WO2006014762A1 - Substituted amide beta secretase inhibitors - Google Patents
Substituted amide beta secretase inhibitors Download PDFInfo
- Publication number
- WO2006014762A1 WO2006014762A1 PCT/US2005/025780 US2005025780W WO2006014762A1 WO 2006014762 A1 WO2006014762 A1 WO 2006014762A1 US 2005025780 W US2005025780 W US 2005025780W WO 2006014762 A1 WO2006014762 A1 WO 2006014762A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- substituted
- aryl
- mmol
- Prior art date
Links
- 239000002439 beta secretase inhibitor Substances 0.000 title abstract description 11
- 150000001408 amides Chemical class 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 178
- 150000003839 salts Chemical class 0.000 claims abstract description 53
- 238000000034 method Methods 0.000 claims abstract description 52
- 238000011282 treatment Methods 0.000 claims abstract description 27
- 230000004770 neurodegeneration Effects 0.000 claims abstract description 26
- 208000015122 neurodegenerative disease Diseases 0.000 claims abstract description 26
- 208000024827 Alzheimer disease Diseases 0.000 claims abstract description 23
- 208000010877 cognitive disease Diseases 0.000 claims abstract description 17
- 230000001149 cognitive effect Effects 0.000 claims abstract description 17
- 239000000544 cholinesterase inhibitor Substances 0.000 claims abstract description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 13
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims abstract description 10
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims abstract description 9
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 claims abstract description 8
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 claims abstract description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 105
- -1 -C(O)-alkyl Chemical group 0.000 claims description 67
- 125000003118 aryl group Chemical group 0.000 claims description 52
- 125000001072 heteroaryl group Chemical group 0.000 claims description 47
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 43
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 41
- 125000000623 heterocyclic group Chemical group 0.000 claims description 37
- 229910052739 hydrogen Inorganic materials 0.000 claims description 34
- 125000004475 heteroaralkyl group Chemical group 0.000 claims description 31
- 239000001257 hydrogen Substances 0.000 claims description 30
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 28
- 125000000304 alkynyl group Chemical group 0.000 claims description 23
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 22
- 125000003342 alkenyl group Chemical group 0.000 claims description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 16
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 16
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 16
- 125000003545 alkoxy group Chemical group 0.000 claims description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 14
- 229910052760 oxygen Inorganic materials 0.000 claims description 14
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 13
- 229910052757 nitrogen Inorganic materials 0.000 claims description 11
- 229940122041 Cholinesterase inhibitor Drugs 0.000 claims description 10
- 125000000732 arylene group Chemical group 0.000 claims description 10
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims description 10
- 125000002947 alkylene group Chemical group 0.000 claims description 9
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 125000002993 cycloalkylene group Chemical group 0.000 claims description 8
- 125000005843 halogen group Chemical group 0.000 claims description 8
- 229940099433 NMDA receptor antagonist Drugs 0.000 claims description 7
- 125000002252 acyl group Chemical group 0.000 claims description 7
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 7
- 125000004414 alkyl thio group Chemical group 0.000 claims description 7
- 239000003112 inhibitor Substances 0.000 claims description 7
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 claims description 7
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 6
- 125000004447 heteroarylalkenyl group Chemical group 0.000 claims description 6
- 125000005312 heteroarylalkynyl group Chemical group 0.000 claims description 6
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 6
- 230000002401 inhibitory effect Effects 0.000 claims description 6
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 5
- 125000005213 alkyl heteroaryl group Chemical group 0.000 claims description 5
- 125000005018 aryl alkenyl group Chemical group 0.000 claims description 5
- 125000004659 aryl alkyl thio group Chemical group 0.000 claims description 5
- 125000005015 aryl alkynyl group Chemical group 0.000 claims description 5
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 claims description 5
- 125000005110 aryl thio group Chemical group 0.000 claims description 5
- 125000004104 aryloxy group Chemical group 0.000 claims description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 125000005549 heteroarylene group Chemical group 0.000 claims description 5
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 claims description 5
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 claims description 4
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 4
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 4
- 125000003435 aroyl group Chemical group 0.000 claims description 4
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 4
- 230000015572 biosynthetic process Effects 0.000 claims description 4
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 claims description 4
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 claims description 4
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 claims description 4
- 125000005143 heteroarylsulfonyl group Chemical group 0.000 claims description 4
- 125000005368 heteroarylthio group Chemical group 0.000 claims description 4
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 4
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 claims description 4
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 4
- 125000003107 substituted aryl group Chemical group 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 claims description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 3
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 claims description 3
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 claims description 3
- 125000001153 fluoro group Chemical group F* 0.000 claims description 3
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 3
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 3
- OFPXSFXSNFPTHF-UHFFFAOYSA-N oxaprozin Chemical compound O1C(CCC(=O)O)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 OFPXSFXSNFPTHF-UHFFFAOYSA-N 0.000 claims description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 3
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 claims description 2
- 208000037259 Amyloid Plaque Diseases 0.000 claims description 2
- 102000013455 Amyloid beta-Peptides Human genes 0.000 claims description 2
- 108010090849 Amyloid beta-Peptides Proteins 0.000 claims description 2
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 claims description 2
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 claims description 2
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 claims description 2
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 claims description 2
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 claims description 2
- RVOLLAQWKVFTGE-UHFFFAOYSA-N Pyridostigmine Chemical compound CN(C)C(=O)OC1=CC=C[N+](C)=C1 RVOLLAQWKVFTGE-UHFFFAOYSA-N 0.000 claims description 2
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 claims description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 2
- 229960005370 atorvastatin Drugs 0.000 claims description 2
- 229960000590 celecoxib Drugs 0.000 claims description 2
- 230000008021 deposition Effects 0.000 claims description 2
- 229960001259 diclofenac Drugs 0.000 claims description 2
- 229960000616 diflunisal Drugs 0.000 claims description 2
- 229960003530 donepezil Drugs 0.000 claims description 2
- 229960003765 fluvastatin Drugs 0.000 claims description 2
- 229960003980 galantamine Drugs 0.000 claims description 2
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 claims description 2
- 229960001680 ibuprofen Drugs 0.000 claims description 2
- 229960000905 indomethacin Drugs 0.000 claims description 2
- 229960000991 ketoprofen Drugs 0.000 claims description 2
- 229960004752 ketorolac Drugs 0.000 claims description 2
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 claims description 2
- 229960004844 lovastatin Drugs 0.000 claims description 2
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 claims description 2
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 claims description 2
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical group C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 claims description 2
- 229960004640 memantine Drugs 0.000 claims description 2
- 229960004270 nabumetone Drugs 0.000 claims description 2
- 229960002009 naproxen Drugs 0.000 claims description 2
- 229960002362 neostigmine Drugs 0.000 claims description 2
- LULNWZDBKTWDGK-UHFFFAOYSA-M neostigmine bromide Chemical compound [Br-].CN(C)C(=O)OC1=CC=CC([N+](C)(C)C)=C1 LULNWZDBKTWDGK-UHFFFAOYSA-M 0.000 claims description 2
- 229960002739 oxaprozin Drugs 0.000 claims description 2
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 claims description 2
- 229960002965 pravastatin Drugs 0.000 claims description 2
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 claims description 2
- 229960002290 pyridostigmine Drugs 0.000 claims description 2
- 229960004136 rivastigmine Drugs 0.000 claims description 2
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 claims description 2
- 229960000672 rosuvastatin Drugs 0.000 claims description 2
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 claims description 2
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 claims description 2
- 229960001685 tacrine Drugs 0.000 claims description 2
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical group C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 claims description 2
- 229960001017 tolmetin Drugs 0.000 claims description 2
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 claims description 2
- 150000002431 hydrogen Chemical group 0.000 claims 9
- 102000003914 Cholinesterases Human genes 0.000 claims 1
- 108090000322 Cholinesterases Proteins 0.000 claims 1
- 239000002260 anti-inflammatory agent Substances 0.000 claims 1
- 229940124599 anti-inflammatory drug Drugs 0.000 claims 1
- 229940048961 cholinesterase Drugs 0.000 claims 1
- 229960005293 etodolac Drugs 0.000 claims 1
- XFBVBWWRPKNWHW-UHFFFAOYSA-N etodolac Chemical compound C1COC(CC)(CC(O)=O)C2=N[C]3C(CC)=CC=CC3=C21 XFBVBWWRPKNWHW-UHFFFAOYSA-N 0.000 claims 1
- 229960002390 flurbiprofen Drugs 0.000 claims 1
- SYTBZMRGLBWNTM-UHFFFAOYSA-N flurbiprofen Chemical compound FC1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-UHFFFAOYSA-N 0.000 claims 1
- 230000000926 neurological effect Effects 0.000 claims 1
- 229960002702 piroxicam Drugs 0.000 claims 1
- 229960000371 rofecoxib Drugs 0.000 claims 1
- 230000003637 steroidlike Effects 0.000 claims 1
- 229960000894 sulindac Drugs 0.000 claims 1
- 239000012453 solvate Substances 0.000 abstract description 37
- 239000003540 gamma secretase inhibitor Substances 0.000 abstract description 7
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 abstract description 2
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 abstract 1
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 abstract 1
- 239000002464 receptor antagonist Substances 0.000 abstract 1
- 229940044551 receptor antagonist Drugs 0.000 abstract 1
- 239000000203 mixture Substances 0.000 description 136
- 239000000047 product Substances 0.000 description 126
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 109
- 239000000243 solution Substances 0.000 description 78
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 66
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 64
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 54
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 50
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 46
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 40
- 239000012044 organic layer Substances 0.000 description 38
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 36
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 35
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 33
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical class CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- 125000004432 carbon atom Chemical group C* 0.000 description 26
- 235000019439 ethyl acetate Nutrition 0.000 description 25
- 239000005457 ice water Substances 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical group O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 239000012267 brine Substances 0.000 description 21
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 19
- 238000004440 column chromatography Methods 0.000 description 19
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 18
- 238000005160 1H NMR spectroscopy Methods 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 15
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 15
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 15
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 15
- 239000000651 prodrug Substances 0.000 description 15
- 229940002612 prodrug Drugs 0.000 description 15
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 15
- 241000124008 Mammalia Species 0.000 description 13
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 13
- 239000003937 drug carrier Substances 0.000 description 13
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- 229920006395 saturated elastomer Polymers 0.000 description 12
- 229910052717 sulfur Inorganic materials 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 11
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 10
- 125000004122 cyclic group Chemical group 0.000 description 10
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 125000006413 ring segment Chemical group 0.000 description 10
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- 239000010410 layer Substances 0.000 description 9
- 239000001301 oxygen Substances 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 125000001424 substituent group Chemical group 0.000 description 9
- 101710137189 Amyloid-beta A4 protein Proteins 0.000 description 8
- 101710151993 Amyloid-beta precursor protein Proteins 0.000 description 8
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 8
- 150000001299 aldehydes Chemical class 0.000 description 8
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 8
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 239000011593 sulfur Substances 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 description 5
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 description 5
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 5
- 0 C*OC[C@@](*)Cc1cccc(C#N)c1 Chemical compound C*OC[C@@](*)Cc1cccc(C#N)c1 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 125000004093 cyano group Chemical group *C#N 0.000 description 5
- 229940043279 diisopropylamine Drugs 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- 229940125373 Gamma-Secretase Inhibitor Drugs 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- 101000894895 Homo sapiens Beta-secretase 1 Proteins 0.000 description 4
- 239000012448 Lithium borohydride Substances 0.000 description 4
- 125000005038 alkynylalkyl group Chemical group 0.000 description 4
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 4
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 4
- KFPMLWUKHQMEBU-UHFFFAOYSA-N 3-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC(S(Cl)(=O)=O)=C1 KFPMLWUKHQMEBU-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 206010061818 Disease progression Diseases 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- 239000012614 Q-Sepharose Substances 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 230000003542 behavioural effect Effects 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 230000005750 disease progression Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- ZHUOMTMPTNZOJE-VIFPVBQESA-N (2s)-2-amino-3-(3-cyanophenyl)propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=CC(C#N)=C1 ZHUOMTMPTNZOJE-VIFPVBQESA-N 0.000 description 2
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 2
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- 108010017640 Aspartic Acid Proteases Proteins 0.000 description 2
- 102000004580 Aspartic Acid Proteases Human genes 0.000 description 2
- 102100021257 Beta-secretase 1 Human genes 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 102000003908 Cathepsin D Human genes 0.000 description 2
- 108090000258 Cathepsin D Proteins 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- JNQNZRQXLMEURU-PMERELPUSA-N N#Cc1cc(C[C@@H](C(OCc2ccccc2)=O)N(Cc2ccccc2)Cc2ccccc2)ccc1 Chemical compound N#Cc1cc(C[C@@H](C(OCc2ccccc2)=O)N(Cc2ccccc2)Cc2ccccc2)ccc1 JNQNZRQXLMEURU-PMERELPUSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 241000534944 Thia Species 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 125000003158 alcohol group Chemical group 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- 125000005206 alkoxycarbonyloxymethyl group Chemical group 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 2
- 229940073608 benzyl chloride Drugs 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000003920 cognitive function Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 2
- 102000044297 human BACE1 Human genes 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 230000003551 muscarinic effect Effects 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 239000002461 renin inhibitor Substances 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical class CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- FBUDYESOPLBQIR-NSHDSACASA-N (2s)-3-(3-bromophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC1=CC=CC(Br)=C1 FBUDYESOPLBQIR-NSHDSACASA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- NNYBQONXHNTVIJ-QGZVFWFLSA-N (R)-etodolac Chemical compound C1CO[C@](CC)(CC(O)=O)C2=C1C(C=CC=C1CC)=C1N2 NNYBQONXHNTVIJ-QGZVFWFLSA-N 0.000 description 1
- SYTBZMRGLBWNTM-SNVBAGLBSA-N (R)-flurbiprofen Chemical compound FC1=CC([C@H](C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-SNVBAGLBSA-N 0.000 description 1
- FTBPZRNURKMEFD-ONEGZZNKSA-N (e)-1-bromopent-2-ene Chemical compound CC\C=C\CBr FTBPZRNURKMEFD-ONEGZZNKSA-N 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000004530 1,2,4-triazinyl group Chemical group N1=NC(=NC=C1)* 0.000 description 1
- ASWYHZXKFSLNLN-UHFFFAOYSA-N 1,3-dibromo-5-fluorobenzene Chemical compound FC1=CC(Br)=CC(Br)=C1 ASWYHZXKFSLNLN-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- 125000005846 1-(alkanoyloxy)ethyl group Chemical group 0.000 description 1
- 125000005848 1-(alkoxycarbonyloxy)ethyl group Chemical group 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- JHLKSIOJYMGSMB-UHFFFAOYSA-N 1-bromo-3,5-difluorobenzene Chemical compound FC1=CC(F)=CC(Br)=C1 JHLKSIOJYMGSMB-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical class CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- 125000005847 1-methyl-1-(alkanoyloxy)-ethyl group Chemical group 0.000 description 1
- 125000005849 1-methyl-1-(alkoxycarbonyloxy)ethyl group Chemical group 0.000 description 1
- 125000001088 1-naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 102100027831 14-3-3 protein theta Human genes 0.000 description 1
- IHPYMWDTONKSCO-UHFFFAOYSA-N 2,2'-piperazine-1,4-diylbisethanesulfonic acid Chemical compound OS(=O)(=O)CCN1CCN(CCS(O)(=O)=O)CC1 IHPYMWDTONKSCO-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- KRLKXOLFFQWKPZ-UHFFFAOYSA-N 4-(bromomethyl)pyridine Chemical compound BrCC1=CC=NC=C1 KRLKXOLFFQWKPZ-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- LPNANKDXVBMDKE-UHFFFAOYSA-N 5-bromopent-1-ene Chemical compound BrCCCC=C LPNANKDXVBMDKE-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 102000009091 Amyloidogenic Proteins Human genes 0.000 description 1
- 108010048112 Amyloidogenic Proteins Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 229940125759 BACE1 protease inhibitor Drugs 0.000 description 1
- 238000009010 Bradford assay Methods 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- OTBBDCMPKZSOMD-UHFFFAOYSA-N CC(C)(C)OC(N(CCN1Cc2ccccc2)CC1=O)=O Chemical compound CC(C)(C)OC(N(CCN1Cc2ccccc2)CC1=O)=O OTBBDCMPKZSOMD-UHFFFAOYSA-N 0.000 description 1
- GZYBBUDZNUOGMF-BCRBLDSWSA-N CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(C#N)ccc2)N(Cc2ccccc2)Cc2ccccc2)C1=O)=O Chemical compound CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(C#N)ccc2)N(Cc2ccccc2)Cc2ccccc2)C1=O)=O GZYBBUDZNUOGMF-BCRBLDSWSA-N 0.000 description 1
- WLAIPYWRPVVJBN-GIWKVKTRSA-N CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(O)cc(F)c2)N(Cc2ccccc2)Cc2ccccc2)C1=O)=O Chemical compound CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(O)cc(F)c2)N(Cc2ccccc2)Cc2ccccc2)C1=O)=O WLAIPYWRPVVJBN-GIWKVKTRSA-N 0.000 description 1
- MTUYSHOIUGFHHY-UNMCSNQZSA-N CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(O)cc(F)c2)N)C1=O)=O Chemical compound CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(O)cc(F)c2)N)C1=O)=O MTUYSHOIUGFHHY-UNMCSNQZSA-N 0.000 description 1
- BRAHFTWKYGFIPI-UPVQGACJSA-N CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(O)cc(F)c2)NC(C)=O)C1=O)=O Chemical compound CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(O)cc(F)c2)NC(C)=O)C1=O)=O BRAHFTWKYGFIPI-UPVQGACJSA-N 0.000 description 1
- BSUARKFCSLHTOC-QARUCVQPSA-N CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(OCC=C)cc(F)c2)N(Cc2ccccc2)Cc2ccccc2)C1=O)=O Chemical compound CC(C)(C)OC(N(CCN1Cc2ccccc2)[C@@H](C[C@H](Cc2cc(OCC=C)cc(F)c2)N(Cc2ccccc2)Cc2ccccc2)C1=O)=O BSUARKFCSLHTOC-QARUCVQPSA-N 0.000 description 1
- CRZSQAXGBHQQJU-XMMPIXPASA-N CC(C)(C)OC(N(CC[C@H](Cc1cc(O)cc(F)c1)NC(C)=O)CCN(C)Cc1ccccc1)=O Chemical compound CC(C)(C)OC(N(CC[C@H](Cc1cc(O)cc(F)c1)NC(C)=O)CCN(C)Cc1ccccc1)=O CRZSQAXGBHQQJU-XMMPIXPASA-N 0.000 description 1
- IBCIMTNSTGEAPJ-AREMUKBSSA-N CC(C)(C)OC(NCc1cccc(C[C@@H](CCN(CCNCc2ccccc2)C(OC(C)(C)C)=O)N)c1)=O Chemical compound CC(C)(C)OC(NCc1cccc(C[C@@H](CCN(CCNCc2ccccc2)C(OC(C)(C)C)=O)N)c1)=O IBCIMTNSTGEAPJ-AREMUKBSSA-N 0.000 description 1
- TZKKHHTUEXQGKG-MUUNZHRXSA-N CC(N[C@H](CCNCCN(Cc1ccccc1)C=O)Cc1cccc(-c2cc(OC)ccc2)c1)=O Chemical compound CC(N[C@H](CCNCCN(Cc1ccccc1)C=O)Cc1cccc(-c2cc(OC)ccc2)c1)=O TZKKHHTUEXQGKG-MUUNZHRXSA-N 0.000 description 1
- GFUDFLIQQASJRJ-UIOOFZCWSA-N CC(N[C@H](C[C@@H]1NCCN(Cc2ccccc2)C1=[O-2])Cc1cc(-c2cccnc2)ccc1)=O Chemical compound CC(N[C@H](C[C@@H]1NCCN(Cc2ccccc2)C1=[O-2])Cc1cc(-c2cccnc2)ccc1)=O GFUDFLIQQASJRJ-UIOOFZCWSA-N 0.000 description 1
- QXZWODJQASFZRB-AREMUKBSSA-N CCCCCCOc1cc(F)cc(C[C@@H](CCNCCN(Cc2ccccc2)[I]=O)NC(C)=O)c1 Chemical compound CCCCCCOc1cc(F)cc(C[C@@H](CCNCCN(Cc2ccccc2)[I]=O)NC(C)=O)c1 QXZWODJQASFZRB-AREMUKBSSA-N 0.000 description 1
- CCILAFJQFQFGHX-VXKWHMMOSA-N CCCCCOc1cc(C[C@@H](C[C@@H](CNCC2)N2C(OC(C)(C)C)=O)NC(C)=O)cc(F)c1 Chemical compound CCCCCOc1cc(C[C@@H](C[C@@H](CNCC2)N2C(OC(C)(C)C)=O)NC(C)=O)cc(F)c1 CCILAFJQFQFGHX-VXKWHMMOSA-N 0.000 description 1
- GRJYGLBUQKOXLK-NSOVKSMOSA-N CCCCCOc1cc(F)cc(C[C@@H](C[C@@H](CN(CC2)S(c3cc(C)ccc3)(=O)=O)N2C(OC(C)(C)C)=O)NC(C)=O)c1 Chemical compound CCCCCOc1cc(F)cc(C[C@@H](C[C@@H](CN(CC2)S(c3cc(C)ccc3)(=O)=O)N2C(OC(C)(C)C)=O)NC(C)=O)c1 GRJYGLBUQKOXLK-NSOVKSMOSA-N 0.000 description 1
- SZQZMRSBRGETNX-XMMPIXPASA-N CCCOc1cc(F)cc(C[C@@H](CCNCCN(Cc2ccccc2)C=O)NC(C)=O)c1 Chemical compound CCCOc1cc(F)cc(C[C@@H](CCNCCN(Cc2ccccc2)C=O)NC(C)=O)c1 SZQZMRSBRGETNX-XMMPIXPASA-N 0.000 description 1
- OGAADESWJIXPHB-ZCYQVOJMSA-N CCCOc1cc(F)cc(C[C@@H](C[C@@H]2N(C)CCN(Cc3ccccc3)C2=O)NC(C)=O)c1 Chemical compound CCCOc1cc(F)cc(C[C@@H](C[C@@H]2N(C)CCN(Cc3ccccc3)C2=O)NC(C)=O)c1 OGAADESWJIXPHB-ZCYQVOJMSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 102100032373 Coiled-coil domain-containing protein 85B Human genes 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000868814 Homo sapiens Coiled-coil domain-containing protein 85B Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 208000031888 Mycoses Diseases 0.000 description 1
- HDVZQLFZFDFKKD-DEOSSOPVSA-N N#Cc1cc(C[C@@H](CI)N(Cc2ccccc2)Cc2ccccc2)ccc1 Chemical compound N#Cc1cc(C[C@@H](CI)N(Cc2ccccc2)Cc2ccccc2)ccc1 HDVZQLFZFDFKKD-DEOSSOPVSA-N 0.000 description 1
- 125000005850 N-(alkoxycarbonyl)aminomethyl group Chemical group 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 238000009004 PCR Kit Methods 0.000 description 1
- 239000007990 PIPES buffer Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 241000224016 Plasmodium Species 0.000 description 1
- 102000015499 Presenilins Human genes 0.000 description 1
- 108010050254 Presenilins Proteins 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- QTENRWWVYAAPBI-YZTFXSNBSA-N Streptomycin sulfate Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@H]1[C@H](N=C(N)N)[C@@H](O)[C@H](N=C(N)N)[C@@H](O)[C@@H]1O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@H]1[C@H](N=C(N)N)[C@@H](O)[C@H](N=C(N)N)[C@@H](O)[C@@H]1O QTENRWWVYAAPBI-YZTFXSNBSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- PVVUJMPJDPJDSE-UHFFFAOYSA-N [CH-]1C=CC=C1.C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.Cl.[Fe+2] Chemical compound [CH-]1C=CC=C1.C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.Cl.[Fe+2] PVVUJMPJDPJDSE-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- OXACROJQNCWAJF-UHFFFAOYSA-N acetyl acetate;2,2,2-trifluoroacetic acid Chemical compound CC(=O)OC(C)=O.OC(=O)C(F)(F)F OXACROJQNCWAJF-UHFFFAOYSA-N 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 229940013181 advil Drugs 0.000 description 1
- 230000016571 aggressive behavior Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 1
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000005466 alkylenyl group Chemical group 0.000 description 1
- 108010004469 allophycocyanin Proteins 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000003942 amyloidogenic effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000005251 aryl acyl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 125000003289 ascorbyl group Chemical class [H]O[C@@]([H])(C([H])([H])O*)[C@@]1([H])OC(=O)C(O*)=C1O* 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 230000006741 behavioral dysfunction Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005513 benzoazaindolyl group Chemical group 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- FCDPQMAOJARMTG-UHFFFAOYSA-M benzylidene-[1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichlororuthenium;tricyclohexylphosphanium Chemical compound C1CCCCC1[PH+](C1CCCCC1)C1CCCCC1.CC1=CC(C)=CC(C)=C1N(CCN1C=2C(=CC(C)=CC=2C)C)C1=[Ru](Cl)(Cl)=CC1=CC=CC=C1 FCDPQMAOJARMTG-UHFFFAOYSA-M 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 229940047475 cataflam Drugs 0.000 description 1
- 229940047495 celebrex Drugs 0.000 description 1
- 230000006727 cell loss Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000004976 cyclobutylene group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004980 cyclopropylene group Chemical group 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 229940070230 daypro Drugs 0.000 description 1
- 125000003493 decenyl group Chemical group [H]C([*])=C([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000005070 decynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- LVTYICIALWPMFW-UHFFFAOYSA-N diisopropanolamine Chemical compound CC(O)CNCC(C)O LVTYICIALWPMFW-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940072701 dolobid Drugs 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- 125000004705 ethylthio group Chemical group C(C)S* 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 210000002196 fr. b Anatomy 0.000 description 1
- 210000003918 fraction a Anatomy 0.000 description 1
- 210000000540 fraction c Anatomy 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 125000005643 gamma-butyrolacton-4-yl group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 125000003147 glycosyl group Chemical group 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 150000002373 hemiacetals Chemical group 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 210000004124 hock Anatomy 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical class I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- RCCPEORTSYDPMB-UHFFFAOYSA-N hydroxy benzenecarboximidothioate Chemical compound OSC(=N)C1=CC=CC=C1 RCCPEORTSYDPMB-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 229940089536 indocin Drugs 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- BWHLPLXXIDYSNW-UHFFFAOYSA-N ketorolac tromethamine Chemical compound OCC(N)(CO)CO.OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 BWHLPLXXIDYSNW-UHFFFAOYSA-N 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- SANNKFASHWONFD-ZCFIWIBFSA-N methyl (2r)-3-hydroxy-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound COC(=O)[C@@H](CO)NC(=O)OC(C)(C)C SANNKFASHWONFD-ZCFIWIBFSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- WZZMHOBVLAEJOD-UHFFFAOYSA-N methylsulfanylmethane;hydrobromide Chemical compound [Br-].C[SH+]C WZZMHOBVLAEJOD-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000005858 morpholino(C2-C3)alkyl group Chemical group 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 229940072709 motrin Drugs 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004370 n-butenyl group Chemical group [H]\C([H])=C(/[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- TXTHKGMZDDTZFD-UHFFFAOYSA-N n-cyclohexylaniline Chemical compound C1CCCCC1NC1=CC=CC=C1 TXTHKGMZDDTZFD-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- 125000005029 naphthylthio group Chemical group C1(=CC=CC2=CC=CC=C12)S* 0.000 description 1
- 229940090008 naprosyn Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 210000002682 neurofibrillary tangle Anatomy 0.000 description 1
- 230000003955 neuronal function Effects 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- ODUCDPQEXGNKDN-UHFFFAOYSA-N nitroxyl Chemical compound O=N ODUCDPQEXGNKDN-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000006201 parenteral dosage form Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000006678 phenoxycarbonyl group Chemical group 0.000 description 1
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 125000001476 phosphono group Chemical group [H]OP(*)(=O)O[H] 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 108091005981 phosphorylated proteins Proteins 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- IWELDVXSEVIIGI-UHFFFAOYSA-N piperazin-2-one Chemical compound O=C1CNCCN1 IWELDVXSEVIIGI-UHFFFAOYSA-N 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000005856 piperidino(C2-C3)alkyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- ALDITMKAAPLVJK-UHFFFAOYSA-N prop-1-ene;hydrate Chemical group O.CC=C ALDITMKAAPLVJK-UHFFFAOYSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- KPBSJEBFALFJTO-UHFFFAOYSA-N propane-1-sulfonyl chloride Chemical compound CCCS(Cl)(=O)=O KPBSJEBFALFJTO-UHFFFAOYSA-N 0.000 description 1
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- DNIAPMSPPWPWGF-UHFFFAOYSA-N propylene glycol Substances CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 125000005550 pyrazinylene group Chemical group 0.000 description 1
- STIKETVNLGXQCS-UHFFFAOYSA-N pyridazin-3-ylmethanol Chemical compound OCC1=CC=CN=N1 STIKETVNLGXQCS-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- SHNUBALDGXWUJI-UHFFFAOYSA-N pyridin-2-ylmethanol Chemical compound OCC1=CC=CC=N1 SHNUBALDGXWUJI-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000005551 pyridylene group Chemical group 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000005576 pyrimidinylene group Chemical group 0.000 description 1
- 125000005857 pyrrolidino(C2-C3)alkyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000006085 pyrrolopyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 229940087462 relafen Drugs 0.000 description 1
- 229940086526 renin-inhibitors Drugs 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000004426 substituted alkynyl group Chemical group 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004588 thienopyridyl group Chemical group S1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005556 thienylene group Chemical group 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- QGUALMNFRILWRA-UHFFFAOYSA-M tolmetin sodium Chemical compound [Na+].C1=CC(C)=CC=C1C(=O)C1=CC=C(CC([O-])=O)N1C QGUALMNFRILWRA-UHFFFAOYSA-M 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 229940019127 toradol Drugs 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229940063674 voltaren Drugs 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- BXIZKCIGQKZYGR-UHFFFAOYSA-M zinc;propane;bromide Chemical compound Br[Zn+].CC[CH2-] BXIZKCIGQKZYGR-UHFFFAOYSA-M 0.000 description 1
- 125000005853 β-dimethylaminoethyl group Chemical group 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/06—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members
- C07D241/08—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention relates to substituted amide beta secretase inhibitors, pharmaceutical compositions comprising said compounds, their use in the treatment of cardiovascular diseases, cognitive and neurodegenerative diseases, and their use as inhibitors of the Human Immunodeficiency Virus, plasmepsins, cathepsin D and protozoal enzymes.
- AD Alzheimer's disease
- Behavioral changes including confusion, depression and aggression also manifest as the disease progresses.
- the cognitive and behavioral dysfunction is believed to result from altered neuronal function and neuronal loss in the hippocampus and cerebral cortex.
- the currently available AD treatments are palliative, and while they ameliorate the cognitive and behavioral disorders, they do not prevent disease progression. Therefore there is an unmet medical need for AD treatments that halt disease progression.
- AD extracellular ⁇ -amyloid
- a ⁇ extracellular ⁇ -amyloid
- intracellular neurofibrillary tangles comprised of abnormally phosphorylated protein tau.
- Individuals with AD exhibit characteristic A ⁇ deposits, in brain regions known to be important for memory and cognition. It is believed that A ⁇ is the fundamental causative agent of neuronal cell loss and dysfunction which is associated with cognitive and behavioral decline.
- Amyloid plaques consist predominantly of A ⁇ peptides comprised of 40 - 42 amino acid residues, which are derived from processing of amyloid precursor protein (APP). APP is processed by multiple distinct protease activities.
- APP amyloid precursor protein
- a ⁇ peptides result from the cleavage of APP by ⁇ -secretase at the position corresponding to the N-terminus of A ⁇ , and at the C- terminus by ⁇ -secretase activity.
- APP is also cleaved by ⁇ -secretase activity resulting in the secreted, non-amyloidogenic fragment known as soluble APP.
- BACE-1 An aspartyl protease known as BACE-1 has been identified as the ⁇ -secretase activity responsible for cleavage of APP at the position corresponding to the N- terminus of A ⁇ peptides.
- a ⁇ has been shown to be toxic to neuronal cells in vitro and when injected into rodent brains.
- inherited forms of early-onset AD are known in which well-defined mutations of APP or the presenilins are present.
- Substituted amine BACE-1 inhibitors are disclosed in, WO 04/04396, WO 02/02505, WO 02/02506, WO 02/02512, WP 02/02518 and WO 02/02520.
- Renin inhibitors comprising a (1 -amino-2 hydroxy-2-heterocyclic)ethyl moiety are disclosed in WO 89/03842.
- WO 02/088101 discloses BACE inhibitors functionally described as being comprised of four hydrophobic moieties, as well as series of compounds preferably comprising a heterocyclic or heteroaryl moiety.
- the present invention relates to compounds having the structural formula I
- R 2 is hydrogen, alkyl or cycloalkyl
- R 3 is arylene, heteroarylene, heterocyclylene or cycloalkylene
- R 4 is hydrogen, -C(O)-alkyl, alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, alkoxyalkyl, heterocyclyl, heterocyclylalkyl, heteroaralkyl or heteroaryl;
- R 5 and R 7 are 1 to 4 moieties, each moiety being independently selected from hydrogen, -OH, alkyl, cycloalkyl, aryl, heteroaryl, aralkyl, heteroaralkyl, aralkoxy, heteroaralkoxy and alkoxy, with the proviso that when R 5 and R 7 are -OH, aralkoxy, heteroaralkoxy and alkoxy, R 5 and R 7 are not attached to a ring carbon adjacent to a ring nitrogen;
- R 9 and R 10 are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, aryl, heteroaryl, heterocyclyl, aralkyl, heteroaralkyl, heterocyclylalkyl, alkenyl and alkynyl; or R 9 and R 10 together with the nitrogen to which they are attached, form a 3-7 membered heterocyclyl ring;
- Y 0 or (H, H); m is O, 1 , 2, or 3; and n is 1 , 2, or 3; wherein each alkyl is optionally substituted with 1 to 3 moieties selected from the group consisting of halo, alkyl, aryl, cycloalkyl, cyano, hydroxy, alkoxy, alkylthio, amino, -NH(alkyl), -NH(cycloalkyl), -N(alkyl) 2 , carboxy and -C(O)O-alkyl; and wherein each arylene, heteroarylene, heterocyclylalkyl, heterocyclylene, cycloalkylene, cycloalkyl, cycloalkylalkyl, aryl, heteroaryl, heterocyclyl, aralkyl, heteroaralkyl, aralkoxy or heteroaralkoxy is optionally substituted with 1 to 4 moieties selected from the group consisting of -CF 3 , alkyl,
- Compounds represented by formula I are beta-secretase inhibitors useful for the prevention and treatment of Alzheimer's disease.
- the invention comprises: the method of treating a cardiovascular disease such as hypertension, renal failure, congestive heart failure or another disease modulated by renin inhibition; the method of treating Human Immunodeficiency Virus; the method of treating a cognitive or neurodegenerative disease such as Alzheimer's Disease; the method of inhibiting plasmepsins I and Il for treatment of malaria; the method of inhibiting Cathepsin D for the treatment of Alzheimer's Disease, breast cancer, and ovarian cancer; and the method of inhibiting protozoal enzymes, for example inhibition of Plasmodium falciparnum, for the treatment of fungal infections.
- Said method of treatment comprise administering at least one compound of formula I to a patient in need of such treatment.
- the invention comprises the method of treating Alzheimer's Disease comprising administering at least one compound of formula I to a patient in need of such treatment.
- the invention comprises the method of treating Alzheimer's Disease comprising administering to a patient in need of such treatment a combination of at least one compound of formula I and a cholinesterase inhibitor or a muscarinic mi agonist or m 2 antagonist.
- the invention relates to a kit comprising in separate containers in a single package pharmaceutical compositions for use in combination, in which one container comprises a compound of formula I in a pharmaceutically acceptable carrier and a second container comprises a cholinesterase inhibitor or a muscarinic Im 1 agonist or m 2 antagonist in a pharmaceutically acceptable carrier, the combined quantities being an effective amount to treat a cognitive disease or neurodegenerative disease such as Alzheimer's Disease.
- a cognitive disease or neurodegenerative disease such as Alzheimer's Disease.
- preferred compounds of the invention are those with the following stereochemistry:
- R 1 can be R 7 wherein n is 2 and R 6 and R 7 are defined herein.
- R 2 is preferably is alkyl, more preferably methyl .
- R 3 is arylene or halo-substituted arylene, more preferably phenylene or halo- substituted phenylene, specifically a halo-substituted phenylene, where said halo is preferably fluoride.
- R 4 is preferably hydrogen, methyl, ethyl, propyl, butyl, pentyl, hexyl, -C(O)Oethyl,
- R 5 and R 7 independently are alkyl substituted by a cycloalkyl group alkyl or substituted by an aryl or heteroaryl group.
- R 6 can be aralkyl
- R 6 is -S(O) 2 -R 8 or more preferably, R 6 is
- X is a bond connecting R 3 and R 4 , O, alkylene, -alkyl-NHSO 2 - or -alkyl-NHC(O)-;
- Y is O and n is 2.
- R 2 is alkyl
- R 3 is phenylene or halo-substituted phenylene
- R 4 is hydrogen, -C(O)-alkyl, alkyl, aryl, aralkyl, substituted aryl, substituted aralkyl, alkoxyalkyl, heteroaralkyl, substituted heteroaryl, substituted heteroaralkyl, alkoxyalkyl, heterocyclyl, heterocyclylalkyl, substituted heterocyclyl, substituted heterocyclylalkyl or heteroaryl;
- R 5 and R 7 are hydrogen, alkyl substituted by a cycloalkyl group, alkyl substituted by an aryl or heteroaryl group;
- R 6 is aralkyl or -S(O) 2 R 8 ;
- R 2 is preferably methyl
- R 4 is preferably hydrogen, methyl, ethyl, propyl, butyl, pentyl, hexyl, -C(O)Oethyl,
- alkyl means an aliphatic hydrocarbon group which may be straight or branched and comprising about 1 to about 20 carbon atoms in the chain. Preferred alkyl groups contain about 1 to about 12 carbon atoms in the chain. More preferred alkyl groups contain about 1 to about 6 carbon atoms in the chain. Branched means that one or more lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkyl chain.
- Alkynyl means an aliphatic hydrocarbon group containing at least one carbon-carbon triple bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain.
- Preferred alkynyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 4 carbon atoms in the chain.
- Branched means that one or more lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkynyl chain.
- “Lower alkynyl” means about 2 to about 6 carbon atoms in the chain which may be straight or branched.
- Aralkyl or “arylalkyl” means an aryl-alkyl- group in which the aryl and alkyl are as previously described. Preferred aralkyls comprise a lower alkyl group. Non-limiting examples of suitable aralkyl groups include benzyl, 2-phenethyl and naphthalenylmethyl. The bond to the parent moiety is through the alkyl.
- Alkylaryl means an alkyl-aryl- group in which the alkyl and aryl are as previously described. Preferred alkylaryls comprise a lower alkyl group. Non-limiting example of a suitable alkylaryl group is tolyl. The bond to the parent moiety is through the aryl.
- Non-limiting examples of suitable multicyclic cycloalkyls include 1-decalinyl, norbornyl, adamantyl and the like, as well as partially saturated species such as, for example, indanyl, tetrahydronaphthyl and the like. Further non- limiting examples of cycloalkyl include the following
- Ring system substituent may also mean a single moiety which simultaneously replaces two available hydrogens on two adjacent carbon atoms (one H on each carbon) on a ring system.
- moieties are methylene dioxy, ethylenedioxy, -C(CH 3 ) 2 - and the like which form moieties such as, for example:
- Heterocyclyl (or heterocycloalkyl) means a non-aromatic saturated monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 4 to about 7 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. There are no adjacent oxygen and/or sulfur atoms present in the ring system.
- Preferred heterocyclyls contain about 5 to about 6 ring atoms.
- the prefix aza, oxa or thia before the heterocyclyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom.
- Any -NH in a heterocyclyl ring may exist protected such as, for example, as an -N(Boc), -N(CBz), - N(Tos) group and the like; such protections are also considered part of this invention.
- the heterocyclyl can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein.
- the nitrogen or sulfur atom of the heterocyclyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S.S-dioxide.
- Heterocyclylene means a difunctional group obtained by removal of a hydrogen atom from an heterocyclyl group that is defined above.
- Non-limiting examples of heterocyclylene include piperidylene, pyrrolidinylene, piperazinylene, morpholinylene, thiomorpholinylene, thiazolidinylene, 1 ,4-dioxanylene, tetrahydrofuranylene and tetrahydrothiophenylene.
- hetero-atom containing ring systems of this invention there are no hydroxyl groups on carbon atoms adjacent to a N, O or S, nor is there an N or S group on a carbon adjacent to another heteroatom.
- N, O or S there are no hydroxyl groups on carbon atoms adjacent to a N, O or S, nor is there an N or S group on a carbon adjacent to another heteroatom.
- heteroarylalkyl or heteroaralkyl and “cycloalkylalkyl” mean a heteroaryl- or cycloalkyl- group in which the heteroaryl, cycloalkyl and alkyl are as previously described. Preferred groups contain a lower alkyl group. The bond to the parent moiety is through the alkyl.
- Heterocyclylalkyl means a heterocyclyl-alkyl group in which the heterocyclyl and the alkyl are as previously described. The bond to the parent moiety is through the alkyl.
- Preferred hydroxyalkyls contain lower alkyl.
- suitable hydroxyalkyl groups include hydroxymethyl and 2-hydroxyethyl.
- Aroyl means an aryl-C(O)- group in which the aryl group is as previously described. The bond to the parent moiety is through the carbonyl.
- suitable groups include benzoyl and 1- naphthoyl.
- Alkoxy means an alkyl-O- group in which the alkyl group is as previously described.
- suitable alkoxy groups include methoxy, ethoxy, n-propoxy, isopropoxy and n-butoxy.
- the bond to the parent moiety is through the ether oxygen.
- Alkoxyalkyl means an alkoxy-alkyl group in which the alkoxy and alkyl groups are as previously described.
- suitable alkoxyalkyl groups include ethoxyethyl, methoxymethyl and ethoxymethyl.
- the bond to the parent moiety is through the alkyl group.
- Aryloxy means an aryl-O- group in which the aryl group is as previously described.
- suitable aryloxy groups include phenoxy and naphthoxy.
- the bond to the parent moiety is through the ether oxygen.
- Aralkoxy means an aralkyl-O- group in which the aralkyl group is as previously described.
- suitable aralkoxy groups include benzyloxy and 1- or 2-naphthalenemethoxy. The bond to the parent moiety is through the ether oxygen.
- Alkylheteroaryl means an alkyl-heteroaryl group in which the alkyl and heteroaryl groups are as previously described. The bond to the parent moiety is through the heteroaryl.
- Alkylthio means an alkyl-S- group in which the alkyl group is as previously described.
- suitable alkylthio groups include methylthio and ethylthio.
- the bond to the parent moiety is through the sulfur.
- Arylthio means an aryl-S- group in which the aryl group is as previously described.
- suitable arylthio groups include phenylthio and naphthylthio. The bond to the parent moiety is through the sulfur.
- Alkylthio means an aralkyl-S- group in which the aralkyl group is as previously described.
- Non-limiting example of a suitable aralkylthio group is benzylthio.
- the bond to the parent moiety is through the sulfur.
- Alkoxycarbonyl means an alkyl-O-C(O)- group in which the alkyl group is as previously described.
- suitable alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
- the bond to the parent moiety is through the carbonyl.
- Aryloxycarbonyl means an aryl-O-C(O)- group in which the aryl group is as previously described.
- suitable aryloxycarbonyl groups include phenoxycarbonyl and naphthoxycarbonyl. The bond to the parent moiety is through the carbonyl.
- Alkoxycarbonyl means an aralkyl-O-C(O)- group in which the aralkyl group is as previously described.
- a suitable aralkoxycarbonyl group is benzyloxycarbonyl.
- the bond to the parent moiety is through the carbonyl.
- Alkylsulfonyl means an alkyl-S(O 2 )- group in which the alkyl group is as previously described. Preferred groups are those in which the alkyl group is lower alkyl. The bond to the parent moiety is through the sulfonyl.
- Arylsulfonyl means an aryl-S(O 2 )- group in which the aryl group is as previously described. The bond to the parent moiety is through the sulfonyl.
- Cycloalkylalkyl means a cycloalkyl-alkyl- group in which the cycloalkyl and alkyl group is as previously described. Preferred groups are those in which the alkyl group is lower alkyl. The bond to the parent moiety is through the alkyl.
- Heteroaralkoxy means a heteroaralkyl-O- group in which the heteroaralkyl group is as previously described. The bond to the parent moiety is through the ether oxygen.
- Heteroarylsulfonyl means a heteroaryl-S(O 2 )- group in which the heteroaryl group is as previously described. The bond to the parent moiety is through the sulfonyl.
- Heteroarylthio means a heteroaryl-S- group in which the heteroaryl group is as previously described. The bond to the parent moiety is through the sulfur.
- substituted means that one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- stable compound' or “stable structure” is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- multicyclic divalent groups for example, arylheterocycloalkylene, can be attached to other groups via bonds that are formed on either ring of said group.
- arylheterocycloalkylene can be attached to other groups via bonds that are formed on either ring of said group.
- Substitution on a cycloalkylalkyl, heterocycloalkylalkyl, arylalkyl, or heteroarylalkyl moiety includes substitution on the ring portion and/or on the alkyl portion of the group.
- variables can be the same or different.
- the wavy line 'vw ⁇ as a b onc j generally indicates a mixture of, or either of, the possible isomers, e.g., containing (R)- and (S)- stereochemistry.
- the possible isomers e.g., containing (R)- and (S)- stereochemistry.
- polymorphic forms of the compounds of formula I, and of the salts, solvates and prodrugs of the compounds of formula I, are intended to be included in the present invention.
- isolated or “in isolated form” for a compound refers to the physical state of said compound after being isolated from a synthetic process or natural source or combination thereof.
- purified or “in purified form” for a compound refers to the physical state of said compound after being obtained from a purification process or processes described herein or well known to the skilled artisan, in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan.
- variable e.g., aryl, heterocycle, R 2 , etc.
- its definition on each occurrence is independent of its definition at every other occurrence.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (Ci-C ⁇ jalkyl, (C 2 - Ci 2 )alkanoyloxymethyl, 1 -(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1- methyl-1-(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1- (alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1-methyl-1- (alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N- (alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N-C ⁇ jalkyl, (C 2 - Ci 2 )alkanoyloxymethyl, 1 -(alkanoyloxy)ethyl having from 4 to
- alkoxycarbonyl)amino)ethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4- crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N-(Ci-C 2 )alkylamino(C 2 -C 3 )alkyl (such as ⁇ -dimethylaminoethyl), carbamoyl-(Ci-C 2 )alkyl, N,N-di (C r C 2 )alkylcarbamoyl-(C1-C2)alkyl and piperidino-, pyrrolidino- or morpholino(C 2 - C 3 )alkyl, and the like.
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (CrC ⁇ Jalkanoyloxymethyl, 1-((Cr C 6 )alkanoyloxy)ethyl, 1 -methyl-1 -((CrC 6 )alkanoyloxy)ethyl, (C r C 6 )alkoxycarbonyloxymethyl, N-(CrC 6 )alkoxycarbonylaminomethyl, succinoyl, (Ci- C 6 )alkanoyl, ⁇ -amino(Ci-C 4 )alkanyl, arylacyl and ⁇ -aminoacyl, or ⁇ -aminoacyl- ⁇ - aminoacyl, where each ⁇ -aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)(OH) 2 , -P(
- a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R-carbonyl, RO-carbonyl, NRR'-carbonyl where R and R' are each independently (CrCio)alkyl, (C 3 -C 7 ) cycloalkyl, benzyl, or R-carbonyl is a natural ⁇ -aminoacyl or natural ⁇ -aminoacyl, — C(OH)C(O)OY 1 wherein Y 1 is H, (Ci-C 6 )alkyl or benzyl, — C(OY 2 )Y 3 wherein Y 2 is (C 1 -C 4 ) alkyl and Y 3 is (C r C 6 )alkyl, carboxy (Ci-C 6 )alkyl, amino(CrC 4 )alkyl or mono-N — or di-
- Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates,) and the like.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, aluminum salts, zinc salts, salts with organic bases (for example, organic amines) such as benzathines, diethylamine, dicyclohexylamines, hydrabamines (formed with N,N-bis(dehydroabietyl)ethylenediamine), N-methyl-D- glucamines, N-methyl-D-glucamides, t-butyl amines, piperazine, phenylcyclohexylamine, choline, tromethamine, and salts with amino acids such as arginine, lysine and the like.
- organic bases for example, organic amines
- organic bases for example, organic amines
- benzathines diethylamine, dicyclohexylamines, hydrabamines (formed with N,N-
- Basic nitrogen-containing groups may be quarternized with agents such as lower alkyl halides (e.g. methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g. dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g. decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g. benzyl and phenethyl bromides), and others.
- lower alkyl halides e.g. methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfates e.g. dimethyl, diethyl, dibutyl, and diamyl sulfates
- long chain halides
- Effective amount or “therapeutically effective amount” is meant to describe an amount of compound or a composition of the present invention effective in inhibiting BACE-1 and thus producing the desired therapeutic effect in a suitable patient.
- a preferred dosage is about 0.001 to 1000 mg/kg of body weight/day of the compound of Formula I or a pharmaceutically acceptable salt or solvate thereof.
- An especially preferred dosage is about 0.01 to 30 mg/kg of body weight/day of a compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound.
- This invention is also directed to pharmaceutical compositions for the treatment of neurodegenerative diseases such as Alzheimer's disease which comprise an effective treating amount of at least one compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound and at least one pharmaceutically acceptable carrier.
- Still yet another aspect of this invention is a method of treating a cognitive or neurodegenerative disease, such as Alzheimer's disease, comprising administering to a mammal in need of such treatment a therapeutically effective amount of at least one compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound.
- a further aspect of this invention is a method for treating a cognitive or neurodegenerative disease such as Alzheimer's disease, comprising administering to a mammal a therapeutically effective amount of at least one compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound.
- compositions which comprise at least one compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound and at least one pharmaceutically acceptable carrier.
- This invention is also directed to pharmaceutical compositions for the treatment of neurodegenerative diseases such as Alzheimer's disease which comprise an effective treating amount of at least one compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound and at least one pharmaceutically acceptable carrier.
- Still yet other aspects of this invention are combinations of a compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound and other compounds as described below. Accordingly, included within the invention is a method for treating neurodegenerative diseases such as Alzheimer's, comprising administering to a mammal (e.g., a female or male human) a. an amount of a first compound, said first compound being a compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound; and b. an amount of a second compound, said second compound being a cholinesterase inhibitor.
- a mammal e.g., a female or male human
- anti-inflammatory compounds include but are non limited to non-steroidal anti-inflammatory drugs such as diclofenac (Voltaren, Cataflam), diflunisal (Dolobid), etodolac (Lodine), flurbiprofen (Ansaid), ibuprofen (Motrin, Advil), indomethacin (Indocin), ketoprofen (Orudis, Oruvail), ketorolac
- non-steroidal anti-inflammatory drugs such as diclofenac (Voltaren, Cataflam), diflunisal (Dolobid), etodolac (Lodine), flurbiprofen (Ansaid), ibuprofen (Motrin, Advil), indomethacin (Indocin), ketoprofen (Orudis, Oruvail), ketorolac
- HMG-CoA reductase inhibitors for use in combination with compounds of formula I include the "stains," e.g., atorvastatin, lovastatin, simvistatin, pravastatin, fluvastatin and rosuvastatin. Accordingly, included within the invention is a method for treating neurodegenerative diseases such as Alzheimer's, comprising administering to a mammal (e.g., a female or male human) a. an amount of a first compound, said first compound being a compound of Formula I, or a pharmaceutically acceptable salt or solvate of said compound; and b. an amount of a second compound, said second compound being a N- methyl-D-aspartate receptor antagonist.
- a suitable N-methyl-D-aspartate receptor antagonist is, for example, memantine.
- the pharmaceutical preparation is in a unit dosage form.
- the preparation is subdivided into suitably sized unit doses containing appropriate quantities of the active component, e.g., an effective amount to achieve the desired purpose.
- the actual dosage employed may be varied depending upon the requirements of the patient and the severity of the condition being treated. Determination of the proper dosage regimen for a particular situation is within the skill of the art. For convenience, the total daily dosage may be divided and administered in portions during the day as required.
- inert, pharmaceutically acceptable carriers can be either solid or liquid.
- Solid form preparations include powders, tablets, dispersible granules, capsules, cachets and suppositories.
- the powders and tablets may be comprised of from about 5 to about 95 percent active ingredient.
- Suitable solid carriers are known in the art, e.g.
- Aerosol preparations suitable for inhalation may include solutions and solids in powder form, which may be in combination with a pharmaceutically acceptable carrier, such as an inert compressed gas.
- a pharmaceutically acceptable carrier such as an inert compressed gas.
- transdermal compositions can take the form of creams, lotions, aerosols and/or emulsions and can be included in a transdermal patch of the matrix or reservoir type as are conventional in the art for this purpose.
- the compound is administered orally.
- the pharmaceutical preparation is in unit dosage form.
- the preparation is subdivided into unit doses containing appropriate quantities of the active component, e.g., an effective amount to achieve the desired purpose.
- the active components may be co ⁇ administered simultaneously or sequentially, or a single pharmaceutical composition comprising a compound of formula I and one of the other agents in a pharmaceutically acceptable carrier can be administered.
- the components of the combination can be administered individually or together in any conventional oral or parenteral dosage form such as capsule, tablet, powder, cachet, suspension, solution, suppository, nasal spray, etc.
- the dosage of the ⁇ -secretase inhibitors other than those of formula I, HMG-CoA reductase inhibitor, gamma-secretase inhibitor, non-steroidal anti-inflammatory agent, N-methyl-D-aspartate receptor antagonist, cholinesterase inhibitor or anti-amyloid antibody can be determined from published material, and may range from 0.001 to 100 mg/kg body weight.
- the invention also includes multi-agent compositions, kits and methods of treatment, e.g., a compound of formula I can be administed in combination with an HMG-CoA reductase inhibitor and a non-steroidal anti-inflammatory agent
- a compound of formula I can be administed in combination with an HMG-CoA reductase inhibitor and a non-steroidal anti-inflammatory agent
- Compounds of Formula I can be produced by processes known to those skilled in the art using either solution phase or solid phase synthesis as shown in the following reaction schemes, in the preparations and examples below, but those skilled in the art will recognize that other procedures can also be suitable.
- Triphenylphosphine PPh 3
- Tetrabutylammonium fluoride TBAF Triethylamine: Et 3 N, NEt 3 Or TEA
- Lithium borohydride LiBH 4
- BACE-I Cloning, Protein Expression and Purification A predicted soluble form of human BACE1 (sBACEI , corresponding to amino acids 1-454) was generated from the full length BACE1 cDNA (full length human BACE 1 cDNA in pCDNA4/mycHisA construct; University of Toronto) by PCR using the advantage-GC cDNA PCR kit (Clontech, Palo Alto, CA).
- a Hindlll/Pmel fragment from pCDNA4-sBACE1 myc/His was blunt ended using Klenow and subcloned into the Stu I site of pFASTBACI(A) (Invitrogen).
- IC 50 determinations for inhibitors were determined by measuring the percent change of the relative fluorescence at 665 nm divided by the relative fluorescence at 620 nm, (665/620 ratio), in the presence of varying concentrations of / and a fixed concentration of enzyme and substrate.
- Nonlinear regression analysis of this data was performed using GraphPad Prism 3.0 software selecting four parameter logistic equation, that allows for a variable slope.
- Y Bottom + (Top-Bottom)/ (1 Slope));
- X is the logarithm of concentration of I
- Y is the percent change in ratio and Y starts at bottom and goes to top with a sigmoid shape.
- Compounds of the present invention have an IC 5O range from about 100 to about 10,000 nM, preferably about 100 to about 1000 nM, more preferably about 100 to about 500 nM.
- Compounds of the preferred stereochemistry have IC 5O values in a range of about 2 to about 500 nM, preferably about 2 to about 100 nM.
- the compound of the following formula has a IC 50 Of 186 nM.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Description
























































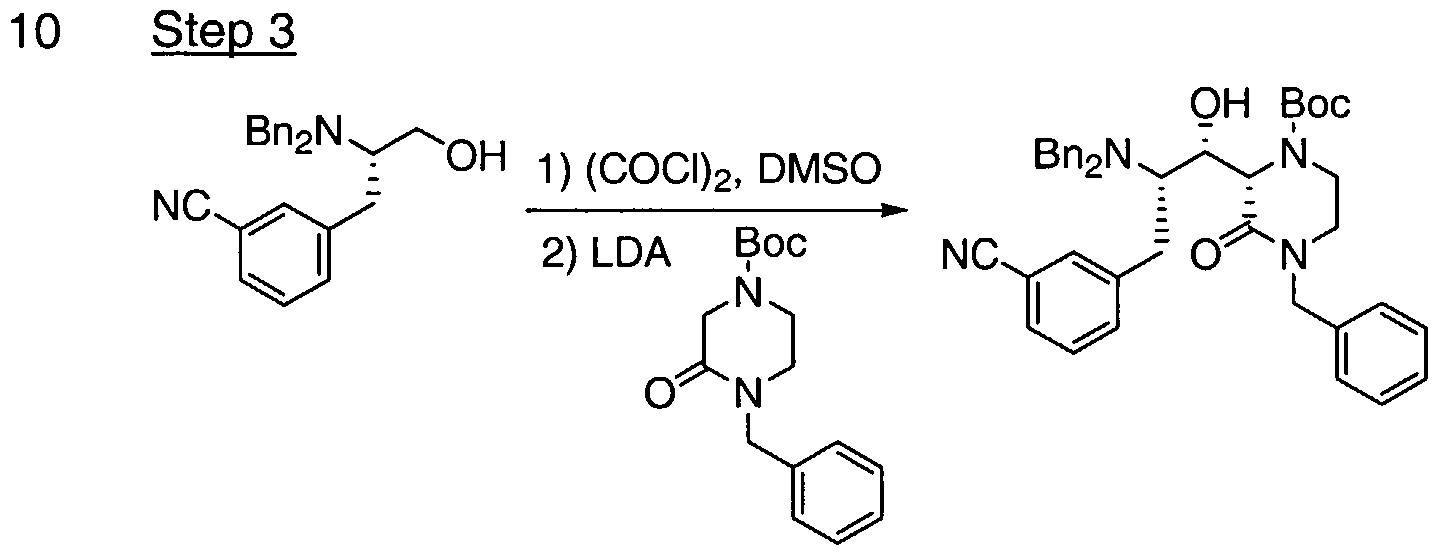
























































Claims


































Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT05773473T ATE491693T1 (en) | 2004-07-22 | 2005-07-20 | SUBSTITUTED AMIDES AS INHIBITORS OF BETA SECRETASE |
| JP2007522711A JP2008507538A (en) | 2004-07-22 | 2005-07-20 | Substituted amide β-secretase inhibitor |
| CA002574218A CA2574218A1 (en) | 2004-07-22 | 2005-07-20 | Substituted amide beta secretase inhibitors |
| EP05773473A EP1781625B1 (en) | 2004-07-22 | 2005-07-20 | Substituted amide beta secretase inhibitors |
| MX2007000760A MX2007000760A (en) | 2004-07-22 | 2005-07-20 | Substituted amide beta secretase inhibitors. |
| DE602005025363T DE602005025363D1 (en) | 2004-07-22 | 2005-07-20 | SUBSTITUTED AMIDS AS INHIBITORS OF BETA-SEKRETASE |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US59010204P | 2004-07-22 | 2004-07-22 | |
| US60/590,102 | 2004-07-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006014762A1 true WO2006014762A1 (en) | 2006-02-09 |
Family
ID=35431616
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/025780 WO2006014762A1 (en) | 2004-07-22 | 2005-07-20 | Substituted amide beta secretase inhibitors |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US7803802B2 (en) |
| EP (1) | EP1781625B1 (en) |
| JP (1) | JP2008507538A (en) |
| CN (1) | CN101014577A (en) |
| AR (1) | AR049726A1 (en) |
| AT (1) | ATE491693T1 (en) |
| CA (1) | CA2574218A1 (en) |
| DE (1) | DE602005025363D1 (en) |
| ES (1) | ES2355733T3 (en) |
| MX (1) | MX2007000760A (en) |
| TW (1) | TW200621249A (en) |
| WO (1) | WO2006014762A1 (en) |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| WO2009032277A1 (en) | 2007-09-06 | 2009-03-12 | Schering Corporation | Gamma secretase modulators |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7700603B2 (en) | 2003-12-15 | 2010-04-20 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2010056849A1 (en) | 2008-11-13 | 2010-05-20 | Schering Corporation | Gamma secretase modulators |
| WO2010075204A2 (en) | 2008-12-22 | 2010-07-01 | Schering Corporation | Gamma secretase modulators |
| WO2010075203A1 (en) | 2008-12-22 | 2010-07-01 | Schering Corporation | Gamma secretase modulators |
| US7759354B2 (en) | 2005-06-14 | 2010-07-20 | Schering Corporation | Bicyclic guanidine derivatives as asparyl protease inhibitors, compositions, and uses thereof |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| WO2010147975A1 (en) | 2009-06-16 | 2010-12-23 | Schering Corporation | Gamma secretase modulators |
| WO2010147969A2 (en) | 2009-06-16 | 2010-12-23 | Schering Corporation | Gamma secretase modulators |
| WO2010147973A1 (en) | 2009-06-16 | 2010-12-23 | Schering Corporation | Gamma secretase modulators |
| US7868000B2 (en) | 2005-06-14 | 2011-01-11 | Schering Corporation | Aspartyl protease inhibitors |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011044181A1 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| WO2011044187A1 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| WO2011044185A2 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Pentafluorosulfur imino heterocyclic compounds as bace-1 inhibitors, compositions, and their use |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| US8093254B2 (en) | 2006-12-12 | 2012-01-10 | Schering Corporation | Aspartyl protease inhibitors |
| US8168641B2 (en) | 2006-06-12 | 2012-05-01 | Schering Corporation | Aspartyl protease inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2012138734A1 (en) | 2011-04-07 | 2012-10-11 | Merck Sharp & Dohme Corp. | C5-c6 oxacyclic-fused thiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| WO2013028670A1 (en) | 2011-08-22 | 2013-02-28 | Merck Sharp & Dohme Corp. | 2-spiro-substituted iminothiazines and their mono-and dioxides as bace inhibitors, compositions and their use |
| US8426595B2 (en) | 2007-12-11 | 2013-04-23 | Xianhai Huang | Gamma secretase modulators |
| US8487099B2 (en) | 2007-11-05 | 2013-07-16 | Merck Sharp & Dohme Corp. | Gamma secretase modulators |
| US8541427B2 (en) | 2008-04-22 | 2013-09-24 | Merck, Sharp & Dohme, Corp. | Phenyl-substituted 2-imino-3-methyl pyrrolo pyrimidinone compounds as BACE-1 inhibitors, compositions, and their use |
| US8557826B2 (en) | 2009-10-08 | 2013-10-15 | Merck Sharp & Dohme Corp. | Pentafluorosulfur imino heterocyclic compounds as BACE-1 inhibitors, compositions, and their use |
| WO2014062549A1 (en) | 2012-10-17 | 2014-04-24 | Merck Sharp & Dohme Corp. | Tricyclic substituted thiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| WO2014062553A1 (en) | 2012-10-17 | 2014-04-24 | Merck Sharp & Dohme Corp. | Tricyclic substituted thiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
| EP3607946A1 (en) | 2012-03-19 | 2020-02-12 | Buck Institute for Research on Aging | App specific bace inhibitors (asbis) and uses thereof |
| EP3653609A1 (en) | 2013-02-12 | 2020-05-20 | Buck Institute for Research on Aging | Hydantoins that modulate bace-mediated app processing |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013036671A1 (en) * | 2011-09-07 | 2013-03-14 | Satori Pharmaceuticals, Inc. | Compounds useful for treating neurodegenerative disorders |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1989003842A1 (en) * | 1987-10-21 | 1989-05-05 | The Upjohn Company | Renin inhibitors containing a (1-amino-2-hydroxy-2-heterocyclic) ethyl moiety |
| US20020019424A1 (en) * | 2000-06-30 | 2002-02-14 | Vincent Mutel | 1-sulfonyl pyrrolidine derivatives |
| EP1299349A2 (en) * | 2000-06-30 | 2003-04-09 | Elan Pharmaceuticals, Inc. | Compounds to treat alzheimer's disease |
| WO2003035613A2 (en) * | 2001-10-22 | 2003-05-01 | Enanta Pharmaceuticals, Inc. | Nitrogen heterocycle inhibitors of aspartyl protease |
| US20040116414A1 (en) * | 2002-05-07 | 2004-06-17 | Tung Jay S. | Succinoyl aminopyrazoles and related compounds |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HRP990246A2 (en) | 1998-08-07 | 2000-06-30 | Du Pont Pharm Co | Succinoylamino benzodiazepines as inhibitors of a beta protein production |
| MXPA01008606A (en) | 1999-02-26 | 2003-05-05 | Merck & Co Inc | Novel sulfonamide compounds and uses thereof. |
| DE19934145C2 (en) * | 1999-07-26 | 2001-05-23 | Deutsche Telekom Mobil | Method for establishing communication connections to a preferably mobile communication terminal device associated with a vehicle or other mobile or stationary device |
| PE20020276A1 (en) | 2000-06-30 | 2002-04-06 | Elan Pharm Inc | SUBSTITUTE AMINE COMPOUNDS AS ß-SECRETASE INHIBITORS FOR THE TREATMENT OF ALZHEIMER |
| US6846813B2 (en) | 2000-06-30 | 2005-01-25 | Pharmacia & Upjohn Company | Compounds to treat alzheimer's disease |
| AU2002256418A1 (en) | 2001-04-27 | 2002-11-11 | Vertex Pharmaceuticals Incorporated | Inhibitors of bace |
| AR035260A1 (en) | 2001-08-03 | 2004-05-05 | Schering Corp | DERIVATIVES OF REPLACED AMINO PIPERIDINE, PHARMACEUTICAL COMPOSITIONS AND THE USE OF SUCH DERIVATIVES FOR THE PREPARATION OF MEDICINES AS INHIBITORS OF THE SECRETASE RANGE, AND FOR THE TREATMENT OF ALZHEIMER'S DISEASE |
| CA2455863C (en) * | 2001-08-03 | 2010-10-12 | Schering Corporation | Tetrahydroquinoline derivatives as gamma secretase inhibitors |
| JP3711131B2 (en) | 2001-08-21 | 2005-10-26 | メルク シャープ エンド ドーム リミテッド | New cyclohexylsulfone |
| TW200302717A (en) | 2002-02-06 | 2003-08-16 | Schering Corp | Novel gamma secretase inhibitors |
| US20050227699A1 (en) | 2002-06-26 | 2005-10-13 | Harmen Schreuder | Method and network element for optimisation of radio resource utilisation in radio access network |
| JP4494212B2 (en) * | 2002-11-12 | 2010-06-30 | メルク・シャープ・エンド・ドーム・コーポレイション | Phenylcarboxamide beta-secretase inhibitor for the treatment of Alzheimer's disease |
| EP1740575A2 (en) * | 2004-04-22 | 2007-01-10 | Eli Lilly And Company | Pyrrolidine derivatives useful as bace inhibitors |
| WO2007058862A2 (en) * | 2005-11-16 | 2007-05-24 | Merck & Co., Inc. | Imidazolidinone compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
-
2005
- 2005-07-20 AR ARP050103000A patent/AR049726A1/en not_active Application Discontinuation
- 2005-07-20 CN CNA2005800300344A patent/CN101014577A/en active Pending
- 2005-07-20 AT AT05773473T patent/ATE491693T1/en not_active IP Right Cessation
- 2005-07-20 MX MX2007000760A patent/MX2007000760A/en active IP Right Grant
- 2005-07-20 EP EP05773473A patent/EP1781625B1/en active Active
- 2005-07-20 DE DE602005025363T patent/DE602005025363D1/en active Active
- 2005-07-20 US US11/185,419 patent/US7803802B2/en active Active
- 2005-07-20 WO PCT/US2005/025780 patent/WO2006014762A1/en active Application Filing
- 2005-07-20 ES ES05773473T patent/ES2355733T3/en active Active
- 2005-07-20 CA CA002574218A patent/CA2574218A1/en not_active Abandoned
- 2005-07-20 JP JP2007522711A patent/JP2008507538A/en active Pending
- 2005-07-21 TW TW094124686A patent/TW200621249A/en unknown
-
2010
- 2010-08-23 US US12/861,347 patent/US8367712B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1989003842A1 (en) * | 1987-10-21 | 1989-05-05 | The Upjohn Company | Renin inhibitors containing a (1-amino-2-hydroxy-2-heterocyclic) ethyl moiety |
| US20020019424A1 (en) * | 2000-06-30 | 2002-02-14 | Vincent Mutel | 1-sulfonyl pyrrolidine derivatives |
| EP1299349A2 (en) * | 2000-06-30 | 2003-04-09 | Elan Pharmaceuticals, Inc. | Compounds to treat alzheimer's disease |
| WO2003035613A2 (en) * | 2001-10-22 | 2003-05-01 | Enanta Pharmaceuticals, Inc. | Nitrogen heterocycle inhibitors of aspartyl protease |
| US20040116414A1 (en) * | 2002-05-07 | 2004-06-17 | Tung Jay S. | Succinoyl aminopyrazoles and related compounds |
Non-Patent Citations (1)
| Title |
|---|
| T. W. GREENE ET AL.: "Protective Groups in organic Synthesis", 1991, WILEY |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8183252B2 (en) | 2003-12-15 | 2012-05-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8242112B2 (en) | 2003-12-15 | 2012-08-14 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8937093B2 (en) | 2003-12-15 | 2015-01-20 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US7973067B2 (en) | 2003-12-15 | 2011-07-05 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8178513B2 (en) | 2003-12-15 | 2012-05-15 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7700603B2 (en) | 2003-12-15 | 2010-04-20 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US9416108B2 (en) | 2003-12-15 | 2016-08-16 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7759354B2 (en) | 2005-06-14 | 2010-07-20 | Schering Corporation | Bicyclic guanidine derivatives as asparyl protease inhibitors, compositions, and uses thereof |
| US7868000B2 (en) | 2005-06-14 | 2011-01-11 | Schering Corporation | Aspartyl protease inhibitors |
| US8629155B2 (en) | 2006-06-12 | 2014-01-14 | Merck Sharp & Dohme, Corp. | Aspartyl protease inhibitors |
| US8168641B2 (en) | 2006-06-12 | 2012-05-01 | Schering Corporation | Aspartyl protease inhibitors |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| US8093254B2 (en) | 2006-12-12 | 2012-01-10 | Schering Corporation | Aspartyl protease inhibitors |
| US8691831B2 (en) | 2007-02-23 | 2014-04-08 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8691833B2 (en) | 2007-02-23 | 2014-04-08 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US8829036B2 (en) | 2007-02-23 | 2014-09-09 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| EP2481408A2 (en) | 2007-03-01 | 2012-08-01 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| WO2009032277A1 (en) | 2007-09-06 | 2009-03-12 | Schering Corporation | Gamma secretase modulators |
| US8487099B2 (en) | 2007-11-05 | 2013-07-16 | Merck Sharp & Dohme Corp. | Gamma secretase modulators |
| US8426595B2 (en) | 2007-12-11 | 2013-04-23 | Xianhai Huang | Gamma secretase modulators |
| US8541427B2 (en) | 2008-04-22 | 2013-09-24 | Merck, Sharp & Dohme, Corp. | Phenyl-substituted 2-imino-3-methyl pyrrolo pyrimidinone compounds as BACE-1 inhibitors, compositions, and their use |
| WO2010056849A1 (en) | 2008-11-13 | 2010-05-20 | Schering Corporation | Gamma secretase modulators |
| WO2010075203A1 (en) | 2008-12-22 | 2010-07-01 | Schering Corporation | Gamma secretase modulators |
| WO2010075204A2 (en) | 2008-12-22 | 2010-07-01 | Schering Corporation | Gamma secretase modulators |
| WO2010147973A1 (en) | 2009-06-16 | 2010-12-23 | Schering Corporation | Gamma secretase modulators |
| WO2010147975A1 (en) | 2009-06-16 | 2010-12-23 | Schering Corporation | Gamma secretase modulators |
| WO2010147969A2 (en) | 2009-06-16 | 2010-12-23 | Schering Corporation | Gamma secretase modulators |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| US9029362B2 (en) | 2009-10-08 | 2015-05-12 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as brace inhibitors, compositions, and their use |
| WO2011044181A1 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| US8557826B2 (en) | 2009-10-08 | 2013-10-15 | Merck Sharp & Dohme Corp. | Pentafluorosulfur imino heterocyclic compounds as BACE-1 inhibitors, compositions, and their use |
| US8563543B2 (en) | 2009-10-08 | 2013-10-22 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| US8569310B2 (en) | 2009-10-08 | 2013-10-29 | Merck Sharp & Dohme Corp. | Pentafluorosulfur imino heterocyclic compounds as BACE-1 inhibitors, compositions and their use |
| US9687494B2 (en) | 2009-10-08 | 2017-06-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| WO2011044185A2 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Pentafluorosulfur imino heterocyclic compounds as bace-1 inhibitors, compositions, and their use |
| WO2011044187A1 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| US9475785B2 (en) | 2009-10-08 | 2016-10-25 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US9428475B2 (en) | 2009-10-08 | 2016-08-30 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| EP3034080A1 (en) | 2009-10-08 | 2016-06-22 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| US8940748B2 (en) | 2009-10-08 | 2015-01-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2012138734A1 (en) | 2011-04-07 | 2012-10-11 | Merck Sharp & Dohme Corp. | C5-c6 oxacyclic-fused thiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| WO2013028670A1 (en) | 2011-08-22 | 2013-02-28 | Merck Sharp & Dohme Corp. | 2-spiro-substituted iminothiazines and their mono-and dioxides as bace inhibitors, compositions and their use |
| EP3607946A1 (en) | 2012-03-19 | 2020-02-12 | Buck Institute for Research on Aging | App specific bace inhibitors (asbis) and uses thereof |
| WO2014062553A1 (en) | 2012-10-17 | 2014-04-24 | Merck Sharp & Dohme Corp. | Tricyclic substituted thiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| WO2014062549A1 (en) | 2012-10-17 | 2014-04-24 | Merck Sharp & Dohme Corp. | Tricyclic substituted thiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| EP3653609A1 (en) | 2013-02-12 | 2020-05-20 | Buck Institute for Research on Aging | Hydantoins that modulate bace-mediated app processing |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2008507538A (en) | 2008-03-13 |
| US7803802B2 (en) | 2010-09-28 |
| EP1781625B1 (en) | 2010-12-15 |
| US20060040994A1 (en) | 2006-02-23 |
| DE602005025363D1 (en) | 2011-01-27 |
| ATE491693T1 (en) | 2011-01-15 |
| CN101014577A (en) | 2007-08-08 |
| AR049726A1 (en) | 2006-08-30 |
| MX2007000760A (en) | 2007-04-09 |
| EP1781625A1 (en) | 2007-05-09 |
| US8367712B2 (en) | 2013-02-05 |
| TW200621249A (en) | 2006-07-01 |
| ES2355733T3 (en) | 2011-03-30 |
| US20110003825A1 (en) | 2011-01-06 |
| CA2574218A1 (en) | 2006-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1781625B1 (en) | Substituted amide beta secretase inhibitors | |
| EP1781644B1 (en) | Macrocyclic beta-secretase inhibitors | |
| EP1945626B1 (en) | Imidazole derivatives as functionally selective alpha2c adrenoreceptor agonists | |
| US8518975B2 (en) | Gamma secretase modulators | |
| JP2010535762A (en) | Gamma secretase modulator | |
| MX2007016183A (en) | The preparation and use of compounds as protease inhibitors. | |
| MX2010011563A (en) | Phenyl-substituted 2-imino-3-methyl pyrrolo pyrimidinone compounds as bace-1 inhibitors, compositions, and their use. | |
| EP2178857A1 (en) | Gamma secretase modulators | |
| US8673900B2 (en) | Gamma secretase modulators | |
| US8759337B2 (en) | Gamma secretase modulators | |
| EP2205567A1 (en) | Gamma secretase modulators | |
| EP2142538B1 (en) | Derivatives and analogs of chroman as functionally selective alpha2c adrenoreceptor agonists | |
| US20120135980A1 (en) | Gamma secretase modulators | |
| US8809318B2 (en) | Gamma secretase modulators | |
| US7879997B2 (en) | Compounds for the treatment of inflammatory disorders | |
| US20120245158A1 (en) | Gamma secretase modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2574218 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/000760 Country of ref document: MX Ref document number: 2007522711 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005773473 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580030034.4 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005773473 Country of ref document: EP |