WO2006044497A2 - Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzhermer’s disease - Google Patents
Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzhermer’s disease Download PDFInfo
- Publication number
- WO2006044497A2 WO2006044497A2 PCT/US2005/036752 US2005036752W WO2006044497A2 WO 2006044497 A2 WO2006044497 A2 WO 2006044497A2 US 2005036752 W US2005036752 W US 2005036752W WO 2006044497 A2 WO2006044497 A2 WO 2006044497A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- optionally substituted
- halogen
- aryl
- cyano
- Prior art date
Links
- 238000011282 treatment Methods 0.000 title abstract description 16
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title abstract description 15
- 201000010099 disease Diseases 0.000 title abstract description 13
- JUXAVSAMVBLDKO-UHFFFAOYSA-N 1-(1-azabicyclo[2.2.2]octan-3-yl)-3-[3-(1h-indol-3-yl)-1-oxo-1-spiro[1,2-dihydroindene-3,4'-piperidine]-1'-ylpropan-2-yl]urea Chemical class C1N(CC2)CCC2C1NC(=O)NC(C(=O)N1CCC2(C3=CC=CC=C3CC2)CC1)CC1=CNC2=CC=CC=C12 JUXAVSAMVBLDKO-UHFFFAOYSA-N 0.000 title abstract description 5
- 239000002439 beta secretase inhibitor Substances 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 135
- 239000000203 mixture Substances 0.000 claims abstract description 48
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 claims abstract description 25
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 claims abstract description 25
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 19
- 208000024827 Alzheimer disease Diseases 0.000 claims abstract description 13
- 125000000217 alkyl group Chemical group 0.000 claims description 287
- 150000002367 halogens Chemical class 0.000 claims description 168
- 229910052736 halogen Inorganic materials 0.000 claims description 137
- 125000003118 aryl group Chemical group 0.000 claims description 97
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 83
- -1 wherein said alky Chemical group 0.000 claims description 69
- 125000001072 heteroaryl group Chemical group 0.000 claims description 67
- 238000000034 method Methods 0.000 claims description 52
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 45
- 150000003839 salts Chemical class 0.000 claims description 45
- 125000002837 carbocyclic group Chemical group 0.000 claims description 39
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 33
- 239000001257 hydrogen Substances 0.000 claims description 30
- 229910052739 hydrogen Inorganic materials 0.000 claims description 30
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 28
- 229910052757 nitrogen Inorganic materials 0.000 claims description 23
- 229910052717 sulfur Inorganic materials 0.000 claims description 22
- 125000005842 heteroatom Chemical group 0.000 claims description 18
- 230000000694 effects Effects 0.000 claims description 16
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 16
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims description 15
- 239000003814 drug Substances 0.000 claims description 15
- 125000000304 alkynyl group Chemical group 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 10
- 125000003342 alkenyl group Chemical group 0.000 claims description 9
- 125000005843 halogen group Chemical group 0.000 claims description 9
- 238000004519 manufacturing process Methods 0.000 claims description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 8
- 230000002401 inhibitory effect Effects 0.000 claims description 7
- 241001465754 Metazoa Species 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 5
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 4
- 239000008024 pharmaceutical diluent Substances 0.000 claims description 2
- 239000003112 inhibitor Substances 0.000 abstract description 13
- JRPDANVNRUIUAB-CMDGGOBGSA-N (e)-dec-3-en-2-one Chemical compound CCCCCC\C=C\C(C)=O JRPDANVNRUIUAB-CMDGGOBGSA-N 0.000 description 132
- 239000001178 (E)-dec-3-en-2-one Substances 0.000 description 108
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 108
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 100
- 239000000243 solution Substances 0.000 description 94
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 78
- 238000006243 chemical reaction Methods 0.000 description 59
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 55
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 50
- 239000000047 product Substances 0.000 description 47
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 46
- 235000019439 ethyl acetate Nutrition 0.000 description 44
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 44
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 41
- 239000007787 solid Substances 0.000 description 41
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 38
- 238000004949 mass spectrometry Methods 0.000 description 37
- 238000005160 1H NMR spectroscopy Methods 0.000 description 36
- 239000012267 brine Substances 0.000 description 31
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 31
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 30
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 29
- 238000003756 stirring Methods 0.000 description 28
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 24
- 239000011541 reaction mixture Substances 0.000 description 23
- 125000001424 substituent group Chemical group 0.000 description 23
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 21
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 21
- 239000000126 substance Substances 0.000 description 21
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 20
- 229940093499 ethyl acetate Drugs 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 239000007832 Na2SO4 Substances 0.000 description 19
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 19
- 239000000543 intermediate Substances 0.000 description 19
- 239000003921 oil Substances 0.000 description 19
- 229910052938 sodium sulfate Inorganic materials 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 150000002081 enamines Chemical group 0.000 description 18
- 150000002466 imines Chemical group 0.000 description 18
- 235000019198 oils Nutrition 0.000 description 18
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 18
- 125000004432 carbon atom Chemical group C* 0.000 description 17
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 17
- 239000000725 suspension Substances 0.000 description 17
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 17
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 16
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 16
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 16
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 239000004480 active ingredient Substances 0.000 description 15
- 239000012044 organic layer Substances 0.000 description 15
- 238000000746 purification Methods 0.000 description 15
- GVRWIAHBVAYKIZ-UHFFFAOYSA-N dec-3-ene Chemical compound CCCCCCC=CCC GVRWIAHBVAYKIZ-UHFFFAOYSA-N 0.000 description 14
- 239000002552 dosage form Substances 0.000 description 13
- 235000019341 magnesium sulphate Nutrition 0.000 description 13
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- 239000000758 substrate Substances 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- 229940079593 drug Drugs 0.000 description 11
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 11
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 11
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- 238000007792 addition Methods 0.000 description 10
- 238000003818 flash chromatography Methods 0.000 description 10
- 238000004128 high performance liquid chromatography Methods 0.000 description 10
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 10
- 102100021257 Beta-secretase 1 Human genes 0.000 description 9
- 102000004190 Enzymes Human genes 0.000 description 9
- 108090000790 Enzymes Proteins 0.000 description 9
- 101000894895 Homo sapiens Beta-secretase 1 Proteins 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- 229910052799 carbon Inorganic materials 0.000 description 9
- 229940088598 enzyme Drugs 0.000 description 9
- 239000000284 extract Substances 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- UGODCLHJOJPPHP-AZGWGOJFSA-J tetralithium;[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-4-hydroxy-2-[[oxido(sulfonatooxy)phosphoryl]oxymethyl]oxolan-3-yl] phosphate;hydrate Chemical compound [Li+].[Li+].[Li+].[Li+].O.C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OS([O-])(=O)=O)[C@@H](OP([O-])([O-])=O)[C@H]1O UGODCLHJOJPPHP-AZGWGOJFSA-J 0.000 description 9
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 8
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- 238000006069 Suzuki reaction reaction Methods 0.000 description 8
- 229960000583 acetic acid Drugs 0.000 description 8
- 125000003545 alkoxy group Chemical group 0.000 description 8
- 150000001721 carbon Chemical group 0.000 description 8
- 239000004615 ingredient Substances 0.000 description 8
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 7
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 125000004122 cyclic group Chemical group 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 238000002953 preparative HPLC Methods 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- YNJCFDAODGKHAV-UHFFFAOYSA-N tert-butyl n-(2-hydroxypropyl)carbamate Chemical compound CC(O)CNC(=O)OC(C)(C)C YNJCFDAODGKHAV-UHFFFAOYSA-N 0.000 description 7
- IAVREABSGIHHMO-UHFFFAOYSA-N 3-hydroxybenzaldehyde Chemical compound OC1=CC=CC(C=O)=C1 IAVREABSGIHHMO-UHFFFAOYSA-N 0.000 description 6
- CLSHIGIZZCVNHG-UHFFFAOYSA-N 4-(cyclohexylamino)-1-(3-fluorophenyl)-1,3,8-triazaspiro[4.5]dec-3-en-2-one Chemical compound FC1=CC=CC(N2C3(CCNCC3)C(NC3CCCCC3)=NC2=O)=C1 CLSHIGIZZCVNHG-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical group CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- PAFZNILMFXTMIY-UHFFFAOYSA-N Cyclohexylamine Natural products NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- WTVNFKVGGUWHQC-UHFFFAOYSA-N decane-2,4-dione Chemical compound CCCCCCC(=O)CC(C)=O WTVNFKVGGUWHQC-UHFFFAOYSA-N 0.000 description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 238000001819 mass spectrum Methods 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- FSDNTQSJGHSJBG-UHFFFAOYSA-N piperidine-4-carbonitrile Chemical compound N#CC1CCNCC1 FSDNTQSJGHSJBG-UHFFFAOYSA-N 0.000 description 6
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 6
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 5
- 241000124008 Mammalia Species 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 150000001299 aldehydes Chemical class 0.000 description 5
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 5
- 239000013058 crude material Substances 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- YNTOKMNHRPSGFU-UHFFFAOYSA-N n-Propyl carbamate Chemical compound CCCOC(N)=O YNTOKMNHRPSGFU-UHFFFAOYSA-N 0.000 description 5
- 235000011056 potassium acetate Nutrition 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 238000004007 reversed phase HPLC Methods 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 4
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 description 4
- ZQAUMGJFUOYCJJ-UHFFFAOYSA-N 4-(cyclohexylamino)-1-(3-fluorophenyl)-8-[[3-(4-methylpyridin-3-yl)phenyl]methyl]-1,3,8-triazaspiro[4.5]dec-3-en-2-one Chemical compound CC1=CC=NC=C1C1=CC=CC(CN2CCC3(CC2)C(=NC(=O)N3C=2C=C(F)C=CC=2)NC2CCCCC2)=C1 ZQAUMGJFUOYCJJ-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 229910002666 PdCl2 Inorganic materials 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 4
- 239000012346 acetyl chloride Substances 0.000 description 4
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 4
- 229910052782 aluminium Inorganic materials 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 230000001276 controlling effect Effects 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 4
- 230000007170 pathology Effects 0.000 description 4
- SRJOCJYGOFTFLH-UHFFFAOYSA-M piperidine-4-carboxylate Chemical compound [O-]C(=O)C1CCNCC1 SRJOCJYGOFTFLH-UHFFFAOYSA-M 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 125000004076 pyridyl group Chemical group 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000011369 resultant mixture Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 0 *N(CC1)CCC1(C(*C1=*)=N*)N1c1ccccc1 Chemical compound *N(CC1)CCC1(C(*C1=*)=N*)N1c1ccccc1 0.000 description 3
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 3
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 3
- WXJFOVDAJMFUBC-UHFFFAOYSA-N 8-[(6-aminopyridin-3-yl)methyl]-4-(cyclohexylamino)-1-(3-fluorophenyl)-1,3,8-triazaspiro[4.5]dec-3-en-2-one Chemical compound C1=NC(N)=CC=C1CN1CCC2(C(=NC(=O)N2C=2C=C(F)C=CC=2)NC2CCCCC2)CC1 WXJFOVDAJMFUBC-UHFFFAOYSA-N 0.000 description 3
- XCCDAZBQOATOMG-UHFFFAOYSA-N 8-[[6-(benzylamino)pyridin-3-yl]methyl]-4-(cyclohexylamino)-1-(3-fluorophenyl)-1,3,8-triazaspiro[4.5]dec-3-en-2-one Chemical compound FC1=CC=CC(N2C3(CCN(CC=4C=NC(NCC=5C=CC=CC=5)=CC=4)CC3)C(NC3CCCCC3)=NC2=O)=C1 XCCDAZBQOATOMG-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 206010012289 Dementia Diseases 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- 238000007059 Strecker synthesis reaction Methods 0.000 description 3
- HMGYUQMBMLLQCL-UHFFFAOYSA-N [3-[[4-(cyclohexylamino)-1-(3-fluorophenyl)-2-oxo-1,3,8-triazaspiro[4.5]dec-3-en-8-yl]methyl]phenyl]boronic acid Chemical compound OB(O)C1=CC=CC(CN2CCC3(CC2)C(=NC(=O)N3C=2C=C(F)C=CC=2)NC2CCCCC2)=C1 HMGYUQMBMLLQCL-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 230000010933 acylation Effects 0.000 description 3
- 238000005917 acylation reaction Methods 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 150000001502 aryl halides Chemical class 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- WRJWRGBVPUUDLA-UHFFFAOYSA-N chlorosulfonyl isocyanate Chemical compound ClS(=O)(=O)N=C=O WRJWRGBVPUUDLA-UHFFFAOYSA-N 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- ZGEBASBUEPAQLU-UHFFFAOYSA-N ethyl 3-(n-(1-benzyl-4-cyanopiperidin-4-yl)-3-fluoroanilino)-3-oxopropanoate Chemical compound C1CN(CC=2C=CC=CC=2)CCC1(C#N)N(C(=O)CC(=O)OCC)C1=CC=CC(F)=C1 ZGEBASBUEPAQLU-UHFFFAOYSA-N 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 125000002541 furyl group Chemical group 0.000 description 3
- 239000008241 heterogeneous mixture Substances 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- IXHBTMCLRNMKHZ-LBPRGKRZSA-N levobunolol Chemical compound O=C1CCCC2=C1C=CC=C2OC[C@@H](O)CNC(C)(C)C IXHBTMCLRNMKHZ-LBPRGKRZSA-N 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- HNQIVZYLYMDVSB-NJFSPNSNSA-N methanesulfonamide Chemical compound [14CH3]S(N)(=O)=O HNQIVZYLYMDVSB-NJFSPNSNSA-N 0.000 description 3
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 3
- 235000019799 monosodium phosphate Nutrition 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 3
- 239000002464 receptor antagonist Substances 0.000 description 3
- 229940044551 receptor antagonist Drugs 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 3
- 229910052979 sodium sulfide Inorganic materials 0.000 description 3
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- MGRVRXRGTBOSHW-UHFFFAOYSA-N (aminomethyl)phosphonic acid Chemical compound NCP(O)(O)=O MGRVRXRGTBOSHW-UHFFFAOYSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- KDDVZGFLGMXKRC-UHFFFAOYSA-N 1-(bromomethyl)-3-propan-2-yloxybenzene Chemical compound CC(C)OC1=CC=CC(CBr)=C1 KDDVZGFLGMXKRC-UHFFFAOYSA-N 0.000 description 2
- KVYKDNGUEZRPGJ-UHFFFAOYSA-N 1-Aminohydantoin Chemical compound NN1CC(=O)NC1=O KVYKDNGUEZRPGJ-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- LPZOCVVDSHQFST-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-3-ethylpyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2)CC LPZOCVVDSHQFST-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- RBQOILDHZGNUQT-UHFFFAOYSA-N 2-bromo-6-propan-2-yloxypyridine Chemical compound CC(C)OC1=CC=CC(Br)=N1 RBQOILDHZGNUQT-UHFFFAOYSA-N 0.000 description 2
- IPOVOSHRRIJKBR-UHFFFAOYSA-N 2-ethylpropanedioyl dichloride Chemical compound CCC(C(Cl)=O)C(Cl)=O IPOVOSHRRIJKBR-UHFFFAOYSA-N 0.000 description 2
- KDSNLYIMUZNERS-UHFFFAOYSA-N 2-methylpropanamine Chemical compound CC(C)CN KDSNLYIMUZNERS-UHFFFAOYSA-N 0.000 description 2
- VSNCJGMWIFUZRG-UHFFFAOYSA-N 3-(3-bromophenyl)-2-methylpropanenitrile Chemical compound N#CC(C)CC1=CC=CC(Br)=C1 VSNCJGMWIFUZRG-UHFFFAOYSA-N 0.000 description 2
- PBKACPWKVGTMTR-UHFFFAOYSA-N 3-(3-formylphenyl)-2-methylpropanenitrile Chemical compound N#CC(C)CC1=CC=CC(C=O)=C1 PBKACPWKVGTMTR-UHFFFAOYSA-N 0.000 description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 208000037259 Amyloid Plaque Diseases 0.000 description 2
- 101710137189 Amyloid-beta A4 protein Proteins 0.000 description 2
- 101710151993 Amyloid-beta precursor protein Proteins 0.000 description 2
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 208000005145 Cerebral amyloid angiopathy Diseases 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- 229910004373 HOAc Inorganic materials 0.000 description 2
- 238000010268 HPLC based assay Methods 0.000 description 2
- 108090000144 Human Proteins Proteins 0.000 description 2
- 102000003839 Human Proteins Human genes 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 2
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- LXNAVEXFUKBNMK-UHFFFAOYSA-N acetic acid;palladium Chemical compound [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- 150000003927 aminopyridines Chemical class 0.000 description 2
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 150000001499 aryl bromides Chemical class 0.000 description 2
- 125000002393 azetidinyl group Chemical group 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 239000010779 crude oil Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 238000006114 decarboxylation reaction Methods 0.000 description 2
- 229960000958 deferoxamine Drugs 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 2
- 238000002451 electron ionisation mass spectrometry Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- WGAVNVOCOWOTSM-UHFFFAOYSA-N ethyl 4-(n-acetyl-3-fluoroanilino)-1-benzylpiperidine-4-carboxylate Chemical compound C1CC(C(=O)OCC)(N(C(C)=O)C=2C=C(F)C=CC=2)CCN1CC1=CC=CC=C1 WGAVNVOCOWOTSM-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000005414 inactive ingredient Substances 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 150000002527 isonitriles Chemical class 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 229940057995 liquid paraffin Drugs 0.000 description 2
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 2
- MKUWVMRNQOOSAT-UHFFFAOYSA-N methylvinylmethanol Natural products CC(O)C=C MKUWVMRNQOOSAT-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- XUWHAWMETYGRKB-UHFFFAOYSA-N piperidin-2-one Chemical class O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical compound [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000002797 proteolythic effect Effects 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 229940124530 sulfonamide Drugs 0.000 description 2
- 150000003456 sulfonamides Chemical class 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 238000006177 thiolation reaction Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- QHFAXRHEKNHTDH-UHFFFAOYSA-N (2-ethenylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC=C1C=C QHFAXRHEKNHTDH-UHFFFAOYSA-N 0.000 description 1
- HJBGZJMKTOMQRR-UHFFFAOYSA-N (3-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(C=O)=C1 HJBGZJMKTOMQRR-UHFFFAOYSA-N 0.000 description 1
- RUGUAVWZSRINHW-UHFFFAOYSA-N (3-propan-2-yloxyphenyl)methanol Chemical compound CC(C)OC1=CC=CC(CO)=C1 RUGUAVWZSRINHW-UHFFFAOYSA-N 0.000 description 1
- VDUKDQTYMWUSAC-UHFFFAOYSA-N (4-methylsulfonylphenyl)boronic acid Chemical compound CS(=O)(=O)C1=CC=C(B(O)O)C=C1 VDUKDQTYMWUSAC-UHFFFAOYSA-N 0.000 description 1
- 125000006652 (C3-C12) cycloalkyl group Chemical group 0.000 description 1
- SYTBZMRGLBWNTM-SNVBAGLBSA-N (R)-flurbiprofen Chemical compound FC1=CC([C@H](C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-SNVBAGLBSA-N 0.000 description 1
- 230000006269 (delayed) early viral mRNA transcription Effects 0.000 description 1
- XNJAYQHWXYJBBD-UHFFFAOYSA-N 1,4-difluoro-2-nitrobenzene Chemical compound [O-][N+](=O)C1=CC(F)=CC=C1F XNJAYQHWXYJBBD-UHFFFAOYSA-N 0.000 description 1
- HMUNWXXNJPVALC-UHFFFAOYSA-N 1-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C(CN1CC2=C(CC1)NN=N2)=O HMUNWXXNJPVALC-UHFFFAOYSA-N 0.000 description 1
- ZYEZCOWVFGHFHO-UHFFFAOYSA-N 1-benzyl-4-(3-fluoroanilino)piperidine-4-carbonitrile Chemical compound FC1=CC=CC(NC2(CCN(CC=3C=CC=CC=3)CC2)C#N)=C1 ZYEZCOWVFGHFHO-UHFFFAOYSA-N 0.000 description 1
- MLEGMEBCXGDFQT-UHFFFAOYSA-N 1-benzylpiperidin-2-one Chemical compound O=C1CCCCN1CC1=CC=CC=C1 MLEGMEBCXGDFQT-UHFFFAOYSA-N 0.000 description 1
- SJZKULRDWHPHGG-UHFFFAOYSA-N 1-benzylpiperidin-4-one Chemical compound C1CC(=O)CCN1CC1=CC=CC=C1 SJZKULRDWHPHGG-UHFFFAOYSA-N 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- 125000004201 2,4-dichlorophenyl group Chemical group [H]C1=C([H])C(*)=C(Cl)C([H])=C1Cl 0.000 description 1
- FEYDZHNIIMENOB-UHFFFAOYSA-N 2,6-dibromopyridine Chemical compound BrC1=CC=CC(Br)=N1 FEYDZHNIIMENOB-UHFFFAOYSA-N 0.000 description 1
- YBAWYTYNMZWMMJ-UHFFFAOYSA-N 2-(6-fluoro-1h-indol-3-yl)-n-[[3-(2,2,3,3-tetrafluoropropoxy)phenyl]methyl]ethanamine Chemical compound FC(F)C(F)(F)COC1=CC=CC(CNCCC=2C3=CC=C(F)C=C3NC=2)=C1 YBAWYTYNMZWMMJ-UHFFFAOYSA-N 0.000 description 1
- FTZILAQGHINQQR-UHFFFAOYSA-N 2-Methylpentanal Chemical compound CCCC(C)C=O FTZILAQGHINQQR-UHFFFAOYSA-N 0.000 description 1
- VWVRASTUFJRTHW-UHFFFAOYSA-N 2-[3-(azetidin-3-yloxy)-4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound O=C(CN1C=C(C(OC2CNC2)=N1)C1=CN=C(NC2CC3=C(C2)C=CC=C3)N=C1)N1CCC2=C(C1)N=NN2 VWVRASTUFJRTHW-UHFFFAOYSA-N 0.000 description 1
- NPHULPIAPWNOOH-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-3-(2,3-dihydroindol-1-ylmethyl)pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2)CN1CCC2=CC=CC=C12 NPHULPIAPWNOOH-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- UMUPQWIGCOZEOY-JOCHJYFZSA-N 2-amino-2-methyl-n-[(2r)-1-(1-methylsulfonylspiro[2h-indole-3,4'-piperidine]-1'-yl)-1-oxo-3-phenylmethoxypropan-2-yl]propanamide Chemical compound C([C@@H](NC(=O)C(C)(N)C)C(=O)N1CCC2(C3=CC=CC=C3N(C2)S(C)(=O)=O)CC1)OCC1=CC=CC=C1 UMUPQWIGCOZEOY-JOCHJYFZSA-N 0.000 description 1
- DUGMCDWNXXFHDE-VZYDHVRKSA-N 2-amino-2-methyl-n-[(2r)-1-(1-methylsulfonylspiro[2h-indole-3,4'-piperidine]-1'-yl)-1-oxo-3-phenylmethoxypropan-2-yl]propanamide;methanesulfonic acid Chemical compound CS(O)(=O)=O.C([C@@H](NC(=O)C(C)(N)C)C(=O)N1CCC2(C3=CC=CC=C3N(C2)S(C)(=O)=O)CC1)OCC1=CC=CC=C1 DUGMCDWNXXFHDE-VZYDHVRKSA-N 0.000 description 1
- YLMFXCIATJJKQL-UHFFFAOYSA-N 2-bromo-4-fluoroaniline Chemical compound NC1=CC=C(F)C=C1Br YLMFXCIATJJKQL-UHFFFAOYSA-N 0.000 description 1
- FWTXFEKVHSFTDQ-UHFFFAOYSA-N 2-bromo-5-fluoroaniline Chemical compound NC1=CC(F)=CC=C1Br FWTXFEKVHSFTDQ-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000006179 2-methyl benzyl group Chemical group [H]C1=C([H])C(=C(C([H])=C1[H])C([H])([H])*)C([H])([H])[H] 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- UGWULZWUXSCWPX-UHFFFAOYSA-N 2-sulfanylideneimidazolidin-4-one Chemical compound O=C1CNC(=S)N1 UGWULZWUXSCWPX-UHFFFAOYSA-N 0.000 description 1
- 125000004189 3,4-dichlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(Cl)C([H])=C1* 0.000 description 1
- KLAIOABSDQUNSA-WUKNDPDISA-N 3-[(e)-octadec-2-enyl]oxolane-2,5-dione Chemical compound CCCCCCCCCCCCCCC\C=C\CC1CC(=O)OC1=O KLAIOABSDQUNSA-WUKNDPDISA-N 0.000 description 1
- NJXPYZHXZZCTNI-UHFFFAOYSA-N 3-aminobenzonitrile Chemical compound NC1=CC=CC(C#N)=C1 NJXPYZHXZZCTNI-UHFFFAOYSA-N 0.000 description 1
- ZBMZOFSLQIPSPW-UHFFFAOYSA-N 3-bis(3-sulfophenyl)phosphanylbenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC(P(C=2C=C(C=CC=2)S(O)(=O)=O)C=2C=C(C=CC=2)S(O)(=O)=O)=C1 ZBMZOFSLQIPSPW-UHFFFAOYSA-N 0.000 description 1
- GSQZOLXWFQQJHJ-UHFFFAOYSA-N 3-bromo-4-methylpyridine Chemical compound CC1=CC=NC=C1Br GSQZOLXWFQQJHJ-UHFFFAOYSA-N 0.000 description 1
- SUISZCALMBHJQX-UHFFFAOYSA-N 3-bromobenzaldehyde Chemical compound BrC1=CC=CC(C=O)=C1 SUISZCALMBHJQX-UHFFFAOYSA-N 0.000 description 1
- CQZIEDXCLQOOEH-UHFFFAOYSA-N 3-bromopropanenitrile Chemical compound BrCCC#N CQZIEDXCLQOOEH-UHFFFAOYSA-N 0.000 description 1
- BHZZWPQWHIIUTB-UHFFFAOYSA-N 3-but-3-en-2-yloxybenzaldehyde Chemical compound C=CC(C)OC1=CC=CC(C=O)=C1 BHZZWPQWHIIUTB-UHFFFAOYSA-N 0.000 description 1
- GPKDGVXBXQTHRY-UHFFFAOYSA-N 3-chloropropane-1-sulfonyl chloride Chemical compound ClCCCS(Cl)(=O)=O GPKDGVXBXQTHRY-UHFFFAOYSA-N 0.000 description 1
- QZVQQUVWFIZUBQ-UHFFFAOYSA-N 3-fluoroaniline Chemical compound NC1=CC=CC(F)=C1 QZVQQUVWFIZUBQ-UHFFFAOYSA-N 0.000 description 1
- MNMXMPLRBXECRC-UHFFFAOYSA-N 3-fluoroaniline;hydrochloride Chemical compound Cl.NC1=CC=CC(F)=C1 MNMXMPLRBXECRC-UHFFFAOYSA-N 0.000 description 1
- 125000006284 3-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(F)=C1[H])C([H])([H])* 0.000 description 1
- NOBKCEXLDDGYID-UHFFFAOYSA-N 3-propan-2-yloxybenzaldehyde Chemical compound CC(C)OC1=CC=CC(C=O)=C1 NOBKCEXLDDGYID-UHFFFAOYSA-N 0.000 description 1
- ZOFYDMUYCAGUPJ-UHFFFAOYSA-N 4,5-diethyl-1H-pyrrole-2,3-dicarboxylic acid Chemical compound CCC=1NC(C(O)=O)=C(C(O)=O)C=1CC ZOFYDMUYCAGUPJ-UHFFFAOYSA-N 0.000 description 1
- RGYXZPFFINZWKN-UHFFFAOYSA-N 4-(3-fluorophenyl)-1-[(3-propan-2-yloxyphenyl)methyl]piperidine-4-carbonitrile Chemical compound CC(C)OC1=CC=CC(CN2CCC(CC2)(C#N)C=2C=C(F)C=CC=2)=C1 RGYXZPFFINZWKN-UHFFFAOYSA-N 0.000 description 1
- XLGLUVXJCDPUGS-UHFFFAOYSA-N 4-(cyclohexylamino)-1-(3-fluorophenyl)-8-[[3-(2-methylpentylamino)phenyl]methyl]-1,3,8-triazaspiro[4.5]dec-3-en-2-one Chemical compound CCCC(C)CNC1=CC=CC(CN2CCC3(CC2)C(=NC(=O)N3C=2C=C(F)C=CC=2)NC2CCCCC2)=C1 XLGLUVXJCDPUGS-UHFFFAOYSA-N 0.000 description 1
- DWWRDPFASOWBEX-UHFFFAOYSA-N 4-[2-[[4-cyclohexylimino-1-(3-fluorophenyl)-2-oxo-1,3,8-triazaspiro[4.5]decan-6-yl]methyl]anilino]butanenitrile Chemical compound C1(CCCCC1)NC1=NC(N(C11C(CNCC1)CC1=C(C=CC=C1)NCCCC#N)C1=CC(=CC=C1)F)=O DWWRDPFASOWBEX-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- NQAUNPZZVCXYEJ-UHFFFAOYSA-N 4-bromo-n,n-dimethylbenzenesulfonamide Chemical compound CN(C)S(=O)(=O)C1=CC=C(Br)C=C1 NQAUNPZZVCXYEJ-UHFFFAOYSA-N 0.000 description 1
- WDFQBORIUYODSI-UHFFFAOYSA-N 4-bromoaniline Chemical compound NC1=CC=C(Br)C=C1 WDFQBORIUYODSI-UHFFFAOYSA-N 0.000 description 1
- KMMHZIBWCXYAAH-UHFFFAOYSA-N 4-bromobenzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=C(Br)C=C1 KMMHZIBWCXYAAH-UHFFFAOYSA-N 0.000 description 1
- TUXYZHVUPGXXQG-UHFFFAOYSA-N 4-bromobenzoic acid Chemical compound OC(=O)C1=CC=C(Br)C=C1 TUXYZHVUPGXXQG-UHFFFAOYSA-N 0.000 description 1
- CQPGDDAKTTWVDD-UHFFFAOYSA-N 4-bromobutanenitrile Chemical compound BrCCCC#N CQPGDDAKTTWVDD-UHFFFAOYSA-N 0.000 description 1
- GXYZREDEYDFJPT-ZMBIFBSDSA-N 4-cyano-n-[(2r)-2-[4-(2,3-dihydro-1,4-benzodioxin-5-yl)piperazin-1-yl]propyl]-n-pyridin-2-ylbenzamide;hydrochloride Chemical compound Cl.C([C@@H](C)N1CCN(CC1)C=1C=2OCCOC=2C=CC=1)N(C=1N=CC=CC=1)C(=O)C1=CC=C(C#N)C=C1 GXYZREDEYDFJPT-ZMBIFBSDSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- KRZCOLNOCZKSDF-UHFFFAOYSA-N 4-fluoroaniline Chemical compound NC1=CC=C(F)C=C1 KRZCOLNOCZKSDF-UHFFFAOYSA-N 0.000 description 1
- LLBZPESJRQGYMB-UHFFFAOYSA-N 4-one Natural products O1C(C(=O)CC)CC(C)C11C2(C)CCC(C3(C)C(C(C)(CO)C(OC4C(C(O)C(O)C(COC5C(C(O)C(O)CO5)OC5C(C(OC6C(C(O)C(O)C(CO)O6)O)C(O)C(CO)O5)OC5C(C(O)C(O)C(C)O5)O)O4)O)CC3)CC3)=C3C2(C)CC1 LLBZPESJRQGYMB-UHFFFAOYSA-N 0.000 description 1
- GRQVZNMSXFEEHD-UHFFFAOYSA-N 6-[(3-cyclobutyl-1,2,4,5-tetrahydro-3-benzazepin-7-yl)oxy]-n-methylpyridine-3-carboxamide;hydrochloride Chemical compound Cl.N1=CC(C(=O)NC)=CC=C1OC1=CC=C(CCN(CC2)C3CCC3)C2=C1 GRQVZNMSXFEEHD-UHFFFAOYSA-N 0.000 description 1
- DFGKGUXTPFWHIX-UHFFFAOYSA-N 6-[2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]acetyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)C1=CC2=C(NC(O2)=O)C=C1 DFGKGUXTPFWHIX-UHFFFAOYSA-N 0.000 description 1
- GUJKANMNMCRQRT-UHFFFAOYSA-N 6-propan-2-yloxypyridine-2-carbaldehyde Chemical compound CC(C)OC1=CC=CC(C=O)=N1 GUJKANMNMCRQRT-UHFFFAOYSA-N 0.000 description 1
- LGYWTSBBMPRAJL-UHFFFAOYSA-N 8-[[6-(benzylamino)pyridin-1-ium-3-yl]methyl]-4-(cyclohexylamino)-1-(3-fluorophenyl)-1,3-diaza-8-azoniaspiro[4.5]dec-3-en-2-one;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.[O-]C(=O)C(F)(F)F.FC1=CC=CC(N2C3(CC[NH+](CC=4C=[NH+]C(NCC=5C=CC=CC=5)=CC=4)CC3)C(NC3CCCCC3)=NC2=O)=C1 LGYWTSBBMPRAJL-UHFFFAOYSA-N 0.000 description 1
- 208000018282 ACys amyloidosis Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 1
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 1
- 102000009091 Amyloidogenic Proteins Human genes 0.000 description 1
- 108010048112 Amyloidogenic Proteins Proteins 0.000 description 1
- 206010002023 Amyloidoses Diseases 0.000 description 1
- 235000003911 Arachis Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 230000007082 Aβ accumulation Effects 0.000 description 1
- 230000007134 Aβ oligomerisation Effects 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- RKDGYYVUOYUHTK-UHFFFAOYSA-O CC(C)(C)OC(Nc1ncc(CN(CC2)CCC2(C(NC2CCCCC2)=NC2=[OH+])N2c2cccc(F)c2)cc1)=O Chemical compound CC(C)(C)OC(Nc1ncc(CN(CC2)CCC2(C(NC2CCCCC2)=NC2=[OH+])N2c2cccc(F)c2)cc1)=O RKDGYYVUOYUHTK-UHFFFAOYSA-O 0.000 description 1
- NVVZZXMKQQVOHZ-UHFFFAOYSA-N CC(C)Oc1cccc(CN(CC2)CCC2(C(NC2CCCCC2)=NC2=O)N2c(cc(cc2)F)c2-c(cc2)ccc2S(N(C)C)(=O)=O)c1 Chemical compound CC(C)Oc1cccc(CN(CC2)CCC2(C(NC2CCCCC2)=NC2=O)N2c(cc(cc2)F)c2-c(cc2)ccc2S(N(C)C)(=O)=O)c1 NVVZZXMKQQVOHZ-UHFFFAOYSA-N 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000010859 Creutzfeldt-Jakob disease Diseases 0.000 description 1
- 102000013717 Cyclin-Dependent Kinase 5 Human genes 0.000 description 1
- 108010025454 Cyclin-Dependent Kinase 5 Proteins 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 201000010374 Down Syndrome Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 208000007487 Familial Cerebral Amyloid Angiopathy Diseases 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 206010019196 Head injury Diseases 0.000 description 1
- 208000032849 Hereditary cerebral hemorrhage with amyloidosis Diseases 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- 101001125071 Homo sapiens Neuromedin-K receptor Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- LHXOCOHMBFOVJS-OAHLLOKOSA-N Ladostigil Chemical compound CCN(C)C(=O)OC1=CC=C2CC[C@@H](NCC#C)C2=C1 LHXOCOHMBFOVJS-OAHLLOKOSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- ULMHMJAEGZPQRY-UHFFFAOYSA-N N-(tert-butoxycarbonyl)piperidin-2-one Chemical compound CC(C)(C)OC(=O)N1CCCCC1=O ULMHMJAEGZPQRY-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 108010040718 Neurokinin-1 Receptors Proteins 0.000 description 1
- 102100029409 Neuromedin-K receptor Human genes 0.000 description 1
- FZRKAZHKEDOPNN-UHFFFAOYSA-N Nitric oxide anion Chemical compound O=[N-] FZRKAZHKEDOPNN-UHFFFAOYSA-N 0.000 description 1
- MJQFIVQQBMJZNH-UHFFFAOYSA-N O=C(N(C12CCN(Cc3c(NCC4CC4)nccc3)CC1)C1=CCCC(F)=C1)N=C2NC1CCCCC1 Chemical compound O=C(N(C12CCN(Cc3c(NCC4CC4)nccc3)CC1)C1=CCCC(F)=C1)N=C2NC1CCCCC1 MJQFIVQQBMJZNH-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229910020667 PBr3 Inorganic materials 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102000029797 Prion Human genes 0.000 description 1
- 108091000054 Prion Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 1
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 description 1
- 239000004280 Sodium formate Substances 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 102100037346 Substance-P receptor Human genes 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- 238000006058 Ugi-reaction Methods 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- PBHFNBQPZCRWQP-QUCCMNQESA-N [(3ar,8bs)-3,4,8b-trimethyl-2,3a-dihydro-1h-pyrrolo[2,3-b]indol-7-yl] n-phenylcarbamate Chemical compound CN([C@@H]1[C@@](C2=C3)(C)CCN1C)C2=CC=C3OC(=O)NC1=CC=CC=C1 PBHFNBQPZCRWQP-QUCCMNQESA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 150000001351 alkyl iodides Chemical class 0.000 description 1
- 230000003281 allosteric effect Effects 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000005219 aminonitrile group Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- MMCPOSDMTGQNKG-UHFFFAOYSA-N anilinium chloride Chemical compound Cl.NC1=CC=CC=C1 MMCPOSDMTGQNKG-UHFFFAOYSA-N 0.000 description 1
- 230000007131 anti Alzheimer effect Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 125000006269 biphenyl-2-yl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C1=C(*)C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000006268 biphenyl-3-yl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C1=C([H])C(*)=C([H])C([H])=C1[H] 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- WLVCAIKDSQAIMG-UHFFFAOYSA-N bromomethyl N-pyridin-2-ylcarbamate Chemical compound N1=CC=CC=C1NC(OCBr)=O WLVCAIKDSQAIMG-UHFFFAOYSA-N 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- KVLLHLWBPNCVNR-SKCUWOTOSA-N capromorelin Chemical compound C([C@@]12CN(CCC1=NN(C2=O)C)C(=O)[C@@H](COCC=1C=CC=CC=1)NC(=O)C(C)(C)N)C1=CC=CC=C1 KVLLHLWBPNCVNR-SKCUWOTOSA-N 0.000 description 1
- 229950004826 capromorelin Drugs 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000000068 chlorophenyl group Chemical group 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 239000000544 cholinesterase inhibitor Substances 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 1
- 125000004802 cyanophenyl group Chemical group 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- XYZMOVWWVXBHDP-UHFFFAOYSA-N cyclohexyl isocyanide Chemical compound [C-]#[N+]C1CCCCC1 XYZMOVWWVXBHDP-UHFFFAOYSA-N 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 125000001887 cyclopentyloxy group Chemical group C1(CCCC1)O* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 210000005220 cytoplasmic tail Anatomy 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 239000002027 dichloromethane extract Substances 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 1
- HXITXNWTGFUOAU-UHFFFAOYSA-N dihydroxy-phenylborane Natural products OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 1
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 1
- HPYNZHMRTTWQTB-UHFFFAOYSA-N dimethylpyridine Natural products CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229960003530 donepezil Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000010410 dusting Methods 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000012156 elution solvent Substances 0.000 description 1
- 125000001240 enamine group Chemical group 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- OPQRBXUBWHDHPQ-UHFFFAOYSA-N etazolate Chemical compound CCOC(=O)C1=CN=C2N(CC)N=CC2=C1NN=C(C)C OPQRBXUBWHDHPQ-UHFFFAOYSA-N 0.000 description 1
- QZJQVQJGYVMBMF-UHFFFAOYSA-N ethyl 4-amino-8-benzyl-1-(5-bromo-2-fluorophenyl)-2-oxo-1,8-diazaspiro[4.5]dec-3-ene-3-carboxylate Chemical compound C=1C(Br)=CC=C(F)C=1N1C(=O)C(C(=O)OCC)=C(N)C1(CC1)CCN1CC1=CC=CC=C1 QZJQVQJGYVMBMF-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000012025 fluorinating agent Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- 239000003540 gamma secretase inhibitor Substances 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000003324 growth hormone secretagogue Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000003395 histamine H3 receptor antagonist Substances 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical group 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- SXWRTZOXMUOJER-UHFFFAOYSA-N hydron;piperidin-4-one;chloride;hydrate Chemical compound O.Cl.O=C1CCNCC1 SXWRTZOXMUOJER-UHFFFAOYSA-N 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960000934 ibutamoren Drugs 0.000 description 1
- 229940076716 ibutamoren mesylate Drugs 0.000 description 1
- 201000008319 inclusion body myositis Diseases 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000006316 iso-butyl amino group Chemical group [H]N(*)C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- RIWNFZUWWRVGEU-UHFFFAOYSA-N isocyanomethylbenzene Chemical compound [C-]#[N+]CC1=CC=CC=C1 RIWNFZUWWRVGEU-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229950008812 ladostigil Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 125000003564 m-cyanobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(C#N)=C1[H])C([H])([H])* 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- RVGDBFWCOWBREZ-UHFFFAOYSA-N methyl 4-amino-8-benzyl-1-(3-fluorophenyl)-2-oxo-1,8-diazaspiro[4.5]dec-3-ene-3-carboxylate Chemical compound C=1C=CC(F)=CC=1N1C(=O)C(C(=O)OC)=C(N)C1(CC1)CCN1CC1=CC=CC=C1 RVGDBFWCOWBREZ-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 208000027061 mild cognitive impairment Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- ULRDYYKSPCRXAJ-KRWDZBQOSA-N n-[(2r)-2-[4-[4-[2-(methanesulfonamido)ethyl]phenyl]phenyl]propyl]propane-2-sulfonamide Chemical compound C1=CC([C@@H](C)CNS(=O)(=O)C(C)C)=CC=C1C1=CC=C(CCNS(C)(=O)=O)C=C1 ULRDYYKSPCRXAJ-KRWDZBQOSA-N 0.000 description 1
- CRPCKOCUUZLLAB-UHFFFAOYSA-N n-[3-[[4-(cyclohexylamino)-1-(3-fluorophenyl)-2-oxo-1,3,8-triazaspiro[4.5]dec-3-en-8-yl]methyl]phenyl]acetamide Chemical compound CC(=O)NC1=CC=CC(CN2CCC3(CC2)C(=NC(=O)N3C=2C=C(F)C=CC=2)NC2CCCCC2)=C1 CRPCKOCUUZLLAB-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000004898 n-terminal fragment Anatomy 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- OGZQTTHDGQBLBT-UHFFFAOYSA-N neramexane Chemical compound CC1(C)CC(C)(C)CC(C)(N)C1 OGZQTTHDGQBLBT-UHFFFAOYSA-N 0.000 description 1
- 229950004543 neramexane Drugs 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 210000002682 neurofibrillary tangle Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000000181 nicotinic agonist Substances 0.000 description 1
- YCWSUKQGVSGXJO-NTUHNPAUSA-N nifuroxazide Chemical group C1=CC(O)=CC=C1C(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 YCWSUKQGVSGXJO-NTUHNPAUSA-N 0.000 description 1
- DLWSRGHNJVLJAH-UHFFFAOYSA-N nitroflurbiprofen Chemical compound FC1=CC(C(C(=O)OCCCCO[N+]([O-])=O)C)=CC=C1C1=CC=CC=C1 DLWSRGHNJVLJAH-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 125000006504 o-cyanobenzyl group Chemical group [H]C1=C([H])C(C#N)=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000004115 pentoxy group Chemical group [*]OC([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- ZNNZYHKDIALBAK-UHFFFAOYSA-M potassium thiocyanate Chemical compound [K+].[S-]C#N ZNNZYHKDIALBAK-UHFFFAOYSA-M 0.000 description 1
- 229940116357 potassium thiocyanate Drugs 0.000 description 1
- YRVIKLBSVVNSHF-JTQLQIEISA-N pozanicline Chemical compound CC1=NC=CC=C1OC[C@H]1NCCC1 YRVIKLBSVVNSHF-JTQLQIEISA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000003672 processing method Methods 0.000 description 1
- 201000002212 progressive supranuclear palsy Diseases 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- SCHKZZSVELPJKU-UHFFFAOYSA-N prx-03140 Chemical compound O=C1N(C(C)C)C=2SC=CC=2C(O)=C1C(=O)NCCCN1CCCCC1 SCHKZZSVELPJKU-UHFFFAOYSA-N 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000002469 receptor inverse agonist Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 1
- 229960000311 ritonavir Drugs 0.000 description 1
- 229960004136 rivastigmine Drugs 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 239000012363 selectfluor Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 1
- 235000019254 sodium formate Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical group ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960001685 tacrine Drugs 0.000 description 1
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical compound C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 1
- CKXZPVPIDOJLLM-UHFFFAOYSA-N tert-butyl n-piperidin-4-ylcarbamate Chemical compound CC(C)(C)OC(=O)NC1CCNCC1 CKXZPVPIDOJLLM-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- NNBZCPXTIHJBJL-UHFFFAOYSA-N trans-decahydronaphthalene Natural products C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 1
- 239000006211 transdermal dosage form Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- WJJYZXPHLSLMGE-UHFFFAOYSA-N xaliproden Chemical compound FC(F)(F)C1=CC=CC(C=2CCN(CCC=3C=C4C=CC=CC4=CC=3)CC=2)=C1 WJJYZXPHLSLMGE-UHFFFAOYSA-N 0.000 description 1
- 229960004664 xaliproden Drugs 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the invention is directed to compounds useful as inhibitors of the beta secretase enzyme, and useful in the treatment of diseases in which the beta secretase enzyme is involved, such as Alzheimer's Disease.
- Alzheimer's disease is characterized by the abnormal deposition of amyloid in the brain in the form of extra-cellular plaques and intra-cellular neurofibrillary tangles.
- the rate of amyloid accumulation is a combination of the rates of formation, aggregation and egress from the brain. It is generally accepted that the main constituent of amyloid plaques is the 4kD amyloid protein ( ⁇ A4, also referred to as A ⁇ , ⁇ -protein and ⁇ AP) which is a proteolytic product of a precursor protein of much larger size.
- the amyloid precursor protein (APP or A ⁇ PP) has a receptor-like structure with a large ectodomain, a membrane spanning region and a short cytoplasmic tail.
- the A ⁇ domain encompasses parts of both extra-cellular and transmembrane domains of APP, thus its release implies the existence of two distinct proteolytic events to generate its NH 2 - and COOH-termini. At least two secretory mechanisms exist which release APP from the membrane and generate soluble, COOH-truncated forms of APP (APP S ). Proteases that release APP and its fragments from the membrane are termed
- secretases Most APP 8 is released by a putative ⁇ -secretase which cleaves within the A ⁇ protein to release ⁇ -APP s and precludes the release of intact A ⁇ . A minor portion of APP 5 is released by a ⁇ - secretase (“ ⁇ -secretase”), which cleaves near the NH 2 -terminus of APP and produces COOH-terminal fragments (CTFs) which contain the whole A ⁇ domain.
- ⁇ -secretase ⁇ -secretase
- CTFs COOH-terminal fragments
- therapeutic agents that can inhibit ⁇ -secretase or BACE may be useful for the treatment of Alzheimer's disease.
- the compounds of the present invention are useful for treating Alzheimer's disease by inhibiting the activity of ⁇ -secretase or BACE, thus preventing the formation of insoluble A ⁇ and arresting the production of A ⁇ .
- the present invention is directed to novel spiropiperidine compounds represented by general formula (I)
- the invention is also directed to pharmaceutical compositions which include an effective amount of a compound of formula (I), or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable carrier.
- the invention is also directed to methods of treating mammals for diseases in which the ⁇ -secretase enzyme is involved, such as Alzheimer's disease, and the use of the compounds and pharmaceutical compositions of the invention in the treatment of such diseases.
- the present invention is directed to methods of treating mammals for diseases in which the ⁇ -secretase enzyme is involved, such as Alzheimer's disease, by administering a compound of formula (I)
- X 1 is CR5H or NH
- Z is O or S
- Rl is selected from the group consisting of
- Rl aryl and heteroaryl moieties are optionally substituted with one or more (a) halogen, (b) -Ci -6 alkyl,
- Q 1 and Q2 are selected from the group consisting of
- n 0, 1 or 2;
- R2 is selected from the group consisting of
- R.2 cycloalkyl moiety is optionally substituted with one or more -C 1-6 alkyl
- R.2 aryl or heteroaryl moiety is optionally substituted with one or more
- Q3 and Q4 are selected from the same group as Ql and Q2;
- R3 is selected from the group consisting of
- R ⁇ alkyl moiety is optionally substituted with one or more
- R.3 aryl and heteroaryl moiety is optionally substituted with one or more
- Rl 2 is selected from the group consisting of (I) hydrogen, (U) -C 1.6 alkyl,
- Rl 2 alkyl, alkenyl and alkynyl moiety is optionally substituted with one or more
- RlO cycloalkyl moiety is optionally substituted with one or more (A) halogen, (B) hydroxyl,
- Rl2 aryl moiety is optionally substituted with one or more
- Q5 and Q6 are selected from the same group as Ql and Q2;
- R4 and R4' are selected from the group consisting of (1) hydrogen, (2) -C 1-8 alkyl, wherein said alkyl is optionally substituted with
- R5 is selected from the group csoonsisting of (1) hydrogen, (2) -C 1-6 alkyl,
- R6, R7, R8 ; R9 ; RlO and Rl 1 are independently selected from the group consisting of: (1) hydrogen, (2) -Ci. 6 alkyl, (3) -C3-8 cycloalkyl,
- the invention is also directed to pharmaceutical compositions which include an effective amount of a compound of formula (I), or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable carrier.
- the present invention is further directed to a method for the manufacture of a medicament or a composition for inhibiting ⁇ -secretase enzyme activity in humans and animals.
- the invention is also directed to a method for the manufacture of a medicament for the treatment of Alzheimer's Disease in humans, comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
- the invention is directed to novel spiropiperidine compounds of formula (I) above, provided that when X is N; Z is O; Rl is unsubstituted cyclohexyl; and R.2 is unsubstituted phenyl, then R ⁇ is not unsubstituted benzyl.
- Z is O.
- the invention is directed to compounds of formula (I) wherein X is N and X 1 is NH .
- X is CR ⁇ , wherein R5 is preferably hydrogen, and X' is CR ⁇ H, wherein R5 is preferably hydrogen.
- Rl is -Ci-6 alkyl or -C ⁇ -3 alkyl-C3_i2 carbocyclic wherein said alkyl or carbocyclic is optionally substituted with one or more (a) -OR4,
- Rl carbocyclic groups include-C ⁇ .g carbocyclic groups, including cyclobutyl, cyclopentyl, cyclohexyl, morpholinyl, tetrahydropyran and pyrrolidinyl.
- Rl is -C ⁇ -3 alkyl -C6-10 ar yl > preferably phenyl or benzyl.
- the Rl aryl moiety is optionally substituted with one or more
- Rl is C ⁇ -3 alkyl-C5-i2 heteroaryl, wherein the heteroaryl moiety is optionally substituted with one or more
- Rl heteroaryl groups include pyridinyl, thienyl, furanyl and imidazolyl.
- R2 is selected from the group consisting of (1) -Ci_6 alkyl
- R2 is selected from -Ci-6 alkyl, phenyl, benzyl, or -C ⁇ -3 alkyl-C3_8 carbocyclic, optionally substituted as described above.
- R2 is -C ⁇ -3 alkyl-C6-lO aryl, preferably the aryl moiety is optionally substituted with one or more
- Rl 5 is selected from the group consisting of (a) -ORlO, (b) halogen,
- R ⁇ is preferably
- R3 is -C ⁇ -3 alkyl-heteroaryl
- the heteroaryl is selected from the group consisting of pyridyl, pyrrolyl, furanyl, thienyl, dihydrobenzofuran, indolyl, isoquinolinyl, imidazolyl, isoxazolyl, quinolinyl, tetrahydroquinolinyl, dihydroindolyl and pyridyl.
- the heteroaryl is substituted with one or more (a) -ORl2,
- X is N and Z is O.
- alkyl by itself or as part of another substituent, means a saturated straight or branched chain hydrocarbon radical having the number of carbon atoms designated (e.g., C ⁇ . 10 alkyl means an alkyl group having from one to ten carbon atoms).
- Preferred alkyl groups for use in the invention are Ci -6 alkyl groups, having from one to six carbon atoms.
- Exemplary alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, pentyl, hexyl, and the like.Co alkyl means a bond.
- alkoxy by itself or as part of another substituent, means the group - O- alkyl, wherein alkyl is defined above, having the number of carbon atoms designated (e.g., Ci-M) alkoxy means an alkoxy group having from one to ten carbon atoms.
- Preferred alkoxy groups for use in the invention are C ⁇ .(, alkoxy groups, having from one to six carbon atoms.
- Exemplary preferred alkoxy groups include methoxy, ethoxy, propoxy, butoxy, sec-butoxy and pentoxy.
- Especially preferred alkoxy groups are Ci .3 alkoxy.
- alkenyl by itself or as part of another substituent, means a straight or branched chain hydrocarbon radical having a single carbon-carbon double bond and the number of carbon atoms designated (e.g., C2-10 alkenyl means an alkenyl group having from two to ten carbon atoms).
- Preferred alkenyl groups for use in the invention are C2-6 alkenyl groups, having from two to six carbon atoms.
- Exemplary alkenyl groups include ethenyl and propenyl.
- alkynyl by itself or as part of another substituent, means a straight or branched chain hydrocarbon radical having a single carbon-carbon triple bond and the number of carbon atoms designated (e.g., C2-10 alkynyl means an alkynyl group having from two to ten carbon atoms).
- Preferred alkynyl groups for use in the invention are C2-6 alkynyl groups, having from two to six carbon atoms.
- Exemplary alkynyl groups include ethynyl and propynyl.
- cycloalkyl by itself or as part of another substituent, means a saturated cyclic hydrocarbon radical having the number of carbon atoms designated (e.g., C3-12 cycloalkyl means a cycloalkyl group having from three to twelve carbon atoms).
- the term cycloalkyl as used herein includes mono-, bi- and tricyclic saturated carbocycles, as well as bridged and fused ring carbocycles, such as spiro fused ring systems.
- Preferred cycloalkyl groups for use in the invention are monocyclic C3.8 cycloalkyl groups, having from three to eight carbon atoms.
- Exemplary monocyclic cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like.
- Exemplary bridged cycloalkyl groups include adamantly and norbornyl.
- Exemplary fused cycloalkyl groups include decahydronaphthalene.
- the term "carbocyclic,” by itself or as part of another substituent, means a cycloalkyl group as defined above, or a non-aromatic heterocyclic group.
- a non-aromatic heterocyclic group, by itself or as part of another substituent means a cycloalkyl group as defined above in which one or more of the ring carbon atoms is replaced with a heteroatom (such as N, S or O).
- Suitable non- aromatic heterocyclic groups for use in the invention include piperidinyl, piperazinyl, morpholinyl, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, pyrazolidinyl, azetidinyl, tetrahydropyranyl and imidazolildinyl.
- Preferred non-aromatic heterocyclic groups are piperidinyl, piperazinyl, tetrahydrofuranyl, tetrahydropyranyl, pyrrolidinyl, morpholinyl and azetidinyl.
- the substituent When a non-aromatic heterocyclic group as defined herein is substituted, the substituent may be bonded to a ring carbon atom of the heterocyclic group, or on a ring heteroatom (i.e., a nitrogen, oxygen or sulfur), which has a valence which permits substitution. Preferably, the substituent is bonded to a ring carbon atom.
- the point of attachment may be at a ring carbon atom of the heterocyclic group or on a ring heteroatom (i.e., a nitrogen, oxygen or sulfur), which has a valence which permits substitution.
- the attachment is at a ring carbon atom.
- aryl by itself or as part of another substituent, means an aromatic cyclic hydrocarbon radical having the number of carbon atoms designated (e.g., C ⁇ -lO aryl means an aryl group having from six to ten carbons atoms).
- aryl includes multiple ring systems as well as single ring systems. Preferred aryl groups for use in the invention include phenyl and naphthyl.
- aryl also includes fused cyclic hydrocarbon rings which are partially aromatic (i.e., one of the fused rings is aromatic and the other is non-aromatic).
- An exemplary aryl group which is partially aromatic is indanyl.
- halo or “halogen” includes fluoro, chloro, bromo and iodo.
- heteroaryl by itself or as part of another substituent, means an aromatic cyclic group having at least one ring heteroatom (O, N or S). ).
- heteroaryl includes multiple ring systems as well as single ring systems.
- heteroaryl groups for use in the invention include furyl, pyranyl, benzofuranyl, isobenzofuranyl, chromenyl, thienyl, benzothiophenyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, indolyl, benzimidazolyl, quinolinyl, isoquinolinyl, tetrazolyl, indazolyl, napthyridinyl, triazolyl, oxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, isoxazolyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl and dihydroindolyl.
- heteroaryl also includes fused aromatic cyclic groups which are partially aromatic (i.e., one of the fused rings is aromatic and the other is non-aromatic).
- exemplary heteroaryl groups which are partially aromatic include tetrahydroquinolyl, dihydrobenzofuran and dihydroindolyl.
- the substituent When a heteroaryl group as defined herein is substituted, the substituent may be bonded to a ring carbon atom of the heteroaryl group, or on a ring heteroatom (i.e., a nitrogen, oxygen or sulfur), which has a valence which permits substitution. Preferably, the substituent is bonded to a ring carbon atom.
- the point of attachment may be at a ring carbon atom of the heteroaryl group, or on a ring heteroatom (i.e., a nitrogen, oxygen or sulfur), which has a valence which permits attachment.
- the attachment is at a ring carbon atom.
- Some of the compounds of the instant invention have at least one asymmetric center. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Compounds with asymmetric centers give rise to enantiomers (optical isomers), diastereomers (configurational isomers) or both, and it is intended that all of the possible enantiomers and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention. The present invention is meant to encompass all such isomeric forms of these compounds.
- tautomer refers to a compound which exists in an equilibrium mixture and which can be isolated in either form and react through either form.
- the tatutomers may differ in linkage, bond, or connections between atoms, and the position or distribution of the atoms in the molecule.
- compounds of formula (I) may be present in the enamine form depicted above, or in the tautomeric imine form (F), as shown below:
- compounds of formula (II) may be present in the enamine form depicted above, or in the tautomeric imine form (II'), as shown below:
- Compounds described herein may contain one or more double bonds, and may thus give rise to cisltrans isomers as well as other conformational isomers.
- the present invention includes all such possible isomers as well as mixtures of such isomers.
- Formulas (I) to (HI) are shown above without a definite stereochemistry at certain positions.
- the present invention includes all stereoisomers of Formulas (I) to (III) and pharmaceutically acceptable salts thereof.
- the independent syntheses of the enantiomerically or diastereomerically enriched compounds, or their chromatographic separations, may be achieved as known in the art by appropriate modification of the methodology disclosed herein.
- Their absolute stereochemistry may be determined by the x-ray crystallography of crystalline products or crystalline intermediates that are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration. If desired, racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diastereomeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods using chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
- Scheme 1 depicts an Ugi four-component coupling reaction between a piperidone derivative, amine, isonitrile, and cyanate, which assembles the core structure 1-1. Further elaboration of 1-1 is possible, for example, removal of a temporary R.3 group to give 1-2, followed by alkylation with a different R.3 to give new structures 1-3.
- substantially pure means that the isolated material is at least 90% pure, and preferably 95% pure, and even more preferably 99% pure as assayed by analytical techniques known in the art.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- the compounds of the invention may be mono, di or tris salts, depending on the number of acid functionalities present in the free base form of the compound.
- Free bases and salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, trifluoroacetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- Particularly preferred are citric, hydrobromic, hydrochloric, trifluoroacetic, maleic, phosphoric, sulfuric, fumaric, and tartaric acids.
- the present invention is directed to the use of the compounds of formulas (I) to (Hl) disclosed herein as inhibitors of ⁇ -secretase enzyme activity or ⁇ -site amyloid precursor protein-cleaving enzyme ("BACE") activity, in a patient or subject such as a mammal in need of such inhibition, comprising the administration of an effective amount of the compound.
- BACE ⁇ -secretase enzyme
- ⁇ -site amyloid precursor protein-cleaving enzyme and “BACE” are used interchangeably in this specification.
- ⁇ -secretase enzyme ⁇ -site amyloid precursor protein-cleaving enzyme
- BACE ⁇ -site amyloid precursor protein-cleaving enzyme
- the compounds of the present invention have utility in treating, ameliorating, controlling or reducing the risk of Alzheimer's disease.
- the compounds may be useful for the prevention of dementia of the Alzheimer's type, as well as for the treatment of early stage, intermediate stage or late stage dementia of the Alzheimer's type.
- the compounds may also be useful in treating, ameliorating, controlling or reducing the risk of diseases mediated by abnormal cleavage of amyloid precursor protein (also referred to as APP), and other conditions that may be treated or prevented by inhibition of ⁇ - secretase.
- APP amyloid precursor protein
- Such conditions include mild cognitive impairment, Trisomy 21 (Down Syndrome), cerebral amyloid angiopathy, degenerative dementia, Hereditary Cerebral Hemorrhage with Amyloidosis of the Dutch-Type (HCHWA-D), Creutzfeld- Jakob disease, prion disorders, amyotrophic lateral sclerosis, progressive supranuclear palsy, head trauma, stroke, pancreatitis, inclusion body myositis, other peripheral amyloidoses, diabetes and atherosclerosis.
- the subject or patient to whom the compounds of the present invention is administered is generally a human being, male or female, in whom inhibition of ⁇ -secretase enzyme activity is desired, but may also encompass other mammals, such as dogs, cats, mice, rats, cattle, horses, sheep, rabbits, monkeys, chimpanzees or other apes or primates, for which inhibition of ⁇ -secretase enzyme activity or treatment of the above noted disorders is desired.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment of diseases or conditions for which the compounds of the present invention have utility, where the combination of the drugs together are safer or more effective than either drug alone. Additionally, the compounds of the present invention may be used in combination with one or more other drugs that treat, prevent, control, ameliorate, or reduce the risk of side effects or toxicity of the compounds of the present invention. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with the compounds of the present invention. Accordingly, the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to the compounds of the present invention. The combinations may be administered as part of a unit dosage form combination product, or as a kit or treatment protocol wherein one or more additional drugs are administered in separate dosage forms as part of a treatment regimen.
- combinations of the compounds of the present invention with other drugs in either unit dose or kit form include combinations with anti-Alzheimer's agents, for example other beta-secretase inhibitors or gamma-secretase inhibitors; tau phpsphorylation inhibitors; Ml receptor positive allosteric modulators; blockers of A ⁇ oligomer formation; 5-HT modulators, such as PRX-03140, GSK 742467, SGS-518, FK-962, SL-65.0155, SRA-333 and xaliproden; p25/CDK5 inhibitors; NK1/NK3 receptor antagonists; COX-2 inhibitors; HMG-CoA reductase inhibitors; NSAIDs including ibuprofen; vitamin E; anti-amyloid antibodies, including anti -amyloid humanized monoclonal antibodies; anti-inflammatory compounds such as (R)-flurbiprofen, nitroflurbiprofen, rosiglitazone, ND-1251, VP
- composition as used herein is intended to encompass a product comprising specified ingredients in predetermined amounts or proportions, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- This term in relation to pharmaceutical compositions is intended to encompass a product comprising one or more active ingredients, and an optional carrier comprising inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active compound which is a compound of formulas (I) to (HI)
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- compositions of the present invention can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient. Further, the compositions can be presented as a powder, as granules, as a solution, as a suspension in an aqueous liquid, as a non-aqueous liquid, as an oil-in-water emulsion or as a water-in-oil liquid emulsion.
- the compounds represented by Formulas (I) to (VII), or pharmaceutically acceptable salts thereof may also be administered by controlled release means and/or delivery devices.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants.
- Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- Each tablet preferably contains from about 0.1 mg to about 500 mg of the active ingredient and each cachet or capsule preferably containing from about 0.1 mg to about 500 mg of the active ingredient.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example peanut oil, liquid paraffin, or olive oil.
- compositions include aqueous suspensions, which contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. Oily suspensions may also contain various excipients.
- the pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions, which may also contain excipients such as sweetening and flavoring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension, or in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions.
- the final injectable form must be sterile and must be effectively fluid for easy syringability.
- the pharmaceutical compositions must be stable under the conditions of manufacture and storage; thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like. Further, the compositions can be in a form suitable for use in transdermal devices. These formulations may be prepared via conventional processing methods. As an example, a cream or ointment is prepared by mixing hydrophilic material and water, together with about 5wt% to about 10wt% of the compound, to produce a cream or ointment having a desired consistency. Pharmaceutical compositions of this invention can also be in a form suitable for rectal administration wherein the carrier is a solid. It is preferable that the mixture forms unit dose suppositories. Suitable carriers include cocoa butter and other materials commonly used in the art.
- administering a should be understood to mean providing a compound of the invention to the individual in need of treatment in a form that can be introduced into that individual's body in a therapeutically useful form and therapeutically useful amount, including, but not limited to: oral dosage forms, such as tablets, capsules, syrups, suspensions, and the like; injectable dosage forms, such as IV, IM, or IP, and the like; transdermal dosage forms, including creams, jellies, powders, or patches; buccal dosage forms; inhalation powders, sprays, suspensions, and the like; and rectal suppositories.
- oral dosage forms such as tablets, capsules, syrups, suspensions, and the like
- injectable dosage forms such as IV, IM, or IP, and the like
- transdermal dosage forms including creams, jellies, powders, or patches
- buccal dosage forms inhalation powders, sprays, suspensions, and the like
- rectal suppositories rectal suppositories.
- an effective amount or “therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- treatment or “treating” means any administration of a compound of the present invention and includes (1) inhibiting the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased (i.e., arresting further development of the pathology and/or symptomatology), or (2) ameliorating the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased (i.e., reversing the pathology and/or symptomatology).
- controlling includes preventing treating, eradicating, ameliorating or otherwise reducing the severity of the condition being controlled.
- compositions containing compounds of the present invention may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
- unit dosage form is taken to mean a single dose wherein all active and inactive ingredients are combined in a suitable system, such that the patient or person administering the drug to the patient can open a single container or package with the entire dose contained therein, and does not have to mix any components together from two or more containers or packages.
- Typical examples of unit dosage forms are tablets or capsules for oral administration, single dose vials for injection, or suppositories for rectal administration.
- compositions containing compounds of the present invention may conveniently be presented as a kit, whereby two or more components, which may be active or inactive ingredients, carriers, diluents, and the like, are provided with instructions for preparation of the actual dosage form by the patient or person adminstering the drug to the patient.
- kits may be provided with all necessary materials and ingredients contained therein, or they may contain instructions for using or making materials or components that must be obtained independently by the patient or person administering the drug to the patient.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 mg to about 100 mg per kg of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 mg to about 2000 mg, preferably from about 0.1 mg to about 20 mg per kg of body weight. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 mg to about 1,400 mg. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- a formulation intended for the oral administration to humans may conveniently contain from about 0.005 mg to about 2.5 g of active agent, compounded with an appropriate and convenient amount of carrier materia.
- Unit dosage forms will generally contain between from about 0.005 mg to about 1000 mg of the active ingredient, typically 0.005, 0.01 mg, 0.05 mg, 0.25 mg, 1 mg, 5 mg, 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg or 1000 mg, administered once, twice or three times a day.
- ECL Assay A homogeneous end point electrochemiluminescence (ECL) assay is employed using a biotinylated BACE substrate.
- the Km of the substrate is greater than 100 ⁇ M and can not be determined due to the limit of solubility of the substrate.
- a typical reaction contains approximately 0.1 nM enzyme, 0.25 ⁇ M of the substrate, and buffer (50 mM NaOAc, pH 4.5 or 6.5, 0.1 mg/ml BSA, 0.2% CHAPS, 15 mM EDTA and 1 mM deferoxamine) in a total reaction volume of 100 ⁇ l. The reaction proceeds for 30 min and is then stopped by the addition of 25 ⁇ L of 1 M Tris-HCl, pH 8.0.
- the resulting enzymatic product is assayed by adding a ruthenylated antibody which specifically recognizes the C- terminal residue of the product. Streptavidin coated magnetic beads are added into the solution and the samples are subjected to M-384 (Igen Inc., Gaithersburg, MD) analysis. Under these conditions, less than 10% of substrate is processed by BACE 1. The enzyme used in these studies is soluble
- HPLC assay A homogeneous end point HPLC assay is used with the substrate (coumarin-CO- REVNFEVEFR), which is cleaved by BACE 1 to release the N-terminal fragment attached with coumarin.
- the Km of the substrate is greater than 100 ⁇ M and can not be determined due to the limit of solubility of the substrate.
- a typical reaction contains approximately 2 nM enzyme, 1.0 ⁇ M of the substrate, and buffer (50 mM NaOAc, pH 4.5 or 6.5, 0.1 mg/ml BSA, 0.2% CHAPS, 15 mM EDTA and 1 mM deferoxamine) in a total reaction volume of 100 ⁇ l.
- the reaction is proceeded for 30 min and the reaction is stopped by the addition of 25 ⁇ L of 1 M Tris-HCl, pH 8.0.
- the resulting reaction mixture is loaded on the HPLC and the product is separated from substrate with 5 min linear gradient. Under these conditions, less than 10% of substrate is processed by BACE 1.
- the enzyme used in these studies is soluble (transmembrane domain and cytoplasmic extension excluded) human protein produced in a baculovirus expression system.
- 12 concentrations of inhibitors are prepared, and the concentration rage is dependent on the potency predicted by ECL. Solutions of inhibitor in DMSO are included in the reaction mixture (final DMSO concentration is 10 %). All experiments are conducted at rt using the standard reaction conditions described above. To determine the IC50 of the compound, four parameters equation is employed for curve fitting. The errors in reproducing the dissociation constants are typically less than two-fold.
- the compounds of the following examples had activity in inhibiting the beta- secretase enzyme in the aforementioned assays, generally with an IC50 from about 1 nM to 500 ⁇ M, preferably 1 nM to 100 ⁇ M. Such a result is indicative of the intrinsic activity of the compounds in use as inhibitors of beta-secretase enzyme activity.
- BSA bovine serum albumin
- TMSCN trimethylsilylnirrile
- PS-DIEA N 5 N (diisopropyl) aminomethylpolystyrene
- N-benzyl piperidinone (833 mg, 4.41 mmol) and benzyl isocyanide (500 mg, 4.2 mmol) were dissolved in MeOH (2.2mL).
- Aniline hydrochloride was added in portions over 5 min. After stirring for Ih the crude product precipitated from solution.
- the resulting solid was isolated via vacuum filtration, rinsed with H 2 O followed by Et 2 O, and dried in vacuo to give 150 mg of a white solid. A portion of this solid was further purified by RP-HPLC.
- Step 1 l-benzyl-4-[(3-fluorophenyl)amino]piperidine-4-carbonitrile (4A-1)
- Step 2 ethyl l-benzyl-4-[(3-fluorophenyl)amino]piperidine-4-carboxylate (4A-2)
- a solution of 5g (16 mmol) l-benzyl-4-[(3-fluorophenyl)amino]piperidine-4-carbonitrile in 20 mL concentrated H 2 SO 4 was stirred vigorously for 18 hr, then cooled in an ice bath. The mixture was neutralized by slow addition of concentrated NaOH and ice chips, and the resulting heterogeneous mixture extracted with 500 mL CH 2 Cl 2 .
- Step 3 ethyl 4-[acetyl(3-fluorophenyl)amino]-l-benzylpiperidrne-4-carboxylate (4A-3)
- a solution of 0.2g (0.56 mmol) ethyl l-benzyl-4-[(3-fluorophenyl)amino]piperidine-4-carboxylate in 2.6 mL (28 mmol) acetic anhydride (neat) was heated to 120 0 C for 20 hr, then poured into 50 mL of a 1: 1 mixture of water and concentrated NH4OH, extracted with 100 mL EtOAc, washed with water then brine, dried over MgSO 4 , filtered, and concentrated.
- Step 4 8-benzyl-l-(3-fluorophenyl)-l,8-diazaspiro[4.5]decane-2,4-dione (4A-4)
- a 0 0 C solution of 0.86g (2.2 mmol) ethyl 4-[acetyl(3-fluorophenyl)amino]-l-benzylpiperidine-4- carboxylate in 1 mL THF was added 3.2 mL (6.5mmol, 2M solution in heptane, THF, ethylbenzene) lithium diisopropylamide solution and the reaction mixture was allowed to slowly warm to rt and stirred for an additional 14 hr.
- Step 6 to give 8-benzyl-l-(3-fluorophenyl)-4-[(4-fluorophenyl)amino]-l,8-diazaspiro[4.5]dec-3-en-2-one.
- Strecker adduct 4A-1 is first acylated to give 4B-2 and then cyclized to 4B-3 using a base such as NaOMe.
- Structures of type 4B-3 and derivatives thereof are also of use for the intended invention described herein.
- Decarboxylation of structures of type 4B-3 using a strong acid, such as aqueous 6N HCl gives initially structures of type 4B-4a, which are also claimed for use in the invention.
- EXAMPLE 4B-7 8-Benzyl-4-[(2-ethylphenyl)amino]-l-(3-fluorophenyl)-l,8-diazaspiro[4.5]dec-3-en-2-one.
- Step 1 Acylation to give Intermediate of type 4B-2.
- ethyl 3-[(l- benzyl-4-cyanopiperidin-4-yl)(3-fluorophenyl)amino]-3-oxopropanoate (intermediate 4A-1 as describe above) in 150 niL CH 2 Cl 2 was added 4.9 mL (37.8 mmol) ethyl malonyl chloride and 5.1 mL (43.6 mmol) 2,6-lutidine. After 3 h, the reaction was diluted with 150 mL CH 2 Cl 2 and washed with water and brine.
- Step 2 8-benzyl-l-(3-fluorophenyl)-l,8-diazaspiro[4.5]decane-2,4-dione
- a rt solution of 14.9 g (35.2 mmol) ethyl 3-[(l-benzyl-4-cyanopiperidin-4-yl)(3-fluorophenyl)amino]- 3-oxopropanoate in 20 mL methanol was added 7.6 mL (42.2 mmol) 30% w/v sodium methoxide in methanol.
- Step C Condensation with amine to give structures of type 4B-4. 8-benzyl-4-[(2-ethylphenyl)amino]-l- (3 -fluorophenyl)- 1 ,8-diazaspiro[4.5]dec-3-en-2-one (4B-7)
- Scheme 5 depicts a method for synthesizing compounds with alternative R.3 groups, such as Example (5-3) below (Example (5-3) is depicted in enamine form, but may also exist in tautomeric imine form).
- Stepl tert-Butyl 4-(cyclohexylamino)-l-(3-fluorophenyl)-2-oxo-l,3,8-triazaspiro[4.5]dec-3-ene-8- carboxylate (5-1)
- Step 2 2: 4-(Cyclohexylamino)-l-(3-fluorophenyl)-2-oxo-l,3,8-triazaspiro[4.5]dec-3-ene dihydrochloride 5-2
- Step 3 8-(3-Bromobenzyl)-4-(cyclohexylamino)-l-(3-fluorophenyl)-l,3,8-triazaspiro[4.5]dec-3-en-2-one (5-3)
- Step 1 3-[(l-methylprop-2-enyl)oxy]benzaldehyde
- Step 2 4-(cyclohexylamino)-l-(3-fluorophenyl)-8- ⁇ 3-[(l-methylprop-2-enyl)oxy]benzyl ⁇ -l,3,8- triazaspiro [4.5] dec-3 -en-2-one
- Step 2 4-(cyclohexylamino)-l-(3-fluorophenyl)-8-(3-isopropoxybenzyl)-l,3,8-triazaspiro[4.5]dec-3-en- 2-one (5-6)
- Scheme 6 depicts a method for preparing compounds wherein Rl is methyl, such as Examples (6-3) - (6-6) below.
- Examples (6-3)-(6-6) are depicted in enamine form, but may also exist in tautomeric imine form as described above.
- Scheme 7 demonstrates another method for preparing compounds with alternate R.3 groups (such as Example (7-2) and (7-3).
- Example (7-2) and (7-3) are depicted in enamine form but may also exist in tautomeric imine form.
- Step 1 3- ⁇ [4-(cyclohexylamino)-l-(3-fluorophenyl)-2 -oxo-1, 3,8-triazaspiro[4.5]dec-3-en-8- yl]methyl ⁇ phenylboronic acid (7-1)
- Step 2 4-(cyclohexylamino)- 1 -(3 -fluorophenyl)-8-[3-(4-methylpyridin-3-yl)benzyl] -1,3,8- triazaspiro[4.5]dec-3-en-2-one (7-3)
- Examples (7-4)- (7-109) listed below in Table 3 were prepared in a manner similar to that described in Schemes 3, 5 and 7.
- Examples (7-4) - (7-109) and their pharmaceutically acceptable salts are depicted in enamine form, but may also exist in tautomeric imine form.
- Table 3- Compounds (or salts thereof) Synthesized According to Schemes 5 and 7, using methods similar to that described for Examples (5-3) - (5-5) and (7-2) - (7-3).
- Scheme 8 describes a method for preparation of compounds containing an aminopyridine R.3 substituent, such as Examples (8-1) - (8-4).
- Examples (8-1) - (8-4) are depicted in enamine form, but may also exist in tautomeric imine form.
- reaction vessel was capped and warmed to 110 0 C overnight.
- the clear toluene layer was decanted and the residue extracted (2x hot toluene).
- the combined organic extracts were concentrated to dryness in vacuo, the residue dissolved in 900 ⁇ L DMF and purified by preparative HPLC (5->95% CH 3 CN/H 2 O over 30m, 0.05% added TFA, C18 PRO YMC 20x150 mm)to afford, after lyophilization, 8- ⁇ [6- (benzylamino)pyridinium-3-yl]methyl ⁇ -4-(cyclohexylamino)-l-(3-fluorophenyl)-2 -oxo-1, 3-diaza-8- azoniaspiro[4.5]dec-3-ene bis(trifluoroacetate) as a white fluffy solid.
- Example (8-5) - (8-25) listed below in Table 4 were prepared in a manner similar to that described in Scheme 8.
- Examples (8-5) - (8-25) and their pharmaceutically acceptable salts are depicted in enamine form, but may also exist in tautomeric imine form.
- Example (8-26) was prepared in the same manner as (8-5)-(8-25) starting with 8-[(6-bromophenyl)methyl]-4- (cyclohexylamino)-l-(3-fluorophenyl)-l,3,8-triazaspiro[4.5]dec-3-en-2-one.
- Step 1 tert-Butyl 2-hydroxypropylcarbamate (9-2): tert-Butyl 2-hydroxypropylcarbamate (2): Biorg. Med Chem., 6, 1998 2405-2419 Added Boc anyhdride (8.19g, 37.4 mmole) in CH 2 Cl 2 (30mL)to a solution of l-amino-2-propanol in 77 mL of 10% Et3N in MeOH. Let stir at rt under Argon. After 6 hr, concentrated in vacuo. Purified on a 12Og redisep coumn eluting with 1-1 EtOAc Hex. Concentrated clean fractions in vacuo.
- Step 2 t-Butyl 2,3-(formylphenoxy)propylcarbamate (9-3) t-Butyl 2,3-(formylphenoxy)propylcarbamate Dissolved triphenylphosphine (0.805g, 3.07 mmole), 3-hydroxybenzaldehyde (0.25g, 2.047 mmole) and t-butyl 2-hydroxypropylcarbamate(0.395g, 2.25 mmole, ) in dry toluene (5 mL) at rt. Added ADDP
- Mass spec (m+1) 508 8-[3-(2-amino-l-(R)- methylethoxy)benzyl]-4-(cyclohexylamino)-l-(3-fluorophenyl)-l,3,8- triazaspiro[4.5]dec-3-en-2-one and 8-[3-(2-amino-l-(S)- methylethoxy)benzyl]-4-(cyclohexylamino)-l- (3-fluorophenyl)-l,3,8-triazaspiro[4.5]dec-3-en-2-one (9-5 and 9-5)
- Methanesulfonyl chloride (.019 mL, 0.245 mmole) was added dropwise to a solution of 8- ⁇ 3-[(6- aminohexyl)oxy]benzyl ⁇ -4-(cyclohexylamino)-l-(3-fluorophenyl)-l,3,8-triazaspiro[4.5]dec-3-en-2-one (0.116 mg, 0.222 mmole)in 5 mL CH2CI2 at rt, and Et ⁇ N (0.034 mL, 0.245 mmole. After stirring for 0.5 hr at rt, the reaction was concentrated in vacuo.
- Table 5 The following compounds (or salts thereof) of Table 5 were prepared by methods similar to that described for 9-4, 9-5, and 9-6, utilizing the appropriate reagents.
- Method Z A 5ml dichloroethane suspension of 0.05g(0.1 lmmol) 4-(cyclohexylamino)-8-[3- aminobenzyl]-l-(3-fluorophenyl)-l,3,8-triazaspiro[4.5]dec-3-en-2-one (Example 11-2), 0.01mL(0.13mmol) 2-methylpentan-l-al, 0.05mL HOAc, and 0.05g (0.22mmol) sodium triacetoxyborohydride was allowed to stir at rt overnight. The reaction was quenched with saturated aqueous NaHCO 3 , diluted with CH 2 CI 2 and allowed to stir vigorously for 15 min.
- Example (12-4) is depicted below in enamine form, but may also exist in tautomeric imine form, as described above Scheme 12
- Step 1 3-(3-bromophenyl)-2-methylpropanenitrile (12-2).
- Step 2 3-(3-formylphenyl)-2-methylpropanenitrile (12-3).
- a 3-neck flask equipped with a condenser and 2 septa was flushed with N 2 , then the flask was charged with 0.6g(2.7mmol) 3-(3-bromophenyl)-2-methylpropionitrile(9-2), 0.275g(4mmol) sodium formate, 0.155g(0.13mmol) tetrakis(triphenylphospine)Palladium and flushed with CO from a balloon for 5 min.
- the solids were suspended in 1OmL DMF and CO continued to bubble into the liquid.
- the flask was heated in an oil bath at 100' for 20min then 110' while stirring.
- Step 3 3-(3- ⁇ [4-(cyclohexylamino)-l-(3-fluorophenyl)-2-oxo-l,3,8-triazaspiro[4.5]dec-3-en-8- yl]methyl ⁇ phenyl)-2-methylpropanenitrile (12-4).
- the compound was prepared in a manner similar to that described in WO 02/076983 (page 80). Dry isopropanol (25 mL, 42.2 mmole) was placed in an oven-dried 250 ml 3 necked flask under nitrogen, sodium spheres (0.5g, 21.1 mmol) were added and the reaction was heated to 80° C to dissolve the sodium. 2,6 dibromopyridine (1Og, 42.2 mmole) was added as a solid and the reaction was heated at 95 0 C . After heating for -2.5 hr the reaction was cooled and partitioned between ether and water.
- ⁇ -isopropoxypyridine ⁇ -carbaldehyde was prepared in a manner similar to that described in D. L Comins, M.O. Killpack, J. Org. Chem. 1990 55 69-73 for the methoxy analog.
- 2-bromo-6-isopropoxypyridine (Ig, 4.6 mmole) was dissolved in 10ml dry THF under argon, cooled to -78° C and treated with added 2.5 M n-Butyl lithium ( 1.9 mL, 4.8 mmole) keeping the temperature of the reaction below -65° C.
- Step 3 4-(cyclohexylamino)-l-(3-fluorophenyl)-8-[(6-isopropoxypyridin-2-yl)methyl]-l,3,8- triazaspiro[4.5]dec-3-en-2-one.
- This compound was prepare in a manner similar to that described for example (5-6) using the intermediate from scheme 13 step 2 and compound (5-2).
- LCMS (m+l) 494.1
- Scheme 14A depicts a method for preparing 8-Benzyl-l-phenyl-4-thioxo-l,3,8-triaza- spiro[4.5]decan-2-one 14A-5 intermediates useful for making compounds of the invention.
- the commercially available 14A-1 may be alkylated with an appropriate alkyl halide like benzyl chloride using a suitable base like potassium carbonate in a suitable solvent like acetonitrile to afford 14A-2.
- a similar method is described by M. E. Kopach et. al. J. Org .Chem. 2003 68 5739-5741.
- 14A-3 A modified Strecker reaction of A-2 yields 14A-3; and followed by the treatment of chlorosulfonyl isocyanate in methylene chloride to afford 14A-4.
- 14A-5 is obtained by the thiolation of 14A-4 by Na 2 S.
- Step 2 l-(3-Isopropoxy-benzyl)-piperidin-4-one monohydrate (14-4)
- Step 3 4-(3-Fluoro-phenyl)-l-(3-isopropoxy-benzyl)-piperidine-4-carbomtrile (14-5)
- TMSCN 2.98g, 30 mmol
- LCMS (MH+ 368.3)
- Step 4 l-(3-Fluoro-phenyl)-8-(3-isopropoxy-benzyl)-4-thioxo-l,3,8-triaza-spiro[4.5]decan-2-one (14-7)
- the 4-(3-Fluoro-phenyl)-l-(3-isopropoxy-benzyl)-piperidine-4-carbonitrile 14-5 (5.28 g, 15.0 mmole) was dissolved into chloroform (15 mL).
- Step 6 l-(3-Fluorophenyl)-4-isobutylamino-8-(3-isopropoxy-benzyl)-l,3,8-triaza-spiro[4.5]dec-3-en-2- one (14-8).
- SNS-I 21.4 mg, 0.05mmol
- isobutyl amine 36.5 mg, 0.5 mmol
- the reaction mixture was heated at 110 0 C for 12 hr.
- the crude was purified by preparative HPLC to provide
- Table 6 The following compounds (or salts thereof) of table 6 were prepared in a manner similar to that described for scheme 14.
- R2 elaboration can be prepared by the methods described in Scheme 3, Scheme 14-A with various substituted aniline or by Suzuki coupling of 15-1 with a various of boronic acid or boronate ester.
- a representative procedure for elaboration of the R.2 substituent, as depicted in Scheme 15 below, can be used in the synthesis of various compounds of the invention, including Examples (15-2) - (15-42) below.
- the boronate ester can be prepared as describe in Scheme 15-A. Examples (15-2) - (15-42) are depicted below in enamine form, but may also exist in tautomeric imine form, as described above.
- the crude 15-B-2 (0.05Og, 0.20 mmol ), potassium acetate (0.059g O. ⁇ Ommol), bis(pinacolato)diboron (0.046g, 0.18 mmol) and PdCl 2 (dppf) (0.016g 0.020mmol) were added to a round bottom flask. Dry DMF (3 ml) was then added, and the flask was flushed with nitrogen, then capped. The resultant mixture was stirred and placed into an 8O 0 C oil bath. The reaction was followed by LCMS. Upon consumption of starting material, the reaction mixture was extracted with ethyl acetate / water 3-4 times, then washed with brine twice.
- Example 15-35 The Suzuki coupling was carried out using the standard conditions already described with the Boykin catalyst.
- Step 1 Step 2: 2'-[4-Cyclohexylamino-8-(3-isopropoxy-benzyl)-2-oxo-l,3,8-triaza-spiro[4.5]dec-3-en-l-yl]-4'- fluoro-biphenyl-4-sulfonic acid dimethylamide (15-2)
- the vial was capped, and placed to stir in a 90° C aluminum block over a hot plate.
- the reaction showed black palladium precipitate (approximately 3 hr)
- the reaction was cooled to rt.
- the reaction was then filtered through celite, and the resulting solution was purified directly by reverse phase HPLC in acetonitrile: water with 0.1% TFA, to provide the desired diaryl product as the TFA salt.
- N-(t-butyloxycarbonyl)-4-piperidone (10.13 g, 50.8 mmol, 1.0 eq) and the 2-bromo-5-fluoroaniline (11.6Og, 61.0 mmol, 1.2 eq.) were weighed into a flask.
- Acetic acid 75 mL was added, followed by trimethylsilyl cyanide (7.75 mL, 58.1 mmol, 1.14 eq.). The mixture was stirred at rt for 24 hr, and was then added to a mixture of 200 mL ice and 200 mL saturated ammonium hydroxide solution.
- the resulting solution was diluted with diethyl ether, and washed with 1 N hydrochloric acid, then dried over magnesium sulfate, filtered and concentrated.
- the crude aminohydantoin was purified by filtration through a 600 mL silica plug in 2 L fritted funnel, using 70% ethyl acetate in hexanes, followed by 100% ethyl acetate, as the elution solvents. This procedure yielded (2.0971g, 23%)of the desired product as a dark green semi-solid.
- a combined solution of A (6.54g, 30mmol), 50% w/w aqueous hydroxylamine (3.6mL, ⁇ Ommol), and 6OmL ethanol was refluxed for three hours. Ethanol was removed under high vacuum to yield B (7.3g, 96.9%) as a brittle foam.
- the reaction mixture was stirred for another one hr at rt, then added to a 0° C aqueous solution Of H 2 S (622.5mg Na 2 S + 313.OmL H 2 O + 622.5-650.0 ⁇ L acetic acid), which was prepared immediately before use.
- the resulting mixture was stirred at room temperature for 24 hr, then made alkaline with saturated sodium bicarbonate solution until pH>7, then extracted two times with ethyl acetate.
- the combined extracts was washed with brine, dried over magnesium sulfate and concentrated.
- the crude material was purified by preparative TLC (40% ethylacetate/60% hexane) to provide D (98.8mg, 38.0%) as an oil residue.
- Boc removal of E (30mg, 0.43mmol) was carried out using 0.5 ml 25%TFA/DCM at rt for 10 min.
- the TFA amine salt was dried in vacuo and redissolved in 0.5mL tetrahydrofuran.
- Triethylamine (0.09mmol, 12.5 ⁇ L) was added to the reaction solution, followed by acetyl chloride ( ⁇ 4.0 ⁇ L, 0.052mmol), and the reaction was then stirred at rt for 15 min.
- the crude reaction was purified by preparative reverse phase HPLC and lyophilized to provide 15-45 (6.4mg, 23.6%) MS (MH+) 633.3, as a fine yellow powder.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description




































































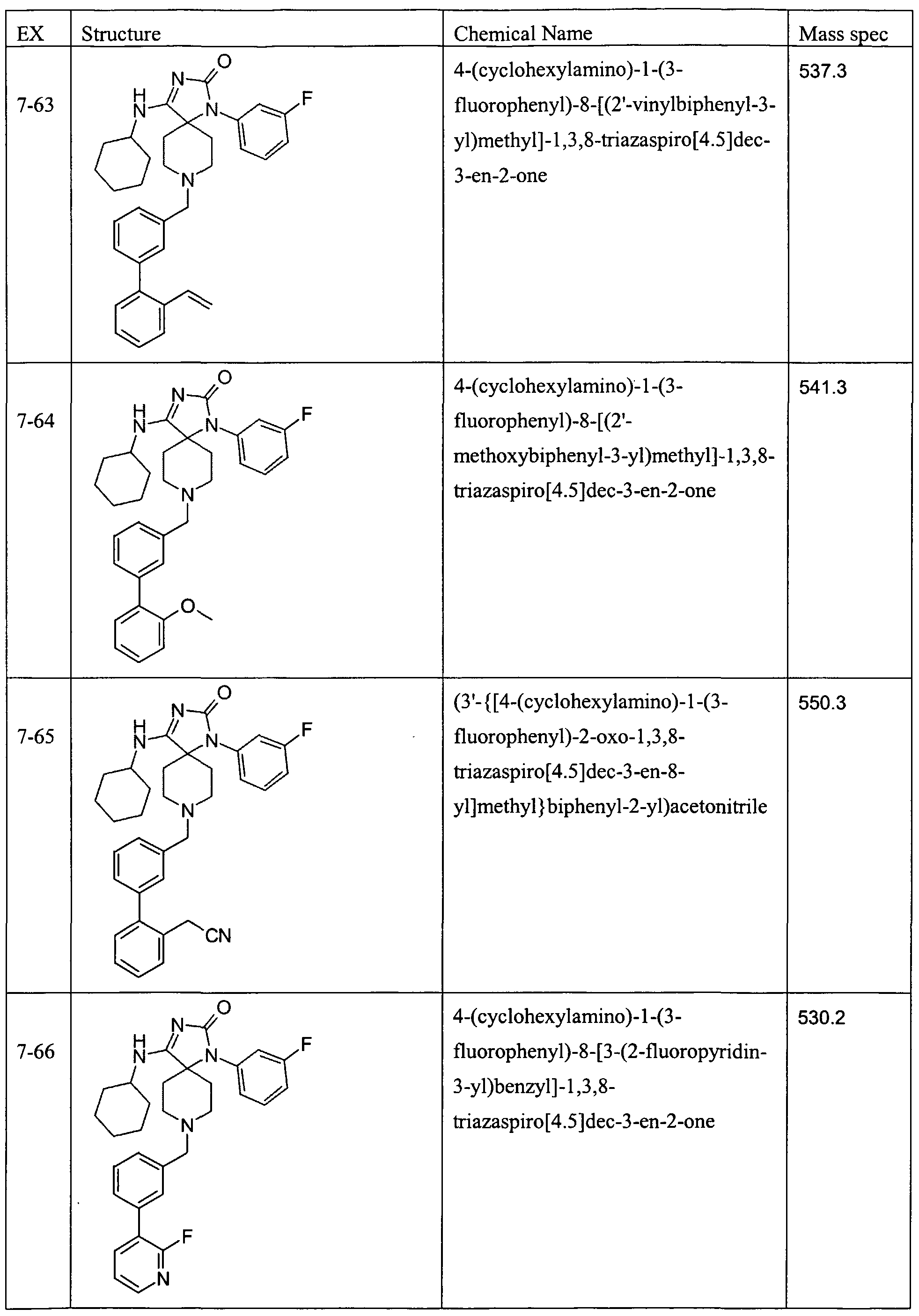














































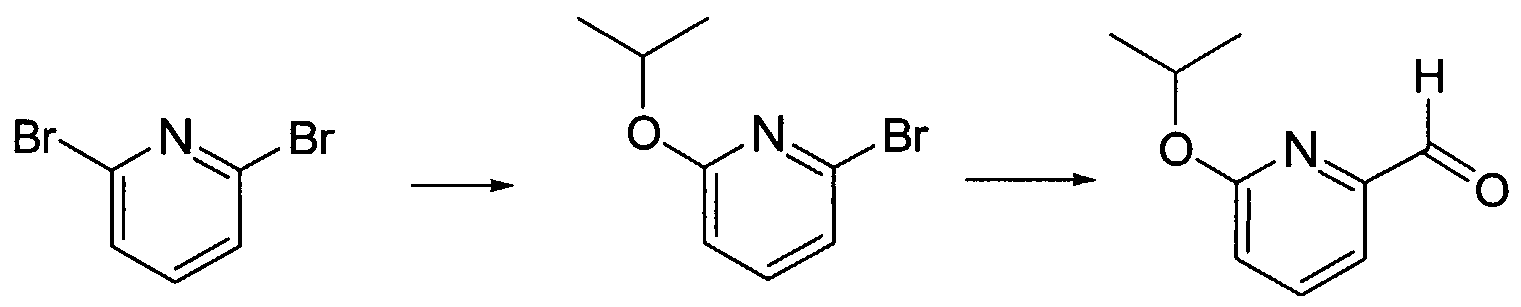










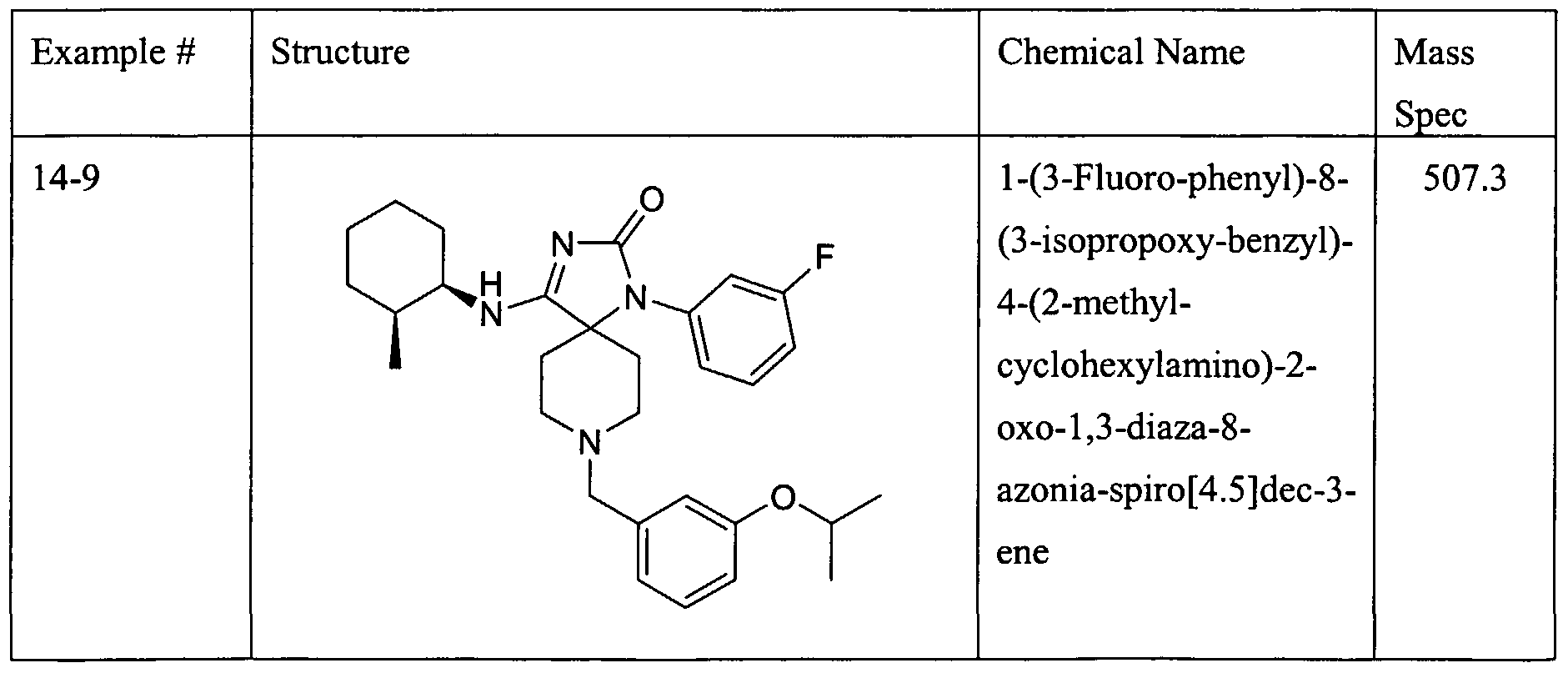












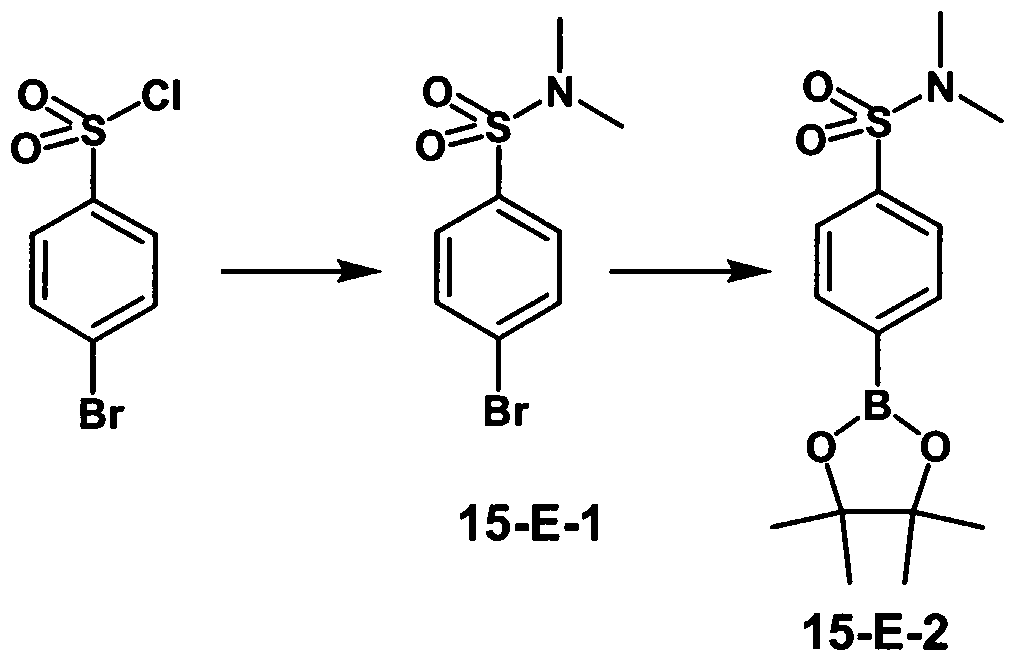




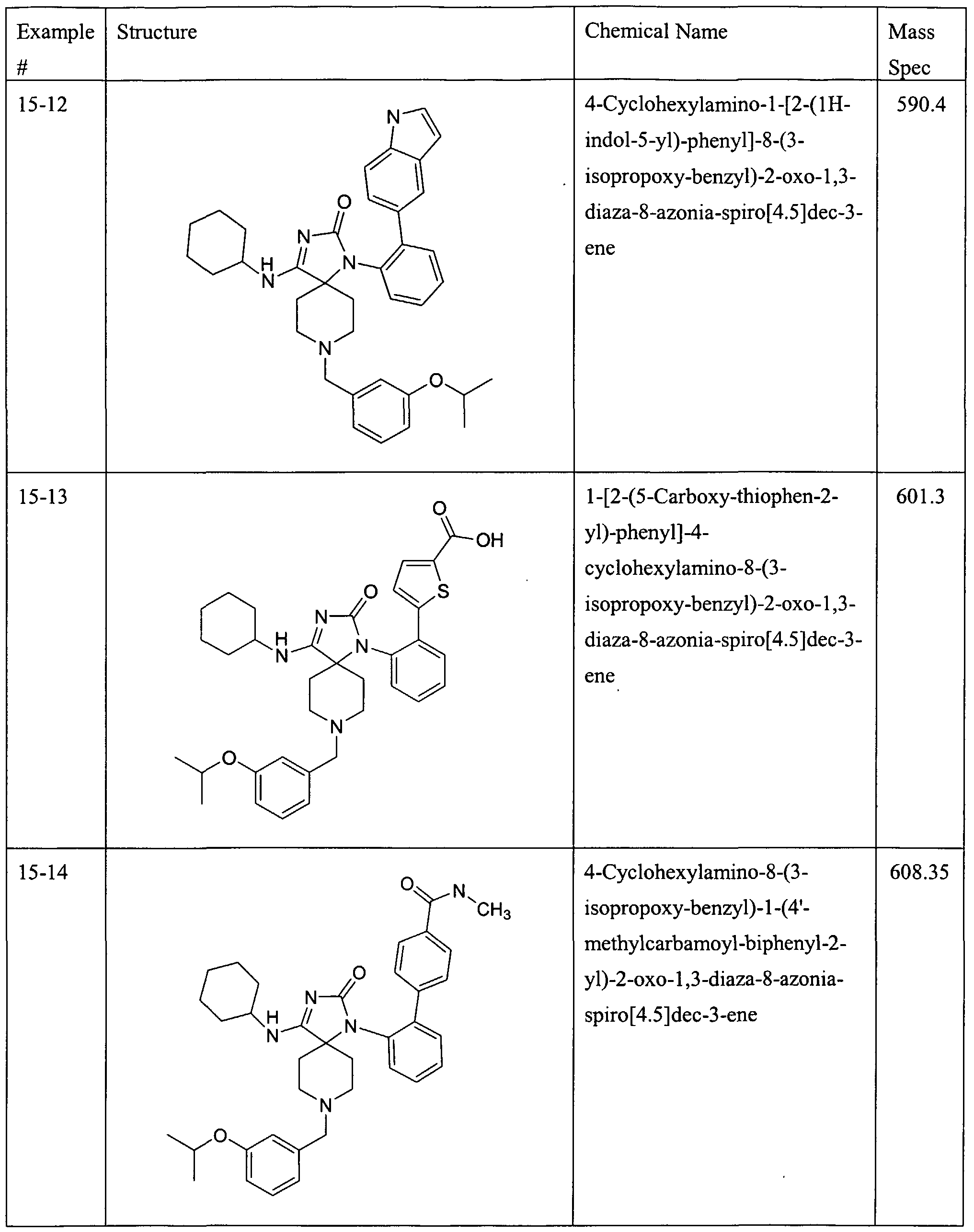
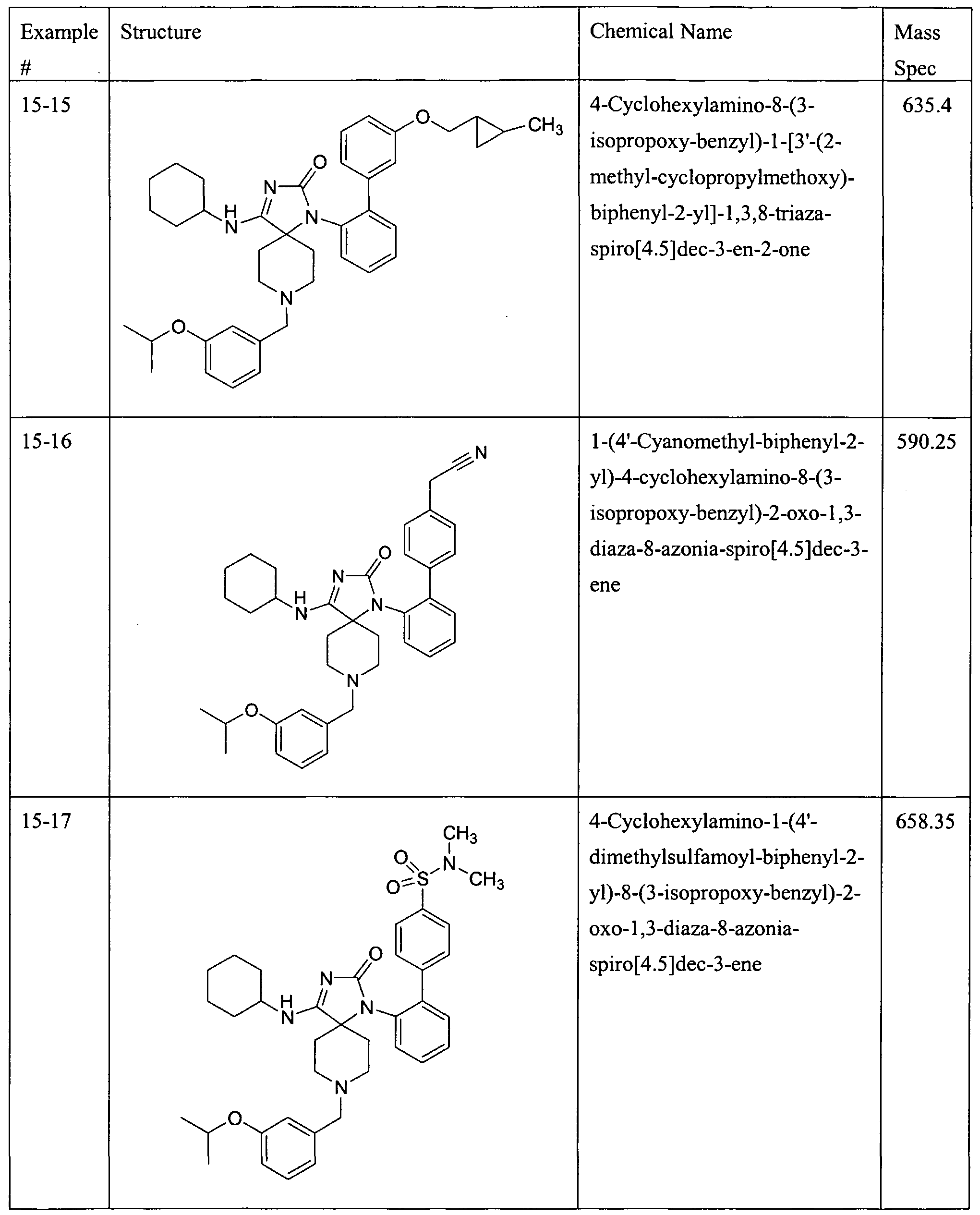



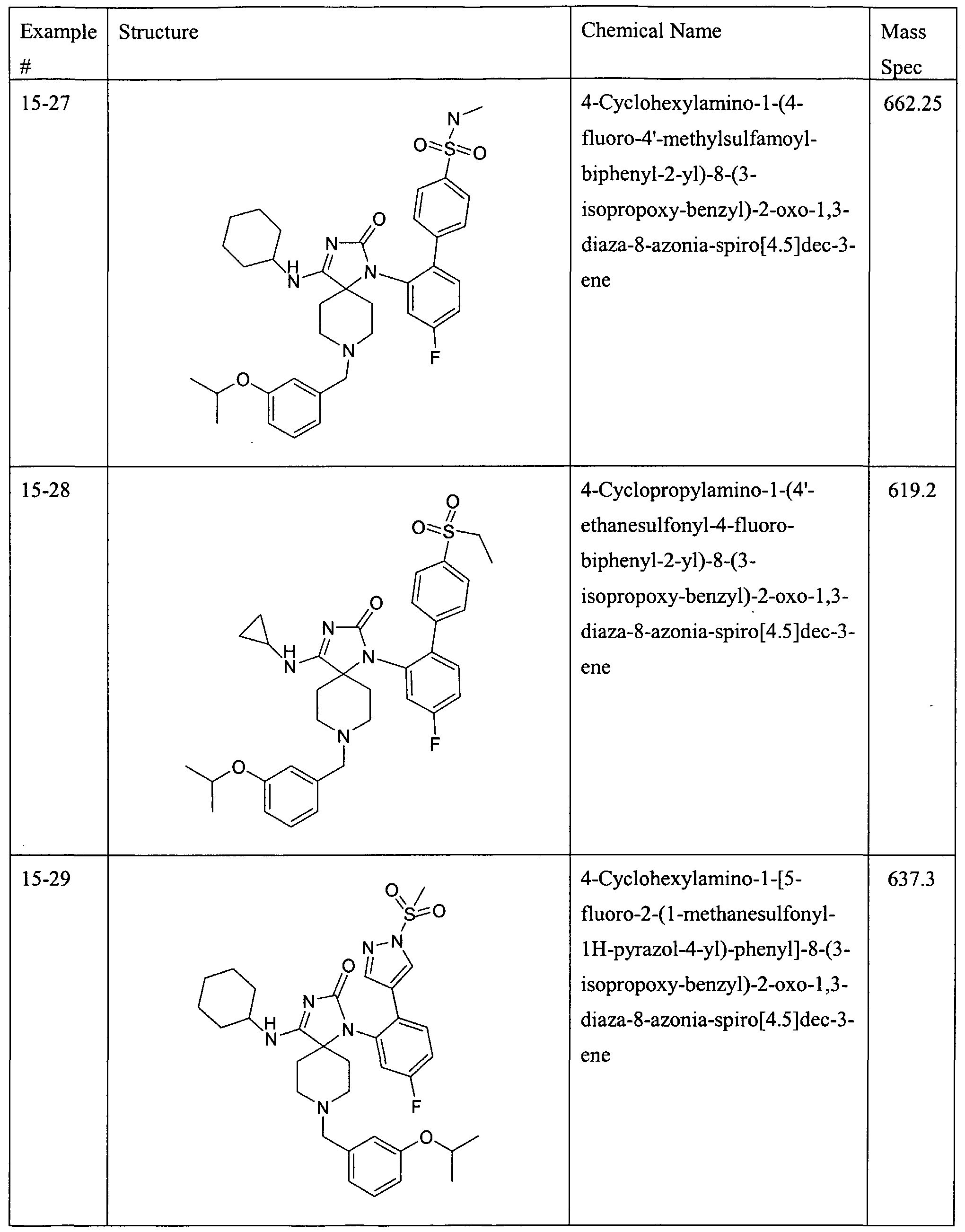












Claims










Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007536860A JP2008515989A (en) | 2004-10-13 | 2005-10-12 | Spiropiperidine compounds useful as β-secretase inhibitors for the treatment of Alzheimer's disease |
| EP05812233.4A EP1804794B1 (en) | 2004-10-13 | 2005-10-12 | Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzhermer s disease |
| AU2005295814A AU2005295814A1 (en) | 2004-10-13 | 2005-10-12 | Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of Alzhermer's disease |
| CA002583342A CA2583342A1 (en) | 2004-10-13 | 2005-10-12 | Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzhermer's disease |
| US11/663,388 US8114887B2 (en) | 2004-10-13 | 2005-10-12 | Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US61842004P | 2004-10-13 | 2004-10-13 | |
| US60/618,420 | 2004-10-13 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006044497A2 true WO2006044497A2 (en) | 2006-04-27 |
| WO2006044497A3 WO2006044497A3 (en) | 2006-09-08 |
Family
ID=36203499
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/036752 WO2006044497A2 (en) | 2004-10-13 | 2005-10-12 | Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzhermer’s disease |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8114887B2 (en) |
| EP (1) | EP1804794B1 (en) |
| JP (1) | JP2008515989A (en) |
| CN (1) | CN101068545A (en) |
| AU (1) | AU2005295814A1 (en) |
| CA (1) | CA2583342A1 (en) |
| WO (1) | WO2006044497A2 (en) |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007011810A1 (en) * | 2005-07-18 | 2007-01-25 | Merck & Co., Inc. | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2007130383A2 (en) * | 2006-04-28 | 2007-11-15 | Northwestern University | Compositions and treatments using pyridazine compounds and secretases |
| WO2008045250A1 (en) * | 2006-10-06 | 2008-04-17 | Merck & Co., Inc. | Macrocyclic spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008055947A1 (en) * | 2006-11-09 | 2008-05-15 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008054698A3 (en) * | 2006-10-30 | 2008-06-19 | Merck & Co Inc | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2008085509A1 (en) * | 2007-01-04 | 2008-07-17 | Merck & Co., Inc. | Bicyclic spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2009136350A1 (en) * | 2008-05-05 | 2009-11-12 | Pfizer Inc. | Novel class of spiro piperidines for the treatment of neurodegenerative diseases |
| JP2010502705A (en) * | 2006-09-07 | 2010-01-28 | メルク エンド カムパニー インコーポレーテッド | Spiropiperidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| US7700603B2 (en) | 2003-12-15 | 2010-04-20 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7759353B2 (en) | 2005-06-14 | 2010-07-20 | Schering Corporation | Substituted spiro iminopyrimidinones as aspartyl protease inhibitors, compositions, and methods of treatment |
| US7759354B2 (en) | 2005-06-14 | 2010-07-20 | Schering Corporation | Bicyclic guanidine derivatives as asparyl protease inhibitors, compositions, and uses thereof |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2011125006A3 (en) * | 2010-04-09 | 2011-12-29 | Pfizer Inc. | Sultam compounds |
| US8093254B2 (en) | 2006-12-12 | 2012-01-10 | Schering Corporation | Aspartyl protease inhibitors |
| US8110682B2 (en) | 2005-06-14 | 2012-02-07 | Schering Corporation | Preparation and use of compounds as aspartyl protease inhibitors |
| US20120041039A1 (en) * | 2007-11-01 | 2012-02-16 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2014071298A1 (en) | 2012-11-05 | 2014-05-08 | Nant Holdings Ip, Llc | Cyclic sulfonamide containing derivatives as inhibitors of hedgehog signaling pathway |
| US8722708B2 (en) | 2005-06-14 | 2014-05-13 | Merck Sharp & Dohme Inc. | Substituted isoindolines as aspartyl protease inhibitors |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US9018391B2 (en) | 2012-08-27 | 2015-04-28 | Boehringer Ingelheim International Gmbh | Inhibitors of beta-secretase |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| EP2776437A4 (en) * | 2011-11-07 | 2015-06-24 | Sunovion Pharmaceuticals Inc | Modulators of opioid receptors and methods of use thereof |
| US9145426B2 (en) | 2011-04-07 | 2015-09-29 | Merck Sharp & Dohme Corp. | Pyrrolidine-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9181236B2 (en) | 2011-08-22 | 2015-11-10 | Merck Sharp & Dohme Corp. | 2-spiro-substituted iminothiazines and their mono-and dioxides as bace inhibitors, compositions and their use |
| US9212153B2 (en) | 2009-03-13 | 2015-12-15 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| US9221839B2 (en) | 2011-04-07 | 2015-12-29 | Merck Sharp & Dohme Corp. | C5-C6 oxacyclic-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9259319B2 (en) | 2005-08-19 | 2016-02-16 | Bioventrix, Inc. | Method and device for treating dysfunctional cardiac tissue |
| US9290477B2 (en) | 2012-09-28 | 2016-03-22 | Vitae Pharmaceuticals, Inc. | Inhibitors of β-secretase |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102317289A (en) * | 2008-11-23 | 2012-01-11 | 辉瑞大药厂 | Lactams as beta secretase inhibitors |
| JP5828848B2 (en) | 2010-02-24 | 2015-12-09 | ヴァイティー ファーマシューティカルズ,インコーポレイテッド | Beta-secretase inhibitor |
| TWI557112B (en) | 2012-03-05 | 2016-11-11 | 百靈佳殷格翰國際股份有限公司 | Inhibitors of beta-secretase |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3639409A (en) | 1967-06-22 | 1972-02-01 | Sankyo Co | 1 3 8-triaza-2-oxo- or thioxo-3-substituted-4-oxo or imin 7 9 9-tetraalkyl spiro(4 o-75)decanes |
| US3542729A (en) | 1968-03-19 | 1970-11-24 | Sankyo Co | Stabilization of synthetic polymers |
| JPS4920973B1 (en) | 1970-05-28 | 1974-05-29 | ||
| US4066615A (en) | 1971-06-05 | 1978-01-03 | Sankyo Company Limited | Polymer compositions containing piperidine derivatives as stabilizers |
| DE3643890A1 (en) | 1986-12-22 | 1988-06-30 | Basf Ag | NEW POLYALKYLPIPERIDE INDEXIVES WITH ALKYLENE BRIDGES, THEIR USE AS A STABILIZER AND INTERMEDIATE PRODUCTS |
| US5221675A (en) * | 1989-12-15 | 1993-06-22 | Abbott Laboratories | Aza-spirocyclic compounds that enhance cholinergic neurotransmission |
| JP2893097B2 (en) | 1989-12-20 | 1999-05-17 | 富士写真フイルム株式会社 | Silver halide color photographic materials |
| US5852029A (en) | 1990-04-10 | 1998-12-22 | Israel Institute For Biological Research | Aza spiro compounds acting on the cholinergic system with muscarinic agonist activity |
| US5534520A (en) * | 1990-04-10 | 1996-07-09 | Fisher; Abraham | Spiro compounds containing five-membered rings |
| EP1087770A4 (en) | 1998-06-15 | 2001-11-14 | Merck & Co Inc | Inhibitors of prenyl-protein transferase |
| US20040067950A1 (en) | 1998-07-27 | 2004-04-08 | Schering-Plough Corporation | High affinity ligands for nociceptin receptor ORL-1 |
| DE10012859A1 (en) | 1999-03-19 | 2000-09-21 | Ciba Sc Holding Ag | New (poly)ethers prepared by reacting hydroxy-2,2,6,6-tetramethyl-piperidine or -piperazine hindered amine light stabilizer with methylene halide are used for stabilizing organic material, e.g. polymer or lacquer |
| KR100974901B1 (en) | 2001-12-28 | 2010-08-10 | 아카디아 파마슈티칼스 인코포레이티드 | Spiroazacyclic compounds as monoamine receptor modulators |
| AU2003223093B2 (en) | 2002-05-03 | 2010-02-04 | Israel Institute For Biological Research | Methods and compositions for treatment of central and peripheral nervous system disorders and novel compounds useful therefor |
| AU2003274053A1 (en) | 2002-10-22 | 2004-05-13 | Glaxo Group Limited | Aryloxyalkylamine derivates as h3 receptor ligands |
-
2005
- 2005-10-12 US US11/663,388 patent/US8114887B2/en active Active
- 2005-10-12 WO PCT/US2005/036752 patent/WO2006044497A2/en active Application Filing
- 2005-10-12 CN CNA2005800343960A patent/CN101068545A/en active Pending
- 2005-10-12 CA CA002583342A patent/CA2583342A1/en not_active Abandoned
- 2005-10-12 AU AU2005295814A patent/AU2005295814A1/en not_active Abandoned
- 2005-10-12 JP JP2007536860A patent/JP2008515989A/en active Pending
- 2005-10-12 EP EP05812233.4A patent/EP1804794B1/en not_active Not-in-force
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1804794A4 * |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8937093B2 (en) | 2003-12-15 | 2015-01-20 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7700603B2 (en) | 2003-12-15 | 2010-04-20 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8178513B2 (en) | 2003-12-15 | 2012-05-15 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US9416108B2 (en) | 2003-12-15 | 2016-08-16 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8722708B2 (en) | 2005-06-14 | 2014-05-13 | Merck Sharp & Dohme Inc. | Substituted isoindolines as aspartyl protease inhibitors |
| US9382242B2 (en) | 2005-06-14 | 2016-07-05 | Merck Sharp & Dohme Corp. | Preparation and use of compounds as protease inhibitors |
| US8110682B2 (en) | 2005-06-14 | 2012-02-07 | Schering Corporation | Preparation and use of compounds as aspartyl protease inhibitors |
| US7759353B2 (en) | 2005-06-14 | 2010-07-20 | Schering Corporation | Substituted spiro iminopyrimidinones as aspartyl protease inhibitors, compositions, and methods of treatment |
| US7759354B2 (en) | 2005-06-14 | 2010-07-20 | Schering Corporation | Bicyclic guanidine derivatives as asparyl protease inhibitors, compositions, and uses thereof |
| AU2006270084B2 (en) * | 2005-07-18 | 2011-08-25 | Merck Sharp & Dohme Corp. | Spiropiperidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2007011810A1 (en) * | 2005-07-18 | 2007-01-25 | Merck & Co., Inc. | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| JP2009502786A (en) * | 2005-07-18 | 2009-01-29 | メルク エンド カムパニー インコーポレーテッド | Spiropiperidine β-secretase inhibitors for treating Alzheimer's disease |
| US8211904B2 (en) | 2005-07-18 | 2012-07-03 | Merck, Sharp & Dohme Corp. | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2007011833A3 (en) * | 2005-07-18 | 2007-03-29 | Merck & Co Inc | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| US9259319B2 (en) | 2005-08-19 | 2016-02-16 | Bioventrix, Inc. | Method and device for treating dysfunctional cardiac tissue |
| WO2007130383A3 (en) * | 2006-04-28 | 2008-06-19 | Neuromedix Inc | Compositions and treatments using pyridazine compounds and secretases |
| WO2007130383A2 (en) * | 2006-04-28 | 2007-11-15 | Northwestern University | Compositions and treatments using pyridazine compounds and secretases |
| JP2010502705A (en) * | 2006-09-07 | 2010-01-28 | メルク エンド カムパニー インコーポレーテッド | Spiropiperidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| US8399473B2 (en) | 2006-10-06 | 2013-03-19 | Merck, Sharp & Dohme, Corp. | Macrocyclic spiropiperidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2008045250A1 (en) * | 2006-10-06 | 2008-04-17 | Merck & Co., Inc. | Macrocyclic spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2008054698A3 (en) * | 2006-10-30 | 2008-06-19 | Merck & Co Inc | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| US8293759B2 (en) | 2006-10-30 | 2012-10-23 | Merck, Sharp & Dohme, Corp. | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2008055947A1 (en) * | 2006-11-09 | 2008-05-15 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| US8093254B2 (en) | 2006-12-12 | 2012-01-10 | Schering Corporation | Aspartyl protease inhibitors |
| US8377954B2 (en) | 2007-01-04 | 2013-02-19 | Merck, Sharp & Dohme, Corp | Bicyclic spiropiperidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2008085509A1 (en) * | 2007-01-04 | 2008-07-17 | Merck & Co., Inc. | Bicyclic spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| EP2481408A2 (en) | 2007-03-01 | 2012-08-01 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| US8716529B2 (en) * | 2007-11-01 | 2014-05-06 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| US9056849B2 (en) | 2007-11-01 | 2015-06-16 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| JP2013216665A (en) * | 2007-11-01 | 2013-10-24 | Acucela Inc | Amine derivative compound for treating ophthalmic disease and disorder |
| US20120041039A1 (en) * | 2007-11-01 | 2012-02-16 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| US9452153B2 (en) | 2007-11-01 | 2016-09-27 | Acucela Inc. | Amine derivative compounds for treating ophthalmic diseases and disorders |
| AU2009245322B2 (en) * | 2008-05-05 | 2011-07-14 | Pfizer Inc. | Novel class of spiro piperidines for the treatment of neurodegenerative diseases |
| CN102083841A (en) * | 2008-05-05 | 2011-06-01 | 美国辉瑞有限公司 | Novel class of spiro piperidines for the treatment of neurodegenerative diseases |
| WO2009136350A1 (en) * | 2008-05-05 | 2009-11-12 | Pfizer Inc. | Novel class of spiro piperidines for the treatment of neurodegenerative diseases |
| US10336717B2 (en) | 2009-03-13 | 2019-07-02 | Vitae Pharmaceuticals, Llc | Inhibitors of beta-secretase |
| US9212153B2 (en) | 2009-03-13 | 2015-12-15 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US9029362B2 (en) | 2009-10-08 | 2015-05-12 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as brace inhibitors, compositions, and their use |
| US8940748B2 (en) | 2009-10-08 | 2015-01-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9687494B2 (en) | 2009-10-08 | 2017-06-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9475785B2 (en) | 2009-10-08 | 2016-10-25 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US9428475B2 (en) | 2009-10-08 | 2016-08-30 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011125006A3 (en) * | 2010-04-09 | 2011-12-29 | Pfizer Inc. | Sultam compounds |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| US9145426B2 (en) | 2011-04-07 | 2015-09-29 | Merck Sharp & Dohme Corp. | Pyrrolidine-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9221839B2 (en) | 2011-04-07 | 2015-12-29 | Merck Sharp & Dohme Corp. | C5-C6 oxacyclic-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9181236B2 (en) | 2011-08-22 | 2015-11-10 | Merck Sharp & Dohme Corp. | 2-spiro-substituted iminothiazines and their mono-and dioxides as bace inhibitors, compositions and their use |
| EP2776437A4 (en) * | 2011-11-07 | 2015-06-24 | Sunovion Pharmaceuticals Inc | Modulators of opioid receptors and methods of use thereof |
| US9580409B2 (en) | 2011-11-07 | 2017-02-28 | Sunovion Pharmaceuticals, Inc. | Modulators of opioid receptors and methods of use thereof |
| AU2012335980B2 (en) * | 2011-11-07 | 2017-08-17 | Sunovion Pharmaceuticals Inc. | Modulators of opioid receptors and methods of use thereof |
| AU2012335980C1 (en) * | 2011-11-07 | 2017-12-14 | Sunovion Pharmaceuticals Inc. | Modulators of opioid receptors and methods of use thereof |
| US9018391B2 (en) | 2012-08-27 | 2015-04-28 | Boehringer Ingelheim International Gmbh | Inhibitors of beta-secretase |
| US9290477B2 (en) | 2012-09-28 | 2016-03-22 | Vitae Pharmaceuticals, Inc. | Inhibitors of β-secretase |
| WO2014071298A1 (en) | 2012-11-05 | 2014-05-08 | Nant Holdings Ip, Llc | Cyclic sulfonamide containing derivatives as inhibitors of hedgehog signaling pathway |
| US9499539B2 (en) | 2012-11-05 | 2016-11-22 | Nantbioscience, Inc. | Cyclic sulfonamide containing derivatives as inhibitors of hedgehog signaling pathway |
| US10183013B2 (en) | 2012-11-05 | 2019-01-22 | Nant Holdings Ip, Llc | Cyclic sulfonamide containing derivatives as inhibitors of hedgehog signaling pathway |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2008515989A (en) | 2008-05-15 |
| EP1804794A2 (en) | 2007-07-11 |
| WO2006044497A3 (en) | 2006-09-08 |
| EP1804794A4 (en) | 2010-03-31 |
| CN101068545A (en) | 2007-11-07 |
| AU2005295814A1 (en) | 2006-04-27 |
| CA2583342A1 (en) | 2006-04-27 |
| US20070197571A1 (en) | 2007-08-23 |
| US8114887B2 (en) | 2012-02-14 |
| EP1804794B1 (en) | 2013-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1804794B1 (en) | Spiropiperidine compounds useful as beta-secretase inhibitors for the treatment of alzhermer s disease | |
| EP1910364B1 (en) | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP2091328B1 (en) | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| WO2008030412A2 (en) | Spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP1817312B1 (en) | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of alzheimer's disease | |
| WO2010094242A1 (en) | Spiropyrrolidine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| WO2007019078A2 (en) | Tricyclic beta-secretase inhibitors for the treatment of alzheimer's disease | |
| AU2009330234A1 (en) | Gamma secretase modulators | |
| CN108148060B (en) | Substituted heterocyclic compound and derivative thereof, pharmaceutical composition, preparation method and application thereof | |
| EP2117309B1 (en) | Bicyclic spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| WO2008045250A1 (en) | Macrocyclic spiropiperidine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP2443119A1 (en) | Gamma secretase modulators | |
| EP1912963B1 (en) | Cyclic ketal beta-secretase inhibitors for the treatment of alzheimer's disease | |
| NZ621092B2 (en) | COMPOUNDS AND COMPOSITIONS AS c-KIT KINASE INHIBITORS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 11663388 Country of ref document: US Ref document number: 2007197571 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1217/CHENP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005295814 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580034396.0 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005812233 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2583342 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007536860 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2005295814 Country of ref document: AU Date of ref document: 20051012 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005295814 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005812233 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 11663388 Country of ref document: US |