WO2004073630A2 - Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors - Google Patents
Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors Download PDFInfo
- Publication number
- WO2004073630A2 WO2004073630A2 PCT/US2004/004494 US2004004494W WO2004073630A2 WO 2004073630 A2 WO2004073630 A2 WO 2004073630A2 US 2004004494 W US2004004494 W US 2004004494W WO 2004073630 A2 WO2004073630 A2 WO 2004073630A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- secretase inhibitor
- secretase
- group
- analogue
- disorders
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/05—Dipeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
Definitions
- This invention relates generally to methods for treating angiogenic-related disorders. More specifically, this invention relates to methods of treating tumors or proliferative disorders associated with angiogenesis, by administering compositions that inhibit such angiogenesis. Description of Related Art
- Angiogenesis the formation of new capillaries from pre-existing blood vessels, is a fundamental process needed for normal growth, primarily in embryo development, during wound healing and in response to ovulation.
- Angiogenesis is stimulated when hypoxic, diseased, or injured tissues produce and release angiogenic promoters such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)-l, angiogenin, epidermal growth factor (EGF), placental growth factor (PGF), platelet-derived growth factor (PDGF), and tumor necrosis factor alpha (TNF-alpha).
- VEGF vascular endothelial growth factor
- FGF fibroblast growth factor
- angiogenin angiogenin
- EGF epidermal growth factor
- PPF placental growth factor
- PDGF platelet-derived growth factor
- TNF-alpha tumor necrosis factor alpha
- Pathological angiogenesis is a hallmark of cancer, but also occurs in various ischemic and inflammatory diseases, such as rheumatoid arthritis, psoriasis and asthma. Indeed, pathological angiogenesis is implicated in over twenty diseases. Additionally, obesity may be accompanied by angiogenesis because of the cellular hypoxia that usually accompanies the obese state. Obesity-related angiogenesis is believed to facilitate the deposition of fat. [0006] Cancer represents the most extreme, life-threatening disease process in which blood vessel growth plays an important role. Without the formation of new blood vessels, solid tumors will not grow beyond a few millimeters, but with an enriched environment provided by the new blood vessels, tumors thrive.
- Tumor angiogenesis is the proliferation of a network of blood vessels that penetrates into cancerous growths, supplying nutrients and oxygen and removing waste products.
- Solid tumors smaller than 1 to 2 cubic millimeters are not vascularized and persist in situ for a long period of time (from months to years) in an avascular, quiescent status, hi this phase the tumor may contain a few million cells.
- the tumor needs to be supplied by blood vessels that bring oxygen and nutrients and remove metabolic wastes. Beyond the critical volume of 2 cubic millimeters, oxygen and nutrients have difficulty diffusing to the cells in the center of the tumor, causing a state of cellular hypoxia that marks the onset of tumor angiogenesis.
- angiogenesis there is the potential to treat a wide variety of cancers, as well as other angiogenic-related disorders, by inhibiting the process of angiogenesis in the patient.
- treatment should only affect the pathological formation of new blood vessels associated with angiogenic-related diseases, because once a person has stopped growing, their blood vessel system is basically stable and only grows to repair an injury.
- Some of the naturally occurring inhibitors of angiogenesis include angiostatin, endostatin, interferons, platelet factor 4, thrombospondin, transforming growth factor beta, and tissue inhibitor of metalloproteinase.
- angiogenic inhibitors for cancer treatment over standard chemotherapy are the lack of resistance to therapy and the lack of significant side effects on other normal tissues. These angiogenic inhibitors, however, may not necessarily kill tumors, but rather hold them in check indefinitely, making it necessary to continue therapy with angiogenic inhibitors for the life of the individual or use in combination with other standard chemotherapy drugs.
- APP Amyloid precursor protein
- vascular cells including endothelial cells. Indeed, APP is expressed very early during fetal life in the endothelia of neovascularized tissue, and particularly in cerebral endothelia (Ott, M.O.
- APP is a single- transmembrane protein with a 590-680 amino acid extracellular amino terminal domain and an approximately 55 amino acid cytoplasmic tail.
- APP mRNA from the APP gene on chromosome 21 undergoes alternative splicing to yield eight possible iso forms, three of which (the 695, 751 and 770 amino acid isoforms) predominate in the brain.
- APP undergoes proteolytic processing via three enzymatic activities, termed -, ⁇ -and ⁇ -secretase.
- Alpha- secretase cleaves APP at amino acid 17 of the amyloid-beta (A ⁇ ) domain, thus releasing the large soluble amino-terminal fragment ⁇ -APP for secretion. Because ⁇ -secretase cleaves within the A ⁇ domain, this cleavage precludes A ⁇ formation.
- APP can be cleaved by ⁇ -secretase to define the amino terminus of A ⁇ and to generate the soluble amino- terminal fragment ⁇ -APP.
- Amyloid plaques (invariably associated with Alzheimer's Disease (AD), as well as vascular amyloid deposits in cerebral amyloid angiopathy), contain A ⁇ , which is believed to play a key role in AD pathophysiology.
- AD Alzheimer's Disease
- vascular amyloid deposits in cerebral amyloid angiopathy contain A ⁇ , which is believed to play a key role in AD pathophysiology.
- the normal physiological functions of APP still remain unknown.
- Beta-secretase also called BACE; ⁇ -site APP-cleaving enzyme
- BACE ⁇ -site APP-cleaving enzyme
- BACE mediates the primary amyloidogenic cleavage of ⁇ APP and generates a membrane-bound ⁇ APP C-terminal fragment (APP CTF ⁇ ), which is the immediate precursor for the intramembranous ⁇ -secretase cleavage.
- Gamma-secretase activity is associated with a protein complex composed of presenilins (PSI or PS2), nicastrin (Net), PEN-2, APH-la, and APH-lb (Yu, G. et al., J. Biol. Chem., 273, 16470-16475, 1998; Capell, A. et al., J. Biol. Chem., 273, 3205-3211, 1998).
- the expression of these complex components is coordinately regulated, and ⁇ -secretase activity is only detected in the presence of all subunits.
- the catalytic activity of ⁇ -secretase is most likely contributed by the presenilins.
- the presenilins are polytopic transmembrane proteins, which together with signal peptide peptidases and type-4 prepilin peptidases may belong to a novel family of aspartyl proteases.
- the presenilins are essential components of an intramembranous proteolytic activity known as ⁇ -secretase (Wolfe, M.S. et al, J. Biol. Chem., 276, 5413-5416, 2001).
- ⁇ -secretase Several type-I integral membrane proteins have been identified as substrates for ⁇ - secretase, including the Notch receptor (Notch) and APP (De Strooper, B. et al., Nature (London), 398, 518-522, 1999).
- Notch is a signaling molecule that regulates cell-fate determination during development (Artavanis-Tsakonas, S. et al., Science, 284, 770-776 1999).
- Notch Signaling through Notch is triggered by the binding of ligands, such as Delta and Jagged, which induce cleavage of Notch by TACE (Brou, C. et al., Mol. Cell, 5, 207-216, 2000). Subsequent cleavage by ⁇ -secretase releases the Notch intracellular domain, which binds to transcription factors and translocates to the nucleus, where it regulates transcription of Enhancer of Split complex genes (Greenwald, I., Genes Dev., 12, 1751-1762, 1998).
- ligands such as Delta and Jagged
- Notch signaling has been implicated as a regulatory feature of the angiogenic process (Zhong, T. et al., Nature, 414(6860), 216-220, 2001). Additionally, presenilin knockout mice (mice lacking one or both of the presenilin genes, thus displaying varying degrees of impairment in ⁇ -secretase activity) suffer from abnormal vessel formation (Herreman A. et al., PNAS, 12, 11872-11877, 1999; Shen, J. et al., Cell, 89, 629-639, 1997), suggesting that ⁇ -secretase activity may play a role during angiogenesis.
- the present invention provides for the first time the discovery that compounds which inhibit ⁇ -secretase and ⁇ -secretase, referred to as ⁇ -secretase and ⁇ -secretase inhibitors, exhibit potent anti-angiogenic activity, and that administration of these inhibitors to animals or humans afflicted with disorders associated with pathological angiogenesis, such as cancer, proliferative disorders, or inflammatory disorders, inhibits the pathological angiogenesis observed in the afflicted animals or humans.
- the present invention provides a method of treating tumors or proliferative disorders in animals or humans in need of such treatment by administering to the animal or human therapeutically effective amounts in unit dosage form of a composition containing a carrier and at least one ⁇ -secretase or ⁇ -secretase inhibitor that inhibits secretase amyloid precursor protein (APP) processing.
- a composition containing a carrier and at least one ⁇ -secretase or ⁇ -secretase inhibitor that inhibits secretase amyloid precursor protein (APP) processing APP
- the present invention also provides a method of inhibiting angiogenesis associated with tumors, proliferative or inflammatory disorders in animals or humans in need of such inhibition by administering to the animal or human therapeutically effective amounts in unit dosage form of a composition containing a carrier and at least one ⁇ -secretase or ⁇ -secretase inhibitor that inhibits secretase APP processing.
- Gamma-secretase inhibitors that are administered according to the method of the present invention can include, without limitation, aspartyl protease transition-state inhibitors, such as L-685,458; dipeptide protease inhibitors, such as DAPT and DAPM; or isocoumarin- based serine protease inhibitors, such as JLK-6.
- aspartyl protease transition-state inhibitors such as L-685,458
- dipeptide protease inhibitors such as DAPT and DAPM
- isocoumarin- based serine protease inhibitors such as JLK-6.
- Beta-secretase inhibitors that are administered according to the method of the present invention can include, without limitation, peptidomimetic tight binding transition- state analogue inhibitors, such as OM99-2, or substrate analogue peptide inhibitors, such as Z-VLL-CHO GL189, or P10-P4'statV.
- peptidomimetic tight binding transition- state analogue inhibitors such as OM99-2
- substrate analogue peptide inhibitors such as Z-VLL-CHO GL189, or P10-P4'statV.
- Tumors that can be treated according to the method of the present invention include, without limitation, malignant brain tumors, such as glioblastomas; malignant lung tumors, such as adenocarcinomas; or malignant tumors of the breast, colon, kidney, bladder, head or neck.
- Proliferative disorders that can be treated according to the method of the present invention include, without limitation, hematopoietic disorders, such as leukemias, lymphomas or polycythemias; ocular disorders, such as diabetic retinopathy, macular degeneration, glaucoma or retinitis pigmentosa.
- hiflammatory disorders that can be treated according to the method of the present invention include, without limitation, rheumatoid arthritis, osteoarthritis, pulmonary fibrosis, sarcoid granulomas, psoriasis or asthma.
- Fig la Effect of ⁇ and ⁇ secretase inhibitors on the viability of human brain endothelial cells. The potential toxicity of various doses of ⁇ and ⁇ secretase inhibitors was estimated by measuring LDH activity in the culture medium. ANOVA revealed no main effect for L-685,458 but a significant main effect for Z-VVL-CHO (PO.03), OM99-2 (P ⁇ 0.006) and DAPT (P ⁇ 0.009).
- Fig. lb Effect of ⁇ and ⁇ secretase inhibitors on the proliferation of human brain endothelial cells. The amount of viable cells following 24 hours of treatment with various doses of b and g secretase inhibitors was measured using the Quick cell proliferation assay kit. ANOVA revealed a significant main effect of L-685,458 dose (PO.001), of Z-VVL- CHO dose (PO.001), of OM99-2 dose (PO.001) and of DAPT (PO.001).
- Fig 2a Representative pictures showing the effect of L-685,458 and Z-VVL-CHO on capillary morphogenesis.
- Fig. 2b Quantification of network length by Image analysis. The numbers in parenthesis on the x-axis represent the number of 4X fields analyzed. ANOVA revealed significant main effects of Z-VVL-CHO (PO.001) and L-685,458. Post-hoc testing showed significant difference between control and Z-VVL-CHO for all the doses tested (PO.001) and between control and L-685,458 for all the doses tested (PO.001).
- Fig 3a-b Representative pictures showing the effect of various DAPT doses and of various OM99-2 doses on capillary morphogenesis.
- Fig. 3c Quantification of network length by Image analysis. The numbers in parenthesis on the x-axis represent the number of 4X fields analyzed. ANOVA revealed significant main effects of DAPT dose and OM99-2 dose (PO.001). Post-hoc testing showed significant difference between control and DAPT 1 ⁇ M (PO.005), control and OM99-2 1 ⁇ M (PO.005), control and DAPT 5 ⁇ M (PO.001), control and OM99-2 2 ⁇ M (PO.001) and between control and OM99-2 5 ⁇ M (PO.001).
- Fig. 4a-c Effects of ⁇ and ⁇ secretase inhibitors on the metabolism of APP in human brain endothelial cells.
- Fig 4a Media were analyzed by immunoblotting to measure secreted ⁇ -sAPP molecules.
- Fig. 4b Carboxyl-terminal APP fragments and full length APP from human brain endothelial cell lysates.
- Fig. 4c Quantification of ⁇ -sAPP molecules secreted in the culture media of human brain endothelial cells.
- ANOVA revealed a significant main effect for Z-WL-CHO (PO.003) but not for L-685,458 (P 0.13) and post-hoc analysis showed significant differences between control and Z-VVL-CHO (PO.006) showing that Z- VVL-CHO significantly inceases the secretion of ⁇ -sAPP. Quantification of carboxyl- terminal APP fragments generated by human brain endothelial cells. ANOVA revealed significant main effects of OM99-2 (PO.002), L-685,458 (PO.001) and DAPT (PO.001).
- Fig 5a-b The ⁇ -secretase inhibitor Z-VVL-CHO dose-dependently inhibits the formation of microvessel outgrowths by explants of rat aortae.
- Fig. 5a Representative pictures of rat aortic rings embedded in Matrigel showing the progressive sprouting of capillaries with time in function of the dose of Z-WL-CHO used.
- Fig. 5b Quantification by image analysis of the area covered by microvessel outgrowths. ANOVA revealed a significant main effect of Z-VVL-CHO dose (PO.001) and time (PO.001) as well as an interactive term between them (PO.001).
- Fig 6a-b Effect of the ⁇ -secretase inhibitors OM99-2 and P10-P4'statV on the sprouting of micro vessels by explants of rat aortae.
- Fig. 6a Representative pictures showing the formation of microvessel outgrowths in function of time for control aortic rings, for aortic rings treated with 20 ⁇ M of P10-P4'statV and aortic rings treated with 20 ⁇ M of OM99-2.
- Fig. 6b Quantification by image analysis of the area covered by microvessel outgrowths.
- Figure 7a-c Effects of ⁇ -secretase inhibitors on the formation of microvessel outgrowths by explants of rat aortae.
- Fig. 7a Representative pictures depicting the effect of DAPT on microvessel outgrowths.
- Fig. 7b Quantification by image analysis of the area covered by microvessel outgrowths following DAPT treatment. ANOVA revealed significant main effects of DAPT dose (PO.OOI) and time (PO.OOI) as well as an interactive term between them (PO.OOI).
- Fig. 7c Quantification by image analysis of the area covered by microvessel outgrowths following L685,458 treatment. ANOVA revealed significant main effects of L685,458 (PO.OOI) and time (PO.OOI) as well as an interactive term between them (PO.005). Post-hoc testing showed significant differences between control and 1 ⁇ M L685,458 (PO.002) and between control and 5 ⁇ M L685,458 (PO.OOI).
- Fig 8a Anti-tumoral effect of the ⁇ -secretase inhibitor DAPT and of the ⁇ -secretase inhibitor Z-VLL-CHO on human glioblastoma (U-87 MG) xenografts growth rates.
- U-87 MG cells (6xl0 6 ) were injected subcutaneously into both flanks of 8-10 weeks-old nude mice. Mice were injected intraperitoneally with either the vehicle, 5 mg/Kg of body weight of the ⁇ -secretase inhibitor Z-VLL-CHO or 10 mg/Kg of body weight of the ⁇ -secretase inhibitor DAPT, starting when tumors had reached a mean tumor volume of approximately 140 mm (day 8 post implantation).
- FIG. 8b Representative pictures of sections of glioblastoma tumors immunostained with CD31 antibodies.
- Fig. 8c Histogram depicting the estimation of glioblastoma tumor vascularization.
- ANOVA reveals significant main effects of DAPT ( PO.OOI) and Z-VLL- CHO (PO.02).
- Post-hoc analysis shows significant differences between the vascular index of veliicle treated mice and DAPT treated mice (PO.002) and between the vascular index of vehicle treated mice and Z-VLL-CHO treated animals (PO.03) showing that Z-VLL-CHO and DAPT significantly reduce the vascularization of human glioblastoma xenografts.
- Figure 9a Anti-tumoral effect of the ⁇ -secretase inhibitor DAPT and of the ⁇ - secretase inhibitor Z-VLL-CHO on human lung adenocarcinoma (A-549) xenografts growth rates.
- A-549 cells (8.5xl0 6 ) were injected subcutaneously into both flanks of 8-10 weeks-old nude mice. Mice were injected intraperitoneally with either the vehicle, 5 mg/Kg of body weight of the ⁇ -secretase inhibitor Z-VLL-CHO or 10 mg/Kg of body weight of the ⁇ - secretase inliibitor DAPT, starting when tumors had reached a mean tumor volume of approximately 200 mm 3 (day 21 post implantation).
- Fig. 9c Histogram depicting the quantification of the vascularization of lung adenocarcinoma tumors. ANOVA reveals significant main effects of DAPT (PO.01) and Z-VLL-CHO (PO.01). Post-hoc analysis shows significant differences between the vascular index of vehicle treated mice and DAPT treated mice (PO.03) and between the vascular index of vehicle treated mice and Z-VLL-CHO treated animals (P .03), showing that Z-VLL-CHO and DAPT significantly reduce the vascularization of human lung adenocarcinoma xenografts.
- FIG. 10 Anti-tumoral effect of the ⁇ -secretase inhibitor JLK-6 on human lung adenocarcinoma (A-549). xenografts growth rates. A-549 cells (8.5xl0 6 ) were injected subcutaneously into both flanks of 8-10 weeks-old nude mice. Mice were injected intraperitoneally everyday from Day 16 (when the tumors reached a volume of approximately 150 mm 3 ) with either the vehicle or 5 mg/Kg of body weight of the ⁇ -secretase inhibitor JLK- 6. Data are expressed as mean tumor volume (mm 3 ) ⁇ S.E.
- ANOVA reveals significant main effects of JLK-6 (PO.OOI) and time (PO.OOI), showing inhibition of tumor growth in the JLK-6 treated group of animals compared to animals treated with the vehicle, showing that JLK-6 significantly inhibits the growth of human lung adenocarcinoma xenografts.
- the present invention provides methods for treating tumors or proliferative disorders in animals or humans in need of such treatment, comprising the administration of therapeutically effective amounts in unit dosage form of a composition comprised of a carrier and at least one ⁇ -secretase or ⁇ -secretase inhibitor that inhibits ⁇ -secretase or ⁇ -secretase APP processing.
- the present invention also provides methods for inhibiting angiogenesis in animals or humans in need of such inhibition, comprising the administration of therapeutically effective amounts in unit dosage form of a composition comprised of a carrier and at least one ⁇ -secretase or ⁇ -secretase inhibitor that inhibits ⁇ -secretase or ⁇ -secretase APP processing.
- the ⁇ -secretase inhibitors administered according to the method of the present invention can include, without limitation, aspartyl protease transition state analogue ⁇ - secretase inhibitors, having a general backbone structure, illustrated below. ("R" refers to analogue substitutions.)
- L-685,458 functions as a transition state analogue at the catalytic site of an aspartyl protease, and exhibits a similar potency toward A ⁇ 40 and A ⁇ 42. L-685,458 exhibits over a hundred-fold greater selectivity for ⁇ -secretase than for the aspartyl protease cathepsin. L-685,458 also binds to presenilin and blocks Notch intracellular domain production.
- Another class of ⁇ -secretase inhibitors administered according to the method of the present invention can include, without limitation, dipeptide protease ⁇ -secretase inhibitors, having the general backbone structures illustrated below. (R refers to analogue substitutions.)
- DAPM N-[N-3,5-difluorophenacetyl]-L-alanyl-S-phenylglycine
- DAPM dipeptide protease inhibitor of ⁇ -secretase (IC 50 Ab ⁇ 10 nM in 7PA2 cells) with anti-aggregation properties.
- DAPM prevents early A ⁇ oligomerization by selectively blocking A ⁇ dimer and trimer formation.
- Another dipeptide protease ⁇ -secretase inhibitor is DAPT (N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S- phenylglycine t-butyl ester).
- Still another class of ⁇ -secretase inhibitors administered according to the method of the present invention can include isocoumarin-based serine protease ⁇ -secretase inhibitors, having the general backbone structure illustrated below. (R refers to analogue substitutions.)
- an isocoumarin-based serine protease ⁇ -secretase inhibitor administered according to the method of the present invention is JLK-6 (7-amino-4-chloro-3- methoxyisocoumarin), a cell-permeable, active site-directed, irreversible serine protease ⁇ - secretase inhibitor that belongs to the class of isocoumarin analogues. JLK-6 acts as a potent and selective inhibitor of ⁇ -secretase and blocks the production of both A ⁇ 40 and A ⁇ 42 (IC 50
- JLK-6 does not affect either the processing of Notch or the proteolysis of presenilin 1 and 2.
- the ⁇ -secretase inhibitors administered according to the method of the present invention can include, without limitation, peptidomimetic tight binding transition-state analogue ⁇ -secretase inhibitors, which all contain a similar peptidomimetic structural backbone, illustrated below. ("R" refers to analogue substitutions.)
- OM99-2 (Glu-Val-Asn-Leu- ⁇ -Ala-Ala-Glu-Phe [ ⁇ denotes replacement of CONH by (S)-CH(OH)CH 2 ]) is an aspartyl protease inhibitor that acts as a peptidomimetic tight binding transition-state analogue ⁇ -secretase inhibitor.
- OM99-2 is designed from the template of the ⁇ -secretase site of Swedish APP with an Asp to Ala replacement.
- the OM99-2 compound also includes a nonhydrolyzable hydroxyethylene isostere between the amino acids leucine and alanine (above-described ⁇ replacement).
- Z-VLL-CHO N-benzyloxycarbony- val-leu-leucinal
- Z-VLL-CHO N-benzyloxycarbony- val-leu-leucinal
- Z-VLL-CHO is a peptide aldehyde protease inhibitor that exhibits potent, cell-permeable and reversible inhibition of ⁇ -secretase.
- GL189 H-Glu-Val-Asn-Statine- Val-Ala-Glu-Phe-NH
- Stat refers to the unusual amino acid statine ((3S,4S)-4-amino- 3-hydroxy-6-methylheptanoic acid), which has become a prototypical hydroxymethylene isostere, and is contained in pepstatin, the naturally occurring peptide produced by various Streptomyces species.
- R analogue substitutions
- R can be greatly variable as known in the art.
- Proliferative disorders that can be treated according to the method of the present invention include, without limitation, hematopoietic disorders, such as leukemias, lymphomas or polycythemias; and ocular disorders, such as diabetic retinopathy, macular degeneration, glaucoma or retinitis pigmentosa.
- mflammatory disorders that can be treated according to the method of the present invention include, without limitation, rheumatoid arthritis, osteoartliritis, pulmonary fibrosis, sarcoid granulomas, psoriasis or asthma.
- compositions containing ⁇ -secretase or ⁇ -secretase inhibitors can be administered to a patient via various routes including parenterally, orally or intraperitoneally.
- Parenteral administration includes the following routes: intravenous; intramuscular; interstitial; intra- arterial; subcutaneous; intraocular; intracranial; intraventricular; intrasynovial; transepithelial, including transdermal, pulmonary via inhalation, ophthalmic, sublingual and buccal; topical, including ophthalmic, dermal, ocular, rectal, or nasal inhalation via insufflation or nebulization.
- Compounds containing ⁇ -secretase or ⁇ -secretase inhibitors that are orally administered can be enclosed in hard or soft shell gelatin capsules, or compressed into tablets. Compounds also can be incorporated with an excipient and used in the form of ingestible tablets, buccal tablets, troches, capsules, sachets, lozenges, elixirs, suspensions, syrups, wafers, and the like.
- compositions containing ⁇ -secretase or ⁇ -secretase inhibitors can be in the form of a powder or granule, a solution or suspension in an aqueous liquid or non- aqueous liquid, or in an oil-in- water or water-in-oil emulsion.
- the tablets, troches, pills, capsules and the like also can contain, for example, a binder, such as gum tragacanth, acacia, corn starch; gelating excipients, such as dicalcium phosphate; a disintegrating agent, such as corn starch, potato starch, alginic acid and the like; a lubricant, such as magnesium stearate; a sweetening agent, such as sucrose, lactose or saccharin; or a flavoring agent.
- a binder such as gum tragacanth, acacia, corn starch
- gelating excipients such as dicalcium phosphate
- a disintegrating agent such as corn starch, potato starch, alginic acid and the like
- a lubricant such as magnesium stearate
- a sweetening agent such as sucrose, lactose or saccharin
- a flavoring agent such as sucrose, lactose or saccharin.
- tablets, pills, or capsules can be coated with shellac, sugar or both.
- a syrup or elixir can contain the active compound, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavoring. Any material used in preparing any dosage unit form should be pharmaceutically pure and substantially non-toxic. Additionally, the ⁇ -secretase or ⁇ -secretase inhibitors can be incorporated into sustained-release preparations and formulations.
- the ⁇ -secretase or ⁇ -secretase inhibitors can be administered to the CNS, parenterally or intraperitoneally.
- Solutions of the compound as a free base or a pharmaceutically acceptable salt can be prepared in water mixed with a suitable surfactant, such as hydroxypropylcellulose.
- Dispersions also can be prepared glycerol, liquid polyethylene glycols, and mixtures thereof, and in oils. Under ordinary conditions of storage and use, these preparations can contain a preservative and/or antioxidants to prevent the growth of microorganisms or chemical degeneration.
- the pharmaceutical forms suitable for injectable use include, without limitation, sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability .exists. It can be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms, such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium which contains, for example, and without limitation, water, ethanol, polyol (such as glycerol, propylene glycol, and liquid polyethylene glycol), suitable mixtures thereof, or vegetable oils.
- the proper fluidity can be maintained, for example, by the use of a coating, such as lecithin, by the maintenance of the required particle size (in the case of a dispersion) and by the use of surfactants.
- a coating such as lecithin
- surfactants for example, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium bicarbonate, sodium sulfate, sodium sulfate, sodium sulfate, sodium sulfate, sodium sorbic acid, thimerosal, and the like.
- isotonic agents for example, sugars or sodium chloride.
- Sterile injectable solutions are prepared by incorporating the ⁇ -secretase or ⁇ - secretase inhibitor(s) in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filtered sterilization.
- dispersions are prepared by incorporating the various sterilized ⁇ -secretase or ⁇ -secretase inhibitor(s) into a sterile vehicle that contains the basic dispersion medium and any of the other ingredients from those enumerated above.
- the preferred methods of preparation are vacuum drying and freeze drying.
- compositions which are suitable for administration to the nose or buccal cavity include, without limitation, self-propelling and spray formulations, such as aerosol, atomizers and nebulizers.
- the therapeutic ⁇ -secretase or ⁇ -secretase inhibitors of the present invention can be administered to an animal or human alone or in combination with pharmaceutically acceptable carriers or as pharmaceutically acceptable salts, the proportion of which is determined by the solubility and chemical nature of the compound, chosen route of administration, and standard pharmaceutical practice.
- pharmaceutically acceptable carriers or as pharmaceutically acceptable salts include, without limitation, mammalian and non-mammalian animals and vertebrates and invertebrates.
- ⁇ -secretase and ⁇ -secretase inliibitors dose dependently affect the proliferation and differentiation of human brain endothelial cells into capillaries, the formation of microvessel outgrowths in a rat aortic ring model of angiogenesis, and the growth and vascularization of human lung adenocarcinomas. This suggests, without being bound by any particular theory, that ⁇ -secretase and ⁇ -secretase activities are required during the angiogenic process.
- the ⁇ - secretase inhibitor Z-VLL-CHO appears to be profoundly anti-angiogenic, both in a capillary morphogenesis assay and in a rat aortic ring model of angiogenesis. Additionally, it has now been demonstrated that the above-identified ⁇ -secretase and ⁇ -secretase inhibitors can completely inhibit the growth of human brain and human lung adenocarcinoma tumors xenografted into nude mice, which are dependent on angiogenesis for their growth.
- the ⁇ -secretase inhibitor, JLK-6 appears to reduce angiogenesis in vitro and to inhibit the growth of human lung tumor xenografts into nude mice, suggesting that the anti-angiogenic activity of ⁇ -secretase inhibitors are independent of Notch cleavage, because this ⁇ -secretase inhibitor does not affect Notch processing. It is believed, without being bound by any particular theory, that in both human brain and human lung tumor models, the anti-tumor activity of the ⁇ -secretase and ⁇ -secretase inhibitors is mediated by inhibition of angiogenesis, because microvessel density values in the treated tumors have been shown to be significantly decreased.
- Example 1- Effect of ⁇ -secretase and ⁇ -secretase Inhibitors on Capillary Morphogenesis
- L-685,458 the dipeptide protease ⁇ -secretase inhibitors DAPM and DAPT
- JLK-6 dipeptide protease ⁇ -secretase inhibitors DAPM and DAPT
- JLK-6 the substrate analogue peptide ⁇ -secretase inhibitors Z-VLL-CHO and GL189
- the peptidomimetic tight binding transition-state analogue ⁇ -secretase inhibitor OM99-2 on the proliferation and differentiation of primary cultures of human brain endothelial cells, on capillary morphogenesis, and on the processing of APP in human brain endothelial cells, in order to determine the potential role of the APP processing pathway in angiogenesis.
- Matrigel-containing microvessel outgrowths from human middle cerebral arteries isolated following rapid autopsies (2 to 4 hours post-mortem delay) were dissected with the aid of an inverted microscope and dissociated several times in endothelial basal medium (EBM) through a sterile pipette tip. Matrigel fragments were then plated on plastic culture flasks, and incubated in EBM (supplemented with 2% fetal bovine serum and IX penicillin-streptomycin-fungizone mixture) at 37°C, 5% CO 2 with medium changed every 3 days.
- EBM endothelial basal medium
- Confluent human brain endothelial cells (grown on 75 cm2 flasks with EBM 4% FBS medium) were treated for 24 hours with 5 ⁇ M of Z-VVL-CHO, 5 ⁇ M of L-685,458, 5 ⁇ M of OM99-2, 5 ⁇ M of DAPT or went untreated (control). Experiments were done in quadriplicate for each treatment condition. 6E10 (Signet), a niAb that recognizes residues 1- immunoprecipitate soluble APP generated following cleavage by ⁇ -secretase from cell culture medium.
- Immunoprecipitated material was resolved on a 4-20% gradient SDS- PAGE, transferred to PVDF membranes and immunodetected with mAb 22C11 (Roche Diagnostics) that recognizes the amino acids 66-81 of the N-terminal portion of APP (Hibich, C, J. Biol. Chem., 268, 26571-26577, 1993).
- Human brain endothelial cells were lysed on ice using MPERTM reagent (Pierce) supplemented with 1 mM PMSF and 1 mM of sodium- orthovanadate. Samples were sonicated and centrifuged at 10,000 g for 30 min at 4°C. The protein content of the lysates was determined using the BCA Protein assay kit (Pierce).
- Total lysates 50 mg of protein/sample) were separated on a 4-20% gradient SDS-PAGE and transferred to PVDF membranes and immunoprobed with mAb 22C11 in order to detect full length APP and also immunoprobed with an Anti-APP-CT20 (Calbiochem) antibody which recognizes the amino acid residues 751-770 of the carboxyl terminal region of APP (Pinnix et al. 2001; J. Biol. Chem., 276, 481).
- ⁇ -secretase inhibitors Z-VLL-CHO, OM99-2 and GL189 all dose dependently inhibited the proliferation of human brain endothelial cells without affecting their viability (Fig. 1 and 3). Additionally, these ⁇ -secretase inhibitors also potently and dose dependently inhibited the formation of capillary structures in the capillary morphogenesis assay (Fig. 2), suggesting that ⁇ -secretase activity also contributes to the angiogenic process. [0068] The ⁇ -secretase inhibitor, Z-VLL-CHO, stimulated the secretion of ⁇ -sAPP, suggesting an inhibition of ⁇ -secretase activity.
- ⁇ -secretase inhibitors DAPT AND L- 685-485 promoted the accumulation of APP CTF in human brain endothelial cells, modeling the accumulation of APP CTF habitually observed in PSI knockout cells deficient in ⁇ - secretase activity (Fig. 4).
- angiogenesis is a self-limited process, triggered by injury and regulated by well-defined autocrine-paracrine mechanisms (Nicosia et al., Amer. J. Path., 151, 1379-1385, (1997).
- the rat aortic endothelium When the rat aortic endothelium is exposed to a three- dimensional matrix, it switches to a microvascular phenotype that generates branching networks of microvessels (Nicosia et al., Atherosclerosis, 95, 191-199,1992).
- the Matrigel was covered with 1 mL of EBM (4% FBS) containing various doses of Z-VVL-CHO, OM99-2, P10-P4'statV, DAPT, or L- 685,458 as indicated in the figure legends (the culture medium was changed every 3 days). Pictures were taken at day 4, 5 and 6 using a 4X objective. Microvessel outgrowth area was quantified using the Image Pro Plus software. Briefly, ring cultures were photographed using a digital video camera linked to an Olympus BX60 microscope. The outgrowth area was delineated and measured with the Image Pro Plus software by using a strategy of microvessel outgrowth detection based on difference in color intensities between the outgrowths, the Matrigel and the artery ring.
- the artery rings were manually selected and excluded from the area of measurement and the color intensity threshold was adjusted to selectively measure the area occupied by the microvessel outgrowths. Results were expressed as a percentage of the area occupied by microvessel outgrowths at day 4 in control condition. Results [0071]
- the ⁇ -secretase inhibitors, Z-VLL-CHO, OM99-2 and P10-P4'stat all dose dependently and potently inhibited the sprouting of microvessel outgrowths from explants of rat aortae (Fig. 5-6), suggesting the involvement of ⁇ -secretase-like activity during the angiogenic process.
- the ⁇ -secretase inhibitor DAPT appeared to stimulate the sprouting of microvessels at 5 ⁇ M and to inhibit the sprouting of microvessels at 20 ⁇ M (Fig. 7). Additionally, the ⁇ - secretase inhibitor L-685,458 inhibited the sprouting at 1 and 5 ⁇ M, further supporting the involvement of a ⁇ -secretase-like activity during angiogenesis (Fig. 7).
- the human glioblastoma U-87 MG and human lung adenocarcinoma A-549 cell lines were obtained from American Tissue Culture Type Collection (Manassas, VA) and were grown in DMEM containing IX penicilline-streptomycine-fungizone and 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO 2 .
- Tumor cells (6xl0 6 ) in 100 ⁇ l of PBS were inoculated subcutaneously into both flanks of 8-10-week-old female nude mice (Harland).
- Tumor volume (in mm 3 ) was determined using the formula (length x width 2 )/2, where length was the longest axis and width the measurement at right angles to the length (Clarke et al. 2000).
- animals were treated intraperitoneally everyday with 100 ⁇ l of 50% DMSO/H O (vehicle group), 5mg/Kg of body weight of Z-VLL-CHO ( ⁇ -secretase inhibitor), 5mg/Kg of JLK-6 ( ⁇ - secretase inhibitor) or with 10 mg/Kg of body weight of DAPT. Data were expressed as mean tumor volume ⁇ S.E for each treatment group.
- Sections were blocked and then immunostained with a 1:40 dilution of a PECAM-1 antibody (BD- Pharmingen, CA) overnight at 4°C in a humidification chamber.
- Vector ABC Kits Vector Laboratories Inc, CA
- Quantification of tumor vascularization was performed using the stereological dissector method. Briefly, forty consecutive sections were taken from a randomly chosen starting point in each tumor. Five sections for each tumor were selected for stereology by taking one section every eight sections. A dissector counting frame was used with inclusion and exclusion lines throughout the reference area. Vessel count was performed at X400 magnification with the use of an Olympus microscope connected to a digital video camera.
- Microvessels were counted in the dissector frame by an experimenter unaware of the different treatment conditions. For each tumor an average vessel count per area of dissector frame was determined. A vascular index was calculated by expressing the vessel count as a percentage of the vessel count in the vehicle treatment condition.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Ophthalmology & Optometry (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Diabetes (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Description

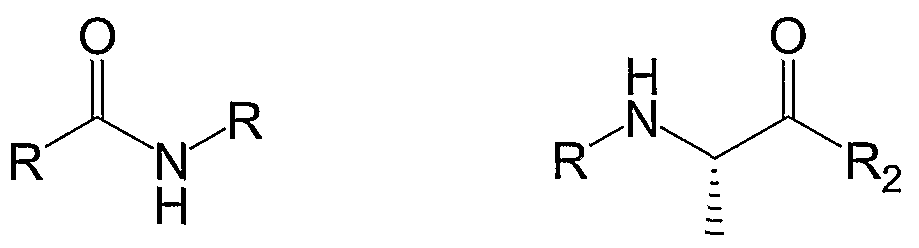


Claims








Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2004212965A AU2004212965A1 (en) | 2003-02-18 | 2004-02-18 | Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors |
| JP2006503611A JP2006517979A (en) | 2003-02-18 | 2004-02-18 | Anti-angiogenic and anti-tumor properties of β- and γ-secretase inhibitors |
| BRPI0407597-8A BRPI0407597A (en) | 2003-02-18 | 2004-02-18 | Anti-angiogenic and anti-tumor properties of beta and gamma secretase inhibitors |
| EP04712274A EP1596878A4 (en) | 2003-02-18 | 2004-02-18 | Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors |
| CA002516259A CA2516259A1 (en) | 2003-02-18 | 2004-02-18 | Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors |
| NO20054221A NO20054221L (en) | 2003-02-18 | 2005-09-12 | Anti-angiogenic and anti-tumor properties of beta and gamma secretase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US31995403P | 2003-02-18 | 2003-02-18 | |
| US60/319,954 | 2003-02-18 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004073630A2 true WO2004073630A2 (en) | 2004-09-02 |
| WO2004073630A3 WO2004073630A3 (en) | 2005-04-28 |
Family
ID=32907558
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/004494 WO2004073630A2 (en) | 2003-02-18 | 2004-02-18 | Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20040229816A1 (en) |
| EP (1) | EP1596878A4 (en) |
| JP (1) | JP2006517979A (en) |
| CN (1) | CN1777436A (en) |
| AU (1) | AU2004212965A1 (en) |
| BR (1) | BRPI0407597A (en) |
| CA (1) | CA2516259A1 (en) |
| NO (1) | NO20054221L (en) |
| WO (1) | WO2004073630A2 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005027969A1 (en) * | 2003-09-24 | 2005-03-31 | Santen Pharmaceutical Co., Ltd. | Remedy for eye diseases accompanied by optic nerve injuries |
| WO2006051405A2 (en) * | 2004-11-12 | 2006-05-18 | Cambridge University Technical Services Ltd. | Methods and means related to cancer stem cells |
| WO2006052128A1 (en) * | 2004-11-10 | 2006-05-18 | Hubrecht Laboratorium | Treatment of an intestinal adenoma and/or adenocarcinoma by inhibition of notch pathway activation |
| EP1671129A1 (en) * | 2003-07-21 | 2006-06-21 | Angiogenetics Sweden AB | Compounds and methods for promoting angiogenesis by using a gamma-secretase inhibitor or inhibiting the gamma-secretase pathway |
| WO2006123185A2 (en) * | 2005-05-19 | 2006-11-23 | Merck Sharp & Dohme Limited | Sulphamides for treatment of cancer |
| WO2006123184A2 (en) * | 2005-05-17 | 2006-11-23 | Merck Sharp & Dohme Limited | Sulphonamido-substituted cyclohexyl sulphones for treatment of cancer |
| WO2006123182A2 (en) | 2005-05-17 | 2006-11-23 | Merck Sharp & Dohme Limited | Cyclohexyl sulphones for treatment of cancer |
| WO2007129457A1 (en) * | 2006-04-25 | 2007-11-15 | The University Of Tokyo | Therapeutic agents for alzheimer's disease and cancer |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008061673A2 (en) * | 2006-11-24 | 2008-05-29 | Synthon B.V. | Drug discovery for cancer |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| WO2008122794A2 (en) * | 2007-04-05 | 2008-10-16 | Imperial Innovations Limited | Breast cancer methods, medicaments and agents |
| EP1996182A2 (en) * | 2006-02-27 | 2008-12-03 | The Johns Hopkins University | Cancer treatment with gamma-secretase inhibitors |
| WO2009024346A2 (en) * | 2007-08-21 | 2009-02-26 | Merz Pharma Gmbh & Co. Kgaa | The use of substances for the treatment of loss of eyesight in humans with glaucoma and other degenerative eye diseases |
| WO2009100218A2 (en) * | 2008-02-05 | 2009-08-13 | Uab Research Foundation | Kruppel-like transcription factor klf4/gklf and uses thereof |
| WO2010063718A1 (en) * | 2008-12-02 | 2010-06-10 | ETH Zürich | Screening assay for metabolic disease therapeuticals |
| US20100222283A1 (en) * | 2007-09-14 | 2010-09-02 | Katalin Susztak | Use of gamma secretase inhibitors and notch pathway inhibitors for treatment and prevention of renal desease |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| CN104987328A (en) * | 2012-05-23 | 2015-10-21 | 复旦大学 | 7-oxygen, sulphur or aza-substituent coumarin and derivative and application thereof |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
| KR20200077480A (en) * | 2020-06-19 | 2020-06-30 | 성균관대학교산학협력단 | A pharmaceutical composition for preventing or treating a human cytomegalovirus disease comprising a gamma secretase inhibitor, and a method for screening a therapeutic agent for a human cytomegalovirus disease using gamma secretase |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1888078A2 (en) * | 2005-05-17 | 2008-02-20 | Merck Sharp & Dohme Ltd. | Sulphone derivatives for treatment of cancer |
| US8110557B2 (en) * | 2006-07-05 | 2012-02-07 | The Trustees Of The University Of Pennsylvania | Gamma secretase inhibitor for treatment of herpesvirus infection |
| KR100881747B1 (en) * | 2007-06-08 | 2009-02-06 | 한양대학교 산학협력단 | A pharmaceutical composition for prevention and treating airway inflammation, overproduction of mucin, and airway hypersensitiveness symptoms in respiratory inflammatory diseases |
| US20110059114A1 (en) * | 2009-08-05 | 2011-03-10 | Duke University | Compositions and Methods for the Treatment of Radioresistant Glioma Stem Cells |
| CA3143449A1 (en) * | 2013-03-14 | 2014-09-25 | The Regents Of The University Of California | In vitro production of medial ganglionic eminence precursor cells |
| US9558394B2 (en) * | 2014-03-10 | 2017-01-31 | Case Western Reserve University | Histogram of hosoya index (HoH) features for quantitative histomorphometry |
| CN108474723A (en) * | 2015-12-02 | 2018-08-31 | 克莱尔莱特诊断有限责任公司 | Prepare and analyze the method that neoplasmic tissue sample is used to detecting and monitoring cancer |
| CN107349414A (en) * | 2016-05-10 | 2017-11-17 | 北京市神经外科研究所 | Purposes of the protein function inhibitor DAPT in the medicine for preparing treatment tumour |
| EP3541408B1 (en) * | 2016-11-15 | 2024-11-13 | The Schepens Eye Research Institute, Inc. | Compositions and methods for the treatment of aberrant angiogenesis |
| CA3082643A1 (en) | 2017-11-14 | 2019-05-23 | The Schepens Eye Research Institute, Inc. | Runx1 inhibition for treatment of proliferative vitreoretinopathy and conditions associated with epithelial to mesenchymal transition |
| US20210309726A1 (en) * | 2018-05-21 | 2021-10-07 | New York University | Treatment of melanoma brain metastasis by inhibition of amyloid precursor protein cleavage |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0008710D0 (en) * | 2000-04-07 | 2000-05-31 | Merck Sharp & Dohme | Therapeutic compounds |
| US20020151487A1 (en) * | 2000-08-31 | 2002-10-17 | Loyola University Chicago | Method and reagents for epithelial barrier formation and treatment of malignant and benign skin disorders by modulating the notch pathway |
| US6436629B1 (en) * | 2000-10-27 | 2002-08-20 | The Regents Of The University Of California | Modulating angiogenesis |
| US20050232927A1 (en) * | 2004-02-03 | 2005-10-20 | The Regents Of The University Of Michigan | Compositions and methods for characterizing, regulating, diagnosing, and treating cancer |
-
2004
- 2004-02-18 BR BRPI0407597-8A patent/BRPI0407597A/en not_active IP Right Cessation
- 2004-02-18 CN CNA2004800104804A patent/CN1777436A/en active Pending
- 2004-02-18 AU AU2004212965A patent/AU2004212965A1/en not_active Abandoned
- 2004-02-18 WO PCT/US2004/004494 patent/WO2004073630A2/en active Application Filing
- 2004-02-18 US US10/780,905 patent/US20040229816A1/en not_active Abandoned
- 2004-02-18 EP EP04712274A patent/EP1596878A4/en not_active Withdrawn
- 2004-02-18 CA CA002516259A patent/CA2516259A1/en not_active Abandoned
- 2004-02-18 JP JP2006503611A patent/JP2006517979A/en active Pending
-
2005
- 2005-09-12 NO NO20054221A patent/NO20054221L/en not_active Application Discontinuation
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1596878A4 * |
Cited By (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1671129A1 (en) * | 2003-07-21 | 2006-06-21 | Angiogenetics Sweden AB | Compounds and methods for promoting angiogenesis by using a gamma-secretase inhibitor or inhibiting the gamma-secretase pathway |
| WO2005027969A1 (en) * | 2003-09-24 | 2005-03-31 | Santen Pharmaceutical Co., Ltd. | Remedy for eye diseases accompanied by optic nerve injuries |
| WO2006052128A1 (en) * | 2004-11-10 | 2006-05-18 | Hubrecht Laboratorium | Treatment of an intestinal adenoma and/or adenocarcinoma by inhibition of notch pathway activation |
| JP2008519829A (en) * | 2004-11-10 | 2008-06-12 | ヒュブレヒト ラボラトリウム | Treatment of intestinal adenoma and / or adenocarcinoma by inhibition of Notch pathway activation |
| WO2006051405A2 (en) * | 2004-11-12 | 2006-05-18 | Cambridge University Technical Services Ltd. | Methods and means related to cancer stem cells |
| WO2006051405A3 (en) * | 2004-11-12 | 2006-10-19 | Univ Cambridge Tech | Methods and means related to cancer stem cells |
| US10538742B2 (en) | 2004-11-12 | 2020-01-21 | Cambridge Enterprise Limited | Methods and means related to cancer stem cells |
| WO2006123184A3 (en) * | 2005-05-17 | 2007-04-19 | Merck Sharp & Dohme | Sulphonamido-substituted cyclohexyl sulphones for treatment of cancer |
| US8362075B2 (en) | 2005-05-17 | 2013-01-29 | Merck Sharp & Dohme Corp. | Cyclohexyl sulphones for treatment of cancer |
| WO2006123182A2 (en) | 2005-05-17 | 2006-11-23 | Merck Sharp & Dohme Limited | Cyclohexyl sulphones for treatment of cancer |
| WO2006123184A2 (en) * | 2005-05-17 | 2006-11-23 | Merck Sharp & Dohme Limited | Sulphonamido-substituted cyclohexyl sulphones for treatment of cancer |
| WO2006123182A3 (en) * | 2005-05-17 | 2007-01-11 | Merck Sharp & Dohme | Cyclohexyl sulphones for treatment of cancer |
| WO2006123185A3 (en) * | 2005-05-19 | 2007-04-19 | Merck Sharp & Dohme | Sulphamides for treatment of cancer |
| WO2006123185A2 (en) * | 2005-05-19 | 2006-11-23 | Merck Sharp & Dohme Limited | Sulphamides for treatment of cancer |
| US8242103B2 (en) | 2005-05-19 | 2012-08-14 | Merck Sharp & Dohme Limited | Sulphamides for treatment of cancer |
| EP1996182A4 (en) * | 2006-02-27 | 2009-08-12 | Univ Johns Hopkins | Cancer treatment with gamma-secretase inhibitors |
| EP2198863A1 (en) * | 2006-02-27 | 2010-06-23 | The Johns Hopkins University | Cancer treatment with gamma-secretase inhibitors |
| EP1996182A2 (en) * | 2006-02-27 | 2008-12-03 | The Johns Hopkins University | Cancer treatment with gamma-secretase inhibitors |
| JP5311303B2 (en) * | 2006-04-25 | 2013-10-09 | 国立大学法人 東京大学 | Alzheimer's disease and cancer treatment |
| US8394376B2 (en) | 2006-04-25 | 2013-03-12 | The University Of Tokyo | Therapeutic agents for alzheimer's disease and cancer |
| US8398981B2 (en) | 2006-04-25 | 2013-03-19 | The University Of Tokyo | Therapeutic agents for alzheimer's disease and cancer |
| WO2007129457A1 (en) * | 2006-04-25 | 2007-11-15 | The University Of Tokyo | Therapeutic agents for alzheimer's disease and cancer |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008061673A2 (en) * | 2006-11-24 | 2008-05-29 | Synthon B.V. | Drug discovery for cancer |
| WO2008061673A3 (en) * | 2006-11-24 | 2008-10-02 | Synthon Bv | Drug discovery for cancer |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| EP2481408A2 (en) | 2007-03-01 | 2012-08-01 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| WO2008122794A3 (en) * | 2007-04-05 | 2009-04-02 | Imp Innovations Ltd | Breast cancer methods, medicaments and agents |
| WO2008122794A2 (en) * | 2007-04-05 | 2008-10-16 | Imperial Innovations Limited | Breast cancer methods, medicaments and agents |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| WO2009024346A2 (en) * | 2007-08-21 | 2009-02-26 | Merz Pharma Gmbh & Co. Kgaa | The use of substances for the treatment of loss of eyesight in humans with glaucoma and other degenerative eye diseases |
| US10525097B2 (en) | 2007-08-21 | 2020-01-07 | Ramot At Tel Aviv University Ltd | Methods for the treatment of glaucoma and age-related macular degeneratiion by a peptide D-TRP-AIB |
| AU2008290825B2 (en) * | 2007-08-21 | 2012-07-12 | Merz Pharma Gmbh & Co. Kgaa | The use of substances for the treatment of loss of eyesight in humans with glaucoma and other degenerative eye diseases |
| US10987400B2 (en) | 2007-08-21 | 2021-04-27 | Ramot At Tel-Aviv University Ltd | Methods for the treatment of glaucoma and age-related macular degeneration by a peptide D-TRP-AIB |
| WO2009024346A3 (en) * | 2007-08-21 | 2009-06-04 | Merz Pharma Gmbh & Co Kgaa | The use of substances for the treatment of loss of eyesight in humans with glaucoma and other degenerative eye diseases |
| US8377886B2 (en) * | 2007-09-14 | 2013-02-19 | Albert Einstein College Of Medicine Of Yeshiva University | Use of gamma secretase inhibitors and notch pathway inhibitors for treatment and prevention of renal disease |
| US20100222283A1 (en) * | 2007-09-14 | 2010-09-02 | Katalin Susztak | Use of gamma secretase inhibitors and notch pathway inhibitors for treatment and prevention of renal desease |
| WO2009100218A3 (en) * | 2008-02-05 | 2009-10-22 | Uab Research Foundation | Kruppel-like transcription factor klf4/gklf and uses thereof |
| WO2009100218A2 (en) * | 2008-02-05 | 2009-08-13 | Uab Research Foundation | Kruppel-like transcription factor klf4/gklf and uses thereof |
| WO2010063718A1 (en) * | 2008-12-02 | 2010-06-10 | ETH Zürich | Screening assay for metabolic disease therapeuticals |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| CN104987328A (en) * | 2012-05-23 | 2015-10-21 | 复旦大学 | 7-oxygen, sulphur or aza-substituent coumarin and derivative and application thereof |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
| KR20200077480A (en) * | 2020-06-19 | 2020-06-30 | 성균관대학교산학협력단 | A pharmaceutical composition for preventing or treating a human cytomegalovirus disease comprising a gamma secretase inhibitor, and a method for screening a therapeutic agent for a human cytomegalovirus disease using gamma secretase |
| KR102130281B1 (en) | 2020-06-19 | 2020-07-08 | 성균관대학교산학협력단 | A pharmaceutical composition for preventing or treating a human cytomegalovirus disease comprising a gamma secretase inhibitor, and a method for screening a therapeutic agent for a human cytomegalovirus disease using gamma secretase |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0407597A (en) | 2006-02-21 |
| US20040229816A1 (en) | 2004-11-18 |
| CN1777436A (en) | 2006-05-24 |
| CA2516259A1 (en) | 2004-09-02 |
| EP1596878A2 (en) | 2005-11-23 |
| JP2006517979A (en) | 2006-08-03 |
| EP1596878A4 (en) | 2008-05-28 |
| WO2004073630A3 (en) | 2005-04-28 |
| AU2004212965A1 (en) | 2004-09-02 |
| NO20054221L (en) | 2005-11-11 |
| NO20054221D0 (en) | 2005-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20040229816A1 (en) | Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors | |
| US10022419B2 (en) | Methods for treating spinal cord injury | |
| JP5426175B2 (en) | Pharmaceutical composition comprising GHRP-6 capable of preventing and eliminating fibrosis and other forms of pathological deposition in tissue | |
| RU2457853C2 (en) | Lipid raft modulation | |
| JP6162759B2 (en) | Therapeutic agent composition for vascular related diseases comprising peptide as active ingredient | |
| JP2004531466A (en) | Therapeutic agents for modulating angiogenesis and methods of using the same | |
| CN106860855A (en) | The application of polypeptide and polypeptide derivative in fibrotic disease is prevented and treated | |
| TW201904990A (en) | Plasminogen treatment for conditions associated with PAI-1 overexpression | |
| CN110025765B (en) | Extracellular matrix-derived peptide of chondrocyte | |
| CN111093748A (en) | Gas mixtures containing low concentrations of xenon and argon provide neuroprotection without inhibiting the catalytic activity of thrombolytic agents | |
| KR101189655B1 (en) | Composition for treating pulmonary disease comprising mesenchymal stem cell-conditioned media | |
| AU2016373364B2 (en) | Short synthetic peptide and uses thereof | |
| US20210128685A1 (en) | Pedf-derived peptides for promoting meibomian gland regeneration and uses thereof | |
| KR101976633B1 (en) | Pharmaceutical composition for preventing or treating pulmonary fibrosis | |
| Dai et al. | Astragalus (Astragalus mongholicus) improves ventricular remodeling via ESR1 downregulation RhoA/ROCK pathway | |
| US20120093776A1 (en) | Methods for treating diseases using a bone morphogenetic protein | |
| KR102115557B1 (en) | Pharmaceutical composition for preventing or treating pulmonary fibrosis | |
| US8106009B2 (en) | Pharmaceutical composition for preventing or treating ischemic diseases | |
| US8093258B2 (en) | Use of urokinase inhibitors for the treatment and/or prevention of neuropathological diseases | |
| WO2004058296A1 (en) | Remedy for degenerative intervertebral discs | |
| Chen et al. | Role of lysosomal cathepsins in post-myocardial infarction remodeling | |
| US7252835B2 (en) | Agent for preventing and/or treating sinusitis | |
| JP2024530029A (en) | Composition for preventing or treating fibrotic diseases containing HAPLN1 | |
| CN111163793A (en) | Angiotensin receptor agonists and uses thereof | |
| Duong | Exploration of Therapeutic Agents Targeting the Blood-Brain Barrier Integrity and Function and Alzheimer's Disease Pathology |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2516259 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006503611 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004712274 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004212965 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2004212965 Country of ref document: AU Date of ref document: 20040218 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004212965 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4091/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048104804 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004712274 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0407597 Country of ref document: BR |