WO2005042692A2 - A method of inhibiting the expression of genes which mediate cellular cholesterol influx in animal cells and inhibiting the production of proteins resulting from the expression of such genes using cholesterol absorption inhibitors - Google Patents
A method of inhibiting the expression of genes which mediate cellular cholesterol influx in animal cells and inhibiting the production of proteins resulting from the expression of such genes using cholesterol absorption inhibitors Download PDFInfo
- Publication number
- WO2005042692A2 WO2005042692A2 PCT/CA2004/002028 CA2004002028W WO2005042692A2 WO 2005042692 A2 WO2005042692 A2 WO 2005042692A2 CA 2004002028 W CA2004002028 W CA 2004002028W WO 2005042692 A2 WO2005042692 A2 WO 2005042692A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- cholesterol
- cholesterol absorption
- hydroxy
- azetidinone
- Prior art date
Links
- 0 CC(CC1*)(C(*)C(*)[C@@]2*)[C@]2(*)C2C1C(C)(CCC(*)C1)[C@@]1CC2* Chemical compound CC(CC1*)(C(*)C(*)[C@@]2*)[C@]2(*)C2C1C(C)(CCC(*)C1)[C@@]1CC2* 0.000 description 3
- HNHDWTQXYXFHHG-UHFFFAOYSA-N CC(C(OC(C(C(CO)O)OC1=O)=C1O)=O)=O Chemical compound CC(C(OC(C(C(CO)O)OC1=O)=C1O)=O)=O HNHDWTQXYXFHHG-UHFFFAOYSA-N 0.000 description 1
- RBAYNTRCUMDCAP-UHFFFAOYSA-N CC(OC(C(C(CO)O)OC1=O)=C1O)=O Chemical compound CC(OC(C(C(CO)O)OC1=O)=C1O)=O RBAYNTRCUMDCAP-UHFFFAOYSA-N 0.000 description 1
- FRZYBXOUKQMDBZ-UHFFFAOYSA-N CC(OC(C(C(CO)O)OC1=O)C1O)=O Chemical compound CC(OC(C(C(CO)O)OC1=O)C1O)=O FRZYBXOUKQMDBZ-UHFFFAOYSA-N 0.000 description 1
- MCBLWBYKPHUPQS-UHFFFAOYSA-N COP(O)(OC(C(C(CO)O)OC1=O)=C1O)=O Chemical compound COP(O)(OC(C(C(CO)O)OC1=O)=C1O)=O MCBLWBYKPHUPQS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/397—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having four-membered rings, e.g. azetidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/565—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol
- A61K31/568—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids not substituted in position 17 beta by a carbon atom, e.g. estrane, estradiol substituted in positions 10 and 13 by a chain having at least one carbon atom, e.g. androstanes, e.g. testosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/661—Phosphorus acids or esters thereof not having P—C bonds, e.g. fosfosal, dichlorvos, malathion or mevinphos
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/665—Phosphorus compounds having oxygen as a ring hetero atom, e.g. fosfomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
Definitions
- TITLE A METHOD OF INHIBITING THE EXPRESSION OF GENES WHICH
- This present invention relates to the field of focused therapies useful in treating the underlying causes of hypercholesterolemia.
- CVD cardiovascular disease
- the primary cause of CVD is atherosclerosis, a disease characterized by the deposition of lipids, including cholesterol, in the arterial vessel wall resulting in a narrowing of the vessel passages and ultimately a hardening of the vascular system.
- Atherosclerosis is a degenerative process resulting from an interplay of inherited (genetic) factors and environmental factors such as diet and lifestyle.
- Research to date suggest that cholesterol may play a role in atherosclerosis by forming atherosclerotic plaques in blood vessels, ultimately cutting off blood supply to the heart muscle or alternatively to the brain or limbs, depending on the location of the plaque in the arterial tree 1 ' 2 .
- a total cholesterol in excess of 225-250 mg/dl is associated with significantly elevated risk of CVD, including vascular disease.
- Cholesteryl esters are a major component of atherosclerotic lesions and the major storage form of cholesterol in arterial wall cells. Formation of cholesteryl esters is also a step in the intestinal absorption of dietary cholesterol through homeostatic control mechanisms. These control mechanisms involve the inter-related regulation of dietary cholesterol, cholesterol biosynthesis and catabolism of cholesterol-containing plasma lipoproteins. Cholesterol biosynthesis and catabolism occur primarily in the liver and hence, it is a prime determinant of plasma cholesterol levels.
- Lipoproteins are complexes of lipids and proteins held together by non-covalent bonds. Each type of lipoprotein class has a characteristic mass, chemical composition, density and physiological role. Irrespective of density or particle size, circulating lipids consist of a core of cholesteryl esters and triglycerides, and an envelope of phospholipids, free cholesterol and apolipoproteins. The apolipoproteins are involved in the assembly and secretion of the lipoprotein, provide structural integrity, activate lipoprotein-modifying enzymes, and are the ligand for a large assortment of receptors and membrane proteins. Lipoprotein classes found in plasma include HDL, LDL, intermediate density lipoproteins (IDL) and very low density lipoproteins (VLDL).
- HDL high density lipoproteins
- IDL intermediate density lipoproteins
- VLDL very low density lipoproteins
- Each type of lipoprotein has a characteristic apolipoprotein composition or ratio.
- the most prominent apolipoprotein in HDL is apolipoprotein-AI (apo-AI), which accounts for approximately 70% of the protein mass, with apo-AII accounting for another 20%.
- the ratio of apoA-l to apoA-ll may determine HDL functional and anti-atherogenic properties.
- Circulating HDL particles consist of a heterogeneous mixture of discoidal and spherical particles with a mass of 200 to 400 kilo-daltons and a diameter of 7 to 10 nm.
- HDL is one of the major classes of lipoproteins that function in the transport of lipids in plasma, and has multiple functions within the body, including reverse cholesterol transport, providing the cholesterol molecule substrate for bile acid synthesis, transport of clusterin, transport of paraoxanase, prevention of lipoprotein oxidation and selective uptake of cholesterol by adrenal cells.
- the major lipids associated with HDL include cholesterol, cholesteryl ester, triglycerides, phospholipids and fatty acids.
- HDL is anti- atherogenic.
- the atherosclerotic process begins when LDL becomes trapped within the vascular wall. Oxidation of this LDL results in the binding of monocvtes to the endothelial cells lining the vessel wall. These monocytes are activated and migrate into the endothelial space where they are transformed into macrophages, leading to further oxidation of the LDL. The oxidized LDL is taken up through the scavenger receptor on the macrophage, leading to the formation of foam cells. A fibrous cap is generated through the proliferation and migration of arterial smooth muscle cells, thus creating an atherosclerotic plaque.
- HDL is essential for the transport of cholesterol from extra-hepatic tissues to the liver, where it is excreted into bile as free cholesterol or as bile acids that are formed from cholesterol.
- the process requires several steps. The first is the formation of nascent or pre-beta HDL particles in the liver and intestine. Excess cholesterol moves across cell membranes into the nascent HDL through the action of the ABCA transporter. Lecithin cholesterol acyl transferase (LCAT) converts the cholesterol to cholesteryl ester and the subsequent conversion of nascent HDL to mature HDL.
- LCAT Lecithin cholesterol acyl transferase
- Esterified cholesterol is then transferred by cholesteryl ester transfer protein (CETP) from HDL to apolipoprotein-B containing lipoproteins, which are taken up by numerous receptors in the liver. Nascent HDL is regenerated via hepatic triglyceride lipase and phospholipid transfer protein and the cycle continues. In addition to the cholesterol removed from peripheral cells, HDL accepts cholesterol from LDL and erythrocyte membranes. Another mechanism of reverse cholesterol transport may involve passive diffusion of cholesterol between cholesterol-poor membranes and HDL or other acceptor molecules.
- CETP cholesteryl ester transfer protein
- HDL protects against the development of atherosclerosis both through its role in reverse cholesterol transport and possibly by impeding LDL oxidation.
- Several HDL- associated enzymes are involved in the process. Paroxonase (PON1), LCAT, and platelet activating factor acetylhydrolase (PAFAH) all participate by hydrolyzing phospholipid hydroperoxides generated during LDL oxidation and act in tandem to prevent the accumulation of oxidized lipid in LDL. These enzymes are responsible for the anti-oxidative and anti-inflammatory properties of HDL. Studies have shown that a low plasma concentration of HDL cholesterol is a significant risk factor for the development of atherosclerosis and that high levels are protective.
- the liver is the major organ responsible for synthesis and secretion of VLDLs, which, as noted above, are metabolized to LDL in circulation.
- LDLs are the predominant cholesterol carrying lipoproteins in plasma and hence an increase in their concentration is directly correlated with atherosclerosis. Simply put, when intestinal cholesterol absorption is reduced, by any means, less cholesterol is delivered to the liver. As a result, VLDL production is reduced and there is a concomitant increase in hepatic clearance of plasma cholesterol, mostly in the form of LDL.
- cholesterol acts on three different levels to regulate its own synthesis. Firstly, it suppresses endogenous cholesterol synthesis by inhibiting the enzyme HMG CoA reductase. Secondly, it activates LCAT. Thirdly, it regulates the synthesis of the LDL- receptor ensuring that a cell already having a sufficient amount of cholesterol will not take up additional cholesterol.
- ABC ATP-binding cassette
- MDR1 multidrug-resistance export proteins
- P-glycoprotein a 170 kDa multispanning transmembrane protein belonging to the above-mentioned ABC superfamily 2 . It is suggested that one mechanism by which tumor cells acquire resistance to drugs is by over expression of P-glycoprotein (also referred to as "P-gp" or "Pgp")) 6,7 ' 8,9
- P-glycoprotein likely acts by rapidly pumping hydrophobic chemotherapeutic agents out of the tumor cells, thereby decreasing intracellular accumulation of certain chemotherapeutic agents below their cytostatic concentrations.
- One in vitro solution is to increase the chemo drug concentration.
- cancer chemotherapeutic agents are already administered at their maximally tolerated range in vivo, increasing the doses is an unacceptable solution leading, on most cases to extreme toxicity 10,11 .
- P-glycoprotein is structurally similar to the cystic fibrosis transporter protein, the major histocompatibility complex-linked peptide transporter, and a non-P-glycoprotein-related multidrug resistance protein (MRP) 12,13,14,15 .
- MRP multidrug resistance protein
- P-glycoprotein is expressed in diverse sites including the normal human adrenal cortex, the luminal aspect of bile canaliculi and colonic epithelium, the renal tubular epithelium and the endothelial cell of the blood-brain and blood testicular barriers.
- the function of the P-glycoproteins at these sites is unclear but appears to function as an energy dependent pump of broad specificity possibly related to secretion of hormones and protection against toxins.
- P- glycoprotein can actively efflux a large number of hydrophobic, and heterocyclic cancer chemotherapeutic agents including adriamycin (doxorubicin), colchicine, colcemid, etoposide, paclitaxel, vincristine, vinblastine as well as others.
- adriamycin doxorubicin
- colchicine colchicine
- colcemid etoposide
- paclitaxel paclitaxel
- vincristine vinblastine as well as others.
- P-glycoproteins are encoded by a highly conserved family of genes 16
- the MDR-1 gene encodes class I P-glycoprotein that confers multidrug resistance in humans 17 .
- the pgp-1 and pgp-2 genes in hamsters and the mdr-3 and MDR-1 genes in mice encode the class and II proteins, both of which confer multidrug resistance in rodents 18 .
- results from Garrigues et aP 2 show that P-glycoprotein mediates the flux of cholesterol from the cytosolic leaflet to the exoplasmic leaflet suggesting that P-glycoprotein may be involved in the cholesterol enrichment of the exoplasmic leaflet of plasma membrane at the level of rafts and caveolae. Further, cholesterol redistribution within plasma membrane induced by P-glycoprotein may affect cellular cholesterol trafficking, which involves both the endogenous biosynthesis and esterification of cholesterol in the endoplasmic reticulum, the import of exogenous cholesterol from the LDL by endocytosis, and the export of cholesterol to HDL. In addition, it was suggested that the enrichment of cholesterol in the exoplasmic leaflet of the plasma membrane may facilitate the efflux of cholesterol out of the cell to the HDL, possibly in cooperation with ABCA1.
- MDR1 -transfected IEC-18 rat intestinal epithelial cells exhibited increased expression of MDR1 protein, reduced accumulation of vinblastine and increased uptake of [3H]cholesterol from cholesterol/monolein/taurochoiate micelles.
- Verapamil an inhibitor of MDR1 and UIC2, an antibody against MDR1 , both diminished cholesterol absorption in these cells, but did not completely inhibit cholesterol cellular uptake.
- MDR1 inhibitors like verapamil, interfere with the ability of MDR1 to extrude substances from the cell by acting as competitive inhibitors; they do not inactivate the protein but act as competing substances for the xenobiotic whose accumulation is being evaluated.
- UIC2 is a monoclonal antibody against an external epitope of the protein.
- the present invention provides, in one aspect, a method of decreasing or inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
- the present invention provides, in another aspect, a method of decreasing or inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
- the present invention provides, in yet another aspect, a method of decreasing the level of serum LDL cholesterol in an animal, by inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
- the present invention provides, in yet another aspect, a method of decreasing the level of serum LDL cholesterol in an animal, by inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in the animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
- the crux of the present invention is the provision and administration of compounds which inhibit the protein mediated uptake of cellular cholesterol through the lowering of specific gene expression and activity.
- cholesterol absorption inhibitors effectively inhibit the expression of multi-drug resistance genes and/or inhibit the uptake of cellular cholesterol which is mediated by the proteins, specifically P-glycoprotein, which are produced by such genes.
- the results presented herein suggest that the reduced production of proteins expressed by these genes prevents the influx of cholesterol into cells.
- some the preferred cholesterol absorption inhibitors of the present invention (those depicted below including those in formulae (i) through (iv) comprise an ascorbyl moiety. These particular compounds have numerous additional advantages. In particular, solubility in aqueous solutions such as water is improved by the ascorbyl moiety thereby allowing oral administration perse. Likewise, other modes of administration are facilitated.
- these selected compounds of the present invention can be prepared and used as such or they can be easily incorporated into pharmaceutical preparations, optionally in conjunction with other therapeutic agents, regardless of whether such preparations are water-based.
- This enhanced solubility generally translates into lower administration dosages of the compounds in order to achieve the desired therapeutic effect.
- Figure 1 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR- 1/GAPDH; "GAPDH” or glyceraldehyde-3-phosphate dehydrogenase) in CaCo2 cells after treatment for one week with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
- Figure 2 is a graph showing the titration for primer drop of GAPDH
- Figure 3 depicts a polymerase chain reaction (1.5% agarose gel) gel electrophoresis results for MDR-1 , GAPDH;
- Figure 4 is a bar graph showing an MTS- and LDH-Assay of cell viability after treatment of CaCo2 cells with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
- Figure 5 a bar graph showing a BCA-Assay of protein concentration after treatment of CaCo2 with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
- Figure 6 a bar graph showing the level of ABCC1 (MRP-1) expression (normalized ratio of MRP-1/GAPDH) in CaCo2 cells after treatment for one week with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM- VP4";
- Figure 7 depicts a polymerase chain reaction (1.5% agarose gel) gel electrophoresis results for MDR-1 , GAPDH; RNA isolation with TRIZOL ⁇ RT-PCR ⁇ PCR after treatment of CaCo2 cells with liposomal formulations of one of cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4;
- Figure 8 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR- 1 /GAPDH) in CaCo2 cells after treatment for one week with one of the cholesterol absorption inhibitors described herein: a liposomal formulation of an ascorbyl stanyl phosphate ester called "FM-VP4" at 2.5, 5 and 10um as compared to a control and empty liposomes;
- FM-VP4 liposomal formulation of an ascorbyl stanyl phosphate ester
- FIG. 9 is a Western Blot analysis of P-glycoprotein in CaCo2 cells after incubation with one of cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
- Figure 10 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR-1/GAPDH; "GAPDH” or glyceraldehyde-3-phosphate dehydrogenase) in CaCo2 cells after treatment for one week with varying concentrations of cholesterol;
- Figure 11 depicts a polymerase chain reaction (1.5% agarose gel) gel electrophoresis results for MDR-1, GAPDH;
- Figure 12 is a bar graph showing cell viability and cytotoxicity after treatment with Cholesterol and effect on Caco-2 cells; MTS-Assay and LDH-Assay after incubation with Cholesterol for 24 hours. Each bar represents the mean ⁇ SD of four experiments;
- Figure 13 is a bar graph showing cell viability and cytotoxicity after treatment with FM- VP4. Effect on Caco-2 cells. MTS-Assay and LDH-Assay after incubation with FM-VP4 for 24 hours. Each bar represents the mean ⁇ SD of four experiments;
- Figure 14 is a bar graph showing cell viability after treatment with FM-VP4. Effect on LLC-PKi cells. Each bar represents the mean ⁇ SD of three experiments;
- Figure 15 is a bar graph showing cell viability after treatment with cholesterol micelles. Effect on LLC-PKi cells. Each bar represents the mean ⁇ SD of three experiments;
- Figure 16 shows the expression profile of mdr-1 gene in Caco-2 cells was examined as an effect of 1 week treatment with FM-VP4.
- a sample from each PCR product was subjected to electrophoresis on a 1.5% agarose gel (A).
- the fluorescent bands were imaged under UV light (UV-Epi Chem II) and quantified with the UVP-Labworks software (B).
- Each value represents the mean ⁇ standard deviation of n experiments (see table C).
- Figure 17 shows mdr-1 expression in Caco-2 cells after treatment with FM-VP4 cholesterol-free Liopsomes.
- A 1.5% agarose gel. 1: Control, 2: 2.5 ⁇ M, 3: 5 ⁇ M, 4: 10 ⁇ M and 4: empty liposomes
- B Bands were quantified with the UVP-Labworks software. *P ⁇ 0.009 versus control (Student T-test);
- Figure 18 shows mdr-1 gene expression in Caco-2 cells after treatment with cholesterol micelles for 1 week. *P ⁇ 0.04 versus control (Student T-test);
- FIG 19 Western Blot, shows expression of P-gp in Caco2-cells after incubation for 1 week with 10 and 50uM FM-VP4.
- 1 °Ab 1:300 dilution of C219 (Signet Pathology Syst.) in Blotto
- 2°Ab 1 :2000 dilution of mouse IgG in Blotto (HRP-conjugated);
- Figure 20 is one representative graph showing TEER values measured before and after the transport studies
- Figure 21 shows time-dependent secretory Rhodamine 123 transport (B to A) after pre- treatment with FM-VP4 for one week;
- Figure 22 shows the effect of pre-incubation with FM-VP4 for 1 week on P-gp transport of Rh123 across Caco-2 monolayer (basolateral to apical). Data represents the average of 3 experiments ⁇ SD. *P ⁇ 0.07; **P ⁇ 0.009;
- Figure 23 shows transport of Rhodamine 123 from basolateral to apical side after 2 days pre-incubation with FM-VP4 on the apical side;
- Figure 24 shows secretory transport of Rhodamine 6G (B to A) after pre-incubation with FM-VP4 for 7 days on apical and basolateral side;
- Figure 25 shows P app for secretory transport of Rhodamine 6G in Caco-2 cells
- Figure 26 shows mdr-1 A gene expression in liver tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight); Figure 27 shows mdr-1 B gene expression in liver tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight);
- Figure 28 shows mdr-1 A gene expression in duodenum tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight). Internal control with GAPDH was run in a different experiment; and
- Figure 29 shows mdr-1 B gene expression in duodenum tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight). Internal control with GAPDH was run in a different experiment. Mdr-1 B was not detectable in these tissues.
- animal means any member of the animal kingdom, including all mammals and most preferably humans. Veterinary use is also contemplated.
- ileal bile acid transporter or "IBAT” is synonymous with apic l sodium co-dependent bile acid transporter, or ASBT.
- benzothiepine IBAT inhibitor means an ileal bile acid transport inhibitor which comprises a therapeutic compound comprising a 2,3,4,5-tetrahydro-1- benzothiepine 1,1 -dioxide structure.
- a multiple drug resistance gene refers to one or more of the following genes: ACB1 (MDR-1), ABCC1 (MRP-1); and ABCC3 (MRP-3).
- prodrug refers to compounds that are drug precursors, which, following administration to a patient, release the drug in vivo via some chemical or physiological process (for example, a prodrug, on being brought to physiological pH or through enzyme action is converted to the desired drug form).
- solvate refers to a molecular or ionic complex of molecules or ions of solvent with those of solute (for example the compounds of formulae a) to f) or prodrugs of compounds a) to f)).
- useful solvents include polar, protic solvents such as water and/or alcohols (for example, methanol).
- the terms "effective” or “therapeutically effective”, are intended to qualify the amount of the compound(s) or composition administered to an animal, in particular a human, in order to elicit a biological or medical response of a cell, tissue, system, animal or mammal that is being sought by the person administering the compound(s) or composition and which amount achieves one or more of the following goals: a) inhibiting or decreasing the expression of a gene which mediates cellular cholesterol influx in an animal cell; b) inhibiting or decreasing the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell; thereby, by one or both means, achieving one or more of the following therapeutic goals: c) decreasing serum LDL levels; d) increasing serum HDL levels; e) preventing, reducing, eliminating or ameliorating a dyslipidemic condition or disorder; f) preventing, reducing, eliminating or ameliorating hypercholesterolemia or hypoalphalipoproteinemia; g) preventing, reducing, eliminating or ameliorating the
- sterol includes all sterols without limitation, for example: (from any source and in any form: ⁇ , ⁇ and y) sitosterol, campesterol, stigmasterol, brassicasterol (including dihydrobrassicasterol), desmosterol, chalinosterol, poriferasterol, clionasterol, ergosterol, coprosterol, codisterol, isofucosterol, fucosterol, clerosterol, nervisterol, lathosterol, stellasterol, spinasterol, chondrillasterol, peposterol, avenasterol, isoavenasterol, fecosterol, pollinastasterol, cholesterol and all natural or synthesized forms and derivatives thereof, including isomers.
- stanol refers to, for example: (from any source and in any form: ⁇ , ⁇ and y) saturated or hydrogenated sterols including all natural or synthesized forms and derivatives thereof, and isomers, including sitostanol, campestanol, stigmastanol, brassicastanol (including dihydrobrassicastanol), desmostanol, chalinostanol, poriferastanol, clionastanol, ergostanol, coprostanol, codistanol, isofucostanol, fucostanol, clerostanol, nervistanol, lathostanol, stellastanol, spinastanol, chondrillastanol, pepostanol, avenastanol, isoavenastanol, fecostanol, and pollinastastanol.
- the sterols and stanols for use in forming derivatives in accordance with this invention may be procured from a variety of natural sources or they may be artificially synthesized. For example, they may be obtained from the processing of plant oils (including aquatic plants) such as corn oil and other vegetable oils, wheat germ oil, soy extract, rice extract, rice bran, rapeseed oil, sunflower oil, sesame oil and fish (and other marine-source) oils. They may also be derived from yeasts and fungi, for example ergosterol. Accordingly, the present invention is not to be limited to any one source of sterols.
- US Patent Serial No. 4,420,427 teaches the preparation of sterols from vegetable oil sludge using solvents such as methanol.
- phytosterols and phytostanols may be obtained from tall oil pitch or soap, by-products of forestry practises as described in US Patent Serial No.5,770,749, incorporated herein by reference.
- a further method of extracting sterols and stanols from tall oil pitch is described in Canadian Patent Application Serial No. 2,230,373 which was filed on February 20, 1998 (corresponding to PCT/CA99/00150 which was filed on February 19, 1999) and US Patent Application Serial No 10/060,022 which was filed on January 28, 2002 the contents of all of which are incorporated herein by reference.
- sterol and "stanol” as used herein, including, but not limited to: free sterols and stanols, esterified sterols and stanols with aliphatic or aromatic acids (thereby forming aliphatic or aromatic esters, respectively), phenolic acid esters, cinnamate esters, ferulate esters, phytosterol and phytostanol glycosides and acylated glycosides or acylglycosides.
- sterols and “stanols” encompasses all analogues, which may further have a double bond at the 5-position in the cyclic unit as in most natural sterols, or one or more double bonds at other positions in the rings (for example, 6, 7, 8(9), 8(14), 14 5/7) or no double bonds in the cyclic unit as in stanols. Further, there may be additional methyl groups as, for example, in ⁇ i-sitosterol.
- Sterols are naturally occurring compounds that perform many critical cellular functions. Sterols such as campesterol, stigmasterol and beta-sitosterol in plants, ergosterol in fungi and cholesterol in animals are each primary components of cellular and sub-cellular membranes in their respective cell types. Phytosterols, in particular, have received a great deal of attention due to their ability to decrease serum cholesterol levels when fed to a number of mammalian species, including humans.
- the compounds of the present invention are formed of naturally-derived or artificially synthesized beta-sitosterol, campestanol, sitostanol, and campesterol and each of these compounds so formed is then admixed in a pharmaceutical composition prior to delivery in various ratios.
- the compound of the present invention comprises a chemical linkage between one or more disodium ascorbyl phytostanyl phosphates (referred to herein as "FM-VP4") which comprises two major components: disodium ascorbyl campestanyl phosphate (“DACP”) and disodium ascorbyl sitostanyl phosphate (“DASP").
- cholesterol absorption inhibitor 3 refers to any compound having a negative effect on cholesterol transport, uptake or absorption, by whatever mechanism and includes any compound which inhibits bile acid reabsorption or transport.
- the cholesterol absorption inhibitor comprises one or more sterols, stanols or mixtures thereof or derivatives thereof as described herein.
- this includes all free sterols and stanols, and all sterol and stanol aliphatic and aromatic esters, sterol and stanol phenolic acid esters, sterol and stanol cinnamate esters, sterol and stanol ferulate esters, sterol and stanol glycosides, sterol and both stanol acylated glycosides or acylglycosides.
- the cholesterol absorption inhibitor comprises one or more derivatives or compounds comprising a sterol or stanol, including biologically acceptable salts thereof, having one or more of the following formulae:
- R is a sterol or stanol moiety
- the cholesterol absorption inhibitor is a disodium ascorbyl stanyl phosphate composition which comprises a mixture of disodium ascorbyl campestanyl phosphate and disodium ascorbyl sitostanyl phosphate.
- the compounds of formulae i) to iv) can be prepared by known methods, for example those described below and in PCT/CAOO/00730, which was filed on June 20, 2000 and claims priority back to US Patent Application 09/339,903 filed on June 23, 1999, the entire contents of which are incorporated herein by reference..
- compounds of formulae i) to iv) can be prepared as follows: the selected sterol or stanol (or halophosphate, halocarbonate or halo-oxalate derivatives thereof) and ascorbic acid are mixed together under reaction conditions to permit condensation of the "acid" moiety with the "alcohol” (sterol).
- reaction conditions are the same as those used in other common esterification reactions such as the Fisher esterification process in which the acid component and the alcohol component are allowed to react directly or in the presence of a suitable acid catalyst such as mineral acid, sulfuric acid, phosphoric acid, p-toluenesulfonic acid.
- organic solvents generally employed in such esterification reactions are ethers such as diethyl ether, tetrahydrofuran, or benzene, toluene or similar aromatic solvents and the temperatures can vary from room to elevated temperatures depending on the reactivity of the reactants undergoing the reaction.
- the process to form the ester comprises "protecting" the hydroxyl groups of the ascorbic acid or derivatives thereof as esters (for example, as acetate esters) or ethers (for example, methyl ethers) and then condensing the protected ascorbic acid with the sterol/stanol halophospahte, halocarbonate or halo-oxalate under suitable reaction conditions.
- condensation reactions are conducted in an organic solvent such as diethyl ether, tetrahydrofuran, or benzene, toluene or similar aromatic solvents.
- the reaction temperatures may vary from low (-15°C) to elevated temperatures.
- ascorbic acid is initially protected from decomposition by the formation of 5,6-isopropylidene-ascorbic acid. This can be achieved by mixing acetone with ascorbic acid and an acidic catalyst such as sulfuric acid or hydrochloric acid under suitable reaction conditions.
- Phytostanol chlorophosphate is prepared by forming a solution of phytostanol in toluene and pyridine (although other nitrogen bases such as aliphatic and aromatic amines may alternatively be used) and treating this solution with a phosphorus derivative such as phosphorus oxychloride.
- the residue so formed after filtration and concentration of the mother liquor is phytostanol chlorophosphate.
- the latter is then mixed with 5,6-isopropylidene-ascorbic acid and, after the addition of a suitable alcohol such as ethanol and HCl, concentrated.
- a suitable alcohol such as ethanol and HCl
- pyridine/THF may be added and the product concentrated. After final washing and drying, the resultant novel product a stanol-phosphate-ascorbate.
- ascorbic acid is protected at the hydroxyl sites not as 5,6-isopropylidene-ascorbic acid but as esters (for example as acetates, phosphates and the like..).
- esters for example as acetates, phosphates and the like..
- the latter may then be condensed with sterols or stanols, derivatized as described above, using known esterification methods ultimately to produce the compounds.
- the formation of mono and diphosphates of ascorbic acid is described thoroughly in the literature. For example, US Patent Serial No. 4,939,128 to Kato et al., the contents of which are incorporated herein by reference, teaches the formation of phosphoric acid esters of ascorbic acid.
- the cholesterol absorption inhibitor of the present invention comprises one or more disodium ascorbyl phytostanyl phosphates (referred to as "FM- VP4") which comprises two major components: disodium ascorbyl campestanyl phosphate (“DACP”) and disodium ascorbyl sitostanyl phosphate (“DASP").
- FM- VP4 disodium ascorbyl phytostanyl phosphates
- DASP disodium ascorbyl sitostanyl phosphate
- the cholesterol absorption inhibitor comprises a compound from the family of hydroxy substituted azetidinones.
- this azetidonone is a hydroxy substituted azetidinone compound represented by the formul
- Ari and Ar2 are independently selected from the group consisting of aryl and R 4 -substituted aryl;
- Ar 3 is aryl or R 5 -substituted aryl
- X, Y and Z are independently selected from the group consisting of -CH 2 -, -CH ⁇ ower alkyl)- and -C(dilower alkyl)-;
- R and R 2 are independently selected from the group consisting of -OR 6 , -O(CO)R 6 , — 0(CO)OR 9 and -O(CO)NR 6 R 7 ;
- R1 and R 3 are independently selected from the group consisting of hydrogen, lower alkyl and aryl; q is 0 or 1 ; r is 0 or 1 ; m, n and p are independently 0, 1 , 2, 3 or 4; provided that at least one of q and r is 1 , and the sum of m, n, p, q and r is 2, 3, 4, 5 or 6; and provided that when p is 0 and r is 1 , the sum of m, q and n is 1 , 2, 3, 4 or 5;
- R 4 is 1-5 substituents independently selected from consisting of lower alkyl, -ORe, - O(CO)R 6l -O(CO)OR 9 , -O(CH 2 )i- 5 OR 6 , -O(CO)NR 6 R 7 , -NR 6 R 7 , ⁇ NR 6 (CO)R 7 , -NR 6 (CO)ORg, -NR 6 (CO)NR 7 R 8 , -NR 6 SO 2 R 9 , -COOR 6 , ⁇ CONR 6 R 7 , -COR 6 , -SO 2 NR 6 R 7 , S(O) 0- 2 Rg, -O(CH 2 ) ⁇ - ⁇ o -COOR 6 , ⁇ O(CH 2 ) ⁇ . ⁇ o CONR 6 R , -(lower alkylene)COOR 6 , -CH.dbd.CH-COOR 6 , -CF 3 , -CN, -NO 2 and halogen;
- R5 is 1-5 substituents independently selected from the group consisting of -ORe, - O(CO)R 6 , -O(CO)OR 9 , -O(CH 2 )i- 5 OR 6 , -O(CO)NR 6 R 7 , -NR 6 R 7 , -NR 6 (CO)R 7 , -NR 6 (CO)ORg, -NR 6 SO 2 R 9 , -COOR 6 , -CONR 6 R 7 , -COR 6 , -SO 2 NR 6 R 7 , S(O) 0- 2 R 9 , -O(CH 2 ) ⁇ - ⁇ o -COOR 6 , -O(CH 2 ) 1-10 CONR 6 R 7 , -(lower alkylene)COOR 6 and -CH.dbd.CH-COORe ;
- R 6 , R 7 and R 8 are independently selected from the group consisting of hydrogen, lower alkyl, aryl and aryl-substituted lower alkyl;
- R 9 is lower alkyl, aryl or aryl-substituted lower alkyl.
- An is phenyl or R 4 -substituted phenyl
- Ar 2 is phenyl or R 4 -substituted phenyl
- Ar 3 is R 5 -substituted phenyl.
- An is R 4 -substituted phenyl wherein R 4 is halogen;
- Ar 2 is R 4 -substituted phenyl wherein R 4 is halogen or -OR 6 , wherein R 6 is lower alkyl or hydrogen; and
- Ar 3 R 5 -substituted phenyl, wherein R 5 is -OR 6 , wherein R6 is lower alkyl or hydrogen.
- the compound is selected from the group consisting of:
- the cholesterol absorption inhibitor within the scope of the present invention may be an inhibitor of bile acid transport or reabsorption, including, but not limited to ileal, apical and hepatic transport inhibitors.
- IBATs ileal bile acid transport inhibitors
- IBAT inhibitors useful in the present invention are disclosed in PCT/US95/10863, the contents of which are incorporated herein by reference. More IBAT inhibitors are described in PCT/US97/04076, herein incorporated by reference. Still further IBAT inhibitors useful in the present invention are described in U.S. application Ser. No. 08/816,065, herein incorporated by reference. More IBAT inhibitor compounds useful in the present invention are described in WO 98/40375, herein incorporated by reference. An array of additional IBAT inhibitor compounds useful in the present invention are described in U.S. Pat. No. 5,994,391 , also incorporated herein by reference.
- bile acid transport inhibitors include selected benzothiepines, such as those disclosed in WO 93/321146, PCT/US97/04076, PCT/US95/10863, EP 508425, FR 2661676, WO 99/35135, WO 92/18462, and U.S. Patent No. 5,994,391 (Lee et al.).
- WO 92/16055 to The Wellcome Foundation Limited describes a number of suitable benzothiazepine compounds. Additional hypolipidemic benzothiazepine compounds (particularly 2, 3,4,5-tetrahydrobenzo-1-thi-4-azepine compounds) are disclosed in patent application Nos. WO 96/05188, WO 96/05188 and WO 96/16051.
- IBAT inhibitor compounds include a class of naphthalene compounds, described by lchihashi 29 .
- S-8921 methyl 1-(3,4-dimethoxyphenyl)-3-(3- ethylvaleryl)-4-hydroxy-6,7,8-trimethoxy-2-na phthoate
- the structure of S-8921 is shown in formula B-20 of this publication.
- Further naphthalene compounds or lignin derivatives are described in PCT Patent Application No. WO 94/24087.
- IBATs include, for example, SC-635 developed by Pharmacia Corporation, IL and Monsanto, MO; 264W94 developed by GlaxoSmithKline; S-8921 developed by Shiongi) and (3R,5R)-3-butyl-3-ethyl-2,3,4,5-tetrahydro-,7,8-dimethoxy-5-phenyl-1-4-benz othiazepine 1,1 -dioxide.
- the cholesterol absorption inhibitor within the scope of the present invention may be an androstane and/or androstene derivative, wherein androstane and/or androstene are coupled with ascorbic acid and represented by one or more of the general formulae:
- R-i, R 2 , R 3 , R 4 , R 5 , Re may individually be chosen from hydrogen, OH, carbonyl, and an ascorbyl moiety; and R may be hydrogen or any halogen.
- the ascorbyl moiety which is coupled to the compound from the androstane or androstene family is selected individually from one or more of the following structures:
- M+ represents any metal, alkali earth metal, or alkali metal.
- the androstane/androstene compounds of formulae vi) to viii), incorporating one or more of the ascorbyl linkers of formulae ix) to xx), can be prepared by known methods, for example those described in PCT/CA03/00824, the contents of which are incorporated herein by reference or can be prepared by the methods described above for the preparation of compounds of formulae i) to iv), adjusted accordingly.
- biologically acceptable salts refers any salts that retain the desired biological and/or physiological activity of the compounds as described herein and exhibit minimal undesired toxicological effects. Accordingly, reference to all compounds herein thereby includes reference to respective acidic and/or base salts thereof, formed with inorganic and/or organic acids and bases.
- Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example trifluroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclpentanepropionates, digluconates, dodecylsulfates, heptanoates, hexanoates, hydrochlorideshyrobromides, hydroiodides, 2- hydroethanesulfonates, lactates, maleates, methanesulfonates, 2-naphthalenesulfonates, nicotinates, nitrates, oxalates, pectinates, persulfonates, 3-phenylpropionates, phosphates, picrates, pivalates, propionates
- Those compounds which contain an acid moiety may form salts with a variety or organic and inorganic bases.
- the present invention encompasses not only the parent compounds comprising, for example, the selected sterol and/or stanol a but also, where possible (i.e. where the parent contains a free hydroxyl group), the present invention encompasses the biologically acceptable metal, alkali earth metal, or alkali metal salts of the disclosed compounds.
- the salts, as described herein, are even more water soluble than the corresponding parent compounds and therefore their efficacy and evaluation both in vitro and in vivo may be enhanced.
- Salt formation of the compounds of the present invention can be readily performed, for example, by treatment of any parent compound containing a free OH group with a series of bases (for example, sodium methoxide or other metal alkoxides) to produce the corresponding alkali metal salts.
- bases for example, sodium methoxide or other metal alkoxides
- Other metal salts of calcium, magnesium, manganese, copper, zinc, and the like can be generated by reacting the parent with suitable metal alkoxides.
- stereoisomers of the compounds of the present invention such as those which may exist due to asymmetric carbons on various constituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons) and diastereomeric forms, are contemplated within the scope of the present invention.
- Individual stereoisomers of the compounds of the present invention may, for example, be admixed as racemates or with all other, or other selected sterioisomers.
- the chiral centres of the compounds can have the S or R configuration as defined by the IUPAC 1974 Recommendations.
- Such stereoisomers can be prepared using conventional techniques, either by reacting enantiomeric starting materials, or by separating isomers of compounds of the present invention. When diastereomeric or enantomeric products are prepared, they can be separated by conventional methods, for example, chromatographic or fractional crystallization.
- Isomers may include geometric isomers, for example cis-isomers or trans-isomers across a double bond. All such isomers are contemplated among the compounds useful in the present invention.
- the compounds useful in the present invention also include tautomers.
- the present invention fully covers the inhibition of expression of any gene which mediates cellular cholesterol influx in an animal cell by use of cholesterol absorption inhibitors
- the cholesterol absorption inhibitors are particularly useful in inhibiting the expression of MDR-1 (ABCB1), which has P-glycoprotein as its gene product, as well as inhibiting the expression of ABCC1 (MRP- 1) and ABCC3 (MRP-3).
- MDR-1 MDR-1
- MRP- 1 MRP-1
- MRP-3 ABCC3
- the cholesterol absorption inhibitors, as described herein are particularly useful in inhibiting the production of the protein products of these genes, for example P-glycoprotein, as expressed by the MDR-1 gene.
- the finding that cholesterol absorption inhibitors exhibit the aforementioned effects on, in particular both the expression of the multiple drug resistance gene (for example the MDR- 1 gene) and the production of the gene product (for example P-glycoprotein), has enormous therapeutic potential.
- the "gene product" of a gene which mediates cellular cholesterol influx may be an influx transporter of cholesterol.
- concentration of the protein is decreased through reduced gene expression, the amount of cholesterol taken up by an animal cell is decreased.
- a method of decreasing or inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
- a method of decreasing or inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
- a method of and pharmaceutical composition for decreasing the level of serum LDL cholesterol in an animal by inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
- a method of and pharmaceutical composition for decreasing the level of serum LDL cholesterol in an animal by inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in the animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
- the present invention provides means to decrease or inhibit the expression of a gene which mediates cellular cholesterol influx in an animal cell and/or to decrease or inhibit the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell in order, ultimately, to achieve one or more of the following therapeutic goals: a) decreasing serum LDL levels; b) increasing serum HDL levels; c) preventing, reducing, eliminating or ameliorating a dyslipidemic condition or disorder; d) preventing, reducing, eliminating or ameliorating hypercholesterolemia or hypoalphalipoproteinemia; e) preventing, reducing, eliminating or ameliorating the development of atherosclerotic lesions; and f) preventing, reducing, eliminating or ameliorating any condition, disease or disorder which has as its basis or which is exacerbated by a deficiency in plasma HDL, or excess of either LDL, VLDL, Lp(a), beta-VLDL, IDL or remnant lipoproteins which comprises administering to an animal or a cell derived
- cholesterol absorption inhibitors As described herein.
- Some of the cholesterol absorption inhibitors ⁇ compounds depicted above having formulae i) through iv)) comprise an ascorbyl moiety.
- These particular compounds have numerous added advantages.
- solubility of the compounds is greatly enhanced, both in aqueous solutions and non-aqueous media such as oils and fats. With this greater solubility, effective therapeutic dosages and concomitantly costs, can be reduced.
- these derivatives are heat stable (stable to oxidation and hydrolysis) which is essential for some processing mechanisms.
- the desired effects described herein may be achieved in a number of different ways.
- the cholesterol absorption inhibitor may be administered by any conventional means available for use in conjunction with pharmaceuticals i.e. with a pharmaceutically acceptable carrier.
- the pharmaceutical compositions can comprise from about 1% to 99% of the "active" components and preferably from about 5% to 95% of the active components.
- compositions and pharmaceutical compositions can be prepared using conventional, pharmaceutically available excipients, and additives and by conventional techniques.
- pharmaceutically acceptable excipients and additives include non-toxic compatible fillers, binders, disintegrants, buffers, preservatives, anti-oxidants, lubricants, flavourings, thickeners, colouring agents, emulsifiers and the like.
- the exact amount or dose of the cholesterol absorption inhibitors which is required to achieve the desired effects will, of course, depend on a number of factors such as the particular compound or composition chosen, the potency of the compound or composition administered, the formulation in which it is administered, the mode of administration and the age, weight, condition and response of the patient. All of these factors, among others, will be considered by the attending clinician with respect to each individual or patient.
- a total daily dose the cholesterol absorption inhibitor having one of formulae i)-viii) and comprising sterols and/or stanols may be administered in a daily dosage range of from 10mg to about 20 g, more preferably 10mg to 1.5g, and most preferably up to 80O mg per day in single or multiple divided doses.
- the chosen a cholesterol absorption inhibitor is a sterol or stanol, whether free or as part of a compound or derivative, it may be administered in a form comprising up to 6 grams sterols and/or stanols per day. It should be recognized that the provision of much larger daily doses of sterols, stanols and their derivatives are not harmful to the animal host, as excess will simply pass through normal excretory channels.
- the cholesterol absorption inhibitor is a substituted azetidinone
- it may be administered in a daily dose of from about 0.1 to about 30 mg/kg of body weight, preferably about 0.1 to about 15 mg/kg, and most preferably up to 10mg/kg per day.
- the dosage level is therefore from about 5 mg to about 1000 mg of drug per day, given preferably in a single dose or 2-4 divided doses.
- the cholesterol absorption inhibitor is any compound which inhibits bile acid reabsorption
- it may be administered in a dose from about 0.003mg — 20mg per kilogram body weight of the individual animal.
- a total daily dose of an IBAT inhibitor can be in the range of from about 0.01 to about 1000 mg/day, preferably from about 0.1 mg to about 50 mg/day, more preferably from about 1 to about 10 mg/day.
- the daily dose of these cholesterol absorption inhibitors can be administered to an individual in a single dose or in multiple doses, as required. Sustained release dosages can be used.
- compositions of the present invention may be administered parenterally, such as by intravenous injection.
- pharmaceutically acceptable carriers well known in the art into dosages suitable for oral administration.
- Such carriers enable the compounds and compositions of the invention to be formulated as tablets, pills, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a patient to be treated.
- compositions comprising one or more of the compounds of the present invention, include compositions wherein the active ingredients are contained in an effective amount to achieve their intended purpose. Determination of the effective amounts is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- compositions may contain suitable pharmaceutically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically.
- suitable pharmaceutically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically.
- the preparations formulated for oral administration may be in the form of tablets, dragees, capsules, or solutions.
- the cholesterol absorption inhibitor is administered in the form of a liposome.
- Liposomes are hollow microspheres composed of one or more double lipid layers. They were first used more than 30 years ago as vehicles for various drug substances, and since then knowledge of their behavior in vitro has allowed a more rational design focused on the specific treatment of certain diseases.
- Liposomes occurs formed when thin lipid films are hydrated.
- the hydrated lipid sheets detach during agitation and self-close to form multi-lamellar vesicles.
- Chemotherapeutic agents such as doxorubicin
- doxorubicin are often encapsulated in liposomes using the established methods.
- 100 nm diameter liposomes are prepared by exposing chloroformic solution of various lipid mixtures to high vacuum and subsequently hydrating the resulting lipid films (DSPC/CHOL, EPC/CHOL, DSPC/PEG-PE/CHOL) with pH 4 buffers, and extruding them through polycarbonated filters, after a freezing and thawing procedure.
- a transmembrane pH gradient is then created by adjusting the pH of the extravesicular medium to 7.5 by addition of an alkalinization agent.
- the selected drug is then entrapped by addition of the drug solution in small aliquots to the vesicle solution, at an elevated temperature, to allow drug accumulation inside the liposomes.
- Trapping efficiencies are determined by separating free from liposome encapsulated drug on gel filtration columns and quantifying the two fractions for lipid and drug content by liquid scintillation counting, fluorescence spectroscopy or UV-VIS spectroscopy. These liposomes are then evaluated for size distribution (quasielastic light scattering, scanning electron microscopy), drug uptake and release studies, stability, and in vivo tumor targeting efficiency.
- compositions of the present invention may be manufactured in any manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes.
- compositions for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
- compositions for oral use can be obtained by combining the active compounds with solid excipient, optionally grinding a resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores.
- suitable excipients include lactose, sucrose, mannitol, sorbitol, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and polyvinylpyrrolidone (PVP).
- disintegrating agents may be added, such as the cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- Dragee cores are provided with suitable coatings.
- suitable coatings For this purpose, concentrated sugar solutions may be used, which may optionally contain gum Dhosph, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
- Oral liquid preparations may be in the form of, for example, emulsions, syrups, or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose, carboxymethylcellulose, aluminium stearate gel, hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non- aqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as esters of glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid; and if desired conventional flavouring or colouring agents.
- suspending agents for example sorbitol, syrup, methyl cellulose
- kits for such purpose.
- a kit is contemplated wherein two separate units are combined: a pharmaceutical composition comprising at last one cholesterol absorption inhibitor, as described herein, and a separate pharmaceutical composition comprising at least one cancer chemotherapeutic agent.
- the kit will preferably include directions for the administration of the separate components. This type of kit arrangement is particularly useful when separate components must be administered in different dosage forms (for example, oral vs. parenteral vs. intravenous) or are administered at different dosage intervals or are administered at different dosage amounts.
- EXAMPLE 1 Formation of one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester refered to herein as "FM-VP4";
- EXAMPLE 2 Formation of one of the cholesterol absorption inhibitors described herein: Disodium ascorbyl phosphate ester of dehydroisoandrosterone
- a dry three neck round bottom flask was fitted with a stirring bar, argon inlet and an addition funnel.
- a solution of dehydroisoandrosterone (1.73 g, 6 mmol) in anhydrous THF (15 ml) and pyridine (2.4 ml) was added dropwise to the mixture of anhydrous THF (12 ml) and POCI 3 (0.7 ml, 7.5 mmol) at 0 °C over a period of 10 minutes.
- a white precipitate formed immediately.
- the suspension was stirred at 0 °C for 40 minutes, and at room temperature for 1 hour and 40 minutes.
- Ascorbyl phosphate ester of dehydroisoandrosterone (0.5 g, 0.95 mmol) was dissolved in methanol (3 ml) at room temperature, and then sodium methoxide in methanol (1ml, 20%) was added. The suspension was stirred at room temperature for 30 minutes. The precipitated solid was filtered out, washed with methanol, acetone and hexanes. The mother liquor was concentrated to 2 ml, acetone was added to precipitate the product. An additional white solid was obtained. The combined solid was dried under vacuum at room temperature. Disodium ascorbyl phosphate ester of dehydroisoandrosterone (0.49 g, yield 91%) was obtained.
- Ascorbyl phosphate ester of 5 -androstan-3 ⁇ -ol-17-one (0.5 g, 0.95 mmol) was dissolved in methanol (3 ml) at room temperature, and then sodium methoxide in methanol (1.5 ml, 20%) was added. The suspension was stirred at room temperature for 25 minutes. The precipitated solid was filtered out, washed with methanol, acetone and hexanes. The mother liquid was concentrated to 2 ml, and then acetone was added to precipitate the product. An additional product was obtained. The combined solid was dried under a reduced pressure at room temperature to give disodium ascorbyl phosphate ester of 5 ⁇ -androstan-3 ⁇ -ol-17-one (0.38 g). The overall yield was 57% (based on 5 ⁇ -androstan-3 ⁇ -ol-17-one).
- EXAMPLE 4 Synthesis of another of the cholesterol absorption inhibitors described herein: Disodium Ascorbyl Phosphate Ester of Androst-5-ene-3 ⁇ ,17 ⁇ -diol
- Instrument is Waters Delta Preparative 4000 HPLC system. Column is Waters Symmetry C18, 5 ⁇ m, 30x100 mm. Mobile phases are 0.1 % H 3 P0 in water and acetonitrile. Water and acetonitrile are HPLC grade or equivalent.
- the crude phosphate ester was dissolved in methylene chloride (25 ml) , and treated with iodine (1.27 g) for 4 hours at room temperature.
- the reaction mixture was diluted with methylene chloride (75 ml), washed with 1N NaOH (2x50 ml) and water (2x50 ml), and dried over Na 2 SO .
- the solvent was removed, and the product (1.4 g, yield 71%) was crystallized from methylene chloride and methanol.
- 3 ⁇ -lodoandrost-5-ene-17-one (1.27 g, 3.19 mmol) was dissolved in glacial acetic acid (40 ml) at 50-55 °C, the activated zinc dust (2.7 g) was added in one portion. The mixture was stirred at 50 °C ⁇ 55 °C for 2 hours, the zinc dust was filtered out and washed with methylene chloride. The solution was diluted with methylene chloride (120 ml), washed with water (2x100 ml), 1 N NaOH (2x100 ml) and water (100 ml), and dried over Na 2 SO 4 . The solvent was removed to afford a white powder.
- the crude was dissolved in THF/1N HCl (1:1, 150 ml), and the hydrolysis was carried out at room temperature under vigorous stirring. After 12 hours of reaction, a TLC test indicated that the hydrolysis was complete.
- the THF in the reaction mixture was removed under a reduced pressure at room temperature, and n-butanol and ethyl acetate (1:1, 100 ml) was used for the extraction.
- the organic layer was washed with water (2x20 ml), and then concentrated to afford the crude product of diascorbyl diphosphate ester of androst-5-ene-3 ⁇ ,17 ⁇ -diol (3.0 g).
- the crude diascorbyl diphosphate ester of androst-5-ene-3 ⁇ ,17 ⁇ -diol (400 mg) was dissolved in methanol (5 ml). To this solution was added 2 ml of sodium methoxide in methanol (20%, w/v) under magnetic stirring. White precipitate was observed upon the addition of sodium methoxide methanol solution. The suspension was stirred for half an hour before it was filtered and washed with methanol and acetone. The solid product was dried under high vacuum, and tetrasodium diascorbyl diphosphate ester of androst- 5-ene-3 ⁇ ,17 ⁇ -diol (330 mg) was obtained.
- EXAMPLE 8 Measuring MDR-1 Expression in CaCo2 cells after treatment with FM- VP4 or Cholesterol for one week
- Confluent CaCo2 cells (P33-39) in T-75 flasks were treated with varying amounts of FM-VP4 (0.5um, LOurn, 5.0um and 10.0uM) or cholesterol (0.5um, LOum, 5.0um, 10.0uM, 25uM) for one week. The media and treatment were changed every other day. After seven days, cells were harvested and total RNA isolated with TRIZOLTM. Analysis conducted by reverse transcription ⁇ cDNA using polymerase chain reaction.
- FM-VP4 0.5um, LOurn, 5.0um and 10.0uM
- cholesterol 0.5um, LOum, 5.0um, 10.0uM, 25uM
- PCR Program 94°C for 5 minutes, 94°C for 1 minute, 55°C for 1 minutes, 72°C for I minute 30 seconds, repeat from step 2 for 29 more times, 72°C for 10 minutes, 4°C.
- Primer drop at cycle 0,6,9,12,16,20,23,26,28 and 30.
- Figure 1 is a graph showing the results, from which it is clear that with increasing concentrations of the cholesterol absorption inhibitor, MDR-1 gene expression is significantly reduced. Further supporting these results are shown in Figure 2, a graph showing the titration for primer drop of GAPDH and Figure 3 depicting polymerase chain reaction (1.5% agarose gel) electrophoretic plate results for MDR-1 and GAPDH.
- Figures 10 and 11 indicate that, at increasing concentrations of cholesterol, a point of saturation is reached wherein in response, the expression of MDR-1 decreases. Including this data is important as it is suspected that cholesterol is taken up by cells by a mechanism involving the gene product of MDR-1.
- Example 8 The entire protocol as provided in Example 8 with respect to the MDR-1 gene was used to confirm the effects of a cholesterol absorption inhibitor on the expression of another multiple drug resistance gene: MRP-1.
- MTS-Assay is a colorimetric method for determining the number of viable cells (cytotoxicity) after treatment.
- MTS reagent a tetrazolium salt
- a color compound formazan
- BCA Protein Assay This particular colorimetric assay measures total protein levels by identifying specific peptide bonds. The protein concentration is calculated from a calibration curve constructed with a protein standard (bovine serum albumin or "BSA").
- the LDH-assay looks at the integrity of cell membranes and can be used for cytotoxicity mediated by chemicals or other agents.
- Cell damage is associated with leakage of intracellular, cytoplasmic contents and lactate dehydrogenase or "LDH" (a stable cytosolic enzyme), can be used as a reported molecule for this event.
- LDH lactate dehydrogenase
- LDH released from cells into the culture medium, was measured using a kit (Cytotox96 Non- Radioactive Cytotoxicity Assay) from PromegaTM. This method is based on a series of linked enzyme reactions, the final reaction being the reduction of a tetrazolium salt to a coloured, insoluble, formazen product which can be measured at 492nm. Background absorbance from media alone and media including the treatment was substracted from the reading to correct the values.
- Figure 4 is a bar graph of the MTS- and LDH-Assay of cell viability after treatment of CaCo2 with "FM-VP4".
- Figure 5 a bar graph showing a BCA-Assay of protein concentration after treatment of CaCo2 with "FM-VP4".
- the cholesterol absorption inhibitor tested, FM-VP4 does not show any toxicity (mitochondrial activity) for a concentration range up to 100mM regardless of whether the treatment was for 24 hours (cell viability is 90.9 +/- 9.8%) or 96 hours (94.6 +/- 4.4%). At 250mM for 24 hour treatment it drops down to 59.2+/- 8.1% and to 19.3+/-7.6% for 96 hours.
- the highest tested concentration of FM-VP4 75mM showed a cell viability of 18.5+/-5.8% after 24 hours and 22.1+/-1.6%.
- EXAMPLE 11 Western Blot analysis of P-glycoprotein in Caco2-cells after incubation with FM-VP4 ( ⁇ Protein expression)
- FIG. 9 is a Western Blot analysis of P-glycoprotein in CaCo2 cells after incubation with the selected cholesterol absorption inhibitor: "FM-VP4".
- the objective was to confirm the influence of a cholesterol-free liposomal formulation of a cholesterol absorption inhibitor ("FM-VP4") on gene expression of a multiple drug resistance gene.
- FM-VP4 cholesterol absorption inhibitor
- FM-VP4 liposomes were utilized at three different concentrations (2.5, 5 and 10 ⁇ M FM-VP4).
- FIG. 7 depicts a polymerase chain reaction (1.5% agarose gel) electrophoresis results for MDR-1, GAPDH; RNA isolation with TRIZOL ⁇ RT-PCR ⁇ PCR after treatment of CaCo2 cells with liposomal formulations of one of cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4;
- Figure 8 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR- 1/GAPDH) in CaCo2 cells after treatment for one week with liposomal FM-VP4 at 2.5, 5 and 10um as compared to a control and empty liposomes.
- MDR-1 gene expression in Caco2-cells after treatment with liposomal FM- VP4 at lower concentrations is significantly reduced and at lower concentrations than non-liposomal formulations.
- the dry lipid film was stored by capping it and placing it in the freezer.
- the water bath was set to 55°C. 2. Warmed up some HBS or selected cholesterol absorption inhibitor (ex: FM-VP4) dissolved in HBS in the water bath.
- FM-VP4 selected cholesterol absorption inhibitor
- Examples 14 through 20 look at mdr-1 gene and P-glycoprotein expression in Caco-2 cells, human intestinal carcinoma cells, after administration with FM-VP4. It was demonstrated that changes in the mdr-1 gene expression levels lead to changes in the P-glycoprotein. These changes then have an influence on the functionality of P-gp. Accordingly, within the scope of this invention, we have found a different unique way of inactivating P-gp by down-regulation of mdr-1 mRNA. Down-regulation of mdr-1 A & B could also be shown in the rat model. We also show that cholesterol can be transported through an active influx mechanism via P-gp. EXAMPLE 14- MDR-1 and P-Glycoprotein Expression in CaCo-2 cells, human intestinal carcinoma cells, after administration of compounds in accordance with present invention
- Caco-2 cells and LLC-PKi (Pig kidney cells) were purchased from American Type Culture Collection (ATCC) (Rockville, MD and Wanassas, VA, USA). Cell culture media were purchased from Gibco BRL (Grand Island, NY, USA). Sterile steritop 0.22 ⁇ m express membrane bottle top filters were purchased from Millipore (Bedford, MA, USA). Sterile 50ml centrifuge tubes, disposable 10 and 25ml serological pipettes were purchased from Starstedt (Montreal, PQ, Canada). Culture flasks and plates were obtained from Corning-Costar (Cambridge, MA). Chemicals like Triton X-100, Verapamil, Rhodamine 123 were purchased from SIGMA.
- TRIzol® reagent Agarose, 10xTBE, RNAse/DNAse free water were purchased from Invitrogen (Vancouver, BC).
- the monoclonal antibody C219 was from Signet Pathology System Dedlam, antimouse IgG rabbit HRP-conjugated antibody from Jackson Immuno Research Laboratories Inc. (West Grove, PA, USA).
- Caco-2 cells were cultured in Dulbecco's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 292 ⁇ g/mL glutamine, 0.1 mM non-essential amino acids, 100U/mL penicillin and 100 ⁇ g/mL streptomycin at 37°C in humidified air containing 5% CO 2 in either 96 well plates or T75 flasks depending on the type of experiment. Media was changed every other day. Cells were used for experiments when they reached between 80 to 90% confluency.
- DMEM Dulbecco's minimal essential medium
- FBS heat-inactivated fetal bovine serum
- FBS heat-inactivated fetal bovine serum
- 292 ⁇ g/mL glutamine 292 ⁇ g/mL glutamine
- 0.1 mM non-essential amino acids 100U/mL penicillin and 100 ⁇ g/mL streptomycin
- Cytotoxicity For cytotoxicity experiments cells were incuabated in media or treated with 0.1% Triton X-100 (as a positive control for toxicity), a cholesterol absorption inhibitor "FM-VP4" or cholesterol micelles and incubated for 24 and 96 hours. Following, LDH- and MTS-assay were performed to determine the integrity of cell membranes and mitochondrial respiration respectively indicating the cytotoxicity and cell viability post treatment as well as BCA-Assays to determine the protein concentration of the surviving cells. After the incubation period 50 ⁇ l from each well were transferred into a new 96-well plate to perform the LDH-Assay. The MTS-Assay (CellTiter 96® AQueous One Solution kit) was done on the original plate.
- MTS reagent a tetrazolium salt
- a color compound formazan
- PBS PBS
- BCA Protein Assay Total protein levels can be measured in a colorimetric assay that identifies specific peptide bonds.
- the protein concentration is calculated from a calibration curve constructed with a protein standard (bovine serum albumin). The LDH-assay looks at the integrity of cell membranes.
- Lactate dehydrogenase released from cells into the culture medium is measured using a CytoTox96 kit from Promega. This method is based on a series of linked enzyme reactions, the final reaction being the reduction of a tetrazolium salt to a coloured, insoluble, formazen product which can be measured at 492nm.
- RNA-lsolation from Caco-2 cells Total RNA from Caco-2 cells was isolated using TRIzol® Reagent according to the manufactures instruction.
- RNA-isolation from rat tissues RNA-isolation from rat tissues:
- the rat tissues were kept at -80°C.
- RNA isolation an RNeasy Mini Kit from Qiagen (74104) was used.
- a slice of the tissue was weighted and lysate buffer was added.
- a Rotor-stator homogenizer has been used to disrupt and simultaneously homogenize the tissue.
- the tissue lysate was centrifuged for 3 minutes at maximum speed and the supernatant carefully transferred into a new 2ml tube. 600 ⁇ l of 70% ethanol were added to the cleared lysate and mixed by pipetting. Up to 700 ⁇ l of the sample were then applied to an RNeasy mini column placed in a 2 ml collection tube. It was then centrifuged for 15 sec at 10,000 rpm and the flow-through discarded by using a pipette to avoid spills on the tube.
- washing buffer 700 ⁇ l was added, the tube was again centrifuged for 15 sec at 10,000 rpm and the flow-through discarded.
- a second washing step is performed by adding 500 ⁇ l of washing buffer 2 and centrifugation for 15 sec at 10,000 rpm. The last step was repeated and the tube was centrifuged for 2 min to dry the RNeasy silica-gel ⁇ p ⁇ v ⁇ . To elute the RNA 50 ⁇ l RNase-free water was directly added onto the RNeasy silica-gel membrane and then centrifuged for 1 min at 10,000 rpm.
- RNA samples that fulfilled the criteria of purity were used for RT-PCR.
- Two to four microgram purified RNA was treated with DNAse I to remove residual DNA and then used for first strand cDNA synthesis with Superscript II, oligo (dT) ⁇ 2- i 8 random primers according to the manufacturer's instruction (Invitrogen).
- the concentration of cDNA was determined with Oligreen Oligonucleotide Quantitation Reagent (Molecular Probe). PCR reactions were performed with the specific primers. All primers were synthesized at the Oligonucleotide Synthesis Laboratory at UBC.
- Caco-2 cells were grown in T-75 flasks and treated regarding the type of experiment. Procedures for protein extraction were followed as directed by the instruction manual for MEM-PER® Eukaryotic Membrane Protein Extraction Reagent kit by Pierce Biotechnology (Rockford, IL, USA). Briefly, cells were rinsed 3 times with 10ml of 1X PBS. Cells were washed with PBS and harvested into microcentrifuge tubes to pellet down. The supernatant was carefully removed and discarded. About 150 ⁇ l of Reagent A was added to the cell pellet and pipetted up and down in order to obtain a homogenous cell suspension. At this stage, 5 ⁇ l of protease inhibitor was added to the mixture and vortexed.
- BCA Bicinchoninic Acid
- the MINI-PROTEAN 3 Cell apparatus with glass plates and power supply purchased from Biorad (Hercules, CA, USA) was used to run the SDS-PAGE (3.5% stacking, 7.5% resolving).
- 1x transfer buffer prior to use The unit was run at settings of constant 70 V for approximately 2 hours and 30 to 50 V for 2 to 4 hours. After the run, the membrane was stained with Ponceau S solution as a control for proper transfer. The membrane was incubated in blocking buffer (1x PBS, 1% nonfat dried milk, 0.1% Tween-20) overnight at 4°C. The blocking buffer was discarded and the membrane was incubated at 4°C overnight with the primary antibody for P-gp (C219) at a dilution 1:300. The membrane was washed 3 times with blocking buffer and incubated for 1 hour in the 2° antibody solution (1: 2000 -3000 anti-mouse IgG rabbit HRP-conjugated antibody ). The membrane was then washed 3 times with blocking buffer and once with 1x PBS and
- the objective of the first part of the study was to determine the limiting and toxic concentrations of Cholesterol and cholesterol absorption inhibitor FM-VP4 in a Caco2- cell system as well as in LLC-PKi cells (Pig kidney cells).
- An in vitro cytotoxicity assay which looks at several aspects was developed.
- One advantage is that it can be carried out in one model system and results in 3 different parameters.
- MTS-, LDH- and BCA- Assays were used to measure cellular metabolic activity, membrane integrity and protein concentrations respectively.
- the aim of this study was to analyze the influence of cholesterol absorption inhibitor
- FM-VP4 on the gene expression level of mdr-1 in Caco-2 cells.
- the primers were synthesized at the Oligonucleotide Synthesis Laboratory at UBC. Parameters and conditions for the tested primers were optimized. A sample from each PCR product was subjected to electrophoresis on a 1.5% agarose gel (containing Ethidium bromide). A 10O bp ladder was used to identify the size of PCR products. The fluorescent bands were imaged under UV light (UV-Epi Chemi II) and quantified with UVP-labworks software.
- the results of Figure 16 are a summary of about 10 repeats for concentrations of 25 and 50 ⁇ M, 11 repeats for 10 ⁇ M FM-VP4 and 12 repeats for Control. A significant decrease is observed for 5 and 10 ⁇ M FM-VP4. At 10 ⁇ M FM-VP4 mdr-1 is significantly down-regulated to 38.5 ⁇ 17%, at 5 ⁇ M FM-VP4 to 61.2 ⁇ 25.1% both compared to control (100 ⁇ 30%).
- the purpose of the study was to determine the influence of liposomal FM-VP4 in a cholesterol-free liposomal preparation on gene expression of ABC-transporter especially mdr-1. Down-regulation of mdr-1 after treatment with 10 ⁇ M FM-VP4 had been shown in previous experiments. For those a dilution of FM-VP4 in water was applied to Caco-2 cells. Of interest following these observations was the influence of liposomal FM-VP4 in cholesterol-free liposomal preparation (DMPC 100:0, ratio 10:1 DMPC:FM-VP4). FM-VP4 liposomes with three different concentrations (2.5, 5 and 10 ⁇ M FM-VP4) were tested.
- Caco-2 cells were seeded at 10,000 cells/cm 2 in T-75 flasks (Corning). The growth media was changed every other day. The cells were kept at 37°C in a humidified atmosphere of 5% CO 2 .
- the treatment groups contained 2.5, 5 and 10 ⁇ M cholesterol-free liposomal FM-VP4 as well as empty cholesterol-free liposomes without FM-VP4. Cells were harvested after one week of treatment, total RNA isolated with TRIzol ® according to the protocol. RT-PCR followed that step and specific primers were used to evaluate the expression level of mdr-1. PCR products were resolved by electrophoresis on a 1.5% Agarose gel (containing Ethidiumbromide) and the fluorescent bands were imaged under UV light and quantified using the UVP-Labworks software.
- the aim of this study was to analyze the influence of Cholesterol micelles on the gene expression level of mdr-1 in Caco-2 cells.
- EXAMPLE 18 rotein expression of P-glycoprotein in Caco-2 cells after incubation with FM-VP4 for 1 week
- the aim of the experiment was to analyze the protein expression of P-gp (which is encoded by mdr-1) after incubation with FM-VP4 as well as Cholesterol over a period of 7 days. Both gene expression and protein level were determined in the same experiment.
- Caco-2 cells were seeded at 10,000 cells/cm 2 in T-75 flasks (Corning). The growth media was changed every 2-3 days. The cells were kept at 37°C in a humidified atmosphere of 5% CO 2 . Treatment was added and done in duplicates (one flask for RNA-isolation and one for protein isolation). Cells from group I were harvested and total cellular RNA was extracted with TRIzol ® according the protocol. RNA was reverse transcripted into cDNA and specific primers were used to evaluate the expression level of mdr-1. Internal control was run by using GAPDH (primer drop). A sample from each PCR product was subjected to electrophoresis on a 1.5% Agarose gel (containing Ethidiumbromide).
- a 100 bp standard was run to identify the size of the product.
- the fluorescent bands were imaged under UV light (UVP-Epi Chemi II Darkroom) and quantified with the UVP-Labworks software.
- Cells from group II underwent a protein extraction procedure. Concentration was measured using the PIERCE BCA Protein Assay Reagent Kit. 15 ⁇ g total protein from each sample was electrophoresed on a 11 % SDS Page Gel and transferred to a PVDF membrane (wet transfer). Identification of P-gp was done using the immunoblot technique.
- the monoclonal antibody used for P-gp was C219 and the secondary was horseradish peroxide rabbit anti-mouse antibody (Jackson Immuno Research Laboratories, Inc.).
- the best dilutions for the antibody solution turned out to be 1:300 for the primary and 1 :2000 for the secondary.
- the protein bands were visualized using a chemiluminescence kit from PerkinElmer and an X-Ray film was placed on the membranes. The film was exposed for about 1 minute and then developed.
- FIG 19 shows that a seven day incubation with FM-VP4 10 ⁇ M leads to a lesser expression of P-gp. Down-regulation of mdr-1 gene expression was already shown and the open question was if this might influence its protein level as well.
- Western Blot experiments could show that mdr-1 gene expression and P-gp protein level are influenced by FM-VP4 and correlated. A lower level of P-Glycoprotein which is encoded by mdr-1 was detectable. Whereas a higher concentration of FM-VP4 (50 ⁇ M) causes no obvious effect on mdr-1 gene.
- Western Blots also show no significant change in protein expression compared to untreated Control cells. These data show that that changes in the mdr-1 gene expression lead to correlating changes in the protein level
- Rhodamine 6G is another fluorescent probe for P-gp.
- the apparent permeability P app can be determined by using the following equation:
- Rhodamine was added into apical chamber. Rhodamine samples from basolateral chamber were analyzed by measuring fluorescence A (Secretory Transport):
- Rhodamine was added into basolateral chamber
- Rhodamine samples from apical chamber were analyzed by measuring fluorescence The above is a schematic of absorptive and secretory transport studies. Rhodamine 123 is either added into the apical or basolateral chamber. Samples are taken out of the receiver side.
- Caco-2 cells were seeded in polycarbonate membrane Transwell plates (Corning, Costar Corp.) at a densitiy of 40.000 cells/cm 2 and grown in a humidified chamber (at 37°C, of 5% CO 2 .) with media changes every 2 days.
- the growth media (Dulbecco's minimal essential medium-DMEM) contained 10% heat- activated fetal bovine serum, 292 ⁇ g/ml glutamine, 0.1 mM non-essential amino acids, 100U/ml penicillin and 100mg/ml glutamine.
- FM-VP4 in different concentrations was added to the media and either applied onto apical and/or the basolateral side of the Transwell plate.
- Transepithelial electrical resistance (TEER) of the monolayers was measured to confirm monolayer integrity. Caco-2 cells with TEER Values above 400 ⁇ /cm 2 were then used for transport studies.
- Transwell Inserts were transferred into a new sterile 12-well plate containing media, media with FM-VP4 or the P-gp inhibitor Verapamil or C219 and all contained 5 ⁇ M Rhodamine 123.
- samples 50 ⁇ l were taken out of the apical chamber, transferred into a 96-well plate and the fluorescence was measured. The excitation wavelength is 485nm, emission wavelength 520nm. Immediately the samples were replaced by fresh receiver medium (media, media plus FM-VP4 or media plus P-gp inhibitor).
- FM-VP4 has an influence on the expression of mdr-1.
- a decrease of the m-RNA level lead to a decrease in P-glycoprotein expression.
- a lower P-gp activity can then be observed.
- Less activity of P-gp was demonstrated by using Rhodamine 123 and Rhodamine 6G which are common substrates for P-gp.
- Goal was to investigate mdr-1 A and mdr-1 B gene expression in rats after one week treatment with cholesterol absorption inhibitor FM-VP4.
- FM-VP4 influences the gene expression of mdr-1 in Caco-2 cells.
- Ten ⁇ M FM-VP4 show a significant decrease in mRNA levels of mdr-1 (38 ⁇ 17% versus 100% for untreated control cells).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides, in one aspect, a method of inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor. In another aspect, it provides a method of inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
Description
TITLE: A METHOD OF INHIBITING THE EXPRESSION OF GENES WHICH
MEDIATE CELLULAR CHOLESTEROL INFLUX IN ANIMAL CELLS AND INHIBITING THE PRODUCTION OF PROTEINS RESULTING FROM THE EXPRESSION OF SUCH GENES
FIELD OF THE INVENTION
This present invention relates to the field of focused therapies useful in treating the underlying causes of hypercholesterolemia.
BACKGROUND OF THE INVENTION
While recent advances in science and technology are helping to improve quality and add years to human life, the prevention of atherosclerosis, the underlying cause of cardiovascular disease ("CVD") has not been sufficiently addressed. In fact, cardiovascular diseases account for more deaths annually than any other disease, including all forms of cancer combined1. In the USA alone, more than one million heart attacks occur each year and more than half a million people die as a result. This enormous toll has necessitated continued research to determine the causes of CVD and means by which it can be prevented and treated.
The primary cause of CVD is atherosclerosis, a disease characterized by the deposition of lipids, including cholesterol, in the arterial vessel wall resulting in a narrowing of the vessel passages and ultimately a hardening of the vascular system. Atherosclerosis is a degenerative process resulting from an interplay of inherited (genetic) factors and environmental factors such as diet and lifestyle. Research to date suggest that cholesterol may play a role in atherosclerosis by forming atherosclerotic plaques in blood vessels, ultimately cutting off blood supply to the heart muscle or alternatively to the brain or limbs, depending on the location of the plaque in the arterial tree 1'2. A total cholesterol in excess of 225-250 mg/dl is associated with significantly elevated risk of CVD, including vascular disease. Overviews have indicated that a 1% reduction in a person's total serum cholesterol yields a 2% reduction in risk of a coronary artery event4. Statistically, a 10%
decrease in average serum cholesterol (e.g. from 6.0 mmol/L to 5.3 mmol/L) may result in the prevention of 100,000 deaths in the United States annually5.
Cholesteryl esters are a major component of atherosclerotic lesions and the major storage form of cholesterol in arterial wall cells. Formation of cholesteryl esters is also a step in the intestinal absorption of dietary cholesterol through homeostatic control mechanisms. These control mechanisms involve the inter-related regulation of dietary cholesterol, cholesterol biosynthesis and catabolism of cholesterol-containing plasma lipoproteins. Cholesterol biosynthesis and catabolism occur primarily in the liver and hence, it is a prime determinant of plasma cholesterol levels.
Lipoproteins are complexes of lipids and proteins held together by non-covalent bonds. Each type of lipoprotein class has a characteristic mass, chemical composition, density and physiological role. Irrespective of density or particle size, circulating lipids consist of a core of cholesteryl esters and triglycerides, and an envelope of phospholipids, free cholesterol and apolipoproteins. The apolipoproteins are involved in the assembly and secretion of the lipoprotein, provide structural integrity, activate lipoprotein-modifying enzymes, and are the ligand for a large assortment of receptors and membrane proteins. Lipoprotein classes found in plasma include HDL, LDL, intermediate density lipoproteins (IDL) and very low density lipoproteins (VLDL).
Each type of lipoprotein has a characteristic apolipoprotein composition or ratio. The most prominent apolipoprotein in HDL is apolipoprotein-AI (apo-AI), which accounts for approximately 70% of the protein mass, with apo-AII accounting for another 20%. The ratio of apoA-l to apoA-ll may determine HDL functional and anti-atherogenic properties. Circulating HDL particles consist of a heterogeneous mixture of discoidal and spherical particles with a mass of 200 to 400 kilo-daltons and a diameter of 7 to 10 nm.
HDL is one of the major classes of lipoproteins that function in the transport of lipids in plasma, and has multiple functions within the body, including reverse cholesterol transport, providing the cholesterol molecule substrate for bile acid synthesis, transport of clusterin, transport of paraoxanase, prevention of lipoprotein oxidation and selective uptake of cholesterol by adrenal cells. The major lipids associated with HDL include cholesterol, cholesteryl ester, triglycerides, phospholipids and fatty acids. HDL is anti- atherogenic.
The atherosclerotic process begins when LDL becomes trapped within the vascular wall. Oxidation of this LDL results in the binding of monocvtes to the endothelial cells lining the vessel wall. These monocytes are activated and migrate into the endothelial space where they are transformed into macrophages, leading to further oxidation of the LDL. The oxidized LDL is taken up through the scavenger receptor on the macrophage, leading to the formation of foam cells. A fibrous cap is generated through the proliferation and migration of arterial smooth muscle cells, thus creating an atherosclerotic plaque.
HDL is essential for the transport of cholesterol from extra-hepatic tissues to the liver, where it is excreted into bile as free cholesterol or as bile acids that are formed from cholesterol. The process requires several steps. The first is the formation of nascent or pre-beta HDL particles in the liver and intestine. Excess cholesterol moves across cell membranes into the nascent HDL through the action of the ABCA transporter. Lecithin cholesterol acyl transferase (LCAT) converts the cholesterol to cholesteryl ester and the subsequent conversion of nascent HDL to mature HDL. Esterified cholesterol is then transferred by cholesteryl ester transfer protein (CETP) from HDL to apolipoprotein-B containing lipoproteins, which are taken up by numerous receptors in the liver. Nascent HDL is regenerated via hepatic triglyceride lipase and phospholipid transfer protein and the cycle continues. In addition to the cholesterol removed from peripheral cells, HDL accepts cholesterol from LDL and erythrocyte membranes. Another mechanism of
reverse cholesterol transport may involve passive diffusion of cholesterol between cholesterol-poor membranes and HDL or other acceptor molecules.
HDL protects against the development of atherosclerosis both through its role in reverse cholesterol transport and possibly by impeding LDL oxidation. Several HDL- associated enzymes are involved in the process. Paroxonase (PON1), LCAT, and platelet activating factor acetylhydrolase (PAFAH) all participate by hydrolyzing phospholipid hydroperoxides generated during LDL oxidation and act in tandem to prevent the accumulation of oxidized lipid in LDL. These enzymes are responsible for the anti-oxidative and anti-inflammatory properties of HDL. Studies have shown that a low plasma concentration of HDL cholesterol is a significant risk factor for the development of atherosclerosis and that high levels are protective.
The liver is the major organ responsible for synthesis and secretion of VLDLs, which, as noted above, are metabolized to LDL in circulation. LDLs are the predominant cholesterol carrying lipoproteins in plasma and hence an increase in their concentration is directly correlated with atherosclerosis. Simply put, when intestinal cholesterol absorption is reduced, by any means, less cholesterol is delivered to the liver. As a result, VLDL production is reduced and there is a concomitant increase in hepatic clearance of plasma cholesterol, mostly in the form of LDL.
Accordingly, cholesterol acts on three different levels to regulate its own synthesis. Firstly, it suppresses endogenous cholesterol synthesis by inhibiting the enzyme HMG CoA reductase. Secondly, it activates LCAT. Thirdly, it regulates the synthesis of the LDL- receptor ensuring that a cell already having a sufficient amount of cholesterol will not take up additional cholesterol.
The transport of specific molecules across lipid membranes is an essential function of all living organisms and a large number of specific transporters have evolved to carry
out this function. The largest transporter gene family s the ATP-binding cassette (ABC) transporter superfamily. These proteins translocate a wide variety of substrates including sugars, amino acids, metal ions, peptides, and proteins, and a large number of hydrophobic compounds and metabolites across extra- and intracellular membranes. ABC genes are essential for many processes in the cell, and mutations in these genes cause or contribute to several human genetic disorders including cystic fibrosis, neurological disease, retinal degeneration, cholesterol and bile transport defects, anemia, and drug response.
To date, two genes encoding multidrug-resistance export proteins have been identified in the human genome. The first of these, MDR1 , encodes P-glycoprotein, a 170 kDa multispanning transmembrane protein belonging to the above-mentioned ABC superfamily2. It is suggested that one mechanism by which tumor cells acquire resistance to drugs is by over expression of P-glycoprotein (also referred to as "P-gp" or "Pgp"))6,7'8,9
P-glycoprotein likely acts by rapidly pumping hydrophobic chemotherapeutic agents out of the tumor cells, thereby decreasing intracellular accumulation of certain chemotherapeutic agents below their cytostatic concentrations. One in vitro solution is to increase the chemo drug concentration. However, since cancer chemotherapeutic agents are already administered at their maximally tolerated range in vivo, increasing the doses is an unacceptable solution leading, on most cases to extreme toxicity10,11.
P-glycoprotein is structurally similar to the cystic fibrosis transporter protein, the major histocompatibility complex-linked peptide transporter, and a non-P-glycoprotein-related multidrug resistance protein (MRP)12,13,14,15. P-glycoprotein is expressed in diverse sites including the normal human adrenal cortex, the luminal aspect of bile canaliculi and colonic epithelium, the renal tubular epithelium and the endothelial cell of the blood-brain and blood testicular barriers. The function of the P-glycoproteins at these sites is unclear but appears to function as an energy dependent pump of broad specificity possibly
related to secretion of hormones and protection against toxins. Expression of the P- glycoprotein can actively efflux a large number of hydrophobic, and heterocyclic cancer chemotherapeutic agents including adriamycin (doxorubicin), colchicine, colcemid, etoposide, paclitaxel, vincristine, vinblastine as well as others.
P-glycoproteins are encoded by a highly conserved family of genes16 The MDR-1 gene encodes class I P-glycoprotein that confers multidrug resistance in humans17. The pgp-1 and pgp-2 genes in hamsters and the mdr-3 and MDR-1 genes in mice encode the class and II proteins, both of which confer multidrug resistance in rodents18.
Several lines of evidence support cause-effect association between increased P- glycoprotein and multidrug resistance in vitro. Structural features of the protein are characteristic of an energy-dependent efflux pump19. Over expression of the protein is associated with multidrug resistance20. There has also been discovered a positive correlation between the degree of expression of P-glycoprotein and a drug-resistant phenotype21.
In addition to the role of such genes in cancer drug or chemotherapy resistance, there is now evidence suggesting that p-glycoprotein, perhaps through MDR-1 expression, modifies cholesterol cellular uptake and disposition. A number of these studies are summarized below:
Results from Garrigues et aP2 show that P-glycoprotein mediates the flux of cholesterol from the cytosolic leaflet to the exoplasmic leaflet suggesting that P-glycoprotein may be involved in the cholesterol enrichment of the exoplasmic leaflet of plasma membrane at the level of rafts and caveolae. Further, cholesterol redistribution within plasma membrane induced by P-glycoprotein may affect cellular cholesterol trafficking, which involves both the endogenous biosynthesis and esterification of cholesterol in the endoplasmic reticulum, the import of exogenous cholesterol from the LDL by
endocytosis, and the export of cholesterol to HDL. In addition, it was suggested that the enrichment of cholesterol in the exoplasmic leaflet of the plasma membrane may facilitate the efflux of cholesterol out of the cell to the HDL, possibly in cooperation with ABCA1.
In Tessner and Stenson23, MDR1 -transfected IEC-18 rat intestinal epithelial cells exhibited increased expression of MDR1 protein, reduced accumulation of vinblastine and increased uptake of [3H]cholesterol from cholesterol/monolein/taurochoiate micelles. Verapamil, an inhibitor of MDR1 and UIC2, an antibody against MDR1 , both diminished cholesterol absorption in these cells, but did not completely inhibit cholesterol cellular uptake. It should be noted that most MDR1 inhibitors, like verapamil, interfere with the ability of MDR1 to extrude substances from the cell by acting as competitive inhibitors; they do not inactivate the protein but act as competing substances for the xenobiotic whose accumulation is being evaluated. UIC2, is a monoclonal antibody against an external epitope of the protein.
Wang et al.24 reported that increasing cholesterol concentrations dramatically inhibits active transport of the P-glycoprotein marker substrate daunorubicin yet does not effect rhodamine 123 transport. The authors also reported a cholesterol concentration- dependent hydrolysis of ATP, the required energy source for P-gp function. Furthermore, it was found that cholesterol supplementation did not impact rhodamine 123 transport suggesting that cholesterol does not significantly affect the permeability or membrane/protein interface of P-glycoprotein. Accordingly, cholesterol appears to have a direct effect on the substrate-binding site of P-glycoprotein, a result consistent with cholesterol being transported by MDR1 P-glycoprotein.
Luker (2001)25 describe the effects of MDR-1a and MDR-1b deficiency on absorption and distribution of cholesterol administered orally and intravenously to mice fed a standard chow diet. Data showed that intestinal absorption of cholesterol was not affected by the
absence of class I P-glycoprotein. However in the liver, P-glycoprotein deficiency enhanced the kinetics of cholesterol esterification following oral dosing but not after IV dosing.
Luker (1999)26 reported that in NIH 3T3 fibroblasts, P-glycoprotein increases cholesterol esterification by facilitating cholesterol movement from the plasma membrane to the ER. It was suggested that the effects of P-glycoprotein on transport were not simply the result of the general expression of an ABC transporter because esterification was not increased in fibroblasts transfected with MRP. Furthermore, the effects on cholesterol trafficking occurred without differences in plasma membrane cholesterol as measured using cholesterol oxidase with or without glutaraldehyde fixation.
In Field et al.27, it was found that progesterone, trifluoperazine and verapamil (inhibitors of P-glycoprotein) interfered with the transport of cholesterol and the secretion of apoB, apoA-l and lipids. However, methotrexate, which does not inhibit P-glycoprotein did not alter cholesterol trafficking and/or lipoprotein secretion. Thus it is postulated that P- glycoprotein or a member of the this family may be involved in cholesterol trafficking and lipoprotein secretion in intestinal cells. P-glycoprotein may function to maintain the acidification of endosomes by acting as a proton pump and chloride channel.
It is an object of the present invention to create new modalities for addressing cholesterol homeostasis, at the cellular level.
SUMMARY OF THE INVENTION
The present invention provides, in one aspect, a method of decreasing or inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
The present invention provides, in another aspect, a method of decreasing or inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
The present invention provides, in yet another aspect, a method of decreasing the level of serum LDL cholesterol in an animal, by inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
The present invention provides, in yet another aspect, a method of decreasing the level of serum LDL cholesterol in an animal, by inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in the animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
The crux of the present invention is the provision and administration of compounds which inhibit the protein mediated uptake of cellular cholesterol through the lowering of specific gene expression and activity.
More specifically, it has been found that cholesterol absorption inhibitors effectively inhibit the expression of multi-drug resistance genes and/or inhibit the uptake of cellular cholesterol which is mediated by the proteins, specifically P-glycoprotein, which are produced by such genes. The results presented herein suggest that the reduced production of proteins expressed by these genes prevents the influx of cholesterol into cells.
In addition, some the preferred cholesterol absorption inhibitors of the present invention (those depicted below including those in formulae (i) through (iv) comprise an ascorbyl moiety. These particular compounds have numerous additional advantages. In particular, solubility in aqueous solutions such as water is improved by the ascorbyl moiety thereby allowing oral administration perse. Likewise, other modes of administration are facilitated. Accordingly, these selected compounds of the present invention can be prepared and used as such or they can be easily incorporated into pharmaceutical preparations, optionally in conjunction with other therapeutic agents, regardless of whether such preparations are water-based. This enhanced solubility generally translates into lower administration dosages of the compounds in order to achieve the desired therapeutic effect.
These effects and other significant advantages will become apparent herein below.
BRIEF DESCRIPTION OF THE DRAWINGS
The present invention is illustrated by way the following non-limiting drawings in which:
Figure 1 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR- 1/GAPDH; "GAPDH" or glyceraldehyde-3-phosphate dehydrogenase) in CaCo2 cells after treatment for one week with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
Figure 2 is a graph showing the titration for primer drop of GAPDH;
Figure 3 depicts a polymerase chain reaction (1.5% agarose gel) gel electrophoresis results for MDR-1 , GAPDH;
Figure 4 is a bar graph showing an MTS- and LDH-Assay of cell viability after treatment of
CaCo2 cells with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
Figure 5 a bar graph showing a BCA-Assay of protein concentration after treatment of CaCo2 with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
Figure 6 a bar graph showing the level of ABCC1 (MRP-1) expression (normalized ratio of MRP-1/GAPDH) in CaCo2 cells after treatment for one week with one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM- VP4";
Figure 7 depicts a polymerase chain reaction (1.5% agarose gel) gel electrophoresis results for MDR-1 , GAPDH; RNA isolation with TRIZOL→ RT-PCR→ PCR after treatment of CaCo2 cells with liposomal formulations of one of cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4;
Figure 8 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR- 1 /GAPDH) in CaCo2 cells after treatment for one week with one of the cholesterol absorption inhibitors described herein: a liposomal formulation of an ascorbyl stanyl phosphate ester called "FM-VP4" at 2.5, 5 and 10um as compared to a control and empty liposomes;
Figure 9 is a Western Blot analysis of P-glycoprotein in CaCo2 cells after incubation with one of cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4";
Figure 10 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR-1/GAPDH; "GAPDH" or glyceraldehyde-3-phosphate dehydrogenase) in CaCo2
cells after treatment for one week with varying concentrations of cholesterol;
Figure 11 depicts a polymerase chain reaction (1.5% agarose gel) gel electrophoresis results for MDR-1, GAPDH; and
Figure 12 is a bar graph showing cell viability and cytotoxicity after treatment with Cholesterol and effect on Caco-2 cells; MTS-Assay and LDH-Assay after incubation with Cholesterol for 24 hours. Each bar represents the mean ± SD of four experiments;
Figure 13 is a bar graph showing cell viability and cytotoxicity after treatment with FM- VP4. Effect on Caco-2 cells. MTS-Assay and LDH-Assay after incubation with FM-VP4 for 24 hours. Each bar represents the mean ± SD of four experiments;
Figure 14 is a bar graph showing cell viability after treatment with FM-VP4. Effect on LLC-PKi cells. Each bar represents the mean ± SD of three experiments;
Figure 15 is a bar graph showing cell viability after treatment with cholesterol micelles. Effect on LLC-PKi cells. Each bar represents the mean ± SD of three experiments;
Figure 16 shows the expression profile of mdr-1 gene in Caco-2 cells was examined as an effect of 1 week treatment with FM-VP4. A sample from each PCR product was subjected to electrophoresis on a 1.5% agarose gel (A). The fluorescent bands were imaged under UV light (UV-Epi Chem II) and quantified with the UVP-Labworks software (B). Each value represents the mean ± standard deviation of n experiments (see table C). *P<0.025; **P<0.0001 versus control (Student T-test);
Figure 17 shows mdr-1 expression in Caco-2 cells after treatment with FM-VP4 cholesterol-free Liopsomes. (A) 1.5% agarose gel. 1: Control, 2: 2.5μM, 3: 5μM, 4: 10μM and 4: empty liposomes (B) Bands were quantified with the UVP-Labworks
software. *P<0.009 versus control (Student T-test);
Figure 18 shows mdr-1 gene expression in Caco-2 cells after treatment with cholesterol micelles for 1 week. *P<0.04 versus control (Student T-test);
Figure 19, Western Blot, shows expression of P-gp in Caco2-cells after incubation for 1 week with 10 and 50uM FM-VP4. 1 °Ab: 1:300 dilution of C219 (Signet Pathology Syst.) in Blotto, 2°Ab: 1 :2000 dilution of mouse IgG in Blotto (HRP-conjugated);
Figure 20 is one representative graph showing TEER values measured before and after the transport studies;
Figure 21 shows time-dependent secretory Rhodamine 123 transport (B to A) after pre- treatment with FM-VP4 for one week;
Figure 22 shows the effect of pre-incubation with FM-VP4 for 1 week on P-gp transport of Rh123 across Caco-2 monolayer (basolateral to apical). Data represents the average of 3 experiments ± SD. *P<0.07; **P<0.009;
Figure 23 shows transport of Rhodamine 123 from basolateral to apical side after 2 days pre-incubation with FM-VP4 on the apical side;
Figure 24 shows secretory transport of Rhodamine 6G (B to A) after pre-incubation with FM-VP4 for 7 days on apical and basolateral side;
Figure 25 shows Papp for secretory transport of Rhodamine 6G in Caco-2 cells;
Figure 26 shows mdr-1 A gene expression in liver tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight);
Figure 27 shows mdr-1 B gene expression in liver tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight);
Figure 28 shows mdr-1 A gene expression in duodenum tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight). Internal control with GAPDH was run in a different experiment; and
Figure 29 shows mdr-1 B gene expression in duodenum tissues of untreated (1) and treated (2) rats. Rats were treated for 1 week with FM-VP4 (20mg/kg body weight). Internal control with GAPDH was run in a different experiment. Mdr-1 B was not detectable in these tissues.
PREFERRED EMBODIMENTS OF THE INVENTION
The following detailed description is provided to aid those skilled in the art in practising the present invention. However, this detailed description should not be construed so as to unduly limit the scope of the present invention. Modifications and variations to the embodiments discussed herein may be made by those with ordinary skill in the art without departing from the spirit or scope of the present invention.
As used herein, "animal" means any member of the animal kingdom, including all mammals and most preferably humans. Veterinary use is also contemplated.
As used herein, the term "compound" is interchangeable with the terms "derivative", "structure" and "analogue".
As used herein, the term "ileal bile acid transporter" or "IBAT" is synonymous with apic l sodium co-dependent bile acid transporter, or ASBT.
As used herein, "benzothiepine IBAT inhibitor" means an ileal bile acid transport inhibitor which comprises a therapeutic compound comprising a 2,3,4,5-tetrahydro-1- benzothiepine 1,1 -dioxide structure.
As used herein, the term "a multiple drug resistance gene" or any abbreviation thereof, refers to one or more of the following genes: ACB1 (MDR-1), ABCC1 (MRP-1); and ABCC3 (MRP-3).
As used herein, the term "prodrug" refers to compounds that are drug precursors, which, following administration to a patient, release the drug in vivo via some chemical or physiological process (for example, a prodrug, on being brought to physiological pH or through enzyme action is converted to the desired drug form).
As used herein, the term "solvate" refers to a molecular or ionic complex of molecules or ions of solvent with those of solute (for example the compounds of formulae a) to f) or prodrugs of compounds a) to f)). Non-limiting examples of useful solvents include polar, protic solvents such as water and/or alcohols (for example, methanol).
As used herein, the terms "effective" or "therapeutically effective", are intended to qualify the amount of the compound(s) or composition administered to an animal, in particular a human, in order to elicit a biological or medical response of a cell, tissue, system, animal or mammal that is being sought by the person administering the compound(s) or composition and which amount achieves one or more of the following goals: a) inhibiting or decreasing the expression of a gene which mediates cellular cholesterol influx in an animal cell; b) inhibiting or decreasing the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell; thereby, by one or both means, achieving one or more of the following therapeutic goals: c) decreasing serum LDL levels;
d) increasing serum HDL levels; e) preventing, reducing, eliminating or ameliorating a dyslipidemic condition or disorder; f) preventing, reducing, eliminating or ameliorating hypercholesterolemia or hypoalphalipoproteinemia; g) preventing, reducing, eliminating or ameliorating the development of atherosclerotic lesions; and h) preventing, reducing, eliminating or ameliorating any condition, disease or disorder which has as its basis or which is exacerbated by a deficiency in plasma HDL, or by an excess of either LDL, VLDL, Lp(a), beta-VLDL, IDL or remnant lipoproteins.
As used herein, the term "sterol" includes all sterols without limitation, for example: (from any source and in any form: α, β and y) sitosterol, campesterol, stigmasterol, brassicasterol (including dihydrobrassicasterol), desmosterol, chalinosterol, poriferasterol, clionasterol, ergosterol, coprosterol, codisterol, isofucosterol, fucosterol, clerosterol, nervisterol, lathosterol, stellasterol, spinasterol, chondrillasterol, peposterol, avenasterol, isoavenasterol, fecosterol, pollinastasterol, cholesterol and all natural or synthesized forms and derivatives thereof, including isomers.
The term "stanol" refers to, for example: (from any source and in any form: α, β and y) saturated or hydrogenated sterols including all natural or synthesized forms and derivatives thereof, and isomers, including sitostanol, campestanol, stigmastanol, brassicastanol (including dihydrobrassicastanol), desmostanol, chalinostanol, poriferastanol, clionastanol, ergostanol, coprostanol, codistanol, isofucostanol, fucostanol, clerostanol, nervistanol, lathostanol, stellastanol, spinastanol, chondrillastanol, pepostanol, avenastanol, isoavenastanol, fecostanol, and pollinastastanol.
It is to be understood that modifications to the sterols and stanols i.e. to include side chains also falls within the purview of this invention. It is also to be understood that,
when in doubt throughout the specification, and unless otherwise specified, the term "sterol" encompasses both sterol and stanol. The terms "phytosterol" and "phytostanol" may also be used and refer to all plant-derived sterols or stanols respectively.
The sterols and stanols for use in forming derivatives in accordance with this invention may be procured from a variety of natural sources or they may be artificially synthesized. For example, they may be obtained from the processing of plant oils (including aquatic plants) such as corn oil and other vegetable oils, wheat germ oil, soy extract, rice extract, rice bran, rapeseed oil, sunflower oil, sesame oil and fish (and other marine-source) oils. They may also be derived from yeasts and fungi, for example ergosterol. Accordingly, the present invention is not to be limited to any one source of sterols. US Patent Serial No. 4,420,427 teaches the preparation of sterols from vegetable oil sludge using solvents such as methanol. Alternatively, phytosterols and phytostanols may be obtained from tall oil pitch or soap, by-products of forestry practises as described in US Patent Serial No.5,770,749, incorporated herein by reference. A further method of extracting sterols and stanols from tall oil pitch is described in Canadian Patent Application Serial No. 2,230,373 which was filed on February 20, 1998 (corresponding to PCT/CA99/00150 which was filed on February 19, 1999) and US Patent Application Serial No 10/060,022 which was filed on January 28, 2002 the contents of all of which are incorporated herein by reference.
Accordingly, it is to be understood that the widest possible definition is to be accorded to the terms "sterol" and "stanol" as used herein, including, but not limited to: free sterols and stanols, esterified sterols and stanols with aliphatic or aromatic acids (thereby forming aliphatic or aromatic esters, respectively), phenolic acid esters, cinnamate esters, ferulate esters, phytosterol and phytostanol glycosides and acylated glycosides or acylglycosides. Thus, the terms "sterols" and "stanols" encompasses all analogues, which may further have a double bond at the 5-position in the cyclic unit as in most natural sterols, or one or more double bonds at other positions in the rings (for example, 6, 7,
8(9), 8(14), 14 5/7) or no double bonds in the cyclic unit as in stanols. Further, there may be additional methyl groups as, for example, in άi-sitosterol.
Sterols are naturally occurring compounds that perform many critical cellular functions. Sterols such as campesterol, stigmasterol and beta-sitosterol in plants, ergosterol in fungi and cholesterol in animals are each primary components of cellular and sub-cellular membranes in their respective cell types. Phytosterols, in particular, have received a great deal of attention due to their ability to decrease serum cholesterol levels when fed to a number of mammalian species, including humans.
Optionally, the compounds of the present invention are formed of naturally-derived or artificially synthesized beta-sitosterol, campestanol, sitostanol, and campesterol and each of these compounds so formed is then admixed in a pharmaceutical composition prior to delivery in various ratios. In the most preferred form, the compound of the present invention comprises a chemical linkage between one or more disodium ascorbyl phytostanyl phosphates (referred to herein as "FM-VP4") which comprises two major components: disodium ascorbyl campestanyl phosphate ("DACP") and disodium ascorbyl sitostanyl phosphate ("DASP").
Within the scope of the present invention, "cholesterol absorption inhibitor3' refers to any compound having a negative effect on cholesterol transport, uptake or absorption, by whatever mechanism and includes any compound which inhibits bile acid reabsorption or transport.
Preferably, the cholesterol absorption inhibitor comprises one or more sterols, stanols or mixtures thereof or derivatives thereof as described herein. Without limiting the generality of the foregoing, this includes all free sterols and stanols, and all sterol and stanol aliphatic and aromatic esters, sterol and stanol phenolic acid esters, sterol and stanol cinnamate esters, sterol and stanol ferulate esters, sterol and stanol glycosides, sterol and both stanol acylated glycosides or acylglycosides.
In a most preferred form, the cholesterol absorption inhibitor comprises one or more derivatives or compounds comprising a sterol or stanol, including biologically acceptable salts thereof, having one or more of the following formulae:
o II R4 — P — O-R I OH
ϋ) O O II II R4 — C — C-O-R
F — R
iv)
O R4 _(CH2)n — C -O-R
wherein R is a sterol or stanol moiety, R4 is derived from ascorbic acid and n=1-5, including all biologically acceptable salts or solvates or prodrugs of at least one such compound or of the salts or of the solvates thereof.
Preferably, the cholesterol absorption inhibitor is a disodium ascorbyl stanyl phosphate composition which comprises a mixture of disodium ascorbyl campestanyl phosphate and disodium ascorbyl sitostanyl phosphate.
The compounds of formulae i) to iv) can be prepared by known methods, for example those described below and in PCT/CAOO/00730, which was filed on June 20, 2000 and
claims priority back to US Patent Application 09/339,903 filed on June 23, 1999, the entire contents of which are incorporated herein by reference..
In general, compounds of formulae i) to iv) can be prepared as follows: the selected sterol or stanol (or halophosphate, halocarbonate or halo-oxalate derivatives thereof) and ascorbic acid are mixed together under reaction conditions to permit condensation of the "acid" moiety with the "alcohol" (sterol). These conditions are the same as those used in other common esterification reactions such as the Fisher esterification process in which the acid component and the alcohol component are allowed to react directly or in the presence of a suitable acid catalyst such as mineral acid, sulfuric acid, phosphoric acid, p-toluenesulfonic acid. The organic solvents generally employed in such esterification reactions are ethers such as diethyl ether, tetrahydrofuran, or benzene, toluene or similar aromatic solvents and the temperatures can vary from room to elevated temperatures depending on the reactivity of the reactants undergoing the reaction.
In a preferred embodiment, the process to form the ester comprises "protecting" the hydroxyl groups of the ascorbic acid or derivatives thereof as esters (for example, as acetate esters) or ethers (for example, methyl ethers) and then condensing the protected ascorbic acid with the sterol/stanol halophospahte, halocarbonate or halo-oxalate under suitable reaction conditions. In general, such condensation reactions are conducted in an organic solvent such as diethyl ether, tetrahydrofuran, or benzene, toluene or similar aromatic solvents. Depending on the nature and reactivity of the reactants, the reaction temperatures may vary from low (-15°C) to elevated temperatures.
In more detail, the following is one preferred mode of preparing the compounds of formulae i) to iv) and in particular formula i): ascorbic acid is initially protected from decomposition by the formation of 5,6-isopropylidene-ascorbic acid. This can be achieved by mixing acetone with ascorbic acid and an acidic catalyst such as sulfuric acid
or hydrochloric acid under suitable reaction conditions. Phytostanol chlorophosphate is prepared by forming a solution of phytostanol in toluene and pyridine (although other nitrogen bases such as aliphatic and aromatic amines may alternatively be used) and treating this solution with a phosphorus derivative such as phosphorus oxychloride. The residue so formed after filtration and concentration of the mother liquor is phytostanol chlorophosphate. The latter is then mixed with 5,6-isopropylidene-ascorbic acid and, after the addition of a suitable alcohol such as ethanol and HCl, concentrated. Alternatively, pyridine/THF may be added and the product concentrated. After final washing and drying, the resultant novel product a stanol-phosphate-ascorbate.
In another preferred form of the process to prepare compounds of formulae i) to iv), ascorbic acid is protected at the hydroxyl sites not as 5,6-isopropylidene-ascorbic acid but as esters (for example as acetates, phosphates and the like..). The latter may then be condensed with sterols or stanols, derivatized as described above, using known esterification methods ultimately to produce the compounds. The formation of mono and diphosphates of ascorbic acid is described thoroughly in the literature. For example, US Patent Serial No. 4,939,128 to Kato et al., the contents of which are incorporated herein by reference, teaches the formation of phosphoric acid esters of ascorbic acid. Similarly, US Patent Serial No. 4,999,437 to Dobler et al., the contents of which are also fully incorporated herein by reference, describes the preparation of ascorbic acid 2-phosphate. In Dobler et al., the core reaction of phosphorylating ascorbic acid or ascorbic acid derivatives with POCI3 in the presence of tertiary amines (described in German Laid Open Application DOS 2,719,303) is improved by adding to the reaction solution a magnesium compound, preferably an aqueous solution of a magnesium compound. Any of these known ascorbic acid derivatives can be used.
In more detail, the following is another preferred mode of preparing the compounds of formulae i) to iv) and in particular formula ii): prepare the "protected" ascorbic acid and follow the same process outlined in detail above; however, the phosphorus oxylchloride is
replaced by oxalyl chloride thereby yielding a stanoi-oxaiaie-ascoroaie.
In a preferred form, the cholesterol absorption inhibitor of the present invention comprises one or more disodium ascorbyl phytostanyl phosphates (referred to as "FM- VP4") which comprises two major components: disodium ascorbyl campestanyl phosphate ("DACP") and disodium ascorbyl sitostanyl phosphate ("DASP").
Alternatively, and in another preferred form of the invention, the cholesterol absorption inhibitor comprises a compound from the family of hydroxy substituted azetidinones. Most preferably this azetidonone is a hydroxy substituted azetidinone compound represented by the formul

Ar3 is aryl or R5 -substituted aryl;
X, Y and Z are independently selected from the group consisting of -CH2-, -CHøower alkyl)- and -C(dilower alkyl)-;
R and R2 are independently selected from the group consisting of -OR6, -O(CO)R6, — 0(CO)OR9 and -O(CO)NR6 R7 ;
R1 and R3 are independently selected from the group consisting of hydrogen, lower alkyl and aryl;
q is 0 or 1 ; r is 0 or 1 ; m, n and p are independently 0, 1 , 2, 3 or 4; provided that at least one of q and r is 1 , and the sum of m, n, p, q and r is 2, 3, 4, 5 or 6; and provided that when p is 0 and r is 1 , the sum of m, q and n is 1 , 2, 3, 4 or 5;
R4 is 1-5 substituents independently selected from consisting of lower alkyl, -ORe, - O(CO)R6l -O(CO)OR9, -O(CH2)i-5 OR6, -O(CO)NR6 R7, -NR6 R7, ~NR6 (CO)R7, -NR6 (CO)ORg, -NR6 (CO)NR7 R8, -NR6 SO2 R9, -COOR6, ~CONR6 R7, -COR6, -SO2 NR6 R7, S(O)0-2 Rg, -O(CH2)ι-ιo -COOR6, ~O(CH2)ι.ιo CONR6 R , -(lower alkylene)COOR6, -CH.dbd.CH-COOR6, -CF3, -CN, -NO2 and halogen;
R5 is 1-5 substituents independently selected from the group consisting of -ORe, - O(CO)R6, -O(CO)OR9, -O(CH2)i-5 OR6, -O(CO)NR6 R7, -NR6 R7, -NR6 (CO)R7, -NR6 (CO)ORg, -NR6 (CO)NR7 R8, ~NR6 SO2 R9, -COOR6, -CONR6 R7, -COR6, -SO2 NR6 R7, S(O)0-2 R9, -O(CH2)ι-ιo -COOR6, -O(CH2)1-10 CONR6 R7, -(lower alkylene)COOR6 and -CH.dbd.CH-COORe ;
R6, R7 and R8 are independently selected from the group consisting of hydrogen, lower alkyl, aryl and aryl-substituted lower alkyl; and
R9 is lower alkyl, aryl or aryl-substituted lower alkyl.
More preferably, in the compound of formula V, An is phenyl or R4 -substituted phenyl, Ar2 is phenyl or R4 -substituted phenyl and Ar3 is R5 -substituted phenyl.
In alternative embodiments, within formula V, An is R4 -substituted phenyl wherein R4 is halogen; Ar2 is R4 -substituted phenyl wherein R4 is halogen or -OR6, wherein R6 is lower alkyl or hydrogen; and Ar3 R5 -substituted phenyl, wherein R5 is -OR6, wherein R6 is lower alkyl or hydrogen.
In alternative embodiments within formula V, wherein in the compound X, Y, and Z are each -CH2 -; Ri and R3 are each hydrogen; R and R2 are each -ORe, wherein R6 is hydrogen; and the sum of m, n, p, q and r is 2, 3 or 4.
In alternative embodiments within formula V, wherein in the compound, m, n and r are each zero, q is 1 and p is 2.
In alternative embodiments within formula V, wherein in the compound, p, q and n are each zero, r is 1 and m is 2 or 3.
In preferred embodiments within formula V, the compound is selected from the group consisting of:
3(R)-(2(R)-hydroxy-2-phenylethyl)-4(R)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(R)-(2(R)-hydroxy-2-phenylethyl)-4(S)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(S)-(1 (S)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
3(S)-(1 (R)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
3(R)-(1 (R)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone; rel-3(R)->(S)-hydroxy-(2-naphthalenyl)methyl!-4(3S)-(4-methoxyphenyl)-1-phe nyl)-1 - phenyl-2-azetid inone; rel-3(R)->(R)-hydroxy-(2-naphthalenyl)methyl!-4(S)-(4-methoxyphenyl)-1-phen yl-2- azetidinone;
3(R)-(3(R)-hydroxy-3-phenylpropyl)-1,4(S)-bis-(4-methoxyphenyl)-2-azetidino ne;
3(R)-(3(S)-hydroxy-3-phenylpropyl)-1,4(S)-bis-(4-methoxyphenyl)-2-azetidino ne;
4(S)-(4-hydroxyphenyl)-3(R)-(3(R)-hydroxy-3-phenylpropyl-1-(4-methoxyphenyl )-2- azetidinone;
4(S)-(4-hydroxyphenyl)-3(R)-(3(S)-hydroxy-3-phenylpropyl-1-(4-methoxyphenyl )-2- azetidinone; rel 3(R)->3(RS)-hydroxy-3->4-(methoxymethoxy)-phenyl!-1 ,4(S)-bis-(4-methoxyphe nyl)-
2-azetidinone;
1-(4-fluorophenyl)-3(R)->3(S)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)-(.sup .4 - hyd roxyphenyl)-2-azetid i none;
1-(4-fluorophenyl)-3(R)->3(R)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)-(4-hy droxyphenyl)-2-azetidinone;
4(S)->4-(acetyloxy)phenyl!-3(R)-(3(R)-hydroxy-3-phenylpropyl)-1-(4-methoxyp henyl)-2- azetidinone;
4(S)->4-(acetyloxy)phenyl!-3(R)-(3(S)-hydroxy-3-phenylpropyl)-1(4-methoxyph enyl)-2- azetid inone;
1-(.sup.4 -fluorophenyl)-3(R)->3(S)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)->4-(phen ylmethoxy)phenyl!-2-azetidinone;
3(R)->3(R)-acetyloxy)-3-phenylpropyl!-1,4(S)-bis-(4-methoxyphenyl)-2-azetid inone;
3(R)->3(S)-acetyloxy)-3-phenylpropyl!-1,4(S)-bis-(4-methoxyphenyl)-2-azetid inone;
3(R)->3(R)-(acetyloxy)-3-(4-fluorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- fluorophenyl)-2-azetidinone;
3(R)->3(S)-(acetyloxy)-3-(4-fluorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- fluorophenyl)-2-azetidinone;
3(R)->3(R)-(acetyloxy)-3-(4-chlorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- chlorophenyl)-2-azetidinone;
3(R)->3(S)-(acetyloxy)-3-(4-chlorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- chlorophenyl)-2-azetidinone; and rel 1-(4-fluorophenyl)-4(S)-(4-hydroxyphenyl)-3(1 R)-(1 (R)-hydroxy-3-phenylprop yl)-2- azetidinone.
Such hydroxy substituted azetidinones are described and claimed in US Patent Serial Nos. 5,846,966 and 5,767,115 to the Schering Corporation, the entire contents of both of which are incorporated herein by reference. In particular, 2-azetidinone derived inhibitors referred to as "Ezetimibe" and sold by Schering under the trade-mark Zetia™ are preferred for use herein.
In an alternative embodiment, the cholesterol absorption inhibitor within the scope of the present invention may be an inhibitor of bile acid transport or reabsorption, including, but not limited to ileal, apical and hepatic transport inhibitors.
There having been found a causal relationship between the recirculation of bile acids from the lumen of the intestinal tract and the reduction of serum cholesterol, ileal bile acid transport inhibitors "IBATs" are now are being extensively investigated for their role in lowering cholesterol and treating atherosclerosis. Stedronsky28 discusses the biochemistry, physiology and known active agents surrounding bile acids and cholesterol.
Some IBAT inhibitors useful in the present invention are disclosed in PCT/US95/10863, the contents of which are incorporated herein by reference. More IBAT inhibitors are described in PCT/US97/04076, herein incorporated by reference. Still further IBAT inhibitors useful in the present invention are described in U.S. application Ser. No. 08/816,065, herein incorporated by reference. More IBAT inhibitor compounds useful in the present invention are described in WO 98/40375, herein incorporated by reference. An array of additional IBAT inhibitor compounds useful in the present invention are described in U.S. Pat. No. 5,994,391 , also incorporated herein by reference.
Further IBATs are disclosed by various Hoechst Aktiengesellschaft patent applications which disclose bile acid transport inhibiting compounds and are each separately listed below:
H Canadian Patent Application No. 2,025,294.
H2. Canadian Patent Application No. 2,078,588.
H3. Canadian Patent Application No. 2,085,782.
H4. Canadian Patent Application No. 2,085,830.
H5. EP Application No. 0 379 161.
H6. EP Application No. 0 549 967. H7. EP Application No. 0 559 064. H8. EP Application No. 0 563 731.
Within the scope of the present invention, other bile acid transport inhibitors include selected benzothiepines, such as those disclosed in WO 93/321146, PCT/US97/04076, PCT/US95/10863, EP 508425, FR 2661676, WO 99/35135, WO 92/18462, and U.S. Patent No. 5,994,391 (Lee et al.).
WO 92/16055 to The Wellcome Foundation Limited describes a number of suitable benzothiazepine compounds. Additional hypolipidemic benzothiazepine compounds (particularly 2, 3,4,5-tetrahydrobenzo-1-thi-4-azepine compounds) are disclosed in patent application Nos. WO 96/05188, WO 96/05188 and WO 96/16051.
Further IBAT inhibitor compounds include a class of naphthalene compounds, described by lchihashi29. In this class, S-8921 (methyl 1-(3,4-dimethoxyphenyl)-3-(3- ethylvaleryl)-4-hydroxy-6,7,8-trimethoxy-2-na phthoate) is particularly useful. The structure of S-8921 is shown in formula B-20 of this publication. Further naphthalene compounds or lignin derivatives are described in PCT Patent Application No. WO 94/24087.
Other IBATs include, for example, SC-635 developed by Pharmacia Corporation, IL and Monsanto, MO; 264W94 developed by GlaxoSmithKline; S-8921 developed by Shiongi) and (3R,5R)-3-butyl-3-ethyl-2,3,4,5-tetrahydro-,7,8-dimethoxy-5-phenyl-1-4-benz othiazepine 1,1 -dioxide.
In an alternative embodiment, the cholesterol absorption inhibitor within the scope of the present invention may be an androstane and/or androstene derivative, wherein androstane and/or androstene are coupled with ascorbic acid and represented by one or more of the general formulae:

VI

VII

Most preferably, the ascorbyl moiety which is coupled to the compound from the androstane or androstene family is selected individually from one or more of the following structures:


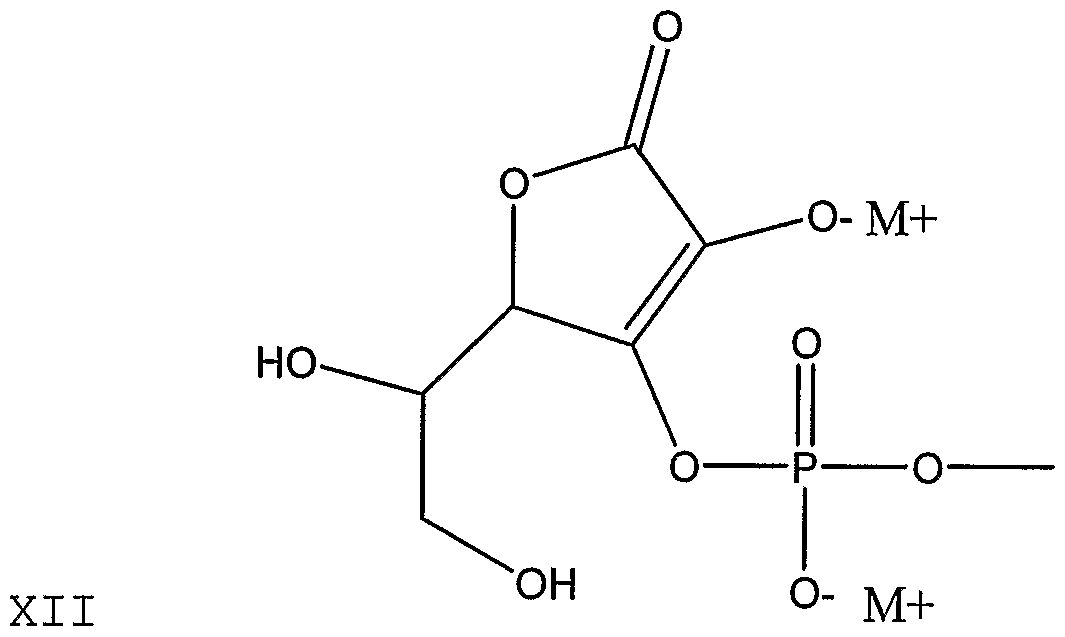





XVIII



wherein M+ represents any metal, alkali earth metal, or alkali metal.
The androstane/androstene compounds of formulae vi) to viii), incorporating one or more of the ascorbyl linkers of formulae ix) to xx), can be prepared by known methods, for example those described in PCT/CA03/00824, the contents of which are incorporated herein by reference or can be prepared by the methods described above for the preparation of compounds of formulae i) to iv), adjusted accordingly.
Salts
As used herein, the term "biologically acceptable salts" refers any salts that retain the desired biological and/or physiological activity of the compounds as described herein and exhibit minimal undesired toxicological effects. Accordingly, reference to all compounds herein thereby includes reference to respective acidic and/or base salts thereof, formed with inorganic and/or organic acids and bases.
Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example trifluroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclpentanepropionates, digluconates, dodecylsulfates, heptanoates, hexanoates, hydrochlorideshyrobromides, hydroiodides, 2- hydroethanesulfonates, lactates, maleates, methanesulfonates, 2-naphthalenesulfonates,
nicotinates, nitrates, oxalates, pectinates, persulfonates, 3-phenylpropionates, phosphates, picrates, pivalates, propionates, salicylates, succinates, sulfates, sulfonates, tartrates, thiocyantes, toluenesulfonates, undecanoates and the like.
Those compounds which contain an acid moiety may form salts with a variety or organic and inorganic bases.
Accordingly, the present invention encompasses not only the parent compounds comprising, for example, the selected sterol and/or stanol a but also, where possible (i.e. where the parent contains a free hydroxyl group), the present invention encompasses the biologically acceptable metal, alkali earth metal, or alkali metal salts of the disclosed compounds.
The salts, as described herein, are even more water soluble than the corresponding parent compounds and therefore their efficacy and evaluation both in vitro and in vivo may be enhanced.
Salt formation of the compounds of the present invention can be readily performed, for example, by treatment of any parent compound containing a free OH group with a series of bases (for example, sodium methoxide or other metal alkoxides) to produce the corresponding alkali metal salts. Other metal salts of calcium, magnesium, manganese, copper, zinc, and the like can be generated by reacting the parent with suitable metal alkoxides.
With respect to the formation of these derivatives, it is to be appreciated that, while selected synthesis processes are described, there are a number of other means by which the variety of derivatives disclosed and claimed can be made. It is well within the purview of a skilled person in this chemical field, once a particular derivative is chosen, to undertake the synthesis using commonly available techniques in the art. For this reason,
the complete synthesis of each and every claimed derivative is not described.
To the extent that the compounds as described herein and salts thereof may exist in their tautomeric form, all such tautomeric forms are contemplated herein as part of the present invention.
All stereoisomers of the compounds of the present invention, such as those which may exist due to asymmetric carbons on various constituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons) and diastereomeric forms, are contemplated within the scope of the present invention. Individual stereoisomers of the compounds of the present invention may, for example, be admixed as racemates or with all other, or other selected sterioisomers. The chiral centres of the compounds can have the S or R configuration as defined by the IUPAC 1974 Recommendations. Such stereoisomers can be prepared using conventional techniques, either by reacting enantiomeric starting materials, or by separating isomers of compounds of the present invention. When diastereomeric or enantomeric products are prepared, they can be separated by conventional methods, for example, chromatographic or fractional crystallization.
Isomers may include geometric isomers, for example cis-isomers or trans-isomers across a double bond. All such isomers are contemplated among the compounds useful in the present invention.
The compounds useful in the present invention also include tautomers.
While the present invention fully covers the inhibition of expression of any gene which mediates cellular cholesterol influx in an animal cell by use of cholesterol absorption inhibitors, it has been found that the cholesterol absorption inhibitors, as described herein, are particularly useful in inhibiting the expression of MDR-1 (ABCB1), which has
P-glycoprotein as its gene product, as well as inhibiting the expression of ABCC1 (MRP- 1) and ABCC3 (MRP-3). In addition, it has been found that the cholesterol absorption inhibitors, as described herein, are particularly useful in inhibiting the production of the protein products of these genes, for example P-glycoprotein, as expressed by the MDR-1 gene.
The finding that cholesterol absorption inhibitors exhibit the aforementioned effects on, in particular both the expression of the multiple drug resistance gene (for example the MDR- 1 gene) and the production of the gene product (for example P-glycoprotein), has enormous therapeutic potential. While not intending to be bound by any one mechanism of action, it is possible that the "gene product" of a gene which mediates cellular cholesterol influx (for example a multiple drug resistance gene) may be an influx transporter of cholesterol. As concentration of the protein is decreased through reduced gene expression, the amount of cholesterol taken up by an animal cell is decreased. The cholesterol uptake data in the examples suggests that cholesterol and the selected cholesterol absorption inhibitor, as defined herein, compete for some type of influx mechanism mediated by this gene product.
Methods of Use
According to one aspect of the present invention, there is provided a method of decreasing or inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
According to another aspect of the present invention, there is provided a method of decreasing or inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
According to yet another aspect of the present invention, there is provided a method of and pharmaceutical composition for decreasing the level of serum LDL cholesterol in an animal, by inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
According to yet another aspect of the present invention, there is provided a method of and pharmaceutical composition for decreasing the level of serum LDL cholesterol in an animal, by inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in the animal cell said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
Accordingly, the present invention provides means to decrease or inhibit the expression of a gene which mediates cellular cholesterol influx in an animal cell and/or to decrease or inhibit the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell in order, ultimately, to achieve one or more of the following therapeutic goals: a) decreasing serum LDL levels; b) increasing serum HDL levels; c) preventing, reducing, eliminating or ameliorating a dyslipidemic condition or disorder; d) preventing, reducing, eliminating or ameliorating hypercholesterolemia or hypoalphalipoproteinemia; e) preventing, reducing, eliminating or ameliorating the development of atherosclerotic lesions; and f) preventing, reducing, eliminating or ameliorating any condition, disease or disorder which has as its basis or which is exacerbated by a deficiency in plasma HDL, or excess of either LDL, VLDL, Lp(a), beta-VLDL, IDL or remnant lipoproteins
which comprises administering to an animal or a cell derived from or within said animal, a non-toxic and therapeutically effective amount of at least one cholesterol absorption inhibitor as described herein. This invention further comprises the use of any of the disclosed compounds and compositions for any indications described herein, more specifically, for use in achieving one or more of the therapeutic goals as defined above.
Heretofore, there has been no recognition of the significant effect of cholesterol absorption inhibitors on genes and their protein products which mediate cellular cholesterol influx.
There are additional advantages to the use of cholesterol absorption inhibitors as described herein. Some of the cholesterol absorption inhibitors {compounds depicted above having formulae i) through iv)) comprise an ascorbyl moiety. These particular compounds have numerous added advantages. First and foremost, solubility of the compounds is greatly enhanced, both in aqueous solutions and non-aqueous media such as oils and fats. With this greater solubility, effective therapeutic dosages and concomitantly costs, can be reduced. Secondly, these derivatives are heat stable (stable to oxidation and hydrolysis) which is essential for some processing mechanisms.
The desired effects described herein may be achieved in a number of different ways. The cholesterol absorption inhibitor may be administered by any conventional means available for use in conjunction with pharmaceuticals i.e. with a pharmaceutically acceptable carrier. The pharmaceutical compositions can comprise from about 1% to 99% of the "active" components and preferably from about 5% to 95% of the active components.
The formulations and pharmaceutical compositions can be prepared using conventional, pharmaceutically available excipients, and additives and by conventional techniques.
Such pharmaceutically acceptable excipients and additives include non-toxic compatible fillers, binders, disintegrants, buffers, preservatives, anti-oxidants, lubricants, flavourings, thickeners, colouring agents, emulsifiers and the like.
The exact amount or dose of the cholesterol absorption inhibitors which is required to achieve the desired effects will, of course, depend on a number of factors such as the particular compound or composition chosen, the potency of the compound or composition administered, the formulation in which it is administered, the mode of administration and the age, weight, condition and response of the patient. All of these factors, among others, will be considered by the attending clinician with respect to each individual or patient.
Generally, a total daily dose the cholesterol absorption inhibitor having one of formulae i)-viii) and comprising sterols and/or stanols may be administered in a daily dosage range of from 10mg to about 20 g, more preferably 10mg to 1.5g, and most preferably up to 80O mg per day in single or multiple divided doses.
Wherein the chosen a cholesterol absorption inhibitor is a sterol or stanol, whether free or as part of a compound or derivative, it may be administered in a form comprising up to 6 grams sterols and/or stanols per day. It should be recognized that the provision of much larger daily doses of sterols, stanols and their derivatives are not harmful to the animal host, as excess will simply pass through normal excretory channels.
Wherein the cholesterol absorption inhibitor is a substituted azetidinone, it may be administered in a daily dose of from about 0.1 to about 30 mg/kg of body weight, preferably about 0.1 to about 15 mg/kg, and most preferably up to 10mg/kg per day. For an average body weight of 70 kg, the dosage level is therefore from about 5 mg to about 1000 mg of drug per day, given preferably in a single dose or 2-4 divided doses.
When the cholesterol absorption inhibitor is any compound which inhibits bile acid
reabsorption, it may be administered in a dose from about 0.003mg — 20mg per kilogram body weight of the individual animal. In general, a total daily dose of an IBAT inhibitor can be in the range of from about 0.01 to about 1000 mg/day, preferably from about 0.1 mg to about 50 mg/day, more preferably from about 1 to about 10 mg/day.
The daily dose of these cholesterol absorption inhibitors can be administered to an individual in a single dose or in multiple doses, as required. Sustained release dosages can be used.
Use of pharmaceutically acceptable carriers to formulate the compounds and compositions herein disclosed for the practice of the invention into dosages suitable for systemic administration is within the scope of the invention. With proper choice of carrier and suitable manufacturing practice, the compounds and compositions of the present invention, in particular, those formulated as solutions, may be administered parenterally, such as by intravenous injection. The compounds and compositions can be formulated readily using pharmaceutically acceptable carriers well known in the art into dosages suitable for oral administration. Such carriers enable the compounds and compositions of the invention to be formulated as tablets, pills, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a patient to be treated.
Pharmaceutical compositions, comprising one or more of the compounds of the present invention, include compositions wherein the active ingredients are contained in an effective amount to achieve their intended purpose. Determination of the effective amounts is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
In addition to the active ingredients these pharmaceutical compositions may contain suitable pharmaceutically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be
used pharmaceutically. The preparations formulated for oral administration may be in the form of tablets, dragees, capsules, or solutions.
In a preferred form, the cholesterol absorption inhibitor is administered in the form of a liposome. Liposomes are hollow microspheres composed of one or more double lipid layers. They were first used more than 30 years ago as vehicles for various drug substances, and since then knowledge of their behavior in vitro has allowed a more rational design focused on the specific treatment of certain diseases.
Formation of liposomes occurs formed when thin lipid films are hydrated. The hydrated lipid sheets detach during agitation and self-close to form multi-lamellar vesicles.
There are a number of methods known and widely practiced in the in the art to prepare liposomes. Chemotherapeutic agents, such as doxorubicin, are often encapsulated in liposomes using the established methods.
Typically, 100 nm diameter liposomes are prepared by exposing chloroformic solution of various lipid mixtures to high vacuum and subsequently hydrating the resulting lipid films (DSPC/CHOL, EPC/CHOL, DSPC/PEG-PE/CHOL) with pH 4 buffers, and extruding them through polycarbonated filters, after a freezing and thawing procedure. A transmembrane pH gradient is then created by adjusting the pH of the extravesicular medium to 7.5 by addition of an alkalinization agent. The technique exploits the ability of weak bases to redistribute across membranes exhibiting pH gradients where [drug]in/[drug]out=[H-«-]in/[H+]out.
The selected drug is then entrapped by addition of the drug solution in small aliquots to the vesicle solution, at an elevated temperature, to allow drug accumulation inside the liposomes. Trapping efficiencies are determined by separating free from liposome encapsulated drug on gel filtration columns and quantifying the two fractions for lipid and
drug content by liquid scintillation counting, fluorescence spectroscopy or UV-VIS spectroscopy. These liposomes are then evaluated for size distribution (quasielastic light scattering, scanning electron microscopy), drug uptake and release studies, stability, and in vivo tumor targeting efficiency.
One preferred, but non-limiting method for use in preparing liposomes is described in the examples below.
For greater certainty, the pharmaceutical compositions of the present invention may be manufactured in any manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes.
Pharmaceutical formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
Pharmaceutical preparations for oral use can be obtained by combining the active compounds with solid excipient, optionally grinding a resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores. Suitable excipients include lactose, sucrose, mannitol, sorbitol, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and
polyvinylpyrrolidone (PVP). If desired, disintegrating agents may be added, such as the cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain gum Dhosph, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
Pharmaceutical preparations which can be used orally include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. The push-fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition, stabilizers may be added.
Oral liquid preparations may be in the form of, for example, emulsions, syrups, or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose, carboxymethylcellulose, aluminium stearate gel, hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non- aqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as esters of glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid; and if desired conventional flavouring or colouring agents.
Since the present invention relates to compositions with a combination of active ingredients which may be administered together or separately, there are also provided herein kits for such purpose. A kit is contemplated wherein two separate units are combined: a pharmaceutical composition comprising at last one cholesterol absorption inhibitor, as described herein, and a separate pharmaceutical composition comprising at least one cancer chemotherapeutic agent. The kit will preferably include directions for the administration of the separate components. This type of kit arrangement is particularly useful when separate components must be administered in different dosage forms (for example, oral vs. parenteral vs. intravenous) or are administered at different dosage intervals or are administered at different dosage amounts.
Without further elaboration, the foregoing so fully illustrates the present invention that others may, by applying current or future knowledge, adapt the same for use under the various conditions described and claimed herein.
EXAMPLES
EXAMPLE 1 — Formation of one of the cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester refered to herein as "FM-VP4";
Step 1 Protection of ascorbic Acid
Oleum (24%, 8.3g) was added dropwise to acetone (50ml). Ascorbic acid (12g) was introduced to the mixture at 0°C and the reaction mixture was stirred at 0°C for 6 hours. The obtained crystals were filtered off under suction, the filtered cake was pressed to dryness and then washed with acetone (30ml). The product, 5,6-isopropylidene ascorbic acid (14g) was obtained.
Step 2: Attachment to Phytostanols
A solution of phytostanol mixture (24g) (campestanol: 36.4%; sitostanol: 62.3%) in toluene (500ml) and pyridine (25ml) was added dropwise to a mixture of phosphorous oxychloride (9ml) in toluene (200ml) at 0°C. The mixture was stirred at room temperature for 3 hours. The pyridine hydrochloride was filtered off and the mother liquor was concentrated to recover the toluene. The residue was dissolved in dry THF (100ml) and a solution of the above prepared protected ascorbic acid (14g) in dry THF (400ml) was added dropwise at 0°C. The stirring at room temperature was maintained for 1 hr. The solution was concentrated to remove the solvent. Ethanol (400ml) and 3N HCl (200ml) were added, the mixture was heated to 50°C for 30 min and concentrated. Ethyl acetate (600ml) was added, the resultant solution was washed with water (3X300ml), dried over sodium phosphate, concentrated and the product (phytostanol-phosphate-ascorbate) was obtained as a white powder 22g.
Step 3: Conversion to Sodium Salt
The above prepared acid (17g) was dissolved in ethanol (100ml) and a solution of sodium methoxide (2.7g) in ethanol (50ml) was added at stirring and at room temperature. The stirring was maintained for 30 min. after the addition. The resultant white cake was filtered off, dried and weighed, to afford a white powder 20g (phytostanol-phosphate- ascorbate sodium).
EXAMPLE 2:- Formation of one of the cholesterol absorption inhibitors described herein: Disodium ascorbyl phosphate ester of dehydroisoandrosterone
To a dry round bottom flask, acetone (150 ml) and L-ascorbic acid (50 g) were added at 0 °C. Acetyl chloride (7.5 ml) was added dropwise through an addition funnel in 10 minutes. The reaction mixture was stirred at 0 °C for 24 hours. The precipitate was filtered off and washed with acetone (3x20 ml). The white product, 5,6-isopropylidine ascorbic acid, was dried under vacuum for 1.5 hours to give a dry powder (52 g), yield
85%.
A dry three neck round bottom flask was fitted with a stirring bar, argon inlet and an addition funnel. A solution of dehydroisoandrosterone (1.73 g, 6 mmol) in anhydrous THF (15 ml) and pyridine (2.4 ml) was added dropwise to the mixture of anhydrous THF (12 ml) and POCI3 (0.7 ml, 7.5 mmol) at 0 °C over a period of 10 minutes. A white precipitate formed immediately. The suspension was stirred at 0 °C for 40 minutes, and at room temperature for 1 hour and 40 minutes.
To the above suspension, a solution of 5,6-isopropylidine ascorbic acid (3.6 g, 16.67 mmol) in anhydrous pyridine (3 ml) and THF (30 ml) was added dropwise at 0 °C over a period of 20 minutes. The suspension was stirred at 0 °C for 30 minutes, and at room temperature for 1.5 hours. The formed pyridinium chloride was filtered out and washed with THF twice. The solvents were evaporated under reduced pressure at 40 °C to afford a residue.
The residue was dissolved in THF (40 ml), and 2N HCl (30 ml) was added in one portion. The mixture was stirred at room temperature for 8 hours. THF was evaporated under a reduced pressure. The water layer was extracted with ethyl acetate (4x50 ml). The combined ethyl acetate solution was washed with brine (100 ml), and dried over Na2SO4. The solvent was evaporated to give a residue. The residue was dissolved in CHCI3, and then hexanes was added to precipitate the product. The precipitated solid was filtered out, washed with hexanes and dried under vacuum (2.43 g, crude product, yield: 77%). The purification of phosphate ester was done by reverse phase C-18 chromatography (Waters, water/methanol = 90/10 to 60/40). Pure compound 4 (Figure 1 , 39 mg) was isolated from 50 mg of the crude product. The overall yield (base on dehydroisoandrosterone) was 60 %.
Ascorbyl phosphate ester of dehydroisoandrosterone (0.5 g, 0.95 mmol) was dissolved
in methanol (3 ml) at room temperature, and then sodium methoxide in methanol (1ml, 20%) was added. The suspension was stirred at room temperature for 30 minutes. The precipitated solid was filtered out, washed with methanol, acetone and hexanes. The mother liquor was concentrated to 2 ml, acetone was added to precipitate the product. An additional white solid was obtained. The combined solid was dried under vacuum at room temperature. Disodium ascorbyl phosphate ester of dehydroisoandrosterone (0.49 g, yield 91%) was obtained.
EXAMPLE 3-- Synthesis of another of the cholesterol absorption inhibitors described herein: Disodium Ascorbyl Phosphate Ester of 5α-Androstan-3β-ol-17-one
To a dry round bottom flask, 5α-androstan-3β-ol-17-one (1.0 g, 3.4 mmol), THF (8.6 ml) and pyridine (1.38 ml) were added. The mixture was stirred at room temperature until a clear solution was obtained. To another dry round bottom flask, THF (6.9 ml) and POCI3 (0.4 ml, 4.25 mmol) were added, stirred at 0 °C for 5 minutes. To this mixture, the above prepared 5α-androstan-3β-ol-17-one solution was added drop-wise under argon atmosphere over a period of 10 minutes. After the addition, the white suspension was stirred at 0 °C for 35 minutes, and at room temperature for 2 hours. The reaction was stopped and the white suspension was used for the coupling reaction without filtration.
5,6-isopropylidine ascorbic acid (2.0 g, 9.52 mmol) was dissolved in pyridine (1.71 ml) and THF (17 ml). The round bottom flask which contained previously prepared white suspension was immersed in an ice-water bath. To this mixture, the above prepared THF solution of the 5,6-isopropylidine ascorbic acid was added dropwise under stirring at 0 °C over a period of 15 minutes. After the addition, the mixture was stirred at 0 °C for 25 minutes, and at room temperature for 2 hours. The white solid of pyridinium chloride was filtered out and washed with THF (8 ml). The filtrate was concentrated to remove THF and excess pyridine to give a residue (2.38 g).
The residue was dissolved in THF (30 ml), and 1N HCl (30 ml) was added in one portion. The mixture was stirred at room temperature for 16 hours and 45 minutes. 12N HCl (4 ml) was added to the reaction mixture at room temperature. The reaction mixture was stirred at room temperature for an additional 4 hours and 45 minutes. THF was evaporated under a reduced pressure. The water layer was extracted with ethyl acetate (3x60 ml). The combined ethyl acetate solution was washed with brine (60 ml), and dried over Na2SO4. The extract was concentrated to about 3 ml. Hexanes (15 ml) was added to precipitate the product. The precipitated solid was filtered out, washed with hexanes and dried under a reduced pressure (1.48 g).
Ascorbyl phosphate ester of 5 -androstan-3β-ol-17-one (0.5 g, 0.95 mmol) was dissolved in methanol (3 ml) at room temperature, and then sodium methoxide in methanol (1.5 ml, 20%) was added. The suspension was stirred at room temperature for 25 minutes. The precipitated solid was filtered out, washed with methanol, acetone and hexanes. The mother liquid was concentrated to 2 ml, and then acetone was added to precipitate the product. An additional product was obtained. The combined solid was dried under a reduced pressure at room temperature to give disodium ascorbyl phosphate ester of 5α-androstan-3β-ol-17-one (0.38 g). The overall yield was 57% (based on 5α-androstan-3β-ol-17-one).
EXAMPLE 4 — Synthesis of another of the cholesterol absorption inhibitors described herein: Disodium Ascorbyl Phosphate Ester of Androst-5-ene-3β,17β-diol
To a dry round bottom flask, 3β-acetoxyandrost-5-ene-17β-ol (1.0 g, 3.0 mmol), anhydrous THF (6.3 ml) and pyridine (0.73 ml) were added. The mixture was stirred at room temperature until a clear solution was obtained. To another dry round bottom flask, THF (2 ml) and POCI3 (0.35 ml, 3.22 mmol) were added, stirred at -5 °C ~ -10 °C for 5 minutes. To this mixture, the above prepared 3β-acetoxyandrost-5-ene-17β-ol solution was added drop-wise under argon atmosphere over a period of 20 minutes. After the
addition, the white suspension was stirred at room temperature for 1 hour. The mixture was concentrated to remove THF and excess POCI3 to give a residue.
5,6-isopropylidine ascorbic acid (0.98 g, 4.55 mmol) was dissolved in anhydrous pyridine (0.70 ml) and THF (6.2 ml). The residue was dissolved in dry THF (4 ml). To this mixture, the above prepared THF solution of the 5,6-isopropylidine ascorbic acid added dropwise under stirring at 0 °C over a period of 20 minutes. After the addition, the mixture was stirred at room temperature for 1 hour and 25 minutes. The white solid of pyridinium chloride was filtered out and washed with THF (6 ml). The filtrate was concentrated to remove THF and excess pyridine to give a residue.
The residue was dissolved in a mixture of ethanol (12.5 ml) and 1 N HCl (12.5 ml). The mixture was kept stirring at 50 °C ~ 55 °C for additional 3 hours and 45 minutes (TLC monitoring). The mixture was extracted with ethyl acetate (60 ml), washed with 10% aqueous NaCl twice (30 ml, 20 ml) and dried over Na2SO (10 g) for 1.5 hours. After the filtration, the filtrate was concentrated to 5 ml. Hexanes (10 ml) was added to precipitate the product. The precipitate was collected, washed with hexanes (10 ml) and dried under the reduced pressure to give a slightly yellow powder (0.95 g, crude product, yield 60%). The pure product was obtained by preparative HPLC.
Instrument is Waters Delta Preparative 4000 HPLC system. Column is Waters Symmetry C18, 5μm, 30x100 mm. Mobile phases are 0.1 % H3P0 in water and acetonitrile. Water and acetonitrile are HPLC grade or equivalent.
The crude product was purified by preparative HPLC. The product was collected and evaporated on a rotary evaporator to remove acetonitrile. The water solution was extracted with ethyl acetate twice. The ethyl acetate layer was dried over Na2SO , concentrated and dried under a reduced pressure to give a white powder product. This product was submitted for NMR and mass spectra. Both spectra indicated the product is
ascorbyl phosphate ester of androst-5-ene-3β,17β-diol.
Preparation of disodium ascorbyl phosphate ester of androst-5-ene-3β,17β-diol (5, Figure 3) was similar to the process described in Example 3.
EXAMPLE 5 — Synthesis of another of the cholesterol absorption inhibitors described herein: Disodium Ascorbyl Phosphate Ester of Androst-5-ene-17β-ol
To a solution of pyridine (0.41 ml) and 1 ,2-phenylenephosphorochloridite (0.6 ml, 5 mmol) in anhydrous THF (10 ml) at 0 °C was added dropwise dehydroiso- androsterone (1.44 g, 5 mmol) in anhydrous THF (10 ml) over a period of 10 minutes. The reaction mixture was stirred at 0 °C for 30 minutes, and at room temperature for 4 hours. The reaction was monitored with TLC (hexanes/EtOAc = 2/1). The formed pyridinium chloride was filtered off and washed with THF. The solvents were evaporated at 40 °C to give a white powder.
The crude phosphate ester was dissolved in methylene chloride (25 ml) , and treated with iodine (1.27 g) for 4 hours at room temperature. The reaction mixture was diluted with methylene chloride (75 ml), washed with 1N NaOH (2x50 ml) and water (2x50 ml), and dried over Na2SO . The solvent was removed, and the product (1.4 g, yield 71%) was crystallized from methylene chloride and methanol.
3β-lodoandrost-5-ene-17-one (1.27 g, 3.19 mmol) was dissolved in glacial acetic acid (40 ml) at 50-55 °C, the activated zinc dust (2.7 g) was added in one portion. The mixture was stirred at 50 °C ~ 55 °C for 2 hours, the zinc dust was filtered out and washed with methylene chloride. The solution was diluted with methylene chloride (120 ml), washed with water (2x100 ml), 1 N NaOH (2x100 ml) and water (100 ml), and dried over Na2SO4. The solvent was removed to afford a white powder. The white powder was dried under vacuum to give and rost-5-ene-17-one (0.83 g, yield: 95%).
Androst-5-ene-17-one (0.65 g, 2.34 mmol) was dissolved in methanol (25 ml) at room temperature. The solution was cooled down to 0 °C, and NaBH4 (50 mg) was added in one portion. The mixture was stirred at 0 °C for 3 hours, and monitored with TLC (hexanes/EtOAc = 3/1). After 3 hours, another portion of NaBH4 (20 mg) was added, and the reaction mixture was stirred at 0 °C for additional half an hour. Aqueous NH4CI (5%, 25 ml) and HCl (6N, 5 ml) were added slowly at 0 °C, and stirred for 1 hour. Water (100 ml) was added to completely precipitate the product. The precipitated solid was filtered out and washed with water, and dried under vacuum. The pure product (0.62 g, yield: 95%) was obtained by column chromatography.
A solution of androst-5-ene-17β-ol (0.63 g, 2.3 mmol) in anhydrous THF (8 ml) and pyridine (1 ml) was added drop-wise to the mixture of anhydrous THF (6 ml) and POCI3 (0.28 ml, 3 mmol) at 0 °C over a period of 5 minutes. The suspension was stirred at 0 °C for 50 minutes, and then at room temperature for one hour.
To the above suspension, a solution of 5,6-isopropylidine ascorbic acid (1.38 g) in anhydrous pyridine (1.2 ml) and THF (12 ml) was added drop-wise at 0 °C over a period of 15 minutes. The suspension was stirred for 1.5 hours at 0 °C, and then overnight at room temperature. The formed pyridine hydrochloride was filtered out and washed with THF twice. The solvents were evaporated under reduced pressure at 40 °C to afford a residue.
The residue was then dissolved in THF (35 ml), and 2N HCl (30 ml) was added as one portion. The mixture was stirred overnight at room temperature. THF was evaporated under reduced pressure. The water layer was extracted with ethyl acetate (3x100 ml). The combined ethyl acetate solution was washed with brine (100 ml), and dried over Na2SO4. The solvent was evaporated to give a residue. The residue was dissolved in acetone, and hexanes was added to precipitate the product. The white precipitated
solid was filtered out, washed with hexanes and dried under vacuum (0.82 g, crude product, yield: 70%).
Preparation of disodium ascorbyl phosphate ester of androst-5-ene-17β-ol was similar to example 2.
EXAMPLE 6 — Synthesis of another of the cholesterol absorption inhibitors described herein: Tetra-sodium Monoascorbyl Diphosphate Ester of 3β-Acetoxyandrost-5-ene- 7β,17β-diol
To a dry round bottom flask, 3β-acetoxyandrost-5-ene-7β,17β-diol (0.5 g, 1.43 mmol), pyridine (0.83 ml) and THF (4 ml) were added. The mixture was stirred at room temperature until a clear solution was obtained. To another dry round bottom flask, THF (5 ml) and POCI3 (0.33 ml) were added, stirred at -5 °C ~ 0 °C for 5 minutes. To this mixture, the above prepared 3β-acetoxyandrost-5-ene-7β,17β-diol solution was added dropwise under argon atmosphere over a period of 15 minutes. After the addition, the white suspension was stirred at room temperature for 2 hours and 45 minutes. The reaction was stopped and the white suspension was used for the coupling reaction without filtration.
5,6-isopropylidine ascorbic acid (1.30 g, 6.02 mmol) was dissolved in pyridine (1 .16 ml) and THF (5.8 ml). The round bottom flask which contained previously prepared white suspension was immersed in an ice-water bath. To this mixture, the above prepared THF solution of the 5,6-isopropylidine ascorbic acid was added dropwise under stirring at 0 °C over a period of 15 minutes. After the addition, the mixture was stirred at 0 °C for 40 min and at room temperature for 17 hours. The white solid of pyridinium chloride was filtered out and washed with THF (5 ml). The filtrate was concentrated to remove THF and excess pyridine to give a residue (2.76 g).
The crude of this compound was dissolved in a mixture of THF (30 ml) and 1N HCl (30
ml). The mixture was kept stirring at room temperature for 3.5 hours (TLC monitoring). The second portion of 1 N HCl (10 ml) were added. The mixture was stirred for an additional 18.5 hours. The THF in the reaction mixture was removed under a reduced pressure. The water suspension was extracted with ethyl acetate and n-butanol (1:1, 110 ml). The organic layer was washed with distilled water (11 ml). The organic layer was concentrated on a rotary evaporator to give a residue. This residue was washed with hexanes (2x10 ml) and dried under the reduced pressure to give a crude product (1.15 g).
Preparation of sodium salt of this compound was similar to Example 3.
EXAMPLE 7 — Synthesis of another of the cholesterol absorption inhibitors described herein: Tetrasodium Diascorbyl Diphosphate Ester of Androst-5-ene-3β,17β-diol
In a dry round bottom flask, androst-5-ene-3β,17β-diol (1.5 g, 5.17 mmol) was dissolved in pyridine (3.0 ml) and THF (15 ml). Into another dry round bottom flask was added THF (20 ml) and POCI3 (1.17 ml, 12.56 mmol). The latter was stirirred at -5 °C for 5 minutes before the addition of androst-5-ene-3β,17β-diol over a period of 20 minutes. White precipitate was observed shortly after this addition and after the initial 20 minutes of reaction at -5 °C, the reaction was allowed to continue at room temperature for 2.5 hours.
The flask was then cooled to 0 °C, and a solution of 5,6-isopropylidene ascorbic acid (3.19 g, 14.78 mmol) in pyridine (3 ml) and THF (15 ml) was added drop-wise over a period of 20 minutes under vigorous stirring. The reaction was allowed to continue for another two hours. Then, the reaction mixture was filtered, and the filtrate was concentrated to a thick syrup. Heptane was added and the mixture was distilled under a reduced pressure. A solid crude was obtained.
The crude was dissolved in THF/1N HCl (1:1, 150 ml), and the hydrolysis was carried out at room temperature under vigorous stirring. After 12 hours of reaction, a TLC test
indicated that the hydrolysis was complete. The THF in the reaction mixture was removed under a reduced pressure at room temperature, and n-butanol and ethyl acetate (1:1, 100 ml) was used for the extraction. The organic layer was washed with water (2x20 ml), and then concentrated to afford the crude product of diascorbyl diphosphate ester of androst-5-ene-3β,17β-diol (3.0 g).
The crude diascorbyl diphosphate ester of androst-5-ene-3β,17β-diol (400 mg) was dissolved in methanol (5 ml). To this solution was added 2 ml of sodium methoxide in methanol (20%, w/v) under magnetic stirring. White precipitate was observed upon the addition of sodium methoxide methanol solution. The suspension was stirred for half an hour before it was filtered and washed with methanol and acetone. The solid product was dried under high vacuum, and tetrasodium diascorbyl diphosphate ester of androst- 5-ene-3β,17β-diol (330 mg) was obtained.
EXAMPLE 8 — Measuring MDR-1 Expression in CaCo2 cells after treatment with FM- VP4 or Cholesterol for one week
Confluent CaCo2 cells (P33-39) in T-75 flasks were treated with varying amounts of FM-VP4 (0.5um, LOurn, 5.0um and 10.0uM) or cholesterol (0.5um, LOum, 5.0um, 10.0uM, 25uM) for one week. The media and treatment were changed every other day. After seven days, cells were harvested and total RNA isolated with TRIZOL™. Analysis conducted by reverse transcription→ cDNA using polymerase chain reaction.
Titration for primer drop of GAPDH procedure:
50ng cDNA (CaCo2 cells)
0.5 uM MDDR_1 forward and 0.5uM MDR-1 reverse
PCR Program: 94°C for 5 minutes, 94°C for 1 minute, 55°C for 1 minutes, 72°C for I minute 30 seconds, repeat from step 2 for 29 more times, 72°C for 10 minutes, 4°C.
Primer drop at cycle: 0,6,9,12,16,20,23,26,28 and 30.
Figure 1 is a graph showing the results, from which it is clear that with increasing concentrations of the cholesterol absorption inhibitor, MDR-1 gene expression is significantly reduced. Further supporting these results are shown in Figure 2, a graph showing the titration for primer drop of GAPDH and Figure 3 depicting polymerase chain reaction (1.5% agarose gel) electrophoretic plate results for MDR-1 and GAPDH.
Figures 10 and 11 indicate that, at increasing concentrations of cholesterol, a point of saturation is reached wherein in response, the expression of MDR-1 decreases. Including this data is important as it is suspected that cholesterol is taken up by cells by a mechanism involving the gene product of MDR-1.
Incubation with 10 mM FM-VP4 leads clearly to a significant downregulation of MDR-1.
EXAMPLE 9- Measuring MRP-1 Expression in CaCo2 cells after treatment with FM- VP4 for one week
The entire protocol as provided in Example 8 with respect to the MDR-1 gene was used to confirm the effects of a cholesterol absorption inhibitor on the expression of another multiple drug resistance gene: MRP-1.
The results in Figure 6, a bar graph showing the level of ABCC1 (MRP-1) expression (normalized ratio of MRP-1/GAPDH) in CaCo2 cells after treatment for one week with FM- VP4 clearly demonstrate the inhibitory effect on gene expression.
EXAMPLE 10- Cytotoxicity Studies: The non-toxicity of the cholesterol absorption inhibitor tested was verified by MTS, LDH and BCA assays
Caco2-cells were seeded into 48- or 96-well plates. The growth media was aspirated every 2-3 days and replaced with fresh media. The cells were kept at 37°C in a humidified atmosphere of 5% C02. Cells then were treated with media, 0.1% Triton X- 100 (as positive control for toxicity), FM-VP4, the selected cholesterol absorption inhibitor.
After 24 hours 50ml of each well were transferred into a new 96-well plate to perform the LDH-Assay. The MTS-Assay (CellTiter 96 AQueous One Solution kit (Promega™) was done on the original plate.
Cell viability can be reflected by the integrity of the mitochondria. MTS-Assay is a colorimetric method for determining the number of viable cells (cytotoxicity) after treatment. When the MTS reagent (a tetrazolium salt) is applied to living cells, it is converted to a color compound (formazan) with the emission of light at 492nm. Cell viability can be reflected by the integrity of mitochondria.
After performing this assay media and solution was aspirated, cells were washed 3 times with PBS and lysed for BCA Protein Assay (PIERCE). This particular colorimetric assay measures total protein levels by identifying specific peptide bonds. The protein concentration is calculated from a calibration curve constructed with a protein standard (bovine serum albumin or "BSA").
The LDH-assay looks at the integrity of cell membranes and can be used for cytotoxicity mediated by chemicals or other agents. Cell damage is associated with leakage of intracellular, cytoplasmic contents and lactate dehydrogenase or "LDH" (a stable cytosolic enzyme), can be used as a reported molecule for this event. LDH, released from cells into the culture medium, was measured using a kit (Cytotox96 Non- Radioactive Cytotoxicity Assay) from Promega™.This method is based on a series of
linked enzyme reactions, the final reaction being the reduction of a tetrazolium salt to a coloured, insoluble, formazen product which can be measured at 492nm. Background absorbance from media alone and media including the treatment was substracted from the reading to correct the values.
Figure 4 is a bar graph of the MTS- and LDH-Assay of cell viability after treatment of CaCo2 with "FM-VP4". Figure 5 a bar graph showing a BCA-Assay of protein concentration after treatment of CaCo2 with "FM-VP4".
The cholesterol absorption inhibitor tested, FM-VP4, does not show any toxicity (mitochondrial activity) for a concentration range up to 100mM regardless of whether the treatment was for 24 hours (cell viability is 90.9 +/- 9.8%) or 96 hours (94.6 +/- 4.4%). At 250mM for 24 hour treatment it drops down to 59.2+/- 8.1% and to 19.3+/-7.6% for 96 hours. The highest tested concentration of FM-VP4 75mM showed a cell viability of 18.5+/-5.8% after 24 hours and 22.1+/-1.6%.
These findings are consistent with the Protein concentration after treatment. It shows the same trend found with the MTS-Assay.
EXAMPLE 11 — Western Blot analysis of P-glycoprotein in Caco2-cells after incubation with FM-VP4 (→ Protein expression)
Experiment:
> 10 flasks of Caco2-cells (T75) > ~ 28 days post-seeding cells were divided into 2 groups (I and II) > both groups received the same treatment for 1 week and then divided for analysis > treatment group I: RNA isolation with TRIZOL → RT-PCR → PCR ^mdr-1 expression
> treatment group II: Protein isolation → SDS-Page Gel → Western Blot ■=> P- glycoprotein expression
Cells from group I were harvested and total cellular RNA was extracted with TRiZOLI® according the protocol. RNA was reverse transcripted into cDNA and specific primers were used to evaluate the expression level of MDR-1. Internal control was run by using GAPDH (primer drop). A sample from each PCR product was subjected to electrophoresis on a 1.5% Agarose gel (containing Ethidiumbromide). A 100 bp standard was run to identify the size of the product. The fluorescent bands were imaged under UV light (UVP-Epi Chemi II Darkroom) and quantified with the UVP-Labworks software.
Cells from group II underwent a protein extraction procedure. Concentration was measured using PIERCE BCA Protein Assay Reagent Kit. 15 μg total protein from each sample was electrophoresed on a 11% SDS Page Gel and transferred to a PVDF membrane (wet transfer). Identification of P-gp was done using the immunoblot technique. The monoclonal antibody used for P-gp was JSB-1 and the secondary was horseradish peroxide rabbit anti-mouse antibody (Jackson Immuno Research Laboratories, Inc.). After optimizing the assay the best dilutions for the antibody solution turned out to be 1 :200 for the primary and 1 :1000 for the secondary. The protein bands were visualized using a chemiluminescence kit from PerkinElmer and an X-Ray film was placed on the membranes. The film was exposed for about 1 minute and then developed.
Figure 9 is a Western Blot analysis of P-glycoprotein in CaCo2 cells after incubation with the selected cholesterol absorption inhibitor: "FM-VP4".
Incubation with FM-VP4 clearly leads to a down-regulation of MDR-1. In other words, a lower level of P-Glycoprotein which is encoded by mdr-1 was detectable. A seven day
incubation with FM-VP4 10μM led to a down-regulation of mdr-1 (confirming earlier experiments). The level of protein also correlates with that finding. A lower level of P- Glycoprotein which is encoded by MDR-1 was detectable.
These data suggest that changes in the mdr-1 gene expression lead to correlating changes in the protein level P-gp which is encoded by mdr-1.
EXAMPLE 12-MDR-1 gene expression in Caco2-cells after treatment with FM-VP4 Liposomes
The objective was to confirm the influence of a cholesterol-free liposomal formulation of a cholesterol absorption inhibitor ("FM-VP4") on gene expression of a multiple drug resistance gene.
A dilution of FM-VP4 in water was applied to Caco2-cells. Of interest was the influence of liposomal FM-VP4 in cholesterol-free liposomal preparation (DMPC 100:0, ratio 10:1 DMPC:FM-VP4). FM-VP4 liposomes were utilized at three different concentrations (2.5, 5 and 10μM FM-VP4).
Procedure: Caco2-cells were seeded at 10,000 cells/cm2 in T-75 flasks (Corning). The growth media was changed every other day. The cells were kept at 37°C in a humidified atmosphere of 5% CO2. The treatment groups contained 2.5, 5 and 10μM cholesterol- free liposomal FM-VP4 as well as empty cholesterol-free liposomes without FM-VP4. Cells were harvested after one week of treatment, total RNA isolated with TRIZOL® according to the protocol. RT-PCR followed that step and specific primers were used to evaluate the expression level of MDR-1. PCR products were resolved by electrophoresis on a 1.5% Agarose gel (containing Ethidiumbromide) and the fluorescent bands were imaged under UV light and quantified using the UVP-Labworks software.
Figure 7 depicts a polymerase chain reaction (1.5% agarose gel) electrophoresis results for MDR-1, GAPDH; RNA isolation with TRIZOL→ RT-PCR→ PCR after treatment of CaCo2 cells with liposomal formulations of one of cholesterol absorption inhibitors described herein: an ascorbyl stanyl phosphate ester called "FM-VP4;
Figure 8 is a bar graph showing the level of MDR-1 expression (normalized ratio of MDR- 1/GAPDH) in CaCo2 cells after treatment for one week with liposomal FM-VP4 at 2.5, 5 and 10um as compared to a control and empty liposomes.
All 3 tested concentrations lead to a down-regulation of MDR-1 gene. Incubation with empty liposomes does not show any effect as hypothesized and can be used as control. The down-regulation is markedly more effective than when the cholesterol absorption inhibitor is solubilized in water.
Accordingly, MDR-1 gene expression in Caco2-cells after treatment with liposomal FM- VP4 at lower concentrations is significantly reduced and at lower concentrations than non-liposomal formulations.
EXAMPLE 13- Preparation of Liposomes comprising cholesterol absorption inhibitor General Protocol
Making dry lipid film
1. Prepared stock solution of Lipids in Chloroform (DM PC/cholesterol 55:45 molar ratio).
2. Transfered appropriate amount of stock solution to test tubes.
3. Dried Solvent was dried under stream of nitrogen gas
4. down with nitrogen
5. When the lipids were almost dried down, placed it in a vacuum apparatus to get rid of the residual chloroform for at least 2 hours.
6. The dry lipid film was stored by capping it and placing it in the freezer.
Hydrating lipid
1. The water bath was set to 55°C.
2. Warmed up some HBS or selected cholesterol absorption inhibitor (ex: FM-VP4) dissolved in HBS in the water bath.
3. Added appropriate amount of HBS or FM-VP4 in HBS to the test tubes.
4. Vortexed.
5. Warmed it in the water bath.
Freeze thaw Cycles
1. Filled a Dewar with liquid Nitrogen
2. Transfered the lipid mixture into a plastic cryo tube.
3. Attached the cryo tube to a crane.
4. Freezed the sample in liquid nitrogen, and then thawed in water bath. Repeated this 5 times.
5. Stored the sample in the freezer.
Extrusion/ Adjusting size of Vesicles
1. Water bath was set to 55°C.
2. Assembled the extruder.
3. Tested the extruder with the nitrogen and then HBS.
4. Increased the pressure to 300 - 600 psi.
5. Before the sample was extruded, aliquoted 500uL, transfered it to an eppendorf, and labelled it BE.
6. Lipid suspension was forced through 2 polycarbonate filters
7. using 0.1 μm filter to extrude sample to a size of 10Onm
8. Extruded sample for 10 times.
9. Aliquoted 500uL, and labelled it AE.
10. After dialysis, aliquoted 500uL, and labelled it AD.
The following examples show further development in this area in support of the invention. Examples 14 through 20 look at mdr-1 gene and P-glycoprotein expression in Caco-2 cells, human intestinal carcinoma cells, after administration with FM-VP4. It was demonstrated that changes in the mdr-1 gene expression levels lead to changes in the P-glycoprotein. These changes then have an influence on the functionality of P-gp. Accordingly, within the scope of this invention, we have found a different unique way of inactivating P-gp by down-regulation of mdr-1 mRNA. Down-regulation of mdr-1 A & B could also be shown in the rat model. We also show that cholesterol can be transported through an active influx mechanism via P-gp.
EXAMPLE 14- MDR-1 and P-Glycoprotein Expression in CaCo-2 cells, human intestinal carcinoma cells, after administration of compounds in accordance with present invention
Caco-2 cells and LLC-PKi (Pig kidney cells) were purchased from American Type Culture Collection (ATCC) (Rockville, MD and Wanassas, VA, USA). Cell culture media were purchased from Gibco BRL (Grand Island, NY, USA). Sterile steritop 0.22μm express membrane bottle top filters were purchased from Millipore (Bedford, MA, USA). Sterile 50ml centrifuge tubes, disposable 10 and 25ml serological pipettes were purchased from Starstedt (Montreal, PQ, Canada). Culture flasks and plates were obtained from Corning-Costar (Cambridge, MA). Chemicals like Triton X-100, Verapamil, Rhodamine 123 were purchased from SIGMA. TRIzol® reagent, Agarose, 10xTBE, RNAse/DNAse free water were purchased from Invitrogen (Vancouver, BC). The monoclonal antibody C219 was from Signet Pathology System Dedlam, antimouse IgG rabbit HRP-conjugated antibody from Jackson Immuno Research Laboratories Inc. (West Grove, PA, USA). Western Lightning Chemiluminescence Reagent was purchased at Perkin Elmer Life Sciences (Mississauga, Ontario), Mem-PER® Eukaryotic Membrane Extraction Kit from PIERCE (Rockford, IL, USA), CellTiter 96® Aqueous One Solution Assay and CytoTox 96 were from Promega (Madison, Wl), BCA protein assay kit from PIERCE (Rockford, IL).
Cell culture: Caco-2 cells were cultured in Dulbecco's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 292μg/mL glutamine, 0.1 mM non-essential amino acids, 100U/mL penicillin and 100μg/mL streptomycin at 37°C in humidified air containing 5% CO2 in either 96 well plates or T75 flasks depending on the type of experiment. Media was changed every other day. Cells were used for experiments when they reached between 80 to 90% confluency.
Cytotoxicity: For cytotoxicity experiments cells were incuabated in media or treated with 0.1% Triton X-100 (as a positive control for toxicity), a cholesterol absorption inhibitor "FM-VP4" or cholesterol micelles and incubated for 24 and 96 hours. Following, LDH- and MTS-assay were performed to determine the integrity of cell membranes and mitochondrial respiration respectively indicating the cytotoxicity and cell viability post treatment as well as BCA-Assays to determine the protein concentration of the surviving cells. After the incubation period 50μl from each well were transferred into a new 96-well plate to perform the LDH-Assay. The MTS-Assay (CellTiter 96® AQueous One Solution kit) was done on the original plate. Cell viability can be reflected by the integrity of the mitochondria. When the MTS reagent (a tetrazolium salt) is applied to living cells, it is converted to a color compound (formazan) with the emission of light at 492nm. After performing this assay media and solution was aspirated, cells were washed 3 times with PBS and lysed for BCA Protein Assay.Total protein levels can be measured in a colorimetric assay that identifies specific peptide bonds. The protein concentration is calculated from a calibration curve constructed with a protein standard (bovine serum albumin).The LDH-assay looks at the integrity of cell membranes. Cell damage is associated with leakage of intracellular, cytoplasmic contents and lactate dehydrogenase can be used as a reported molecule for this event. Lactate dehydrogenase, released from cells into the culture medium is measured using a CytoTox96 kit from Promega. This method is based on a series of linked enzyme reactions, the final reaction being the reduction of a tetrazolium salt to a coloured, insoluble, formazen product which can be measured at 492nm.
Animal Studies: Adult Sprague-Dawley rats were treated with 20mg cholesterol absorption inhibitor "FM-VP4" per kg body weight. After 7 days rats were sacrificed and samples of the following tissues were immediately removed; liver, duodenum, jejunum and ileum and were then transferred to tubes for flash freezing in liquid nitrogen. All rats were cared for in accordance with the Canadian Council on Animal Care and the UBC
guidelines.
RNA-Isolation and RT-PCR:
RNA-lsolation from Caco-2 cells: Total RNA from Caco-2 cells was isolated using TRIzol® Reagent according to the manufactures instruction.
RNA-isolation from rat tissues:
The rat tissues were kept at -80°C. For the RNA isolation an RNeasy Mini Kit from Qiagen (74104) was used.
A slice of the tissue was weighted and lysate buffer was added. A Rotor-stator homogenizer has been used to disrupt and simultaneously homogenize the tissue. The tissue lysate was centrifuged for 3 minutes at maximum speed and the supernatant carefully transferred into a new 2ml tube. 600μl of 70% ethanol were added to the cleared lysate and mixed by pipetting. Up to 700μl of the sample were then applied to an RNeasy mini column placed in a 2 ml collection tube. It was then centrifuged for 15 sec at 10,000 rpm and the flow-through discarded by using a pipette to avoid spills on the tube. 700μl of washing buffer was added, the tube was again centrifuged for 15 sec at 10,000 rpm and the flow-through discarded. A second washing step is performed by adding 500μl of washing buffer 2 and centrifugation for 15 sec at 10,000 rpm. The last step was repeated and the tube was centrifuged for 2 min to dry the RNeasy silica-gel μεμβpαvε. To elute the RNA 50μl RNase-free water was directly added onto the RNeasy silica-gel membrane and then centrifuged for 1 min at 10,000 rpm.
RT-PCR:
The concentration of RNA was determined by measuring the absorption at 260nm (A260) in a specfrophotometer. The ratio of the readings at 260 nm and 280 nm provides an estimate of the RNA purity. A ratio of A2eo/A28o between 1.8 and 2.0 is a sign for pure RNA. RNA samples that fulfilled the criteria of purity were used for RT-PCR. Two to four microgram purified RNA was treated with DNAse I to remove residual DNA and
then used for first strand cDNA synthesis with Superscript II, oligo (dT)ι2-i8 random primers according to the manufacturer's instruction (Invitrogen). The concentration of cDNA was determined with Oligreen Oligonucleotide Quantitation Reagent (Molecular Probe). PCR reactions were performed with the specific primers. All primers were synthesized at the Oligonucleotide Synthesis Laboratory at UBC.
Table 1: Primers with sequences used for this study

A sample from each PCR product was then subjected to electrophoresis on a 1.5% agarose gel. The fluorescent bands were visualised under UV light (UV-Epi Chemi II) and quantified with UVP-labworks software.
Western Blotting
Membrane Extraction:
Caco-2 cells were grown in T-75 flasks and treated regarding the type of experiment. Procedures for protein extraction were followed as directed by the instruction manual for MEM-PER® Eukaryotic Membrane Protein Extraction Reagent kit by Pierce Biotechnology (Rockford, IL, USA). Briefly, cells were rinsed 3 times with 10ml of 1X PBS. Cells were washed with PBS and harvested into microcentrifuge tubes to pellet down. The supernatant was carefully removed and discarded. About 150μl of Reagent A was added to the cell pellet and pipetted up and down in order to obtain a homogenous cell suspension. At this stage, 5μl of protease inhibitor was added to the mixture and vortexed. It was incubated at room temperature for 10 mins with occasional vortexing. White debris was noted upon addition of Reagent A. Lysed cells were placed on ice. Two parts of Reagent C were diluted with one part Reagent B in order to make sufficient mixture to add 450μl in each sample. 450μl of Reagent B/C mixture was added to each tube of lysed cells and vortexed. Tubes were incubated on ice for 30 minutes, vortexing every 5 minutes. Tubes were centrifuged at 10000g for 3 mins at 4°C. The supernatant was transferred to new tubes and incubated for 10 minutes at 37°C to separate the membrane protein fraction. Tubes were then centrifuged again at room temperature for 2 mins at 10000g to isolate the hydrophobic fraction. The hydrophilic layer (top) was carefully removed from the hydrophobic (bottom) and saved in a new tube. The separated fractions were placed on ice. Protein assay was conducted in order to determine the protein content. Protein Concentration:
Protein content was determined in lysed cells with the Bicinchoninic Acid (BCA) Protein Assay, which utilizes a detergent-compatible formulation for the colorimetric detection and quantitation of total protein. A standard curve with BSA at the range from 25μg/ml to 2mg/ml was prepared. 25μl of both standards and cell samples (in triplicates) were aliquoted into a 96-well microtiter plate and 200μl of Reagent (Mix A:B + 50:1) was added to each well. Absorbance at 540nm was measured. Protein concentration of each sample was determined against the standard curve.
SDS-PAGE (3.5% stacking. 7.5% resolving)
The MINI-PROTEAN 3 Cell apparatus with glass plates and power supply purchased from Biorad (Hercules, CA, USA) was used to run the SDS-PAGE (3.5% stacking, 7.5% resolving).
Cell samples diluted in Laemmli sample buffer containing β-mercaptoethanol in a 1:2 ratio (sample: buffer were boiled at 95°C for 5 minutes.
Total membrane protein (equivalent to 30μg for each sample) was loaded onto 10%
SDS-page. 10μl of prestained protein standards (Kaleidoskop) was loaded onto the first lane.
Western Blotting:
Filter papers and fiber pads were soaked in 1x Transfer buffer for 30 minutes. PVDF membrane was soaked in 100% methanol for 1 minute, rinsed with dH2O and soaked in
1x transfer buffer prior to use. The unit was run at settings of constant 70 V for approximately 2 hours and 30 to 50 V for 2 to 4 hours. After the run, the membrane was stained with Ponceau S solution as a control for proper transfer. The membrane was incubated in blocking buffer (1x PBS, 1% nonfat dried milk, 0.1% Tween-20) overnight at 4°C. The blocking buffer was discarded and the membrane was incubated at 4°C overnight with the primary antibody for P-gp (C219) at a dilution 1:300. The membrane was washed 3 times with blocking buffer and incubated for 1 hour in the 2° antibody solution (1: 2000 -3000 anti-mouse IgG rabbit HRP-conjugated antibody ). The membrane was then washed 3 times with blocking buffer and once with 1x PBS and
0.1% Tween-20. The membrane was incubated in Western Lightning
Chemiluminescence reagent and exposed to Hyperfilm ECL.
Rationale:
The objective of the first part of the study was to determine the limiting and toxic concentrations of Cholesterol and cholesterol absorption inhibitor FM-VP4 in a Caco2- cell system as well as in LLC-PKi cells (Pig kidney cells). An in vitro cytotoxicity assay which looks at several aspects was developed. One advantage is that it can be carried
out in one model system and results in 3 different parameters. MTS-, LDH- and BCA- Assays were used to measure cellular metabolic activity, membrane integrity and protein concentrations respectively.
Experimental Procedure: Caco-2 cells and LLC-PKi cells were seeded into 96-well plates. The growth media was replaced with fresh media every other day. After reaching 90% confluency cells were treated with media, 0.1% Triton X-100 (as positive control for toxicity), FM-VP4 or Cholesterol micelles and incubated for 24 and 96 hours. LDH- and MTS-assay were performed to measure the cytoxicity and cell viability respectively post treatment. 50μl of each well were transferred into a new 96-well plate to perform the LDH-Assay. The MTS-Assay was done on the original plate. At the end cells were washed 3 times with PBS and lysed for BCA Protein Assay. Results:
Figures 12 through 15 show these results.
Discussion:
Treatment with Cholesterol (0.5μM - 200μM) and FM-VP4 (1μM - 500μM) does not show toxic effects on Caco2-cells for 24 hour incubation. Four day incubation with 500μM FM-VP4 results in ~ 60% cytotoxicity. Lower concentrations don't show an increase in cytotoxicity compared to 24 hour treatment. Four day incubation with Cholesterol increases the cytotoxicity at about 10-15%. This assay is now well established in the lab. The advantage is that we have obtained information about 3 paramaters (Cell viability, Cytotoxcity and protein concentration) in one experiment.
EXAMPLE 15 — MDR-1 gene expression after treatment with Cholesterol Absorption Inhibitor
The aim of this study was to analyze the influence of cholesterol absorption inhibitor
"FM-VP4" on the gene expression level of mdr-1 in Caco-2 cells.
Experimental procedure: Caco-2 cells were seeded at 10,000 cells/cm2 in T-75 flasks
(Corning). The growth media was changed every second day. The cells were kept at
37°C in a humidified atmosphere of 5% CO2. For experiments the cells were left for about three to four weeks until they reached about 80-90% confluency. Cells were then treated for one week with different concentrations of FM-VP4 and Cholesterol. Media alone was used as control. Media and media which contained the treatment was changed every other day (resulting in continuous adding of freshly prepared treatment). After 7 days cells were harvested and total RNA was isolated with TRIzol®Reagent (Invitrogen). RNA was reverse transcribed into cDNA. The concentration of cDNA reaction product was measured by using Oligreen-Assay (Molecular Probes). The primers were synthesized at the Oligonucleotide Synthesis Laboratory at UBC. Parameters and conditions for the tested primers were optimized. A sample from each PCR product was subjected to electrophoresis on a 1.5% agarose gel (containing Ethidium bromide). A 10O bp ladder was used to identify the size of PCR products. The fluorescent bands were imaged under UV light (UV-Epi Chemi II) and quantified with UVP-labworks software.
The results of Figure 16 are a summary of about 10 repeats for concentrations of 25 and 50μM, 11 repeats for 10μM FM-VP4 and 12 repeats for Control. A significant decrease is observed for 5 and 10μM FM-VP4. At 10μM FM-VP4 mdr-1 is significantly down-regulated to 38.5 ± 17%, at 5μM FM-VP4 to 61.2 ± 25.1% both compared to control (100 ± 30%).
EXAMPLE 16— DR-7 gene expression after treatment with cholesterol-free liposomal formulation of cholesterol absorption inhibitor "FM-VP4"
The purpose of the study was to determine the influence of liposomal FM-VP4 in a cholesterol-free liposomal preparation on gene expression of ABC-transporter especially mdr-1. Down-regulation of mdr-1 after treatment with 10μM FM-VP4 had been shown in previous experiments. For those a dilution of FM-VP4 in water was applied to Caco-2 cells. Of interest following these observations was the influence of liposomal FM-VP4 in cholesterol-free liposomal preparation (DMPC 100:0, ratio 10:1 DMPC:FM-VP4). FM-VP4 liposomes with three different concentrations (2.5, 5 and
10μM FM-VP4) were tested.
Experimental Procedure: Caco-2 cells were seeded at 10,000 cells/cm2 in T-75 flasks (Corning). The growth media was changed every other day. The cells were kept at 37°C in a humidified atmosphere of 5% CO2. The treatment groups contained 2.5, 5 and 10μM cholesterol-free liposomal FM-VP4 as well as empty cholesterol-free liposomes without FM-VP4. Cells were harvested after one week of treatment, total RNA isolated with TRIzol® according to the protocol. RT-PCR followed that step and specific primers were used to evaluate the expression level of mdr-1. PCR products were resolved by electrophoresis on a 1.5% Agarose gel (containing Ethidiumbromide) and the fluorescent bands were imaged under UV light and quantified using the UVP-Labworks software.
All 3 tested concentrations, as shown in Figure 17, lead to a down-regulation of mdr-1 gene. Incubation with empty liposomes does not show any effect as we hypothesized and can be used as control. The down-regulation in the low concentration range is much stronger than FM-VP4 solubilized in water. Mdr-1 gene expression in Caco-2 cells after treatment with liposomal FM-VP4 at lower concentrations is significantly reduced.
EXAMPLE 17 — DR-f gene expression after treatment with Cholesterol micelles
The aim of this study was to analyze the influence of Cholesterol micelles on the gene expression level of mdr-1 in Caco-2 cells.
Experimental procedure: Caco-2 cells were seeded at 10,000 cells/cm2 in T-75 flasks (Corning). The growth media was changed every second day. The cells were kept at 37°C in a humidified atmosphere of 5% CO2. For experiments the cells were left for about three to four weeks until they reached about 80-90% confluency. Cells were then treated for one week with different concentrations of Cholesterol micelles. Media alone was used as control. Media and media which contained cholesterol micelles and micelle buffer was changed every second day. After one week cells were harvested and RNA following the previously mentioned protocol isolated.
Figure 18 shows cholesterol concentrations up to 2.5μM do not affect the expression level of mdr-1. 5, 10, 25 and 50μM lead to down-regulation of mdr-1. These findings might contribute to our proposed mechanism of an active influx transport via P-gp. In higher concentrations cells don't need much more of cholesterol and therefore down-regulate the possible "influx transporter".
EXAMPLE 18 — rotein expression of P-glycoprotein in Caco-2 cells after incubation with FM-VP4 for 1 week
The aim of the experiment was to analyze the protein expression of P-gp (which is encoded by mdr-1) after incubation with FM-VP4 as well as Cholesterol over a period of 7 days. Both gene expression and protein level were determined in the same experiment.
Experimental Procedure: Caco-2 cells were seeded at 10,000 cells/cm2 in T-75 flasks (Corning). The growth media was changed every 2-3 days. The cells were kept at 37°C in a humidified atmosphere of 5% CO2. Treatment was added and done in duplicates (one flask for RNA-isolation and one for protein isolation). Cells from group I were harvested and total cellular RNA was extracted with TRIzol® according the protocol. RNA was reverse transcripted into cDNA and specific primers were used to evaluate the expression level of mdr-1. Internal control was run by using GAPDH (primer drop). A sample from each PCR product was subjected to electrophoresis on a 1.5% Agarose gel (containing Ethidiumbromide). A 100 bp standard was run to identify the size of the product. The fluorescent bands were imaged under UV light (UVP-Epi Chemi II Darkroom) and quantified with the UVP-Labworks software. Cells from group II underwent a protein extraction procedure. Concentration was measured using the PIERCE BCA Protein Assay Reagent Kit. 15 μg total protein from each sample was electrophoresed on a 11 % SDS Page Gel and transferred to a PVDF membrane (wet transfer). Identification of P-gp was done using the immunoblot technique. The
monoclonal antibody used for P-gp was C219 and the secondary was horseradish peroxide rabbit anti-mouse antibody (Jackson Immuno Research Laboratories, Inc.). After optimizing the assay the best dilutions for the antibody solution turned out to be 1:300 for the primary and 1 :2000 for the secondary. The protein bands were visualized using a chemiluminescence kit from PerkinElmer and an X-Ray film was placed on the membranes. The film was exposed for about 1 minute and then developed.
Figure 19 shows that a seven day incubation with FM-VP4 10μM leads to a lesser expression of P-gp. Down-regulation of mdr-1 gene expression was already shown and the open question was if this might influence its protein level as well. Western Blot experiments could show that mdr-1 gene expression and P-gp protein level are influenced by FM-VP4 and correlated. A lower level of P-Glycoprotein which is encoded by mdr-1 was detectable. Whereas a higher concentration of FM-VP4 (50μM) causes no obvious effect on mdr-1 gene. Western Blots also show no significant change in protein expression compared to untreated Control cells. These data show that that changes in the mdr-1 gene expression lead to correlating changes in the protein level
P-gp-
EXAMPLE 19-Effect of FM-VP4 on functional activity of P-gp in Caco-2 cells
Experiments were performed to measure the activity of P-gp in Caco-2 cells after treatment with FM-VP4. Previous experiments showed that FM-VP4 decreases the gene expression of mdr-1 (which encodes for P-gp) after a one week administration of FM-VP4 (10μM). The down-regulation of mdr-1 resulted in a lower protein expression level of P-gp as shown in Western Blot analysis. Human intestinal epithelia monolayers Caco-2 cells express P-gp on their apical membranes. A good system to measure apical and basolateral transport across monolayers and to determine the permeability of a certain substance is a Transwell Plate. Cells are growing on a semi-permeable membrane which is placed between two chambers. Caco-2 cells differentiate into highly functionalized epithelial barrier. Morphological and biochemical is similar to the small
intestinal columnar epithelium.The kind of experiment determines the donor and acceptor chamber. To measure the drug absorption like the intestinal environment, the substance is given into the apical side and the concentration in the basolateral side is measured. This represents the intestinal absorption. Secretory transport can be determined if the substance is added onto the basolateral side and the transport into the apical chamber is measured. It is a P-gp mediated transport if the basolateral to apical transport is higher than the apical to basolateral transport. Rhodamine 123 is a P-gp substrate and has been used as a probe substrate to measure the functional activity of P-gp. It is a fluorescent dye. Rhodamine 123 has a molar extinction coefficient of 85,200 M"1cm"1 at 511nm.

Rhodamine 6G is another fluorescent probe for P-gp.


The apparent permeability Papp can be determined by using the following equation:
Pa aDpDp= dQ . I
Caco-2 cells

A→B (Absorptive Transport) : Rhodamine was added into apical chamber. Rhodamine samples from basolateral chamber were analyzed by measuring fluorescence
A (Secretory Transport):
Rhodamine was added into basolateral chamber
Rhodamine samples from apical chamber were analyzed by measuring fluorescence The above is a schematic of absorptive and secretory transport studies. Rhodamine 123 is either added into the apical or basolateral chamber. Samples are taken out of the receiver side.
Experimental Procedures: Caco-2 cells were seeded in polycarbonate membrane Transwell plates (Corning, Costar Corp.) at a densitiy of 40.000 cells/cm2 and grown in a humidified chamber (at 37°C, of 5% CO2.) with media changes every 2 days. The growth media (Dulbecco's minimal essential medium-DMEM) contained 10% heat- activated fetal bovine serum, 292 μg/ml glutamine, 0.1 mM non-essential amino acids, 100U/ml penicillin and 100mg/ml glutamine. For the treatment experiments, FM-VP4 in different concentrations was added to the media and either applied onto apical and/or the basolateral side of the Transwell plate. Transepithelial electrical resistance (TEER) of the monolayers was measured to confirm monolayer integrity. Caco-2 cells with TEER Values above 400 Ω/cm2 were then used for transport studies.
Secretory Transport B → A: Cells were treated with different concentrations of FM- VP4 on the basolateral and the apical side. After one week of treatment cells were washed with HBSS with 10mM Hepes, pH 7.4 several times and preincubated in FM- VP4 in HBSS and 10mM Hepes for 1 hour. TEER Values were measured to check the integrity of the monolayer. Transwells with a TEER Value below 400 Ω/cm2 were excluded from the experiment. The Control Transwell contained only media. A positive control for inhibition of P-gp was Verapamil (calcium channel blocker) or C219 a P-gp antibody. Transwell Inserts were transferred into a new sterile 12-well plate containing media, media with FM-VP4 or the P-gp inhibitor Verapamil or C219 and all contained 5μM Rhodamine 123. In a time dependent manner samples (50μl) were taken out of the apical chamber, transferred into a 96-well plate and the fluorescence was measured. The excitation wavelength is 485nm, emission wavelength 520nm. Immediately the samples were replaced by fresh receiver medium (media, media plus
FM-VP4 or media plus P-gp inhibitor).
Results:
As shown in Figure 20 transport of Rhodamine 123 from basolateral into apical chamber after treatment with FM-VP4 is inhibited.
In summary, an effect of FM1-VP4 on the apical uptake of Rhodamine 123 in a Caco-2 cell Transwell system could be observed. Significant P-gp inhibition occurs at concentrations of 25μM FM-VP4 when incubated at both sides (apical as well as basolateral side).
Inhibition of the P-gp activity could be observed at FM-VP4 concentrations higher than 5μM. One concern was that there might be some interaction between FM-VP4 and Rhodamine123. The following experiment was then performed: FM-VP4 was added only to the apical side. After preincubation for 2 days the transfer of Rhodamine 123 from basolateral to apical was measured following the above protocol.
The positive control for inhibition, Verapamil, showed a significant decrease, independently from the side it was added. 10, 50 and 100μM FM-VP4 tend to decrease the efflux of Rhodamine (basolateral to apical) when added into the apical side for two days. Refer to Figures 21-23.
More Efflux studies were performed with a different P-gp substrate, Rhodamine 6G. Again, referring to Figures 24 and 25 and a significant inhibition of P-gp mediated transport can be seen with Verapamil as a positive control. 10, 50 and 100 μM FM-VP4 tend to decrease the efflux of Rhodamine 6G.
In summary it can clearly be seen that FM-VP4 has an influence on the expression of mdr-1. A decrease of the m-RNA level lead to a decrease in P-glycoprotein expression. A lower P-gp activity can then be observed. Less activity of P-gp was demonstrated by using Rhodamine 123 and Rhodamine 6G which are common substrates for P-gp.
EXAMPLE 20— Animal studies
Goal was to investigate mdr-1 A and mdr-1 B gene expression in rats after one week treatment with cholesterol absorption inhibitor FM-VP4.
Experimental Procedure: Adult Sprague-Dawley rats were treated with 20mg FM-VP4 per kg body weight. After 7 days rats were sacrificed and samples of liver and the small intestines (duodenum, jejunum and ileum) were removed and transferred to tubes for flash freezing in liquid nitrogen. RNA was isolated and reverse transcribed.
Discussion:
In earlier experiments we could show that FM-VP4 influences the gene expression of mdr-1 in Caco-2 cells. Ten μM FM-VP4 show a significant decrease in mRNA levels of mdr-1 (38 ± 17% versus 100% for untreated control cells).
Animal experiments were designed to investigate the in vivo effect of FM-VP4. Therefore rats were treated for one week with FM-VP4 (20mg/kg body weight). The level of gene expression of mdr-1 A & B in liver and duodenum was then measured. A decrease in mdr-1 A mRNA and even more in mdr-1 B mRNA was observed in liver tissues in FM-VP4 treated rats. In the duodenum a lower level of mdr-1 A mRNA was measured in rats that were treated with FM-VP4. Mdr-1 B was not detectable in these tissues.
These results, provided in Figures 26 through 29, show that FM-VP4 influences the gene expression of mdr-1 A & B in liver and duodenum tissues in rats.
REFERENCES
1. Levi Rl Declining Mortality in Coronary Hear Diseases Atherosclerosis 1981 1 312-325 2. Law M.R., Wald N.J., Wu., Hacksaw ZA., Bailey A.; Systemic underestimation of association between serum cholesterol concentration and ischemic heart disease in observational studies: Data from BUPA Study; Br. Med. J. 1994; 308:363-366 3. Law M.R., Wald N.J., Thompson S.G.; By how much and how quickly does reduction in serum cholesterol concentration lower risk of ischemic heart disease? Br. Med. J. 1994; 308:367-373 4. La Rosa J.C., Hunninghake D.. Bush D. et al.; The cholesterol facts: A summary of the evidence relating to dietary fats, serum cholesterol and coronary heart disease: A joint statement by the American Heart Association and the National Heart, Lung and Blood Institute. Circulation 1990; 81:1721- 1733 5. Havel R.J., Rapaport E.. Drug Therapy: Management of Primary Hyperlipidemia. New England Journal of Medicine, 1995; 332:1491-1498 6. Endicott et al. , Annu. Rev. Biochem. 59:137, 1989 7. Gerlach et al. , Cancer Surv. 5:25, 1986 8. Look et al., N. Engl. J. Med. 311:231 , 1984 9. Riordan et al., Nature 316:817, 1985) 10. Bellamy et al. , Cancer Invest. 8,547, 1990 11. Moscow et al., Cancer Chemotherapy Biol. Response Modifiers Ann. 81 ,844, 1990) 12. Rommens et al., Proc. Natl. Acad. Sci. USA 88:7500, 1991 13. Juranka et al. , FASE J. 3:2583, 1989 14. Cole et al. Science 258:1650, 1992 80
15. Barrand et al., J. Natl. Cancer Inst. 86:110-117, 1994
16. Georges et al., Proc. Natl. Acad. Sci. USA 87:152, 1990
17. Ng et al., Mol. Cell. Biol. 9:1224, 1989;
18. Van Der Bliek et al. EMBO J., 6:3325, 1987; and Arias et al., "Structure and function of P-glycoprotien and normal liver intestine" in Xenobiotics and Cancer, L Ernster et al (eds.), Japan Sci. Soc. Press, Tokyo/Taylor and Francis, LTD., London, pp. 229-239, 1991
19. Gros et al., Cell 47:371 , 1986
20. Gerlach et al., Cancer Surv. 5:25, 1986
21. Fojo et al., Proc. Natl. Acad. Sci. USA, 84:7735-7738, 1987
22. Garrigues et al., The multi-drug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. PNAS, 99(16): 10347-10352, 2002.
23. Tessner TG & Stenson Wf. Overexpression of MDR1 in an intestinal cell line results in increased cholesterol uptake from micelles. Biochem Biophys Res Comm. 267:565-571,2000.
24. Wang et al., Cholesterol interaction with the Daunorubicin binding site of P- Glycoprotien. Biochem. Biophys. Res. Comm. 276:909-916,2000
25. Luker GD et al., Decreased hepatic accumulation and enhanced esterification of cholesterol in mice deficient in mdrla and mdrlb P-glycoproteins. J. Lipid Res. 42:1389-1394,2001.
26. Luker GD et al., Multidrug resistance (MDR1) p-glycoprotein enhances esterification of plasma membrane cholesterol. J. Biol. Chem. 21 A (11): 6979- 6991 ,1999.
27. Field FJ et al., Esterification of plasma membrane cholesterol and triacylglycerol-rich lipoprotein secretion in CaCo-2 cells: possible role of p- glycoprotein. J. Lipid RΘS. 36:1533-1543,1995.
28. Stedronsky, Biochimica et Biophysica Ada, 1210, 255-287 (1994)
29. Ichihashi et al. in J. Pharmacol. Exp. Ther., 284(1), 43-50 (1998).
Claims
1. A method of decreasing or inhibiting the expression of a gene which mediates cellular cholesterol influx in an animal cell which comprises administering to an animal an effective amount of at least one cholesterol absorption inhibitor.
2. The method of claim 1 wherein the cholesterol absorption inhibitor is a sterol or stanol, or mixture thereof, in a natural or artificially synthesized form.
3. The method of claim 1 wherein the cholesterol absorption inhibitor is a sterol or stanol in any one of their isomeric forms.
4. The method of claim 1 wherein the cholesterol absorption inhibitor is a sterol selected from the group consisting of sitosterol, campesterol, stigmasterol, brassicasterol (including dihydrobrassicasterol), desmosterol, chalinosterol, poriferasterol, clionasterol, ergosterol, coprosterol, codisterol, isofucosterol, fucosterol, clerosterol, nervisterol, lathosterol, stellasterol, spinasterol, chondrillasterol, peposterol, avenasterol, isoavenasterol, fecosterol, and pollinastasterol.
5. The method of claim 1 wherein the cholesterol absorption inhibitor is a stanol selected from the group consisting of selected from the group consisting of sitostanol, campestanol, stigmastanol, brassicastanol (including dihydrobrassicastanol), desmostanol, chalinostanol, poriferastanol, clionastanol, ergostanol, coprostanol, codistanol, isofucostanol, fucostanol, clerostanol, nervistanol, lathostanol, stellastanol, spinastanol, chondrillastanol, pepostanol, avenastanol, isoavenastanol, fecostanol, and pollinastastanol.
6. The method of claim 1 wherein the cholesterol absorption inhibitor is a sterol 83 derivative or a stanol derivative selected from the group consisting of aliphatic esters, aromatic esters, phenolic acid esters, cinnamate esters, ferulate esters, glycosides, acylated glycosides and acylglycosides.
7. The method of claim 1 wherein the cholesterol absorption inhibitor is one or more compounds comprising a sterol or stanol, including biologically acceptable salts thereof, having one or more of the following formulae:

ϋ) O O II II Ft, — C — C -O-R
iii)
R4 — R
iv)
O R4 — (CH2)n —C -O-R
wherein R is a sterol or stanol moiety, R is derived from ascorbic acid and n=1-5, including all biologically acceptable salts or solvates or prodrugs of at least one such compound or of the salts or of the solvates thereof.
8. The method of claim 7 wherein the cholesterol absorption inhibitor is a disodium ascorbyl stanyl phosphate composition which comprises disodium ascorbyl campestanyl phosphate and disodium ascorbyl sitostanyl phosphate. 84
9. The method of claim 1 wherein the cholesterol absorption inhibitor is a hydroxy substituted azetidinone.
10. The method of claim 1 wherein the cholesterol absorption inhibitor is a hydroxy substituted azetidinone compound represented by the formula:

Ar3 is aryl or R5 -substituted aryl;
X, Y and Z are independently selected from the group consisting of -CH2 -, -CH(lower alkyl)- and -C(dilower alkyl)-;
R and R2 are independently selected from the group consisting of -ORe, -O(CO)R6, - O(CO)OR9 and ~0(CO)NR6 R7 ;
Ri and R3 are independently selected from the group consisting of hydrogen, lower alkyl and aryl;
q is 0 or 1 ; r is 0 or 1 ; m, n and p are independently 0, 1 , 2, 3 or 4; provided that at least one of q and r is 1 , and the sum of m, n, p, q and r is 2, 3, 4, 5 or 6; and provided that when p is 0 and r is 1 , the sum of m, q and n is 1 , 2, 3, 4 or 5; 85 R4 is 1-5 substituents independently selected from consisting of lower alkyl, -OR6, - O(CO)R6, -O(CO)OR9, -O(CH2)ι-5 OR6, ~O(CO)NR6 R7, ~NR6 R7, -NR6 (CO)R7, -NR6 (CO)OR9, --NRe (CO)NR7 R8, -NR6 SO2 R9, -COOR6, -CONR6 R7, -COR6, -S02 NR6 R7, S(O)0-2 R9, -O(CH2)ι-ιo -COORe, -O(CH2)ι-ιo CONR6 R7, -(lower alkylene)COOR6, -CH=CH-COOR6, -CF3, -CN, -NO2 and halogen;
R5 is 1-5 substituents independently selected from the group consisting of ~OR6, - O(CO)R6, -O(CO)OR9) -O(CH2)i-5 OR6, -O(CO)NR6 R7, -NR6 R7, -NR6 (CO)R7, -NR6 (CO)OR9, -NR6 (CO)NR7 R8, ~NR6 SO2 Rg, -COOR6, -CONR6 R7, -COR6, -SO2 NR6 R7, S(O)0-2 Rg, -O(CH2)ι-10 -COOR6, -O(CH2)ι-ιo CONR6 R7, -(lower alkylene)COOR6 and -CH=CH-COOR6 ;
Re, R7 and Rs are independently selected from the group consisting of hydrogen, lower alkyl, aryl and aryl-substituted lower alkyl; and
R9 is lower alkyl, aryl or aryl-substituted lower alkyl.
11. The method of claim 10 wherein, in the compound, Aη is phenyl or R4 -substituted phenyl, Ar2 is phenyl or R4 -substituted phenyl and Arβ is R5 -substituted phenyl.
12. The method of claim 10, wherein in the compound, An is R4 -substituted phenyl wherein R4 is halogen; Ar2 is R4 -substituted phenyl wherein R4 is halogen or -ORe, wherein Re is lower alkyl or hydrogen; and Ar3 R5 -substituted phenyl, wherein R5 is - ORε, wherein R6 is lower alkyl or hydrogen.
13. The method of claim 10 wherein in the compound X, Y, and Z are each -CH2 --; R1 and R3 are each hydrogen; R and R2 are each ~OR6, wherein Re is hydrogen; and the sum of m, n, p, q and r is 2, 3 or 4. 86
14. The method of claim 10 wherein in the compound, m, n and r are each zero, q is 1 and p is 2.
15. The method of claim 10 wherein in the compound, p, q and n are each zero, r is 1 and m is 2 or 3.
16. The method of claim 10 wherein the compound is selected from the group consisting of
3(R)-(2(R)-hydroxy-2-phenylethyl)-4(R)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(R)-(2(R)-hydroxy-2-phenylethyl)-4(S)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(S)-(1 (S)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
3(S)-(1 (R)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
3(R)-(1 (R)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
rel-3(R)->(S)-hydroxy-(2-naphthalenyl)methyl!-4(3S)-(4-methoxyphenyl)-1-phe nyl)-1 - phenyl-2-azetidinone;
rel-3(R)->(R)-hydroxy-(2-naphthalenyl)methyl!-4(S)-(4-methoxyphenyl)-1-phen yl-2- azetidinone;
3(R)-(3(R)-hydroxy-3-phenylpropyl)-1 ,4(S)-bis-(4-methoxyphenyl)-2-azetidino ne;
3(R)-(3(S)-hydroxy-3-phenylpropyl)-1 ,4(S)-bis-(4-methoxyphenyl)-2-azetidino ne; 87 4(S)-(4-hydroxyphenyl)-3(R)-(3(R)-hydroxy-3-phenylpropyl-1-(4-methoxyphenyl )-2- azetidinone;
4(S)-(4-hydroxyphenyl)-3(R)-(3(S)-hydroxy-3-phenylpropyl-1-(4-methoxyphenyl )-2- azetidinone;
rel 3(R)->3(RS)-hydroxy-3->4-(methoxymethoxy)-phenyl!-1 ,4(S)-bis-(4-methoxyphe nyl)- 2-azetidinone;
1-(4-fluorophenyl)-3(R)->3(S)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)-(.sup .4 - hydroxyphenyl)-2-azetidinone;
1-(4-fluorophenyl)-3(R)->3(R)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)-(4-hy droxyphenyl)-2-azetidinone;
4(S)->4-(acetyloxy)phenyl!-3(R)-(3(R)-hydroxy-3-phenylpropyl)-1-(4-methoxyp henyl)-2- azetidinone;
4(S)->4-(acetyloxy)phenyl!-3(R)-(3(S)-hydroxy-3-phenylpropyl)-1(4-methoxyph enyl)-2- azetidinone;
1-(.sup.4 -fluorophenyl)-3(R)->3(S)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)->4-(phen ylmethoxy)phenyl!-2-azetidinone;
3(R)->3(R)-acetyloxy)-3-phenylpropyl!-1,4(S)-bis-(4-methoxyphenyl)-2-azetid inone;
3(R)->3(S)-acetyloxy)-3-phenylpropyl!-1,4(S)-bis-(4-methoxyphenyl)-2-azetid inone; 3(R)->3(R)-(acetyloxy)-3-(4-fluorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- fluorophenyl)-2-azetidinone;
3(R)->3(S)-(acetyloxy)-3-(4-fluorophenyl)propyi!-4(S)->4-(acetyloxy)phenyl! -1-(4- fluorophenyl)-2-azetidinone;
3(R)->3(R)-(acetyloxy)-3-(4-chlorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- chlorophenyl)-2-azetidinone;
3(R)->3(S)-(acetyloxy)-3-(4-chlorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- chlorophenyl)-2-azetidinone; and
rel 1-(4-fluorophenyl)-4(S)-(4-hydroxyphenyl)-3(1 R)-(1 (R)-hydroxy-3-phenylprop yl)-2- azetidinone.
17. The method of claim 1 wherein the cholesterol absorption inhibitor is an androstane and/or androstene derivative, wherein androstane and/or androstene are coupled with ascorbic acid and represented by one or more of the general formulae:

VI
89

VII

VIII wherein Ri, R2, R3, R4, R5, Re may individually be chosen from hydrogen, OH, carbonyl, and an ascorbyl moiety; and R7 may be hydrogen or any halogen.
18. The method of claim 17 wherein the ascorbyl moiety which is coupled to the compound from the androstane or androstene family is selected individually from one or more of the following structures:
90 

91


92
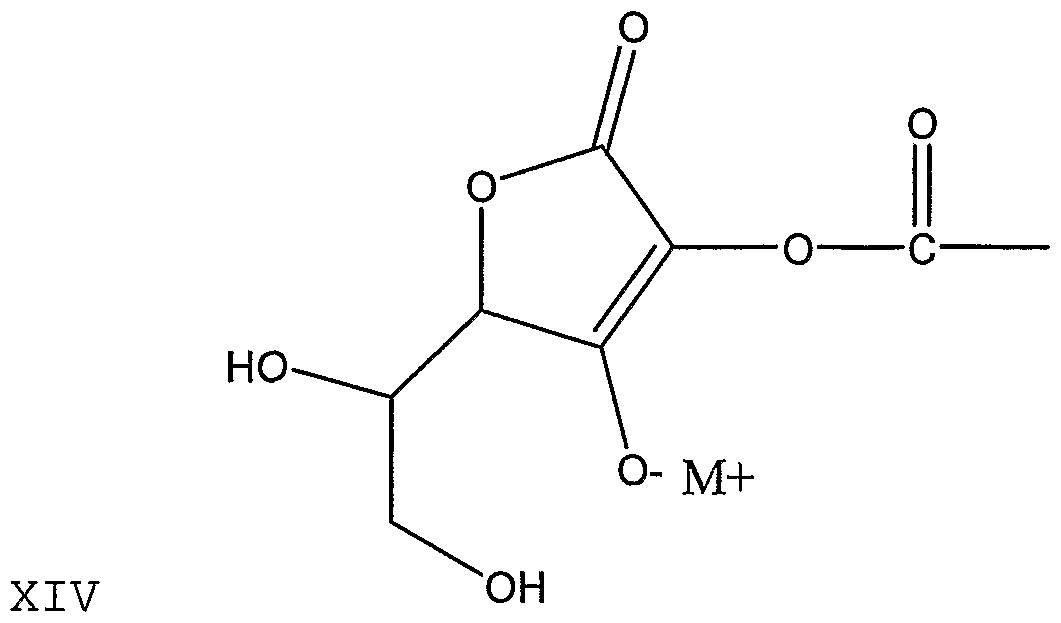



XVIII 
94


wherein M+ represents any metal, alkali earth metal, or alkali metal.
19. The method of claim 1 wherein the cholesterol absorption inhibitor is an inhibitor of bile acid transport or reabsorption and is selected from the group consisting of all ileal, apical and hepatic transport inhibitors.
20. A method of decreasing or inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in an animal cell comprises administering to 95 an animal an effective amount of at least one cholesterol absorption inhibitor.
21. The method of claim 20 wherein the cholesterol absorption inhibitor is a sterol or stanol, or mixture thereof, in a natural or artificially synthesized form.
22. The method of claim 20 wherein the cholesterol absorption inhibitor is a sterol or stanol in any one of their isomeric forms.
23. The method of claim 20 wherein the cholesterol absorption inhibitor is a sterol selected from the group consisting of sitosterol, campesterol, stigmasterol, brassicasterol (including dihydrobrassicasterol), desmosterol, chalinosterol, poriferasterol, clionasterol, ergosterol, coprosterol, codisterol, isofucosterol, fucosterol, clerosterol, nervisterol, lathosterol, stellasterol, spinasterol, chondrillasterol, peposterol, avenasterol, isoavenasterol, fecosterol, and pollinastasterol.
24. The method of claim 20 wherein the cholesterol absorption inhibitor is a stanol selected from the group consisting of selected from the group consisting of sitostanol, campestanol, stigmastanol, brassicastanol (including dihydrobrassicastanol), desmostanol, chalinostanol, poriferastanol, clionastanol, ergostanol, coprostanol, codistanol, isofucostanol, fucostanol, clerostanol, nervistanol, lathostanol, stellastanol, spinastanol, chondrillastanol, pepostanol, avenastanol, isoavenastanol, fecostanol, and pollinastastanol.
25. The method of claim 20 wherein the cholesterol absorption inhibitor is a sterol derivative or a stanol derivative selected from the group consisting of aliphatic esters, aromatic esters, phenolic acid esters, cinnamate esters, ferulate esters, glycosides, acylated glycosides and acylglycosides.
26. The method of claim 20 wherein the cholesterol absorption inhibitor is one or more 96 compounds comprising a sterol or stanol, including biologically acceptable salts thereof, having one or more of the following formulae:
i)

ϋ) O O II II R4 —C — C -O-R
iϋ) F^ — R
iv)
O R4 — (CH2)n —C -O-R
wherein R is a sterol or stanol moiety, R4 is derived from ascorbic acid and n=1-5, including all biologically acceptable salts or solvates or prodrugs of at least one such compound or of the salts or of the solvates thereof.
27. The method of claim 26 wherein the cholesterol absorption inhibitor is a disodium ascorbyl stanyl phosphate composition which comprises disodium ascorbyl campestanyl phosphate and disodium ascorbyl sitostanyl phosphate.
28. The method of claim 20 wherein the cholesterol absorption inhibitor is a hydroxy substituted azetidinone.
29. The method of claim 20 wherein the cholesterol absorption inhibitor is a hydroxy 97 substituted azetidinone compound represented by the formula:

Ar3 is aryl or R5 -substituted aryl;
X, Y and Z are independently selected from the group consisting of -CH2 --, -CH(lower alkyl)- and -C(dilower alkyl)-;
R and R2 are independently selected from the group consisting of -ORe, -0(CO)R6, - 0(CO)OR9 and -0(CO)NR6 R7 ;
Ri and R3 are independently selected from the group consisting of hydrogen, lower alkyl and aryl;
q is 0 or 1 ; r is 0 or 1 ; m, n and p are independently 0, 1 , 2, 3 or 4; provided that at least one of q and r is 1 , and the sum of m, n, p, q and r is 2, 3, 4, 5 or 6; and provided that when p is 0 and r is 1 , the sum of m, q and n is 1 , 2, 3, 4 or 5;
R4 is 1-5 substituents independently selected from consisting of lower alkyl, -ORe, - O(CO)R6, -O(CO)OR9, -O(CH2)ι-5 OR6, -O(CO)NR6 R7, -NR6 R7, -NR6 (CO)R7, -NRe (CO)ORg, -NR6 (CO)NR7 Rs, -NR6 SO2 R9, -COOR6, -CONR6 R7, -COR6, -SO2 NR6 R7, S(O)0-2 Rg, ~0(CH2)ι-ιo -COOR6, -O(CH2)ι-ιo CONR6 R , -(lower alkylene)COOR6, 98 -CH=CH-COOR6, -CF3, -CN, -NO2 and halogen;
Rs is 1-5 substituents independently selected from the group consisting of --ORe, - O(CO)R6, -O(CO)OR9, -O(CH2)ι-5 OR6, -O(CO)NR6 R7, -NR6 R7, -NR6 (CO)R7, -NR6 (CO)ORg, -NR6 (CO)NR7 Rs, -NR6 SO2 Rg, -COOR6, -CONR6 R7, -COR6, -SO2 NR6 R7, S(O)0-2 Rg, -0(CH2)ι-ιo -COOR6, -O(CH2)ι-ιo CONR6 R7, -(lower alkylene)COOR6 and -CH=CH~COOR6 ;
Re, R7 and R8 are independently selected from the group consisting of hydrogen, lower alkyl, aryl and aryl-substituted lower alkyl; and
Rg is lower alkyl, aryl or aryl-substituted lower alkyl.
30. The method of claim 29 wherein, in the compound, An is phenyl or R4 -substituted phenyl, Ar2 is phenyl or R -substituted phenyl and Ar3 is R5 -substituted phenyl.
31. The method of claim 29, wherein in the compound, An is R4 -substituted phenyl wherein R4 is halogen; Ar2 is R4 -substituted phenyl wherein R4 is halogen or -ORe, wherein Re is lower alkyl or hydrogen; and Ar3 R5 -substituted phenyl, wherein R5 is - ORe, wherein R6 is lower alkyl or hydrogen.
32. The method of claim 29 wherein in the compound X, Y, and Z are each -CH2 --; Ri and R3 are each hydrogen; R and R2 are each -OR6, wherein R6 is hydrogen; and the sum of m, n, p, q and r is 2, 3 or 4.
33. The method of claim 29 wherein in the compound, m, n and r are each zero, q is 1 and p is 2.
34. The method of claim 29 wherein in the compound, p, q and n are each zero, r is 1 99 and m is 2 or 3.
35. The method of claim 29 wherein the compound is selected from the group consisting of
3(R)-(2(R)-hydroxy-2-phenylethyl)-4(R)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(R)-(2(R)-hydroxy-2-phenylethyl)-4(S)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(S)-(1 (S)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
3(S)-(1(R)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1-phenyl-2-azetid inone;
3(R)-(1 (R)-hydroxy-3-phenylpropyl)-4(S)-(4-methoxyphenyl)-1 -phenyl-2-azetid inone;
rel-3(R)->(S)-hydroxy-(2-naphthalenyl)methyl!-4(3S)-(4-methoxyphenyl)-1 -phe nyl)-1 - phenyl-2-azetidinone;
rel-3(R)->(R)-hydroxy-(2-naphthalenyl)methyl!-4(S)-(4-methoxyphenyl)-1-phen yl-2- azetidinone;
3(R)-(3(R)-hydroxy-3-phenylpropyl)-1,4(S)-bis-(4-methoxyphenyl)-2-azetidino ne;
3(R)-(3(S)-hydroxy-3-phenylpropyl)-1,4(S)-bis-(4-methoxyphenyl)-2-azetidino ne;
4(S)-(4-hydroxyphenyl)-3(R)-(3(R)-hydroxy-3-phenylpropyl-1-(4-methoxyphenyl )-2- azetidinone;
4(S)-(4-hydroxyphenyl)-3(R)-(3(S)-hydroxy-3-phenylpropyl-1-(4-methoxyphenyl )-2- 100 azetidinone;
rel 3(R)->3(RS)-hydroxy-3->4-(methoxymethoxy)-phenyl!-1 ,4(S)-bis-(4-methoxyphe nyl)- 2-azetidinone;
1-(4-fluorophenyl)-3(R)->3(S)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)-(.sup .4 - hydroxyphenyl)-2-azetidinone;
1-(4-fluorophenyl)-3(R)->3(R)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)-(4-hy droxyphenyl)-2-azetidinone;
4(S)->4-(acetyloxy)phenyl!-3(R)-(3(R)-hydroxy-3-phenylpropyl)-1-(4-methoxyp henyl)-2- azetidinone;
4(S)->4-(acetyloxy)phenyl!-3(R)-(3(S)-hydroxy-3-phenylpropyl)-1(4-methoxyph enyl)-2- azetid inone;
1-(.sup.4 -fluorophenyl)-3(R)->3(S)-(4-fluorophenyl)-3-hydroxypropyl)!-4(S)->4-(phen ylmethoxy)phenyl!-2-azetidinone;
3(R)->3(R)-acetyloxy)-3-phenylpropyl!-1,4(S)-bis-(4-methoxyphenyl)-2-azetid inone;
3(R)->3(S)-acetyloxy)-3-phenylpropyl!-1,4(S)-bis-(4-methoxyphenyl)-2-azetid inone;
3(R)->3(R)-(acetyloxy)-3-(4-fluorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- fluorophenyl)-2-azetidinone;
3(R)->3(S)-(acetyloxy)-3-(4-fluorophenyl)propyl!-4(S)->4-(acetyIoxy)phenyl! -1-(4- fluorophenyl)-2-azetidinone; 101 3(R)->3(R)-(acetyloxy)-3-(4-chlorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- chlorophenyl)-2-azetidinone;
3(R)->3(S)-(acetyloxy)-3-(4-chlorophenyl)propyl!-4(S)->4-(acetyloxy)phenyl! -1-(4- chlorophenyl)-2-azetidinone; and
rel 1-(4-fluorophenyl)-4(S)-(4-hydroxyphenyl)-3(1 R)-(1 (R)-hydroxy-3-phenylprop yl)-2- azetidinone.
36. The method of claim 20 wherein the cholesterol absorption inhibitor is an androstane and/or androstene derivative, wherein androstane and/or androstene are coupled with ascorbic acid and represented by one or more of the general formulae:

VI
102
VII

VIII wherein R ( R2, R3, R4, R5, ε may individually be chosen from hydrogen, OH, carbonyl, and an ascorbyl moiety; and R may be hydrogen or any halogen.
37. The method of claim 36 wherein the ascorbyl moiety which is coupled to the compound from the androstane or androstene family is selected individually from one or more of the following structures:
103


104


105




XVIII 
107


wherein MI+ represents any metal, alkali earth metal, or alkali metal.
38. The method of claim 20 wherein the cholesterol absorption inhibitor is an inhibitor of bile acid transport or reabsorption and is selected from the group consisting of all ileal, apical and hepatic transport inhibitors.
39. The method of claim 1 wherein the gene which mediates cellular cholesterol influx in an animal cell is ABCBI (MDR-1). 108
40. The method of claim 20 wherein the protein is P-glycoprotein.
41. The method of claim 1 wherein the cholesterol absorption inhibitor is provided in the form of a liposome.
42. The method of claim 20 wherein the cholesterol absorption inhibitor is provided in the form of a liposome.
43. A composition for use in decreasing the level of serum LDL cholesterol in an animal, by inhibiting the expression of a gene which mediates cellular cholesterol influx in a cell within said animal and/or by inhibiting the production of a protein expressed by a gene which mediates cellular cholesterol influx in a cell within said animal, said method comprising administering to an animal a therapeutically effective amount of at least one cholesterol absorption inhibitor.
44. The composition of claim 43 wherein the cholesterol absorption inhibitor is a sterol or stanol, or mixture thereof, in a natural or artificially synthesized form.
45. The composition of claim 43 wherein the cholesterol absorption inhibitor is a sterol or stanol, or mixture thereof, in any one of their isomeric forms.
46. The composition of claim 43 wherein the cholesterol absorption inhibitor is a sterol selected from the group consisting of sitosterol, campesterol, stigmasterol, brassicasterol (including dihydrobrassicasterol), desmosterol, chalinosterol, poriferasterol, clionasterol, ergosterol, coprosterol, codisterol, isofucosterol, fucosterol, clerosterol, nervisterol, lathosterol, stellasterol, spinasterol, chondrillasterol, peposterol, avenasterol, isoavenasterol, fecosterol, and pollinastasterol.
109
47. The composition of claim 43 wherein the cholesterol absorption inhibitor is a stanol selected from the group consisting of selected from the group consisting of sitostanol, campestanol, stigmastanol, brassicastanol (including dihydrobrassicastanol), desmostanol, chalinostanol, poriferastanol, clionastanol, ergostanol, coprostanol, codistanol, isofucostanol, fucostanol, clerostanol, nervistanol, lathostanol, stellastanol, spinastanol, chondrillastanol, pepostanol, avenastanol, isoavenastanol, fecostanol, and pollinastastanol.
48. The composition of claim 43 wherein the cholesterol absorption inhibitor is a sterol derivative or a stanol derivative selected from the group consisting of aliphatic esters, aromatic esters, phenolic acid esters, cinnamate esters, ferulate esters, glycosides, acylated glycosides and acylglycosides.
49. The composition of claim 43 wherein the cholesterol absorption inhibitor is one or more compounds comprising a sterol or stanol, including biologically acceptable salts thereof, having one or more of the following formulae:
i) o II R4 — P- O-R I OH
ϋ) O O II II R4 — C— C-O — R
iii)
F^ — R
iv)
110 O 4 — (CH2)n C -O-R
wherein R is a sterol or stanol moiety, R4 is derived from ascorbic acid and n=1-5, including all biologically acceptable salts or solvates or prodrugs of at least one such compound or of the salts or of the solvates thereof.
50. The composition of claim 43 wherein the cholesterol absorption inhibitor is a disodium ascorbyl stanyl phosphate composition which comprises disodium ascorbyl campestanyl phosphate and disodium ascorbyl sitostanyl phosphate.
51. The composition of claim 43 wherein the cholesterol absorption inhibitor is a hydroxy substituted azetidinone.
52. The composition of claim 43 wherein the cholesterol absorption inhibitor is provided in the form of a liposome.
53. The composition of claim 43 wherein the gene which mediates cellular cholesterol influx in an animal cell is ABCB1 (MDR-1).
54. The composition of claim 43 wherein the protein is P-glycoprotein.
Ill
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US69794903A | 2003-10-31 | 2003-10-31 | |
| US10/697,949 | 2003-10-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005042692A2 true WO2005042692A2 (en) | 2005-05-12 |
| WO2005042692A3 WO2005042692A3 (en) | 2005-09-09 |
Family
ID=34550501
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2004/002028 WO2005042692A2 (en) | 2003-10-31 | 2004-11-01 | A method of inhibiting the expression of genes which mediate cellular cholesterol influx in animal cells and inhibiting the production of proteins resulting from the expression of such genes using cholesterol absorption inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2005042692A2 (en) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| DE102007005045A1 (en) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | New phenothiazine derivative for use in preparing medicine for blood sugar lowering and for treatment of diabetes, nicotine dependence, alcohol dependence, central nervous system disorders, schizophrenia, and Alzheimer's disease |
| US7470678B2 (en) | 2002-07-05 | 2008-12-30 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| DE102007063671A1 (en) | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | New crystalline diphenylazetidinone hydrates, medicaments containing these compounds and their use |
| US7635705B2 (en) | 2005-06-20 | 2009-12-22 | Schering Corporation | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US7842684B2 (en) | 2006-04-27 | 2010-11-30 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265B2 (en) | 2005-06-20 | 2011-01-04 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998B2 (en) | 2003-12-23 | 2011-01-18 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048B2 (en) | 2005-06-22 | 2011-02-22 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US7906502B2 (en) | 2005-06-22 | 2011-03-15 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN114716491A (en) * | 2021-11-25 | 2022-07-08 | 广西民族大学 | Three compounds derived from Tougo lignin, and preparation method and application thereof |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0665238A1 (en) * | 1994-01-28 | 1995-08-02 | Senju Pharmaceutical Co., Ltd. | Corticoid derivatives and pharmaceutical and cosmetic compositions |
| WO1998001759A1 (en) * | 1996-07-03 | 1998-01-15 | Forbes Medi-Tech Inc. | A method of assessing risk for cardiovascular disease and other disorders and phytosterol-based compositions useful in preventing and treating cardiovascular disease and other disorders |
| US5767115A (en) * | 1993-09-21 | 1998-06-16 | Schering-Plough Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| US5804576A (en) * | 1983-08-02 | 1998-09-08 | Research Corporation Technologies, Inc. | Derivatives of 5-androsten-17-ones and 5-androstan-17-ones |
| US5994391A (en) * | 1994-09-13 | 1999-11-30 | G.D. Searle And Company | Benzothiepines having activity as inhibitors of ileal bile acid transport and taurocholate uptake |
| WO2000045648A1 (en) * | 1999-02-03 | 2000-08-10 | Forbes Medi-Tech Inc. | Method of preparing microparticles of phytosterols or phytostanols |
| WO2001000653A1 (en) * | 1999-06-23 | 2001-01-04 | Forbes Medi-Tech Inc. | Conjugates of phytosterol or phytostanol with ascorbic acid and use thereof in treating or preventing cardiovascular disease |
| US20030130296A1 (en) * | 2001-08-13 | 2003-07-10 | Ulrike Bauer | FXR NR1H4 nuclear receptor binding compounds |
| WO2003059360A1 (en) * | 2002-01-11 | 2003-07-24 | Medical Isotopes, Inc. | Novel cholesterol absorption lowering agents |
| WO2003104254A2 (en) * | 2002-06-06 | 2003-12-18 | Forbes Medi-Tech Inc. | Novel derivatives of androstane and androstene with ascorbic a cid and use thereof in treating or preventing various conditions, diseases, and disorders |
-
2004
- 2004-11-01 WO PCT/CA2004/002028 patent/WO2005042692A2/en active Application Filing
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5804576A (en) * | 1983-08-02 | 1998-09-08 | Research Corporation Technologies, Inc. | Derivatives of 5-androsten-17-ones and 5-androstan-17-ones |
| US5767115A (en) * | 1993-09-21 | 1998-06-16 | Schering-Plough Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| EP0665238A1 (en) * | 1994-01-28 | 1995-08-02 | Senju Pharmaceutical Co., Ltd. | Corticoid derivatives and pharmaceutical and cosmetic compositions |
| US5994391A (en) * | 1994-09-13 | 1999-11-30 | G.D. Searle And Company | Benzothiepines having activity as inhibitors of ileal bile acid transport and taurocholate uptake |
| WO1998001759A1 (en) * | 1996-07-03 | 1998-01-15 | Forbes Medi-Tech Inc. | A method of assessing risk for cardiovascular disease and other disorders and phytosterol-based compositions useful in preventing and treating cardiovascular disease and other disorders |
| WO2000045648A1 (en) * | 1999-02-03 | 2000-08-10 | Forbes Medi-Tech Inc. | Method of preparing microparticles of phytosterols or phytostanols |
| WO2001000653A1 (en) * | 1999-06-23 | 2001-01-04 | Forbes Medi-Tech Inc. | Conjugates of phytosterol or phytostanol with ascorbic acid and use thereof in treating or preventing cardiovascular disease |
| US20030130296A1 (en) * | 2001-08-13 | 2003-07-10 | Ulrike Bauer | FXR NR1H4 nuclear receptor binding compounds |
| WO2003059360A1 (en) * | 2002-01-11 | 2003-07-24 | Medical Isotopes, Inc. | Novel cholesterol absorption lowering agents |
| WO2003104254A2 (en) * | 2002-06-06 | 2003-12-18 | Forbes Medi-Tech Inc. | Novel derivatives of androstane and androstene with ascorbic a cid and use thereof in treating or preventing various conditions, diseases, and disorders |
Non-Patent Citations (2)
| Title |
|---|
| DE JONG A. ET AL: 'Metabolic effects of plant sterol and stanols(review).' J.NUTR.BIOCHEM. vol. 14, no. 7, July 2003, pages 362 - 369 * |
| KOSUBEK ET AL: 'Liposomal Drug Delivery, A novel Approach: PLARosomes.' ACTA BIOCHIMICA POLONICA. vol. 47, no. 3, 2000, pages 639 - 649 * |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7470678B2 (en) | 2002-07-05 | 2008-12-30 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US7871998B2 (en) | 2003-12-23 | 2011-01-18 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7635705B2 (en) | 2005-06-20 | 2009-12-22 | Schering Corporation | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists |
| US7846946B2 (en) | 2005-06-20 | 2010-12-07 | Schering Plough Corporation | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists |
| US7863265B2 (en) | 2005-06-20 | 2011-01-04 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7893048B2 (en) | 2005-06-22 | 2011-02-22 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502B2 (en) | 2005-06-22 | 2011-03-15 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7842684B2 (en) | 2006-04-27 | 2010-11-30 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| DE102007005045A1 (en) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | New phenothiazine derivative for use in preparing medicine for blood sugar lowering and for treatment of diabetes, nicotine dependence, alcohol dependence, central nervous system disorders, schizophrenia, and Alzheimer's disease |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| DE102007063671A1 (en) | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | New crystalline diphenylazetidinone hydrates, medicaments containing these compounds and their use |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN114716491A (en) * | 2021-11-25 | 2022-07-08 | 广西民族大学 | Three compounds derived from Tougo lignin, and preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005042692A3 (en) | 2005-09-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2005042692A2 (en) | A method of inhibiting the expression of genes which mediate cellular cholesterol influx in animal cells and inhibiting the production of proteins resulting from the expression of such genes using cholesterol absorption inhibitors | |
| AU2004255285A1 (en) | Novel compounds and compositions comprising sterols and/or stanols and cholesterol biosynthesis inhibitors and use thereof in treating or preventing a variety of diseases and conditions. | |
| ES2200587T3 (en) | COMBINATIONS OF ILEON BILIAR ACID TRANSPORTATION INHIBIDORS AND ESTER CHOLESTERILE TRANSFER PROTEIN INHIBITORS FOR CARDIOVASCULAR INDICATIONS. | |
| AU2022221410A1 (en) | Compositions for improving cell viability and methods of use thereof | |
| KR20010102963A (en) | Combinations of ileal bile acid transport inhibitors and nicotinic acid derivatives for cardiovascular indications | |
| US20030153541A1 (en) | Novel anticholesterol compositions and method for using same | |
| IL294063B1 (en) | Berberine salts, ursodeoxycholic salts and combinations, methods of preparation and application thereof | |
| EP1140190A1 (en) | Combinations of ileal bile acid transport inhibitors and bile acid sequestering agents for cardiovascular indications | |
| US8361999B2 (en) | Methods of treating cholesterol gallstone disease with ezetimibe | |
| EP1906972B1 (en) | LPA2 receptor agonist for treating diarrhea | |
| AU2007222282A1 (en) | Use of 3,5-seco-4-norcholestane derivatives for obtaining a cytoprotective medicament | |
| AU2007239395A1 (en) | Use of cholest-4-in-3-in-one derivatives to obtain a cytoprotective medicine | |
| US20120245190A1 (en) | Autophagy inducing compound and the uses thereof | |
| WO2012068539A1 (en) | Use and composition of quercetin-3'-o-sulfate for therapeutic treatment | |
| CA2540364A1 (en) | Method of inhibiting the expression of a multi-drug resistance genes and inhibiting the production of proteins resulting from the expression of such genes thereby enhancing the effectiveness of chemotherapeutic agents to treat cancers | |
| NO328733B1 (en) | Phospholipid complexes of proantocyanidin A2, pharmaceutical composition containing the complexes and use thereof | |
| JP2007530528A (en) | Use of serine palmitoyltransferase (SPT) inhibitors for the treatment of atherosclerosis and lipid metabolism disorders | |
| KR20010099937A (en) | Combinations of cholesteryl ester transfer protein inhibitors and fibric acid derivatives for cardiovascular indications | |
| US20210244686A1 (en) | Novel Systems And Methods For The Therapeutic Use of Cannabinoids or Cannabinoid Analogs to Increase The Lipid Order of Cholesterol Containing Cell Membranes | |
| EP1704857B1 (en) | Probucol spiroquinone or probucol diphenoquinone for use in the treatment of low hdl cholesterolemia | |
| WO2000038670A1 (en) | Propanolol metabolites useful for antioxidant activities | |
| DE69908644T2 (en) | COMBINATIONS OF CHOLESTERYL ESTER TRANSFER PROTEIN INHIBITORS AND GALLIC ACID SEQUESTRING AGENTS FOR CARDIOVASCULAR INDICATIONS | |
| Snyder | Investigation of the effects of α-TEA, MSA and t-RES alone and in combination of human MDA-MB-435 breast cancer cells in vitro and in vivo | |
| Dayspring | 14 Phytosterolemia | |
| MXPA06011060A (en) | Use of a serine palmitoyltransferase (spt) inhibitor to treat atherosclerosis and dyslipidemia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| 122 | Ep: pct application non-entry in european phase |