamino)acetylamino.
- R 9 is 2-(N,N-dmiethylamino)ethoxy.
- R 10 is 2-(N,N-dimethylainino)ethoxy.
- R 8 , R 9 , R 10 , or R 11 is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N- methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl-N-(2-methoxy-2-oxoethyl)amino)- ethoxy.
- R 8 , R 9 , R 10 , or R 11 is selected from 2-(morpholin-4 ⁇ yl)ethoxy, ( 1 -methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(NN-dimethylamino)- acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl-N-(2-methoxy-2-oxoethyl)amino)ethoxy.
- R 8 , R 9 , or R 10 is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N- methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl-N-(2-methoxy-2-oxoethyl)amino)- ethoxy.
- R 8 is selected from 2-(methylamino)ethoxy., 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-methyl-N-
- R 8 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4- yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(N,N-dimethylamino)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl-N-(2 ⁇ methoxy-2-o ⁇ oethyl)amino)ethoxy.
- R 9 is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino,
- R 10 is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino,
- R 8 is 2-(methylamino)ethoxy. In yet another embodiment, R 8 is 2-aminoethoxy.
- R 8 is 2-(N-methyl-N-(carboxymethyl)amino)ethoxy.
- R 8 is 2-(N-methyl-N-(2-methoxy-2-oxoethyl)amino)ethoxy.
- R 8 , R 9 , R 10 , or R 11 contains a water-soluble group
- the other substituents may each independently either be hydrogen, a substituent containing another water-soluble group or a substituent containing no water-soluble group.
- R 9 , R 10 , and R 11 are each hydrogen.
- R 8 contains a water-soluble group and R 9 , R 10 , and R 11 are each hydrogen.
- R 8 contains a water-soluble group and R 9 is methoxy.
- R 9 is methoxy and R 10 and R 11 are each, hydrogen.
- R 8 contains a water-soluble group and R 9 is methoxy and R 10 and R 11 are each hydrogen.
- R 8 is 2-(N,N-dimethylamino)ethoxy
- R 9 is methoxy
- R 10 and R 11 are each hydrogen.
- R 9 or R 10 is a substituent containing a water-soluble group and R 8 is not hydrogen.
- R 9 is a substituent containing a water-soluble group and R 8 is not hydrogen.
- R 10 is a substituent containing a water-soluble group and R 8 is not hydrogen.
- R 8 is selected from
- X 33 is selected from O, S, and NR 15 , wherein R 15 is as defined hereinabove, R a and R b are as defined hereinabove, and NR a R b is connected to the phenyl ring through any one of the carbon atoms 1-4. In another embodiment, NR a R b is connected to the phenyl ring through carbon atom 2 or 4.
- R 8 may also be a substituent of the formula:
- X 3 ', X 4 ', X 5 ', R 8' , R 9' , R 10 ', and R 1 r have the same meaning as defined above for X 3 , X 4 , X s , R 8 , R 9 , R 10 , and R 11 , respectively, except that R 8' , R 9> , R 10 ', and R 11' do not have to contain a water- soluble group when there is already another water-soluble group present in R , R 9 , R 10 , or R , and wherein R 8 may be selected from H and optionally substituted C 1 - S alkyl or C 1- S heteroalkyl and optionally be joined with R 9 or R 11 to form an optionally substituted aliphatic or aromatic heterocycle.
- R 8 or R 8 and R 11 , and/or R 8 and R 11' may be joined to form together with the linking atoms an optionally substituted dihydropyrrole.
- Compounds of formulae (I) and (II) have as one advantage that they have an increased water solubility with respect to similar compounds from the prior art.
- the presence of for example a carboxylic acid group or a secondary aliphatic amino group may significantly contribute to water solubility.
- such a group may prevent a compound of formula (I) or (II) from crossing a biological barrier, especially when it is an apolar barrier such as a cell membrane. This may be advantageous, especially when a compound of formula (I) or (II) is delivered into a targeted cell through conjugation to a targeting moiety before it is released from the conjugate.
- R 2 is not hydrogen
- R 2 is H and at least one of R 3 , R 3 , R 4 , R 4 , R 5 , and R 5 present is not H.
- R 2 is H, X 1 is O, and at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5 ' present is not H.
- R 2 in a compound of formula (Ib) or (lib), R 2 is H, X 1 is O, X 2 is CR 14 , and at least one of R 3 , R 3' , R 4 , R 4' , R 5 , and R 5' present is not H.
- R 2 is H, X 1 is O, X 2 is CR 14 , X 5 is O, and at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present is not H.
- R 2 is H, X 1 is O, X 2 is CR 14 , X s is O, X 3 is NR 15 , and at least one of R 3 , R 3 ', R 4 , R 4' , R 5 , and R 5 ' present is not H.
- R 2 is H, X 1 is O, X 2 is CH, X 5 is O, X 3 is NH, and at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5 ' present is not H.
- R 2 is H, X 1 is O, X 2 is CR 14 , X s is O, X 3 is NR 15 , at least one of R 3 , R 3 ', R 4 , R 4' , R 5 , and R 5' present is not H, and R 8 , R 9 , R 10 , or R 11 is selected from -0-C ]-6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R 100 ) 2 , (l-(R 100 )piperidin-4-yl)- Ci-5 alkylene-O-, and (morpholin-4-yl)-Ci -8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being Ci -4 alkyl.
- R 2 is H, X 1 is O, X 2 is CR 14 , X 5 is O, X 3 is NR 15 , at least one of R 3 , R 3" , R 4 , R 4 ', R 5 , and R 5 ' present is not H, and R 8 , R 9 , or R 10 is selected from -0-Ci -6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R 100 ) 2 , (l-(R 100 ) ⁇ iperidin-4-yl)- Ci -5 alkylene-O-, and (mo ⁇ holin-4-yl)-Ci_ 8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being Ci -4 alkyl.
- R 2 is H, X 1 is O, X 2 is CR 14 , X s is O, X 3 is NR 15 , at least one of R 3 , R 3' , R 4 , R 4' , R 5 , and R 5 ' present is not H, and R 8 is selected from -0-C 1-6 alkylene-N(R I00 ) 2 , -NC(O)-Ci -5 alkylene-NtR 100 ) 2 , (l-(R 100 )piperidin-4-yl)-Ci -5 alkylene-O- , and (mo ⁇ holin-4-yl)-Ci -8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C 1-4 alkyl.
- R 2 is H, X 1 is O, X 2 is CR 14 , X 5 is O, X 3 is NR 15 , at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present is not H, and R 9 is selected from -0-C 1-6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R I00 ) 2 , (l-(R 100 )piperidin-4-yI)-Ci -5 alkylene-O- , and (morpholin-4-yl)-Ci_ 8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being Ci A alkyl.
- R 2 is H, X 1 is O, X 2 is CR 14 , X 5 is O, X 3 is NR 15 , at least one of R 3 , R 3' , R 4 , R 4 ', R 5 , and R 5' present is not H, and R 10 is selected from -O-Ci- 6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R 100 ) 2 , (l-(R 100 )piperidin-4-yl)-Ci -5 alkylene-O- , and (morpholin-4-yl)-Ci -8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being Ci -4 alkyl.
- R 2 is H, X 1 is O, X 2 is CR 14 , X 5 is O, X 3 is NR !S , at least one of R 3 , R 3' , R 4 , R 4 ', R 5 , and R 5 ' present is not H, and R 8 , R 9 , R 10 , or R 11 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N 5 N- dimethylamino)ethoxy, 2-(N,N ⁇ dunethylarnino)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N- methyl-N-(carboxymethyl)amino)eth
- R 2 is H
- X 1 is O
- X 2 is CR 14
- X 5 is O
- X 3 is ⁇ R !5
- at least one of R 3 , R 3 ', R 4 , R 4' , R 5 , and R 5 ' present is not H
- R 8 , R 9 , or R 10 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N,N- dimethylamino)ethoxy, 2-(N,N-dimethylamino)acetylamino, 2-(methylamino)ethoxy,
- R 2 is H, X 1 is O, X 2 is CR 14 , X 5 is O, X 3 is NR 15 , at least one of R 3 , R 3' , R 4 , R 4> , R 5 , and R 5> present is not H, and R 8 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(N,N-dimethylamino)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-methyl-N- (carboxymethyl)amino)ethoxy, and 2-(N-methyl-N-(2-methoxy-2-
- R 2 is H, X 1 is O, X 2 is CR 14 , X 4 is CR 16 , X 5 is O, X 3 is NR 15 , at least one of R 3 , R 3 ", R 4 , R 4' , R 5 , and R 5 ' present is not H, and R 8 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N 5 N- dimethylamino)ethoxy, 2-(N,N-dimemylamino)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N- methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl-methyl-
- R 2 is H, X 1 is O, X 2 is CR 14 , X 4 is CR 16 , X s is O, X 3 is NR 15 , at least one of R 3 , R 3 ', R 4 , R 4' , R 5 , and R 5' present is not H, R 10 and R 11 are both H, and R 8 is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(N,N-dimethylamino)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N- methyl-N-(carboxymethyl)amino)
- R 2 is H
- X 1 is O
- X 2 is CR 14
- X 4 is CR 16
- X 5 is O
- X 3 is NR 15
- at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present is not H
- R 10 and R H are both H
- R 9 is selected from H and OMe
- R 8 is selected from 2-(morpholin-4-yl)ethoxy, ( 1 -methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(N,N-dimethylamino)- acetylamino, 2-(methylamino)ethoxy, 2-(inethylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-
- R 2 is H, at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present is not H, and R 8 is selected from -O-Q- ⁇ alkylene- ⁇ (R 100 ) 2 , -NC(O)-Ci -5 alkyIene-N(R 100 ) 2 , (l-(R 100 )piperidin-4-yl)- Ci -5 alkylene-O-, and (morpholm-4-yl)-Ci -8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C 1-4 alkyl.
- R 2 is H, at least one of R 3 , R 3' , R 4 , R 4' , R 5 , and R 5 ' present is not H, and R 9 is selected from -O-C 1-6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R 100 ) 25 (l-(R 100 )piperidm-4-yl)- Ci -5 alkylene-O-, and (morpholin-4-yl)-C I - 8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C 1-4 alkyl.
- R 2 is H, at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present is not H, and R 10 is selected from -O-Ci -5 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R 100 ) 2 , (l-(R 100 ) ⁇ iperidin-4-yl)- Ci -5 alkylene-O-, and (morpholin-4-yl)-Ci -8 alkylene-O-, wherein each R 100 is independently selected from H and Ci -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C 1-4 alkyl.
- R 2 is H, at least one of R 3 , R 3 ', R 4 , R 4 ', R 5 , and R 5' present is not H, and R 8 is selected from 2-(mo ⁇ holin-4-yi)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N 5 N- dimethylamino)ethoxy, 2-(N,N-dimethylamino)acetylamino, 2-(methylamino)ethoxy,
- R 5 is not NO 2 .
- At least one of the substituents R 1 , R 2 , R 3 , R 3 ', R 4 , R 4' , R 5 , R 5' , R 6 , R 6' , R 7 , R 7' , R 8 , R 9 , R 10 , R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , and R 17 contains or is the moiety COOH.
- At least one of the substituents R 1 , R 2 , R 3 , R 3 ', R 4 , R 4' , R 5 , R 5' , R 6 , R 6' , R 7 , R 7' , R 8 , R 9 , R 10 , R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , and R 17 contains or is the moiety COOH, and if there is only one COOH moiety and said COOH moiety is contained within R 8 , R 9 , R 10 , or R 11 , there is at least another water-soluble group present in R 8 , R 9 , R 10 , or R 11 .
- at least one of the substituents R 8 , R 9 , R 10 , and R 11 contains or is the moiety COOH.
- At least one of the substituents R 8 , R 9 , R 10 , and R 11 contains or is the moiety COOH and there is at least another water-soluble group present in R 8 , R 9 , R 10 , or R 11 .
- at least one of the water-soluble groups in R 8 , R 9 , R 10 , and R 11 is an aliphatic secondary amine moiety not being conjugated to an aromatic moiety or a carbonyl group.
- R is selected from
- At least one of the water-soluble groups in R 8 , R 9 , R 10 , and R 11 is an aliphatic secondary amine moiety not being conjugated to an aromatic moiety or a carbonyl group and at least one of the substituents R 1 , R 2 , R 3 , R 3' , R 4 , R 4' , R 5 , R 5' , R 6 , R 6' , R 7 , R 7' , R 8 , R 9 , R 10 , R 11 ,
- R 12 , R 13 , R 14 , R 15 , R 16 , and R 17 contains or is the moiety COOH.
- At least one of the water-soluble groups in R 8 , R 9 , R 10 , and R 11 is an aliphatic secondary amine moiety not being conjugated to an aromatic moiety or a carbonyl group and at least one of the substituents R 8 , R 9 , R 10 , and R 11 contains or is the moiety COOH.
- At least one of the substituents R 8 , R 9 , R 10 , and R 11 contains a COOH moiety and an aliphatic secondary amine moiety not being conjugated to an aromatic moiety or a carbonyl group.
- a compound of this invention is represented by formula (IbI):
- a compound of this invention is represented by formula (Ib2):
- R 8a ' is a substituent containing a water-soluble group and R 9a is H or optionally substituted Ci -I5 alkoxy.
- this invention relates to a compound of formula (IbI) or (Ib2) wherein R 8a or R 8a ', respectively, is selected from -0-C 1-6 alkylene-N(R 100 ) 2 , -NC(O)-Ci -5 alkylene-N(R 100 ) 2 ,
- each R 100 is independently selected from H and Q -3 alkyl, the latter being optionally substituted with COOH or COOR 300 , R 300 being C M alkyl.
- this invention relates to a compound of formula (IbI) or (Ib2) wherein R 8a or R 8a ', respectively, is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4-yl)methoxy,
- this invention relates to a compound of formula (IbI) wherein R 8a is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, and
- this invention relates to a compound of formula (IbI) wherein R 8a is selected from 2-(methylamino)ethoxy, 2-(methylammo)acetylamino, 2-aminoethoxy,
- this invention relates to a compound of formula (IbI) wherein R 8a is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl-iV-(2-methoxy-2-oxoethyl)amino)ethoxy and R 9a is methoxy.
- a compound of this invention is represented by the formula:
- R la is chloro (Cl) or bromo (Br)
- R 2a and R 5b are methyl and H, respectively, or H and methyl
- R 8d is selected from 2-(morpholin-4 ⁇ yl)ethoxy, (l-methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(N, ⁇ V " -dimethylamino)- acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, (piperidin-4-yl)methoxy, 2-(N-methyl-N-(carboxymethyl)amino)ethoxy, and 2-(N-methyl
- a compound of this invention is represented by the formula:
- a compound of this invention is represented by the formula: or by its (Ii?, 105) isomer, its (Ii?, 1Oi?) isomer, its (IS 1 , 105) isomer, or by a mixture of two or more of said isomers.
- a compound of this invention is represented by formula (Ib3):
- R 5a has the same meaning as R 5 defined above except that it cannot be hydrogen.
- R 5a in a compound of formula (Ib3) is selected from nitro, halogen, amino, hydroxy, optionally substituted alkylamino, optionally substituted alkylcarbonylamino, optionally substituted aUcoxycarbonylamino, optionally substituted alkyloxy, optionally substituted alkylcarbonyloxy, . optionally substituted alkylaminocarbonyloxy, and optionally substituted C 1-4 alkyl.
- R 5a in a compound of formula (Ib3) is optionally substituted linear Ci 4 alkyl.
- R 5a in a compound of formula (Ib3) is unsubstituted linear Ci -4 alkyl.
- R 5a in a compound of formula (Ib3) is methyl.
- a compound of this invention is represented by formula (Ib3-1):
- each R loOa is independently methyl, carboxymethyl, 2-methoxy-2-oxoethyl, or hydrogen.
- this invention relates to a compound of formula (I') or (II'):
- this invention relates to a compound of formula (I') or (H'), said compound containing a cyclopropyl group, which results from rearrangement of and concomitant elimination of H-R 1 from a compound of formula (I) or (II).
- this invention relates to a compound of formula (VII) or (VUI):
- R 1 is selected from halogen and OSC ⁇ R" , wherein R u is selected from optionally substituted
- R 2 is selected from H and optionally substituted Ci-S alkyl
- R 3 , R 3' , R 4 , and R 4 ' are independently selected from H and optionally substituted Ci -8 alkyl, wherein two or more of R 2 , R 3 , R 3 , R 4 , and R 4' are optionally joined to form one or more optionally substituted carbocycles or heterocycles;
- X 2 is selected from O, C(R 14 )(R 14 '), and NR 14 ', wherein R 14 is selected from H and optionally substituted Ci -8 alkyl or acyl and wherein R 14 ' may be absent or be selected from H and optionally substituted Ci -8 alkyl or acyl;
- Each R 5 , R 5' , R 6 , R 6' , R 7 , and R 7' is independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO,
- X 1 is selected from O, S, and NR 13 , wherein R 13 is selected from H and optionally substituted C 1-8 alkyl;
- X 3 is selected from O, S, and NR 15 , wherein R 15 is selected from H and optionally substituted Ci -8 alkyl or acyl, or -X 3 - represents -X 3a and X 3b - wherein X 3a is connected to the carbon to which X 4 is attached and X 3b is connected to the phenyl ring ortho to R 10 , wherein X 3a is independently selected from H and optionally substituted Ci -8 alkyl or acyl and X 3b is selected from the same group of substituents as R 8 ;
- X 4 is selected from N and CR 16 , wherein R 16 is selected from H and optionally substituted C 1- S alkyl or acyl;
- X 5 is selected from O, S, and NR 17 , wherein R 17 is selected from H and optionally substituted Ci- 8 allcyl or acyl;
- R 8 , R 9 , R 10 , and R 11 are each independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO, CF 3 , CN, C(O)NH 2 , C(O)H, C(O)OH, halogen, R x , SR X , S(O)R X , S(O) 2 R X , S(O)OR X , S(O) 2 OR X , OS(O)R X , OS(O) 2 R X , OS(O)OR X , OS(O) 2 OR X , OR X , NHR X , N(R x )R y , 4 N(R ⁇ (R y )R 2 , P(O)(OR x )(OR
- R 9 is OMe and R 8 is OCH 2 CH 2 N(CH 3 )2.
- Conjugates of compounds of formulae (VII) and (VIII) may therefore have improved properties with respect to conjugates of similar compounds from the prior art.
- Conjugates of formulae (VII) and (VIII) may for example have improved water solubility, show less aggregation, and show less side effects as premature liberation of a compound of formula
- this invention relates to conjugates of a compound of formula (I) or (II) that can be converted in vivo and in one or more steps to a compound of formula (I) or (II), respectively. These conjugates may favorably affect the pharmacokinetic properties and other characteristics of a compound of formula (I) or (II).
- this invention relates to a conjugate comprising a compound of formula (I) or (II) conjugated to at least one promoiety, i.e., a moiety that can be removed in vivo to release a compound of formula (I) or (II).
- this invention relates to a conjugate comprising a compound of formula (I) or (II) conjugated to one promoiety.
- this invention relates to a compound of formula (III):
- V 2 is either absent or a functional moiety
- Each L 2 is independently absent or a linking group linking V 2 to L or to V 1 or Y when L is absent;
- Each L is independently absent or a linking group linking L 2 or V 2 when L 2 is absent to one or more V 1 and/or Y;
- Each V 1 is independently H or a conditionally-cleavable or conditionally-transformable moiety, which can be cleaved or transformed by a chemical, photochemical, physical, biological, or enzymatic process;
- Each Y is independently absent or a self-eliminating spacer system which is comprised of 1 or more self-elimination spacers and is linked to V 1 , optionally L, and one or more Z;
- Each p and q are numbers representing a degree of branching and are each independently a positive integer;
- z is a positive integer equal to or smaller than the total number of attachment sites for Z in the one or more V 1 -Y moieties;
- Each Z is independently a compound of formula (I) or (II) as defined hereinabove wherein one or more of X 1 , R 6 , R 7 , R 8 , R 9 , R 10 , and R 11 may optionally in addition be substituted by a substituent of formula (V):
- each V 2 ', L 2 ', L', V 1 ', Y', Z', p', q', and z' have the same meaning as defined for V 2 , L 2 , L, V 1 , Y, Z, p, q, and z, the one or more substituents of formula (V) being independently connected to one or more of X 1 , R 6 , R 7 , R 8 , R 9 , R 10 , and R 11 via Y' or V 1 ' when Y' is absent, each Z being connected to Y or V 1 when Y is absent through either X 1 or an atom in R 6 , R 7 , R 8 , R 9 , R 10 , or R 11 ; with the proviso that at least one of the one or more V 1 and the one or more V 1 ' is not H.
- L can either be connected to V 1 and/or to Y. If L is connected to Y, this means that both V 1 and L, as well as one or more Z, are connected to Y. IfY is absent, L must be connected to V 1 ; L cannot be directly connected to Z.
- V 2 (-L 2 -L(-V 1 -Y)p)q(Z) z _i and one or more V 2 '(-L 2 '-L'(-V 1 '-Y%Oq ⁇ Z') 2' -i moieties connected to a compound of formula (I) or (II) are herein referred to as promo ieties.
- the present invention also relates to a compound of formula (IV):
- RM is a reactive moiety and L, V 1 , Y, Z 5 p, and z are as defined above, except that L is now linking RM to one or more V 1 and/or Y, and V 1 , Y, and Z may contain protecting groups and the one or more V 2 -L 2 ' moieties optionally present in Z as defined hereinabove may optionally and independently be replaced by RM', which is a reactive moiety, and wherein, if there is more than 1 reactive moiety in (IV), the reactive moieties are the same or different.
- These linker-agent conjugates may or may not be considered intermediates for compounds of formula (III).
- RM-L(Y- Y) 5 (Z) 2-1 and one or more RM'-L'(-V 1 '-Y')p ⁇ Z I )z ' -i moieties connected to a compound of formula (I) or (II) are herein referred to as promoieties.
- this invention relates to enantiomerically pure and/or diastereomerically pure compounds of formulae (III) and (IV) as well as to enantiomeric and/or diastereomeric mixtures of compounds of formulae (III) and (IV).
- Y and/or V 1 moieties in a compound of formula (III) or (IV) contain attachment sites for Z that are not coupled to Z, for instance as a consequence of an incomplete coupling reaction during synthesis, these attachment sites are considered to be attached to H, OH, or a leaving group instead. If all of said attachment sites are connected to Z, then z equals the number of said attachment sites; otherwise, z is lower.
- Compounds of this invention may exist as a mixture, wherein each component of the mixture has a different z value.
- the compound may exist as a mixture of two separate compounds, one compound wherein z is 4 and another compound wherein z is 3.
- the compound may exist as a mixture of isomers as Z may be connected to distinct sets of attachment sites in the one or more Y and/or V 1 moieties.
- Z may be connected to distinct sets of attachment sites in the one or more Y and/or V 1 moieties.
- the comiections of one first moiety to other moieties within formula (III) or (IV) in general only those said other moieties are mentioned that are directly next to said first moiety in formula (III) or (IV). It should be understood that if one of said other moieties is not present, said first moiety is actually connected to the moiety first in line that is present, unless explicitly stated otherwise.
- a compound of formula (I) or (II) may be conjugated to a promoiety through its water-soluble group.
- the water-soluble group may contribute less to the water solubility of the compound of formula (III) or (IV), but may contribute again to the water solubility of Z upon removal of said promoiety.
- V 2 , L 2 , L, V 1 , Y, Z, RM, p, q, or z is mentioned, it should be understood that the same can apply for each V 2' , L 2 ', L', V 1 ', Y', Z', RM', p', q', or z', respectively.
- the V 1 moiety can be a group that is conditionally cleavable or transformable. In other words, it is designed to be transformed and/or cleaved from Y by a chemical, photochemical, physical, biological, or enzymatic process upon being brought in or under a certain condition.
- This condition may for example be bringing a compound of the invention in an aqueous environment, which leads to hydrolysis of V 1 , or bringing a compound .of the invention in an environment that contains an enzyme that recognizes and cleaves V 1 , or bringing a compound of the invention under reducing conditions, which leads to reduction and/or removal of V 1 , or bringing a compound of the invention in contact with radiation, e.g., UV light, which leads to transformation and/or cleavage, or bringing a compound of the invention in contact with heat, which leads to transformation and/or cleavage, or bringing a compound of the invention under reduced pressure, which leads to transformation, e.g., a retrocycloaddition, and/or cleavage, or bringing a compound of the invention under elevated or high pressure, which leads to transformation and/or cleavage.
- radiation e.g., UV light
- heat which leads to transformation and/or cleavage
- This condition may be met after administrating a compound of this invention to an animal, e.g., a mammal, for example a human: the condition may be met when the compound localizes to for example a specific organ, tissue, cell, subcellular target, or microbial target, for example by the presence of internal factors (e.g., target-specific enzymes or hypoxia) or application of external factors (e.g., radiation, magnetic fields) or the condition may already be met directly upon administration (e.g., ubiquitous enzymes).
- transformation of V 1 will directly or indirectly lead to cleavage of V 1 from Y.
- a compound of this invention may contain more than one V 1 moiety per promo iety (p and/or q > 1). These V 1 moieties may or may not be the same and may or may not require the same conditions for transformation and/or cleavage.
- a conjugate is used to target one or more moieties Z to target cells.
- a V 1 moiety may for example contain a substrate molecule that is cleaved by an enzyme present in the vicinity of the target cells or inside the target cells, for example tumor cells.
- V 1 can for example contain a substrate that is cleaved by an enzyme present at elevated levels in the vicinity of or inside the target cells as compared to other parts of the body, or by an enzyme that is present only in the vicinity of or inside the target cells.
- the condition causing the cleavage should preferably, at least to a certain degree, be target cell-specific, whereas the presence of another target-specific moiety in the compound of the invention, for instance in a V 2 moiety, reduces or takes away this requirement.
- V 2 causes selective internalization into a target cell
- an enzyme also present in other cells may transform and/or cleave V 1 .
- transformation and/or cleavage of V 1 occur intracellularly.
- transformation and/or cleavage of V 1 occur extracellularly.
- V 1 contains a di-, tri-, tetra-, or oligopeptide which consists of an amino acid sequence recognized by a proteolytic en2yme, for example plasmin, a cathepsin, cathepsin B, prostate-specific antigen (PSA), urokinase-type plasminogen activator (u-PA), or a member of the family of matrix metalloproteinases, present in the vicinity of or inside the target cells, for example tumor cells.
- a proteolytic en2yme for example plasmin, a cathepsin, cathepsin B, prostate-specific antigen (PSA), urokinase-type plasminogen activator (u-PA), or a member of the family of matrix metalloproteinases, present in the vicinity of or inside the target cells, for example tumor cells.
- V 1 is a peptide.
- V 1 is a dipeptide.
- V 1 is a tripeptide.
- V 1 contains a ⁇ -glucuronide that is recognized by ⁇ -glucuronidase present in the vicinity of or inside tumor cells.
- V 1 contains a substrate for an enzyme. In one embodiment, V 1 contains a substrate for an extracellular enzyme.
- V 1 contains a substrate for an intracellular enzyme.
- V 1 contains a substrate for a lysosomal enzyme.
- V 1 contains a substrate for the serine protease plasmin.
- V 1 contains a substrate for one or more of the cathepsins, for example cathepsin B.
- V 1 contains a substrate for a galactosidase.
- the one or more Z moieties may be released extracellularly.
- these Z moieties are not only able to affect the cell(s) directly surrounding the site of activation (e.g., target-positive cells), but also cells somewhat further away from the site of activation (e.g., target-negative cells) due to diffusion (bystander effect).
- An enzyme to cleave V 1 can also be transported to the vicinity of or inside target cells or target tissue via for example antibody-directed enzyme prodrug therapy (ADEPT), polymer-directed enzyme prodrug therapy (PDEPT) or macromolecular-directed enzyme prodrug therapy (MDEPT), virus-directed enzyme prodrug therapy (VDEPT), or gene-directed enzyme prodrug therapy (GDEPT).
- ADPT antibody-directed enzyme prodrug therapy
- PDEPT polymer-directed enzyme prodrug therapy
- MDEPT macromolecular-directed enzyme prodrug therapy
- VDEPT virus-directed enzyme prodrug therapy
- GDEPT gene-directed enzyme prodrug therapy
- transformation and/or cleavage of V 1 occur through an enzyme linked to an antibody.
- V 1 contains a moiety, for example a nitro(hetero)aromatic moiety, that can be transformed and/or cleaved by reduction under hypoxic conditions or by reduction by a nitroreductase. After reduction of the nitro group and cleavage of the resulting moiety, elimination of the spacer system Y, if present, leads to release of one or more moieties Z.
- the invention relates to a compound wherein V 1 is a monosaccharide, disaccharide, or oligosaccharide of hexoses or pentoses or heptoses that may also be included among the group of desoxy sugars or amino sugars and belong to the D-series or L-series and in the disaccharides or oligosaccharides are either identical or different.
- V 1 is typically cleaved by a glycosidase.
- V 1 has the formula C(OR a )R b CHR c R d , wherein R a ' is a hydroxy protecting group, an optionally substituted Ci -8 alkyl group that optionally forms a ring with R 5 , R 5 , R 6 , R 6 , R 7 , R 7 , R 14 , or R 14 ', or a monosaccharide, a disaccharide, or an oligosaccharide residue optionally substituted with a methyl group, hydroxymethyl group, and/or a radical of the formula -NR e R f , wherein R e and R f are identical or different and are selected from hydrogen, Ci -6 alkyl, or an amino protecting group, and wherein the hydroxy groups of the saccharide residues are optionally protected; R b , R°, and R d are independently selected from hydrogen, optionally substituted Ci -8 alkyl or Ci -8 cycloalkyl, or a group of the
- the invention relates to a conjugate wherein V 1 is a dipeptide, tripeptide, tetrapeptide, or oligopeptide moiety comprised of natural L amino acids, unnatural D amino acids, or synthetic amino acids, or a peptidomimetic, or any combination thereof.
- the invention relates to a compound wherein V 1 comprises a tripeptide.
- the tripeptide may be linked via its C-terminus to Y.
- the C-terminal amino acid residue of the tripeptide is selected from arginine, citrulline, and lysine
- the middle amino acid residue of the tripeptide is selected from alanine, valine, leucine, isoleucine, methionine, phenylalanine, cyclohexylglycine, tryptophan and proline
- the N-tenninal amino acid residue of the tripeptide is selected from any natural or unnatural amino acid.
- the invention relates to a compound wherein V 1 comprises a dipeptide.
- the dipeptide may be linked via its C-terminus to Y.
- the C-terminal amino acid residue of the dipeptide is selected from alanine, arginine, citrulline, and lysine
- the N-terminal amino acid residue of the dipeptide is selected from any natural or unnatural amino acid.
- this amino acid when the ⁇ -amino group of the N-terminal amino acid of V 1 is not coupled to L, this amino acid may be functionalized with a suitable blocking group coupled to the ⁇ -amino group or may be an unnatural amino acid such that undesired premature degradation of V 1 by for example ubiquitous enzymes or exopeptidases is prevented.
- embodiment V 1 is selected . from D-alanylphenylalanyllysine, D-valylleucyllysine, D-alanylleucyllysine, D-valylphenylalanyllysine, D-valyltryptophanyllysine, D-alanyltrypto- phanyllysine, alanylphenylalanyllysine, valylleucyllysine, alanylleucyllysine, valylphenyl- alanyllysine, valyltryptophanyllysine, alanyltryptophanyllysine, D-alanylphenylalanylcitrulline, D-valylleucylcitrulline, D-alanylleucylcitrulline, D-valylphenylalanylcitrulline, D-valyl- tryptophanylcitrulline, D-alanyltryptophanyllys
- V 1 is selected from phenylalanyllysine, valyllysine, valylalanine, D-phenylalanylphenylalanyllysine, phenylalanylphenylalanyllysine, glycylphenylalanyllysine, alanyllysine, valylcitrulline, N-methylvalylcitrulline, phenylalanylcitrulline, isoleucylcitrulline, tryptophanyllysine, tryptophanylcitrulline, phenylalanylarginine, phenylalanylalanine, glycylphenylalanylleucylglycine, alanylleucylalanylleucine, alanylarginylarginine, phenylalanyl- N 9 -tosylarginine, phenylalanyl-N 9 -nitroarginine, le
- V 1 is selected from phenylalanyllysine, valyllysine, and- valylcitrulline. Therefore, in one embodiment this invention relates to a compound wherein V 1 contains a substrate that can be cleaved by a proteolytic enzyme, plasmin, a cathepsin, cathepsin B, ⁇ -glucuronidase, a galactosidase, prostate-specific antigen (PSA), urokinase-type plasminogen activator (u-PA), a member of the family of matrix metalloproteinases, or an enzyme localized by means of directed enzyme prodrug therapy, such as ADEPT, VDEPT, MDEPT, GDEPT, or PDEPT, or wherein V 1 contains a moiety that can be cleaved or transformed through reduction under hypoxic conditions or through reduction by a nitroreductase.
- ADEPT ADEPT
- VDEPT MDEPT
- GDEPT GDEPT
- a conjugate of this invention is used to (also) improve the (pharmacokinetic) properties of Z.
- V 1 of said promoiety may for example be or contain a group that is cleaved by ubiquitous enzymes, e.g., esterases that are present in the circulation or intracellular enzymes, such as for example proteases and phosphatases, by pH-controlled intramolecular cyclization, or by acid- catalyzed, base-catalyzed, or non-catalyzed hydrolysis, or V 1 may for example be or contain a disulfide or form a disulfide with a neighboring moiety.
- V 1 may therefore, optionally together with the connecting atom(s) of L and/or Y, for example form a carbonate, carbamate, ureum, ester, amide, imine, hydrazone, oxime, disulfide, acetal, or ketal group.
- V may in this case for example be selected from R g -[O(R n O)P(O)] pp -, R s -C(0)-, R g -OC(O)-, and R g -N(R n )C(0)-, wherein pp is selected from 1 to 3 and each R g and R n are independently selected from H and optionally substituted Ci- 15 alkyl, C M5 heteroalkyl, Ci -I5 cycloalkyl, Ci -I5 heterocycloalkyl, C 4-I5 aryl, or C 4-I5 heteroaryl, R g and R n optionally being joined to form an optionally substituted carbocycle or heterocycle.
- V 1 is selected from phosphono, phenylaminocarbonyl, 4-(piperidino)piperidinocarbonyl, piperazinocarbonyl, and 4-methylpiperazinocarbonyl.
- V 1 may contain protecting groups.
- Compounds of the invention comprising such a protected V 1 may not release any Z moiety when put under conditions that will transform and/or cleave the corresponding unprotected V 1 . However, when said compounds are deprotected, such compounds will release one or more Z moieties when put under the appropriate conditions.
- Compounds comprising such a protected V also fall under the scope of this invention. In particular the above can be envisioned for compounds of formula (IV).
- Suitable protecting groups for functional groups, in particular for amino acids are well-known to the organic chemist and may for example be found in T. W. Greene, Protective Groups in Organic Synthesis, JoIm Wiley & Sons, New York, 1981.
- the compounds of formulae (III) and (IV) are designed to eventually release a compound of formula (I) or (II), or a compound of formula (I') or (II 1 ), after transformation and/or cleavage of the one or more V 1 and V 1 ' moieties. Release of a compound of formula (I) or (II), a compound of formula (I 1 ) or (II 1 ), or a derivative thereof, from a conjugate of this invention via another mechanism is however not excluded from this invention.
- a compound of formula (IH) represents an intermediate for the preparation of a compound of formula (I) or (II) or another compound of formula (III).
- V 2 , L 2 , L, and Y are absent, p, q, and z all are 1, and the V 1 moiety may be a protecting group.
- a compound of formula (III) is a compound of formula (I) or (II) to which a V 1 moiety is attached.
- a compound of formula (III) is a compound of formula (I) or (II) to which a V 1 moiety and a V 2' (-L 2 '-L'(-V r -Y')p ' )q ⁇ Z') z '-i moiety are attached.
- a compound of formula (III) is a compound of formula (I) or (II) to which a V 1 moiety and a V 1 ' moietyare attached.
- V 1 is not a protecting group.
- V 2 , L 2 , L, and Y are absent, and p, q, and z all are 1.
- V 1 is a chemically removable group.
- V 1 is a chemically removable group connected to Z via X 1 .
- V 1 is a benzyl group connected to Z via X 1 .
- V 1 is connected to L via more than one functional group on V 1 .
- V 1 is connected to L via one functional group on V 1 .
- V 1 is connected to L via a functional group in the side chain of one of the natural or unnatural amino acids of V .
- the N-terminal amino acid of V 1 is connected via its ⁇ amino group to L.
- the self-elimination spacer system Y when present, links V 1 and optionally L to one or more moieties Z.
- a self-elimination spacer system Y may be incorporated in a conjugate of this invention to for example improve the properties of Z or the conjugate in general, to provide for suitable coupling chemistries, and/or to create space between V 1 and Z.
- a compound of this invention may contain more than one spacer system Y per promoiety. These moieties Y may or may not be the same. After cleavage or transformation of V 1 , the left-hand side of Y may become unblocked, which results in eventual release of one or more moieties Z.
- the self-elimination spacer systems may for example be those described in WO 02/083180 and WO 2004/043493, which are incorporated herein by reference in their entirety, as well as other self-elimination spacers known to a person skilled in the art.
- the invention is related to compounds wherein Y is selected from (W-) W (X-) X (A-) S (W-) w (X-) x C((A) s -) r or (W-) w (X-) x C(D((A) s -) d ) r or
- W and X are each a single-release 1 ,2+2 « electronic cascade spacer (n > 1), being the same or different;
- A is an ⁇ -amino aminocarbonyl cyclization spacer that forms a cyclic ureum derivative upon cyclization
- C, D, E, and F are each a self-eliminating multiple-release spacer or spacer system that upon activation can maximally release r, d, e, and f groups, respectively; s is 0 or 1; r, d, e, and f are numbers representing degree of branching; w and x are numbers representing degree of polymerization; r, d, e, and fare independently an integer from 2 (included) to 24 (included); w and x are independently an integer from 0 (included) to 5 (included).
- the self-elimination multiple-release spacer systems C, D, E, and F are independently selected from a moiety having the formula:
- B is selected from NR 21 , O, and S;
- P is C(R 22 )(R 23 )Q-(W-) W (X-) X ;
- Q is absent or is -O-CO-;
- W and X are each a single-release 1,2+2 « electronic cascade spacer (n > 1), being the same or different;
- G, H, I, J, K, L, M, N, and O are independently selected from moieties having the formula: R 24
- G, J, and M may in addition be selected from the group of P and hydrogen with the proviso that if two of G, J, and M are hydrogen, the remaining group must be
- R 21 is selected from H and optionally substituted Ci -6 allcyl
- R 22 , R 23 , R 24 , and R 25 are independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO, CF 3 , CN, C(O)NH 2 , C(O)H, C(O)OH, halogen, R x , SR x , S(O)R x , S(O) 2 R x , S(O)OR x , S(O) 2 OR x , OS(O)R x , OS(O) 2 R x , OS(O)OR x , OS(O) 2 OR x , OR x , NHR x , N(R x )R x 1 , 4 N(R x )(R x ⁇ R x 2 , P(O)(OR x )(OR x 1 ), OP(O)(OR x )(OR x 1 ), C(O)R x , C(O)OR
- the l,2+2 ⁇ electronic cascade spacers W and X are independently selected from a moiety having the formula:
- R 26 , R 27 , B, and (T-) t (T-) t (T"-) t ..P are connected to C a , C b , C c , and C d in such a way that B and
- T-) t (T'-)t ' (T M -) t" P are connected to two adjacent carbon atoms or to C a and C d ;
- Q is absent or is -O-CO-; t, t 1 , and t" are numbers representing degree of polymerization and are independently an integer from 0 (included) to 5 (included);
- T, T', and T" are independently selected from moieties having the formula:
- R 26 , R 27 , R 28 , R 29 , R 30 , R 31 , R 32 , R 33 , and R 34 are independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO, CF 3 , CN, C(O)NH 2 , C(O)H, C(O)OH, halogen, R x , SR x , S(O)R x , S(O) 2 R x , S(O)OR x , S(O) 2 OR x , OS(O)R x , OS(O) 2 R x , OS(O)OR x , OS(O) 2 OR x , OR x , NHR x , N(R x )R x 1 , + N(R X )(R X 1 )R X 2 , P(O)(OR x )(OR x '), OP(O)(OR x )(
- R x , R x 1 and R x 2 are independently selected from H and optionally substituted Cue alkyl, Cj -6 heteroalkyl, C 3-2 O cycloalkyl, C 3-20 heterocycloalkyl, C 4-2 O aryl, or C 4-2 O heteroaryl, R x , R x 1 , and R x 2 optionally being joined to form one or more optionally substituted aliphatic or aromatic carbocycles or heterocycles, two or more of the substituents R 26 , R 27 , R 28 , R 29 , R 30 , R 31
- Q may be O-CO, but it may also be absent.
- Q may be O-CO, but it may also be absent.
- a compound with a benzyl ether linkage between self-elimination spacer and the group that leaves, the oxycarbonyl function being absent (Q is absent) has been reported to undergo self-elimination 11 .
- the ⁇ -amino aminocarbonyl cyclization elimination spacer A is a moiety having the formula:
- R 37 , R 38 , R 39 , R 40 , R 41 , and R 42 are independently selected from H, OH, SH, NH 2 , N 3 , NO 2 , NO, CF 3 , CN, C(O)NH 2 , C(O)H, C(O)OH, halogen, R x , SR x , S(O)R x , S(O) 2 R x , S(O)OR x , S(O) 2 OR x , OS(O)R x , OS(O) 2 R x , OS(O)OR x , OS(O) 2 OR x , OR x , NHR x , N(R x )R x 1 , + N(R x )(R x ⁇ R x 2 , P(O)(OR x )(OR x 1 ), OP(O)(OR x )(OR x 1 ), C(O)R x
- R x , R x 1 and R x 2 are independently selected from H and optionally substituted Ci -6 alkyl, C 1 .
- R x , R x 1 , and R x 2 optionally being joined to form one or more optionally substituted aliphatic or aromatic carbocycles or heterocycles
- R 35 , R 36 , R 37 , R 38 , R 39 , R 40 , R 41 , aiid R 42 are optionally part of one or more optionally substituted aliphatic or aromatic cyclic structures, two or more of the substituents R 35 , R 36 , R 37 , R 38 , R 39 , R 40 , R 41 , or R 42 optionally being connected to one another to form one or more aliphatic or aromatic carbocycles or heterocycles.
- this invention relates to a compound wherein X 1 is O and Y is connected to X 1 via an ⁇ -amino aminocarbonyl cyclization spacer being part of Y.
- the spacer system Y is selected from
- the spacer system Y is
- the spacer system Y is
- self-eliminating spacers include, but are not limited to, spacers that can undergo cyclization 12 , such as optionally substituted 4-aminobutyric acid amides, appropriately substituted bicyclo[2.2.1] and bicyclo[2.2.2] ring systems, and 2-aminophenylpropionic acid amides and "trimethyl-lock" cyclization spacers 13 .
- a glycine spacer in which an amine-containing leaving group is connected at the ⁇ -position is another useful spacer for the compounds of the invention. 14
- a spacer system Y may be connected to more than one V 1 moiety. In this case, transformation and/or cleavage of one of these V 1 moieties may trigger the release of one or more Z moieties.
- V 1 moieties that are transformed or cleaved under different conditions are connected to the same Y, release of one or more Z moieties may occur when a conjugate of this invention is brought under one of several different conditions.
- a spacer system Y may be used that requires to be triggered twice or even more times in order to self- eliminate.
- An example of such a self-elimination spacer is a bicine spacer. 15 When such a spacer is used in combination with different, selectively cleavable V 1 moieties connected to said spacer, selectivity of release of Z may be increased as two different conditions must be met before Z is released.
- the Linking Group L links one or more V 1 and/or Y moieties to L 2 or RM. Synthesis may be more straightforward when L is connected to V 1 and the compound may be less prone to premature degradation. Connection of L to Y may have the advantage that V may be transformed and/or cleaved with more ease. Other reasons to connect L to Y may for example be that (part of) Y remains bound to L upon cleavage of V 1 , which prevents the release of reactive small molecules, or that the compound displays improved (pharmacokinetic) properties, solubility, or aggregation behavior. L may be a bond connecting V 1 or Y directly to either L 2 or RM.
- L is a linking group that functionally links or spaces the one or more moieties VVY and the L 2 or RM moiety.
- spacing may make the reactive moiety RM more accessible to the reaction partner, for example when the functional moiety is being coupled.
- spacing may provide for a better accessibility of V 1 , because V 2 is further removed, which, especially in the case of enzymatic cleavage or transformation of V 1 , may improve the rate at which V 1 is transformed and/or cleaved.
- the linking group L may be a water- soluble moiety or contain one or more water-soluble moieties, such that L contributes to the water solubility of a compound of formula (III) or (IV).
- L may also be a moiety or contain one or more moieties that reduce(s) aggregation of a compound of formula (III) or (IV), which may or may not be a moiety/moieties that also increase(s) the water solubility of a compound of formula (III) or (IV).
- the linking group L must contain suitable functional groups at both of its ends to provide for selective coupling with the one or more V 1 and/or Y moieties and L 2 or RM.
- the L moiety is a linear, branched, or dendritic moiety, so that it can optionally be connected to more than one V 1 and/or Y moiety.
- Branching can occur via one or more cyclic structures or at one or more branching atoms that may for example be carbon, nitrogen, silicon, or phosphorus.
- the number of branches in L that are connected to V 1 and/or Y does not necessarily equal the total number of branches as in the coupling reaction with VVY not all branches may be coupled to V 1 and/or Y moieties due to incomplete chemical conversion. This means that L may contain branches that are not coupled to V 1 or Y, but instead end in for example a functional group, H, OH, or a leaving group.
- compounds of this invention may exist as a mixture, wherein each component of the mixture has a different p value.
- the compound may exist as a mixture of two separate compounds, one compound wherein p is 2 and another compound wherein p is 3.
- the compound may exist as a mixture of isomers as VVY may be connected to distinct sets of branches on L.
- L is a bond.
- L is a linear linker
- L is a linear linker built up through a cycloaddition reaction between a molecule containing an azide group and one containing an acetylene group.
- L is a branched linker.
- L is a dendritic linker.
- the dendritic structure may for example be built up through cycloaddition reactions between molecules containing an azide group and ones containing an acetylene group.
- p is 1.
- p is 2 or 3 or 4 or 6 or 8 or 9.
- L is represented by the formula:
- X 81 and X 82 are each independently O, NR 85 , or S;
- Each X 83 and X 84 are each independently O, NR 86 , or S; Each x81, x82, x83, and x84 are independently 0 or 1; p" is a number representing a degree of branching and is an integer selected from 1 (included) to
- p'" is a number representing a degree of branching and is an integer selected from 0 (included) to
- Each DD is independently H, OH, or a leaving group
- R 80 is absent or is either a dendritic, branched or unbranched moiety and selected from optionally substituted alkylene or polyalkylene, optionally substituted heteroalkylene or polyheteroalkylene, optionally substituted arylene or polyarylene, optionally substituted heteroarylene or polyheteroarylene, optionally substituted cycloalkylene or polycycloalkylene, optionally substituted heterocycloalkylene or polyheterocycloalkylene, -(CH 2 CH 2 O) V -, -alkylene-(CH2CH2 ⁇ ) v -,
- R 85 and R 86 are independently selected from H and C] -8 alkyl; v is selected from 1 (included) to 500 (included).
- L may be selected from optionally substituted C 1-1 O alkylene, Ci -I0 alkylenecarbonyl, C 1-12 alkyleneoxycarbonyl, C 1-12 carbonylalkylene, Ci -I2 carbonylalkyleneoxycarbonyl, and (CH 2 CH 2 O) v -carbonyl.
- L is selected from
- the Reactive Moiety RM and the Linking Group L 2 The reactive moiety RM in a compound of formula (IV) is connected to the linking group L and is able to react with a suitable functional group on a reaction partner.
- the reactive moiety RM is designed to react with a functional group on the moiety V 2 , which results in formation of a compound of formula (III). In this reaction, the moiety RM is transformed into the moiety L 2 . In another embodiment, the reactive moiety RM is designed to react with a complementary moiety in situ, e.g., in vivo, to give a compound that may or may not be a compound of formula (III).
- the reactive moiety RM contains an electrophilic group that reacts with a nucleophilic group on the reaction partner, for example V 2 , e.g., a thiol group, an amino group, or a hydroxy group.
- the reactive moiety RM contains a nucleophilic group that reacts with an electrophilic group on the reaction partner, for example V 2 , e.g., an aldehyde group.
- the reactive moiety RM contains a cycloaddition partner moiety, e.g., an alkene, a diene, a 1,3-dipole, or a 1,3-dipolarophile, that reacts with a suitable complementary cycloaddition partner moiety on the reaction partner, for example V 2 , e.g., a diene, an alkene, a 1,3-dipolarophile, or a 1,3-dipole.
- a cycloaddition partner moiety e.g., an alkene, a diene, a 1,3-dipole, or a 1,3-dipolarophile.
- the reactive moiety RM contains a group that can be coupled with a suitable complementary group on the reaction partner, for example V 2 , under metal- catalyzed, biocatalyzed, or enzyme-catalyzed conditions, e.g., palladium-catalyzed conditions.
- a suitable complementary group on the reaction partner for example V 2
- the reactive moiety RM is, without limitation,
- X 8 is selected from -Cl, -Br, -I, -F, -OH, -O-N-succinimide, -O-(4-nitrophenyl), -O-pentafluorophenyl, -O-tetrafluorophenyl, -0-C(O)-R 50 , and -0-C(O)-OR 50 ;
- X 9 is selected from -Cl, -Br, -I, -0-mesyl, -O-triflyl, and-O-tosyl;
- R 50 is Ci-Cio allcyl or aryl.
- the moiety RM is chosen from
- the moiety RM is
- the moiety RM is chosen from
- the linking group L 2 in compounds of formula (III) represents the remainder of RM when the reactive moiety RM has reacted with V 2 . This group then links the moiety V 2 with L.
- the group that remains may be a bond.
- L 2 is a linking group.
- L 2 does not represent the remainder of RM, but may represent a similar or the same moiety and in addition be selected from for example optionally substituted Q-g alkylene, C ⁇ heteroalkylene, C 3 - 7 cycloalkylene, C 3-7 heterocycloalkylene, C 5- ] 0 arylene, and Cs -10 heteroarylene.
- the moiety L 2 is a bond.
- the moiety L 2 is, without limitation,
- the moiety L is
- V 2 is a functional moiety, which means that it adds additional functionality to a compound of the invention.
- V 2 is a targeting moiety.
- the V 2 moiety is a moiety that improves the pharmacokinetic properties of a compound of the invention.
- the V 2 moiety is a moiety that causes accumulation of compounds of the invention at a target site.
- the V 2 moiety is a moiety that improves the aqueous solubility of a compound of the invention.
- the V 2 moiety is a moiety that increases the hydrophobicity of a compound of the invention.
- the V 2 moiety is a moiety that reduces extravasation of a compound of the invention. In yet another embodiment, the V 2 moiety is a moiety that reduces excretion of a compound of the invention. In yet another embodiment, the V 2 moiety is a moiety that reduces the immunogenicity of a compound of the invention. In yet another embodiment, the V 2 moiety is a moiety that enhances the circulation time of a compound of the invention. In yet another embodiment, the V 2 moiety is a moiety that enhances the ability of a compound of the invention to cross a biological barrier, e.g., a membrane, cell wall, or the blood-brain barrier.
- a biological barrier e.g., a membrane, cell wall, or the blood-brain barrier.
- the V 2 moiety is a moiety that enhances the ability of a compound of the invention to internalize. In yet another embodiment, the V 2 moiety is a moiety that causes the compounds of the invention to aggregate. In yet another embodiment, the V 2 moiety is a moiety that reduces aggregation of compounds of the invention. In yet another embodiment, the V 2 moiety is a moiety that causes the compounds of the invention to form micelles or liposomes. In yet another embodiment, the V 2 moiety is a moiety that causes complexation of a compound of the invention to another molecule, e.g., a biomolecule.
- the V 2 moiety is a polynucleotide moiety that complexes with a complementary nucleotide sequence, for example RNA or DNA.
- the V 2 moiety is a moiety that causes a compound of the invention to bind, associate, interact, or complex to another moiety, for example a (functionalized) surface or solid support.
- V 2 exhibits two or more different functions.
- the moiety V 2 includes within its scope any unit that binds or reactively associates or complexes with a receptor, a receptor complex, antigen, or other receptive moiety associated with a given target cell population.
- V 2 can be any molecule that binds to, complexes with, or reacts with a moiety of a cell population sought to be therapeutically or otherwise biologically modified.
- the V 2 moiety acts to deliver the one or more moieties Z to the particular target cell population with which V 2 reacts or to which V 2 binds.
- V 2 moieties include, but are not limited to, aptamers, full-length antibodies and antibody fragments, lectins, biologic response modifiers, enzymes, vitamins, growth factors, steroids, nutrients, sugar residues, oligosaccharide residues, hormones, and any derivatives thereof, or any combination of any of these.
- the compounds of the invention may or may not be internalized. If internalization occurs, transformation and/or cleavage of V 1 preferably occur inside the target cell. ⁇
- Useful non-immunoreactive protein, polypeptide, or peptide V 2 moieties include, but are not limited to, transferrin, epidermal growth factors ("EGF"), bombesin, gastrin and its derivatives, particularly those containing the tetrapeptide sequence Trp-Met-Asp-Phe-NH 2 , gastrin-releasing peptide, platelet-derived growth factor, IL-2, IL-6, transforming growth factors ("TGF”), such as TGF-a and TGF-P, tumor growth factors, vaccinia growth factor (“VGF”), insulin and insulin-like growth factors I and II, lectins, and apoprotein from low density lipoprotein.
- Useful polyclonal antibody V 2 moieties are heterogeneous populations of antibody molecules. Various procedures well-known in the art may be used for the production of polyclonal antibodies to an antigen-of-interest.
- Useful monoclonal antibody V 2 moieties are homogeneous populations of antibodies to a particular antigen (e.g., a cancer cell antigen).
- a monoclonal antibody (mAb) to an antigen-of-interest can be prepared by using any technique known in the art which provides for the production of monoclonal antibody molecules.
- Useful monoclonal antibody V 2 moieties include, but are not limited to, human monoclonal antibodies, humanized monoclonal antibodies, or chimeric human-mouse (or other species) monoclonal antibodies.
- Human monoclonal antibodies may be made by any of numerous techniques known in the art.
- the V 2 moiety can also be a bispecific antibody.
- Methods for making bispecific antibodies are known in the art.
- the V moiety can be a functionally active fragment, derivative, or analog of an antibody that immunospecifically binds to antigens on the target cells, e.g., cancer cell antigens.
- functionally active means that the fragment, derivative, or analog is able to elicit anti-anti- idiotype antibodies that recognize the same antigen that the antibody from which the fragment, derivative, or analog is derived, recognizes.
- V 2 moieties comprise fragments of antibodies including, but not limited to, F(ab') 2 fragments, which contain the variable region, the light chain constant region and the CHl domain of the heavy chain, which can be produced by pepsin digestion of the antibody molecule, and Fab fragments, which can be generated by reducing the disulfide bridges of the F(ab') 2 fragments.
- Other useful V 2 moieties are heavy chain and light chain dimers of antibodies, or any minimal fragment thereof such as Fvs or single chain antibodies (SCAs), domain antibodies, anticalins, affibodies, nanobodies, or any other molecule with the same, similar, or comparable specificity as the antibody.
- recombinant antibodies such as chimeric and humanized monoclonal antibodies, comprising both human and non-human portions, which can be made using standard recombinant DNA techniques, are useful V 2 moieties.
- a chimeric antibody is a molecule in which different portions are derived from different animal species, such as those having a variable region derived from a murine monoclonal and a human k ⁇ munoglobulin constant region.
- Humanized antibodies are antibody molecules from non-human species having one or more complementarity detennining regions (CDRs) from the non-human species and a framework region from a human immunoglobulin molecule. Completely human antibodies are particularly desirable as V 2 moieties.
- the V 2 moiety is a fusion protein of an antibody, or a functionally active fragment or derivative thereof, for example in which the antibody is fused via a covalent bond (e.g., a peptide bond), at either the N-terminus or the C-terminus to an amino acid sequence of another protein (or portion thereof, preferably at least a 10, 20, or 50 amino acid portion of the protein) that is not the antibody.
- a covalent bond e.g., a peptide bond
- the antibody or fragment thereof is covalently linked to the other protein at the N-terminus of the constant domain.
- V 2 moiety antibodies include analogs and derivatives that are modified, i.e., by the covalent attachment of any type of molecule as long as such covalent attachment permits the antibody to retain its antigen binding immunospecificity.
- derivatives and analogs of antibodies include those that have been further modified, e.g., by glycosylation, acetylation, pegylation, disulfide reduction, phosphylation, amidation, derivatization by known protecting or blocking groups, proteolytic cleavage, linkage to an other protein, etc.
- the analog or derivative can contain one or more unnatural amino acids.
- the V 2 moiety antibodies include antibodies having modifications (e.g., substitutions (for example cysteine to serine), deletions, or additions) in amino acid residues that interact with Fc receptors.
- modifications e.g., substitutions (for example cysteine to serine), deletions, or additions
- they include antibodies having modifications in amino acid residues identified as involved in the interaction between the Fc domain and the FcRn receptor. Modifications may also be introduced to be able to couple the antibody to linker-agent conjugates at specific positions on the antibody.
- an antibody immunospecific for a cancer or tumor antigen is used as a V 2 moiety in accordance with the compounds, compositions, and methods of the invention.
- Antibodies immunospecific for a cancer cell antigen can be obtained commercially or produced by any method known to one of skill in the art such as, e.g., chemical synthesis or recombinant expression techniques.
- the nucleotide sequences encoding antibodies immunospecific for a cancer cell antigen can be obtained, e.g., from the GenBank database or a database like it, a commercial or other source, the literature publications, or by routine cloning and sequencing.
- antibodies available for the treatment of cancer include, but are not limited to, HERCEPTIN (trastuzumab; Genentech, CA) which is a humanized anti-HER2 monoclonal antibody for the treatment of patients with metastatic breast cancer; RITUXAN (rituximab; Genentech, CA), which is a chimeric anti-CD20 monoclonal antibody for the treatment of patients with non-Hodgkii ⁇ s lymphoma; OvaRex (oregovomab; AltaRex Corporation, MA) which is a murine antibody for the treatment of ovarian cancer; Panorex (edrecolomab; Glaxo Wellcome, NC) which is a murine IgG 2a antibody for the treatment of colorectal cancer; IMC-BEC2 (initumomab; ImClone Systems, NY) which is a murine IgG antibody for the treatment of lung cancer; IMC-C225 (erbitux; Imclone Systems, NY) which is
- antibodies useful in the treatment of cancer include, but are not limited to, antibodies against the following antigens: CA125, CA15-3, CA19-9, L6, Lewis Y, Lewis X, alpha fetoprotein, CA 242, placental alkaline phosphatase, prostate specific antigen, prostatic acid phosphatase, epidermal growth factor receptors, HER2, EGFR, VEGF, MAGE-I, MAGE-2, MAGE-3, MAGE-4, anti-transferrin receptor, p97, MUCl-KLH, MUC18, PSMA, CTLA4, CEA 3 gplOO, MARTl, PSA, IL-2 receptor, CD2, CD4, CD20, CD30, CD52, CD56, CD74, CD33, CD22, HLA-DR, HLA-DRlO, human chorionic gonadotropin, CD38, CD40, CD70, mucin, P21, MPG, and Neu oncogene product.
- the antibody is an anti-nuclear antibody or an antibody that can bind to a receptor or receptor complex expressed on a target cell.
- the receptor or receptor complex can comprise an immunoglobulin gene superfamily member, an integrin, a chemokine receptor, a TNF receptor superfamily member, a cytokine receptor, a major histocompatibility protein, a complement control protein, or a lectin.
- an antibody immunospecif ⁇ c for an antigen associated with an autoimmune disease is used as a V 2 moiety in accordance with the compounds, compositions, and methods of the invention.
- an antibody immunospecific for a viral or microbial antigen is used as a V 2 moiety in accordance with the compounds, compositions, and methods of the invention.
- the term "viral antigen” includes, but is not limited to, any viral peptide or polypeptide protein that is capable of eliciting an immune response.
- microbial antigen includes, but is not limited to, any microbial peptide, polypeptide, protein, saccharide, polysaccharide, or lipid that is capable of eliciting an immune response.
- New antibodies are continually being discovered and developed, and the present invention provides that these new antibodies may also be incorporated into a compound of this invention.
- V 2 can react with the reactive moiety RM via for example a heteroatom on V 2 .
- Heteroatoms that may be present on V 2 include, without limitation, sulfur (in one embodiment, from a sulfhydryl group), oxygen (in one embodiment, from a carboxyl or hydroxyl group), and nitrogen (in one embodiment, from a primary or secondary amino group).
- V 2 may also react via for example a carbon atom (in one embodiment, from a carbonyl group). These atoms can be present on V 2 in V 2 's natural state, for example a naturally occurring antibody, or can be introduced into V 2 via chemical modification.
- Free sulfhydryl groups can be generated in an antibody or antibody fragment by reduction of the antibody (fragment) with a reducing agent such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP).
- a reducing agent such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP).
- DTT dithiothreitol
- TCEP tris(2-carboxyethyl)phosphine
- modified antibodies can be obtained that can have from 1 to about 20 sulfhydryl groups, but typically between about 1 and about 9 sulfhydryl groups.
- V 2 can have one or more carbohydrate groups that can be chemically modified to have one or more sulfhydryl groups.
- sulfhydryl groups can be generated by reaction of amino groups, for example from lysine moieties, on V 2 with 2-iminothiolane (Traut's reagent) or another sulfhydryl-generating reagent.
- the V 2 moiety is a receptor-binding moiety.
- the V 2 moiety is an antibody or an antibody fragment.
- the V 2 moiety is a monoclonal antibody or a fragment thereof.
- V 2 has one or more sulfhydryl groups and V 2 reacts with one or more RM moieties of compounds of formula (IV) via one or more of these sulfhydryl groups' sulfur atoms to form a compound of formula (III).
- V 2 contains disulfide bonds that can be chemically reduced to sulfhydryl groups (two for each disulfide bond), which can then be reacted with one or more reactive moieties RM. In another embodiment, V 2 contains about 1 to about 3 sulfhydryl groups, which can be reacted with one or more reactive moieties RM.
- V 2 contains about 3 to about 5 sulfhydryl groups, which can be reacted with one or more reactive moieties RM.
- V 2 contains about 7 to about 9 sulfhydryl groups, which can be reacted with one or more reactive moieties RM.
- V 2 can have one or more carbohydrate groups that can be chemically modified to have one or more sulfhydryl groups. V 2 reacts with RM moieties via these one or more sulfhydryl groups' sulfur atoms.
- V 2 can have one or more lysine groups that can be chemically modified to have one or more sulfhydryl groups, which can be reacted with one or more reactive moieties RM.
- Reactive moieties that can react with a sulfhydryl group include, but are not limited to, carbamoyl halide, acyl halide, ⁇ -haloacetamide, halomethyl ketone, vinyl sulfone, maleimide, and
- V 2 can have one or more carbohydrate groups that can be oxidized to provide one or more aldehyde groups.
- the corresponding aldehyde(s) can then react with one or more reactive moieties RM.
- Reactive moieties that can react with a carbonyl group on V 2 include, but are not limited to, hydrazine, hydrazide, amine, and hydroxylamine.
- V 2 can have one or more amino groups, e.g., from lysine residues, which can be reacted with one or more reactive moieties RM.
- Reactive moieties that can react with an amino group include, but are not limited to, carbamoyl halide, ⁇ -haloacetamide, acyl halide, aldehyde, isocyanate, and isothiocyanate.
- a conjugate of formula (III) may exist as a mixture, wherein each component of the mixture has a different q value.
- the compound may exist as a mixture of two separate compounds, one compound wherein q is 3 and another compound wherein q is 4.
- q may be the (rounded) average number of L 2 -L(-V 1 -Y-) p (Z) z / q units per V 2 moiety.
- the compound may exist as a mixture of isomers as the q L 2 -L(-V 1 -Y-) p (Z) 2/q moieties may be connected to distinct sets of functional groups on V 2 . It should be noted that the number of Z moieties in each unit only equals z/q if all units are the same and/or contain the same number of Z moieties.
- the V 2 moiety is connected to L 2 via a sulfur atom.
- V 2 moiety is connected to L 2 via a sulfur atom and q ranges from about 1 1 ttoo aabboouutt 2200..
- the V 2 moiety is connected to L 2 via a sulfur atom and q ranges from about 1 1 ttoo aabboouutt 99.. In another embodiment, the V 2 moiety is connected to L 2 via a sulfur atom and q ranges from about
- V 2 moiety is connected to L 2 via a sulfur atom and q is about 2.
- V 2 moiety is connected to L 2 via a sulfur atom and q ranges from about
- V 2 moiety is connected to L 2 via a sulfur atom and q is about 4.
- V 2 moiety is connected to L 2 via a sulfur atom and q ranges from about
- V 2 moiety is connected to L 2 via a sulfur atom and q is about 8.
- a compound of formula (III) exists as a mixture of separate compounds.
- a compound of formula (III) exists as a mixture of separate compounds wherein q for three compounds is 1, 2, and 3, respectively.
- a compound of formula (III) exists as a mixture of separate compounds wherein q for three compounds is 3, 4, and 5, respectively.
- a compound of formula (III) exists as a mixture of separate compounds wherein q for three compounds is 5, 6, and 7, respectively.
- a compound of formula (III) exists as a mixture of separate compounds wherein q for three compounds is 7, 8, and 9, respectively.
- the V 2 moiety is connected to L 2 via a nitrogen atom.
- the V 2 moiety is connected to L 2 via a carbon atom.
- the V 2 moiety includes any unit that causes accumulation of compounds of the invention at the target site or in the vicinity thereof by a mechanism other than binding or reactively associating or complexing with a receptor, antigen, or other receptive moiety associated with a given target site, e.g., a target cell population.
- a receptor, antigen, or other receptive moiety associated with a given target site, e.g., a target cell population.
- a target cell population e.g., a target cell population.
- One way to achieve this is for example to use a large macromolecule as a V 2 moiety, which targets to solid tumor tissue through the enhanced permeability and retention (EPR) effect. Ringsdorf reported use of polymers to target antitumor agents to tumors.
- EPR enhanced permeability and retention
- the V 2 moiety may for example be a branched or unbranched polymer, such as for example poly[N-(2-hydroxypropyl)methacrylamide] (HPMA), poly(2-hydroxyethyl methacrylate) (HEMA), polyglutamic acid or poly-L-glutamic acid (PG), carboxymethyldextran (CMDex), a polyacetal, chitosan, a polypeptide, an oligoethylene glycol or polyethylene glycol (PEG), or a copolymer, such as an HPMA copolymer, an HPMA-methacrylic acid copolymer, a HEMA-methacrylic acid copolymer, a CMDex copolymer, a ⁇ -cyclodextrin copolymer, a PEG copolymer, or a poly(lactic-co-glycolic) acid copolymer. 18 In this document both polymer and copolymer are referred to as polymer.
- the polymer may be connected to L 2 via any suitable functional group, which can be located at one or both ends of the polymer, meaning that in the conjugate q ranges from 1 to 2, or alternatively, the functional groups may (also) be located on groups pendant on the polymer such that L is (also) connected to the polymer via these pendant groups with q typically ranging from 1 to about 1000.
- the polymer may also contain an additional targeting group that can bind or reactively associate or complex with a receptive moiety, e.g., an antibody or antibody derivative, bonded to the polymer either via a pendant group or end group, such that improved targeting to the target site is achieved.
- the V 2 moiety may be a dendrimer or a protein or protein fragment, e.g., albumin, which has no targeting properties except for its ability to accumulate at the target site because of its size or molecular weight.
- the V 2 moiety is a polymer.
- the V 2 moiety is a polymer and q ranges from 1 to about 1000. In other embodiments, the V 2 moiety is a polymer and q ranges from 1 to about 500 or 400 or 300 or 200 or 100 or less than 100. In another embodiment, the V 2 moiety is a polymer and q ranges from 1 to 2. In a specific embodiment, the V 2 moiety is an oligoethylene glycol or a polyethylene glycol or a derivative thereof.
- the V 2 moiety is a dendrimer, a protein, or a protein fragment.
- the .V 2 moiety is a moiety that is able to transport the .conjugate across a biological barrier, e.g., a cell membrane, either with or without prior binding, associating, or complexing with a receptor or receptor complex.
- the V 2 moiety is a Tat peptide or a derivative, fragment, or analog thereof, or a moiety that has similar transmembrane delivery properties.
- the V 2 moiety is protein or protein fragment, an antibody or an antibody fragment, a receptor-binding or peptide vector moiety, or a polymeric or dendritic moiety, or any combination thereof, to which is attached a Tat peptide or a derivative, fragment, or analog thereof, or a moiety that has similar transmembrane delivery properties.
- the moiety V 2 is a targeting moiety and is for example selected from the group consisting of a protein or protein fragment, an antibody or an antibody fragment, a receptor-binding or peptide vector moiety, and a polymeric or dendritic moiety, or any combination thereof.
- the V 2 moiety is a moiety that improves the pharmacokinetic properties of a conjugate of the invention.
- the moiety V can be chosen such that the water solubility of the conjugate is (further) improved. This can be achieved by choosing V to be a hydrophilic moiety.
- the V 2 moiety can be used to for example increase the residence time of the compound in the circulation, to reduce extravasation and excretion, to reduce aggregation, and/or to reduce the immunogenicity of the compound. This can for example be achieved by choosing V 2 to be a polyethylene glycol or oligoethylene glycol or derivative thereof.
- V 2 is a moiety that improves the pharmacokinetic properties of the compound of the invention
- V 1 is a moiety that can be cleaved or transformed aspecifically, and there are no V and V 2 ' moieties, the compound solely serves to improve the (pharmacokinetic) properties of the one or more Z moieties.
- V 2 is a moiety that improves the pharmacokinetic properties and V 1 is a moiety that can be cleaved or transformed specifically.
- V 2 is an oligoethylene glycol or a polyethylene glycol or a derivative thereof and V 1 is a moiety that can be cleaved or transformed specifically. In one embodiment, V 2 is a moiety that improves the pharmacokinetic properties and V 1 is a moiety that can be cleaved or transformed aspecifically.
- V 2 is an oligoethylene glycol or a polyethylene glycol or a derivative thereof and V 1 is a moiety that can be cleaved or transformed aspecifically.
- V 2 is an oligoethylene glycol or a polyethylene glycol or a derivative thereof and V 1 is a moiety that can be cleaved by ubiquitous enzymes.
- V 2 is an oligoethylene glycol or a polyethylene glycol or a derivative thereof and V 1 is a hydrolysable moiety.
- V 2 moiety is represented by formula (VI):
- V 2* , L 2* , L*, V 1* , Y*, p*, q*, and z* have the same meaning as V 2 , L 2 , L, V 1 , Y, p, q, and z as defined in this document, except that Y* is connected to L 2 . It should be noted that z* actually equals q.
- a compound of formula (HI) contains a V 2 moiety represented by formula (VI), the one or more L 2 moieties are thus connected to Y*.
- V 2 , L 2 , L, V 1 , Y, p, q, or z the same can apply for each V 2* , L 2* , L*, V 1* , Y*, p*, q*, or z*, respectively.
- V 2 moiety of formula (VI) in a conjugate of formula (III) implicates that two conditionally-cleavable or conditionally-transformable moieties may be present in the same promoiety, and therefore two separate cleavages/transformations may be required to completely remove the promoiety.
- the requirement that two different conditions need to have been met before one or more Z are released might favorably affect the properties of the conjugate. For instance, it may increase the targeting efficiency of the conjugate.
- the two transformations/cleavages may occur at different extracellular/intracellular locations.
- the moiety to be removed by the second cleavage or as a consequence of the second transformation may be used in this instance to help transport Z from a first extracellular or intracellular location to a second extracellular or intracellular location.
- V 2 can be a moiety that improves the pharmacokinetic properties of a compound of this invention and at the same time be or contain a targeting moiety.
- Conjugates of the invention may contain one or more promo ieties. These promoieties may be the same or different. The presence of two or more promoieties may favorably affect the properties of the conjugate. For instance, it may improve the water solubility and/or increase the targeting efficiency of the conjugate. In one embodiment, when there are two or more promoieties, said promoieties are different from each other. The two or more different promoieties may have different functions and may be removed under different conditions and at different extracellular/intracellular locations.
- promoiety linked to Z via X there is one promoiety linked to Z via X . In another embodiment, there are two promoieties linked to Z.
- promoieties linked to Z of which one is connected via X 1 .
- a compound of formula (III) is represented by a compound of formula (IH-I) or (III-2):
- a compound of formula (III) is represented by a compound of formula (i ⁇ -3) or (III-4):
- Y' is connected to an atom being part of R 8 , R 9 , R 10 , or R ⁇ .
- p is an integer from 1 (included) to 128 (included).
- q is an integer from 1 (included) to 1000 (included).
- p is an integer from 1 (included) to 64 (included) or 32 (included) or 16 (included) or 8 (included) or 4 (included) or 2 (included).
- q is an integer from 1 (included) to 500 (included) or 400 (included) or 300 (included) or 200 (included) or 100 (included) or 16 (included) or 8 (included) or 6 (included) or 4 (included) or 2 (included).
- a compound of formula (III) is represented by
- p in a compound of formula (Ilia) is 1.
- R la is chloro (Cl) or bromo (Br)
- (AA) aa is selected from valylcitrulline, valyllysine, phenylalanyllysine, alanylphenylalanyllysine, and D-alanylphenylalanyllysine
- ss is 1 or 2
- R 2a and R 5b are methyl and H, respectively, or H and methyl, respectively
- R 8d is selected from 2-(morpholin-4-yl)ethoxy, (1 -methylpiperidin-4-yl)methoxy, 2-(N,N-dimethylamino)ethoxy, 2-(N,N-dimethylairimo)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)ace
- R 9d is selected from H and methoxy
- LL is selected from qq ranges from 1 to about 20
- rr and rr' each independently range from 1 to about 4
- Ab is an antibody or a fragment or derivative thereof.
- p* in a compound of formula (Ilia*) is 1.
- p in a compound of formula (IHb) is 1.
- p* in a compound of formula (HIb*) is 1.
- p* is 1 and z* equals q*.
- V 1 in a compound of formula (HIb*) is an enzyme-cleavable substrate.
- V 1 can be cleaved by an intracellular enzyme.
- V 1 is an optionally substituted diallcylaminocarbonyl group wherein the two alkyl groups may be the same or different and optionally be connected to each other to form an optionally substituted carbocycle or heterocycle.
- V 1 is piperazinocarbonyl.
- a compound of formula (IHb*) is represented by
- R la is chloro (Cl) or bromo (Br)
- (AA) aa is selected from valylcitrulline, valyllysine, phenylalanyllysine, alanylphenylalanyllysine, and D-alanylphenylalanyllysine
- ss is 1 or 2
- R 2a and R 5b are methyl and H, respectively, or H and methyl, respectively
- R 8d is selected from 2 ⁇ (morpholin-4-yl) ethoxy, ( 1 -methy lpiperidin-4-yl)methoxy , 2-(N,N-dimethylamino)ethoxy , 2-(NN-dimethylamino)acetylamino, 2-(methylamino)ethoxy, 2-(methylamino)ethoxy, 2-(methyla
- R 9d is selected from H and methoxy
- LL is selected from
- qq ranges from 1 to about 20
- rr and rr' each independently range from 1 to about 4
- Ab is an antibody or a fragment or derivative thereof.
- a compound of formula (III) is represented by V 1 z (Hid)
- a compound of formula (HId) is represented by
- R la is chloro (Cl) or bromo (Br)
- (AA) aa is selected from valylcitrulline, valyllysine, phenylalanyllysine, alanylpheiiylalanyllysine, and D-alanylphenylalanyllysine
- ss is 1 or 2
- R 2a and R 5b are methyl and H, respectively, or H and methyl, respectively
- R 8e is selected from 2-(methylamino)ethoxy, 2-(methylamino)acetylamino, 2-aminoethoxy, 2-aminoacetylamino, and (piperidin-4-yl)methoxy, in which the carbonyl connected to R 8e is attached to the nitrogen atom of R 8e , R 9d
- the invention relates to a compound of formula (IHdI) or (IIId2):
- V 1 is a monosaccharide, disaccharide or oligosaccharide of hexoses or pentoses or heptoses that may also be included among the group of desoxy sugars or amino sugars and belong to the D-series or L-series and in the disaccharides or oligosaccharides are either identical or different, or wherein V 1 has the formula -C(OR a' )R b' CHR c' R d' , wherein R a' , R b ', R 0 ', and R d' have the same meaning as defined hereinabove.
- a compound of this invention is represented by
- R 8c is selected from 2-(morpholin-4-yl)ethoxy, (l-methylpiperidin-4- yl)methoxy, 2-(N,N-dimethylamino)ethoxy, and 2-(N,N-dimethylamino)acetylamino, and R 9c is selected from H and OCH 3 .
- a compound of this invention is represented by or by its (IR,WS) isomer, its (Ii?, 1Oi?) isomer, its (1>S,1O5) isomer, or by a mixture of two or more of said isomers.
- a compound of this invention is represented by
- a compound of this invention is represented by
- a compound of formula (I) or (II) is used to prepare a compound of formula (III).
- a compound of formula (I) or (II) is used to prepare a compound of formula (IV).
- a compound of formula (IV) is used to prepare a compound of formula (III).
- a compound of formula (III) wherein V 1 is a protecting group is used to prepare another compound of formula (III) wherein V 1 is an in vivo removable moiety.
- Figure 5 describes the synthesis of DNA binder 19. Starting from aldehyde 14, alkylation to give 15 and subsequent aldol condensation afforded azide 16. Ring closure gave indole 17, which was saponif ⁇ cated to give 18. Substitution of chloride by dimethylamine finally afforded 19 in 50% overall yield.
- Figure 6 describes the synthesis of DNA binder 27 starting from ethyl isonipecotate (20). Methylation to give 21 was followed by reduction of the ester group to give 22. This was coupled to l-chloro-4-nitrobenzene (23) to give 24. Reduction of the nitro group afforded 25. Fisher indole synthesis afforded 26; saponification finally gave 27 in 40% overall yield.
- the preparation of DNA binder 30 is depicted in Figure 7. Starting from ester 28, reduction of the nitro group and subsequent coupling with N,N-dimethylglycine gave 29. Saponification of the ester group finally gave 30 in 68% yield.
- Figure 8 depicts D ⁇ A binder 41 and its use for the preparation of Boc-protected D ⁇ A binder 37, D ⁇ A binder 38, and fert-butyl-protected D ⁇ A binder 39.
- Figure 2 depicts the partial deprotection of doubly protected D ⁇ A alkylator 1 followed by coupling of the amino group with D ⁇ A binders 2, 13, 19, 27, 30, 37, 38, and 39, respectively. This provided protected compounds 3a-h, which were deprotected to provide agents 4a-h.
- Figure 9 depicts the synthesis of agent 45 from compound 44 and D ⁇ A binder 2 following a similar protocol as for the preparation of agents 4a-h.
- Compound 44 was prepared analogously to the preparation of compound I 10 , o-tolualdehyde instead of benzaldehyde being used as a starting material.
- Figure 10 depicts the synthesis of agent 33.
- Agent 33 Starting from 31, partial deprotection followed by coupling with D ⁇ A binder 2 gave 32. Final deprotection afforded agent 33.
- Figure 3 depicts the synthesis of several galactose conjugates. Starting from partially protected alkylating unit 5, coupling of O-(2,3,4,6-tetra-O-acetyl- ⁇ ;-D-galactopyranosyl)trichloracetimidate (6), subsequent deprotection, and coupling with a D ⁇ A binder moiety provided protected conjugates 7a-e. The synthesis of conjugates 7b and 7d required an additional reduction step using PtO 2 -H 2 CVH 2 .
- Figure 13 describes the synthesis of linker-agent conjugates 48c and 48d from 47c and 47d, respectively, by means of a Cu(I)-catalyzed reaction with a maleimide-containing acetylene.
- Figure 14 describes the synthesis of linker-agent conjugates 50a - 50c starting from 49.
- Compound 49 was prepared from 5 by first introducing the methylpiperazine moiety analogously to the route depicted in Figure 12, followed by coupling to D ⁇ A binder 37 according to the scheme depicted in Figure 2. Deprotection of 49 followed by coupling with p-nitrophenyl carbonate-activated linker constructs 59a, 59b, and 60 provided compounds 50a - 50b and protected 50c.
- Compound 50c was obtained after subsequent palladium-catalyzed deprotection.
- Compounds 47e, 47f, 50b, and 50c can be converted to compounds similar to 48c by Cu(I)-catalyzed cycloaddition with acetylene 40.
- Conjugates (liS,10R)-8a and (lR,10»S)-8a' were tested in an in vitro cytotoxicity assay against the human lung carcinoma cell line A549 ( Figure 16).
- Conjugate (15,10R)-Sa showed an IC50 in the absence of enzyme of 3600, whereas the IC 50 decreased to 0.75 nM in the presence of the enzyme ⁇ -D-galactosidase, the cytotoxicity quotient amounting to 4800.
- Conjugates 8b-e were tested in the same cell line and shown to have a cytotoxicity quotient similar to or substantially greater than the ones reported before (cf. ref. 10), agents 4b-e showing nanomolar or subnanomolar activity.
- Compound (+)-8a was evaluated in an orthotopic breast tumor SCID mouse model using the ADEPT concept.
- mice Female SCID mice were inoculated with 1 x 10 6 MDA-MB-231 cells (estrogen- independent human breast cancer cell line) suspended in 25 ⁇ l sterile PBS in the mammary fad pat of the 4 th mammary complex. Tumors were allowed to grow and ADEPT therapy was started after tumor volumes reached a detectable size. Mice were treated according to the treatment schedule in Figure 17 and received two treatment cycles of a monoclonal anti-human urokinase plasminogen activator receptor (uPAR) antibody conjugated with ⁇ -Galactosidase, uPAR* ⁇ -Gal, in PBS followed by three injections with conjugate (+)-8a in 1% DMSO/NaCl solution.
- uPAR monoclonal anti-human urokinase plasminogen activator receptor
- Control groups received either antibody-enzyme and 1% DMSO/NaCl solution (antibody-enzyme only group), PBS and conjugate (conjugate only group), or PBS and 1% DMSO/NaCl solution (control group).
- primary tumors in ADEPT-treated mice reached an average volume of 0.23 ⁇ 0.07 cm , while the average primary tumor volume of vehicle-treated mice was 0.27 ⁇ 0.10 cm 3 .
- mean tumor volumes of 0.33 ⁇ 0.13 cm 3 and 0.52 ⁇ 0.29 cm 3 were found for the ADEPT-treated group and the control group, respectively ( Figure 18).
- the inhibitory effect of the ADEPT concept on the tumor growth rate seems thus somewhat more prominent after the second cycle of therapy.
- mice that had relatively small tumors at the start of therapy had relatively small tumors at the start of therapy (data not shown).
- tumors treated with ADEPT therapy showed more necrotic tissue within tumors than tumors from vehicle-treated mice.
- Treatment with either uPAR* ⁇ -Gal or conjugate alone showed no inhibitory effect on tumor growth with respect to vehicle control.
- ADEPT therapy was well tolerated in all animals. The general condition of the animals at the day of sacrifice was similar to the mice receiving vehicle only. Blood parameters were not altered in treated mice compared to controls.
- lymphomas treated with ADEPT therapy showed an inhibition of tumor growth.
- this invention relates to use of a compound of formula (I) or (II) for the preparation of a compound of formula (III).
- this invention relates to use of a compound of formula (IV) for the preparation of a compound of formula (III). In yet another aspect, this invention relates to use of a compound of fo ⁇ nula (I) or (II) for the preparation of a compound of formula (IV).
- this invention relates to use of a compound of formula (III) wherein V 1 is a protecting group for the preparation of another compound of formula (III) wherein V 1 is an in vivo removable moiety.
- the invention relates to the use of any of the compounds defined above for the manufacture of a pharmaceutical composition for the treatment of a mammal being in need thereof.
- the invention relates to the use of any of the compounds defined above for the manufacture of a pha ⁇ naceutical composition for the treatment of a tumor in a mammal.
- the invention also relates to any of the compounds defined above as a medicament or an active component or active, substance in a. medicament.
- the invention relates to a process for preparing a pharmaceutical composition containing a compound as defined above, to provide a solid or a liquid formulation for administration orally, topically, or by injection.
- a method or process at least comprises the step of mixing the compound with a pharmaceutically acceptable carrier.
- a compound of the invention is used to treat an illness characterized by undesired proliferation. In another embodiment, a compound of the invention is used to treat an illness characterized by undesired cell proliferation. In another embodiment, a compound of the invention is used to treat a tumor. In yet another embodiment, a compound of the invention is used to treat an inflammatory disease. In yet another embodiment a compound of the invention is used to treat an autoimmune disease. In yet another embodiment a compound of the invention is used to treat a bacterial or microbial infection.
- this invention relates to a method of treating a mammal having an illness characterized by undesired (cell) proliferation with a compound of this invention.
- this invention relates to a method of treating a mammal carrying a tumor with a compound of this invention.
- this invention relates to a method of treating a mammal having an inflammatory disease with a compound of this invention.
- this invention relates to a method of treating a mammal having an autoimmune disease with a compound of this invention.
- this invention relates to a method of treating a mammal having a bacterial or microbial infection with a compound of this invention.
- the invention relates to a method of treating a mammal being in need thereof, whereby the method comprises the administration of a pharmaceutical composition comprising a compound of this invention to the mammal in a therapeutically effective dose.
- the invention relates to a method of treating or preventing a tumor in a mammal, whereby the method comprises the administration of a pharmaceutical composition comprising a compound of this invention to the mammal in a therapeutically effective dose.
- the invention relates to a method of treating or preventing an inflammatory disease in a mammal, whereby the method comprises the administration of a pharmaceutical composition comprising a compound of this invention to the mammal in a therapeutically effective dose.
- the invention in another embodiment, relates to a method of treating or preventing an autoimmune disease in a mammal, whereby the method comprises the administration of a pharmaceutical composition comprising a compound of this invention to the mammal in a therapeutically effective dose. . .
- the invention in another embodiment, relates to a method of treating or preventing a bacterial or microbial infection in a mammal, whereby the method comprises the administration of a pharmaceutical composition comprising a compound of this invention to the mammal in a therapeutically effective dose.
- the invention also relates to pharmaceutical compositions comprising the compounds of the invention as defined above.
- a compound of the invention may be administered in purified form together with a pharmaceutical carrier as a pharmaceutical composition.
- the preferred form depends on the intended mode of administration and therapeutic application.
- the pharmaceutical carrier can be any compatible, nontoxic substance suitable to deliver the compounds of the invention to the patient.
- Pharmaceutically acceptable carriers are well known in the art and include, for example, aqueous solutions such as (sterile) water or physiologically buffered saline or other solvents or vehicles such as glycols, glycerol, oils such as olive oil or injectable organic esters, alcohol, fats, waxes, and inert solids.
- a pharmaceutically acceptable carrier may further contain physiologically acceptable compounds that act for example to stabilize or to increase the absorption of the compounds of the invention.
- physiologically acceptable compounds include, for example, carbohydrates, such as glucose, sucrose or dextrans, antioxidants, such as ascorbic acid or glutathione, chelating agents, low molecular weight proteins, or other stabilizers or excipients.
- carbohydrates such as glucose, sucrose or dextrans
- antioxidants such as ascorbic acid or glutathione
- chelating agents such ascorbic acid or glutathione
- low molecular weight proteins or other stabilizers or excipients.
- Pharmaceutically acceptable adjuvants, buffering agents, dispersing agents, and the like may also be incorporated into the pharmaceutical compositions.
- the active ingredient can be administered in solid dosage forms, such as ' capsules, tablets, and powders, or in liquid dosage forms, such as elixirs, syrups, and suspensions.
- Active component(s) can be encapsulated in gelatin capsules together with inactive ingredients and powdered carriers, such as glucose, lactose, sucrose, mannitol, starch, cellulose or cellulose derivatives, magnesium stearate, stearic acid, sodium saccharin, talcum, magnesium carbonate, and the like.
- inactive ingredients examples include red iron oxide, silica gel, sodium lauryl sulfate, titanium dioxide, edible white ink, and the like.
- Similar diluents can be used to make compressed tablets. Both tablets and capsules can be manufactured as sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar-coated or film-coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric-coated for selective disintegration in the gastrointestinal tract.
- Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance.
- the compounds of the invention are however preferably administered parenterally. Preparations of the compounds of the invention for parenteral administration must be sterile. Sterilization is readily accomplished by filtration through sterile filtration membranes, optionally prior to or following lyophilization and reconstitution.
- the parenteral route for administration of compounds of the invention is in accord with known methods, e.g. injection or infusion by intravenous, intraperitoneal, intramuscular, intraarterial, or intralesional routes.
- the compounds of the invention may be administered continuously by infusion or by bolus injection.
- a typical composition for intravenous infusion could be made up to contain 100 to 500 ml of sterile 0.9% NaCl or 5% glucose optionally supplemented with a 20% albumin solution and 1 mg to 10 g of the compound of the invention, depending on the particular type of compound of the invention and its required dosing regime.
- Methods for preparing parenterally administrable compositions are well known in the art and described in more detail in various sources, including, for example, Remington's Pharmaceutical Science 19 .
- Example 1 rac- ⁇ (140)-fl «ri-5-Benzyloxy-3-[(5-(2-( ⁇ yV-dimethylamino)ethoxy)-indol-2-yI)carbonyl]-l-(10- chloroethyl)-l,2-dihydro-3H-benz[e]indole ⁇ (rac-3a): Racemic 1 (100 mg, 228 ⁇ mol) was suspended in 4 M HCl / EtOAc and stirred at room temperature for 3 h. The reaction mixture was concentrated and dried in vacuo for 1 h. The residue was dissolved in DMF (10 mL).
- Example 2 r «c- ⁇ (l,10)- ⁇ nri-l-(10-CMoroethyl)-5-hydroxy-3-[(5-(2-(iV,iV-dimethyIamino)ethoxy)indol-2- yl)carbonyI]-l,2-dihydro-3H-benz[e]indole-hydrochloride ⁇ (r ⁇ c-4a): Racemic 3a (200 mg, 352 ⁇ mol) was dissolved 4 M HCl / ethyl acetate (15 mL) and stirred for 2 h at room temperature. The solution was concentrated. The residue was dried in vacuo for 1 h and then suspended in THF (15 mL).
- Example 11 [(+)-(l,10)- ⁇ n ⁇ -l-(10-Chloroethyl)-3-[(5-(2-(morphoKn-4-yI)ethoxy)indoI-2-yl)carbonyl]-l,2- dihydro-3H-benz[e]indol-5-yl]-2,3,4,6-tetra-0-acetyl- ⁇ -D-gaIactopyranoside ((+)-7b): A solution of (+)-5 (142 mg, 408 ⁇ mol) in dry CH 2 Cl 2 (18 mL) and molecular sieves 4 A (0.8 g) was stirred for 30 min at room temperature.
- Example 12 [(+)-(l,10)-fl «ft-l-(10-Chloroethyl)-3-[(5-(2-(morphoIin-4-yI)ethoxy)-indol-2-yl)carbonyl]-l,2- dihydro-3H-benz[e]indol-5-yI]-/?-D-galactopyranoside ((+)-8b): To a solution of 7a (51 mg, 60 ⁇ mol) in dry MeOH (6 mL) was added NaOMe in methanol (22 ⁇ L of a ca. 5.4 M solution, 120 ⁇ mol, 2.0 equiv.).
- Example 13 3-(2-Chloroethoxy)-4-methoxybenzaIdehyde (15): A mixture of 3-hydroxy-4-methoxy- benzaldehyde (14) (10.0 g, 65.7 mmol), K 2 CO 3 (45.4 g, 329 mmol), 1,2-dichloroethane (104 mL, 1.31 mol), and DMF (300 mL) was stirred at 65-70 0 C for 16 h. After cooling, 1,2-dichloroethane was removed under reduced pressure, the remaining slurry was poured onto ice and the mixture was extracted with Et 2 O (3 x 250 mL) and EtOAc (4 x 250 mL).
- Example 14 2-Azido-3-[3-(2-chloroethoxy)-4-methoxyphenyl] acrylic acid methyl ester (16): NaN 3 (22.2 g, 341 mmol) was added slowly to a solution of methyl chloroacetate (20.0 mL, 227 mmol) in DMSO (100 mL). After stirring at room temperature for 24 h, water (150 mL) was added and the mixture was extracted with Et 2 O (3 x 100 mL). The combined organic fractions were dried (MgSO ⁇ and concentrated in vacuo to 50 mL.
- Example 16 5-(2-Chloroethoxy)-6-methoxy-U ⁇ -indole-2-carboxyIic acid (18): A suspension of ester 17 (2.00 g, 7.05 mmol), Cs 2 CO 3 (3.45 g, 10.6 mmol), 95% EtOH (40 mL), and water (20 mL) was heated at reflux for 8 h. After cooling to room temperature the solvent was removed in vacuo, the residue was treated with water (50 mL), and the resulting solution was acidified with 2 M HCl. The formed precipitate was isolated by filtration, washed with water (100 mL), and dried in vacuo to give 18 (1.80 g, 95%) as a beige solid.
- (+)- ⁇ (140)-a «//-l-(10-Chloroethyl)-5-hydroxy-3-[(5-(2-(i ⁇ VV-dimethylamino)ethoxy)-6- methoxy-indol-2-yl)carbonyl]-l,2-dihydro-3H-benz[e]indole-hydrochloride ⁇ ((+)-4c):
- Example 22 l-MethyIpiperidine-4-carboxylic acid ethyl ester (21): Ethyl isonipecotate (20) (5.00 g, 31.8 nnno 1) was dissolved in an ice-cold mixture of glacial acetic acid (3.80 g, 63.6 mmol) and water (11 mL). Then, a 37% aqueous formaldehyde solution (2.85 niL, 38.2 mmol) was added and the reaction mixture was hydrogenated over Pd/C (10%, 338 mg) at 58 psi H 2 for 3.5 h at room temperature.
- Example 24 l-Methyl-4-(4-nitrophenoxymethyl)piperidine (24): A mixture of l-chloro-4-nitrobenzene (23) (2.37 g, 15.0 mmol), alcohol 22 (1.94 g, 15.0 mmol), and DMSO (25 mL) was treated portionwise with NaH (60% in mineral oil, 660 mg, 16.5 mmol) at 40 0 C. The mixture was stirred at 70 0 C for 3 h, poured into water (15O mL), and extracted with Et 2 O (5 x 10O mL). The combined organic fractions were washed with water (250 mL) and brine (250 mL), dried (MgSQ 4 ), and the solvent was removed in vacuo.
- Example 30 [(+)-(l,10)- ⁇ «ft-l-(10-Chloroethyl)-3-[(5-((l-methylpiperidin-4-yl)methoxy)indol-2- y ⁇ carbonyy-l j l-dihydro-SH-benz ⁇ indol-S-ylJ-Z ⁇ -tetra-O-acetyl- ⁇ -D-galactopyranoside ((+)-7d): A solution of (+)-5 (134 mg, 385 ⁇ mol) in dry CH 2 Cl 2 (17 mL) and molecular sieves 4 A (0.8 g) was stirred for 30 min at room temperature.
- Example 31 [(+)-(l,10)- ⁇ nrf-l-(10-ChIoroethyl)-3-[(5-((l-methylpiperidin-4-yl)methoxy)-indol-2- yl)carbonyl]-l,2-dihydro-3H-benz[e]indol-5-yl]- j ⁇ -D-galactopyranoside ((+)-8d): To a solution of 7d (74 mg, 87 ⁇ mol) in dry MeOH (6 mL) was added NaOMe in methanol (32 ⁇ L of a ca. 5.4 M solution, 173 ⁇ mol, 2.0 equiv.).
- Example 34 (4 ⁇ )- ⁇ (l,10)- ⁇ n ⁇ 5-Benzyloxy-3-[(5-(2-(iV r /V-Dimethylamino)acetylammo)indol-2-yl)carbonyl]- l-(10-chloroethyl)-l,2 ⁇ dihydro-3H-benz[e]indole ⁇ ((+)-3e): Compound (+)-l (150 mg, 342 ⁇ mol) was suspended in 4 M HCl / EtOAc (14 mL) and stirred at room temperature for 3.5 h. The reaction mixture was concentrated and dried in vacuo for 1 h. The residue was dissolved in DMF (10 mL).
- Example 36 [(+)-(15 r ,10i?)-l-(10-Chloroethyl)-3-[(5-(2-(7V, ⁇ r -Dimethylamino)acetyLamino)indol-2- yI)carbonyl]-l,2-dihydro-3H-benz[ ⁇ ]indoI-5-yI]-2,3,4,6-tetra-(?-acetyl-y&-D-gaIactopyranoside ((+)-7e): A solution of (+)-5 (137 mg, 394 ⁇ mol) in dry CH 2 Cl 2 (17 mL) and molecular sieves 4 A (0.8 g) was stirred for 30 min at room temperature.
- Example 39 l-(10-Chloromethyl)-5-hydroxy-3-[(5-(2-(2-(iV ⁇ V-Dimethylammo)acetylamino)indol-2- yl)carbonyI]-l,2-dihydro-3H-benz[e]indole-hydrochloride ⁇ (r ⁇ c-33): Compound r ⁇ c-32 (100 mg, 180 ⁇ mol) was dissolved in 4 M HCl / ethyl acetate (10 mL) and stirred for 2 h at room temperature. The solution was concentrated. The residue was dried in vacuo for 1 h and then suspended in THF (15 mL).
- Compound 46b was prepared from (+)-4a according to the procedure used for 46a, except that N-Boc-piperazine was used instead of N-Boc-N,N'-dimethylethylenediamine. Compound 46b was obtained quantitatively as an off-white solid.
- Example 48 Generalized procedure for the preparation of 48c-48d from 47c-47d: A solution of 47c or 47d (25 ⁇ mol), sodium ascorbate (3.0 mg, 15 ⁇ mol), 40 (10.1 mg, 38 ⁇ r ⁇ o.l), and CuS ⁇ 4 (1.9 mg, 7.5 ⁇ mol) in THF/water was stirred at room temperature. The reaction was followed by HPLC. Upon completion of the reaction, the reaction mixture was diluted with THF/water and the product was purified by preparative HPLC (acetonitrile / aqueous ammonium formate) to afford the product as an off-white solid after freeze-drying.
- preparative HPLC acetonitrile / aqueous ammonium formate
- Herceptin antibody 40 mg was treated with 1.5 molar equivalents of dithiothreitol (DTT) in 0.025 M sodium borate pH 8, 0.025 M NaCl, ImM DTPA for 2 h at 37 0 C. Excess DTT was removed using a Sephadex G-25 column (0.025 M sodium borate pH 8, 0.025 M NaCl, ImM DTPA). Thiol determination with Ellman's reagent indicated that there were approximately three thiol groups per Herceptin.
- DTT dithiothreitol
- AU substances were used as freshly prepared solutions in DMSO (Merck, Darmstadt, Germany) diluted with incubation medium to a final concentration of 1 % DMSO in the wells. After 24 h of exposure the test substance was removed and the cells were washed with fresh medium. Cultivation was done at 37 0 C and 7.5% CO 2 in air for 12 days. The medium was removed and the clones were dried and stained with L ⁇ ffler's methylene blue (Merck, Darmstadt, Germany). They were then counted macroscopically.
- mice were randomized in groups of 5-12 mice (control, antibody-enzyme only, prodrug only, and ADEPT therapy).
- mice were injected intravenously with PBS (control, prodrug only groups) or 50 ⁇ g of a monoclonal anti- human urokinase plasminogen activator receptor (uPAR) antibody conjugated with ⁇ -Galactosidase, uPAR* ⁇ -Gal, in PBS (antibody-enzyme only, ADEPT therapy groups).
- PBS control, prodrug only groups
- uPAR monoclonal anti- human urokinase plasminogen activator receptor
- mice On days 24, 26, 28, 32, 34, and 36, mice were injected intravenously with 1% DMSO/NaCl solution (control, antibody-enzyme only groups) or 104 ⁇ g of (+)-8a in 1% DMSO/NaCl solution (prodrug only, ADEPT therapy groups). Mice were sacrificed on day 38 and tumors were excised, weighed, placed in phosphate-buffered 4 % formalin for 16 hours at room temperature, and embedded in paraffin. Tissue sections (2.5 ⁇ m) were obtained, stained with hematoxylin and eosin (H & E), and inspected by routine microscopic examinations.
- H & E hematoxylin and eosin
- Tumor size was determined at distinct time points during the therapy experiment (days 21, 29, and 38) with a flat panel volume computer tomograph prototype (fpVCT GE Global Research, Niskayuna NY, US). Mice were anesthetized with vaporized isoflurane at 0.8 - 1 % concentration throughout the imaging session, centered on the fpVCT gantry axis of rotation, and placed perpendicular to the z-axis of the system, so that it was possible to scan the whole mouse with one rotation. Contrast medium, 150 ⁇ l Isovist ® 300 (Schering, Berlin, Germany), was applied intravenously 20 seconds before scanning.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Plural Heterocyclic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
















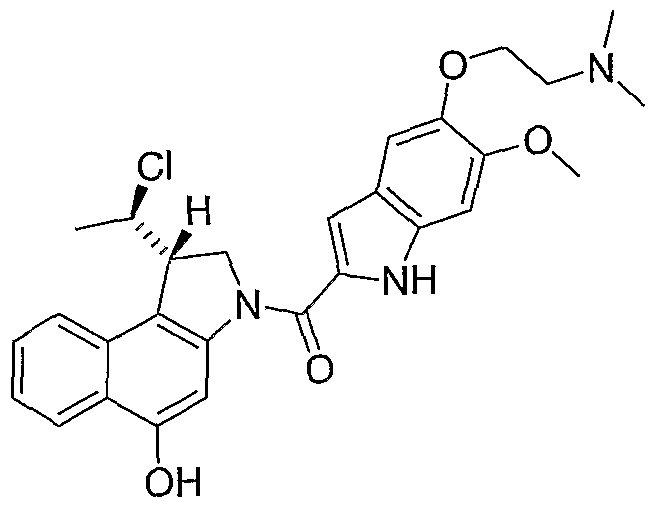

































































Claims




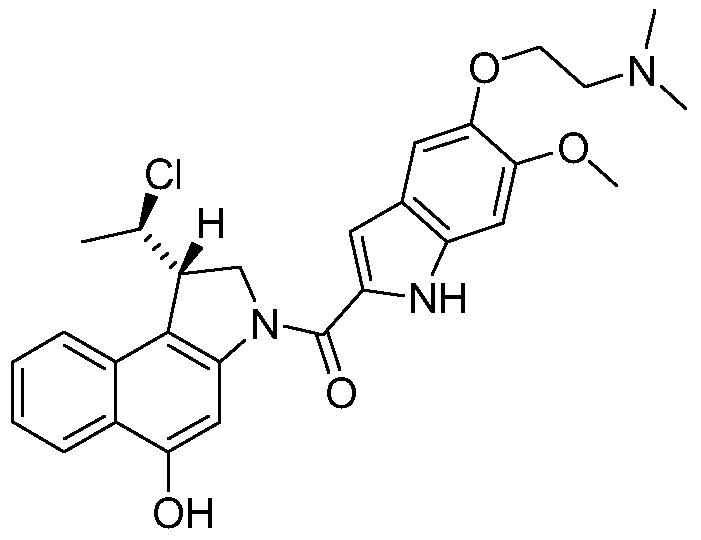







































Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0707426-3A BRPI0707426A2 (en) | 2006-02-02 | 2007-02-02 | compound, conjugate, use of a compound, pharmaceutical composition, process for preparing a pharmaceutical composition, and methods of treating a mammal in need thereof, and treating or preventing a tumor in a mammal |
| AU2007210377A AU2007210377B2 (en) | 2006-02-02 | 2007-02-02 | Water-soluble CC-1065 analogs and their conjugates |
| EP07715863.2A EP1994000B1 (en) | 2006-02-02 | 2007-02-02 | Water-soluble cc-1065 analogs and their conjugates |
| US12/223,682 US8940784B2 (en) | 2006-02-02 | 2007-02-02 | Water-soluble CC-1065 analogs and their conjugates |
| RU2008135440/04A RU2489423C2 (en) | 2006-02-02 | 2007-02-02 | Water-soluble analogues cc-1065 and their conjugates |
| CA002641899A CA2641899A1 (en) | 2006-02-02 | 2007-02-02 | Water-soluble cc-1065 analogs and their conjugates |
| JP2008553192A JP5346589B2 (en) | 2006-02-02 | 2007-02-02 | Water-soluble CC-1065 analog and its conjugate |
| MX2008009956A MX2008009956A (en) | 2006-02-02 | 2007-02-02 | Water-soluble cc-1065 analogs and their conjugates. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NL2006050020 | 2006-02-02 | ||
| NLPCT/NL2006/050020 | 2006-02-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007089149A2 true WO2007089149A2 (en) | 2007-08-09 |
| WO2007089149A3 WO2007089149A3 (en) | 2007-09-27 |
Family
ID=38227758
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/NL2007/050043 WO2007089149A2 (en) | 2006-02-02 | 2007-02-02 | Water-soluble cc-1065 analogs and their conjugates |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US8940784B2 (en) |
| EP (1) | EP1994000B1 (en) |
| JP (2) | JP5346589B2 (en) |
| KR (1) | KR20080114709A (en) |
| CN (1) | CN101415679A (en) |
| AU (1) | AU2007210377B2 (en) |
| BR (1) | BRPI0707426A2 (en) |
| CA (1) | CA2641899A1 (en) |
| MX (1) | MX2008009956A (en) |
| RU (1) | RU2489423C2 (en) |
| WO (1) | WO2007089149A2 (en) |
Cited By (109)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008103693A2 (en) * | 2007-02-21 | 2008-08-28 | Medarex, Inc. | Chemical linkers with single amino acids and conjugates thereof |
| WO2009017394A1 (en) * | 2007-08-01 | 2009-02-05 | Syntarga B.V. | Substituted cc-1065 analogs and their conjugates |
| WO2010062171A2 (en) * | 2008-11-03 | 2010-06-03 | Syntarga B.V. | Novel cc-1065 analogs and their conjugates |
| WO2011051484A1 (en) | 2009-10-30 | 2011-05-05 | Georg-August-Universität Göttingen Stiftung Öffenlichen Rechts | Fluorescence dyes |
| DE102009051799A1 (en) | 2009-11-03 | 2011-05-05 | Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts | Bifunctional prodrugs and drugs |
| US8034959B2 (en) | 2001-05-31 | 2011-10-11 | Medarex, Inc. | Methods of treating cancer with an antibody-drug conjugate |
| WO2011133039A2 (en) | 2010-04-21 | 2011-10-27 | Syntarga B.V. | Novel conjugates of cc-1065 analogs and bifunctional linkers |
| WO2011147982A2 (en) | 2010-05-27 | 2011-12-01 | Genmab A/S | Monoclonal antibodies against her2 epitope |
| WO2011147986A1 (en) | 2010-05-27 | 2011-12-01 | Genmab A/S | Monoclonal antibodies against her2 |
| WO2011157741A2 (en) | 2010-06-15 | 2011-12-22 | Genmab A/S | Human antibody drug conjugates against tissue factor |
| WO2012016227A2 (en) | 2010-07-29 | 2012-02-02 | Xencor, Inc. | Antibodies with modified isoelectric points |
| US8124738B2 (en) | 2005-09-26 | 2012-02-28 | Medarex, Inc. | Human monoclonal antibodies to CD70 |
| WO2012045085A1 (en) | 2010-10-01 | 2012-04-05 | Oxford Biotherapeutics Ltd. | Anti-rori antibodies |
| WO2012092616A1 (en) | 2010-12-30 | 2012-07-05 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| WO2012103165A2 (en) | 2011-01-26 | 2012-08-02 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies and uses thereof |
| US8268970B2 (en) | 2007-10-01 | 2012-09-18 | Bristol-Myers Squibb Company | Human antibodies that bind mesothelin, and uses thereof |
| WO2012143523A1 (en) | 2011-04-20 | 2012-10-26 | Genmab A/S | Bispecifc antibodies against her2 |
| WO2012162482A1 (en) * | 2011-05-26 | 2012-11-29 | Bristol-Myers Squibb Company | Immunoconjugates, compositions containing them, and methods of making and use |
| WO2013004842A2 (en) | 2011-07-06 | 2013-01-10 | Genmab A/S | Antibody variants and uses thereof |
| WO2013022855A1 (en) | 2011-08-05 | 2013-02-14 | Xencor, Inc. | Antibodies with modified isoelectric points and immunofiltering |
| US8399403B2 (en) | 2004-05-19 | 2013-03-19 | Medarex, Inc. | Chemical linkers and conjugates thereof |
| WO2013055809A1 (en) | 2011-10-10 | 2013-04-18 | Xencor, Inc. | A method for purifying antibodies |
| US8461117B2 (en) | 2006-12-28 | 2013-06-11 | Medarex, Inc. | Chemical linkers and cleavable substrates and conjugates thereof |
| WO2014006217A1 (en) | 2012-07-06 | 2014-01-09 | Genmab B.V. | Dimeric protein with triple mutations |
| WO2014018625A1 (en) | 2012-07-25 | 2014-01-30 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies and uses thereof |
| WO2014108198A1 (en) | 2013-01-10 | 2014-07-17 | Genmab B.V. | Human igg1 fc region variants and uses thereof |
| WO2014113510A1 (en) | 2013-01-15 | 2014-07-24 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| WO2014145806A2 (en) | 2013-03-15 | 2014-09-18 | Xencor, Inc. | Heterodimeric proteins |
| WO2015052537A1 (en) | 2013-10-11 | 2015-04-16 | Oxford Biotherapeutics Ltd | Conjugated antibodies against ly75 for the treatment of cancer |
| US9072798B2 (en) | 2009-02-18 | 2015-07-07 | Ludwig Institute For Cancer Research Ltd. | Specific binding proteins and uses thereof |
| US9090693B2 (en) | 2007-01-25 | 2015-07-28 | Dana-Farber Cancer Institute | Use of anti-EGFR antibodies in treatment of EGFR mutant mediated disease |
| WO2015149077A1 (en) | 2014-03-28 | 2015-10-01 | Xencor, Inc. | Bispecific antibodies that bind to cd38 and cd3 |
| WO2016001485A1 (en) | 2014-06-30 | 2016-01-07 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| WO2016005593A1 (en) | 2014-07-11 | 2016-01-14 | Genmab A/S | Antibodies binding axl |
| WO2016014984A1 (en) | 2014-07-24 | 2016-01-28 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| US9283276B2 (en) | 2007-08-14 | 2016-03-15 | Ludwig Institute For Cancer Research Ltd. | Monoclonal antibody 175 targeting the EGF receptor and derivatives and uses thereof |
| WO2016070089A2 (en) | 2014-10-31 | 2016-05-06 | Abbvie Biotherapeutics Inc. | Anti-cs1 antibodies and antibody drug conjugates |
| US9421278B2 (en) | 2014-01-10 | 2016-08-23 | Synthon Biopharmaceuticals B.V. | Duocarmycin ADCS showing improved in vivo antitumor activity |
| US9427480B2 (en) | 2014-01-10 | 2016-08-30 | Synthon Biopharmaceuticals B.V. | Duocarmycin ADCs for use in treatment of endometrial cancer |
| WO2016141387A1 (en) | 2015-03-05 | 2016-09-09 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US9493578B2 (en) | 2009-09-02 | 2016-11-15 | Xencor, Inc. | Compositions and methods for simultaneous bivalent and monovalent co-engagement of antigens |
| US9562102B2 (en) | 2001-05-11 | 2017-02-07 | Ludwig Institute For Cancer Research | Specific binding proteins and uses thereof |
| US9605084B2 (en) | 2013-03-15 | 2017-03-28 | Xencor, Inc. | Heterodimeric proteins |
| DE102016105449A1 (en) | 2015-10-29 | 2017-05-04 | Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts | Bifunctional prodrugs |
| US9650446B2 (en) | 2013-01-14 | 2017-05-16 | Xencor, Inc. | Heterodimeric proteins |
| WO2017095805A1 (en) | 2015-11-30 | 2017-06-08 | Abbvie Inc. | ANTI-huLRRC15 ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
| WO2017095808A1 (en) | 2015-11-30 | 2017-06-08 | Abbvie Inc. | ANTI-huLRRC15 ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
| US9701759B2 (en) | 2013-01-14 | 2017-07-11 | Xencor, Inc. | Heterodimeric proteins |
| WO2017121877A1 (en) | 2016-01-13 | 2017-07-20 | Genmab A/S | Axl-specific antibody-drug conjugates for cancer treatment |
| WO2017201204A1 (en) | 2016-05-17 | 2017-11-23 | Abbvie Biotherapeutics Inc. | ANTI-cMet ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
| US9850320B2 (en) | 2014-11-26 | 2017-12-26 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD20 |
| US9856327B2 (en) | 2014-11-26 | 2018-01-02 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD123 |
| US9901567B2 (en) | 2007-08-01 | 2018-02-27 | Syntarga B.V. | Substituted CC-1065 analogs and their conjugates |
| WO2018083126A1 (en) | 2016-11-01 | 2018-05-11 | Genmab B.V. | Polypeptide variants and uses thereof |
| US10106624B2 (en) | 2013-03-15 | 2018-10-23 | Xencor, Inc. | Heterodimeric proteins |
| US10131710B2 (en) | 2013-01-14 | 2018-11-20 | Xencor, Inc. | Optimized antibody variable regions |
| EP3421495A2 (en) | 2013-03-15 | 2019-01-02 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US10227410B2 (en) | 2015-12-07 | 2019-03-12 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| US10233212B2 (en) | 2015-11-03 | 2019-03-19 | Industrial Technology Research Institute | Compounds, linker-drugs and ligand-drug conjugates |
| US10266606B2 (en) | 2014-01-10 | 2019-04-23 | Synthon Biopharmaceuticals B.V. | Method for purifying Cys-linked antibody-drug conjugates |
| US10316088B2 (en) | 2016-06-28 | 2019-06-11 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| WO2019145455A1 (en) | 2018-01-24 | 2019-08-01 | Genmab B.V. | Polypeptide variants and uses thereof |
| US10428155B2 (en) | 2014-12-22 | 2019-10-01 | Xencor, Inc. | Trispecific antibodies |
| US10487155B2 (en) | 2013-01-14 | 2019-11-26 | Xencor, Inc. | Heterodimeric proteins |
| WO2019229701A2 (en) | 2018-06-01 | 2019-12-05 | Novartis Ag | Binding molecules against bcma and uses thereof |
| US10501543B2 (en) | 2016-10-14 | 2019-12-10 | Xencor, Inc. | IL15/IL15Rα heterodimeric Fc-fusion proteins |
| WO2019238843A1 (en) | 2018-06-14 | 2019-12-19 | Berlin-Chemie Ag | Pharmaceutical combinations |
| US10519242B2 (en) | 2013-03-15 | 2019-12-31 | Xencor, Inc. | Targeting regulatory T cells with heterodimeric proteins |
| US10526417B2 (en) | 2014-11-26 | 2020-01-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| WO2020010079A2 (en) | 2018-07-02 | 2020-01-09 | Amgen Inc. | Anti-steap1 antigen-binding protein |
| EP3632462A1 (en) | 2012-07-06 | 2020-04-08 | Genmab B.V. | Dimeric protein with triple mutations |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| US10787518B2 (en) | 2016-06-14 | 2020-09-29 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US10793632B2 (en) | 2016-08-30 | 2020-10-06 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| WO2020236792A1 (en) | 2019-05-21 | 2020-11-26 | Novartis Ag | Cd19 binding molecules and uses thereof |
| WO2020236797A1 (en) | 2019-05-21 | 2020-11-26 | Novartis Ag | Variant cd58 domains and uses thereof |
| US10851178B2 (en) | 2011-10-10 | 2020-12-01 | Xencor, Inc. | Heterodimeric human IgG1 polypeptides with isoelectric point modifications |
| US10858417B2 (en) | 2013-03-15 | 2020-12-08 | Xencor, Inc. | Heterodimeric proteins |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| US10918735B2 (en) | 2012-12-04 | 2021-02-16 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-diones for cancer treatment |
| US10968276B2 (en) | 2013-03-12 | 2021-04-06 | Xencor, Inc. | Optimized anti-CD3 variable regions |
| US10982006B2 (en) | 2018-04-04 | 2021-04-20 | Xencor, Inc. | Heterodimeric antibodies that bind fibroblast activation protein |
| US10981992B2 (en) | 2017-11-08 | 2021-04-20 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| US11053316B2 (en) | 2013-01-14 | 2021-07-06 | Xencor, Inc. | Optimized antibody variable regions |
| WO2021142086A1 (en) | 2020-01-08 | 2021-07-15 | Synthis Therapeutics, Inc. | Alk5 inhibitor conjugates and uses thereof |
| US11084863B2 (en) | 2017-06-30 | 2021-08-10 | Xencor, Inc. | Targeted heterodimeric Fc fusion proteins containing IL-15 IL-15alpha and antigen binding domains |
| WO2021178896A1 (en) | 2020-03-06 | 2021-09-10 | Go Therapeutics, Inc. | Anti-glyco-cd44 antibodies and their uses |
| US11312770B2 (en) | 2017-11-08 | 2022-04-26 | Xencor, Inc. | Bispecific and monospecific antibodies using novel anti-PD-1 sequences |
| US11319355B2 (en) | 2017-12-19 | 2022-05-03 | Xencor, Inc. | Engineered IL-2 Fc fusion proteins |
| US11358999B2 (en) | 2018-10-03 | 2022-06-14 | Xencor, Inc. | IL-12 heterodimeric Fc-fusion proteins |
| US11365258B2 (en) | 2017-03-10 | 2022-06-21 | Berlin-Chemie Ag | Pharmaceutical combinations comprising an anti-LY75 antibody |
| WO2022187591A1 (en) | 2021-03-05 | 2022-09-09 | Go Therapeutics, Inc. | Anti-glyco-cd44 antibodies and their uses |
| US11472890B2 (en) | 2019-03-01 | 2022-10-18 | Xencor, Inc. | Heterodimeric antibodies that bind ENPP3 and CD3 |
| US11505595B2 (en) | 2018-04-18 | 2022-11-22 | Xencor, Inc. | TIM-3 targeted heterodimeric fusion proteins containing IL-15/IL-15RA Fc-fusion proteins and TIM-3 antigen binding domains |
| WO2022248835A1 (en) | 2021-05-26 | 2022-12-01 | Oxford Biotherapeutics Ltd | Pharmaceutical combination comprising an anti-cd205 antibody and an immune checkpoint inhibitor |
| US20220380360A1 (en) * | 2020-01-30 | 2022-12-01 | Aimed Bio Inc. | Benzoselenophene-based compound, pharmaceutical composition containing same, and antibody-drug conjugate |
| US11524991B2 (en) | 2018-04-18 | 2022-12-13 | Xencor, Inc. | PD-1 targeted heterodimeric fusion proteins containing IL-15/IL-15Ra Fc-fusion proteins and PD-1 antigen binding domains and uses thereof |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| WO2023014863A1 (en) | 2021-08-05 | 2023-02-09 | Go Therapeutics, Inc. | Anti-glyco-muc4 antibodies and their uses |
| US11591401B2 (en) | 2020-08-19 | 2023-02-28 | Xencor, Inc. | Anti-CD28 compositions |
| WO2023034569A1 (en) | 2021-09-03 | 2023-03-09 | Go Therapeutics, Inc. | Anti-glyco-cmet antibodies and their uses |
| WO2023034571A1 (en) | 2021-09-03 | 2023-03-09 | Go Therapeutics, Inc. | Anti-glyco-lamp1 antibodies and their uses |
| WO2023089314A1 (en) | 2021-11-18 | 2023-05-25 | Oxford Biotherapeutics Limited | Pharmaceutical combinations |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| US11859012B2 (en) | 2021-03-10 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and GPC3 |
| US11919956B2 (en) | 2020-05-14 | 2024-03-05 | Xencor, Inc. | Heterodimeric antibodies that bind prostate specific membrane antigen (PSMA) and CD3 |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
| US12030888B2 (en) | 2021-02-24 | 2024-07-09 | Massachusetts Institute Of Technology | Himastatin derivatives, and processes of preparation thereof, and uses thereof |
| WO2024153716A1 (en) * | 2023-01-17 | 2024-07-25 | University Of Bradford | Cancer treating compounds |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110313230A1 (en) | 2001-05-11 | 2011-12-22 | Terrance Grant Johns | Specific binding proteins and uses thereof |
| US7838542B2 (en) * | 2006-06-29 | 2010-11-23 | Kinex Pharmaceuticals, Llc | Bicyclic compositions and methods for modulating a kinase cascade |
| ES2542152T3 (en) | 2007-03-15 | 2015-07-31 | Ludwig Institute For Cancer Research Ltd. | Treatment method using EGFR antibodies and Src inhibitors and related formulations |
| US20110076232A1 (en) | 2009-09-29 | 2011-03-31 | Ludwig Institute For Cancer Research | Specific binding proteins and uses thereof |
| ES2661574T3 (en) * | 2010-12-23 | 2018-04-02 | Alkermes Pharma Ireland Limited | Multiple PAF loading prodrugs |
| CN104220442B (en) | 2012-04-05 | 2016-11-23 | 内尔维阿诺医学科学有限公司 | Alkylating agent |
| EP2836493B1 (en) * | 2012-04-05 | 2018-06-20 | Nerviano Medical Sciences S.r.l. | Functionalized thieno-indole derivatives for the treatment of cancer |
| CN104736179B (en) * | 2012-05-07 | 2017-09-26 | 日东电工株式会社 | Polymer conjugate with joint |
| EP3049420B1 (en) * | 2013-09-25 | 2019-05-15 | Nerviano Medical Sciences S.r.l. | Thieno[2,3-e]indole derivatives as new antitumor agents |
| CA2934030A1 (en) | 2013-10-15 | 2015-04-23 | Sorrento Therapeutics Inc. | Drug-conjugates with a targeting molecule and two different drugs |
| WO2016090050A1 (en) * | 2014-12-03 | 2016-06-09 | Genentech, Inc. | Quaternary amine compounds and antibody-drug conjugates thereof |
| CN113209306A (en) | 2014-12-09 | 2021-08-06 | 艾伯维公司 | Antibody drug conjugates with cell permeable BCL-XL inhibitors |
| SG11201704710PA (en) | 2014-12-09 | 2017-07-28 | Abbvie Inc | Bcl xl inhibitory compounds having low cell permeability and antibody drug conjugates including the same |
| EP3230283A1 (en) | 2014-12-09 | 2017-10-18 | AbbVie Inc. | Bcl-xl inhibitory compounds and antibody drug conjugates including the same |
| WO2017214339A1 (en) | 2016-06-08 | 2017-12-14 | Abbvie Inc. | Anti-b7-h3 antibodies and antibody drug conjugates |
| EP3835322A3 (en) | 2016-06-08 | 2021-10-06 | AbbVie Inc. | Anti-b7-h3 antibodies and antibody drug conjugates |
| CN109562169A (en) | 2016-06-08 | 2019-04-02 | 艾伯维公司 | Anti-CD 98 antibody and antibody drug conjugates |
| AU2017277916A1 (en) | 2016-06-08 | 2019-01-03 | Abbvie Inc. | Anti-CD98 antibodies and antibody drug conjugates |
| US20200002421A1 (en) | 2016-06-08 | 2020-01-02 | Abbvie Inc. | Anti-b7-h3 antibodies and antibody drug conjugates |
| BR112018075645A2 (en) | 2016-06-08 | 2019-04-09 | Abbvie Inc. | anti-egfr drug antibody conjugates |
| MX2018015284A (en) | 2016-06-08 | 2019-09-18 | Abbvie Inc | Anti-egfr antibody drug conjugates. |
| CN116059202A (en) | 2016-06-08 | 2023-05-05 | 艾伯维公司 | anti-EGFR antibody drug conjugates |
| AU2017277920A1 (en) | 2016-06-08 | 2019-01-03 | Abbvie Inc. | Anti-CD98 antibodies and antibody drug conjugates |
| EA037848B1 (en) | 2016-07-14 | 2021-05-27 | Общество С Ограниченной Ответственностью "Биохимический Агент" | Fusion protein, polynucleotide, genetic construct, producer, preparation for regeneration of cartilage (variants) |
| UY39610A (en) | 2021-01-20 | 2022-08-31 | Abbvie Inc | ANTI-EGFR ANTIBODY-DRUG CONJUGATES |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5739350A (en) * | 1990-04-25 | 1998-04-14 | Pharmacia & Upjohn Company | CC-1065 analogs |
| WO2000064864A1 (en) * | 1999-04-26 | 2000-11-02 | Cancer Research Campaign Technology Limited | N-protected amines and their use as prodrugs |
| WO2001083448A2 (en) * | 2000-05-02 | 2001-11-08 | Tietze Lutz F | Novel prodrugs von 6-hydroxy-2,3-dihydro-1h-indoles, 5-hydroxy-1,2-dihydro-3h-pyrrolo[3,2-e]indoles and 5-hydroxy-1,2-dihydro-3h-benzo(e)indoles as well as of 6-hydroxy-1,2,3,4-tetrahydro-benzo[f]quinoline derivatives for use in selective cancer therapy |
| WO2002067937A1 (en) * | 2001-02-22 | 2002-09-06 | School Of Pharmacy, University Of London | Indoline and tetrahydro-quinolines as prodrugs for tumour treatment |
| WO2005112919A2 (en) * | 2004-05-19 | 2005-12-01 | Medarex, Inc. | Self-immolative linkers and drug conjugates |
| WO2006043839A1 (en) * | 2004-10-22 | 2006-04-27 | Auckland Uniservices Limited | Nitrobenzindoles and their use in cancer therapy |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1238907A (en) * | 1984-02-21 | 1988-07-05 | Robert C. Kelly | 1,2,8,8a-tetrahydrocyclopropa¬c|pyrrolo(3,2-e)- indol-4(5h)-ones and related compounds |
| JPH0784377B2 (en) * | 1984-04-10 | 1995-09-13 | 花王株式会社 | Weakly acidic bath salt |
| JP2562935B2 (en) * | 1988-04-28 | 1996-12-11 | 明治製菓株式会社 | Antitumor substance SF2582 derivative |
| JP2612649B2 (en) * | 1990-07-26 | 1997-05-21 | 協和醗酵工業株式会社 | DC-89 derivative |
| EP0563475B1 (en) * | 1992-03-25 | 2000-05-31 | Immunogen Inc | Cell binding agent conjugates of derivatives of CC-1065 |
| GB9516943D0 (en) * | 1995-08-18 | 1995-10-18 | Cancer Soc Auckland Div Nz Inc | Novel cyclopropylindoles and their secoprecursors,and their use as prodrugs |
| US5843937A (en) * | 1996-05-23 | 1998-12-01 | Panorama Research, Inc. | DNA-binding indole derivatives, their prodrugs and immunoconjugates as anticancer agents |
| AU721037B2 (en) * | 1996-09-12 | 2000-06-22 | Auckland Uniservices Limited | Condensed N-acylindoles as antitumor agents |
| CA2448319C (en) * | 2001-05-31 | 2010-07-27 | Medarex, Inc. | Cytotoxins, prodrugs, linkers and stabilizers useful therefor |
| AU2004210136C1 (en) * | 2003-01-27 | 2010-10-07 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| JP2008505144A (en) * | 2004-06-30 | 2008-02-21 | ノバルティス アクチエンゲゼルシャフト | Compositions and methods for delivery of antineoplastic agents |
-
2007
- 2007-02-02 KR KR1020087021441A patent/KR20080114709A/en not_active Application Discontinuation
- 2007-02-02 JP JP2008553192A patent/JP5346589B2/en not_active Expired - Fee Related
- 2007-02-02 EP EP07715863.2A patent/EP1994000B1/en active Active
- 2007-02-02 US US12/223,682 patent/US8940784B2/en active Active
- 2007-02-02 AU AU2007210377A patent/AU2007210377B2/en not_active Ceased
- 2007-02-02 CN CNA2007800120073A patent/CN101415679A/en active Pending
- 2007-02-02 WO PCT/NL2007/050043 patent/WO2007089149A2/en active Application Filing
- 2007-02-02 MX MX2008009956A patent/MX2008009956A/en active IP Right Grant
- 2007-02-02 RU RU2008135440/04A patent/RU2489423C2/en not_active IP Right Cessation
- 2007-02-02 BR BRPI0707426-3A patent/BRPI0707426A2/en not_active IP Right Cessation
- 2007-02-02 CA CA002641899A patent/CA2641899A1/en not_active Abandoned
-
2013
- 2013-06-25 JP JP2013132225A patent/JP2013227326A/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5739350A (en) * | 1990-04-25 | 1998-04-14 | Pharmacia & Upjohn Company | CC-1065 analogs |
| WO2000064864A1 (en) * | 1999-04-26 | 2000-11-02 | Cancer Research Campaign Technology Limited | N-protected amines and their use as prodrugs |
| WO2001083448A2 (en) * | 2000-05-02 | 2001-11-08 | Tietze Lutz F | Novel prodrugs von 6-hydroxy-2,3-dihydro-1h-indoles, 5-hydroxy-1,2-dihydro-3h-pyrrolo[3,2-e]indoles and 5-hydroxy-1,2-dihydro-3h-benzo(e)indoles as well as of 6-hydroxy-1,2,3,4-tetrahydro-benzo[f]quinoline derivatives for use in selective cancer therapy |
| WO2002067937A1 (en) * | 2001-02-22 | 2002-09-06 | School Of Pharmacy, University Of London | Indoline and tetrahydro-quinolines as prodrugs for tumour treatment |
| WO2005112919A2 (en) * | 2004-05-19 | 2005-12-01 | Medarex, Inc. | Self-immolative linkers and drug conjugates |
| WO2006043839A1 (en) * | 2004-10-22 | 2006-04-27 | Auckland Uniservices Limited | Nitrobenzindoles and their use in cancer therapy |
Non-Patent Citations (2)
| Title |
|---|
| L. F. TIETZE ET AL: "Highly Selective Glycosylated Prodrugs of Cytostatic CC-1065 Analogues for Antibody-Directed Enzyme Tumor Therapy" CHEMBIOCHEM, vol. 2, no. 10, 2001, pages 758-765, XP002442494 * |
| L. F. TIETZE ET AL: "Synthesis and Biological Evaluation of Novel Analogues and Prodrugs of the Cytotoxic Antibiotic CC-1065 for Selective Cancer Therapy" EUROPEAN JOURNAL OF ORGANIC CHEMISTRY, 2002, pages 1634-1645, XP002442495 * |
Cited By (203)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9562102B2 (en) | 2001-05-11 | 2017-02-07 | Ludwig Institute For Cancer Research | Specific binding proteins and uses thereof |
| US8034959B2 (en) | 2001-05-31 | 2011-10-11 | Medarex, Inc. | Methods of treating cancer with an antibody-drug conjugate |
| US8399403B2 (en) | 2004-05-19 | 2013-03-19 | Medarex, Inc. | Chemical linkers and conjugates thereof |
| US8124738B2 (en) | 2005-09-26 | 2012-02-28 | Medarex, Inc. | Human monoclonal antibodies to CD70 |
| US8461117B2 (en) | 2006-12-28 | 2013-06-11 | Medarex, Inc. | Chemical linkers and cleavable substrates and conjugates thereof |
| US9090693B2 (en) | 2007-01-25 | 2015-07-28 | Dana-Farber Cancer Institute | Use of anti-EGFR antibodies in treatment of EGFR mutant mediated disease |
| WO2008103693A3 (en) * | 2007-02-21 | 2008-12-11 | Medarex Inc | Chemical linkers with single amino acids and conjugates thereof |
| WO2008103693A2 (en) * | 2007-02-21 | 2008-08-28 | Medarex, Inc. | Chemical linkers with single amino acids and conjugates thereof |
| US8680293B2 (en) | 2007-08-01 | 2014-03-25 | Syntarga B.V. | Substituted CC-1065 analogs and their conjugates |
| US9901567B2 (en) | 2007-08-01 | 2018-02-27 | Syntarga B.V. | Substituted CC-1065 analogs and their conjugates |
| US20110065767A1 (en) * | 2007-08-01 | 2011-03-17 | Patrick Henry Beusker | Substituted CC-1065 Analogs and Their Conjugates |
| WO2009017394A1 (en) * | 2007-08-01 | 2009-02-05 | Syntarga B.V. | Substituted cc-1065 analogs and their conjugates |
| AU2007357156B2 (en) * | 2007-08-01 | 2013-01-10 | Syntarga B.V. | Substituted CC-1065 analogs and their conjugates |
| US9283276B2 (en) | 2007-08-14 | 2016-03-15 | Ludwig Institute For Cancer Research Ltd. | Monoclonal antibody 175 targeting the EGF receptor and derivatives and uses thereof |
| US8268970B2 (en) | 2007-10-01 | 2012-09-18 | Bristol-Myers Squibb Company | Human antibodies that bind mesothelin, and uses thereof |
| US8425904B2 (en) | 2007-10-01 | 2013-04-23 | Bristol-Myers Squibb Company | Human antibodies that bind mesothelin, and uses thereof |
| US8399623B2 (en) | 2007-10-01 | 2013-03-19 | Bristol-Myers Squibb Company | Human antibodies that bind mesothelin, and uses thereof |
| US8383779B2 (en) | 2007-10-01 | 2013-02-26 | Bristol-Myers Squibb Company | Human antibodies that bind mesothelin, and uses thereof |
| JP2015096500A (en) * | 2008-11-03 | 2015-05-21 | シンタルガ・ビーブイ | Novel cc-1065 analogs and their conjugates |
| US9815784B2 (en) | 2008-11-03 | 2017-11-14 | Syntarga B.V. | CC-1065 analogs and their conjugates |
| US8889868B2 (en) | 2008-11-03 | 2014-11-18 | Syntarga Bv | CC-1065 analogs and their conjugates |
| WO2010062171A3 (en) * | 2008-11-03 | 2013-03-14 | Syntarga B.V. | Cc-1065 analogs and their conjugates |
| WO2010062171A2 (en) * | 2008-11-03 | 2010-06-03 | Syntarga B.V. | Novel cc-1065 analogs and their conjugates |
| CN106967062A (en) * | 2008-11-03 | 2017-07-21 | 辛塔佳股份有限公司 | The analogs of novel C C 1065 and its conjugate |
| JP2012513954A (en) * | 2008-11-03 | 2012-06-21 | シンタルガ・ビーブイ | Novel CC-1065 analogs and complexes thereof |
| US9072798B2 (en) | 2009-02-18 | 2015-07-07 | Ludwig Institute For Cancer Research Ltd. | Specific binding proteins and uses thereof |
| US9493578B2 (en) | 2009-09-02 | 2016-11-15 | Xencor, Inc. | Compositions and methods for simultaneous bivalent and monovalent co-engagement of antigens |
| DE102009051438A1 (en) | 2009-10-30 | 2011-05-05 | Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts | fluorescent dyes |
| WO2011051484A1 (en) | 2009-10-30 | 2011-05-05 | Georg-August-Universität Göttingen Stiftung Öffenlichen Rechts | Fluorescence dyes |
| DE102009051799A1 (en) | 2009-11-03 | 2011-05-05 | Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts | Bifunctional prodrugs and drugs |
| WO2011054837A2 (en) | 2009-11-03 | 2011-05-12 | Georg-August-Universität Göttingen Stiftung Öffenlichen Rechts | Bifunctional prodrugs and drugs |
| WO2011133039A2 (en) | 2010-04-21 | 2011-10-27 | Syntarga B.V. | Novel conjugates of cc-1065 analogs and bifunctional linkers |
| US9629924B2 (en) | 2010-04-21 | 2017-04-25 | Syntarga Bv | Conjugates of CC-1065 analogs and bifunctional linkers |
| EP3056203A1 (en) | 2010-04-21 | 2016-08-17 | Syntarga B.V. | Conjugates of cc-1065 analogs and bifunctional linkers |
| US11052155B2 (en) | 2010-04-21 | 2021-07-06 | Syntarga Bv | Conjugates of CC-1065 analogs and bifunctional linkers |
| EP3108886A2 (en) | 2010-04-21 | 2016-12-28 | Syntarga B.V. | Conjugates of cc-1065 analogs and bifunctional linkers |
| WO2011147982A2 (en) | 2010-05-27 | 2011-12-01 | Genmab A/S | Monoclonal antibodies against her2 epitope |
| WO2011147986A1 (en) | 2010-05-27 | 2011-12-01 | Genmab A/S | Monoclonal antibodies against her2 |
| EP3539988A2 (en) | 2010-05-27 | 2019-09-18 | Genmab A/S | Monoclonal antibodies against her2 |
| WO2011157741A2 (en) | 2010-06-15 | 2011-12-22 | Genmab A/S | Human antibody drug conjugates against tissue factor |
| EP4434550A2 (en) | 2010-06-15 | 2024-09-25 | Genmab A/S | Human antibody drug conjugates against tissue culture |
| EP3281956A2 (en) | 2010-06-15 | 2018-02-14 | Genmab A/S | Human antibody drug conjugates against tissue factor |
| US8637641B2 (en) | 2010-07-29 | 2014-01-28 | Xencor, Inc. | Antibodies with modified isoelectric points |
| US9605061B2 (en) | 2010-07-29 | 2017-03-28 | Xencor, Inc. | Antibodies with modified isoelectric points |
| WO2012016227A2 (en) | 2010-07-29 | 2012-02-02 | Xencor, Inc. | Antibodies with modified isoelectric points |
| EP3029066A2 (en) | 2010-07-29 | 2016-06-08 | Xencor, Inc. | Antibodies with modified isoelectric points |
| EP3828205A1 (en) | 2010-10-01 | 2021-06-02 | Oxford BioTherapeutics Ltd | Anti-ror1 antibodies |
| WO2012045085A1 (en) | 2010-10-01 | 2012-04-05 | Oxford Biotherapeutics Ltd. | Anti-rori antibodies |
| EP3219731A1 (en) | 2010-10-01 | 2017-09-20 | Oxford BioTherapeutics Ltd | Anti-ror1 antibodies |
| WO2012092616A1 (en) | 2010-12-30 | 2012-07-05 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| EP3284755A1 (en) | 2010-12-30 | 2018-02-21 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| EP3798231A1 (en) | 2010-12-30 | 2021-03-31 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| EP3763740A1 (en) | 2011-01-26 | 2021-01-13 | Celldex Therapeutics, Inc. | Anti-kit antibodies and uses thereof |
| WO2012103165A2 (en) | 2011-01-26 | 2012-08-02 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies and uses thereof |
| WO2012143523A1 (en) | 2011-04-20 | 2012-10-26 | Genmab A/S | Bispecifc antibodies against her2 |
| US9186416B2 (en) | 2011-05-26 | 2015-11-17 | Bristol-Myers Squibb Company | Immunoconjugates, compositions for making them, and methods of making and use |
| WO2012162482A1 (en) * | 2011-05-26 | 2012-11-29 | Bristol-Myers Squibb Company | Immunoconjugates, compositions containing them, and methods of making and use |
| US8852599B2 (en) | 2011-05-26 | 2014-10-07 | Bristol-Myers Squibb Company | Immunoconjugates, compositions for making them, and methods of making and use |
| EA024844B1 (en) * | 2011-05-26 | 2016-10-31 | Бристол-Майерс Сквибб Компани | Immunoconjugate and use thereof for treating cancer |
| EP3970746A2 (en) | 2011-07-06 | 2022-03-23 | Genmab B.V. | Polypeptide variants and uses thereof |
| WO2013004842A2 (en) | 2011-07-06 | 2013-01-10 | Genmab A/S | Antibody variants and uses thereof |
| WO2013022855A1 (en) | 2011-08-05 | 2013-02-14 | Xencor, Inc. | Antibodies with modified isoelectric points and immunofiltering |
| EP3611187A1 (en) | 2011-10-10 | 2020-02-19 | Xencor, Inc. | A method for purifying antibodies |
| US10851178B2 (en) | 2011-10-10 | 2020-12-01 | Xencor, Inc. | Heterodimeric human IgG1 polypeptides with isoelectric point modifications |
| WO2013055809A1 (en) | 2011-10-10 | 2013-04-18 | Xencor, Inc. | A method for purifying antibodies |
| EP3632462A1 (en) | 2012-07-06 | 2020-04-08 | Genmab B.V. | Dimeric protein with triple mutations |
| WO2014006217A1 (en) | 2012-07-06 | 2014-01-09 | Genmab B.V. | Dimeric protein with triple mutations |
| EP4063391A1 (en) | 2012-07-25 | 2022-09-28 | Celldex Therapeutics, Inc. | Anti-kit antibodies and uses thereof |
| EP3381943A1 (en) | 2012-07-25 | 2018-10-03 | Celldex Therapeutics, Inc. | Anti-kit antibodies and uses thereof |
| WO2014018625A1 (en) | 2012-07-25 | 2014-01-30 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies and uses thereof |
| US10918735B2 (en) | 2012-12-04 | 2021-02-16 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-diones for cancer treatment |
| EP4434543A2 (en) | 2013-01-10 | 2024-09-25 | Genmab B.V. | Human igg1 fc region variants and uses thereof |
| WO2014108198A1 (en) | 2013-01-10 | 2014-07-17 | Genmab B.V. | Human igg1 fc region variants and uses thereof |
| US9650446B2 (en) | 2013-01-14 | 2017-05-16 | Xencor, Inc. | Heterodimeric proteins |
| US9701759B2 (en) | 2013-01-14 | 2017-07-11 | Xencor, Inc. | Heterodimeric proteins |
| US10738133B2 (en) | 2013-01-14 | 2020-08-11 | Xencor, Inc. | Heterodimeric proteins |
| US10738132B2 (en) | 2013-01-14 | 2020-08-11 | Xencor, Inc. | Heterodimeric proteins |
| US11053316B2 (en) | 2013-01-14 | 2021-07-06 | Xencor, Inc. | Optimized antibody variable regions |
| US10487155B2 (en) | 2013-01-14 | 2019-11-26 | Xencor, Inc. | Heterodimeric proteins |
| US10472427B2 (en) | 2013-01-14 | 2019-11-12 | Xencor, Inc. | Heterodimeric proteins |
| US11634506B2 (en) | 2013-01-14 | 2023-04-25 | Xencor, Inc. | Heterodimeric proteins |
| US11718667B2 (en) | 2013-01-14 | 2023-08-08 | Xencor, Inc. | Optimized antibody variable regions |
| US10131710B2 (en) | 2013-01-14 | 2018-11-20 | Xencor, Inc. | Optimized antibody variable regions |
| WO2014113510A1 (en) | 2013-01-15 | 2014-07-24 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| US9738722B2 (en) | 2013-01-15 | 2017-08-22 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| US10968276B2 (en) | 2013-03-12 | 2021-04-06 | Xencor, Inc. | Optimized anti-CD3 variable regions |
| US10106624B2 (en) | 2013-03-15 | 2018-10-23 | Xencor, Inc. | Heterodimeric proteins |
| US11299554B2 (en) | 2013-03-15 | 2022-04-12 | Xencor, Inc. | Heterodimeric proteins |
| EP3421495A2 (en) | 2013-03-15 | 2019-01-02 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US9605084B2 (en) | 2013-03-15 | 2017-03-28 | Xencor, Inc. | Heterodimeric proteins |
| US10858417B2 (en) | 2013-03-15 | 2020-12-08 | Xencor, Inc. | Heterodimeric proteins |
| US10544187B2 (en) | 2013-03-15 | 2020-01-28 | Xencor, Inc. | Targeting regulatory T cells with heterodimeric proteins |
| EP3936521A1 (en) | 2013-03-15 | 2022-01-12 | Xencor, Inc. | Heterodimeric proteins |
| EP3587448A1 (en) | 2013-03-15 | 2020-01-01 | Xencor, Inc. | Heterodimeric proteins |
| US10519242B2 (en) | 2013-03-15 | 2019-12-31 | Xencor, Inc. | Targeting regulatory T cells with heterodimeric proteins |
| US10287364B2 (en) | 2013-03-15 | 2019-05-14 | Xencor, Inc. | Heterodimeric proteins |
| WO2014145806A2 (en) | 2013-03-15 | 2014-09-18 | Xencor, Inc. | Heterodimeric proteins |
| US11814423B2 (en) | 2013-03-15 | 2023-11-14 | Xencor, Inc. | Heterodimeric proteins |
| WO2015052537A1 (en) | 2013-10-11 | 2015-04-16 | Oxford Biotherapeutics Ltd | Conjugated antibodies against ly75 for the treatment of cancer |
| US10266606B2 (en) | 2014-01-10 | 2019-04-23 | Synthon Biopharmaceuticals B.V. | Method for purifying Cys-linked antibody-drug conjugates |
| US9427480B2 (en) | 2014-01-10 | 2016-08-30 | Synthon Biopharmaceuticals B.V. | Duocarmycin ADCs for use in treatment of endometrial cancer |
| US9421278B2 (en) | 2014-01-10 | 2016-08-23 | Synthon Biopharmaceuticals B.V. | Duocarmycin ADCS showing improved in vivo antitumor activity |
| US10092659B2 (en) | 2014-01-10 | 2018-10-09 | Synthon Biopharmaceuticals B.V. | Duocarmycin ADCs for use in treatment of endometrial cancer |
| US10603387B2 (en) | 2014-01-10 | 2020-03-31 | Synthon Biopharmaceuticals B.V. | Duocarmycin ADCs showing improved in vivo antitumor activity |
| US11382982B2 (en) | 2014-01-10 | 2022-07-12 | Byondis B.V. | Duocarmycin ADCs showing improved in vivo antitumor activity |
| EP3954713A2 (en) | 2014-03-28 | 2022-02-16 | Xencor, Inc. | Bispecific antibodies that bind to cd38 and cd3 |
| EP3699195A2 (en) | 2014-03-28 | 2020-08-26 | Xencor, Inc. | Bispecific antibodies that bind to cd38 and cd3 |
| US9822186B2 (en) | 2014-03-28 | 2017-11-21 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| US10858451B2 (en) | 2014-03-28 | 2020-12-08 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| WO2015149077A1 (en) | 2014-03-28 | 2015-10-01 | Xencor, Inc. | Bispecific antibodies that bind to cd38 and cd3 |
| US11840579B2 (en) | 2014-03-28 | 2023-12-12 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| EP3682904A1 (en) | 2014-06-30 | 2020-07-22 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| US10973920B2 (en) | 2014-06-30 | 2021-04-13 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| WO2016001485A1 (en) | 2014-06-30 | 2016-01-07 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| EP3763738A1 (en) | 2014-07-11 | 2021-01-13 | Genmab A/S | Antibodies binding axl |
| WO2016005593A1 (en) | 2014-07-11 | 2016-01-14 | Genmab A/S | Antibodies binding axl |
| WO2016014984A1 (en) | 2014-07-24 | 2016-01-28 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| US10308713B2 (en) | 2014-10-31 | 2019-06-04 | Abbvie Biotherapeutics Inc. | Anti-CS1 antibodies and antibody drug conjugates |
| US10011657B2 (en) | 2014-10-31 | 2018-07-03 | Abbvie Biotherapeutics Inc. | Anti-CS1 antibodies and antibody drug conjugates |
| WO2016070089A2 (en) | 2014-10-31 | 2016-05-06 | Abbvie Biotherapeutics Inc. | Anti-cs1 antibodies and antibody drug conjugates |
| US11352442B2 (en) | 2014-11-26 | 2022-06-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| US9856327B2 (en) | 2014-11-26 | 2018-01-02 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD123 |
| US9850320B2 (en) | 2014-11-26 | 2017-12-26 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD20 |
| US11945880B2 (en) | 2014-11-26 | 2024-04-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11111315B2 (en) | 2014-11-26 | 2021-09-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10526417B2 (en) | 2014-11-26 | 2020-01-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| US11225528B2 (en) | 2014-11-26 | 2022-01-18 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10913803B2 (en) | 2014-11-26 | 2021-02-09 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10259887B2 (en) | 2014-11-26 | 2019-04-16 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11859011B2 (en) | 2014-11-26 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US12129309B2 (en) | 2014-11-26 | 2024-10-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| US11673972B2 (en) | 2014-11-26 | 2023-06-13 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10889653B2 (en) | 2014-11-26 | 2021-01-12 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10428155B2 (en) | 2014-12-22 | 2019-10-01 | Xencor, Inc. | Trispecific antibodies |
| WO2016141387A1 (en) | 2015-03-05 | 2016-09-09 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US10227411B2 (en) | 2015-03-05 | 2019-03-12 | Xencor, Inc. | Modulation of T cells with bispecific antibodies and FC fusions |
| US11091548B2 (en) | 2015-03-05 | 2021-08-17 | Xencor, Inc. | Modulation of T cells with bispecific antibodies and Fc fusions |
| EP3730520A1 (en) | 2015-07-10 | 2020-10-28 | Genmab A/S | Axl-specific antibody-drug conjugates for cancer treatment |
| DE102016105449A1 (en) | 2015-10-29 | 2017-05-04 | Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts | Bifunctional prodrugs |
| US10618935B2 (en) | 2015-11-03 | 2020-04-14 | Industrial Technology Research Institute | Antibody-drug conjugate (ADC) and method for forming the same |
| US10683327B2 (en) | 2015-11-03 | 2020-06-16 | Industrial Technology Research Institute | Compounds, linker-drugs and ligand-drug conjugates |
| US10233212B2 (en) | 2015-11-03 | 2019-03-19 | Industrial Technology Research Institute | Compounds, linker-drugs and ligand-drug conjugates |
| US10188660B2 (en) | 2015-11-30 | 2019-01-29 | Abbvie Inc. | Anti-huLRRC15 antibody drug conjugates and methods for their use |
| US11045480B2 (en) | 2015-11-30 | 2021-06-29 | Abbvie Inc. | Anti-huLRRC15 antibody drug conjugates and methods for their use |
| WO2017095808A1 (en) | 2015-11-30 | 2017-06-08 | Abbvie Inc. | ANTI-huLRRC15 ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
| WO2017095805A1 (en) | 2015-11-30 | 2017-06-08 | Abbvie Inc. | ANTI-huLRRC15 ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
| US10195209B2 (en) | 2015-11-30 | 2019-02-05 | Abbvie Inc. | Anti-huLRRC15 antibody drug conjugates and methods for their use |
| US11623957B2 (en) | 2015-12-07 | 2023-04-11 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| US10227410B2 (en) | 2015-12-07 | 2019-03-12 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| WO2017121877A1 (en) | 2016-01-13 | 2017-07-20 | Genmab A/S | Axl-specific antibody-drug conjugates for cancer treatment |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| WO2017201204A1 (en) | 2016-05-17 | 2017-11-23 | Abbvie Biotherapeutics Inc. | ANTI-cMet ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
| EP3626273A1 (en) | 2016-05-17 | 2020-03-25 | AbbVie Biotherapeutics Inc. | Anti-cmet antibody drug conjugates and methods for their use |
| EP4233909A2 (en) | 2016-05-17 | 2023-08-30 | AbbVie Biotherapeutics Inc. | Anti-cmet antibody drug conjugates and methods for their use |
| EP3804765A1 (en) | 2016-05-17 | 2021-04-14 | AbbVie Biotherapeutics Inc. | Anti-cmet antibody drug conjugates and methods for their use |
| US11492407B2 (en) | 2016-06-14 | 2022-11-08 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US10787518B2 (en) | 2016-06-14 | 2020-09-29 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US11236170B2 (en) | 2016-06-14 | 2022-02-01 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US10316088B2 (en) | 2016-06-28 | 2019-06-11 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US12054545B2 (en) | 2016-06-28 | 2024-08-06 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US11225521B2 (en) | 2016-06-28 | 2022-01-18 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US10793632B2 (en) | 2016-08-30 | 2020-10-06 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| US10550185B2 (en) | 2016-10-14 | 2020-02-04 | Xencor, Inc. | Bispecific heterodimeric fusion proteins containing IL-15-IL-15Rα Fc-fusion proteins and PD-1 antibody fragments |
| US10501543B2 (en) | 2016-10-14 | 2019-12-10 | Xencor, Inc. | IL15/IL15Rα heterodimeric Fc-fusion proteins |
| WO2018083126A1 (en) | 2016-11-01 | 2018-05-11 | Genmab B.V. | Polypeptide variants and uses thereof |
| US11365258B2 (en) | 2017-03-10 | 2022-06-21 | Berlin-Chemie Ag | Pharmaceutical combinations comprising an anti-LY75 antibody |
| EP4257614A2 (en) | 2017-03-10 | 2023-10-11 | Berlin-Chemie AG | Pharmaceutical combinations comprising an anti-ly75 antibody |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
| US11084863B2 (en) | 2017-06-30 | 2021-08-10 | Xencor, Inc. | Targeted heterodimeric Fc fusion proteins containing IL-15 IL-15alpha and antigen binding domains |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| US10981992B2 (en) | 2017-11-08 | 2021-04-20 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| US11312770B2 (en) | 2017-11-08 | 2022-04-26 | Xencor, Inc. | Bispecific and monospecific antibodies using novel anti-PD-1 sequences |
| US11319355B2 (en) | 2017-12-19 | 2022-05-03 | Xencor, Inc. | Engineered IL-2 Fc fusion proteins |
| WO2019145455A1 (en) | 2018-01-24 | 2019-08-01 | Genmab B.V. | Polypeptide variants and uses thereof |
| US10982006B2 (en) | 2018-04-04 | 2021-04-20 | Xencor, Inc. | Heterodimeric antibodies that bind fibroblast activation protein |
| US11524991B2 (en) | 2018-04-18 | 2022-12-13 | Xencor, Inc. | PD-1 targeted heterodimeric fusion proteins containing IL-15/IL-15Ra Fc-fusion proteins and PD-1 antigen binding domains and uses thereof |
| US11505595B2 (en) | 2018-04-18 | 2022-11-22 | Xencor, Inc. | TIM-3 targeted heterodimeric fusion proteins containing IL-15/IL-15RA Fc-fusion proteins and TIM-3 antigen binding domains |
| WO2019229701A2 (en) | 2018-06-01 | 2019-12-05 | Novartis Ag | Binding molecules against bcma and uses thereof |
| WO2019238843A1 (en) | 2018-06-14 | 2019-12-19 | Berlin-Chemie Ag | Pharmaceutical combinations |
| US11530274B2 (en) | 2018-07-02 | 2022-12-20 | Amgen Inc. | Anti-STEAP1 antigen-binding protein |
| WO2020010079A2 (en) | 2018-07-02 | 2020-01-09 | Amgen Inc. | Anti-steap1 antigen-binding protein |
| US11358999B2 (en) | 2018-10-03 | 2022-06-14 | Xencor, Inc. | IL-12 heterodimeric Fc-fusion proteins |
| US11472890B2 (en) | 2019-03-01 | 2022-10-18 | Xencor, Inc. | Heterodimeric antibodies that bind ENPP3 and CD3 |
| WO2020236797A1 (en) | 2019-05-21 | 2020-11-26 | Novartis Ag | Variant cd58 domains and uses thereof |
| WO2020236792A1 (en) | 2019-05-21 | 2020-11-26 | Novartis Ag | Cd19 binding molecules and uses thereof |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| WO2021142086A1 (en) | 2020-01-08 | 2021-07-15 | Synthis Therapeutics, Inc. | Alk5 inhibitor conjugates and uses thereof |
| US20220380360A1 (en) * | 2020-01-30 | 2022-12-01 | Aimed Bio Inc. | Benzoselenophene-based compound, pharmaceutical composition containing same, and antibody-drug conjugate |
| US11938115B2 (en) * | 2020-01-30 | 2024-03-26 | Aimed Bio Inc. | Benzoselenophene-based compound, pharmaceutical composition containing same, and antibody-drug conjugate |
| WO2021178896A1 (en) | 2020-03-06 | 2021-09-10 | Go Therapeutics, Inc. | Anti-glyco-cd44 antibodies and their uses |
| US11919956B2 (en) | 2020-05-14 | 2024-03-05 | Xencor, Inc. | Heterodimeric antibodies that bind prostate specific membrane antigen (PSMA) and CD3 |
| US11591401B2 (en) | 2020-08-19 | 2023-02-28 | Xencor, Inc. | Anti-CD28 compositions |
| US11919958B2 (en) | 2020-08-19 | 2024-03-05 | Xencor, Inc. | Anti-CD28 compositions |
| US12030888B2 (en) | 2021-02-24 | 2024-07-09 | Massachusetts Institute Of Technology | Himastatin derivatives, and processes of preparation thereof, and uses thereof |
| WO2022187591A1 (en) | 2021-03-05 | 2022-09-09 | Go Therapeutics, Inc. | Anti-glyco-cd44 antibodies and their uses |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| US11859012B2 (en) | 2021-03-10 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and GPC3 |
| WO2022248835A1 (en) | 2021-05-26 | 2022-12-01 | Oxford Biotherapeutics Ltd | Pharmaceutical combination comprising an anti-cd205 antibody and an immune checkpoint inhibitor |
| WO2023014863A1 (en) | 2021-08-05 | 2023-02-09 | Go Therapeutics, Inc. | Anti-glyco-muc4 antibodies and their uses |
| WO2023034571A1 (en) | 2021-09-03 | 2023-03-09 | Go Therapeutics, Inc. | Anti-glyco-lamp1 antibodies and their uses |
| WO2023034569A1 (en) | 2021-09-03 | 2023-03-09 | Go Therapeutics, Inc. | Anti-glyco-cmet antibodies and their uses |
| WO2023089314A1 (en) | 2021-11-18 | 2023-05-25 | Oxford Biotherapeutics Limited | Pharmaceutical combinations |
| WO2024153716A1 (en) * | 2023-01-17 | 2024-07-25 | University Of Bradford | Cancer treating compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009525322A (en) | 2009-07-09 |
| EP1994000A2 (en) | 2008-11-26 |
| JP5346589B2 (en) | 2013-11-20 |
| CA2641899A1 (en) | 2007-08-09 |
| US8940784B2 (en) | 2015-01-27 |
| AU2007210377A1 (en) | 2007-08-09 |
| US20090318668A1 (en) | 2009-12-24 |
| CN101415679A (en) | 2009-04-22 |
| EP1994000B1 (en) | 2017-08-23 |
| KR20080114709A (en) | 2008-12-31 |
| WO2007089149A3 (en) | 2007-09-27 |
| RU2489423C2 (en) | 2013-08-10 |
| AU2007210377B2 (en) | 2012-08-09 |
| MX2008009956A (en) | 2008-12-12 |
| BRPI0707426A2 (en) | 2011-05-03 |
| JP2013227326A (en) | 2013-11-07 |
| RU2008135440A (en) | 2010-03-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8940784B2 (en) | Water-soluble CC-1065 analogs and their conjugates | |
| EP2173739B1 (en) | Substituted cc-1065 analogs and their conjugates | |
| EP2560645B1 (en) | Conjugates of cc-1065 analogs and bifunctional linkers | |
| AU2009320481B9 (en) | Novel CC-1065 analogs and their conjugates | |
| US9815784B2 (en) | CC-1065 analogs and their conjugates | |
| US9901567B2 (en) | Substituted CC-1065 analogs and their conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/009956 Country of ref document: MX Ref document number: 2641899 Country of ref document: CA Ref document number: 2008553192 Country of ref document: JP |
|
| REEP | Request for entry into the european phase |
Ref document number: 2007715863 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007210377 Country of ref document: AU Ref document number: 2007715863 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 6937/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087021441 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2008135440 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780012007.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12223682 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0707426 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080801 |