WO2006120544A1 - β-AMINO ACID DERIVATIVES - Google Patents
β-AMINO ACID DERIVATIVES Download PDFInfo
- Publication number
- WO2006120544A1 WO2006120544A1 PCT/IB2006/001209 IB2006001209W WO2006120544A1 WO 2006120544 A1 WO2006120544 A1 WO 2006120544A1 IB 2006001209 W IB2006001209 W IB 2006001209W WO 2006120544 A1 WO2006120544 A1 WO 2006120544A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- formula
- compound
- crc
- alkyl
- Prior art date
Links
- GCWYZLPOJZXQGJ-GCJKJVERSA-N CC(C)(C)[C@@H](N(C)C([C@H](Cc1cccc(Br)c1)C1)=O)N1C(c1ccccc1)=O Chemical compound CC(C)(C)[C@@H](N(C)C([C@H](Cc1cccc(Br)c1)C1)=O)N1C(c1ccccc1)=O GCWYZLPOJZXQGJ-GCJKJVERSA-N 0.000 description 1
- YSJTXBKDIUSLGN-UHFFFAOYSA-N CC(C)Cc1cc(Br)ccc1 Chemical compound CC(C)Cc1cc(Br)ccc1 YSJTXBKDIUSLGN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/62—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
- C07C323/63—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton the carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/34—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/60—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Definitions
- This invention relates to ⁇ -amino acid derivatives derivatives. More particularly, this invention relates to ⁇ -arylmethyl- ⁇ -amino acid derivatives and to processes for the preparation of, intermediates used in the preparation of, compositions containing, and the uses of such derivatives.
- alpha-2-delta receptor ligands also known as alpha-2-delta ligands
- An alpha-2-delta receptor ligand is a molecule which binds to any sub-type of the human calcium channel alpha-2-delta subunit.
- the calcium channel alpha-2-delta subunit comprises a number of sub-types which have been described in the literature (e.g. type 1 , J. Biol. Chem., 1996, 271 (10), 5768-76; types 2 and 3, J. Membr. Biol., 2001 , 184(1), 35-43 and MoI. Pharmacol., 2001 , 59(5), 1243-1248, 2001 ; and type 4, MoI. Pharmacol., 2002, 62(3), 485-496).
- Alpha-2-delta receptor ligands are also sometimes known as GABA analogues.
- alpha-2-delta ligands are marketed drugs such as gabapentin (sold under the trade mark Neurontin) and pregabalin (sold under the trade mark Lyrica).
- Gabapentin is an anti-convulsant which is marketed for the treatment of epilepsy.
- Pregabalin is marketed for the treatment of neuropathic pain.
- R 1 and R 2 are each independently H, cyano, halo or -X-Y-R 4 , or R 1 and R 2 , taken together with two adjacent carbon atoms to which they are attached, form a fused 5- or 6-membered, saturated, partially unsaturated or aromatic ring optionally containing one or two nitrogen, oxygen or sulphur hereroatoms, said ring being optionally substituted by one ore more groups selected from cyano, halo and -X-Y-R 4 ;
- -X- is a direct bond, CrC 6 alkylene, C 3 -C 8 cycloalkylene, C 2 -C 6 alkenylene or C 2 - C 6 alkynylene, said CrC 6 alkylene, 0-3-C 8 cycloalkylene, C 2 -C 6 alkenylene and C 2 -C 6 alkynylene being optionally substituted by one or more halo (preferably fluoro) groups;
- -Y- is a direct bond, -O-, -S-, -SO-, -SO 2 -, -NR 5 -, -CO-, -OCO-, -C(O)O-, -NR 5 CO-, -C(O)NR 5 -, -NR 5 SO 2 - or -SO 2 NR 5 -;
- R 3 is H, cyano, halo, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, CrC 6 alkoxy or C 3 - C 8 cycloalkyl, said CrC 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, CrC 6 alkoxy and C 3 - C 8 cycloalkyl being optionally substituted by one or more halo (preferably fluoro) groups;
- R 4 is CrC 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 8 cycloalkyl, aryl, Het 1 or Het 2 , said CrC 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 8 cycloalkyl being optionally substituted by one or more halo (preferably fluoro) groups;
- R 5 is H, CrC 6 alkyl or C 3 -C 8 cycloalkyl
- Het 1 is a 3- to 8-membered, saturated or partially unsaturated heterocyclic group comprising one or two ring members selected from -NR 6 -, -O-, -S-, -SO- and -SO 2 - said heterocyclic group being optionally substituted on a ring carbon atom by one or more substituents selected from oxo, halo, -R 5 or -OR 5 and optionally benzo-fused, said benzo-fused portion being optionally substituted by one or more substituents selected from halo, CrC 6 alkyl, C 3 -C 8 cycloalkyl, CrC 6 alkoxy and cyano;
- R 6 is H, C 1 -C 6 alkyl, C 3 -C 8 cycloalkyl, -COR 7 , -SO 2 R 7 or a bond to the group which is substituted with Het 1 ;
- R 7 is CrC 6 alkyl or C 3 -C 8 cycloalkyl
- Het 2 is a 5-membered aromatic heterocyclic group comprising either (a) 1 to 4 nitrogen atoms, (b) one oxygen or one sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms or a 6-membered aromatic heterocyclic group comprising 1 or 2 nitrogen atoms, said 5- or 6-membered heterocyclic group being optionally substituted by one or more substituents selected from halo, -NR 5 R 5 , C r C 6 alkyl, C 3 -C 8 cycloalkyl, C 1 -C 6 alkoxy and cyano;
- aryl is phenyl or naphthyl optionally substituted by one or more substituents sseelleecctteedd ffrr ⁇ om halo, -NR 5 R 5 , C 1 -C 6 alkyl, C 3 -C 8 cycloalkyl, C 1 -C 6 alkoxy and cyano; and
- R 8 and R 9 are each independently H or a group which is converted to H following administration of the compound to a mammal;
- halo means fluoro, chloro or bromo and is preferably fluoro or chloro.
- Alkyl, alkenyl, alkynyl, alkylene, alkenylene, alkynylene and alkoxy groups containing the requisite number of carbon atoms can be unbranched or branched chain. Examples of alkyl include methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl and t-butyl.
- alkenyl examples include ethenyl, propen-1-yl, propen-2-yl, propen-3-yl, but-1-en-1-yl, but-1-en-2-yl, but-1 - en-3-yl, but-1-en-4-yl, but-2-en-1-yl and but-2-en-2-yl.
- alkynyl examples include ethynyl, propyn-3-yl, but-1-yn-1-yl, but-1-yn-3-yl, but-1-yn-4-yl and but-2- yn-1-yl.
- alkoxy examples include methoxy, ethoxy, n-propoxy, i-propoxy, n- butoxy, i-butoxy, sec-butoxy and t-butoxy.
- alkylene examples include methylene, 1 ,1 -ethylene, 1 ,2-ethylene, 1 ,1 -propylene, 1 ,2-propylene, 1 ,3- propylene and 2,2-propylene.
- alkenylene include 1 ,1-ethenylene, 1 ,2-ethenylene, 1 ,1-propenylene, 1 ,2-propenylene and 1 ,3-propenylene.
- alkynylene examples include 1 ,2-ethynylene, 1 ,3-propynylene and 3,3- propynylene.
- cycloalkyl examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- the R 1 , R 2 and R 3 groups may be attached to any of the five free positions on the phenyl ring.
- Y is -O-, -S-, -NR 5 -, -C(O)O-, -C(O)NR 5 -, or - SO 2 NR 5 - and R 4 is Het 1 or Het 2
- the Het 1 or Het 2 group may not be attached to the Y group through a ring heteroatom.
- Het 1 are oxiranyl, aziridinyl, oxetanyl, azetidinyl, tetrahydrofuranyl, pyrrolidinyl, tetrahydropyranyl, piperidinyl, morpholinyl, thiomorpholinyl, piperazinyl, azepanyl, oxapanyl, oxazepanyl and diazepanyl (optionally substituted as specified above).
- Het 2 are thienyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, tetrazolyl, pyridyl, pyrimidinyl, pyrazinyl and pyridazinyl (optionally substituted as specified above).
- the invention provides a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, wherein:
- R 8 is H, C 1 -C 6 alkyl, aryl, indanyl or (CrC 8 alkyl)OC(O)O(C r C 6 alkyl);and
- R 9 is H, -CO(CrC 6 alkyl), -CO(aryl), or a natural ⁇ -amino acid joined through its carboxy group; or
- the invention provides a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, wherein R 8 and R 9 are as defined above in embodiment A or embodiment B and R 3 is H.
- the invention provides a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, wherein R 8 and R 9 are as defined above in embodiment A or embodiment B, R 3 is as defined above in embodiment A or embodiment C and R 1 and R 2 are independently either: (a) H, halo or -X-Y-R 4 ; or (b) H, halo or -Y-R 4 ; or
- compositions of a compound of formula (I) include the acid addition and base salts thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, saccharate, stearate, succinate, tartrate, tosylate and trifluor
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
- Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
- a compound of formula (I) may exist in both unsolvated and solvated forms.
- the term 'solvate' is used herein to describe a molecular complex comprising the compound of formula (I) and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- the term 'hydrate' is employed when said solvent is water.
- complexes such as clathrates, drug-host inclusion complexes wherein, in contrast to the aforementioned solvates, the drug and host are present in stoichiometric or non-stoichiometric amounts.
- complexes of the drug containing two or more organic and/or inorganic components which may be in stoichiometric or non- stoichiometric amounts.
- the resulting complexes may be ionised, partially ionised, or non-ionised.
- references to a compound of formula (I) include references to salts, solvates and complexes thereof and to solvates and complexes of salts thereof.
- a compound of formula (I), as hereinbefore defined, may exist in one or more crystalline (polymorphic) or isomeric forms (including optical, geometric and tautomeric isomers), in an isotopically labelled form or as a prodrug. All such crystalline/isomeric forms and prodrugs are within the scope of the present invention and are further described below. All references to a compound of formula (I) should be interpreted accordingly.
- prodrugs Compounds of the formula (I) wherein R 8 and/or R 9 is a group which is converted to H following administration of the compound to a mammal (preferably a human) are known as prodrugs.
- these derivatives which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) wherein R 8 and R 9 are both H, such compounds having the desired activity as alpha-2-delta ligands.
- prodrugs can be converted, for example, by hydrolytic cleavage.
- R 8 is an alkyl group, preferably a CrC 6 alkyl group. Specific examples of suitable alkyl groups are ethyl, isopropyl and n-butyl. Alternatively, R 8 can be an aryl group (wherein aryl is as defined above), such as phenyl, or an indanyl group.
- R 8 can be an alkylocycarbonyloxyalkyl group, such as -CH 2 OC(O)O 4 Bu, -CH(CH 3 )OC(O)OEt or CH(CH 3 )OC(O)O 1 Pr (see Journal of Pharmcology and Experimental Therapeutics, 311 , 1 , 324-335) or a cyclic carbonate linked via a methylene group.
- alkylocycarbonyloxyalkyl group such as -CH 2 OC(O)O 4 Bu, -CH(CH 3 )OC(O)OEt or CH(CH 3 )OC(O)O 1 Pr (see Journal of Pharmcology and Experimental Therapeutics, 311 , 1 , 324-335) or a cyclic carbonate linked via a methylene group.
- R 9 is an amide-forming group such as -CO(CrC 6 alkyl) or -CO(aryl) (wherein aryl is as defined above). Specific examples are methylcarbonyl, isopropylcarbonyl and phenylcarbonyl. Alternatively, R 9 may be an ⁇ -amino acid residue joined through its carboxyl group to form an amide.
- the naturally occurring amino acids, particularly glycine, alanine and valine are preferred.
- Prodrug hydrolysis can be characterised in vitro using a range of tissue fractions including simple homogenates and microsomes: see, for example, Journal of Pharmacology and Experimental Therapeutics, 294, 2, 580-587; Life ScL, 62, 14, 1231-124; International Journal of Pharmaceutics, 166, 1 , 45-53; and Toxicol. Lett, 82-83, 439-445. Rat liver microsome homogenates are particularly useful in this regard. In vivo assays can also be used to investigate prodrug properties.
- Intravenous and oral pharmacokinetics with both the active principle and the prodrug provides information about the relative bioavailability of the prodrug, the ability of the body to hydrolyse the prodrug and the rate of hydrolysis to the active species (see Antimicrob. Agents. Chemother. 42, 3, 647-653).
- a proposed screening strategy for assaying prodrugs has been given in a recent review (Current Drug Metabolism, 2003, vol 4, no. 6, p 483).
- prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association).
- Prodrugs of compounds of the formula (I) other than those involving R 8 and R 9 groups are also within the scope of the invention and can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
- prodrugs in accordance with the invention include:
- metabolites of compounds of formula (I), that is, compounds formed in vivo upon administration of the drug are also included within the scope of the invention.
- Some examples of metabolites in accordance with the invention include
- Compounds of formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a compound of formula (I) contains an alkenyl or alkenylene group, geometric cis/trans (or Z/E) isomers are possible. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in compounds of formula (I) containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
- Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
- Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral compounds of the invention may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine.
- Stereoisomeric conglomerates may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, New York, 1994).
- a preferred compound of formula (I) is a compound according to any of embodiments A-D above wherein the stereochemistry at the carbon atom alpha to the carboxylic acid is in the S configuration, i.e. a compound of formula (Ia).
- the present invention includes all pharmaceutically acceptable isotopically- labelled compounds of formula (I) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and
- C chlorine, such as 36, Cl 1 fluorine, such as 18r F, iodine, such as 123, I and 125 ⁇ I, nitrogen, such as 13
- isotopically-labelled compounds of formula (I), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes tritium, i.e. 3 H, and carbon-14, i.e. 14 C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
- substitution with heavier isotopes such as deuterium, i.e. 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
- solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 O, de-acetone, d e -DMSO.
- the compounds of formula (I), being alpha-2-delta receptor ligands, are potentially useful in the treatment of a wide range of disorders.
- the treatment of pain, particularly neuropathic, is a preferred use.
- Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment.
- the system operates through a specific set of, primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review).
- These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities. Nociceptors encode the intensity, duration and quality of noxious stimulus and by virtue of their topographically organised projection to the spinal cord, the location of the stimulus.
- nociceptive nerve fibres of which there are two main types, A-delta fibres (myelinated) and C fibres (non-myelinated).
- A-delta fibres myelinated
- C fibres non-myelinated.
- the activity generated by nociceptor input is transferred, after complex processing in the dorsal horn, either directly, or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the cortex, where the sensation of pain is generated.
- Pain may generally be classified as acute or chronic. Acute pain begins suddenly and is short-lived (usually in twelve weeks or less). It is usually associated with a specific cause such as a specific injury and is often sharp and severe. It is the kind of pain that can occur after specific injuries resulting from surgery, dental work, a strain or a sprain. Acute pain does not generally result in any persistent psychological response. In contrast, chronic pain is long-term pain, typically persisting for more than three months and leading to significant psychological and emotional problems. Common examples of chronic pain are neuropathic pain (e.g. painful diabetic neuropathy, postherpetic neuralgia), carpal tunnel syndrome, back pain, headache, cancer pain, arthritic pain and chronic postsurgical pain.
- neuropathic pain e.g. painful diabetic neuropathy, postherpetic neuralgia
- carpal tunnel syndrome e.g. painful diabetic neuropathy, postherpetic neuralgia
- back pain e.g. painful diabetic neuropathy, postherpetic neuralgia
- Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13-44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
- Nociceptive pain is induced by tissue injury or by intense stimuli with the potential to cause injury. Pain afferents are activated by transduction of stimuli by nociceptors at the site of injury and activate neurons in the spinal cord at the level of their termination. This is then relayed up the spinal tracts to the brain where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44). The activation of nociceptors activates two types of afferent nerve fibres. Myelinated A-delta fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain.
- Moderate to severe acute nociceptive pain is a prominent feature of pain from central nervous system trauma, strains/sprains, burns, myocardial infarction and acute pancreatitis, post-operative pain (pain following any type of surgical procedure), posttraumatic pain, renal colic, cancer pain and back pain.
- Cancer pain may be chronic pain such as tumour related pain (e.g. bone pain, headache, facial pain or visceral pain) or pain associated with cancer therapy (e.g. postchemotherapy syndrome, chronic postsurgical pain syndrome or post radiation syndrome). Cancer pain may also occur in response to chemotherapy, immunotherapy, hormonal therapy or radiotherapy.
- Back pain may be due to herniated or ruptured intervertabral discs or abnormalities of the lumber facet joints, sacroiliac joints, paraspinal muscles or the posterior longitudinal ligament. Back pain may resolve naturally but in some patients, where it lasts over 12 weeks, it becomes a chronic condition which can be particularly debilitating.
- Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies. These include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain is pathological as it has no protective role.
- neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp., 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
- the inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56).
- Arthritic pain is the most common inflammatory pain.
- Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407).
- Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain.
- Gl gastrointestinal
- FBD functional bowel disorder
- IBD inflammatory bowel disease
- Gl disorders include a wide range of disease states that are currently only moderately controlled, including, in respect of FBD, gastroesophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain.
- Other types of visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
- musculoskeletal disorders including myalgia, fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-articular rheumatism, dystrophinopathy, glycogenosis, polymyositis and pyomyositis; • heart and vascular pain, including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
- head pain such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
- orofacial pain including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
- the compounds of formula (I) are potentially useful in the treatment of all kinds of pain but are particularly useful in the treatment of neuropathic pain.
- the compounds of formula (I) are also potentially useful in the treatment of any disease or condition which is treatable using an alpha-2-delta ligand.
- diseases include epilepsy, gastrointestinal disorders, premature ejaculation, burning mouth syndrome, bladder disorders (such as over active bladder), faintness attacks, fibromyalgia, hypokinesia, cranial disorders, hot flashes, essential tremor, chemical dependencies and addictions, withdrawal symptoms associated with dependencies or addictions, addictive behaviours, spasticity, arthritis, inflammatory disorders (e.g.
- rheumatoid arthritis osteoarthritis, psoriasis
- diuresis premenstrual syndrome
- premenstrual dysphoric disorder tinnitus
- gastric damage Down's syndrome
- demyelinating diseases e.g. multiple sclerosis and amylolateral sclerosis
- cerebral vascular disorders due to acute or chronic cerebrovascular damage e.g. cerebral infarction, subarachnoid haemorrhage or cerebral oedema
- head trauma spinal cord trauma and neuronal damage that occurs, for instance, during stroke, in cardiac bypass surgery, in incidents of intracranial hemorrhage, in perinatal asphyxia, in cardiac arrest and in status epilepticus.
- Alpha-2-delta ligands may also be useful in the treatment of delirium, dementia and amnestic and other cognitive or neurodegenerative -disorders (e.g. Parkinson's disease, Huntington's disease, Alzheimer's disease, senile dementia, memory disorder, vascular dementia). They may be useful in the treatment of movement disorders such as akinesias, dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, palsys, akinetic-rigid syndrome and extra-pyramidal movement disorders. They may also be useful in the treatment of sleep disorders, mood disorders, depression, depressive disorders, bipolar disorders, anxiety disorders, panic, borderline personality disorder, schizophrenia, psychotic disorders, behavioural disturbances associated with mental retardation, autistic disorder and conduct disorders.
- Parkinson's disease Huntington's disease
- Alzheimer's disease senile dementia
- memory disorder e.g. Parkinson's disease
- vascular dementia e.g. Parkinson's disease, Huntington's disease, Alzheimer's
- All of the compounds of formula (I) can be prepared by conventional routes such as by the procedures described in the general methods presented below or by the specific methods described in the Examples section and the Preparations section, or by similar methods thereto.
- the present invention also encompasses any one or more of these processes for preparing the compounds of formula (I), in addition to any novel intermediates used therein.
- R 1 , R 2 , R 3 , R 8 and R 9 are as previously defined for a compound of formula (I) unless otherwise stated.
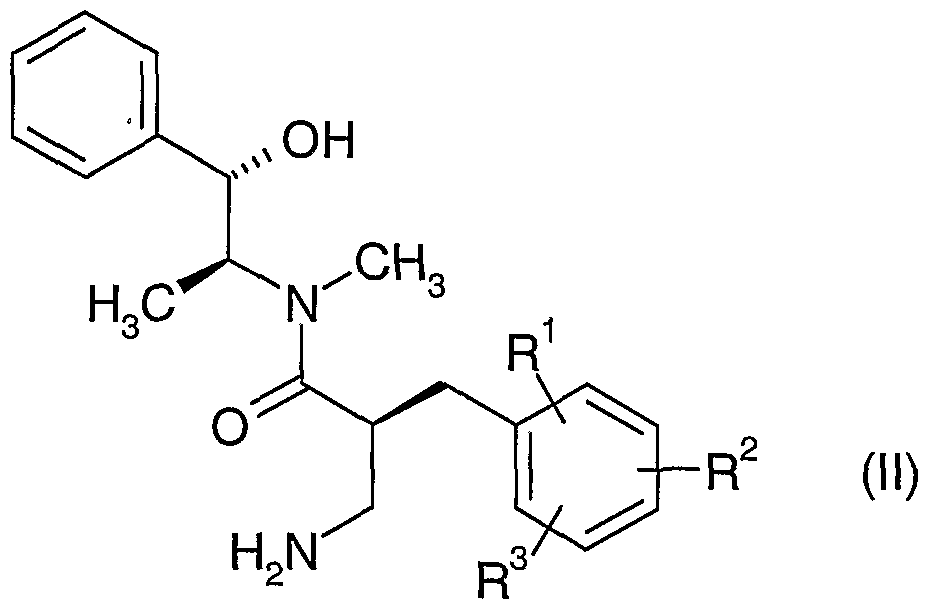
- a compound of formula (Ia), wherein R 8 and R 9 are H, may be prepared by the hydrolysis of a compound of formula (II)
- R »1 , n R2 and R are as defined above.
- the reaction may be achieved under neutral conditions or with basic or acidic catalysis, , most typically under neutral conditions.
- the reaction is typically performed in water, optionally in the presence of a co-solvent (e.g. 1 ,4-dioxane or tetrahydrofuran), at elevated temperature for about 4 days.
- a co-solvent e.g. 1 ,4-dioxane or tetrahydrofuran
- a solution of the compound of formula (II) in a 1 :1 (by volume) mixture of water and 1 ,4- dioxane is heated under at reflux for 4 days.
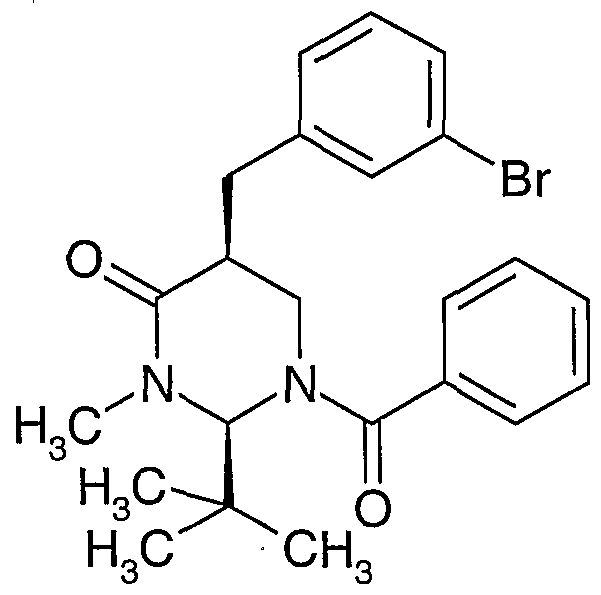
- a compound of formula (II) may be prepared by the alkylation of a compound of formula (III)
- L 1 is a suitable leaving group.
- L 1 is typically a bromide or trifluoromethanesulphonate (triflate) group, preferably a bromide group.
- the compound of formula (III) is de-protonated using a suitable base, and the resulting anion is quenched by the addition of a compound of formula (IV).
- a solution of a compound of formula (III) in a suitable solvent such as tetrahydrofuran or ether
- a strong base such as lithium diisopropylamide or lithium hexamethyldisilazide
- an additive such as lithium chloride
- a solution of the compound of formula (III) in tetrahydrofuran is treated with 3.2 equivalents of lithium hexamethyldisilazide and 4 equivalents of lithium chloride at a temperature of between -5° and 0°C for about 1 hour, followed by the addition of 1 equivalent of the compound of formula (IV) at 0 0 C.
- a compound of formula (III) may be prepared using the methods described in Organic Letters, 2000, 2(22), 3527-3529.
- Compounds of formula (IV) are either commercially available or may be prepared from commercially available starting materials using standard chemical transformations well known to the skilled ⁇ ; person, e.g. the treatment of a corresponding alcohol with hydrogen bromide (see 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.) for details of such standard transformations).
- R 1 , R 2 and R 3 are as defined above.
- This reaction may be achieved with basic or acidic catalysis, acidic catalysis being preferred.
- a compound of formula (V) in a mixture of water and a mineral acid e.g. hydrochloric acid or sulphuric acid
- an organic co- solvent such as tetrahydrofuran
- a solution of a compound of formula (V) in 6M hydrochloric acid is heated under reflux for about 24 hours.
- Compounds of formula (V) may be prepared by epimerisation of a compound of formula (Vl)
- the epimerisation is accomplished by treating a compound of formula (Vl) with suitable base, and quenching the resulting anion with aqueous acid.
- a solution of a compound of formula (Vl) in a suitable solvent e.g. tetrahydrofuran or ether
- a strong base e.g. lithium diisopropylamide, lithium hexamethyldisilazide or sodium hexamethyldisilazide
- very low temperature for example between about -78° and -6O 0 C, for about 3 hours
- the resulting anion is quenched (e.g.
- a compound of formula (IV) as defined above.
- the compound of formula (VII) is de-protonated using a suitable base, and the resulting anion is quenched by addition of the compound of formula (IV).
- a solution of a compound of formula (VII) in a suitable solvent such as tetrahydrofuran or ether
- a strong base such as lithium diisopropylamide or lithium hexamethyldisilazide
- a solution of the compound of formula (VII) in tetrahydrofuran is treated with 1.2 equivalents of lithium diisopropylamide at a temperature of about -78°C for about 80 minutes, followed by the addition of 1.2 equivalents of the compound of formula (IV) at -78 0 C, warming slowly to room temperature.
- a compound of formula (VII) may be prepared by the method of Juaristi et al in Tetrahedron Asymmetry, 1996, 2233-2246.
- a compound of formula (Vila) may also be prepared using the method of Juaristi et al in Tetrahedron Asymmetry, 1996, 2233-2246.
- compounds of formula (I) having the alternative (R) stereochemistry may be prepared directly from a compound of formula (Vl) applying the hydrolysis described above in relation to a compound of formula (V).
- compounds of formula (Ia) may be prepared by a sequence of alkylation and hydrolysis performed on a compound of formula (Vila).
- Compounds of formula (I) may also be prepared using methods analogous to those described in WO-A-03/082807 and the references therein or by the method of Lavielle et al in European Journal of Organic Chemistry 2000, 1 , 83-89.
- Compounds of formula (I) can also be prepared by using the reactions described above to construct a compound wherein R 1 , R 2 , R 3 , R 8 or R 9 are partially formed or protected and then completing the synthesis by functional group manipulation.
- Suitable protecting groups are described in 'Protective Groups in Organic Synthesis 1 by Theodora Greene and Peter Wuts (third edition, 1999, John Wiley and Sons).
- Suitable functional group transformations are described in 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.).
- Compounds of formula (I) may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
- excipients may be administered alone or in combination with one or more other compounds of formula (I) or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients.
- excipient' is used herein to describe any ingredient other than a compound of formula (I). The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- compositions suitable for the delivery of compounds of formula (I) and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences. 19th Edition (Mack Publishing Company, 1995).
- a compound of formula (I) may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid formulations such as tablets, capsules containing particulates, liquids, powders, lozenges (including liquid-filled lozenges), chews, multi- and nano-particulates, gels, solid solutions, liposomes, films, ovules, sprays and liquid formulations.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- a compound of formula (I) may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
- a compound of formula (I) may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl- substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose.
- Tablets may also contain diluents, such as lactose (as, for example, the monohydrate, spray-dried monohydrate or anhydrous form), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose as, for example, the monohydrate, spray-dried monohydrate or anhydrous form
- mannitol mannitol
- xylitol dextrose
- sucrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
- ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
- Exemplary tablets contain up to about 80% of a compound of formula (I), from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
- a compound of formula (I) from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
- the formulation of tablets is discussed in Pharmaceutical Dosage Forms: Tablets, Vol. 1 , by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
- Consumable oral films for human or veterinary use are typically pliable water- soluble or water-swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula (I), a film- forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent.
- Some components of the formulation may perform more than one function.
- a compound of formula (I) for use in a film may be water-soluble or insoluble.
- a water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes.
- a compound of formula (I) may be used in the form of multiparticulate beads.
- the film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
- ingredients in such a film include anti-oxidants, colorants, flavourings, flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
- Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
- Solid formulations for oral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed, sustained, pulsed, controlled, targeted and programmed release formulations.
- Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Pharmaceutical Technology On-line. 25(2), 1-14, by Verma et al (2001). The use of chewing gum to achieve controlled release is described in WO 00/35298.
- a compound of formula (I) may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable routes for such parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous delivery.
- Suitable means for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably at a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably at a pH of from 3 to 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of a compound of formula (I) used in the preparation of a parenteral formulation may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed, sustained, pulsed, controlled, targeted and programmed release formulations.
- a compound of formula (I) may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug- coated stents and poly( ⁇ f/-lactic-coglycolic)acid (PGLA) microspheres.
- PGLA poly( ⁇ f/-lactic-coglycolic)acid
- a compound of formula (I) may also be administered topically to the skin or mucosa, i.e. dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Penetration enhancers may be incorporated - see, for example, J. Pharm. Sci., 88 (10), 955-958, by Finnin and
- topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. PowderjectTM, BiojectTM, etc.) injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed, sustained, pulsed, controlled, targeted and programmed release formulations.
- a compound of formula (I) can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane.
- the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
- the pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of a compound of formula (I) comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a compound of formula (I) comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a drug product Prior to use in a dry powder or suspension formulation, a drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- Capsules made, for example, from gelatin or hydroxypropylmethylcellulose
- blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of a compound of formula (I), a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 ⁇ g to 20mg of a compound of formula (I) per actuation and the actuation volume may vary from 1 ⁇ l to 100 ⁇ l.
- a typical formulation may comprise a compound of formula (I), propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavours such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA.
- Modified release formulations include delayed, sustained, pulsed, controlled, targeted and programmed release formulations.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or "puff".
- the overall daily dose will be administered in a single dose or, more usually, as divided doses throughout the day.
- a compound of formula (I) may be administered rectally or vaginally, e.g. in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed, sustained, pulsed, controlled, targeted and programmed release formulations.
- a compound of formula (I) may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- Formulations for ocular/aural administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed, sustained, pulsed, controlled, targeted, or programmed release formulations.
- a compound of formula (I) may be combined with a soluble macromolecular entitiy, such as a cyclodextrin or a suitable derivative thereof or a polyethylene glycol-containing polymer, in order to improve its solubility, dissolution rate, taste- masking, bioavailability and/or stability in any of the aforementioned modes of administration.
- Drug-cyclodextrin complexes are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Applications Nos. WO-A-91/11172, WO-A-94/02518 and WO-A-98/55148.
- the total daily dose of a compound of formula (I) is typically in the range of from 1 mg to 1000 mg (preferably between 10mg and 500mg) depending, of course, on the mode of administration and the potency of the selected compound.
- the total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
- These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
- references herein to "treatment” include references to curative, palliative and prophylactic treatment.
- the biological activity of the alpha-2-delta ligands of the invention may be measured in a radioligand binding assay using [ 3 H]gabapentin and the ⁇ 2 ⁇ subunit derived from porcine brain tissue based on the method given in J. Biol. Ch ⁇ m., 1996, 271(10), 5768-5776). This assay is reproduced below. [ 3 H]Gabapentin binding assay
- Pig brain cortex (up to 50 g) (fresh or frozen) was homogenised in 10 volumes of Buffer A (0.32 M Sucrose/1 mM EDTA/1 mM EGTA/10 mM Hepes/KOH, pH 7.4) by six strokes of a glass/teflon homogeniser at 600 r.p.m. After removal of the 1000 g x 10 minute pellet, the supernatant was centrifuged at 40,000 g for 20 minutes and the resulting pellet was resuspended in 10 volumes of Buffer B (1 mM EDTA/1 mM EGTA/10 mM Hepes/KOH, pH 7.4).
- Buffer A 0.32 M Sucrose/1 mM EDTA/1 mM EGTA/10 mM Hepes/KOH, pH 7.4
- membranes were pelleted as above twice more by centrifugation with Buffer B, before a final re-suspension in approximately 3 volumes of storage buffer (1.25 mM EDTA/1.25 mM EGTA/25% Glycerol/12.5 mM Hepes/KOH, pH 7.4) to give a concentration of about 3 milligrams of protein per millilitre. Aliquots were stored at -8O 0 C until required.
- Binding of [ 3 H]gabapentin to pig cerebral cortex membranes was carried out at 22 0 C in 10 mM Hepes/KOH, pH 7.4 for 60 minutes.
- Non-specific binding (nsb) was defined as the binding obtained in the presence of 10 ⁇ M pregabalin.
- An assay volume of 250 ⁇ l was employed, comprising 200 ⁇ l of membranes, 25 ⁇ l test compound/buffer/nsb, 25 ⁇ l [ 3 H]gabapentin (final assay concentration ⁇ 10nM).
- an alpha-2-delta receptor ligand may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain.
- an alphas- delta receptor ligand particularly a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, as defined above, may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
- an opioid analgesic e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
- NSAlD nonsteroidal antiinflammatory drug
- NSAlD nonsteroidal antiinflammatory drug
- diclofenac diflusinal, etodolac
- fenbufen fenoprofen
- flufenisal flurbiprofen
- ibuprofen indomethacin
- ketoprofen ketorolac
- meclofenamic acid mefenamic acid
- nabumetone naproxen
- oxaprozin phenylbutazone
- piroxicam sulindac, tolmetin or zomepirac
- a barbiturate sedative e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal or thiopental; • a benzodiazepine having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
- an Hi antagonist having a sedative action e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine; • a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
- a skeletal muscle relaxant e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N- methylmorphinan), ketamine, memantine, pyrroloquinoline quinone or cis- 4-(phosphonomethyl)-2-piperidinecarboxylic acid
- an alpha-adrenergic e.g.
- a tricyclic antidepressant e.g. desipramine, imipramine, amytriptiline or nortriptiline
- an anticonvulsant e.g. carbamazepine or valproate
- a tachykinin (NK) antagonist particularly an NK-3, NK-2 or NK-1 antagonist, e.g. ( ⁇ R,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11- tetrahydro-9-methyl-5-(4-methylphenyl)-7H-[1 ,4]diazocino[2, 1 - g][1 ,7]naphthridine-6-13-dione (TAK-637), 5-[[(2R,3S)-2-[(1 R)-1 -[3,5- bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4-morpholinyl]methyl]-
- a tachykinin (NK) antagonist particularly an NK-3, NK-2 or NK-1 antagonist
- a muscarinic antagonist e.g oxybutin, tolterodine, propiverine, tropsium chloride or darifenacin;
- a selective COX-2 inhibitor e.g. celecoxib, rofecoxib or valdecoxib;
- a non-selective COX inhibitor e.g. nitroflurbiprofen (HCT-1026);
- a vanilloid receptor agonist e.g. resinferatoxin
- antagonist e.g. capsazepine
- a beta-adrenergic such as propranolol
- a local anaesthetic such as mexiletine
- a corticosteriod such as dexamethasone
- a 5-HT receptor agonist or antagonist particularly a 5-HTIB/ID agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan;
- a PDEV inhibitor such as sildenafil, vardenafil, taladafil, 5-[2-ethoxy-5-(4- ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6- dihydro-7H-pyrazolo[4,3- ⁇ (]pyrimidin-7-one, 5-(5-acetyl-2-butoxy-3- pyridinyl)-3-ethyl-2-(1-ethyl-3-azetidinyl)-2,6-dihydro-7/-/-pyrazolo[4,3- ⁇ yrimidin-7-one, 1- ⁇ 6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7- oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyr
- mGluRI metabotropic glutamate subtype 1 receptor
- a serotonin reuptake inhibitor such as sertraline
- a noradrenaline reuptake inhibitor especially a selective noradrenaline reuptake inhibitor such as (S,S)-reboxetine; • a dual serotonin/noradrenaline reuptake inhibitor such as duloxetine;
- an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1- iminoethyl)amino]ethyl]-2-methyl-L-cysteine or (2S,5Z)-2-amino-2-methyl- 7-[(1 -iminoethyl)amino]-5-heptenoic acid;
- iNOS inducible nitric oxide synthase
- an acetylcholine esterase inhibitor such as donepezil
- a dopamine type 2 (D2) antagonist such as ziprazidone
- an prostaglandin E 2 subtype 4 (EP4) antagonist such as /V-[( ⁇ 2-[4-(2-ethyl- 4,6-dimethyl-1 H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl ⁇ amino)carbonyl]-4- methylbenzenesulfonamide or 4-[(1 S)-1-( ⁇ [5-chloro-2-(3- fluorophenoxy)pyridin-3-yl]carbonyl ⁇ amino)ethyl]benzoic acid;
- two or more pharmaceutical compositions may conveniently be combined in the form of a kit suitable for co-administration of the compositions.
- Such a kit comprises two or more separate pharmaceutical compositions, at least one of which contains an alpha-2-delta receptor antagonist, particularly a compound of formula (I), and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- an alpha-2-delta receptor antagonist particularly a compound of formula (I)
- means for separately retaining said compositions such as a container, divided bottle, or divided foil packet.
- An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
- kit is particularly suitable for administering different dosage forms, for example, oral and parenteral formulations, for administering separate compositions at different dosage intervals, or for titrating separate compositions against one another.
- the kit typically comprises directions for administration and may be provided with a so-called memory aid.
- a pharmaceutical composition including a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, together with a pharmaceutically acceptable excipient;
- a compound of formula (I) or a pharmaceutically acceptable salt, solvate or composition thereof, for use as a medicament for use as a medicament;
- TLC thin layer chromatography
- the tetrahydropyrimidinone of Preparation 8 (144mg, 0.34mmol) was suspended in 6M aqueous hydrochloric acid (15ml) and the reaction mixture was heated at reflux for twenty four hours. Upon cooling, the reaction was diluted with water (10ml) and then extracted with ethyl acetate (2x25ml). The organic phases were discarded and the aqueous layer was concentrated under reduced pressure to provide the title compound (58mg, 62%) as a pale brown solid, containing methylamine hydrochloride as an impurity (1 :1 ratio).
- the tetrahydropyrimidinone of Preparation 15 (200mg, 0.45mmol) was suspended in 6M aqueous hydrochloric acid (20ml) and dioxane (3ml). The reaction mixture was heated at reflux for 18 hours. The cooled mixture was concentrated under reduced pressure and the residue was dissolved in water and filtered. The filtrate was extracted with ethyl acetate. The organic phases were discarded and the aqueous layer was concentrated under reduced pressure. The crude product was then purified by ion-exchange chromatography eluting with water then water:ammonia:methanol (60:20:20, by volume) to provide the title compound (71 mg, 61 %) as a pale white solid.
- the title compound was prepared by a similar method to that of Example 1 using the compound of Preparation 16 as starting material.
- n-Butyl lithium (2.5M in hexanes, 6.4ml, 16.0mmol) was added dropwise over 10 minutes at 0 0 C to a solution of the bromobenzene of Preparation 4 (3.11 g, 14.6mmol) in diethyl ether (30ml).
- the reaction mixture was stirred at O 0 C for 15 minutes and then dry N,N-dimethylformamide (1.13ml, 14.6mmol) was added at O 0 C.
- the reaction was stirred at O 0 C for ten minutes, at room temperature for ten minutes and then heated under reflux for four hours. After cooling, the reaction was quenched with water (25ml).
- the organic phase was separated and the aqueous phase was extracted with more diethyl ether (25ml). The organic phases were combined, washed with water (25ml), dried over magnesium sulphate and concentrated under reduced pressure.
- the crude product was purified by flash chromatography on silica gel eluting with ethyl acetate: heptane (0:100 to 10:90, by volume) to provide the title compound (1.35g, 57%) as a yellow oil.
- the reaction mixture was stirred at -78°C for eighty minutes and then the benzyl bromide of Preparation 7 (199mg, 0.875mmol) was added at -78°C.
- the reaction mixture was stirred at -78 0 C for two hours, then warmed to room temperature over three and a half hours and stirred at room temperature for thirty minutes.
- the reaction was quenched by addition of an aqueous ammonium chloride solution.
- the mixture was stirred at room temperature for 18 hours and then concentrated under reduced pressure.
- the aqueous phase was extracted with dichloromethane.
- the dichloromethane layer was filtered through a hydrophobic membrane and concentrated under reduced 1 pressure.
- the crude product was purified by flash chromatography on silica gel eluting with ethyl acetate:heptane (0:100 to 60:40, by volume) to provide the title compound (149mg, 49%) as a yellow solid.
- Lithium diisopropylamide (1.5M in tetrahydrofuran, 4.11 ml, 6.2mmol) was added dropwise at -78°C, under nitrogen, to a solution of the tetrahydropyrimidinone of Preparation 14 (2.05g, 4.75mmol) in tetrahydrofuran (40ml).
- the reaction mixture was stirred at -60 0 C for three hours. It was then quenched by addition of an aqueous ammonium chloride solution. The mixture was stirred at room temperature and then concentrated under reduced pressure. The reaction was extracted with ethyl acetate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
































Claims

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008509534A JP2008542198A (en) | 2005-05-06 | 2006-04-24 | β-amino acid derivatives |
| EP06744676A EP1883620A1 (en) | 2005-05-06 | 2006-04-24 | ß-AMINO ACID DERIVATIVES |
| CA002606254A CA2606254A1 (en) | 2005-05-06 | 2006-04-24 | .beta.-amino acid derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US67852305P | 2005-05-06 | 2005-05-06 | |
| US60/678,523 | 2005-05-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006120544A1 true WO2006120544A1 (en) | 2006-11-16 |
Family
ID=36960801
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/001209 WO2006120544A1 (en) | 2005-05-06 | 2006-04-24 | β-AMINO ACID DERIVATIVES |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1883620A1 (en) |
| JP (1) | JP2008542198A (en) |
| CA (1) | CA2606254A1 (en) |
| WO (1) | WO2006120544A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2025672A1 (en) * | 2006-05-31 | 2009-02-18 | Asubio Pharma Co., Ltd. | Seven-membered ring compound, production method thereof and pharmaceutical use thereof |
| JP2011502983A (en) * | 2007-11-01 | 2011-01-27 | アキュセラ インコーポレイテッド | Amine derivative compounds for the treatment of eye diseases and disorders |
| US9096558B2 (en) | 2010-07-09 | 2015-08-04 | Pfizer Limited | N-sulfonylbenzamide compounds |
| IT202100011237A1 (en) * | 2021-05-03 | 2022-11-03 | Univ Degli Studi Di Torino | NLRP3 INFLAMMASOME INHIBITOR COMPOUNDS AND THEIR USE |
-
2006
- 2006-04-24 JP JP2008509534A patent/JP2008542198A/en not_active Withdrawn
- 2006-04-24 EP EP06744676A patent/EP1883620A1/en not_active Withdrawn
- 2006-04-24 WO PCT/IB2006/001209 patent/WO2006120544A1/en not_active Application Discontinuation
- 2006-04-24 CA CA002606254A patent/CA2606254A1/en not_active Abandoned
Non-Patent Citations (16)
| Title |
|---|
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399446, retrieved from XFIRE Database accession no. BNR 10158765 * |
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399447, retrieved from XFIRE Database accession no. BRN 6399363 * |
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399448, retrieved from XFIRE Database accession no. BRN 6513133 * |
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399449, retrieved from XFIRE Database accession no. BRN 8260461 * |
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399450, retrieved from XFIRE Database accession no. 5377846 * |
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399451, retrieved from XFIRE Database accession no. BRN 3279771 * |
| DATABASE BEILSTEIN Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; XP002399452, retrieved from XFIRE Database accession no. BRN 2807401 * |
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; SHEN, TAO ET AL: "Preparation of .alpha.-substituted .beta.-amino acid by one step hydrogenation method", XP002399453, retrieved from STN Database accession no. 142:447408 * |
| EUR J ORG CHEM, vol. 1, 1999, pages 335 - 360 * |
| J MED CHEM, vol. 32, no. 7, 1989, pages 1497 - 1503 * |
| J MED CHEM, vol. 32, no. 7, 1989, pages 1607 - 1611 * |
| J ORG CHEM, vol. 22, 1957, pages 1521 - 1526 * |
| J ORG CHEM, vol. 64, no. 9, 1999, pages 3060 - 3065 * |
| ORG LETT, vol. 13, 2005, pages 2571 - 2573 * |
| PHYTOCHEMISTRY, vol. 27, no. 3, 1988, pages 711 - 714 * |
| VAUGHT J L ET AL: "A COMPARISON OF THE ANTINOCICEPTIVE RESPONSES TO THE GABA-RECEPTOR AGONISTS THIP AND BACLOFEN", NEUROPHARMACOLOGY, PERGAMON PRESS, OXFORD, GB, vol. 24, no. 3, March 1985 (1985-03-01), pages 211 - 216, XP008037569, ISSN: 0028-3908 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2025672A1 (en) * | 2006-05-31 | 2009-02-18 | Asubio Pharma Co., Ltd. | Seven-membered ring compound, production method thereof and pharmaceutical use thereof |
| EP2025672A4 (en) * | 2006-05-31 | 2009-12-09 | Asubio Pharma Co Ltd | Seven-membered ring compound, production method thereof and pharmaceutical use thereof |
| US8049006B2 (en) | 2006-05-31 | 2011-11-01 | Daiichi Sankyo Company, Limited | 7-membered ring compound and method of production and pharmaceutical application thereof |
| JP2011502983A (en) * | 2007-11-01 | 2011-01-27 | アキュセラ インコーポレイテッド | Amine derivative compounds for the treatment of eye diseases and disorders |
| US8716529B2 (en) | 2007-11-01 | 2014-05-06 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| US9056849B2 (en) | 2007-11-01 | 2015-06-16 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| US9452153B2 (en) | 2007-11-01 | 2016-09-27 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| US9096558B2 (en) | 2010-07-09 | 2015-08-04 | Pfizer Limited | N-sulfonylbenzamide compounds |
| IT202100011237A1 (en) * | 2021-05-03 | 2022-11-03 | Univ Degli Studi Di Torino | NLRP3 INFLAMMASOME INHIBITOR COMPOUNDS AND THEIR USE |
| WO2022234447A1 (en) * | 2021-05-03 | 2022-11-10 | Universita' Degli Studi Di Torino | Nlrp3 inflammasome-inhibiting compounds and the use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1883620A1 (en) | 2008-02-06 |
| CA2606254A1 (en) | 2006-11-16 |
| JP2008542198A (en) | 2008-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20070191462A1 (en) | Combination of A 5-HT(1) Receptor Agonist and an Alpha-2-Delta Ligand for the Treatment of Migraine | |
| JP4555263B2 (en) | Proline derivative having affinity for calcium channel α2δ subunit | |
| MX2007000885A (en) | Pyridine derivatives. | |
| CA2606353C (en) | Alpha-methyl amino acid derivatives | |
| JP2007533723A (en) | Combinations containing α2δ ligands | |
| WO2012120398A1 (en) | Aryl substituted carboxamide derivatives as trpm8 modulators | |
| US7053122B2 (en) | Therapeutic use of aryl amino acid derivatives | |
| WO2006120544A1 (en) | β-AMINO ACID DERIVATIVES | |
| US20090227680A1 (en) | Amino Acid Derivatives | |
| US20040092498A1 (en) | Substituted glycine derivatives for use as medicaments | |
| US20050038047A1 (en) | Azaquinazoline derivatives | |
| WO2004016583A1 (en) | Substituted glycine derivatives for use as medicaments | |
| WO2007057756A2 (en) | Isocystene derivatives for the treatment of pain | |
| WO2004014357A2 (en) | Therapeutic use of aryl amino acid derivatives | |
| WO2007052134A1 (en) | (2s)-2-aminomethyl-5-ethyl heptanoic acid its pharmaceutical use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2606254 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008509534 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006744676 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006744676 Country of ref document: EP |