THERMOFORMABLE OPHTHALMIC LENS
BACKGROUND OF THE INVENTION
This invention relates to the preparation of ophthalmic devices through a molding process. More particularly, the invention relates to a thermoformable hydrogel polymer, and to ophthalmic lenses, in particular, contact lenses, made by the melt processing of that polymer.
The use of contact lenses as corrective ophthalmic devices, as well as for cosmetic purposes, is well known. Various materials have been utilized in making contact lenses, but these materials have been found less than ideal. To be effective as a material for a contact lens, the material must possess certain key properties: (a) sufficient oxygen permeability, (b) good wettability by tear fluid, (c) resistance to protein deposition, (d) dehydration resistance (for hydrogels), (e) sufficient durability for its intended use (e.g. disposable, extended wear, etc.), (f) optical clarity, (g) sufficient light transmission, (h) appropriate manufacturing costs, and (i) comfortable to wear.
Various materials possess some but not all of these properties. For example, contact lenses derived from hydrophobic materials often possess good durability and oxygen permeability, but are not comfortable to wear. Some hydrophobic materials used to make contact lens include methacrylate and acrylate monomers containing large bulky pendant groups containing substituted silicones, styrene monomers containing bulky substituents and the like. However, these lack wettability and comfort.
On the other hand, contact lenses derived from hydrophilic material permit wettability and oxygen permeability. Examples of hydrophilic monomers commonly used include hydroxyalkyl methacrylates, N-vinyl pyrrolidone and polyvinyl alcohol. But these materials lack such properties as durability and dehydration resistance.
Accordingly, there is a need for an ophthalmic lens material that has the benefits of both hydrophobic and hydrophilic materials without the attendant
disadvantages of those materials. Substantial research efforts have focused on developing such new materials. These new materials are in the form of crosslinked hydrogel polymers that have improved physical properties. A hydrogel polymer is generally considered to be a polymer having substantial hydrophilic character that is plasticized by the water it absorbs, and can be described as a soft, elastic, water- containing gel. Materials which contain both hydrophilic and hydrophobic components can form hydrogels. In some cases these materials can be considered amphiphilic, that is exhibiting either hydrophilic or hydrophobic characteristics, depending on the conditions. In addition to the desirability of good physical properties found in hydrogel polymers, it is likewise desirable to be able to efficiently manufacture the polymer and/or convert it into an ophthalmic lens.
Historically, contact lenses have been manufactured by one of the three processes: lathing, spin-casting, and cast molding. Lathing is not able to meet demands of cheap, high- volume, fast production. Efforts to reduce the inherent cost disadvantages of lathing have produced a process that is a hybrid of lathing and cast molding. For example, lenses may be prepared by casting one side of the lens and lathing the other side. This process is cheaper than lathing, but not as cheap as a complete cast molding process. Spin-casting, on the other hand, often results in lenses with optical quality and fitting problems because the back surface of the lens is determined by centrifugal force and not the requirements of an optimized lens mold design.
In contrast, cast molding requires the use of two complementary molds. These molds are often disposable, and the cost to replace the mold for each new lens is a significant part of the total cost of the final lens. Furthermore, lenses made by cast molding also suffer a large number of quality defects during in situ polymerization due to shrinkage. For example, shrinkage may cause surface voids and the non-adherence of the final product to the lens design. Others have attempted to eliminate shrinkage and thereby improve cast molding techniques. For example, U.S. Patent No. 5,039,459 to Kindt-Larsen et al. discloses a replaceable diluent in the monomer mixture polymerized in the casting cup.
However, the disposable casting cup with all of its costs and complications, as well as the complexities of removing the replaceable diluent, are still present. The advantage of the invention described below is that the need for the casting cup and replaceable diluent are eliminated. Accordingly, there is room for improvement in developing suitable processes to make lenses from new polymers with improved properties. Due to processing difficulties of crosslinked polymers, however, the complete polymerization is typically done in the final molding step in making contact lenses. A crossliked polymer is a polymer having linkages between the polymer chains, resulting in a network structure. But in that final molding/polymerization step, numerous quality control issues are present due to process variations. Accordingly, it would be desirable to use a process where the polymerization is performed before the final molding step.
In the contact lens industry, attempts have been made to use injection molding processes to make contact lenses from polymethylmethacrylate (PMMA).
PMMA lenses are hard and not oxygen permeable, i.e., they do not compare to the quality of hydrogel lenses. Thus, while injection molded processes, such as typically used in the plastics industry, are capable of high-speed, high-volume, consistent-quality mass production, there have not been good contact lens materials that possess the key properties outlined above that could take advantage of those plastics manufacturing processes.
Crosslinked hydrophilic polymers for use as hydrogels that may be first prepared in large batches, however, cannot generally be later melt processed in injection molding equipment. Once crosslinked, the polymer cannot be dissolved. Crosslinked polymer can be molded and/or recycled only under special circumstances. Partially, or lightly, crosslinked polymers can be somewhat molded to form a more highly crosslinked thermoset shape through the process of heat stabilization. Also, some crosslinks can be broken during processing under conditions of heat and shear stress. Because of the difficulties in processing crosslinked polymers, there is a need for a thermoformable or melt processable polymer that can be made in large batches of a consistent quality, and processed
using high speed, high volume injection molding equipment to make a soft contact lens with desirable physical qualities. The present invention addresses these needs, and also provides other advantages as will be evident from the following description.
SUMMARY OF THE INVENTION
In one embodiment, the present invention is directed to a melt processable hydrophilic polymer prepared by polymerizing monomers comprising between about 20 and 90% by weight of total monomer of at least one hydrophilic monomer, between about 5 and 80% by weight of total monomer of at least one copolymerizable hydrophobic monomer and no crosslinking agents. In the unhydrated state, the polymer is melt processable at a temperature between about 50°C and 300°C. In the hydrated state, the polymer is a hydrogel that includes a water content between about 35% and about 90% by weight of the total hydrated hydrogel. Preferably, the water content is between about 40% and about 80% by weight. The polymer is optically transparent after it has been formed into an ophthalmic lens and hydrated.
In a second embodiment, this invention includes an ophthalmic lens comprising the polymer in accordance with the first embodiment, and sufficient water absorbed by the polymer to become a hydrogel. In a third embodiment of the invention, a melt processable block copolymer without substantial chemical crosslinks is provided. The uncrosslinked copolymer includes blocks that are self-segregating into immiscible phases. The major volume component in the copolymer includes blocks of hydrophilic units, and the minor volume component includes blocks of hydrophobic units. At temperatures above at least about 80°C or higher, the blocks made from the hydrophobic monomer sufficiently disassociate to allow the polymer to be thermally processed and formed into the desired shape of the ophthalmic device. The uncrosslinked copolymer has sufficient amount of internal hydrophobic association within the hydrophobic domains to be dimensionally stable when
hydrated into a hydrogel polymer, and subject to temperatures up to at least about 60°C or higher.
In a fourth embodiment of the invention, a method is provided for making an ophthalmic lens comprising the steps of providing a polymer prepared by copolymerizing monomers comprising at least one hydrophilic monomer and at least one hydrophobic monomer without the formation of substantial amounts of chemical crosslinks, raising the temperature of the polymer above either the glass transition temperature or the melting temperature, introducing said polymer into an ophthalmic lens mold, forming the polymer into a lens, allowing said lens to solidify in the mold, removing the lens from the mold, and hydrating the lens in a saline solution to form a hydrogel lens having a water content of between about 35 and 90% by weight.
In a fifth embodiment of the invention, a method is provided for molding an ophthalmic lens comprising the steps of copolymerizing a hydrophilic monomer and a hydrophobic monomer in the presence of a monomer with a latent reactive functional group to form a melt processable polymer, then introducing the polymer into a mold, molding said polymer into a lens, reacting said latent functional group to form covalently bonded crosslinks in the polymer, and hydrating the lens to form a hydrogel material. In a sixth embodiment of the invention, a method is provided for molding an ophthalmic lens comprising the steps of providing a solid crosslinkable polymer that is substantially uncrosslinked, introducing the polymer into a lens mold, thermally processing the polymer to allow it to solidify in the lens mold, crosslinking the polymer while it is in the shape of the lens mold, and removing the lens from the mold and hydrating the lens to form a hydrogel with more than
35 weight percent water content of the hydrated lens.
In a seventh embodiment of this invention, a melt processable graft copolymer without substantial chemical crosslinks is provided. The uncrosslinked copolymer includes a backbone chain and grafted side chains that are self- segregating into immiscible phases, or domains. The major volume component in the copolymer is hydrophilic, and the minor volume component is hydrophobic.
At temperatures above at least about 80°C or higher, the domains containing the hydrophobic component sufficiently disassociate to allow the polymer to be thermally processed and formed into the desired shape of the ophthalmic device. The uncrosslinked copolymer has sufficient internal hydrophobic association within the hydrophobic domains to be dimensionally stable when hydrated into a hydrogel polymer, and subjected to temperatures up to at least about 60°C or higher.
In an eighth embodiment of this invention, a melt processable polymer is provided. The polymer includes at least one of a variety of repeating units. In the unhydrated state, the polymer is melt processable at a temperature between about
50°C and 300°C. In the hydrated state, the polymer is a hydrogel that includes a water content between about 35% and about 90% by weight of the total hydrated hydrogel. Preferably, the water content is between about 40% and about 80% by weight. The polymer is optically transparent after it has been formed into an ophthalmic lens and hydrated.
Various embodiments of this invention provide the advantage of a material that can be made into a contact lens that combines excellent key properties otherwise found in either hydrophilic materials or hydrophobic materials. Moreover, various embodiments of this invention provide a material that can be made into a contact lens taking advantage of high speed, high volume thermal molding equipment that uses reusable molds, and eliminates the need for single- use molds. Those advantages and other advantages of the invention will be more readily apparent from the detailed description of the preferred embodiments of the invention that follows.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention may take the form of several different embodiments. One embodiment of the present invention includes a melt processable hydrophilic polymer prepared by polymerizing monomers comprising between about 20 and 90%) by weight monomer of at least one hydrophilic monomer, and between about
5 and 80% by weight of at least one copolymerizable hydrophobic monomer. Preferably, the polymer comprises substantially no crosslinking agents.
In the unhydrated state, the hydrophilic polymer (i.e., xerogel) is melt processable or thermoformable at a temperature between about 50°C and about 300°C. Melt processing is a general method of processing polymers and includes, but is not limited to, such techniques as thermoforming, injection molding, compression molding, resin transfer molding, and extrusion. Melt processing can be carried out in more than one stage, such as extruding the polymer into an intermediate form followed by compression molding into the final shape. Preferably, the xerogel is melt processable at a temperature between about 80°C and 250°C. More preferably, the xerogel is melt processable at a temperature between about 115°C and about 200°C. At these temperatures, the polymer preferably has a viscosity of less than about 100,000 centipoises (cps), or more preferably between about l,000cps and 50,000cps. For some melt processing techniques, such as compression molding, however, the working melt viscosity may be greater than the preferred viscosity ranges stated above. The temperature at which the xerogel is melt processable is preferably greater than the glass transition temperature (Tg) or melt temperature (Tm) of the xerogel, but less than the temperature at which the xerogel degrades. The xerogel has a Tg preferably between about 30°C and 200°C, and a creep stability preferably of at least about
30 minutes at about 120°C. The preferred composition and parameters will depend on the manufacturing process conditions and required characteristics (e.g., oxygen permeability, optical clarity, and water content) of the lens.
In the hydrated state, the hydrophilic polymer forms a hydrogel that includes a water content between about 35% and about 90% by weight of the hydrated polymer (unless stated otherwise herein, water content is expressed by weight percent of the total weight of the hydrated lens), an oxygen permeability (Dk) between about 8 and about 50 barrers (Dk units), a tear strength greater than about 1 g/mm, a tensile modulus between about 20 and about 140 g/mm2. Preferably, the hydrogel polymer is mechanically stable and dimensionally stable after it has been formed into an ophthalmic lens, and is optically transparent with a
visible light transmission of at least 90%, more preferably at least 95%, most preferably at least 99%.
The terms "mechanically stable" and "mechanical stability" are used herein to mean that when the polymer is hydrated into a hydrogel, it maintains its coherent structural integrity and does not dissolve, or become so fragile that it cannot support its own hydrated weight. For example, the fragility or lack of mechanical stability may be demonstrated by the hydrogel tearing apart when attempting to pick up a lens size portion with tweezers. Of course, to be useful as an ophthalmic device, a hydrogel material must have certain mechanical stability to withstand the normal forces associated with handling the device.
The terms "dimensionally stable" and "dimensional stability" are used herein to mean that the change in linear dimension and/or curvature of the polymeric hydrogel material is relatively consistent in any direction (i.e., isotropic) when the material, after being shaped into a lens and hydrated, is subject to a one-time change in temperature. That is to say that the diameter of the lens does not change significantly more in one direction compared to another direction in the plane of the lens, and that the curved shape of the lens does not distort to a degree that it is no longer suitable for its intended use.
A second embodiment of this invention includes an ophthalmic lens that comprises the polymer in accordance with the first embodiment, which becomes a hydrogel material that incorporates between about 35% and 90% by weight water based on the total hydrated weight of the lens, is optically clear, and is dimensionally stable. Preferably, the ophthalmic lens hydrogel material has substantially no covalently bonded crosslinks, which, in other words, means that the crosslink density is sufficiently low so that the hydrogel, if it were dehydrated, would be melt processable at a temperature below where it begins to degrade.
In a third embodiment of the invention, a melt processable block copolymer without substantial chemical crosslinks is provided. The uncrosslinked copolymer includes blocks that are self-segregating into immiscible phases. The
major volume component in the copolymer includes blocks of hydrophilic units, and the minor volume component includes blocks of hydrophobic units. The term "unit" is defined as a moiety linked through at least two bonds to other units to form a polymer. Monomers are transformed into units of a polymer during the polymerization process, thus units are derived or formed from monomers.
At temperatures above at least about 80°C, the blocks of hydrophobic units sufficiently disassociate to allow the polymer to be thermally processed and formed into the desired shape of the ophthalmic device. The uncrosslinked copolymer has sufficient amount of internal hydrophobic association within the domains of hydrophobic units to be mechanically stable when hydrated into a hydrogel polymer, and dimensionally stable when the hydrogel is subject to temperatures up to at least about 60°C or higher.
The copolymer may optionally undergo subsequent treatment to chemically crosslink the polymer and enhance its dimensional stability after being formed into the shape of an ophthalmic device. Alternatively, to enhance dimensional stability, the uncrosslinked polymer may optionally incorporate long side chains pendant to the main polymer chain that can form entanglements or self- segregating immiscible phases. Typically, those long side chains have a weight average molecular weight between about 300 and about 100,000. Preferably, the long side chains have a weight average molecular weight between about 500 and about 50,000. More preferably, the long side chains have a weight average molecular weight between about 1,000 and 10,000. For example, long-chain polysiloxanes or poly(ethylene glycol) acrylates may be incoφorated into the polymer for this purpose. Further, the ophthalmic device of the present invention may take the form of a contact lens, a corneal implant, or an intraocular lens.
When the copolymer is an amorphous polymer without crystalline ordering of the polymer chains, the uncrosslinked copolymer preferably should have a glass transition temperature (Tg) high enough so that it is not soft or tacky at ambient temperatures when it may be handled, stored or transported. Also, the Tg should preferably be low enough so that it does not require a melt processing temperature
to be used near the thermal decomposition temperature, to avoid degradation of the polymer during processing. Accordingly, the Tg may be -40°C to 200°C. For practical purposes, it is desirable that the Tg be between about 30°C and about 200°C. Preferably, the Tg is between about 50°C and 150°C. More preferably, the Tg is between about 60°C and about 125°C. The Tg is the temperature below which the polymer is a rigid glass and above which the polymer is a more flexible material. For example, as the temperature of a polymer is increased through the glass transition region, the polymer may be transformed from a rubbery material to a gum and eventually into a liquid. The Tg is conveniently measured by differential scanning calorimetry (DSC) as the temperature at which there is a change in heat capacity of the polymer. Also, an amorphous polymer will not yield a sharp pattern when analyzed by X-ray diffraction techniques.
Likewise, when the copolymer is a crystalline polymer, the uncrosslinked copolymer should have a melting temperature (Tm) high enough so that it is not soft or tacky at ambient temperatures when it may be handled, stored or transported. Also, the Tm should be low enough so that it does not require a melt processing temperature to be used near the thermal decomposition temperature, to avoid degradation of the polymer during processing. Accordingly, for practical purposes, it is desirable for the polymer to have a Tm between about 100°C or the Tg (whichever is greater) and 250°C. More preferably, the Tm is at least about 5°C above steam sterilization temperature, which is typically about 120°C, but less than about 250°C. Still more preferably, for practical operation in a variety of thermal processing equipment, the Tm is desired to be about 20°C above steam sterilization temperature, that is, about 140°C, but less than about 200°C. The Tm is the temperature at which the crystalline domains in the polymer become disordered. The Tm is conveniently measured by DSC as the temperature at which there is an endothermic transition. Also, a crystalline polymer will yield a pattern of rings, spots, or arcs when analyzed by X-ray diffraction techniques.
In addition, the copolymer may include a combination of amorphous hydrophilic domain, and a crystalline hydrophobic domain. As the temperature of a semicrystalline polymer is increased through the glass transition region, the
polymer may be transformed from a glassy state to an elastomeric state. Further increases in temperature may transform the material to a liquid at its Tm. For this copolymer, the Tg of the amorphous domain and the Tm of the crystalline domain may be independently optimized to attain beneficial advantages associated with the morphological and rheological attributes imparted on the copolymer by each domain. Both the Tg and the Tm should be measurable by DSC.
Optimizing the Tg and Tm has certain advantages. Decreasing the Tg and Tm is expected to allow the polymer to be melt processed at lower temperatures. This, in turn, will improve processability by extending the processable lifetime of the polymers, especially those containing heat-activated latent crosslinking functional groups. Also, lowering the Tg is expected to minimize any crosslinking and/or curing of the polymer before it reaches a lens mold. The Tg and Tm may be varied by adjusting the amount of hydrophobic units in the polymer. Also, greater amounts of units that contain flexible (long) side chains will lower the Tg of the polymer. The degree of crystallinity in a polymer can be lowered by incorporating unsymmetrical units or side chains into the structure. In extreme cases of this embodiment, Tg values well below room temperature could be achieved. Poly(alkyl acrylates) can exhibit Tg values of about -50°C, and polysiloxanes can exhibit values of about -120°C. Incorporation of large amounts of units derived from alkyl acrylates or siloxanes into the overall material could be expected to decrease the Tg values below the temperature ranges specified above.
Preferably, a melt processed hydrogel block copolymer in accordance with this embodiment of the invention would comprise the block copolymer of this embodiment and a water content greater than about 35 weight percent of the hydrated lens. More preferably, the water content is between 35 weight percent and 90 weight percent. Even more preferably, the water content is between 40 weight percent and 80 weight percent. Desirably, the hydrogel block copolymer is optically clear and has a refractive index between about 1.3 and 1.5, preferably about 1.4.
In a fourth embodiment of the invention, a method is provided for making an ophthalmic lens comprising the steps of providing a polymer prepared by copolymerizing monomers comprising at least one hydrophilic monomer and at least one hydrophobic monomer without forming substantial amounts of chemical crosslinks (i.e., less than about 0.25 percent by weight solids of crosslinking agents, preferably about zero percent), introducing said polymer into a lens mold, raising the temperature of the polymer above its glass transition temperature and/or melting temperature, forming a lens, allowing said lens to solidify in the mold, removing the lens from the mold, and hydrating the lens in an aqueous solution to form a hydrogel lens having a water content of between about 35 and
90% by weight of the hydrated lens.
The temperature of the polymer is raised either before or after it is introduced into the mold, or in both steps, depending on the type of processing equipment used. For example, for typical thermoplastic injection molding equipment, the temperature of the polymer would be raised before injecting it into the mold. On the other hand, for compression molding, or vacuum-assisted molding, the polymer being shaped in a thin film, likely need not be heated up until introduced into the mold. Depending on whether the polymer is amorphous or crystalline, the temperature is raised above the Tg or the Tm of the polymer, respectively. The desirable ranges for the Tg or the Tm are consistent with the desirable ranges described above for the third embodiment of the invention.
Optionally, a mold release agent may be used to prevent the polymer from adhering to the mold. A typical mold release agent is a food-grade silicon. One skilled in the art may select any suitable mold release agent that is commercially available and compatible for use with the polymer materials of this invention.
In a fifth embodiment of the invention, a method is provided for molding an ophthalmic lens comprising the steps of copolymerizing a hydrophilic monomer and a hydrophobic monomer in the presence of a monomer with a latent reactive functional group to form a melt processable polymer, then introducing the
polymer into a mold, shaping said polymer into a lens, reacting said latent functional group to form covalently bonded crosslinks in the polymer, and hydrating the lens into a clear hydrogel having a water content greater than about 35 weight percent. The latent reaction of the functional groups may be performed during or after the shaping, and before, during or after the hydrating step. To obtain sufficient latent crosslinking it is desirable to use up to about 40% by weight of monomer having latent reactive functional groups. Preferably between about 2% and about 25% by weight of total monomer is used. More preferably, between about 5% and about 20% by weight of such monomer is used.
The amount of latent reactive functional group-containing units used to obtain a certain level of crosslink density in this embodiment of the invention is believed to be appreciably more than the amount of crosslinking agent used to obtain a like amount of crosslink density in a typical crosslinking polymerization reaction of conventional hydrogel polymers. For example, about 0.5 weight percent of EGDMA is typically used during the polymerization of HEMA-based hydrogels to obtain suitable crosslink densities. Likely, the low amounts of EGDMA needed is believed to be attributable to the more efficient mobility of crosslinking agents in the polymerization mixture compared to the limited mobility of pendant latent functional groups already tied to a fixed position in the polymer chain, or the limited access to crosslinking sites by pendant latent functional groups on monomer compounded with the uncrosslinked polymer of an alternative embodiment of the invention.
Because of the perceived inefficiency in crosslinking by this invention, however, it is expected that one skilled in the art may be able to determine whether a crosslinked hydrogel has been crosslinked by use of conventional crosslinking agents during polymerization, or crosslinked by use of the methods of this invention. Simple analytical measurements should be able to provide a ratio of crosslink density per units of functional crosslinking groups. It is believed that for conventionally crosslinked hydrogels, this ratio would be relatively high, and for
hydrogels crosslinked in accordance with this invention, the ratio would be relatively low.
In a sixth embodiment of the invention, an ophthalmic lens is made by a process that includes the steps of providing a solid crosslinkable polymer that is substantially uncrosslinked, introducing the polymer into a lens mold, thermally processing (i.e., heating) the polymer to allow it to solidify in the lens mold, crosslinking the polymer while it is in the shape of the lens mold, and removing the lens from the mold. The crosslinking may be initiated or activated by the heat provided when thermally processing the polymer. Also, a curing agent could be used to crosslink the polymer. The curing agent may be compounded with the polymer before it is introduced into the mold, or the curing agent may be applied to the polymer after it has been shaped into the form of a lens. Preferably, a mold release agent is applied to the lens mold before introducing the polymer into the mold. The mold release agent should prevent the lens from adhering to the mold surface, thereby allowing its easy removal. After removal, the lens may be hydrated to form a hydrogel polymer with more than 35 weight percent water content of the hydrated lens.
In one variant of this sixth embodiment, the polymer is placed in a heated mold along with an agent capable of abstracting a part of the polymer to form a reactive species. For example, a suitable peroxide may be formulated with the polymer. The heat decomposes the peroxide, which abstracts a hydrogen from the polymeric material leaving a polymer radical that continues to cause other reactions and crosslinking. If the polymer contains alkenyl groups, the peroxy radicals might add to the alkenyl groups and form polymer radicals. The polymer radicals can combine or add to other alkenyl groups and form crosslinks. Similar effects should result from the use of azides or photosensitive compounds.
In a seventh embodiment of this invention, a melt processable graft copolymer without substantial chemical crosslinks is provided. The uncrosslinked copolymer includes a backbone chain and grafted side chains that are self-
segregating into immiscible phases, or domains. The major volume component in the copolymer is preferably hydrophilic, and the minor volume component is perferably hydrophobic. At temperatures above at least about 80°C or higher, the domains containing the hydrophobic component sufficiently disassociate to allow the polymer to be thermally processed and formed into the desired shape of the ophthalmic device. The uncrosslinked copolymer has sufficient internal hydrophobic association within the hydrophobic domains to be dimensionally stable when hydrated into a hydrogel polymer, and subjected to temperatures up to at least about 60°C or higher.
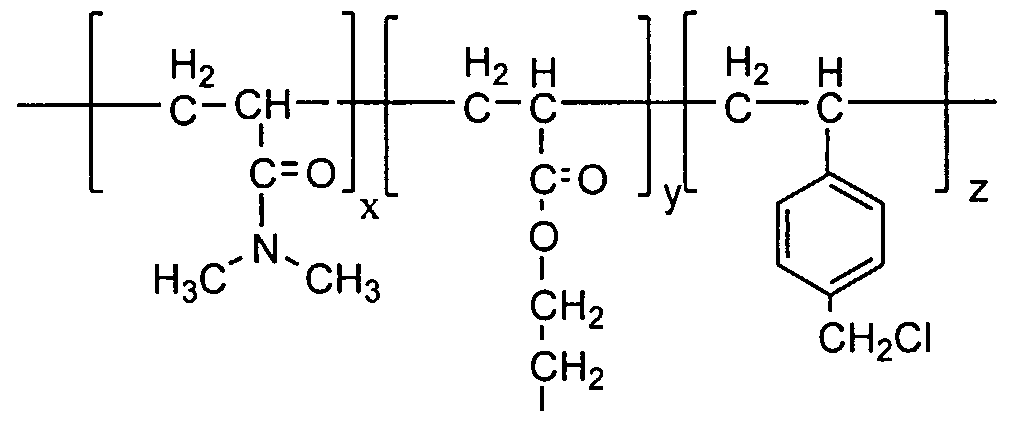
In an eighth embodiment of this invention, a melt processable polymer is provided. The polymer comprises units of formula I and formula II as illustrated below, wherein a unit is a moiety linked through at least two bonds to other units to form a polymer. The dissimilar units are not necessarily linked to each other as is suggested in the illustration. Also, more than one species according to each formula may be present in the polymer.
In formula I, R1 is H or CH3 and R2 is NR5R6 or OR7, wherein R5 is H, CH3, C1-C500 alkyloxy, C1-C500 alkenyloxy, CrC5oo alkylamino, or Cι-C50o alkenylamino; R6 is H, CH3, CH2CH3, Cι-C500 alkyloxy, -C500 alkenyloxy, Cr C500 alkylamino, or Cι-C50o alkenylamino; and R7 is H, CpCsoo alkyloxy, C1-C500 alkenyloxy, Cι-C50o alkylamino, or C1-C500 alkenylamino.
In formula II, R3 is H or CH3 and R4 is Cι-C500 alkyl, C6-C500 aryl, C(O)NR8R9 or C(O)OR10, wherein R8 is H, CH3, C,-C5o0 alkyl, or C6-C500 aryl; R9
is C3-C5o0 alkyl, C6-C50o aryl, polysiloxane, or a chain comprising units of formula II; and R10 is H, C C5o0 alkyl, -C500 fluoroalkoxy optionally containing a linking group of -(CH2CH2N(CH2CH3)SO2)-, or a chain comprising units of formula II. Alkyl is a hydrocarbon chain which may be straight, branched, or cyclic. Alkyloxy is an alkyl group having at least one oxygen (O) present as hydroxy group or ether linkage, for example -(CH2CH(OH)CH3) and -(CH2CH2OCH2CH3). Alkylamino is an alkyl group having at least one nitrogen (N) present as an amino or amido groups or linkages, for example -(CH2CH(NH2)CH3) and - (CH2CH2N(H)CH2CH3). Alkenyl is a hydrocarbon chain which may be straight, branched, or cyclic, containing at least one multiple carbon-carbon bond, but which is not aromatic. Alkenyloxy is an alkyl group having at least one oxygen (O) present as hydroxy group or ether linkage, for example - (CH2CH2OCH2CH=CHCH3). Alkenylamino is an alkyl group having at least one nitrogen (N) present as an amino or amido groups or linkages, for example - (CH2CH2N(H)CH2CH=CHCH3). Aryl is a hydrocarbon comprising at least one aromatic carbocyclic group, optionally containing alkyl or alkoxy substituents or linkages, optionally containing halogen substituents. Polysiloxane is comprised of at least one unit of -(Si(CH3)O)-, optionally containing alkyl or alkoxy linkages or end groups, for example -(CH2CH2CH2O(Si(CH3)2O)xC(CH3)3). Fluoroalkoxy is an alkyl group having at least one fluorine (F) present, for example -
(CH2CH2(CF2)7CF3).
In the unhydrated state, the polymer is melt processable at a temperature between about 50°C and 300°C. In the hydrated state, the polymer is a hydrogel that includes a water content between about 35% and about 90% by weight of the total hydrated hydrogel. Preferably, the water content is between about 40% and about 80% by weight. The polymer is optically transparent after it has been formed into an ophthalmic lens and hydrated.
To practice the various embodiments of this invention, one may prepare the polymer by polymerizing at least one hydrophilic monomer and at least one hydrophobic monomer that are copolymerizable. Such monomers are generally
copolymerizable by free-radical-initiated polymerization at the vinylic bond to create a single unbranched polymer backbone. Preferably, vinylic monomers having either hydrophilic or hydrophobic substituents are used. A polymer prepared completely from a hydrophilic monomer will exhibit hydrophilic characteristics, whereas a polymer prepared completely from a hydrophobic monomer will exhibit hydrophobic characteristics. Hydrophilicity is determined by measuring the contact angle formed between the polymer surface and a drop of water on the surface. The angle is measured with a goniometer. Preferably, a hydrophilic polymer has a contact angle of 0-90°, more preferably 0-45°. Preferably, a hydrophobic polymer has a contact angle of 91 - 180°, more preferably 135-180°.
Generally, for all the embodiments disclosed herein, the hydrophilic monomer may be selected from the group which includes, but is not limited to N,N-dimethyl acrylamide (DMA), 2-hydroxyl ethylmethacrylate (HEMA), 1,2- dihydroxy-ethyl methacrylate, 2-hydroxyl ethyl acrylate (2-HEA), 2-ethoxy ethyl methacrylate (EOEMA), N-vinylpyrrolidone (NVP), glycidyl methacrylate (GMA), glycidyl acrylate, glycerol monomethacrylate, glycerol monoacrylate, 2,3- dihydroxypropylmethacrylate (DHPMA), 2,3-dihydroxypropyl acrylate (DHPA), 3-hydroxypropylmethacrylate, 3-hydroxypropylacrylate, polyhydroxy sucryl alkyl acrylates, acrylamide, acrylic acid, methacrylic acid, 4-hydroxybutyl methacrylate,
4-hydroxybutyl acrylate, N-(2-hydroxypropyl) methacrylamide, N- methylmethacrylamide, poly(ethyleneglycol)monomethacrylate, poly(ethylene glycol)monomethyl ethermonomethacryl, N-vinyl-N-methyl acetamide, vinylmethyl sulfone, N-acryloylmorpholine, N-methyacryloylmorpholine, N- methacryloylpiperidine, N-alkyl acryloyl amide, acid functional monomers (and salts, thereof), and the like, and combinations thereof. More than one hydrophilic monomer may be incorporated into the polymerization mixture.
The preferred hydrophilic monomers are DMA, HEMA, and NVP. The more preferred hydrophilic monomers are: DMA, and HEMA. If too little hydrophilic monomer is used, then the resulting polymer may possess insufficient water content and stiffness, the lens may not be compatible
with eye tissue, and the oxygen permeability may drop. If too much hydrophilic monomer is used, then the resulting polymer may not form a distinct hydrogel. In the preferred embodiment, the hydrophilic monomer is used in the amount between about 20% and about 90% by weight of the total monomeric mixture, preferably in the amount between about 35% and about 80% by weight, and more preferably in the amount between about 45% and about 65% by weight. The exact amount depends on the selected monomeric mixture, processing conditions, desired lens characteristics, and molecular weights of the monomers. The precise amounts of hydrophilic monomers used may be optimized by one of ordinary skill in the art to obtain the desired polymer qualities without undue experimentation based on the teachings contained herein.
Generally, for all embodiments disclosed herein, the hydrophobic monomers are selected from a broad range of monomers that are copolymerizable with the desired hydrophilic monomer. For example, the hydrophilic monomers and hydrophobic monomers may both be vinylic monomers. Such monomers are generally copolymerizable by free-radical-initiated polymerization at the vinylic bond to create a single unbranched polymer backbone. Examples of suitable hydrophobic monomers for use in this invention include, but are not limited to, methylmethacrylate and other alkylated alkyl acrylates, styrene, alkyl substituted styrenes, such as t-butyl styrene, substituted acrylamides and methacrylamides such as: N-isopropylacrylamide (N-IPA), N-(t-butyl)acrylamide, N-(n- octyl)acrylamide, N-(n-octadecyl)acrylamide, N-benzylmethacrylamide, N- diphenylmethacrylamide, N,N-diphenylmethacrylamide and the like, and combinations thereof.
Preferably, the hydrophilic monomer is copolymerized with a hydrophobic fluorinated monomer. Such fluorinated monomers are preferred because of their relatively stronger hydrophobicity compared to non- fluorinated hydrophobic monomers. Preferred fluorinated monomers include 2-(N-ethylperfluorooctane- sulfonamido)ethylacrylate (FX-13), 2-(N-ethylperfluorooctane- sulfonamido)ethylmethacrylate (FX-14), hexafiuoro isopropyl acrylate,
lH,lH,2H,2H-heptadeca fluorodecylacrylate, pentafluoro styrene, trifluoromethyl styrene, fluorostyrene, pentafluoro acrylate, pentafluoro methacrylate, and the like. The preferred hydrophobic monomers are FX-13, FX-14 and 1H,1H,2H,2H- heptadeca fluorodecylacrylate. The most preferred hydrophobic monomers are FX-13 and FX-14.
Also, the polymer may be prepared by copolymerizing a hydrophilic monomer with both a fluorinated hydrophobic monomer and a non- fluorinated hydrophobic monomer. But, it is preferred that the fluorinated hydrophobic monomers are not fluorinated silicon-containing monomer or fluorinated polysiloxane monomer. Such monomers are often used for making materials for rigid gas permeable lenses that often require surface treatments to obtain wetting qualities that are compatible with the cornea. Therefore, such qualities are not favorably disposed on hydrogel materials.
Further, amphiphilic monomers may also be used to prepare the polymer. Such monomers have both hydrophilic and hydrophobic characteristics on different portions of the molecule, and are known in the art. Depending upon which characteristic dominates, these monomers may be considered to be either a hydrophilic monomer or hydrophobic monomer.
The total sum of hydrophobic monomers, or monomers with hydrophobic characteristics, are desirably used in such amounts as to obtain a sufficient degree of association between the resulting hydrophobic units in the polymer chains. The resulting "blocky copolymers" or "block copolymers" have mechanical stability and dimensional stability at storage and use temperatures, and preferably at steam sterilization temperatures. The terms "blocky copolymer" or "block copolymer," unless stated otherwise, are used herein to mean a copolymer with a degree of aggregation of the hydrophobic regions sufficient to form distinct phases apart from the hydrophilic regions to a much greater extent than expected to be found in polymers having a random statistical distribution, in a strict mathematical sense, of a like ratio of hydrophobic and hydrophilic units. Use temperatures are approximately the surface temperature of the human eye, or about 35°C. Storage temperatures may typically range from about 0°C to 50°C, and may be for a
relatively unlimited duration of months, or a few years, and may entail large temperature swings. Heat or steam sterilization temperature is about 120°C, and is for a relatively short duration of about 30 minutes.
Sufficient amounts of hydrophobic monomers are used to achieve transitory pseudo-crosslinking properties. In other words, the hydrophobic units derived from hydrophobic monomers, distributed throughout the polymer have the ability to cause the characteristically hydrophobic portions of the polymer to aggregate and, by internal hydrophobic association, to hold the material together in the absence of covalent bond crosslinks. This domain may be either amorphous or crystalline in structure. The need for this aggregation is lessened as more covalently bonded crosslinking is employed in the material. If too much hydrophobic monomer is used, then lens made from the hydrogel may not be compatible with eye tissue, or segregation, cloudiness, and leatherlike characteristics may occur. If too little hydrophobic monomer is used, then the lens made from the hydrogel may not have sufficient strength, or the polymer may dissolve and the hydrogel not form at all when the dry polymer is hydrated.
In the preferred embodiments of the invention, the hydrophobic monomer is used in the amount between about 5% and about 80% by weight of the starting monomeric mixture, preferably in the amount between about 15% and about 50% by weight, and more preferably between about 20 and 40% by weight. The exact amount depends on the selected monomeric mixture, processing conditions, desired lens characteristics, and molecular weights of the monomers used. The precise amounts of hydrophobic monomers used may be optimized by one of ordinary skill in the art to obtain the desired polymer qualities without undue experimentation based on the teachings contained herein.
By using larger amounts of the same hydrophobic monomer, the water content of the resulting hydrated polymers may be lowered. Alternatively, amounts of additional different hydrophobic monomers may be used, such as methyl methacrylate and N-isopropyl acrylamide, to control water uptake in the resulting swollen hydrogel polymer.
Also, as noted above, the Tm and Tg of the polymer may be lowered by incorporating greater amounts of hydrophobic units or units with flexible side chains or units with unsymmetrical structure into the polymer. It should also be possible to alter Tg and/or Tm by varying the structure of the hydrophilic units. The following list of monomers are examples of some, but not all, of the monomers that are useful to lower the Tg and/or Tm of the polymer upon conversion into units: methyl acrylate, ethyl acrylate, propyl acrylate, butyl acrylate, 2-methoxyethyl acrylate, 2-ethoxyethyl acrylate, 2-propoxyethyl acrylate, 2-butoxyethyl acrylate, pentyl acrylate, hexyl acrylate, hexadecyl acrylate, octadecyl acrylate, poly(ethylene glycol) methyl ether acrylate, poly(ethylene glycol) alkyl ether acrylates, poly(ethylene glycol) phenyl ether acrylates, poly(ethylene glycol) mono acrylate, poly(propylene glycol) methyl ether acrylate, poly(propylene glycol) alkyl ether acrylates, poly(propylene glycol), poly(propylene glycol) mono acrylate, and perfluoro alkyl ether acrylates. In addition, corresponding methacrylate versions of these acrylate monomers could also be used. However, acrylates are more desirable, because their resulting polymers are known to have lower Tg values than the polymers formed from their corresponding methacrylates.
The polymer may also be prepared using small amounts of one or more high refractive index monomers to obtain a material which, when hydrated, will have a desirable refractive index of about 1.4. The presence of the resulting high refractive index units in a polymer can increase the refractive index of that polymer relative to a similar polymer without high refractive index units. High refractive index monomers include pentabromophenyl methacrylate, pentachlorophenyl methacrylate, 2,4,6-tribromophenyl acrylate, 2-phenoxyethyl acrylate, 2-phenoxyethyl methacrylate, phenyl methacrylate, phenyl acrylate, benzyl methacrylate, benzyl acrylate, and the like. In the case when fluorinated hydrophobic monomers are used, these aromatic monomers may be necessary to offset a possible reduction in refractive index of the resulting polymer caused by the presence of the fluorinated hydrophobic units.
In addition, one or more units comprising UV absorbing functional groups may be incoφorated into the polymer. Such groups include, but are not limited to, 2-hydroxy benzophenone groups, benzotriazole groups, and combinations thereof. The presence of UV absorbing units in a polymer results in that polymer having UV transmittance lower than that of a polymer not containing UV absorbing units.
The polymerization technique is not critical to this invention. Solution, bulk or emulsion polymerization methods may be used. Living radical (or controlled radical) polymerization techniques can be used to control block sizes if desired. Chain transfer agents, other than the reaction solvent, may be used to control molecular weight. Most any free radical initiator may be used. Preferred initiators are Vazo®64 (also known as AIBN) or Vazo®52, which are commercially availble from E.I. du Pont de Nemours & Co., Wilmington, Delaware, USA. Alternatively, the polymer system may be designed to rely on other types of initiation and polymerization, such as for example, thermal initiation, photoinitiation, cationic or anionic polymerization.
As noted above, at least one embodiment of this invention includes a polymer with pendant functional groups capable of latent crosslinking. Indeed, the latent crosslinking of the polymer may be combined with most embodiments of this invention by a number of means. In general, covalently bonded crosslinking in a material that has been polymerized can be brought about by (1) vulcanization; (2) free radical reactions caused by ionizing radiations; (3) actinic radiation induced reactions involving photosensitive functional groups; or (4) chemical reactions involving labile functional groups. The particular means selected depends on the chemical system and polymerization methods used to make the polymer. No matter which crosslinking means is selected, the crosslinkable functionality must be latent, that is, it cannot be activated during the polymerization process, but must be initiated during or after thermoforming the polymer into the desired shape of the ophthalmic lens. Of course, the crosslinking means selected should not detrimentally alter the chemical or physical properties of the lens.
Photocrosslinking is one of the preferred means for latent crosslinking of the polymer after it has been melt processed into its desired shape. The photocrosslinkers, or photoinitiators, or photosensitive chromophores, may be incoφorated into the polymer in several ways. For example, vinylic monomers having photosensitive functional groups may be readily incoφorated into a mixture comprised of other vinylic monomers during the initial polymerization process. Alternatively, the photosensitive chromophores may be compounded or blended into a polymer for subsequent crosslinking. Benzophenones are one such class of photosensitive chromophores that may be useful for crosslinking the polymer. Some examples of polymerizable latent photo-crosslinkers are as follows: 4-(2-acryloxyethoxy)-2-hydroxybenzophenone; 1 ,3-bis(4-benzoy-3- hydroxyphenoxy)-2-propyl acrylate (cinnamyl methacrylate); and 2-cinnamoyloxyethyl acrylate. Other latent photo-crosslinkers that can be used in the present invention are described in J. Macromol. Sci-Chem., A28(9), pp. 925-947 (1991) and include: l-2-diphenyl-2-hydroxy-2-(4-vinylphenyl)ethan-
1 -one; 1 -methoxy- 1 ,2-diphenyl-2-(4-vinylphenyl)ethan-2-ol; 1 , 1 -dimethoxy- 1 ,2- diphenyl-2-(4-vinylphenyl)ethan- 1 -one; 1 ,2-diphenyl-2-hydroxy-2-(4- vinylphenyl)ethan- 1 -one; 1 , 1 -dimethoxy- 1 -phenyl-2-(4-vinylphenyl)ethan-2-ol; 1 - phenyl-2-hydroxy-2-(4-vinylphenyl)ethan- 1 -one; 2-Hydroxy-2-methyl- 1 -(4- vinylphenyl)propan- 1 -one; 2-Hydroxy-2-methyl- 1 -(4-vinylphenyl)butan- 1 -one; and 2-methyl-2-methoxy-l-(4-vinylphenyl)propan-l-one. After a melt processing step, the lens can be subjected to UV radiation that cures the polymer, i.e., causes the photosensitive functional groups to react and induce crosslinking of the polymer. This UV curing may be performed with the polymer in a dry or wet state, either before, during or after hydration of the lens.
As an alternative to incoφorating photoinitiators into the polymer during the polymerization step, it is proposed that melt processable polymers be compounded just prior to or during the melt processing step with the photoinitiators such as those listed below. These photoinitiators could induce crosslinking after molding is complete. It is possible to photocrosslink either non- hydrated polymer or the corresponding hydrated polymer. The photoinitiators
listed below may be used in this invention and are commercially available from Ciba Specialty Chemicals Additives Division, Tarrytown, New York, USA. The commercial designation is in parentheses following the chemical name of each compound: 1-hydroxycyclohexyl phenyl ketone (Irgacure® 184); 2,2-dimethoxy- 2-phenylacetophenone (Irgacure® 651); 2-hydroxy-2-methyl- 1 -phenyl propanl- one (Darocur® 1173); 1-hydroxycyclohexyl phenyl ketone (Irgacure® 500); 2- methyl- 1 -[4-(methylthio)phenyl-2-moφholino]propan- 1 -one (Irgacure® 907); 2,4,6-trimethyl benzoyl diphenyl phosphineoxide (Darocur® 4265); 4-(2- hydroxyethoxy)phenyl-(2-hydroxy propyl)ketone (Irgacure® 2959); 2-benzyl-2- N,N-dimethylamino- 1 -(4-moφholinophenyl) 1 -butanone (η 5-2,4-cyclopentadien- l-yl)[(l, 2, 3,4,5,6-η)-(l -methyl ethyl)benzen]iron(+) hexafluorophosphate(l) (Irgacure® 369); 25% bis(2,6-dimethoxybenzoyl)-2,4,4-trimethylpentyl phosphine oxide plus 75% 2-hydroxy-2-methyl-l-phenyl-propan-l-one (Irgacure® 261); and bis((η5-2,4-cyclopentadien-l-yl)bis[2,6-difluoro-3-(lH-pyrrol-l- yl)phenyl]titanium (Irgacure® 784 DC).
Also, it is believed that the photoinitiators may be incoφorated into the polymer of the present invention after it has been shaped into the form of an ophthalmic lens during the hydration step of the process. The hydration solution may contain water soluble photoinitiators that would be incoφorated into the polymer as it uptakes water and swells into a hydrogel. For example, it might be possible to induce crosslinking by soaking the polymers in hydrogen peroxide solution. Crosslinking reactions could be promoted by UV light or heat. Alternatively, a reducing agent such as a ferrous salt (e.g., Iron(II) acetate, Iron(II) chloride, Iron(II) bromide) could be added to peroxide-containing hydration solution.
Another preferred means for latent crosslinking of the polymer is the secondary reaction of labile functional groups. For example, vinylic monomers having secondary reactive functional groups may be readily incoφorated into a mixture comprised of other vinylic monomers during the polymerization process. Peroxy, isocyanato, epoxy, hydroxyl, anhydride and more particularly N-
hydroxymethyl functional groups are capable of latent crosslinking activated by the heat used during the thermoforming step.
As noted above, epoxy containing units may be incoφorated into the polymer to provide latent crosslinking functionality. Epoxy-containing monomers may be polymerized by free radical polymerization, and so may be copolymerized when making the polymers of this invention. These monomers that contain epoxy functionality include, but are not limited to: glycidyl methacrylate, glycidyl methacrylate plus glycerol monomethacrylate*, glycidyl acrylate, glycerol monovinylbenzyl ether, glycerol monovinylether, glycidyl acrylate plus glycerol monoacrylate*, glycidyl vinylbenzyl ether, glycidyl vinyl ether, allylglycidyl ether, l,2-epoxy-3-butene, l,2-epoxy-4-pentene, and l,2-epoxy-5-hexene, etc. (* denotes that glycerol monomethacrylate or glycerol monoacrylate should promote crosslinking reactions of epoxy groups). Most epoxy (or glycidyl) monomers might contain diol impurities. The diol impurities (if present) are expected to promote/enhance crosslinking reactions.
Further, as noted above, functionalized acrylamides and methacrylamides, such as monomers listed below, are capable of providing for latent crosslinking, and may be incoφorated into the polymerization mixture of the present invention. These monomers include, but are not limited to: acrylamide, N- (hydroxymethyl)acrylamide, N-(hydroxymethyl)methacrylamide, N-(iso- butoxymethyl)acrylamide, N-(iso-butoxymethyl)methacrylamide, N-(2- hydroxypopyl)methacrylamide, and N-(2-hydroxypropyl)acrylamide.
Also, as noted above, polymerizable isocyanates are useful for providing units with latent crosslinking groups. These isocyanate monomers include, for example, at least 2-isocyanatamethacrylate, and α,α-dimethyl-3- isopropenylbenzyl isocyanate.
Likewise, as noted above anhydrides are useful for latent crosslinking. Anhydride-containing monomers include, for example, at least 4- methacryloxyethyl trimellitic anhydride, and itaconic anhydride.
Moreover, monomers, such as those listed below, could be used to introduce units functionalized with hydroxy groups into polymers. Crosslinking of hydroxy-containing groups within the polymer can be activated by treating the polymers with diisocyanates (one could also use tri-, tetra-, etc., isocyanates), diepoxides (or tri-, tetra, etc., epoxides) or anhydrides (or dianhydrides). The hydroxy group-containing monomers that may be used include, for example, at least 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate, 3-hydroxypropyl methacrylate, 3-hydroxypropyl acrylate, 4-hydroxybutyl methacrylate, 4- hydroxybutyl acrylate, and the like, glycerol monomethacrylate, glycerol monoacrylate, vinyl benzyl alcohol, various isomers thereof, glycerol methacrylate, and other functionally similar generalized structures.
In addition, the polymer may be compounded with materials such as diols, triols or diamines to enhance the crosslinking of those functional groups. The curing agents for polymers containing pendant hydroxy groups include, for example, at least 1,6-hexamethylenediisocyanate, toluenediisocyanate, methylenediphenyl diisocyanate, 4,4"-methylenebis(N,N-diglycidylaniline), N,N-diglycidylaniline, N,N-diglycidylaniline, diglycidyl 1,2- cyclohexanedicarboxylate, diglycolic anhydride, ethylene glycol diglycidyl ether, 3 ,4-epoxycyclohexylmethy 1 3 ,4-epoxycyclohexanecarboxylate, glycerol triglycidyl ether, succinic anhydride, phthalic anhydride, and pyromellitic anhydride. The possible curing agents for epoxy- and isocyanate-containing polymers include, for example, at least ethylene glycol, glycerol, 1 ,2-propanediol, 1,3-propanediol, 1 ,4-butanediol, 1,2-butanediol, 1 ,3 -butanediol, pentane diols (and various isomers thereof), hexane diols (and various isomers thereof), 1 ,4- diaminobutane, 1,3-diaminobutane, butylamine (and various isomers thereof), bisphenol-A, ethanol amine, 3-amino-l-propanol, l-amino-2-propanol, 3-amino-l- butanol, 4,4"-methylenedianiline, water, and sodium hydroxide. Also, those curing agents may be used to treat the lens made after the thermoforming process step. In addition, the crosslinking may be carried out by reactive processing of the polymer with a number of reagents. For example, benzyl alcohol groups could
be incoφorated into the polymer by using vinyl benzyl alcohol in the polymerization step. This hydroxyl group may be reacted with diisocyanates, diepoxides, dianhydrides, or anhydrides, for example, during an injection molding process. If needed, additional heat or UV radiation may be applied to initiate the reaction to carry out the crosslinking.
Another similar example of reactive latent crosslinking would involve an alkenyl (i.e., olefinically unsaturated) pendant functional group on the polymer. Pendant alkenyl functionality can be incoφorated into copolymers by copolymerizations involving monomers such as dicyclopentenyl methacrylate, dicyclopentenyloxyethyl methacrylate, dicyclopentenyl acrylate and dicyclopentenyloxyethyl acrylate. The double bond in the dicyclopentenyl group does not participate in polymerization, but can be post-reacted (e.g., by oxidative crosslinking).
For latent crosslinking of an alkenyl-containing polymer during a molding or shaping process, the polymer is first compounded with a peroxide to induce crosslinking of the polymer. Several types of peroxy compounds could be used to promote crosslinking. Some examples are given below.
• Acyl peroxides such as diacetyl peroxide and dibenzoyl peroxide
• Alkyl peroxides such as dicumylperoxide and di- tertiarybutylperoxide
• Hydroperoxides such as t-butyl hydroperoxide and cumyl hydroperoxide
• Peresters such as t-butyl perbenzoate, t-butyl peracetate and Lupersol® 256 (2,5-dimethyl-2,5-di-(2-ethylhexanoyl peroxyhexane)) commercially available from Elf-Atochem North
America, Inc., Philadelphia, PA, USA
• Diperoxycarbonates such as OO-t-butyl O-isopropyl monoperoxycarbonate
• Di-Peroxyketals such as Ethyl-3,3-di(t-butylperoxy)butyrate and 2,2-di-(t-butylperoxy)butane
Some of these above-listed peroxy compounds are described in Polymer Engineering and Science, July, 1979, Vol. 19, No. 9, pp. 597-606.
Similarly, rather than activating the latent crosslinking reaction with heat, the crosslinking reaction may be activated by moisture, acid or base catalysis, or combinations thereof. To allow for moisture-activated crosslinking, incoφorating one or more of the following into the polymer is desirable: alkoxysilane, silanol, acetoxysilane, silanes, or halosilane groups. Alkoxysilane, acetoxysilane, silanes, or halosilane form silanols upon exposure to moisture. The silanols react with each other to form siloxane bonds. Therefore, polymer chains containing pendant silanol groups (or precursors) can combine to form crosslinks through the formation of siloxane linkages. Accordingly, polymers prepared by copolymerizing such siloxane-functionalized monomers may be melt processed before the crosslinks are formed. Being moisture activated, the crosslinks would form during hydration of the polymer.
Crosslinking reactions involving organosilicon functional groups could also occur by non-hydrolytic processes. Halosilanes can form siloxane bonds by being exposed to alkoxy silane, metal oxides (e.g., calcium calcium oxide, magnesium oxide, zinc oxide, copper oxide, etc.) and alcohol + carboxylic acid. Alkoxysilanes can combine with each other to form siloxane bonds. Crosslinking and curing of silicones is well known (see W. Noll, Chemistry and Technology of Silicones, Academic Press, Inc., London). In addition, alkoxysilanes also form siloxane bonds when exposed to silanols, acetoxysilanes, carboxylic acids, and acids such as HCl. Hydrolytic and non-hydrolytic reactions in which siloxane bonds are formed could be used to introduce crosslinks into melt processed contact lens materials. If moisture or other crosslinking initiators were mixed with polymers containing silanol (or silanol precursors), then crosslinking reactions could be initiated and gellation might occur in the lens mold under appropriate processing conditions. Alternatively, placing the melt-processed lens in hydrating solution could induce gellation. Catalysts could be added to the hydration solution if needed.
Possible organosilicon monomers for introducing moisture-activated crosslinks into a polymer by way of copolymerization with other monomers are given below. The monomers contain carbon-carbon double bonds that participate
in initial polymerization reactions and silicon-containing groups that can be converted siloxane bonds. Examples of these monomers include, but are not limited to: methacryloxy ethyltrimethoxysilane, methacryloxyethylmethyldimethoxysilane, methacryloxyethyldimethyl- methoxysilane, methacryloxyethyltriethoxysilane, methacryloxyethylmethyl- diethoxysilane, methacryloxyethyldimethylethoxysilane, methacryloxyethyltrichlorosilane, methacryloxypropyltrimethoxysilane, methacryloxy propylmethyldimethoxysilane, methacryloxy propyldimethylmethoxysilane, methacryloxy propyltriethoxysilane, methacryloxy propylmethyldiethoxysilane, methacryloxy propyldimethylethoxysilane, methacryloxy propyltrichlorosilane, styrylethyltrimethoxysilane, and 3- (N-styrlmethyl-2-aminoethylamino)propyltrimethoxysilane hydrochloride.
To carry out the latent crosslinking of the polymer, a specific thermoforming process may be selected to facilitate that crosslinking. For example, if only heat is needed to activate the crosslinking, thermal compression molding of the uncrosslinked polymer film may be the most efficient processing technique. On the other hand, if it is necessary to compound the uncrosslinked polymer with a curing agent, then a thermoplastic injection molding process may be suitable. Likewise, the uncrosslinked polymer could be dissolved in an organic solvent and mixed with the curing agent in a typical reaction injection molding
(RIM) process.
A polymer for this application, whether undergoing latent crosslinking or not, should preferably possess, in addition to the usual properties of contact lenses, transparency, oxygen transmissibility, mechanical and geometric integrity, structural and dimensional stability during sterilization conditions, biocompatibility, softness (comfort), nontoxicity, etc. Also, for use in injection molding equipment, the polymer may require special properties, such as low viscosity at elevated processing temperatures, thermal stability of the material at high temperature without degradation, and short cycle time implying little or no chemical reactions during processing.
Without being bound by any theory of operation, it is believed that suitable polymer systems in accordance with at least one embodiment of this invention will aggregate in a certain way at room temperature. By heating the polymer up to elevated processing temperatures, those aggregates will melt out to provide a flowable and processable thermoplastic material. When the polymer cools to room temperature, the domains will reaggregate, and will maintain those aggegrations when the lens formed of that polymer is hydrated to a swollen gel state. Those aggregates may be crystalline domains with high melting temperature, Tm, amoφhous domains with high glass transition temperature, Tg, highly hydrophobic domains, or domains with high degree of ionic interactions or chain entanglements. These domains remain structurally stable at low temperatures because of the cohesive forces between the segments of the polymers in those domains. For example, high Tg blocks are known to have high transition temperatures, resulting in strong cohesive force at temperatures below Tg. This may impart enough structural integrity to a hydrogel to withstand osmotic forces tending to pull the polymers apart.
Likewise, hydrophobic blocks may form due to the immiscibility of long sequences of hydrophobic units in a copolymer with a majority of hydrophilic units. Fluorinated units, in particular, are more immiscible with the hydrophilic domain than are their corresponding hydrocarbon analogs, thereby enhancing the cohesive forces, or hydrophobic association, within the hydrophobic blocks. Thus, dimensional stability of the hydrated hydrogel may be enhanced with lesser amounts of fluorinated hydrophobic units as compared to their hydrocarbon counteφarts incoφorated into the polymer. It is believed that certain preferred embodiments of the present invention take advantage of the transitory, virtual, physical, thermally-reversible, non- covalently bonded pseudo-crosslinks in the aggregate elements. These aggregates hold the polymer together at low temperature, and under hydrated conditions, yet can be melted out at elevated temperatures to permit thermoforming. In contrast, prior art polymers for contact lenses typically relied upon the formation of
covalently bonded crosslinks during polymerization that are permanent and do not allow the polymers to be thermoformed into molded shapes.
In addition, it is believed that this invention may take the form of several alternate embodiments. In one alternate embodiment, a block copolymer is employed in which the blocks are self segregating into immiscible phases. The volume fraction of the major component consists of blocks of known hydrophilic material: e.g. poly(2-hydroxy ethyl methacrylate), polyethylene oxide, polyacrylamide, polystyrene-maleic acid copolymer, and Zwitterionic phosphate containing units as described in U.S. Patent No. 5,658,561 to Nakabayashi et al. Such materials may be polar, cationic, anionic, or Zwitterionic and have only their hydrophilic nature in common. The minor component consists of hard hydrophobic blocks having high glass transition temperature (Tg) or high melting temperature (Tm) relative to the sterilization or use temperature, but low Tg or Tm compared to the degradation temperature. Such high temperature hard blocks are exemplified by various types of nylons, perfluoroalkyl chains, perfluoroalkyl ether chains, polyacrylonitrile units, poly(ethyleneterephthalate) units, polyvinylcarbazole units, suitable polymeric liquid crystalline blocks of all chemistries, etc.
The types of linkage between the hard and soft blocks is not critical to the invention. However, the domain size of the hard blocks must be small enough so as not to provoke significant light scattering, and thereby obtain optical quality light transmission. The block copolymer can be of any moφhology enabling this to occur, i.e. di-block, tri-block or multi-block, all of which are known. Importantly, optical clarity must be attained in any case. Therefore, it is believed that the domain size of the hydrophobic blocks should be smaller than the wavelength of visible light, that is on the order of 100 nm or smaller. Alternatively, if the hydrophilic and hydrophobic blocks exhibit substantially similar refractive indices, the domain size of the hydrophobic blocks may be larger than 100 nm, yet optical clarity can be maintained. When processing these polymers in injection molding equipment, one would expect the polymer to have the following properties. At high temperatures,
the polymer flows as expected according to the rheological behavior experienced by block copolymers. The blocks melt out at elevated temperatures to provide a thermoformable material. Preferably, there is no chemical reaction taking place during lens formation. Cycle time is short due to the thin walled nature of the ophthalmic lens and the fast cure time of the material. After molding and during solidification, the heterogeneous structure forms and is stable at use and sterilization temperatures. The structure is also stable under the osmotic stresses generated in hydrating the hydrophilic portion of the system. The hydration and solidification process must produce reproducible mechanical and optical outcomes. Crosslinking must be controlled so as to not change the flow characteristics of the material significantly. Once the lens is formed, any crosslinking should have reproducible effects on the lens and optics.
A second alternate form of the present invention relies on the fact that moφhologies of block copolymers are known to form discrete patterns as they are constrained by thermodynamics and chain statistics and entropy. One of the well- recognized patterns is a closed packed (possibly hexagonal, but not necessarily so) arrangement of cylinders embedded in a matrix phase. The cylinders will probably be the minority component, that is, less than about 30 percent. At about 70 percent, phase inversion will occur. The moφhology of the intermediate concentrations remains to be seen. The ability of rods to be aligned by external fields is well known. Extensional flow fields are particularly effective. The orientation of a rod moφhology in the X- Y plane along a principal director axis would in general provide a highly anisotropic and birefringent film. Rod size must be sufficiently small such as to avoid light scattering, that is, not to degrade visible light transmission. A lens such as described would represent an innovative bifocal for simultaneous vision.
A third alternate form of the present invention achieves short processing cycle time through the use of polyurethane chemistry in which, for example, isocyanates react with hydrophilic macromonomers in a steel mold cavity to directly form a lens. Such chemistry is known to be rapid and the cycle time will be further reduced by the use of the macromonomers. Urethane linkages are
known to be good transmitters of oxygen so a reduced water content may be applicable. Previous workers in the area of polyurethane lenses lamented the lack of mechanical strength, but this is now not perceived as an issue in the era of disposable lenses. The process described above can utilize the RIM (Reaction Injection Molding) technique in which liquid reactive components are injected into a mold for reaction to form the final part. Efficient manufacturing processes may take advantage of multiple RIM molds placed on a rotating table, i.e. "Lazy Susan" device.
The above embodiments of the present invention are suited to take advantage of high-speed, high-volume, screw extrusion thermoplastic injection molding equipment, thermal stamping, vacuum-assisted thermal molding, as well as RIM injection molding equipment. The direct molding or injection molding of a lens requires some attention to the placement of the gates and possible discomfort to the lens-wearer due to the vestige in the final lens. There are a number of advanced approaches which can be developed to obviate this effect.
Hot runner molding is well known to process technologists. Additional effects which can be called upon to minimize gate effects are rotating or vibrating the mold halves with respect to each other while the lens body is in the fluid state. The application of an ultrasonic beam to the gate area is an extension of the above mentioned concept. RIM technology may be employed so as to reduce or eliminate the vestiges of a gate.
In addition, other molding processes may be used to take advantage of the thermoformable or melt-processable polymers made in accordance with this invention. Such processes include resin transfer molding, compression molding, extrusion, vacuum assisted plug thermoforming, and stamped thermoforming.
Those processes may be adapted by those skilled in the art to manufacture ophthalmic lenses.
The methods of this invention also include a method for making a hydrogel contact lens comprising forming a polymeric material into a thin film, introducing the film between heated mold halves, closing the mold halves onto the film for a sufficient period of time, with a sufficient force, and with a sufficient amount of
heat for the film to form into the shape of a contact lens precursor, opening the mold halves, removing the lens precursor from the mold halves, and hydrating the lens precursor to form a hydrogel contact lens.
Another embodiment of the invention includes a method for making a hydrogel contact lens comprising applying heat to melt a polymeric material; injecting the melted polymer into a cavity of a lens mold; allowing the melted polymer to solidify in the lens mold for a sufficient period to retain the shape of the lens mold; opening the lens mold; removing the lens from the mold halves; and hydrating the lens precursor to form a hydrogel contact lens. Also, the lens precursor can be hydrated before removing from the mold halves.
Another embodiment of the invention includes a method for making a hydrogel contact lens comprising dissolving a polymeric material in an organic solvent; mixing said polymeric material with a curing agent; dispensing said mixture of polymer and agent into the cavity of a lens mold; allowing the mixture to remain in the cavity for a crosslinking reaction to occur such that the reacted mixture solidifies in the shape of the lens mold; opening the lens mold; removing the lens from the mold halves; and hydrating the lens to form a hydrogel contact lens. Also, the lens can be hydrated before removing from the mold halves.
The above processes have the advantage of using re-useable lens molds. Thus, the present cost disadvantage of single-use plastic lens molds is eliminated.
These re-useable molds may be comprised of metal or a transparent durable material such as quartz. The transparent molds would be advantageous for processes requiring UV curing of the polymer in the mold.
The methods for making ophthalmic lenses of the present invention may be modified by those skilled in the art to allow for the incoφoration of additives into or onto the lenses. These additives may include colorants and handling tints, printed indicia, wetting agents and surface treatments, pharmaceutical or other active ingredients. Also, the additive may be a plasticizer that makes processing of the polymer easier. The plasticizer may be reactive, such as glycerol, or non- reactive, such as phthalate esters. Examples of possible phthalate esters are di(2-
ethylhexyl)phthalate and diethylphthalate. Depending on the nature of the additive, it may be added during the initial polymerization, added during the melt processing, added before, during or after the hydration of the polymer, added by dissolving with the polymer, or compounded or blended with dry ground polymer particles.
Accordingly, one of the advantages of this invention may be found in the adaptability of manufacturing methods to the particular designed characteristics of the polymer and processing constraints of the ingredients of the finished ophthalmic lens. Thus, one of a variety of melt processing methods may be selected depending on the constraints, such as a temperature limit, established by a particular additive desired to be incoφorated into the lens.
One skilled in the art may appreciate that this invention may have broader application than for use in making ophthalmic devices. For example, hydrogel polymers made in accordance with this invention, or the methods of this invention, may be useful for making thin film wound dressings, subcutaneous drug delivery devices, or coatings for catheters (via coextrusion).
The following examples illustrate just a few embodiments in accordance with this invention, and are not intended to limit the invention. Unless otherwise expressly stated, amounts are by weight, percentages are by weight percentage, and temperatures are by degrees Celsius.
Examples
Several new polymers have been synthesized, and characterized. These evaluations are summarized in Table 1. The polymers have been prepared from N,N-dimethylacrylamide and one or more of the following: a perfluoroacrylate known as FX-13, a perfluoromethacrylate known as FX-14, N-isopropyl- acrylamide (N-IPA) and methylmethacrylate (MMA). It has been possible to melt-press clear films from polymers made from these monomers. These films were clear and formed hydrogels with water contents ranging from about 55 to about 90 weight percent by total weight of the hydrated lens. The oxygen
permeability, i.e., Dk values for these materials ranged from about 45 to 31 barrers.
The dehydration behaviors of the new polymers for Examples 1 through 4 were tested. The dehydration behavior of these materials is similar to that of netrafilcon contact lenses manufactured and sold by Wesley-Jessen Coφ.,
Des Plaines, Illinois, USA under the trademark Gentle Touch™.
Characteristics of some of the moldable materials are given in Table 1. Although none of the hydrogel polymers in Table 1 were dimensionally stable at the high temperatures seen during autoclave sterilization (e.g., 120°C), alternative sterilization techniques are available, such as irradiation, that do not subject the lenses to high temperatures. Moreover, sterilization steps may not be necessary if contact lenses made from these polymers are thermoformed under sterile conditions, and thereafter, while being maintained in a sterile environment, placed in sterile packaging.
Experimental Materials and Methods
2-(N-ethylperfluorooctanesulfonamido) ethyl acrylate (FX-13) was supplied by 3M Coφoration of St. Paul, Minnesota, USA. when noted herein as being purified, the FX-13 was recrystallized twice from methanol before use. The following materials were used as received from Aldrich Chemical: N,N-dim ethyl acrylamide, N-isopropyl acrylamide, 2,2'-azobisisobutyronitrile (AIBN), 1,4- diaminobutane and 1 ,4-dioxane.
For examples 1 through 4, the following general polymerization procedure was used. A round bottom flask was charged with the noted proportional amounts of monomers, and the noted solvent, either toluene or dioxane. After the monomers dissolved, the flask was charged with a polymerization initiator, AIBN, and solvent. The flask was lowered into a 60°C oil bath. The reaction mixture was stirred for about 24 hours under nitrogen. Most of the solvent was removed from the reaction mixture by rotary evaporation. The viscous solution was poured into stirred diethyl ether, and the polymer precipitated. The precipitated polymer
was separated from the diethyl ether and dried in a vacuum oven (65-70°C, pressure was less than or equal toabout 0.1 mm of Hg) for at least 24 hours.
The dried polymer material was placed in a hydraulic laboratory Carver press, Model C, commercially available from Carver Press, Wabash, Indiana, USA. The press had heating platens covered with sheet metal plates lined with aluminum foil on the contact surface. When indicated, a mold release agent (food grade silicone) was applied to the aluminum foil. The heating platens were heated up to the molding temperature of about 150°C (unless otherwise noted) for pressing the polymer material of the following examples into a clear film having a thickness of either 75 μm or 100 μm. To obtain these thicknesses, brass spacer gaskets having a thickness of either 75 μm or 100 μm were used. The molding time was about 10 minutes applying force between about seven and nine metric tons, unless otherwise noted. The film formed a clear hydrogel after it was allowed to soak in 0.9% NaCl irrigation solution. Circular disks about 13 mm in diameter were punched out of the hydrogel and subject to autoclave sterilization to determine dimensional stability at those conditions.
DSC measurements were made with a Dupont 910 Differential Scanning Calorimeter. The DSC cell was flushed with nitrogen during experiments. Samples were encapsulated in aluminum pans. The instrument was calibrated with an indium standard and an empty aluminum pan was used as a reference.
The observed onset of melting for the indium was 156.6°C +/- 0.5°C. Tg values were taken as inflection points in the thermograms (2nd or higher scan number). Thermogravimetric analyses (TGA) were made with a Dupont 951 Thermogravimetric analyzer in a nitrogen atmosphere. A 10°C/min. heating rate was used for DSC and TGA experiments.
FT-IR spectra were recorded with a Biorad FTS 175 Spectrometer to determine whether certain monomers were incoφorated into the polymer, and what the composition was to confirm the polymer structure. For FT-IR analysis, samples were typically dissolved in chloroform and then cast onto a NaCl disk. The films were then allowed to dry in a vacuum oven at about 80°C. Hydrogels
were prepared by allowing compression molded polymer films to hydrate in 0.9% USP NaCl solution for about 24 hours or more. Hydrogel water contents were determined with the aide of an ATAGO hand held refractometer and or gravimetrically with an analytical balance. The calibration of the ATAGO refractometer was checked with a saturated NaCl solution.
Oxygen permeability was measured polarographically with a Delta Scientific Products Model 2110 (D.O.) monitor or equivalent. Refractive index was measured with an Abbe Refractometer (Mark II model) at 25°C.
Weight average molecular weight (Mw)and number average molecular weight (M„) were determined with a Perkin Elmer ISS 200 HPLC/SEC. The ISS
200 was equipped with a UV-VIS detector and a Model 250 pump. Ultra styragel columns (1 x 104, 1 x 103, 5 x 102 A) connected in series were used with THF as the eluent at a flow rate of 1 mL/min. Narrow molecular weight poly(methyl methacrylate) standards were used. Elemental analyses were carried out. Prior to elemental analysis, samples were dried in a vacuum oven at 80°C (about 25 inches Hg) for about 17 hours and then further dried at 100-105°C (about 25 inches Hg) for about 48 hours. The weight percent of various monomers incoφorated into polymer samples were calculated from sulfur and nitrogen elemental analysis results. The molecular weight of FX-13 was estimated to be about 593. Chemicals were used as received unless otherwise indicated.
Table 1
* Chemicals were used as received unless otherwise specified, Rxn indicates the noted solvent was used of the polymerization reaction.
Detailed information on each of these polymers in Table 1 is described in the below examples.
Example No. 1
Copolymerization of DMA, N-IPA, and FX-14 in Toluene was carried out in this example. Relative weight percent of monomers does not take into account initiator.
Polymer Characteristics a. FT-IR: Spectrum was consistent for material containing functional groups from amide, ester, and sulfonamide functional units. b. Glass Transition Temperature: 124°C c. Thermal decomposition: about 325 °C significant weight loss began to occur. d. Molding Characteristics: Clear films were formed @ 155°C. Total molding time about 18 minutes e. Dimensional changes: Area of hydrated film / Area of dry film about 2.59 f. Dimensional stability of hydrogel during heat sterilization: The sample distorted during autoclaving. g. Refractive Index: 1.37 @ 25.1°C h. Dk = 35 barrers
i. Water Content: 75% j. Molecular Weight: Mw = 88,000 Mw/Mn - 2.41
Example No. 2
Copolymerization of DMA, MMA, and FX-14 in Dioxane was carried out in this example. Relative weight percent of monomers does not take into account initiator.
Polymer Characteristics a. FT-IR: Spectrum was consistent for material containing functional groups from amide, ester, and sulfonamide functional units. b. Glass Transition Temperature: 104°C c. Thermal decomposition: about 315°C significant weight loss began to occur.
Molding Characteristics: Clear films were formed ® 160°C. Total molding time about 22 minutes, Hydrogel is slightly cloudy e. Dimensional changes: Area of hydrated film / Area of dry film about 1.9 f. Dimensional stability of hydrogel during heat sterilization: The sample distorted and became opaque during autoclaving. g- Refractive Index: 1.38 (2} 25. C
h. Dk = 35 barrers i. Water Content: 72% j. Molecular Weight: Mw = 300,000, Mw/Mn = 2.70
Example No. 3
Copolymerization of DMA, MMA, and FX-14 in Toluene was carried out in this example. Relative weight percent of monomers does not take into account initiator.
Polymer Characteristics a. FT-IR: Spectrum was consistent for material containing functional groups from amide, ester, and sulfonamide functional units. b. Glass Transition Temperature: 102°C c. Thermal Decomposition: about 315°C significant weight loss began to occur. d. Molding Characteristics: Clear films were formed @ 160°C. Total molding time about 16-20 minutes e. Dimensional changes: Area of hydrated film / Area of dry film about 2.48
Dimensional stability of hydrogel during heat sterilization: The sample distorted and became opaque during autoclaving.
Refractive Index: 1.38 @ 25.1 °C h. Dk = 31 barrers i. Water Content: 70 % j- Molecular Weight: Mw = 150,000, Mw/Mn = 2.6
Example No. 4
Copolymerization of DMA, MMA, and FX-14 in Toluene was carried out in this example. Relative weight percent of monomers does not take into account initiator.
Polymer Characteristics a. FT-IR: Spectrum was consistent for material containing functional groups from amide, ester, and sulfonamide functional units. b. Glass Transition Temperature: 103°C c. Thermal decomposition: about 315°C significant weight loss began to occur.
d. Molding Characteristics: Clear films were formed @ 160°C. Total molding time about 22 minutes e. Dimensional changes: Area of hydrated film / Area of dry film about 2.76 f. Dimensional stability of hydrogel during heat sterilization: The sample distorted during autoclaving. g. Refractive Index: 1.38 @ 25.1°C h. Dk = 34 barrers i Water Content: 75 % j. Molecular Weight: Mw = 78,000 Mw/Mn = 1.82
The following methods were used for Examples 5 and 6.
DSC measurements were made with a Dupont 2920 Differential Scanning Calorimeter. The DSC cell was flushed with nitrogen during experiments. Samples were encapsulated in aluminum pans. An empty aluminum pan was used as a reference. Thermogravimetric analyses (TGA) were made with a Dupont 951 Thermogravimetric analyzer. The performance of the instrument was checked with calcium oxalate monohydrate. A 10°C/minute heating rate was used for all DSC and TGA measurements. FT-IR spectra were obtained with a Nicolet infrared spectrometer. Proton NMR spectra were obtained with a Bruker ADVANCE DMX500 high resolution digital NMR spectrometer. Water contents were calculated using weights of dry and hydrated polymer films. Oxygen permeability measurements were determined by polarographic method using 2110 (D.O.) monitor or equivalent.
Example No. 5
Copolymerization of DMA with FX-13 was carried out in this example.
AIBN Dioxane 60 "C
A round bottom flask containing a magnetic stir bar was charged with N,N-dimethyl acrylamide (20.0765 g), FX-13 (4.5208 g) and 1 ,4-dioxane (110 mL). After the FX-13 dissolved, 0.0408 g of AIBN dissolved in 20 mL of 1 ,4-dioxane was added to the reaction flask. The flask was fitted with a rubber septum and flushed with nitrogen at room temperature for 30 minutes. AIBN (0.083 g) dissolved in 15 mL of 1 ,4-dioxane was added to the reaction mixture and the flask was lowered into a 60°C oil bath. The reaction mixture was stirred under nitrogen for 24 hours.
After 24 hours, most of the dioxane was stripped off by rotary evaporation. Approximately 30 mL of dioxane was added the reaction mixture. The resulting solution was then poured into about 300 mL of stirred diethyl ether. The ether was decanted from the precipitated polymer and an additional 100 ml. of fresh ether was poured over the polymer. The polymer was separated from the ether and dried in a vacuum oven (65-70°C, pressure was less than or equal to 0.1 mm Hg).
After 24 hours of drying, a small portion of the sample was removed for FT-IR, DSC, and NMR analysis. After 48 hours of drying, the polymer sample weighed 19.805 grams. FT-IR (Film cast on NaCl): 3532, 2927, 1734, 1641, 1497, 1398, 1355, 1255, 1216, 1143, 1096, 1058, 926 cm"1.
Η NMR (CDCI3): The spectrum showed evidence that both monomers were incorporated into the polymer. Polymer that was dried for 24 hours (65-70°C) under vacuum still showed some dioxane as evidenced by the singlet at 3.7 ppm. Differential Scanning Calorimetry (DSC): The material showed a glass transition at 109°C. Significant weight loss begins to occurs near 325°C with a 10 deg./min. heating rate during thermal gravimetric analysis in nitrogen indicating the thermal decomposition temperature (Decomp Temp.).
Films were hydrated in 0.9% NaCl irrigation solution. The hydrated material had a water content of 84% and a refractive index of 1.3545 (25.1°C).
Molding of Films: The sample used for molding a film was dried for about 48 hours. Clear films were formed in a Carver Press at 155 - 160°C. The molding time was about 10 minutes. This material formed a clear hydrogel after it was allowed to soak in 0.9% NaCl irrigation solution.
Polymer Characteristics a. Glass Transition: 109°C b. Decomposition during 10°C/min. heat up: Onset of significant weight loss occurs near 325°C c. FT-IR: Spectrum was consistent for material containing N,N-disubstituted amide, ester, and sulfonamide functional groups. d. Proton NMR: Spectrum showed some residual 1,4-dioxane (singlet, 3.7 ppm), peak near 2.9 ppm due to methyl groups derived from DMA units. Resonances near 2.6 - 2.4 ppm might be due to ester methylene groups derived from the FX-13. Methylene protons in polymer backbone occur near 1.3 ppm e. Dk = 45 barrers f. Water content after (hydration in 0.9% NaCl): about 84% g. Dimensional changes: Area of hydrated film / Area of dry film about 3.57 h. Refractive Index (R.I.) @ 25.1°C: 1.3545
i. Molding Characteristics: Clear films were molded at 155 to 160°C
(molding time was about 10 minutes), j. Molecular Weight: Mw = 100,000 Mw/Mn = 3.0
Example Nos. 5(a) through 5(0 A further study of the DMA/FX-13 copolymer was conducted. The copolymer was made with the amounts of DMA and FX-13 in feed ratios varying inversely from 0 to 100 percent at roughly 10 percent intervals. The physical properties of the resultant material were measured and are set forth below in Table 2. A representative procedure for the preparation of the polymer is as follows:
A three neck round bottom flask equipped with an overhead stirrer balloon and gas inlet was charged with DMA (15.149 g), FX-13 (6.063 g), VAZO 64 (0.102 g) and toluene (150 mL). The flask was lowered into a 60°C water bath and vacuum was applied until the flask contents just began to boil. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen an additional three times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at 60°C. Solvent was removed from the reaction mixture by rotary evaporation and the resulting polymer was dissolved in about 100 mL of THF. The THF solution was added into about 400 mL of stirring water (Milli Q). The precipitated polymer was separated from the THF/water mixture and placed in fresh Milli Q water. The polymer was then allowed to stir in boiling Milli Q water for about 2 hours. The polymer was then separated from the water and washed with several 200 mL portions of fresh Milli Q water. The polymer was dried in a vacuum oven at about 80°C (about 25 in. Hg) for about 48 hours. Approximately 27.0 grams of product were obtained.
Table 2
* Parameters measured for hydrated polymer.
** Actual composition was estimated from sulfur and nitrogen elemental analysis results. FX-13 is sold as a mixture. The molecular weight of FX-13 was estimated to be about 593.
Example No. 6
Copolymerization of DMA, N-isopropylacrylamide, and FX-13 was carried out in this example.
A round bottom flask containing a magnetic stir bar was charged with N,N-dimethyl acrylamide (8.0198 g), N-Isopropyl acrylamide (2.0715 g), FX-13 (2.2625 g) and 1 ,4-dioxane (60 mL). After the FX-13 dissolved, 0.0408 g of AIBN dissolved in 20 mL of 1 ,4-dioxane was added to the reaction flask. The flask was fitted with a rubber septum and flushed with nitrogen at room temperature for 30 minutes. The reaction flask was immersed into a 60°C oil bath and the reaction mixture was stirred for 24 hours.
After 24 hours, the reaction mixture became noticeably more viscous. The viscous solution was poured into 300 mL of stirring diethyl ether. The precipitated polymer was separated from the diethyl ether and dried in a vacuum oven (65 -70°C, pressure was less than or equal to 0.1 mm of Hg). The weight of the polymer after more than 24 hours in the vacuum oven was 10.30 grams.
Samples for FT-IR, NMR, and DSC were dried for about 24 hours. FT-IR (Film cast on NaCl): 3488, 3313, 2929, 1734, 1643, 1498, 1458, 1436, 1355, 1244, 1216, 1146, 1097, 1059 cm"1.
Η NMR (CDC13): The spectrum showed evidence that all three monomer components were incorporated into the polymer. For example, the small, broad peak between 3.9 - 3.8 ppm is probably due to an N-H proton derived from N-isopropyl acrylamide units. The large peak centered near 2.67 ppm is from the methyl groups derived from DMA. The peak centered at 2.40 ppm is probably due to ester methylene units derived from the FX-13 component. The spectrum showed some residual dioxane (sharp singlet 3.7 ppm).
Differential Scanning Calorimetry (DSC): The material showed a glass transition at 105°C. Significant weight loss began to occur near 300°C with a 10 deg/min heating rate during thermal gravimetric analysis in nitrogen.
Films were hydrated in 0.9% NaCl irrigation solution. The hydrated material had a water content of about 80% and refractive index of 1.36 (25.1°C).
Molding of Films: The sample used for molding a film was dried for about 28 hours. Clear films were formed at about 150°C in a Carver Press. The molding time was about 9 minutes. This material formed a clear hydrogel after it was allowed to soak in 0.9% NaCl irrigation solution.
Polymer Characteristics a. Glass Transition: 105°C b. Thermal decomposition during 10°C/min heat up: Onset of significant weight loss occurs near 300°C c. FT-IR: Spectrum was consistent for material containing functional groups from ester, and sulfonamide and di- and mono substituted amide units. d. Proton NMR: Spectrum showed some residual 1,4-dioxane (singlet, 3.7 ppm), peak at 2.89 ppm presumably due to methyl groups from DMA units. The peak near 2.6 - 2.4 ppm might be due to ester methylene groups (derived from the FX-13). Methylene protons in polymer backbone peak occured near 1.3 ppm. e. Dk = 44 barrers f. Water content after (hydration in 0.9% NaCl) about 80 % g. Dimensional changes: Area of hydrated film / Area of dry film about 3.06 h. Refractive Index (R.I.) = 1.36 @ 25.1 °C i. Molding Characteristics: Clear films were formed at a molding temperature of about 150°C (molding time was about 9 minutes), j. Molecular Weight: Mw = 110,000 Mw/Mn = 1.8
The results of the following Examples 7 through 1 1 are summarized below in Table 4.
Example No. 7
Copolymerization of DMA, GMA, MMA and FX-13 was carried out in this example.
A three neck round bottom flask equipped with an overhead stirrer (teflon blade), balloon and gas inlet was charged with DMA (16.515 g), GMA (3.184 g), FX-13 (6.036 g), MMA (4.535 g), VAZO 52 (0.150 g) and toluene (150 mL). The flask was lowered into a 55°C water bath and filled with nitrogen until the balloon (9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the flask contents just began to bubble. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen an additional three times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at 55°C. Solvent was removed from the reaction mixture by rotary evaporation and the resulting viscous solution was diluted with about 50 mL of THF. The THF solution was poured into about 500 mL of stirring hexanes. The precipitated, polymer was separated from the THF/hexanes mixture and washed with 3 x 100 mL of hexanes. The sample was dried under vacuum (about 25 in Hg) at about 70°C for 72 hours to yield 20.249 grams of product. The sample
was analyzed by FT-IR, DSC, TGA and GPC. FT-IR (Film cast on NaCl from chloroform, selected peaks): 3324.7, 3210.8, 2932.6, 1727.8, 1682.7, 1643.2, 1497.7, 1455.5, 1397.8, 1354.9, 1243.5, 1214.1, 1139.2, 1058.1 cm"1. Differential scanning calorimetry: The polymer showed a glass transition temperature of 96°C (2nd scan). Thermal gravimetric analysis: Significant weight loss began to occur near 359°C when the sample was heated at 10°C/min in nitrogen. Molecular weight: Mw = 111,300, Mw/Mn= 3.85.
Polymer Characteristics a. Monomer Feed: DMA(54.5%)/FX-13(19.9%)/MMA(15%)/GMA(10.6%) (initiator level not taken into account) b. Tg = 96°C by DSC c. FT-IR consistent with structure d. Decomposition Temp.: about 383°C by TGA
Molding Example 7A A transparent film with Tg of about 96°C was obtained when the polymer of Example 7 was compression molded for about 20 minutes at about 150°C. Force was gradually increased to about 9-10 metric tons. This material formed a clear hydrogel with a water content of about 69% and a Dk of 44 barrers. In addition, it was stable to autoclave sterilization. In order to evaluate dimensional stability, circular disks (13 mm) were cut from hydrogel sheet and then sterilized in Wesley- Jessen Corp. packaging solution, which is an isotonic borate buffered saline solution containing 0.0005% poloxamer. The diameters increased to 14 mm after the first sterilization. However, the disks remained circular (shape was preserved) and did not undergo further increases after a second sterilization. The slight increase in diameter after the first sterilization might be due to hydrolysis of residual epoxy groups. These results suggest that gelation occurred in about 20 minutes or less during compression molding at 150°C in the absence of added curing agent.
Molding Example 7B
A film obtained by compression molding the polymer of Example 7 in the presence of glycerol was rather soft and flexible. This material was obtained by first compounding ground polymer with about 20% (by wt) glycerol to make a paste, followed by compression molding the paste for about 40 minutes at 150°C to yield a transparent film with Tg of about 50-60°C. This value is about 46-56°C lower than the glass transition temperature of compression molded sample that did not contain added glycerol. In addition to being a curing agent, the glycerol also acts as plasticizer. This property might allow glycerol to be used for optimizing crosslink density and durability. The GMA might contain small amounts of glycerol monomethacrylate. Such a material should promote crosslinking of the epoxy groups.
The molded film formed a clear hydrogel with a water content of 70% and a Dk of 42 barrers. A 13 mm circular disk of the hydrogel expanded to 14 mm upon heat sterilization. However, the shape of the disk was preserved. The diameter and shape of the disk remained unchanged after a second sterilization.
Molding Example 7C
Treating compression-molded film of Example 7A with a solution of diamine curing agent (1,4-diaminobutane) induced additional crosslinking. As previously mentioned, treatment of poly(DMA/FX13/MMA/GMA) at 150°C caused some crosslinking. Film that was compression molded (150°C/about 20 min / about 9 metric tons) in the absence of glycerol was dipped into a 20% solution of 1,4-diaminobutane in isopropanol. The sample was removed from the diamine solution after 3 minutes and then heated for 45 minutes at 70°C and 5 minutes at 135°C. This material showed a Tg of 112°C (2nd scan) and a Tg of
120°C during a third DSC scan. These Tg. values are 16 and 24°C higher than the Tg of compression molded film that was not treated with diamine. The increase in Tg from 112 to 120°C was attributed to additional crosslinking caused by reaction of amine groups with epoxy groups during DSC heating. These results suggest
that the diamine treated film is more highly crosslinked than the glycerol and non- glycerol treated polymers. The amine treated film formed a clear hydrogel with a water content of about 72%. The shape of the hydrogel remained intact after two sterilizations. The diameter of the hydrogel decreased from 13 to 12 mm after two sterilization cycles.
The films described in "Molding Examples 7A-C" above formed clear hydrogels with water contents in the range of 69% to 72%. The oxygen permeability of hydrogels obtained by molding with or without glycerol was about 43 barrers.
Example No. 8
Copolymerization of DMA, GMA, MMA and FX-14 was carried out in this example.
A three neck round bottom flask equipped with an overhead stirrer (teflon blade), balloon and gas inlet was charged with DMA (16.507 g), GMA (3.018 g), FX-14 (6.084 g), MMA (4.614 g), VAZO 52 (0.159 g) and toluene (150 mL). The flask was lowered into a 55°C water bath and filled with nitrogen until the balloon
(9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the flask contents just began to boil. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen an additional three times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at 55°C. Solvent was removed from the reaction mixture by rotary evaporation and the resulting solid was dissolved in about 50 mL of THF. The THF solution was poured into about 500 mL of stirring hexanes. The precipitated polymer was separated from the THF/hexanes mixture and washed with 3 x
100 mL of hexanes. The sample was dried under vacuum (about 25 in Hg) at room temperature for about six days to yield 23.894 grams of product. The sample was analyzed by FT-IR, DSC, TGA and GPC. FT-IR (Film cast on NaCl from chloroform, selected peaks): 3438, 3266, 2984.1, 2947.1, 1731.7, 1644.8, 1504.0, 1455.2 1397.4, 1357,2, 1243.5, 1143.9, 1059.9, 987.9, 909.7, 1397.cn!*1.
Differential scanning calorimetry: the polymer showed a glass transition temperature of 98°C (2nd scan). Thermal gravimetric analysis: significant weight loss began to occur near 383°C when the sample was heated at 10°C/min in nitrogen. Molecular weight: Mw = 118,300, Mw/Mn = 2.99.
Molding Example 8A
A glycerol-free film was obtained by compression molding the polymer of Example 8 in the Carver press at 150°C and coated with a food grade silicone as a mold release agent. This film was transparent and displayed a Tg at 91°C. This is about 7°C lower than the Tg of polymer that was not compression molded. The decrease in glass transition temperature was likely due to plasticization caused by the mold release agent. A film prepared in the absence of glycerol had a water content of 71% and a Dk of 38 barrers. Although this film was crosslinked, a hydrogel of this material did distort during autoclave sterilization. Its shape was converted from a 13 mm circular disk to a 16 x 12 mm oval. This result suggests
that the material was lightly crosslinked and that glycerol is an effective curing agent for such materials. Further more, these results indicate that gelation occurs in 40 minutes or less at about 150°C without added curing agent.
Molding Example 8B
A film obtained by compression molding the polymer of Example 8 in the presence of glycerol was rather soft and flexible. The glass transition temperature of the glycerol-containing material was about 72°C (3rd scan). This value is about 26°C lower than the glass transition temperature of glycerol free, non-molded polymer. Again, the depression of Tg is attributed to glycerol acting as a plasticizer. The glycerol-containing material formed a clear hydrogel that was stable to autoclave sterilization. This hydrogel had a 68% water content and a Dk of 43 barrers. These results also indicate that gelation occurs in 40 minutes or less for this material when it is heated at about 150°C with the presence of glycerol.
Example No. 9
Copolymerization of DMA, GMA and FX-14 was carried out in this example.
A three neck round bottom flask equipped with an overhead stirrer (teflon blade), balloon and gas inlet was charged with DMA (16.503 g), FX-14 (7.555 g), GMA (ca. 6.25 g), VAZO 52 (0.124 g) and toluene (150 mL). The flask was lowered into a 55°C water bath and filled with nitrogen until the balloon (9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the flask contents just began to boil. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen an additional three times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at 55°C.
Solvent was removed from the reaction mixture by rotary evaporation and the resulting viscous solution was diluted with about 50 mL of THF. The THF solution was poured into about 500 mL of stirring hexanes. The precipitated polymer was separated from the THF/hexanes mixture and washed with 3 x 100 mL of hexanes. The sample was dried under vacuum (~ 25 in Hg) at room temperature for about 20 hours. The sample was further dried under vacuum (~ 25 in Hg) at 70°C for about 7 days to yield 18.386 grams of product. The sample was analyzed by FT-IR, DSC, TGA and GPC. FT-IR (Film cast on NaCl from chloroform, selected peaks): 2933.5, 1728.4, 1643.2, 1496.0, 1396.2, 1355.5, 1244.5, 1212.9, 11373, 1059.9, 1000.2, 909.5 cm. Differential scanning calorimetry: the polymer showed a glass transition temperature of 91°C (2nd scan). Thermal gravimetric analysis: significant weight loss began to occur near 368°C when the sample was heated at 10°C/min in nitrogen. Molecular weight: Mw = 195,500, Mw/Mn = 4.06.
Polymer Characteristics a. Monomer feed: DMA(54.4)/FX-14(24.9%)/GMA(20.6%) (initiator level not taken into account) b. Tg = 91°C by DSC c. FT-IR consistent with structure
d. Decomposition Temp.: about 368°C by TGA e. Compression molded @ about 155°C for about 40 min f. A transparent slightly yellow film was obtained g. The film was judged to be crosslinked by its lack of solubility in THF h. Film formed a clear hydrogel i. Water content of hydrogel = 56% j. Refractive Index (R.I.) = 1.41 k. Dk = 41 barrers
1. Dimensional stability of hydrogel: Before sterilization diameter of disk = 13 mm
After 1st sterilization diameter of disk = 14 mm After 2nd sterilization diameter of disk = 14 mm
Poly(DMA/FX-14/GMA) was compression molded at about 150°C for about 40 minutes to yield transparent film. The film was judged to be crosslinked by its lack of solubility in organic solvent. Upon hydration, it yielded a clear hydrogel that was stable to autoclave sterilization. The hydrogel had a water content of about 56% and a Dk of about 41 barrers. The lower water content of this material as compared to other examples is attributed to the higher epoxy content in the unmolded polymer. A higher epoxy content should result in higher crosslink density which, in turn, might result in a lower water content hydrogel. A
13 mm circular disk of the hydrogel increased to 14 mm (shape was preserved) upon sterilization. A second sterilization cycle did not result in further increases of diameter. The increase in diameter after the first sterilization cycle might be due to hydrolysis of residual epoxy groups. The dimensional stability of the hydrogel and the insolubility of the zerogel in THF suggests that crosslinking during compression molding was successful.
The tensile properties of poly(DMA/FX-14/GMA) under various compression molding conditions were examined. These conditions included compression molding at either 150°C or 160°C, for either 20 or 40 minutes, and with or without the addition of glycerol to the polymer prior to molding, to determine the tensile properties, a Lloyd Instruments Model 500 Tensile Tester
(available from Chatillon, Greensboro, NC, USA) was used. The tensile tester was operated with a five Newton load cell and had a strain rate set at 100 mm/minute. The results are shown below in Table 3.
Table 3
Example No. 10
Copolymerization of DMA, NHMA, MMA and FX-14 was carried out in this example.
A three neck round bottom flask equipped with an overhead stirrer (teflon blade), balloon and gas inlet was charged with DMA (16.519 g), FX-14 (6.103 g), MMA (4.517 g), NHMA (6.267 g of a 48% solution in water), VAZO 52 (0.142 g)
and THF (150 mL). The flask was lowered into a 55°C water bath and filled with nitrogen until the balloon (9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the flask contents just began to boil. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen an additional three times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at 55°C. Solvent was removed from the reaction mixture by rotary evaporation and the resulting solid was dissolved in 50 mL of THF. The THF solution was poured into about 500 mL of stirring hexanes. The precipitated polymer was separated from the THF/hexanes mixture and washed with 3 x 100 mL of hexanes. The sample was dried under vacuum (~ 25 in Hg) at about 65°C for about 23 hours. The sample was allowed to further dry under vacuum for about six days. About 30 grams of product was obtained. The sample was analyzed by FT-IR, DSC and TGA and GPC. FT-IR (Film cast on NaCl from chloroform, selected peaks): 3318,2, 2933.5, 1727.76, 1638.7, 1498.3, 1397.8, 1355.5, 1243,6, 1213.5, 1 139.2, 1056.6, cm"1. Differential scanning calorimetry: the polymer showed a glass transition temperature of 117°C (2nd scan). Thermal gravimetric analysis: significant weight loss began to occur near 380°C when the sample was heated at 10°C/min in nitrogen. Molecular weight (based on entire GPC peak): Mw = 65,200, Mw/Mn = 5.5.
Polymer Characteristics a. Approximate monomer feed: DMA(49.2%) FX-14(18.2%) MMA(13.5%)/ NHMA(18.7%) b. Tg = 117°C by DSC c. FT-IR consistent with structure d. Decomposition Temperature about 380°C by TGA e. Compression molded @ about 160°C for about 30 min f. A transparent slightly yellow film was obtained
g. The film was judged to be crosslinked by its lack of solubility in THF h. Film formed a clear hydrogel i. Water content of hydrogel = 76% j. Refractive Index (R. I. ) = 1.37 k. Dk = 54 barrers (std dev. = 7.9)
1. Dimensional stability of hydrogel:
Before sterilization diameter of disk = 13 mm After 1st sterilization diameter of disk = 14 mm After 2nd sterilization diameter of disk = 14 mm Poly (DMA/FX- 14/MMA/ NHMA) was compression molded to yield transparent film. The film was judged to be crosslinked by its lack of solubility in organic solvent. Upon hydration, it yielded a clear hydrogel that was stable to autoclave sterilization. The hydrogel had a water content of about 76% and a Dk of about 53 barrers.
Example No. 11
Copolymerization of DMA, NHMA, MMA and FX-14 was carried out in this example.
A three neck round bottom flask equipped with an overhead stirrer (teflon blade), balloon and gas inlet was charged with DMA (13.430 g), FX-14 (6.241 g), MMA (4.540 g), NHMA (12.508 g of a 48% solution in water), VAZO 52
(0.149 g) and THF (150 mL). The flask was lowered into a 55°C water bath and filled with nitrogen until the balloon (9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the flask contents just began to boil. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen an additional three times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at 55°C. Solvent was removed from the reaction mixture by rotary evaporation and the resulting solid was dissolved in 50 mL of THF. The THF solution was poured into about 500 mL of
stirring hexanes. The precipitated polymer was separated from the THF/hexanes mixture and washed with 3 x 100 mL of hexanes The sample was dried under vacuum (about 25 in Hg) at about 65°C for 15 hours. The sample was allowed to further dry under vacuum for about six days at room temperature. About 28 grams of product was obtained. The sample became insoluble in organic solvent after drying. The polymer apparently crosslinked prematurely. The sample was analyzed by DSC and TGA.
Table 4
* Initiator level not taken into account.
** Water content partly depends on molding conditions.
*** Based on total peak and shoulder area of chromatograph.
Example No. 12
Copolymerization of DMA, FX-13, GMA and RA (alkyl acrylates) ws carried out in this example. The purpose of this example was to demonstrate that the use of flexible side chains (R) incorporated into the polymer lowers the glass transition temperature. Following the general polymerization procedures of the preceding examples, DMA, FX-13 and GMA were copolymerized with various alkyl acrylates (RA). R was selected from an n-butyl, sec-butyl, n-hexyl and n- butoxy-ethyl group. Table 5 below shows the resulting Tg of the various polymers as determined by DSC. These numbers demonstrate that the Tg of the unhydrated
polymer can be altered by varying the monomer side chains. It is well known that flexible pendant groups reduce glass transition temperatures of polymers by acting as internal diluents. This lowers the frictional interaction between the chains (see L.H. Sperling, Introduction to Polymer Science, 2nd edition, John Wiley & Sons). It is expected that a polymer with a lower Tg will have a longer melt processable life time.
Table 5
DMA/FX-13/GMA/RA: 55 / 20 / 10 / 15 Wt%
The polymer of Example 12D was compression molded in a Carver Press at about 160°C for about 40 minutes. A soft clear flexible film was obtained. The film was judged to be crosslinked by its lack of solubility in THF. Upon hydration, the film formed a clear hydrogel. The water content was about 75 weight percent based on the total weight of the hydrated film. The hydrated film was then subject to a heat sterilization cycle and judged to be dimensionally stable at heat sterilization conditions. Both the diameter (about 13 mm) and the shape of the film was preserved after being subject to sterilization.
The polymer of Example 12D was also compression molded at lower temperatures. In one case, the polymer was molded at 110°C for about 12 minutes. A clear flexible film was obtained. The film was judged to be not crosslinked from its solubility in THF. Upon hydration, the film formed a clear hydrogel having a water content of about 76 weight percent. In a second case, the polymer was molded at 100°C for about 20 minutes, after being compounded with about six weight percent of glycerol. A very soft clear film was obtained. The
film was judged to be not crosslinked from its solubility in THF. Upon hydration, the film formed a clear hydrogel.
Example No. 13
Copolymerizations of DMA, FX-13, GMA and RA (Alkyl Acrylates) were carried out in this example. The purpose of this example was to further illustrate the effect of flexible side chains (R) on the properties of the copolymer and resultant hydrogel. Following the general polymerization procedures of the preceding examples, DMA, FX- 13 and GMA were copolymerized with various alkyl acrylates (RA). R was selected from an n- butyl, iso-butyl, n-hexyl, n- butoxy-ethyl and n-octadecyl group.
The following general polymerization procedure was used. A round bottom flask was charged with the noted proportional amounts of monomers, Vazo52 initiator (approximately 0.5 wt% relative to monomer plus initiator), and toluene. The approximate relative monomer contents in the reaction were DMA (54.5 wt%), FX-13 (20 wt%), GMA(10.0 wt%), RA (15 wt%). After the monomers dissolved, the flask was lowered into a 55°C water bath and purged with nitrogen. The reaction mixture was then stirred for about 24 hours under nitrogen. The solvent was removed from the reaction mixture by rotary evaporation. The resulting sample was diluted with 75 mL of THF. The viscous solution was poured into stirred hexanes, and the polymer precipitated. The precipitated polymer was separated from the hexanes and dried in a vacuum oven (65-70°C, pressure was less than or equal to 0.1 mm of Hg) for at least 24 hours. The FT-IR spectrum of each copolymer is consistent with a material containing amide, ester, and sulfonamide functional units. The results of characterization of both dry copolymers and hydrogels are summarized in Table 6, below.
The polymer of Example 13A employed n-butoxyethyl acrylate (BEA) as the alkyl acrylate. Clear flexible films were obtained by molding the copolymer in a Carver press. Upon molding at 100°C for 20 minutes, the film was uncrosslinked as indicated by solubility in organic solvent. Molding at 150°C or at 160°C produced crosslinked films, which were hydrated to produce hydrogels.
The polymer of Example 13B employed n-hexyl acrylate (HA) as the alkyl acrylate. Clear flexible films were obtained by molding the copolymer in a Carver press. Molding at 160°C or at 180°C produced crosslinked films, which were hydrated to produce translucent hydrogels. Copolymer samples compounded with about 6 wt% glycerol produced clear films which were smooth and very flexible.
Hydration of these films resulted in hydrogels which were optically clear and were stable to heat sterilization.
The polymer of Example 13C employed n-butyl acrylate (BA) as the alkyl acrylate. Clear flexible films were obtained by molding the copolymer in a Carver press. Molding at 160°C for 20 or 40 minutes produced crosslinked films which were clear and slightly brittle. Molding at 180°C for 10 minutes produced clear flexible films. Films pressed at either 160°C or 180°C were hydrated to yield clear hydrogels. In addition, this copolymer can be compounded with about 6 wt% glycerol before compression molding at either 160°C or 180°C. Hydration of these films resulted in clear hydrogels with water contents and mechanical stabilities lower than those formed from copolymer not containing glycerol.
The polymer of Example 13D employed iso-butyl acrylate (IB A) as the alkyl acrylate. Clear flexible films were obtained by molding the copolymer in a Carver press. Molding at 160°C or at 180°C produced crosslinked films, which were hydrated to produce clear hydrogels. These hydrogels became cloudy and adhesive upon heat sterilization. Copolymer samples compounded with about 6 wt% glycerol produced clear films which were smooth and flexible. Hydration of these films resulted in hydrogels which were optically clear both before and after heat sterilization. The polymer of Example 13E employed n-octadecyl acrylate (ODA) as the alkyl acrylate. Clear flexible films were obtained by molding the copolymer in a Carver press. Upon molding at 100°C for 12 minutes, the film was uncrosslinked as indicated by solubility in organic solvent. Molding at 160°C or at 175°C produced crosslinked films, which were hydrated to produce hydrogels. The hydrogel of the polymer molded at 175°C became slightly cloudy upon heat
sterilization, whereas the hydrogel of the polymer molded at 160°C remained clear.
Table 6
*Compounded with glycerol before molding
♦♦Conditions required to produce a hydrogel with dimensional stability and optical clarity after autoclave
Example No. 14
A two step molding process using the copolymers from Examples 13 A, 13B, 13C, and 13D was carried out in this example. These copolymers were individually converted into uncrosslinked sheet when molded in a Carver press at 90°C for 20 minutes. Solubility tests in THF indicated that films molded under these conditions were not chemically crosslinked. These same copolymers also formed uncrosslinked sheet when they were compounded with about 6 wt% glycerol prior to molding under these conditions. Pieces of molded film underwent crosslinking reactions when placed in a 200°C air-circulating oven. The gelation was be monitored by removing pieces of the films at specified time intervals and determining their solubilities in THF. Gelation of these copolymers
occured in approximately 3-5 minutes at 200°C. Although gelation occured without glycerol, the gelation process appears to be accelerated when glycerol is present. Without the presence of glycerol, the copolymer from Example 13A was substantially crosslinked after 5-7 minutes at 160°C or after 10 minutes at 145°C.
Example No. 15
A two step molding process using the copolymer from Examples 13E was carried out in this example. The copolymer compounded with 5 wt% glycerol was converted into crosslinked sheet when molded in a Carver press at 170°C for 20 minutes. Solubility tests in THF indicated that the film molded under these conditions was chemically crosslinked. A circular disk with a diameter of 12 mm was cut from the molded sheet and further molded in a Carver press at 170°C under a load of 0.5 metric tons for 30 minutes. Hydration of this sample produced a lens like object.
This demonstrates that a partially cured polymer sheet or disk can still be formed into a lens like object. The low extent of crosslinking allows for additional processing to be carried out, during which the unreacted latent crosslinking sites may be reacted to produce an object with an increased crosslink density relative to that of the partially cured polymer.
Example No. 16 Copolymerization of DMA, FX-14, MMA and NIB MA was carried out in this example. The purpose of this example was to demonstrate the use of a functionalized acrylamide monomer as a latent crosslinker. Following the general polymerization procedures of the preceding examples, DMA (55 weight percent), FX-14 (20 weight percent) and MMA (15 weight percent) were copolymerized with about 10 weight percent NIBMA (N-isobutoxymethylacrylamide). The
NIBMA acts as a latent crosslinker. The resulting polymer was compression molded at 160°C for about 40 minutes to obtain a clear flexible film. The film was judged to be crosslinked by its insolubility in THF. Upon hydration, the film
formed a hydrogel with a water content of about 73%. Some distortion of the hydrogel occurred during a heat sterilization process.
Example No. 17
Copolymerization of DMA, FX-14, MMA and DCPMA (dicyclopentylmethacrylate) was carried out in this example. The purpose of this example was to demonstrate the use of an alkenyl-containing methacrylate monomer that is incoφorated into a polymer to undergo latent crosslinking by the addition of a peroxide prior to compression molding. Following the general polymerization procedures of the preceding examples, DMA (54.5 weight percent), FX-14 (19.9 weight percent) and MMA (14.9 weight percent) were copolymerized with 10.2 weight percent of DCPMA (dicyclopentenyl methacrylate). A polymer was obtained with a Tg of 71°C.
The polymer underwent three separate molding experiments. In one case, the polymer was compression molded at 155°C for about 20 minutes to obtain a transparent yellow-tinted film. The film dissolved in organic solvent indicating that it is not crosslinked. Upon hydration, the film formed a slightly cloudy hydrogel having a water content of about 71 weight percent. The hydrogel was not dimensionally stable under heat sterilization conditions. Under such conditions, the hydrogel shape distorted from round to oblong. In a second case, the polymer was compounded with about six weight percent peroxide. The compounding was accomplished by dissolving the polymer in an organic solvent, such as THF, acetone, or chloroform, and then mixing in the peroxide. The solvent was then allowed to evaporate, and the resultant dry polymer/peroxide blend was compression molded at 155°C for about 22 minutes to obtain a transparent yellow-tinted film. The film was not soluble in an organic solvent indicating that it is substantially crosslinked. Upon hydration, the film formed a clear hydrogel having a water content of 72 weight percent. The hydrogel was dimensionally stable under heat sterilization conditions maintaining both shape and size after two heat sterilization cycles.
In a third case, the polymer was compounded with about six weight percent peroxide, and then is compression molded at 175°C for about 22 minutes to obtain a transparent yellow-tinted film. The film was not soluble in an organic solvent indicating that it was substantially crosslinked. Upon hydration, the film formed a clear hydrogel having a water content of about 71 weight percent. The hydrogel was dimensionally stable under heat sterilization conditions maintaining both size and shape after three heat sterilization cycles.
Example No. 18
Copolymerization of DMA, FX-14, MMA, DCPMA and HMABP was carried out in this example. The puφose of this example was to demonstrate a thermoprocesssable, photocrosslinkable polymer. Following the general polymerization procedures of the preceding examples (except that the reaction flask was wrapped in aluminum foil to avoid light causing premature reaction of the HMABP photoinitiator, DMA (54.15 weight percent), FX-14 (13.34 weight percent), MMA (1 1.7 weight percent), and DCPMA (9.87 weight percent) were copolymerized with 6.68 weight percent of HMABP (3-hydroxy-l-methacryloxy- benzophenone). A polymer was obtained with a Tg of 72°C. The resulting polymer was compression molded at 140°C for about 20 minutes to obtain a clear film with a yellowish tint. The film dissolved in an organic solvent indicating that it is not crosslinked.
Upon hydration, the film formed a clear hydrogel with a yellow tint having a water content of 56 weight percent.
A portion of the dry film was exposed to UV radiation to activate the HMABP photocrosslinker and "cure" the polymer. The film was exposed under a Blak-ray Lamp Model XX- 15 with a GE-F 1518 BLB blacklight, a short wavelenght source. The film was positioned about 5.5 cm away from the light and exposed for a period of about six hours. After this curing, the film was found to be insoluble in organic solvent indicating that the film was substantially crosslinked. Upon hydration, the UV cured film formed a clear hydrogel having a
water content of 51 weight percent. This hydrogel was dimensionally stable under heat sterilization conditions.
Example No. 19
Copolymerization of DMA, FX-13, BE A, and MPMDES (methyl propyl diethoxysilyl methacrylate) was carried out in this example. The puφose of this example was to demonstrate a thermoprocesssable, moisture curable polymer. Following the general polymerization procedures of the preceding examples, DMA (55 weight percent), FX-13 (20 weight percent), BEA (20 weight percent), and MPMDES (5 weight percent) were copolymerized with Vazo52 initiator (0.54 weight percent). The resulting polymer was compression molded at 100°C for about 12 minutes to obtain a clear film. The film dissolved in an organic solvent indicating that it was not crosslinked.
A portion of the dry film was hydrated and then dehydrated. The dehydrated film was found to be insoluble in organic solvent indicating that the film was substantially crosslinked. Upon hydration, the moisture cured film formed a clear hydrogel having a water content of 74 weight percent. This hydrogel was not dimensionally stable under heat sterilization conditions and became opaque. Alternatively, the polymer could be compression molded at 160°C for about 20 minutes to obtain a clear film. The film was found to be insoluble in an organic solvent indicating that the film was substantially crosslinked. Upon hydration, the thermally cured film formed a clear hydrogel having a water content of 70 weight percent. This hydrogel was dimensionally stable under heat sterilization conditions.
In another manner to carry out this invention, thermoprocessable coplymer materials containing latent crosslinking groups may be synthesized such that the crosslinking can be reversed after the processing is complete. This may be accomplished by copolymerizing monomers such as N,N'-cystamine-bis- acrylamide (CBA) with the formulations listed above. See for example H. Lee and T. Park, Polymer Journal, vol 30., No. 12, pp. 976-980. Copolymerization of
CBA with monomers such as DMA should result in crosslinked hydrogel forming polymer. The disulfide linkages in the crosslinks can be subjected to chemical reduction to form thiol groups, resulting in a loss of the chemical crosslinks. Oxidation of the thiols would result in a reformation of the disulfide groups, resulting in the formation of chemical crosslinks. An example of a reducing agent includes, but is not limited to, dithiothreitol, and example of oxidizing agents include, but are not limited to, cystamine and molecular oxygen (O2). Therefore, copolymers of this nature which contain pendant thiol groups could be melt processed and then crosslinked through the oxidation to disulfide linkages. It should also be possible to use disulfide containing dimethacrylate or diacrylate monomers to achieve a similar effect.
In yet another manner to carry out this invention, thermoprocessable copolymer materials may be synthesized such that the latent crosslinking is a result of the thermal decomposition of peroxy or azo species contained in the side chains of the copolymer. These peroxy or azo groups will decompose at elevated temperatures to form radicals. This elevated temperature is preferably above the temperature of the polymerization reaction. The radicals formed from the decomposition will induce crosslinking reactions in the polymer through the processes described above.
From the above examples and the previous discussion, it should be apparent to one skilled in the art that this copolymers made in accordance with this invention, or that may be suitable for thermal processing into ophthalmic lenses in accordance with this invention, may take the form of various embodiments. In addition, based on these teachings, one skilled in the art should be able to carry out this invention using a variety of monomers, including monomers not used in the above examples.
To further illustrate this invention, additional embodiments building upon the above disclosure will be provided. This section of the discussion relates particularly to uncrosslinked hydrogels comprising branch copolymers (also called graft copolymers or comb copolymers) containing a polymeric hydrophilic main
chain (backbone) with polymeric hydrophobic side chains (branches) suitable for use in the manufacture of medical devices including contact lenses. Alternatively, the main chain can be hydrophobic, and the side chain can be hydrophilic. The copolymer is uncrosslinked, that is, it does not have chemical covalently bonded crosslinks. However, the copolymer attains mechanical stability as a hydrogel due to the pseudo-crosslinks (i.e., physical crosslinks, such as due to internal hydrophobic association of hydrophobic branches across different polymer molecules) that were described in detail in the previous section of the discussion above. The puφose of designing a branch copolymer with both hydrophilic and hydrophobic segments is to fabricate a physically crosslinked hydrogel with both high water content and distinguishable physical strength. In general, the presence of hydrophilic main chain in a copolymer can strongly enhance the absoφtion of moisture to give the bulk materials with hydrogel property. On the other hand, after the material is hydrated, the hydrophobic branches may aggregate with each other to form a separated phase, which make a significant improvement on the physical properties of this hydrogel. The described branch copolymer usually could be synthesized by several different methods, which will be discussed in detail later, and than purified and dried in subsequent processes. The dry copolymers may be molded to a desired shape, such as a film, at a preferred temperature and then hydrated to a hydrogel.
Even though the hydrogel films made from the branch copolymers with the structure mentioned above do not include chemical crosslinking, these hydrogels still show remarkable mechanical strength for handling and dimensional stability for thermal sterilization. The unique physical characteristics of these hydrogel films are mainly due to the existence of physical crosslinking associated with the hydrophobic side chains. Since this type of copolymer may be made without thermally reactive functional groups that build chemical crosslinking, the materials can be molded with broader choice of processing temperature and time. The polymeric branch copolymers useful in the methods of this invention may encompass a broad range of sizes and compositions. The copolymers may
have a weight average molecular weight preferably in the range from about 10,000 grams/mole to about 1 ,000,000 grams/mole. More preferably, the molecular weight is in the range from about 50,000 grams/mole to about 500,000 grams/mole. The individual side chains on the branch copolymers have a weight average molecular weight preferable in the range from about 300 grams/mole to
100,000 grams/mole. The polymeric side chains are covalently bonded to the backbone. The macromer units with the side chains make up a fraction preferably in the range from about 0% to about 95% by weight of the total branch copolymer. More preferably, the macromer units make up a fraction from about 5% to about 60% by weight of the total copolymer. As used herein, a macromer is considered to be a macromolecule with an active group, such as, but not limited to, a vinyl group, for incoφoration into a main polymer chain, and a side group made of a long polymeric repeating chain or peptide mimetic chain.
Generally, the branch copolymers containing a hydrophilic main chain with hydrophobic side chains may be prepared by three possible routes listed below.
These methods, however, may not be the only methods to construct a copolymer with branches.
(A) Copolymerization of monomer(s) with macromer(s): A branch copolymer can be directly synthesized from a mixture of monomer(s) and macromer(s) with initiator(s). The monomer(s) suitable for use herein can be hydrophilic, or a mixture of hydrophilic and hydrophobic. The macromer(s) suitable for use herein can be hydrophobic or a mixture of hydrophobic and hydrophilic. In this embodiment, the selected macromers are polydimethylsiloxane monomethacrylate (a hydrophobic macromer) and poly(ethylene glycol) methyl ether acrylate macromers (a hydrophilic macromer) because of their convenient availability from a commercial catalog supplier, such as Gelest or Aldrich. Synthesis of other hydrophobic and hydrophilic macromers with desired composition are available using the methods which are described in the published literature, such as K. Ito, N. Usami, and Y. Yamashita,
Macromolecules, 13, 216 (1980). For example, one such proposed reaction scheme for one of the potential methods to prepare hydrophobic or hydrophilic macromers is as shown below.
H2C=CH
C= 0 O
I
CH2 FX-13 CH,
C?H " S09- CnF2n+l
HS-CH2COOH
AIBN
60°C / 20 hours
The monomer used for this synthesis could be any type of hydrophilic or hydrophobic monomer, or even a blend of both. It is not limited to 2-(N-ethyl perfluorooctane-sulfonamido) ethyl acrylate (FX-13) used in this particular example.
Example No. 20
Copolymerization of dimethylacrylamide, FX- 13 and polydimethylsiloxane monomethacrylate (MW = 5,000) was carried out in this example. The puφose of this example was to illustrate a branched copolymer made through a single step chemically initiated free radical polymerization reaction using a vinylic monomer having a long, hydrophobic side chain.
To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (64.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (20 wt%), and monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 5,000 (15 wt%) with Vazo64 (0.3 wt%). The total mass of monomers is 30 g. The solution was purged with nitrogen for 10 minutes, and then was polymerized with thermal initiation at 60°C for 20 hours. Alternatively, the monomers could also be polymerized with photo initiation in a Rayonet-600 reaction chamber (4 watts x 4, λ= 365 nm) at room temperature for about 20 hours. After the branch copolymer is collected and dried, the material was compression molded to a film at 160°C and was then hydrated into a hydrogel. The expected polymeric structure is presented below and the results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
CH3
2-(n-ethylperfluorooctanesulfonamido) Monoacryloxypropyl terminated ethyl acrylate polydimethyl siloxane
Toluene
AIBN
UV / 20 hours
Poly(dimethylacrylamide-co-FX- 13-co-polydimethylsiloxane monomethacrylate)
The following examples 21-24 illustrate the effect of the hydrophobic side chain on the properties of both the dry copolymer and the hydrogel. The structures of these copolymers are similar to that presented above.
Example No. 21
Copolymerization of dimethylacrylamide, FX-13 and polydimethylsiloxane monomethacrylate (MW = 1 ,000) was carried out in this example.
To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (200 mL), dimethylacrylamide (DMA) (54.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (20 wt%), and monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 1,000 (25 t%) with Vazo64 (0.3 t%). The total mass of monomers is 50 g. The solution was purged with nitrogen for 10 minutes, and then was polymerized with photo initiation in a Rayonet-600 reaction chamber
(4 watts x 4, λ= 365 nm) at room temperature for about 20 hours. (Alternatively, the monomers could also be polymerized by thermal initiation at 60°C for 20 hours). After the branch copolymer was collected and dried, the material was compression molded to a film at 160°C and was then hydrated into a hydrogel. The results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
Example No. 22
Copolymerization of dimethylacrylamide, FX-13 and polydimethylsiloxane monomethacrylate (MW = 10,000) was carried out in this example. The branch copolymer was prepared following the procedure of Example
20 except the hydrophobic macromer employed had a larger number of repeating units. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (64.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (20 wt%), and monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 10,000 (15 wt%) with Vazo64 (0.3 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 10 minutes, and was then polymerized with thermal initiation at 60°C for 20 hours. The branch copolymer was collected and dried. Molecular weight (based on
GPC): Mw = 160,000. Differential scanning calorimetry: the copolymer showed a glass transition temperature of 110°C. After compression molding to a film at 160°C the material was hydrated into a hydrogel. The dry film was translucent and exhibits a rough surface. The water content of the hydrogel was 74%.
Example No. 23
Copolymerization of dimethylacrylamide, FX-13 and polydimethylsiloxane monomethacrylate (MW = 10,000) was carried out in this example.
The branch copolymer was prepared following the procedure of Example 22 except that a larger percentage of the same hydrophobic macromer was employed. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (49.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (20 wt%), and monoacryloxypropyl terminated polydimethyl siloxane, M.W .= 10,000 (30 t%) with Vazo64 (0.3 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 10 minutes, and then was polymerized with thermal initiation at 60°C for 20 hours. After the branch copolymer was collected and dried, the material was compression molded to a film at 160°C and then hydrated into a hydrogel. The branch copolymer was collected and dried. Molecular weight (based on GPC): Mw = 166,000. Differential scanning calorimetry: the copolymer showed a glass transition temperature of 104°C. After compression molding to a film at 160°C the material was hydrated into a hydrogel. The dry film was slightly hazy and exhibited a slightly rough surface. The water content of the hydrogel was 68%.
Example No. 24 Copolymerization of dimethylacrylamide, FX- 13 and polydimethylsiloxane monomethacrylate (MW = 5,000) was carried out in this example.
The branch copolymer was prepared following the procedure of Example 20 except that a larger percentage of the hydrophobic macromer was employed. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux
condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (49.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX- 13) (20 wt%), and monoacryloxypropyl terminated polydimethyl siloxane, M.W .= 5,000 (30 wt%) with Vazo64 (0.3 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 10 minutes, and then was polymerized with thermal initiation at 60°C for 20 hours. After the branch copolymer was collected and dried, the material was compression molded to a film at 160°C and then was hydrated into a hydrogel. The results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
Example No. 25
Copolymerization of dimethylacrylamide (DMA), FX-13, polydimethylsiloxane monomethacrylate, and polyethyleneglycol methyl ether acrylate was carried out in this example. The puφose of this example was to illustrate a branch copolymer having both hydrophobic and hydrophilic long side chains. The hydrophilic side chains may be useful to improve the surface wettability of a contact lens made from this material.
The branch copolymer was prepared following the procedure of Example 20 except that the formula included both hydrophilic and hydrophobic macromers in the polymerization flask. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (14.16 g, 142.89 mmole), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (6.00 g, 10.24 mmole), monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 5,000 (7.50 g, 1.50 mmole) and poly(ethylene glycol) methyl ether acrylate, M.W.= 454 (2.25 g, 4.96 mmole) with Vazo64 (0.3 wt%). The solution was purged with nitrogen for
10 minutes and then polymerized with photo initiation in a Rayonet-600 reaction chamber (4 watts x 4, λ= 365 nm) at room temperature for about 20 hours. (Alternatively, the monomers could also be polymerized by thermal initiation at 60°C for 20 hours). After the branch copolymer was collected and dried, the material is compression molded to a film at 160°C and then was hydrated into a
hydrogel. The expected polymeric structure is presented below and the results of characterization of both dry copolymer and hydrogel are also summarized in Table 7, below.
The following examples 26-28 illustrate the effect of both the hydrophobic side chain and the hydrophilic side chain on the properties of both the dry copolymer and the hydrogel. The structures of these copolymers are similar to that presented below.
Example No. 26 Copolymerization of dimethylacrylamide (DMA), FX- 13 , polydimethylsiloxane monomethacrylate, and polyethyleneglycol methyl ether acrylate is carried out in this example.
The branch copolymer was prepared following the procedure of Example 25 except that a larger percentage of the hydrophilic monomer is employed. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, is charged toluene (180 mL), dimethylacrylamide (DMA) (39.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX- 13) (20 wt%), monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 5,000 (25.0 wt%) and polyethylene glycol) methyl ether acrylate, M.W.= 454 (15.0 wt%) with Vazo64 (0.3 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 10 minutes and then polymerized with photo initiation in a Rayonet-600 reaction chamber (4 watts x 4, λ= 365 nm) at room temperature for about 20 hours. (Alternatively, the monomers could also be polymerized by thermal initiation at 60°C for 20 hours). After the branch copolymer was collected and dried, the material is compression molded to a film at 160°C and then was hydrated into a hydrogel. The results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.

Dimethylacrylamide Poly(ethylene glycol) methyl ether acrylate
CH,
H2C= = CH
1 H2C=C
C:0
+ + C:0 ό 1 O
I
CH2 CH2
Toluene
AIBN
UV / 20 hours
Poly(dimethylacrylamide-co-FX- 13-co-polydimethylsiloxane monomethacrylate- co-poly(ethylene glycol) methyl ethyl acrylate)
Example No. 27
Copolymerization of dimethylacrylamide (DMA), FX-13, polydimethylsiloxane monomethacrylate, and polyethyleneglycol methyl ether acrylate was carried out in this example. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (54.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (20.0 wt%), monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 5,000 (15.0 wt%) and poly(ethylene glycol) methyl ether acrylate, M.W.= 1100 (10 wt%) with Vazo64 (0.3 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 10 minutes, and then was polymerized with thermal initiation at 60°C for 20 hours. After the branch copolymer was collected and dried, the material was compression molded to a film at 160°C and then was hydrated into a hydrogel. The results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
Example No. 28
Copolymerization of dimethylacrylamide (DMA), FX-13, polydimethylsiloxane monomethacrylate, and polyethyleneglycol methyl ether acrylate was carried out in this example.
The branch copolymer is prepared following the procedure of Example 27 except that a larger amount of the hydrophobic macromer was employed. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (180 mL), dimethylacrylamide (DMA) (39.7 wt%), 2-(N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-
13) (20.0 wt%), monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 5,000 (30.0 wt%) and polyethylene glycol) methyl ether acrylate, M.W.= 1100 (10 wt%) with Vazo 64 (0.3 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 10 minutes, and then was polymerized with thermal initiation at 60°C for 20 hours. After the branch copolymer was collected
and dried, the material was compression molded to a film at 160°C and then was hydrated into a hydrogel. The expected polymeric structure is presented below and the results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
Example No. 29
Copolymerization of dimethylacrylamide (DMA), FX-13, polydimethylsiloxane monomethacrylate, and polyethyleneglycol methyl ether acrylate was carried out in this example.
The branch copolymer is prepared following the procedure of Example 27 except that a different initiator (Vazo52) was employed. To a 500-mL four neck cylinder flask, equipped with mechanical stirring, reflux condenser, and nitrogen inlet, was charged toluene (150 mL), dimethylacrylamide (DMA) (54.5 wt%), 2- (N-ethyl perfluorooctanesulfonamido) ethyl acrylate (FX-13) (20.0 wt%), monoacryloxypropyl terminated polydimethyl siloxane, M.W.= 5,000 (15.0 wt%) and polyethylene glycol) methyl ether acrylate, M.W .= 1 100 (10 wt%) with
Vazo52 (0.5 wt%). The total mass of monomers was 30 g. The solution was purged with nitrogen for 20 minutes, and then was polymerized with thermal initiation at 45°C for 20 hours. The branch copolymer was collected and dried. The results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
(B) Grafting of hydrophobic side chain onto a hydrophilic main chain
In this grafting polymerization technique, the branch copolymer is prepared by two consecutive polymerization reactions. In the first step, the polymeric main chain may be synthesized using either hydrophilic or hydrophobic monomers, or a mixture of hydrophilic and hydrophobic monomers, or macromers, or a mixture of both, with a small fraction, preferable in the range from about 0.5% to about 30%, of functional monomer(s) by well-known polymerization techniques which
include chain growth, step growth, Ziegler-Natta, and group transfer polymerization etc. The functional monomer(s) defined herein contains any type of active functionality that can be used to graft side chains.
The polymeric side chains grafted from the active site may be polymerized from hydrophobic or hydrophilic monomers, or a mixture of hydrophobic and hydrophilic monomers, or macromer, or a mixture of both monomers and macromers, up to the desired chain length. The chain length can be controlled by the concentration ratio between polymerizable units used for making the side chain versus the active sites on the main chain. One kind of active site used in this invention is represented by the organic halide moiety which can be grafted with monomer(s) through Atom Transfer Radical Polymerization (ATRP) technique. This technique is described in U.S. Patent No. 5,763,548, to Matyjaszewski et al., which is incoφorated herein by reference.
In the following examples, the main chain copolymer was prepared by copolymerizing a monomer mixture containing between 1% and 5% by weight of the total monomer content of organic halide-containing monomer. The halogen atom in this monomer does not have to be chlorine, as used in these examples, but may also be bromine, iodine, or fluorine. The final graft copolymer is composed of between about 50% and 75% by weight hydrophilic main chain and between about 25% and 50% by weight hydrophobic side chains. The ratio between hydrophilic main chain and hydrophobic side chain may be possible to make with a range as wide as possible. Alternatively, the main chain may be hydrophobic and the side chains may be hydrophilic. The resulting hydrogel of the polymer should be characterized by a hydrophilic continuous phase. A measure of the extent of grafting may be obtained by taking the FT-IR peak ratio of carbonyl groups from ester and amide groups. The ratio of ester to amide increases with the extent of grafting.
Example No. 30
Copolymerization of dimethylacrylamide, FX-13, and vinylbenzyl chloride, with subsequent grafting of poly(methyl methacrylate) was carried out in this
example. The puφose of this example was to illustrate a multiple-step polymerization technique for building a branch copolymer that was suitable for thermally processing into an ophthalmic lens.
To a 500-mL three-neck round bottom flask, equipped with mechanical stirring, reflux condenser and nitrogen inlet, was charged with toluene(180 mL), 2-
(N-ethylperfluorooctanesulfonamido) ethyl acrylate (FX-13, 6.00 g, 10.24 mmole), dimethylacrylamide (DMA, 23.61 g, 238.24 mmole), vinylbenzyl chloride (0.30 g, 1.97 mmole), and AIBN (azobisisobutyronitrile, 0.09 g, 0.55 mmole). After purging with nitrogen for 10 minutes, the mixture was copolymerized with a slow nitrogen flow at 60°C for about 20 hours, and then was further heated to about 100°C for about one hour. When the solution was cooled to the room temperature, the grafting reagents including toluene (60 mL), methyl methacrylate (10.00 g, 100 mmole), CuCl (0.1948 g, 1.97 mmole), and 2,2'- dipyridine (0.9218 g, 5.90 mmole) were charged to the round bottom flask and then are heated to 120°C for about 6 hours. After cooling to room temperature, the catalyst was removed by filtration, and then the filtrate was precipitated in hexanes. The isolated solid was washed with hot water several times followed by drying in vacuum oven. The dry material was compression molded to a film at 160°C, and then was hydrated to hydrogel. The expected polymeric structure is presented below, and the results of characterization of both dry copolymer and hydrogel are summarized in Table 7, below.
Table 7
l Chloride
2-(n-ethylperfluorooctanesulfonamido) ethyl acrylate
Toluene
AIBN
60°C / 20 hours
Methyl methacrylate CuCl / 2,2'-dipyridyl I 120°C / 6 hours
Poly(dimethylacrylamide-co-FX-13-co-vinbenzylchloride)-g-poly(methyl methacrylate)
Example No. 31
Copolymerization of dimethylacrylamide and vinylbenzyl chloride was carried out in this example. The resulting polymer was used for subsequent graft polymerization. A three neck flask 500 mL glass flask equipped with a condenser, mechanical stirring unit, balloon and a gas inlet line was charged with dimethyl acrylamide (DMA, 95.0 g), vinyl benzyl chloride (VBC, 5.0 g), VAZO-52 (0.51 g) and toluene (500 mL). The flask was filled with nitrogen until the balloon (9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the balloon deflated and the flask contents just began to bubble. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9-inch capacity balloon was inflated. The reaction mixture was de-gassed and then filled with nitrogen about four times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 24 hours at about 45°C. Pouring the reaction mixture into about
500 mL of stirring hexanes isolated the copolymer. After soaking for about 30 minutes, the solvent was decanted away from the precipitated copolymer. The copolymer was washed with about 300 mL of fresh hexanes and then vacuum dried for about one day at about 60°C. The sample was analyzed by UV-VIS, FT-IR, and DSC. UV-VIS (Sample in chloroform): peak centered at 244 nm.
FT-IR (Film cast on NaCl from chloroform, selected peaks): 2928, 2863, 1643, 1496, 1463, 1399, 1355, 1259, 1139, 1096, 1058 cm"1. Differential scanning calorimetry: The polymer showed a glass transition temperature of 100°C.
Example No. 32 Copolymerization of dimethylacrylamide, n-octadecyl acrylate, and vinylbenzyl chloride was carried out in this example. The resulting polymer was used for subsequent graft polymerization.
A glass kettle reactor equipped with an overhead stirrer (Teflon blade), balloon, condenser and gas inlet was charged with dimethyl acrylamide (DMA,
79.32 g, 79.20 wt%), vinyl benzylchloride(VBC, 5.14 g, 5.13 wt%), n-octadecyl acrylate (ODA, 14.85 g, 14.82 wt%), VAZO 64 (0.85 g, 0.85 wt%) and butylacetate (1300 mL). The flask was lowered into a water bath and filled with nitrogen until the balloon (9-inch capacity) was inflated. Vacuum was applied to the reaction vessel until the flask contents just began to bubble. At this point the vacuum was turned off and nitrogen was bled into the flask until the 9 inch capacity balloon was inflated. The reaction mixture was de-gassed and the balloon was filled with nitrogen about six times. The reaction mixture was left under nitrogen and the overhead stirrer was started. The reaction mixture was allowed to stir under nitrogen for about 20 hours at about 60°C. At this point, about 0.5 mL of sample was removed from the reaction flask and mixed with about 20 mL of hexanes. The resulting precipitate was filtered, washed with about 20 mL of hexanes. A few milligrams of the isolated product were dissolved in chloroform and then analyzed by UV-VIS spectroscopy. The sample displayed a peak centered at about 244 nm. The temperature of the bath was then raised to about 70°C for about one hour. The polymer solution was stored in a sealed container to be used at a later time to prepare graft copolymers by atom transfer radical polymerization (ATRP).
The precipitated sample from above was dried at about 60°C under vacuum and then analyzed analyzed by FT-IR and DSC. FT-IR (Film cast on NaCl from chloroform, selected peaks): 2924.3, 2854.0, 1728, 1643, 1494, 1463, 1398, 1354, 1259, 1139, 1096, 1059 cm"1. Differential scanning calorimetry: The polymer showed a glass transition temperature of 95°C. Copolymer formed a clear brittle film when it was compression molded under a force of 8 metric tons at 130°C for 5 minutes The film formed a clear hydrogel that was unable to support its own weight.
Example No. 33
Graft copolymerization of poly(n-butyl acrylate) with the polymer from Example 32 is carried out in this example. The puφose of this example was to
illustrate the second step of a polymerization technique for building a branch copolymer that was suitable for thermally processing into an ophthalmic lens.
A three neck flask equipped with a condenser, mechanical stirring unit, and a gas inlet line was charged with about 20 grams of poly(DMA/VBC/ODA) in about 245 mL of butylacetate (from Example 32). To the flask was added
N,N,N',N',N"-pentamethyldiethylenetriamine (PMDETA, 3.53 g), copper (I) chloride (0.67 g), and n-butylacrylate (BA, 20.3 g). Nitrogen was bubbled through the reaction mixture for 10 minutes. The nitrogen flow was then decreased and the mixture was heated to 100°C over 22 minutes. A balloon was then attached to the reaction flask and inflated with nitrogen. The reaction mixture was then heated for 24 hours at 100°C. The reaction mixture was diluted with 100 mL of THF and 200 mL of water. Addition of water caused copolymer to precipitate. The aqueous layer was removed and 700 mL of fresh water was added to the copolymer. The sample was washed a few more times with 700 mL portions of water and then dissolved in 250 mL of acetone. The acetone solution was stirred with 50 grams of neutral aluminum oxide. The aluminum oxide was removed by filtration and the resulting acetone solution was then added to 700 mL of stirring hexanes. The resulting precipitate was separated, washed twice with 100 mL of hexanes, and vacuum dried at 60°C for about 24 hours. The polymer was analyzed by FT-IR and DSC. FT-IR (Film cast on NaCl from THF, selected peaks): 2926,
2856, 1732, 1643, 1497, 1465, 1398, 1259, 1140, 1096, 1065 cm"1. Differential scanning calorimetry: A glass transition temperature of 74°C was detected.
The polymer was compression molded under an applied load of 8 metric tons) at 150°C for 30 minutes to yield a transparent, yellow tinted, flexible film. The material remained thermoplastic as demonstrated by its ability to be remolded.
The film formed a hydrogel with water content of 73%. The stress at break for the hydrogel was 0.2 N/mm2 while the elongation at break was 186%. Circular disks (13 mm) cut from hydrogel sheet did not maintain their shape and dimensions when subjected to heat sterilization in an autoclave at 120°C for 45 minutes. Circular disks which were cut from a hydrogel that had been heat treated by
boiling in Wesley lessen lens packaging solution (100°C for 5 minutes) maintained their shape and dimensions when sterilized in the autoclave.
Example No. 34
Graft copolymerization of poly(methyl methacrylate) with the polymer from Example 32 was carried out in this example.
Polymerization was carried out according to the procedure of Example 33 except using 20 grams of poly(DMA/VBC/ODA) in about 245 mL of butylacetate (from Example 32), 3.51 grams of N,N,N',N',N"-pentamethyldiethylenetriamine (PMDETA), 0.68 grams of copper (I) chloride, and 20.7 grams of methyl methacrylate (MMA). After being purged with nitrogen, the mixture was heated to 105°C over 22 minutes. The reaction mixture was then heated for 20 hours at 105°C. Pouring the reaction mixture into 700 mL of stirring hexanes caused copolymer to precipitate. The resulting precipitate was filtered, washed three times with 500 mL of water, and soaked in 700 mL of water for 48 hours. The solid polymer was vacuum dried at 75°C for about 9 hours. The polymer was analyzed by FT-IR and DSC. FT-IR (Film cast on NaCl from toluene, selected peaks): 2925, 2857, 1730, 1643, 1486, 1451, 1398, 1354, 1265, 1191, 1147, 1097, 1059 cm"1. Differential scanning calorimetry: A glass transition temperature of 69°C was detected. The polymer was compression molded under an applied load of 8 metric tons) at 130°C for 7 minutes to yield a transparent, brown tinted, brittle film. The material remained thermoplastic as demonstrated by its ability to be remolded. The film formed a hydrogel with water content of 57%. The stress at break for the hydrogel was 0.4 N/mm2 while the elongation at break was 60%. Circular disks (13 mm) cut from hydrogel sheet did not maintain their shape and dimensions when subjected to heat treatement by boiling at 100°C for 10 minutes in Wesley Jessen lens packaging solution. However, further heat treatment for 10 minutes or 30 minutes did not result in more loss of shape. Circular disks which were cut from a hydrogel that had been heat treated by boiling in Wesley Jessen lens
packaging solution (100°C for 5 minutes) did not maintain their shape and dimensions when sterilized in the autoclave at 120°C for 45 minutes.
Example No. 35
Graft copolymerization of poly(n-octadecylacrylate) with the polymer from Example 32 was carried out in this example.
Polymerization was carried out according to the procedure of Example 33 except using 20 grams of poly(DMA/VBC/ODA) in about 245 mL of butylacetate (from Example 32), 3.51 grams of N,N,N',N',N"-pentamethyldiethylenetriamine (PMDETA), 0.67 grams of copper (I) chloride, and 20.0 grams of n- octadecylacrylate (ODA). After being purged with nitrogen, the mixture was heated to 100°C over 22 minutes. A balloon was attached to the flask and filled with nitrogen. The reaction mixture was then heated for 48 hours at 100°C. Pouring the reaction mixture into 700 mL of stirring hexanes caused copolymer to precipitate. The resulting precipitate was filtered and washed five times with 300 mL of water. The solid polymer was vacuum dried at 75°C for 24 hours. The polymer was dissolved in 600 mL THF, and 150 mL of water were added. The mixture was filtered, passed through 200 grams of neutral aluminum oxide, and then concentrated by rotary evaporation. The polymer was filtered from the remaining water and washed twice with 250 mL of water. The polymer was vacuum dried at 75°C for 9 hours. The polymer was analyzed by FT-IR and DSC.
FT-IR (Film cast on NaCl from THF, selected peaks): 2924, 2853, 1729, 1642, 1496, 1465, 1398, 1354, 1259, 1139, 1100, 1060 cm"1. Differential scanning calorimetry: A glass transition temperature of 74°C was detected.
The polymer was compression molded under an applied load of 8 metric tons) at 150°C for 30 minutes to yield a transparent, brown tinted, flexible film.
The material remained thermoplastic as demonstrated by its ability to be remolded. The film formed a hydrogel with water content of 65%. The stress at break for the hydrogel was 0.3 N/mm2 while the elongation at break was 106%. Circular disks (13 mm) cut from hydrogel sheet did not maintain their shape and dimensions when subjected to heat sterilization in an autoclave at 120°C for 45 minutes.
Circular disks which were cut from a hydrogel that had been heat treated by boiling in Wesley Jessen lens packaging solution (100°C for 5 minutes) maintained their shape and dimensions when sterilized in the autoclave.
The graft copolymers prepared in accordance with the invention may vary from the preceding examples. For instance, the molecular weights of the precursor polymers such as those described in Examples 31 and 32 can be altered by modifying the reaction conditions in the polymerization step. These modifications include, but are not limited to, changing the reaction temperature, changing the concentration of initiator, and adding a chain transfer agent (such as, but not limited to 1-decanethiol). Another characteristic dependent on reaction conditions and monomer feed is the microstructure of the grafted chains. The grafted chains can be polymerized such that the chains have di-block, tri-block, or multi-block structures or alternatively random or alternating structures. Any of the monomers listed herein can be useful as components of the polymerization mixture for the graft chains.
Further variations of graft copolymers are within the scope of the invention. The halocarbon end group on the grafted chains can be made to function as latent crosslinkers for the polymer if the polymer is subjected to appropriate heat, light, or chemical reagent. Nucleophilic substitution of halocarbons is well known. Possible reagents for nucleophilic substitution include, but are not limited to, multifunctional primary or secondary amines (such as 1,4-diaminobutane and various isomers thereof), diols, triols, and polyols, such as ethyleneglycol, glycerol, poly(ethyleneglycol), and poly(vinyl alcohol). The halogen group can also be replaced with hydroxyl, alkoxide, or aryloxide. The hydroxyl groups can be obtained by treatment of the polymer with water- containing solutions under acidic, basic, or neutral conditions. The alkoxide or aryloxide groups can be obtained by heating the polymer in the presence of alcohol, alkoxide, or aryloxide. Also, photolysis of a solution containing the polymer and reagents such as chloroform or 1-decanethiol can result in exchange of the halogen with a hydrogen atom.
(C) Chemical bonding of hydrophobic side chain to hydrophilic main chain:
The branch copolymer can also be prepared by another potential method by attaching the polymeric hydrophobic side chain to the polymeric hydrophilic main chain through a post chemical reaction after the main chain and side chain copolymers have been prepared from the separated reaction. A proposed method that can be used to synthesize the branch copolymer is described below. For example, a polymeric hydrophilic main chain can be prepared by copolymerizing a small fraction of monomer(s) containing primary or secondary amino group; on the other hand, the polymeric hydrophobic side chain can be prepared through ATRP such that there is an organic halide moiety at the chain end. Mixing of these two pre-synthesized main chain and side chain copolymers followed by a nucleophilic substitution reaction can give a branch copolymer by attaching the polymeric branches to the backbone via a chemical bonding.
Blend Copolymers
In another manner to carry out this invention, melt processable polymer materials may be developed from composite (blend) materials. The composite materials may be composed of two or more different kinds of copolymers which are somewhat miscible. The copolymers must be sufficiently compatible for a homogenous blending to avoid segregation that would result in an opaque or optically unclear ophthalmic lens.
Polymers with various compositions may be prepared in the separated polymerization reactions, and then blended to each other in different ratios. The composite can comprise at least two different copolymers, each containing hydrophilic and hydrophobic units. Alternatively, the composite can comprise a hydrophobic polymer or copolymer and a copolymer containing hydrophilic and hydrophobic units. The purified composites can also be molded to the desired shape as discussed for the multiple embodiments above. The type of composite materials that may be made include both thermoplastic (linear copolymers and
branch copolymers) and thermoset (chemically crosslinked copolymers) materials. All of the curing techniques (including thermo curing, photo curing, moisture curing, redox curing, etc.) discussed earlier are also suitable for the thermoset blend copolymers described here. It is believed that this manner of the invention can provide more easily for melt processable polymers capable of forming hydrogels.
The above preferred embodiments and examples are given to illustrate the scope and spirit of the present invention. These embodiments are not intended to limit the invention, but will make apparent to those skilled in the art other embodiments and examples within the contemplated scope of the invention. Therefore, the present invention should be limited only by the following claims.