WO2005051391A1 - Quinazolinone compounds with reduced bioaccumulation - Google Patents
Quinazolinone compounds with reduced bioaccumulation Download PDFInfo
- Publication number
- WO2005051391A1 WO2005051391A1 PCT/US2004/039020 US2004039020W WO2005051391A1 WO 2005051391 A1 WO2005051391 A1 WO 2005051391A1 US 2004039020 W US2004039020 W US 2004039020W WO 2005051391 A1 WO2005051391 A1 WO 2005051391A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- compound
- groups
- ethyl
- unsubstituted
- Prior art date
Links
- 230000002829 reductive effect Effects 0.000 title description 20
- 231100000693 bioaccumulation Toxicity 0.000 title description 10
- AVRPFRMDMNDIDH-UHFFFAOYSA-N 1h-quinazolin-2-one Chemical class C1=CC=CC2=NC(O)=NC=C21 AVRPFRMDMNDIDH-UHFFFAOYSA-N 0.000 title description 7
- 150000001875 compounds Chemical class 0.000 claims abstract description 192
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 41
- 201000010099 disease Diseases 0.000 claims abstract description 23
- 230000001404 mediated effect Effects 0.000 claims abstract description 11
- 102100023724 Melanocortin receptor 4 Human genes 0.000 claims abstract description 4
- 101710085775 Melanocortin receptor 4 Proteins 0.000 claims abstract 3
- -1 4-substituted phenylethyl group Chemical group 0.000 claims description 264
- 239000000203 mixture Substances 0.000 claims description 100
- 238000000034 method Methods 0.000 claims description 99
- 125000000217 alkyl group Chemical group 0.000 claims description 39
- 125000001072 heteroaryl group Chemical group 0.000 claims description 33
- 125000003118 aryl group Chemical group 0.000 claims description 30
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 28
- 125000000623 heterocyclic group Chemical group 0.000 claims description 27
- 150000003839 salts Chemical class 0.000 claims description 19
- 208000008589 Obesity Diseases 0.000 claims description 17
- 235000020824 obesity Nutrition 0.000 claims description 17
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 239000008194 pharmaceutical composition Substances 0.000 claims description 13
- 125000003545 alkoxy group Chemical group 0.000 claims description 12
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 12
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 12
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 12
- 210000004556 brain Anatomy 0.000 claims description 11
- IWELDVXSEVIIGI-UHFFFAOYSA-N piperazin-2-one Chemical compound O=C1CNCCN1 IWELDVXSEVIIGI-UHFFFAOYSA-N 0.000 claims description 11
- 125000003342 alkenyl group Chemical group 0.000 claims description 10
- 125000000304 alkynyl group Chemical group 0.000 claims description 10
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 10
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims description 10
- 125000003367 polycyclic group Chemical group 0.000 claims description 7
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- 210000004185 liver Anatomy 0.000 claims description 6
- 229910052794 bromium Inorganic materials 0.000 claims description 5
- 229910052801 chlorine Inorganic materials 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 229910052740 iodine Inorganic materials 0.000 claims description 5
- 125000003070 2-(2-chlorophenyl)ethyl group Chemical group [H]C1=C([H])C(Cl)=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000002856 2-fluorophenylethyl group Chemical group [H]C1=C([H])C(F)=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000000940 2-methoxyphenyl ethyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 4
- KBSDLBVPAHQCRY-UHFFFAOYSA-N 307496-19-1 Chemical group C1CC=CCC1CC[Si](O1)(O2)O[Si](O3)(C4CCCC4)O[Si](O4)(C5CCCC5)O[Si]1(C1CCCC1)O[Si](O1)(C5CCCC5)O[Si]2(C2CCCC2)O[Si]3(C2CCCC2)O[Si]41C1CCCC1 KBSDLBVPAHQCRY-UHFFFAOYSA-N 0.000 claims description 4
- 230000008081 blood perfusion Effects 0.000 claims description 4
- 210000002216 heart Anatomy 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 125000003282 alkyl amino group Chemical group 0.000 claims description 3
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 3
- 125000002541 furyl group Chemical group 0.000 claims description 3
- 125000002883 imidazolyl group Chemical group 0.000 claims description 3
- 125000001041 indolyl group Chemical group 0.000 claims description 3
- 125000002757 morpholinyl group Chemical group 0.000 claims description 3
- 125000004193 piperazinyl group Chemical group 0.000 claims description 3
- 125000003386 piperidinyl group Chemical group 0.000 claims description 3
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 claims description 3
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 3
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 3
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 claims description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 3
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 claims description 3
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 claims description 3
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 3
- 125000000335 thiazolyl group Chemical group 0.000 claims description 3
- 125000001544 thienyl group Chemical group 0.000 claims description 3
- 125000004568 thiomorpholinyl group Chemical group 0.000 claims description 3
- 125000001425 triazolyl group Chemical group 0.000 claims description 3
- 239000000556 agonist Substances 0.000 abstract description 16
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 abstract description 8
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 abstract description 4
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 abstract description 4
- 150000003384 small molecules Chemical class 0.000 abstract description 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 187
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 171
- 230000015572 biosynthetic process Effects 0.000 description 132
- 238000003786 synthesis reaction Methods 0.000 description 131
- 239000000243 solution Substances 0.000 description 123
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 104
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 96
- 238000006243 chemical reaction Methods 0.000 description 93
- 235000019439 ethyl acetate Nutrition 0.000 description 70
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 68
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 65
- 239000000047 product Substances 0.000 description 48
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 45
- 239000007787 solid Substances 0.000 description 45
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 44
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 43
- 108010021436 Type 4 Melanocortin Receptor Proteins 0.000 description 41
- 102000008316 Type 4 Melanocortin Receptor Human genes 0.000 description 41
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 38
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 36
- 239000012044 organic layer Substances 0.000 description 32
- 241000699670 Mus sp. Species 0.000 description 31
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 31
- 238000005160 1H NMR spectroscopy Methods 0.000 description 29
- 239000011541 reaction mixture Substances 0.000 description 29
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 26
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 26
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 26
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 26
- 238000000746 purification Methods 0.000 description 26
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 26
- 239000008103 glucose Substances 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical class CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 22
- 239000003921 oil Substances 0.000 description 21
- 235000019198 oils Nutrition 0.000 description 21
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 20
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 19
- 239000012043 crude product Substances 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- 238000010992 reflux Methods 0.000 description 17
- 239000000741 silica gel Substances 0.000 description 17
- 229910002027 silica gel Inorganic materials 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- 238000004128 high performance liquid chromatography Methods 0.000 description 16
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 15
- 208000035475 disorder Diseases 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 239000007924 injection Substances 0.000 description 15
- 238000002347 injection Methods 0.000 description 15
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 14
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 14
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 14
- 229910052938 sodium sulfate Inorganic materials 0.000 description 14
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 13
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 13
- 150000001718 carbodiimides Chemical class 0.000 description 13
- 235000019253 formic acid Nutrition 0.000 description 13
- 239000000725 suspension Substances 0.000 description 13
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 12
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical group [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 12
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- 238000004440 column chromatography Methods 0.000 description 12
- 229910052757 nitrogen Inorganic materials 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- ASFOHWKEKBODLZ-BYPYZUCNSA-N (6s)-6-methylpiperazin-2-one Chemical compound C[C@H]1CNCC(=O)N1 ASFOHWKEKBODLZ-BYPYZUCNSA-N 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Chemical class 0.000 description 11
- 230000037396 body weight Effects 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 238000003818 flash chromatography Methods 0.000 description 11
- 235000012631 food intake Nutrition 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- 235000011152 sodium sulphate Nutrition 0.000 description 11
- 239000013058 crude material Substances 0.000 description 10
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 10
- 230000037406 food intake Effects 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 239000001257 hydrogen Substances 0.000 description 10
- 239000010410 layer Substances 0.000 description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 10
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- 102000004877 Insulin Human genes 0.000 description 9
- 108090001061 Insulin Proteins 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 9
- 229940125396 insulin Drugs 0.000 description 9
- 210000002966 serum Anatomy 0.000 description 9
- 210000001519 tissue Anatomy 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- QRXAIIWZPWKFGE-UHFFFAOYSA-N 2-chloro-n-[2-(4-fluorophenyl)ethyl]acetamide Chemical compound FC1=CC=C(CCNC(=O)CCl)C=C1 QRXAIIWZPWKFGE-UHFFFAOYSA-N 0.000 description 8
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 8
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 206010012601 diabetes mellitus Diseases 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 235000021588 free fatty acids Nutrition 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 8
- 210000002381 plasma Anatomy 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- UEALKTCRMBVTFN-UHFFFAOYSA-N 4-nitroanthranilic acid Chemical class NC1=CC([N+]([O-])=O)=CC=C1C(O)=O UEALKTCRMBVTFN-UHFFFAOYSA-N 0.000 description 7
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- 229910052799 carbon Chemical group 0.000 description 7
- 150000003857 carboxamides Chemical class 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000012230 colorless oil Substances 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 150000002357 guanidines Chemical class 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 239000002002 slurry Substances 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 7
- ONUCCUGDWPXAJG-YFKPBYRVSA-N (2r)-2-(fluoromethyl)piperazine Chemical compound FC[C@H]1CNCCN1 ONUCCUGDWPXAJG-YFKPBYRVSA-N 0.000 description 6
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 6
- 229920000742 Cotton Polymers 0.000 description 6
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 6
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 235000013305 food Nutrition 0.000 description 6
- 125000005842 heteroatom Chemical group 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- CODOMZDOWFAELZ-UHFFFAOYSA-N methyl 2-[benzyl-[2-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]amino]acetate Chemical compound CC(C)(C)OC(=O)NC(C)CN(CC(=O)OC)CC1=CC=CC=C1 CODOMZDOWFAELZ-UHFFFAOYSA-N 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- YOWDLUNPZVDDRV-JTQLQIEISA-N (6s)-4-benzyl-6-methylpiperazin-2-one Chemical compound C1C(=O)N[C@@H](C)CN1CC1=CC=CC=C1 YOWDLUNPZVDDRV-JTQLQIEISA-N 0.000 description 5
- ZJSQZQMVXKZAGW-UHFFFAOYSA-N 2H-benzotriazol-4-ol hydrate Chemical compound O.OC1=CC=CC2=C1N=NN2 ZJSQZQMVXKZAGW-UHFFFAOYSA-N 0.000 description 5
- DNTFDAYESNOOAY-UHFFFAOYSA-N 7-amino-3-[2-(2,4-dichlorophenyl)ethyl]-1h-quinazoline-2,4-dione Chemical compound C=1C(N)=CC=C(C2=O)C=1NC(=O)N2CCC1=CC=C(Cl)C=C1Cl DNTFDAYESNOOAY-UHFFFAOYSA-N 0.000 description 5
- 102100034216 Melanocyte-stimulating hormone receptor Human genes 0.000 description 5
- 101710161100 Melanocyte-stimulating hormone receptor Proteins 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 229910002092 carbon dioxide Inorganic materials 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 150000002825 nitriles Chemical group 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 238000002953 preparative HPLC Methods 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 238000010898 silica gel chromatography Methods 0.000 description 5
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 238000011282 treatment Methods 0.000 description 5
- LTHOXYJTFXCVRG-LELQROKWSA-N (3r,5s)-n-[3-[2-(2,4-dichlorophenyl)ethyl]-2,4-dioxo-1h-quinazolin-7-yl]-3,5-dimethyl-n'-[(1r,3s,4s,5r)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]piperazine-1-carboximidamide Chemical compound N(/[C@@H]1[C@@H](C)[C@]2(C[C@](C1)(C2(C)C)[H])[H])=C(N1C[C@@H](C)N[C@@H](C)C1)\NC(C=1)=CC=C(C2=O)C=1NC(=O)N2CCC1=CC=C(Cl)C=C1Cl LTHOXYJTFXCVRG-LELQROKWSA-N 0.000 description 4
- MVDLQUQTSVGZBF-VGOBXOCWSA-N 1-[3-[2-(2,4-dichlorophenyl)ethyl]-2,4-dioxo-1h-quinazolin-7-yl]-3-[(1r,3s,4s,5r)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]thiourea Chemical compound N([C@@H]1[C@@H](C)[C@]2(C[C@](C1)(C2(C)C)[H])[H])C(=S)NC(C=1)=CC=C(C2=O)C=1NC(=O)N2CCC1=CC=C(Cl)C=C1Cl MVDLQUQTSVGZBF-VGOBXOCWSA-N 0.000 description 4
- WUZWLNZTNAVJMN-UHFFFAOYSA-N 2-amino-n-[2-(2,4-dichlorophenyl)ethyl]-4-nitrobenzamide Chemical compound NC1=CC([N+]([O-])=O)=CC=C1C(=O)NCCC1=CC=C(Cl)C=C1Cl WUZWLNZTNAVJMN-UHFFFAOYSA-N 0.000 description 4
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 108010008364 Melanocortins Proteins 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- LJVDCHZGALIYPV-RIHPBJNCSA-N [(2s,3s)-3-methylpyrrolidin-2-yl]methanol;hydrochloride Chemical compound Cl.C[C@H]1CCN[C@@H]1CO LJVDCHZGALIYPV-RIHPBJNCSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 150000001408 amides Chemical group 0.000 description 4
- 229940024606 amino acid Drugs 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- 235000019577 caloric intake Nutrition 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- FHUKGWIMPWBXLF-WDSKDSINSA-N ethyl (2s,3s)-3-hydroxypyrrolidine-2-carboxylate Chemical compound CCOC(=O)[C@H]1NCC[C@@H]1O FHUKGWIMPWBXLF-WDSKDSINSA-N 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 125000001153 fluoro group Chemical group F* 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000002865 melanocortin Substances 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 238000002390 rotary evaporation Methods 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 125000003107 substituted aryl group Chemical group 0.000 description 4
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- FPUBEXOAVGKKCY-DTWKUNHWSA-N tert-butyl (2s,3s)-2-(hydroxymethyl)-3-methylpyrrolidine-1-carboxylate Chemical compound C[C@H]1CCN(C(=O)OC(C)(C)C)[C@@H]1CO FPUBEXOAVGKKCY-DTWKUNHWSA-N 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 4
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 4
- 229920002554 vinyl polymer Polymers 0.000 description 4
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 3
- VPTSZLVPZCTAHZ-KZVJFYERSA-N (1r,3s,4s,5s)-4,6,6-trimethylbicyclo[3.1.1]heptan-3-amine Chemical compound C1[C@H](N)[C@@H](C)[C@H]2C(C)(C)[C@@H]1C2 VPTSZLVPZCTAHZ-KZVJFYERSA-N 0.000 description 3
- GNCDPUYXABYJAU-SCSAIBSYSA-N (2r)-2-(difluoromethyl)piperazine Chemical compound FC(F)[C@H]1CNCCN1 GNCDPUYXABYJAU-SCSAIBSYSA-N 0.000 description 3
- VADWFRGDDJHKNB-VKKIDBQXSA-N (3r,4r)-pyrrolidine-3,4-diol;hydrochloride Chemical compound Cl.O[C@@H]1CNC[C@H]1O VADWFRGDDJHKNB-VKKIDBQXSA-N 0.000 description 3
- CURKBIZVHWYYEN-PZAFXMOQSA-N (3s)-n-[3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-4-oxo-2-pyridin-3-ylquinazolin-7-yl]-3-methyl-5-oxo-n'-[(1r,3s,4s,5s)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]piperazine-1-carboximidamide Chemical compound N([C@@H]1[C@@H](C)[C@@]2(C[C@](C1)(C2(C)C)[H])[H])=C(N1CC(=O)N[C@@H](C)C1)NC(C=1)=CC=C(C(N2CCC=3C(=CC(OC)=CC=3)F)=O)C=1N=C2C1=CC=CN=C1 CURKBIZVHWYYEN-PZAFXMOQSA-N 0.000 description 3
- XKEIPRRJHYNJLO-QKHKRXKCSA-N (3s,5r)-n-[3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-4-oxoquinazolin-7-yl]-3,5-dimethyl-n'-[(1r,4s,5s)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]piperazine-1-carboximidamide Chemical compound C1([C@@H](C)[C@@]2(C[C@](C1)(C2(C)C)[H])[H])N=C(N1C[C@@H](C)N[C@@H](C)C1)NC(C=1)=CC=C(C2=O)C=1N=CN2CCC1=CC=C(OC)C=C1F XKEIPRRJHYNJLO-QKHKRXKCSA-N 0.000 description 3
- DEIBXAPEZDJDRC-UHFFFAOYSA-M (dimethylaminomethylideneamino)methylidene-dimethylazanium;chloride Chemical compound [Cl-].CN(C)C=NC=[N+](C)C DEIBXAPEZDJDRC-UHFFFAOYSA-M 0.000 description 3
- BCVZHHLJPZIBCC-UHFFFAOYSA-N 1-benzhydryl-3-methylazetidin-3-ol Chemical compound C1C(C)(O)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 BCVZHHLJPZIBCC-UHFFFAOYSA-N 0.000 description 3
- LYTNNHXGUOKXFI-UHFFFAOYSA-N 1-benzhydrylazetidin-3-amine Chemical compound C1C(N)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 LYTNNHXGUOKXFI-UHFFFAOYSA-N 0.000 description 3
- MMAJXKGUZYDTHV-UHFFFAOYSA-N 1-benzhydrylazetidin-3-ol Chemical compound C1C(O)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 MMAJXKGUZYDTHV-UHFFFAOYSA-N 0.000 description 3
- WJMVAMUASKTIQZ-IUCAKERBSA-N 1-o-tert-butyl 2-o-ethyl (2s,3s)-3-hydroxypyrrolidine-1,2-dicarboxylate Chemical compound CCOC(=O)[C@@H]1[C@@H](O)CCN1C(=O)OC(C)(C)C WJMVAMUASKTIQZ-IUCAKERBSA-N 0.000 description 3
- IQWOMDHVSKITEF-UWVGGRQHSA-N 1-o-tert-butyl 2-o-ethyl (2s,3s)-3-methylpyrrolidine-1,2-dicarboxylate Chemical compound CCOC(=O)[C@@H]1[C@@H](C)CCN1C(=O)OC(C)(C)C IQWOMDHVSKITEF-UWVGGRQHSA-N 0.000 description 3
- UOFDVMLPYHVIRI-UHFFFAOYSA-N 2-chloro-3-[2-(2,4-dichlorophenyl)ethyl]-7-nitroquinazolin-4-one Chemical compound C=1C([N+](=O)[O-])=CC=C(C2=O)C=1N=C(Cl)N2CCC1=CC=C(Cl)C=C1Cl UOFDVMLPYHVIRI-UHFFFAOYSA-N 0.000 description 3
- FAHCBGKMOGWEQX-UHFFFAOYSA-N 3-[2-(2,4-dichlorophenyl)ethyl]-7-nitro-1h-quinazoline-2,4-dione Chemical compound C=1C([N+](=O)[O-])=CC=C(C2=O)C=1NC(=O)N2CCC1=CC=C(Cl)C=C1Cl FAHCBGKMOGWEQX-UHFFFAOYSA-N 0.000 description 3
- MQOFXVWAFFJFJH-UHFFFAOYSA-N 4,4-difluorocyclohexan-1-amine Chemical compound NC1CCC(F)(F)CC1 MQOFXVWAFFJFJH-UHFFFAOYSA-N 0.000 description 3
- 108700034262 4-Nle-7-Phe-alpha- MSH Proteins 0.000 description 3
- ASFOHWKEKBODLZ-UHFFFAOYSA-N 6-methylpiperazin-2-one Chemical compound CC1CNCC(=O)N1 ASFOHWKEKBODLZ-UHFFFAOYSA-N 0.000 description 3
- 102000006822 Agouti Signaling Protein Human genes 0.000 description 3
- 108010072151 Agouti Signaling Protein Proteins 0.000 description 3
- 102000054930 Agouti-Related Human genes 0.000 description 3
- 101710127426 Agouti-related protein Proteins 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 208000024172 Cardiovascular disease Diseases 0.000 description 3
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 3
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 102000004378 Melanocortin Receptors Human genes 0.000 description 3
- 108090000950 Melanocortin Receptors Proteins 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 206010036049 Polycystic ovaries Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- UAHFGYDRQSXQEB-LEBBXHLNSA-N afamelanotide Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 UAHFGYDRQSXQEB-LEBBXHLNSA-N 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 3
- 229960000258 corticotropin Drugs 0.000 description 3
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 3
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 150000003949 imides Chemical group 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 239000012948 isocyanate Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- RUZLIIJDZBWWSA-INIZCTEOSA-N methyl 2-[[(1s)-1-(7-methyl-2-morpholin-4-yl-4-oxopyrido[1,2-a]pyrimidin-9-yl)ethyl]amino]benzoate Chemical group COC(=O)C1=CC=CC=C1N[C@@H](C)C1=CC(C)=CN2C(=O)C=C(N3CCOCC3)N=C12 RUZLIIJDZBWWSA-INIZCTEOSA-N 0.000 description 3
- CIMGUABKISQZNN-UHFFFAOYSA-N n-(1-benzhydrylazetidin-3-yl)methanesulfonamide Chemical compound C1C(NS(=O)(=O)C)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 CIMGUABKISQZNN-UHFFFAOYSA-N 0.000 description 3
- 238000007410 oral glucose tolerance test Methods 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 230000019612 pigmentation Effects 0.000 description 3
- 201000010065 polycystic ovary syndrome Diseases 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- VWSBNWIPICCWAM-UHFFFAOYSA-N tert-butyl 3-methyl-4-oxopiperidine-1-carboxylate Chemical compound CC1CN(C(=O)OC(C)(C)C)CCC1=O VWSBNWIPICCWAM-UHFFFAOYSA-N 0.000 description 3
- YYMDJYBUKLXHPU-UHFFFAOYSA-N tert-butyl 4-methoxyiminopiperidine-1-carboxylate Chemical compound CON=C1CCN(C(=O)OC(C)(C)C)CC1 YYMDJYBUKLXHPU-UHFFFAOYSA-N 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- 238000003828 vacuum filtration Methods 0.000 description 3
- BJBUEDPLEOHJGE-UHFFFAOYSA-N (2R,3S)-3-Hydroxy-2-pyrolidinecarboxylic acid Natural products OC1CCNC1C(O)=O BJBUEDPLEOHJGE-UHFFFAOYSA-N 0.000 description 2
- QOJCEWYVJOLFHN-JBUOLDKXSA-N (2r,3s)-2-(hydroxymethyl)pyrrolidin-3-ol;hydrochloride Chemical compound Cl.OC[C@H]1NCC[C@@H]1O QOJCEWYVJOLFHN-JBUOLDKXSA-N 0.000 description 2
- CFFLXBUMALVKSD-JBUOLDKXSA-N (2r,3s)-2-methylpyrrolidin-3-ol;hydrochloride Chemical compound Cl.C[C@H]1NCC[C@@H]1O CFFLXBUMALVKSD-JBUOLDKXSA-N 0.000 description 2
- SGTINJIFHCXYDU-KNCHESJLSA-N (2r,6s)-1-hydroxy-2,6-dimethylpiperazine;hydrochloride Chemical compound Cl.C[C@H]1CNC[C@@H](C)N1O SGTINJIFHCXYDU-KNCHESJLSA-N 0.000 description 2
- IFNWESYYDINUHV-OLQVQODUSA-N (2s,6r)-2,6-dimethylpiperazine Chemical compound C[C@H]1CNC[C@@H](C)N1 IFNWESYYDINUHV-OLQVQODUSA-N 0.000 description 2
- XBFBOFLWOYMVSX-TYSVMGFPSA-N (3r,5r)-5-(hydroxymethyl)pyrrolidin-3-ol;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC[C@H]1C[C@@H](O)CN1 XBFBOFLWOYMVSX-TYSVMGFPSA-N 0.000 description 2
- YLLYVDKTPNOANT-VQHVLOKHSA-N (e)-n-methoxy-3-methylpiperidin-4-imine Chemical compound CO\N=C1/CCNCC1C YLLYVDKTPNOANT-VQHVLOKHSA-N 0.000 description 2
- 0 *C(Nc(c(*)c1N=C(*)N2*)c(*)c(*)c1C2=O)=N* Chemical compound *C(Nc(c(*)c1N=C(*)N2*)c(*)c(*)c1C2=O)=N* 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- AUDQMCULSZOJII-UHFFFAOYSA-N 1-[3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-4-oxoquinazolin-7-yl]-3-(4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl)thiourea Chemical compound FC1=CC(OC)=CC=C1CCN1C(=O)C2=CC=C(NC(=S)NC3C(C4CC(C4(C)C)C3)C)C=C2N=C1 AUDQMCULSZOJII-UHFFFAOYSA-N 0.000 description 2
- AVUDXLOVIBJFQA-UHFFFAOYSA-N 1-benzhydrylazetidin-3-one Chemical compound C1C(=O)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 AVUDXLOVIBJFQA-UHFFFAOYSA-N 0.000 description 2
- MZMNEDXVUJLQAF-HTQZYQBOSA-N 1-o-tert-butyl 2-o-methyl (2r,4r)-4-hydroxypyrrolidine-1,2-dicarboxylate Chemical compound COC(=O)[C@H]1C[C@@H](O)CN1C(=O)OC(C)(C)C MZMNEDXVUJLQAF-HTQZYQBOSA-N 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- ZCGYIRXIAQVDFM-UHFFFAOYSA-N 2-(2-fluoro-4-methoxyphenyl)ethanamine Chemical compound COC1=CC=C(CCN)C(F)=C1 ZCGYIRXIAQVDFM-UHFFFAOYSA-N 0.000 description 2
- CKLFJWXRWIQYOC-UHFFFAOYSA-N 2-(4-fluorophenyl)ethanamine Chemical compound NCCC1=CC=C(F)C=C1 CKLFJWXRWIQYOC-UHFFFAOYSA-N 0.000 description 2
- VLLLPYCROULMJP-UHFFFAOYSA-N 2-(diaminomethylideneamino)benzamide Chemical class NC(N)=NC1=CC=CC=C1C(N)=O VLLLPYCROULMJP-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- QIRPRJMKAIYQQX-UHFFFAOYSA-N 2-amino-n-[2-(2-fluoro-4-methoxyphenyl)ethyl]-4-nitrobenzamide Chemical compound FC1=CC(OC)=CC=C1CCNC(=O)C1=CC=C([N+]([O-])=O)C=C1N QIRPRJMKAIYQQX-UHFFFAOYSA-N 0.000 description 2
- 125000004352 2-phenylcyclohexyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 2
- ZSDQBDWWGGBJFY-UHFFFAOYSA-N 3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-7-isothiocyanatoquinazolin-4-one Chemical compound FC1=CC(OC)=CC=C1CCN1C(=O)C2=CC=C(N=C=S)C=C2N=C1 ZSDQBDWWGGBJFY-UHFFFAOYSA-N 0.000 description 2
- OAKUSEOVCNLQPS-UHFFFAOYSA-N 3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-7-nitroquinazolin-4-one Chemical compound FC1=CC(OC)=CC=C1CCN1C(=O)C2=CC=C([N+]([O-])=O)C=C2N=C1 OAKUSEOVCNLQPS-UHFFFAOYSA-N 0.000 description 2
- JZGFCVZHKXATNJ-UHFFFAOYSA-N 3-methyl-2h-azet-3-ol;hydrochloride Chemical compound Cl.CC1(O)CN=C1 JZGFCVZHKXATNJ-UHFFFAOYSA-N 0.000 description 2
- KSDASUMDLOLQEB-UHFFFAOYSA-N 7-amino-3-[2-(2-fluoro-4-methoxyphenyl)ethyl]quinazolin-4-one Chemical compound FC1=CC(OC)=CC=C1CCN1C(=O)C2=CC=C(N)C=C2N=C1 KSDASUMDLOLQEB-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 2
- DYDCUQKUCUHJBH-UWTATZPHSA-N D-Cycloserine Chemical compound N[C@@H]1CONC1=O DYDCUQKUCUHJBH-UWTATZPHSA-N 0.000 description 2
- DYDCUQKUCUHJBH-UHFFFAOYSA-N D-Cycloserine Natural products NC1CONC1=O DYDCUQKUCUHJBH-UHFFFAOYSA-N 0.000 description 2
- 108010016626 Dipeptides Proteins 0.000 description 2
- 208000010228 Erectile Dysfunction Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 2
- 206010022489 Insulin Resistance Diseases 0.000 description 2
- 239000012448 Lithium borohydride Substances 0.000 description 2
- 102100023723 Melanocortin receptor 5 Human genes 0.000 description 2
- 101710085771 Melanocortin receptor 5 Proteins 0.000 description 2
- 102400000740 Melanocyte-stimulating hormone alpha Human genes 0.000 description 2
- 101710200814 Melanotropin alpha Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- VVBXKASDRZXWON-UHFFFAOYSA-N N=[PH3] Chemical compound N=[PH3] VVBXKASDRZXWON-UHFFFAOYSA-N 0.000 description 2
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 2
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- 206010037660 Pyrexia Diseases 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical group C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 125000004849 alkoxymethyl group Chemical group 0.000 description 2
- 230000008485 antagonism Effects 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 150000004982 aromatic amines Chemical class 0.000 description 2
- CUFNKYGDVFVPHO-UHFFFAOYSA-N azulene Chemical compound C1=CC=CC2=CC=CC2=C1 CUFNKYGDVFVPHO-UHFFFAOYSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- XTXJPZGMVKIPHE-SECBINFHSA-N benzyl n-[(2r)-1-oxopropan-2-yl]carbamate Chemical compound O=C[C@@H](C)NC(=O)OCC1=CC=CC=C1 XTXJPZGMVKIPHE-SECBINFHSA-N 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000008238 biochemical pathway Effects 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- FUSUHKVFWTUUBE-UHFFFAOYSA-N buten-2-one Chemical compound CC(=O)C=C FUSUHKVFWTUUBE-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- DCFKHNIGBAHNSS-UHFFFAOYSA-N chloro(triethyl)silane Chemical compound CC[Si](Cl)(CC)CC DCFKHNIGBAHNSS-UHFFFAOYSA-N 0.000 description 2
- KQIADDMXRMTWHZ-UHFFFAOYSA-N chloro-tri(propan-2-yl)silane Chemical compound CC(C)[Si](Cl)(C(C)C)C(C)C KQIADDMXRMTWHZ-UHFFFAOYSA-N 0.000 description 2
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- WDECIBYCCFPHNR-UHFFFAOYSA-N chrysene Chemical compound C1=CC=CC2=CC=C3C4=CC=CC=C4C=CC3=C21 WDECIBYCCFPHNR-UHFFFAOYSA-N 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 2
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 2
- 125000006317 cyclopropyl amino group Chemical group 0.000 description 2
- 229960003077 cycloserine Drugs 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000001856 erectile effect Effects 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- QUPDWYMUPZLYJZ-UHFFFAOYSA-N ethyl Chemical compound C[CH2] QUPDWYMUPZLYJZ-UHFFFAOYSA-N 0.000 description 2
- YJXMGRSSLHPGTO-BQBZGAKWSA-N ethyl (2s,3s)-3-methylpyrrolidine-2-carboxylate Chemical compound CCOC(=O)[C@H]1NCC[C@@H]1C YJXMGRSSLHPGTO-BQBZGAKWSA-N 0.000 description 2
- 125000005348 fluorocycloalkyl group Chemical group 0.000 description 2
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 2
- 201000008980 hyperinsulinism Diseases 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 201000001881 impotence Diseases 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000336 melanocortin receptor agonist Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- BTJJYUVKHQRVOB-LLVKDONJSA-N methyl 2-[[(2r)-2-(phenylmethoxycarbonylamino)propyl]amino]acetate Chemical compound COC(=O)CNC[C@@H](C)NC(=O)OCC1=CC=CC=C1 BTJJYUVKHQRVOB-LLVKDONJSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- JHKHDIBLFHELCU-UHFFFAOYSA-N n-(azetidin-3-yl)methanesulfonamide Chemical compound CS(=O)(=O)NC1CNC1 JHKHDIBLFHELCU-UHFFFAOYSA-N 0.000 description 2
- MMNHRHMPBOEBAS-UHFFFAOYSA-N n-methoxypiperidin-4-imine;hydrochloride Chemical compound Cl.CON=C1CCNCC1 MMNHRHMPBOEBAS-UHFFFAOYSA-N 0.000 description 2
- 230000001537 neural effect Effects 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 238000003305 oral gavage Methods 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- 239000003002 pH adjusting agent Substances 0.000 description 2
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 2
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000001814 pectin Substances 0.000 description 2
- 235000010987 pectin Nutrition 0.000 description 2
- 229920001277 pectin Polymers 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 230000010287 polarization Effects 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 208000020016 psychiatric disease Diseases 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 238000002821 scintillation proximity assay Methods 0.000 description 2
- 230000009329 sexual behaviour Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- MGNXUUUQPBTDNG-OLZOCXBDSA-N tert-butyl (2r,3s)-3-[tert-butyl(dimethyl)silyl]oxy-2-(hydroxymethyl)pyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@H](O[Si](C)(C)C(C)(C)C)[C@H]1CO MGNXUUUQPBTDNG-OLZOCXBDSA-N 0.000 description 2
- PBMBXMVFAQFPHO-KGLIPLIRSA-N tert-butyl (2r,3s)-3-[tert-butyl(dimethyl)silyl]oxy-2-(methylsulfonyloxymethyl)pyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@H](O[Si](C)(C)C(C)(C)C)[C@H]1COS(C)(=O)=O PBMBXMVFAQFPHO-KGLIPLIRSA-N 0.000 description 2
- PUYIXRSWSUZEPO-OLZOCXBDSA-N tert-butyl (2r,3s)-3-[tert-butyl(dimethyl)silyl]oxy-2-methylpyrrolidine-1-carboxylate Chemical compound C[C@@H]1[C@@H](O[Si](C)(C)C(C)(C)C)CCN1C(=O)OC(C)(C)C PUYIXRSWSUZEPO-OLZOCXBDSA-N 0.000 description 2
- RLHMCXQTIPFLRQ-SFYZADRCSA-N tert-butyl (2r,3s)-3-hydroxy-2-methylpyrrolidine-1-carboxylate Chemical compound C[C@@H]1[C@@H](O)CCN1C(=O)OC(C)(C)C RLHMCXQTIPFLRQ-SFYZADRCSA-N 0.000 description 2
- UFJNFQNQLMGUTQ-HTQZYQBOSA-N tert-butyl (2r,4r)-4-hydroxy-2-(hydroxymethyl)pyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C[C@H](O)C[C@@H]1CO UFJNFQNQLMGUTQ-HTQZYQBOSA-N 0.000 description 2
- IDSQAKWJQJMPMD-AOOOYVTPSA-N tert-butyl (3r,5s)-4-cyano-3,5-dimethylpiperazine-1-carboxylate Chemical compound C[C@H]1CN(C(=O)OC(C)(C)C)C[C@@H](C)N1C#N IDSQAKWJQJMPMD-AOOOYVTPSA-N 0.000 description 2
- NUZXPHIQZUYMOR-DTORHVGOSA-N tert-butyl (3s,5r)-3,5-dimethylpiperazine-1-carboxylate Chemical compound C[C@H]1CN(C(=O)OC(C)(C)C)C[C@@H](C)N1 NUZXPHIQZUYMOR-DTORHVGOSA-N 0.000 description 2
- HEENFWLXSOVOBA-JLHYYAGUSA-N tert-butyl (4e)-4-methoxyimino-3-methylpiperidine-1-carboxylate Chemical compound CO\N=C1/CCN(C(=O)OC(C)(C)C)CC1C HEENFWLXSOVOBA-JLHYYAGUSA-N 0.000 description 2
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 2
- NEMXVXVJGXZDRR-UHFFFAOYSA-N tert-butyl n-(azetidin-3-yl)carbamate Chemical compound CC(C)(C)OC(=O)NC1CNC1 NEMXVXVJGXZDRR-UHFFFAOYSA-N 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 231100001274 therapeutic index Toxicity 0.000 description 2
- 150000003568 thioethers Chemical group 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- BJBUEDPLEOHJGE-IMJSIDKUSA-N trans-3-hydroxy-L-proline Chemical compound O[C@H]1CC[NH2+][C@@H]1C([O-])=O BJBUEDPLEOHJGE-IMJSIDKUSA-N 0.000 description 2
- TYLFLHPQWQQWRD-UHFFFAOYSA-N trans-3-hydroxy-L-prolinol Natural products OCC1NCCC1O TYLFLHPQWQQWRD-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- WHNFPRLDDSXQCL-UAZQEYIDSA-N α-msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 WHNFPRLDDSXQCL-UAZQEYIDSA-N 0.000 description 2
- MSVZMUILYMLJCF-UHFFFAOYSA-N (1-benzhydrylazetidin-3-yl) methanesulfonate Chemical compound C1C(OS(=O)(=O)C)CN1C(C=1C=CC=CC=1)C1=CC=CC=C1 MSVZMUILYMLJCF-UHFFFAOYSA-N 0.000 description 1
- VVLNFOCXNDDLEU-JXUBOQSCSA-N (1r,3s,4s,5s)-3-isocyanato-4,6,6-trimethylbicyclo[3.1.1]heptane Chemical compound C1[C@@]2([H])[C@H](C)[C@@H](N=C=O)C[C@]1([H])C2(C)C VVLNFOCXNDDLEU-JXUBOQSCSA-N 0.000 description 1
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 1
- QSLVRADSLBPUHL-UHNVWZDZSA-N (2R,3S)-3-hydroxy-2-(hydroxymethyl)pyrrolidine-1-carboxylic acid Chemical compound OC[C@@H]1[C@@H](O)CCN1C(O)=O QSLVRADSLBPUHL-UHNVWZDZSA-N 0.000 description 1
- XFMCMQYCRUJAAQ-UHNVWZDZSA-N (2R,3S)-3-hydroxy-2-methylpyrrolidine-1-carboxylic acid Chemical compound C[C@@H]1[C@@H](O)CCN1C(O)=O XFMCMQYCRUJAAQ-UHNVWZDZSA-N 0.000 description 1
- SDEMZAXQOIWPPI-IBGZPJMESA-N (2r)-1,4-dibenzyl-2-(fluoromethyl)piperazine Chemical compound C([C@@H](N(CC1)CC=2C=CC=CC=2)CF)N1CC1=CC=CC=C1 SDEMZAXQOIWPPI-IBGZPJMESA-N 0.000 description 1
- YPISUIDLTZBHPM-PGMHMLKASA-N (2r)-2-(difluoromethyl)piperazine;guanidine Chemical class NC(N)=N.FC(F)[C@H]1CNCCN1 YPISUIDLTZBHPM-PGMHMLKASA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- BENKAPCDIOILGV-RNFRBKRXSA-N (2r,4r)-4-hydroxy-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1C[C@H](O)C[C@@H]1C(O)=O BENKAPCDIOILGV-RNFRBKRXSA-N 0.000 description 1
- ONUCCUGDWPXAJG-RXMQYKEDSA-N (2s)-2-(fluoromethyl)piperazine Chemical compound FC[C@@H]1CNCCN1 ONUCCUGDWPXAJG-RXMQYKEDSA-N 0.000 description 1
- QVHJQCGUWFKTSE-YFKPBYRVSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound OC(=O)[C@H](C)NC(=O)OC(C)(C)C QVHJQCGUWFKTSE-YFKPBYRVSA-N 0.000 description 1
- JOMNTHCQHJPVAZ-YFKPBYRVSA-N (2s)-2-methylpiperazine Chemical compound C[C@H]1CNCCN1 JOMNTHCQHJPVAZ-YFKPBYRVSA-N 0.000 description 1
- CNPSFBUUYIVHAP-WHFBIAKZSA-N (2s,3s)-3-methylpyrrolidin-1-ium-2-carboxylate Chemical compound C[C@H]1CCN[C@@H]1C(O)=O CNPSFBUUYIVHAP-WHFBIAKZSA-N 0.000 description 1
- IFNWESYYDINUHV-WDSKDSINSA-N (2s,6s)-2,6-dimethylpiperazine Chemical compound C[C@H]1CNC[C@H](C)N1 IFNWESYYDINUHV-WDSKDSINSA-N 0.000 description 1
- QJRIUWQPJVPYSO-GHMZBOCLSA-N (3r,4r)-1-benzylpyrrolidine-3,4-diol Chemical compound C1[C@@H](O)[C@H](O)CN1CC1=CC=CC=C1 QJRIUWQPJVPYSO-GHMZBOCLSA-N 0.000 description 1
- QJRIUWQPJVPYSO-QWRGUYRKSA-N (3s,4s)-1-benzylpyrrolidine-3,4-diol Chemical compound C1[C@H](O)[C@@H](O)CN1CC1=CC=CC=C1 QJRIUWQPJVPYSO-QWRGUYRKSA-N 0.000 description 1
- VADWFRGDDJHKNB-MMALYQPHSA-N (3s,4s)-pyrrolidine-3,4-diol;hydrochloride Chemical compound Cl.O[C@H]1CNC[C@@H]1O VADWFRGDDJHKNB-MMALYQPHSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- FMCUPJKTGNBGEC-UHFFFAOYSA-N 1,2,4-triazol-4-amine Chemical compound NN1C=NN=C1 FMCUPJKTGNBGEC-UHFFFAOYSA-N 0.000 description 1
- LRANPJDWHYRCER-UHFFFAOYSA-N 1,2-diazepine Chemical compound N1C=CC=CC=N1 LRANPJDWHYRCER-UHFFFAOYSA-N 0.000 description 1
- SDEMZAXQOIWPPI-UHFFFAOYSA-N 1,4-dibenzyl-2-(fluoromethyl)piperazine Chemical compound C1CN(CC=2C=CC=CC=2)C(CF)CN1CC1=CC=CC=C1 SDEMZAXQOIWPPI-UHFFFAOYSA-N 0.000 description 1
- UOMTVKMKHZMFMQ-UHFFFAOYSA-N 1,4-thiazinane 1,1-dioxide;hydrochloride Chemical compound Cl.O=S1(=O)CCNCC1 UOMTVKMKHZMFMQ-UHFFFAOYSA-N 0.000 description 1
- JUXAVSAMVBLDKO-UHFFFAOYSA-N 1-(1-azabicyclo[2.2.2]octan-3-yl)-3-[3-(1h-indol-3-yl)-1-oxo-1-spiro[1,2-dihydroindene-3,4'-piperidine]-1'-ylpropan-2-yl]urea Chemical class C1N(CC2)CCC2C1NC(=O)NC(C(=O)N1CCC2(C3=CC=CC=C3CC2)CC1)CC1=CNC2=CC=CC=C12 JUXAVSAMVBLDKO-UHFFFAOYSA-N 0.000 description 1
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 1
- WLCZOVFHWMKAJC-ZKRGBOFFSA-N 1-[3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-4-oxoquinazolin-7-yl]-3-(1,2,4-triazol-4-yl)-2-[(1s,3s,4s,5r)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]guanidine;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.N([C@@H]1[C@@H](C)[C@]2(C[C@@](C1)(C2(C)C)[H])[H])=C(NN1C=NN=C1)NC(C=1)=CC=C(C2=O)C=1N=CN2CCC1=CC=C(OC)C=C1F WLCZOVFHWMKAJC-ZKRGBOFFSA-N 0.000 description 1
- HDXFCPZKMFTOLH-UHFFFAOYSA-N 1-amino-2,3-dihydroinden-1-ol Chemical compound C1=CC=C2C(N)(O)CCC2=C1 HDXFCPZKMFTOLH-UHFFFAOYSA-N 0.000 description 1
- OVQAJYCAXPHYNV-UHFFFAOYSA-N 1-benzyl-3-methylpiperidin-4-one Chemical compound C1CC(=O)C(C)CN1CC1=CC=CC=C1 OVQAJYCAXPHYNV-UHFFFAOYSA-N 0.000 description 1
- PPJVXZVTPWQOQS-UHFFFAOYSA-N 1-ethoxy-1-(1-ethoxyethoxy)ethane Chemical compound CCOC(C)OC(C)OCC PPJVXZVTPWQOQS-UHFFFAOYSA-N 0.000 description 1
- GPAAEZIXSQCCES-UHFFFAOYSA-N 1-methoxy-2-(2-methoxyethoxymethoxymethoxy)ethane Chemical compound COCCOCOCOCCOC GPAAEZIXSQCCES-UHFFFAOYSA-N 0.000 description 1
- DRSBBLXKXAITOR-KBPBESRZSA-N 1-o-tert-butyl 2-o-ethyl (2s,3s)-3-[tert-butyl(dimethyl)silyl]oxypyrrolidine-1,2-dicarboxylate Chemical compound CCOC(=O)[C@@H]1[C@@H](O[Si](C)(C)C(C)(C)C)CCN1C(=O)OC(C)(C)C DRSBBLXKXAITOR-KBPBESRZSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical compound NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- HGUFODBRKLSHSI-UHFFFAOYSA-N 2,3,7,8-tetrachloro-dibenzo-p-dioxin Chemical compound O1C2=CC(Cl)=C(Cl)C=C2OC2=C1C=C(Cl)C(Cl)=C2 HGUFODBRKLSHSI-UHFFFAOYSA-N 0.000 description 1
- NVWVWEWVLBKPSM-UHFFFAOYSA-N 2,4-difluorophenol Chemical compound OC1=CC=C(F)C=C1F NVWVWEWVLBKPSM-UHFFFAOYSA-N 0.000 description 1
- NTBWWUHBVFUMLG-UHFFFAOYSA-N 2,4-difluoroquinazoline Chemical compound C1=CC=CC2=NC(F)=NC(F)=C21 NTBWWUHBVFUMLG-UHFFFAOYSA-N 0.000 description 1
- VHJKDOLGYMULOP-UHFFFAOYSA-N 2-(2,4-dichlorophenyl)ethanamine Chemical compound NCCC1=CC=C(Cl)C=C1Cl VHJKDOLGYMULOP-UHFFFAOYSA-N 0.000 description 1
- YNEGKGZLMMSZTD-UHFFFAOYSA-N 2-(2-oxo-1h-quinazolin-4-yl)guanidine Chemical class C1=CC=C2C(NC(=N)N)=NC(=O)NC2=C1 YNEGKGZLMMSZTD-UHFFFAOYSA-N 0.000 description 1
- ZVVUOCJSVSACNG-UHFFFAOYSA-N 2-(chloromethyl)-3-[2-(4-fluorophenyl)ethyl]-7-nitroquinazolin-4-one Chemical compound C=1C([N+](=O)[O-])=CC=C(C2=O)C=1N=C(CCl)N2CCC1=CC=C(F)C=C1 ZVVUOCJSVSACNG-UHFFFAOYSA-N 0.000 description 1
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical class O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 1
- XGAKPGATOOROOT-UHFFFAOYSA-N 2-(phenylmethoxycarbonylamino)propanethioic S-acid Chemical compound SC(=O)C(C)NC(=O)OCC1=CC=CC=C1 XGAKPGATOOROOT-UHFFFAOYSA-N 0.000 description 1
- UMGZWLHVYMGKKF-UHFFFAOYSA-N 2-amino-4,5-difluoro-n-[2-(4-fluorophenyl)ethyl]benzamide Chemical compound NC1=CC(F)=C(F)C=C1C(=O)NCCC1=CC=C(F)C=C1 UMGZWLHVYMGKKF-UHFFFAOYSA-N 0.000 description 1
- DGOZIZVTANAGCA-UHFFFAOYSA-N 2-amino-4,5-difluorobenzoic acid Chemical compound NC1=CC(F)=C(F)C=C1C(O)=O DGOZIZVTANAGCA-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical class NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- WXHHOJFTHYWFLG-UHFFFAOYSA-N 2-amino-n-[2-(4-fluorophenyl)ethyl]-4-nitrobenzamide Chemical compound NC1=CC([N+]([O-])=O)=CC=C1C(=O)NCCC1=CC=C(F)C=C1 WXHHOJFTHYWFLG-UHFFFAOYSA-N 0.000 description 1
- UXGVMFHEKMGWMA-UHFFFAOYSA-N 2-benzofuran Chemical compound C1=CC=CC2=COC=C21 UXGVMFHEKMGWMA-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- QKGYKRACHNWPCA-UHFFFAOYSA-N 2-chloro-2-(4-chlorophenyl)ethanamine Chemical compound NCC(Cl)C1=CC=C(Cl)C=C1 QKGYKRACHNWPCA-UHFFFAOYSA-N 0.000 description 1
- SVDDJQGVOFZBNX-UHFFFAOYSA-N 2-chloroethyl carbonochloridate Chemical compound ClCCOC(Cl)=O SVDDJQGVOFZBNX-UHFFFAOYSA-N 0.000 description 1
- QCFAKTACICNQGT-UHFFFAOYSA-N 2-methyl-2-[(2-methylpropan-2-yl)oxymethoxymethoxy]propane Chemical compound CC(C)(C)OCOCOC(C)(C)C QCFAKTACICNQGT-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 1
- PNAVVYQFNJPUKU-UHFFFAOYSA-N 3-(2-fluoro-4-methoxyphenyl)-n-methylpropanamide Chemical compound CNC(=O)CCC1=CC=C(OC)C=C1F PNAVVYQFNJPUKU-UHFFFAOYSA-N 0.000 description 1
- DNRXTZKGJNXJIR-UHFFFAOYSA-N 3-[2-(2-fluoro-4-methoxyphenyl)ethyl]-7-nitro-1,2,3-benzotriazin-4-one Chemical compound FC1=CC(OC)=CC=C1CCN1C(=O)C2=CC=C([N+]([O-])=O)C=C2N=N1 DNRXTZKGJNXJIR-UHFFFAOYSA-N 0.000 description 1
- BRQQEHKNUWOUHL-YCULYRLHSA-N 3-amino-n-[3-[2-(4-fluorophenyl)ethyl]-4-oxoquinazolin-7-yl]-n'-[(1r,3s,4s,5s)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]azetidine-1-carboximidamide Chemical compound N([C@@H]1[C@@H](C)[C@@]2(C[C@](C1)(C2(C)C)[H])[H])=C(N1CC(N)C1)NC(C=1)=CC=C(C2=O)C=1N=CN2CCC1=CC=C(F)C=C1 BRQQEHKNUWOUHL-YCULYRLHSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- BWCDLEQTELFBAW-UHFFFAOYSA-N 3h-dioxazole Chemical compound N1OOC=C1 BWCDLEQTELFBAW-UHFFFAOYSA-N 0.000 description 1
- KWIVRAVCZJXOQC-UHFFFAOYSA-N 3h-oxathiazole Chemical compound N1SOC=C1 KWIVRAVCZJXOQC-UHFFFAOYSA-N 0.000 description 1
- RELAJOWOFXGXHI-UHFFFAOYSA-N 3h-oxathiole Chemical compound C1SOC=C1 RELAJOWOFXGXHI-UHFFFAOYSA-N 0.000 description 1
- CAUXQOLTFGCRKD-UHFFFAOYSA-N 4,4-dimethylcyclohexan-1-amine Chemical compound CC1(C)CCC(N)CC1 CAUXQOLTFGCRKD-UHFFFAOYSA-N 0.000 description 1
- PXQMSTLNSHMSJB-UHFFFAOYSA-N 4,4-dimethylcyclohexan-1-one Chemical compound CC1(C)CCC(=O)CC1 PXQMSTLNSHMSJB-UHFFFAOYSA-N 0.000 description 1
- NXBWFXMEJVOABP-UHFFFAOYSA-N 4-amino-1h-quinazolin-2-one Chemical class C1=CC=C2C(N)=NC(=O)NC2=C1 NXBWFXMEJVOABP-UHFFFAOYSA-N 0.000 description 1
- KFAMTQFKYUXQKV-UHFFFAOYSA-N 4-benzyl-1,4-thiazinane 1,1-dioxide Chemical compound C1CS(=O)(=O)CCN1CC1=CC=CC=C1 KFAMTQFKYUXQKV-UHFFFAOYSA-N 0.000 description 1
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- AYXAEOAVYSHGSB-UHFFFAOYSA-N 4-methylpiperidin-4-ol;hydrochloride Chemical compound Cl.CC1(O)CCNCC1 AYXAEOAVYSHGSB-UHFFFAOYSA-N 0.000 description 1
- ZESWUEBPRPGMTP-UHFFFAOYSA-N 4-nitrobenzamide Chemical compound NC(=O)C1=CC=C([N+]([O-])=O)C=C1 ZESWUEBPRPGMTP-UHFFFAOYSA-N 0.000 description 1
- RUCHWTKMOWXHLU-UHFFFAOYSA-N 5-nitroanthranilic acid Chemical compound NC1=CC=C([N+]([O-])=O)C=C1C(O)=O RUCHWTKMOWXHLU-UHFFFAOYSA-N 0.000 description 1
- SPKGMANCFQFUGS-UHFFFAOYSA-N 7-azido-1h-quinazolin-4-one Chemical compound N1=CNC(=O)C=2C1=CC(N=[N+]=[N-])=CC=2 SPKGMANCFQFUGS-UHFFFAOYSA-N 0.000 description 1
- KCNIWFFVWBXWAV-UHFFFAOYSA-N 7-nitro-1h-3,1-benzoxazine-2,4-dione Chemical compound N1C(=O)OC(=O)C=2C1=CC([N+](=O)[O-])=CC=2 KCNIWFFVWBXWAV-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102100022455 Adrenocorticotropic hormone receptor Human genes 0.000 description 1
- 101710115814 Adrenocorticotropic hormone receptor Proteins 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 241001120493 Arene Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- LPZUTQDBEUCNOY-KBPBESRZSA-N CCOC([C@H]([C@H](CC1)O[Si+](C)(C)C(C)(C)C)N1C(OC(C)(C)C)=O)=O Chemical compound CCOC([C@H]([C@H](CC1)O[Si+](C)(C)C(C)(C)C)N1C(OC(C)(C)C)=O)=O LPZUTQDBEUCNOY-KBPBESRZSA-N 0.000 description 1
- GRXVKHFAPSGQDJ-UHFFFAOYSA-N CON=C1CCNCC1 Chemical compound CON=C1CCNCC1 GRXVKHFAPSGQDJ-UHFFFAOYSA-N 0.000 description 1
- AXJSPSVTVRIEFT-YFKPBYRVSA-N C[C@@H](CN(C)C1)NC1=O Chemical compound C[C@@H](CN(C)C1)NC1=O AXJSPSVTVRIEFT-YFKPBYRVSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102000011632 Caseins Human genes 0.000 description 1
- 108010076119 Caseins Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 208000002705 Glucose Intolerance Diseases 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000978431 Homo sapiens Melanocortin receptor 3 Proteins 0.000 description 1
- 101000978418 Homo sapiens Melanocortin receptor 4 Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102000016267 Leptin Human genes 0.000 description 1
- 108010092277 Leptin Proteins 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102000030612 Melanocortin 5 receptor Human genes 0.000 description 1
- 108010088565 Melanocortin 5 receptor Proteins 0.000 description 1
- 102100023726 Melanocortin receptor 3 Human genes 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- 102400000747 Melanocyte-stimulating hormone beta Human genes 0.000 description 1
- 101710129905 Melanotropin beta Proteins 0.000 description 1
- 102400000744 Melanotropin gamma Human genes 0.000 description 1
- 101800000520 Melanotropin gamma Proteins 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- 101100189356 Mus musculus Papolb gene Proteins 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical group 0.000 description 1
- 102000008299 Nitric Oxide Synthase Human genes 0.000 description 1
- 108010021487 Nitric Oxide Synthase Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- JHHZLHWJQPUNKB-SCSAIBSYSA-N O[C@H]1CNCC1 Chemical compound O[C@H]1CNCC1 JHHZLHWJQPUNKB-SCSAIBSYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010069820 Pro-Opiomelanocortin Proteins 0.000 description 1
- 239000000683 Pro-Opiomelanocortin Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 125000000066 S-methyl group Chemical group [H]C([H])([H])S* 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- SLGBZMMZGDRARJ-UHFFFAOYSA-N Triphenylene Natural products C1=CC=C2C3=CC=CC=C3C3=CC=CC=C3C2=C1 SLGBZMMZGDRARJ-UHFFFAOYSA-N 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 108010021433 Type 3 Melanocortin Receptor Proteins 0.000 description 1
- 102000008318 Type 3 Melanocortin Receptor Human genes 0.000 description 1
- SOUPGWGAPDMYFM-LJQANCHMSA-N [(2r)-1,4-dibenzylpiperazin-2-yl]methanol Chemical compound C([C@@H](N(CC1)CC=2C=CC=CC=2)CO)N1CC1=CC=CC=C1 SOUPGWGAPDMYFM-LJQANCHMSA-N 0.000 description 1
- KJCSLIIJEPKARF-UHFFFAOYSA-N [O-][N+](c(cc1)cc(NCN2CCc(ccc(Cl)c3)c3Cl)c1C2=O)=O Chemical compound [O-][N+](c(cc1)cc(NCN2CCc(ccc(Cl)c3)c3Cl)c1C2=O)=O KJCSLIIJEPKARF-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 239000003570 air Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000003973 alkyl amines Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000005107 alkyl diaryl silyl group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- HAMNKKUPIHEESI-UHFFFAOYSA-N aminoguanidine Chemical compound NNC(N)=N HAMNKKUPIHEESI-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001262 anti-secretory effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000004019 antithrombin Substances 0.000 description 1
- 239000003699 antiulcer agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000002567 autonomic effect Effects 0.000 description 1
- XYOVOXDWRFGKEX-UHFFFAOYSA-N azepine Chemical compound N1C=CC=CC=C1 XYOVOXDWRFGKEX-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960002433 cysteine Drugs 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 230000004300 dark adaptation Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000005265 dialkylamine group Chemical group 0.000 description 1
- 125000005105 dialkylarylsilyl group Chemical group 0.000 description 1
- 125000005266 diarylamine group Chemical group 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000012971 dimethylpiperazine Substances 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical group [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 125000004119 disulfanediyl group Chemical group *SS* 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 150000002081 enamines Chemical group 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940095399 enema Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethanethiol Chemical compound CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 210000003499 exocrine gland Anatomy 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- RMBPEFMHABBEKP-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2C3=C[CH]C=CC3=CC2=C1 RMBPEFMHABBEKP-UHFFFAOYSA-N 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 239000007897 gelcap Substances 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000014101 glucose homeostasis Effects 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002314 glycerols Chemical class 0.000 description 1
- PGLSRXLRLNOCQA-UHFFFAOYSA-N guanidine;1h-quinazolin-2-one Chemical class NC(N)=N.C1=CC=C2NC(=O)N=CC2=C1 PGLSRXLRLNOCQA-UHFFFAOYSA-N 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229940083094 guanine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- DDTGNKBZWQHIEH-UHFFFAOYSA-N heptalene Chemical compound C1=CC=CC=C2C=CC=CC=C21 DDTGNKBZWQHIEH-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000002390 heteroarenes Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 150000007857 hydrazones Chemical group 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- COQRGFWWJBEXRC-UHFFFAOYSA-N hydron;methyl 2-aminoacetate;chloride Chemical compound Cl.COC(=O)CN COQRGFWWJBEXRC-UHFFFAOYSA-N 0.000 description 1
- RGZRSLKIOCHTSI-UHFFFAOYSA-N hydron;n-methylhydroxylamine;chloride Chemical compound Cl.CNO RGZRSLKIOCHTSI-UHFFFAOYSA-N 0.000 description 1
- XNXVOSBNFZWHBV-UHFFFAOYSA-N hydron;o-methylhydroxylamine;chloride Chemical compound Cl.CON XNXVOSBNFZWHBV-UHFFFAOYSA-N 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 230000003345 hyperglycaemic effect Effects 0.000 description 1
- 230000000910 hyperinsulinemic effect Effects 0.000 description 1
- 230000002218 hypoglycaemic effect Effects 0.000 description 1
- 150000002466 imines Chemical group 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 150000002467 indacenes Chemical class 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- ZBKFYXZXZJPWNQ-UHFFFAOYSA-N isothiocyanate group Chemical group [N-]=C=S ZBKFYXZXZJPWNQ-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 1
- 229940039781 leptin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000013227 male C57BL/6J mice Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000845 maltitol Substances 0.000 description 1
- 235000010449 maltitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 1
- 229940035436 maltitol Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- FBBDOOHMGLLEGJ-UHFFFAOYSA-N methane;hydrochloride Chemical compound C.Cl FBBDOOHMGLLEGJ-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- NSPJNIDYTSSIIY-UHFFFAOYSA-N methoxy(methoxymethoxy)methane Chemical compound COCOCOC NSPJNIDYTSSIIY-UHFFFAOYSA-N 0.000 description 1
- GMZGWPPEZCREPP-JTQLQIEISA-N methyl (2s)-2-(benzylamino)-3-hydroxypropanoate Chemical compound COC(=O)[C@H](CO)NCC1=CC=CC=C1 GMZGWPPEZCREPP-JTQLQIEISA-N 0.000 description 1
- QKFSNPDEINFHRS-NSHDSACASA-N methyl (2s)-2-[benzyl-(2-chloroacetyl)amino]-3-hydroxypropanoate Chemical compound COC(=O)[C@H](CO)N(C(=O)CCl)CC1=CC=CC=C1 QKFSNPDEINFHRS-NSHDSACASA-N 0.000 description 1
- NDBQJIBNNUJNHA-DFWYDOINSA-N methyl (2s)-2-amino-3-hydroxypropanoate;hydrochloride Chemical compound Cl.COC(=O)[C@@H](N)CO NDBQJIBNNUJNHA-DFWYDOINSA-N 0.000 description 1
- HRGFWZFYDVIMBC-NSHDSACASA-N methyl (2s)-3-hydroxy-2-[(4-methoxyphenyl)methylamino]propanoate Chemical compound COC(=O)[C@H](CO)NCC1=CC=C(OC)C=C1 HRGFWZFYDVIMBC-NSHDSACASA-N 0.000 description 1
- UAWVZBDVSNLPDT-UHFFFAOYSA-N methyl 2-(benzylamino)acetate Chemical compound COC(=O)CNCC1=CC=CC=C1 UAWVZBDVSNLPDT-UHFFFAOYSA-N 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- TYRGLVWXHJRKMT-QMMMGPOBSA-N n-benzyloxycarbonyl-l-serine-betalactone Chemical compound OC(=O)[C@H](C)NC(=O)OCC1=CC=CC=C1 TYRGLVWXHJRKMT-QMMMGPOBSA-N 0.000 description 1
- SAVQQRYWWAGSQW-UHFFFAOYSA-N n-methyl-n-(trifluoro-$l^{4}-sulfanyl)methanamine Chemical compound CN(C)S(F)(F)F SAVQQRYWWAGSQW-UHFFFAOYSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 230000000422 nocturnal effect Effects 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N o-biphenylenemethane Natural products C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- AZHVQJLDOFKHPZ-UHFFFAOYSA-N oxathiazine Chemical compound O1SN=CC=C1 AZHVQJLDOFKHPZ-UHFFFAOYSA-N 0.000 description 1
- CQDAMYNQINDRQC-UHFFFAOYSA-N oxatriazole Chemical compound C1=NN=NO1 CQDAMYNQINDRQC-UHFFFAOYSA-N 0.000 description 1
- ATYBXHSAIOKLMG-UHFFFAOYSA-N oxepin Chemical compound O1C=CC=CC=C1 ATYBXHSAIOKLMG-UHFFFAOYSA-N 0.000 description 1
- 150000002923 oximes Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- FAQJJMHZNSSFSM-UHFFFAOYSA-N phenylglyoxylic acid Chemical compound OC(=O)C(=O)C1=CC=CC=C1 FAQJJMHZNSSFSM-UHFFFAOYSA-N 0.000 description 1
- TYZYRCHEVXXLSJ-UHFFFAOYSA-N phenylmethoxymethoxymethoxymethylbenzene Chemical compound C=1C=CC=CC=1COCOCOCC1=CC=CC=C1 TYZYRCHEVXXLSJ-UHFFFAOYSA-N 0.000 description 1
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 150000003053 piperidines Chemical class 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000003571 reporter gene assay Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000009666 routine test Methods 0.000 description 1
- SDYRZPPZUQAFHG-SNVBAGLBSA-N s-ethyl (2r)-2-(phenylmethoxycarbonylamino)propanethioate Chemical compound CCSC(=O)[C@@H](C)NC(=O)OCC1=CC=CC=C1 SDYRZPPZUQAFHG-SNVBAGLBSA-N 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000010009 steroidogenesis Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000001174 sulfone group Chemical group 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 125000003375 sulfoxide group Chemical group 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- GIIDZEBNRLNGMS-SFYZADRCSA-N tert-butyl (2r,3s)-3-hydroxy-2-(hydroxymethyl)pyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@H](O)[C@H]1CO GIIDZEBNRLNGMS-SFYZADRCSA-N 0.000 description 1
- WYHQDHOSMANRCP-TXEJJXNPSA-N tert-butyl (3s,5r)-3,5-dimethyl-4-(3-oxobutyl)piperazine-1-carboxylate Chemical compound C[C@H]1CN(C(=O)OC(C)(C)C)C[C@@H](C)N1CCC(C)=O WYHQDHOSMANRCP-TXEJJXNPSA-N 0.000 description 1
- YPVKDSYMBCAQJR-QKBGKSAYSA-N tert-butyl n-[1-[n-[3-[2-(4-fluorophenyl)ethyl]-4-oxoquinazolin-7-yl]-n'-[(1r,3s,4s,5s)-4,6,6-trimethyl-3-bicyclo[3.1.1]heptanyl]carbamimidoyl]azetidin-3-yl]carbamate Chemical compound N(/[C@@H]1[C@@H](C)[C@@]2(C[C@](C1)(C2(C)C)[H])[H])=C(N1CC(C1)NC(=O)OC(C)(C)C)\NC(C=1)=CC=C(C2=O)C=1N=CN2CCC1=CC=C(F)C=C1 YPVKDSYMBCAQJR-QKBGKSAYSA-N 0.000 description 1
- 150000003511 tertiary amides Chemical class 0.000 description 1
- 150000003512 tertiary amines Chemical group 0.000 description 1
- IFLREYGFSNHWGE-UHFFFAOYSA-N tetracene Chemical compound C1=CC=CC2=CC3=CC4=CC=CC=C4C=C3C=C21 IFLREYGFSNHWGE-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- 229930192474 thiophene Chemical group 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000004665 trialkylsilyl group Chemical group 0.000 description 1
- 125000005106 triarylsilyl group Chemical group 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 229940066528 trichloroacetate Drugs 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 239000005051 trimethylchlorosilane Substances 0.000 description 1
- 125000005580 triphenylene group Chemical group 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- SFVVQRJOGUKCEG-OPQSFPLASA-N β-MSH Chemical compound C1C[C@@H](O)[C@H]2C(COC(=O)[C@@](O)([C@@H](C)O)C(C)C)=CCN21 SFVVQRJOGUKCEG-OPQSFPLASA-N 0.000 description 1
- GZWUQPQBOGLSIM-VOOUCTBASA-N γ msh Chemical compound C([C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(O)=O)C(C)C)C1=CC=C(O)C=C1 GZWUQPQBOGLSIM-VOOUCTBASA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/24—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/626—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B
- C04B35/63—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B using additives specially adapted for forming the products, e.g.. binder binders
- C04B35/632—Organic additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/91—Oxygen atoms with aryl or aralkyl radicals attached in position 2 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/95—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in positions 2 and 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/95—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in positions 2 and 4
- C07D239/96—Two oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D253/00—Heterocyclic compounds containing six-membered rings having three nitrogen atoms as the only ring hetero atoms, not provided for by group C07D251/00
- C07D253/08—Heterocyclic compounds containing six-membered rings having three nitrogen atoms as the only ring hetero atoms, not provided for by group C07D251/00 condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- This invention relates to melanocortin-4 receptor (MC4-R) agonists and methods of their preparation. More specifically, the invention relates to quinazolinone compounds that exhibit reduced bipaccumulation properties when administered to a subject.
- M4-R melanocortin-4 receptor
- Melanocortins are peptide products resulting from post- translational processing of pro-opiomelanocortin and are known to have a broad array of physiological activities.
- the natural melanocortins include the different types of melanocyte stimulating hormone ( ⁇ -MSH, ⁇ -MSH, ⁇ -MSH) and ACTH. Of these, ⁇ -MSH and ACTH are considered to be the main endogenous melanocortins.
- MC-Rs melanocortin receptors
- MC1-R mediates pigmentation of the hair and skin.
- MC2-R mediates the effects of ACTH on steroidogenesis in the adrenal gland.
- MC3-R and MC4-R are predominantly expressed in the brain.
- MC5-R is considered to have a role in the exocrine gland system.
- the melanocortin-4 receptor is a seven- transmembrane receptor. MC4-R may participate in modulating the flow of visual and sensory information, coordinate aspects of somatomotor control, and/or participate in the modulation of autonomic outflow to the heart.
- Significantly, inactivation of this receptor by gene targeting has resulted in mice that develop a maturity onset obesity syndrome associated with hyperphagia, hyperinsulinemia, and hyperglycemia.
- MC4-R has also been implicated in other disease states including erectile disorders, cardiovascular disorders, neuronal injuries or disorders, inflammation, fever, cognitive disorders, and sexual behavior disorders.
- M. E. Hadley and C. Haskell-Luevano The proopiomelanocortin system, Ann. N. Y. Acad. Sci., 885:1 (1999).
- R antagonists indicate that MC4-R is implicated in endogenous energy regulation.
- an agouti protein is normally expressed in the skin and is an antagonist of the cutaneous MC receptor involved in pigmentation, MC1-R. M. M. Ollmann et al, Science, 278:135-138 (1997).
- agouti protein in mice leads to a yellow coat color due to antagonism of MC1-R and increased food intake and body weight due to antagonism of MC4-R.
- L. L. Kiefer et al Biochemistry, 36: 2084-2090 (1997); D. S. Lu et al, Nature, 371 :799-802 (1994).
- Agouti related protein an agouti protein homologue, antagonizes MC4-R but not MC1-R.
- AGRP Agouti related protein
- M. Fong et al Biochem. Biophys. Res. Commun. 237:629-631 (1997).
- Administration of AGRP in mice increases food intake and causes obesity but does not alter pigmentation.
- M. Rossi et al Endocrinology, 739:4428-4431 (1998). Together, this research indicates that MC4-R participates in energy regulation, and therefore, identifies this receptor as a target for a rational drug design for the treatment of obesity.
- MC4-R agonists that are piperidine compounds and derivatives
- WO 01/70337 and WO 99/64002 disclose MC-R agonists that are spiropiperidine derivatives.
- Other known melanocortin receptor agonists include aromatic amine compounds containing amino acid residues, particularly tryptophan residues, as disclosed in WO 01/55106. Similar agonists are disclosed in WO 01/055107 which comprise aromatic amine compounds containing tertiary amide or tertiary amine groups.
- WO 01/055109 discloses melanocortin receptor agonists comprising aromatic amines which are generally bisamides separated by a nitrogen-containing alkyl linker.
- Guanidine-containing compounds having a variety of biological activities are also known in the prior art.
- U.S. patent No. 4,732,916 issued to Satoh et al. discloses guanidine compounds useful as antiulcer agents
- U.S. Patent No. 4,948,901 issued to Schnur et al. and EP 0343 894 disclose guanidino compounds useful as protease inhibitors and as anti-plasmin and anti-thrombin agents
- U.S. Patent No. 5,352,704 issued to Okuyama et al. discloses a guanidino compound useful as an antiviral agent.
- Guanidine- containing compounds are also disclosed in other references.
- U.S. Patent No. 6,030,985 issued to Gentile et al. discloses guanidine compounds useful for treating and preventing conditions in which inhibition of nitric oxide synthetase is beneficial such as stroke, schizophrenia, anxiety, and pain.
- U.S. Patent No. 5,952,381 issued to Chen et al. discloses certain guanidine compounds for use in selectively inhibiting or antagonizing ⁇ v ⁇ 3 integrins.
- MC4-R agonist activity a need remains for new compounds and pharmaceutical compositions that may be used to treat MC4-R mediated diseases and disease states.
- a need also remains for compounds that exhibit desirable pharmacological properties such as compounds that have reduced bioaccumulation properties in subjects to which they are administered.
- the instant invention provides potent and specific agonists of
- R is selected from substituted or unsubstituted arylalkyl, heteroarylalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, heterocyclylalkyl, cycloalkylalkyl, alkenyl, alkynyl, or alkyl groups;
- R 2 is selected from H or substituted or unsubstituted arylalkyl, heteroarylalkyl, alkoxy, alkylamino, dialkylamino, aryl, heteroaryl, heterocyclyl, cycloalkyl, heterocyclylalkyl, cycloalkylalkyl, alkenyl, alkynyl, or alkyl groups;
- R 3 , R 4 , and R 5 are independently selected from H, Cl, I, F, Br, OH, NH 2 , CN, NO 2 , or substituted or unsubstituted alkoxy or alkyl groups;
- R 3 ' is selected from H or substituted or unsubstituted aryl, alkyl, alkenyl, alkynyl, cycloalkyl, heteroaryl, heterocyclyl, heterocyclylalkyl, arylalkyl, heteroarylalkyl, or cycloalkylalkyl groups;
- Z is selected from a piperazinone of formula
- Compounds provided by the invention further include prodrugs of the compounds of formula IA and IB, pharmaceutically acceptable salts thereof, stereoisomers thereof, tautomers thereof, hydrates thereof, or solvates thereof.
- the invention further provides compounds of formula IA and IB in which R 3 , R 4 , and R 5 are all H.
- the invention further provides compounds of formula IA and IB in which R 3' is selected from the group consisting of substituted and unsubstituted cyclohexyl, 2-alkylcyclohexyl, 2,2-dialkylcyclohexyl, 2,3- dialkylcyclohexyl, 2,4-dialkylcyclohexyl, 2,5-dialkylcyclohexyl, 2,6- dialkylcyclohexyl, 3,4-dialkylcyclohexyl, 3-alkylcyclohexyl, 4-alkylcyclohexyl, 3,3,5-trialkylcyclohexyl, 2-aminocyclohexyl, 3-aminocyclohexyl, 4- aminocyclohexyl, 2,3-diaminocyclohexyl, 2,4-diaminocyclohexyl, 3,4- diaminocyclohexyl, 2,5-diaminocycl
- R 3' is selected from the group consisting of substituted and unsubstituted cyclohexyl, 2-methylcyclohexyl, 2,2-dimethylcyclohexyl, 2,3- dimethylcyclohexyl, 2,4-dimethylcyclohexyl, 2,5-dimethylcyclohexyl, 2,6- dimethylcyclohexyl, 3,4-dimethylcyclohexyl, 3-methylcyclohexyl, 4- methylcyclohexyl, 3,3,5-trimethylcyclohexyl, 4- -butylcyclohexyl, isopinocampheyl, 7,7-dimethylnorbomyl, 4-isopropylcyclohexyl, 3- methylcycloheptyl groups, 2-fluoro-4-methylcyclohexyl, 4-fluoro-2- methylcyclohexyl, 4,4-difluoro-2-methylcyclohexyl,
- the invention further provides compounds of formula IA and IB in which R 3 ' is a substituted or unsubstituted polycyclic cycloalkyl group.
- R 3 ' is a substituted or unsubstituted polycyclic cycloalkyl group having the formula II
- the invention further provides compounds of formula IA and IB in which R 1 is a substituted or unsubstituted arylalkyl group such as a substituted or unsubstituted phenylethyl group.
- R 1 is a substituted phenylethyl group such as a 4-substituted phenylethyl group or a 2,4-disubstituted phenylethyl group.
- R 1 is selected from phenylethyl, 2,4-dichlorophenylethyl, 4-methoxyphenylethyl, 4- phenoxyphenylethyl, 4-bromophenylethyl, 4-methylphenylethyl, 4- chlorophenylethyl, 4-fluorophenylethyl, 4-ethylphenylethyl, cyclohexenylethyl, 2-methoxyphenylethyl, 2-chlorophenylethyl, 2-fluorophenylethyl, 3- methoxyphenylethyl, 3-fluorophenylethyl, thienylethyl, indolylethyl, 4- hydroxyphenylethyl, 3,4-dimethoxyphenylethyl, 2-chloro-4-iodophenylethyl, 2- fluoro-4-methylphenylethyl, 4-chloro-2-fluor
- R 1 is selected from 2-fluoro-4-methoxyphenylethyl, 2- chloro-4-methoxyphenylethyl, 4-fluorophenylethyl, 4-chlorophenylethyl, 4- chloro-2-fluorophenylethyl, 2,4-dichlorophenylethyl, 4-bromophenylethyl, or 4- bromo-2-fluorophenylethyl groups.
- R 2 is selected from substituted or unsubstituted heterocyclyl groups or substituted or unsubstituted heteroaryl groups.
- R 2 is selected from substituted or unsubstituted pyridinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, tetrahydrofuranyl, furanyl, pyrrolidinyl, pyrrolyl, thiophenyl, tetrahydrothiophenyl, pyranyl, tetrahydropyranyl, tetrahydrothiopyranyl, pyrazinyl, thiazolyl, pyrimidinyl, quinuclidinyl, indolyl, imidazolyl, triazolyl, tetrazolyl, or pyridazinyl groups.
- R 2 is selected from heteroary
- the invention further provides compounds of formula IA and IB in which R 2 is selected from substituted or unsubstituted aryl or cycloalkyl groups.
- R 2 is selected from aryl or cycloalkyl groups of formula which may be additionally substituted or may be unsubstituted.
- R 2 is selected from substituted or unsubstituted heterocyclylalkyl, or cycloalkylamino groups.
- R 2 is selected from a group such as a substituted or unsubstituted cyclopropylamino group; a substituted or unsubstituted piperazinylalkyl group such as a piperazinylmethyl group or an N-methylpiperazinylmethyl group; or a piperidinylalkyl group such as a piperidinylmethyl group or a piperidinylethyl group.
- the invention further provides compounds of formula IA and IB in which Z is a piperazinone having the following formula
- Z is a piperazinone having the following formula
- the invention provides compounds in which the fy 2 value for the compound is less than 35, 30, 25, 20, 15, 10, or 5 hours in a tissue with high blood perfusion such as brain, liver, kidney, and heart.
- the t a is less than or about 4 hours and in some embodiments is less than or about 3 hours in a subject to which the compound(s) have been administered.
- compositions such as a pharmaceutical formulation or medicament comprising a compound according to the instant invention and a pharmaceutically acceptable carrier.
- the invention further provides the use of the compounds of the invention in preparing a medicament or pharmaceutical formulation for use in treating an MC4-R mediated disease.
- a disease is obesity or type II diabetes.
- a method of treating an MC4-R mediated disease comprising administering to a subject in need thereof, a compound or composition of the instant invention.
- the compounds of the invention exhibit reduced bioaccumulation in the tissue and plasma of the subject.
- a disease to be treated by those methods of the instant invention is obesity or type II diabetes.
- a compound or composition of the invention is intranasally administered.
- a compound or composition of the invention is administered to a human subject.
- the instant invention relates to novel classes of small molecule melanocortin-4 receptor (MC4-R) agonists. These compounds can be formulated into compositions and are useful in activating MC4-R, or in the treatment of MC4-R-mediated diseases, such as obesity, type II diabetes, erectile dysfunction, polycystic ovary disease, complications resulting from or associated with obesity and diabetes, and Syndrome X.
- MC4-R melanocortin-4 receptor
- Alkyl groups include straight chain and branched alkyl groups having from 1 to about 8 carbon atoms.
- straight chain alkyl groups include methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, and octyl groups.
- branched alkyl groups include, but are not limited to, isopropyl, sec-butyl, t-butyl, and isopentyl groups.
- Representative substituted alkyl groups may be substituted one or more times with, for example, amino, thio, alkoxy, or halo groups such as F, Cl, Br, and I groups.
- Cycloalkyl groups are cyclic alkyl groups such as, but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl groups. Cycloalkyl groups also includes rings that are substituted with straight or branched chain alkyl groups as defined above, and further include cycloalkyl groups that are substituted with other rings including fused rings such as, but not limited to, decalinyl, tetrahydronaphthyl, and indanyl.
- Cycloalkyl groups also include polycyclic cycloalkyl groups such as, but not limited to, norbornyl, adamantyl, bornyl, camphenyl, isocamphenyl, and carenyl groups.
- Representative substituted cycloalkyl groups may be mono- substituted or substituted more than once, such as, but not limited to, 2,2-, 2,3-, 2,4- 2,5- or 2,6-disubstituted cyclohexyl groups or mono-, di- or tri- substituted norbornyl or cycloheptyl groups, which may be substituted with, for example, alkyl, alkoxy, amino, thio, cyano, or halo groups.
- Alkenyl groups are straight chain, branched or cyclic lower alkyl groups having 2 to about 8 carbon atoms, and further including at least one double bond, as exemplified, for instance, by vinyl, propenyl, 2-butenyl, 3- butenyl, isobutenyl, cyclohexenyl, cyclopentenyl, cyclohexadienyl, butadienyl, pentadienyl, and hexadienyl groups among others.
- Alkynyl groups are straight chain or branched lower alkyl groups having 2 to about 8 carbon atoms, and further including at least one triple bond, as exemplified by groups, including, but not limited to, ethynyl, propynyl, and butynyl groups.
- Aryl groups are cyclic aromatic hydrocarbons that do not contain heteroatoms.
- aryl groups include, but are not limited to, phenyl, azulene, heptalene, biphenylene, indacene, fluorene, phenanthrene, triphenylene, pyrene, naphthacene, chrysene, biphenyl, anthracenyl, and naphthenyl groups.
- aryl groups includes groups containing fused rings, such as fused aromatic-aliphatic ring systems, it does not include aryl groups that have other groups, such as alkyl or halo groups, bonded to one of the ring members.
- substituted aryl groups groups such as tolyl are referred to as substituted aryl groups.
- Representative substituted aryl groups may be mono-substituted or substituted more than once, such as, but not limited to, 2- , 3-, 4-, 5-, or 6-substituted phenyl or benzyl groups, which may be substituted with groups including, but not limited to, amino, alkoxy, alkyl, cyano, or halo.
- aryl groups have from 6 to 14 ring member carbon atoms.
- Cycloalkylalkyl groups are alkyl groups as defined above in which a hydrogen or carbon bond of an alkyl group is replaced with a bond to a cycloalkyl group as defined above.
- Arylalkyl groups are alkyl groups as defined above in which a hydrogen or carbon bond of an alkyl group is replaced with a bond to an aryl group as defined above.
- Heterocyclyl groups are nonaromatic ring compounds containing
- heterocyclyl group includes fused ring species including those comprising fused aromatic and nonaromatic groups.
- the phrase also includes polycyclic ring systems containing a heteroatom such as, but not limited to, quinuclidyl.
- the phrase does not include heterocyclyl groups that have other groups, such as alkyl or halo groups, bonded to one of the ring members. Rather, these are referred to as "substituted heterocyclyl groups”.
- Heterocyclyl groups include, but are not limited to, piperazino, morpholino, thiomorpholino, pyrrolidino, piperidino and homopiperazino groups.
- Representative substituted heterocyclyl groups may be mono-substituted or substituted more than once, such as, but not limited to, morpholino or piperazino groups, which are 2-, 3-, 4-, 5-, or 6-substituted, or disubstituted with groups including, but not limited to, amino, alkoxy, alkyl, cyano, or halo.
- Heteroaryl groups are aromatic ring compounds containing 3 or more ring members, of which, one or more is a heteroatom such as, but not limited to, N, O, and S.
- Heteroaryl groups include, but are not limited to, groups such as furan, thiophene, pyrrole, isopyrrole, diazole, imidazole, isoimidazole, triazole, dithiole, oxathiole, isoxazole, oxazole, thiazole, isothiazole, oxadiazole, oxatriazole, dioxazole, oxathiazole, pyran, dioxin, pyridine, pyrimidine, pyridazine, pyrazine, triazine, oxazine, isoxazine, oxathiazine, azepin, oxepin, thiepin, diazepine, benzo
- heteroaryl groups includes fused ring compounds, the phrase does not include heteroaryl groups that have other groups bonded to one of the ring members, such as alkyl groups. Rather, heteroaryl groups with such substitution are referred to as "substituted heteroaryl groups". Representative substituted heteroaryl groups may be substituted one or more times with groups including, but not limited to, amino, alkoxy, alkyl, cyano, or halo. In some embodiments, heteroaryl groups include from 5 to 14 ring members.
- Heterocyclylalkyl groups are alkyl groups as defined above in which a hydrogen or carbon bond of an alkyl group is replaced with a bond to a heterocyclyl group as defined above.
- Heteroarylalkyl groups are alkyl groups as defined above in which a hydrogen or carbon bond of an alkyl group is replaced with a bond to a heteroaryl group as defined above.
- Aminocarbonyl groups are groups of the formula RR'NC(O)-, wherein R or R' may be the same or different, and each is independently selected from H, or substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl or heteroaryl groups, as defined above.
- substituted refers to a group as defined above in which one or more bonds to a hydrogen atom contained therein are replaced by a bond to non-hydrogen or non-carbon atoms such as, but not limited to, a halogen atom such as F, Cl, Br, and I; an oxygen atom in groups such as hydroxyl groups, alkoxy groups, aryloxy groups, and ester groups; a sulfur atom in groups such as thiol groups, alkyl and aryl sulfide groups, sulfone groups, sulfonyl groups, and sulfoxide groups; a nitrogen atom in groups such as amines, amides, alkylamines, dialkylamines, arylamines, alkylarylamines, diarylamines, N-oxides, imides, and enamines; a silicon atom in groups such as in trialkylsilyl groups, dialkylarylsilyl groups, alky
- Substituted alkyl groups and also substituted cycloalkyl groups and others also include groups in which one or more bonds to a carbon(s) or hydrogen(s) atom is replaced by a bond to a heteroatom such as oxygen in carbonyl, carboxyl, and ester groups; nitrogen in groups such as imines, oximes, hydrazones, and nitriles.
- Substituted cycloalkyl, substituted aryl, substituted heterocyclyl and substituted heteroaryl also include rings and fused ring systems in which a bond to a hydrogen atom is replaced with a bond to a carbon atom. Therefore, substituted cycloalkyl, substituted aryl, substituted heterocyclyl and substituted heteroaryl groups may also be substituted with alkyl groups as defined above.
- Pharmaceutically acceptable salts include a salt with an inorganic base, organic base, inorganic acid, organic acid, or basic or acidic amino acid.
- the invention includes, for example, alkali metals such as sodium or potassium, alkali earth metals such as calcium and magnesium or aluminum, and ammonia.
- the invention includes, for example, trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine.
- the instant invention includes, for example, hydrochloric acid, hydroboric acid, nitric acid, sulfuric acid, and phosphoric acid.
- the instant invention includes, for example, formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, lactic acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, and p-toluenesulfonic acid.
- salts of basic amino acids the instant invention includes, for example, arginine, lysine and ornithine.
- Acidic amino acids include, for example, aspartic acid and glutamic acid.
- protected with respect to hydroxyl groups, amine groups, and sulfhydryl groups refers to forms of these functionalities which are protected from undesirable reaction with a protecting group known to those skilled in the art such as those set forth in Protective Groups in Organic Synthesis, Greene, T.W.; Wuts, P. G. M., John Wiley & Sons, New York, NY, (3rd Edition, 1999) which can be added or removed using the procedures set forth therein.
- protected hydroxyl groups include, but are not limited to, silyl ethers such as those obtained by reaction of a hydroxyl group with a reagent such as, but not limited to, t-butyldimethyl-chlorosilane, trimethylchlorosilane, triisopropylchlorosilane, triethylchlorosilane; substituted methyl and ethyl ethers such as, but not limited to methoxymethyl ether, methythiomethyl ether, benzyloxymethyl ether, t-butoxymethyl ether, 2- methoxyethoxymethyl ether, tetrahydropyranyl ethers, 1-ethoxyethyl ether, allyl ether, benzyl ether; esters such as, but not limited to, benzoylformate, formate, acetate, trichloroacetate, and trifluoracetat ⁇ r
- protected amine groups include, but are not limited to,
- protected sulfhydryl groups include, but are not limited to, thioethers such as S-benzyl thioether, and S-4- picolyl thioether; substituted S-methyl derivatives such as hemithio, dithio and aminothio acetals; and others.
- Prodrugs as used in the context of the instant invention, includes those derivatives of the instant compounds which undergo in vivo metabolic biotransformation, by enzymatic or nonenzymatic processes, such as hydrolysis, to form a compound of the invention.
- Prodrugs can be employed to improve pharmaceutical or biological properties, as for example solubility, melting point, stability and related physicochemical properties, absorption, pharmacodynamics and other delivery-related properties.
- the instant invention provides potent and specific agonists of
- the invention provides compounds of formula IA, IB, mixtures thereof, and pharmaceutically acceptable salts thereof.
- Compounds provided by the invention further include prodrugs of the compound of formula IA and IB, pharmaceutically acceptable salts thereof, stereoisomers thereof, tautomers thereof, hydrates thereof, and solvates thereof.
- Compounds of formula IA or IB have the following structures:
- R 1 is selected from substituted or unsubstituted arylalkyl, heteroarylalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl, heterocyclylalkyl, cycloalkylalkyl, alkenyl, alkynyl, or alkyl groups.
- R 1 is a substituted or unsubstituted arylalkyl group such as a substituted or unsubstituted phenylethyl group.
- R is a substituted phenylethyl group such as a 4- substituted phenylethyl group or a 2,4-disubstituted phenylethyl group such as 4-halophenylethyl, 2-halo-4-alkoxyphenylethyl, and 2,4-dihalophenylethyl groups.
- R 1 is selected from phenylethyl, 2,4- dichlorophenylethyl, 4-methoxyphenylethyl, 4-phenoxyphenylethyl, 4- bromophenylethyl, 4-methylphenylethyl, 4-chlorophenylethyl, 4- fluorophenylethyl, 4-ethylphenylethyl, cyclohexenylethyl, 2- methoxyphenylethyl, 2-chlorophenylethyl, 2-fluorophenylethyl, 3- methoxyphenylethyl, 3-fluorophenylethyl, thienylethyl, indolylethyl, 4- hydroxyphenylethyl, 3,4-dimethoxyphenylethyl, 2-chloro-4-iodophenylethyl, 2- fluoro-4-methylphenylethyl, 4-chloro-2-fluorophenyleth
- R 1 is selected from 2-fluoro-4-methoxyphenylethyl, 2- chloro-4-methoxyphenylethyl, 4-fluorophenylethyl, 4-chlorophenylethyl, 4- chloro-2-fluorophenylethyl, 2,4-dichlorophenylethyl, 4-bromophenylethyl, or 4- bromo-2-fluorophenylethyl group.
- R 1 is selected from phenylethyl, '2,4-dichlorophenylethyl, 4-methoxyphenylethyl, 4- phenoxyphenylethyl, 4-bromophenylethyl, 4-methylphenylethyl, 4- chlorophenylethyl, 4-ethylphenylethyl, cyclohexenylethyl, 2- methoxyphenylethyl, 2-chlorophenylethyl, 2-fluorophenylethyl, 3- methoxyphenylethyl, 3-fluorophenylethyl, thienylethyl, 4-hydroxyphenylethyl, 3,4-dimethoxyphenylethyl, 2-chloro-4-iodophenylethyl, 2-fluoro-4- methylphenylethyl, 2-fluoro-4-chlorophenylethyl, 2-fluoro-4-bromoph
- R 2 is selected from H or substituted or unsubstituted arylalkyl, heteroarylalkyl, alkoxy, alkylamino, dialkylamino, aryl, heteroaryl, heterocyclyl, cycloalkyl, heterocyclylalkyl, cycloalkylalkyl, alkenyl, alkynyl, or alkyl groups.
- Compounds of formula IA and IB with R 2 values such as those set forth above have been found to exhibit reduced bioaccumulation properties as evidenced by lower t 2 blood plasma values in test subjects to which the compounds have been administered. Generally, such compounds also provide improved plasma Cmax values and may also provide improved brain C max values.
- R 2 is selected from substituted or unsubstituted heterocyclyl groups or substituted or unsubstituted heteroaryl groups. In other embodiments, R 2 is selected from substituted or unsubstituted pyridinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, tetrahydrofuranyl, furanyl, pyrrolidinyl, pyrrolyl, thiophenyl, tetrahydrothiophenyl, pyranyl, tetrahydropyranyl, tetrahydrothiopyranyl, pyrazinyl, thiazolyl, pyrimidinyl, quinuclidinyl, indolyl, imidazolyl, triazolyl, tetrazolyl, or pyridazinyl groups. In some such embodiments, R 2 is selected from heteroaryl or heterocyclyl group of formula
- R 2 is selected from substituted or unsubstituted heterocyclylalkyl, or cycloalkylamino groups.
- R 2 is selected from a group such as a substituted or unsubstituted cyclopropylamino group; a substituted or unsubstituted piperazinylalkyl group such as a piperazinylmethyl group or an N-methylpiperazinylmethyl group; or a piperidinylalkyl group such as a piperidinylmethyl group or a piperidinylethyl group.
- R 2 may be selected from a substituted or unsubstituted aryl or cycloalkyl group. Examples include compounds of the following formula which may be additionally substituted.
- R 3 , R 4 , and R 5 are independently selected from H, Cl, I, F, Br, OH, NH 2 , CN, NO 2 , or substituted or unsubstituted alkoxy or alkyl groups. In some embodiments, each of R 3 , R 4 , and R 5 are H.
- Z is a piperazinone of formula
- Z is a piperazinone of formula
- Z is a piperazinone of formula
- R 3' is selected from H or substituted or unsubstituted aryl, alkyl, alkenyl, alkynyl, cycloalkyl, heteroaryl, heterocyclyl, heterocyclylalkyl, arylalkyl, heteroarylalkyl, or cycloalkylalkyl groups. In some embodiments, R 3 ' is selected from substituted or unsubstituted cycloalkyl groups.
- R 3 ' is selected from the group consisting of substituted and unsubstituted cyclohexyl, 2- alkylcyclohexyl, 2,2-dialkylcyclohexyl, 2,3-dialkylcyclohexyl, 2,4- dialkylcyclohexyl, 2,5-dialkylcyclohexyl, 2,6-dialkylcyclohexyl, 3,4- dialkylcyclohexyl, 3-alkylcyclohexyl, 4-alkylcyclohexyl, 3,3,5-trialkylcyclohexyl, 2-aminocyclohexyl, 3-aminocyclohexyl, 4-aminocyclohexyl, 2,3- diaminocyclohexyl, 2,4-diaminocyclohexyl, 3,4-diaminocyclohexyl, 2,5- diaminocyclohexyl, 2,6-di
- R 3 is selected from the group consisting of substituted and unsubstituted cyclohexyl, 2-methylcycIohexyl, 2,2- dimethylcyclohexyl, 2,3-dimethylcyclohexyl, 2,4-dimethylcyclohexyl, 2,5- dimethylcyclohexyl, 2,6-dimethylcyclohexyl, 3,4-dimethylcyclohexyl, 3- methylcyclohexyl, 4-methylcyclohexyl, 3,3,5-trimethylcyclohexyl, A-t- butylcyclohexyl, isopinocampheyl, 7,7-dimethylnorbornyl, 4- isopropylcyclohexyl, 3-methylcycloheptyl groups, 2-fluoro-4-methylcyclohexyl, 4-fluoro-2-methylcyclohexyl, 4,4-difluoro-2-methylcyclohexyl,
- R 3 ' is a substituted or unsubstituted polycyclic cycloalkyl group. In some such embodiments, R 3' is a substituted or unsubstituted polycyclic cycloalkyl group having the formula II
- Compounds of formula IA and IB may exhibit reduced bioaccumulation properties in animal subjects to which they are administered. Such subjects may include human and non-human animal subjects. Examples of mammalian subjects include, but are not limited to, rodents such as mice and rats, bovines, equines, canines, felines, rabbits, guinea pigs, porcines, primates such as humans and monkeys, and the like.
- the invention provides compounds in which the ty value for the compound is less than 35, 30, 25, 20, 15, 10, or 5 hours in a tissue with high blood perfusion such as brain, liver, kidney, and heart. In some such embodiments, the t V2 value for the compound is less than 4 hours and in some embodiments is less than or about 3 hours in a tissue of a subject to which the compound has been administered.
- One or more compounds of the invention may be included in pharmaceutical formulations or medicaments.
- Such compositions include at least one compound of formula IA and/or IB and a pharmaceutically acceptable carrier. Therefore, the compounds of formula IA and IB may be used to prepare medicaments and pharmaceutical formulations for use in treating an MC4-R mediated disease such as, but not limited to, obesity, type II diabetes, erectile dysfunction, polycystic ovary disease, and Syndrome X.
- the MC4-R mediated disease is obesity or type II diabetes.
- Methods for treating MC4-R mediated diseases include administering to a subject in need thereof, a compound or composition of the instant invention.
- the compounds of the invention exhibit reduced bioaccumulation in the tissues such as in the brain or blood plasma of a subject.
- Administration of the compounds and compositions of the invention may be accomplished using various methods such as those described herein.
- the compound or composition is administered intranasally.
- the instant compounds may exist as one or more stereoisomers.
- the various stereoisomers include enantiomers, diastereomers, atropisomers and geometric isomers.
- one stereoisomer may be more active and/or may exhibit beneficial effects in comparison to other stereoisomer(s) or when separated from the other stereoisomer(s).
- stereoisomers of the instant invention necessarily includes mixtures of stereoisomers, individual stereoisomers, or optically active forms.
- compositions which may be prepared by mixing one or more compounds of the instant invention, or pharmaceutically acceptable salts or tautomers thereof, with pharmaceutically acceptable carriers, excipients, binders, diluents or the like, to treat or ameliorate a variety of disorders.
- disorders include, but are not limited to obesity, erectile disorders, cardiovascular disorders, neuronal injuries or disorders, inflammation, fever, cognitive disorders, sexual behavior disorders.
- a therapeutically effective dose further refers to that amount of one or more compounds of the instant invention sufficient to result in amelioration of symptoms of the disorder.
- compositions of the instant invention can be manufactured by methods well known in the art such as conventional granulating, mixing, dissolving, encapsulating, lyophilizing, emulsifying or levigating processes, among others.
- the compositions can be in the form of, for example, granules, powders, tablets, capsules, syrup, suppositories, injections, emulsions, elixirs, suspensions or solutions.
- compositions can be formulated for various routes of administration, for example, by oral administration, by intranasal administration, by transmucosal administration, by rectal administration, or subcutaneous administration as well as intrathecal, intravenous, intramuscular, intraperitoneal, intranasal, intraocular or intraventricular injection.
- the compound or compounds of the instant invention can also be administered in a local rather than a systemic fashion, such as injection as a sustained release formulation.
- the following dosage forms are given by way of example and should not be construed as limiting the instant invention.
- powders, suspensions, granules, tablets, pills, capsules, gelcaps, and caplets are acceptable as solid dosage forms. These can be prepared, for example, by mixing one or more compounds of the instant invention, or pharmaceutically acceptable salts or tautomers thereof, with at least one additive or excipient such as a starch or other additive.
- Suitable additives or excipients are sucrose, lactose, cellulose sugar, mannitol, maltitol, dextran, sorbitol, starch, agar, alginates, chitins, chitosans, pectins, tragacanth gum, gum arabic, gelatins, collagens, casein, albumin, synthetic or semi-synthetic polymers or glycerides, methyl cellulose, hydroxypropylmethyl-cellulose, and/or polyvinylpyrrolidone.
- oral dosage forms can contain other ingredients to aid in administration, such as an inactive diluent, or lubricants such as magnesium stearate, or preservatives such as paraben or sorbic acid, or anti-oxidants such as ascorbic acid, tocopherol or cysteine, a disintegrating agent, binders, a thickeners, buffers, a sweeteners, flavoring agents or perfuming agents. Additionally, dyestuffs or pigments may be added for identification. Tablets and pills may be further treated with suitable coating materials known in the art.
- Liquid dosage forms for oral administration may be in the form of pharmaceutically acceptable emulsions, syrups, elixirs, suspensions, slurries and solutions, which may contain an inactive diluent, such as water.
- Pharmaceutical formulations may be prepared as liquid suspensions or solutions using a sterile liquid, such as, but not limited to, an oil, water, an alcohol, and combinations of these.
- Pharmaceutically suitable surfactants, suspending agents, emulsifying agents may be added for oral or parenteral administration.
- suspensions may include oils.
- oils include, but are not limited to, peanut oil, sesame oil, cottonseed oil, corn oil and olive oil.
- Suspension preparation may also contain esters of fatty acids such as ethyl oleate, isopropyl myristate, fatty acid glycerides and acetylated fatty acid glycerides.
- Suspension formulations may include alcohols, such as, but not limited to, ethanol, isopropyl alcohol, hexadecyl alcohol, glycerol and propylene glycol.
- Ethers such as but not limited to, poly(ethyleneglycol), petroleum hydrocarbons such as mineral oil and petrolatum; and water may also be used in suspension formulations.
- the pharmaceutical formulations may be a solution, a spray, a dry powder, or aerosol containing any appropriate solvents and optionally other compounds such as, but not limited to, stabilizers, antimicrobial agents, antioxidants, pH modifiers, surfactants, bioavailability modifiers and combinations of these.
- examples of intranasal formulations and methods of administration can be found in WO 01/41782, WO 00/33813, WO 91/97947, U.S. Patent No. 6,180,603, U.S. Patent No. 5,624,898; Published U.S. Patent Application No.
- a propellant for an aerosol formulation may include compressed air, nitrogen, carbon dioxide, or a hydrocarbon based low boiling solvent.
- the compound or compounds of the instant invention are conveniently delivered in the form of an aerosol spray presentation from a nebulizer or the like.
- Injectable dosage forms generally include aqueous suspensions or oil suspensions which may be prepared using a suitable dispersant or wetting agent and a suspending agent. Injectable forms may be in solution phase or in the form of a suspension, which is prepared with a solvent or diluent.
- Acceptable solvents or vehicles include sterilized water, Ringer's solution, or an isotonic aqueous saline solution.
- sterile oils may be employed as solvents or suspending agents.
- the oil or fatty acid is non-volatile, including natural or synthetic oils, fatty acids, mono-, di- or tri-glycerides.
- the pharmaceutical formulation may be a powder suitable for reconstitution with an appropriate solution as described above. Examples of these include, but are not limited to, freeze dried, rotary dried or spray dried powders, amorphous powders, granules, precipitates, or particulates.
- the formulations may optionally contain stabilizers, pH modifiers, surfactants, bioavailability modifiers and combinations of these.
- the compounds may be formulated for parenteral administration by injection such as by bolus injection or continuous infusion.
- a unit dosage form for injection may be in ampoules or in multi-dose containers.
- the pharmaceutical formulations may be in the form of a suppository, an ointment, an enema, a tablet or a cream for release of compound in the intestines, sigmoid flexure and/or rectum.
- Rectal suppositories are prepared by mixing one or more compounds of the instant invention, or pharmaceutically acceptable salts or tautomers of the compound, with acceptable vehicles, for example, cocoa butter or polyethylene glycol, which is present in a solid phase at normal storing temperatures, and present in a liquid phase at those temperatures suitable to release a drug inside the body, such as in the rectum. Oils may also be employed in the preparation of formulations of the soft gelatin type and suppositories.
- suspension formulations which may also contain suspending agents such as pectins, carbomers, methyl cellulose, hydroxypropyl cellulose or carboxymethyl cellulose, as well as buffers and preservatives.
- suspending agents such as pectins, carbomers, methyl cellulose, hydroxypropyl cellulose or carboxymethyl cellulose, as well as buffers and preservatives.
- excipients and carriers are generally known to those skilled in the art and are thus included in the instant invention. Such excipients and carriers are described, for example, in "Remingtons Pharmaceutical Sciences” Mack Pub. Co., New Jersey (1991), which is incorporated herein by reference.
- the formulations of the invention may be designed for to be short-acting, fast-releasing, long-acting, and sustained-releasing as described below.
- the pharmaceutical formulations may also be formulated for controlled release or for slow release.
- compositions may also comprise, for example, micelles or liposomes, or some other encapsulated form, or may be administered in an extended release form to provide a prolonged storage and/or delivery effect. Therefore, the pharmaceutical formulations may be compressed into pellets or cylinders and implanted intramuscularly or subcutaneously as depot injections or as implants such as stents. Such implants may employ known inert materials such as silicones and biodegradable polymers.
- a therapeutically effective dose refers to that amount of the compound that results in amelioration of symptoms. Specific dosages may be adjusted depending on conditions of disease, the age, body weight, general health conditions, sex, diet of the subject, dose intervals, administration routes, excretion rate, and combinations of drugs. Any of the above dosage forms containing effective ⁇ amounts are well within the bounds of routine experimentation and therefore, well within the scope of the instant invention.
- a therapeutically effective dose may vary depending upon the route of administration and dosage form.
- the preferred compound or compounds of the instant invention is a formulation that exhibits a high therapeutic index.
- the therapeutic index is the dose ratio between toxic and therapeutic effects which can be expressed as the ratio between LD 50 and ED 50 .
- the LD 50 is the dose lethal to 50% of the population and the ED 50 is the dose therapeutically effective in 50% of the population.
- the LD50 and ED 50 are determined by standard pharmaceutical procedures in animal cell cultures or experimental animals.
- the present invention also provides methods of enhancing
- MC4-R activity in a human or non-human animal comprises administering an effective amount of a compound, or composition, of the instant invention to said mammal or non-human animal.
- Effective amounts of the compounds of the instant invention include those amounts that activate MC4-R which are detectable, for example, by an assay described below in the illustrative Examples, or any other assay known by those skilled in the art that a detect signal transduction, in a biochemical pathway, through activation of G-protein coupled receptors, for example, by measuring an elevated cAMP level as compared to a control model. Accordingly, "activating" means the ability of a compound to initiate a detectable signal. Effective amounts may also include those amounts which alleviate symptoms of a MC4-R disorder treatable by activating MC4-R.
- An MC4-R disorder, or MC4-R-mediated disease which may be treated by those methods provided, include any biological disorder or disease in which MC4-R is implicated, or which inhibition of MC4-R potentiates a biochemical pathway that is defective in the disorder or disease state.
- diseases are obesity, erectile disorders, cardiovascular disorders, neuronal injuries or disorders, inflammation, fever, cognitive disorders, type II diabetes, polycystic ovary disease, Syndrome X, complications from obesity and diabetes, and sexual behavior disorders.
- the instant invention provides compounds, compositions, and methods effective for reducing energy intake and body weight; reducing serum insulin and glucose levels; alleviating insulin resistance; and reducing serum levels of free fatty acids. Accordingly, the instant invention is particularly effective in treating those disorders or diseases associated with obesity or type II diabetes.
- Treating within the context of the instant invention, therefore, means an alleviation of symptoms associated with a disorder or disease, or halt of further progression or worsening of those symptoms, or prevention or prophylaxis of the disease or disorder.
- successful treatment may include an alleviation of symptoms or halting the progression of the disease, as measured by reduction in body weight, or a reduction in amount of food or energy intake.
- successful treatment of type I or type II diabetes may include an alleviation of symptoms or halting the progression of the disease, as measured by a decrease in serum glucose or insulin levels in, for example, hyperinsulinemic or hyperglycemic patients.
- Scheme 1a illustrates a general synthetic route that may be used to synthesize various guanidinyl-substituted quinazolinone compounds.
- nitro and amino quinazolinone compounds such as (d) and (e) may be readily converted into a plethora of guanidinyl quinazolinones by converting the amino functionality to an isothiocyanate functionality such as possessed by compound (f). This may be accomplished reacting the amine group with thiophosgene.
- Isothiocyanate compounds such as (f) may then be readily converted into a thiourea such as compound (g) by reaction with a suitable amine compound such as (1S,2S,3S,5R)-(+)- isopinocampheylamine.
- a suitable amine compound such as (1S,2S,3S,5R)-(+)- isopinocampheylamine.
- Preparation of the desired guanidinylamine such as compound (h) may then be accomplished by reacting the thiourea with a compound such as 1-[3-(dimethylamino)-propyl]-3-ethylcarbodiimide hydrochloride and then with a suitable amine such as cis-2,6- dimethylpiperazine, (S)-2-(fluoromethyl)piperazine, or the like.
- Scheme 1 b illustrates another generally applicable method that may be employed to synthesize a large number of guanidinyl-substituted quinazolinones and heterocyclic derivatives of such compounds where a carbon of the benzene ring of the quinazolinone is replaced with a nitrogen atom.
- conversion of compound (d) to (e) may be accomplished by initially adding trimethylphosphine to form a reactive iminophosphorane intermediate, adding a substituted isocyanate such as a cycloalkyl isocyanate for example, a polycyclic isocyanate to produce a carbodiimide, and finally forming (e) by addition of and reaction with an amine such as, but not limited to a substituted piperazine.
- a substituted isocyanate such as a cycloalkyl isocyanate for example, a polycyclic isocyanate to produce a carbodiimide
- an amine such as, but not limited to a substituted piperazine.
- Scheme 2a illustrates another general procedure that may be used to prepare a wide variety of guanidinyl-substituted quinazolinone compounds.
- Scheme 2b shows yet another alternative route that may be used to prepare various compounds of of the invention.
- DIBAL Diisobutylaluminum hydride
- NMO N-Morpholine oxide
- THF Tetrahydrofuran SYNTHESIS OF 6-METHYLPIPERAZIN-2-ONE
- Ester (2) was stirred in a 50:50 solution of TFA:CH 2 CI 2 for 1 hour. The solvent was then removed, and the residue was redissolved in methylene chloride and washed with a saturated solution of Na 2 CO 3 . The organic layer was then separated and dried over MgSO 4 giving desired piperazine compound (3) as a white solid (87%).
- 6(S)-Methyl piperazin-2-one guanidine compounds of the invention were prepared according to the methods described herein by EDC activation of the thiourea intermediate to provide the carbodiimide followed by coupling with 6(S)-Methyl piperazin-2-one.
- Trimethylsilyl diazomethane (3.89 mmol) was slowly added to an ice cooled solution of (4R)-1- ⁇ [(1 ,1-dimethylethyl)oxy]carbonyl ⁇ -4-hydroxy-D- proline (0.75 g, 3.25 mmol), 60 mL toluene and 20 mL of methanol. The reaction was stirred for 2 hours on the ice bath, was warmed to room temperature, and was concentrated to give 0.89 g of the title compound as a clear yellow oil.
- Step 1 Synthesis of cis-3,5-Dimethyl-4-(3-oxo-butyl)-piperazine-1 - carboxylic acid tert-butyl ester
- Cis-3,5-Dimethyl-4-(3-oxo-butyi)-piperazine-1 -carboxylic acid tert-butyl ester (1.00 g) in chloroform (40 mL) was treated with m-chloroperbenzoic acid (77%, 0.97g ) at 0° C. The solution was allowed to warm to room temperature and stirred overnight. The reaction mixture was then cooled to 0° C, filtered to remove precipitates, washed with saturated aqueous sodium bicarbonate, filtered through a plug of silica gel and concentrated. The residue was taken up in a 1 :1 mixture of DCM and MeOH and treated with an excess of 4.0 M HCI/dioxane.
- MeOH (10 mL) was treated with 4.0 N HCI/dioxane (1.0 mL) followed by an excess of palladium hydroxide on carbon (wet, Degusa type).
- the solution was then reacted with hydrogen on a Parr hydrogenator over night at 45 psi.
- the reaction mixture was then filtered through Celite, concentrated, diluted to a known concentration and used without further purification.
- Step 2 Synthesis of 1 -(1 ,1 -Dimethylethyl) 2-ethyl (2S,3S)-3- ⁇ [(1 ,1 dimethylethy
- Step 4 Synthesis of 1,1 -Dimethylethyl (2R,3S)-3- ⁇ [(1,1- dimethylethyl)(dimethyl)silyl]oxy ⁇ -2- ⁇ [(methylsulfonyl)oxy]methyl ⁇ -1 -pyrrolidinecarboxylate
- Methanesulfonyl chloride (3.85 mL, 39.29 mmol) was added to an ice cold methylene chloride solution of 1 ,1 -dimethylethyl (2R,3S)-3- ⁇ [(1 ,1- dimethylethyl) (dimethyl)silyl]oxy ⁇ -2-(hydroxymethyl)-1 -pyrrolidinecarboxylate (6.05 g, 26.19 mmol) and triethylamine (7.28 mL, 52.38 mmol). The reaction was warmed to room temperature, stirred for 16 hours then concentrated under reduced pressure.
- Tetrabutyl ammonium fluoride (16.7 mL, 1 N in THF, 16.73 mmol) was added to a solution of 1 ,1 -dimethylethyl (2R,3S)-3- ⁇ [(1 ,1- dimethylethyl)(dimethyl)silyl]oxy ⁇ -2-methyl-1-pyrrolidinecarboxylate in THF and stirred for 16 hours at room temperature.
- the reaction mixture was concentrated under reduced pressure and purified by silica gel column chromatography to yield the title compound (1.55g, 7.8 mmol).
- a THF solution of a carbodiimide such as Carbodiimide A or Carbodiimide B was stirred with approximately 1.5 equivalents of primary or secondary amine at room temperature for 10 minutes.
- the amine was dissolved in minimal methanol and 2 equivalents of triethylamine was added.
- the reaction were concentrated under a stream of nitrogen, dissolved in methanol and purified by preparative HPLC.
- Step 1 Synthesis of (2-amino-4,5-difluorophenyl)-N-[2-(4- fluorophenyl)ethyl]-carboxamide
- Step 3 Synthesis of 7-(azadiazomvinyl)-6-fluoro-3-[2-(4- fluorophenyl)ethyl]-3-hydroquinazolin-4-one
- the aqueous layer was extracted with two more portions of ethyl acetate. The organic layers were then combined and concentrated in vacuo. The crude mixture was dissolved in DMSO and purified via preparative HPLC using water (0.1% TFA) /acetonitrile (0.1% TFA). The pure fractions were combined and concentrated in vacuo to remove the majority of acetonitrile. Sodium carbonate (15 equivalents) was then added to the resulting aqueous solution and the slurry was allowed to sit at room temperature for 1 hour with occasional swirling. The basic aqueous solution was then extracted with 3 separate portions of ethyl acetate.
- the mixture was then diluted with ethyl acetate and washed with water. After the organic layer was isolated, the aqueous layer was extracted with two more portions of ethyl acetate. The organic layers were then combined and concentrated in vacuo.
- the crude mixture was dissolved in DMSO/acetonitrile and purified via preparative HPLC using water (0.1% TFA) /acetonitrile (0.1% TFA). The pure fractions were combined and concentrated in vacuo to remove the majority of acetonitrile.
- Sodium hydroxide (10 equivalents) was then added to the resulting aqueous solution and the slurry was allowed to sit at room temperature for 1 hour with occasional swirling.
- Example 1 (1 equivalent) in THF (0.5 M in (a)) was added 4,4- difluorocyclohexylamine prepared as described above (1.5 equivalents). The reaction was stirred at room temperature for 10 hours. The crude product mixture was then concentrated in vacuo, dissolved in methylene chloride, and purified via flash chromatography using hexanes/ethyl acetate.
- Nitrile 1 shown above (0.9 g, 2.4 mmoles) was dissolved in dry
- Example 11 was prepared using the methodology described in Scheme 1a and the appropriate amino indanol.
- Examples 14 and 18 which include hydroxymethyl-substituted arylalkyl groups were also prepared using the general methodology of Scheme 1a with the appropriate amino alcohol.
- Example 36 was prepared by: first, mono-Boc protecting 2,6-trans dimethylpiperazine; second, treating the mono-Boc protected compound with cyanogen bromide (2.5 equivalents) and Hunig's base (1.1 equivalent); third, purifying the resulting nitrile piperazine compound on silica gel; fourth, deprotecting the purified compound; and fifth, reacting the resulting purified nitrile trans dimethyl piperazine compound using the methods described herein to produce Example 36.
- Compounds such as Examples 73 and 76 were prepared using the procedure of Method 2 with the appropriated amides of methacrylic acid and acetic acid.
- This method was accomplished by injection of 3 ⁇ L of sample onto a SynergiMax-RP (50 x 2.0 mm) column (4 ⁇ m particle size). Elution was with 15% methanol to 100% methanol over 3.5 minutes, then 1 minute at 100% methanol. Column was heated at 50°C and the flow rate was 1.5 mL/minute. The water contained 0.1 % formic acid, and the methanol contained 0.075% formic acid by volume. DAD scans were collected from 220 to 400 nm.
- This method was accomplished by injection of 3 ⁇ L of sample onto a SynergiMax-RP (50 x 2.0 mm) column (4 ⁇ m particle size). Elution was with 15% methanol to 100% methanol over 5 minutes, then 1 minute at 100% methanol. Column was at room temperature and the flow rate was 1 mL/minute. The water contained 0.1% formic acid, and the methanol contained 0.075% formic acid by volume. DAD scans were collected from 220 to 400 nm.
- This method was accomplished by injection of 3 ⁇ L of sample onto a SynergiHydro-RP (50 x 2.0 mm) column (4 ⁇ m particle size). Elution was with 2% methanol to 100% methanol over 5 minutes, then 1 minute at 100% methanol. Column was at room temperature and the flow rate was 1 mL/minute. The water contained 0.1% formic acid, and the methanol contained 0.075% formic acid by volume. DAD scans were collected from 220 to 400 nm.
- This method was accomplished by injection of 3 ⁇ L of sample onto a SynergiHydro-RP (50 x 2.0 mm) column (4 ⁇ m particle size). Elution was with 2% methanol to 100% methanol over 3.5 minutes, then 0.5 minutes at 100% methanol. Column was at room temperature and the flow rate was 1.5 mL/minute. The water contained 0.1% formic acid, and the methanol contained 0.075% formic acid by volume. DAD scans were collected from 220 to 400 nm.
- This method was accomplished by injection of 3 ⁇ L of sample onto a SynergiHydro-RP (50 x 2.0 mm) column (4 ⁇ m particle size). Elution was with 2% methanol to 100% methanol over 5 minutes, then 1 minute at 100% methanol. Column was at room temperature and the flow rate was 0.8 mL/minute. The water contained 0.1% formic acid, and the methanol contained 0.075% formic acid by volume. DAD scans were collected from 220 to 400 nm.
- This method was accomplished by injection of 3 ⁇ L of sample onto a SynergiHydro-RP (50 x 2.0 mm) column (4 ⁇ m particle size). Elution was with 10% methanol to 100% methanol over 3 minutes, then 1 minute at 100% methanol. Column was at room temperature and the flow rate was 2.0 mL/minute. The water contained 0.1% formic acid, and the methanol contained 0.075% formic acid by volume. DAD scans were collected from 220 to 400 nm.
- EC50 values of test compounds were determined by treating cells expressing MC4-R with test compound and lysing the cells and measuring intercellular cAMP concentration with an Amersham-Pharmacia RPA-559 cAMP Scintillation Proximity Assay (SPA) kit. EC 50 values of test compounds were also determined using the following method reported by Goetz, et al. which is hereby incorporated by reference in its entirety and for all purposes as if fully set forth herein. Goetz, A.G.; Andrews J.L.; Littleton, T.R.; Ignar, D.M. DEVELOPMENT OF A FACILE METHOD FOR HIGH THROUGHPUT SCREENING WITH REPORTER GENE ASSAYS J.
- SPA Amersham-Pharmacia RPA-559 cAMP Scintillation Proximity Assay
- CHO-6xCRE-luc+ reporter cell lines expressing human MC1 R, MC3R, MC4R, and MC5R (GenBank accession numbers X65634, LO6155, S77415 and U08353) and the CHO host reporter gene cell line were propagated in complete medium in T225 flasks. Forty-eight hours prior to assay, cells were harvested with 2 mL of 0.05% trypsin, washed with complete medium and plated at a concentration of 4000 cells/well in complete medium. Sixteen hours prior to the assay, the medium was removed from the cells and replaced with 90 ⁇ L/well of serum-free DMEM/F12.
- agonists were added in a 10 ⁇ L volume and plates were incubated for 4 hours at 37°C in a cell culture incubator.
- the medium was aspirated followed by the addition of 50 ⁇ L of a 1 :1 mixture of LucLiteTM and dPBS containing 1 imM CaCI 2 and 1 mM MgCI 2 .
- the plates were then sealed and subjected to dark adaptation at room temperature for 10 minutes before luciferase activity was quantitated using a TopCountTM microplate scintillation counter (Packard) using 3 second/well count time.
- the NDP- ⁇ MSH concentration-response curve data were expressed as a percentage of the fold stimulation in the NDP- ⁇ MSH control for each receptor subtype.
- the control value is the average of duplicate wells treated with 1 *10 "7 M NDP- ⁇ MSH.
- mice In vivo studies are conducted to observe the effect of MCR-4 agonists on energy intake, body weight, hyperinsulinemia, and glucose levels. All studies are conducted with male 9-10 week old ob/ob mice which display early onset of obesity, insulin resistance and diabetes due to leptin deficiency. Mice are acclimated in the facility for 1 week before studies and are caged individually. Vehicle-treated (control) and drug treated mice studies are always run in parallel. In multi-day studies, mice (8-15 per group) are monitored for baseline body weight, fasting levels of glucose, insulin, blood lipids and energy expenditure and then injected twice daily (9 a.m.
- a MC4-R agonist of the present invention for 4 weeks.
- Body weight as well as food and water intake are monitored daily. Animals are fasted overnight for measurements of fasting levels of glucose, insulin, and lipids once a week until the end of the study.
- Energy expenditure resting metabolic rate, i.e., O 2 consumption and CO 2 production
- O 2 consumption and CO 2 production are monitored using Oxymax systems (Columbus Instruments).
- Oral glucose tolerance test (OGTT - a routine test for diabetes and glucose intolerance) is performed on overnight fasted mice at the end of the study.
- Blood glucose and oral glucose tolerance are measured using a glucose monitor (Onetouch sold by Lifescan).
- Free fatty acids are measured using an non-esterified free fatty acids enzymatic assay (Waco Chemicals).
- Serum insulin levels are measured by immunoassay (Alpco).
- the effect of the compounds of the present invention on blood glucose levels is determined by measuring blood glucose levels as represented as mg of glucose/dL of blood. Mice are fasted overnight and glucose levels are measured the following morning. Vehicle treated mice show an increase in blood glucose consistent with the rapid progression of diabetes in this mouse strain whereas, diabetes is slowed down considerably in drug treated mice. A significant reduction in fasting glucose levels is demonstrated in those mice treated IP with the compounds of this invention.
- the effect of the compounds of the present invention on glucose levels during oral glucose tolerance test is determined by measuring blood glucose in overnight fasted mice. Blood glucose is represented as mg of glucose/dL of blood. Glucose levels are measured the following morning. Orally administered glucose quickly elevates blood glucose, similar to a meal, and the response to this exogenous glucose gives a measure of how well the body regulated glucose homeostasis. Vehicle treated mice show an elevated response to glucose consistent with their diabetic state, whereas drug treated mice show a very much improved glucose disposal.
- FFA free fatty acid
- the effect of the compounds of the present invention on serum insulin levels is determined by measuring serum insulin levels one hour after single IP dosing of 1 and 3 mg/kg in overnight fasted ob/ob mice. .Serum insulin levels are represented as ng of insulin/mL of serum. Drug treated mice show a dose dependent decrease relative to vehicle.
- mice Male CD-1 mice, body weight of 20 grams at arrival, were used in these studies. Mice were given 30 mg/kg of compound in HPMC/Tween solution or suspension via oral gavage. Plasma, brain, and liver samples were collected at time periods of 1 , 2, 4, 8, and 24 hours post dosing. One mouse was used per time point. Thus, a total of 5 mice were used for each compound tested. For sample collection, mice were euthanized with CO 2 . Blood samples were taken by cardiac puncture and kept on ice. Brain and liver samples were collected immediately after bleeding and the samples were kept on dry ice.
- tissue half- lives (ty 2 s)
- the terminal rate constant k was estimated by the absolute value of the slope of a log-linear regression of the terminal phase of the tissue concentration-time profile.
- the tissue half-life fX 2 is ln(2)/ .
- mice of 6-9 weeks age were used in these studies.
- the mice were singly housed at least 5 days prior to the study. Two and a half hours before the onset of the dark cycle, food was removed from the cage top.
- Mice were dosed with a compound of the invention (in HPMC/Tween, as vehicle) or vehicle via oral gavage two hours before the onset of the dark cycle.
- pre-weighed food was given to each mouse. Food was weighed at 16 and 24 hours after the introduction of food to obtain cumulative food intake values. Mice were then euthanized with CO 2 followed by cervical dislocation.
- the following table includes ty 2 data for plasma, brain, kidney, and liver obtained after oral administration.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Manufacturing & Machinery (AREA)
- Ceramic Engineering (AREA)
- Reproductive Health (AREA)
- Biomedical Technology (AREA)
- Endocrinology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Hematology (AREA)
- Materials Engineering (AREA)
- Obesity (AREA)
- Gynecology & Obstetrics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Structural Engineering (AREA)
- Inorganic Chemistry (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Psychiatry (AREA)
- Pregnancy & Childbirth (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description














































































































































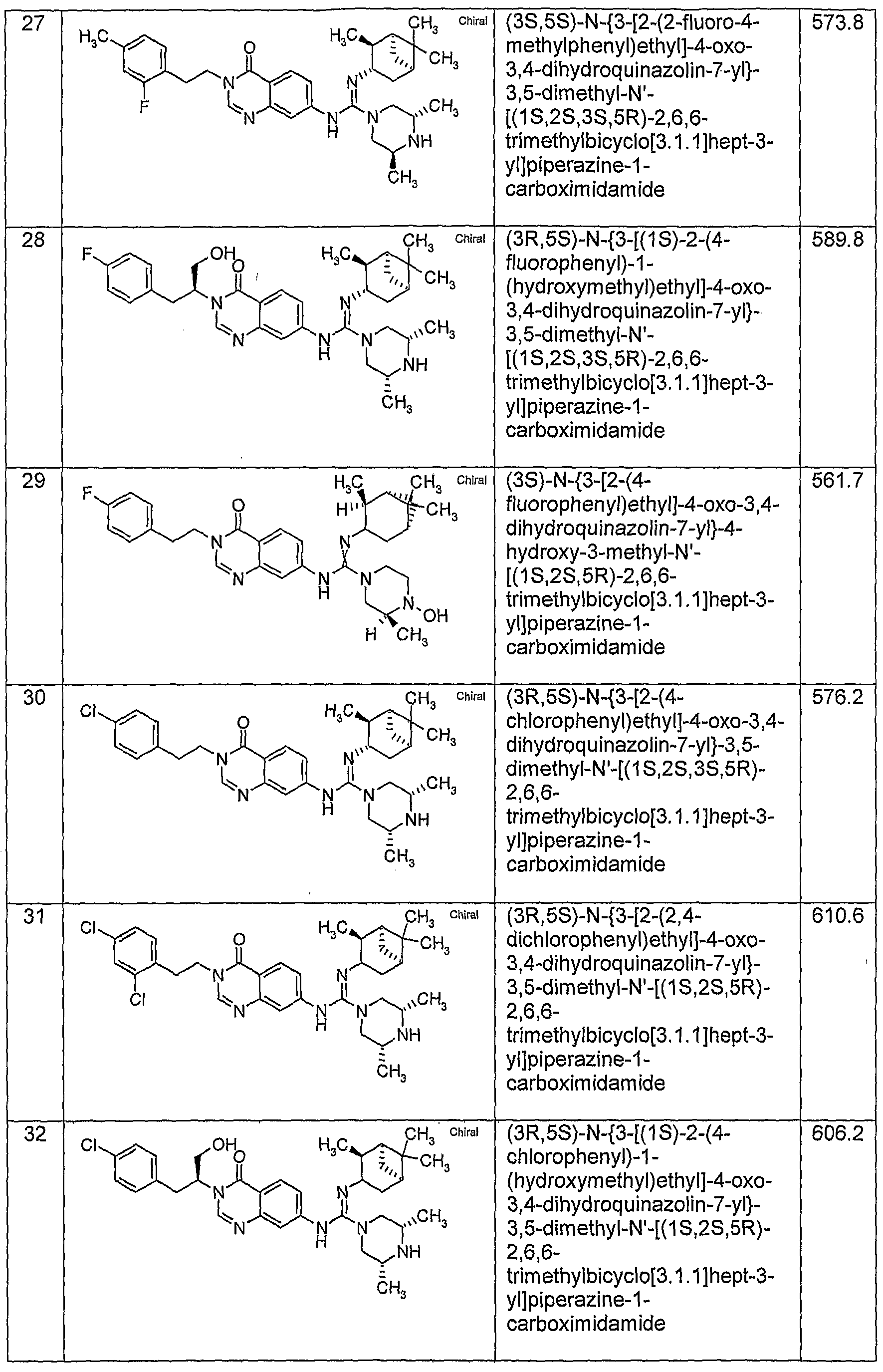










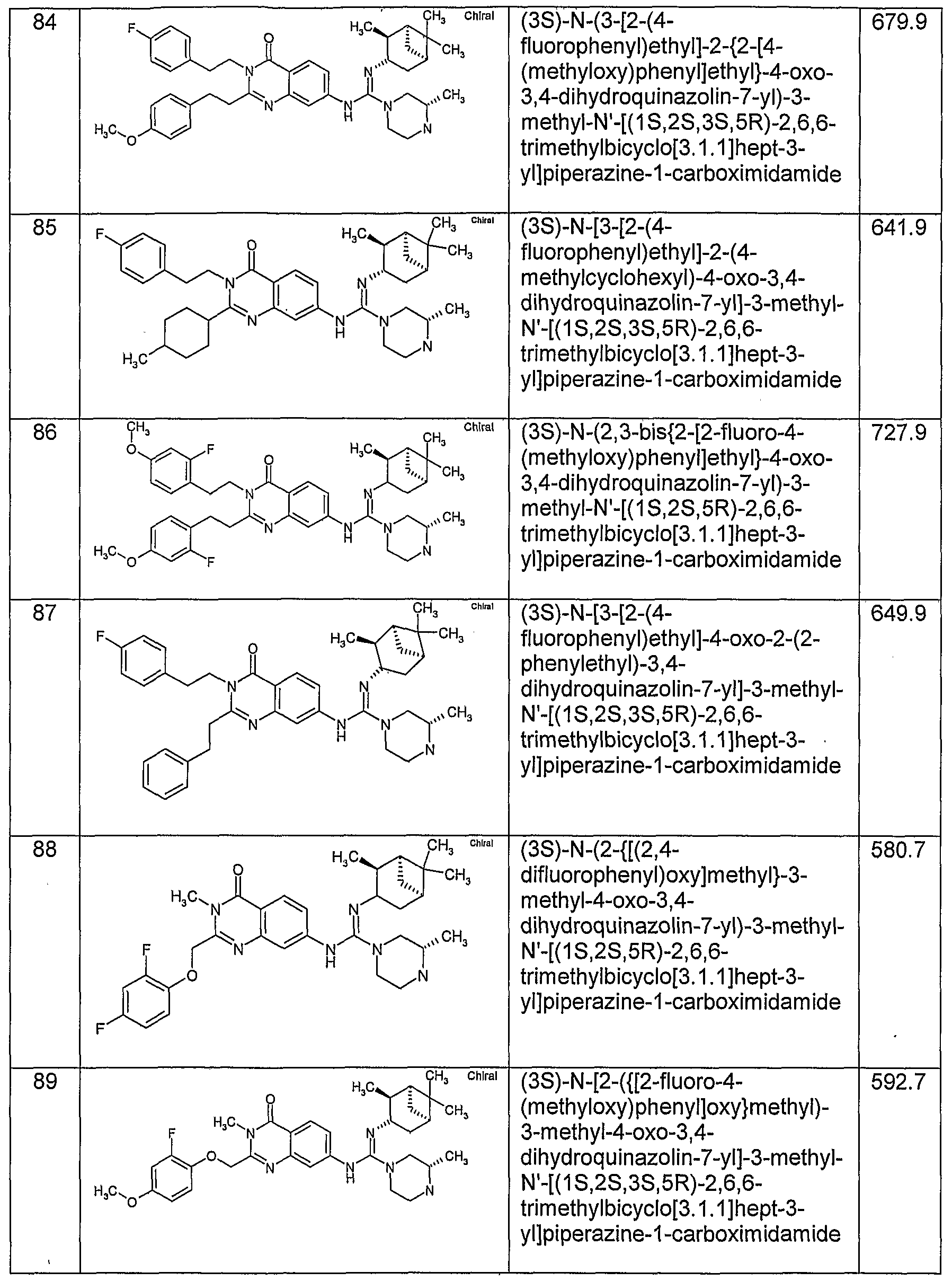



























































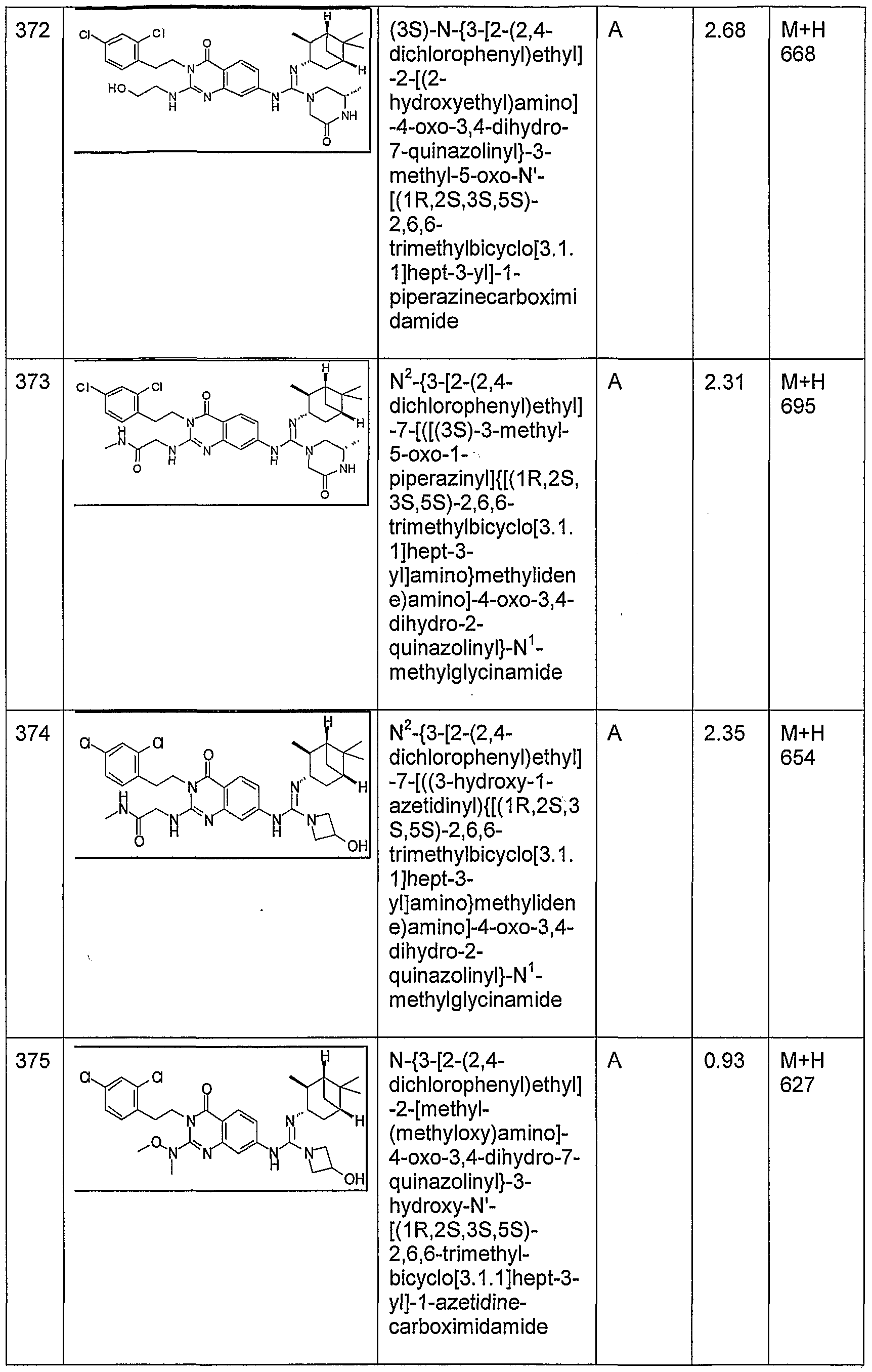








Claims







Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04811698A EP1686996A4 (en) | 2003-11-19 | 2004-11-19 | Quinazolinone compounds with reduced bioaccumulation |
| AU2004293012A AU2004293012A1 (en) | 2003-11-19 | 2004-11-19 | Quinazolinone compounds with reduced bioaccumulation |
| CA002545601A CA2545601A1 (en) | 2003-11-19 | 2004-11-19 | Quinazolinone compounds with reduced bioaccumulation |
| JP2006541565A JP2007511612A (en) | 2003-11-19 | 2004-11-19 | Quinazolinone compounds with reduced bioaccumulation |
| MXPA06005736A MXPA06005736A (en) | 2003-11-19 | 2004-11-19 | Quinazolinone compounds with reduced bioaccumulation. |
| IL175715A IL175715A0 (en) | 2003-11-19 | 2006-05-17 | Quinazolinone compounds with reduced bioaccumulation |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US52333603P | 2003-11-19 | 2003-11-19 | |
| US60/523,336 | 2003-11-19 | ||
| US52449203P | 2003-11-24 | 2003-11-24 | |
| US60/524,492 | 2003-11-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005051391A1 true WO2005051391A1 (en) | 2005-06-09 |
Family
ID=34636462
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/039020 WO2005051391A1 (en) | 2003-11-19 | 2004-11-19 | Quinazolinone compounds with reduced bioaccumulation |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7368453B2 (en) |
| EP (1) | EP1686996A4 (en) |
| JP (1) | JP2007511612A (en) |
| KR (1) | KR20060105785A (en) |
| AU (1) | AU2004293012A1 (en) |
| CA (1) | CA2545601A1 (en) |
| IL (1) | IL175715A0 (en) |
| MX (1) | MXPA06005736A (en) |
| WO (1) | WO2005051391A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1608317A2 (en) * | 2003-03-25 | 2005-12-28 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| EP1698375A1 (en) * | 2003-12-25 | 2006-09-06 | Ono Pharmaceutical Co., Ltd. | Azetidine ring compounds and drugs comprising the same |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7625909B2 (en) | 2003-05-23 | 2009-12-01 | Novartis Vaccines And Diagnostics, Inc. | Substituted quinazolinone compounds |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US7858641B2 (en) | 2002-05-23 | 2010-12-28 | Novartis Vaccines And Diagnostics, Inc. | Substituted dihydroisoquinolinone compounds |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2009003673A (en) | 2006-10-04 | 2009-04-22 | Pfizer Prod Inc | Pyrido[4,3-d]pyrimidin-4(3h)-one derivatives as calcium receptor antagonists. |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3177218A (en) * | 1962-01-17 | 1965-04-06 | Monsanto Chemicals | Methylene-bis(2-guanidino-4-methylquinazoline) |
| US4128643A (en) * | 1976-05-28 | 1978-12-05 | Hoechst Aktiengesellschaft | 4-Quinazolinyl-guanidines |
| US4287341A (en) * | 1979-11-01 | 1981-09-01 | Pfizer Inc. | Alkoxy-substituted-6-chloro-quinazoline-2,4-diones |
| US4496571A (en) * | 1981-02-27 | 1985-01-29 | Ici Americas Inc. | Histamine H2-antagonists |
| US4748165A (en) * | 1981-05-18 | 1988-05-31 | Imperial Chemical Industries Plc | Amidine derivatives |
Family Cites Families (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4211867A (en) * | 1976-03-19 | 1980-07-08 | Mcneil Laboratories, Incorporated | Nitrogen heterocyclic carboximidamide compounds |
| DE3108322A1 (en) | 1980-03-18 | 1981-12-24 | Immuno Aktiengesellschaft für chemisch-medizinische Produkte, 1220 Wien | Chromogenic enzyme substrate |
| JPS6229566A (en) * | 1985-07-30 | 1987-02-07 | Taiyo Yakuhin Kogyo Kk | Novel guanidinomthylbenzoic acid derivative |
| US4948901A (en) * | 1988-05-24 | 1990-08-14 | Pfizer Inc. | Benzamide protease inhibitors |
| US4874864A (en) * | 1988-05-24 | 1989-10-17 | Pfizer Inc. | Benzamide protease inhibitors |
| US4948891A (en) * | 1988-05-24 | 1990-08-14 | Pfizer Inc. | Benzamide protease inhibitors |
| JPH0276880A (en) * | 1988-06-16 | 1990-03-16 | Sankyo Co Ltd | Cachexia-improving remidy |
| GB8926512D0 (en) * | 1989-11-23 | 1990-01-10 | Pfizer Ltd | Therapeutic agents |
| US5624898A (en) * | 1989-12-05 | 1997-04-29 | Ramsey Foundation | Method for administering neurologic agents to the brain |
| CA2032420A1 (en) * | 1989-12-22 | 1991-06-23 | Akira Okuyama | Guanidinobenzene derivatives |
| US5124328A (en) * | 1990-10-11 | 1992-06-23 | Merck & Co., Inc. | Morpholine derivatives compositions and use |
| EP0690851B1 (en) | 1993-03-23 | 1999-05-19 | Astra Aktiebolag | Guanidine derivatives useful in therapy |
| SK16396A3 (en) * | 1993-08-12 | 1996-09-04 | Astra Ab | Amidine derivatives with nitric oxide synthetase activities |
| US5547966A (en) * | 1993-10-07 | 1996-08-20 | Bristol-Myers Squibb Company | Aryl urea and related compounds |
| US5599984A (en) * | 1994-01-21 | 1997-02-04 | The Picower Institute For Medical Research | Guanylhydrazones and their use to treat inflammatory conditions |
| US5637439A (en) * | 1994-11-07 | 1997-06-10 | Mitsubishi Paper Mills Ltd. | Photographic silver halide photosensitive material and method for developing the same |
| AU4534596A (en) | 1995-02-09 | 1996-08-27 | Novo Nordisk A/S | Compounds with growth hormone releasing properties |
| US5731408A (en) * | 1995-04-10 | 1998-03-24 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Peptides having potent antagonist and agonist bioactivities at melanocortin receptors |
| US6054556A (en) * | 1995-04-10 | 2000-04-25 | The Arizona Board Of Regents On Behalf Of The University Of Arizona | Melanocortin receptor antagonists and agonists |
| DE19544687A1 (en) | 1995-11-30 | 1997-06-05 | Thomae Gmbh Dr K | Amino acid derivatives, medicaments containing these compounds and processes for their preparation |
| DE19544685A1 (en) * | 1995-11-30 | 1997-06-05 | Thomae Gmbh Dr K | Amino acid derivatives, medicaments containing these compounds and processes for their preparation |
| AU2536097A (en) * | 1996-03-29 | 1997-10-22 | G.D. Searle & Co. | Para-substituted phenylpropanoic acid derivatives as integrin antagonists |
| ZA973850B (en) * | 1996-05-06 | 1997-12-02 | Reddy Research Foundation | Novel antidiabetic compounds having hypolipidaemic, anti-hypertensive properties, process for their preparation and pharmaceutical compositions containing them. |
| US5766877A (en) * | 1996-05-10 | 1998-06-16 | Amgen Inc. | Genes encoding art, an agouti-related transcript |
| US6127343A (en) * | 1996-05-14 | 2000-10-03 | Novo Nordisk A/S | Somatostatin agonists and antagonists |
| DE69713402T2 (en) | 1996-08-23 | 2002-11-07 | Agouron Pharma | LIGANDS OF THE NEUROPEPTID Y |
| WO1998018786A1 (en) * | 1996-10-31 | 1998-05-07 | Novo Nordisk A/S | Constrained somatostatin agonists and antagonists |
| CZ184099A3 (en) * | 1996-11-25 | 1999-11-17 | The Procter & Gamble Company | Compound, pharmaceutical composition containing thereof and prevention or treatment method |
| EP0981526B1 (en) | 1997-05-02 | 2004-02-25 | Dr. Reddy's Laboratories Ltd. | Novel antidiabetic compounds having hypolipidaemic, antihypertensive properties, process for their preparation and pharmaceutical compositions containing them |
| EP1085869A4 (en) | 1998-06-11 | 2001-10-04 | Merck & Co Inc | Spiropiperidine derivatives as melanocortin receptor agonists |
| DE19831710A1 (en) | 1998-07-15 | 2000-01-20 | Merck Patent Gmbh | New diacyl-hydrazine derivatives, are integrin inhibitors useful for treating e.g. thrombosis, cardiac infarction, tumors, osteoporosis, inflammation or infection |
| FR2784678B1 (en) | 1998-09-23 | 2002-11-29 | Sod Conseils Rech Applic | NOVEL N- (IMINOMETHYL) AMINE DERIVATIVES, THEIR PREPARATION, THEIR USE AS MEDICAMENTS AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| AU760174B2 (en) * | 1999-02-09 | 2003-05-08 | Bristol-Myers Squibb Company | Lactam inhibitors of FXa and method |
| EP1187614A4 (en) | 1999-06-04 | 2005-06-22 | Merck & Co Inc | Substituted piperidines as melanocortin-4 receptor agonists |
| MXPA02001160A (en) | 1999-08-04 | 2002-07-02 | Millennium Pharm Inc | Melanocortin-4 receptor binding compounds and methods of use thereof. |
| AU2001228681A1 (en) | 2000-01-28 | 2001-08-07 | Melacure Therapeutics Ab | Novel aromatic amines and amides acting on the melanocortin receptors |
| EP1254114A2 (en) | 2000-01-28 | 2002-11-06 | Melacure Therapeutics AB | Melanocortin receptor agonists and antagonists |
| GB0002059D0 (en) | 2000-01-28 | 2000-03-22 | Melacure Therapeutics Ab | Novel aromatic amines |
| EP1268449A4 (en) | 2000-03-23 | 2004-09-15 | Merck & Co Inc | Substituted piperidines as melanocortin receptor agonists |
| CA2403631A1 (en) | 2000-03-23 | 2001-09-27 | Brenda L. Palucki | Spiropiperidine derivatives as melanocortin receptor agonists |
| DZ3415A1 (en) | 2000-08-31 | 2002-03-07 | Chiron Corp | GUANIDINOBENZAMIDES AS MC4-R AGONISTS. |
| GB0102668D0 (en) | 2001-02-02 | 2001-03-21 | Glaxo Group Ltd | Compounds |
| PT1385823E (en) * | 2001-04-09 | 2007-01-31 | Novartis Vaccines & Diagnostic | Guanidino compounds as melanocortin-4 receptor (mc4-r) agonists |
| US20030207814A1 (en) * | 2002-02-04 | 2003-11-06 | Chiron Corporation | Novel guanidinyl derivatives |
| US20030195187A1 (en) * | 2002-02-04 | 2003-10-16 | Chiron Corporation | Guanidino compounds |
| US20030229025A1 (en) * | 2002-02-25 | 2003-12-11 | Chiron Corporation | Intranasal administration of MC4-R agonists |
| AU2003245325A1 (en) | 2002-05-23 | 2003-12-12 | Chiron Corporation | Substituted quinazolinone compounds |
| US7625909B2 (en) * | 2003-05-23 | 2009-12-01 | Novartis Vaccines And Diagnostics, Inc. | Substituted quinazolinone compounds |
-
2004
- 2004-11-19 WO PCT/US2004/039020 patent/WO2005051391A1/en active Application Filing
- 2004-11-19 EP EP04811698A patent/EP1686996A4/en not_active Withdrawn
- 2004-11-19 AU AU2004293012A patent/AU2004293012A1/en not_active Abandoned
- 2004-11-19 CA CA002545601A patent/CA2545601A1/en not_active Abandoned
- 2004-11-19 KR KR1020067012089A patent/KR20060105785A/en not_active Application Discontinuation
- 2004-11-19 MX MXPA06005736A patent/MXPA06005736A/en active IP Right Grant
- 2004-11-19 JP JP2006541565A patent/JP2007511612A/en active Pending
- 2004-11-19 US US10/993,147 patent/US7368453B2/en not_active Expired - Fee Related
-
2006
- 2006-05-17 IL IL175715A patent/IL175715A0/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3177218A (en) * | 1962-01-17 | 1965-04-06 | Monsanto Chemicals | Methylene-bis(2-guanidino-4-methylquinazoline) |
| US4128643A (en) * | 1976-05-28 | 1978-12-05 | Hoechst Aktiengesellschaft | 4-Quinazolinyl-guanidines |
| US4287341A (en) * | 1979-11-01 | 1981-09-01 | Pfizer Inc. | Alkoxy-substituted-6-chloro-quinazoline-2,4-diones |
| US4496571A (en) * | 1981-02-27 | 1985-01-29 | Ici Americas Inc. | Histamine H2-antagonists |
| US4748165A (en) * | 1981-05-18 | 1988-05-31 | Imperial Chemical Industries Plc | Amidine derivatives |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1686996A4 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7858631B2 (en) | 2002-05-23 | 2010-12-28 | Novartis Vaccines And Diagnostics, Inc. | Substituted pyrido [2,3-d] pyrimidinone compounds |
| US7858641B2 (en) | 2002-05-23 | 2010-12-28 | Novartis Vaccines And Diagnostics, Inc. | Substituted dihydroisoquinolinone compounds |
| EP1608317A2 (en) * | 2003-03-25 | 2005-12-28 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| EP1608317B1 (en) * | 2003-03-25 | 2012-09-26 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7625909B2 (en) | 2003-05-23 | 2009-12-01 | Novartis Vaccines And Diagnostics, Inc. | Substituted quinazolinone compounds |
| US7511159B2 (en) | 2003-12-25 | 2009-03-31 | Ono Pharmaceutical Co., Ltd. | Azetidine ring compounds and drugs comprising the same |
| EP1698375A4 (en) * | 2003-12-25 | 2007-11-07 | Ono Pharmaceutical Co | Azetidine ring compounds and drugs comprising the same |
| EP1698375A1 (en) * | 2003-12-25 | 2006-09-06 | Ono Pharmaceutical Co., Ltd. | Azetidine ring compounds and drugs comprising the same |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA06005736A (en) | 2006-12-14 |
| EP1686996A4 (en) | 2008-11-12 |
| IL175715A0 (en) | 2006-09-05 |
| EP1686996A1 (en) | 2006-08-09 |
| JP2007511612A (en) | 2007-05-10 |
| KR20060105785A (en) | 2006-10-11 |
| CA2545601A1 (en) | 2005-06-09 |
| US20050192297A1 (en) | 2005-09-01 |
| AU2004293012A1 (en) | 2005-06-09 |
| US7368453B2 (en) | 2008-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7625909B2 (en) | Substituted quinazolinone compounds | |
| EP1551834B1 (en) | Substituted quinazolinone compounds | |
| US7368453B2 (en) | Quinazolinone compounds with reduced bioaccumulation | |
| US6995269B2 (en) | Guanidinobenzamides | |
| ES2442518T3 (en) | Derivatives of 2-adamantyl-butyramine as selective inhibitors of 11 beta-HSD1 | |
| KR100883184B1 (en) | CXCR3 antagonists | |
| KR20130032863A (en) | Hematopoietic growth factor mimetic small molecule compounds and their uses | |
| SK1192002A3 (en) | Caspase inhibitors and uses thereof | |
| US20030195187A1 (en) | Guanidino compounds | |
| KR20150118152A (en) | Quinazolines as kinase inhibitors | |
| US20050124652A1 (en) | Guanidino compounds | |
| WO2005111020A2 (en) | Pyrimidine hydantoin analogues which inhibit leukocyte adhesion mediated by vla-4 | |
| EP0315112A2 (en) | Novel amide compounds | |
| MXPA05012483A (en) | Guanidino-substituted quinazolinone compounds as mc4-r agonists | |
| ES2353214T3 (en) | SUBSTITUTED QUINAZOLINONA COMPOUNDS. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 175715 Country of ref document: IL Ref document number: 2545601 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006541565 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/005736 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004293012 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004811698 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1610/KOLNP/2006 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2004293012 Country of ref document: AU Date of ref document: 20041119 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004293012 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067012089 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480039762.7 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004811698 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067012089 Country of ref document: KR |