相關申請案之交叉引用
本申請案主張2016年10月18日申請之美國臨時申請案第62/409,756號與2016年10月18日申請之美國臨時申請案第62/409,768號的優先權與權益,該等臨時申請案中之每一者以全文引用的方式併入本文中。 如本文大體描述,本發明提供適用於預防及/或治療大範圍之病症的經取代氧固醇,該等病症包括(但不限於)NMDA介導性病症。預期此等化合物展示與其他氧固醇相比之經改良的活體內效能、藥物動力學(PK)特性、口服生物可用性、可調配性、穩定性及/或安全性。
化合物
在一個態樣中,本文提供根據式(I-63)之化合物:
或其醫藥學上可接受之鹽,其中:R
1
為烷基(例如C
1
-C
6
烷基);R
2
及R
3
中之各者獨立地為氫、烷基(例如C
1
-C
6
烷基)、碳環基、雜環基、芳基或雜芳基,或R
2
及R
3
與其所附接之碳原子一起形成3員至8員環;R
4
及R
5
中之各者獨立地為氫、鹵基或-OR
C
,其中R
C
為氫或C
1
-C
6
烷基(例如C
1
-C
3
烷基),或R
4
及R
5
與其所附接之碳原子一起形成側氧基;R
6
不存在或為氫;且
表示單鍵或雙鍵,其中當
中之一者為雙鍵時,另一
為單鍵;當
兩者均為單鍵時,則R
6
為氫;且當
中之一者為雙鍵時,R
6
不存在。 在一些實施例中,R
1
為烷基(例如C
1
-C
6
烷基)。在一些實施例中,R
1
為C
1
-C
6
烷基(例如-CH
3
、-CH
2
CH
3
、-CH
2
OCH
3
或-CF
3
)。在一些實施例中,R
1
為-CH
3
、-CF
3
或-CH
2
CH
3
。在一些實施例中,R
1
為-CH
2
OR
A
,其中R
A
為C
1
-C
6
烷基(例如C
1
-C
3
烷基)。 在一些實施例中,R
2
為氫或烷基(例如C
1
-C
6
烷基)。 在一些實施例中,R
2
及R
3
中之各者獨立地為氫或烷基(例如C
1
-C
6
烷基)。在一些實施例中,R
2
及R
3
中之各者獨立地為氫或C
1
-C
6
鹵烷基(例如,-CF
3
)。在一些實施例中,R
2
及R
3
中之各者獨立地為C
5
烷基(例如經取代或未經取代之異戊基)或氫。在一些實施例中,R
2
及R
3
中之各者獨立地為異戊基(例如經取代或未經取代之異戊基)或氫。在一些實施例中,R
2
及R
3
中之各者獨立地為氫、-CF
3
或-CH
3
。 在一些實施例中,R
4
為-OH或鹵基(例如-F)。 在一些實施例中,R
4
及R
5
與其所附接之碳原子一起形成側氧基。在一些實施例中,R
4
為氫且R
5
為鹵基(例如-F)。 在一些實施例中,R
4
及R
5
為鹵基(例如-F)。在一些實施例中,R
4
及R
5
為氫。 在一些實施例中,R
2
為芳基或雜芳基,且R
3
為氫。在一些實施例中,R
2
為碳環基或雜環基且R
3
為氫。在一些實施例中,R
2
及R
3
為氫。在一些實施例中,R
2
為異戊基(例如經取代或未經取代之異戊基)且R
3
為氫。在一些實施例中,R
2
為-CF
3
或-CH
3
且R
3
為氫或-CH
3
。在一些實施例中,R
2
及R
3
與其所附接之碳原子一起形成3員至8員碳環或雜環。 在一些實施例中,R
1
為-CH
3
或-CH
2
CH
3
,R
2
為異戊基(例如經取代或未經取代之異戊基),且R
3
為氫。在一些實施例中,R
1
為-CH
3
或-CH
2
CH
3
,R
2
為未經取代之異戊基,且R
3
為氫。 在一些實施例中,式(I-63)之化合物選自式(I-A63)、(I-B63)或(I-C63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-A63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-C63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-D63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-E63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-D-i63)或(I-D-ii63)之化合物:
或
。 在一些實施例中,式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物:
或
。 在一個態樣中,本文提供根據式(I-67)之化合物:
或其醫藥學上可接受之鹽,其中:R
1
為氫或烷基(例如C
1
-C
6
烷基);R
2
及R
3
中之各者獨立地為氫、烷基(例如C
1
-C
6
烷基)、碳環基、雜環基、芳基或雜芳基,或R
2
及R
3
與其所附接之碳原子一起形成3員至8員環;R
4
及R
5
中之各者獨立地為氫、鹵基或-OR
C
,其中R
C
為氫或C
1
-C
6
烷基(例如C
1
-C
3
烷基),或R
4
及R
5
與其所附接之碳原子一起形成側氧基;R
6
不存在或為氫;且
表示單鍵或雙鍵,其中當
中之一者為雙鍵時,另一
為單鍵;當
兩者均為單鍵時,則R
6
為氫;且當
中之一者為雙鍵時,R
6
不存在。 在一些實施例中,R
1
為烷基(例如C
1
-C
6
烷基)。在一些實施例中,R
1
為C
1
-C
6
烷基(例如-CH
3
、-CH
2
CH
3
、-CH
2
OCH
3
或-CF
3
)。在一些實施例中,R
1
為-CH
3
、-CF
3
或-CH
2
CH
3
。在一些實施例中,R
1
為-CH
2
OR
A
,其中R
A
為C
1
-C
6
烷基(例如C
1
-C
3
烷基)。 在一些實施例中,R
2
為氫或烷基(例如C
1
-C
6
烷基)。 在一些實施例中,R
2
及R
3
中之各者獨立地為氫或烷基(例如C
1
-C
6
烷基)。在一些實施例中,R
2
及R
3
中之各者獨立地為氫或C
1
-C
6
鹵烷基(例如,-CF
3
)。在一些實施例中,R
2
及R
3
中之各者獨立地為氫、-CF
3
或-CH
3
。 在一些實施例中,R
4
為-OH或鹵基(例如-F)。 在一些實施例中,R
4
及R
5
與其所附接之碳原子一起形成側氧基。在一些實施例中,R
4
為氫且R
5
為鹵基(例如-F)。在一些實施例中,R
4
及R
5
為鹵基(例如-F)。在一些實施例中,R
4
及R
5
為氫。 在一些實施例中,R
2
為芳基或雜芳基,且R
3
為氫。在一些實施例中,R
2
為碳環基或雜環基且R
3
為氫。在一些實施例中,R
2
及R
3
為氫。在一些實施例中,R
2
及R
3
與其所附接之碳原子一起形成3員至8員碳環或雜環。 在一些實施例中,式(I-67)之化合物選自式(I-A67)、(I-B67)或(I-C67)之化合物:
。 在一些實施例中,式(I-67)之化合物選自式(I-A67)之化合物:
。 在一些實施例中,式(I-67)之化合物選自式(I-C67)之化合物:
。 在替代實施例中,本文所描述之化合物亦可包含一或多種同位素取代。舉例而言,氫可為
2
H (D或氘)或
3
H (T或氚);碳可為(例如)
13
C或
14
C;氧可為(例如)
18
O;氮可為(例如)
15
N,等等。在其他實施例中,特定同位素(例如
3
H、
13
C、
14
C、
18
O或
15
N)可表示佔據化合物特異位點之元素的總同位素豐度的至少1%、至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少99%或至少99.9%。 本發明之例示性化合物包括:
及
。
替代實施例
在替代實施例中,本文所描述之化合物亦可包含一或多種同位素取代。舉例而言,氫可為
2
H (D或氘)或
3
H (T或氚);碳可為(例如)
13
C或
14
C;氧可為(例如)
18
O;氮可為(例如)
15
N,等等。在其他實施例中,特定同位素(例如
3
H、
13
C、
14
C、
18
O或
15
N)可表示佔據化合物特異位點之元素的總同位素豐度的至少1%、至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少99%或至少99.9%。
醫藥組合物
在另一態樣中,本發明提供一種醫藥組合物,其包含醫藥學上可接受之載劑及有效量的本文所描述之化合物或其醫藥學上可接受之鹽。 在用作藥物時,本文提供之化合物通常以醫藥組合物之形式投與。此等組合物可以醫藥技術中熟知之方式來製備且包含至少一種活性化合物。 在一個實施例中,關於醫藥組合物,載劑為非經腸載劑、口服或局部載劑。 本發明亦係關於本文所描述之化合物,或其用作藥物或藥劑之醫藥組合物。 大體而言,本文所提供之化合物按治療有效量投與。實際投與之化合物之量通常將由醫師鑒於相關情形確定,該等相關情形包括待治療之病狀、所選擇之投與途徑、所投與之實際化合物、個別患者之年齡、體重及反應、患者症狀之嚴重程度及其類似者。 本文所提供之醫藥組合物可藉由各種途徑投與,包括經口、經直腸、經皮、皮下、靜脈內、肌內及鼻內。視預期傳遞途徑而定,本文所提供之化合物較佳調配為可注射或口服組合物,或調配為均用於經皮投與之油膏、洗劑或貼片。 用於經口投與之組合物可採取散裝液體溶液或懸浮液或散裝粉末之形式。然而,組合物更通常以單位劑型呈現以便於準確給藥。術語「單位劑型」係指適合以單位劑量形式用於人類個體及其他哺乳動物的物理上不連續之單元,各單元含有經計算以產生所需治療作用的預定量之活性材料,其與適合之醫藥賦形劑結合。典型單位劑型包括液體組合物之預填充、預量測安瓿或注射器或在固體組合物情況中之丸劑、錠劑、膠囊或其類似物。在此類組合物中,化合物通常為次要組分(約0.1重量%至約50重量%,或較佳約1重量%至約40重量%),其餘部分為有助於形成所需給藥形式之各種媒劑或載劑及加工助劑。 適於經口投與之液體形式可包括適合水性或非水性媒劑與緩衝劑、懸浮劑及分散劑、著色劑、調味劑及其類似物。固體形式可包括例如以下成分或具有類似性質之化合物中之任一者:黏合劑,諸如微晶纖維素、黃蓍膠或明膠;賦形劑,諸如澱粉或乳糖;崩解劑,諸如褐藻酸、澱粉羥基乙酸鈉(Primogel)或玉米澱粉;潤滑劑,諸如硬脂酸鎂;滑動劑,諸如膠態二氧化矽;甜味劑,諸如蔗糖或糖精;或調味劑,諸如胡椒薄荷、水楊酸甲酯或橙味調味劑。 可注射組合物通常係基於可注射無菌生理食鹽水或磷酸鹽緩衝生理食鹽水或此項技術中已知的其他可注射載劑。如前所述,此類組合物中之活性化合物通常為次要組分(常為約0.05重量%至10重量%),其餘部分為可注射載劑及其及類似物。 經皮組合物通常調配為含有活性成分之局部軟膏或乳霜,其量之範圍大體上為約0.01重量%至約20重量%,較佳約0.1重量%至約20重量%,較佳約0.1重量%至約10重量%,且更佳約0.5重量%至約15重量%。當調配成軟膏時,活性成分通常將與石蠟或水可混溶性軟膏基劑合併。或者,活性成分可使用例如水包油乳霜基劑調配成乳霜。此類經皮調配物為此項技術中所熟知且大體上包括額外成分以增強活性成分或調配物之真皮穿透穩定性。所有此類已知之經皮調配物及成分均包括在本文提供之範疇內。 本文提供之化合物亦可藉由經皮裝置投與。因此,經皮投與可使用儲集層型或多孔膜型或固體基質類貼片來實現。 上文所描述之用於可經口投與、可注射或可局部投與組合物之組分僅為代表性的。其他材料以及加工技術及其類似者闡述於
Remington's Pharmaceutical Sciences
之第8部分,第17版,1985, Mack Publishing Company, Easton, Pennsylvania,其以引用的方式併入本文中。 上文所描述之用於可經口投與、可注射或可局部投與組合物之組分僅為代表性的。其他材料以及加工技術及其類似者闡述於
Remington's The Science and Practice of Pharmacy
之第8部分,第21版,2005, 出版商:Lippincott Williams & Wilkins,其以引用的方式併入本文中。 本發明之化合物亦可以持續釋放形式或自持續釋放藥物傳遞系統投與。代表性持續釋放材料之描述可見於
Remington's Pharmaceutical Sciences
。 本發明亦係關於本文所描述之化合物的醫藥學上可接受之調配物。在一個實施例中,該調配物包含水。在另一實施例中,該調配物包含環糊精衍生物。最常見環糊精為分別由6、7及8個α-l,4-鍵聯葡糖單元組成之α-環糊精、β-環糊精及γ-環糊精,其視情況包含在鍵聯糖部分上之一或多個取代基,該一或多個取代基包括(但不限於)甲基化、羥基烷基化、醯基化及磺基烷基醚取代。在某些實施例中,環糊精為磺基烷基醚β-環糊精,例如磺基丁基醚β-環糊精,亦稱為Captisol®。參見(例如) U.S. 5,376,645。在某些實施例中,調配物包含六丙基-β-環糊精。在更特定實施例中,調配物包含六丙基-β-環糊精(10-50%於水中)。 本發明亦係關於本文所描述之化合物的醫藥學上可接受之酸加成鹽。可用於製備醫藥學上可接受之鹽的酸為形成無毒酸加成鹽,亦即,含有藥理學上可接受之陰離子之鹽的酸,該鹽諸如鹽酸鹽、氫碘酸鹽、氫溴酸鹽、硝酸鹽、硫酸鹽、硫酸氫鹽、磷酸鹽、乙酸鹽、乳酸鹽、檸檬酸鹽、酒石酸鹽、丁二酸鹽、順丁烯二酸鹽、反丁烯二酸鹽、苯甲酸鹽、對甲苯磺酸鹽及其類似物。 以下調配物實例說明可根據本發明製備之代表性醫藥組合物。然而,本發明不限於以下醫藥組合物。
例示性調配物 1— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為240 mg至270 mg錠劑(每一錠劑80 mg至90 mg活性化合物)。
例示性調配物 2— 膠囊
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與澱粉稀釋劑按大致1:1重量比摻混。混合物填充成250 mg膠囊(每一錠劑125 mg活性化合物)。
例示性調配物 3— 液體 :
本文所描述之化合物或其醫藥學上可接受之鹽(125 mg)可與蔗糖(1.75 g)及三仙膠(4 mg)摻混,且所得混合物可經摻合,經過10號目U.S.篩,並接著與先前製得的微晶纖維素與羧甲基纖維素鈉(11:89,50 mg)於水中之溶液混合。苯甲酸鈉(10 mg)、調味劑及著色劑用水稀釋且在攪拌下添加。接著可再添加足夠水產生5 mL之總體積。
例示性調配物 4— 錠劑 :
本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為450 mg至900 mg錠劑(150 mg至300 mg活性化合物)。
例示性調配物 5— 注射劑 :
本文所描述之化合物或其醫藥學上可接受之鹽可溶解或懸浮於緩衝無菌生理食鹽水可注射水性介質中,達到大致5mg/mL之濃度。
例示性調配物 6— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為90 mg至150 mg錠劑(每一錠劑30 mg至50 mg活性化合物)。
例示性調配物 7— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為30 mg至90 mg錠劑(每一錠劑10 mg至30 mg活性化合物)。
例示性調配物 8— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為0.3 mg至30 mg錠劑(每一錠劑0.1 mg至10 mg活性化合物)。
例示性調配物 9— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為150 mg至240 mg錠劑(每一錠劑50 mg至80 mg活性化合物)。
例示性調配物 10— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為270 mg至450 mg錠劑(每一錠劑90 mg至150 mg活性化合物)。 注射劑量在約0.1 mg/kg/hr至至少10 mg/kg/hr範圍內,均歷時約1至約120小時且尤其24至96小時。亦可投與約0.1 mg/kg至約10 mg/kg或10 mg/kg以上之預負載大丸劑以達到充分穩態水準。對於40至80 kg人類患者,最大總劑量預期不超過約2公克/天。 為了預防及/或治療長期病狀,治療療程通常延續多月或多年,因此對於患者便利性及耐受性而言,經口給藥係較佳的。在經口給藥下,每天一個至五個且尤其兩個至四個及通常三個經口劑量為代表性方案。使用此等給藥模式,各劑量提供約0.01至約20 mg/kg本文所提供之化合物,其中較佳劑量各提供約0.1至約10 mg/kg及尤其約1至約5 mg/kg。 通常選擇經皮給藥以提供與使用注射給藥所達成的血液含量相比類似或較低的血液含量。 當用於預防CNS病症發作時,應通常根據醫師之建議且在醫師之監督下,以上文所描述之劑量向在出現該病狀之風險下的個體投與本文所提供之化合物。處於出現特定病狀之風險下的個體一般包括具有該病狀之家族病史之彼等個體,或已藉由遺傳測試或篩檢而鑑別為尤其易於出現該病狀之彼等個體。
治療及使用方法
如本文所描述,本發明化合物(例如,式(A)、(I-63)或(I-67)之化合物)及其醫藥學上可接受之鹽,一般設計成調節NMDA功能,且因此充當用於治療及預防個體中之(例如) CNS相關病況的氧固醇。在一些實施例中,如本文所描述,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)及其醫藥學上可接受之鹽,一般設計成穿透血腦障壁(例如,設計成跨越血腦障壁傳輸)。如本文所使用,調節係指(例如)抑制或增強NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽充當NMDA之負向異位調節劑(negative allosteric modulator,NAM)並抑制NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽充當NMDA之正向異位調節劑(positive allosteric modulator,PAM)並增強NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽藉由天然產生之基質阻斷或減少NMDA受體功能之增強或抑制。此類化合物並不充當NMDA之負向異位調節劑(NAM)或正向異位調節劑(PAM)。在一些實施例中,病症為癌症。在一些實施例中,病症為糖尿病。在一些實施例中,病症為固醇合成障礙。在一些實施例中,該病症為腸胃(GI)病症,例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病)、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌症或結腸炎。在一些實施例中,病症為發炎性腸病。 與NMDA-調節有關之例示性病況包括(但不限於)腸胃(GI)病症,例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病)、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌症、結腸炎及CNS病狀,(例如)如本文中所描述。 與NMDA-調節有關之例示性病況(例如CNS病況)包括(但不限於)失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質濫用相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病 、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症及耳鳴。 在某些實施例中,本發明化合物,例如,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽,可用於誘導鎮靜或麻醉。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質濫用相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病 、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症及耳鳴。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、物質濫用相關病症、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)或分娩後精神病。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症或耳鳴。 在一些實施例中,本發明化合物(充當NMDA受體功能之PAM的本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、多發性硬化、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病及與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)。 在一些實施例中,充當NMDA受體功能之NAM之本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、人格障礙(包括強迫性人格障礙)、神經發育性障礙(包括雷特症候群)、疼痛(包括急性及疼痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)及結節性硬化症(TSC))、中風、創傷性腦損傷、失調症、神經精神性狼瘡及耳鳴。 在一些實施例中,充當NMDA受體功能之PAM或NAM之本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、固醇合成障礙及進食障礙。 在另一態樣中,提供一種治療或預防易感或罹患與腦興奮相關之病狀的個體的腦興奮的方法,其包含向該個體投與有效量之本發明化合物(例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)或其醫藥學上可接受之鹽。 在又一態樣中,本發明提供本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)或其醫藥學上可接受之鹽與另一藥理活性劑的組合。本文所提供之化合物可作為唯一活性劑投與或其可與其他藥劑組合投與。組合投與可藉由熟習此項技術者清楚之任何技術進行,包括(例如)分開、連續、同時及交替投與。 在另一態樣中,本文提供一種誘導個體中之NMDA受體之負向異位調節之方法,其包含向該個體投與本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物。
運動障礙
本文中亦描述用於治療動作障礙之方法。如本文所使用,「運動障礙」係指與過動性運動障礙及肌肉控制相關異常相關之多種疾病及病症。例示性運動障礙包括(但不限於)帕金森氏病及巴金森氏症(具體由動作遲緩定義)、肌張力障礙、舞蹈症及亨廷頓氏病、運動失調、左旋多巴誘導之運動困難、震顫(例如特發性震顫)、肌陣攣及驚跳、抽搐及妥瑞症候群、腿不寧症候群、僵人症候群及步態障礙。
震顫
為非自主性、有時節律性、肌肉收縮及鬆弛,其可涉及一或多個身體部分(例如手、臂、眼睛、臉、頭部、聲帶褶、軀幹、腿)之振盪或抽搐。震顫包括:遺傳性、退化性及特發性病症,分別諸如威爾森氏病、帕金森氏病及特發性震顫;代謝障礙(例如甲狀腺-副甲狀腺病症、肝病及低血糖);周邊神經病(與恰克-馬利-杜斯氏病、魯西-利維病、糖尿病、複雜區域疼痛症候群相關);毒素(尼古丁、汞、鉛、CO、錳、砷、甲苯);藥物性(發作性睡眠病、三環化合物、鋰、可卡因、酒精、腎上腺素、支氣管擴張劑、茶鹼、咖啡鹼、類固醇、丙戊酸鹽、胺碘酮、甲狀腺激素、長春新鹼);及精神性病症。臨床震顫可分為生理震顫、增強型生理震顫、特發性震顫症候群(包括經典特發性震顫、原發直立性震顫、及任務特異性及位置特異性震顫)、肌張力障礙性震顫、巴金森式震顫、小腦震顫、霍氏震顫(亦即紅核震顫)、上顎震顫、神經病性震顫、中毒性或藥物性震顫及心因性震顫。其他形式之震顫包括小腦震顫或意向震顫、肌張力障礙性震顫、特發性震顫、直立性震顫、巴金森式震顫、生理性震顫、心因性震顫或紅核震顫。
小腦震顫
或
意向震顫
為在有目的之運動之後發生的肢端緩慢而廣泛之震顫。小腦震顫由因例如腫瘤、中風、疾病(例如多發性硬化症,一種遺傳性退化性病症)而產生之小腦病變或損傷引起。
肌張力障礙性震顫
發生在受肌張力障礙(一種持續非自主性肌肉收縮引起扭動及重複動作及/或疼痛及異常姿勢或位置之運動障礙)影響之個體中。肌張力障礙性震顫可影響體內任何肌肉。肌張力障礙性震顫不規則地發生且常常可藉由徹底休息而減輕。
特發性震顫
或良性特發性震顫為最常見類型的震顫。特發性震顫可為輕度的且在一些情況下無進展,且可緩慢進展,起始於身體一側,但在3年內影響兩側。手最常受影響,但頭部、聲音、舌、腿及軀幹亦可涉及。震顫頻率可隨人變老而降低,但嚴重程度可增加。高漲之情緒、壓力、發熱、筋疲力盡或低血糖可引發震顫及/或增加其嚴重程度。症狀一般隨時間推移發展且可均為發病後可見與持久。
直立性震顫
之特徵為在站立之後腿及軀幹中很快發生的快速(例如超過12 Hz)節律性肌肉收縮。大腿及腿中感覺到絞痛且患者可在要求站立在一點時不受控制地震盪。直立性震顫可發生在患有特發性震顫之患者中。
帕金森式震顫
由對大腦內控制運動之結構之破壞引起。帕金森式震顫常常為帕金森氏病之前兆且通常表現為亦可影響下頜、嘴唇、腿及軀幹之手的「搓丸樣」動作。帕金森式震顫之發病通常在60歲之後開始。運動始於一肢或身體一側且可進展至包括另一側。
生理性震顫
可出現在正常個體中且無臨床意義。其在所有隨意肌群中均可見。生理性震顫可由某些藥物、戒酒或包括過度活躍甲狀腺及低血糖之醫學病狀引起。震顫通常具有約10 Hz之頻率。
心因性震顫
或癔病性震顫可在休息時或在姿勢或動態運動期間出現。患有心因性震顫之患者可具有轉換性障礙或另一精神疾病。
紅核震顫
之特徵為可在休息時、在擺姿勢時及刻意下存在之粗緩慢震顫。震顫與影響中腦中紅核之病狀(經典異常中風)相關。
帕金森氏病
影響大腦中產生多巴胺的神經細胞。症狀包括肌強直、震顫及語音及步態變化。
巴金森氏症
之特徵為震顫、動作遲緩、強直及姿勢不穩定。巴金森氏症共用帕金森氏病中發現之症狀,但其為症候群而非進行性神經退化性疾病。
肌張力障礙
為一種運動障礙,其特徵為引起異常(常常為反覆)的運動或姿勢的持續性或間斷肌肉收縮。肌張力障礙性運動可為模式化的,帶扭動,且可為震顫樣的。肌張力障礙常由自主行動起始或惡化,並與溢出肌肉活化相關。
舞蹈症
為一種神經性病症,其特徵為通常影響肩部、髖部及臉的急抽式非自主運動。
亨廷頓氏病
為一種引起大腦中神經細胞日益衰弱的遺傳性疾病。症狀包括不受控運動、動作笨拙及平衡問題。亨廷頓氏病可能妨礙步行、講話及吞咽。
運動失調
係指喪失對整體運動的完全控制,且可影響手指、手、手臂、腿、身體、語音及眼球運動。
肌陣痙及驚跳
係對於突然及未預期刺激的反應,刺激可為聲學、觸感、可見或前庭刺激。
抽搐
為非自主運動,一般突然發病,係短暫、反覆的,但係非節律性的,其通常模仿正常行為且常常發生在正常活動背景中。抽搐可分類為運動或發聲抽搐,運動抽搐與運動相關,而發聲抽搐與聲音有關。抽搐的特徵可為簡單或複雜的。舉例而言,簡單運動抽搐僅涉及受限於特定身體部分的一些肌肉。
妥瑞症候群
為一種在兒童中發病之遺傳性神經精神異常,其特徵為多種運動抽搐及至少一種發聲抽搐。
腿不寧症候群
為一種神經感覺運動障礙,其特徵為在休息時有壓倒性的移動腿之衝動。
僵人症候群
為一種進行性運動障礙,其特徵為非自主疼痛痙攣及肌肉僵直,一般涉及下背及腿。通常產生具有加重之腰椎過度凸出的僵腿式步態。通常觀察具有脊椎旁軸肌之連續運動單元活動的EMG記錄上的特徵性異常。變體包括產生通常會影響遠端腿部及腳之病灶性僵硬的「僵肢症候群」。
步態障礙
係指步行方式或風格之異常,其起因於神經肌肉變化、關節炎變化或其他身體變化。步態係根據負責用於異常移動之系統而分類,且包括偏癱步態、兩側癱瘓步態、神經病性步態、肌病步態、巴金森式步態、舞蹈病狀步態、運動失調步態及感官步態。
情緒障礙
本文亦提供用於治療例如以下之情緒障礙的方法:臨床抑鬱、產後抑鬱或分娩後抑鬱、圍產期抑鬱、非典型抑鬱、憂鬱型抑鬱、精神病性嚴重抑鬱症、緊張型抑鬱、季節性情緒失調症、輕鬱症、雙重抑鬱、抑鬱性人格障礙、復發性短暫抑鬱、輕度抑鬱障礙、躁鬱症或躁狂抑鬱性障礙、由慢性醫學病狀引起之抑鬱、耐治療性抑鬱、難治性抑鬱、自殺傾向、自殺觀念或自殺行為。
臨床抑鬱
亦稱為嚴重抑鬱症、重度抑鬱症(MDD)、嚴重抑鬱、單極抑鬱、單極病症及復發性抑鬱,且係指特徵為普遍及持久情緒低落,伴隨有較低自尊心及正常娛樂活動之興趣或快樂感喪失的精神病症。一些患有臨床抑鬱之人入睡困難,體重減輕且一般感到焦躁及易受刺激。臨床抑鬱影響個體之感覺、思想及行為,且可導致許多情感及身體問題。患有臨床抑鬱之個體每天的活動可能困難,且使個體感到生活不值得過。
產後抑鬱
(PND)亦稱為
分娩後抑鬱 (PPD)
,且係指在分娩之後影響女性之一種類型的臨床抑鬱。症狀可包括悲傷、疲乏、睡眠及飲食習慣變化、性慾減少、哭喊事件、焦慮及易怒。在一些實施例中,PND為耐治療性抑鬱(例如,如本文所描述之耐治療性抑鬱)。在一些實施例中,PND為難治性抑鬱(例如,如本文所描述之難治性抑鬱)。 在一些實施例中,具有PND之個體在懷孕期間亦經歷抑鬱或抑鬱症狀。此抑鬱在本文中被稱作
圍產期抑鬱
。在實施例中,經歷圍產期抑鬱之個體經歷PND之風險增加。
非典型抑鬱 (AD)
之特徵為情緒反應性(例如反常快感缺乏)及積極性、體重顯著增加或食慾增加。患有AD之患者亦可具有過度睡眠或嗜睡(睡眠過度)、四肢沉重之感覺及由於對所感知人際排斥超敏感而有顯著社會功能損傷。
憂鬱型抑鬱
之特徵為在大部分或所有活動中喪失快樂(快感缺乏)、無法對快樂刺激作出反應、比哀傷或喪失更明顯之憂鬱情緒、體重過度減輕或過度內疚。
精神病性嚴重抑鬱症 (PMD)
或精神病性抑鬱係指嚴重抑鬱事件,尤其具有憂鬱性,其中個體經歷諸如妄想及幻覺之精神病症狀。
緊張型抑鬱
係指涉及運動行為紊亂及其他症狀之嚴重抑鬱症。個體可變得沉默且麻木,且不能動或展現無目的或古怪動作。
季節性情緒失調症 (SAD)
係指其中個體具有在秋季或冬季來到之抑鬱事件季節性模式的一種類型的季節性抑鬱。
輕鬱症
係指與單極抑鬱相關之病狀,其中相同身體及認知問題顯而易見。其不如單極抑鬱嚴重且傾向於持續較長時間(例如至少2年)。
雙重抑鬱
係指持續至少2年之極其憂鬱情緒(輕鬱症),且間雜有嚴重抑鬱症時期。
抑鬱性人格障礙 (DPD)
係指具有抑鬱特徵之人格障礙。
復發性短暫抑鬱 (RBD)
係指其中個體約每月發生一次抑鬱事件,各事件持續2週或更少時間且通常小於2-3天的病狀。
輕度抑鬱障礙
或輕度抑鬱係指其中至少2種症狀存在2週之抑鬱。
躁鬱症或躁狂抑鬱性障礙
引起極端情緒波動,包括情緒高漲(躁症或輕躁症)及低落(抑鬱)。在躁症期期間,個體可感到或行為異常歡樂、高能或易受刺激。其常常無法作出考慮周到之決定,對結果幾乎不作考慮。睡眠需求通常減少。在抑鬱期期間,可存在哭喊,眼睛與他人缺乏接觸,且對生活之觀點負面。在患有該病症之患者中自殺風險高,20年間超過6%,同時自殘發生率為30%-40%。諸如焦慮症之其他精神健康問題及物質使用障礙通常與躁鬱症相關。
由慢性醫學病狀引起之抑鬱
係指由諸如癌症之慢性醫學病狀或慢性疼痛、化學療法、慢性壓力引起的抑鬱。
耐治療性抑鬱
係指個體已針對抑鬱進行治療,但症狀未改善的病狀。舉例而言,抗抑鬱劑或心理諮詢(心理療法)未減輕患有耐治療性抑鬱之個體的抑鬱症狀。在一些情況下,患有耐治療性抑鬱之個體改善症狀,但又恢復。
難治性抑鬱
發生在罹患抑鬱之對包括三環抗抑鬱劑、MAOI、SSRI及雙重及三重吸收抑制劑及/或抗焦慮劑藥物之標準藥理學治療以及非藥理學治療(例如心理療法、電驚厥療法、迷走神經刺激及/或經顱磁刺激)具抗性的患者中。
自殺傾向、自殺觀念、自殺行為
係指個體自殺之傾向。自殺觀念涉及關於自殺之想法或對自殺異常專注。自殺觀念之範圍變化很大,例如稍縱即逝的念頭至深入的想法、詳細計劃、角色扮演、著手未遂。症狀包括討論自殺、獲得進行自殺之方式、退出社會接觸、一心想著死亡、對某事感到被困或無望、增加酒精或藥物之使用、作出有風險或自毀事情、向人們說再見,好像其不會再見面一樣。 抑鬱
症狀
包括持久焦慮或悲傷感、感到無助、無望、悲觀、無價值、低能、坐立不安、入睡困難、不眠、易怒、疲乏、動作攻擊、喪失娛樂活動或愛好之興趣、注意力缺乏、缺乏能量、自尊心差、無積極思想或計劃、睡眠過度、飲食過量、食慾喪失、失眠、自殘、自殺想法及自殺嘗試。症狀之存在、嚴重程度、頻率及持續時間可隨病例而變化。抑鬱症狀及其減輕可藉由醫師或心理學家(例如藉由精神狀態檢查)來確定。
焦慮症 本文提供治療焦慮症之方法。焦慮症
為覆蓋異常及病理學恐懼及焦慮之若干不同形式的總稱。當前精神診斷標準辨認各種焦慮症。
廣泛性焦慮症
為一種常見慢性病症,其特徵為持久焦慮,不集中於任一個目標或情況。罹患廣泛性焦慮之彼等個體經歷非特定的持久恐懼及擔憂,且變得過度擔心每日事情。廣泛性焦慮症為影響老年人之最常見焦慮症。 在
恐慌症
中,個人遭受強烈恐懼及憂懼之短暫發作,特徵常常為戰慄、搖晃、意識模糊、眩暈、噁心、呼吸困難。此等恐慌發作藉由APA定義為突然出現且在不到十分鐘內達到峰值之恐懼或不適,可持續若干小時且可由壓力、害怕或甚至運動觸發;不過具體病因始終不清楚。除復發性意外恐慌發作之外,診斷恐慌症亦需要該等發作具有慢性後果:擔憂發作之潛在影響,持久性害怕將來發作或與發作相關之行為變化顯著。因此,罹患恐慌症之個體經歷甚至在特定恐慌事件外之症狀。常常,恐慌患者注意到心跳之正常變化,導致其認為其心臟出問題或其將再具有恐慌發作。在一些情況下,身體功能之感知加強(警覺過度)在恐慌發作期間發生,其中任何所感知之生理改變均解釋為可能威脅生命之疾病(亦即極端疑病症)。
強迫症
為主要特徵為重複困擾(令人苦惱、持久性及侵入性想法或影像)及強迫(進行特定動作或儀式之衝動)之一種類型的焦慮症。OCD思想模式可類似於迷信,到其將信念與實際上不存在之因果關係相連的程度。常常,該過程全部為不合邏輯的;舉例而言,強迫以一定模式步行可用以緩解對即將發生損害之困擾。且在許多情況下,強迫性全部不可解釋,簡單而言,完成由緊張觸發之儀式的衝動。在少數情況下,OCD患者可僅僅經歷困擾,無明顯強迫症;數目少得多之患者僅僅經歷強迫症。 單一最大類別之焦慮症為
恐懼症
,其包括由特定刺激或情況觸發害怕及焦慮之所有情況。患者通常由遇到其害怕之目標來預計恐怖後果,其害怕目標可為自動物至體液位置之任何東西。
創傷後壓力症
或
PTSD
為一種由創傷經歷產生之焦慮症。創傷後壓力可由極端情況引起,諸如打鬥、強姦、人質情況或甚至嚴重事故。其亦可由長期(慢性)暴露於嚴重應激物引起,例如經受個體戰鬥,但無法應付連續打鬥之士兵。常見症狀包括閃回、回避行為及抑鬱。
癲癇症
癲癇症為一種特徵為隨時間推移的重複癲癇發作之大腦病症。癲癇症之類型可包括(但不限於)廣泛性癲癇症,例如兒童失神癲癇症、幼年肌陣攣癲癇症、喚醒時癲癇大發作之癲癇症、韋斯特症候群(West syndrome)、倫-加症候群(Lennox-Gastaut syndrome);部分性癲癇症,例如兒童之顳葉性癲癇症、額葉性癲癇症、良性病灶性癲癇症。
癲癇形成
癲癇形成為正常大腦產生癲癇症的漸進過程(癲癇發作發生之慢性病狀)。癲癇形成因藉由初始損傷促成之神經元損害而引起(例如持續性癲癇)。
持續性癲癇 (SE)
持續性癲癇(SE)可包括例如痙攣性持續性癲癇,例如早期持續性癲癇、現有持續性癲癇、難治性持續性癲癇、超難治性持續性癲癇;非痙攣性持續性癲癇,例如廣泛性持續性癲癇、複雜部分持續性癲癇;廣泛性週期性癲癇樣放電;及週期性偏側性癲癇樣放電。痙攣性持續性癲癇之特徵為存在痙攣性持續性癲癇發作,且可包括早期持續性癲癇、現有持續性癲癇、難治性持續性癲癇、超難治性持續性癲癇。早期持續性癲癇用一線療法治療。現有持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法治療其仍持續且投與二線療法。難治性持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法及二線療法治療其仍持續且通常投與一般麻醉劑。超難治性持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法、二線療法及一般麻醉劑治療24小時或24小時以上,其仍持續。 非痙攣性持續性癲癇可包括例如病灶性非痙攣性持續性癲癇,例如複雜部分非痙攣性持續性癲癇、簡單部分非痙攣性持續性癲癇、隱微非痙攣性持續性癲癇;廣泛性非痙攣性持續性癲癇,例如晚發性失神非痙攣性持續性癲癇、非典型失神非痙攣性持續性癲癇或典型失神非痙攣性持續性癲癇。
癲癇發作
癲癇發作為大腦中異常電活動事件後發生之生理發現或行為改變。術語「癲癇發作」通常可與「痙攣」互換使用。痙攣為當人的身體快速且不受控制地搖晃時。在痙攣期間,人的肌肉反覆收縮及放鬆。 基於行為及大腦活動之類型,將癲癇發作分成兩種較寬廣類別:廣泛性及部分(亦稱為局部或病灶性)。將癲癇發作類型進行分類有助於醫生診斷患者是否患有癲癇症。 廣泛性癲癇由整個大腦中之電脈衝產生,而部分癲癇由相對較小部分大腦中之電脈衝產生(至少起初)。產生癲癇之大腦部分有時稱為病灶。 存在六種類型之廣泛性癲癇。最常見且嚴重的且因此最熟知的為廣泛性痙攣,亦稱為大發作癲癇。在此類型癲癇中,患者失去意識且通常虛脫。失去意識後廣泛性身體僵硬(稱為癲癇之「強直性」階段)30 至60秒,隨後劇烈抽動(「陣攣性」階段)30至60秒,其後患者進入深度睡眠(「發作後」或癲癇後階段)。在大發作癲癇期間,可能發生損傷及事故,諸如咬舌及尿失禁。 失神癲癇導致短暫意識喪失(僅幾秒),幾乎無症狀。患者(最常為兒童)通常中斷活動且目直。此等癲癇突然開始及結束,且可一日發生若干次。患者通常未意識到其癲癇發作,但其可能意識到「時間流失」。 肌陣攣癲癇由偶發性急抽組成,通常在身體兩側。患者有時將急抽描述為短暫電擊。劇烈時,此等癲癇可能導致物體掉落或無意地投擲物體。 陣攣性癲癇為重複的節律性急抽,其同時涉及身體兩側。 強直性癲癇之特徵為肌肉僵硬。 失張性癲癇由尤其在臂及腿中,肌肉張力之突然及一般損失組成,其通常導致跌倒。 本文所描述之癲癇可包括癲癇發作;急性重複癲癇;密集式癲癇;連續癲癇;無間斷癲癇;長期癲癇;反覆性癲癇;持續性癲癇發作,例如難治性痙攣性持續性癲癇、非痙攣性持續性癲癇發作;難治性癲癇;肌陣攣性癲癇;強直性癲癇;強直性陣攣性癲癇;簡單部分癲癇;複雜部分癲癇;繼發廣泛性癲癇;非典型失神癲癇;失神癲癇;失張性癲癇;良性羅蘭多(Rolandic)癲癇;發熱性癲癇;情緒性癲癇;病灶性癲癇;癡笑性癲癇;廣泛性發作癲癇;嬰兒痙攣;傑克遜氏癲癇(Jacksonian seizures);大規模雙側肌陣攣癲癇;多灶性癲癇;新生兒發作癲癇;夜間癲癇;枕葉性癲癇;創傷後癲癇;微小癲癇;西爾萬癲癇(Sylvan seizures);視覺反射癲癇或撤藥癲癇。在一些實施例中,癲癇為與德拉韋症候群、倫-加症候群、結節性硬化症、雷特症候群或PCDH19女嬰兒癲癇症相關之廣泛性癲癇。
縮寫
PCC:氯鉻酸吡啶;t-BuOK:第三丁醇鉀;9-BBN:9-硼雙環[3.3.1]壬烷;Pd(
t
-Bu
3
P)
2
:雙(三-第三丁基膦)鈀(0);AcCl:氯化乙醯基;
i
-PrMgCl:異丙基氯化鎂;TBSCl:第三丁基(氯)二甲基矽烷;(
i
-PrO)
4
Ti:四異丙醇鈦;BHT:2,6-二-第三丁基-4-甲基苯酚;Me:甲基;
i
-Pr:異丙基;
t
-Bu:第三丁基;Ph:苯基;Et:乙基;Bz:苯甲醯基;BzCl:氯化苯甲醯;CsF:氟化銫;DCC:二環己基碳化二亞胺;DCM:二氯甲烷;DMAP:4-二甲胺基吡啶;DMP:戴斯-馬丁高碘烷;EtMgBr:乙基溴化鎂;EtOAc:乙酸乙酯;TEA:三乙胺;AlaOH:丙胺酸;Boc:第三丁氧羰基。Py:吡啶;TBAF:氟化四正丁基銨;THF:四氫呋喃;TBS:第三丁基二甲基矽烷基;TMS:三甲基矽烷基;TMSCF
3
:(三氟甲基)三甲基矽烷;Ts:對甲苯磺醯基;Bu:丁基;Ti(OiPr)
4
:四異丙氧基鈦;LAH:氫化鋰鋁;LDA:二異丙胺基鋰;LiOH.H
2
O:氫氧化鋰水合物;MAD:甲基鋁雙(2,6-二-第三丁基-4-甲基苯酚);MeCN:乙腈;NBS:N-溴代丁二醯亞胺;Na
2
SO
4
:硫酸鈉;Na
2
S
2
O
3
:硫代硫酸鈉;PE:石油醚;MeCN:乙腈;MeOH:甲醇;Boc:第三丁氧羰基;MTBE:甲基第三丁基醚;DIAD:偶氮二甲酸二異丙酯。
實例
為了能更全面地理解本文中所描述之發明,闡述以下實例。提供本申請案中所描述之合成及生物實例以說明本文提供之化合物、醫藥組合物及方法,且並不理解為以任何方式限制其範疇。在以下合成實例中,按序號列出反應序列內之實驗程序之描述。
物質及方法
本文提供之化合物可使用以下通用方法及程序自容易獲得之起始物質製備。應瞭解,除非另行說明,否則在給定典型或較佳製程條件(亦即,反應溫度、時間、反應物之莫耳比、溶劑、壓力等)的情況下,亦可使用其他製程條件。最佳反應條件可隨所用特定反應物或溶劑而變,但此類條件可由熟習此項技術者藉由常規最佳化來確定。 此外,如熟習此項技術者將顯而易知,可能必需習知保護基來防止某些官能基經歷非所需反應。對於特定官能團適合的保護基以及保護及去保護之適合的條件之選擇為此項技術中熟知的。舉例而言,諸多保護基及其引入及移除描述於T. W. Greene及P. G. M. Wuts,
Protecting Groups in Organic Synthesis
,第二版, Wiley, New York, 1991及其中所引用之參考文獻中。 本文提供之化合物可藉由已知標準程序分離及純化。此類程序包括(但不限於)再結晶、管柱層析、HPLC或超臨界流體層析(SFC)。關於製備本文中已列出之代表性吡唑的細節呈現以下流程。本文提供之化合物可由熟習有機合成技術者自已知或市售起始物質及試劑製備。可用於分離/純化本文提供之對映異構體/非對映異構體之例示性對掌性管柱包括(但不限於) CHIRALPAK® AD-10。CHIRALCEL® OB、CHIRALCEL® OB-H、CHIRALCEL® OD、CHIRALCEL® OD-H、CHIRALCEL® OF、CHIRALCEL® OG、CHIRALCEL® OJ及CHIRALCEL® OK。 製備型HPLC之例示性通用方法:管柱:Waters Rbridge製備型10 μm C18,19 × 250 mm。移動相:乙腈、水(NH
4
HCO
3
) (30 L水、24 g NH
4
HCO
3
、30 mL NH
3
.H
2
O)。流速:25 mL/min。 分析型HPLC之例示性通用方法:移動相:A:水 (10 mM NH
4
HCO
3
),B:乙腈梯度:在1.6或2 min內,5%-95% B;流速:1.8或2 mL/min;管柱:XBridge C18,4.6×50mm,在45℃為3.5 μm。
NMDA 調節
使用自動箝膜系統來評定表現NMDA受體之哺乳動物細胞中之NMDA增強,該自動箝膜系統可用於測定如下文所描述之化合物之NAM活性。全細胞箝膜系統可用於測定如下文所描述之化合物之PAM活性。
自動化箝膜系統 (QPatch HTX) :
在此研究中,經GRIN1/2A次型之麩胺酸激活通道穩定轉染之HEK 293細胞將與次極大NMDA濃度(300 μM NMDA,與8 μM甘胺酸共施加)一起用來研究測試化合物之負向異位調節。
細胞培養物
大體而言,細胞將以約80%至90%之會合率傳代。對於電生理量測,將以約80%至90%之會合率自含有完全培養基之無菌培養瓶收集細胞。細胞將作為PBS中之懸浮液傳送至QPatch 16X或QPatch HTX系統,直接傳送至離心機/洗滌器。 標準實驗室條件:細胞將在具有5% CO
2
之潮濕氛圍(相對濕度約95%)中在37℃下培育。
培養基
:細胞將在含有補充有10%胎牛血清、1%青黴素/鏈黴素溶液及50 μM AP-5阻斷劑之達爾伯克氏改良伊格爾培養基與營養混合物F-12 (D-MEM/F-12 1×,液體與L-麩醯胺酸)之1:1混合物的無菌培養瓶中不斷保持及傳代。
抗生素
:如上所指出之完全培養基補充有100 μg/mL潮黴素、15 μg/mL殺稻瘟菌素及1 μg/mL嘌呤黴素。
表現之誘導
:在開始實驗之前24 h添加2.5 μg/mL四環素。
劑量調配
劑量係就供應時之測試化合物而言。將添加媒劑以達成10 mM之儲備濃度(在-10℃至-30℃下儲存)。將在DMSO中製備另外的1.0 mM之儲備溶液。儲備溶液利用之細節(解凍、劑量調配)將記錄在原始資料中。儲備溶液利用之時段將詳述於報告中。
測試化合物濃度
劑量係就供應時之測試化合物而言。將添加媒劑以達成10 mM之儲備濃度(在-10℃至-30℃下儲存)。將在DMSO中製備另外的1.0 mM之儲備溶液。儲備溶液利用之細節(解凍、劑量調配)將記錄在原始資料中。儲備溶液利用之時段將詳述於報告中。 將測試一個測試濃度1.0 μM。 將藉由在電生理實驗前不久僅用不含Mg之電解液或含有NMDA (300 µM)及甘胺酸(8.0 µM)的不含Mg之電解液稀釋儲備溶液來製備所有測試溶液,且在使用時將其保持在室溫(19℃至30℃)下。0.1%DMSO將被用作媒劑。
製備頻率
:對於各測試濃度,每日將製備測試化合物之新鮮溶液。
劑量調配之穩定性
:所有製備時間將記錄在原始資料中。關於測試化合物之不穩定性之任何觀察結果將在原始資料中提及。
劑量調配物之儲存
:在實驗之日,劑量調配物在使用時將維持在室溫(19℃至30℃)下。
電解液
為了準備實驗及為了形成千兆歐姆密封,將使用以下
標準電解液 :
氯化鈉:137 mM;氯化鉀:4 mM;氯化鈣:1.8 mM;氯化鎂:1 mM;HEPES:10 mM;D-葡糖:10 mM;十六醇聚氧乙烯醚:0.02%;pH (NaOH):7.4 將藉由至少每7天用水稀釋不含葡糖之10×電解液及100×葡糖溶液來製備1×電解液。在當前研究之實驗開始之前已經製備兩種儲備溶液且將其儲存在1℃至9℃下(10×電解液)或-10℃至-30℃下(100×葡糖溶液)。實驗中所使用之電解液之批號將記錄在原始資料中。當使用時,1×電解液將保持在室溫(19℃至30℃)下。當未使用時,1×電解液將儲存於1℃至9℃下。 在形成千兆密封後,將使用以下
不含 Mg 的電解液
: 氯化鈉:137 mM;氯化鉀:4 mM;氯化鈣:2.8 mM;HEPES:10 mM;D-葡糖:10 mM;
十六醇聚氧乙烯醚
:0.02%;pH (NaOH):7.4 此不含Mg的電解液將製備為1×溶液且儲存於1℃至9℃下。其將至少每10天新鮮製備。
胞內溶液
1×胞內溶液將每日自冷凍的1×胞內溶液解凍,其已在當前研究之實驗開始之前製備、取等分及儲存於-10℃至-30℃下。當使用時,1×胞內溶液將保持在室溫(19℃至30℃)下。剩餘的1×胞內溶液將儲存於冰箱(1℃至9℃)中。1×胞內溶液將包括下文概述之組分: 氯化鉀:130 mM;氯化鎂:1 mM;Mg-ATP:5 mM;HEPES:10 mM;EGTA:5 mM;pH (KOH):7.2
細胞處理
對於此研究,細胞將不斷灌注有NMDA/甘胺酸、測試化合物或測試化合物/NMDA/甘胺酸。 在各情況下,將在施加之間進行利用測試化合物之至少30秒預洗滌步驟。細節請參見下表A。 將在至少n=3個分離細胞中分析各實驗類型。將在當前研究之實驗開始之前製備NMDA及甘胺酸儲備溶液,冷凍儲存(-10℃至-30℃),直至實驗當天為止。在電生理實驗前不久,冷凍儲備溶液將經解凍及稀釋。 對照:每兩週將在三個細胞處量測媒劑(0.1% DMSO)及D-(-)-2-胺基-5-膦醯基戊酸(AP-5) (100 μM)之效果,以便確保NMDA受體之成功表現。 已在當前研究之實驗開始之前製備AP-5之50 mM儲備溶液,取等分試樣且冷凍儲存(-10℃至-30℃),直至實驗當天為止。在電生理實驗前不久,冷凍儲備溶液將經解凍且接著在含有NMDA (300 μM)及甘胺酸(8.0 μM)之不含Mg的電解液中稀釋,得到最終灌注濃度100 µM。
實驗程序
細胞作為無血清培養基中之懸浮液傳送至QPatch HTX系統且在實驗期間保持在細胞儲槽/攪拌器中。施加至細胞之所有溶液(包括胞內溶液)將維持在室溫下(19℃至30℃)下。 在密封製程期間,將使用上文所描述之
標準電解液
。施加至細胞之所有溶液(包括移液管溶液)將維持在室溫下(19℃至30℃)下。在膜片電極與經轉染個別HEK293細胞之間形成千兆歐姆密封之後,將僅灌注
不含 Mg 的電解液
且細胞膜將會破裂以確保電氣進入細胞內部(全細胞膜片組態)。將在向膜片鉗位細胞施加300 µM NMDA (及8.0 µM甘胺酸)後量測內向電流5秒。在整個實驗期間,細胞將在-80 mV之保持電位下經電壓鉗位。 為了分析測試化合物,NMDA受體將由下文所描述之300 µM NMDA及8.0 µM甘胺酸及測試化合物組合刺激。將在施加之間進行利用測試化合物之三十二個預洗滌步驟。 表A:施加協議;測試化合物之使用依賴性
表B:施加協議;對照實驗
哺乳動物細胞之全細胞膜片鉗 (Ionworks Barracuda (IWB))
全細胞膜片鉗技術用於研究測試化合物之正向異位調節活性對表現於哺乳動物細胞中之GlunN1/GluN2A及GluN2B麩胺酸受體的作用。結果展示於
表 1
中。 HEK293細胞經腺病毒5 DNA轉型且經編碼人GRIN1/GRIN2A基因之cDNA轉染。使用併入至表現質體中之G418及吉歐黴素-抗性基因以及在培養基中以G418及吉歐黴素維持的選擇壓力選擇穩定轉染子。在補充有10%胎牛血清、100 µg/ml青黴素G鈉、100 µg/ml硫酸鏈黴素、100 µg/ml吉歐黴素、5 µg/ml殺稻瘟菌素及500 µg/ml G418的達爾伯克氏改良伊格爾培養基/營養混合物(D-MEM/F-12)中培養細胞。 以8點濃度反應格式(4個複製槽孔/濃度)評估測試物效果。所有測試及對照溶液含有0.3% DMSO及0.01% Kolliphor®EL (C5135,Sigma)。使用自動化液體處理系統(SciClone ALH3000,Caliper LifeScienses)將測試物調配物加至384-槽孔複板中。在此程序之後使用Ion Works Barracuda平台進行量測: 電生理程序: a) 胞內溶液(mM):50 mM CsCl、90 mM CsF、2 mM MgCl
2
、5 mM EGTA、10 mM HEPES。以CsOH調整至pH 7.2。 b) 胞外溶液HB-PS (以mM為單位之組合物):NaCl,137;KCl,1.0;CaCl
2
,5;HEPES,10;葡糖,10;pH經NaOH調整至7.4 (冷藏直至使用為止)。 c) 保持電位:-70 mV,促效劑/PAM施加期間之電位:-40 mV。 記錄程序: a) 胞外緩衝液將經加至PPC培養盤槽孔中(每槽孔11 μL)。細胞懸浮液將經吸入至PPC平面電極之槽孔中(每槽孔9 μL) 。 b) 將經由使用由機載膜片鉗放大器記錄之膜電流的膜片穿孔來確定全細胞記錄組態。 c) 將進行兩個記錄(掃描)。首先,在僅預施加測試物期間(預施加之持續時間-5 min),且其次,在測試物及促效劑(EC
20
L-麩胺酸及30 µM甘胺酸)共施加期間,偵測測試物之正調節效果。 測試物投與:第一次預施加將由添加20 µL 2×濃縮測試物溶液組成,且第二次預施加將由以10 µL/s (總施加時間2秒)添加20 µL 1×濃縮測試物及促效劑組成。
正向異位調節劑 (PAM) 對通道之增強效果
正向異位調節劑(PAM)對通道之增強效果將計算為 活化% = (I
PAM
/ I
EC10-30
) x 100% - 100% 其中I
PAM
將為在各種濃度之測試物之存在下的L-麩胺酸EC
10-30
引發電流,且I
EC20
將為以L-麩胺酸EC
20
引發之平均電流。 PAM濃度反應資料將與以下形式之方程式擬合: 激活% = % L-麩胺酸EC
20
+ {(MAX% - L-麩胺酸EC
20
%) / [1 + ([Test]/EC
50
)
N
]}, 其中[測試]將為PAM (測試物)之濃度,EC
50
將為產生半最大激活之PAM之濃度,N將為希爾係數,L-麩胺酸EC
20
%將為經L-麩胺酸EC
20
引發之電流的百分比,MAX%為經與L-麩胺酸EC
20
共投與之最高劑量之PAM激活的電流的百分比,且激活%將為在各PAM濃度下經L-麩胺酸EC
10-30
引發之電流的百分比。 誘發電流之最大振幅經量測且經定義為峰值電流振幅(PCA)。
縮寫
PCC:氯鉻酸吡啶;t-BuOK:第三丁醇鉀;9-BBN:9-硼雙環[3.3.1]壬烷;Pd(
t
-Bu
3
P)
2
:雙(三-第三丁基膦)鈀(0);AcCl:氯化乙醯基;
i
-PrMgCl:異丙基氯化鎂;TBSCl:第三丁基(氯)二甲基矽烷;(
i
-PrO)
4
Ti:四異丙醇鈦;BHT:2,6-二-第三丁基-4-甲基苯酚;Me:甲基;
i
-Pr:異丙基;
t
-Bu:第三丁基;Ph:苯基;Et:乙基;Bz:苯甲醯基;BzCl:氯化苯甲醯;CsF:氟化銫;DCC:二環己基碳化二亞胺;DCM:二氯甲烷;DMAP:4-二甲胺基吡啶;DMP:戴斯-馬丁高碘烷;EtMgBr:乙基溴化鎂;EtOAc:乙酸乙酯;TEA:三乙胺;AlaOH:丙胺酸;Boc:第三丁氧羰基。Py:吡啶;TBAF:氟化四正丁基銨;THF:四氫呋喃;TBS:第三丁基二甲基矽烷基;TMS:三甲基矽烷基;TMSCF
3
:(三氟甲基)三甲基矽烷;Ts:對甲苯磺醯基;Bu:丁基;Ti(OiPr)
4
:四異丙氧基鈦;LAH:氫化鋰鋁;LDA:二異丙胺基鋰;LiOH.H
2
O:氫氧化鋰水合物;MAD:甲基鋁雙(2,6-二-第三丁基-4-甲基苯酚);MeCN:乙腈;NBS:N-溴代丁二醯亞胺;Na
2
SO
4
:硫酸鈉;Na
2
S
2
O
3
:硫代硫酸鈉;PE:石油醚;MeCN:乙腈;MeOH:甲醇;Boc:第三丁氧羰基;MTBE:甲基第三丁基醚;hr/hrs:一或更多小時;min/mins:一或更多分鐘。
實例 1 : (3S,8S,9S,10R,13S,14S,17R)-10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-3-( 三氟甲基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (1) 之合成 1.在0℃下向TBAF (3.04 mL,1 M於THF中,3.04 mmol,Aldrich)於THF (100 mL)中之溶液,接著向
S-200-INT-2
(19 g,60.8 mmol)於THF (100 mL)中之溶液中逐滴添加TMSCF
3
(25.8 g,182 mmol)。在0℃下攪拌混合物30 min。在0℃下向混合物添加TBAF (200 mL,1 M於THF中,200 mmol,國內的)。在0℃下再攪拌混合物30 min。向混合物添加NH
4
Cl (100 mL,飽和水溶液)。真空濃縮混合物。向殘餘物添加PE/EtOAc (400 mL,1:1),分離有機層,其與其他兩個批次(2×10 g
S200-INT-2
)合併。合併之有機層用水(300 mL)、鹽水(300 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到油狀物。使殘餘物溶解於DCM (150 mL)中且用PE (750 mL)稀釋。將溶液倒入矽膠管柱(500 g,100至200目)且用PE:DCM:EtOAc=5:1:0.05至5:1:0.1溶離,得到呈油狀物之
S200-CF3_1B
(12 g,70%純度,17%產量)及不純
S200-CF3_1A
。該不純自MeCN (250 mL)再結晶,得到呈固體之
S200-CF3_1A
(6.5 g)。自MeCN過濾藉由矽膠管柱(PE:DCM:EtOAc=50:1:1至20:1:1)純化得到粗產物,該粗產物自MeCN (20 mL)再結晶,得到呈固體之
S-200-CF3_1A
(1 g,16%總產量)。
注意 :
自
3 JH,CF3
(FDCS)鑑別
200-CF3_1A
及
200-CF3_1B
。
(J. Org. Chem. 2015, 80, 1754
.
S-200-CF3_1A: 1 H NMR
(400 MHz, CDCl
3
)δ 5.43-5.33 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H); 2.49 (s, 2H); 2.11-1.97 (m, 4H), 1.95-1.32 (m, 14H), 1.30-0.98 (m, 7H), 0.59 (s, 3H)。
S-200-CF3_1B: 1 H NMR
(400 MHz, CDCl
3
)δ 5.54-5.41 (m, 1H), 4.86 (s, 1H), 4.72 (s, 1H); 2.78-2.65 (m, 1H); 2.18-1.97 (m, 3H), 1.95-1.35 (m, 16H), 1.32-0.98 (m, 7H), 0.59 (s, 3H)。 2.向
S-200-CF3_1A
(8 g,20.9 mmol)於THF (80 mL)中之溶液中添加9-BBN二聚體(5.85 g,24 mmol)。在40℃下攪拌混合物1 h。將混合物冷卻至0℃。向該混合物逐滴添加EtOH (12 mL)、NaOH (41.8 mL,5 M水溶液)及H
2
O
2
(20.9 mL,10 M水溶液)。在50℃下攪拌混合物1 h。在冷卻後向混合物添加Na
2
SO
3
(100 mL,25%,水溶液)。用EtOAc (300 mL)萃取混合物。有機層經分離且藉由矽膠管柱(PE:EtOAc=10:1至5:1)純化,得到呈固體之
S-200-CF3_2A
(7.1 g,85%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.42-5.32 (m, 1H), 3.64 (dd,
J
= 3.2, 10.4 Hz, 1H), 3.37 (dd,
J
= 6.8, 10.4 Hz, 1H), 2.49 (s, 2H), 2.32-1.92 (m, 4H), 1.92-1.70 (m, 4H), 1.70-1.29 (m, 8H), 1.29-0.91 (m, 11H), 0.71 (s, 3H)。 3.在25℃下向
S-200-CF3_5A
(3g,7.49 mmol)於DCM (50 mL)中之溶液中添加DMP (6.31 g,14.9 mmol),在25℃下攪拌30 min之後,反應混合物用飽和NaHCO
3
(100 mL)淬滅且添加DCM (100 mL)且攪拌10 min。DCM相經分離且用Na
2
S
2
O
3
飽和水溶液(2×100 mL)洗滌。合併之有機層用飽和鹽水(2×100 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(5%至20% EtOAc/PE)純化,得到呈固體之
N-004-027_1
(1.5 g,50%)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.58-9.55 (m, 1H), 5.38-5.36 (m, 1H), 2.49 (s, 1H), 2.40-2.25 (m, 1H), 2.23-1.60 (m, 10H), 1.53-1.20 (m, 9H), 1.15-1.00 (m, 7H), 0.78-0.64 (m, 3H)。 4.在0℃下向
N-004-027_1
(1.5 g,3.76 mmol)於無水THF (40 mL)中之溶液中添加CsF (1.42 g,9.40 mmol)。在0℃下攪拌20 min之後,在0℃下添加TMSCF
3
(1.33 g,9.40 mmol)且攪拌30 min。顏色變成淡黃色。添加TBAF.3H
2
O (4.74 g,15.0 mmol)且在50℃下攪拌30 min。將反應混合物倒入冰水(100 mL)中。用EtOAc (2×100 mL)萃取水相。合併之有機相用飽和鹽水(2×100 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,得到呈黃色固體之異構體混合物(1.45 g,粗產物),其藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之
53
(340 mg,24%)及呈白色固體之
1
(200 mg,14%)。
1: 1 H NMR
(400 MHz, CDCl
3
) δ 5.38-5.36 (m, 1H), 4.10-4.00 (m, 1H), 2.49 (s, 2H), 2.19-2.12 (m, 1H), 2.06-1.61 (m, 10H), 1.53-1.29 (m, 6H), 1.27-0.98 (m, 10H), 0.71 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.121 min,30-90AB_2MIN_E,純度100%,
MS
50-100_1_4min.m,C
24
H
33
F
6
O[M+H-H
2
O]
+
451,實驗值451。
1: 1 H NMR
(400 MHz, CDCl
3
) δ 5.38-5.36 (m, 1H), 4.10-4.00 (m, 1H), 2.49 (s, 2H), 2.19-2.12 (m, 1H), 2.06-1.61 (m, 10H), 1.53-1.29 (m, 6H), 1.27-0.98 (m, 10H), 0.71 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.121 min,30-90AB_2MIN_E,純度100%,
MS
50-100_1_4min.m,C
24
H
33
F
6
O[M+H-H
2
O]
+
451,實驗值451。
實例 2 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (2) 之合成 1.在20℃下向
N-4-4_1
(50 g,157 mmol)於無水甲醇(500 mL)中之懸浮液一次性添加無水TsOH (2.84 g,15.7 mmol)。使混合物升溫直至60℃且攪拌1 h。反應混合物用Et
3
N (1.58 g,15.7 mmol)淬滅且攪拌另外30 min。濾出沈澱之固體,用甲醇(250 mL)洗滌且於空氣中乾燥,得到呈固體之
N-4-1_2
(51 g,90%)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.18 (s, 3H), 3.14 (s, 3H); 2.54-2.48 (m, 1H); 2.10-2.00 (m, 4H); 1.95-1.75 (m, 2H), 1.65-1.50 (m, 7H), 1.48-0.80 (m, 11H), 0.78-0.75 (m, 4H), 0.59 (s, 3H)。 2.在20℃下於N
2
下向Ph
3
PMeBr (75 g,210 mmol)於無水THF (500 mL)中之懸浮液逐份添加t-BuOK (23.5 g,210 mmol)。混合物變成深橙色且在20℃下攪拌30 min。隨後添加
N-4-1_2
(51 g,140 mmol)。使混合物升溫至40℃且攪拌1 h。將反應混合物冷卻且逐份倒入NH
4
Cl水溶液(冰) (400mL)中。分離所得混合物;用THF (200 mL)萃取含水層。合併之有機層直接用作
N-4-1_3
之溶液而無需進一步純化。 3.在20℃下向
N-4-1_3
(50.4 g,139 mmol)於THF (700 mL)中之溶液中添加HCl水溶液(1 M,208 mL,208 mmol)。在20℃下攪拌混合物1小時,且固體沈澱。將水(200 mL)添加至混合物中,且濾出沈澱之固體,用水洗滌且乾燥,得到呈固體之
N-4-1_4
(41 g,94%)。
1 H NMR
(400 MHz, CDCl
3
)δ 4.85 (s, 1H), 4.70 (s, 1H); 2.38-2.25 (m, 3H); 2.10-1.98 (m, 3H), 1.88-1.49 (m, 10H), 1.40-1.08 (m, 11H), 0.97-0.72 (m, 2H), 0.58 (s, 3H)。 4.在25℃下於N
2
下向Me
3
SI (101 g,496 mmol)於無水THF (400 mL)中之溶液中逐份添加t-BuOK (58.3 g,520 mmol)且攪拌30 min。添加
N-4-1_4
(39 g,124 mmol)於無水THF (300 mL)中之溶液。使反應混合物升溫至50℃且攪拌2小時。將反應混合物冷卻至25℃且用NH
4
Cl水溶液(500 mL)處理。用EtOAc (2×500 mL))萃取水相。合併之有機相用鹽水(2×300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至10/1)純化,得到呈固體之
N-4-3_1
(35 g,不純)。
1 H NMR
(400 MHz, CDCl
3
)δ 4.84 (s, 1H), 4.70 (s, 1H); 2.65-2.55 (m, 2H); 2.10-1.98 (m, 2H), 1.92-1.49 (m, 13H), 1.40-1.13 (m, 8H), 0.99-0.69 (m, 6H), 0.57 (s, 3H)。 5.在25℃下向
N-4-3_1
(35 g,647 mmol)於無水MeOH (500 mL)中之溶液中添加MeONa (57.2 g,1.06 mol)且在N
2
下攪拌混合物30 min。使反應混合物升溫至70℃且於N
2
下攪拌回流3小時。將反應混合物冷卻至25℃且用水(500 mL)處理。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮,以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc=10/1至6/1)純化,得到呈固體之
N-4-3_2
(25 g,不純)。粗產物在25℃下自PE (250 mL)濕磨1 h。過濾懸浮液且真空乾燥濾餅以獲得呈固體之
N-4-3_2
(15 g,25%)。
1 H NMR
(400 MHz, CDCl
3
)δ 4.86 (s, 1H), 4.72 (s, 1H), 3.46-3.37 (m, 5H), 2.54 (s, 1H), 2.07-1.99 (m, 1H), 1.89-1.52 (m, 15H), 1.41-1.06 (m, 10H), 0.86 (s, 3H); 0.58 (s, 3H) 6.在0℃下向
N-4-3_2
(15 g,41.6 mmol)於無水THF (200 mL)中之溶液中添加9-BBN二聚體(27.7 g,124 mmol)且於N
2
下攪拌30 min。使反應混合物升溫至50℃且攪拌1 h。將反應混合物冷卻至0℃且添加EtOH (50 mL),隨後在0℃下極緩慢地添加NaOH (41.6 mL,5M,208 mmol)。緩慢地添加H
2
O
2
(23.5 g,208 mmol,30%於水中)同時使內部溫度保持在10℃以下。使混合物升溫至50℃且再攪拌1小時。將反應混合物冷卻,逐份倒入冰水(500 mL)中且過濾。真空濃縮濾液以提供呈油狀物之
N-4-3_3
(14 g,粗產物)。粗殘餘物直接用於下一步驟。 7.在25℃下將DMP (3.35 g,7.92 mmol)添加至
N-4_3
(1 g,2.64 mmol)於DCM (20 mL)中之混合物中。使反應混合物升溫至40℃且攪拌1 h。在pH 7至8及10℃以下用NaHCO
3
飽和水溶液淬滅反應混合物。過濾懸浮液。濾液中之DCM相經分離且用NaHCO
3
/Na
2
S
2
O
3
飽和水溶液(1:1,2×50 mL)、鹽水(2×50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮以獲得固體。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之
N-4-3_4
(0.6 g,60%)。
1 H NMR
(400 MHz, CDCl
3
)δ 9.57 (s, 1H), 3.40-3.34 (m, 5H); 2.38-2.28 (m, 1H); 1.94-1.76 (m, 2H), 1.74-1.35 (m, 16H), 1.06-0.82 (m, 10H), 0.73-0.64 (m, 5H)。 8.在0℃下於N
2
下向
N-4-3_4
(0.6 g,1.59 mmol)於無水THF (10 mL)中之溶液中添加異戊基溴化鎂(4.37 mL,8.74 mmol,2 M於二乙醚中)。使反應混合物升溫至25℃且攪拌1小時。向反應混合物中添加NH
4
Cl 飽和水溶液(50 mL)。用EtOAc (3×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,得到呈固體之
N-4-4A
(0.5g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.64-3.60 (m, 1H), 3.40-3.37 (m, 5H); 2.02-1.79 (m, 3H); 1.75-1.50 (m, 11H), 1.25-1.10 (m, 14H), 0.99-0.75 (m, 14H), 0.70-0.64 (m, 4H)。 9.在25℃下向
N-4-4A
(0.5 g,粗產物)於DCM (20 mL)中之溶液中添加DMP (1.88 g,4.44 mmol)。使反應混合物升溫至40℃且攪拌1小時。在pH 7至8及10℃以下用NaHCO
3
飽和水溶液淬滅反應混合物。過濾懸浮液。DCM相經分離且用NaHCO
3
/Na
2
S
2
O
3
飽和水溶液(1:1,2×50 mL)、鹽水(2×50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-4-4O
(0.4 g,粗產物),其直接用於下一步驟。 在25℃下向
N-4-4O
(0.4 g,0.895 mmol)於MeOH (4 mL)中之溶液中緩慢地添加NaBH
4
(0.340 g,8.95 mmol)且攪拌2小時。用DCM (2 × 20 mL)萃取水相。合併之有機相用飽和鹽水(2×20 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮,以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc=8/1至5/1)純化,得到呈固體之
35
(150 mg,不純)及
2
(130 mg,不純)。在82
o
C下回流1小時自MeCN (3 mL)再結晶
2
(130 mg,不純)。攪拌混合物且冷卻至25℃。過濾懸浮液且真空濃縮濾液以提供呈固體之
2
(50 mg,12%)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.63-3.61 (m, 1H), 3.41-3.38 (m, 5H); 2.51 (s, 1H); 1.97-1.81 (m, 2H), 1.71-1.31 (m, 15H), 1.26-1.03 (m, 10H), 0.97-0.78 (m, 14H), 0.71-0.59 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.350 min,30-90 AB,純度99%,C
29
H
48
O [M+H-2H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 3 : (3S,8R,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (3) 之合成 1.
在35℃下於氮氣下將t-BuOH (350 mL)裝入三頸圓底燒瓶且於氮氣下攪拌10 min。將t-BuOK (90.5 g,807 mmol)添加至混合物中且於氮氣下攪拌15 min。將
S-310-B9_1
(20 g, 73.4 mmol)添加至以上混合物且在35℃下於氮氣下攪拌1.5小時。將反應混合物倒入至10%乙酸水溶液(500 mL)中且在35℃以下攪拌15 min。添加水(500 mL)且攪拌混合物30 min。用碳酸氫鈉(500 mL)將混合物之pH調節為7至8且攪拌30 min。用PE (2×500 mL)萃取混合物。有機層經分離,用鹽水(500 mL)洗滌,經無水硫酸鈉乾燥,過濾且在35℃以下濃縮,得到呈油狀之
S-200-N19-3_1
(17 g,粗產物)。粗殘餘物直接用於下一步驟。
2.
在0℃下向2,6-二第三丁基-4-甲苯酚(100 g,453 mmol)於甲苯(300 ml)中之溶液中逐滴添加AlMe
3
(113 mL,226 mmol,2 M於甲苯中)。在25℃下攪拌混合物1小時以產生MAD。在-70℃下將
S-200-N19-3_1
(10 g,36.7 mmol)於甲苯(50 mL)中之溶液逐滴添加至MAD溶液。在-70℃下攪拌1小時之後,在-70℃下逐滴添加MeMgBr (36.6 mL,110 mmol,3M於乙基醚中)。在-70℃下攪拌所得溶液1小時。在-70℃下用飽和檸檬酸(400 ml)淬滅反應混合物。在25℃下攪拌10 min後,過濾所得混合物且用EtOAc (2×200 ml)洗滌。合併之有機層經分離,用鹽水(2×200 ml)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(PE/EtOAc=10/1至5/1)純化以產生呈固體之
S-200-N19-3_2
(7.6 g,不純)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.45-5.40 (m, 1H), 2.51-2.38 (m, 1H), 2.49-2.21 (m, 1H), 2.14-1.88 (m, 5H), 1.86-1.77 (m, 2H), 1.73-1.38 (m, 8H), 1.34-1.22 (m, 4H), 0.95-0.81 (m, 8H)。 3.在40℃下於N
2
下向PPh
3
EtBr (37.1 g,100 mmol)於THF (200 mL)中之懸浮液中添加t-BuOK (11.2 g,100 mmol)。在20℃下攪拌10 min後,添加
S-200-N19-3_2
(7.6 g, 25.1 mmol)。在40℃下攪拌反應混合物1小時。反應物在0℃下用NH
4
Cl水溶液(200 mL)淬滅,用EtOAc (3×200 mL)萃取。合併之有機相用鹽水(200 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由Combi-flash (0%至30% EtOAc/PE)純化,得到呈固體之
S-200-N19-3_3
(5 g,63%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.45-5.35 (m, 1H), 5.20-5.00 (m, 1H), 2.41-2.30 (m, 1H), 2.29-2.12 (m, 3H), 2.09-1.76 (m, 6H), 1.69-1.38 (m, 15H), 1.35-0.94 (m, 7H)。
4.
在0℃下於N
2
下向
S-200-N19-3_3
(2 g,6.35 mmol)於THF (20 mL)中之溶液中添加9-BNN二聚體(3.09 g,12.7 mmol)。在60℃下攪拌溶液1小時。冷卻至0℃後,極緩慢地添加EtOH (20 ml)及NaOH (12.7 ml,5M,63.5 mmol)之溶液。添加後,緩慢地添加H
2
O
2
(2.15 mg,6.35 mmol,30%於水中),且內部溫度維持在10℃以下。在60℃下於N
2
下攪拌混合物1小時。將混合物重新冷卻至30℃。將水(100 mL)添加至溶液且用EtOAc (100 mL)萃取含水層。有機層用(2×100 mL)鹽水洗滌。合併之有機層經無水Na
2
SO
4
乾燥,且藉由矽膠層析(PE/EtOAc=2/1)純化,得到呈固體之
S-200-N19-4_1
(1.6 g,不純)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.45-5.35 (m, 1H), 3.75-3.62 (m, 1H), 2.28-2.19 (m, 1H), 2.10-1.75 (m, 7H), 1.71-0.97 (m, 19H), 0.92-0.75 (m, 4H), 0.68 (s, 3H)。
5.
向
S-200-N19-4_1
(1.6 g,4.81 mmol)於DCM (20 mL)中之溶液中添加矽膠(2 g)及PCC (2.07 g,9.62 mmol)。在25℃下攪拌混合物3小時。向混合物添加PE (50 mL)。混合物經由矽膠墊過濾,且固體用PE/DCM (30 mL/30 mL)洗滌。過濾混合物且真空濃縮濾液。殘餘物藉由矽膠層析(PE/EtOAc=10/1至5/1)純化,得到呈固體之
S-200-N19-4_2
(1.2 g,不純),其在回流下自MeCN (10 mL)再結晶以提供呈固體之
S-200-N19-4_2
(1.0 g,84.0%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.40-5.35 (m, 1H), 2.61-2.45 (m, 1H), 2.30-2.10 (m, 5H), 2.00-1.75 (m, 6H), 1.70-1.10 (m, 14H), 0.90-0.75 (m, 4H); 0.633 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.058 min,30-90 AB,純度100%,C
22
H
34
[M+H-H
2
O]
+
之MS ESI計算值313,實驗值313。 6.在40℃下於N
2
下向Ph
3
PMeBr (11.1 g,31.4 mmol)於THF (50 mL)中之懸浮液中添加t-BuOK (3.51 g,31.4 mmol)。在25℃下攪拌10 min之後,添加
S-200-N19-4_2
(2.6 g,7.86 mmol)。在40℃下攪拌反應混合物1 h。反應物在0℃下用NH
4
Cl水溶液(100 mL)淬滅,其用EtOAc (2×100 mL)萃取。合併之有機相用鹽水(2 × 100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由Combi-flash (0%至30% EtOAc/PE)純化,得到呈固體之
S-200-N19-4_3
(2.4 g,93%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.45-5.35 (m, 1H), 4.86-4.83 (m, 1H), 8.70-4.65 (m, 1H), 2.27-2.20 (m, 1H), 2.10-1.90 (m, 4H), 1.89-1.50 (m, 11H), 1.49-1.30 (m, 3H), 1.28-1.00 (m, 6H), 0.80-0.60 (m, 5H), 0.59 (s, 3H)。 7.在0℃下於N
2
下將9-BBN二聚體(9.27 g,38.0 mmol)添加至
S-200-N19-4_3
(5 g,15.2 mmol)於THF (60 mL)中之溶液中。在60℃下攪拌溶液1 h。冷卻至0℃後,極緩慢地添加EtOH (60 ml)及NaOH (30.4 ml,5M,152 mmol)之溶液。添加後,緩慢地添加H
2
O
2
(15.2 ml,152 mmol,30%於水中),且內部溫度維持在10℃以下。在60℃下於N
2
下攪拌混合物1小時。將混合物重新冷卻至30℃。將水(100 mL)與EtOH (100 ml)添加至溶液。獲得懸浮液,其經過濾且真空濃縮以產生呈固體之
S-200-N19-4_4
(5 g,粗產物)。
1 H NMR
(400MHz, CDCl3) δ 5.44-5.32 (m, 1H), 3.68-3.59 (m, 1H), 3.39-3.35 (m, 1H), 2.29-2.19 (m, 1H), 2.08-1.89 (m, 4H), 1.88-1.75 (m, 3H), 1.62-1.60 (m, 2H), 1.56-1.39 (m, 6H), 1.36-1.24 (m, 3H), 1.23-1.11 (m, 4H), 1.08-0.98 (m, 4H), 0.92-0.75 (m, 5H), 0.70 (s, 3H)。 8.在25℃下將戴斯-馬丁高碘烷(2.44 g,5.76 mmol)添加至
S-200-N19-4_4
(1 g,2.88 mmol)於DCM (150 mL)中之溶液中。在25℃下攪拌反應物1小時。在25℃下攪拌反應物30 min。在0℃下將混合物倒入飽和Na
2
S
2
O
3
(100 ml)中,其用DCM (3×100 ml)萃取。合併之有機層用飽和NaHCO
3
(100 mL×2)、鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物,藉由矽膠管柱(PE/EtOAc=10:1)將粗產物純化得到呈固體之
S-500-15-2_1
(800 mg,80%)。
1 H NMR
(400MHz, CDCl3) δ 9.58-9.57 (m, 1H), 5.40-5.38 (m, 1H), 2.37-2.35 (m, 1H), 2.25-2.23 (m, 1H), 2.08-1.76 (m, 7H), 1.65-1.63 (m, 2H), 1.53-1.37 (m, 5H), 1.31-1.21 (m, 4H), 1.19-1.00 (m, 6H), 0.90-0.80 (m, 5H), 0.73 (s, 3H)。 9.在60℃下將1-溴-3-甲基丁烷(4 g,26.4 mmol)於THF (27 mL)中之溶液逐滴添加至Mg (947 mg,39.5 mmol)及I
2
(33.5 mg,0.132 mmol)於THF (3 mL)中之懸浮液。在60℃下攪拌混合物1小時。在0℃下於N
2
下將新鮮製備的異戊基溴化鎂(30 mL,0.88 M於THF中,26.4 mmol)添加至
S-500-15-2_1
(800 mg,2.32 mmol)於THF (2 mL)中之溶液中。在0℃下攪拌混合物1小時。向混合物添加NH
4
Cl (50 mL,飽和水溶液)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物,藉由矽膠層析(PE/EtOAc=10/1至5/1)將粗產物純化得到呈固體之
44
(720 mg,75%)。
1 H NMR
(400MHz, CDCl3) δ 5.40-5.38 (m, 1H), 3.63-3.61 (m, 1H), 2.23-2.21 (m, 1H), 2.10-1.74 (m, 7H), 1.69-1.58 (m, 2H), 1.54-1.34 (m, 8H), 1.33-1.00 (m, 11H), 0.95-0.75 (m, 14H), 0.70 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.289 min,30-90 AB,純度100%,C
28
H
45
[M+H-2H
2
O]+之MS ESI計算值381,實驗值381。 10a.在25℃下於N
2
下向
44
(300 mg,0.720 mmol)於THF (14 mL)中之溶液中添加苯甲酸(348 mg,2.85 mmol)及三苯基膦(1.11 g,4.27 mmol)。在25℃下攪拌20 min之後,在0℃下於N
2
下添加DIAD (780 mg,3.86 mmol)。在0℃下攪拌混合物20 min隨後升溫至25℃且在25℃下攪拌17小時。添加水(100 mL)且用EtOAc (2×100 mL)萃取混合物。有機相用鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾,真空濃縮,得到待純化之粗產物(1.5 g,粗產物)。 10b.在25℃下於N
2
下向
44
(1.9 g,4.55 mmol)於THF (70 mL)中之溶液中添加苯甲酸(2.19 g,18.0 mmol)及三苯膦(7.07 g,27.0 mmol)。在25℃下攪拌20 min之後,在0℃下於N
2
下添加DIAD (4.93 g,24.4 mmol)。在0℃下攪拌混合物20 min隨後升溫至25℃且在25℃下攪拌17小時。添加水(250 mL)且用EtOAc (2×250 mL)萃取混合物。有機相用鹽水(2×300 mL)洗滌,經Na
2
SO
4
乾燥,過濾,真空濃縮,得到粗產物。與另一批次之300 mg之
44
合併,粗產物藉由矽膠管柱(PE/EtOAc=8/1)純化,得到呈油狀物之
S-500-15-1_1
(1.2 g,不純),其直接用於下一步驟。 11.在25℃下向
S-500-15-1_1
(1.2 g,不純)於THF/MeOH (2 mL/2 mL)中之溶液中添加NaOH (400 mg)及H
2
O (2 mL)。在50℃下攪拌反應物16 h。冷卻後,反應混合物用H
2
O (20 mL)稀釋且用EtOAc (2×30 mL)萃取。合併之有機層經Na
2
SO
4
乾燥,過濾且真空濃縮。粗產物藉由矽膠管柱(PE/EtOAc=4/1)純化,得到產物
3
(150 mg,不純),其藉由在25℃下用MeCN (5 mL)濕磨純化,得到呈固體之
3
(30 mg,純,及100 mg,不純)。
1 H NMR
(400MHz, CDCl3) δ 5.39-5.37 (m, 1H), 3.63-3.59 (m, 1H), 2.26-2.21 (m, 1H), 2.09-1.88 (m, 4H), 1.86-1.76 (m, 2H), 1.75-1.61 (m, 3H), 1.54-1.32 (m, 7H), 1.32-1.08 (m, 10H), 1.07-0.96 (m, 1H), 0.95-0.74 (m, 14H), 0.95-0.74 (m, 1H), 0.70 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.281 min,30-90 AB,純度98%,C
28
H
47
O [M+H-H
2
O]+之MS ESI計算值399,實驗值399。
實例 4 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己 基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (4) 之合成 1.在0℃下將t-BuLi (90.7 mL,118 mmol,1.3 M於正己烷中,3.0當量)添加至氯基(甲氧基甲基)三苯基磷烷(40.4 g,118 mmol,3.0當量)於THF (200 mL)之溶液中。添加之後,在0℃下攪拌反應混合物1小時。在0℃下將混合物添加至
S-500-6-29_2A
(5 g,39.6 mmol,1.0當量)於THF (50 mL)中之溶液中且在15℃下攪拌反應混合物2 h。混合物用NH
4
Cl(100 mL,10%)處理且用EtOAc (2×200 mL)萃取。有機相經分離且真空濃縮,得到
S-500-6-29_2B
(18.0 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.74 (s, 1H), 3.52 (s, 3H), 2.20-2.15 (m, 2H), 1.95-1.90 (m, 2H), 1.26-1.16 (m, 4H), 0.90 (s, 6H)。 2.在15℃下將TFA (21.4 mL,290 mmol)添加至
S-500-6-29_2B
(5.6 g,不純)於DCM (25 mL)中之攪拌溶液中且在15℃下攪拌1.5 h。反應混合物用NaHCO
3
飽和水溶液(10 mL)淬滅且用EtOAc (2×20 mL)萃取。合併之有機層經無水Na
2
SO
4
乾燥且在減壓下濃縮,得到呈油狀物之
S-500-6-29_2C
(5.0 g,粗產物),其用於下一步驟中而無需純化。
1 H NMR
(400 MHz, CDCl
3
) δ 9.64 (s, 1H), 2.15-2.05 (m, 1H), 1.80-1.60 (m, 2H), 1.70-1.35 (m, 4H), 1.25-1.15 (m, 2H), 0.91 (s, 3H), 0.87 (s, 3H)。
3.
在15℃下於N
2
下將NaBH
4
(1.61 g,42.7 mmol)添加至
S-500-6-29_2C
(5.0 g,35.6 mmol)於MeOH (50 mL)中之溶液中。在15℃下攪拌混合物1小時。將混合物倒入水(50 mL)中且攪拌20分鐘。用EtOAc (3×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,得到呈油狀物之
S-500-6-29_2D
(5.6 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.47-3.42 (m, 2H), 1.60-1.50 (m, 2H), 1.42-1.30 (m, 4H), 1.25-1.0 (m, 4H), 0.91 (s, 3H), 0.87 (s, 3H)。 4.在15℃下於N
2
下將TsCl (8.23 g,43.2 mmol)添加至
S-500-6-29_2D
(5.6 g,39.3 mmol)於吡啶(50 mL)中之溶液中。在15℃下攪拌混合物16小時。將混合物倒入水(50 mL)中且攪拌20分鐘。用DCM (3×40 mL)萃取水相。合併之有機相用飽和鹽水(2×200 mL)、HCl (0.5 M,50 ml)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,得到油狀物,其在68
o
C下自己烷(50 mL)再結晶得到呈固體之
S-500-6-29_2E
(4.2 g,61%)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.80-7.76 (m, 2H), 7.35-7.25 (m, 2H), 3.86-3.80 (m, 2H), 2.45 (s, 3H), 1.60-1.45 (m, 3H), 1.40-1.30 (m, 2H), 1.20-1.05 (m, 4H), 0.88 (s, 3H), 0.82 (s, 3H)。
5.
將LiBr (2.33 g,26.9 mmol)添加至
S-500-6-29_2E
(2 g,6.74 mmol)於丙酮(50 mL)中之溶液中。在65℃下攪拌混合物12小時。混合物用水(50 mL)淬滅且用MTBE (3×20 mL)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈液體之
S-500-6-29_2
(1.3 g,粗產物)。與另一批次之2.2 g之
S-500-6-29_2E
合併,合併之粗產物經由小矽膠過濾且用PE (100 mL)洗滌且濃縮,得到呈油狀物之
S-500-6-29_2
(2.6 g,90%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.34-3.28 (m, 2H), 1.72-1.64 (m, 2H), 1.60-1.48 (m, 1H), 1.42-1.35 (m, 2H), 1.28-1.18 (m, 4H), 0.91 (s, 3H), 0.87 (s, 3H)。 6.在25℃下向Ph
3
PMeBr (167 g,470 mmol)於THF (900 mL)中之溶液中添加
t
-BuOK (52.7 g,470 mmol)。將反應混合物加熱至60℃且攪拌1小時。添加
孕烯醇酮
(50 g,157 mmol)。在60℃下攪拌反應混合物1小時。添加飽和NH
4
Cl (900 mL)。用EtOAc (2×1000 mL)萃取混合物。合併之有機層用鹽水(2×2000 mL)洗滌,經Na
2
SO
4
乾燥且真空濃縮,得到呈油狀物之粗產物,其藉由矽膠管柱層析(PE:EtOAc=20:1至5:1)純化,得到呈固體之
S-200-INT_1
(45 g,91.2%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.40-5.30 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 3.60-3.40 (m, 1H), 2.40-2.20 (m, 2H), 2.05-1.90 (m, 2H), 1.85-1.60 (m, 9H), 1.53-1.40 (m, 5H), 1.25-0.90 (m, 9H), 0.59 (s, 3H)。 7.在20℃下向
S-200-INT_1
(45 g,143 mmol)於DCM (1500 mL)中之溶液中添加DMP (108 g,257 mmol)。在20℃下攪拌混合物2小時。添加水(800 mL)且添加NaHCO
3
(200 g固體)。過濾混合物。濾液用飽和Na
2
S
2
O
3
(2×2000 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到
S-200-INT_2
於DCM (100 mL)中之溶液,其直接用於下一步驟。 8.在10℃下向BHT (191 g,866 mmol)於甲苯(500 mL)中之溶液中添加AlMe
3
(2 M於甲苯中,216 mL,433 mmol)且攪拌1小時。在-78℃下向混合物添加
S-200-INT_2
(理論質量:44.6 g)於DCM (100 mL)中之溶液。在-78℃下攪拌混合物1小時。在-78℃下添加EtMgBr (141 mL,426 mmol)。在-78℃下攪拌混合物20 min。添加飽和檸檬酸(1 L)。有機相經分離,用鹽水(600 mL)洗滌,經Na
2
SO
4
乾燥且真空濃縮,得到粗產物,其藉由矽膠管柱層析(PE:EtOAc=50:1至30:1)純化,得到呈固體之
S-200-INT_3a
(27 g,55%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.35-5.25 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.40-2.30 (m, 1H), 2.10-1.60 (m, 14H), 1.50-0.75 (m, 17H), 0.58 (s, 3H)。 9.將9-BBN二聚體(17.6 g,72.5 mmol)添加至
S-200-INT_3E
(5 g,14.5 mmol)於THF (40 mL)中之溶液中。在60℃下於N
2
下攪拌混合物3小時,且形成固體。向反應混合物添加乙醇(8.33 mL,145 mmol)及NaOH (28.9 mL,5 M,145 mmol)。混合物變得澄清。在25℃下逐滴添加H
2
O
2
(14.4 mL,10 M,145 mmol)且使內部溫度升至回流(75℃)。混合物冷卻,在添加且攪拌1小時之後,形成固體。在25℃下向混合物添加Na
2
SO
3
(20 mL,20%水溶液)。用EtOAc (2×100 mL)萃取混合物。合併之有機相用鹽水(2×200 mL)洗滌,經Na
2
SO
4
乾燥,真空濃縮,且藉由矽膠管柱(PE/EtOAc=10/1至3/1)純化,以提供呈固體之
S-200-INT_4E
(3.5 g,67%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.68-3.60 (m, 1H), 3.41-3.32 (m, 1H), 2.40-2.32 (m, 1H), 2.03-1.93 (m, 2H), 1.92-1.65 (m, 4H), 1.58-1.16 (m, 13H), 1.16-0.90 (m, 11H), 0.90-0.81 (m, 3H), 0.73-0.62 (s, 3H)。 10.在25℃下將DMP (4.66 g,11.0 mmol)添加至
S-200-INT_4E
(2 g,5.54 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌反應混合物10 min。在25℃下用NaHCO
3
飽和水溶液(30 mL)淬滅反應混合物。分離DCM層且用DCM (30 mL)萃取水相。合併之有機相用Na
2
SO
3
飽和水溶液(3×50 mL)、鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
S-200-INT_5E
(2.0 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.59-9.56 (m, 1H), 5.31-5.26 (m, 1H), 2.42-2.10 (m, 2H), 2.10-1.80 (m, 4H), 1.79-1.54 (m, 7H), 1.54-1.31 (m, 7H), 1.28-0.90 (m, 9H), 0.90-0.81 (m, 4H), 0.73 (s, 3H)。 11.在75℃下將
S-500-6-29_2
(2.56 g,12.5 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (600 mg,25.0 mmol)及I
2
(63.4 mg,0.25 mmol)於THF (3 mL)中之懸浮液中。在75℃下攪拌混合物1小時。冷卻後,在15℃下緩慢地添加
S-500-6-1_1
(1 g,2.78 mmol)於THF (30 mL)中之溶液。添加之後,在15℃下攪拌混合物2小時,用飽和NH
4
Cl (40 mL)及飽和檸檬酸(20 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之
S-500-6-29_1
(800 mg,60%)之混合物。
1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.19 (m, 1H), 3.88-3.71 (m, 1H), 2.42-2.29 (m, 1H), 2.07-1.86 (m, 4H), 1.78-1.59 (m, 4H), 1.54-1.31 (m, 13H), 1.29-1.13 (m, 8H), 1.12-0.99 (m, 8H), 0.94-0.79 (m, 13H), 0.68 (s, 3H)。 12.將DMP (1.39 g,3.30 mmol)添加至
S-500-6-29_1
(800 mg,1.65 mmol)於DCM (30 mL)中之溶液中。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO
3
飽和水溶液(50 mL)淬滅直至含水層之pH為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3×40 mL)、飽和NaHCO
3
(40 mL)、鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈固體之粗產物
S-500-6-29_2
(800 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.25 (m, 1H), 2.54-2.43 (m, 1H), 2.40-2.21 (m, 3H), 2.07-1.87 (m, 3H),1.81-1.57 (m, 7H), 1.53-1.39 (m, 7H), 1.38-1.29 (m, 3H), 1.27-1.16 (m, 4H), 1.15-1.04 (m, 8H), 1.03 (s, 3H), 1.00-0.92 (m, 2H), 0.91-0.80 (m, 9H), 0.69 (s, 3H)。 13.將NaBH
4
(2.80 g,82.5 mmol)每五分鐘添加五次至
S-500-6-29_2
(800 mg,1.65 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘。混合物用飽和NH
4
Cl (50 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到
49
(290 mg,36%)及呈固體之
12
(120 mg,45%)。
49: 1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.07-1.87 (m, 4H), 1.76-1.69 (m, 1H), 1.66-1.55 (m, 5H), 1.53-1.42 (m, 7H), 1.41-1.31 (m, 5H), 1.30-1.12 (m, 8H), 1.11-1.05 (m, 3H), 1.03 (s, 3H), 1.01-0.92 (m, 2H), 0.91-0.82 (m, 12H), 0.68 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.718 min,30-90AB_E,純度98%,C
33
H
53
[M+H-2H
2
O]
+
之MS ESI計算值449,實驗值449。
12: 1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.06-1.95 (m, 3H), 1.77-1.58 (m, 7H), 1.54-1.28 (m, 12H), 1.27-1.06 (m, 11H), 1.03 (s, 3H), 1.00-0.95 (m, 2H), 0.93-0.82 (m, 12H), 0.69 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.708 min,30-90AB_E,純度100%,C
33
H
53
[M+H-2H
2
O]
+
之MS ESI計算值449,實驗值449。 14.將Pd(OH)
2
(200 mg,無水)添加至
49
(140 mg,0.288 mmol)於MeOH (30 mL)中之溶液中。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到
59
(27 mg,19%)及呈固體之
4
(42 mg,30%)。
4: 1 H NMR
(400 MHz, CDCl
3
) δ 3.84-3.76 (m, 1H), 1.98-1.85 (m, 2H), 1.69-1.54 (m, 9H), 1.53-1.46 (m, 3H), 1.45-1.28 (m, 9H), 1.27-1.20 (m, 4H), 1.19-1.13 (m, 5H), 1.12-1.02 (m, 4H), 1.01-0.92 (m, 2H), 0.91-0.85 (m, 12H), 0.82 (s, 3H), 0.70-0.61 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.799 min,30-90AB_E,純度100%,C
33
H
55
[M+H-H
2
O]
+
之MS ESI計算值451,實驗值451。
實例 5 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (5) 之合成 1.在50℃至55℃下於N
2
下將1-溴-3,3-二甲基丁烷(3.68 g,22.3 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (1.08 g,44.6 mmol)及I
2
(1 mg)於THF (2 mL)中之懸浮液中。在55℃下攪拌混合物1小時。隨後在0℃下將
S-500-6-1_1
(0.8 g,2.23 mmol)於THF (5 mL)中之溶液添加至新鮮製備的(3,3-二甲基丁基)溴化鎂(22.3 mmol於10 mL之THF中)。在15℃下攪拌混合物2小時。向混合物添加檸檬酸(20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到
5
(580 mg,P1,58%)及
54
(50 mg,5%,不純)。
5: 1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.24 (m, 1H), 3.65-3.54 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.84 (m, 4H), 1.76-1.38 (m, 15H), 1.38-1.00 (m, 12H), 0.93-0.80 (m, 15H), 0.70 (s, 3H)。
54: 1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.24 (m, 1H), 3.62-3.52 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.90 (m, 3H), 1.75-1.00 (m, 28H), 1.00-0.75 (m, 18H), 0.70 (s, 3H)。 2.在20℃下將DMP (1.1 g,2.6 mmol)及水(1滴)添加至
5
(580 mg,1.3 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌混合物2 h。將NaHCO
3
飽和溶液(20 mL)及Na
2
S
2
O
3
(20 mL,飽和)添加至混合物中。用EtOAc (50 mL)萃取混合物。有機層用NaHCO
3
/Na
2
S
2
O
3
(20+20 mL,飽和)洗滌兩次,經Na
2
SO
4
乾燥,過濾,真空濃縮,得到呈固體之
S-500-6-1_3
(520 mg,90%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.38-5.18 (m, 1H), 2.62-2.22 (m, 4H), 2.11-1.85 (m, 3H), 1.78-1.57 (m, 7H), 1.57-1.32 (m, 8H), 1.32-1.21 (m, 2H), 1.19-1.09 (m, 5H), 1.08-1.01 (m, 4H), 1.00-0.91 (m, 1H), 0.90-0.80 (m, 12H), 0.70 (s, 3H)。 3.在15℃下將NaBH
4
(1.77 g,46.8 mmol)逐份添加至
S-500-6-1_3
(520 mg,1.17 mmol)於THF (5 mL)及MeOH (10 mL)中之溶液中。在15℃下攪拌混合物20 min。混合物用NH
4
Cl (20 mL,飽和水溶液)淬滅且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到
5
(300 mg,不純)及
54
(170 mg,不純)。 4.不純
5
(300 mg,不純)藉由閃蒸塔(0%至12% EtOAc/PE)純化,得到固體。在60℃下使固體溶解於MeCN (50 mL)中且真空濃縮,得到呈固體之
5
(270 mg,52%)。
5: 1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.24 (m, 1H), 3.67-3.54 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.84 (m, 4H), 1.78-1.57 (m, 5H), 1.55-1.38 (m, 12H), 1.38-1.07 (m, 7H), 1.03 (s, 3H), 0.93-0.89 (m, 12H), 0.85 (t,
J
= 7.6 Hz, 3H), 0.70 (s, 3H)。 在7.0 min層析中,
LCMS
Rt=5.587 min,30-90_AB_E,純度96.5%,C
30
H
49
[M+H-2H
2
O]
+
之MS ESI計算值409,實驗值409。
實例 6 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-5- 環丙基 -3- 羥基戊 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (6) 之合成 1.在50℃至55℃下於N
2
下將(2-溴乙基)環丙烷(1.8 g,12 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (641 mg,26.4 mmol)及I
2
(1 mg)於THF (2 mL)中之懸浮液中。在55℃下攪拌1小時之後,用THF (10 mL)稀釋混合物。在0℃下將格林納溶液添加至
S-500-6-1_1
(0.8 g,2.23 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌4小時之後,反應物用NH
4
Cl (20 mL,10%水溶液)淬滅且用EtOAc (30 mL)萃取。有機層經分離且真空濃縮,得到呈固體之混合物(1g,粗產物),該混合物藉由閃蒸塔(0%至25% DCM/EtOAc (1/1)/PE)分離,得到
S-500-6-20
(700 mg,73%,不純)及呈固體之
S-500-6-19
(70 mg,7%,不純)。
2.
在20℃下將DMP (1.38 g,3.26 mmol)及水(1滴)添加至
S-500-6-20
(700 mg,1.63 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌2 h之後,混合物用NaHCO
3
(20 mL,飽和)及Na
2
S
2
O
3
(20 mL,飽和)處理且用EtOAc (50 mL)萃取。有機層用飽和NaHCO
3
/Na
2
S
2
O
3
(2× (20 mL/20 mL))洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
S-500-6-19_3
(700 mg,100%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.35-5.20 (m, 1H), 2.72-2.26 (m, 4H), 2.17-1.87 (m, 3H), 1.82-1.35 (m, 13H), 1.35-1.20 (m, 2H), 1.20-0.91 (m, 12H), 0.85 (t,
J
= 7.2 Hz, 3H), 0.80-0.62 (m, 4H), 0.53-0.33 (m, 2H), 0.12-0.00 (m, 2H)。 3.在15℃下將NaBH
4
(2.46 g,65.1 mmol)逐份添加至
S-500-6-1_3
(700 mg,1.63 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌20 min之後,用NH
4
Cl (20 mL,飽和水溶液)淬滅混合物且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到760 mg呈固體之混合物,其藉由閃蒸塔(0%至35% DCM/EtOAc (1/1)/PE)分離,得到
69
(330 mg,47%)及呈固體之
6
(250 mg,35%,不純)。不純
6
(250 mg)進一步藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到呈固體之
6
(170 mg,23%)。
6: 1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.24 (m, 1H), 3.77-3.66 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.91 (m, 3H), 1.79-1.59 (m, 6H), 1.55-1.21 (m, 14H), 1.21-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.95 (m, 1H), 0.93 (d,
J
= 6.8 Hz, 3H) 0.85 (t,
J
= 7.6 Hz, 3H), 0.70 (s, 3H), 0.68-0.62 (m, 1H), 0.49-0.38 (m, 2H), 0.11-0.02 (m, 2H)。 在2.0 min層析中,
LCMS
Rt=1.380 min,30-90_AB_E,純度100%,C
29
H
47
O [M+H-H
2
O]
+
之MS ESI計算值411,實驗值411。
實例 7 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1R,2S)-1- 羥基 -1-( 吡啶 -3- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (7) 之合成 1.在0℃下於N
2
下向BHT (416 g,1.88 mol)於甲苯(1500 mL)中之溶液逐滴添加三甲基鋁(2 M於甲苯中,469 mL,939 mmol)。在0℃下攪拌混合物30 min且直接用作
MAD
(0.47 M於甲苯中)之溶液而無需進一步純化。在-70℃下於N
2
下向
MAD
(0.47 M於甲苯中,2.01 L,945 mmol)之溶液逐滴添加
N-005 _1
(100 g,315 mmol)於甲苯(800 mL)中之溶液。在-70℃下攪拌混合物30 min。向以上混合物逐滴添加EtMgBr (3 M於乙基醚中,315 mL,945 mmol)。在-70℃下攪拌所得混合物1小時。將反應混合物倒入冰冷卻之檸檬酸水溶液中(1000 mL),用EtOAc (2×600 mL)萃取。合併之有機層用鹽水(500 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠層析(0%至20% EtOAc/PE)純化,得到85 g呈固體之
N-005_2
(78%產量)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.55-2.46 (m, 1H), 2.19-2.12 (m, 1H), 2.11-2.09 (m, 3H), 2.08-1.96 (m, 1H), 1.71-1.48 (m, 10H), 1.47-1.31 (m, 5H), 1.30-1.09 (m, 7H), 1.06-0.94 (m, 2H), 0.92-0.87 (m, 3H), 0.86-0.79 (m, 3H), 0.75-0.64 (m, 1H), 0.60(s, 3H)。
2.
在15℃下於N
2
下向MePPh
3
Br (174 g,0.49 mol)於THF (1000 mL)中之懸浮液中添加t-BuOK (54.9 g,0.49 mol)。在50℃下攪拌30 min之後,在65℃以下逐份添加
N-005_2
(85 g,245 mmol)於THF (800 mL)中之溶液。在50℃下攪拌混合物1小時,用NH
4
Cl (1000 mL)淬滅,用EtOAc (2×900 mL)萃取。有機層經分離,真空濃縮,得到粗產物,該粗產物在50℃下自MeOH/水(1.5 L,1:1)濕磨。冷卻之後過濾混合物且濾餅用MeOH/水(2×500 mL,1:1)洗滌,真空濃縮,得到呈固體之
N-005_3
(75 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.85-4.82 (m, 1H), 4.71-4.68 (m, 1H), 2.06-1.94 (m, 1H), 1.86-1.78 (m, 1H), 1.76-1.71 (m, 4H), 1.70-1.62 (m, 4H), 1.61-1.48 (m, 6H), 1.47-1.30 (m, 3H), 1.29-1.05 (m, 8H), 1.04-0.92 (m, 1H), 0.91-0.82 (m, 6H), 0.76-0.63 (m, 1H), 0.56 (s, 3H)。 3.於N2下向
N-005_3
(75 g,217 mmol)於THF (1800 mL)中之溶液中添加9-BBN二聚體(105 g,434 mmol)。在60℃下攪拌混合物3小時。向反應混合物逐份添加乙醇(124 mL,2.17 mol)及NaOH水溶液(434 mL,5 M,2.17 mmol)。隨後在0℃下逐滴添加H
2
O
2
(217 mL,10 M,2.17 mol)。使混合物升溫至65℃且攪拌1小時且用水(1.5 L)稀釋。用EtOAc (2×800 mL)萃取反應混合物。合併之有機層添加飽和水溶液Na
2
S
2
O
3
(600 mL)且攪拌1小時。反應物藉由碘化鉀澱粉測試紙檢查以確認過量的H
2
O
2
經破壞。隨後有機相用飽和鹽水(2×500 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-005_4
(78 g,粗產物)。在15℃下粗產物
N-005_4
(78 g,不純)自MeOH/H2O=10/1濕磨,得到呈固體之
N-005_4
(70 g,不純)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.68-3.60 (m, 1H), 3.41-3.32 (m, 1H), 1.99-1.92 (m, 1H), 1.88-1.75 (m, 1H), 1.69-1.45 (m, 10H), 1.44-1.29 (m, 4H), 1.28-1.15 (m, 6H), 1.14-0.91 (m, 8H), 0.90-0.79 (m, 7H), 0.67 (s, 3H)。 4.向
N-005_4
(70 g,193 mmol)於DCM (800 mL)中之溶液中添加DMP (122 g,289 mmol)。此後,在15℃下攪拌反應物30 min。反應混合物添加NaHCO
3
飽和水溶液(500 ml)且在15℃下攪拌20 min。添加Na
2
S
2
O
3
飽和水溶液(600 mL)且在15℃下再攪拌混合物1小時。反應物藉由碘化鉀澱粉測試紙檢查以確認過量的DMP經破壞。用DCM (2×400 mL)萃取水相。合併之有機層用NaHCO
3
飽和水溶液(400 mL)及鹽水(400 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-005_5
(70 g,不純)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.58-9.55 (m, 1H), 2.39-2.30 (m, 1H), 1.95-1.78 (m, 2H), 1.69-1.42 (m, 10H), 1.41-1.30 (m, 4H), 1.29-1.14 (m, 5H), 1.13-0.95 (m, 6H), 0.94-0.86 (m, 4H), 0.85-0.81 (m, 3H), 0.69 (m, 4H)。
5.
將i-PrMgCl (2.49 mL,4.98 mmol,2 M於乙醚中)逐滴添加至3-溴吡啶(875 mg,5.54 mmol)於THF (5 mL)中之溶液中。在25℃下攪拌1 h之後,添加
N-8-7_1
(200 mg,0.554 mmol)於THF (5 mL)中之溶液。在25℃下攪拌16小時之後,反應混合物用NH
4
Cl (50 mL,10%水溶液)淬滅且用EtOAc (2×20 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由閃蒸塔(0%至50% EtOAc/DCM)純化,得到呈固體之
N-8-19_1
(100 mg,41%)。
6.N-8-19_1
(100 mg, 0.227 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:50-50% B (A= 0.05% NH
3
/H
2
O,B= MeOH),流速:80 mL/min)分離,得到
7
(峰值1,57 mg,57%)及呈固體之
89
(峰值2,8 mg,8%)。 在3 min層析中,
SFC
峰值1:Rt=1.798 min且峰值2 Rt=1.985 min,AD-H_3UM_4_5_40_4ML (「Chiralpak AD-3 50×4.6mm I.D.,3um 移動相:A:CO
2
,B:異丙醇(0.05% DEA),梯度:在1.4 min內B為5%至40%且保持40% 1.05 min,隨後B為5%持續0.35 min,流速:4 mL/min,管柱溫度:40℃」)。
7: 1 H NMR
(400 MHz, CDCl
3
) δ 8.56-8.52 (m,1H), 8.49-8.45 (m, 1H), 7.68-7.62 (m, 1H), 7.29-7.24 (m, 1H), 5.01-4.95 (m,1H), 2.11-2.01 (m, 1H), 1.96-1.89 (m, 1H), 1.83-1.76 (m, 1H), 1.73-1.63 (m, 4H), 1.59-1.47 (m, 6H),1.43-1.29 (m, 4H), 1.27-1.20 (m, 4H), 1.19-1.06 (m, 4H), 1.03-0.92 (m, 1H), 0.91-0.85 (m, 4H), 0.83 (s, 3H), 0.77-0.73 (m, 3H), 0.70-0.64 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.017 min,10-80AB_E,純度100%,C
29
H
46
NO
2
[M+H]
+
之MS ESI計算值440,實驗值440。 在3 min層析中,
SFC
Rt=1.780 min,AD-H_3UM_4_5_40_4ML,100%de。
實例 8 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (8) 之合成 1.
在25℃以下於N
2
氛圍下向BHT (1.97 kg,8.94 mol)於甲苯(1 L)中之溶液中逐滴添加AlMe
3
(2.14 L,2.0 M於甲苯中,4.28 mol)。在25℃下攪拌所得混合物1小時。在-70℃下添加於DCM (3 L)中之
S-200-INT_2
(794 g,85%重量百分比,2.16 mol)。在-70℃下攪拌混合物1小時。在-70℃下添加MeMgBr (862 mL,3.0 M於二乙醚中,2.59 mol)。在-70℃下攪拌反應混合物10 min。混合物用飽和檸檬酸(3 L)淬滅,用EtOAc (2×2 L)萃取。合併之有機相用鹽水(2 L)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到殘餘物,其在25℃下自MeCN (3 L)濕磨得到呈固體之
S-200-INT_3
(340 g,43%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.34
-
5.26 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.50-2.35 (m, 1H), 2.07
-
1.94 (m, 3H), 1.91
-
1.84 (m, 1H), 1.83
-
1.63 (m, 8H), 1.58
-
1.33 (m, 6H), 1.27
-
1.13 (m, 3H), 1.12 (s, 3H), 1.10
-
1.05 (m, 1H), 1.02 (s, 3H), 1.00
-
0.92 (m, 1H), 0.58 (s, 3H)。
2.
在15℃下於N
2
下向
S-200-INT_3
(149 g,453 mmol)及9-BBN二聚體(127 g,520 mmol)之混合物添加THF (1 L)。在60℃下攪拌反應混合物1小時。將混合物冷卻至15℃。在15℃下添加EtOH (208 g,4.53 mol)。在15℃下逐滴添加NaOH水溶液(906 mL,5 M,4.53 mol)。在15℃下逐滴添加H
2
O
2
(514 g,30%,4.53 mol)。在60℃下攪拌所得混合物1小時。生產一種固體。用乙醇(200 mL)洗滌該固體,得到固體,其在80℃下在回流下用EtOH (2.3 L)及水(2.5 L)連續濕磨,得到呈固體之
15-3b
(131 g,84%)。自乙醇之濾液真空濃縮,得到呈固體之
15-
3b
(30 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
)δ5.35
-
5.24 (m, 1H), 3.67
-
3.61 (m, 1H), 3.42
-
3.33 (m, 1H), 2.50-2.35 (m, 1H), 2.07
-
1.92 (m, 3H), 1.88
-
1.65 (m, 3H), 1.60
-
1.38 (m, 9H), 1.37
-
1.26 (m, 1H), 1.26
-
1.12 (m, 4H), 1.11 (s, 3H), 1.08 (s, 1H), 1.05 (d,
J
= 6.8 Hz, 3H), 1.01 (s, 3H), 1.00
-
0.91 (m, 1H), 0.70 (s, 3H)。 3.將DMP (2.44 g,5.76 mmol)添加至
15-3b
(1 g,2.88 mmol)於DCM (10 mL)中之溶液中。此後,在25℃下攪拌反應物10 min。藉由添加NaHCO
3
飽和水溶液(20 mL)及Na
2
S
2
O
3
飽和水溶液(20 mL)來淬滅反應混合物,用DCM (2×50 mL)萃取。合併之有機層用NaHCO
3
飽和水溶液(3×50 mL)及鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
S-500-2-9_1
(1 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.57 (brs, 1H), 5.35-5.25 (m, 1H), 2.50-2.30 (m, 2H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.75-1.65 (m, 1H), 1.65-1.60 (m, 3H), 1.55-1.50 (m, 2H), 1.50-1.40 (m, 2H), 1.40-1.30 (m, 1H), 1.25-1.20 (m, 2H), 1.20-1.15 (m, 2H), 1.15-1.10 (m, 6H), 1.05-0.95 (m, 5H), 0.90-0.70 (m, 1H), 0.68 (s, 3H)。 4.在60℃下攪拌鎂(641 mg,26.4 mmol)及I
2
(33.5 mg,0.132 mmol)之混合物,且於N
2
下逐滴添加異戊基溴化鎂(2 g,13.2 mmol)於THF (20 mL)中之溶液。此後,在60℃下攪拌反應混合物1小時。反應混合物直接用作異戊基溴化鎂溶液而無需任何純化。在0℃下於N
2
下向
S-500-2-9_1
(1 g,2.90 mmol)於THF (10 mL)中之溶液添加格林納溶液。此後,在25℃下攪拌反應混合物1小時。反應混合物添加NH
4
Cl飽和水溶液(50 mL),用EtOAc (2 × 50 mL)萃取,用鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(EtOAc/ PE=1/4)純化,得到呈固體之不純
S-500-2-9-1A
(560 mg)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.28-5.25 (m, 1H), 3.90-3.80 (m, 0.25H), 3.68-3.58 (m, 0.75H), 2.48-2.36 (m, 1H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.80-1.75 (m, 1H), 1.75-1.52 (m, 6H) 1.52-1.42 (m, 6H), 1.42-1.32 (m, 3H), 1.32-1.22 (m, 3H), 1.22-1.12 (m, 3H), 1.12-1.02 (m, 2H), 1.01 (s, 3H), 1.00-0.92 (m, 1H), 0.92-0.85 (m, 9H), 0.85-0.77 (m, 1H), 0.69 (s, 3H)。 5.
S-500-2-9-1A
(560 mg)藉由SFC (管柱:Chiralcel OD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃)純化,得到呈固體之不純
30
(160 mg)及呈固體之
75
(265 mg,47%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.35-5.30 (m, 1H), 3.70-3.60 (m, 1H), 2.50-2.40 (m, 1H), 2.05-1.90 (m, 4H), 1.85-1.75 (m, 2H), 1.75-1.60 (m, 1H), 1.55-1.45 (m, 8H), 1.45-1.25 (m, 8H), 1.25-1.10 (m, 4H), 1.10-1.05 (m, 2H), 1.02 (s, 3H), 0.99-0.91 (m, 3H), 0.91-0.89 (m, 4H), 0.88 (s, 3H), 0.69 (s, 3H)。 在1.5 min層析中,
LCMS
Rt=1.162 min,5-95 AB,純度99%,C
28
H
45
[M+H-2H
2
O]
+
之MS ESI計算值381,實驗值381。 6.將無水Pd(OH)
2
/C (100 mg)添加至
75
(230 mg,0.551 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液中。在50℃下於H
2
及50 Psi下攪拌反應混合物24 h。此後,HNMR顯示反應完成。反應混合物用濾紙過濾且真空濃縮,得到不純產物。不純產物用MeCN (3 mL)再結晶得到呈灰白色固體之
8
(68 mg,30%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.65-3.55 (m, 1H), 2.00-1.80 (m, 2H), 1.76-1.60 (m, 3H), 1.55-1.48 (m, 3H), 1.48-1.38 (m, 4H), 1.38-1.26 (m, 7H), 1.26-1.23 (m, 4H), 1.23-1.06 (m, 5H), 1.06-1.02 (m, 3H), 1.02-095 (m, 1H), 0.95-0.85 (m, 10H), 0.81 (s, 3H), 0.70-0.60 (m, 4H)。 在1.5 min層析中,
LCMS
Rt=1.171 min,5-95 AB,純度100%。 C
28
H
47
[M+H-2H
2
O]
+
之MS ESI計算值383,實驗值383。
實例 9 : (3S,5R,8R,9S,10S,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 - 17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (9) 之合成 1.將Pd/C (5 g,<1%水)添加至
N-004-022_1
(50 g,151 mmol)於MeOH (100 mL)及THF (100 mL)中之溶液中。隨後在25℃下於30 psi氫氣下氫化溶液48小時。混合物經由矽藻土墊過濾且真空濃縮濾液,得到呈固體之
N-004-022_2
(50 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.65-3.55 (m, 1H), 3.40-3.3 (m, 1H), 2.80-2.60 (m, 1H), 2.40-2.30 (m, 1H), 2.25-2.10 (m, 1H), 2.10-1.95 (m, 3H), 1.80-1.65 (m, 3H), 1.65-1.53 (m, 1H), 1.53-1.40 (m, 4H), 1.40-1.01 (m, 17H), 0.70 (s, 3H)。 2.在60℃下於N
2
下加熱三甲基氧化鋶碘(19.8 g,90.2 mmol)及t-BuOK (10.1 g,90.2 mmol)於DMSO (200 mL)中之經攪拌溶液1小時。將
N-004-022_2
(15 g,45.1 mmol)添加至反應混合物中且在60℃下攪拌10 min。用水(1000 mL)處理反應物且用EtOAc (2×500 mL)萃取。合併之有機相用水(2×500 mL)、鹽水(300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾,真空濃縮,得到呈固體之
N-004-022_3
(15.5 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.18-4.08 (m, 1H), 3.67-3.60 (m, 1H), 3.40-3.30 (m, 1H), 2.70-2.50 (m, 3H), 2.40-2.30 (m, 1H), 2.01-1.50 (m, 14H), 1.40-0.65 (m, 14H), 0.68 (s, 3H)。 3.在25℃下於N
2
下將MeONa (12.0 g,223 mmol)添加至
N-004-022_3
(15.5 g,44.7 mmol)於MeOH (500 mL)中之溶液中。在70℃回流下於N
2
下攪拌混合物16小時。用水(500 mL)處理反應物。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,得到呈固體之
N-004-022_4
(15 g,粗產物)。粗產物
N-004-022_4
(15 g)藉由矽膠層析(PE/EtOAc=10/1至5/1)純化,得到呈固體之
N-004-022_4
(7.4 g,50%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.76-3.73 (m, 1H), 3.64-3.60 (m, 1H), 3.40-3.33 (m, 4H), 3.22-3.16 (m, 2H), 2.01-1.69 (m, 6H), 1.62-1.51 (m, 4H), 1.44-1.31 (m, 13H), 1.10-0.99 (m, 5H), 0.97 (s, 3H), 0.67 (s, 3H)。 4.將DMP (1.56 g,3.69 mmol)添加至
N-004-022_4
(1.4 g,3.69 mmol)於DCM (15 mL)中之溶液中。此後,在25℃下攪拌反應混合物10 min。反應混合物用NaHCO
3
飽和水溶液(20 mL)淬滅直至pH=9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3×10 mL)、NaHCO
3
飽和溶液(10 mL)、鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物用MeCN (10 mL)濕磨,得到呈固體之
N-004-022_5
(700,不純)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.56-9.58 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.40-2.31 (m, 1H), 2.01-1.50 (m, 11H), 1.47-1.01 (m, 16H), 0.97 (s, 3H), 0.70 (s, 3H)。
5.在0℃下於N
2
下向
N-004-022_5
(200 mg,0.531 mmol)、CsF (40.2 mg,0.265 mmol)於THF (5 mL))於中之溶液添加TMSCF
3
(187 mg,1.32 mmol)。在25℃下攪拌混合物1小時。向混合物添加TBAF.3H2O (836 mg,2.65 mmol)。在25℃下攪拌2小時之後,用50% NH
4
Cl (20 mL)淬滅混合物且用EtOAc (2 × 10 mL)萃取。合併之有機相用鹽水(20 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA=10/1)純化,得到
9
(56 mg,24%)及呈白色固體之
71
(30 mg,不純)。
9: 1 H NMR
(400 MHz, CDCl
3
) δ 4.05-3.95 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.00-1.83 (m, 5H), 1.77-1.68 (m, 2H), 1.64-1.47 (m, 8H), 1.43-1.35 (m, 5H), 1.31-1.08 (m, 6H), 1.06-1.00 (m, 3H), 0.97 (s, 3H), 0.70 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.156 min,30-90AB_2min.lcm,純度100%,C
25
H
41
F
3
O
3
[M+Na]
+
之MS ESI計算值469,實驗值469。
實例 10 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1S,2S)-1- 環丙基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (10) 之合成 1.
在0℃下將於THF (5 mL)中之
N-8-7_1
(500 mg,1.38 mmol)添加至環丙基溴化鎂(1 g,13.7 mL,0.5M於THF中)於THF (5 mL)中之溶液中且在25℃下攪拌4小時。混合物添加NH
4
Cl (20 mL,10%水溶液)且用EtOAc (2×30 mL)萃取。有機層經分離且真空濃縮,得到殘餘物。殘餘物藉由矽膠層析純化用PE/EtOAc=1/1溶離,得到呈固體之
N-8-13_1
(140 mg,25%)。 在2.0 min層析中,
LCMS
Rt=1.192 min,30-90AB_2MIN_E,純度99%,C
27
H
43
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 2.將DMP (294 mg,0.694 mmol)添加至
N-8-13_1
(140 mg,0.347 mmol)於DCM (5 mL)中之溶液中。此後,在25℃下攪拌反應混合物1 h。反應混合物用NaHCO
3
飽和水溶液(50 mL)淬滅直至含水層之pH變成約9為止。過濾混合物。分離DCM層且用DCM (100 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3×100 mL)、飽和NaHCO
3
(100 mL)、鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈固體之
N-8-13_2
(140 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.65-2.55 (m, 1H), 1.95-1.90 (m, 2H), 1.50-1.15 (m, 19H), 1.14-0.95 (m, 7H), 0.94-0.80 (m, 12H), 0.69 (s, 3H)。 3.向N-8-13_2 (140 mg,0.347 mmol)於MeOH (1 mL)及THF (1 mL)中之溶液每五分鐘添加五次NaBH4 (1.18 g,17.4 mmol)。在15℃下攪拌混合物30分鐘。混合物用飽和NH
4
Cl (50 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之
10
(26 mg,19%)及呈固體之
90
(12 mg,9%)。
10: 1 H NMR
(400 MHz, CDCl
3
) δ 2.85-2.80 (m, 1H), 2.00-1.95 (m, 1H), 1.90-1.80 (m, 1H), 1.55-1.10 (m, 16H), 1.09-0.80 (m, 17H), 0.70-0.60 (m, 5H), 0.58-0.43 (m, 3H), 0.32-0.34 (m, 1H), 0.13-0.06 (m, 1H)。 在7.0 min層析中,
LCMS
Rt=3.840 min,30-90AB_7MIN_E,純度97%,C
27
H
43
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 在30 min層析中,
HPLC
Rt=13.470 min,70-90AB_1_30MIN.M,純度97%。
實例 11 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (11) 之合成 1.在0℃下將TMSCF
3
(493 mg,3.47mmol)添加至
S-500-6-1_1
(500 mg,1.39 mmol)及CsF (105 mg,695 μmol)於THF (5 mL)中之溶液中。在25℃下攪拌混合物1小時且用TBAF.3H
2
O (1.09 g,3.47 mmol)處理。在25℃下攪拌混合物2小時,用水(100 mL)淬滅且用EtOAc (2 × 50 mL)萃取。合併之有機相用鹽水(100 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA=10/1)純化,得到呈固體之
S-500-6-1_2
(400 mg,67%)。
1 H NMR
(400 MHz, CDCl3) δ 5.33-5.24 (m, 1H), 4.06-4.00 (m, 1H), 2.38-2.35 (m, 1H), 2.08-1.82 (m, 6H), 1.77-1.69 (m, 1H), 1.62-1.20 (m, 13H), 1.16-1.00 (m, 8H), 0.99-0.92 (m, 1H), 0.87-0.83 (m, 4H), 0.74-0.64 (m, 3H)。
2.S-500-6-1_2
(350 mg)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH
3
.H
2
O ETOH,梯度:35%至35%,流速(ml/min):60 mL/min,25℃)純化,得到
81
(峰值1,130 mg,37%)及呈固體之
62
(峰值2,180 mg,52%)。
81: 1 H NMR
(400 MHz, CDCl3) δ 5.34-5.24 (m, 1H), 4.09-4.00 (m, 1H), 2.43-2.33 (m, 1H), 2.14 (d,
J
= 4Hz, 1H), 2.07-1.80 (m, 5H), 1.77-1.55 (m, 5H), 1.53-1.30 (m, 7H), 1.28-1.00 (m, 11H), 1.00-0.91 (m, 1H), 0.85 (t,
J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.220 min,30-90 AB,純度100%,C
25
H
38
F
3
O [M+H-H
2
O]
+
之MS ESI計算值411,實驗值411。 在10 min層析中,
SFC
峰值1:Rt=4.561 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),100%de。
3.
將Pd(OH)
2
(0.2 g,<1%水)添加至
81
(110 mg,0.256 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中。在50℃下於50 psi氫氣下氫化溶液48小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到
56
(38 mg,35%)及呈固體之
11
(42 mg,38%)。
11: 1 H NMR
(400 MHz, CDCl3) δ 4.09-3.99 (m, 1H), 2.11 (d,
J
= 6.0 Hz, 1H), 1.99-1.80 (m, 3H), 1.70-1.55 (m, 6H), 1.53-1.30 (m, 8H), 1.27-0.96 (m, 11H), 0.95-0.81 (m, 7H), 0.70-0.61 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.247 min,30-90 AB,純度100%,C
25
H
40
F
3
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 12 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (12) 之合成 1.將NaBH
4
(2.80 g,82.5 mmol)每五分鐘添加五次至
S-500-6-29_2
(800 mg,1.65 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘。混合物用飽和NH
4
Cl (50 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到
S-500-6-30
(290 mg,36%)及呈固體之
12
(120 mg,45%)。
12: 1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.06-1.95 (m, 3H), 1.77-1.58 (m, 7H), 1.54-1.28 (m, 12H), 1.27-1.06 (m, 11H), 1.03 (s, 3H), 1.00-0.95 (m, 2H), 0.93-0.82 (m, 12H), 0.69 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.708 min,30-90AB_E,純度100%,C
33
H
53
[M+H-2H
2
O]
+
之MS ESI計算值449,實驗值449。
實例 13 : (1S,3R,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (13) 之合成 1.向
N-4-1_4
(27 g,85.8 mmol)於THF (200 mL)中之溶液中添加CsF (25.9 g,171 mmol)及TMSCF
3
(24.3 g,171 mmol)。在10℃下攪拌混合物1小時。向混合物中添加水(10 mL)及TBAF.3H
2
O (30 g)。在30℃下再攪拌混合物2小時。真空濃縮混合物。使殘餘物溶解於EtOAc (500 ml)中,用水(2×500 mL)洗滌,經Na
2
SO
4
乾燥,過濾,真空濃縮且藉由閃蒸塔(DCM/EtOAc(1:1)於PE中,0%至10%)純化,得到
N-4-1_5
(27 g,82%)及呈固體之
N-4-1_5A
(3.5 g,11%)。
N-4-1_5: 1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.12-1.94 (m, 3H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.60 (m, 5H), 1.58-1.48 (m, 2H), 1.45-1.09 (m, 10H), 1.01-0.89 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.56 (s, 3H)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.09-1.99 (m, 1H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.52 (m, 9H), 1.45-1.06 (m, 10H), 1.00-1.81 (m, 2H), 0.79 (s, 3H), 0.56 (s, 3H)。 2.向
N-4-1_5
(23 g,59.8 mmol)於THF (250 mL)中之溶液中添加9-BBN二聚體(29 g,119 mmol),在40℃下於N
2
下攪拌16小時。向反應混合物添加乙醇(34.3 mL,598 mmol)及NaOH (119 mL,5 M,598 mmol)。混合物變得澄清。在25℃下逐滴添加H
2
O
2
(59.8 mL,10 M,598 mmol)且使內部溫度升至回流(70℃)。添加之後將混合物冷卻至30℃。向該混合物添加Na
2
SO
3
(100 mL,20%水溶液)。有機層經分離且倒入水(800 mL)中。形成固體。過濾混合物且固體用水洗滌,真空乾燥且用MeCN (250 mL)濕磨,得到固體。該固體在60℃下自MeOH/水(250 mL/12.5 mL)濕磨且在冷卻至15℃之後過濾。真空乾燥固體,得到呈固體之
N-4-1_6
(16.4 g,68%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.69-3.60 (m, 1H), 3.39-3.29 (m, 1H), 2.09-2.01 (m, 1H), 1.99-1.92 (m, 1H), 1.87-1.75 (m, 2H), 1.72-1.43 (m, 7H), 1.42-1.07 (m, 11H), 1.03 (d,
J
= 6.8 Hz, 3H), 1.01-0.86 (m, 3H), 0.85 (s, 3H), 0.73-0.69 (m, 1H), 0.67 (s, 3H)。
3.
向
N-4-1_6
(5 g,12.4 mmol)於DCM (200 mL)中之懸浮液中添加水(223 mg,12.4 mmol)及DMP (10.5 g,24.8 mmol)。在15℃下攪拌混合物15 min。混合物用NaHCO
3
/Na
2
S
2
O
3
(200 mL/200 mL,飽和)洗滌兩次,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-4-1_7
(4.5 g,90%)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.60-9.51 (m, 1H), 2.40-2.30 (m, 1H), 2.12-1.78 (m, 5H), 1.75-1.59 (m, 4H), 1.57-1.15 (m, 11H), 1.14-0.84 (m, 8H), 0.78-0.63 (m, 5H)。 4.在0℃下將MeLi (7.75 mL,1.6 M,12.4 mmol)添加至
N-4-1_7
(1 g,2.49 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌混合物1 h。向混合物添加NH
4
Cl (10%,20 mL)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈膠狀之混合物(1 g)。混合物
N-3-2_1
(1 g)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到
91
(450 mg)及
22
(460 mg)及130 mg混合物。
91
(450 mg)自MeCN (10 mL)再結晶得到呈固體之
91
(50 mg),
22
(460 mg)自MeCN (10 mL)再結晶兩次得到呈固體之
22
(50 mg)。
91: 1 H NMR
(400 MHz, CDCl
3
) δ 3.98-3.88 (m, 1H), 2.11-2.02 (m, 1H), 2.00 (s, 1H), 1.98-1.88 (m, 2H), 1.85-1.79 (m, 1H), 1.73-1.58 (m, 4H), 1.52-1.20 (m, 11H), 1.19-1.11 (m, 4H), 1.10-1.00 (m, 3H), 0.97-0.89 (m, 4H), 0.85 (s, 3H), 0.75-0.68 (m, 1H), 0.66 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.155 min,30-90_AB_E,純度100%,C
24
H
38
F
3
O [M+H-H
2
O]
+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,
HPLC
Rt=5.23 min,30-90_AB_E,純度98.88%,d.e. 100%。
22: 1 H NMR
(400 MHz, CDCl
3
) δ 3.97-3.82 (m, 1H), 2.10-1.92 (m, 3H), 1.85-1.78 (m, 1H), 1.77-1.60 (m, 5H), 1.59-1.06 (m, 13H), 1.05-0.81 (m, 12H), 0.74-0.62 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.136 min,30-90_AB_E,純度100%,C
24
H
38
F
3
O [M+H-H
2
O]
+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,
HPLC
Rt=5.05 min,30-90_AB_E,純度100%,d.e. 100%。
5.
向
N-3-2_1
(0.88 g,2.11 mmol)於DCM (20 mL)中之溶液中添加水(2滴)及DMP (1.78 g,4.22 mmol)。在25℃下攪拌混合物30 min。混合物用NaHCO
3
/Na
2
S
2
O
3
(30 mL/30 mL,飽和)洗滌兩次,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-3-2_2
(0.85 g,97%)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.53-2.42 (m, 1H), 2.13-2.00 (m, 4H), 1.97-1.78 (m, 2H), 1.75-1.45 (m, 9H), 1.43-1.13 (m, 9H), 1.11 (d,
J
= 8.4 Hz, 3H), 1.07-1.00 (m, 1H), 0.98-0.88 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.67 (s, 3H)。 6.在0℃下向
N-3-2_2
(0.85 g,2.05 mmol)於DCM (5 mL)中之溶液中添加咪唑(279 mg,4.10 mmol)及TMSCl (333 mg,3.07 mmol)。在0℃下攪拌混合物0.5 h。添加混合物,藉由NaHCO
3
(20 mL,飽和)淬滅且用PE (15 mL)萃取。有機層經分離,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-3-2_2A
(0.98 g,98%)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.53-2.42 (m, 1H), 2.13-2.03 (m, 4H), 1.97-1.78 (m, 2H), 1.75-1.31 (m, 11H), 1.31-1.00 (m, 10H), 1.00-0.88 (m, 1H), 0.83 (s, 3H), 0.75-0.61 (m, 4H), 0.15 (s, 9H)。 7.在-70℃下將BuLi (0.384 mL,2.5 M,0.615 mmol)添加至於THF (0.5 mL)中之i-Pr
2
NH (62.2 mg,0.615 mmol)中且在0℃下攪拌10 min。在-70℃下添加於THF (1 mL)中之
N-3-2_2A
(0.2 g,0.41 mmol)且在-70℃下攪拌30 min。在-70℃下添加苯甲醛(91.3 mg,0.861 mmol)於THF (0.5 mL)中之溶液且在-70℃下攪拌15 min。將NH
4
Cl (1 mL,飽和水溶液)添加至混合物且用EtOAc (10 mL)萃取。有機層經分離,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈油狀物之
N-3-2_3C
(250 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.46-7.30 (m, 5H), 5.70-5.55 (m, 1H), 3.60-3.25 (m, 1H), 2.90-2.70 (m, 2H), 2.55-2.41 (m, 1H), 2.16-2.00 (m, 2H), 1.96-1.75 (m, 3H), 1.50-1.15 (m, 9H), 1.13-1.05 (m, 4H), 1.05-0.88 (m, 4H), 0.87-0.80 (m, 5H), 0.73-0.62 (m, 5H), 0.15 (s, 9H)。
8.
在0℃下將LiAlH
4
(159 mg,4.20 mmol)添加至
N-3-2_3C
(250 mg,0.421 mmol)於THF (10 mL)中之溶液中。在0℃下攪拌混合物5 min。將水(0.16 mL)、NaOH (0.16 mL,15%水溶液)及水(0.48 mL)以本文中所撰寫之順序添加至混合物。過濾混合物且用THF (30 mL)洗滌固體。真空濃縮合併之濾液,得到呈固體之
N-3-2_4C
(250 mg,100%)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.42-7.28 (m, 5H), 5.70-5.30 (m, 1H), 4.15-3.65 (m, 1H), 2.18-1.55 (m, 9H), 1.53-1.00 (m, 15H), 1.00-0.75 (m, 9H), 0.75-0.50 (m, 4H), 0.15 (s, 9H)。 9.將TBAF (219 mg,0.84 mmol)添加至
N-3-2_4C
(250 mg,0.42 mmol)於THF (2mL)中之溶液中。在25℃下攪拌混合物3 h。真空濃縮混合物。使殘餘物溶解於EtOAc (10 mL)中且用水(3×10 mL)洗滌,藉由閃蒸塔(10%至25% EtOAc/PE)純化,得到呈固體之
N-3-2_5C
(150 mg,68%)。
10.
混合物
N-3-2_5C
(150 mg)藉由SFC (儀器:MG-II;管柱:IC (250mm×30mm,10 um);條件:0.1%NH
3
H
2
O MeOH;開始B:30%;結束B:30%;流速(mL/min):60;注射:300)分離,得到不純
79
(35 mg,不純)、
31
與
25
之混合物(55 mg)及
13
(28 mg,不純)。 不純
79
(35 mg)藉由閃蒸塔(10%至30% EtOAc/PE)純化,真空濃縮溶離劑。使殘餘物溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到呈固體之
79
(12 mg)。
25
及
13
(55 mg)藉由SFC(儀器:MG-II;管柱:AD (250mm×30mm,5um);條件:0.1%NH
3
H
2
O MeOH;開始B:35%;結束B:35%;流速(mL/min):60;注射70)分離。溶離劑中之每一者分別真空濃縮,溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到均呈固體之
25
(28 mg)及
13
(7 mg)。 不純
31
(28 mg)藉由SFC (儀器:SFC 17;管柱:AS (250mm×30mm,5um),條件:0.1%NH
3
H
2
O EtOH;開始B:30%;結束B:30%;流速(mL/min):50;注射:60)純化,得到固體。使殘餘物溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到呈固體之
31
(9 mg)。
四種異構體之 SFC :在
10 min層析中,峰值1:Rt=1.501 min,峰值2:Rt=1.730 min及峰值3:Rt=1.943 min,IC-3_MeOH(DEA)_40_2.5ML (「管柱:ChiralPak IC-3 150×4.6mm I.D.,3um;梯度:40%甲醇(0.05% DEA)/CO
2
;流速:2.5mL/min;管柱溫度:40℃」)。
25 及 13 之 SFC :在
8 min層析中,峰值1:Rt=4.411 min及峰值2:Rt=4.920 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN (「管柱:Chiralpak AD-3 100×4.6mm I.D.,3um;移動相:A:CO
2
,B:甲醇(0.05% DEA);梯度:在4.5min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續1 min;流速:2.8mL/min;管柱溫度:40℃」)。
79: 1 H NMR
(400 MHz, CDCl
3
) δ 7.43-7.28 (m, 5H), 5.05-4.94 (m, 1H), 4.04-3.91 (m, 1H), 2.51 (brs, 1H), 2.07-1.78 (m, 6H), 1.70-1.61 (m, 4H), 1.51-1.41 (m, 3H), 1.39-1.12 (m, 11H), 1.05-0.98 (m, 2H), 0.91-0.81 (m, 7H), 0.71-0.60 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.298 min,10-80AB_2MIN_E,純度96.7%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在10 min層析中,
SFC
Rt=1.483 min,IC-3_MeOH(DEA)_40_2.5ML,100%de。
25: 1 H NMR
(400 MHz, CDCl
3
) δ 7.42-7.28 (m, 5H), 4.97-4.81 (m, 1H), 4.12-3.92 (m, 1H), 3.23 (brs, 1H), 2.69 (brs, 1H), 2.10-1.88 (m, 3H), 1.82-1.62 (m, 7H), 1.48-1.18 (m, 10H), 1.10-0.88 (m, 8H), 0.87-0.78 (m, 4H), 0.70-0.58 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.319 min,10-80AB_2MIN_E,純度97.0%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在5 min層析中,
SFC
Rt=1.718 min,IC-3_MeOH(DEA)_40_2.5ML,98.26%de。 在8 min層析中,
SFC
Rt=4.367 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,100%de。
31: 1 H NMR
(400 MHz, CDCl
3
) δ 7.45-7.28 (m, 5H), 5.02-4.81 (m, 1H), 4.18-3.98 (m, 1H), 3.35 (brs, 1H), 2.47 (brs, 1H), 2.15-1.72 (m, 8H), 1.53-1.31 (m, 8H), 1.30-1.03 (m, 8H), 0.99-0.89 (m, 4H), 0.89-0.78 (m, 4H), 0.75-0.60 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.327 min,10-80AB_2MIN_E,純度100%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在10 min層析中,
SFC
Rt=1.929 min,IC-3_MeOH(DEA)_40_2.5ML,98.4%de。
13: 1 H NMR
(400 MHz, CDCl
3
) δ 7.40-7.28 (m, 5H), 5.12-5.07 (m, 1H), 3.95-3.88 (m, 1H), 2.76 (brs, 1H), 2.08-1.78 (m, 6H), 1.75-1.60 (m, 5H), 1.51-1.38 (m, 4H), 1.36-1.09 (m, 9H), 1.00-0.89 (m, 6H), 0.83 (s, 3H), 0.71-0.64 (m, 1H), 0.63 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.309 min,10-80AB_2MIN_E,純度100%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在5 min層析中,
SFC
Rt=1.683 min,IC-3_MeOH(DEA)_40_2.5ML,98.94%de。 在8 min層析中,
SFC
Rt=4.785 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,94.03%de。
實例 14 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R,E)-3- 羥基 -5- 苯基戊 -4- 烯 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (14) 之合成 1.在-70℃下將BuLi (0.384 mL,2.5 M,0.615 mmol)添加至於THF (0.5 mL)中之i-Pr
2
NH (62.2 mg,0.615 mmol)中且在0℃下攪拌10 min。在-70℃下添加於THF (1 mL)中之N-3-2_2A (0.2 g,0.41 mmol)且在-70℃下攪拌1 h。在-70℃下添加苯甲醛(91.3 mg,0.861 mmol)於THF (0.5 mL)中之溶液且在20℃下攪拌4 h。將NH
4
Cl (1 mL,飽和水溶液)添加至混合物且用EtOAc (10 mL)萃取。有機層經分離,經Na
2
SO
4
乾燥,過濾且真空濃縮,藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之
N-3-2_3D
(150 mg,64%)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.64-7.55 (m, 3H), 7.43-7.39 (m, 3H), 6.79 (d,
J
= 16.0 Hz, 1H), 2.86-2.73 (m, 1H), 2.15-2.08 (m, 1H), 2.00-1.90 (m, 1H), 1.88-1.80 (m, 1H), 1.72-1.59 (m, 5H), 1.53-1.22 (m, 9H), 1.21-1.03 (m, 7H), 0.99-0.88 (m, 1H), 0.84 (s, 3H), 0.75-0.61 (m, 4H), 0.15 (s, 9H)。
2.
將TBAF (135 mg,0.52 mmol)添加至
N-3-2_3D
(150 mg,0.26 mmol)於THF (1 mL)中之溶液中。在20℃下攪拌混合物20 h。向混合物添加EtOAc (5 mL)。混合物用水(2×5 mL)、鹽水(5 mL)洗滌,真空濃縮,得到呈固體之
N-003-005_1
(140 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.64-7.53 (m, 3H), 7.43-7.35 (m, 3H), 6.79 (d,
J
= 16.0 Hz, 1H), 2.88-2.73 (m, 1H), 2.13-1.90 (m, 3H), 1.88-1.78 (m, 1H), 1.77-1.90 (m, 5H), 1.60-1.22 (m, 8H), 1.21-0.88 (m, 9H), 0.86 (s, 3H), 0.75-0.61 (m, 4H)。
3.
在20℃下向
N-003-005_1
(140 mg,0.278 mmol)於THF (2 mL)及MeOH (1 mL)中之溶液逐份添加NaBH
4
(419 mg,11.1 mmol)。在20℃下攪拌混合物10 min。反應物用水(20 mL)及NH
4
Cl (20mL,飽和)淬滅。用EtOAc (50 mL)萃取混合物。真空濃縮有機層且藉由prep-TLC (PE/EtOAc=4/1)純化,得到均呈固體之
N-003-005
(50 mg,不純)及
N-003-006
(50 mg)。 使14 (50 mg)溶解於MeCN (20 mL)中且真空濃縮,得到呈固體之14 (29 mg)。
14: 1 H NMR
(400 MHz, CDCl
3
) δ 7.44-7.38 (m, 2H), 7.37-7.29 (m, 2H), 7.25-7.18 (m, 1H), 6.59 (d,
J
= 16.0 Hz, 1H), 6.24 (dd,
J
= 7.2, 16.0 Hz, 1H), 4.40-4.30 (m, 1H), 2.08-1.92 (m, 3H), 1.89-1.77 (m, 3H), 1.68-1.60 (m, 3H), 1.50-1.08 (m, 13H), 1.03-0.82 (m, 9H), 0.72-0.62 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.236 min,30-90AB_2MIN_E,純度99.0%,C
31
H
42
F
3
O [M+H-H
2
O]
+
之MS ESI計算值487,實驗值487。 在8 min層析中,
HPLC
Rt=5.89 min,30-90_AB_1.2ml,98.1% d.e. (220 nm)
實例 15 : 合成 (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (15) 1.將Pd(OH)
2
/C (100 mg)添加至
44
(100 mg)於MeOH/THF (2 mL/2 mL)中之溶液中。在50℃下於H
2
(50 psi)下攪拌混合物20 h。過濾混合物。濃縮濾液得到100 mg固體。NMR顯示9%
54
保留。在相同條件下再氫化不純樣本3次。過濾混合物。濃縮濾液且藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到呈固體之
5
(7 mg)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.66-3.48 (m, 1H), 2.00-1.55 (m, 9H), 1.50-1.22 (m, 15H), 1.19-1.03 (m, 8H), 0.96 (s, 3H), 0.91-0.81 (m, 15H), 0.67 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.492 min,30-90_AB_E,純度100%,C
30
H
51
[M+H-2H
2
O]
+
之MS ESI計算值411,實驗值411。
實例 16 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (16) 之合成 1.將Pd(OH)
2
(0.2 g,<1%水)添加至
62
(160 mg,0.373 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中。在50℃下於50 psi氫氣下氫化溶液16小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到呈固體之
44
(27 mg,17%)及
16
(117 mg,73%)。
16: 1 H NMR
(400 MHz, CDCl3) δ 4.04-3.96 (m, 1H), 1.98-1.83 (m, 4H), 1.69-1.59 (m, 3H), 1.56-1.20 (m, 13H), 1.17-0.95 (m, 8H), 0.91-0.83 (m, 8H), 0.70-0.62 (m, 4H)。 在2.0 min層析中,
LCMS
Rt = 1.240 min,30-90 AB,純度100%,C
25
H
40
F
3
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 17 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (17) 之合成 1.將Pd(OH)
2
/C (100 mg)添加至
5
(250 mg)於MeOH/THF (2 mL/2 mL)中之溶液中。在50℃下於H
2
(50 psi)下攪拌混合物20 小時。過濾混合物。濃縮濾液得到250 mg固體。NMR顯示70%
5
保留。在相同條件下再氫化不純樣本3次。過濾混合物。濃縮濾液且藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到呈固體之
17
(3 mg)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.63-3.50 (m, 1H), 1.98-1.55 (m, 8H), 1.49-1.37 (m, 8H), 1.35-1.21 (m, 8H), 1.19-1.01 (m, 8H), 0.97 (s, 3H), 0.91-0.82 (m, 15H), 0.66 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.529 min,30-90_AB_E,純度95.6%,C
30
H
51
[M+H-2H
2
O]
+
之MS ESI計算值411,實驗值411。
實例 18 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4-( 四氫 -2H- 哌喃 -4- 基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (18) 之合成 1.
在20℃下於氮氣下向Me
3
SI (8.44 g,41.4 mmol)於無水THF (50 mL)中之懸浮液中添加t-BuOK (4.64 g,41.4 mmol)。在20℃下攪拌混合物1小時,且添加
N-005_5
(5 g,13.8 mmol)。使所得混合物升溫至45℃且攪拌4小時。將反應混合物冷卻至室溫,用水(50 mL)淬滅且用EtOAc (2×50 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之
N-005_001
(2.7 g,52%)。 在2.0 min層析中,
LCMS
Rt=1.324 min,30-90AB_E,純度92%,C
25
H
41
O [M+H-H
2
O]
+
之MS ESI計算值357,實驗值357。 2.在60℃下於N
2
下將4-氯四氫-2H-哌喃(1 g,8.29 mmol)於無水THF (8 mL)中之溶液逐滴添加至Mg (401 mg,16.5 mmol)及I
2
(105 mg,0.414 mmol)於無水THF (2 mL)中之混合物。在60℃下攪拌混合物10 min。溫度升至66℃。再攪拌反應混合物30 min,冷卻至室溫,其直接用作(四氫-2H-哌喃-4-基)氯化鎂(0.83 M於THF中)之溶液。 3.在20℃下於氮氣下將(四氫-2H-哌喃-4-基)-氯化鎂(0.83 M於THF中,6.38 mL,5.30 mmol)之溶液逐滴添加至
N-005_001
(400 mg,1.06 mmol)及CuI (20.1 mg,0.106 mmol)於無水THF (10 mL)中之懸浮液中。在20℃下攪拌混合物18小時。反應混合物用水(10 mL)及飽和NH
4
Cl (10 mL)淬滅,用EtOAc (2×15 mL)萃取。合併之有機層用鹽水(15 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮,得到呈固體之
N-6-9/10
(760 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.01-3.92 (m, 3H), 3.83-3.62 (m, 1H), 3.42-3.32 (m, 3H), 1.97-1.87 (m, 1H), 1.68-1.58 (m, 7H), 1.57-1.45 (m, 6H), 1.43-1.29 (m, 8H), 1.24-0.95 (m, 10H), 0.91-0.79 (m, 9H), 0.73-0.56 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.332 min,10-80AB,純度93%,C
30
H
52
NaO
3
[M+Na]
+
之MS ESI計算值483,實驗值483。 4.將BzCl (691 mg,4.92 mmol)及DMAP (20 mg,0.164 mmol)添加至
N-6-9/10
(760 mg,1.64 mmol)於吡啶(10 mL)中之溶液中。在20℃下攪拌混合物2小時。反應混合物用水(15 mL)淬滅,用EtOAc (2×20 mL)萃取。合併之有機層用10% HCl水溶液(2×20 mL)、飽和NaHCO
3
(40 mL)及鹽水(20 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之
N-6-9_1
(400 mg,43%)。
1 H NMR
(400 MHz, CDCl
3
) δ 8.08-8.01 (m, 1H), 8.08-8.01 (m, 1H), 7.61-7.42 (m, 3H), 5.42-5.29 (m, 1H), 3.99-3.85 (m, 2H), 3.41-3.24 (m, 2H), 2.06-1.65 (m, 5H), 1.65-1.57 (m, 5H), 1.54-1.42 (m, 6H), 1.42-1.14 (m, 11H), 1.14-0.90 (m, 8H), 0.89-0.77 (m, 7H), 0.69-0.51 (m, 4H)。
N-6-9_1
(400 mg,0.708 mmol)經分離且藉由SFC (管柱:C2 250mm×30mm,10 um,梯度:45-45% B (A= 0.1%NH
3
/H
2
O,B= MeOH ),流速:60 mL/min)純化,得到呈固體之
N-6-9_2
(峰值1,Rt=3.926 min,80 mg,20%)及呈固體之
N-6-10_1
(峰值2,Rt=4.893 min,180 mg,45%)。
N-6-9_2: 1 H NMR
(400 MHz, CDCl
3
) δ 8.06-8.00 (m, 2H), 7.60-7.53 (m, 1H), 7.49-7.42 (m, 2H), 5.35-5.28 (m, 1H), 3.98-3.91 (m, 2H), 3.41-3.31 (m, 2H), 1.88-1.67 (m, 5H), 1.66-1.57 (m, 4H), 1.54-1.36 (m, 10H), 1.35-1.16 (m, 8H), 1.08-0.88 (m, 8H), 0.88-0.82 (m, 4H), 0.80 (s, 3H), 0.64 (s,3H), 0.61-0.54 (m, 1H)。 在2.0 min層析中,
LCMS
Rt=1.540 min,30-90AB,純度96%,C
30
H
49
O [M-BzOH-H
2
O+H]
+
之MS ESI計算值425,實驗值425。 在8 min層析中,
SFC
Rt=3.789 min,管柱:Lux Cellulose-2 150×4.6mm I.D.,3 μm;移動相:40% 甲醇(0.05% DEA)/CO2;流速:2.5mL/min;管柱溫度:40℃,97%de。
N-6-10_1: 1 H NMR
(400 MHz, CDCl
3
) δ 8.06-8.01 (m, 2H), 7.61-7.53 (m, 1H), 7.49-7.42 (m, 2H), 5.41-5.33 (m, 1H), 3.98-3.86 (m, 2H), 3.40-3.27 (m, 2H), 2.05-1.91 (m, 2H), 1.84-1.72 (m, 2H), 1.66-1.59 (m, 3H), 1.55-1.38 (m, 9H), 1.37-1.16 (m, 11H), 1.13-1.00 (m, 6H), 1.00-0.90 (m, 2H), 0.89-0.79 (m, 7H), 0.67 (s, 3H), 0.63-0.54 (m, 1H)。 在2.0 min層析中,
LCMS
Rt=1.507 min,30-90AB,純度97%,C
30
H
49
O [M-BzOH-H
2
O+H]
+
之MS ESI計算值425,實驗值425。 在8 min層析中,
SFC
Rt=4.699 min,管柱:Lux Cellulose-2 150×4.6mm I.D.,3 μm;移動相:40% 甲醇(0.05% DEA)/CO2;流速:2.5mL/min;管柱溫度:40℃,97%de。 5.將水(1 mL)及KOH (78.5 mg,1.40 mmol)添加至
N-6-9_2
(80 mg,0.141 mmol)於THF (2 mL)及甲醇(1 mL)中之溶液中。在50℃下攪拌混合物18小時。將反應混合物冷卻,用水(5 mL)稀釋,用10% HCl (0.2 mL)酸化且用EtOAc (3 × 5 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(10%至30% EtOAc/PE)純化,得到呈固體之
18
(13 mg,20%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.05-3.89 (m, 3H), 3.45-3.34 (m, 2H), 1.94-1.87 (m, 1H), 1.86-1.76 (m, 1H), 1.72-1.59 (m, 6H), 1.54-1.45 (m, 4H), 1.44-1.28 (m, 9H), 1.28-1.15 (m, 7H), 1.13-0.92 (m, 5H), 0.91-0.85 (m, 4H), 0.84-0.79 (m, 6H), 0.70-0.62 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.213 min,30-90AB,純度100%。 C
30
H
49
O [M-2H
2
O+H]
+
之MS ESI計算值425,實驗值425。
實例 19 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1- 苯基丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (19) 之合成 1.在0℃下將
N-8-7_1
(300 mg,0.832 mmol)於THF (5 mL)中之溶液添加至PhMgBr (1.38 mL,3 M於乙醚中,4.15 mmol)於THF (10 mL)中之溶液中,隨後在0℃下攪拌反應混合物3小時。接下來,在25℃下攪拌反應混合物5小時。在0℃下用水(10 mL)淬滅反應混合物。過濾溶液且用EtOAc (10 mL)洗滌濾餅。用EtOAc (3×15 mL)萃取水相。合併之有機相用飽和鹽水(2×10 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至1/1)純化,得到呈固體之粗產物(200 mg)。粗產物藉由SFC (管柱:AD (250mm×30mm,5um),梯度:25-25% B (A= 0.1%NH3/H2O,B= EtOH),流速:60 mL/min)純化,得到
55
(峰值2,55 mg, 15%)及呈固體之
19
(峰值1,21 mg,6%)。
19: 1 H NMR
(400MHz, CDCl
3
) δ 7.40-7.20 (m, 5H), 4.85- 4.80 (m, 1H), 2.10-1.60 (m, 5H), 1.55-1.05 (m, 17H), 0.95-0.75 (m, 14H), 0.71 (s, 3H), 0.60-0.50 (m, 1H)。 在2.0 min層析中,
LCMS
Rt=1.208 min,30-90AB_2 min.,純度100%,C
30
H
43
[M-2H
2
O+H]
+
之MS ESI計算值403,實驗值403。 在3 min層析中,
SFC
Rt=1.047 min,OJ_3_EtOH_DEA_5_40_25ML,100%de。
實例 20 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (20) 之合成 1.
向
12
(100 mg,0.206 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)
2
(150 mg,無水)。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
87
(12 mg,12%)及
20
(11 mg,11%)。
20: 1 H NMR
(400 MHz, CDCl
3
) δ 3.83-3.75 (m, 1H), 1.99-1.92 (m, 1H), 1.71-1.57 (m, 8H), 1.52-1.43 (m, 3H), 1.41-1.29 (m, 8H), 1.27-1.13 (m, 11H), 1.12-1.04 (m, 4H), 1.03-0.94 (m, 3H), 0.91-0.86 (m, 12H), 0.82 (s, 3H), 0.68-0.59 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.748 min,30-90AB_E,純度100%,C
33
H
55
[M+H-H
2
O]
+
之MS ESI計算值451,實驗值451。
實例 21 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (21) 之合成 1.在25℃下於N
2
下將EtMgBr (0.553 mL,3 M於乙醚中,1.66 mmol)逐滴添加至
N-8_1
(250 mg,0.8320 mmol)於THF (3 mL)中之溶液中。在25℃下攪拌混合物1小時,用飽和NH
4
Cl (10 mL)淬滅且用EtOAc (3×15 mL)萃取。有機層用鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,過濾,真空濃縮,得到粗產物
N-8-24_1
,其藉由閃蒸塔(0%至15% EtOAc/PE)純化得到呈固體之
21
(130 mg,不純)。不純
N-8-24
(130 mg,0.3327 mmol)在85℃下自MeCN (3 mL)再結晶,得到呈固體之純
21
(111 mg,86%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.62-3.50 (m, 1H), 2.02-1.81 (m, 2H), 1.72-1.59 (m, 3H), 1.56-1.46 (m, 4H), 1.45-1.17 (m, 12H), 1.16-1.00 (m, 5H), 0.99-0.85 (m, 11H), 0.84-0.78 (m, 4H), 0.66 (s, 4H)。 在10 min層析中,
HPLC
Rt=5.73 min,30-90_AB_1.2 mL_E,純度100%。 C
26
H
43
[M+H-2H
2
O]
+
之MS ESI計算值355,實驗值355。
實例 22 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥丁 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (22) 之合成 1.在0℃下將MeLi (7.75 mL,1.6 M,12.4 mmol)添加至
N-4-1_7
(1 g,2.49 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌混合物1小時。向混合物添加NH
4
Cl (10%,20 mL)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈膠狀之混合物(1 g)。混合物(1 g)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到
91
(450 mg)及
22
(460 mg)及130 mg之混合物。
91
(450 mg)自MeCN (10 mL)再結晶得到呈固體之
91
(50 mg)。
22
(460 mg)自MeCN (10 mL)再結晶兩次得到呈固體之
22
(50 mg)。
22: 1 H NMR
(400 MHz, CDCl
3
) δ 3.97-3.82 (m, 1H), 2.10-1.92 (m, 3H), 1.85-1.78 (m, 1H), 1.77-1.60 (m, 5H), 1.59-1.06 (m, 13H), 1.05-0.81 (m, 12H), 0.74-0.62 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.136 min,30-90_AB_E,純度100%,C
24
H
38
F
3
O [M+H-H
2
O]
+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,
HPLC
Rt=5.05 min,30-90_AB_E,純度100%,d.e. 100%。
實例 23 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (23) 之合成 1.
在0℃下向
S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液緩慢地添加丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)。添加之後,在15℃下攪拌混合物1小時。用飽和NH
4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
72
(500 mg,56%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,
LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C
27
H
43
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 2.向
72
(150 mg,0.372 mmol)於MeOH (20 mL)中之溶液中添加Pd(OH)
2
(300 mg,無水)。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之
23
(9 mg,6%)及
38
(43 mg,29%)。
23: 1 H NMR
(400 MHz, CDCl
3
) δ 3.71-3.62 (m, 1H), 2.01-1.83 (m, 3H), 1.82-1.72 (m, 1H), 1.69-1.57 (m, 3H), 1.51-1.37 (m, 9H), 1.36-1.22 (m, 9H), 1.20-1.00 (m, 8H), 0.97 (s, 3H), 0.94-0.87 (m, 8H), 0.66 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.440 min,30-90AB_E,純度98.8%,C
27
H
45
[M+H-2H
2
O]
+
之MS ESI計算值369,實驗值369。
實例 24 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (24) 之合成 1.在60℃下向Mg (4.35 g,179 mmol)及I
2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。混合物用THF (10 mL)稀釋並直接使用。在0℃下於N
2
下向
S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。將NH
4
Cl (20 mL,飽和水溶液)添加至混合物。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH
3
CN (10 mL)再結晶,得到呈固體之
77
(255 mg,21%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.306 min,30-90 AB,純度100%,C
29
H
49
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。 2.在25℃下於N
2
下將苯甲酸(508 mg,4.16 mmol)及三苯基膦(1.63 g,6.24 mmol)添加至
77
(900 mg,2.08 mmol)於THF (30 mL)中之溶液中。在25℃下攪拌混合物20 min。在0℃下於N
2
下添加DIAD (1.26 g,6.24 mmol)。在0℃下攪拌混合物20 min,升溫至25℃且在25℃下攪拌16小時。反應混合物用水(60 mL)淬滅且用MTBE (3×30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na
2
SO
4
乾燥,過濾,真空濃縮,得到粗產物,該粗產物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈油狀物之不純產物
S-500-2-15_1
(900 mg),其直接用於下一步驟。
3.
將NaOH溶液(974 mg於6 mL H
2
O中,16.8 mmol)添加至
S-500-2-15_1
(900 mg,1.68 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在50℃下加熱混合物16小時。反應混合物用飽和NH
4
Cl (60 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na
2
SO
4
乾燥,過濾濃縮,且藉由combi-flash (0%至15% EtOAc/PE)純化,得到210 mg固體,其藉由SFC (管柱:AD (250mm×30mm,5um),梯度:35-35% B (A= 0.1%NH
3
/H
2
O,B= MeOH),流速:80 mL/min)純化,得到呈固體之
52
(150 mg,68%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.30-5.26 (m, 1H), 3.64-3.58 (m, 1H), 2.40-2.30 (m, 1H), 2.02-1.92 (m, 3H), 1.80-1.58 (m, 7H), 1.56-1.31 (m, 9H), 1.30-1.05 (m, 8H), 1.03 (s, 3H), 1.02-0.96 (m, 2H), 0.95-0.86 (m, 9H), 0.85-0.80 (m, 3H), 0.69 (s, 3H)。 在2 min層析中,
LCMS
t
R
=1.335 min,30-90AB_ELSD,純度100.0%,C
29
H
47
[M+H-2H
2
O]
+
之MS ESI計算值395,實驗值395。 4.向
52
(50 mg,0.116 mmol)於MeOH (10 mL)中之溶液添加Pd(OH)
2
(200 mg)。在50℃下於H
2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之
24
(15 mg,30%)及呈固體之
96
(1.2 mg,3%)。
24: 1 H NMR
(400 MHz, CDCl
3
) δ 3.66-3.52 (m, 1H), 2.02-1.91 (m, 1H), 1.74-1.57(m, 7H), 1.52-1.44 (m, 2H), 1.43-1.29 (m, 7H), 1.28-1.04 (m, 11H), 1.03-0.94 (m, 3H), 0.94-0.85 (m, 13H), 0.82 (s, 3H), 0.71-0.60 (m, 4H)。 在2 min層析中,
LCMS
t
R
=1.342 min,30-90AB_ELSD,純度100.0%,C
29
H
49
[M+H-2H
2
O]
+
之MS ESI計算值397,實驗值397。
實例 25 : (1S,3S,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (25) 之合成 25
之製備可見於實例13中。
25: 1 H NMR
(400 MHz, CDCl
3
) δ 7.42-7.28 (m, 5H), 4.97-4.81 (m, 1H), 4.12-3.92 (m, 1H), 3.23 (brs, 1H), 2.69 (brs, 1H), 2.10-1.88 (m, 3H), 1.82-1.62 (m, 7H), 1.48-1.18 (m, 10H), 1.10-0.88 (m, 8H), 0.87-0.78 (m, 4H), 0.70-0.58 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.319 min,10-80AB_2MIN_E,純度97.0%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在5 min層析中,
SFC
Rt=1.718 min,IC-3_MeOH(DEA)_40_2.5ML,98.26%de。 在8 min層析中,
SFC
Rt=4.367 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,100%de。
實例 26 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((S)-3- 羥基 -3- 甲基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (26) 之合成 1.在0℃下於N
2
下將MeMgBr (0.83 mL,2.49 mmol,3 M於乙醚中)逐滴添加至於
N-8-7_1
(300 mg,0.832 mmol)於THF (20 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應混合物用飽和NH
4
Cl (50 mL)淬滅且用EtOAc (2×10 mL)萃取。合併層用鹽水(10 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到殘餘物,其藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之
N-8-22_1
(100 mg,31%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.99-3.88 (m, 1H), 1.98-1.84 (m, 2H), 1.69-1.57 (m, 6H), 1.52-1.45 (m, 2H), 1.44-1.28 (m, 3H), 1.26-1.17 (m, 5H), 1.16-1.11 (m, 5H), 1.10-0.95 (m, 5H), 0.93-0.86 (m, 7H), 0.84-0.80 (m, 4H), 0.69-0.62 (m, 4H)。
2.
將DMP (224 mg,0.53 mmol)添加至
N-8-22_1
(100 mg,0.265 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌10 min之後,用NaHCO
3
飽和溶液(30 mL)淬滅反應混合物直至含水層之pH為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3×40 mL)、飽和NaHCO
3
(40 mL)、鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0%至20% EtOAc/DCM)純化,得到呈固體之
N-8-22_2
(80mg,80%)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.54-2.42 (m, 1H), 2.09 (s, 3H), 1.94-1.87 (m, 1H), 1.71-1.59 (m, 4H), 1.54-1.45 (m, 3H), 1.44-1.30 (m, 4H), 1.29-1.16 (m, 6H), 1.15-1.07 (m, 5H), 1.06-0.92 (m, 4H), 0.91-0.79 (m, 7H), 0.74-0.61 (m, 4H)。 3.於N
2
下將MeMgBr (0.353 mL,1.06 mmol,3 M於乙醚中)添加至
N-8-22_2
(80 mg,0.213 mmol)於THF (5 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應混合物用NH
4
Cl飽和水溶液(30 mL)淬滅且用EtOAc (20 mL×3)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物,該粗產物藉由矽膠管柱(0%至10% EtOAc/PE)純化,得到呈固體之
26
(7 mg,8%)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.08-2.01 (m, 1H), 1.97-1.86 (m, 1H), 1.69-1.57 (m, 6H), 1.53-1.45 (m, 3H), 1.40-1.27 (m, 5H), 1.26-1.17 (m, 8H), 1.14 (s, 3H), 1.13-1.01 (m, 3H), 0.99-0.92 (m, 5H), 0.91-0.85 (m, 4H), 0.82 (s, 3H), 0.70 (s, 3H), 0.67-0.60 (m, 1H)。 在2.0 min層析中,
LCMS
Rt=1.240 min,30-90AB_E,純度100%,C
26
H
43
[M+H-2H
2
O]
+
之MS ESI計算值355,實驗值355。
實例 27 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (27) 之合成 1.在0℃下於N
2
下經30 min之時段向
N-014-012_4
(300 mg,0.8366 mmol)於THF (20 mL)中之溶液逐滴添加異丙基氯化鎂(1.25 mL,2.50 mmol,2 M)之溶液,在此期間將溫度維持在0℃以下。在20℃下再攪拌反應混合物2小時,得到懸浮液。反應混合物添加NH
4
Cl飽和水溶液(15 mL)且攪拌20 min,隨後用EtOAc (3×10mL)萃取混合物。合併之有機相用鹽水(2×10 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,以獲得呈固體之
N-014-001_1
(360 mg,粗產物),
1
H NMR顯示所需產物,且直接用於下一步驟。 2.
X1
(150 mg,0.37 mmol)藉由SFC (管柱:Chiralpak AS-H 250×30 5u;條件:0.1% NH
3
H
2
O EtOH;開始B:20%;結束B:20%;流速(ml/min):65)純化,以獲得
37
(峰值2,46 mg,31%)及呈固體之
27
(峰值1,27 mg,18%)。 37: 27:
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.26 (m, 1H), 3.42-3.34 (m, 1H), 2.43-2.34 (m, 1H), 2.06-1.91 (m, 3H), 1.90-1.75 (m, 2H), 1.74-1.66 (m, 2H), 1.63-1.58 (m, 3H), 1.54-1.26 (m, 11H), 1.22-1.04 (m, 3H), 1.03-0.99 (m, 3H), 0.97-0.93 (m, 7H), 0.92-0.87 (m, 3H), 0.86-0.77 (m, 3H), 0.70 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.228 min,30-90AB_2MIN_E,純度100%,C
27
H
45
O [M+H-H
2
O]
+
之MS ESI計算值385,實驗值385。 在10 min層析中,
SFC
Rt=2.440 min,OJ_3_EtOH_DEA_5_40_25ML (「管柱:Chiralcel OJ-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),97.38%de。
實例 28 : (3S,5S,8R,9R,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 ) -13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (28) 之合成 1.
在Ar下向
44
(200 mg,0.480 mmol)於MeOH/THF (10 mL/10 mL)中之溶液添加Pd/C (無水,200 mg)。將懸浮液真空脫氣且用H
2
淨化三次。在50℃下於H
2
(50 psi)下攪拌混合物48小時,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用THF (100 mL)洗滌。濃縮濾液,得到呈固體之
28
(30 mg,15%)及呈固體之
82
(30 mg,15%)。
28: 1 H NMR
(400MHz, CDCl3) δ 3.63-3.61 (m, 1H), 1.98-1.76 (m, 4H), 1.72-1.55 (m, 7H), 1.55-1.47 (m, 4H), 1.46-1.23 (m, 6H), 1.22-0.97 (m, 11H), 0.92-0.78 (m, 12H), 0.76-0.54 (m, 5H)。 在2 min層析中,
LCMS
Rt=1.298 min,30-90AB,純度100%,C
28
H
47
[M+H-2H
2
O]+之MS ESI計算值383,實驗值383。
實例 29 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (29) 之合成 1.在60℃下向Mg (4.35 g,179 mmol)及I
2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。混合物用THF (10 mL)稀釋並直接使用。在0℃下於N
2
下向
S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。將NH
4
Cl (20 mL,飽和水溶液)添加至混合物。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH
3
CN (10 mL)再結晶,得到呈固體之
77
(255 mg,21%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.306 min,30-90 AB,純度100%,C
29
H
49
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。
2.
於Ar下將Pd(OH)
2
(無水,20%,50.0 mg)添加至
77
(100 mg,232 umol)於THF(10 mL)及MeOH (10 mL)中之溶液中。將懸浮液真空脫氣且用H
2
淨化三次。在50℃下於H
2
(50 psi)下攪拌混合物16 小時。反應混合物經由矽藻土墊過濾且用THF (3×10 mL)洗滌。濃縮濾液。殘餘物藉由矽膠層析(PE/EtOAc=20/1)純化,得到呈固體之
29
(7.00 mg,7%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.68-3.60 (m, 1H), 1.96-1.88 (m, 2H), 1.68-1.60 (m, 3H), 1.53-1.47 (m, 7H), 1.39-1.23 (m, 13H), 1.16-0.95 (m, 7H), 0.90-0.86 (m, 12H), 0.83 (s, 3H), 0.66-0.63 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.603 min,30-90 AB_ELSD,純度97%,C
29
H
49
[M+H-H
2
O]
+
之MS ESI計算值397,實驗值397。
實例 30 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (30) 之合成 1.
在25℃下於N
2
下將苯甲酸(2.03 g,16.7 mmol)及三苯基膦(6.57 g,25.1 mmol)添加至
S-500-2-10
(3.5 g,8.39 mmol)於THF (30 mL)中之溶液中。在25℃下攪拌混合物20 min。在0℃下於N
2
下添加DIAD (5.07 g,25.1 mmol)。在0℃下攪拌混合物20 min,隨後升溫至25℃且攪拌1 h。反應混合物用水(100 mL)淬滅且用MTBE (3×30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na
2
SO
4
乾燥,過濾,真空濃縮,得到粗產物,該粗產物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到300 mg呈油狀物之粗產物
S-500-2-9_1
,其直接用於下一步驟。 2.將NaOH (1.14 g於3 mL H
2
O中,28.7 mmol)添加至
S-500-2-9_1
(300 mg,0.576 mmol)於THF (5 mL)及MeOH (3 mL)中之溶液中。在50℃下攪拌混合物16小時。混合物用飽和NH
4
Cl (20 mL)淬滅,且用EtOAc (3×10 mL)萃取。合併之有機相用鹽水(30 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮,且藉由combi-flash (0%至10% EtOAc/PE)純化,得到呈固體之
30
(12 mg,5%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.28 (m, 1H), 3.63-3.59 (m, 1H), 2.44-2.40 (m, 1H), 2.05-1.90 (m, 3H), 1.80-1.62 (m, 4H), 1.61-1.58 (m, 3H), 1.56-1.30 (m, 9H), 1.28-1.03 (m, 10H), 1.01 (s, 3H), 0.99-0.85 (m, 10H), 0.69 (s, 3H)。 在2 min層析中,
LCMS
t
R
=1.260 min,30-90AB_ELSD,純度100.0%,C
28
H
45
[M+H-2H
2
O]
+
之MS ESI計算值381,實驗值381。
實例 31 : (1R,3R,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (31) 之合成 31
之合成可見於實例13中。
31: 1 H NMR
(400 MHz, CDCl
3
) δ 7.45-7.28 (m, 5H), 5.02-4.81 (m, 1H), 4.18-3.98 (m, 1H), 3.35 (brs, 1H), 2.47 (brs, 1H), 2.15-1.72 (m, 8H), 1.53-1.31 (m, 8H), 1.30-1.03 (m, 8H), 0.99-0.89 (m, 4H), 0.89-0.78 (m, 4H), 0.75-0.60 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.327 min,10-80AB_2MIN_E,純度100%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在10 min層析中,
SFC
Rt=1.929 min,IC-3_MeOH(DEA)_40_2.5ML,98.4%de。
實例 32 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 苯基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (32) 之合成 1.
在20℃下於氮氣下將t-BuOK (4.64 g,41.4 mmol)添加至Me
3
SI (8.44 g,41.4 mmol)於無水THF (50 mL)中之懸浮液中。在20℃下攪拌混合物1小時,且添加
N-005_5
(5 g,13.8 mmol)。使所得混合物升溫至45℃且攪拌4小時。使反應混合物冷卻至室溫,用水(50 mL)淬滅,用EtOAc (2×50 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之
N-005_001
(2.7 g,52%)。 在2.0 min層析中,
LCMS
Rt=1.324 min,30-90AB_E,純度92%,C
25
H
41
O [M+H-H
2
O]
+
之MS ESI計算值357,實驗值357。 2.在0℃下於氮氣下將CuI (10.1 mg,0.0534 mmol)及PhMgBr (1 M於THF中,2.66 mL,2.66 mmol)添加至
N-005_001
(200 mg,0.534 mmol)於無水THF (20 mL)中之溶液中。使混合物逐漸升溫至15℃且攪拌16小時。反應混合物用NH
4
Cl水溶液(20 mL)淬滅,用EtOAc (2×10 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0-5% EtOAc/PE)純化,得到呈固體之
NA-6-5/6
(190 mg,79%)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.35-7.27 (m, 2H), 7.25-7.18 (m, 3H), 3.95-3.81 (m, 1H), 2.87-2.39 (m, 2H), 2.04-1.92 (m, 1H), 1.89-1.80 (m, 1H), 1.71-1.58 (m, 4H), 1.56-1.43 (m, 6H), 1.41-1.27 (m, 5H), 1.26-1.18 (m, 4H), 1.18-1.08 (m, 2H), 1.06-0.96 (m, 5H), 0.92-0.79 (m, 8H), 0.73-0.55 (m, 4H)。
3.N-6-5/6
(190 mg,0.420 mmol)藉由製備型HPLC (管柱:YMC-Actus Triart C18 100×30mm×5um;條件:水(0.05%HCl)-ACN;梯度:90-100%B;流速:25 mL/min)分離,得到呈固體之
93
(56 mg,30%)及呈固體之
32
(12 mg,6%)。
32: 1 H NMR
(400 MHz, CDCl
3
) δ 7.36-7.29 (m, 2H), 7.25-7.19 (m, 3H), 3.89-3.83 (m, 1H), 2.79-2.72 (m, 1H), 2.49-2.40 (m, 1H), 2.05-1.98 (m, 1H), 1.92-1.79 (m, 2H), 1.72-1.51 (m, 9H), 1.44-1.31 (m, 5H), 1.30-1.09 (m, 7H), 1.08-0.96 (m, 5H), 0.92-0.81 (m, 7H), 0.74-0.63 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.343 min,30-90AB,純度100%,C
31
H
47
O [M+H-H
2
O]
+
之MS ESI計算值435,實驗值435。
實例 33 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4-(3- 甲基氧雜環丁 -3- 基 ) 丁 -2- 基 )-10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (33) 之合成 1.在25℃下向
N-014-005_1
(10 g,97.9 mmol)於DCM (100 mL)中之溶液中添加1-甲基-1H-咪唑(16.0 g,195 mmol)及TEA (19.7 g,195 mmol)。將TsCl (37.1 g,195 mmol)添加至溶液中。在25℃下攪拌反應混合物2小時。混合物用水(2×100 mL)、鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈淡黃色固體之
N-014-005_2
(25 g,粗產物),其藉由矽膠管柱層析(0%至15% EtOAc/PE)純化,得到呈白色固體之
N-014-005_2
(23.6 g,95%)。
1 H NMR
(400 MHz, CDCl
3
) δ 7.80-7.68 (m, 2H), 7.41-7.26 (m, 2H), 3.40-3.29 (m, 4H), 4.12-4.00 (s, 2H), 2.44 (s, 3H), 1.28 (s, 3H)。 2.向
N-014-005_2
(10 g,39.0 mmol)於丙酮(100 mL)中之溶液中添加LiBr (13.5 g,156 mmol)。在65℃下攪拌混合物1小時。混合物在0℃下用水(200 mL)淬滅且用己烷(3×200 mL)萃取。合併之有機相用鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈黃色液體之
N-014-005_3
(2.54 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.50-4.30 (m, 4H), 3.64 (s, 2H), 1.58 (s, 1H), 1.43 (s, 3H)。
3.在50℃至55℃下於N
2
下向Mg (807 mg,33.2 mmol)及I
2
(1 mg)於THF (2 mL)中之懸浮液逐滴添加
N-014-005_3
(2.5 g,15.1 mmol)於THF (8 mL)中之溶液。在55℃下攪拌混合物1 h。用THF (10 mL)稀釋混合物,且直接用於下一步驟中而無需監測。在0℃下向
N-14-12_4
(1.01 g,2.83 mmol)於THF (10 mL)中之溶液添加新鮮製備的3-[(溴鎂)甲基]-3-甲基環氧丙烷(15 mmol於20 mL THF中)。在15℃下攪拌混合物4 h。向混合物添加NH
4
Cl (20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。分離有機層且真空濃縮。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈白色固體之混合物(190 mg,15%),其藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3H2O ETOH,梯度:50%至50%,流速(ml/min):60mL/min,25℃)純化,得到
33
(峰值1,110 mg,9%)及呈白色固體之
70
(峰值2,30mg,不純)。不純
70
(30 mg,不純)藉由矽膠管柱層析(15% EtOAc/PE)純化,得到呈白色固體之
70
(10 mg,5%)。
33: 1 H NMR
(400 MHz, CDCl
3
) δ 5.30-5.26 (m, 1H), 4.59-4.70 (m, 1H), 4.50-4.48 (m, 1H),4.36-4.33 (m, 1H), 3.83 (s, 1H), 2.40-2.33 (m, 1H), 2.10-1.50 (m, 17H), 1.49-1.35 (m, 9H), 1.30-0.80 (m, 13H), 0.68 (s, 3H)。 在3 min層析中,
LCMS
Rt=1.069 min,30-90AB_2MIN_E.M,純度100%,C
29
H
49
O
3
[M+H]
+
之MS ESI計算值445,實驗值445。
實例 34 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (34) 之合成 1.
在0℃下將於THF (5 mL)中之
N-8-7_1
(500 mg,1.38 mmol)添加至異丙基氯化鎂(708 mg,3.44 mL,2 M於THF中)於THF (5 mL)中之溶液中。在25℃下攪拌混合物4小時。向混合物添加NH
4
Cl (20 mL,10%水溶液)。用EtOAc (2 × 30 mL)萃取混合物。有機層經分離且真空濃縮,得到殘餘物。殘餘物藉由矽膠層析純化用PE/EtOAc=3/1溶離,得到呈固體之
N-8-11_1
(170 mg,30%)。該不純產物(120 mg)藉由矽膠層析進一步純化用PE/EtOAc=3/1溶離,得到呈固體之
N-8-11_1
(50 mg,42%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.20-3.10 (m, 1H), 2.00-1.80 (m, 2H), 1.70-1.60 (m, 2H), 1.55-1.40 (m, 4H), 1.39-0.95 (m, 19H), 0.90-0.80 (m, 15H), 0.70-0.60 (m, 5H)。 2.將DMP (457 mg,1.08 mmol)添加至
N-8-11_1
(220 mg,0.543 mmol)於DCM (5 mL)中之溶液中。在25℃下攪拌10 min之後,反應混合物用NaHCO
3
飽和水溶液(50 mL)淬滅直至含水層之pH變成約9為止。過濾混合物。分離DCM層,且用DCM (100 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3×100 mL)、飽和NaHCO
3
(100 mL)、鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈固體之
N-8-11_2
(140 mg,64%)。 在2.0 min層析中,
LCMS
Rt=1.300 min,30-90AB_2MIN_E,純度100%,C
27
H
45
O [M+H-H
2
O]
+
之MS ESI計算值385,實驗值385。 3.將NaBH
4
(1.17 g,17.3 mmol)每五分鐘添加五次至
N-8-11_2
(140 mg,0.347 mmol)於MeOH (2 mL)及THF (2 mL)中之溶液中。在15℃下攪拌混合物30分鐘。用飽和NH
4
Cl (50 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到
N-8-11_1
(140 mg,不純)。
N-8-11_1
藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之
34
(50 mg,不純)及呈固體之
65
(10 mg,不純)。
4.N-8-11_1
(50 mg,0.123 mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之
34
(30 mg,不純)。
N-8-11_1
(30 mg,0.0741mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之
34
(9 mg,30%)。
34: 1 H NMR
(400 MHz, CDCl
3
) δ 3.18-3.07 (m, 1H), 1.98-1.81 (m, 2H), 1.71-1.58 (m, 6H), 1.53-1.31 (m, 7H), 1.30-0.98 (m, 14H), 0.97-0.78 (m, 14H), 0.70-0.60 (m, 4H)。 在7.0 min層析中,
LCMS
Rt=4.387 min,30-90AB_7MIN_E,純度97.6%,C
27
H
45
[M+H-2H
2
O]
+
之MS ESI計算值369,實驗值369。
實例 35 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (35) 之合成 1.在25℃下將NaOH (71.9 mg,180 mmol)添加至
N-4-4B
(20 mg,0.0361 mmol)於THF/MeOH (2 mL)中之溶液中。使反應混合物升溫至50℃且攪拌1 h。使反應混合物冷卻且添加水(20 mg)。用EtOAc (3 × 10 mL)萃取水相。合併之有機相用飽和鹽水(2×20 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化以提供呈白色固體之
35
(8 mg,50%)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.63-3.61 (m, 1H), 3.41-3.38 (m, 5H); 2.51 (s, 1H); 1.97-1.81 (m, 1H), 1.71-1.54 (m, 8H), 1.51-1.48 (m, 4H), 1.25-1.10 (m, 15H), 0.99-0.80 (m, 9H), 0.78-0.75 (m, 4H),0.71-0.59 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.301 min,30-90 AB,純度96%,C
29
H
48
O [M+H-2H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 36 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (36) 之合成 1.將Pd(OH)
2
(200 mg)添加至
30
(100 mg,0.239 mmol)於MeOH (10 mL)中之溶液中。在50℃下於H
2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0%至10% EtOAc/PE)純化,得到
47
(21 mg,21%)及呈固體之
36
(1 mg,1%)。
37: 1 H NMR
(400 MHz, CDCl
3
) δ 3.66-3.55 (m, 1H), 2.05-1.77 (m, 3H), 1.72-1.63 (m, 3H), 1.55-1.48 (m, 3H), 1.47-1.31 (m, 9H), 1.29-1.12 (m, 13H), 1.11-1.00 (m, 3H), 0.96 (s, 3H), 0.93-0.87 (m, 9H), 0.67 (s, 3H)。 在2 min層析中,
LCMS
t
R
=1.296 min,30-90AB_ELSD,純度100.0%,C
28
H
47
[M+H-2H
2
O]
+
之MS ESI計算值383,實驗值383。
實例 37 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (37) 之合成 1.
N-014-001_1
(150 mg,0.37 mmol)藉由SFC (管柱:Chiralpak AS-H 250×30 5u;條件:0.1% NH
3
H
2
O EtOH;開始B:20%;結束B:20%;流速(ml/min):65)純化,以獲得
37
(峰值2,46 mg,31%)及呈固體之
27
(峰值1,27 mg,18%)。 014-001A:
1 H NMR
(400 MHz, CDCl
3
) δ 5.35-5.28 (m, 1H), 3.18-3.09 (m, 1H), 2.39-2.35 (m, 1H), 2.06-1.81 (m, 4H), 1.73-1.57 (m, 6H), 1.54-1.41 (m, 8H), 1.40-1.26 (m, 3H), 1.24-1.11 (m, 3H),1.10-0.97 (m, 6H), 0.96-0.92 (m, 1H), 0.90-0.85 (m, 5H), 0.84-0.76 (m, 4H), 0.69 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.207 min,30-90AB_2MIN_E,純度100%,C
27
H
45
O [M+H-H
2
O]
+
之MS ESI計算值385,實驗值385。 在10 min層析中,
SFC
Rt=2.635 min,OJ_3_EtOH_DEA_5_40_25ML (「管柱:Chiralcel OJ-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),98.66%de。
實例 38 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (38) 之合成 1.在0℃下將丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)緩慢地添加至
S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液中。添加之後,在15℃下攪拌混合物1小時。混合物用飽和NH
4
Cl (40 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
72
(500 mg,56%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,
LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C
27
H
43
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 2.向
72
(150 mg,0.372 mmol)於MeOH (20 mL)中之溶液中添加Pd(OH)
2
(300 mg,無水)。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到
23
(9 mg,6%)及呈固體之
38
(43 mg,29%)。
38: 1 H NMR
(400 MHz, CDCl
3
) δ 3.71-3.62 (m, 1H), 1.99-1.82 (m, 2H), 1.70-1.56 (m, 6H), 1.54-1.45 (m, 3H), 1.44-1.38 (m, 3H), 1.37-1.17 (m, 10H), 1.16-1.01 (m, 5H), 1.00-0.85 (m, 11H), 0.82 (s, 3H), 0.70-0.60 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.397 min,30-90AB_E,純度100%,C
27
H
45
[M+H-2H
2
O]
+
之MS ESI計算值369,實驗值369。
實例 39 : (3S,8S,9S,10R,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (39) 之合成 1. 在35℃下於N
2
下將t-BuOH (600 mL)饋入至三頸圓底燒瓶中,隨後添加t-BuOK (101 g,905 mmol)。在35℃下攪拌30分鐘之後,將
N-004-029_1
(50 g,151 mmol)添加至上述混合物且在35℃下攪拌1小時。將反應混合物倒入至5%乙酸水溶液(2 L)中,在此期間將溫度維持在低於10℃。添加冰水(1 L)。用NaHCO
3
將混合物之pH調節至約7至8且過濾。將濾餅溶解於DCM (1.5 L)中。合併之有機相用水(2×500 ml)、鹽水(2×500 ml)洗滌,經Na
2
SO
4
乾燥,過濾且在低於35℃下於真空中濃縮,得到呈固體之
N-004-029_2
(45 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.35-5.32 (m, 1H), 3.71-3.58 (m, 1H), 3.38-3.25 (m, 2H), 2.90-2.78 (m, 1H), 2.55-2.20 (m, 2H), 2.13-1.92 (m, 3H), 1.90-1.59 (m, 5H), 1.46-1.14 (m, 10H), 1.12-0.96 (m, 6 H), 0.72 (s, 3H)。
2.
在0℃下於N
2
下將n-BuLi (108 mL,272 mmol,2.5 M於己烷中)逐滴添加至Me
3
SI (73.8 g,362 mmol)於無水THF (300 mL)中之混合物中。在0℃下攪拌30分鐘之後,在-40℃下添加
N-004-029_2
(30 g,90.7 mmol)於無水THF (600 mL)中之溶液。在-40℃下攪拌混合物2小時且在25℃下攪拌16小時。將反應混合物倒入至冰水(1 L)中。用EtOAc (2×500 mL))萃取水相。合併之有機相用鹽水(2×500 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之
N-004-029_3
(1.8 g,6%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.25 (m, 1H), 3.66-3.61 (m, 1H), 3.39-3.31 (m, 1H), 2.93-2.86 (m, 1H), 2.59-2.53 (m, 1H), 2.20-1.93 (m, 4H), 1.89-1.14 (m, 15H), 1.12-0.90 (m, 9H), 0.71 (s, 3H)。 3. 在25℃下於N
2
下將MeONa (5.61 g,104 mmol)添加至
N-004-029_3
(1.8 g,5.22 mmol)於MeOH (20 mL)中之溶液中。在50℃下攪拌12小時之後,將水(100 mL)添加至混合物中且攪拌10分鐘。用EtOAc (2×80 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之
N-004-029_4
(1.5 g,76%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.28 (m, 1H), 3.70-3.54 (m, 1H), 3.40-3.35 (m, 6H), 3.28-3.16 (m, 2H), 2.40-2.35 (m, 1H), 2.09-1.90 (m, 5H), 1.87-1.57 (m, 11H), 1.34-1.06 (m, 10H), 0.70 (s, 3H)。 4. 在25℃下將DMP (2.53 g,5.97 mmol)添加至
N-004-029_4
(1.5 g,3.98 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌30 min之後,在25℃下用飽和NaHCO
3
(50 mL)淬滅反應混合物。將DCM (50 mL)添加至混合物中且攪拌10 min。分離DCM相且用飽和Na
2
S
2
O
3
水溶液(2×50 mL)、鹽水(2×50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(5%至25% EtOAc/PE)純化,得到呈固體之
N-004-029_5
(0.6 g,40%)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.59-9.57 (m, 1H), 5.32-5.29 (m, 1H), 3.37 (s, 3H), 3.30-3.15 (m, 2H), 2.44-2.31 (m, 2H), 2.13-1.40 (m, 16H), 1.27-1.02 (m, 10H), 0.73 (s, 3H)。 5. 在0℃下將CsF (607 mg,4.00 mmol)添加至
N-004-029_5
(0.6 g,1.60 mmol)於無水THF (20 mL)中之溶液中。在攪拌20 min之後,在0℃下添加TMSCF
3
(568 mg,4.00 mmol)且攪拌混合物1小時。將TBAF.3H
2
O (2.02 g,6.40 mmol)添加至混合物中,在50℃下攪拌該混合物1小時。將反應混合物倒入至冰水(50 mL)中。用EtOAc (2×80 mL)萃取水相。將合併之有機相用飽和鹽水(2×80 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之
N-004-029A
(450 mg,63%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.29 (m, 1H), 4.11-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.43-2.36 (m, 1H), 2.26-1.82 (m, 7H), 1.78-1.61 (m, 5H), 1.34-0.80 (m, 15H), 0.75-0.67 (m, 3H)。 6.
N-004-029A
(0.45 g,1.01 mmol)藉由SFC (管柱:AD (250 mm×30 mm,5 um),條件:0.1% NH
3
H
2
O ETOH,開始B:30%,結束B:30%)純化,得到呈白色固體之
39
(PK1:120 mg,26.7%)及呈白色固體之
95
(PK2:200 mg,44.5%)。 藉由NOE確認
39 之
結構。
39 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.29 (m, 1H), 4.06-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.43-2.36 (m, 1H), 2.25-2.19 (m, 1H), 2.15-2.07 (m, 1H), 2.04-1.60 (m, 9H), 1.55-1.34 (m, 5H), 1.25-0.88 (m, 11H), 0.70 (s, 3H)。
LCMS
Rt=2 min層析中1.078 min,30-90AB_2MIN_E,純度100%,C
24
H
34
F
3
O[M-CH
5
O
2
]
+
之MS ESI計算值395,實驗值395。
實例 40 : (3S,5S,8R,9R,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (40) 之合成 1. 在25℃下於Ar下,將Pd(OH)
2
(100 mg,無水)添加至
3
(30 mg,0.072 mmol)於MeOH/THF (5 mL/5mL)中之溶液中。在50℃下於H
2
(50 Psi)下攪拌反應物48 h。過濾混合物且在真空中濃縮濾液,得到粗產物,其藉由矽膠管柱(PE/EtOAc=10/1至5/1)純化,得到呈固體之
41
(10 mg,33%)。
1 H NMR
(400MHz, CDCl3) δ 3.62-3.59 (m, 1H), 1.99-1.91 (m, 1H), 1.88-1.76 (m, 2H), 1.74-1.61 (m, 6H), 1.46-1.29 (m, 6H), 1.26-1.09 (m, 9H), 1.08-0.99 (m, 6H), 0.95-0.78 (m, 14H), 0.74-0.58 (m, 5H)。 在2 min層析中,
LCMS
Rt=1.491 min,30-90 AB,純度99%,C
28
H
47
[M+H-2H
2
O]
+
之MS ESI計算值383,實驗值383。
實例 41 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基庚 -5- 炔 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (41) 之合成 1. 在-20℃下將n-BuLi (13.8 mL,34.5 mmol,2.5 M於己烷中)逐滴添加至丁-2-炔(1.86 g,34.5 mmol)於於THF (100 mL)中之溶液中。-20℃下攪拌溶液2.5小時,且接著將其冷卻至-78℃,添加含
N-8-7_1
(5.0 g,13.8 mmol)之THF (100 mL)。在此溫度下攪拌溶液30分鐘,接著在-20℃下攪拌1小時,且隨後20℃下攪拌18小時。所得凝膠藉由倒入至飽和NH
4
Cl (100 mL)中淬滅,隨後用EtOAc (2×150 mL)萃取。合併之有機層用水(40 mL)及鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,經過濾且藉由閃蒸塔純化用含0至20%EtOAc之PE溶離,得到呈油狀物之
N-8-7_2B
(1 g,粗產物)及
N-8-7_2B
(1.7 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.90-3.75 (m, 1H), 2.45-2.35 (m, 1H), 2.25-2.05 (m, 3H), 1.95-1.85 (m, 2H), 1.85-1.40 (m, 8H), 1.40-1.20 (m, 9H), 1.20-0.75 (m, 18H), 0.65 (s, 4H)。 在10 min層析中,SFC 峰值1:Rt = 3.008 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6 mm I.D.,3 um行動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。 2. 粗產物
N-8-7_2B
(250 mg,0.868 mmol)藉由SFC (管柱:AD (250 mm×30 mm,10 um),梯度:35%至35% B (A= 0.1% NH
3
/H
2
O,B = EtOH),流速:60 mL/min)進一步純化,得到呈固體之
41
(峰值2,81 mg,33%)及呈固體之
68
(峰值1,78 mg,31%)。
41 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.82-3.70 (m, 1H), 2.79-2.08 (m, 2H), 2.00-1.90 (m, 1H), 1.80 (s, 4H), 1.78-1.69 (m, 1H), 1.69-1.42 (m, 10H), 1.40-1.31 (m, 4H), 1.31-1.18 (m, 4H), 1.18-0.92 (m, 6H), 0.92-0.85 (m, 7H),0.82 (s, 3H), 0.66 (s, 3H)。
LCMS
Rt=2 min層析中之1.206 min,30-90AB_2MIN_E,純度100%,C
28
H
45
O [M+H-H
2
O]
+
之MS ESI計算值397,實驗值397。 在10 min層析中,
SFC
Rt=5.823 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
實例 42 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (42) 之合成 :
1. 在25℃下將4-甲基苯磺酸(2.70 g,15.7 mmol)添加至
N-4-1_1
(50 g,157 mmol)於MeOH (500 mL)中之溶液中。在65℃下攪拌混合物1小時。使反應混合物冷卻至25℃且添加TEA (2.16 mL,15.7 mmol)。攪拌混合物0.5 h。沈澱物藉由過濾採集且用甲醇(2×100 mL)洗滌,得到呈固體之
N-4-1_2
(50 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.25-3.05 (m, 6H), 2.60-2.40 (m, 1H), 2.20-2.05 (m, 4H), 2.00-1.95 (m, 1H), 1.90-1.80 (m, 1H), 1.75-1.50 (m, 6H), 1.49-1.05 (m, 12H), 1.04-0.95 (m, 1H), 0.78 (s, 3H), 0.59 (s, 3H)。 2. 在25℃下將t-BuOK (23.0 g,205 mmol)添加至溴基(甲基)三苯基磷烷(73.2 g,205 mmol)於THF (500 mL)中之溶液中。將混合物加熱至45℃且攪拌1小時。添加
N-4-1_2
(50 g,137 mmol)。在45℃下攪拌混合物2小時。混合物用NH
4
Cl (200 mL)淬滅且用THF (3×100 mL)萃取。有機層經鹽水(200 mL)洗滌,經Na
2
SO
4
乾燥且過濾,得到混合物(50 g,500 mL),該混合物不經進一步純化即用於下一步驟中。
3.
將HCl水溶液(207 mL,1 M於水中)添加至
N-4-1_3
(50 g,138 mmol)於THF (500 mL)中之溶液中。在25℃下攪拌混合物0.5 h。過濾混合物且將濾餅溶解於DCM (200 mL)中並用鹽水(100 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到呈固體之
N-4-1_4
(39 g,90%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.45-2.20 (m, 3H), 2.15-2.00 (m, 3H), 1.90-1.65 (m, 8H), 1.60-1.50 (m, 2H), 1.45-1.05 (m, 8H), 1.00 (s, 3H) 0.90-0.85 (m, 1H), 0.80-0.75 (m, 1H), 0.58 (s, 3H)。 4. 將CsF (25.9 g,171 mmol)及TMSCF
3
(24.3 g,171 mmol)添加至
N-4-1_4
(27 g,85.8 mmol)於THF (200 mL)中之溶液中。在10℃下攪拌混合物1小時。將水(10 mL)及TBAF.3H
2
O (30 g)添加至混合物中。在30℃下再攪拌混合物2小時。真空濃縮混合物。將殘餘物溶解於EtOAc (500 mL)中,用水(2×500 mL)洗滌,經Na
2
SO
4
乾燥,經過濾,在真空中濃縮且藉由閃蒸塔(DCM/EtOAc (1:1)/PE,0%至10%)純化,得到呈固體之
N-4-1_5
(27 g,82%)及
N-4-1_5A
(3.5 g,11%) 。
N-4-1_5 : 1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.12-1.94 (m, 3H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.60 (m, 5H), 1.58-1.48 (m, 2H), 1.45-1.09 (m, 10H), 1.01-0.89 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.56 (s, 3H)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.09-1.99 (m, 1H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.52 (m, 9H), 1.45-1.06 (m, 10H), 1.00-1.81 (m, 2H), 0.79 (s, 3H), 0.56 (s, 3H)。 5. 將9-BBN二聚體(29 g,119 mmol)添加至
N-4-1_5
(23 g, 59.8 mmol)於THF (250 mL)中之溶液中且在40℃下於N
2
下攪拌混合物16小時。將乙醇(34.3 mL,598 mmol)及NaOH (119 mL,5 M,598 mmol)添加至反應混合物。使混合物變得澄清。在25℃下逐滴添加H
2
O
2
(59.8 mL,10 M,598 mmol)且使內部溫度升至回流(70℃)。在添加之後,使混合物冷卻至30℃。向混合物添加Na
2
SO
3
(100 mL,20%水溶液)。有機層經分離且倒入水(800 mL)中。形成固體。過濾混合物且該固體用水洗滌,在真空下乾燥且用MeCN (250 mL)濕磨,得到固體。該固體在60℃下自MeOH/水(250 mL/12.5 mL)濕磨且在冷卻至15℃之後過濾。在真空下乾燥固體,得到呈固體之
N-4-1_6
(16.4 g,68%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.69-3.60 (m, 1H), 3.39-3.29 (m, 1H), 2.09-2.01 (m, 1H), 1.99-1.92 (m, 1H), 1.87-1.75 (m, 2H), 1.72-1.43 (m, 7H), 1.42-1.07 (m, 11H), 1.03 (d,
J
= 6.8 Hz, 3H), 1.01-0.86 (m, 3H), 0.85 (s, 3H), 0.73-0.69 (m, 1H), 0.67 (s, 3H)。
6.
將水(223 mg,12.4 mmol)及DMP (10.5 g,24.8 mmol)添加至
N-4-1_6
(5 g,12.4 mmol)於DCM (200 mL)中之懸浮液中。在15℃下攪拌混合物15 min。混合物用NaHCO
3
/Na
2
S
2
O
3
(200 mL/200 mL,飽和)洗滌兩次,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-4-1_7
(4.5 g,90%)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.60-9.51 (m, 1H), 2.40-2.30 (m, 1H), 2.12-1.78 (m, 5H), 1.75-1.59 (m, 4H), 1.57-1.15 (m, 11H), 1.14-0.84 (m, 8H), 0.78-0.63 (m, 5H)。 7. 在50℃至55℃下於N
2
下將1-溴-3-甲基丁烷(2.79 g,18.5 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (899 mg,37 mmol)及I
2
(1 mg)於THF (2 mL) 於中之懸浮液中。在55℃下攪拌混合物1小時,得到異戊基溴化鎂溶液。在0℃下將新鮮製備的異戊基溴化鎂(18.5 mmol於10 mL THF中)添加至
N-4-1_7
(0.5 g,1.24 mmol)於THF (5 mL)之溶液中。在15℃下攪拌混合物2小時。向混合物中添加NH
4
Cl (20 mL,10%水溶液)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到呈固體之
N-4-2
(0.6 g,粗產物)。 8. 在15℃下將水(1滴)及DMP (1.06 g,2.52 mmol)添加至N-4-2 (0.6 g,1.26 mmol)於DCM (20 mL)中之溶液中。在15℃下攪拌混合物1 h。混合物用NaHCO
3
/Na
2
S
2
O
3
(20 mL/20 mL,飽和)洗滌兩次,經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到呈固體之
N-4-1_8
(0.6 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.59-2.30 (m, 3H), 2.11-1.78 (m, 4H), 1.75-1.36 (m, 13H), 1.35-0.98 (m, 11H), 0.91-0.82 (m, 10H), 0.78-0.70 (m, 1H), 0.67 (s, 3H)。 9. 在15℃下將NaBH
4
(0.96 g,25.4 mmol)逐份添加至
N-4-1_8
(0.6 g,1.27 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌混合物30 min。向混合物中添加NH
4
Cl (50 mL,10%)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機層經Na
2
SO
4
乾燥,過濾且真空濃縮,且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到不純
42
及
85
。
N42
在15℃下自MeCN (10 mL)濕磨且在真空中乾燥,得到呈固體之
42
(153 mg,25%)。
85
藉由閃蒸塔(0%至15% EtOAc於PE中)純化,得到油狀物,用MeCN (5 mL)及水(5 mL)處理該油狀物,且真空濃縮,得到呈固體之
85
(70 mg,12%)。
42 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.66-3.55 (m, 1H), 2.01-1.78 (m, 6H), 1.71-1.59 (m, 4H), 1.51-1.15 (m, 16H), 1.09-1.02 (m, 3H), 0.92-0.81 (m, 13H), 0.72-0.61 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.378 min,30-90_AB_E,純度100%,C
28
H
46
F
3
O [M+H-H
2
O]
+
之MS ESI計算值 455,實驗值455。 在10.0 min層析中,
HPLC
Rt=5.38 min,50-100_AB_E,純度99.58%。
實例 43 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (43) 之合成 1. 向
62
(160 mg,0.373 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中添加Pd(OH)
2
(0.2 g,<1%水)。在50℃下於50 psi氫氣下氫化溶液16小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到
43
(27 mg,17%)及呈白色固體之
16
(117 mg,73%)。
43 : 1 H NMR
(400 MHz, CDCl3) δ.4.05-3.99 (m, 1H), 1.99-1.81 (m, 5H), 1.79-1.72 (m, 1H), 1.70-1.56 (m, 3H), 1.53-1.35 (m, 7H), 1.35-1.07 (m, 12H), 1.04-1.02 (m, 3H), 0.97 (s, 3H), 0.92 (t,
J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt = 1.271 min,30-90 AB,純度100%,C
25
H
40
F
3
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 44 : (3S,8R,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (44) 之合成 1.
在60℃下將1-溴-3-甲基丁烷(4 g,26.4 mmol)於THF (27 mL)中之溶液逐滴添加至Mg (947 mg,39.5 mmol)及I
2
(33.5 mg,0.132 mmol)於THF (3 mL)中之懸浮液中。在60℃下攪拌混合物1小時。在0℃下於N
2
下將新鮮製備的異戊基溴化鎂(30 mL,0.88 M於THF中,26.4 mmol)添加至
S-500-15-2_1
(800 mg,2.32 mmol)於THF (2 mL)中之溶液中。在0℃下攪拌混合物1 h。將NH
4
Cl (50 mL,飽和水溶液)添加至混合物,用EtOAc (2 × 50 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到粗產物,其藉由矽膠(PE/EtOAc=10/1至5/1)純化,得到呈白色固體之
44
(720 mg,75%)。 1H NMR (400MHz, CDCl3) δ 5.40-5.38 (m, 1H), 3.63-3.61 (m, 1H), 2.23-2.21 (m, 1H), 2.10-1.74 (m, 7H), 1.69-1.58 (m, 2H), 1.54-1.34 (m, 8H), 1.33-1.00 (m, 11H), 0.95-0.75 (m, 14H), 0.70 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.289 min,30-90 AB,純度100%,C28H45 [M+H-2H2O]
+
之MS ESI計算值381,實驗值381。
實例 45 : (3S,5R,8R,9R,10S,13S,14S,17R)-3-( 甲氧基甲基 )-13- 甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (45) 之合成 1.
向
G-020-004_1
(100 g,364 mmol)於無水甲醇(1 L)中之溶液中添加TsOH (6.26 g,36.4 mmol)。在60℃下攪拌混合物18小時。反應混合物經濃縮以移除大部分溶劑,用Et
3
N (3.7 g)中和,用EtOAc (600 mL)稀釋,用水(500 mL)及鹽水(500 mL)洗滌。有機層經濃縮,得到呈油狀物之
G-020-004_2
(133 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.20 (s, 3H), 3.14 (s, 3H), 2.63-2.39 (m, 2H), 2.14-2.03 (m, 2H), 1.97-1.89 (m, 2H), 1.86-1.77 (m, 3H), 1.64-1.60 (m, 2H), 1.56-1.49 (m, 3H), 1.47-1.42 (m, 2H), 1.40-1.32 (m, 2H), 1.29-1.23 (m, 3H), 1.16-1.06 (m, 2H), 0.87 (s, 3H)。
2.
向Ph
3
PEtBr (308 g,830 mmol)於20℃下氮氣下之無水THF (700 mL)中之懸浮液中添加t-BuOK (93.1 g,830 mmol)。在20℃下攪拌1小時之後,將
G-020-004_2
(133 g,415 mmol)於無水THF (300 mL)中之溶液添加至混合物。使所得混合物升溫至50℃且攪拌4小時。反應混合物經冷卻,用水(400 mL)及飽和NH
4
Cl (300 mL)淬滅,攪拌30 min。分離有機層,且用THF (300 mL)萃取水相。合併之有機層直接用於下一步驟中。 3. 向
G-020-004_3
(137 g,412 mmol,理論上)於THF (1.3 L)中之溶液中添加HCl水溶液(1 M,618 mL,618 mmol)。在20℃下攪拌1小時之後,反應混合物用飽和NaHCO
3
(800 mL)淬滅且用EtOAc (2×500 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮,得到固體(300 g)。自石油醚(800 mL)濕磨固體18小時。過濾出固體,且用石油醚(400 mL)洗滌濾餅。濃縮濾液,得到殘餘物(117 g)。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之
N-004-023_5
(70 g)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.17-5.09 (m, 1H), 2.65-2.55 (m, 1H), 2.43-2.34 (m, 1H), 2.33-2.15 (m, 6H), 2.11-2.05 (m, 1H), 1.83-1.70 (m, 2H), 1.68-1.64 (m, 4H), 1.63-1.59 (m, 2H), 1.58-1.46 (m, 3H), 1.42-1.25 (m, 3H), 1.25-1.14 (m, 3H), 0.91 (s, 3H)。
4.
在60℃下於N
2
下加熱三甲基氧化鋶碘(30.5 g,139 mmol)及t-BuOK (15.5 g,139 mmol)於DMSO (200 mL)中之經攪拌溶液1.0 h;將
N-004-023_5
(20 g,69.8 mmol)添加至反應混合物且在60℃下攪拌10分鐘。用水(1000 mL)處理反應物。用EtOAc (2×500 mL))萃取水相。合併之有機相用水(2×500 mL)、鹽水(300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
N-004-023_6
(20.5 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.13-5.10 (m, 1H), 2.62-2.60 (m, 2H), 2.25-2.20 (m, 5H), 2.00-1.48 (m, 12H), 1.46-1.00 (m, 8H), 0.98-0.89 (m, 4H)。 5. 在25℃下於N
2
下將MeONa (18.4 g,341 mmol)添加至
N-004-023_6
(20.5 g,68.2 mmol)於MeOH (500 mL)中之溶液中。在70℃回流下於N
2
下攪拌混合物16 h。用水(500 mL)處理反應物。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na
2
SO
4
乾燥,經過濾且濃縮物藉由矽膠層析(PE/EtOAc=10/1至6/1)純化,得到呈固體之
N-004-023_7
(20 g,88%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.12-5.06 (m, 1H), 3.38 (s, 3H), 3.19 (s, 2H), 2.25-2.22 (m, 1H), 2.20-2.09 (m, 3H), 1.66-1.63 (m, 3H), 1.60-1.24 (m, 14H), 1.22-1.00 (m, 6H), 0.87 (s, 3H)。 6. 在0℃下於N
2
下將9-BBN二聚體(29.2 g,120 mmol)添加至
N-004-023_7
(20 g,60.1 mmol)於THF (100 mL)中之溶液中。在65℃下攪拌溶液2小時。冷卻至0℃後,添加EtOH (34.9 mL,601 mmol)。接著極緩慢地添加NaOH (120 mL,5 M,601 mmol)之溶液。添加後,緩慢添加H
2
O
2
(68.0 g,601 mmol,30%於水中),且將內部溫度維持在低於10℃。在75℃下於N
2
下攪拌混合物1小時。將混合物再冷卻至25℃。將混合物添加至H
2
O (2 L)。攪拌混合物30分鐘。沈澱物藉由過濾採集且用H
2
O (2×500 mL)洗滌,得到呈固體之
N-004-023_8
(17.8 g,85%) 。
1 H NMR
(400 MHz, CDCl
3
) δ 3.70-3.55 (m, 1H), 3.38 (s, 3H), 3.19 (m, 2H), 2.11-1.86 (m, 4H), 1.80-1.25 (m, 13H), 1.23-0.88 (m, 12H), 0.68 (s, 3H)。
7. 在25℃下將矽膠(24 g)及PCC (24.5 g,114 mmol)添加至
N-004-023_8
(20 g,57.0 mmol)於DCM (500 mL)中之懸浮液中。在25℃下攪拌混合物2小時。過濾混合物且用DCM (2×100 mL)洗滌濾餅。真空濃縮合併之濾液。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之
NA-004-023_9
(19 g,95%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.39 (s, 3H), 3.20 (s, 2H), 2.60-2.52 (m, 1H), 2.20-2.10 (m, 5H), 1.99-1.80 (m, 3H), 1.75-1.40 (m, 12H), 1.30-1.04 (m, 7H), 0.61 (s, 3H)。 8. 在25℃下將t-BuOK (12.2 g,54.5 mmol)添加至MePPh
3
Br (38.9 g,109 mmol)於THF (300 mL)中之懸浮液中。在添加之後,將反應混合物加熱至45℃且攪拌1小時。接著添加N-004-023_9 (19 g,35.9 mmol)且在45℃下攪拌反應混合物16小時。用NH
4
Cl (100 mL,飽和水溶液)處理混合物。分離有機層。用EtOAc (2×300 mL)萃取水相。將合併之有機相用飽和鹽水(2×200 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。在25℃下自MeOH/H
2
O (100 mL/100 mL)濕磨殘餘物,得到呈固體之
N-004-023_10
(17 g,90%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.68 (s, 1H), 3.39 (s, 3H), 3.19 (s, 2H), 2.10-2.04 (m, 1H), 2.03-1.90 (m, 3H), 1.75-1.56 (m, 12H), 1.49-1.25 (m, 4H), 1.22-0.89 (m, 8H), 0.57 (s, 3H)。 9. 將9-BBN二聚體(29.5 g,122 mmol)添加至
N-004-023_10
(17 g,49.0 mmol)於無水THF (300 mL)中之溶液中且在0℃下於N
2
下攪拌30 min。使反應混合物升溫至25℃ (室溫)且攪拌2小時。冷卻反應混合物。在0℃下藉由EtOH (100 mL)淬滅混合物。極緩慢地添加NaOH (98.0 mL,490 mol,5 M於水中)。在添加之後,添加緩慢H
2
O
2
(44.5 mL,490 mmol,11 M)直至內部溫度不再上升且在此期間將溫度維持在低於30℃。在50℃下再攪拌混合物1小時。接著混合物經冷卻,用水(2 L)處理且攪拌30 min。真空中過濾懸浮液,得到呈固體之
N-004-023_11
(17 g, 粗產物)。在25℃下自MeOH/H
2
O (100/100 mL)濕磨
N-004-023_11
(17 g,46.6 mmol)且攪拌1小時。在真空下過濾懸浮液,以獲得呈固體之
N-004-023_11
(14 g,不純)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.79-3.66 (m, 1H), 3.50-3.37 (m, 4H), 3.28 (s, 2H), 2.43 (s, 1H), 2.26-1.98 (m, 3H), 2.18-2.12 (m, 1H), 1.95-1.60 (m, 11H), 1.34-1.04 (m, 14H), 0.76 (s, 3H)。 10. 在25℃下將DMP (9.24 g,21.8 mmol)添加至
N-004-023_11
(4 g,10.9 mmol)於DCM (80 ml)中之溶液中。將一滴水添加至混合物且攪拌30分鐘。在低於10℃下用NaHCO
3
飽和水溶液(pH=7至8)淬滅反應混合物。分離濾液中之DCM相位且用飽和NaHCO
3
/Na2S
2
O
3
水溶液(1:1,2×50 mL)洗滌。有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到呈油狀物之
N-004-023_12
(1.8 g,46%)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.57-9.55 (m, 1H), 3.38 (s, 3H), 3.20 (s, 2H), 2.39-2.26 (m, 1H), 2.17-2.06 (m, 1H), 2.05-1.75 (m, 4H), 1.74-1.53 (m, 8H), 1.85-1.00 (m, 15H), 0.74 (s, 3H)。 11. 在0℃下將CsF (1.86 g,12.3 mmol)添加至
N-004-023_12
(1.8 g,4.96 mmol)於無水THF (20 mL)中之溶液中。在0℃下攪拌20 min之後,在0℃下添加TMSCF
3
(1.74 g,12.3 mmol)且攪拌1小時,接著添加TBAF.3H
2
O (6.25 g,19.8 mmol) 。使混合反應物升溫至50℃且再攪拌1小時。將反應混合物倒入至冰水(50 mL)中且攪拌10 min。用EtOAc (2×80 mL)萃取水相。將合併之有機相用飽和鹽水(2×80 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之
N-004-023_13
(1.2 g,56%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.03-3.98 (m, 1H), 3.39 (s, 3H), 3.20 (s, 2H), 2.17-1.80 (m, 7H), 1.73-1.41 (m, 10H), 1.28-0.95 (m, 13H), 0.71 (s, 3H)。 12. 在25℃下將DMP (2.34 g,5.54 mmol)添加至
N-004-023_13
(1.2 g,2.77 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌30分鐘之後,在低於10℃下用飽和NaHCO
3
水溶液(30 mL) (pH=7至8)淬滅反應混合物。接著添加DCM (30 mL)且攪拌混合物10 min。過濾懸浮液。分離濾液中之DCM相位且用飽和NaHCO
3
/Na
2
S
2
O
3
水溶液(1:1,2×50 mL)洗滌。有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到呈油狀物之
N-004-023_13A
(1.2 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.39 (s, 3H), 3.20 (s, 2H), 3.05-2.95 (m, 1H), 1.91-1.51 (m, 10H), 1.46-1.20 (m, 10H), 1.17-0.96 (m, 8H), 0.72 (s, 3H)。
13.
在0℃將NaBH
4
(210 mg,5.56 mmol)添加至
N-004-023_13A
(1.2 g,2.78 mmol)於MeOH (5 mL)之溶液中且攪拌30 min。在用MeOH/H
2
O (20/20 mL)處理之後,攪拌混合物10 min。用EtOAc (2×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之
48
(57 mg,5% )及呈固體之
45
(200 mg,不純)。
45
(200 mg,0.462 mmol)藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之
45
(120 mg,10% )。
45
:
1 H NMR
(400 MHz, CDCl
3
) δ 4.01-3.98 (m, 1H), 3.38 (s, 3H), 3.19 (s, 2H), 2.15-2.10 (m, 1H), 2.05-1.80 (m, 5H), 1.72-1.55 (m, 5H), 1.54-1.34 (m, 6H), 1.31-1.20 (m, 4H), 1.16-0.95 (m, 9H), 0.71 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.129 min,30-90AB_2MIN_E,純度100%,C
24
H
38
F
3
[M -HO
3
]
+
之MS ESI計算值383,實驗值383。
實例 46 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (46) 之合成 1.
在75℃下將(溴甲基)環戊烷(2.25 g,13.8 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (662 mg,27.6 mmol)及I
2
(70 mg,0.276 mmol)於THF (3 mL)中之懸浮液中。在75℃下攪拌混合物1小時。冷卻後,在15℃下緩慢地添加
S-500-6-1_1
(1 g,2.78 mmol)於THF (30 mL)中之溶液。在添加之後,在15℃下攪拌混合物2小時,用飽和NH
4
Cl (40 mL)及飽和檸檬酸(20 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之
S-500-6-13_1
及22位置處之異構體的混合物(900 mg,73%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.23 (m, 1H), 3.77-3.67 (m, 1H), 2.40-2.30 (m, 1H), 2.07-1.89 (m, 4H), 1.88-1.69 (m, 4H), 1.67-1.59 (m, 4H), 1.55-1.26 (m, 15H), 1.16-1.05 (m, 5H), 1.05-1.00 (m, 4H), 0.99-0.81 (m, 8H), 0.68 (s, 3H)。 2.將DMP (1.72 g,4.06 mmol)添加至
S-500-6-13_1
(900 mg,2.03 mmol)於DCM (30 mL)中之溶液中。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO
3
飽和水溶液(50 mL)淬滅直至水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3×40 mL)、飽和NaHCO
3
(40 mL)、鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈固體之粗產物
S-500-6-13_
2 (900 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.30-5.25 (m, 1H), 2.54-2.42 (m, 2H), 2.41-2.33 (m, 1H), 2.30-2.17 (m, 1H), 2.06-1.90 (m, 3H), 1.87-1.78 (m, 2H), 1.73-1.66 (m, 2H), 1.65-1.35 (m, 15H), 1.33-1.21 (m, 2H), 1.17-0.92 (m, 13H), 0.88-0.82 (m, 3H), 0.69 (s, 3H)。 3. 將NaBH
4
(3.46 g,102 mmol)每五分鐘添加五次至
S-500-6-13_2
(900 mg,2.04 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘,用飽和NH
4
Cl (50 mL) 淬滅且用EtOAc (3×20 mL)萃取。將合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之不純
46
(120 mg),其再次藉由SFC (管柱:AD (250 mm×30 mm,5um),梯度:45%至45% B (A= 0.05% NH
3
/H
2
O,B= MeOH),流速:60 mL/min)純化,得到呈固體之純
46
(100 mg,84%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.76-3.67 (m, 1H), 2.40-2.33 (m, 1H), 2.08-1.91 (m, 4H), 1.90-1.78 (m, 2H), 1.77-1.55 (m, 10H), 1.54-1.31 (m, 9H), 1.26-1.22 (m, 2H), 1.22-1.05 (m, 6H), 1.03 (s, 3H), 1.01-0.89 (m, 5H), 0.89-0.82 (m, 3H), 0.69 (s, 3H)。
LCMS
Rt=2.0 min層析中1.474 min,30-90AB_E,純度99%,C
30
H
49
O [M+H-H
2
O]
+
之MS ESI計算值425,實驗值425。
實例 47 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (47) 之合成 1.將Pd(OH)
2
(200 mg)添加至
30
(100 mg,0.239 mmol)於MeOH (10 mL)中之溶液中。在50℃下於H
2
(50 Psi)下攪拌混合物。混合物經過濾,濃縮且藉由combi-flash (0%至10% EtOAc/PE)純化,得到
47
(21 mg,21%)及呈白色固體之
36
(1 mg,1%)。
47 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.68-3.54 (m, 1H), 2.02-1.90 (m, 1H), 1.76-1.57 (m, 6H), 1.54-1.27 (m, 10H), 1.26-1.21 (m, 7H), 1.20-1.08 (m, 5H), 1.07-0.95 (m, 3H), 0.94-0.83 (m, 10H), 0.81 (s, 3H), 0.72-0.60 (m, 4H)。
LCMS
t
R
=2 min層析中1.290 min,30-90AB_ELSD,純度100.0%,C
28
H
47
[M+H-2H
2
O]
+
之MS ESI計算值383,實驗值383。
實例 48 : (3S,5R,8R,9R,10S,13S,14S,17R)-3-( 甲氧基甲基 )-13- 甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (48) 之合成 在0℃下將NaBH
4
(210 mg,5.56 mmol)添加至
N-004-023_13A
(1.2 g,2.78 mmol)於1. MeOH (5 mL)中之溶液中攪拌且30 min。在用MeOH/H
2
O (20/20 mL)處理之後,攪拌混合物10 min。用EtOAc (2×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之
48
(57 mg,5% )及呈固體之
45
(200 mg,不純)。
45
(200 mg,0.462 mmol)藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之
45
(120 mg,10%)。
48 : 1 H NMR
(400 MHz, CDCl
3
) δ 4.07-4.01 (m, 1H), 3.39 (s, 3H), 3.19 (s, 2H), 2.30-2.22 (m, 1H), 2.14-2.05 (m, 1H), 2.00-1.80 (m, 4H), 1.72-1.57 (m, 6H), 1.49-1.20 (m, 9H), 1.18-0.95 (m, 9H), 0.68 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.085 min,30-90AB_2MIN_E,純度100%,C
24
H
38
F
3
[M -HO
3
]
+
之MS ESI計算值383,實驗值383。
實例 49 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (49) 之合成 49
之合成描述於實例4中。
49 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.07-1.87 (m, 4H), 1.76-1.69 (m, 1H), 1.66-1.55 (m, 5H), 1.53-1.42 (m, 7H), 1.41-1.31 (m, 5H), 1.30-1.12 (m, 8H), 1.11-1.05 (m, 3H), 1.03 (s, 3H), 1.01-0.92 (m, 2H), 0.91-0.82 (m, 12H), 0.68 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.718 min,30-90AB_E,純度98%,C
33
H
53
[M+H-2H
2
O]
+
之MS ESI計算值449,實驗值449。
實例 50 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (50) 之合成 1.
在0℃下於N
2
下將MeMgBr (0.83 mL,2.49 mmol,3 M於乙醚中)逐滴添加至
N-8-7_1
(300 mg,0.832 mmol)於THF (20 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應物用飽和NH
4
Cl (50 mL)淬滅且用EtOAc (2×10 mL)萃取。合併之相位用鹽水洗滌(10 mL),經Na
2
SO
4
乾燥,經過濾,濃縮且藉由閃蒸塔(步驟2) (0%至10% EtOAc/PE)純化,得到呈固體之
50
(40 mg,29%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.98-3.89 (m, 1H), 1.99-1.84 (m, 2H), 1.69-1.56 (m, 6H), 1.54-1.45 (m, 2H), 1.43-1.29 (m, 6H), 1.28-1.17 (m, 4H), 1.17-1.12 (m, 4H), 1.12-0.94 (m, 5H), 0.92-0.84 (m, 7H), 0.82 (s, 3H), 0.68-0.61 (m, 4H)。 在7.0 min層析中,
LCMS
Rt = 3.428 min,30-90AB_E,純度100%,C
25
H
41
[M+H-2H
2
O]
+
之MS ESI計算值341,實驗值341。
實例 51 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (51) 之合成 1.
在25℃下於N
2
下將
S-500-6-29_2
(999 mg,1.22 M於THF中,4.87 mmol)逐滴添加至
N-8-1_1
(210 mg,05576 mmol)於THF (2 mL)中之溶液中。在25℃下攪拌16小時之後,反應混合物用飽和NH
4
Cl (10 mL)淬滅且用乙酸乙酯(3×10 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到粗產物,其藉由閃蒸塔(0%至15% EtOAc/PE)純化2次,得到不純產物(30 mg)。不純產物藉由ELSD製備型HPLC (管柱:Phenomenex Synergi C18 150×30 mm×4 um,梯度:90%至95% B (A=水(0.05% HCl),B=MeCN),流速:25 mL/min)進一步純化,得到呈固體之純
51
(4 mg,1.4%產率)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.84-3.76 (m, 1H), 3.45-3.32 (m, 5H), 2.62-2.39 (m, 1H), 1.99-1.85 (m, 2H), 1.73-1.62 (m, 4H), 1.53-1.40 (m, 7H), 1.39-1.31 (m, 5H), 1.30-1.21 (m, 7H), 1.20-1.13 (m, 4H), 1.12-1.10 (m, 5H), 0.99-0.93 (m, 1H), 0.89-0.86 (m, 6H), 0.85 (s, 3H), 0.83 (s, 3H), 0.68-0.61 (m, 4H)。 在7.0 min層析中,
LCMS
Rt = 5.669 min,30-90AB_E,純度100%,C
33
H
55
O [M+H-2H
2
O]
+
之MS ESI計算值467,實驗值467。
實例 52 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (52) 之合成 1. 將NaOH溶液(974 mg於6 mL H
2
O中,16.8 mmol)添加至
S-500-2-15_1
(900 mg,1.68 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在50℃下加熱混合物16小時。反應混合物用飽和NH
4
Cl (60 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到210 mg固體,其藉由SFC (管柱:AD (250 mm×30 mm,5 um),梯度:35%至35% B (A= 0.1% NH
3
/H
2
O,B= MeOH),流速:80 mL/min)純化,得到呈固體之
52
(150 mg,68%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.30-5.26 (m, 1H), 3.64-3.58 (m, 1H), 2.40-2.30 (m, 1H), 2.02-1.92 (m, 3H), 1.80-1.58 (m, 7H), 1.56-1.31 (m, 9H), 1.30-1.05 (m, 8H), 1.03 (s, 3H), 1.02-0.96 (m, 2H), 0.95-0.86 (m, 9H), 0.85-0.80 (m, 3H), 0.69 (s, 3H). 在2 min層析中,
LCMS
t
R
=1.335 min,30-90AB_ELSD,純度100.0%,C
29
H
47
[M+H-2H
2
O]
+
之MS ESI計算值395,實驗值395。
實例 53 : (3S,8S,9S,10R,13S,14S,17R)-10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-3-( 三氟甲基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (53) 之合成 1. 在0℃下向
N-004-027_1
(1.5 g,3.76 mmol)於無水THF (40 mL)中之溶液中添加CsF (1.42 g,9.40 mmol)。在0℃下攪拌20 min之後,在0℃下添加TMSCF
3
(1.33 g,9.40 mmol)且攪拌30 min。顏色變為淡黃色。添加TBAF.3H
2
O (4.74 g,15.0 mmol)且在50℃下攪拌30 min。將反應混合物倒入至冰水(100 mL)中。用EtOAc (2×100 mL)萃取水相。合併之有機相用飽和鹽水(2×100 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮,得到呈黃色固體之異構體混合物(1.45 g,粗產物),其藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之
53
(340 mg,24%)及呈白色固體之
1
(200 mg,14%)。
53 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.38-5.36 (m, 1H), 4.06-3.94 (m, 1H), 2.49 (s, 2H), 2.09-1.58 (m, 13H), 1.48-0.85 (m, 14H), 0.73 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.134 min,30-90AB_2MIN_E,純度99%。
MS
50-100_1_4min.m,C
24
H
33
F
6
O[M+H-H
2
O]
+
之MS ESI計算值451,實驗值451。
實例 54 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (54) 之合成 1. 在15℃下將NaBH
4
(1.77 g,46.8 mmol)逐份添加至
S-500-6-1_3
(520 mg,1.17 mmol)於THF (5 mL)及MeOH (10 mL)中之溶液中。在15℃下攪拌混合物20 min。混合物用NH
4
Cl (20 mL,飽和水溶液)淬滅且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到
S-500-6-1
(300 mg,不純)及
54
(170 mg,不純)。 不純
54
(220 mg,不純)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到固體。在60℃下將固體溶解於MeCN (50 mL)中且真空濃縮,得到呈固體之
54
(120 mg,23%)。
54 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.24 (m, 1H), 3.62-3.52 (m, 1H), 2.42-2.31 (m, 1H), 2.11-1.90 (m, 3H), 1.72-1.35 (m, 15H), 1.29-1.08 (m, 8H), 1.03 (s, 3H), 1.01-0.96 (m, 2H), 0.93 (d,
J
= 6.8 Hz, 3H), 0.90 (s, 9H), 0.85 (t,
J
= 7.6 Hz, 3H), 0.70 (s, 3H)。 在7.0 min層析中,
LCMS
Rt = 5.463 min,30-90_AB_E,純度100%,C
30
H
49
[M+H-2H
2
O]
+
之MS ESI計算值409,實驗值409。
實例 55 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1R,2S)-1- 羥基 -1- 苯基丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇
(55)
之合成 1. 在0℃下將
N-8-7_1
(300 mg,0.832 mmol)於THF (5 mL)中之溶液添加至PhMgBr (1.38 mL,3 M於乙醚中,4.15 mmol)於THF (10 mL)中之溶液中,隨後在0℃下攪拌反應混合物3小時。接著,在25℃下攪拌反應混合物5小時。在0℃下用水(10 mL)淬滅反應混合物。過濾溶液且用EtOAc (10 mL)洗滌濾餅。用EtOAc (3×15 mL)萃取水相。合併之有機相用飽和鹽水(2×10 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至1/1)純化,得到呈固體之
59
及
19
(200 mg,粗產物)。粗產物藉由SFC (管柱:AD (250 mm×30 mm,5um),梯度:25%至25% B (A= 0.1% NH
3
/H
2
O,B= EtOH ),流速:60 mL/min)純化,得到
55
(峰值2,55 mg, 15%)及呈固體之
19
(峰值1,21 mg,6%)。
55 : 1 H NMR
(400MHz, CDCl
3
) δ 7.38-7.28 (m, 4H), 7.25-7.20 (m, 1H), δ4.95- 4.90 (m, 1H), 2.13-2.01 (m, 1H), 1.98-1.88 (m, 1H), 1.77-1.59 (m, 6H) , 1.57-1.43 (m, 6H), 1.43-0.93 (m, 13H), 0.91-0.85 (m, 3H), 0.83 (s, 3H), 0.76-0.72 (m, 3H), 0.68 (s, 4H)。 在2.0層析層析中,
LCMS
Rt=1.239 min,30-90AB_2 min.,純度100%,C
30
H
43
[M-2H
2
O+H]
+
之MS ESI計算值403,實驗值403。 在3 min層析中,SFC Rt=1.192 min,OJ_3_EtOH_DEA_5_40_25ML,99%de。
實例 56 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (56) 之合成 1. 向
81
(1 g,3.26 mmol)於MeOH (30 mL)及THF (10 mL)中之溶液中添加Pd(OH)
2
(1 g,<1%水)。接著在50℃下於50 psi下氫化混合物48小時。在無監測之情況下經由矽藻土墊過濾混合物且在真空中濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到呈白色固體之
56
(331 mg,33%)。
1 H NMR
(400 MHz, CDCl3) δ 4.09-3.99 (m, 1H), 2.18-2.13 (m, 1H), 1.99-1.78 (m, 4H), 1.75-1.59 (m, 3H), 1.50-1.3 (m, 7H), 1.34-1.22 (m, 6H), 1.21-1.00 (m, 10H), 0.96 (s, 3H), 0.94-0.89 (m, 3H), 0.67 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.184 min,30-90AB_E,純度100%,C
25
H
40
F
3
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 57 : (3S,5R,8R,9S,10S,13S,14S,17R)-3,10,13- 三甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (57) 之合成 1. 在10℃下於N
2
下攪拌LiCl (13.9 g,329 mmol,無水)於THF (500 mL,無水)中之懸浮液30分鐘。在10℃下添加FeCl
3
(27.8 g,172 mmol,無水)。使混合物冷卻至-30℃。在-30℃下向混合物中滴落添加MeMgBr (209 mL,3M於二乙醚中)。在-30℃下攪拌10分鐘之後,在-30℃下添加
S-500-2-12_1
(50 g,157 mmol)。在-15℃下攪拌混合物2小時且用檸檬酸(500 mL,10%水溶液)淬滅。用EtOAc (3×800 mL)萃取混合物。合併之有機相用飽和鹽水(300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物,其藉由矽膠管柱(PE/DCM/EtOAc=1/1/1)純化,得到呈固體之
S-500-2-12_2
(50 g,86%)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.57-2.48 (m, 1H), 2.23-2.13 (m, 1H), 2.06-1.78 (m, 3H), 1.64-1.25 (m, 14H), 1.24-1.01 (m, 10H), 0.96 (s, 3H), 0.74 (s, 1H), 0.60 (s, 3H)。 2. 在20℃下向PPh
3
MeBr (79.7 g,244 mmol)於THF (400 mL)中之懸浮液中添加t-BuOK (25.1 g,224 mmol)。在40℃下攪拌30 min之後,在40℃下添加
S-500-2-12_2
(50 g,150 mmol)於THF (100 mL)中之溶液且在40℃下攪拌反應混合物1小時。將反應混合物倒入至50 g冰中且攪拌15分鐘。分離有機層且用用EtOAc (3×50 mL)萃取水相。在真空中濃縮合併之有機相,得到油狀物。粗產物在MeOH/H
2
O (200 mL/200 mL)中經濕磨且過濾,得到呈固體之
S-500-2-12_3
(55 g,88%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.69 (s, 1H), 2.06-1.79 (m, 4H), 1.75 (s, 3H), 1.73-1.58 (m, 4H), 1.56-1.25 (m, 9H), 1.22 (s, 3H), 1.21-1.02 (m, 6H), 1.01-0.94 (s, 3H), 0.93-0.74 (m, 1H), 0.55 (s, 3H)。
3.
向
S-500-2-12_3
(55 g,166 mmol)於THF (500 mL)中之溶液中添加9-BBN二聚體(60.7 g,249 mmol)且在25℃下於N
2
下攪拌1小時,形成固體。向反應混合物添加乙醇(95.3 mL,1.66 mmol)及NaOH (166 mL,5 M,830 mmol)。使混合物變得澄清。在25℃下逐滴添加H
2
O
2
(132 mL,10 M,1.32 mmol)且使內部溫度升至回流(75℃)。添加且攪拌16小時之後使混合物冷卻,形成固體。在25℃下向混合物中添加Na
2
S2O
3
(500 mL,20%水溶液)及水(1 L)。攪拌混合物1小時。在關閉攪拌器之後,形成澄清底層及上懸浮液層。丟棄澄清底層。向上懸浮液層添加水(2 L)。攪拌混合物15分鐘。過濾混合物,得到呈固體之
S-500-2-12_4
(50 g,不純)。在100℃下在EtOH/H
2
O (90 mL/10 mL)中濕磨
S-500-2-12_4
(50 g,143 mmol,不純)2小時,接著使其冷卻至15℃且過濾,得到呈固體之
S-500-2-12_4
(38 g,不純)。在100℃下在EtOH/H
2
O (45 mL/5 mL)中濕磨
S-500-2-12_4
(38 g,109 mmol,不純)2小時,接著使其冷卻至15℃且過濾,得到呈固體之
S-500-2-12_4
(28 g,43%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.67-3.59 (m, 1H), 3.39-3.32 (m, 1H), 2.01-1.75 (m, 4H), 1.69-1.58 (m, 3H), 1.54-1.24 (m, 10H), 1.23-1.14 (m, 9H), 1.09-1.02 (m, 5H), 0.96 (s, 3H), 0.74 (m, 1H), 0.67 (s, 3H)。 4. 向
N-004-016_1
(10.0 g,28.6 mmol)於DCM (100 mL)中之溶液中添加DMP (24.2 g,57.2mmol)。接著將H
2
O (0.2 mL)添加至混合物。此後,在25℃下攪拌反應物1小時。向反應混合物中添加飽和NaHCO
3
水溶液 (100 mL)。過濾混合物且用DCM (2×100 mL)洗滌濾餅。分離混合物,且用DCM (2×100 mL)萃取水相。合併之有機層用飽和NaHCO
3
/Na
2
S2O
3
水溶液(100 mL/100 mL)及鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到白色固體。殘餘物藉由矽膠層析(PE/EtOAc=0%至20%)純化,得到呈白色固體之
N-004-016_2
(3.5 g,35%)。
1 H NMR
(400 MHz, CDCl3) δ 9.58-9.54 (m, 1H), 2.39-2.32 (m, 1H), 1.96-1.77 (m, 4H), 1.69-1.31 (m, 14H), 1.23-1.16 (m, 6H), 1.14-1.02 (m, 5H), 0.96 (s, 3H), 0.76-0.59 (m, 4H)。 5. 在0℃下向
N-004-016_2
(1.5 g,4.32 mmol)、CsF (328 mg,2.16 mmol)於THF (10 mL)中之溶液中添加TMSCF
3
(1.53 g,10.8 mmol) 。在25℃下攪拌混合物1小時。向混合物中添加TBAF.
3
H
2
O (3.4 g,10.8 mmol)。在25℃下攪拌混合物2小時。混合物用水(20 mL)淬滅且用EtOAc (2×30 mL)萃取。合併之有機相用鹽水(50 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且在真空中濃縮。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之
N-004-017_1
(700 mg,39%)。
1 H NMR
(400 MHz, CDCl3) δ 4.07-3.96 (m, 1H), 2.21-2.09 (m, 1H), 1.99-1.77 (m, 5H), 1.72-1.29 (m, 15H), 1.24-1.20 (m, 4H), 1.13-1.01 (m, 5H), 0.96 (s, 3H), 0.89-0.84 (m, 1H), 0.76-0.64 (m, 3H), 0.60 (s, 1H)。 6. 在25℃下向
N-004-017_1
(700 mg,1.68 mmol)於吡啶(5 mL)中之溶液中添加苯甲醯氯(354 mg,2.52 mmol)及DMAP (102 mg,0.84 mmol)。將混合物加熱至60℃且攪拌10小時。反應混合物用EtOAc (10 mL)稀釋,接著用水(10 mL)淬滅。用EtOAc (2×20 mL)萃取水溶液。合併之有機層用鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到白色固體。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之
N-004-017_2
(600 mg,68%)。 在2 min層析中,
LCMS
R
t
=1.464 min,30-90AB_2MIN_E.M,純度92%。 SFC條件:
峰值 1
:Rt=2.434 min且在8 min層析中
峰值 2
:Rt=2.541 min,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M (管柱:Chiralcel OD -3 100×4.6 mm I.D.,3um行動相:A:CO2 B:甲醇(0.05% DEA),梯度:在4.5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續1 min,流速:2.8 mL/min,min管柱溫度:40℃)。
7. N-004-017_2
(600 mg,1.15 mmol) 藉由SFC (管柱:Chiralcel OD (250 mm×30 mm,5 um),梯度:20%至20% B(A=0.1% NH
3
/H
2
O,B=MeOH),流速:60 mL/min)純化,得到
N-004-017_3
(峰值2,190 mg,不純,31%)、呈白色固體之
N-004-018_1
(峰值1,180 mg,30%)。不純
N-004-017_3
(190 mg,0.36 mmol) 藉由SFC (管柱:OD (250 mm×30 mm,5 um),梯度:20%至20% B (A=0.1% NH
3
/H
2
O,B=MeOH),流速:60 mL/min)純化,得到呈白色固體之
N-004-017_3
(100 mg,53%)。
N-004-018_1 : 1 H NMR
(400 MHz, CDCl3) δ 8.12-8.06 (m, 2H), 7.65-7.59 (m, 1H), 7.53-7.46 (m, 2H), 5.68-5.58 (m, 1H), 2.15-2.03 (m, 2H), 1.97-1.69 (m, 3H), 1.67-1.57 (m, 3H), 1.44-1.24 (m, 9H), 1.23-1.13 (m, 11H), 1.12-1.97 (m, 3H), 0.94 (s, 3H), 0.72 (s, 3H)。 在2 min層析中,
LCMS
Rt= 1.525 min,30-90AB_2min_220&254.lcm,純度100%。 在8 min層析中,
SFC _D1
Rt = 2.450 min,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M,100%de。
N-004-017_3 : 1 H NMR
(400 MHz, CDCl3) δ 8.14-8.06 (m, 2H), 7.64-7.57 (m, 1H), 7.52-7.44 (m, 2H), 5.63-5.52 (m, 1H), 2.11 (s, 1H), 2.06-1.77 (m, 6H), 1.72-1.62 (m, 3H), 1.44-1.32 (m, 8H), 1.28-1.18 (m, 12H), 0.99-0.93 (m, 4H), 0.65 (s, 3H)。 在2 min層析中,
LCMS
Rt = 1.529 min,30-90AB_2min_220&254.lcm,純度100%。 在8 min層析中,
SFC
Rt = 2.544 mi,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M,98%de。 8. 向
N-004-018_1
(180 mg,0.34 mmol)於THF (3 mL)及MeOH (1.5 mL)及水(1.5 mL)中之溶液中添加KOH (96.5 mg,1.72 mmol)。在60℃下攪拌混合物16小時。將混合物倒入水(20 mL)中且用EtOAc (2×40 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之
57
(114 mg,79%)。
1 H NMR
(400 MHz, CDCl3) δ 4.00 (brs, 1H), 2.03-1.77 (m, 8H), 1.69-1.61 (m, 2H), 1.54-1.49 (m, 1H), 1.47-1.24 (m, 10H), 1.22 (s, 3H), 1.21-1.10 (m, 4H), 1.08-1.01 (m, 4H), 0.96 (s, 3H), 0.69 (s, 3H)。 在2 min層析中,
LCMS
Rt = 1.169 min,30-90AB_2MIN_E.M,純度95%,C
24
H
38
F
3
O[M+H-H
2
O]
+
之MS ESI計算值399,實驗值399。 在10 min最終C18 3×50 mm 3 um中,
HPLC
Rt = 5.44 min,30-90_AB_1.2ML_E.MET,純度100%。
實例 58 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1R,2S)-1- 環戊基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (58) 之合成 1.
在0℃下於N
2
下將含
N-8-7_1
(500 mg, 1.38 mmol)之THF (5 mL)添加至環戊基溴化鎂(1.38 mL,3 M於THF中)。在15℃下攪拌18小時之後,反應混合物用飽和NH
4
Cl (10 mL)淬滅,且用EtOAc (2×10 mL)萃取。合併之有機層用鹽水(10 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之
N-8-15_1
(170 mg,29%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.39-3.22 (m, 1H), 2.00-1.81 (m, 4H), 1.70-1.41 (m, 12H), 1.41-1.13 (m, 13H), 1.13-0.95 (m, 6H), 0.95-0.79 (m, 11H), 0.65 (s, 3H)。 2. 將DMP (0.881 g,2.08 mmol)添加至
N-8-15_1
(300 mg,0.696 mmol)於DCM (20 mL)中之溶液中。在15℃下攪拌10 min之後,反應混合物用飽和NaHCO
3
(10 mL)淬滅。混合物用DCM (3×20 mL)萃取。合併之有機相用飽和Na
2
S
2
O
3
(3×20 mL)及鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0%至20% EtOAc/PE)純化,得到呈固體之
N-8-15_2
(240 mg)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.98-2.89 (m, 1H), 2.62-2.55 (m, 1H), 1.98-1.87 (m, 1H), 1.81-1.72 (m, 4H), 1.71-1.49 (m, 10H), 1.41-1.29 (m, 4H), 1.29-1.19 (m, 6H), 1.14-0.98 (m, 9H), 0.94-0.87 (m, 4H), 0.82 (s, 3H), 0.67 (m, 5H)。
3.
向
N-8-15_2
(240 mg,0.559 mmol)於MeOH (3 mL)及THF (2 mL)中之混合物添加NaBH4 (550 mg,14.5 mmol)。在15℃下攪拌混合物0.5 h。添加另一批NaBH
4
(550 mg,14.5 mmol)。再攪拌反應混合物1 h。向反應混合物中添加水(5 mL)。用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(10 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之
58
(7 mg,5%),且
78
(50 mg,不純)藉由閃蒸塔(0-5% EtOAc/PE)進一步純化,得到呈固體之
78
(17 mg,12%)。
58 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.64-3.59 (m, 1H), 2.09-1.90 (m, 2H), 1.89-1.70 (m, 4H), 1.70-1.45 (m, 11H), 1.45-1.32 (m, 5H), 1.32-1.19 (m, 9H), 1.19-1.08 (m, 3H),1.08-0.98 (m, 5 H), 0.98-0.89 (m, 4 H), 0.84 (s, 3H), 0.68 (s, 3H)。 在7 min層析中,
LCMS
Rt=4.832 min,30-90AB_7MIN_E,純度100%,C
29
H
47
[M+H-2H
2
O]
+
之MS ESI計算值395,實驗值395。 在10 min層析中,
HPLC
Rt=6.338 min,50-100AB_10MIN.M,純度98%。
實例 59 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (59) 之合成 1. 將Pd(OH)
2
(200 mg,無水)添加至
S-500-6-30
(140 mg,0.288 mmol)於MeOH (30 mL)中之溶液中。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到
S-500-6-25
(27 mg,19%)及呈固體之
S-500-6-26
(42 mg,30%)。
S-500-6-25 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.84-3.77 (m, 1H), 1.99-1.84 (m, 2H), 1.81-1.72 (m, 1H), 1.68-1.56 (m, 4H), 1.53-1.43 (m, 5H), 1.42-1.32 (m, 9H), 1.31-1.23 (m, 5H), 1.22-.12 (m, 7H), 1.12-1.00 (m, 5H), 0.99-0.95 (m, 4H), 0.94-0.85 (m, 12H), 0.66 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.797 min,30-90AB_E,純度100%,C
33
H
55
[M+H-2H
2
O]
+
之MS ESI計算值451,實驗值451。
實例 60 : (3S,5S,8R,9S,10S,13S,14S,17R)-3 乙基 -17-((2S,3R)-3- 羥基 -4-((1R,2S)-2- 甲基環丙基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (60) 之合成 1.
在0℃下向
N-8-7_2
(1 g,2.41 mmol)於THF (50 mL)中之溶液添加LiAlH
4
(914 mg,24.1 mmol)。在66℃下加熱灰色懸浮液18小時。將反應混合物冷卻至0℃,藉由冰水(914 mg)、接著15% w/w NaOH水溶液(914 mg)及水(2.74 g)淬滅。過濾混合物且濾餅用DCM (3 × 50 mL)洗滌。濃縮濾液,得到殘餘物,其藉由急驟層析(乙酸乙酯,10%於PE中)純化兩次,得到
N-8-7_3
(192 mg,19%)及呈油狀物之
N-8-7_3A
(397 mg,39%)。
N-8-7_3 : 1 H NMR
(400 MHz, CDCl3) δ 5.59 - 5.36 (m, 2H), 3.69 - 3.61 (m, 1H), 2.25 - 2.12 (m, 1H), 2.08 - 1.81 (m, 3H), 1.68 (d, J = 10.0 Hz, 3H), 1.64 - 1.54 (m, 9H), 1.53 - 1.15 (m, 11H), 1.14 - 0.92 (m, 5H), 0.92 - 0.85 (m, 5H), 0.83 (s, 4H), 0.69 - 0.60 (m, 4H)。
N-8-7_3A : 1 H NMR
(400 MHz, CDCl
3
) δ 5.62 - 5.37 (m, 2H), 3.62 (br d,
J
=10.0 Hz, 1H), 2.20 - 2.06 (m, 1H), 1.99 - 1.61 (m, 6H), 1.61 - 1.44 (m, 11H), 1.43 - 1.18 (m, 5H), 1.16 - 0.94 (m, 6H), 0.94 - 0.85 (m, 5H), 0.82 (s, 5H), 0.70 - 0.58 (m, 6H)。
2.
在0℃下經15 min之時段向二乙基鋅(1 M於甲苯中,4.31 mL,4.31 mmol)於0℃下之DCM (15 ml )中之溶液添加CH
2
I
2
(2.31 g,8.63 mmol)。在0℃下攪拌乳白色懸浮液10 min,且經由注射器快速添加Charette配位體((4R,5R)-2-(第三丁基)-N4,N4,N5,N5-四甲基-1,3,2-二氧硼戊環-4,5-二甲醯胺) (233 mg,0.8638 mmol)及
N-8-7_3A
(300 mg,0.7199 mmol)於DCM (20 ml)中之預製溶液,隨即反應混合物變為澄清。使溶液達至25℃且在此溫度下攪拌16 h。接著藉由添加飽和NH
4
Cl水溶液(150 ml)淬滅反應,分離各相且用DCM (3×100 ml)萃取水相。合併之有機相用飽和NaHCO
3
水溶液(150 mL)、飽和Na
2
S
2
O
3
水溶液(150 mL)及鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且在減壓下濃縮,且殘餘物藉由閃蒸塔層析(11%乙酸乙酯/PE)純化,得到呈固體之
N-8-7_4A
(140 mg,45%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.81-3.72 (m, 1H), 1.97-1.90 (m, 1H), 1.76-1.58 (m, 4H), 1.53-1.44 (m, 4H), 1.43 - 1.15 (m, 10H), 1.13-0.91 (m, 8H), 0.90-0.78 (m, 12H), 0.66 (s, 3H), 0.54-0.34 (m, 4H), 0.32-0.22 (m, 2H), 0.19-0.12 (m, 1H)。 3. 在25℃下向
N-8-7_4A
(140 mg,0.325 mmol)於吡啶(5 mL)中之溶液中添加苯甲醯氯(91.3 mg,0.65 mmol),隨後添加DMAP (15.8 mg,0.13 mmol)。在60℃下攪拌反應混合物16小時。用DCM (80 mL)稀釋反應混合物。DCM相位用水(100 mL)、1.0 M HCl水溶液(2×100 mL)、10% NaHCO
3
(2×100 mL)水溶液、鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且在真空中濃縮,得到油狀物,其藉由閃蒸塔(1%乙酸乙酯/PE)純化,得到呈油狀物之
N-8-7_5A
(180 mg,不純),其藉由閃蒸塔(PE)進一步純化,得到呈固體之
N-8-7_5A
(110 mg,61%)。 在2 min層析中,
LCMS
Rt=1.439 min,5-95AB_220&254,純度93%,C
36
H
53
O
2
[M-H
2
O+H]
+
之MS ESI計算值517.8,實驗值517.8。
SFC
峰值1:Rt=4.079 min,且在10 min層析中峰值2:Rt=4.345 min,AD_3_EtOH_DEA_5_40_25ML (「Chiralpak AD-3 150×4.6 mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。
4. N-8-7A_5
(110 mg,0.206 mmol)藉由SFC (管柱:AD (250 mm×30 mm,5 um),梯度:30%至30% B (A=0.1% NH
3
/H
2
O,B=EtOH),流速:60 mL/min)純化,得到呈固體之不純
N-8-7A_6
(峰值1,54 mg,50%)及呈固體之不純
N-8-8A_1
(峰值2,23 mg,不純)。 在10 min層析中,
SFC
Rt=4.088 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
5.
向
N-8-7A_6
(54 mg,0.101 mmol)於THF/MeOH (1.5 mL/1.5 mL)中之溶液添加含KOH (45.2 mg,0.807 mmol)之水(0.5 mL)。在50℃下攪拌反應混合物16小時。向混合物中添加HCl (0.2 M,50 mL)。用DCM (2 × 60 mL)萃取懸浮液。合併之有機相用3% NaHCO
3
水溶液(80 mL)、鹽水(80 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到固體,其藉由快速層析(15%乙酸乙酯/PE)純化,得到呈固體之
60
(21 mg,48%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.80-3.75 (m, 1H), 1.97-1.91 (m, 1H), 1.76-1.58 (m, 7H), 1.54-1.27 (m, 7H), 1.26-1.06 (m, 7H), 1.04 (d,
J
= 6.0 Hz, 4H), 0.95 (s, 3H), 0.90-0.84 (m, 8H), 0.82 (s, 4H), 0.66 (s, 3H), 0.64-0.59 (m, 1H), 0.52-0.37 (m, 2H), 0.32-0.22 (m, 2H)。 在2 min層析中,
LCMS
Rt=1.327 min,30-90AB_2MIN_E,純度100%,C
29
H
49
O [M-H
2
O+H]
+
之MS ESI計算值413.4,實驗值413.4。
實例 61 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (61) 之合成 1.向
S-500-6-11_1
(580 mg,1.44 mmol)於DCM (30 mL)中之溶液添加DMP (1.22 g,2.88 mmol)。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO
3
飽和水溶液(50 mL)淬滅直至含水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3 × 40 mL)、飽和NaHCO
3
(40 mL)、鹽水(40 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到呈白色固體之粗產物
S-500-6-11_1A
(550 mg,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.34-5.25 (m, 1H), 2.58-2.29 (m, 4H), 2.08-1.90 (m, 3H), 1.78-1.56 (m, 9H), 1.54-1.35 (m, 6H), 1.31-1.21 (m, 2H), 1.19-1.08 (m, 5H), 1.06-0.99 (m, 5H), 0.93-0.82 (m, 6H), 0.69 (s, 3H)。 2.以五個兩分鐘間隔向
S-500-6-11A
(550 mg,1.37 mmol)於THF (4 mL)及MeOH (2 mL)中之溶液添加NaBH
4
(1.39 g,41.1 mmol)。在15℃下攪拌混合物30分鐘。混合物用飽和NH
4
Cl (20 mL)淬滅,且用EtOAc (3 × 6 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈白色固體之不純
61
(120 mg),其再藉由combi-flash (0-15% EtOAc/PE)進一步純化,得到呈白色固體之純
61
(150 mg,75%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.25 (m, 1H), 3.73-3.59 (m, 1H), 2.44-2.29 (m, 1H), 2.08-1.92 (m, 3H),1.76-1.57 (m, 6H), 1.54-1.26 (m, 10H), 1.25-1.18 (m, 3H), 1.17-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.88 (m, 8H), 0.87-0.82 (m, 3H), 0.69 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.345 min,30-90AB_E,純度100%,C
27
H
45
O [M+H-H
2
O]
+
之MS ESI計算值385,實驗值385。
實例 62 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (62) 之合成 1.在0℃下向
S-500-6-1_1
(500 mg,1.39 mmol)及CsF (105 mg,695 umol)於THF (5 mL)中之溶液添加TMSCF
3
(493 mg,3.47mmol)。在25℃下攪拌混合物1小時且用TBAF.3H
2
O (1.09 g,3.47 mmol)處理。在25℃下攪拌混合物2小時,用水(100 mL)淬滅且用EtOAc (2 × 50 mL)萃取。合併之有機相用鹽水(100 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE:EA=10:1)純化,得到呈白色固體之
S-500-6-1_2
(400 mg,67%)。
1 H NMR
(400 MHz, CDCl3) δ 5.33-5.24 (m, 1H), 4.06-4.00 (m, 1H), 2.38-2.35 (m, 1H), 2.08-1.82 (m, 6H), 1.77-1.69 (m, 1H), 1.62-1.20 (m, 13H), 1.16-1.00 (m, 8H), 0.99-0.92 (m, 1H), 0.87-0.83 (m, 4H), 0.74-0.64 (m, 3H)。 2. 3.5 g
S-500-6-1_2
藉由SFC (管柱:AD (250mm×30mm,5um),梯度:40-40% B (A=0.05% NH
3
/H
2
O,B=MeOH),流速:200 mL/min)分離,得到純
81
(1 g,28%,峰值1)及呈白色固體之
62
(1871 mg,53%,峰值2)。
62 : 1 H NMR
(400 MHz, CDCl3) δ 5.30-5.28 (m, 1H), 4.03-3.99 (m, 1H), 2.38-2.34 (m, 1H), 2.10-1.83 (m, 6H), 1.78-1.55 (m, 5H), 1.52-1.32 (m, 6H), 1.31-1.01 (m, 12H), 0.98-0.92 (s, 1H), 0.85 (t,
J
= 8 Hz, 3H), 0.73 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.219 min,30-90 AB,純度100%,C
25
H
38
F
3
O [M+H-H
2
O]
+
之MS ESI計算值411,實驗值411。 在10 min層析中,
SFC
峰值2:Rt=5.262 min,AD_3_EtOH_DEA_5_40_25ML,99%de。
實例 63 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4-( 四氫 -2H- 哌喃 -4- 基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (63) 之合成 1.向
N-6-10_1
(180 mg,0.318 mmol)於THF (2 mL)及甲醇(1 mL)中之溶液添加水(1 mL)及KOH (177 mg,3.17 mmol)。在50℃下攪拌混合物18小時。將反應混合物冷卻,用水(5 mL)稀釋,用10% HCl (0.2 mL)酸化且用EtOAc (3 × 5 mL)萃取。合併之有機層經無水硫酸鈉乾燥、過濾且濃縮。殘餘物藉由矽膠管柱層析(10-30% EtOAc/PE)純化,得到呈固體之
63
(108 mg,74%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.98-3.90 (m, 2H), 3.85-3.77 (m, 1H), 3.44-3.33 (m, 2H), 1.99-1.82 (m, 2H), 1.69-1.58 (m, 6H), 1.57-1.45 (m, 6H), 1.43-1.29 (m, 7H), 1.28-1.14 (m, 7H), 1.13-0.95 (m, 5H), 0.93-0.84 (m, 7H), 0.83 (s, 3H), 0.69-0.61 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.167 min,30-90AB,純度100%。 C
30
H
49
O [M-2H
2
O+H]
+
之
MS
ESI計算值425,實驗值425。
實例 64 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (64) 之合成 1.在-70℃下於N
2
下向
E-3_1
(20.0 g,63.1 mmol)於THF (300 mL)中之溶液緩慢添加EtMgBr (42 mL,126 mmol,3 M於醚中)。添加之後,在-70℃下攪拌混合物2小時。混合物用飽和NH
4
Cl (500 mL)淬滅,且用EtOAc (3 ×500 mL)萃取。合併之有機相用鹽水(500 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
E-3_2
(6.50 g,30%)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.57-2.49 (m, 1H), 2.23-1.80 (m, 6H), 1.78-1.52 (m, 4H), 1.50-1.02 (m, 17H), 0.97 (s, 3H), 0.95-0.80 (m, 4H), 0.60 (s, 3H)。 2.在15℃下於N
2
下向MePPh
3
Br (13.3 g,37.4 mmol)於THF (200 mL)中之懸浮液添加t-BuOK(4.19 g,37.4 mmol)。在50℃下攪拌混合物30 min。在50℃以下向混合物逐份添加
E-3_2
(6.50 g,18.7 mmol)。在50℃下攪拌混合物1小時。向混合物添加NH
4
Cl (400 mL)。有機層經分離且真空濃縮,得到粗產物,其在50℃下自MeOH/水(200 mL,1:1)濕磨而得。冷卻之後過濾混合物,且固體用MeOH/水(2 × 30 mL,1:1)洗滌並真空濃縮,得到呈固體之
E-3_3
(5.8 g,不純)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.06-1.97 (m, 1H), 1.94-1.55 (m, 12H), 1.53-1.05 (m, 16H), 0.97 (s, 3H), 0.95-0.85 (m, 3H), 0.55 (s, 3H)。 3.在15℃下於N
2
下向
E-3_3
(5.80 g,16.8 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(8.19 g,33.6 mmol)。在60℃下攪拌反應混合物1小時。將混合物冷卻至15℃。在15℃下添加乙醇(7.72 g,168 mmol)。在15℃下逐滴添加NaOH水溶液(33.6 mL,5 M,168 mmol)。在15℃下逐滴添加H
2
O
2
(16.8 mL,10.0 M,168 mmol)。在60℃下攪拌所得混合物1小時。用EtOAc (3 × 100 mL)萃取水相。合併之有機相用鹽水(2 × 100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。在65℃下自CH
3
OH/H
2
O = 1/1 (150 mL)濕磨殘餘物,得到呈固體之
E-3_4
(2.80 g,46%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.66-3.61 (m, 1H), 3.38-3.32 (m, 1H), 2.03-1.56 (m, 4H), 1.56-1.51 (m, 5H), 1.51-1.10 (m, 16H), 1.10-1.02 (m, 6H), 0.97 (s, 3H), 0.96-0.88 (m, 3H), 0.67 (s, 3H)。 4.向
E-3_4
(2.50 g,6.89 mmol)於DCM (50 mL)中之溶液添加DMP (5.80 g,13.7 mmol)。此後,在20℃下攪拌反應物30 min。向反應混合物添加NaHCO
3
飽和水溶液(50 mL)、Na
2
S
2
O
3
飽和水溶液(30 mL),用DCM (2 × 20mL)萃取。合併之有機層用NaHCO
3
飽和水溶液(3 × 10 mL)及鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
E-3_5
(2.45 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.60-9.55 (m, 1H), 2.26 (s, 1H), 1.98-1.70 (m, 6H), 1.70-1.51 (m, 6H), 1.51-1.00 (m, 12H), 1.00-0.89 (m, 10H), 1.75-0.65 (m, 4H)。 5.在0℃下於N
2
下向
E-3_5
(2.45 g,6.79 mmol)於THF (10 mL)中之溶液添加異丁基溴化鎂(33.9 mL,2 M於THF中,67.9 mmol)。在20℃下攪拌混合物16小時。向混合物添加NH
4
Cl (20 mL,飽和水溶液),用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,真空濃縮,且藉由閃蒸塔(0-20% EtOAc/PE)純化,得到呈固體之
E-3_6
(1.6 g,55%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.65-3.55 (m, 1H), 2.01-1.85 (m, 3H), 1.85-1.49 (m, 3H), 1.49-1.36 (m, 12H), 1.36-1.22 (m, 10H), 1.22-1.02 (m, 9H), 1.02-0.98 (m, 4H), 0.98-0.80 (m, 7H), 0.66 (s, 3H)。
6.
向
E-3_6
(1.6 g,3.69 mmol)於DCM (30 mL)中之溶液添加DMP (3.12 g,7.38 mmol)。此後,在20℃下攪拌反應物30 min。向反應混合物添加NaHCO
3
飽和水溶液(20 mL)、Na
2
S
2
O
3
飽和水溶液(20 mL),接著用DCM (2 × 20 mL)萃取。合併之有機層用NaHCO
3
飽和水溶液(3 × 10 mL)及鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
E-3_7
(1.5 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 2.55-2.80 (m, 3H), 2.80-2.23 (m, 1H), 1.98-1.81 (m, 2H), 1.81-1.1.69 (m, 1H), 1.69-1.25 (m, 16H), 1.25-1.01 (m, 10H), 1.01-0.81 (m, 14H), 0.67 (s, 3H)。 7.在0℃下向
E-3_7
(1.45 g,3.36 mmol)於MeOH (20 mL)中之溶液一次性添加NaBH
4
(255 mg,6.72 mmol)。添加之後,在20℃下攪拌混合物1小時且用NH
4
Cl (20 mL,飽和水溶液)淬滅。用DCM (2 × 20 mL)萃取混合物。合併之有機相用(2 × 10 mL)鹽水洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由閃蒸塔(0-20% EtOAc/PE)純化,得到呈固體之
E-3_8
(1.2 g,83%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.80-3.70 (m, 1H), 3.70-3.55 (m, 1H), 2.05-1.82 (m, 3H), 1.82-1.55 (m, 4H), 1.55-1.35 (m, 5H), 1.35-1.00 (m, 18H), 1.00-0.79 (m, 17H), 0.66 (s, 3H)。 8.向
E-3_8
(1.15 g,2.65 mmol)於吡啶(20 mL)中之溶液添加苯甲醯氯(1.85 g,13.2 mmol)。在20℃下攪拌反應混合物4小時。將反應混合物倒入水(20 mL)中。用EtOAc (2 × 20 mL)萃取混合物。合併之有機相用飽和鹽水(2 × 10 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾濃縮且藉由閃蒸塔(0-10% EtOAc/PE)純化,得到呈固體之混合物產物(1.45 g,不純)。混合物產物(1.45 g,不純)藉由SFC (管柱:AD (250mm×50mm,10um),梯度:30-30% B (A=0.1% NH3/H2O,B=EtOH),流速:200 mL/min)純化,得到呈固體之
E-3_9
(峰值2,470 mg,33%,DE%=100%)及呈固體之
E-3_9A
(峰值1,600 mg,42%,DE%=99.1%)。
E-3_9A : 1 H NMR
(400 MHz, CDCl
3
) δ 8.10-7.99 (m, 2H), 7.60-7.50 (m, 1H), 7.50-7.38 (m, 2H), 5.15-5.05 (m, 1H), 2.05-1.70 (m, 6H), 1.70-1.35 (m, 5H), 1.35-1.05 (m, 19H), 1.05-0.82 (m, 17H), 0.65 (s, 3H)。 在10 min層析中,
SFC
Rt=3.344 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
E-3_9 : 1 H NMR
(400 MHz, CDCl
3
) δ 8.10-7.99 (m, 2H), 7.60-7.50 (m, 1H), 7.50-7.38 (m, 2H), 5.25-5.15 (m, 1H), 2.05-1.80 (m, 3H), 1.80-1.45 (m, 15H), 1.45-1.09 (m, 13H), 1.09-0.85 (m, 16H), 0.68 (s, 3H)。 在10 min層析中,
SFC
Rt=3.851 min,AD_3_EtOH_DEA_5_40_25ML,99.1%de。 9.在25℃下向
E-3_9A
(600 mg,1.11 mmol)於THF (2 mL)及MeOH (2 mL)中之溶液添加NaOH(531 mg,13.3 mmol)及H
2
O (0.5 mL)。在50℃下攪拌溶液48小時。添加水(10 mL)。用EtOAc (2 × 10 mL)萃取混合物。合併之有機層經Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物,其用MeCN (10 mL)濕磨,得到呈固體之所需產物
69
(473 mg,99%) 。
1 H NMR
(400 MHz, CDCl
3
) δ 3.68-3.55 (m, 1H), 2.01-1.85 (m, 3H), 1.85-1.70 (m, 1H), 1.70-1.45 (m, 8H), 1.45-1.22 (m, 13H), 1.22-1.05 (m, 8H), 1.05-1.86 (m, 15H), 0.66 (s, 3H)。 在2 min層析中,
LCMS
tR=1.403 min,30-90AB_ELSD,純度100.0%,C
29
H
49
[M+H-2H
2
O]
+
之MS ESI計算值397,實驗值397。
實例 65 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (65) 之合成 N-8-11_1
(30 mg,0.0741 mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之
65
(9 mg,30%)。
65 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.18-3.07 (m, 1H), 1.98-1.81 (m, 2H), 1.71-1.58 (m, 6H), 1.53-1.31 (m, 7H), 1.30-0.98 (m, 14H), 0.97-0.78 (m, 14H), 0.70-0.60 (m, 4H)。 在7.0 min層析中,
LCMS
Rt=4.387 min,30-90AB_7MIN_E,純度97.6%,C
27
H
45
[M+H-2H
2
O]
+
之MS ESI計算值369,實驗值369。
實例 66 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- (2- 甲基環丙基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (66) 之合成 1.
在0℃下向
N-8-7_2
(1 g,2.41 mmol)於THF (50 mL)中之溶液添加LiAlH
4
(914 mg,24.1 mmol)。在66℃下加熱灰色懸浮液18小時。將反應混合物冷卻至0℃,藉由冰水(914 mg)、接著15% w/w NaOH水溶液(914 mg)、水(2.74 g)淬滅。過濾混合物且濾餅用DCM (3 × 50 mL)洗滌。濃縮濾液,得到殘餘物,其藉由急驟層析(乙酸乙酯,10%於PE中)純化兩次,得到
N-8-7_3
(192 mg,19%)及呈油狀物之
N-8-7_3A
(397 mg,39%)。
N-8-7_3 :
1H NMR (400 MHz, CDCl3) δ 5.59 - 5.36 (m, 2H), 3.69 - 3.61 (m, 1H), 2.25 - 2.12 (m, 1H), 2.08 - 1.81 (m, 3H), 1.68 (d, J = 10.0 Hz, 3H), 1.64 - 1.54 (m, 9H), 1.53 - 1.15 (m, 11H), 1.14 - 0.92 (m, 5H), 0.92 - 0.85 (m, 5H), 0.83 (s, 4H), 0.69 - 0.60 (m, 4H)。
N-8-7_3A : 1 H NMR
(400 MHz, CDCl
3
) δ 5.62 - 5.37 (m, 2H), 3.62 (br d,
J
=10.0 Hz, 1H), 2.20 - 2.06 (m, 1H), 1.99 - 1.61 (m, 6H), 1.61 - 1.44 (m, 11H), 1.43 - 1.18 (m, 5H), 1.16 - 0.94 (m, 6H), 0.94 - 0.85 (m, 5H), 0.82 (s, 5H), 0.70 - 0.58 (m, 6H)。 2.在0℃下經15 min之時段向二乙基鋅(1 M於甲苯中,2.59 mL,2.59 mmol)於DCM (10 ml )中之溶液添加CH
2
I
2
(1.38 g,5.18 mmol)。在0℃下攪拌乳白色懸浮液10 min,且經由注射器快速添加Charette配位體((4R,5R)-2-(第三丁基)-N4,N4,N5,N5-四甲基-1,3,2-二氧硼戊環-4,5-二甲醯胺) (139 mg,0.5182 mmol)及
N-8-7_3
(180 mg,0.4319 mmol)於DCM (15 mL)中之預製溶液,隨即反應混合物變為澄清。將溶液升溫至25℃且在25℃下攪拌16 h。接著藉由添加NH
4
Cl飽和水溶液(150 ml)來淬滅反應物。分離各相且用DCM (3 × 60 ml)萃取水相。合併之有機相用NaHCO
3
飽和水溶液(150 mL)、Na
2
S
2
O
3
飽和水溶液(150 mL)及鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且減壓濃縮,且殘餘物藉由閃蒸塔層析(11% 乙酸乙酯/PE)純化,得到呈固體之
N-8-7_4
(60 mg,32%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.77 (br s, 1H), 1.99-1.87 (m, 2H), 1.70-1.56 (m, 6H), 1.54-1.34 (m, 6H), 1.31-1.16 (m, 5H), 1.15-1.05 (m, 1H), 1.15-0.97 (m, 7H), 0.99-0.94 (m, 1H), 0.92-0.77 (m, 10H), 0.69-0.58 (m, 6H), 0.52-0.13 (m, 5H)。 3.在25℃下向
N-8-7_4A
(60 mg,0.1393 mmol)於吡啶(3 mL)中之溶液添加苯甲醯氯(39.1 mg,0.2786 mmol)隨後添加DMAP (6.79 mg,0.05572 mmol)。在60℃下攪拌反應混合物16小時。用DCM (80 mL)稀釋反應混合物。DCM相用水(100 mL)、1.0 M HCl水溶液(2 × 100 mL)、10% NaHCO
3
水溶液(2 × 100 mL)、鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到油狀物,其藉由閃蒸塔(1% 乙酸乙酯/PE)純化,得到呈油狀物之
N-8-7_5
(24 mg,32%)。 在2 min層析中,
LCMS
Rt=1.431 min,5-95AB_220&254,純度90%,C
36
H
53
O
2
[M-H
2
O+H]
+
之MS ESI計算值517.3,實驗值517.3。 在10 min層析中,
SFC
峰值1:Rt = 5.703 min,AD_3_EtOH_DEA_5_40_25ML (「Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。
4. N-8-7_5
(24 mg,0.04487 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:40-40% B (A=0.1% NH
3
/H
2
O,B=EtOH),流速:50 mL/min)純化,得到呈固體之不純
N-8-7_6
(RT:5.732,19 mg,不純)。未獲得異構體。 在2 min層析中,
LCMS
Rt=1.435 min,5-95AB_220&254,純度98%,C
36
H
53
O
2
[M-H
2
O+H]
+
之MS ESI計算值517.3,實驗值517.3。 在10 min層析中,
SFC
Rt=5.732 min,AD_3_EtOH_DEA_5_40_25ML,97.76%de。 5.向
N-8-7_6
(19 mg,0.0355 mmol)於THF/MeOH (0.5 ml/0.5 mL)中之溶液添加含KOH (19.8 mg,0.3552 mmol)之水(0.2 mL)。在55℃下攪拌反應混合物16小時。向混合物添加HCl (0.2 M,50 mL)。用DCM (2 × 60 mL)萃取懸浮液。合併之有機相用3% NaHCO
3
水溶液(80 mL)及鹽水(80 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到固體,其藉由急驟層析(乙酸乙酯/PE,15%)純化,得到呈固體之
66
(2 mg,13%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.77 (br s, 1H), 1.98-1.87 (m, 2H), 1.70-1.57 (m, 7H), 1.54 - 1.28 (m, 11H), 1.26 - 0.94 (m, 12H), 0.91 - 0.84 (m, 7H), 0.83 (s, 3H), 0.72 - 0.60 (m, 4H), 0.51 - 0.15 (m, 3H)。 在2 min層析中,
LCMS
Rt=1.315 min,30-90AB_2MIN_E,純度100%,C
29
H
49
O [M-H
2
O+H]
+
之MS ESI計算值395.3,實驗值395.3。
實例 67 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (67) 之合成 1.向
32
(80 mg,0.18 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)
2
(160 mg,無水)。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到
67
(10 mg,12%)及呈固體之
84
(30 mg,37%)。
67 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.76-3.66 (m, 1H), 2.01-1.78 (m, 5H), 1.76-1.58 (m, 7H), 1.52-1.31 (m, 13H), 1.28-1.10 (m, 10H), 1.09-0.99 (m, 4H), 0.96 (s, 3H), 0.93-0.86 (m, 6H), 0.67 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.508 min,30-90AB_E,純度100%,C
30
H
49
[M+H-2H
2
O]
+
之MS ESI計算值409,實驗值409。
實例 68 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基庚 -5- 炔 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (68) 之合成 1.粗產物
N-8-7_2B
(250 mg,0.868 mmol)藉由SFC (管柱:AD (250 mm×30 mm,10 um),梯度:35-35% B (A=0.1% NH
3
/H
2
O,B=EtOH),流速:60 mL/min)進一步純化,得到呈固體之
41
(峰值2,81 mg,33%)及呈固體之
68
(峰值1,78 mg,31%)。
68 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.87-3.78 (m, 1H), 2.21-2.12 (m, 1H), 1.99-1.86 (m, 2H), 1.80 (s, 3H), 1.73-1.51 (m, 8H), 1.51-1.42 (m, 4H), 1.42-1.20 (m, 8H), 1.20-0.95 (m,7H), 0.95-0.79 (m, 8H), 0.95 (s, 4H)。 在2 min層析中,
LCMS
Rt=1.188 min,30-90AB_2MIN_E,純度100%,C
28
H
45
O [M+H-H
2
O]
+
之MS ESI計算值397,實驗值397。 在10 min層析中,
SFC
Rt=6.465 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
實例 69 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-5- 環丙基 -3- 羥基戊 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (69) 之合成 1.在15℃下向
S-500-6-19_3
(700 mg,1.63 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液逐份添加NaBH
4
(2.46 g,65.1 mmol)。在15℃下攪拌20 min之後,用NH
4
Cl (20 mL,飽和水溶液)淬滅混合物且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到呈固體之760 mg混合物,其藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到
69
(330 mg,47%)及呈固體之
6
(250 mg,35%,不純)。不純
6
(250 mg)進一步藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到呈固體之
6
(170 mg,23%)。
69 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.33-5.23 (m, 1H), 3.75-3.63 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.85 (m, 4H), 1.78-1.59 (m, 5H), 1.53-1.38 (m, 9H), 1.38-1.05 (m, 9H), 1.03 (s, 3H), 1.00-0.91 (m, 1H), 0.91 (d,
J
= 6.4 Hz, 3H) 0.85 (t,
J
= 7.6 Hz, 3H), 0.69 (s, 3H), 0.68-0.60 (m, 1H), 0.45-0.36 (m, 2H), 0.09--0.08 (m, 2H)。 在2.0 min層析中,
LCMS
Rt=1.387 min,30-90_AB_E,純度98.1%,C
29
H
47
O [M+H-H
2
O]
+
之MS ESI計算值411,實驗值411。
S-500-6-19 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.24 (m, 1H), 3.77-3.66 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.91 (m, 3H), 1.79-1.59 (m, 6H), 1.55-1.21 (m, 14H), 1.21-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.95 (m, 1H), 0.93 (d,
J
= 6.8 Hz, 3H) 0.85 (t,
J
= 7.6 Hz, 3H), 0.70 (s, 3H), 0.68-0.62 (m, 1H), 0.49-0.38 (m, 2H), 0.11-0.02 (m, 2H)。 在2.0 min層析中,
LCMS
Rt=1.380 min,30-90_AB_E,純度100%,C
29
H
47
O [M+H-H
2
O]
+
之MS ESI計算值411,實驗值411。
實例 70 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4-(3- 甲基氧雜環丁 -3- 基 ) 丁 -2- 基 )-10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (70) 之合成 1.在50℃至55℃下於N
2
下向Mg (807 mg,33.2 mmol)及I
2
(1 mg)於THF (2 mL)中之懸浮液逐滴添加
N-014-005_3
(2.5 g,15.1 mmol)於THF (8 mL)中之溶液。在55℃下攪拌混合物1 h。用THF (10 mL)稀釋混合物,且在不經監測的情況下直接用於下一步驟中。在0℃下向
N-14-12_4
(1.01 g,2.83 mmol)於THF (10 mL)中之溶液添加新鮮製備的3-[(溴鎂)甲基]-3-甲基環氧丙烷(15 mmol於20 ml THF中)。在15℃下攪拌混合物4 h。向混合物添加NH
4
Cl (20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。分離有機層且真空濃縮。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈白色固體之
混合物
(190 mg,15%),其藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3H2O ETOH,梯度:50%至50%,流速(ml/min):60mL/min,25℃)純化,得到
33
(峰值1,110 mg,9%)及呈白色固體之
70
(峰值2,30mg,不純)。不純
70
(30 mg,不純)藉由矽膠管柱層析(15% EtOAc/PE)純化,得到呈白色固體之
70
(10 mg,5%)。
33 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.30-5.26 (m, 1H), 4.59-4.70 (m, 1H), 4.50-4.48 (m, 1H),4.36-4.33 (m, 1H), 3.83 (s, 1H), 2.40-2.33 (m, 1H), 2.10-1.50 (m, 17H), 1.49-1.35 (m, 9H), 1.30-0.80 (m, 13H), 0.68 (s, 3H)。 在3 min層析中,
LCMS
Rt=1.069 min,30-90AB_2MIN_E.M,純度100%,C
29
H
49
O
3
[M+H]
+
之MS ESI計算值445,實驗值445。
70 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.30-5.26 (m, 1H), 4.10-4.02 (m, 1H), 3.81-3.70 (m, 1H), 3.50-3.41 (m, 2H), 3.34-3.30 (m, 1H), 2.35-2.31 (m, 1H), 2.10-1.50 (m, 18H), 1.49-1.05 (m, 13H), 1.05-0.90 (m, 4H), 0.90-0.80 (m, 3H), 0.67 (s, 3H)。 在3 min層析中,
LCMS
Rt=1.115 min,30-90AB_2MIN_E.M,純度100%,C
29
H
49
O
3
[M+H]
+
之MS ESI計算值445,實驗值445。
實例 71 : (3S,5R,8R,9S,10S,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 - 17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (71) 之合成 1.在0℃下於N
2
下向
N-004-022_5
(200 mg,0.531 mmol)、CsF (40.2 mg,0.265 mmol)於THF (5 mL)中之溶液添加TMSCF
3
(187 mg,1.32 mmol)。在25℃下攪拌混合物1小時。向混合物添加TBAF.3H2O (836 mg,2.65 mmol)。在25℃下攪拌2小時之後,用50% NH
4
Cl (20 mL)淬滅混合物且用EtOAc (2 × 10 mL)萃取。合併之有機相用鹽水(20 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA = 10/1)純化,得到
9
(56 mg,24%)及呈白色固體之
71
(30 mg,不純)。 在25℃下自正己烷(2 mL)再結晶
71
(30 mg,0.067 mmol),得到呈白色固體之
71
(24 mg,10%)。
71 : 1 H NMR
(400 MHz, CDCl
3
) δ 4.08-4.00 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.22-2.15 (m, 1H), 2.02-1.77 (m, 5H), 1.75-1.68 (m, 2H), 1.64-1.52 (m, 5H), 1.47-1.31 (m, 6H), 1.28-1.01 (m, 10H), 0.97 (s, 3H), 0.67 (s, 3H)。 在2 min層析中,
LCMS
Rt = 1.105 min,30-90AB_POS_E.M,純度100%,C
25
H
41
F
3
O
3
[M+Na]
+
之MS ESI計算值469,實驗值469。
實例 72 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (72) 之合成 1.在0℃下向
S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液緩慢地添加丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)。添加之後,在15℃下攪拌混合物1小時。用飽和NH
4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2 × 30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
72
(500 mg,56%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,
LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C
27
H
43
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。
實例 73 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (73) 之合成。 1.在N
2
下向
G-21-1
(40 g,110 mmol)於無水二噁烷(1 L)中之溶液添加甲醇鈉(29.7 g,550 mmol)。在110℃下攪拌混合物16小時。TLC顯示起始物質幾乎完全耗盡。移除溶劑至1/3體積,且用2 M HCl酸化混合物至pH=5-6,用DCM (500 mL × 3)萃取,用碳酸氫鈉水溶液(500 mL)及鹽水(500 mL)洗滌,經硫酸鈉乾燥且濃縮。殘餘物藉由矽膠管柱層析(PE:EA:MeOH = 3:1:0.1)純化,得到呈白色固體之
G-21-4A
(11 g,33.1%)。 2.在-70℃下向液態銨(1000 mL,製備於13-601中歷時1.5小時)逐份添加Li (2.54 g,363 mmol)之溶液。在-70℃下攪拌混合物30 min直至所有Li溶解為止。逐滴添加
G-21-4A
(11 g,36.3 mmol)及第三-BuOH (5.38 g,72.6 mmol)於400 ml無水四氫呋喃中之溶液,且攪拌90 min直至反應混合物變成淺黃色為止。TLC (PE:EA= 1:1,PMA)顯示大部分STM經消耗。添加氯化銨(15 g)且使過量氨蒸發。用0.5N HCl (500 mL)及二氯甲烷(500 mL × 2)萃取殘餘物。合併之有機層用NaHCO
3
飽和溶液洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到
G-21-5A
及
G-21-5B
(10 g,不純)之混合物,其未經進一步純化即直接用於下一步驟中。 3.向
G-21-5A
及
G-21-5B
(10 g,27.9 mmol)於100 mL無水二氯甲烷中之溶液添加PCC (16.6 g,65.6 mmol)及矽膠(16.6 g)。在25℃下攪拌2 h之後,TLC (PE:EA = 1:1,PMA)顯示STM經消耗。濃縮所得溶液且藉由矽膠層析(石油醚/乙酸乙酯= 5:1至2:1)純化,得到呈白色固體之
G-21-6A
(4.6 g,46.4%)。 4.在0℃下於氮氣下向BHT (34.8 g,158 mmol)於甲苯(120 mL)中之溶液逐滴添加三甲基鋁(2 M於甲苯中,39.5 mL,79.1 mmol)。在20℃下攪拌30分鐘之後,在-70℃下於氮氣下逐滴添加
G-21-6A
(8 g,26.4 mmol)於甲苯(80 mL)中之溶液。在-70℃下攪拌30 min之後,逐滴添加MeMgBr (3 M於乙醚中,26.3 mL,79.1 mmol,3 M於醚中)。在-70℃下攪拌所得混合物1小時,緩慢地倒入冰冷的檸檬酸水溶液(300 mL)且用EtOAc (3 × 100 mL)萃取。合併之有機層用鹽水(300 mL)洗滌,經無水硫酸鈉乾燥,過濾,濃縮且藉由combi-flash (0-40% EtOAc/PE)純化,得到呈白色固體之
N-4-10_1
(6.5 g,77%)。
1 H NMR
(400 MHz, CDCl
3
)δ 2.60-2.49 (m, 1H), 2.47-2.37 (m, 2H), 2.34-2.19 (m, 3H), 2.14-2.03 (m, 1H), 2.00-1.82 (m, 3H), 1.73-1.58 (m, 3H), 1.56-1.46 (m, 2H), 1.36-1.26 (m, 3H), 1.24 (s, 3H), 1.21-1.07 (m, 2H), 1.04 (s, 3H), 1.00-0.84 (m, 1H), 0.82 (s, 3H)。 5.在20℃下於N
2
下向溴(乙基)三苯基磷烷(22.6 g,61.1 mmol)於無水THF (200 mL)中之懸浮液添加2-甲基丙-2-醇化鉀(6.84 g,61.1 mmol)。在40℃下攪拌30分鐘之後,緩慢添加
N-4-10_1
(6.5 g,20.4 mmol)於無水THF (50 mL)中之溶液。在40℃下攪拌所得混合物10分鐘,接著用NH
4
Cl水溶液(400 mL)淬滅且用EtOAc (2 × 150 mL)萃取。合併之有機相經硫酸鈉乾燥,過濾,濃縮且藉由矽膠管柱層析(0-25% EtOAc/PE)純化,得到呈白色固體之
N-4-10_2
(5.5 g,82%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.22-5.13 (m, 1H), 2.91-2.81 (m, 1H), 2.62-2.51 (m, 1H), 2.50-2.39 (m, 2H), 2.38-2.24 (m, 1H), 1.91-1.81(m, 1H), 1.80-1.70 (m, 4H), 1.55-1.41 (m, 4H), 1.36-1.25 (m, 5H), 1.23 (s, 3H), 1.21-1.04 (m, 3H), 1.01 (s, 3H), 0.98-0.84 (m, 2H), 0.81 (s, 3H)。 6.在15℃下於N
2
下向
N-4-10_2
(5.5 g,16.6 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(8.10 g,33.2 mmol)。在50℃下攪拌1小時之後,將混合物冷卻至15℃。在15℃以下逐滴添加NaOH水溶液(33.2 mL,5 M,166 mmol),接著在15℃以下逐滴添加H
2
O
2
(18.8 g,30%,166 mmol)。用EtOAc (3 × 100 mL)萃取混合物。合併之有機相用飽和Na
2
S
2
O
3
(5 × 100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到7 g粗產物,其直接用於下一步驟中。 7.向
N-4-10_3
(7 g,19.9 mmol)於DCM (300 mL)中之溶液添加DMP (25.2 g,59.6 mmol)。在20℃下攪拌10 min之後,用NaHCO
3
飽和溶液(500 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層,且用DCM (200 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3 × 400 mL)、飽和NaHCO
3
(400 mL)、鹽水(400 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-20% EtOAc/DCM)純化,得到呈白色固體之
N-4-10_4
(4 g,58%)。
1 H NMR
(400 MHz, CDCl
3
)δ 2.77-2.67 (m, 1H), 2.65-2.38 (m, 3H), 2.32-2.17 (m, 1H), 2.09 (s, 3H), 1.88-1.63 (m, 7H), 1.59-1.49 (m, 3H), 1.35-1.21 (m, 7H), 1.19-1.09 (m, 2H), 1.01 (s, 3H), 0.96-0.84 (m, 1H), 0.57 (s, 3H)。 8.向MePh
3
PBr (8.18 g,23.0 mmol)於THF (100 mL)中之懸浮液添加t-BuOK (2.57 g,23.0 mmol)。在40℃下攪拌10分鐘之後,在20℃下向
N-4-10_4
(4 g,11.5 mmol)於THF (50 mL)中之溶液緩慢地逐滴添加混合物。添加之後,用NH
4
Cl (200 mL)淬滅混合物且用EtOAc (3 × 80 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-25% EtOAc/PE)純化,得到呈白色固體之
N-4-10_5
(3.2 g,80%)。
1 H NMR
(400 MHz, CDCl
3
)δ 4.88 (s, 1H), 4.70 (s, 1H), 2.48-2.37 (m, 2H), 2.31-2.22 (m, 2H), 1.89-1.77 (m, 4H), 1.75-1.61 (m, 7H), 1.54-1.45 (m, 2H), 1.34-1.29 (m, 2H), 1.28-1.24 (m, 3H), 1.23 (s, 3H), 1.17-1.05 (m, 2H), 1.01 (s, 3H), 0.93-0.83 (m, 1H), 0.51 (s, 3H)。 9.在15℃下於N
2
下向
N-4-10_5
(3.2 g,9.28 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(4.51 g,18.5 mmol)。在50℃下攪拌1小時之後,將混合物冷卻至15℃。在15℃以下逐滴添加NaOH水溶液(18.5 mL,5 M,92.8 mmol),接著在15℃以下逐滴添加H
2
O
2
(10.5 g,30%,92.8 mmol)。用EtOAc (3 × 100 mL)萃取混合物。合併之有機相用飽和Na
2
S
2
O
3
(5 × 100 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到5 g粗產物,其直接用於下一步驟中。 10.向
N-4-10_5A
(5 g,13.7 mmol)於DCM (300 mL)中之溶液添加DMP (11.6 g,27.4 mmol)。在20℃下攪拌10 min之後,用NaHCO
3
飽和溶液 (300 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (100 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3 × 300 mL)、飽和NaHCO
3
(300 mL)、鹽水(300 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-10% 丙酮/DCM)純化,得到呈白色固體之
N-4-10_7
(1 g,20%)。
1 H NMR
(400 MHz, CDCl
3
)δ 9.58-9.55 (m, 1H), 2.52-2.27 (m, 4H), 2.08-1.96 (m, 1H), 1.84-1.62 (m, 8H), 1.51-1.39 (m, 3H), 1.32-1.21 (m, 7H), 1.17-1.06 (m, 5H), 1.01 (s, 3H), 0.94-0.83 (m, 1H), 0.66 (s, 3H)。 11.在0℃下於N
2
下向
N-4-10_6
(400 mg,0.832 mmol)於THF (20 mL)中之溶液逐滴添加異戊基溴化鎂(1.65 mL,3.30 mmol,2 M於醚中)。在0℃下攪拌10分鐘之後,用飽和NH
4
Cl (60 mL)淬滅混合物且用EtOAc (2 × 30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-10% 丙酮/DCM)純化,得到呈白色固體之
73
(200 mg,42%)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.65-3.56 (m, 1H), 2.55-2.49 (m, 1H), 2.46-2.38 (m, 1H), 2.32-2.25 (m, 1H), 2.10-1.98 (m, 1H), 1.83-1.62 (m, 7H), 1.57-1.44 (m, 4H), 1.42-1.25 (m, 7H), 1.24-1.20 (m, 4H), 1.19-1.04 (m, 5H), 1.01 (s, 3H), 0.94-0.82 (m, 10H), .0.63 (s, 3H)。 在7.0 min層析中,
LCMS
Rt=3.381 min,30-90AB_E,純度100%,C
28
H
47
O
2
[M+H-H
2
O]
+
之MS ESI計算值415,實驗值415。
實例 74 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (74) 之合成 1.在35℃下於N
2
下將t-BuOH (1.7 L)裝入三頸圓底燒瓶且攪拌10 min。向混合物添加t-BuOK (292 g,2.61 mol)且攪拌直至反應物變得澄清。此後,向以上混合物添加
S-310-B9_1
(65 g, 238 mmol)且在35℃下於N
2
下攪拌1.5 h。將反應混合物倒入10%乙酸水溶液(2 L)中且攪拌30 min,在此期間將溫度維持在10℃以下。接著混合物用水(1.5 L)處理且用NaHCO
3
將pH值調整至7至8,且攪拌混合物30 min。水相用MTBE (3 L)萃取。分離有機層,用鹽水(3 × 1 L)洗滌,經無水Na
2
SO
4
乾燥,過濾並在35℃以下濃縮,得到呈油狀物之
S-200-N19-3_1
(65 g,粗產物)。粗殘餘物直接用於下一步驟。 2.在0℃下向2,6-二第三丁基-4-甲苯酚(340 g,1.54 mol)於甲苯(700 mL)中之溶液逐滴添加AlMe
3
(385 mL, 770 mmol, 2 M於甲苯中)。在25℃下攪拌混合物1小時且直接用作MAD溶液。在-70℃下於N
2
下經30 min之時段向MAD溶液添加
200-N19-3_1
(60 g,220 mmol)於無水甲苯(200 mL)及無水DCM (200 mL)中之溶液。在-70℃下攪拌反應混合物1 h。接著在-70℃下逐滴添加MeMgBr (220 mL, 660 mmol, 3 M於乙基醚中)且攪拌1 h。在0℃下將反應物倒入檸檬酸飽和水溶液(2 L)中且攪拌30 min,用EtOAc萃取(2 × 1 L)。合併之有機相用飽和鹽水(2×1 L)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc = 10/1至5/1)純化,得到呈固體之
200-N19-M22_1
(33 g,52%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.46-5.42 (m, 1H), 2.25-2.40 (m, 1H), 2.21-1.60 (m, 13H),1.35-1.21 (m, 4H), 1.13 (s, 3H), 0.98-0.83 (m, 6H)。 3.在25℃下於N
2
下向Ph
3
PEtBr (102 g,277 mmol)於無水THF (500 mL)中之懸浮液一次性添加t-BuOK (31.0 g,277 mmol)。在25℃下攪拌30 min之後,添加
200-N19-M22_1
(20 g, 69.3 mmol)且在25℃下攪拌2 h。在0℃下用NH
4
Cl水溶液(800 mL)淬滅反應物,用EtOAc (2 × 500 mL)萃取。合併之有機相用鹽水(2 × 500 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc = 10/1至5/1)純化,得到呈固體之
N-4-14_1
(15 g,72%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.43-5.40 (m, 1H), 5.16-5.10 (m, 1H), 2.41-2.33 (m, 1H), 2.28-1.86 (m, 8H), 1.78-1.71 (m, 1H), 1.69-1.50 (m, 11H), 1.41-1.10 (m, 6H), 0.94-0.81 (s, 3H)。
4.
向
N-4-14_1
(30 g, 99.8 mmol)於無水THF (500 mL)中之溶液添加9-BNN二聚體(66.9 g, 299 mmol)且在0℃下於N
2
下攪拌30 min。使反應混合物升溫至50℃且攪拌1小時。冷卻至0℃之後,添加EtOH (100 mL)。極緩慢地添加NaOH水溶液(99.8 mL, 5 M, 499 mmol)。緩慢添加H
2
O
2
(53.0 g,499 mmol, 30%於水中)且將內部溫度維持在30℃以下。將混合物升溫至50℃且攪拌1小時。冷卻反應混合物且添加冰水(1 L)並攪拌30 min。過濾且真空濃縮,得到呈固體之
N-4-11_2
(30 g,粗產物)。粗殘餘物直接用於下一步驟。 5.向
N-4-14_2
(30 g,粗產物)於DCM (500 mL)中之溶液添加矽膠(150 g)及PCC (81.0 g,376 mmol)。使反應混合物升溫至40℃且攪拌1小時。冷卻反應混合物,用PE (500 mL)處理,經由矽膠墊過濾,且固體用PE/DCM (500/500 mL)洗滌。母液經過濾且真空濃縮,得到粗產物。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之
N-4-14_3
(20 g,不純)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.43-5.40 (m, 1H), 2.57-2.50 (m, 1H), 2.21-2.08 (m, 6H), 1.77-1.43 (m, 10H), 1.37-1.12 (m, 9H), 1.00-0.82 (m, 2H), 0.64 (s, 3H)。 6.在0℃下於N
2
下向Ph
3
PMeBr (44.8 g,126 mmol)於無水THF (300 mL)中之懸浮液一次性添加t-BuOK (14.1 g,126 mmol)。在25℃下攪拌反應混合物30 min。添加
N-4-14_3
(20 g,63.1 mmol)。使反應混合物升溫至40℃且攪拌1小時。在0℃下將反應物倒入冰水(500 mL)中。用EtOAc (2 × 400 mL)萃取水相。合併之有機相用鹽水(2 × 300 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由矽膠管柱(PE/EtOAc = 8/1-5/1)純化,得到呈固體之
N-4-14_4
(19 g,不純)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.43-5.40 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.23-2.13 (m, 2H), 1.87-1.64 (m, 11H), 1.42-1.40 (m, 2H), 1.29-1.08 (m, 8H), 0.97-0.80 (m, 3H), 0.59 (s, 3H)。 7.在0℃下於N
2
下向
N-4-14_4
(19 g,60.4 mmol)於無水THF (300 mL)中之溶液一次性添加9-BNN二聚體 (40.5 g,181 mmol)。將混合物升溫至50℃且攪拌1 h。冷卻反應混合物且添加EtOH (100 mL)。極緩慢添加NaOH水溶液(60.3 mL,5 M,302 mmol)。緩慢添加H
2
O
2
(34.0 g,302 mmol, 30%於水中) 且將內部溫度維持在10℃以下。將混合物升溫至50℃且攪拌1小時。冷卻之後,添加冰水(1 L)且攪拌30 min。濾出沈澱固體。濾餅在空氣中乾燥,得到呈固體之
N-4-11_5
(17 g,粗產物),其直接用於下一步驟。 8.在25℃下向
N-4-14_5
(17 g,粗產物)於DCM (300 mL)中之溶液一次性添加矽膠(60 g)及PCC (43.9 g,204 mmol)。使反應混合物升溫至40℃且攪拌1小時。冷卻反應混合物,且添加PE (200 mL)。混合物經由矽膠墊過濾,且固體用PE/DCM (200/200 mL)洗滌。過濾且真空濃縮,得到固體。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之
N-4-14_6
(5.5 g,不純)。在82℃下自MeCN (50 mL)再結晶殘餘物,得到呈固體之
N-4-14_6
(5 g,91%)。
1 H NMR
(400 MHz, CDCl
3
)δ 9.60-9.56 (m, 1H), 5.42-5.40 (m, 1H), 2.58-2.51 (m, 1H),2.40-1.85 (m, 9H), 1.44-1.04 (m, 16H), 1.00-0.80 (m, 3H), 0.75-0.71 (m, 3H)。 9.在25℃下於N
2
下向Mg (3.96 g,165 mmol)及I
2
(1 mg)於無水THF (20 mL)中之懸浮液逐滴添加
1
(12.5 g,82.7 mmol)於無水THF (63 mL)中之溶液,且將內部溫度升高至65℃且攪拌2小時。混合物直接用於下一步驟中。在0℃下於N
2
下向
N-4-14_6
(5 g,15.1 mmol)於無水THF (50 mL)中之溶液一次性添加異戊基溴化鎂(83.0 mL,1 M於THF中)。使反應混合物升溫至15℃且攪拌1小時。向反應混合物添加NH
4
Cl 飽和水溶液(100 mL)。用EtOAc (3 × 100 mL)萃取水相。合併之有機相用飽和鹽水(2 × 200 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(0-20% EtOAc/PE)純化,得到呈固體之
N-4-14_7
(2.5 g,不純)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.40-5.37 (m, 1H), 3.67-3.61 (m, 1H), 2.19-1.71 (m, 9H), 1.64-1.28 (m, 13H), 1.18-1.03 (m, 7H), 0.95-0.78 (m, 12H), 0.69 (s, 3H)。 10.在25℃下向DMP (10.5 g,24.8 mmol)添加含
N-4-14_7
(2.5 g,6.20 mmol)之DCM (40 mL)。使反應混合物升溫至40℃且攪拌1 h。在pH 7至8且低於10℃下用NaHCO
3
飽和水溶液淬滅反應混合物。過濾懸浮液。分離濾液中之DCM相且用NaHCO
3
/Na
2
S
2
O
3
飽和水溶液(1:1,2 × 30 mL)洗滌,合併之有機相用飽和鹽水(2 × 30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到固體。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈固體之
N-4- 14_7O
(1.5 g,60%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.41-5.39 (m, 1H), 2.56-2.31 (m, 3H), 2.19-1.83 (m, 5H), 1.84-1.42 (m, 12H), 1.30-0.97 (m, 12H), 0.96-0.77 (m, 8H), 0.74-0.70 (m, 3H)。 11.在25℃下向
N-4-14_7O
(1.5 g,3.74)於MeOH (10 mL)中之溶液緩慢添加NaBH
4
(1.42 g,37.4 mmol),且攪拌2小時。添加冰水(100 mL)且攪拌混合物30 min。用DCM (2 × 20 mL)萃取水相。合併之有機相用飽和鹽水(2 × 20 mL)洗滌,經無水Na
2
SO
4
乾燥,過濾且真空濃縮以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之
N-4-14_7
(1 g, 67%)。
1 H NMR
(400 MHz, CDCl
3
)δ 5.41-5.39 (m, 1H), 3.65-3.63 (m, 1H), 2.20-2.16 (m, 1H), 2.11-1.88 (m, 5H), 1.86-1.54 (m, 10H), 1.33-0.99 (m, 14H), 0.95-0.79 (m, 11H), 0.70 (s, 3H)。
SFC
峰值1:Rt=4.644 min,且在10 min層析中峰值2 Rt=5.240 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。 12.
N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH
3
H
2
O ETOH,開始:B:40%,結束B:40%)純化,得到
80
(峰值2,300 mg,不純)及呈固體之
83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶
80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之
80
(150 mg,15%)。在82℃下回流1 小時,自MeCN (3 mL)再結晶
83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之
83
(150 mg,15%)。
83 : 1 H NMR
(400 MHz, CDCl
3
)δ 5.41-5.39 (m, 1H), 3.62-3.60 (m, 1H), 2.22-1.89 (m, 6H), 1.64-1.49 (m, 9H), 1.46-1.11 (m, 16H), 0.98-0.86 (m, 10H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.268 min,30-90 AB,純度100%,C
27
H
42
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 在10 min層析中,
SFC
Rt=4.609 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
80 : 1 H NMR
(400 MHz, CDCl
3
)δ 5.41-5.39 (m, 1H), 3.63-3.62 (m, 1H), 2.22-1.67 (m, 10H), 1.64-1.36 (m, 12H), 1.16-1.03 (m, 8H), 0.98-0.80 (m, 11H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.276 min,30-90 AB,純度99%,C
27
H
42
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 在10 min層析中,
SFC
Rt=5.236 min,AD_3_EtOH_DEA_5_40_25ML,100%de。 13. 在Ar下向
80
(150 mg,0.372 mmol)於THF (3 mL)及MeOH (3 mL)中之溶液添加無水Pd(OH)
2
(200 mg)。將懸浮液真空脫氣且用H
2
淨化三次。在50℃下於H
2
(50 psi)下攪拌混合物12 h,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用DCM (3 × 50 mL)洗滌。真空濃縮濾液以提供油狀物。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之
74
(20 mg,13%)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.63-3.61 (m, 1H), 1.96-1.85 (m, 2H), 1.78-1.50 (m, 8H), 1.45-1.19 (s, 12H), 1.17-1.00 (m, 11H), 0.98-0.83 (m, 11H), 0.72-0.59 (m, 3H)。 在2.0 min層析中,
LCMS
Rt=1.333 min,30-90 AB,純度99%,C
27
H
44
[M+H-2H
2
O]
+
之MS ESI計算值369,實驗值369。
實例 75 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (75) 之合成 1.
向
15_3a
(1 g,2.88 mmol)於DCM (10 mL)中之溶液添加DMP (2.44 g,5.76 mmol)。此後,在25℃下攪拌反應物10 min。反應混合物藉由添加NaHCO
3
飽和水溶液(20 mL)及Na
2
S
2
O
3
飽和水溶液(20 mL)淬滅,用DCM (2×50 mL)萃取。合併之有機層用NaHCO
3
飽和水溶液(3 × 50 mL)及鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到呈固體之
S-500-2-9_1
(1 g,粗產物)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.57 (br. s, 1H), 5.35-5.25 (m, 1H), 2.50-2.30 (m, 2H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.75-1.65 (m, 1H), 1.65-1.60 (m, 3H), 1.55-1.50 (m, 2H), 1.50-1.40 (m, 2H), 1.40-1.30 (m, 1H), 1.25-1.20 (m, 2H), 1.20-1.15 (m, 2H), 1.15-1.10 (m, 6H), 1.05-0.95 (m, 5H), 0.90-0.70 (m, 1H), 0.68 (s, 3H)。 2.在60℃下攪拌鎂(641 mg,26.4 mmol)及I
2
(33.5 mg,0.132 mmol)之混合物,且於N
2
下逐滴添加異戊基溴化鎂(2 g,13.2 mmol)於THF (20 mL)中之溶液。此後,在60℃下攪拌反應混合物1 h。反應混合物直接用作異戊基溴化鎂溶液而無需任何純化。在0℃下於N
2
下向
S-500-2-9_1
(1 g,2.90 mmol)於THF (10 mL)中之溶液添加格林納溶液。此後,在25℃下攪拌反應混合物1 h。反應混合物添加NH
4
Cl飽和水溶液(50 mL),用EtOAc (2 × 50 mL)萃取,用鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(EtOAc/ PE = 1/4)純化,得到呈固體之不純
S-500-2- 9_2
(560 mg)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.28-5.25 (m, 1H), 3.90-3.80 (m, 0.25H), 3.68-3.58 (m, 0.75H), 2.48-2.36 (m, 1H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.80-1.75 (m, 1H), 1.75-1.52 (m, 6H) 1.52-1.42 (m, 6H), 1.42-1.32 (m, 3H), 1.32-1.22 (m, 3H), 1.22-1.12 (m, 3H), 1.12-1.02 (m, 2H), 1.01 (s, 3H), 1.00-0.92 (m, 1H), 0.92-0.85 (m, 9H), 0.85-0.77 (m, 1H), 0.69 (s, 3H)。 3.
S-500-2-9_2
(560 mg)藉由SFC (管柱:Chiralcel OD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃)純化,得到呈固體之不純
30
(160 mg)及呈固體之
75
(265 mg,47%)。
75 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.35-5.30 (m, 1H), 3.70-3.60 (m, 1H), 2.50-2.40 (m, 1H), 2.05-1.90 (m, 4H), 1.85-1.75 (m, 2H), 1.75-1.60 (m, 1H), 1.55-1.45 (m, 8H), 1.45-1.25 (m, 8H), 1.25-1.10 (m, 4H), 1.10-1.05 (m, 2H), 1.02 (s, 3H), 0.99-0.91 (m, 3H), 0.91-0.89 (m, 4H), 0.88 (s, 3H), 0.69 (s, 3H)。 在1.5 min層析中,
LCMS
Rt=1.162 min,5-95 AB,純度99%,C
28
H
45
[M+H-2H
2
O]
+
之MS ESI計算值381,實驗值381。
實例 76 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((S)-1-(1- 羥基環丙基 ) 乙基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (76) 之合成 1.向
N-8-7_1
(500 mg,1.38 mmol)於MeCN (30 mL)中之溶液添加NaClO
2
(374 mg,4.14 mmol),TEMPO (645 mg,4.14 mmol)及NaClO (10 mL,10%於水中)。在50℃下攪拌混合物48小時之後,出現固體。藉由過濾收集固體,且用DCM (5 mL)濕磨,得到呈固體之
N-8-26_1
(180 mg,34%)。
1 H NMR
(400 MHz, MeOD) δ 2.30-2.20 (m, 1H), 1.98-1.91 (m,1H), 1.86-1.72 (m, 1H), 1.71-1.63 (m, 1H), 1.62-1.49 (m, 8H), 1.43-1.31 (m, 4H), 1.30-1.20 (m, 4H), 1.19-1.07 (m, 8H), 1.06-0.86 (m, 8H), 0.75-0.65 (m, 4H)。 2.向
N-8-26_1
(180 mg,0.477 mmol)於DMF (5 mL)中之溶液添加K
2
CO
3
(328 mg,2.38 mmol)及MeI (686 mg,4.77 mmol)。在20℃下攪拌16小時之後,用50% NH
4
Cl (20 mL)淬滅混合物且用EtOAc (3 × 10 mL)萃取。合併之有機相用LiCl (3%於水中,30 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之
N-8-26_2
(160 mg,86%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.63 (s, 3H), 2.45-2.36 (m, 1H), 1.92-1.85 (m, 1H), 1.74-1.58 (m, 6H), 1.56-1.46 (m, 4H), 1.42-1.19 (m, 9H), 1.18-1.15 (m, 3H), 1.13-0.93 (m, 4H), 0.91-0.84 (m, 4H), 0.82 (s, 3H), 0.70-0.61 (m, 4H)。 3.在20℃下向
N-8-26_2
(80 mg,0.204 mmol)於THF (2 mL)中之溶液添加Ti(i-PrO)
4
(57.9 mg,0.204 mmol)及EtMgBr (0.204 mL,3 M於Et
2
O,0.612 mmol)。在20℃下攪拌30分鐘之後,用NH
4
Cl (30 mL)飽和溶液淬滅反應混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空濃縮得到粗產物,其藉由矽膠管柱(0-10% EtOAc/ PE)純化,得到呈固體之
76
(16 mg,20%)。
1 H NMR
(400 MHz, CDCl
3
) δ 1.98-1.86 (m, 2H), 1.69-1.58 (m, 6H), 1.54-1.44 (m, 3H), 1.44-1.29 (m, 4H), 1.28-1.18 (m, 4H), 1.18-1.10 (m, 5H), 1.09-0.93 (m, 4H), 0.91-0.81 (m, 9H), 0.71-0.57 (m, 6H), 0.31-0.24 (m, 1H)。 在2.0 min層析中,
LCMS
Rt=1.184 min,30-90AB_E,純度100%,C
26
H
41
[M+H]
+
之MS ESI計算值353,實驗值353。
實例 77 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (77) 之合成 1.在60℃下向Mg (4.35 g,179 mmol)及I
2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。用THF (10 mL)稀釋混合物且直接使用。在0℃下於N
2
下向
S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。向混合物添加NH
4
Cl (20 mL,飽和水溶液)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na
2
SO
4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH
3
CN (10 mL)再結晶,得到呈固體之
77
(255 mg,21%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.306 min,30-90 AB,純度100%,C
29
H
49
O [M+H-H
2
O]
+
之MS ESI計算值413,實驗值413。
實例 78 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1S,2S)-1- 環戊基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (78) 之合成 1. 向
N-8-15_2
(240 mg,0.559 mmol)於MeOH (3 mL)及THF (2 mL)中之混合物添加NaBH
4
(550 mg,14.5 mmol)。在15℃下攪拌混合物0.5 h。添加另一批次之NaBH
4
(550 mg,14.5 mmol)。再攪拌反應混合物1小時。向反應混合物添加水(5 mL)。用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(10 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之
58
(7 mg,5%),且
78
(50 mg,不純)藉由閃蒸塔(0-5% EtOAc/PE)進一步純化,得到呈固體之
78
(17 mg,12%)。
78 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.38-3.47 (m, 1H), 2.01-1.82 (m, 4H), 1.71-1.53 (m, 11H), 1.53-1.48 (m, 4H), 1.48-1.30 (m, 5H), 1.30-1.11 (m, 7H), 1.11-0.98 (m, 5H),0.98-0.85 (m, 7 H), 0.85-0.80 (m, 3 H), 0.65 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.358 min,30-90AB_7MIN_E,純度100%,C
29
H
47
[M+H-2H
2
O]
+
之MS ESI計算值395,實驗值395。 在10 min層析中,
HPLC
Rt=6.093 min,50-100AB_10 MIN.M,純度98%。
實例 79 : (1R,3S,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (79) 之合成 79
之合成描述於實例13中。
79 : 1 H NMR
(400 MHz, CDCl
3
) δ 7.43-7.28 (m, 5H), 5.05-4.94 (m, 1H), 4.04-3.91 (m, 1H), 2.51 (brs, 1H), 2.07-1.78 (m, 6H), 1.70-1.61 (m, 4H), 1.51-1.41 (m, 3H), 1.39-1.12 (m, 11H), 1.05-0.98 (m, 2H), 0.91-0.81 (m, 7H), 0.71-0.60 (m, 4H)。 在2 min層析中,
LCMS
Rt=1.298 min,10-80AB_2MIN_E,純度96.7%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在10 min層析中,
SFC
Rt=1.483 min,IC-3_MeOH(DEA)_40_2.5ML,100%de。
13 : 1 H NMR
(400 MHz, CDCl
3
) δ 7.40-7.28 (m, 5H), 5.12-5.07 (m, 1H), 3.95-3.88 (m, 1H), 2.76 (brs, 1H), 2.08-1.78 (m, 6H), 1.75-1.60 (m, 5H), 1.51-1.38 (m, 4H), 1.36-1.09 (m, 9H), 1.00-0.89 (m, 6H), 0.83 (s, 3H), 0.71-0.64 (m, 1H), 0.63 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.309 min,10-80AB_2MIN_E,純度100%,C
31
H
45
F
3
O
3
Na [M+Na]
+
之MS ESI計算值545,實驗值545。 在5 min層析中,
SFC
Rt=1.683 min,IC-3_MeOH(DEA)_40_2.5ML,98.94%de。 在8 min層析中,
SFC
Rt=4.785 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,94.03%de。
實例 80 : (3S,8R,9S,10R,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (80) 之合成 1.
N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH
3
H
2
O ETOH,開始:B:40%,結束B:40%)純化,得到
80
(峰值2,300 mg,不純)及呈固體之
83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶
80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空中過濾懸浮液,得到呈固體之
80
(150 mg,15%)。在82℃下回流1 小時,自MeCN (3 mL)再結晶
83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空中過濾懸浮液,得到呈固體之
83
(150 mg,15%)。
80 : 1 H NMR
(400 MHz, CDCl
3
)δ 5.41-5.39 (m, 1H), 3.63-3.62 (m, 1H), 2.22-1.67 (m, 10H), 1.64-1.36 (m, 12H), 1.16-1.03 (m, 8H), 0.98-0.80 (m, 11H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.276 min,30-90 AB,純度99%,C
27
H
42
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 在10 min層析中,
SFC
Rt=5.236 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
實例 81 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (81) 之合成 1. S-500-6-1_2
(350 mg)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH
3
.H
2
O EtOH,梯度:35%至35%,流速 (mL/min):60 mL/min,25℃)純化,得到
81
(峰值1,130 mg,37%)及呈白色固體之
65
(峰值2,180 mg,52%)。
81 : 1 H NMR
(400 MHz, CDCl3) δ 5.34-5.24 (m, 1H), 4.09-4.00 (m, 1H), 2.43-2.33 (m, 1H), 2.14 (d,
J
= 4Hz, 1H), 2.07-1.80 (m, 5H), 1.77-1.55 (m, 5H), 1.53-1.30 (m, 7H), 1.28-1.00 (m, 11H), 1.00-0.91 (m, 1H), 0.85 (t,
J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.220 min,30-90 AB,純度100%,C
25
H
38
F
3
O [M+H-H
2
O]
+
之MS ESI計算值411,實驗值411。 在10 min層析中,
SFC
__E1峰值1:Rt=4.561 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),100%de。
實例 82 : (3S,5R,8R,9R , 10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (82) 之合成 1.在Ar下向
44
(200 mg,0.480 mmol)於MeOH/THF (10 mL/10 mL)中之溶液添加Pd/C (無水,200 mg)。將懸浮液真空脫氣且用H
2
淨化三次。在50℃下於H
2
(50 psi)下攪拌混合物48小時,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用THF (100 mL)洗滌。濃縮濾液,得到呈固體之
28
(30 mg,15%)及呈固體之
82
(30 mg,15%)。
82 : 1 H NMR
(400MHz, CDCl3) δ 3.63-3.61 (m, 1H), 2.13-2.00 (m, 1H), 1.99-1.81 (m, 2H), 1.72-1.57 (m, 6H), 1.54-1.34 (m, 11H), 1.33-1.16 (m, 7H), 1.15-0.96 (m, 5H), 0.92-0.85 (m, 13H), 0.81-0.69 (m, 1H), 0.67 (s, 3H)。 在2 min層析中,
LCMS
Rt=1.348 min,30-90AB,純度100%,C
28
H
47
[M+H-2H
2
O]
+
之MS ESI計算值383,實驗值383。
實例 83 : (3S,8R,9S,10R,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (83) 之合成 1.
N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH
3
H
2
O ETOH,開始:B:40%,結束B:40%)純化,得到
80
(峰值2,300 mg,不純)及呈固體之
83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶
80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之
80
(150 mg,15%)。在82℃下回流1 h,自MeCN (3 mL)再結晶
83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之
83
(150 mg,15%)。
83 : 1 H NMR
(400 MHz, CDCl
3
)δ 5.41-5.39 (m, 1H), 3.62-3.60 (m, 1H), 2.22-1.89 (m, 6H), 1.64-1.49 (m, 9H), 1.46-1.11 (m, 16H), 0.98-0.86 (m, 10H), 0.70 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.268 min,30-90 AB,純度100%,C
27
H
42
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 在10 min層析中,
SFC
Rt=4.609 min,AD_3_EtOH_DEA_5_40_25ML,100%de。
實例 84 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (84) 之合成 1.向
32
(80 mg,0.18 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)
2
(160 mg,無水)。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到
67
(10 mg,12%)及呈固體之
84
(30 mg,37%)。
84 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.75-3.66 (m, 1H), 2.00-1.90 (m, 2H), 1.86-1.75 (m, 2H), 1.73-1.55 (m, 11H), 1.53-1.26 (m, 9H), 1.25-1.15 (m, 6H), 1.14-1.03 (m, 5H), 1.02-0.92 (m, 3H), 0.91-0.85 (m, 6H), 0.82 (s, 3H), 0.72-0.58 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.518 min,30-90AB_E,純度100%,C
30
H
49
[M+H-H
2
O]
+
之MS ESI計算值409,實驗值409。
實例 85 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (85) 之合成 1.在15℃下將NaBH
4
(0.96 g,25.4 mmol)逐份添加至
N-4-1_8
(0.6 g,1.27 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌混合物30 min。向混合物添加NH
4
Cl (50 ml,10%)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機層經Na
2
SO
4
乾燥,過濾且真空濃縮,且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到不純
42
及
85
。在15℃下自MeCN (10 mL)濕磨
42
且在真空中乾燥,得到呈固體之
42
(153 mg,25%)。
85
藉由閃蒸塔(0-15% EtOAc/PE)純化以提供油狀物,用MeCN (5 mL)及水(5 mL)處理該油狀物,真空濃縮,得到呈固體之
85
(70 mg,12%)。
85 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.66-3.55 (m, 1H), 2.10-1.91 (m, 3H), 1.88-1.78 (m, 1H), 1.72-1.55 (m, 6H), 1.50-1.38 (m, 9H), 1.37-0.95 (m, 10H), 0.94-0.79 (m, 13H), 0.75-0.61 (m, 4H)。 在2.0 min層析中,
LCMS
Rt=1.343 min,30-90_AB_E,純度100%,C
28
H
46
F
3
O [M+H-H
2
O]
+
之MS ESI計算值 455,實驗值455。 在10.0 min層析中,
HPLC
Rt=5.14 min,50-100_AB_E,純度98.56%。
實例 86 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1-( 四氫 -2H- 哌喃 -4- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (86) 之合成 1.在70℃下向Mg (486 mg,20 mmol)及I
2
(1 mg)之混合物逐滴添加4-氯四氫-2H-哌喃(1.2 g,10 mmol)於THF (5 mL)中之溶液。在50℃下攪拌混合物0.5 h,用THF (5 mL)稀釋且直接使用。在0℃下於N
2
下向(四氫-2H-哌喃-4-基)氯化鎂(4.14 mL,1 M於THF中)之溶液添加含
N-8-7_1
(500 mg,1.38 mmol)之THF (5 mL)。在15℃下再攪拌混合物18小時。用飽和NH
4
Cl (5 mL)淬滅反應混合物,且用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(5 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈固體之
N-8-17_1
(350mg,57%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.08-3.91 (m, 2H), 3.43-3.31 (m, 2H), 3.31-3.25 (m, 1H), 1.98-1.91 (m, 2H), 1.91-1.80 (m, 1H),1.70-1.50 (m, 10H), 1.50-1.41 (m, 2H), 1.41-1.32 (m, 5H), 1.32-1.15 (m, 9H), 1.15-0.92 (m, 6H), 0.92-0.83 (m, 7H), 0.65 (s, 3H)。
2.
向
N-8-17_1
(300 mg,0.671 mmol)於DCM (5 mL)中之溶液添加DMP (0.852 g,2.01 mmol)。在15℃下攪拌10 min之後,用飽和NaHCO3 (20 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層,且用DCM (2 × 20 mL)萃取水相。合併之有機相用Na
2
S
2
O
3
飽和水溶液(3 × 20 mL)、飽和NaHCO3 (20 mL)、鹽水(50 mL)洗滌,經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (0-30% EtOAc/PE)純化,得到呈固體之
N-8-17_3
(200 mg)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.08-3.91 (m, 2H), 3.50-3.31 (m, 2H), 2.73-2.51 (m, 2H), 1.98-1.79 (m, 1H), 1.79-1.42 (m, 16H), 1.42-1.18 (m, 7H), 1.18-0.93 (m, 8H), 0.93-0.79 (m, 6H), 0.68 (s, 4H)。 3.在0℃下向
N-8-17_3
(200 mg,0.449 mmol)於THF (5 mL)中之混合物添加LiAlH4 (50.9 mg,1.34 mmol)。在15℃下攪拌0.5 h之後,用水(3 mL)淬滅反應混合物,且用EtOAc (2 × 10 mL)萃取。合併之有機層用鹽水(10 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之
86
(23mg,11%)。
1 H NMR
(400 MHz, CDCl
3
) δ 4.08-3.91 (m, 2H), 3.41-3.31 (m, 2H), 3.31-3.22(m, 1H), 2.01-1.79 (m, 3H), 1.70-1.61 (m, 1H), 1.61-1.53 (m, 8H), 1.53-1.51 (m, 1H), 1.51-1.39 (m, 5H), 1.39-1.13 (m, 8H), 1.13-0.92 (m, 5H), 0.92-0.85 (m, 7H), 0.82 (s, 3H), 0.66 (s, 4H)。 在7 min層析中,
LCMS
Rt=4.832 min,30-90AB_7MIN_E,純度100%,C
29
H
47
O [M+H-2H
2
O]
+
之MS ESI計算值411,實驗值411。 在10 min層析中,
HPLC
Rt=6.338 min,30-90AB_1.2 mL e. Met,100%純度。
實例 87 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (87) 之合成 1.向
12
(100 mg,0.206 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)
2
(150 mg,無水)。在50℃下於H
2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
87
(12 mg,12%)及
20
(11 mg,11%)。
87 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.82-3.75 (m, 1H), 2.00-1.83 (m, 2H), 1.80-1.58 (m, 7H), 1.52-1.42 (m,4H), 1.40-1.27 (m, 10H), 1.25-1.14 (m, 10H), 1.13-0.98 (m, 6H), 0.96 (s, 3H), 0.94-0.82 (m, 12H), 0.67 (s, 3H)。 在2.0 min層析中,
LCMS
Rt=1.734 min,30-90AB_E,純度100%,C
33
H
55
[M+H-2H
2
O]
+
之MS ESI計算值451,實驗值451。
實例 88 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (88) 之合成 1.在Ar下向
83
(150 mg,0.372 mmol)於MeOH (3 mL)及THF (3 mL)中之溶液添加Pd(OH)
2
(200 mg)。將懸浮液真空脫氣且用H
2
淨化三次。在50℃下於H
2
(50 psi)下攪拌混合物12 h,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用DCM (3 × 50 mL)洗滌。真空濃縮濾液以提供油狀物。殘餘物藉由矽膠層析(PE/EtOAc=8/1至5/1)純化,得到呈固體之
88
(18 mg,12%)。
1 H NMR
(400 MHz, CDCl
3
)δ 3.62-3.59 (m, 1H), 1.97-1.81 (m, 2H), 1.76-1.50 (m, 12H), 1.46-1.28 (m, 5H), 1.24-1.03 (m, 12H), 0.95-0.83 (m, 11H), 0.74-0.57 (m, 5H)。 在1.317 min層析中,
LCMS
Rt=2.0 min,30-90 AB,純度99%,C
27
H
44
[M+H-2H
2
O]
+
之MS ESI計算值369,實驗值369。
實例 89 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1-( 吡啶 -3- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (89) 之合成 1.
N-8-19_1 (3340)
(100 mg,0.227 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:50-50% B (A=0.05% NH3/H2O,B=MeOH),流速:80 mL/min)分離,得到
7
(峰值1,57 mg,57%)及呈固體之
89
(峰值2,8 mg,8%)。 在3 min層析中,
SFC
峰值1:Rt=1.798 min且峰值2 Rt=1.985 min,AD-H_3UM_4_5_40_4ML (「Chiralpak AD-3 50×4.6mm I.D.,3um 移動相:A:CO
2
,B:異丙醇(0.05% DEA),梯度:在1.4 min內B為5%至40%且保持40% 1.05 min,隨後B為5%持續0.35 min,流速:4 mL/min,管柱溫度:40℃」)。
8
之結構藉由x射線晶體分析法確認。
實例 90 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1R,2S)-1- 環丙基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (90) 之合成 1.向
N-8-13_2
(140 mg,0.347 mmol)於MeOH (1 mL)及THF (1 mL)中之溶液每五分鐘添加五次NaBH
4
(1.18 g,17.4 mmol)。在15℃下攪拌混合物30分鐘。用飽和NH
4
Cl (50 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相經Na
2
SO
4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之
10
(26 mg,19%)及呈固體之
90
(12 mg,9%)。
90 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.00-2.80 (m, 1H), 2.01-1.95 (m, 1H), 1.75-1.60 (m, 5H), 1.47-1.18 (m, 13H), 1.15-0.79 (m, 18H), 0.70-0.60 (m, 4H), 0.58-0.50 (m, 1H), 0.48-0.40 (m, 1H), 0.38-0.30 (m, 1H), 0.24-0.16 (m, 1H)。 在7.0 min層析中,
LCMS
Rt=3.796 min,30-90AB_7MIN_E,純度100%,C
27
H
43
[M+H-2H
2
O]
+
之MS ESI計算值367,實驗值367。 在30 min層析中,
HPLC
Rt= 13.689 min,70-90AB_1_30MIN.M,純度98%。
實例 91 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥丁 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (91) 之合成 91
之合成描述於實例13中。
91 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.98-3.88 (m, 1H), 2.11-2.02 (m, 1H), 2.00 (s, 1H), 1.98-1.88 (m, 2H), 1.85-1.79 (m, 1H), 1.73-1.58 (m, 4H), 1.52-1.20 (m, 11H), 1.19-1.11 (m, 4H), 1.10-1.00 (m, 3H), 0.97-0.89 (m, 4H), 0.85 (s, 3H), 0.75-0.68 (m, 1H), 0.66 (s, 3H)。 在2.0 min層析中,
LCMS
Rt= 1.155 min,30-90_AB_E,純度100%,C
24
H
38
F
3
O[M+H-H
2
O]
+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,
HPLC
Rt= 5.23 min,30-90_AB_E,純度98.88%,d.e. 100%。
實例 92 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (92) 之合成 1.向
N-4-16_1
(1.00 g,2.86 mmol)於DCM (20 mL)中之溶液添加DMP (2.42 g,5.72 mmol)。此後,在15℃下攪拌反應物10 min。向反應混合物添加NaHCO3飽和水溶液(20 mL)及Na
2
S
2
O
3
飽和水溶液(20 mL),接著用DCM (2 × 20 mL)萃取。合併之有機層用NaHCO
3
飽和水溶液(3 × 60 mL)及鹽水(60 mL)洗滌,經Na
2
SO
4
乾燥,過濾且真空下濃縮,得到固體。殘餘物藉由矽膠層析(PE/EtOAc=0%至30%)純化,得到呈固體之
N-4-16_2
(800 mg,81%)。
1 H NMR
(400 MHz, CDCl
3
) δ 9.58-9.53 (m, 1H), 2.41-2.31 (m, 1H), 1.96-1.81 (m, 4H), 1.89-1.34 (m, 10H), 1.32-1.21 (m, 10H), 1.16-1.09 (m, 5H), 0.97 (s, 3H), 0.89-0.84 (m, 1H), 0.69 (s, 3H)。
2.
在25℃下於N
2
下向異戊基溴化鎂(4.32 mL,2 M於醚中,8.65 mmol)之溶液中添加含
N-4-16_2
(300 mg,0.86 mmol)之THF (10 mL)。在25℃下攪拌混合物30分鐘,藉由飽和NH
4
Cl (10 mL)來淬滅且用(3 × 10 mL)乙酸乙酯萃取。有機層用鹽水(20 mL)洗滌,經Na
2
SO
4
乾燥且過濾,真空濃縮且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈固體之
N-4-16
(200 mg,不純),其在25℃下於MeCN (10 mL)中濕磨,得到呈固體之
92
(141 mg,70%)。
1 H NMR
(400 MHz, CDCl
3
) δ 3.67-3.57 (m, 1H), 2.01-1.77 (m, 4H), 1.67-1.57 (m, 4H), 1.55-1.26 (m, 14H), 1.25-1.21 (m, 5H), 1.19-0.99 (m, 7H), 0.96 (s, 3H), 0.93-0.84 (m, 9H), 0.66 (s, 3H)。 在2 min層析中,
LCMS
Rt= 1.367 min,30-90AB_2MIN_E,純度100%,C
28
H
47
[M+H-2H
2
O]
+
之MS ESI計算值383,實驗值383。
實例 93 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 苯基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (93) 之合成 1.
N-6-5 及 N-6-6
(190 mg,0.420 mmol)之混合物藉由製備型HPLC (管柱:YMC-Actus Triart C18 100×30mm×5um;條件:水(0.05% HCl)-ACN;梯度:90-100% B;流速:25 mL/min)分離,得到呈固體之
93
(56 mg,30%)及呈固體之
32
(12 mg,6%)。
93 : 1 H NMR
(400 MHz, CDCl
3
) δ 7.34-7.28 (m, 2H), 7.25-7.18 (m, 3H), 3.95-3.86 (m, 1H), 2.87-2.75 (m, 1H), 2.69-2.58 (m, 1H), 2.01-1.91 (m, 1H), 1.90-1.79 (m, 1H), 1.69-1.58 (m, 4H), 1.55-1.41 (m, 6H), 1.40-1.11 (m, 11H), 1.07-0.95 (m, 6H), 0.91-0.80 (m, 7H), 0.69-0.59 (m, 4H)。 在2.0 min層析中,
LCMS
Rt= 1.334 min,30-90AB,純度100%,C
31
H
48
O
2
Na[M+Na]
+
之MS ESI計算值475,實驗值475。
實例 94 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S
,
3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (94) 之合成 1.向
100
(150 mg,0.338 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)
2
(300 mg,無水)。在50℃下於H
2
(50 psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到
94
(6 mg,4%)及呈固體之
98
(46 mg,30%)。
94 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.78-3.66 (m, 1H), 1.98-1.72 (m, 7H), 1.69-1.59 (m, 4H), 1.48-1.32 (m, 12H), 1.27-1.07 (m, 12H), 1.06-1.00 (m, 3H), 0.97 (s, 3H), 0.94-0.85 (m, 7H), 0.66 (s, 3H)。 在2.0 min層析中,
LCMS
Rt= 1.639 min,30-90AB_E,純度98.8%,C
30
H
49
[M+H-2H
2
O]
+
之MS ESI計算值409,實驗值409。
實例 95 : (3S,8S,9S,10R,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (95) 之合成 1.
N-004-029A
(0.45 g,1.01 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH
3
H
2
O ETOH,開始B:30%,結束B:30%)純化,得到呈白色固體之
39
(
PK1
:120 mg,26.7%)及呈白色固體之
95
(
PK2
:200 mg,44.5%)。
95 : 1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.29 (m, 1H), 4.06-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.42-2.33 (m, 1H), 2.17-2.07 (m, 1H), 2.20-1.85 (m, 5H), 1.77-1.63 (m, 4H), 1.51-0.83 (m, 17H), 0.73 (s, 3H)。 在2 min層析中,
LCMS
Rt= 1.103 min,30-90AB_2MIN_E,純度100%,C
24
H
34
F
3
O[M-CH
5
O
2
]
+
之MS ESI計算值395,實驗值395。
實例 96 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (96) 之合成 1.向
52
(50 mg,0.116 mmol)於MeOH (10 mL)中之溶液添加Pd(OH)
2
(200 mg)。在50℃下於H
2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之
24
(15 mg,30%)及呈固體之
96
(1.2 mg,3%)。
96 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.67-3.55 (m, 1H), 2.01-1.83 (m, 2H), 1.80-1.62 (m, 4H), 1.61-1.56 (m, 2H), 1.55-1.50 (m, 1H), 1.49-1.31 (m, 10H), 1.30-1.10 (m, 11H), 1.09-1.00 (m, 3H), 0.96 (s, 3H), 0.94-0.86 (m, 12H), 0.67(s, 3H)。 在2 min層析中,
LCMS
tR= 1.326 min,30-90AB_ELSD,純度100.0%,C
29
H
49
[M+H-2H
2
O]
+
之MS ESI計算值397,實驗值397。
實例 97 : (3S,5R,8R,9S,10S,13S,14S,17R)-3,10,13- 三甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (97) 之合成 1.向
N-004-017_3
(100 mg,0.19 mmol)於THF (2 mL)及MeOH (1 mL)以及水(1 mL)中之溶液添加KOH (53.8 mg,0.96 mmol)。在60℃下攪拌混合物16小時。將混合物倒入水(20 mL)中且用EtOAc (2 × 40 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈白色固體之
97
(80 mg,不純)。
97
(80 mg,0.19 mmol)藉由閃蒸塔(0-10% EtOAc/PE)純化,得到
97
(60 mg,不純)。在0℃下於N
2
下向
97
(40 mg,0.096 mmol)於甲醇(3 mL)中之溶液一次性添加NaBH
4
(10.8 mg,0.28 mmol)。在0℃下攪拌混合物30 min。將混合物倒入水(10 mL)中並攪拌20 min。用EtOAc (3 × 10 mL)萃取水相。合併之有機相用鹽水(2 × 20 mL)洗滌,經Na
2
SO
4
乾燥,過濾且濃縮,得到粗產物,該粗產物與另一批次的20 mg不純
97
合併,殘餘物藉由閃蒸塔(0-10% EtOAc/PE)純化,得到呈白色固體之
97
(31 mg,31%)。
1 H NMR
(400 MHz, CDCl3) δ 4.11-3.96 (m, 1H), 2.18-2.11 (d,
J ab
= 6.4 Hz, 1H), 2.02-1.77 (m, 5H), 1.68-1.57 (m, 3H), 1.49-1.24 (m, 11H), 1.23-1.19 (m, 5H), 1.18-1.01 (m, 7H), 0.96 (s, 3H), 0.67 (s, 3H)。 在2 min層析中,
LCMS
Rt= 1.124 min,30-90AB_2MIN_E.M,純度100%,C
24
H
38
F
3
O[M+H-H
2
O]
+
之MS ESI計算值399,實驗值399。
實例 98 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (98) 之合成 1.向
100
(150 mg,0.338 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)
2
(300 mg,無水)。在50℃下於H
2
(50 psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到
94
(6 mg,4%)及呈固體之
98
(46 mg,30%)。
98 : 1 H NMR
(400 MHz, CDCl
3
) δ 3.78-3.67 (m, 1H), 1.97-1.74 (m, 6H), 1.68-1.56 (m, 8H), 1.53-1.45 (m, 4H), 1.44-1.31 (m, 10H), 1.28-1.21 (m, 1H), 1.16-0.96 (m, 9H), 0.91-0.85 (m, 6H), 0.82 (s, 3H), 0.69-0.61 (m, 4H)。 在2.0 min層析中,
LCMS
Rt= 1.582 min,30-90AB_E,純度100%,C
30
H
49
[M+H-2H
2
O]
+
之MS ESI計算值409,實驗值409。
實例 99 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S,E)-3- 羥基 -5- 苯基戊 -4- 烯 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (99) 之合成 1.在20℃下向
N-003-005_1
(140 mg,0.278 mmol)於THF (2 mL)及MeOH (1 mL)中之溶液逐份添加NaBH
4
(419 mg,11.1 mmol)。在20℃下攪拌混合物10 min。用水(20 mL)及NH
4
Cl (20 mL,飽和)淬滅反應物。用EtOAc (50 mL)萃取混合物。真空濃縮有機層且藉由prep-TLC (PE/EtOAc=4/1)純化,得到均呈固體之
N-003-005
(50 mg,不純)及
14
(50 mg)。 不純
99
(50 mg)藉由SFC (儀器:SFC 1;管柱:OD (250mm×30mm,5um);條件:0.1% NH
3
H
2
O EtOH;開始B:40%;結束B:40%;流速(mL/min):50;注入:60)純化以提供溶解於MeCN (20 mL)中之固體且真空濃縮,得到呈固體之
99
(17 mg)。
99 : 1 H NMR
(400 MHz, CDCl
3
) δ 7.44-7.36 (m, 2H), 7.36-7.28 (m, 2H), 7.25-7.20 (m, 1H), 6.58 (d,
J
= 16.0 Hz, 1H), 6.24 (dd,
J
= 4.8, 16.0 Hz, 1H), 4.49-4.40 (m, 1H), 2.09-1.91 (m, 4H), 1.86-1.75 (m, 1H), 1.72-1.58 (m, 5H), 1.52-1.04 (m, 14H), 0.94 (d,
J
= 6.4 Hz, 3H), 0.91-0.87 (m, 1H), 0.86 (s, 3H), 0.75-0.70 (m, 1H), 0.69 (s, 3H)。 在2 min層析中,
LCMS
Rt= 1.280 min,30-90AB_2MIN_E,純度98.5%,C
31
H
42
F
3
O [M+H-H
2
O]
+
之MS ESI計算值487,實驗值487。 在8 min層析中,
HPLC
Rt= 6.29 min,30-90_AB_1.2ml,100% d.e.。
實例 100 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (100) 之合成 1. 在75℃下向Mg (528 mg,22.0 mmol)及I
2
(55.8 mg,0.22 mmol)於THF (3 mL)中之懸浮液逐滴添加(溴甲基)環戊烷(1.8 g,11.0 mmol)於THF (11 mL)中之溶液。在75℃下攪拌混合物1小時。在15℃下向於
S-500-6-1_1
(800mg,2.23mmol)於THF (30 mL)中之溶液緩慢添加(環戊基甲基)溴化鎂(11.1mL,11.1mmol,1 M於THF中)之混合物。添加之後,在15℃下攪拌混合物1小時。用飽和NH
4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2 × 30 mL)洗滌,經Na
2
SO
4
乾燥,過濾及濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之
100
(350 mg,35%)。
1 H NMR
(400 MHz, CDCl
3
) δ 5.32-5.26 (m, 1H), 3.77-3.69 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.89 (m, 4H), 1.88-1.69 (m, 4H), 1.68-1.55 (m, 6H), 1.54-1.27 (m, 12H), 1.26-1.15 (m, 2H), 1.14-1.05 (m, 4H), 1.04-0.99 (m, 5H), 0.98-0.88 (m, 4H), 0.87-0.81 (m, 3H), 0.69 (s, 3H)。 在7.0 min層析中,
LCMS
Rt= 5.661 min,30-90AB_E,純度100%,C
30
H
47
[M+H-2H
2
O]
+
之MS ESI計算值407,實驗值407。
表 1. 例示性化合物之資料。
對於
表 1
,「A」指示1至100 nM之EC
50
;「B」指示大於100 nM達至1 µM之EC
50
;「C」指示大於1 µM之EC
50
;「D」指示高達100%之E
Max
;「E」指示在100%與500%之間的E
Max
;「F」指示大於500%之E
Max
;「G」指示在10%與-10%之間且包括10%及-10%的增效作用%;「H」指示低於-10%且大於或等於-40%的增效作用%;且「I」指示低於-40%的增效作用%。
其它實施例
在申請專利範圍中,除非相反地指示或另外從上下文顯而易見,否則諸如「一(a/an)」及「該」之冠詞可意謂一或大於一。除非相反地指示或另外從上下文顯而易見,否則若一個、大於一個或所有群組成員存在於、用於給定產物或方法中或另外與給定產物或方法有關,則在群組的一或多個成員之間包括「或」之申請專利範圍或描述被視為滿足。本發明包括群組中恰好一個成員存在於、用於給定產物或方法中或以其他方式與給定產物或方法相關之實施例。本發明包括多於一個或所有的群組成員存在、用於給定產物或方法中或以其他方式與給定產物或方法相關之實施例。 此外,本發明涵蓋其中來自一或多條所列技術方案之一或多個限制、要素、條款及描述性用語引入另一條技術方案中的所有變化、組合及排列。舉例而言,依附於另一技術方案之任何技術方案可經修改以包括在依附於同一基本技術方案之任何其他技術方案中可見的一或多個限制。在要素如所列,例如呈馬庫什(Markush)組格式呈現時,亦揭示要素之各子組,且可自該組移除任何要素。大體而言,應瞭解,在本發明或本發明之態樣被稱為包含特定要素及/或特徵時,本發明或本發明態樣之某些實施例由此類要素及/或特徵組成或主要由此類要素及/或特徵組成。出於簡單之目的,彼等實施例尚未具體地以詞語闡述在本文中。亦應注意,術語「包含」及「含有」意欲為開放性的且容許包括額外要素或步驟。當給出範圍時,包括端點。此外,除非另外指示或另外自上下文及一般技術者的理解顯而易見,否則表示為範圍之值可在本發明之不同實施例中採用所述範圍內之任何特定值或子範圍,除非上下文另外明確規定,否則達到該範圍下限之單位的十分之一。 本申請案提及各種頒予之專利、公開之專利申請案、期刊文章及其他出版物,以上所有者均以引用之方式併入本文中。若任何併入之參考文獻與本說明書之間存在衝突,則應以本說明書為準。另外,本發明之屬於先前技術之任何特定實施例可明確地自申請專利範圍中之任一或多項排除。因為此類實施例被認為是一般技術者所已知的,其可經排除,即使未在本文中明確地闡述該排除。本發明之任何特定實施例可出於任何原因自任何技術方案排除,無論是否與先前技術之存在相關。 熟習此項技術者最多使用常規實驗將認識到或能夠確定本文所描述之特定實施例之許多等效物。本文所描述之本發明實施例之範疇並不意欲限於以上描述,而是實際上如所附申請專利範圍中所闡述。一般技術者將瞭解,可在不脫離如以下申請專利範圍所定義之本發明之精神或範疇的情況下對本說明書進行各種改變及修改。
 及其醫藥學上可接受之鹽以及其醫藥組合物;其中R1
、R2
、R3
、R4
、R5
、R6
及RG
如本文所定義。預期本發明之化合物適用於預防及治療各種病狀。
及其醫藥學上可接受之鹽以及其醫藥組合物;其中R1
、R2
、R3
、R4
、R5
、R6
及RG
如本文所定義。預期本發明之化合物適用於預防及治療各種病狀。 或其醫藥學上可接受之鹽,其中: R1
為氫或烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;RG
為或烷基;且
或其醫藥學上可接受之鹽,其中: R1
為氫或烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;RG
為或烷基;且 表示單鍵或雙鍵,其中當
表示單鍵或雙鍵,其中當 表示單鍵。 在一些實施例中,RG
為氫或-CH3
。 在一個態樣中,本文提供根據式(I-63)之化合物:
表示單鍵。 在一些實施例中,RG
為氫或-CH3
。 在一個態樣中,本文提供根據式(I-63)之化合物: 或其醫藥學上可接受之鹽,其中:R1
為烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且
或其醫藥學上可接受之鹽,其中:R1
為烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且
 。 在一些實施例中,式(I-63)之化合物選自式(I-A63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-A63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-C63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-C63)之化合物: 。 在一些實施例中,式(I63)之化合物選自式(I-D63)之化合物:
。 在一些實施例中,式(I63)之化合物選自式(I-D63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-E63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-E63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-D-i63)或(I-D-ii63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-D-i63)或(I-D-ii63)之化合物: 或
或 。 在一些實施例中,式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物: 或
或 。 在一個態樣中,本文提供根據式(I-67)之化合物:
。 在一個態樣中,本文提供根據式(I-67)之化合物: 或其醫藥學上可接受之鹽,其中:R1
為氫或烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且
或其醫藥學上可接受之鹽,其中:R1
為氫或烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且 。 在一些實施例中,式(I-67)之化合物選自式(I-A67)之化合物:
。 在一些實施例中,式(I-67)之化合物選自式(I-A67)之化合物: 。 在一些實施例中,式(I-67)之化合物選自式(I-C67)之化合物:
。 在一些實施例中,式(I-67)之化合物選自式(I-C67)之化合物: 。 在一態樣中,本文中提供選自由以下組成之群的化合物:
。 在一態樣中,本文中提供選自由以下組成之群的化合物:









 及
及 。 在一態樣中,本文提供一種醫藥組合物,其包含本文所描述之化合物或其醫藥學上可接受之鹽及醫藥學上可接受之載劑。。 在一態樣中,本文提供一種誘導鎮靜或麻醉之方法,其包含向個體投與有效量的本文所描述之化合物或其醫藥學上可接受之鹽或其醫藥組合物。 在一態樣中,本文提供一種用於治療或預防本文所描述之病症的方法,其包含向有需要之個體投與有效量的本文所描述之化合物或其醫藥學上可接受之鹽或其醫藥組合物。 在一些實施例中,該病症為代謝障礙。 在一些實施例中,該病症為自體免疫病症。 在一些實施例中,該病症為類風濕性關節炎、青少年特發性關節炎、僵直性脊椎炎、牛皮癬性關節炎、克羅恩氏病、潰瘍性結腸炎及斑塊型牛皮癬。 在一些實施例中,該病症為腸胃(GI)病症(例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病))、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌或結腸炎。 在一些實施例中,該病症為發炎性腸病。 在一些實施例中,該病症為癌症、糖尿病或固醇合成障礙。 在一態樣中,本文提供一種用於治療或預防CNS相關病狀的方法,其包含向有需要之個體投與有效量的本文所描述之化合物或其醫藥學上可接受之鹽或其醫藥組合物。在一些實施例中,CNS相關病狀為失調症、焦慮症(包括強迫症、創傷後壓力症及社交恐懼症)、認知障礙(包括阿茲海默病及其他形式的癡呆(例如額顳葉型癡呆))、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群、結節性硬化症)、多發性硬化、固醇合成障礙、疼痛(包括急性及慢性疼痛;頭痛,例如偏頭痛)、繼發於醫學病狀的腦病(包括肝腦病及抗NMDA受體腦炎)、急性肝功能衰竭、甘胺酸腦病、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease))、中風、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、視覺減損、聽覺缺失或耳鳴。 在一些實施例中,該病症為亨廷頓氏病。在一些實施例中,該病症為帕金森氏病。在一些實施例中,該病症為發炎性疾病(例如狼瘡)。 在一些實施例中,該病症為固醇合成障礙。 在一些實施例中,該病症為史密斯-萊姆利-奧皮茨症候群(Smith-Lemli-Opitz Syndrome,SLOS)。在一些實施例中,該病症為鏈固醇病(desmosterolosis)。在一些實施例中,該病症為谷固醇血症(sitosterolemia)。在一些實施例中,該病症為腦腱黃瘤病(cerebrotendinous xanthomatosis,CTX)。在一些實施例中,該病症為甲羥戊酸激酶缺乏(Mevalonate Kinase Deficiency,MKD)。在一些實施例中,該病症為SC4MOL基因突變(SMO缺乏)。在一些實施例中,該病症為尼曼-皮克(Niemann-Pick)病。在一些實施例中,該病症為自閉症譜系障礙(ASD)。在一些實施例中,該病症與苯丙酮糖尿病(phenylketomuria)相關。 在一態樣中,本文提供一種誘導個體中之NMDA受體之負向異位調節之方法,其包含向該個體投與本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物。定義 化學定義
下文更詳細地描述特定官能基及化學術語之定義。化學元素係根據元素週期表(Periodic Table of the Elements), CAS版本, Handbook of Chemistry and Physics, 第75版, 內封面來識別,且特定官能基一般如其中所描述來定義。另外,有機化學之一般原理以及特定官能部分及反應性描述於以下各者中:Thomas Sorrell,Organic Chemistry
, University Science Books, Sausalito, 1999;Smith及March,March's Advanced Organic Chemistry
, 第5版, John Wiley & Sons, Inc., New York, 2001;Larock,Comprehensive Organic Transformations,
VCH Publishers, Inc., New York, 1989;及Carruthers,Some Modern Methods of Organic Synthesis
, 第3版, Cambridge University Press, Cambridge, 1987。 異構體可藉由熟習此項技術者已知之方法(包括對掌性高壓液相層析(HPLC)以及對掌性鹽的形成及結晶)而自混合物中分離;或可藉由不對稱合成來製備較佳異構體。參見(例如) Jacques等人,Enantiomers, Racemates and Resolutions
(Wiley Interscience, New York, 1981);Wilen等人,Tetrahedron
33:2725 (1977);Eliel,Stereochemistry of Carbon Compounds
(McGraw-Hill, NY, 1962);及Wilen,Tables of Resolving Agents and Optical Resolutions
第268頁 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972)。本發明另外涵蓋呈實質上不含其他異構體之個別異構體形式及替代地呈各種異構體之混合物形式的本文所描述之化合物。 如本文中所使用之組合物之「對映異構過量」(「e.e.」)或「對映異構過量%」(「%e.e.」)係指一種對映異構體相對於存在於該組合物中之另一對映異構體的過量。舉例而言,組合物可含有90%一種對映異構體,例如S對映異構體,及10%另一對映異構體,亦即R對映異構體。 e.e. = (90-10)/100 = 80%。 因此,含有90%一種對映異構體及10%其他對映異構體之組合物被稱為具有80%對映異構體過量。 如本文中所使用之組合物之「非對映異構過量」(「d.e.」)或「非對映異構過量%」(「d.e.%」)係指一種非對映異構體相對於存在於該組合物中之一或多種不同的非對映異構體的過量。舉例而言,組合物可含有90%一種對非映異構體,及10%一或多種不同的非對映異構體。 d.e. = (90-10)/100 = 80%。 因此,含有90%一種非對映異構體及10%一或多種不同的非對映異構體之組合物被稱為具有80%非對映異構體過量。 不對稱中心之絕對組態可使用熟習此項技術者已知之方法來測定。在一些實施例中,可自化合物之X射線單晶結構闡明化合物中之不對稱中心之絕對組態。在一些實施例中,藉由化合物之X射線晶體結構闡明之不對稱中心之絕對組態可用來推斷自相同或類似合成方法獲得之另一種化合物中之相對應的不對稱中心的絕對組態。 當列出值範圍時,意欲涵蓋該範圍內之各值及子範圍。舉例而言,「C1-6
烷基」意欲涵蓋C1
、C2
、C3
、C4
、C5
、C6
、C1-6
、C1-5
、C1-4
、C1-3
、C1-2
、C2-6
、C2-5
、C2-4
、C2-3
、C3-6
、C3-5
、C3-4
、C4-6
、C4-5
及C5-6
烷基。 以下術語意欲具有下文提供之意義且適用於理解本發明之描述及預期範疇。當描述本發明時,其可包括化合物、含有此類化合物之醫藥組合物及使用此類化合物及組合物之方法,除非另外指明,否則以下術語若存在則具有以下含義。應理解,當在本文中描述時,下文定義之部分中之任一者可經多種取代基取代,且各別定義意欲將該等經取代之部分包括在其如下文所述之範疇內。除非另外陳述,否則術語「經取代」應如下文所述定義。應進一步理解,術語「基團(group及radical)」在本文中使用時可被視為可互換的。冠詞「一(a及an)」可在本文中用於指該冠詞之一個或多於一個(亦即至少一個)文法對象。舉例而言,「類似物」意謂一個類似物或多於一個類似物。 「脂族」係指烷基、烯基、炔基或碳環基,如本文所定義。 「烷基」係指具有1至20個碳原子的直鏈或分支鏈飽和烴基(「C1-20
烷基」)。在一些實施例中,烷基具有1至12個碳原子(「C1-12
烷基」)。在一些實施例中,烷基具有1至10個碳原子(「C1-10
烷基」)。在一些實施例中,烷基具有1至9個碳原子(「C1-9
烷基」)。在一些實施例中,烷基具有1至8個碳原子(「C1-8
烷基」)。在一些實施例中,烷基具有1至7個碳原子(「C1-7
烷基」)。在一些實施例中,烷基具有1至6個碳原子(「C1-6
烷基」,在本文中亦稱為「低碳烷基」)。在一些實施例中,烷基具有1至5個碳原子(「C1-5
烷基」)。在一些實施例中,烷基具有1至4個碳原子(「C1-4
烷基」)。在一些實施例中,烷基具有1至3個碳原子(「C1-3
烷基」)。在一些實施例中,烷基具有1至2個碳原子(「C1-2
烷基」)。在一些實施例中,烷基具有1個碳原子(「C1
烷基」)。在一些實施例中,烷基具有2至6個碳原子(「C2-6
烷基」)。C1-6
烷基之實例包括甲基(C1
)、乙基(C2
)、正丙基(C3
)、異丙基(C3
)、正丁基(C4
)、第三丁基(C4
)、第二丁基(C4
)、異丁基(C4
)、正戊基(C5
)、3-戊基(C5
)、戊基(C5
)、異戊基(C5
)、新戊基(C5
)、3-甲基-2-丁基(C5
)、第三戊基(C5
)及正己基(C6
)。烷基之其他實例包括正庚基(C7
)、正辛基(C8
)及其類似基團。除非另外規定,否則烷基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之烷基」,或經一或多個取代基(例如,1至5個取代基、1至3個取代基或1個取代基)取代(「經取代烷基」)。在某些實施例中,烷基為未經取代之C1-10
烷基(例如-CH3
)。在某些實施例中,烷基為經取代之C1-10
烷基。常用烷基縮寫包括Me (-CH3
)、Et (-CH2
CH3
)、iPr (-CH(CH3
)2
)、nPr (-CH2
CH2
CH3
)、n-Bu (-CH2
CH2
CH2
CH3
)或i-Bu (-CH2
CH(CH3
)2
)。 「伸烷基」係指其中兩個氫經移除以提供二價基團且該二價基團可經取代或未經取代的烷基。未經取代之伸烷基包括(但不限於)亞甲基(-CH2
-)、伸乙基(-CH2
CH2
-)、伸丙基(-CH2
CH2
CH2
-)、伸丁基(-CH2
CH2
CH2
CH2
-)、伸戊基(-CH2
CH2
CH2
CH2
CH2
-)、伸己基(-CH2
CH2
CH2
CH2
CH2
CH2
-)及類似基團。(例如)經一或多個烷基(甲基)取代之示例性經取代伸烷基包括(但不限於):經取代亞甲基(-CH(CH3
)-、(-C(CH3
)2
-);經取代伸乙基(-CH(CH3
)CH2
-、-CH2
CH(CH3
)-、-C(CH3
)2
CH2
-、-CH2
C(CH3
)2
-);經取代伸丙基(-CH(CH3
)CH2
CH2
-、-CH2
CH(CH3
)CH2
-、-CH2
CH2
CH(CH3
)-、-C(CH3
)2
CH2
CH2
-、-CH2
C(CH3
)2
CH2
-、-CH2
CH2
C(CH3
)2
-);及類似基團。當提供用於特定伸烷基的一定範圍或數目之碳時,應理解,該範圍或數目係指直連碳二價鏈中之碳的數目或範圍。伸烷基可經如本文所描述之一或多個取代基取代或未經取代。 「烯基」係指具有2至20個碳原子、一或多個碳-碳雙鍵(例如,1、2、3或4個碳-碳雙鍵)及視情況選用之一或多個碳-碳參鍵(例如,1、2、3或4個碳-碳參鍵)的直鏈或分支鏈烴基(「C2-20
烯基」)。在某些實施例中,烯基不含任何參鍵。在一些實施例中,烯基具有2至10個碳原子(「C2-10
烯基」)。在一些實施例中,烯基具有2至9個碳原子(「C2-9
烯基」)。在一些實施例中,烯基具有2至8個碳原子(「C2-8
烯基」)。在一些實施例中,烯基具有2至7個碳原子(「C2-7
烯基」)。在一些實施例中,烯基具有2至6個碳原子(「C2-6
烯基」)。在一些實施例中,烯基具有2至5個碳原子(「C2-5
烯基」)。在一些實施例中,烯基具有2至4個碳原子(「C2-4
烯基」)。在一些實施例中,烯基具有2至3個碳原子(「C2-3
烯基」)。在一些實施例中,烯基具有2個碳原子(「C2
烯基」)。一或多個碳-碳雙鍵可在內部(諸如在2-丁烯基中)或末端(諸如在1-丁烯基中)。C2-4
烯基之實例包括乙烯基(C2
)、1-丙烯基(C3
)、2-丙烯基(C3
)、1-丁烯基(C4
)、2-丁烯基(C4
)、丁二烯基(C4
)及其類似基團。C2-6
烯基之實例包括前述C2-4
烯基以及戊烯基(C5
)、戊二烯基(C5
)、己烯基(C6
)及其類似基團。烯基之額外實例包括庚烯基(C7
)、辛烯基(C8
)、辛三烯基(C8
)及其類似基團。除非另外規定,否則烯基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之烯基」,或經一或多個取代基(例如,1至5個取代基、1至3個取代基或1個取代基)取代(「經取代烯基」)。在某些實施例中,烯基為未經取代之C2-10
烯基。在某些實施例中,烯基為經取代之C2-10
烯基。 「炔基」係指具有2至20個碳原子、一或多個碳-碳參鍵(例如,1、2、3或4個碳-碳參鍵)及視情況選用之一或多個碳-碳雙鍵(例如,1、2、3或4個碳-碳雙鍵)的直鏈或分支鏈烴基(「C2-20
炔基」)。在某些實施例中,炔基不含任何雙鍵。在一些實施例中,炔基具有2至10個碳原子(「C2-10
炔基」)。在一些實施例中,炔基具有2至9個碳原子(「C2-9
炔基」)。在一些實施例中,炔基具有2至8個碳原子(「C2-8
炔基」)。在一些實施例中,炔基具有2至7個碳原子(「C2-7
炔基」)。在一些實施例中,炔基具有2至6個碳原子(「C2-6
炔基」)。在一些實施例中,炔基具有2至5個碳原子(「C2-5
炔基」)。在一些實施例中,炔基具有2至4個碳原子(「C2-4
炔基」)。在一些實施例中,炔基具有2至3個碳原子(「C2-3
炔基」)。在一些實施例中,炔基具有2個碳原子(「C2
炔基」)。一或多個碳-碳參鍵可在內部(諸如在2-丁炔基中)或末端(諸如在1-丁炔基中)。C2-4
炔基之實例包括(但不限於)乙炔基(C2
)、1-丙炔基(C3
)、2-丙炔基(C3
)、1-丁炔基(C4
)、2-丁炔基(C4
)及其類似基團。C2-6
烯基之實例包括前述C2-4
炔基以及戊炔基(C5
)、己炔基(C6
)及其類似基團。炔基之額外實例包括庚炔基(C7
)、辛炔基(C8
)及其類似基團。除非另外規定,否則炔基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之炔基」,或經一或多個取代基(例如,1至5個取代基、1至3個取代基或1個取代基)取代(「經取代炔基」)。在某些實施例中,炔基為未經取代之C2-10
炔基。在某些實施例中,炔基為經取代之C2-10
炔基。 如本文所使用,術語「雜烷基」係指如本文所定義之烷基,其進一步包含在母鏈內的1個或多個(例如,1、2、3或4個)雜原子(例如,氧、硫、氮、硼、矽、磷),其中該一或多個雜原子插入在母碳鏈內的相鄰碳原子之間,及/或該一或多個雜原子插入在碳原子與母分子之間,亦即插入在連接點之間。在某些實施例中,雜烷基係指具有1至10個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-10
烷基」)。在一些實施例中,雜烷基係指具有1至9個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-9
烷基」)。在一些實施例中,雜烷基為具有1至8個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-8
烷基」)。在一些實施例中,雜烷基為具有1至7個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-7
烷基」)。在一些實施例中,雜烷基為具有1至6個碳原子及1、2或3個雜原子的基團(「雜C1-6
烷基」)。在一些實施例中,雜烷基為具有1至5個碳原子及1或2個雜原子的飽和基團(「雜C1-5
烷基」)。在一些實施例中,雜烷基為具有1至4個碳原子及1或2個雜原子的飽和基團(「雜C1-4
烷基」)。在一些實施例中,雜烷基為具有1至3個碳原子及1個雜原子的飽和基團(「雜C1-3
烷基」)。在一些實施例中,雜烷基為具有1至2個碳原子及1個雜原子的飽和基團(「雜C1-2
烷基」)。在一些實施例中,雜烷基為具有1個碳原子及1個雜原子的飽和基團(「雜C1
烷基」)。在一些實施例中,雜烷基為具有2至6個碳原子及1或2個雜原子的飽和基團(「雜C2-6
烷基」)。除非另外規定,否則雜烷基之各個例係獨立地未經取代(「未經取代之雜烷基」)或經一或多個取代基取代(「經取代雜烷基」)。在某些實施例中,雜烷基為未經取代之雜C1-10
烷基。在某些實施例中,雜烷基為經取代之雜C1-10
烷基。 「芳基」係指具有芳環系統中所提供之6至14個環碳原子及0個雜原子的單環或多環(例如雙環或三環) 4n+2芳環系統(例如,具有在環狀陣列中共用之6、10或14個π電子)之基團(「C6-14
芳基」)。在一些實施例中,芳基具有六個環碳原子(「C6
芳基」;例如苯基)。在一些實施例中,芳基具有十個環碳原子(「C10
芳基」;例如萘基,諸如1-萘基及2-萘基)。在一些實施例中,芳基具有十四個環碳原子(「C14
芳基」;例如蒽基)。「芳基」亦包括其中如上文所定義之芳基環與一或多個碳環基或雜環基稠合之環系統,其中連接基團或連接點在芳基環上,且在此類情況下,碳原子數目繼續指示芳基環系統中之碳原子的數目。典型芳基包括(但不限於)衍生自以下之基團:乙烯合蒽、乙烯合萘、乙烯合菲、蒽、甘菊環、苯、䓛、六苯并苯、丙二烯合茀、苯并茚、稠六苯、己二吩(hexaphene)、己二烯(hexalene)、as-二環戊二烯并苯、s-二環戊二烯并苯、茚滿、茚、萘、稠八苯、辛二吩(octaphene)、辛二烯(octalene)、卵苯、環戊二烯并-2,4-二烯、稠五苯、并環戊二烯、環戊二烯吩、苝、丙烯合萘、菲、苉、七曜烯、芘、苒、茹(rubicene)、聯伸三苯及三萘。特定芳基包括苯基、萘基、茚基及四氫萘基。除非另外規定,否則芳基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之芳基」,或經一或多個取代基取代(「經取代芳基」)。在某些實施例中,芳基為未經取代之C6-14
芳基。在某些實施例中,芳基為經取代之C6-14
芳基。 在某些實施例中,芳基經選自以下之基團中的一或多者取代:鹵基、C1
-C8
烷基、C1
-C8
鹵烷基、氰基、羥基、C1
-C8
烷氧基及胺基。 代表性的經取代芳基之實例包括以下
。 在一態樣中,本文提供一種醫藥組合物,其包含本文所描述之化合物或其醫藥學上可接受之鹽及醫藥學上可接受之載劑。。 在一態樣中,本文提供一種誘導鎮靜或麻醉之方法,其包含向個體投與有效量的本文所描述之化合物或其醫藥學上可接受之鹽或其醫藥組合物。 在一態樣中,本文提供一種用於治療或預防本文所描述之病症的方法,其包含向有需要之個體投與有效量的本文所描述之化合物或其醫藥學上可接受之鹽或其醫藥組合物。 在一些實施例中,該病症為代謝障礙。 在一些實施例中,該病症為自體免疫病症。 在一些實施例中,該病症為類風濕性關節炎、青少年特發性關節炎、僵直性脊椎炎、牛皮癬性關節炎、克羅恩氏病、潰瘍性結腸炎及斑塊型牛皮癬。 在一些實施例中,該病症為腸胃(GI)病症(例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病))、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌或結腸炎。 在一些實施例中,該病症為發炎性腸病。 在一些實施例中,該病症為癌症、糖尿病或固醇合成障礙。 在一態樣中,本文提供一種用於治療或預防CNS相關病狀的方法,其包含向有需要之個體投與有效量的本文所描述之化合物或其醫藥學上可接受之鹽或其醫藥組合物。在一些實施例中,CNS相關病狀為失調症、焦慮症(包括強迫症、創傷後壓力症及社交恐懼症)、認知障礙(包括阿茲海默病及其他形式的癡呆(例如額顳葉型癡呆))、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群、結節性硬化症)、多發性硬化、固醇合成障礙、疼痛(包括急性及慢性疼痛;頭痛,例如偏頭痛)、繼發於醫學病狀的腦病(包括肝腦病及抗NMDA受體腦炎)、急性肝功能衰竭、甘胺酸腦病、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease))、中風、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、視覺減損、聽覺缺失或耳鳴。 在一些實施例中,該病症為亨廷頓氏病。在一些實施例中,該病症為帕金森氏病。在一些實施例中,該病症為發炎性疾病(例如狼瘡)。 在一些實施例中,該病症為固醇合成障礙。 在一些實施例中,該病症為史密斯-萊姆利-奧皮茨症候群(Smith-Lemli-Opitz Syndrome,SLOS)。在一些實施例中,該病症為鏈固醇病(desmosterolosis)。在一些實施例中,該病症為谷固醇血症(sitosterolemia)。在一些實施例中,該病症為腦腱黃瘤病(cerebrotendinous xanthomatosis,CTX)。在一些實施例中,該病症為甲羥戊酸激酶缺乏(Mevalonate Kinase Deficiency,MKD)。在一些實施例中,該病症為SC4MOL基因突變(SMO缺乏)。在一些實施例中,該病症為尼曼-皮克(Niemann-Pick)病。在一些實施例中,該病症為自閉症譜系障礙(ASD)。在一些實施例中,該病症與苯丙酮糖尿病(phenylketomuria)相關。 在一態樣中,本文提供一種誘導個體中之NMDA受體之負向異位調節之方法,其包含向該個體投與本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物。定義 化學定義
下文更詳細地描述特定官能基及化學術語之定義。化學元素係根據元素週期表(Periodic Table of the Elements), CAS版本, Handbook of Chemistry and Physics, 第75版, 內封面來識別,且特定官能基一般如其中所描述來定義。另外,有機化學之一般原理以及特定官能部分及反應性描述於以下各者中:Thomas Sorrell,Organic Chemistry
, University Science Books, Sausalito, 1999;Smith及March,March's Advanced Organic Chemistry
, 第5版, John Wiley & Sons, Inc., New York, 2001;Larock,Comprehensive Organic Transformations,
VCH Publishers, Inc., New York, 1989;及Carruthers,Some Modern Methods of Organic Synthesis
, 第3版, Cambridge University Press, Cambridge, 1987。 異構體可藉由熟習此項技術者已知之方法(包括對掌性高壓液相層析(HPLC)以及對掌性鹽的形成及結晶)而自混合物中分離;或可藉由不對稱合成來製備較佳異構體。參見(例如) Jacques等人,Enantiomers, Racemates and Resolutions
(Wiley Interscience, New York, 1981);Wilen等人,Tetrahedron
33:2725 (1977);Eliel,Stereochemistry of Carbon Compounds
(McGraw-Hill, NY, 1962);及Wilen,Tables of Resolving Agents and Optical Resolutions
第268頁 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972)。本發明另外涵蓋呈實質上不含其他異構體之個別異構體形式及替代地呈各種異構體之混合物形式的本文所描述之化合物。 如本文中所使用之組合物之「對映異構過量」(「e.e.」)或「對映異構過量%」(「%e.e.」)係指一種對映異構體相對於存在於該組合物中之另一對映異構體的過量。舉例而言,組合物可含有90%一種對映異構體,例如S對映異構體,及10%另一對映異構體,亦即R對映異構體。 e.e. = (90-10)/100 = 80%。 因此,含有90%一種對映異構體及10%其他對映異構體之組合物被稱為具有80%對映異構體過量。 如本文中所使用之組合物之「非對映異構過量」(「d.e.」)或「非對映異構過量%」(「d.e.%」)係指一種非對映異構體相對於存在於該組合物中之一或多種不同的非對映異構體的過量。舉例而言,組合物可含有90%一種對非映異構體,及10%一或多種不同的非對映異構體。 d.e. = (90-10)/100 = 80%。 因此,含有90%一種非對映異構體及10%一或多種不同的非對映異構體之組合物被稱為具有80%非對映異構體過量。 不對稱中心之絕對組態可使用熟習此項技術者已知之方法來測定。在一些實施例中,可自化合物之X射線單晶結構闡明化合物中之不對稱中心之絕對組態。在一些實施例中,藉由化合物之X射線晶體結構闡明之不對稱中心之絕對組態可用來推斷自相同或類似合成方法獲得之另一種化合物中之相對應的不對稱中心的絕對組態。 當列出值範圍時,意欲涵蓋該範圍內之各值及子範圍。舉例而言,「C1-6
烷基」意欲涵蓋C1
、C2
、C3
、C4
、C5
、C6
、C1-6
、C1-5
、C1-4
、C1-3
、C1-2
、C2-6
、C2-5
、C2-4
、C2-3
、C3-6
、C3-5
、C3-4
、C4-6
、C4-5
及C5-6
烷基。 以下術語意欲具有下文提供之意義且適用於理解本發明之描述及預期範疇。當描述本發明時,其可包括化合物、含有此類化合物之醫藥組合物及使用此類化合物及組合物之方法,除非另外指明,否則以下術語若存在則具有以下含義。應理解,當在本文中描述時,下文定義之部分中之任一者可經多種取代基取代,且各別定義意欲將該等經取代之部分包括在其如下文所述之範疇內。除非另外陳述,否則術語「經取代」應如下文所述定義。應進一步理解,術語「基團(group及radical)」在本文中使用時可被視為可互換的。冠詞「一(a及an)」可在本文中用於指該冠詞之一個或多於一個(亦即至少一個)文法對象。舉例而言,「類似物」意謂一個類似物或多於一個類似物。 「脂族」係指烷基、烯基、炔基或碳環基,如本文所定義。 「烷基」係指具有1至20個碳原子的直鏈或分支鏈飽和烴基(「C1-20
烷基」)。在一些實施例中,烷基具有1至12個碳原子(「C1-12
烷基」)。在一些實施例中,烷基具有1至10個碳原子(「C1-10
烷基」)。在一些實施例中,烷基具有1至9個碳原子(「C1-9
烷基」)。在一些實施例中,烷基具有1至8個碳原子(「C1-8
烷基」)。在一些實施例中,烷基具有1至7個碳原子(「C1-7
烷基」)。在一些實施例中,烷基具有1至6個碳原子(「C1-6
烷基」,在本文中亦稱為「低碳烷基」)。在一些實施例中,烷基具有1至5個碳原子(「C1-5
烷基」)。在一些實施例中,烷基具有1至4個碳原子(「C1-4
烷基」)。在一些實施例中,烷基具有1至3個碳原子(「C1-3
烷基」)。在一些實施例中,烷基具有1至2個碳原子(「C1-2
烷基」)。在一些實施例中,烷基具有1個碳原子(「C1
烷基」)。在一些實施例中,烷基具有2至6個碳原子(「C2-6
烷基」)。C1-6
烷基之實例包括甲基(C1
)、乙基(C2
)、正丙基(C3
)、異丙基(C3
)、正丁基(C4
)、第三丁基(C4
)、第二丁基(C4
)、異丁基(C4
)、正戊基(C5
)、3-戊基(C5
)、戊基(C5
)、異戊基(C5
)、新戊基(C5
)、3-甲基-2-丁基(C5
)、第三戊基(C5
)及正己基(C6
)。烷基之其他實例包括正庚基(C7
)、正辛基(C8
)及其類似基團。除非另外規定,否則烷基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之烷基」,或經一或多個取代基(例如,1至5個取代基、1至3個取代基或1個取代基)取代(「經取代烷基」)。在某些實施例中,烷基為未經取代之C1-10
烷基(例如-CH3
)。在某些實施例中,烷基為經取代之C1-10
烷基。常用烷基縮寫包括Me (-CH3
)、Et (-CH2
CH3
)、iPr (-CH(CH3
)2
)、nPr (-CH2
CH2
CH3
)、n-Bu (-CH2
CH2
CH2
CH3
)或i-Bu (-CH2
CH(CH3
)2
)。 「伸烷基」係指其中兩個氫經移除以提供二價基團且該二價基團可經取代或未經取代的烷基。未經取代之伸烷基包括(但不限於)亞甲基(-CH2
-)、伸乙基(-CH2
CH2
-)、伸丙基(-CH2
CH2
CH2
-)、伸丁基(-CH2
CH2
CH2
CH2
-)、伸戊基(-CH2
CH2
CH2
CH2
CH2
-)、伸己基(-CH2
CH2
CH2
CH2
CH2
CH2
-)及類似基團。(例如)經一或多個烷基(甲基)取代之示例性經取代伸烷基包括(但不限於):經取代亞甲基(-CH(CH3
)-、(-C(CH3
)2
-);經取代伸乙基(-CH(CH3
)CH2
-、-CH2
CH(CH3
)-、-C(CH3
)2
CH2
-、-CH2
C(CH3
)2
-);經取代伸丙基(-CH(CH3
)CH2
CH2
-、-CH2
CH(CH3
)CH2
-、-CH2
CH2
CH(CH3
)-、-C(CH3
)2
CH2
CH2
-、-CH2
C(CH3
)2
CH2
-、-CH2
CH2
C(CH3
)2
-);及類似基團。當提供用於特定伸烷基的一定範圍或數目之碳時,應理解,該範圍或數目係指直連碳二價鏈中之碳的數目或範圍。伸烷基可經如本文所描述之一或多個取代基取代或未經取代。 「烯基」係指具有2至20個碳原子、一或多個碳-碳雙鍵(例如,1、2、3或4個碳-碳雙鍵)及視情況選用之一或多個碳-碳參鍵(例如,1、2、3或4個碳-碳參鍵)的直鏈或分支鏈烴基(「C2-20
烯基」)。在某些實施例中,烯基不含任何參鍵。在一些實施例中,烯基具有2至10個碳原子(「C2-10
烯基」)。在一些實施例中,烯基具有2至9個碳原子(「C2-9
烯基」)。在一些實施例中,烯基具有2至8個碳原子(「C2-8
烯基」)。在一些實施例中,烯基具有2至7個碳原子(「C2-7
烯基」)。在一些實施例中,烯基具有2至6個碳原子(「C2-6
烯基」)。在一些實施例中,烯基具有2至5個碳原子(「C2-5
烯基」)。在一些實施例中,烯基具有2至4個碳原子(「C2-4
烯基」)。在一些實施例中,烯基具有2至3個碳原子(「C2-3
烯基」)。在一些實施例中,烯基具有2個碳原子(「C2
烯基」)。一或多個碳-碳雙鍵可在內部(諸如在2-丁烯基中)或末端(諸如在1-丁烯基中)。C2-4
烯基之實例包括乙烯基(C2
)、1-丙烯基(C3
)、2-丙烯基(C3
)、1-丁烯基(C4
)、2-丁烯基(C4
)、丁二烯基(C4
)及其類似基團。C2-6
烯基之實例包括前述C2-4
烯基以及戊烯基(C5
)、戊二烯基(C5
)、己烯基(C6
)及其類似基團。烯基之額外實例包括庚烯基(C7
)、辛烯基(C8
)、辛三烯基(C8
)及其類似基團。除非另外規定,否則烯基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之烯基」,或經一或多個取代基(例如,1至5個取代基、1至3個取代基或1個取代基)取代(「經取代烯基」)。在某些實施例中,烯基為未經取代之C2-10
烯基。在某些實施例中,烯基為經取代之C2-10
烯基。 「炔基」係指具有2至20個碳原子、一或多個碳-碳參鍵(例如,1、2、3或4個碳-碳參鍵)及視情況選用之一或多個碳-碳雙鍵(例如,1、2、3或4個碳-碳雙鍵)的直鏈或分支鏈烴基(「C2-20
炔基」)。在某些實施例中,炔基不含任何雙鍵。在一些實施例中,炔基具有2至10個碳原子(「C2-10
炔基」)。在一些實施例中,炔基具有2至9個碳原子(「C2-9
炔基」)。在一些實施例中,炔基具有2至8個碳原子(「C2-8
炔基」)。在一些實施例中,炔基具有2至7個碳原子(「C2-7
炔基」)。在一些實施例中,炔基具有2至6個碳原子(「C2-6
炔基」)。在一些實施例中,炔基具有2至5個碳原子(「C2-5
炔基」)。在一些實施例中,炔基具有2至4個碳原子(「C2-4
炔基」)。在一些實施例中,炔基具有2至3個碳原子(「C2-3
炔基」)。在一些實施例中,炔基具有2個碳原子(「C2
炔基」)。一或多個碳-碳參鍵可在內部(諸如在2-丁炔基中)或末端(諸如在1-丁炔基中)。C2-4
炔基之實例包括(但不限於)乙炔基(C2
)、1-丙炔基(C3
)、2-丙炔基(C3
)、1-丁炔基(C4
)、2-丁炔基(C4
)及其類似基團。C2-6
烯基之實例包括前述C2-4
炔基以及戊炔基(C5
)、己炔基(C6
)及其類似基團。炔基之額外實例包括庚炔基(C7
)、辛炔基(C8
)及其類似基團。除非另外規定,否則炔基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之炔基」,或經一或多個取代基(例如,1至5個取代基、1至3個取代基或1個取代基)取代(「經取代炔基」)。在某些實施例中,炔基為未經取代之C2-10
炔基。在某些實施例中,炔基為經取代之C2-10
炔基。 如本文所使用,術語「雜烷基」係指如本文所定義之烷基,其進一步包含在母鏈內的1個或多個(例如,1、2、3或4個)雜原子(例如,氧、硫、氮、硼、矽、磷),其中該一或多個雜原子插入在母碳鏈內的相鄰碳原子之間,及/或該一或多個雜原子插入在碳原子與母分子之間,亦即插入在連接點之間。在某些實施例中,雜烷基係指具有1至10個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-10
烷基」)。在一些實施例中,雜烷基係指具有1至9個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-9
烷基」)。在一些實施例中,雜烷基為具有1至8個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-8
烷基」)。在一些實施例中,雜烷基為具有1至7個碳原子及1、2、3或4個雜原子的飽和基團(「雜C1-7
烷基」)。在一些實施例中,雜烷基為具有1至6個碳原子及1、2或3個雜原子的基團(「雜C1-6
烷基」)。在一些實施例中,雜烷基為具有1至5個碳原子及1或2個雜原子的飽和基團(「雜C1-5
烷基」)。在一些實施例中,雜烷基為具有1至4個碳原子及1或2個雜原子的飽和基團(「雜C1-4
烷基」)。在一些實施例中,雜烷基為具有1至3個碳原子及1個雜原子的飽和基團(「雜C1-3
烷基」)。在一些實施例中,雜烷基為具有1至2個碳原子及1個雜原子的飽和基團(「雜C1-2
烷基」)。在一些實施例中,雜烷基為具有1個碳原子及1個雜原子的飽和基團(「雜C1
烷基」)。在一些實施例中,雜烷基為具有2至6個碳原子及1或2個雜原子的飽和基團(「雜C2-6
烷基」)。除非另外規定,否則雜烷基之各個例係獨立地未經取代(「未經取代之雜烷基」)或經一或多個取代基取代(「經取代雜烷基」)。在某些實施例中,雜烷基為未經取代之雜C1-10
烷基。在某些實施例中,雜烷基為經取代之雜C1-10
烷基。 「芳基」係指具有芳環系統中所提供之6至14個環碳原子及0個雜原子的單環或多環(例如雙環或三環) 4n+2芳環系統(例如,具有在環狀陣列中共用之6、10或14個π電子)之基團(「C6-14
芳基」)。在一些實施例中,芳基具有六個環碳原子(「C6
芳基」;例如苯基)。在一些實施例中,芳基具有十個環碳原子(「C10
芳基」;例如萘基,諸如1-萘基及2-萘基)。在一些實施例中,芳基具有十四個環碳原子(「C14
芳基」;例如蒽基)。「芳基」亦包括其中如上文所定義之芳基環與一或多個碳環基或雜環基稠合之環系統,其中連接基團或連接點在芳基環上,且在此類情況下,碳原子數目繼續指示芳基環系統中之碳原子的數目。典型芳基包括(但不限於)衍生自以下之基團:乙烯合蒽、乙烯合萘、乙烯合菲、蒽、甘菊環、苯、䓛、六苯并苯、丙二烯合茀、苯并茚、稠六苯、己二吩(hexaphene)、己二烯(hexalene)、as-二環戊二烯并苯、s-二環戊二烯并苯、茚滿、茚、萘、稠八苯、辛二吩(octaphene)、辛二烯(octalene)、卵苯、環戊二烯并-2,4-二烯、稠五苯、并環戊二烯、環戊二烯吩、苝、丙烯合萘、菲、苉、七曜烯、芘、苒、茹(rubicene)、聯伸三苯及三萘。特定芳基包括苯基、萘基、茚基及四氫萘基。除非另外規定,否則芳基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之芳基」,或經一或多個取代基取代(「經取代芳基」)。在某些實施例中,芳基為未經取代之C6-14
芳基。在某些實施例中,芳基為經取代之C6-14
芳基。 在某些實施例中,芳基經選自以下之基團中的一或多者取代:鹵基、C1
-C8
烷基、C1
-C8
鹵烷基、氰基、羥基、C1
-C8
烷氧基及胺基。 代表性的經取代芳基之實例包括以下 其中R56
及R57
中之一者可為氫,且R56
及R57
中之至少一者係各自獨立地選自C1
-C8
烷基、C1
-C8
鹵烷基、4員至10員雜環基、烷醯基、C1
-C8
烷氧基、雜芳氧基、烷胺基、芳胺基、雜芳基胺基、NR58
COR59
、NR58
SOR59
NR58
SO2
R59
、COO烷基、COO芳基、CONR58
R59
、CONR58
OR59
、NR58
R59
、SO2
NR58
R59
、S-烷基、SO烷基、SO2
烷基、S芳基、SO芳基、SO2
芳基;或R56
及R57
可接合形成5至8個原子之環狀環(飽和或不飽和),其視情況包含選自以上基團之一或多個雜原子N、O或S。R60
及R61
獨立地為氫、C1
-C8
烷基、C1
-C4
鹵烷基、C3
-C10
環烷基、4員至10員雜環基、C6
-C10
芳基、經取代之C6
-C10
芳基、5員至10員雜芳基或經取代之5員至10員雜芳基。 「稠合芳基」係指其兩種環碳與第二芳基或雜芳基環相同或與碳環基或雜環基環相同的芳基。 「雜芳基」係指具有芳環系統中所提供之環碳原子及1至4個環雜原子的5員至10員單環或雙環4n+2芳環系統(例如具有在環狀陣列中共用之6或10個π電子)的基團,其中各雜原子獨立地選自氮、氧及硫(「5員至10員雜芳基」)。在含有一或多個氮原子之雜芳基中,價數允許時,連接點可為碳或氮原子。雜芳基雙環系統可在一個或兩個環中包括一或多個雜原子。「雜芳基」包括其中如上文所定義之雜芳基環與一或多個碳環基或雜環基稠合之環系統,其中連接點在雜芳基環上,且在此類情況下,環成員的數目繼續指示雜芳基環系統中之環成員的數目。「雜芳基」亦包括如上文所定義之雜芳基環與一或多個芳基稠合之環系統,其中連接點在芳基或雜芳基環上,且在此類情況下,環成員的數目指示稠合(芳基/雜芳基)環系統中之環成員的數目。其中一個環不含雜原子之雙環雜芳基(例如,吲哚基、喹啉基、咔唑基及其類似基團),連接點可在任一環上,亦即,攜帶雜原子之環(例如2-吲哚基)或不含雜原子之環(例如5-吲哚基)。 在一些實施例中,雜芳基為具有芳環系統中所提供之環碳原子及1至4個環雜原子之5員至10員芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至10員雜芳基」)。在一些實施例中,雜芳基為具有芳環系統中所提供之環碳原子及1至4個環雜原子之5員至8員芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至8員雜芳基」)。在一些實施例中,雜芳基為具有芳環系統中所提供之環碳原子及1至4個環雜原子之5員至6員芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至6員雜芳基」)。在一些實施例中,5員至6員雜芳基具有1至3個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜芳基具有1至2個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜芳基具有1個選自氮、氧及硫之環雜原子。除非另外規定,否則雜芳基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之雜芳基」,或經一或多個取代基取代(「經取代雜芳基」)。在某些實施例中,雜芳基為未經取代之5員至14員雜芳基。在某些實施例中,雜芳基為經取代之5員至14員雜芳基。 含有一個雜原子之例示性5員雜芳基包括(但不限於)吡咯基、呋喃基及噻吩基。含有兩個雜原子之例示性5員雜芳基包括(但不限於)咪唑基、吡唑基、噁唑基、異噁唑基、噻唑基及異噻唑基。含有三個雜原子之例示性5員雜芳基包括(但不限於)三唑基、噁二唑基及噻二唑基。含有四個雜原子之例示性5員雜芳基包括(但不限於)四唑基。含有一個雜原子之例示性6員雜芳基包括(但不限於)吡啶基。含有兩個雜原子之例示性6員雜芳基包括(但不限於)噠嗪基、嘧啶基及吡嗪基。含有三個或四個雜原子之例示性6員雜芳基分別包括(但不限於)三嗪基及四嗪基。含有一個雜原子之例示性7員雜芳基包括(但不限於)氮雜卓基、氧雜卓基及噻雜卓基。例示性5,6-雙環雜芳基包括(但不限於)吲哚基、異吲哚基、吲唑基、苯并三唑基、苯并噻吩基、異苯并噻吩基、苯并呋喃基、苯并異呋喃基、苯并咪唑基、苯并噁唑基、苯并異噁唑基、苯并噁二唑基、苯并噻唑基、苯并異噻唑基、苯并噻二唑基、吲哚嗪基及嘌呤基。例示性6,6-雙環雜芳基包括(但不限於)㖠啶基、喋啶基、喹啉基、異喹啉基、㖕啉基、喹喏啉基、酞嗪基及喹唑啉基。 代表性雜芳基之實例包括以下:
其中R56
及R57
中之一者可為氫,且R56
及R57
中之至少一者係各自獨立地選自C1
-C8
烷基、C1
-C8
鹵烷基、4員至10員雜環基、烷醯基、C1
-C8
烷氧基、雜芳氧基、烷胺基、芳胺基、雜芳基胺基、NR58
COR59
、NR58
SOR59
NR58
SO2
R59
、COO烷基、COO芳基、CONR58
R59
、CONR58
OR59
、NR58
R59
、SO2
NR58
R59
、S-烷基、SO烷基、SO2
烷基、S芳基、SO芳基、SO2
芳基;或R56
及R57
可接合形成5至8個原子之環狀環(飽和或不飽和),其視情況包含選自以上基團之一或多個雜原子N、O或S。R60
及R61
獨立地為氫、C1
-C8
烷基、C1
-C4
鹵烷基、C3
-C10
環烷基、4員至10員雜環基、C6
-C10
芳基、經取代之C6
-C10
芳基、5員至10員雜芳基或經取代之5員至10員雜芳基。 「稠合芳基」係指其兩種環碳與第二芳基或雜芳基環相同或與碳環基或雜環基環相同的芳基。 「雜芳基」係指具有芳環系統中所提供之環碳原子及1至4個環雜原子的5員至10員單環或雙環4n+2芳環系統(例如具有在環狀陣列中共用之6或10個π電子)的基團,其中各雜原子獨立地選自氮、氧及硫(「5員至10員雜芳基」)。在含有一或多個氮原子之雜芳基中,價數允許時,連接點可為碳或氮原子。雜芳基雙環系統可在一個或兩個環中包括一或多個雜原子。「雜芳基」包括其中如上文所定義之雜芳基環與一或多個碳環基或雜環基稠合之環系統,其中連接點在雜芳基環上,且在此類情況下,環成員的數目繼續指示雜芳基環系統中之環成員的數目。「雜芳基」亦包括如上文所定義之雜芳基環與一或多個芳基稠合之環系統,其中連接點在芳基或雜芳基環上,且在此類情況下,環成員的數目指示稠合(芳基/雜芳基)環系統中之環成員的數目。其中一個環不含雜原子之雙環雜芳基(例如,吲哚基、喹啉基、咔唑基及其類似基團),連接點可在任一環上,亦即,攜帶雜原子之環(例如2-吲哚基)或不含雜原子之環(例如5-吲哚基)。 在一些實施例中,雜芳基為具有芳環系統中所提供之環碳原子及1至4個環雜原子之5員至10員芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至10員雜芳基」)。在一些實施例中,雜芳基為具有芳環系統中所提供之環碳原子及1至4個環雜原子之5員至8員芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至8員雜芳基」)。在一些實施例中,雜芳基為具有芳環系統中所提供之環碳原子及1至4個環雜原子之5員至6員芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至6員雜芳基」)。在一些實施例中,5員至6員雜芳基具有1至3個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜芳基具有1至2個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜芳基具有1個選自氮、氧及硫之環雜原子。除非另外規定,否則雜芳基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之雜芳基」,或經一或多個取代基取代(「經取代雜芳基」)。在某些實施例中,雜芳基為未經取代之5員至14員雜芳基。在某些實施例中,雜芳基為經取代之5員至14員雜芳基。 含有一個雜原子之例示性5員雜芳基包括(但不限於)吡咯基、呋喃基及噻吩基。含有兩個雜原子之例示性5員雜芳基包括(但不限於)咪唑基、吡唑基、噁唑基、異噁唑基、噻唑基及異噻唑基。含有三個雜原子之例示性5員雜芳基包括(但不限於)三唑基、噁二唑基及噻二唑基。含有四個雜原子之例示性5員雜芳基包括(但不限於)四唑基。含有一個雜原子之例示性6員雜芳基包括(但不限於)吡啶基。含有兩個雜原子之例示性6員雜芳基包括(但不限於)噠嗪基、嘧啶基及吡嗪基。含有三個或四個雜原子之例示性6員雜芳基分別包括(但不限於)三嗪基及四嗪基。含有一個雜原子之例示性7員雜芳基包括(但不限於)氮雜卓基、氧雜卓基及噻雜卓基。例示性5,6-雙環雜芳基包括(但不限於)吲哚基、異吲哚基、吲唑基、苯并三唑基、苯并噻吩基、異苯并噻吩基、苯并呋喃基、苯并異呋喃基、苯并咪唑基、苯并噁唑基、苯并異噁唑基、苯并噁二唑基、苯并噻唑基、苯并異噻唑基、苯并噻二唑基、吲哚嗪基及嘌呤基。例示性6,6-雙環雜芳基包括(但不限於)㖠啶基、喋啶基、喹啉基、異喹啉基、㖕啉基、喹喏啉基、酞嗪基及喹唑啉基。 代表性雜芳基之實例包括以下: 其中各Z選自羰基、N、NR65
、O及S;且R65
獨立地為氫、C1
-C8
烷基、C3
-C10
環烷基、4員至10員雜環基、C6
-C10
芳基及5員至10員雜芳基。 「碳環基」或「碳環」係指在非芳環系統中具有3至10個環碳原子(「C3-10
碳環基」)及零個雜原子之非芳族環烴基。在一些實施例中,碳環基具有3至8個環碳原子(「C3-8
碳環基」)。在一些實施例中,碳環基具有3至6個環碳原子(「C3-6
碳環基」)。在一些實施例中,碳環基具有3至6個環碳原子(「C3-6
碳環基」)。在一些實施例中,碳環基具有5至10個環碳原子(「C5-10
碳環基」)。例示性C3-6
碳環基包括(但不限於)環丙基(C3
)、環丙烯基(C3
)、環丁基(C4
)、環丁烯基(C4
)、環戊基(C5
)、環戊烯基(C5
)、環己基(C6
)、環己烯基(C6
)、環己二烯基(C6
)及其類似基團。例示性C3-8
碳環基包括(但不限於)前述C3-6
碳環基以及環庚基(C7
)、環庚烯基(C7
)、環庚二烯基(C7
)、環庚三烯基(C7
)、環辛基(C8
)、環辛烯基(C8
)、雙環[2.2.1]庚基(C7
)、雙環[2.2.2]辛基(C8
)及其類似基團。例示性C3-10
碳環基包括(但不限於)前述C3-8
碳環基以及環壬基(C9
)、環壬烯基(C9
)、環癸基(C10
)、環癸烯基(C10
)、八氫-1H
-茚基(C9
)、十氫萘基(C10
)、螺[4.5]癸基(C10
)及其類似基團。如前述實例所說明,在某些實施例中,碳環基為單環(「單環碳環基」)或含有稠合、橋連或螺環系統,諸如雙環系統(「雙環碳環基」),且可為飽和的或可為部分不飽和的。「碳環基」亦包括其中如上文所定義之碳環基環與一或多個芳基或雜芳基稠合之環系統,其中連接點在碳環基環上,且在此類情況下,碳之數目繼續指示碳環系統中之碳之數目。除非另外規定,否則碳環基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之碳環基」,或經一或多個取代基取代(「經取代碳環基」)。在某些實施例中,碳環基為未經取代之C3-10
碳環基。在某些實施例中,碳環基為經取代之C3-10
碳環基。 在一些實施例中,「碳環基」為具有3至10個環碳原子之單環飽和碳環基(「C3-10
環烷基」)。在一些實施例中,環烷基具有3至8個環碳原子(「C3-8
環烷基」)。在一些實施例中,環烷基具有3至6個環碳原子(「C3-6
環烷基」)。在一些實施例中,環烷基具有5至6個環碳原子(「C5-6
環烷基」)。在一些實施例中,環烷基具有5至10個環碳原子(「C5-10
環烷基」)。C5-6
環烷基之實例包括環戊基(C5
)及環己基(C5
)。C3-6
環烷基之實例包括前述C5-6
環烷基以及環丙基(C3
)及環丁基(C4
)。C3-8
環烷基之實例包括前述C3-6
環烷基以及環庚基(C7
)及環辛基(C8
)。除非另外規定,否則環烷基之各個例獨立地未經取代(「未經取代之環烷基」)或經一或多個取代基取代(「經取代之環烷基」)。在某些實施例中,環烷基為未經取代之C3-10
環烷基。在某些實施例中,環烷基為經取代之C3-10
環烷基。 「雜環基」或「雜環」係指具有環碳原子及1至4個環雜原子的3員至10員非芳環系統之基團,其中各雜原子獨立地選自氮、氧、硫、硼、磷及矽(「3至10員雜環基」)。在含有一或多個氮原子之雜環基中,在價數准許時,連接點可為碳或氮原子。雜環基可為單環(「單環雜環基」)或稠合、橋連或螺環系統,諸如雙環系統(「雙環雜環基」),且可為飽和或部分不飽和的。雜環基雙環系統可在一個或兩個環中包括一或多個雜原子。「雜環基」亦包括其中如上文所定義之雜環基環與一或多個碳環基稠合之環系統,其中連接點在碳環基或雜環基環上;或其中如上文所定義之雜環基與一或多個芳基或雜芳基稠合之環系統,其中連接點在雜環基環上,且在此類情況下,環成員的數目繼續指示雜環基環系統中之環成員的數目。除非另外規定,否則雜環基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之雜環基」,或經一或多個取代基取代(「經取代雜環基」)。在某些實施例中,雜環基為未經取代之3員至10員雜環基。在某些實施例中,雜環基為經取代之3員至10員雜環基。 在一些實施例中,雜環基為具有環碳原子及1至4個環雜原子之5員至10員非芳環系統,其中各雜原子獨立地選自氮、氧、硫、硼、磷及矽(「5員至10員雜環基」)。在一些實施例中,雜環基為具有環碳原子及1至4個環雜原子之5員至8員非芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至8員雜環基」)。在一些實施例中,雜環基為具有環碳原子及1至4個環雜原子的5員至6員非芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至6員雜環基」)。在一些實施例中,5員至6員雜環基具有1至3個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜環基具有1至2個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜環基具有一個選自氮、氧及硫之環雜原子。 含有一個雜原子之例示性3員雜環基包括(但不限於)氮丙啶基、環氧乙烷基及環硫乙基(thiorenyl)。含有一個雜原子之例示性4員雜環基包括(但不限於)氮雜環丁烷基、氧雜環丁烷基及硫雜環丁烷基。含有一個雜原子之例示性5員雜環基包括(但不限於)四氫呋喃基、二氫呋喃基、四氫噻吩基、二氫噻吩基、吡咯啶基、二氫吡咯基及吡咯基-2,5-二酮。含有兩個雜原子之例示性5員雜環基包括(但不限於)二氧戊環基、氧雜硫呋喃基、二硫呋喃基及噁唑啶-2-酮。含有三個雜原子之例示性5員雜環基包括(但不限於)三唑啉基、噁二唑啉基及噻二唑啉基。含有一個雜原子之例示性6員雜環基包括(但不限於)哌啶基、四氫哌喃基、二氫吡啶基及噻烷基。含有兩個雜原子之例示性6員雜環基包括(但不限於)哌嗪基、嗎啉基、二噻烷基、二噁烷基。含有四個雜原子之例示性6員雜環基包括(但不限於)三氮雜環己烷基。含有一個雜原子之例示性7員雜環基包括(但不限於)氮雜環庚烷基、氧雜環庚烷基及硫雜環庚烷基。含有一個雜原子之例示性8員雜環基包括(但不限於)氮雜環辛基、氧雜環辛基及硫雜環辛基。與C6
芳環稠合之例示性5員雜環基(在本文中亦稱為5,6-雙環雜環)包括(但不限於)二氫吲哚基、異吲哚啉基、二氫苯并呋喃基、二氫苯并噻吩基、苯并噁唑啉酮基及其類似基團。與芳環稠合之例示性6員雜環基(在本文中亦稱為6,6-雙環雜環)包括(但不限於)四氫喹啉基、四氫異喹啉基及其類似基團。 「含氮雜環基」意謂含有至少一個氮原子之4員至7員非芳族環基,例如(但不限於)嗎啉、哌啶(例如,2-哌啶基、3-哌啶基及4-哌啶基)、吡咯啶(例如,2-吡咯啶基及3-吡咯啶基)、氮雜環丁烷、吡咯啶酮、咪唑啉、咪唑啶酮、2-吡唑啉、吡唑啶、哌嗪及N-烷基哌嗪,諸如N-甲基哌嗪。特定實例包括氮雜環丁烷、哌啶酮及哌嗪酮。 「雜」當用於描述化合物或化合物上存在之基團時,意謂化合物或基團中之一或多個碳原子已置換為氮、氧或硫雜原子。異可應用於上述烴基中之任一者,諸如具有1至5個且詳言之具有1至3個雜原子的烷基(例如,雜烷基、環烷基)、(例如)雜環基、芳基(例如雜芳基)、環烯基(例如環雜烯基)及其類似基團。 「醯基」係指基團-C(O)R20
,其中R20
為氫、經取代或未經取代之烷基、經取代或未經取代之烯基、經取代或未經取代之炔基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、經取代或未經取代之芳基或經取代或未經取代之雜芳基,如本文所定義。「烷醯基」為其中R20
為除氫外之基團的醯基。代表性醯基包括(但不限於)甲醯基(-CHO)、乙醯基(-C(=O)CH3
)、環己基羰基、環己基甲基羰基、苯甲醯基(-C(=O)Ph)、苯甲基羰基(-C(=O)CH2
Ph)、--C(O)-C1
-C8
烷基、-C(O)-(CH2
)t
(C6
-C10
芳基)、-C(O)-(CH2
)t
(5員至10員雜芳基)、-C(O)-(CH2
)t
(C3
-C10
環烷基)及-C(O)-(CH2
)t
(4員至10員雜環基),其中t為0至4之整數。在某些實施例中,R21
為經鹵基或羥基取代之C1
-C8
烷基;或C3
-C10
環烷基、4員至10員雜環基、C6
-C10
芳基、芳烷基、5員至10員雜芳基或雜芳烷基,其中之各者經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥烷基或未經取代之C1
-C4
鹵烷氧基或羥基。 「烷氧基」係指基團-OR29
,其中R29
為經取代或未經取代之烷基、經取代或未經取代之烯基、經取代或未經取代之炔基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、經取代或未經取代之芳基或經取代或未經取代之雜芳基。特定烷氧基為甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、第三丁氧基、第二丁氧基、正戊氧基、正己氧基及1,2-二甲基丁氧基。特定烷氧基為低碳烷氧基,亦即具有1至6個碳原子。其他特定烷氧基具有1至4個碳原子。 在某些實施例中,R29
為具有1個或多於1個取代基,例如1至5個取代基,且詳言之1至3個取代基,詳言之1個取代基之基團,該取代基選自由以下組成之群:胺基、經取代之胺基、C6
-C10
芳基、芳氧基、羧基、氰基、C3
-C10
環烷基、4員至10員雜環基、鹵素、5員至10員雜芳基、羥基、硝基、硫烷氧基、硫芳氧基、巰基、烷基-S(O)-、芳基-S(O)-、烷基-S(O)2
-及芳基-S(O)2
-。例示性『經取代烷氧基』包括(但不限於)-O-(CH2
)t
(C6
-C10
芳基)、-O-(CH2
)t
(5員至10員雜芳基)、-O-(CH2
)t
(C3
-C10
環烷基)及-O-(CH2
)t
(4員至10員雜環基),其中t為0至4之整數,且所存在之任何芳基、雜芳基、環烷基或雜環基本身可經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥基烷基或未經取代之C1
-C4
鹵烷氧基或羥基。特定例示性『經取代烷氧基』為-OCF3
、-OCH2
CF3
、-OCH2
Ph、-OCH2
-環丙基、-OCH2
CH2
OH及-OCH2
CH2
NMe2
。 「胺基」係指基團-NH2
。 「側氧基」係指-C(=O)-。 「經取代胺基」係指式-N(R38
)2
之胺基,其中R38
為氫、經取代或未經取代之烷基、經取代或未經取代之烯基、經取代或未經取代之炔基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、經取代或未經取代之芳基、經取代或未經取代之雜芳基或胺基保護基,其中R38
中之至少一者不為氫。在某些實施例中,各R38
獨立地選自氫、C1
-C8
烷基、C3
-C8
烯基、C3
-C8
炔基、C6
-C10
芳基、5員至10員雜芳基、4員至10員雜環基或C3
-C10
環烷基或經鹵基或羥基取代之C1
-C8
烷基、經鹵基或羥基取代之C3
-C8
烯基、經鹵基或羥基取代之C3
-C8
炔基或-(CH2
)t
(C6
-C10
芳基)、-(CH2
)t
(5員至10員雜芳基)、-(CH2
)t
(C3
-C10
環烷基)或-(CH2
)t
(4員至10員雜環基),其中t為0與8之間的整數,其中之各者經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥烷基或未經取代之C1
-C4
鹵烷氧基或羥基;或兩個R38
基團接合形成伸烷基。 例示性「經取代胺基」包括(但不限於)-NR39
-C1
-C8
烷基、-NR39
-(CH2
)t
(C6
-C10
芳基)、-NR39
-(CH2
)t
(5員至10員雜芳基)、-NR39
-(CH2
)t
(C3
-C10
環烷基)及-NR39
-(CH2
)t
(4員至10員雜環基),其中t為0至4之整數,例如1或2,各R39
獨立地表示H或C1
-C8
烷基;且所存在之任何烷基本身可經鹵基、經取代或未經取代之胺基或羥基取代;且所存在之任何芳基、雜芳基、環烷基或雜環基本身可經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥基烷基或未經取代之C1
-C4
鹵烷氧基或羥基。為避免疑問,術語『經取代胺基』包括基團烷基胺基、經取代之烷基胺基、烷基芳基胺基、經取代之烷基芳基胺基、芳基胺基、經取代之芳基胺基、二烷胺基及經取代之二烷胺基,如下文所定義。經取代胺基涵蓋單取代胺基及二取代胺基兩者。 「羧基」係指基團-C(O)OH。 「氰基」係指基團-CN。 「鹵基」或「鹵素」係指氟基(F)、氯基(Cl)、溴基(Br)及碘基(I)。在某些實施例中,鹵基為氟基或氯基。 「鹵烷基」係指其中烷基經一或多個鹵素取代之烷基。典型鹵烷基包括(但不限於)三氟甲基(-CF3
)、二氟甲基(-CHF2
)、氟甲基(-CH2
F)、氯甲基(-CH2
Cl)、二氯甲基(-CHCl2
)、三溴甲基(-CH2
Br)及其類似基團。 「羥基」係指基團-OH。 「硝基」係指基團-NO2
。 「硫酮基」係指基團=S。 如本文所定義,烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基係視情況經取代的(例如,「經取代」或「未經取代」烷基、「經取代」或「未經取代」烯基、「經取代」或「未經取代」炔基、「經取代」或「未經取代」碳環基、「經取代」或「未經取代」雜環基、「經取代」或「未經取代」芳基或「經取代」或「未經取代」雜芳基)。大體而言,術語「經取代」無論是否在術語「視情況」之前均意謂基團(例如碳或氮原子)上存在之至少一個氫經容許取代基置換,該容許取代基例如係在取代後產生穩定化合物之取代基,該化合物例如係不會自發地經歷(諸如)藉由重排、環化、消除或其他反應之轉化的化合物。除非另外指明,否則「經取代」基團在該基團之一或多個可取代位置具有取代基,且當任何既定結構中之多於一個位置經取代時,取代基在各位置相同或不同。術語「經取代」預期包括經有機化合物之所有容許取代基、引起穩定化合物形成之本文所描述之任何取代基取代。本發明涵蓋任何及所有此類組合以便獲得穩定化合物。出於本發明之目的,雜原子(諸如氮)可具有氫取代基及/或滿足雜原子價數且使得穩定部分形成之如本文所描述之任何適合取代基。 例示性碳原子取代基包括(但不限於)-CN、-NO2
、-N3
、-SO2
H、-SO3
H、-OH、-ORaa
、-ON(Rbb
)2
、-N(Rbb
)2
、-N(Rbb
)3 +
X-
、-N(ORcc
)Rbb
、-SH、-SRaa
、-SSRcc
、-C(=O)Raa
、-CO2
H、-CHO、-C(ORcc
)2
、-CO2
Raa
、-OC(=O)Raa
、-OCO2
Raa
、-C(=O)N(Rbb
)2
、-OC(=O)N(Rbb
)2
、-NRbb
C(=O)Raa
、-NRbb
CO2
Raa
、-NRbb
C(=O)N(Rbb
)2
、-C(=NRbb
)Raa
、-C(=NRbb
)ORaa
、-OC(=NRbb
)Raa
、-OC(=NRbb
)ORaa
、-C(=NRbb
)N(Rbb
)2
、-OC(=NRbb
)N(Rbb
)2
、-NRbb
C(=NRbb
)N(Rbb
)2
、-C(=O)NRbb
SO2
Raa
、-NRbb
SO2
Raa
、-SO2
N(Rbb
)2
、-SO2
Raa
、-SO2
ORaa
、-OSO2
Raa
、-S(=O)Raa
、-OS(=O)Raa
、-Si(Raa
)3
、-OSi(Raa
)3
-C(=S)N(Rbb
)2
、-C(=O)SRaa
、-C(=S)SRaa
、-SC(=S)SRaa
、-SC(=O)SRaa
、-OC(=O)SRaa
、-SC(=O)ORaa
、-SC(=O)Raa
、-P(=O)2
Raa
、-OP(=O)2
Raa
、-P(=O)(Raa
)2
、-OP(=O)(Raa
)2
、-OP(=O)(ORcc
)2
、-P(=O)2
N(Rbb
)2
、-OP(=O)2
N(Rbb
)2
、-P(=O)(NRbb
)2
、-OP(=O)(NRbb
)2
、-NRbb
P(=O)(ORcc
)2
、-NRbb
P(=O)(NRbb
)2
、-P(Rcc
)2
、-P(Rcc
)3
、-OP(Rcc
)2
、-OP(Rcc
)3
、-B(Raa
)2
、-B(ORcc
)2
、-BRaa
(ORcc
)、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,其中各烷基、烯基、炔基、碳環基、雜環基、芳基、雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代;或碳原子上的兩個成對氫經基團=O、=S、=NN(Rbb
)2
、=NNRbb
C(=O)Raa
、=NNRbb
C(=O)ORaa
、=NNRbb
S(=O)2
Raa
、=NRbb
或=NORcc
置換; Raa
之各個例獨立地選自C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或兩個Raa
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代; Rbb
之各個例獨立地選自氫、-OH、-ORaa
、-N(Rcc
)2
、-CN、-C(=O)Raa
、-C(=O)N(Rcc
)2
、-CO2
Raa
、-SO2
Raa
、-C(=NRcc
)ORaa
、-C(=NRcc
)N(Rcc
)2
、-SO2
N(Rcc
)2
、-SO2
Rcc
、-SO2
ORcc
、-SORaa
、-C(=S)N(Rcc
)2
、-C(=O)SRcc
、-C(=S)SRcc
、-P(=O)2
Raa
、-P(=O)(Raa
)2
、-P(=O)2
N(Rcc
)2
、-P(=O)(NRcc
)2
、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或兩個Rbb
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代; Rcc
之各個例獨立地選自氫、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或兩個Rcc
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代; Rdd
之各個例獨立地選自鹵素、CN、-NO2
、-N3
、-SO2
H、-SO3
H、-OH、-ORee
、-ON(Rff
)2
、-N(Rff
)2
、-N(Rff
)3 +
X-
、-N(ORee
)Rff
、-SH、-SRee
、-SSRee
、-C(=O)Ree
、-CO2
H、-CO2
Ree
、-OC(=O)Ree
、-OCO2
Ree
、-C(=O)N(Rff
)2
、-OC(=O)N(Rff
)2
、-NRff
C(=O)Ree
、-NRff
CO2
Ree
、-NRff
C(=O)N(Rff
)2
、-C(=NRff
)ORee
、-OC(=NRff
)Ree
、-OC(=NRff
)ORee
、-C(=NRff
)N(Rff
)2
、-OC(=NRff
)N(Rff
)2
、-NRff
C(=NRff
)N(Rff
)2
、-NRff
SO2
Ree
、-SO2
N(Rff
)2
、-SO2
Ree
、-SO2
ORee
、-OSO2
Ree
、-S(=O)Ree
、-Si(Ree
)3
、-OSi(Ree
)3
、-C(=S)N(Rff
)2
、-C(=O)SRee
、-C(=S)SRee
、-SC(=S)SRee
、-P(=O)2
Ree
、-P(=O)(Ree
)2
、-OP(=O)(Ree
)2
、-OP(=O)(ORee
)2
、C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、3員至10員雜環基、C6-10
芳基、5員至10員雜芳基,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rgg
基團取代,或兩個成對Rdd
取代基可接合形成=O或=S; Ree
之各個例獨立地選自C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、C6-10
芳基、3員至10員雜環基及3員至10員雜芳基,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rgg
基團取代; Rff
之各個例獨立地選自氫、C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、3員至10員雜環基、C6-10
芳基及5員至10員雜芳基,或兩個Rff
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rgg
基團取代;以及 Rgg
之各個例獨立地為鹵素、-CN、-NO2
、-N3
、-SO2
H、-SO3
H、 -OH、-OC1-6
烷基、-ON(C1-6
烷基)2
、 -N(C1-6
烷基)2
、 -N(C1-6
烷基)3 +
X-
、-NH(C1-6
烷基)2 +
X-
、-NH2
(C1-6
烷基)+
X-
、-NH3 +
X-
、-N(OC1-6
烷基)(C1-6
烷基)、-N(OH)(C1-6
烷基)、-NH(OH)、-SH、-SC1-6
烷基、-SS(C1-6
烷基)、-C(=O)(C1-6
烷基)、-CO2
H、-CO2
(C1-6
烷基)、-OC(=O)(C1-6
烷基)、-OCO2
(C1-6
烷基)、-C(=O)NH2
、-C(=O)N(C1-6
烷基)2
、-OC(=O)NH(C1-6
烷基)、-NHC(=O)( C1-6
烷基)、-N(C1-6
烷基)C(=O)( C1-6
烷基)、-NHCO2
(C1-6
烷基)、-NHC(=O)N(C1-6
烷基)2
、-NHC(=O)NH(C1-6
烷基)、 -NHC(=O)NH2
、 -C(=NH)O(C1-6
烷基)、-OC(=NH)(C1-6
烷基)、-OC(=NH)OC1-6
烷基、-C(=NH)N(C1-6
烷基)2
、-C(=NH)NH(C1-6
烷基)、-C(=NH)NH2
、-OC(=NH)N(C1-6
烷基)2
、-OC(NH)NH(C1-6
烷基)、-OC(NH)NH2
、-NHC(NH)N(C1-6
烷基)2
、-NHC(=NH)NH2
、-NHSO2
(C1-6
烷基)、-SO2
N(C1-6
烷基)2
、-SO2
NH(C1-6
烷基)、-SO2
NH2
、-SO2
C1-6
烷基、-SO2
OC1-6
烷基、-OSO2
C1-6
烷基、-SOC1-6
烷基、-Si(C1-6
烷基)3
、-OSi(C1-6
烷基)3
-C(=S)N(C1-6
烷基)2
、C(=S)NH(C1-6
烷基)、C(=S)NH2
、-C(=O)S(C1-6
烷基)、-C(=S)SC1-6
烷基、-SC(=S)SC1-6
烷基、-P(=O)2
(C1-6
烷基)、-P(=O)(C1-6
烷基)2
、-OP(=O)(C1-6
烷基)2
、-OP(=O)(OC1-6
烷基)2
、C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、C6-10
芳基、3員至10員雜環基、5員至10員雜芳基;或兩個成對Rgg
取代基可接合形成=O或=S,其中X-
為相對離子。 「相對離子」或「陰離子相對離子」為與陽離子四級胺基相關的帶負電基團以便維持電子電中性。例示性相對離子包括鹵化物離子(例如F-
、Cl-
、Br-
、I-
)、NO3 -
、ClO4 -
、OH-
、H2
PO4 -
、HSO4 -
、SO4 -2
磺酸根離子(例如甲烷磺酸根、三氟甲烷磺酸根、對甲苯磺酸根、苯磺酸根、10-樟腦磺酸根、萘-2-磺酸根、萘-1-磺酸-5-磺酸根、乙-1-磺酸-2-磺酸根及其類似物)及羧酸根離子(例如乙酸根、醋酸根、丙酸根、苯甲酸根、甘油酸根、乳酸根、酒石酸根、羥乙酸根及其類似物)。 價數允許時,氮原子可經取代或未經取代,且包括一級、二級、三級及四級氮原子。例示性氮原子取代基包括(但不限於)氫、-OH、-ORaa
、-N(Rcc
)2
、-CN、-C(=O)Raa
、-C(=O)N(Rcc
)2
、-CO2
Raa
、-SO2
Raa
、-C(=NRbb
)Raa
、-C(=NRcc
)ORaa
、-C(=NRcc
)N(Rcc
)2
、-SO2
N(Rcc
)2
、-SO2
Rcc
、-SO2
ORcc
、-SORaa
、-C(=S)N(Rcc
)2
、-C(=O)SRcc
、-C(=S)SRcc
、-P(=O)2
Raa
、-P(=O)(Raa
)2
、-P(=O)2
N(Rcc
)2
、-P(=O)(NRcc
)2
、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或連接至氮原子的兩個Rcc
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代且其中Raa
、Rbb
、Rcc
及Rdd
如上文所定義。 此等及其他例示性取代基更詳細地描述於實施方式
、實例
及申請專利範圍
中。本發明並不意欲以任何方式受取代基之上述例示性清單限制。其他定義
術語「醫藥學上可接受之鹽」係指在合理醫學判斷範疇內適用於與人類及低等動物之組織接觸而無異常毒性、刺激、過敏反應及其類似情況且與合理益處/風險比相稱的彼等鹽。醫藥學上可接受之鹽在此項技術中為所熟知的。舉例而言,Berge等人在J. Pharmaceutical Sciences
(1977) 66:1-19中詳細描述醫藥學上可接受之鹽。本發明化合物之醫藥學上可接受之鹽包括衍生自適合的無機及有機酸及鹼之彼等鹽。醫藥學上可接受之無毒酸加成鹽之實例為胺基與無機酸(諸如鹽酸、氫溴酸、磷酸、硫酸及過氯酸)或有機酸(諸如乙酸、草酸、順丁烯二酸、酒石酸、檸檬酸、丁二酸或丙二酸)形成之鹽,或藉由使用此項技術中所用之其他方法(諸如離子交換)形成之鹽。其他醫藥學上可接受之鹽包括己二酸鹽、海藻酸鹽、抗壞血酸鹽、天冬胺酸鹽、苯磺酸鹽、苯甲酸鹽、硫酸氫鹽、硼酸鹽、丁酸鹽、樟腦酸鹽、樟腦磺酸鹽、檸檬酸鹽、環戊烷丙酸鹽、二葡糖酸鹽、十二烷基硫酸鹽、乙烷磺酸鹽、甲酸鹽、反丁烯二酸鹽、葡庚糖酸鹽、甘油磷酸鹽、葡糖酸鹽、半硫酸鹽、庚酸鹽、己酸鹽、氫碘酸鹽、2-羥基-乙烷磺酸鹽、乳糖酸鹽、乳酸鹽、月桂酸鹽、月桂基硫酸鹽、蘋果酸鹽、順丁烯二酸鹽、丙二酸鹽、甲烷磺酸鹽、2-萘磺酸鹽、菸鹼酸鹽、硝酸鹽、油酸鹽、草酸鹽、棕櫚酸鹽、雙羥萘酸鹽、果膠酸鹽、過硫酸鹽、3-苯基丙酸鹽、磷酸鹽、苦味酸鹽、特戊酸鹽、丙酸鹽、硬脂酸鹽、丁二酸鹽、硫酸鹽、酒石酸鹽、硫氰酸鹽、對甲苯磺酸鹽、十一烷酸鹽、戊酸鹽及其類似鹽。衍生自適當鹼之醫藥學上可接受之鹽包括鹼金屬、鹼土金屬、銨及N+
(C1-4
烷基)4
鹽。代表性鹼金屬鹽或鹼土金屬鹽包括鈉鹽、鋰鹽、鉀鹽、鈣鹽、鎂鹽及其類似鹽。在適當時,其他醫藥學上可接受之鹽包括使用諸如鹵化物、氫氧化物、羧酸鹽、硫酸鹽、磷酸鹽、硝酸鹽、低碳烷基磺酸鹽及芳基磺酸鹽之相對離子形成之無毒銨、四級銨及胺陽離子。 涵蓋投與之「個體」,包括(但不限於)人類(亦即,任何年齡組之男性或女性,例如兒科個體(例如、嬰兒、兒童、青少年)或成人個體(例如年輕人、中年人或老年人))及/或非人類動物,例如哺乳動物,諸如靈長類動物(例如食蟹獼猴、恆河猴)、牛、豬、馬、綿羊、山羊、嚙齒動物、貓及/或狗。在某些實施例中,個體為人類。在某些實施例中,個體為非人類動物。術語「人類」、「患者」及「個體」在本文中可互換使用。 疾病、病症及病狀在本文中可互換使用。 如本文所使用且除非另外說明,否則術語「治療(treat)」、「治療(treating)」及「治療(treatment)」涵蓋在個體正患指定疾病、病症或病狀的同時發生的降低疾病、病症或病狀嚴重程度或延遲或減緩疾病、病症或病狀進展的作用(「治療性治療」),且亦涵蓋在個體開始罹患指定疾病、病症或病狀之前發生的作用(「預防性治療」)。 大體而言,化合物之「有效量」係指足以引出所需生物反應的量。如一般技術者將瞭解,有效量之本發明化合物可視諸如所需生物終點、化合物之藥物動力學、所治療之疾病、投與模式及個體年齡、體重、健康及病狀之因素而變化。有效量涵蓋治療性及預防性治療。 如本文所使用且除非另外說明,否則化合物之「治療有效量」為足夠提供治療疾病、病症或病狀之治療益處或延遲或使與疾病、病症或病狀相關之一或多種症狀減至最少的量。化合物之治療有效量意謂單獨或與其他療法組合的治療劑之量,其提供治療疾病、病症或病狀之治療益處。術語「治療有效量」可涵蓋改良整體療法、減少或避免疾病或病狀的症狀或病因或增強另一治療劑之治療功效的量。 如本文所用且除非另外說明,否則化合物之「預防有效量」為足夠預防疾病、病症或病狀或與疾病、病症或病狀相關之一或多種症狀或防止其復發的量。化合物之預防有效量意謂治療劑單獨或與其他試劑組合提供預防疾病、病症或病狀之預防益處的量。術語「預防有效量」可涵蓋改良整體預防或增強另一預防劑之預防功效的量。
其中各Z選自羰基、N、NR65
、O及S;且R65
獨立地為氫、C1
-C8
烷基、C3
-C10
環烷基、4員至10員雜環基、C6
-C10
芳基及5員至10員雜芳基。 「碳環基」或「碳環」係指在非芳環系統中具有3至10個環碳原子(「C3-10
碳環基」)及零個雜原子之非芳族環烴基。在一些實施例中,碳環基具有3至8個環碳原子(「C3-8
碳環基」)。在一些實施例中,碳環基具有3至6個環碳原子(「C3-6
碳環基」)。在一些實施例中,碳環基具有3至6個環碳原子(「C3-6
碳環基」)。在一些實施例中,碳環基具有5至10個環碳原子(「C5-10
碳環基」)。例示性C3-6
碳環基包括(但不限於)環丙基(C3
)、環丙烯基(C3
)、環丁基(C4
)、環丁烯基(C4
)、環戊基(C5
)、環戊烯基(C5
)、環己基(C6
)、環己烯基(C6
)、環己二烯基(C6
)及其類似基團。例示性C3-8
碳環基包括(但不限於)前述C3-6
碳環基以及環庚基(C7
)、環庚烯基(C7
)、環庚二烯基(C7
)、環庚三烯基(C7
)、環辛基(C8
)、環辛烯基(C8
)、雙環[2.2.1]庚基(C7
)、雙環[2.2.2]辛基(C8
)及其類似基團。例示性C3-10
碳環基包括(但不限於)前述C3-8
碳環基以及環壬基(C9
)、環壬烯基(C9
)、環癸基(C10
)、環癸烯基(C10
)、八氫-1H
-茚基(C9
)、十氫萘基(C10
)、螺[4.5]癸基(C10
)及其類似基團。如前述實例所說明,在某些實施例中,碳環基為單環(「單環碳環基」)或含有稠合、橋連或螺環系統,諸如雙環系統(「雙環碳環基」),且可為飽和的或可為部分不飽和的。「碳環基」亦包括其中如上文所定義之碳環基環與一或多個芳基或雜芳基稠合之環系統,其中連接點在碳環基環上,且在此類情況下,碳之數目繼續指示碳環系統中之碳之數目。除非另外規定,否則碳環基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之碳環基」,或經一或多個取代基取代(「經取代碳環基」)。在某些實施例中,碳環基為未經取代之C3-10
碳環基。在某些實施例中,碳環基為經取代之C3-10
碳環基。 在一些實施例中,「碳環基」為具有3至10個環碳原子之單環飽和碳環基(「C3-10
環烷基」)。在一些實施例中,環烷基具有3至8個環碳原子(「C3-8
環烷基」)。在一些實施例中,環烷基具有3至6個環碳原子(「C3-6
環烷基」)。在一些實施例中,環烷基具有5至6個環碳原子(「C5-6
環烷基」)。在一些實施例中,環烷基具有5至10個環碳原子(「C5-10
環烷基」)。C5-6
環烷基之實例包括環戊基(C5
)及環己基(C5
)。C3-6
環烷基之實例包括前述C5-6
環烷基以及環丙基(C3
)及環丁基(C4
)。C3-8
環烷基之實例包括前述C3-6
環烷基以及環庚基(C7
)及環辛基(C8
)。除非另外規定,否則環烷基之各個例獨立地未經取代(「未經取代之環烷基」)或經一或多個取代基取代(「經取代之環烷基」)。在某些實施例中,環烷基為未經取代之C3-10
環烷基。在某些實施例中,環烷基為經取代之C3-10
環烷基。 「雜環基」或「雜環」係指具有環碳原子及1至4個環雜原子的3員至10員非芳環系統之基團,其中各雜原子獨立地選自氮、氧、硫、硼、磷及矽(「3至10員雜環基」)。在含有一或多個氮原子之雜環基中,在價數准許時,連接點可為碳或氮原子。雜環基可為單環(「單環雜環基」)或稠合、橋連或螺環系統,諸如雙環系統(「雙環雜環基」),且可為飽和或部分不飽和的。雜環基雙環系統可在一個或兩個環中包括一或多個雜原子。「雜環基」亦包括其中如上文所定義之雜環基環與一或多個碳環基稠合之環系統,其中連接點在碳環基或雜環基環上;或其中如上文所定義之雜環基與一或多個芳基或雜芳基稠合之環系統,其中連接點在雜環基環上,且在此類情況下,環成員的數目繼續指示雜環基環系統中之環成員的數目。除非另外規定,否則雜環基之各個例獨立地視情況經取代,亦即,未經取代(「未經取代之雜環基」,或經一或多個取代基取代(「經取代雜環基」)。在某些實施例中,雜環基為未經取代之3員至10員雜環基。在某些實施例中,雜環基為經取代之3員至10員雜環基。 在一些實施例中,雜環基為具有環碳原子及1至4個環雜原子之5員至10員非芳環系統,其中各雜原子獨立地選自氮、氧、硫、硼、磷及矽(「5員至10員雜環基」)。在一些實施例中,雜環基為具有環碳原子及1至4個環雜原子之5員至8員非芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至8員雜環基」)。在一些實施例中,雜環基為具有環碳原子及1至4個環雜原子的5員至6員非芳環系統,其中各雜原子獨立地選自氮、氧及硫(「5員至6員雜環基」)。在一些實施例中,5員至6員雜環基具有1至3個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜環基具有1至2個選自氮、氧及硫之環雜原子。在一些實施例中,5員至6員雜環基具有一個選自氮、氧及硫之環雜原子。 含有一個雜原子之例示性3員雜環基包括(但不限於)氮丙啶基、環氧乙烷基及環硫乙基(thiorenyl)。含有一個雜原子之例示性4員雜環基包括(但不限於)氮雜環丁烷基、氧雜環丁烷基及硫雜環丁烷基。含有一個雜原子之例示性5員雜環基包括(但不限於)四氫呋喃基、二氫呋喃基、四氫噻吩基、二氫噻吩基、吡咯啶基、二氫吡咯基及吡咯基-2,5-二酮。含有兩個雜原子之例示性5員雜環基包括(但不限於)二氧戊環基、氧雜硫呋喃基、二硫呋喃基及噁唑啶-2-酮。含有三個雜原子之例示性5員雜環基包括(但不限於)三唑啉基、噁二唑啉基及噻二唑啉基。含有一個雜原子之例示性6員雜環基包括(但不限於)哌啶基、四氫哌喃基、二氫吡啶基及噻烷基。含有兩個雜原子之例示性6員雜環基包括(但不限於)哌嗪基、嗎啉基、二噻烷基、二噁烷基。含有四個雜原子之例示性6員雜環基包括(但不限於)三氮雜環己烷基。含有一個雜原子之例示性7員雜環基包括(但不限於)氮雜環庚烷基、氧雜環庚烷基及硫雜環庚烷基。含有一個雜原子之例示性8員雜環基包括(但不限於)氮雜環辛基、氧雜環辛基及硫雜環辛基。與C6
芳環稠合之例示性5員雜環基(在本文中亦稱為5,6-雙環雜環)包括(但不限於)二氫吲哚基、異吲哚啉基、二氫苯并呋喃基、二氫苯并噻吩基、苯并噁唑啉酮基及其類似基團。與芳環稠合之例示性6員雜環基(在本文中亦稱為6,6-雙環雜環)包括(但不限於)四氫喹啉基、四氫異喹啉基及其類似基團。 「含氮雜環基」意謂含有至少一個氮原子之4員至7員非芳族環基,例如(但不限於)嗎啉、哌啶(例如,2-哌啶基、3-哌啶基及4-哌啶基)、吡咯啶(例如,2-吡咯啶基及3-吡咯啶基)、氮雜環丁烷、吡咯啶酮、咪唑啉、咪唑啶酮、2-吡唑啉、吡唑啶、哌嗪及N-烷基哌嗪,諸如N-甲基哌嗪。特定實例包括氮雜環丁烷、哌啶酮及哌嗪酮。 「雜」當用於描述化合物或化合物上存在之基團時,意謂化合物或基團中之一或多個碳原子已置換為氮、氧或硫雜原子。異可應用於上述烴基中之任一者,諸如具有1至5個且詳言之具有1至3個雜原子的烷基(例如,雜烷基、環烷基)、(例如)雜環基、芳基(例如雜芳基)、環烯基(例如環雜烯基)及其類似基團。 「醯基」係指基團-C(O)R20
,其中R20
為氫、經取代或未經取代之烷基、經取代或未經取代之烯基、經取代或未經取代之炔基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、經取代或未經取代之芳基或經取代或未經取代之雜芳基,如本文所定義。「烷醯基」為其中R20
為除氫外之基團的醯基。代表性醯基包括(但不限於)甲醯基(-CHO)、乙醯基(-C(=O)CH3
)、環己基羰基、環己基甲基羰基、苯甲醯基(-C(=O)Ph)、苯甲基羰基(-C(=O)CH2
Ph)、--C(O)-C1
-C8
烷基、-C(O)-(CH2
)t
(C6
-C10
芳基)、-C(O)-(CH2
)t
(5員至10員雜芳基)、-C(O)-(CH2
)t
(C3
-C10
環烷基)及-C(O)-(CH2
)t
(4員至10員雜環基),其中t為0至4之整數。在某些實施例中,R21
為經鹵基或羥基取代之C1
-C8
烷基;或C3
-C10
環烷基、4員至10員雜環基、C6
-C10
芳基、芳烷基、5員至10員雜芳基或雜芳烷基,其中之各者經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥烷基或未經取代之C1
-C4
鹵烷氧基或羥基。 「烷氧基」係指基團-OR29
,其中R29
為經取代或未經取代之烷基、經取代或未經取代之烯基、經取代或未經取代之炔基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、經取代或未經取代之芳基或經取代或未經取代之雜芳基。特定烷氧基為甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、第三丁氧基、第二丁氧基、正戊氧基、正己氧基及1,2-二甲基丁氧基。特定烷氧基為低碳烷氧基,亦即具有1至6個碳原子。其他特定烷氧基具有1至4個碳原子。 在某些實施例中,R29
為具有1個或多於1個取代基,例如1至5個取代基,且詳言之1至3個取代基,詳言之1個取代基之基團,該取代基選自由以下組成之群:胺基、經取代之胺基、C6
-C10
芳基、芳氧基、羧基、氰基、C3
-C10
環烷基、4員至10員雜環基、鹵素、5員至10員雜芳基、羥基、硝基、硫烷氧基、硫芳氧基、巰基、烷基-S(O)-、芳基-S(O)-、烷基-S(O)2
-及芳基-S(O)2
-。例示性『經取代烷氧基』包括(但不限於)-O-(CH2
)t
(C6
-C10
芳基)、-O-(CH2
)t
(5員至10員雜芳基)、-O-(CH2
)t
(C3
-C10
環烷基)及-O-(CH2
)t
(4員至10員雜環基),其中t為0至4之整數,且所存在之任何芳基、雜芳基、環烷基或雜環基本身可經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥基烷基或未經取代之C1
-C4
鹵烷氧基或羥基。特定例示性『經取代烷氧基』為-OCF3
、-OCH2
CF3
、-OCH2
Ph、-OCH2
-環丙基、-OCH2
CH2
OH及-OCH2
CH2
NMe2
。 「胺基」係指基團-NH2
。 「側氧基」係指-C(=O)-。 「經取代胺基」係指式-N(R38
)2
之胺基,其中R38
為氫、經取代或未經取代之烷基、經取代或未經取代之烯基、經取代或未經取代之炔基、經取代或未經取代之碳環基、經取代或未經取代之雜環基、經取代或未經取代之芳基、經取代或未經取代之雜芳基或胺基保護基,其中R38
中之至少一者不為氫。在某些實施例中,各R38
獨立地選自氫、C1
-C8
烷基、C3
-C8
烯基、C3
-C8
炔基、C6
-C10
芳基、5員至10員雜芳基、4員至10員雜環基或C3
-C10
環烷基或經鹵基或羥基取代之C1
-C8
烷基、經鹵基或羥基取代之C3
-C8
烯基、經鹵基或羥基取代之C3
-C8
炔基或-(CH2
)t
(C6
-C10
芳基)、-(CH2
)t
(5員至10員雜芳基)、-(CH2
)t
(C3
-C10
環烷基)或-(CH2
)t
(4員至10員雜環基),其中t為0與8之間的整數,其中之各者經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥烷基或未經取代之C1
-C4
鹵烷氧基或羥基;或兩個R38
基團接合形成伸烷基。 例示性「經取代胺基」包括(但不限於)-NR39
-C1
-C8
烷基、-NR39
-(CH2
)t
(C6
-C10
芳基)、-NR39
-(CH2
)t
(5員至10員雜芳基)、-NR39
-(CH2
)t
(C3
-C10
環烷基)及-NR39
-(CH2
)t
(4員至10員雜環基),其中t為0至4之整數,例如1或2,各R39
獨立地表示H或C1
-C8
烷基;且所存在之任何烷基本身可經鹵基、經取代或未經取代之胺基或羥基取代;且所存在之任何芳基、雜芳基、環烷基或雜環基本身可經以下取代:未經取代之C1
-C4
烷基、鹵基、未經取代之C1
-C4
烷氧基、未經取代之C1
-C4
鹵烷基、未經取代之C1
-C4
羥基烷基或未經取代之C1
-C4
鹵烷氧基或羥基。為避免疑問,術語『經取代胺基』包括基團烷基胺基、經取代之烷基胺基、烷基芳基胺基、經取代之烷基芳基胺基、芳基胺基、經取代之芳基胺基、二烷胺基及經取代之二烷胺基,如下文所定義。經取代胺基涵蓋單取代胺基及二取代胺基兩者。 「羧基」係指基團-C(O)OH。 「氰基」係指基團-CN。 「鹵基」或「鹵素」係指氟基(F)、氯基(Cl)、溴基(Br)及碘基(I)。在某些實施例中,鹵基為氟基或氯基。 「鹵烷基」係指其中烷基經一或多個鹵素取代之烷基。典型鹵烷基包括(但不限於)三氟甲基(-CF3
)、二氟甲基(-CHF2
)、氟甲基(-CH2
F)、氯甲基(-CH2
Cl)、二氯甲基(-CHCl2
)、三溴甲基(-CH2
Br)及其類似基團。 「羥基」係指基團-OH。 「硝基」係指基團-NO2
。 「硫酮基」係指基團=S。 如本文所定義,烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基係視情況經取代的(例如,「經取代」或「未經取代」烷基、「經取代」或「未經取代」烯基、「經取代」或「未經取代」炔基、「經取代」或「未經取代」碳環基、「經取代」或「未經取代」雜環基、「經取代」或「未經取代」芳基或「經取代」或「未經取代」雜芳基)。大體而言,術語「經取代」無論是否在術語「視情況」之前均意謂基團(例如碳或氮原子)上存在之至少一個氫經容許取代基置換,該容許取代基例如係在取代後產生穩定化合物之取代基,該化合物例如係不會自發地經歷(諸如)藉由重排、環化、消除或其他反應之轉化的化合物。除非另外指明,否則「經取代」基團在該基團之一或多個可取代位置具有取代基,且當任何既定結構中之多於一個位置經取代時,取代基在各位置相同或不同。術語「經取代」預期包括經有機化合物之所有容許取代基、引起穩定化合物形成之本文所描述之任何取代基取代。本發明涵蓋任何及所有此類組合以便獲得穩定化合物。出於本發明之目的,雜原子(諸如氮)可具有氫取代基及/或滿足雜原子價數且使得穩定部分形成之如本文所描述之任何適合取代基。 例示性碳原子取代基包括(但不限於)-CN、-NO2
、-N3
、-SO2
H、-SO3
H、-OH、-ORaa
、-ON(Rbb
)2
、-N(Rbb
)2
、-N(Rbb
)3 +
X-
、-N(ORcc
)Rbb
、-SH、-SRaa
、-SSRcc
、-C(=O)Raa
、-CO2
H、-CHO、-C(ORcc
)2
、-CO2
Raa
、-OC(=O)Raa
、-OCO2
Raa
、-C(=O)N(Rbb
)2
、-OC(=O)N(Rbb
)2
、-NRbb
C(=O)Raa
、-NRbb
CO2
Raa
、-NRbb
C(=O)N(Rbb
)2
、-C(=NRbb
)Raa
、-C(=NRbb
)ORaa
、-OC(=NRbb
)Raa
、-OC(=NRbb
)ORaa
、-C(=NRbb
)N(Rbb
)2
、-OC(=NRbb
)N(Rbb
)2
、-NRbb
C(=NRbb
)N(Rbb
)2
、-C(=O)NRbb
SO2
Raa
、-NRbb
SO2
Raa
、-SO2
N(Rbb
)2
、-SO2
Raa
、-SO2
ORaa
、-OSO2
Raa
、-S(=O)Raa
、-OS(=O)Raa
、-Si(Raa
)3
、-OSi(Raa
)3
-C(=S)N(Rbb
)2
、-C(=O)SRaa
、-C(=S)SRaa
、-SC(=S)SRaa
、-SC(=O)SRaa
、-OC(=O)SRaa
、-SC(=O)ORaa
、-SC(=O)Raa
、-P(=O)2
Raa
、-OP(=O)2
Raa
、-P(=O)(Raa
)2
、-OP(=O)(Raa
)2
、-OP(=O)(ORcc
)2
、-P(=O)2
N(Rbb
)2
、-OP(=O)2
N(Rbb
)2
、-P(=O)(NRbb
)2
、-OP(=O)(NRbb
)2
、-NRbb
P(=O)(ORcc
)2
、-NRbb
P(=O)(NRbb
)2
、-P(Rcc
)2
、-P(Rcc
)3
、-OP(Rcc
)2
、-OP(Rcc
)3
、-B(Raa
)2
、-B(ORcc
)2
、-BRaa
(ORcc
)、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,其中各烷基、烯基、炔基、碳環基、雜環基、芳基、雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代;或碳原子上的兩個成對氫經基團=O、=S、=NN(Rbb
)2
、=NNRbb
C(=O)Raa
、=NNRbb
C(=O)ORaa
、=NNRbb
S(=O)2
Raa
、=NRbb
或=NORcc
置換; Raa
之各個例獨立地選自C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或兩個Raa
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代; Rbb
之各個例獨立地選自氫、-OH、-ORaa
、-N(Rcc
)2
、-CN、-C(=O)Raa
、-C(=O)N(Rcc
)2
、-CO2
Raa
、-SO2
Raa
、-C(=NRcc
)ORaa
、-C(=NRcc
)N(Rcc
)2
、-SO2
N(Rcc
)2
、-SO2
Rcc
、-SO2
ORcc
、-SORaa
、-C(=S)N(Rcc
)2
、-C(=O)SRcc
、-C(=S)SRcc
、-P(=O)2
Raa
、-P(=O)(Raa
)2
、-P(=O)2
N(Rcc
)2
、-P(=O)(NRcc
)2
、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或兩個Rbb
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代; Rcc
之各個例獨立地選自氫、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或兩個Rcc
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代; Rdd
之各個例獨立地選自鹵素、CN、-NO2
、-N3
、-SO2
H、-SO3
H、-OH、-ORee
、-ON(Rff
)2
、-N(Rff
)2
、-N(Rff
)3 +
X-
、-N(ORee
)Rff
、-SH、-SRee
、-SSRee
、-C(=O)Ree
、-CO2
H、-CO2
Ree
、-OC(=O)Ree
、-OCO2
Ree
、-C(=O)N(Rff
)2
、-OC(=O)N(Rff
)2
、-NRff
C(=O)Ree
、-NRff
CO2
Ree
、-NRff
C(=O)N(Rff
)2
、-C(=NRff
)ORee
、-OC(=NRff
)Ree
、-OC(=NRff
)ORee
、-C(=NRff
)N(Rff
)2
、-OC(=NRff
)N(Rff
)2
、-NRff
C(=NRff
)N(Rff
)2
、-NRff
SO2
Ree
、-SO2
N(Rff
)2
、-SO2
Ree
、-SO2
ORee
、-OSO2
Ree
、-S(=O)Ree
、-Si(Ree
)3
、-OSi(Ree
)3
、-C(=S)N(Rff
)2
、-C(=O)SRee
、-C(=S)SRee
、-SC(=S)SRee
、-P(=O)2
Ree
、-P(=O)(Ree
)2
、-OP(=O)(Ree
)2
、-OP(=O)(ORee
)2
、C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、3員至10員雜環基、C6-10
芳基、5員至10員雜芳基,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rgg
基團取代,或兩個成對Rdd
取代基可接合形成=O或=S; Ree
之各個例獨立地選自C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、C6-10
芳基、3員至10員雜環基及3員至10員雜芳基,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rgg
基團取代; Rff
之各個例獨立地選自氫、C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、3員至10員雜環基、C6-10
芳基及5員至10員雜芳基,或兩個Rff
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rgg
基團取代;以及 Rgg
之各個例獨立地為鹵素、-CN、-NO2
、-N3
、-SO2
H、-SO3
H、 -OH、-OC1-6
烷基、-ON(C1-6
烷基)2
、 -N(C1-6
烷基)2
、 -N(C1-6
烷基)3 +
X-
、-NH(C1-6
烷基)2 +
X-
、-NH2
(C1-6
烷基)+
X-
、-NH3 +
X-
、-N(OC1-6
烷基)(C1-6
烷基)、-N(OH)(C1-6
烷基)、-NH(OH)、-SH、-SC1-6
烷基、-SS(C1-6
烷基)、-C(=O)(C1-6
烷基)、-CO2
H、-CO2
(C1-6
烷基)、-OC(=O)(C1-6
烷基)、-OCO2
(C1-6
烷基)、-C(=O)NH2
、-C(=O)N(C1-6
烷基)2
、-OC(=O)NH(C1-6
烷基)、-NHC(=O)( C1-6
烷基)、-N(C1-6
烷基)C(=O)( C1-6
烷基)、-NHCO2
(C1-6
烷基)、-NHC(=O)N(C1-6
烷基)2
、-NHC(=O)NH(C1-6
烷基)、 -NHC(=O)NH2
、 -C(=NH)O(C1-6
烷基)、-OC(=NH)(C1-6
烷基)、-OC(=NH)OC1-6
烷基、-C(=NH)N(C1-6
烷基)2
、-C(=NH)NH(C1-6
烷基)、-C(=NH)NH2
、-OC(=NH)N(C1-6
烷基)2
、-OC(NH)NH(C1-6
烷基)、-OC(NH)NH2
、-NHC(NH)N(C1-6
烷基)2
、-NHC(=NH)NH2
、-NHSO2
(C1-6
烷基)、-SO2
N(C1-6
烷基)2
、-SO2
NH(C1-6
烷基)、-SO2
NH2
、-SO2
C1-6
烷基、-SO2
OC1-6
烷基、-OSO2
C1-6
烷基、-SOC1-6
烷基、-Si(C1-6
烷基)3
、-OSi(C1-6
烷基)3
-C(=S)N(C1-6
烷基)2
、C(=S)NH(C1-6
烷基)、C(=S)NH2
、-C(=O)S(C1-6
烷基)、-C(=S)SC1-6
烷基、-SC(=S)SC1-6
烷基、-P(=O)2
(C1-6
烷基)、-P(=O)(C1-6
烷基)2
、-OP(=O)(C1-6
烷基)2
、-OP(=O)(OC1-6
烷基)2
、C1-6
烷基、C1-6
鹵烷基、C2-6
烯基、C2-6
炔基、C3-10
碳環基、C6-10
芳基、3員至10員雜環基、5員至10員雜芳基;或兩個成對Rgg
取代基可接合形成=O或=S,其中X-
為相對離子。 「相對離子」或「陰離子相對離子」為與陽離子四級胺基相關的帶負電基團以便維持電子電中性。例示性相對離子包括鹵化物離子(例如F-
、Cl-
、Br-
、I-
)、NO3 -
、ClO4 -
、OH-
、H2
PO4 -
、HSO4 -
、SO4 -2
磺酸根離子(例如甲烷磺酸根、三氟甲烷磺酸根、對甲苯磺酸根、苯磺酸根、10-樟腦磺酸根、萘-2-磺酸根、萘-1-磺酸-5-磺酸根、乙-1-磺酸-2-磺酸根及其類似物)及羧酸根離子(例如乙酸根、醋酸根、丙酸根、苯甲酸根、甘油酸根、乳酸根、酒石酸根、羥乙酸根及其類似物)。 價數允許時,氮原子可經取代或未經取代,且包括一級、二級、三級及四級氮原子。例示性氮原子取代基包括(但不限於)氫、-OH、-ORaa
、-N(Rcc
)2
、-CN、-C(=O)Raa
、-C(=O)N(Rcc
)2
、-CO2
Raa
、-SO2
Raa
、-C(=NRbb
)Raa
、-C(=NRcc
)ORaa
、-C(=NRcc
)N(Rcc
)2
、-SO2
N(Rcc
)2
、-SO2
Rcc
、-SO2
ORcc
、-SORaa
、-C(=S)N(Rcc
)2
、-C(=O)SRcc
、-C(=S)SRcc
、-P(=O)2
Raa
、-P(=O)(Raa
)2
、-P(=O)2
N(Rcc
)2
、-P(=O)(NRcc
)2
、C1-10
烷基、C1-10
鹵烷基、C2-10
烯基、C2-10
炔基、C3-10
碳環基、3員至14員雜環基、C6-14
芳基及5員至14員雜芳基,或連接至氮原子的兩個Rcc
基團接合形成3員至14員雜環基或5員至14員雜芳基環,其中各烷基、烯基、炔基、碳環基、雜環基、芳基及雜芳基獨立地經0、1、2、3、4或5個Rdd
基團取代且其中Raa
、Rbb
、Rcc
及Rdd
如上文所定義。 此等及其他例示性取代基更詳細地描述於實施方式
、實例
及申請專利範圍
中。本發明並不意欲以任何方式受取代基之上述例示性清單限制。其他定義
術語「醫藥學上可接受之鹽」係指在合理醫學判斷範疇內適用於與人類及低等動物之組織接觸而無異常毒性、刺激、過敏反應及其類似情況且與合理益處/風險比相稱的彼等鹽。醫藥學上可接受之鹽在此項技術中為所熟知的。舉例而言,Berge等人在J. Pharmaceutical Sciences
(1977) 66:1-19中詳細描述醫藥學上可接受之鹽。本發明化合物之醫藥學上可接受之鹽包括衍生自適合的無機及有機酸及鹼之彼等鹽。醫藥學上可接受之無毒酸加成鹽之實例為胺基與無機酸(諸如鹽酸、氫溴酸、磷酸、硫酸及過氯酸)或有機酸(諸如乙酸、草酸、順丁烯二酸、酒石酸、檸檬酸、丁二酸或丙二酸)形成之鹽,或藉由使用此項技術中所用之其他方法(諸如離子交換)形成之鹽。其他醫藥學上可接受之鹽包括己二酸鹽、海藻酸鹽、抗壞血酸鹽、天冬胺酸鹽、苯磺酸鹽、苯甲酸鹽、硫酸氫鹽、硼酸鹽、丁酸鹽、樟腦酸鹽、樟腦磺酸鹽、檸檬酸鹽、環戊烷丙酸鹽、二葡糖酸鹽、十二烷基硫酸鹽、乙烷磺酸鹽、甲酸鹽、反丁烯二酸鹽、葡庚糖酸鹽、甘油磷酸鹽、葡糖酸鹽、半硫酸鹽、庚酸鹽、己酸鹽、氫碘酸鹽、2-羥基-乙烷磺酸鹽、乳糖酸鹽、乳酸鹽、月桂酸鹽、月桂基硫酸鹽、蘋果酸鹽、順丁烯二酸鹽、丙二酸鹽、甲烷磺酸鹽、2-萘磺酸鹽、菸鹼酸鹽、硝酸鹽、油酸鹽、草酸鹽、棕櫚酸鹽、雙羥萘酸鹽、果膠酸鹽、過硫酸鹽、3-苯基丙酸鹽、磷酸鹽、苦味酸鹽、特戊酸鹽、丙酸鹽、硬脂酸鹽、丁二酸鹽、硫酸鹽、酒石酸鹽、硫氰酸鹽、對甲苯磺酸鹽、十一烷酸鹽、戊酸鹽及其類似鹽。衍生自適當鹼之醫藥學上可接受之鹽包括鹼金屬、鹼土金屬、銨及N+
(C1-4
烷基)4
鹽。代表性鹼金屬鹽或鹼土金屬鹽包括鈉鹽、鋰鹽、鉀鹽、鈣鹽、鎂鹽及其類似鹽。在適當時,其他醫藥學上可接受之鹽包括使用諸如鹵化物、氫氧化物、羧酸鹽、硫酸鹽、磷酸鹽、硝酸鹽、低碳烷基磺酸鹽及芳基磺酸鹽之相對離子形成之無毒銨、四級銨及胺陽離子。 涵蓋投與之「個體」,包括(但不限於)人類(亦即,任何年齡組之男性或女性,例如兒科個體(例如、嬰兒、兒童、青少年)或成人個體(例如年輕人、中年人或老年人))及/或非人類動物,例如哺乳動物,諸如靈長類動物(例如食蟹獼猴、恆河猴)、牛、豬、馬、綿羊、山羊、嚙齒動物、貓及/或狗。在某些實施例中,個體為人類。在某些實施例中,個體為非人類動物。術語「人類」、「患者」及「個體」在本文中可互換使用。 疾病、病症及病狀在本文中可互換使用。 如本文所使用且除非另外說明,否則術語「治療(treat)」、「治療(treating)」及「治療(treatment)」涵蓋在個體正患指定疾病、病症或病狀的同時發生的降低疾病、病症或病狀嚴重程度或延遲或減緩疾病、病症或病狀進展的作用(「治療性治療」),且亦涵蓋在個體開始罹患指定疾病、病症或病狀之前發生的作用(「預防性治療」)。 大體而言,化合物之「有效量」係指足以引出所需生物反應的量。如一般技術者將瞭解,有效量之本發明化合物可視諸如所需生物終點、化合物之藥物動力學、所治療之疾病、投與模式及個體年齡、體重、健康及病狀之因素而變化。有效量涵蓋治療性及預防性治療。 如本文所使用且除非另外說明,否則化合物之「治療有效量」為足夠提供治療疾病、病症或病狀之治療益處或延遲或使與疾病、病症或病狀相關之一或多種症狀減至最少的量。化合物之治療有效量意謂單獨或與其他療法組合的治療劑之量,其提供治療疾病、病症或病狀之治療益處。術語「治療有效量」可涵蓋改良整體療法、減少或避免疾病或病狀的症狀或病因或增強另一治療劑之治療功效的量。 如本文所用且除非另外說明,否則化合物之「預防有效量」為足夠預防疾病、病症或病狀或與疾病、病症或病狀相關之一或多種症狀或防止其復發的量。化合物之預防有效量意謂治療劑單獨或與其他試劑組合提供預防疾病、病症或病狀之預防益處的量。術語「預防有效量」可涵蓋改良整體預防或增強另一預防劑之預防功效的量。 或其醫藥學上可接受之鹽,其中:R1
為烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且
或其醫藥學上可接受之鹽,其中:R1
為烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且 。 在一些實施例中,式(I-63)之化合物選自式(I-A63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-A63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-C63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-C63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-D63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-D63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-E63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-E63)之化合物: 。 在一些實施例中,式(I-63)之化合物選自式(I-D-i63)或(I-D-ii63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-D-i63)或(I-D-ii63)之化合物: 或
或 。 在一些實施例中,式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物:
。 在一些實施例中,式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物: 或
或 。 在一個態樣中,本文提供根據式(I-67)之化合物:
。 在一個態樣中,本文提供根據式(I-67)之化合物: 或其醫藥學上可接受之鹽,其中:R1
為氫或烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且
或其醫藥學上可接受之鹽,其中:R1
為氫或烷基(例如C1
-C6
烷基);R2
及R3
中之各者獨立地為氫、烷基(例如C1
-C6
烷基)、碳環基、雜環基、芳基或雜芳基,或R2
及R3
與其所附接之碳原子一起形成3員至8員環;R4
及R5
中之各者獨立地為氫、鹵基或-ORC
,其中RC
為氫或C1
-C6
烷基(例如C1
-C3
烷基),或R4
及R5
與其所附接之碳原子一起形成側氧基;R6
不存在或為氫;且
 。 在一些實施例中,式(I-67)之化合物選自式(I-A67)之化合物:
。 在一些實施例中,式(I-67)之化合物選自式(I-A67)之化合物: 。 在一些實施例中,式(I-67)之化合物選自式(I-C67)之化合物:
。 在一些實施例中,式(I-67)之化合物選自式(I-C67)之化合物: 。 在替代實施例中,本文所描述之化合物亦可包含一或多種同位素取代。舉例而言,氫可為2
H (D或氘)或3
H (T或氚);碳可為(例如)13
C或14
C;氧可為(例如)18
O;氮可為(例如)15
N,等等。在其他實施例中,特定同位素(例如3
H、13
C、14
C、18
O或15
N)可表示佔據化合物特異位點之元素的總同位素豐度的至少1%、至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少99%或至少99.9%。 本發明之例示性化合物包括:
。 在替代實施例中,本文所描述之化合物亦可包含一或多種同位素取代。舉例而言,氫可為2
H (D或氘)或3
H (T或氚);碳可為(例如)13
C或14
C;氧可為(例如)18
O;氮可為(例如)15
N,等等。在其他實施例中,特定同位素(例如3
H、13
C、14
C、18
O或15
N)可表示佔據化合物特異位點之元素的總同位素豐度的至少1%、至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少99%或至少99.9%。 本發明之例示性化合物包括:








 及
及 。替代實施例
在替代實施例中,本文所描述之化合物亦可包含一或多種同位素取代。舉例而言,氫可為2
H (D或氘)或3
H (T或氚);碳可為(例如)13
C或14
C;氧可為(例如)18
O;氮可為(例如)15
N,等等。在其他實施例中,特定同位素(例如3
H、13
C、14
C、18
O或15
N)可表示佔據化合物特異位點之元素的總同位素豐度的至少1%、至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少99%或至少99.9%。醫藥組合物
在另一態樣中,本發明提供一種醫藥組合物,其包含醫藥學上可接受之載劑及有效量的本文所描述之化合物或其醫藥學上可接受之鹽。 在用作藥物時,本文提供之化合物通常以醫藥組合物之形式投與。此等組合物可以醫藥技術中熟知之方式來製備且包含至少一種活性化合物。 在一個實施例中,關於醫藥組合物,載劑為非經腸載劑、口服或局部載劑。 本發明亦係關於本文所描述之化合物,或其用作藥物或藥劑之醫藥組合物。 大體而言,本文所提供之化合物按治療有效量投與。實際投與之化合物之量通常將由醫師鑒於相關情形確定,該等相關情形包括待治療之病狀、所選擇之投與途徑、所投與之實際化合物、個別患者之年齡、體重及反應、患者症狀之嚴重程度及其類似者。 本文所提供之醫藥組合物可藉由各種途徑投與,包括經口、經直腸、經皮、皮下、靜脈內、肌內及鼻內。視預期傳遞途徑而定,本文所提供之化合物較佳調配為可注射或口服組合物,或調配為均用於經皮投與之油膏、洗劑或貼片。 用於經口投與之組合物可採取散裝液體溶液或懸浮液或散裝粉末之形式。然而,組合物更通常以單位劑型呈現以便於準確給藥。術語「單位劑型」係指適合以單位劑量形式用於人類個體及其他哺乳動物的物理上不連續之單元,各單元含有經計算以產生所需治療作用的預定量之活性材料,其與適合之醫藥賦形劑結合。典型單位劑型包括液體組合物之預填充、預量測安瓿或注射器或在固體組合物情況中之丸劑、錠劑、膠囊或其類似物。在此類組合物中,化合物通常為次要組分(約0.1重量%至約50重量%,或較佳約1重量%至約40重量%),其餘部分為有助於形成所需給藥形式之各種媒劑或載劑及加工助劑。 適於經口投與之液體形式可包括適合水性或非水性媒劑與緩衝劑、懸浮劑及分散劑、著色劑、調味劑及其類似物。固體形式可包括例如以下成分或具有類似性質之化合物中之任一者:黏合劑,諸如微晶纖維素、黃蓍膠或明膠;賦形劑,諸如澱粉或乳糖;崩解劑,諸如褐藻酸、澱粉羥基乙酸鈉(Primogel)或玉米澱粉;潤滑劑,諸如硬脂酸鎂;滑動劑,諸如膠態二氧化矽;甜味劑,諸如蔗糖或糖精;或調味劑,諸如胡椒薄荷、水楊酸甲酯或橙味調味劑。 可注射組合物通常係基於可注射無菌生理食鹽水或磷酸鹽緩衝生理食鹽水或此項技術中已知的其他可注射載劑。如前所述,此類組合物中之活性化合物通常為次要組分(常為約0.05重量%至10重量%),其餘部分為可注射載劑及其及類似物。 經皮組合物通常調配為含有活性成分之局部軟膏或乳霜,其量之範圍大體上為約0.01重量%至約20重量%,較佳約0.1重量%至約20重量%,較佳約0.1重量%至約10重量%,且更佳約0.5重量%至約15重量%。當調配成軟膏時,活性成分通常將與石蠟或水可混溶性軟膏基劑合併。或者,活性成分可使用例如水包油乳霜基劑調配成乳霜。此類經皮調配物為此項技術中所熟知且大體上包括額外成分以增強活性成分或調配物之真皮穿透穩定性。所有此類已知之經皮調配物及成分均包括在本文提供之範疇內。 本文提供之化合物亦可藉由經皮裝置投與。因此,經皮投與可使用儲集層型或多孔膜型或固體基質類貼片來實現。 上文所描述之用於可經口投與、可注射或可局部投與組合物之組分僅為代表性的。其他材料以及加工技術及其類似者闡述於Remington's Pharmaceutical Sciences
之第8部分,第17版,1985, Mack Publishing Company, Easton, Pennsylvania,其以引用的方式併入本文中。 上文所描述之用於可經口投與、可注射或可局部投與組合物之組分僅為代表性的。其他材料以及加工技術及其類似者闡述於Remington's The Science and Practice of Pharmacy
之第8部分,第21版,2005, 出版商:Lippincott Williams & Wilkins,其以引用的方式併入本文中。 本發明之化合物亦可以持續釋放形式或自持續釋放藥物傳遞系統投與。代表性持續釋放材料之描述可見於Remington's Pharmaceutical Sciences
。 本發明亦係關於本文所描述之化合物的醫藥學上可接受之調配物。在一個實施例中,該調配物包含水。在另一實施例中,該調配物包含環糊精衍生物。最常見環糊精為分別由6、7及8個α-l,4-鍵聯葡糖單元組成之α-環糊精、β-環糊精及γ-環糊精,其視情況包含在鍵聯糖部分上之一或多個取代基,該一或多個取代基包括(但不限於)甲基化、羥基烷基化、醯基化及磺基烷基醚取代。在某些實施例中,環糊精為磺基烷基醚β-環糊精,例如磺基丁基醚β-環糊精,亦稱為Captisol®。參見(例如) U.S. 5,376,645。在某些實施例中,調配物包含六丙基-β-環糊精。在更特定實施例中,調配物包含六丙基-β-環糊精(10-50%於水中)。 本發明亦係關於本文所描述之化合物的醫藥學上可接受之酸加成鹽。可用於製備醫藥學上可接受之鹽的酸為形成無毒酸加成鹽,亦即,含有藥理學上可接受之陰離子之鹽的酸,該鹽諸如鹽酸鹽、氫碘酸鹽、氫溴酸鹽、硝酸鹽、硫酸鹽、硫酸氫鹽、磷酸鹽、乙酸鹽、乳酸鹽、檸檬酸鹽、酒石酸鹽、丁二酸鹽、順丁烯二酸鹽、反丁烯二酸鹽、苯甲酸鹽、對甲苯磺酸鹽及其類似物。 以下調配物實例說明可根據本發明製備之代表性醫藥組合物。然而,本發明不限於以下醫藥組合物。 例示性調配物 1— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為240 mg至270 mg錠劑(每一錠劑80 mg至90 mg活性化合物)。 例示性調配物 2— 膠囊
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與澱粉稀釋劑按大致1:1重量比摻混。混合物填充成250 mg膠囊(每一錠劑125 mg活性化合物)。 例示性調配物 3— 液體 :
本文所描述之化合物或其醫藥學上可接受之鹽(125 mg)可與蔗糖(1.75 g)及三仙膠(4 mg)摻混,且所得混合物可經摻合,經過10號目U.S.篩,並接著與先前製得的微晶纖維素與羧甲基纖維素鈉(11:89,50 mg)於水中之溶液混合。苯甲酸鈉(10 mg)、調味劑及著色劑用水稀釋且在攪拌下添加。接著可再添加足夠水產生5 mL之總體積。 例示性調配物 4— 錠劑 :
本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為450 mg至900 mg錠劑(150 mg至300 mg活性化合物)。 例示性調配物 5— 注射劑 :
本文所描述之化合物或其醫藥學上可接受之鹽可溶解或懸浮於緩衝無菌生理食鹽水可注射水性介質中,達到大致5mg/mL之濃度。 例示性調配物 6— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為90 mg至150 mg錠劑(每一錠劑30 mg至50 mg活性化合物)。 例示性調配物 7— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為30 mg至90 mg錠劑(每一錠劑10 mg至30 mg活性化合物)。 例示性調配物 8— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為0.3 mg至30 mg錠劑(每一錠劑0.1 mg至10 mg活性化合物)。 例示性調配物 9— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為150 mg至240 mg錠劑(每一錠劑50 mg至80 mg活性化合物)。 例示性調配物 10— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為270 mg至450 mg錠劑(每一錠劑90 mg至150 mg活性化合物)。 注射劑量在約0.1 mg/kg/hr至至少10 mg/kg/hr範圍內,均歷時約1至約120小時且尤其24至96小時。亦可投與約0.1 mg/kg至約10 mg/kg或10 mg/kg以上之預負載大丸劑以達到充分穩態水準。對於40至80 kg人類患者,最大總劑量預期不超過約2公克/天。 為了預防及/或治療長期病狀,治療療程通常延續多月或多年,因此對於患者便利性及耐受性而言,經口給藥係較佳的。在經口給藥下,每天一個至五個且尤其兩個至四個及通常三個經口劑量為代表性方案。使用此等給藥模式,各劑量提供約0.01至約20 mg/kg本文所提供之化合物,其中較佳劑量各提供約0.1至約10 mg/kg及尤其約1至約5 mg/kg。 通常選擇經皮給藥以提供與使用注射給藥所達成的血液含量相比類似或較低的血液含量。 當用於預防CNS病症發作時,應通常根據醫師之建議且在醫師之監督下,以上文所描述之劑量向在出現該病狀之風險下的個體投與本文所提供之化合物。處於出現特定病狀之風險下的個體一般包括具有該病狀之家族病史之彼等個體,或已藉由遺傳測試或篩檢而鑑別為尤其易於出現該病狀之彼等個體。治療及使用方法
如本文所描述,本發明化合物(例如,式(A)、(I-63)或(I-67)之化合物)及其醫藥學上可接受之鹽,一般設計成調節NMDA功能,且因此充當用於治療及預防個體中之(例如) CNS相關病況的氧固醇。在一些實施例中,如本文所描述,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)及其醫藥學上可接受之鹽,一般設計成穿透血腦障壁(例如,設計成跨越血腦障壁傳輸)。如本文所使用,調節係指(例如)抑制或增強NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽充當NMDA之負向異位調節劑(negative allosteric modulator,NAM)並抑制NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽充當NMDA之正向異位調節劑(positive allosteric modulator,PAM)並增強NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽藉由天然產生之基質阻斷或減少NMDA受體功能之增強或抑制。此類化合物並不充當NMDA之負向異位調節劑(NAM)或正向異位調節劑(PAM)。在一些實施例中,病症為癌症。在一些實施例中,病症為糖尿病。在一些實施例中,病症為固醇合成障礙。在一些實施例中,該病症為腸胃(GI)病症,例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病)、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌症或結腸炎。在一些實施例中,病症為發炎性腸病。 與NMDA-調節有關之例示性病況包括(但不限於)腸胃(GI)病症,例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病)、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌症、結腸炎及CNS病狀,(例如)如本文中所描述。 與NMDA-調節有關之例示性病況(例如CNS病況)包括(但不限於)失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質濫用相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病 、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症及耳鳴。 在某些實施例中,本發明化合物,例如,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽,可用於誘導鎮靜或麻醉。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質濫用相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病 、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症及耳鳴。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、物質濫用相關病症、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)或分娩後精神病。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症或耳鳴。 在一些實施例中,本發明化合物(充當NMDA受體功能之PAM的本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、多發性硬化、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病及與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)。 在一些實施例中,充當NMDA受體功能之NAM之本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、人格障礙(包括強迫性人格障礙)、神經發育性障礙(包括雷特症候群)、疼痛(包括急性及疼痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)及結節性硬化症(TSC))、中風、創傷性腦損傷、失調症、神經精神性狼瘡及耳鳴。 在一些實施例中,充當NMDA受體功能之PAM或NAM之本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、固醇合成障礙及進食障礙。 在另一態樣中,提供一種治療或預防易感或罹患與腦興奮相關之病狀的個體的腦興奮的方法,其包含向該個體投與有效量之本發明化合物(例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)或其醫藥學上可接受之鹽。 在又一態樣中,本發明提供本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)或其醫藥學上可接受之鹽與另一藥理活性劑的組合。本文所提供之化合物可作為唯一活性劑投與或其可與其他藥劑組合投與。組合投與可藉由熟習此項技術者清楚之任何技術進行,包括(例如)分開、連續、同時及交替投與。 在另一態樣中,本文提供一種誘導個體中之NMDA受體之負向異位調節之方法,其包含向該個體投與本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物。運動障礙
本文中亦描述用於治療動作障礙之方法。如本文所使用,「運動障礙」係指與過動性運動障礙及肌肉控制相關異常相關之多種疾病及病症。例示性運動障礙包括(但不限於)帕金森氏病及巴金森氏症(具體由動作遲緩定義)、肌張力障礙、舞蹈症及亨廷頓氏病、運動失調、左旋多巴誘導之運動困難、震顫(例如特發性震顫)、肌陣攣及驚跳、抽搐及妥瑞症候群、腿不寧症候群、僵人症候群及步態障礙。震顫
為非自主性、有時節律性、肌肉收縮及鬆弛,其可涉及一或多個身體部分(例如手、臂、眼睛、臉、頭部、聲帶褶、軀幹、腿)之振盪或抽搐。震顫包括:遺傳性、退化性及特發性病症,分別諸如威爾森氏病、帕金森氏病及特發性震顫;代謝障礙(例如甲狀腺-副甲狀腺病症、肝病及低血糖);周邊神經病(與恰克-馬利-杜斯氏病、魯西-利維病、糖尿病、複雜區域疼痛症候群相關);毒素(尼古丁、汞、鉛、CO、錳、砷、甲苯);藥物性(發作性睡眠病、三環化合物、鋰、可卡因、酒精、腎上腺素、支氣管擴張劑、茶鹼、咖啡鹼、類固醇、丙戊酸鹽、胺碘酮、甲狀腺激素、長春新鹼);及精神性病症。臨床震顫可分為生理震顫、增強型生理震顫、特發性震顫症候群(包括經典特發性震顫、原發直立性震顫、及任務特異性及位置特異性震顫)、肌張力障礙性震顫、巴金森式震顫、小腦震顫、霍氏震顫(亦即紅核震顫)、上顎震顫、神經病性震顫、中毒性或藥物性震顫及心因性震顫。其他形式之震顫包括小腦震顫或意向震顫、肌張力障礙性震顫、特發性震顫、直立性震顫、巴金森式震顫、生理性震顫、心因性震顫或紅核震顫。小腦震顫
或意向震顫
為在有目的之運動之後發生的肢端緩慢而廣泛之震顫。小腦震顫由因例如腫瘤、中風、疾病(例如多發性硬化症,一種遺傳性退化性病症)而產生之小腦病變或損傷引起。肌張力障礙性震顫
發生在受肌張力障礙(一種持續非自主性肌肉收縮引起扭動及重複動作及/或疼痛及異常姿勢或位置之運動障礙)影響之個體中。肌張力障礙性震顫可影響體內任何肌肉。肌張力障礙性震顫不規則地發生且常常可藉由徹底休息而減輕。特發性震顫
或良性特發性震顫為最常見類型的震顫。特發性震顫可為輕度的且在一些情況下無進展,且可緩慢進展,起始於身體一側,但在3年內影響兩側。手最常受影響,但頭部、聲音、舌、腿及軀幹亦可涉及。震顫頻率可隨人變老而降低,但嚴重程度可增加。高漲之情緒、壓力、發熱、筋疲力盡或低血糖可引發震顫及/或增加其嚴重程度。症狀一般隨時間推移發展且可均為發病後可見與持久。直立性震顫
之特徵為在站立之後腿及軀幹中很快發生的快速(例如超過12 Hz)節律性肌肉收縮。大腿及腿中感覺到絞痛且患者可在要求站立在一點時不受控制地震盪。直立性震顫可發生在患有特發性震顫之患者中。帕金森式震顫
由對大腦內控制運動之結構之破壞引起。帕金森式震顫常常為帕金森氏病之前兆且通常表現為亦可影響下頜、嘴唇、腿及軀幹之手的「搓丸樣」動作。帕金森式震顫之發病通常在60歲之後開始。運動始於一肢或身體一側且可進展至包括另一側。生理性震顫
可出現在正常個體中且無臨床意義。其在所有隨意肌群中均可見。生理性震顫可由某些藥物、戒酒或包括過度活躍甲狀腺及低血糖之醫學病狀引起。震顫通常具有約10 Hz之頻率。心因性震顫
或癔病性震顫可在休息時或在姿勢或動態運動期間出現。患有心因性震顫之患者可具有轉換性障礙或另一精神疾病。紅核震顫
之特徵為可在休息時、在擺姿勢時及刻意下存在之粗緩慢震顫。震顫與影響中腦中紅核之病狀(經典異常中風)相關。帕金森氏病
影響大腦中產生多巴胺的神經細胞。症狀包括肌強直、震顫及語音及步態變化。巴金森氏症
之特徵為震顫、動作遲緩、強直及姿勢不穩定。巴金森氏症共用帕金森氏病中發現之症狀,但其為症候群而非進行性神經退化性疾病。肌張力障礙
為一種運動障礙,其特徵為引起異常(常常為反覆)的運動或姿勢的持續性或間斷肌肉收縮。肌張力障礙性運動可為模式化的,帶扭動,且可為震顫樣的。肌張力障礙常由自主行動起始或惡化,並與溢出肌肉活化相關。舞蹈症
為一種神經性病症,其特徵為通常影響肩部、髖部及臉的急抽式非自主運動。亨廷頓氏病
為一種引起大腦中神經細胞日益衰弱的遺傳性疾病。症狀包括不受控運動、動作笨拙及平衡問題。亨廷頓氏病可能妨礙步行、講話及吞咽。運動失調
係指喪失對整體運動的完全控制,且可影響手指、手、手臂、腿、身體、語音及眼球運動。肌陣痙及驚跳
係對於突然及未預期刺激的反應,刺激可為聲學、觸感、可見或前庭刺激。抽搐
為非自主運動,一般突然發病,係短暫、反覆的,但係非節律性的,其通常模仿正常行為且常常發生在正常活動背景中。抽搐可分類為運動或發聲抽搐,運動抽搐與運動相關,而發聲抽搐與聲音有關。抽搐的特徵可為簡單或複雜的。舉例而言,簡單運動抽搐僅涉及受限於特定身體部分的一些肌肉。妥瑞症候群
為一種在兒童中發病之遺傳性神經精神異常,其特徵為多種運動抽搐及至少一種發聲抽搐。腿不寧症候群
為一種神經感覺運動障礙,其特徵為在休息時有壓倒性的移動腿之衝動。僵人症候群
為一種進行性運動障礙,其特徵為非自主疼痛痙攣及肌肉僵直,一般涉及下背及腿。通常產生具有加重之腰椎過度凸出的僵腿式步態。通常觀察具有脊椎旁軸肌之連續運動單元活動的EMG記錄上的特徵性異常。變體包括產生通常會影響遠端腿部及腳之病灶性僵硬的「僵肢症候群」。步態障礙
係指步行方式或風格之異常,其起因於神經肌肉變化、關節炎變化或其他身體變化。步態係根據負責用於異常移動之系統而分類,且包括偏癱步態、兩側癱瘓步態、神經病性步態、肌病步態、巴金森式步態、舞蹈病狀步態、運動失調步態及感官步態。情緒障礙
本文亦提供用於治療例如以下之情緒障礙的方法:臨床抑鬱、產後抑鬱或分娩後抑鬱、圍產期抑鬱、非典型抑鬱、憂鬱型抑鬱、精神病性嚴重抑鬱症、緊張型抑鬱、季節性情緒失調症、輕鬱症、雙重抑鬱、抑鬱性人格障礙、復發性短暫抑鬱、輕度抑鬱障礙、躁鬱症或躁狂抑鬱性障礙、由慢性醫學病狀引起之抑鬱、耐治療性抑鬱、難治性抑鬱、自殺傾向、自殺觀念或自殺行為。臨床抑鬱
亦稱為嚴重抑鬱症、重度抑鬱症(MDD)、嚴重抑鬱、單極抑鬱、單極病症及復發性抑鬱,且係指特徵為普遍及持久情緒低落,伴隨有較低自尊心及正常娛樂活動之興趣或快樂感喪失的精神病症。一些患有臨床抑鬱之人入睡困難,體重減輕且一般感到焦躁及易受刺激。臨床抑鬱影響個體之感覺、思想及行為,且可導致許多情感及身體問題。患有臨床抑鬱之個體每天的活動可能困難,且使個體感到生活不值得過。產後抑鬱
(PND)亦稱為分娩後抑鬱 (PPD)
,且係指在分娩之後影響女性之一種類型的臨床抑鬱。症狀可包括悲傷、疲乏、睡眠及飲食習慣變化、性慾減少、哭喊事件、焦慮及易怒。在一些實施例中,PND為耐治療性抑鬱(例如,如本文所描述之耐治療性抑鬱)。在一些實施例中,PND為難治性抑鬱(例如,如本文所描述之難治性抑鬱)。 在一些實施例中,具有PND之個體在懷孕期間亦經歷抑鬱或抑鬱症狀。此抑鬱在本文中被稱作圍產期抑鬱
。在實施例中,經歷圍產期抑鬱之個體經歷PND之風險增加。非典型抑鬱 (AD)
之特徵為情緒反應性(例如反常快感缺乏)及積極性、體重顯著增加或食慾增加。患有AD之患者亦可具有過度睡眠或嗜睡(睡眠過度)、四肢沉重之感覺及由於對所感知人際排斥超敏感而有顯著社會功能損傷。憂鬱型抑鬱
之特徵為在大部分或所有活動中喪失快樂(快感缺乏)、無法對快樂刺激作出反應、比哀傷或喪失更明顯之憂鬱情緒、體重過度減輕或過度內疚。精神病性嚴重抑鬱症 (PMD)
或精神病性抑鬱係指嚴重抑鬱事件,尤其具有憂鬱性,其中個體經歷諸如妄想及幻覺之精神病症狀。緊張型抑鬱
係指涉及運動行為紊亂及其他症狀之嚴重抑鬱症。個體可變得沉默且麻木,且不能動或展現無目的或古怪動作。季節性情緒失調症 (SAD)
係指其中個體具有在秋季或冬季來到之抑鬱事件季節性模式的一種類型的季節性抑鬱。輕鬱症
係指與單極抑鬱相關之病狀,其中相同身體及認知問題顯而易見。其不如單極抑鬱嚴重且傾向於持續較長時間(例如至少2年)。雙重抑鬱
係指持續至少2年之極其憂鬱情緒(輕鬱症),且間雜有嚴重抑鬱症時期。抑鬱性人格障礙 (DPD)
係指具有抑鬱特徵之人格障礙。復發性短暫抑鬱 (RBD)
係指其中個體約每月發生一次抑鬱事件,各事件持續2週或更少時間且通常小於2-3天的病狀。輕度抑鬱障礙
或輕度抑鬱係指其中至少2種症狀存在2週之抑鬱。躁鬱症或躁狂抑鬱性障礙
引起極端情緒波動,包括情緒高漲(躁症或輕躁症)及低落(抑鬱)。在躁症期期間,個體可感到或行為異常歡樂、高能或易受刺激。其常常無法作出考慮周到之決定,對結果幾乎不作考慮。睡眠需求通常減少。在抑鬱期期間,可存在哭喊,眼睛與他人缺乏接觸,且對生活之觀點負面。在患有該病症之患者中自殺風險高,20年間超過6%,同時自殘發生率為30%-40%。諸如焦慮症之其他精神健康問題及物質使用障礙通常與躁鬱症相關。由慢性醫學病狀引起之抑鬱
係指由諸如癌症之慢性醫學病狀或慢性疼痛、化學療法、慢性壓力引起的抑鬱。耐治療性抑鬱
係指個體已針對抑鬱進行治療,但症狀未改善的病狀。舉例而言,抗抑鬱劑或心理諮詢(心理療法)未減輕患有耐治療性抑鬱之個體的抑鬱症狀。在一些情況下,患有耐治療性抑鬱之個體改善症狀,但又恢復。難治性抑鬱
發生在罹患抑鬱之對包括三環抗抑鬱劑、MAOI、SSRI及雙重及三重吸收抑制劑及/或抗焦慮劑藥物之標準藥理學治療以及非藥理學治療(例如心理療法、電驚厥療法、迷走神經刺激及/或經顱磁刺激)具抗性的患者中。自殺傾向、自殺觀念、自殺行為
係指個體自殺之傾向。自殺觀念涉及關於自殺之想法或對自殺異常專注。自殺觀念之範圍變化很大,例如稍縱即逝的念頭至深入的想法、詳細計劃、角色扮演、著手未遂。症狀包括討論自殺、獲得進行自殺之方式、退出社會接觸、一心想著死亡、對某事感到被困或無望、增加酒精或藥物之使用、作出有風險或自毀事情、向人們說再見,好像其不會再見面一樣。 抑鬱症狀
包括持久焦慮或悲傷感、感到無助、無望、悲觀、無價值、低能、坐立不安、入睡困難、不眠、易怒、疲乏、動作攻擊、喪失娛樂活動或愛好之興趣、注意力缺乏、缺乏能量、自尊心差、無積極思想或計劃、睡眠過度、飲食過量、食慾喪失、失眠、自殘、自殺想法及自殺嘗試。症狀之存在、嚴重程度、頻率及持續時間可隨病例而變化。抑鬱症狀及其減輕可藉由醫師或心理學家(例如藉由精神狀態檢查)來確定。焦慮症 本文提供治療焦慮症之方法。焦慮症
為覆蓋異常及病理學恐懼及焦慮之若干不同形式的總稱。當前精神診斷標準辨認各種焦慮症。廣泛性焦慮症
為一種常見慢性病症,其特徵為持久焦慮,不集中於任一個目標或情況。罹患廣泛性焦慮之彼等個體經歷非特定的持久恐懼及擔憂,且變得過度擔心每日事情。廣泛性焦慮症為影響老年人之最常見焦慮症。 在恐慌症
中,個人遭受強烈恐懼及憂懼之短暫發作,特徵常常為戰慄、搖晃、意識模糊、眩暈、噁心、呼吸困難。此等恐慌發作藉由APA定義為突然出現且在不到十分鐘內達到峰值之恐懼或不適,可持續若干小時且可由壓力、害怕或甚至運動觸發;不過具體病因始終不清楚。除復發性意外恐慌發作之外,診斷恐慌症亦需要該等發作具有慢性後果:擔憂發作之潛在影響,持久性害怕將來發作或與發作相關之行為變化顯著。因此,罹患恐慌症之個體經歷甚至在特定恐慌事件外之症狀。常常,恐慌患者注意到心跳之正常變化,導致其認為其心臟出問題或其將再具有恐慌發作。在一些情況下,身體功能之感知加強(警覺過度)在恐慌發作期間發生,其中任何所感知之生理改變均解釋為可能威脅生命之疾病(亦即極端疑病症)。強迫症
為主要特徵為重複困擾(令人苦惱、持久性及侵入性想法或影像)及強迫(進行特定動作或儀式之衝動)之一種類型的焦慮症。OCD思想模式可類似於迷信,到其將信念與實際上不存在之因果關係相連的程度。常常,該過程全部為不合邏輯的;舉例而言,強迫以一定模式步行可用以緩解對即將發生損害之困擾。且在許多情況下,強迫性全部不可解釋,簡單而言,完成由緊張觸發之儀式的衝動。在少數情況下,OCD患者可僅僅經歷困擾,無明顯強迫症;數目少得多之患者僅僅經歷強迫症。 單一最大類別之焦慮症為恐懼症
,其包括由特定刺激或情況觸發害怕及焦慮之所有情況。患者通常由遇到其害怕之目標來預計恐怖後果,其害怕目標可為自動物至體液位置之任何東西。創傷後壓力症
或PTSD
為一種由創傷經歷產生之焦慮症。創傷後壓力可由極端情況引起,諸如打鬥、強姦、人質情況或甚至嚴重事故。其亦可由長期(慢性)暴露於嚴重應激物引起,例如經受個體戰鬥,但無法應付連續打鬥之士兵。常見症狀包括閃回、回避行為及抑鬱。癲癇症
癲癇症為一種特徵為隨時間推移的重複癲癇發作之大腦病症。癲癇症之類型可包括(但不限於)廣泛性癲癇症,例如兒童失神癲癇症、幼年肌陣攣癲癇症、喚醒時癲癇大發作之癲癇症、韋斯特症候群(West syndrome)、倫-加症候群(Lennox-Gastaut syndrome);部分性癲癇症,例如兒童之顳葉性癲癇症、額葉性癲癇症、良性病灶性癲癇症。癲癇形成
癲癇形成為正常大腦產生癲癇症的漸進過程(癲癇發作發生之慢性病狀)。癲癇形成因藉由初始損傷促成之神經元損害而引起(例如持續性癲癇)。持續性癲癇 (SE)
持續性癲癇(SE)可包括例如痙攣性持續性癲癇,例如早期持續性癲癇、現有持續性癲癇、難治性持續性癲癇、超難治性持續性癲癇;非痙攣性持續性癲癇,例如廣泛性持續性癲癇、複雜部分持續性癲癇;廣泛性週期性癲癇樣放電;及週期性偏側性癲癇樣放電。痙攣性持續性癲癇之特徵為存在痙攣性持續性癲癇發作,且可包括早期持續性癲癇、現有持續性癲癇、難治性持續性癲癇、超難治性持續性癲癇。早期持續性癲癇用一線療法治療。現有持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法治療其仍持續且投與二線療法。難治性持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法及二線療法治療其仍持續且通常投與一般麻醉劑。超難治性持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法、二線療法及一般麻醉劑治療24小時或24小時以上,其仍持續。 非痙攣性持續性癲癇可包括例如病灶性非痙攣性持續性癲癇,例如複雜部分非痙攣性持續性癲癇、簡單部分非痙攣性持續性癲癇、隱微非痙攣性持續性癲癇;廣泛性非痙攣性持續性癲癇,例如晚發性失神非痙攣性持續性癲癇、非典型失神非痙攣性持續性癲癇或典型失神非痙攣性持續性癲癇。癲癇發作
癲癇發作為大腦中異常電活動事件後發生之生理發現或行為改變。術語「癲癇發作」通常可與「痙攣」互換使用。痙攣為當人的身體快速且不受控制地搖晃時。在痙攣期間,人的肌肉反覆收縮及放鬆。 基於行為及大腦活動之類型,將癲癇發作分成兩種較寬廣類別:廣泛性及部分(亦稱為局部或病灶性)。將癲癇發作類型進行分類有助於醫生診斷患者是否患有癲癇症。 廣泛性癲癇由整個大腦中之電脈衝產生,而部分癲癇由相對較小部分大腦中之電脈衝產生(至少起初)。產生癲癇之大腦部分有時稱為病灶。 存在六種類型之廣泛性癲癇。最常見且嚴重的且因此最熟知的為廣泛性痙攣,亦稱為大發作癲癇。在此類型癲癇中,患者失去意識且通常虛脫。失去意識後廣泛性身體僵硬(稱為癲癇之「強直性」階段)30 至60秒,隨後劇烈抽動(「陣攣性」階段)30至60秒,其後患者進入深度睡眠(「發作後」或癲癇後階段)。在大發作癲癇期間,可能發生損傷及事故,諸如咬舌及尿失禁。 失神癲癇導致短暫意識喪失(僅幾秒),幾乎無症狀。患者(最常為兒童)通常中斷活動且目直。此等癲癇突然開始及結束,且可一日發生若干次。患者通常未意識到其癲癇發作,但其可能意識到「時間流失」。 肌陣攣癲癇由偶發性急抽組成,通常在身體兩側。患者有時將急抽描述為短暫電擊。劇烈時,此等癲癇可能導致物體掉落或無意地投擲物體。 陣攣性癲癇為重複的節律性急抽,其同時涉及身體兩側。 強直性癲癇之特徵為肌肉僵硬。 失張性癲癇由尤其在臂及腿中,肌肉張力之突然及一般損失組成,其通常導致跌倒。 本文所描述之癲癇可包括癲癇發作;急性重複癲癇;密集式癲癇;連續癲癇;無間斷癲癇;長期癲癇;反覆性癲癇;持續性癲癇發作,例如難治性痙攣性持續性癲癇、非痙攣性持續性癲癇發作;難治性癲癇;肌陣攣性癲癇;強直性癲癇;強直性陣攣性癲癇;簡單部分癲癇;複雜部分癲癇;繼發廣泛性癲癇;非典型失神癲癇;失神癲癇;失張性癲癇;良性羅蘭多(Rolandic)癲癇;發熱性癲癇;情緒性癲癇;病灶性癲癇;癡笑性癲癇;廣泛性發作癲癇;嬰兒痙攣;傑克遜氏癲癇(Jacksonian seizures);大規模雙側肌陣攣癲癇;多灶性癲癇;新生兒發作癲癇;夜間癲癇;枕葉性癲癇;創傷後癲癇;微小癲癇;西爾萬癲癇(Sylvan seizures);視覺反射癲癇或撤藥癲癇。在一些實施例中,癲癇為與德拉韋症候群、倫-加症候群、結節性硬化症、雷特症候群或PCDH19女嬰兒癲癇症相關之廣泛性癲癇。縮寫
PCC:氯鉻酸吡啶;t-BuOK:第三丁醇鉀;9-BBN:9-硼雙環[3.3.1]壬烷;Pd(t
-Bu3
P)2
:雙(三-第三丁基膦)鈀(0);AcCl:氯化乙醯基;i
-PrMgCl:異丙基氯化鎂;TBSCl:第三丁基(氯)二甲基矽烷;(i
-PrO)4
Ti:四異丙醇鈦;BHT:2,6-二-第三丁基-4-甲基苯酚;Me:甲基;i
-Pr:異丙基;t
-Bu:第三丁基;Ph:苯基;Et:乙基;Bz:苯甲醯基;BzCl:氯化苯甲醯;CsF:氟化銫;DCC:二環己基碳化二亞胺;DCM:二氯甲烷;DMAP:4-二甲胺基吡啶;DMP:戴斯-馬丁高碘烷;EtMgBr:乙基溴化鎂;EtOAc:乙酸乙酯;TEA:三乙胺;AlaOH:丙胺酸;Boc:第三丁氧羰基。Py:吡啶;TBAF:氟化四正丁基銨;THF:四氫呋喃;TBS:第三丁基二甲基矽烷基;TMS:三甲基矽烷基;TMSCF3
:(三氟甲基)三甲基矽烷;Ts:對甲苯磺醯基;Bu:丁基;Ti(OiPr)4
:四異丙氧基鈦;LAH:氫化鋰鋁;LDA:二異丙胺基鋰;LiOH.H2
O:氫氧化鋰水合物;MAD:甲基鋁雙(2,6-二-第三丁基-4-甲基苯酚);MeCN:乙腈;NBS:N-溴代丁二醯亞胺;Na2
SO4
:硫酸鈉;Na2
S2
O3
:硫代硫酸鈉;PE:石油醚;MeCN:乙腈;MeOH:甲醇;Boc:第三丁氧羰基;MTBE:甲基第三丁基醚;DIAD:偶氮二甲酸二異丙酯。實例
為了能更全面地理解本文中所描述之發明,闡述以下實例。提供本申請案中所描述之合成及生物實例以說明本文提供之化合物、醫藥組合物及方法,且並不理解為以任何方式限制其範疇。在以下合成實例中,按序號列出反應序列內之實驗程序之描述。物質及方法
本文提供之化合物可使用以下通用方法及程序自容易獲得之起始物質製備。應瞭解,除非另行說明,否則在給定典型或較佳製程條件(亦即,反應溫度、時間、反應物之莫耳比、溶劑、壓力等)的情況下,亦可使用其他製程條件。最佳反應條件可隨所用特定反應物或溶劑而變,但此類條件可由熟習此項技術者藉由常規最佳化來確定。 此外,如熟習此項技術者將顯而易知,可能必需習知保護基來防止某些官能基經歷非所需反應。對於特定官能團適合的保護基以及保護及去保護之適合的條件之選擇為此項技術中熟知的。舉例而言,諸多保護基及其引入及移除描述於T. W. Greene及P. G. M. Wuts,Protecting Groups in Organic Synthesis
,第二版, Wiley, New York, 1991及其中所引用之參考文獻中。 本文提供之化合物可藉由已知標準程序分離及純化。此類程序包括(但不限於)再結晶、管柱層析、HPLC或超臨界流體層析(SFC)。關於製備本文中已列出之代表性吡唑的細節呈現以下流程。本文提供之化合物可由熟習有機合成技術者自已知或市售起始物質及試劑製備。可用於分離/純化本文提供之對映異構體/非對映異構體之例示性對掌性管柱包括(但不限於) CHIRALPAK® AD-10。CHIRALCEL® OB、CHIRALCEL® OB-H、CHIRALCEL® OD、CHIRALCEL® OD-H、CHIRALCEL® OF、CHIRALCEL® OG、CHIRALCEL® OJ及CHIRALCEL® OK。 製備型HPLC之例示性通用方法:管柱:Waters Rbridge製備型10 μm C18,19 × 250 mm。移動相:乙腈、水(NH4
HCO3
) (30 L水、24 g NH4
HCO3
、30 mL NH3
.H2
O)。流速:25 mL/min。 分析型HPLC之例示性通用方法:移動相:A:水 (10 mM NH4
HCO3
),B:乙腈梯度:在1.6或2 min內,5%-95% B;流速:1.8或2 mL/min;管柱:XBridge C18,4.6×50mm,在45℃為3.5 μm。NMDA 調節
使用自動箝膜系統來評定表現NMDA受體之哺乳動物細胞中之NMDA增強,該自動箝膜系統可用於測定如下文所描述之化合物之NAM活性。全細胞箝膜系統可用於測定如下文所描述之化合物之PAM活性。自動化箝膜系統 (QPatch HTX) :
在此研究中,經GRIN1/2A次型之麩胺酸激活通道穩定轉染之HEK 293細胞將與次極大NMDA濃度(300 μM NMDA,與8 μM甘胺酸共施加)一起用來研究測試化合物之負向異位調節。細胞培養物
大體而言,細胞將以約80%至90%之會合率傳代。對於電生理量測,將以約80%至90%之會合率自含有完全培養基之無菌培養瓶收集細胞。細胞將作為PBS中之懸浮液傳送至QPatch 16X或QPatch HTX系統,直接傳送至離心機/洗滌器。 標準實驗室條件:細胞將在具有5% CO2
之潮濕氛圍(相對濕度約95%)中在37℃下培育。培養基
:細胞將在含有補充有10%胎牛血清、1%青黴素/鏈黴素溶液及50 μM AP-5阻斷劑之達爾伯克氏改良伊格爾培養基與營養混合物F-12 (D-MEM/F-12 1×,液體與L-麩醯胺酸)之1:1混合物的無菌培養瓶中不斷保持及傳代。抗生素
:如上所指出之完全培養基補充有100 μg/mL潮黴素、15 μg/mL殺稻瘟菌素及1 μg/mL嘌呤黴素。表現之誘導
:在開始實驗之前24 h添加2.5 μg/mL四環素。劑量調配
劑量係就供應時之測試化合物而言。將添加媒劑以達成10 mM之儲備濃度(在-10℃至-30℃下儲存)。將在DMSO中製備另外的1.0 mM之儲備溶液。儲備溶液利用之細節(解凍、劑量調配)將記錄在原始資料中。儲備溶液利用之時段將詳述於報告中。測試化合物濃度
劑量係就供應時之測試化合物而言。將添加媒劑以達成10 mM之儲備濃度(在-10℃至-30℃下儲存)。將在DMSO中製備另外的1.0 mM之儲備溶液。儲備溶液利用之細節(解凍、劑量調配)將記錄在原始資料中。儲備溶液利用之時段將詳述於報告中。 將測試一個測試濃度1.0 μM。 將藉由在電生理實驗前不久僅用不含Mg之電解液或含有NMDA (300 µM)及甘胺酸(8.0 µM)的不含Mg之電解液稀釋儲備溶液來製備所有測試溶液,且在使用時將其保持在室溫(19℃至30℃)下。0.1%DMSO將被用作媒劑。製備頻率
:對於各測試濃度,每日將製備測試化合物之新鮮溶液。劑量調配之穩定性
:所有製備時間將記錄在原始資料中。關於測試化合物之不穩定性之任何觀察結果將在原始資料中提及。劑量調配物之儲存
:在實驗之日,劑量調配物在使用時將維持在室溫(19℃至30℃)下。電解液
為了準備實驗及為了形成千兆歐姆密封,將使用以下標準電解液 :
氯化鈉:137 mM;氯化鉀:4 mM;氯化鈣:1.8 mM;氯化鎂:1 mM;HEPES:10 mM;D-葡糖:10 mM;十六醇聚氧乙烯醚:0.02%;pH (NaOH):7.4 將藉由至少每7天用水稀釋不含葡糖之10×電解液及100×葡糖溶液來製備1×電解液。在當前研究之實驗開始之前已經製備兩種儲備溶液且將其儲存在1℃至9℃下(10×電解液)或-10℃至-30℃下(100×葡糖溶液)。實驗中所使用之電解液之批號將記錄在原始資料中。當使用時,1×電解液將保持在室溫(19℃至30℃)下。當未使用時,1×電解液將儲存於1℃至9℃下。 在形成千兆密封後,將使用以下不含 Mg 的電解液
: 氯化鈉:137 mM;氯化鉀:4 mM;氯化鈣:2.8 mM;HEPES:10 mM;D-葡糖:10 mM;十六醇聚氧乙烯醚
:0.02%;pH (NaOH):7.4 此不含Mg的電解液將製備為1×溶液且儲存於1℃至9℃下。其將至少每10天新鮮製備。胞內溶液
1×胞內溶液將每日自冷凍的1×胞內溶液解凍,其已在當前研究之實驗開始之前製備、取等分及儲存於-10℃至-30℃下。當使用時,1×胞內溶液將保持在室溫(19℃至30℃)下。剩餘的1×胞內溶液將儲存於冰箱(1℃至9℃)中。1×胞內溶液將包括下文概述之組分: 氯化鉀:130 mM;氯化鎂:1 mM;Mg-ATP:5 mM;HEPES:10 mM;EGTA:5 mM;pH (KOH):7.2細胞處理
對於此研究,細胞將不斷灌注有NMDA/甘胺酸、測試化合物或測試化合物/NMDA/甘胺酸。 在各情況下,將在施加之間進行利用測試化合物之至少30秒預洗滌步驟。細節請參見下表A。 將在至少n=3個分離細胞中分析各實驗類型。將在當前研究之實驗開始之前製備NMDA及甘胺酸儲備溶液,冷凍儲存(-10℃至-30℃),直至實驗當天為止。在電生理實驗前不久,冷凍儲備溶液將經解凍及稀釋。 對照:每兩週將在三個細胞處量測媒劑(0.1% DMSO)及D-(-)-2-胺基-5-膦醯基戊酸(AP-5) (100 μM)之效果,以便確保NMDA受體之成功表現。 已在當前研究之實驗開始之前製備AP-5之50 mM儲備溶液,取等分試樣且冷凍儲存(-10℃至-30℃),直至實驗當天為止。在電生理實驗前不久,冷凍儲備溶液將經解凍且接著在含有NMDA (300 μM)及甘胺酸(8.0 μM)之不含Mg的電解液中稀釋,得到最終灌注濃度100 µM。實驗程序
細胞作為無血清培養基中之懸浮液傳送至QPatch HTX系統且在實驗期間保持在細胞儲槽/攪拌器中。施加至細胞之所有溶液(包括胞內溶液)將維持在室溫下(19℃至30℃)下。 在密封製程期間,將使用上文所描述之標準電解液
。施加至細胞之所有溶液(包括移液管溶液)將維持在室溫下(19℃至30℃)下。在膜片電極與經轉染個別HEK293細胞之間形成千兆歐姆密封之後,將僅灌注不含 Mg 的電解液
且細胞膜將會破裂以確保電氣進入細胞內部(全細胞膜片組態)。將在向膜片鉗位細胞施加300 µM NMDA (及8.0 µM甘胺酸)後量測內向電流5秒。在整個實驗期間,細胞將在-80 mV之保持電位下經電壓鉗位。 為了分析測試化合物,NMDA受體將由下文所描述之300 µM NMDA及8.0 µM甘胺酸及測試化合物組合刺激。將在施加之間進行利用測試化合物之三十二個預洗滌步驟。 表A:施加協議;測試化合物之使用依賴性
。替代實施例
在替代實施例中,本文所描述之化合物亦可包含一或多種同位素取代。舉例而言,氫可為2
H (D或氘)或3
H (T或氚);碳可為(例如)13
C或14
C;氧可為(例如)18
O;氮可為(例如)15
N,等等。在其他實施例中,特定同位素(例如3
H、13
C、14
C、18
O或15
N)可表示佔據化合物特異位點之元素的總同位素豐度的至少1%、至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少99%或至少99.9%。醫藥組合物
在另一態樣中,本發明提供一種醫藥組合物,其包含醫藥學上可接受之載劑及有效量的本文所描述之化合物或其醫藥學上可接受之鹽。 在用作藥物時,本文提供之化合物通常以醫藥組合物之形式投與。此等組合物可以醫藥技術中熟知之方式來製備且包含至少一種活性化合物。 在一個實施例中,關於醫藥組合物,載劑為非經腸載劑、口服或局部載劑。 本發明亦係關於本文所描述之化合物,或其用作藥物或藥劑之醫藥組合物。 大體而言,本文所提供之化合物按治療有效量投與。實際投與之化合物之量通常將由醫師鑒於相關情形確定,該等相關情形包括待治療之病狀、所選擇之投與途徑、所投與之實際化合物、個別患者之年齡、體重及反應、患者症狀之嚴重程度及其類似者。 本文所提供之醫藥組合物可藉由各種途徑投與,包括經口、經直腸、經皮、皮下、靜脈內、肌內及鼻內。視預期傳遞途徑而定,本文所提供之化合物較佳調配為可注射或口服組合物,或調配為均用於經皮投與之油膏、洗劑或貼片。 用於經口投與之組合物可採取散裝液體溶液或懸浮液或散裝粉末之形式。然而,組合物更通常以單位劑型呈現以便於準確給藥。術語「單位劑型」係指適合以單位劑量形式用於人類個體及其他哺乳動物的物理上不連續之單元,各單元含有經計算以產生所需治療作用的預定量之活性材料,其與適合之醫藥賦形劑結合。典型單位劑型包括液體組合物之預填充、預量測安瓿或注射器或在固體組合物情況中之丸劑、錠劑、膠囊或其類似物。在此類組合物中,化合物通常為次要組分(約0.1重量%至約50重量%,或較佳約1重量%至約40重量%),其餘部分為有助於形成所需給藥形式之各種媒劑或載劑及加工助劑。 適於經口投與之液體形式可包括適合水性或非水性媒劑與緩衝劑、懸浮劑及分散劑、著色劑、調味劑及其類似物。固體形式可包括例如以下成分或具有類似性質之化合物中之任一者:黏合劑,諸如微晶纖維素、黃蓍膠或明膠;賦形劑,諸如澱粉或乳糖;崩解劑,諸如褐藻酸、澱粉羥基乙酸鈉(Primogel)或玉米澱粉;潤滑劑,諸如硬脂酸鎂;滑動劑,諸如膠態二氧化矽;甜味劑,諸如蔗糖或糖精;或調味劑,諸如胡椒薄荷、水楊酸甲酯或橙味調味劑。 可注射組合物通常係基於可注射無菌生理食鹽水或磷酸鹽緩衝生理食鹽水或此項技術中已知的其他可注射載劑。如前所述,此類組合物中之活性化合物通常為次要組分(常為約0.05重量%至10重量%),其餘部分為可注射載劑及其及類似物。 經皮組合物通常調配為含有活性成分之局部軟膏或乳霜,其量之範圍大體上為約0.01重量%至約20重量%,較佳約0.1重量%至約20重量%,較佳約0.1重量%至約10重量%,且更佳約0.5重量%至約15重量%。當調配成軟膏時,活性成分通常將與石蠟或水可混溶性軟膏基劑合併。或者,活性成分可使用例如水包油乳霜基劑調配成乳霜。此類經皮調配物為此項技術中所熟知且大體上包括額外成分以增強活性成分或調配物之真皮穿透穩定性。所有此類已知之經皮調配物及成分均包括在本文提供之範疇內。 本文提供之化合物亦可藉由經皮裝置投與。因此,經皮投與可使用儲集層型或多孔膜型或固體基質類貼片來實現。 上文所描述之用於可經口投與、可注射或可局部投與組合物之組分僅為代表性的。其他材料以及加工技術及其類似者闡述於Remington's Pharmaceutical Sciences
之第8部分,第17版,1985, Mack Publishing Company, Easton, Pennsylvania,其以引用的方式併入本文中。 上文所描述之用於可經口投與、可注射或可局部投與組合物之組分僅為代表性的。其他材料以及加工技術及其類似者闡述於Remington's The Science and Practice of Pharmacy
之第8部分,第21版,2005, 出版商:Lippincott Williams & Wilkins,其以引用的方式併入本文中。 本發明之化合物亦可以持續釋放形式或自持續釋放藥物傳遞系統投與。代表性持續釋放材料之描述可見於Remington's Pharmaceutical Sciences
。 本發明亦係關於本文所描述之化合物的醫藥學上可接受之調配物。在一個實施例中,該調配物包含水。在另一實施例中,該調配物包含環糊精衍生物。最常見環糊精為分別由6、7及8個α-l,4-鍵聯葡糖單元組成之α-環糊精、β-環糊精及γ-環糊精,其視情況包含在鍵聯糖部分上之一或多個取代基,該一或多個取代基包括(但不限於)甲基化、羥基烷基化、醯基化及磺基烷基醚取代。在某些實施例中,環糊精為磺基烷基醚β-環糊精,例如磺基丁基醚β-環糊精,亦稱為Captisol®。參見(例如) U.S. 5,376,645。在某些實施例中,調配物包含六丙基-β-環糊精。在更特定實施例中,調配物包含六丙基-β-環糊精(10-50%於水中)。 本發明亦係關於本文所描述之化合物的醫藥學上可接受之酸加成鹽。可用於製備醫藥學上可接受之鹽的酸為形成無毒酸加成鹽,亦即,含有藥理學上可接受之陰離子之鹽的酸,該鹽諸如鹽酸鹽、氫碘酸鹽、氫溴酸鹽、硝酸鹽、硫酸鹽、硫酸氫鹽、磷酸鹽、乙酸鹽、乳酸鹽、檸檬酸鹽、酒石酸鹽、丁二酸鹽、順丁烯二酸鹽、反丁烯二酸鹽、苯甲酸鹽、對甲苯磺酸鹽及其類似物。 以下調配物實例說明可根據本發明製備之代表性醫藥組合物。然而,本發明不限於以下醫藥組合物。 例示性調配物 1— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為240 mg至270 mg錠劑(每一錠劑80 mg至90 mg活性化合物)。 例示性調配物 2— 膠囊
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與澱粉稀釋劑按大致1:1重量比摻混。混合物填充成250 mg膠囊(每一錠劑125 mg活性化合物)。 例示性調配物 3— 液體 :
本文所描述之化合物或其醫藥學上可接受之鹽(125 mg)可與蔗糖(1.75 g)及三仙膠(4 mg)摻混,且所得混合物可經摻合,經過10號目U.S.篩,並接著與先前製得的微晶纖維素與羧甲基纖維素鈉(11:89,50 mg)於水中之溶液混合。苯甲酸鈉(10 mg)、調味劑及著色劑用水稀釋且在攪拌下添加。接著可再添加足夠水產生5 mL之總體積。 例示性調配物 4— 錠劑 :
本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為450 mg至900 mg錠劑(150 mg至300 mg活性化合物)。 例示性調配物 5— 注射劑 :
本文所描述之化合物或其醫藥學上可接受之鹽可溶解或懸浮於緩衝無菌生理食鹽水可注射水性介質中,達到大致5mg/mL之濃度。 例示性調配物 6— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為90 mg至150 mg錠劑(每一錠劑30 mg至50 mg活性化合物)。 例示性調配物 7— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為30 mg至90 mg錠劑(每一錠劑10 mg至30 mg活性化合物)。 例示性調配物 8— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為0.3 mg至30 mg錠劑(每一錠劑0.1 mg至10 mg活性化合物)。 例示性調配物 9— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為150 mg至240 mg錠劑(每一錠劑50 mg至80 mg活性化合物)。 例示性調配物 10— 錠劑
:本文所描述之化合物或其醫藥學上可接受之鹽可以乾粉形式與乾明膠黏合劑按大致1:2重量比摻混。添加微量硬脂酸鎂作為潤滑劑。混合物在製錠機中形成為270 mg至450 mg錠劑(每一錠劑90 mg至150 mg活性化合物)。 注射劑量在約0.1 mg/kg/hr至至少10 mg/kg/hr範圍內,均歷時約1至約120小時且尤其24至96小時。亦可投與約0.1 mg/kg至約10 mg/kg或10 mg/kg以上之預負載大丸劑以達到充分穩態水準。對於40至80 kg人類患者,最大總劑量預期不超過約2公克/天。 為了預防及/或治療長期病狀,治療療程通常延續多月或多年,因此對於患者便利性及耐受性而言,經口給藥係較佳的。在經口給藥下,每天一個至五個且尤其兩個至四個及通常三個經口劑量為代表性方案。使用此等給藥模式,各劑量提供約0.01至約20 mg/kg本文所提供之化合物,其中較佳劑量各提供約0.1至約10 mg/kg及尤其約1至約5 mg/kg。 通常選擇經皮給藥以提供與使用注射給藥所達成的血液含量相比類似或較低的血液含量。 當用於預防CNS病症發作時,應通常根據醫師之建議且在醫師之監督下,以上文所描述之劑量向在出現該病狀之風險下的個體投與本文所提供之化合物。處於出現特定病狀之風險下的個體一般包括具有該病狀之家族病史之彼等個體,或已藉由遺傳測試或篩檢而鑑別為尤其易於出現該病狀之彼等個體。治療及使用方法
如本文所描述,本發明化合物(例如,式(A)、(I-63)或(I-67)之化合物)及其醫藥學上可接受之鹽,一般設計成調節NMDA功能,且因此充當用於治療及預防個體中之(例如) CNS相關病況的氧固醇。在一些實施例中,如本文所描述,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)及其醫藥學上可接受之鹽,一般設計成穿透血腦障壁(例如,設計成跨越血腦障壁傳輸)。如本文所使用,調節係指(例如)抑制或增強NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽充當NMDA之負向異位調節劑(negative allosteric modulator,NAM)並抑制NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽充當NMDA之正向異位調節劑(positive allosteric modulator,PAM)並增強NMDA受體功能。在某些實施例中,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽藉由天然產生之基質阻斷或減少NMDA受體功能之增強或抑制。此類化合物並不充當NMDA之負向異位調節劑(NAM)或正向異位調節劑(PAM)。在一些實施例中,病症為癌症。在一些實施例中,病症為糖尿病。在一些實施例中,病症為固醇合成障礙。在一些實施例中,該病症為腸胃(GI)病症,例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病)、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌症或結腸炎。在一些實施例中,病症為發炎性腸病。 與NMDA-調節有關之例示性病況包括(但不限於)腸胃(GI)病症,例如,便秘、大腸急躁症(IBS)、發炎性腸病(IBD) (例如潰瘍性結腸炎、克羅恩氏病)、影響GI之結構性病症、肛門病症(例如痔瘡、內痔、外痔、肛裂、肛周膿腫、肛瘻)、結腸息肉、癌症、結腸炎及CNS病狀,(例如)如本文中所描述。 與NMDA-調節有關之例示性病況(例如CNS病況)包括(但不限於)失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質濫用相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病 、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症及耳鳴。 在某些實施例中,本發明化合物,例如,本文所描述之化合物(例如式(A)、(I-63)或(I-67)之化合物)或其醫藥學上可接受之鹽,可用於誘導鎮靜或麻醉。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、物質濫用相關病症、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病 、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症及耳鳴。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防失調症、焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、物質濫用相關病症、解離性障礙、進食障礙、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、人格障礙(包括強迫性人格障礙)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)或分娩後精神病。 在某些實施例中,本文所描述之化合物或其醫藥學上可接受之鹽適用於治療或預防神經發育性障礙(包括雷特症候群)、多發性硬化、固醇合成障礙、史密斯-萊姆利-奧皮茨症候群、疼痛(包括急性疼痛、慢性疼痛及神經痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)、結節性硬化症(TSC)及嬰幼兒痙攣症)、中風、蛛膜下出血、腦內出血、大腦缺血、創傷性腦損傷、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)、神經退化性病症、神經發炎、神經精神性狼瘡、尼曼-皮克C病症或耳鳴。 在一些實施例中,本發明化合物(充當NMDA受體功能之PAM的本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):精神分裂症或其他精神病性障礙(包括分裂情感性精神障礙)、睡眠障礙(包括失眠)、自閉症譜系障礙(包括涉及蛋白質柄群(例如柄3)突變的彼等障礙)、多發性硬化、運動障礙(包括亨廷頓氏病及帕金森氏病)、注意力不足症、注意力不足過動症、代謝腦病(包括苯酮尿症)、分娩後精神病及與高效價之抗NMDA受體抗體相關的症候群(包括抗NMDA受體腦炎)。 在一些實施例中,充當NMDA受體功能之NAM之本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):焦慮症(包括強迫症、創傷後壓力症、社交恐懼症、廣泛性焦慮症)、情緒障礙(包括抑鬱(例如分娩後抑鬱)、躁鬱症、神經官能性憂鬱症、自殺傾向)、人格障礙(包括強迫性人格障礙)、神經發育性障礙(包括雷特症候群)、疼痛(包括急性及疼痛)、癲癇發作症(包括持續性癲癇及單基因形式的癲癇,諸如德拉韋氏病(Dravet's disease)及結節性硬化症(TSC))、中風、創傷性腦損傷、失調症、神經精神性狼瘡及耳鳴。 在一些實施例中,充當NMDA受體功能之PAM或NAM之本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)可適用於治療或預防包括以下的病狀(例如CNS相關病狀):認知障礙(包括阿茲海默病及其他形式的癡呆,包括皮質基底核癡呆-進行性核上麻痹、額顳葉型癡呆、原發進行性失語、帕金森氏病癡呆及路易體性癡呆)、固醇合成障礙及進食障礙。 在另一態樣中,提供一種治療或預防易感或罹患與腦興奮相關之病狀的個體的腦興奮的方法,其包含向該個體投與有效量之本發明化合物(例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)或其醫藥學上可接受之鹽。 在又一態樣中,本發明提供本發明化合物(例如,式(A)化合物、式(I-63)之化合物或式(I-67)之化合物)或其醫藥學上可接受之鹽與另一藥理活性劑的組合。本文所提供之化合物可作為唯一活性劑投與或其可與其他藥劑組合投與。組合投與可藉由熟習此項技術者清楚之任何技術進行,包括(例如)分開、連續、同時及交替投與。 在另一態樣中,本文提供一種誘導個體中之NMDA受體之負向異位調節之方法,其包含向該個體投與本文所描述之化合物,例如式(A)化合物、式(I-63)之化合物或式(I-67)之化合物。運動障礙
本文中亦描述用於治療動作障礙之方法。如本文所使用,「運動障礙」係指與過動性運動障礙及肌肉控制相關異常相關之多種疾病及病症。例示性運動障礙包括(但不限於)帕金森氏病及巴金森氏症(具體由動作遲緩定義)、肌張力障礙、舞蹈症及亨廷頓氏病、運動失調、左旋多巴誘導之運動困難、震顫(例如特發性震顫)、肌陣攣及驚跳、抽搐及妥瑞症候群、腿不寧症候群、僵人症候群及步態障礙。震顫
為非自主性、有時節律性、肌肉收縮及鬆弛,其可涉及一或多個身體部分(例如手、臂、眼睛、臉、頭部、聲帶褶、軀幹、腿)之振盪或抽搐。震顫包括:遺傳性、退化性及特發性病症,分別諸如威爾森氏病、帕金森氏病及特發性震顫;代謝障礙(例如甲狀腺-副甲狀腺病症、肝病及低血糖);周邊神經病(與恰克-馬利-杜斯氏病、魯西-利維病、糖尿病、複雜區域疼痛症候群相關);毒素(尼古丁、汞、鉛、CO、錳、砷、甲苯);藥物性(發作性睡眠病、三環化合物、鋰、可卡因、酒精、腎上腺素、支氣管擴張劑、茶鹼、咖啡鹼、類固醇、丙戊酸鹽、胺碘酮、甲狀腺激素、長春新鹼);及精神性病症。臨床震顫可分為生理震顫、增強型生理震顫、特發性震顫症候群(包括經典特發性震顫、原發直立性震顫、及任務特異性及位置特異性震顫)、肌張力障礙性震顫、巴金森式震顫、小腦震顫、霍氏震顫(亦即紅核震顫)、上顎震顫、神經病性震顫、中毒性或藥物性震顫及心因性震顫。其他形式之震顫包括小腦震顫或意向震顫、肌張力障礙性震顫、特發性震顫、直立性震顫、巴金森式震顫、生理性震顫、心因性震顫或紅核震顫。小腦震顫
或意向震顫
為在有目的之運動之後發生的肢端緩慢而廣泛之震顫。小腦震顫由因例如腫瘤、中風、疾病(例如多發性硬化症,一種遺傳性退化性病症)而產生之小腦病變或損傷引起。肌張力障礙性震顫
發生在受肌張力障礙(一種持續非自主性肌肉收縮引起扭動及重複動作及/或疼痛及異常姿勢或位置之運動障礙)影響之個體中。肌張力障礙性震顫可影響體內任何肌肉。肌張力障礙性震顫不規則地發生且常常可藉由徹底休息而減輕。特發性震顫
或良性特發性震顫為最常見類型的震顫。特發性震顫可為輕度的且在一些情況下無進展,且可緩慢進展,起始於身體一側,但在3年內影響兩側。手最常受影響,但頭部、聲音、舌、腿及軀幹亦可涉及。震顫頻率可隨人變老而降低,但嚴重程度可增加。高漲之情緒、壓力、發熱、筋疲力盡或低血糖可引發震顫及/或增加其嚴重程度。症狀一般隨時間推移發展且可均為發病後可見與持久。直立性震顫
之特徵為在站立之後腿及軀幹中很快發生的快速(例如超過12 Hz)節律性肌肉收縮。大腿及腿中感覺到絞痛且患者可在要求站立在一點時不受控制地震盪。直立性震顫可發生在患有特發性震顫之患者中。帕金森式震顫
由對大腦內控制運動之結構之破壞引起。帕金森式震顫常常為帕金森氏病之前兆且通常表現為亦可影響下頜、嘴唇、腿及軀幹之手的「搓丸樣」動作。帕金森式震顫之發病通常在60歲之後開始。運動始於一肢或身體一側且可進展至包括另一側。生理性震顫
可出現在正常個體中且無臨床意義。其在所有隨意肌群中均可見。生理性震顫可由某些藥物、戒酒或包括過度活躍甲狀腺及低血糖之醫學病狀引起。震顫通常具有約10 Hz之頻率。心因性震顫
或癔病性震顫可在休息時或在姿勢或動態運動期間出現。患有心因性震顫之患者可具有轉換性障礙或另一精神疾病。紅核震顫
之特徵為可在休息時、在擺姿勢時及刻意下存在之粗緩慢震顫。震顫與影響中腦中紅核之病狀(經典異常中風)相關。帕金森氏病
影響大腦中產生多巴胺的神經細胞。症狀包括肌強直、震顫及語音及步態變化。巴金森氏症
之特徵為震顫、動作遲緩、強直及姿勢不穩定。巴金森氏症共用帕金森氏病中發現之症狀,但其為症候群而非進行性神經退化性疾病。肌張力障礙
為一種運動障礙,其特徵為引起異常(常常為反覆)的運動或姿勢的持續性或間斷肌肉收縮。肌張力障礙性運動可為模式化的,帶扭動,且可為震顫樣的。肌張力障礙常由自主行動起始或惡化,並與溢出肌肉活化相關。舞蹈症
為一種神經性病症,其特徵為通常影響肩部、髖部及臉的急抽式非自主運動。亨廷頓氏病
為一種引起大腦中神經細胞日益衰弱的遺傳性疾病。症狀包括不受控運動、動作笨拙及平衡問題。亨廷頓氏病可能妨礙步行、講話及吞咽。運動失調
係指喪失對整體運動的完全控制,且可影響手指、手、手臂、腿、身體、語音及眼球運動。肌陣痙及驚跳
係對於突然及未預期刺激的反應,刺激可為聲學、觸感、可見或前庭刺激。抽搐
為非自主運動,一般突然發病,係短暫、反覆的,但係非節律性的,其通常模仿正常行為且常常發生在正常活動背景中。抽搐可分類為運動或發聲抽搐,運動抽搐與運動相關,而發聲抽搐與聲音有關。抽搐的特徵可為簡單或複雜的。舉例而言,簡單運動抽搐僅涉及受限於特定身體部分的一些肌肉。妥瑞症候群
為一種在兒童中發病之遺傳性神經精神異常,其特徵為多種運動抽搐及至少一種發聲抽搐。腿不寧症候群
為一種神經感覺運動障礙,其特徵為在休息時有壓倒性的移動腿之衝動。僵人症候群
為一種進行性運動障礙,其特徵為非自主疼痛痙攣及肌肉僵直,一般涉及下背及腿。通常產生具有加重之腰椎過度凸出的僵腿式步態。通常觀察具有脊椎旁軸肌之連續運動單元活動的EMG記錄上的特徵性異常。變體包括產生通常會影響遠端腿部及腳之病灶性僵硬的「僵肢症候群」。步態障礙
係指步行方式或風格之異常,其起因於神經肌肉變化、關節炎變化或其他身體變化。步態係根據負責用於異常移動之系統而分類,且包括偏癱步態、兩側癱瘓步態、神經病性步態、肌病步態、巴金森式步態、舞蹈病狀步態、運動失調步態及感官步態。情緒障礙
本文亦提供用於治療例如以下之情緒障礙的方法:臨床抑鬱、產後抑鬱或分娩後抑鬱、圍產期抑鬱、非典型抑鬱、憂鬱型抑鬱、精神病性嚴重抑鬱症、緊張型抑鬱、季節性情緒失調症、輕鬱症、雙重抑鬱、抑鬱性人格障礙、復發性短暫抑鬱、輕度抑鬱障礙、躁鬱症或躁狂抑鬱性障礙、由慢性醫學病狀引起之抑鬱、耐治療性抑鬱、難治性抑鬱、自殺傾向、自殺觀念或自殺行為。臨床抑鬱
亦稱為嚴重抑鬱症、重度抑鬱症(MDD)、嚴重抑鬱、單極抑鬱、單極病症及復發性抑鬱,且係指特徵為普遍及持久情緒低落,伴隨有較低自尊心及正常娛樂活動之興趣或快樂感喪失的精神病症。一些患有臨床抑鬱之人入睡困難,體重減輕且一般感到焦躁及易受刺激。臨床抑鬱影響個體之感覺、思想及行為,且可導致許多情感及身體問題。患有臨床抑鬱之個體每天的活動可能困難,且使個體感到生活不值得過。產後抑鬱
(PND)亦稱為分娩後抑鬱 (PPD)
,且係指在分娩之後影響女性之一種類型的臨床抑鬱。症狀可包括悲傷、疲乏、睡眠及飲食習慣變化、性慾減少、哭喊事件、焦慮及易怒。在一些實施例中,PND為耐治療性抑鬱(例如,如本文所描述之耐治療性抑鬱)。在一些實施例中,PND為難治性抑鬱(例如,如本文所描述之難治性抑鬱)。 在一些實施例中,具有PND之個體在懷孕期間亦經歷抑鬱或抑鬱症狀。此抑鬱在本文中被稱作圍產期抑鬱
。在實施例中,經歷圍產期抑鬱之個體經歷PND之風險增加。非典型抑鬱 (AD)
之特徵為情緒反應性(例如反常快感缺乏)及積極性、體重顯著增加或食慾增加。患有AD之患者亦可具有過度睡眠或嗜睡(睡眠過度)、四肢沉重之感覺及由於對所感知人際排斥超敏感而有顯著社會功能損傷。憂鬱型抑鬱
之特徵為在大部分或所有活動中喪失快樂(快感缺乏)、無法對快樂刺激作出反應、比哀傷或喪失更明顯之憂鬱情緒、體重過度減輕或過度內疚。精神病性嚴重抑鬱症 (PMD)
或精神病性抑鬱係指嚴重抑鬱事件,尤其具有憂鬱性,其中個體經歷諸如妄想及幻覺之精神病症狀。緊張型抑鬱
係指涉及運動行為紊亂及其他症狀之嚴重抑鬱症。個體可變得沉默且麻木,且不能動或展現無目的或古怪動作。季節性情緒失調症 (SAD)
係指其中個體具有在秋季或冬季來到之抑鬱事件季節性模式的一種類型的季節性抑鬱。輕鬱症
係指與單極抑鬱相關之病狀,其中相同身體及認知問題顯而易見。其不如單極抑鬱嚴重且傾向於持續較長時間(例如至少2年)。雙重抑鬱
係指持續至少2年之極其憂鬱情緒(輕鬱症),且間雜有嚴重抑鬱症時期。抑鬱性人格障礙 (DPD)
係指具有抑鬱特徵之人格障礙。復發性短暫抑鬱 (RBD)
係指其中個體約每月發生一次抑鬱事件,各事件持續2週或更少時間且通常小於2-3天的病狀。輕度抑鬱障礙
或輕度抑鬱係指其中至少2種症狀存在2週之抑鬱。躁鬱症或躁狂抑鬱性障礙
引起極端情緒波動,包括情緒高漲(躁症或輕躁症)及低落(抑鬱)。在躁症期期間,個體可感到或行為異常歡樂、高能或易受刺激。其常常無法作出考慮周到之決定,對結果幾乎不作考慮。睡眠需求通常減少。在抑鬱期期間,可存在哭喊,眼睛與他人缺乏接觸,且對生活之觀點負面。在患有該病症之患者中自殺風險高,20年間超過6%,同時自殘發生率為30%-40%。諸如焦慮症之其他精神健康問題及物質使用障礙通常與躁鬱症相關。由慢性醫學病狀引起之抑鬱
係指由諸如癌症之慢性醫學病狀或慢性疼痛、化學療法、慢性壓力引起的抑鬱。耐治療性抑鬱
係指個體已針對抑鬱進行治療,但症狀未改善的病狀。舉例而言,抗抑鬱劑或心理諮詢(心理療法)未減輕患有耐治療性抑鬱之個體的抑鬱症狀。在一些情況下,患有耐治療性抑鬱之個體改善症狀,但又恢復。難治性抑鬱
發生在罹患抑鬱之對包括三環抗抑鬱劑、MAOI、SSRI及雙重及三重吸收抑制劑及/或抗焦慮劑藥物之標準藥理學治療以及非藥理學治療(例如心理療法、電驚厥療法、迷走神經刺激及/或經顱磁刺激)具抗性的患者中。自殺傾向、自殺觀念、自殺行為
係指個體自殺之傾向。自殺觀念涉及關於自殺之想法或對自殺異常專注。自殺觀念之範圍變化很大,例如稍縱即逝的念頭至深入的想法、詳細計劃、角色扮演、著手未遂。症狀包括討論自殺、獲得進行自殺之方式、退出社會接觸、一心想著死亡、對某事感到被困或無望、增加酒精或藥物之使用、作出有風險或自毀事情、向人們說再見,好像其不會再見面一樣。 抑鬱症狀
包括持久焦慮或悲傷感、感到無助、無望、悲觀、無價值、低能、坐立不安、入睡困難、不眠、易怒、疲乏、動作攻擊、喪失娛樂活動或愛好之興趣、注意力缺乏、缺乏能量、自尊心差、無積極思想或計劃、睡眠過度、飲食過量、食慾喪失、失眠、自殘、自殺想法及自殺嘗試。症狀之存在、嚴重程度、頻率及持續時間可隨病例而變化。抑鬱症狀及其減輕可藉由醫師或心理學家(例如藉由精神狀態檢查)來確定。焦慮症 本文提供治療焦慮症之方法。焦慮症
為覆蓋異常及病理學恐懼及焦慮之若干不同形式的總稱。當前精神診斷標準辨認各種焦慮症。廣泛性焦慮症
為一種常見慢性病症,其特徵為持久焦慮,不集中於任一個目標或情況。罹患廣泛性焦慮之彼等個體經歷非特定的持久恐懼及擔憂,且變得過度擔心每日事情。廣泛性焦慮症為影響老年人之最常見焦慮症。 在恐慌症
中,個人遭受強烈恐懼及憂懼之短暫發作,特徵常常為戰慄、搖晃、意識模糊、眩暈、噁心、呼吸困難。此等恐慌發作藉由APA定義為突然出現且在不到十分鐘內達到峰值之恐懼或不適,可持續若干小時且可由壓力、害怕或甚至運動觸發;不過具體病因始終不清楚。除復發性意外恐慌發作之外,診斷恐慌症亦需要該等發作具有慢性後果:擔憂發作之潛在影響,持久性害怕將來發作或與發作相關之行為變化顯著。因此,罹患恐慌症之個體經歷甚至在特定恐慌事件外之症狀。常常,恐慌患者注意到心跳之正常變化,導致其認為其心臟出問題或其將再具有恐慌發作。在一些情況下,身體功能之感知加強(警覺過度)在恐慌發作期間發生,其中任何所感知之生理改變均解釋為可能威脅生命之疾病(亦即極端疑病症)。強迫症
為主要特徵為重複困擾(令人苦惱、持久性及侵入性想法或影像)及強迫(進行特定動作或儀式之衝動)之一種類型的焦慮症。OCD思想模式可類似於迷信,到其將信念與實際上不存在之因果關係相連的程度。常常,該過程全部為不合邏輯的;舉例而言,強迫以一定模式步行可用以緩解對即將發生損害之困擾。且在許多情況下,強迫性全部不可解釋,簡單而言,完成由緊張觸發之儀式的衝動。在少數情況下,OCD患者可僅僅經歷困擾,無明顯強迫症;數目少得多之患者僅僅經歷強迫症。 單一最大類別之焦慮症為恐懼症
,其包括由特定刺激或情況觸發害怕及焦慮之所有情況。患者通常由遇到其害怕之目標來預計恐怖後果,其害怕目標可為自動物至體液位置之任何東西。創傷後壓力症
或PTSD
為一種由創傷經歷產生之焦慮症。創傷後壓力可由極端情況引起,諸如打鬥、強姦、人質情況或甚至嚴重事故。其亦可由長期(慢性)暴露於嚴重應激物引起,例如經受個體戰鬥,但無法應付連續打鬥之士兵。常見症狀包括閃回、回避行為及抑鬱。癲癇症
癲癇症為一種特徵為隨時間推移的重複癲癇發作之大腦病症。癲癇症之類型可包括(但不限於)廣泛性癲癇症,例如兒童失神癲癇症、幼年肌陣攣癲癇症、喚醒時癲癇大發作之癲癇症、韋斯特症候群(West syndrome)、倫-加症候群(Lennox-Gastaut syndrome);部分性癲癇症,例如兒童之顳葉性癲癇症、額葉性癲癇症、良性病灶性癲癇症。癲癇形成
癲癇形成為正常大腦產生癲癇症的漸進過程(癲癇發作發生之慢性病狀)。癲癇形成因藉由初始損傷促成之神經元損害而引起(例如持續性癲癇)。持續性癲癇 (SE)
持續性癲癇(SE)可包括例如痙攣性持續性癲癇,例如早期持續性癲癇、現有持續性癲癇、難治性持續性癲癇、超難治性持續性癲癇;非痙攣性持續性癲癇,例如廣泛性持續性癲癇、複雜部分持續性癲癇;廣泛性週期性癲癇樣放電;及週期性偏側性癲癇樣放電。痙攣性持續性癲癇之特徵為存在痙攣性持續性癲癇發作,且可包括早期持續性癲癇、現有持續性癲癇、難治性持續性癲癇、超難治性持續性癲癇。早期持續性癲癇用一線療法治療。現有持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法治療其仍持續且投與二線療法。難治性持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法及二線療法治療其仍持續且通常投與一般麻醉劑。超難治性持續性癲癇之特徵為持續性癲癇發作,儘管用一線療法、二線療法及一般麻醉劑治療24小時或24小時以上,其仍持續。 非痙攣性持續性癲癇可包括例如病灶性非痙攣性持續性癲癇,例如複雜部分非痙攣性持續性癲癇、簡單部分非痙攣性持續性癲癇、隱微非痙攣性持續性癲癇;廣泛性非痙攣性持續性癲癇,例如晚發性失神非痙攣性持續性癲癇、非典型失神非痙攣性持續性癲癇或典型失神非痙攣性持續性癲癇。癲癇發作
癲癇發作為大腦中異常電活動事件後發生之生理發現或行為改變。術語「癲癇發作」通常可與「痙攣」互換使用。痙攣為當人的身體快速且不受控制地搖晃時。在痙攣期間,人的肌肉反覆收縮及放鬆。 基於行為及大腦活動之類型,將癲癇發作分成兩種較寬廣類別:廣泛性及部分(亦稱為局部或病灶性)。將癲癇發作類型進行分類有助於醫生診斷患者是否患有癲癇症。 廣泛性癲癇由整個大腦中之電脈衝產生,而部分癲癇由相對較小部分大腦中之電脈衝產生(至少起初)。產生癲癇之大腦部分有時稱為病灶。 存在六種類型之廣泛性癲癇。最常見且嚴重的且因此最熟知的為廣泛性痙攣,亦稱為大發作癲癇。在此類型癲癇中,患者失去意識且通常虛脫。失去意識後廣泛性身體僵硬(稱為癲癇之「強直性」階段)30 至60秒,隨後劇烈抽動(「陣攣性」階段)30至60秒,其後患者進入深度睡眠(「發作後」或癲癇後階段)。在大發作癲癇期間,可能發生損傷及事故,諸如咬舌及尿失禁。 失神癲癇導致短暫意識喪失(僅幾秒),幾乎無症狀。患者(最常為兒童)通常中斷活動且目直。此等癲癇突然開始及結束,且可一日發生若干次。患者通常未意識到其癲癇發作,但其可能意識到「時間流失」。 肌陣攣癲癇由偶發性急抽組成,通常在身體兩側。患者有時將急抽描述為短暫電擊。劇烈時,此等癲癇可能導致物體掉落或無意地投擲物體。 陣攣性癲癇為重複的節律性急抽,其同時涉及身體兩側。 強直性癲癇之特徵為肌肉僵硬。 失張性癲癇由尤其在臂及腿中,肌肉張力之突然及一般損失組成,其通常導致跌倒。 本文所描述之癲癇可包括癲癇發作;急性重複癲癇;密集式癲癇;連續癲癇;無間斷癲癇;長期癲癇;反覆性癲癇;持續性癲癇發作,例如難治性痙攣性持續性癲癇、非痙攣性持續性癲癇發作;難治性癲癇;肌陣攣性癲癇;強直性癲癇;強直性陣攣性癲癇;簡單部分癲癇;複雜部分癲癇;繼發廣泛性癲癇;非典型失神癲癇;失神癲癇;失張性癲癇;良性羅蘭多(Rolandic)癲癇;發熱性癲癇;情緒性癲癇;病灶性癲癇;癡笑性癲癇;廣泛性發作癲癇;嬰兒痙攣;傑克遜氏癲癇(Jacksonian seizures);大規模雙側肌陣攣癲癇;多灶性癲癇;新生兒發作癲癇;夜間癲癇;枕葉性癲癇;創傷後癲癇;微小癲癇;西爾萬癲癇(Sylvan seizures);視覺反射癲癇或撤藥癲癇。在一些實施例中,癲癇為與德拉韋症候群、倫-加症候群、結節性硬化症、雷特症候群或PCDH19女嬰兒癲癇症相關之廣泛性癲癇。縮寫
PCC:氯鉻酸吡啶;t-BuOK:第三丁醇鉀;9-BBN:9-硼雙環[3.3.1]壬烷;Pd(t
-Bu3
P)2
:雙(三-第三丁基膦)鈀(0);AcCl:氯化乙醯基;i
-PrMgCl:異丙基氯化鎂;TBSCl:第三丁基(氯)二甲基矽烷;(i
-PrO)4
Ti:四異丙醇鈦;BHT:2,6-二-第三丁基-4-甲基苯酚;Me:甲基;i
-Pr:異丙基;t
-Bu:第三丁基;Ph:苯基;Et:乙基;Bz:苯甲醯基;BzCl:氯化苯甲醯;CsF:氟化銫;DCC:二環己基碳化二亞胺;DCM:二氯甲烷;DMAP:4-二甲胺基吡啶;DMP:戴斯-馬丁高碘烷;EtMgBr:乙基溴化鎂;EtOAc:乙酸乙酯;TEA:三乙胺;AlaOH:丙胺酸;Boc:第三丁氧羰基。Py:吡啶;TBAF:氟化四正丁基銨;THF:四氫呋喃;TBS:第三丁基二甲基矽烷基;TMS:三甲基矽烷基;TMSCF3
:(三氟甲基)三甲基矽烷;Ts:對甲苯磺醯基;Bu:丁基;Ti(OiPr)4
:四異丙氧基鈦;LAH:氫化鋰鋁;LDA:二異丙胺基鋰;LiOH.H2
O:氫氧化鋰水合物;MAD:甲基鋁雙(2,6-二-第三丁基-4-甲基苯酚);MeCN:乙腈;NBS:N-溴代丁二醯亞胺;Na2
SO4
:硫酸鈉;Na2
S2
O3
:硫代硫酸鈉;PE:石油醚;MeCN:乙腈;MeOH:甲醇;Boc:第三丁氧羰基;MTBE:甲基第三丁基醚;DIAD:偶氮二甲酸二異丙酯。實例
為了能更全面地理解本文中所描述之發明,闡述以下實例。提供本申請案中所描述之合成及生物實例以說明本文提供之化合物、醫藥組合物及方法,且並不理解為以任何方式限制其範疇。在以下合成實例中,按序號列出反應序列內之實驗程序之描述。物質及方法
本文提供之化合物可使用以下通用方法及程序自容易獲得之起始物質製備。應瞭解,除非另行說明,否則在給定典型或較佳製程條件(亦即,反應溫度、時間、反應物之莫耳比、溶劑、壓力等)的情況下,亦可使用其他製程條件。最佳反應條件可隨所用特定反應物或溶劑而變,但此類條件可由熟習此項技術者藉由常規最佳化來確定。 此外,如熟習此項技術者將顯而易知,可能必需習知保護基來防止某些官能基經歷非所需反應。對於特定官能團適合的保護基以及保護及去保護之適合的條件之選擇為此項技術中熟知的。舉例而言,諸多保護基及其引入及移除描述於T. W. Greene及P. G. M. Wuts,Protecting Groups in Organic Synthesis
,第二版, Wiley, New York, 1991及其中所引用之參考文獻中。 本文提供之化合物可藉由已知標準程序分離及純化。此類程序包括(但不限於)再結晶、管柱層析、HPLC或超臨界流體層析(SFC)。關於製備本文中已列出之代表性吡唑的細節呈現以下流程。本文提供之化合物可由熟習有機合成技術者自已知或市售起始物質及試劑製備。可用於分離/純化本文提供之對映異構體/非對映異構體之例示性對掌性管柱包括(但不限於) CHIRALPAK® AD-10。CHIRALCEL® OB、CHIRALCEL® OB-H、CHIRALCEL® OD、CHIRALCEL® OD-H、CHIRALCEL® OF、CHIRALCEL® OG、CHIRALCEL® OJ及CHIRALCEL® OK。 製備型HPLC之例示性通用方法:管柱:Waters Rbridge製備型10 μm C18,19 × 250 mm。移動相:乙腈、水(NH4
HCO3
) (30 L水、24 g NH4
HCO3
、30 mL NH3
.H2
O)。流速:25 mL/min。 分析型HPLC之例示性通用方法:移動相:A:水 (10 mM NH4
HCO3
),B:乙腈梯度:在1.6或2 min內,5%-95% B;流速:1.8或2 mL/min;管柱:XBridge C18,4.6×50mm,在45℃為3.5 μm。NMDA 調節
使用自動箝膜系統來評定表現NMDA受體之哺乳動物細胞中之NMDA增強,該自動箝膜系統可用於測定如下文所描述之化合物之NAM活性。全細胞箝膜系統可用於測定如下文所描述之化合物之PAM活性。自動化箝膜系統 (QPatch HTX) :
在此研究中,經GRIN1/2A次型之麩胺酸激活通道穩定轉染之HEK 293細胞將與次極大NMDA濃度(300 μM NMDA,與8 μM甘胺酸共施加)一起用來研究測試化合物之負向異位調節。細胞培養物
大體而言,細胞將以約80%至90%之會合率傳代。對於電生理量測,將以約80%至90%之會合率自含有完全培養基之無菌培養瓶收集細胞。細胞將作為PBS中之懸浮液傳送至QPatch 16X或QPatch HTX系統,直接傳送至離心機/洗滌器。 標準實驗室條件:細胞將在具有5% CO2
之潮濕氛圍(相對濕度約95%)中在37℃下培育。培養基
:細胞將在含有補充有10%胎牛血清、1%青黴素/鏈黴素溶液及50 μM AP-5阻斷劑之達爾伯克氏改良伊格爾培養基與營養混合物F-12 (D-MEM/F-12 1×,液體與L-麩醯胺酸)之1:1混合物的無菌培養瓶中不斷保持及傳代。抗生素
:如上所指出之完全培養基補充有100 μg/mL潮黴素、15 μg/mL殺稻瘟菌素及1 μg/mL嘌呤黴素。表現之誘導
:在開始實驗之前24 h添加2.5 μg/mL四環素。劑量調配
劑量係就供應時之測試化合物而言。將添加媒劑以達成10 mM之儲備濃度(在-10℃至-30℃下儲存)。將在DMSO中製備另外的1.0 mM之儲備溶液。儲備溶液利用之細節(解凍、劑量調配)將記錄在原始資料中。儲備溶液利用之時段將詳述於報告中。測試化合物濃度
劑量係就供應時之測試化合物而言。將添加媒劑以達成10 mM之儲備濃度(在-10℃至-30℃下儲存)。將在DMSO中製備另外的1.0 mM之儲備溶液。儲備溶液利用之細節(解凍、劑量調配)將記錄在原始資料中。儲備溶液利用之時段將詳述於報告中。 將測試一個測試濃度1.0 μM。 將藉由在電生理實驗前不久僅用不含Mg之電解液或含有NMDA (300 µM)及甘胺酸(8.0 µM)的不含Mg之電解液稀釋儲備溶液來製備所有測試溶液,且在使用時將其保持在室溫(19℃至30℃)下。0.1%DMSO將被用作媒劑。製備頻率
:對於各測試濃度,每日將製備測試化合物之新鮮溶液。劑量調配之穩定性
:所有製備時間將記錄在原始資料中。關於測試化合物之不穩定性之任何觀察結果將在原始資料中提及。劑量調配物之儲存
:在實驗之日,劑量調配物在使用時將維持在室溫(19℃至30℃)下。電解液
為了準備實驗及為了形成千兆歐姆密封,將使用以下標準電解液 :
氯化鈉:137 mM;氯化鉀:4 mM;氯化鈣:1.8 mM;氯化鎂:1 mM;HEPES:10 mM;D-葡糖:10 mM;十六醇聚氧乙烯醚:0.02%;pH (NaOH):7.4 將藉由至少每7天用水稀釋不含葡糖之10×電解液及100×葡糖溶液來製備1×電解液。在當前研究之實驗開始之前已經製備兩種儲備溶液且將其儲存在1℃至9℃下(10×電解液)或-10℃至-30℃下(100×葡糖溶液)。實驗中所使用之電解液之批號將記錄在原始資料中。當使用時,1×電解液將保持在室溫(19℃至30℃)下。當未使用時,1×電解液將儲存於1℃至9℃下。 在形成千兆密封後,將使用以下不含 Mg 的電解液
: 氯化鈉:137 mM;氯化鉀:4 mM;氯化鈣:2.8 mM;HEPES:10 mM;D-葡糖:10 mM;十六醇聚氧乙烯醚
:0.02%;pH (NaOH):7.4 此不含Mg的電解液將製備為1×溶液且儲存於1℃至9℃下。其將至少每10天新鮮製備。胞內溶液
1×胞內溶液將每日自冷凍的1×胞內溶液解凍,其已在當前研究之實驗開始之前製備、取等分及儲存於-10℃至-30℃下。當使用時,1×胞內溶液將保持在室溫(19℃至30℃)下。剩餘的1×胞內溶液將儲存於冰箱(1℃至9℃)中。1×胞內溶液將包括下文概述之組分: 氯化鉀:130 mM;氯化鎂:1 mM;Mg-ATP:5 mM;HEPES:10 mM;EGTA:5 mM;pH (KOH):7.2細胞處理
對於此研究,細胞將不斷灌注有NMDA/甘胺酸、測試化合物或測試化合物/NMDA/甘胺酸。 在各情況下,將在施加之間進行利用測試化合物之至少30秒預洗滌步驟。細節請參見下表A。 將在至少n=3個分離細胞中分析各實驗類型。將在當前研究之實驗開始之前製備NMDA及甘胺酸儲備溶液,冷凍儲存(-10℃至-30℃),直至實驗當天為止。在電生理實驗前不久,冷凍儲備溶液將經解凍及稀釋。 對照:每兩週將在三個細胞處量測媒劑(0.1% DMSO)及D-(-)-2-胺基-5-膦醯基戊酸(AP-5) (100 μM)之效果,以便確保NMDA受體之成功表現。 已在當前研究之實驗開始之前製備AP-5之50 mM儲備溶液,取等分試樣且冷凍儲存(-10℃至-30℃),直至實驗當天為止。在電生理實驗前不久,冷凍儲備溶液將經解凍且接著在含有NMDA (300 μM)及甘胺酸(8.0 μM)之不含Mg的電解液中稀釋,得到最終灌注濃度100 µM。實驗程序
細胞作為無血清培養基中之懸浮液傳送至QPatch HTX系統且在實驗期間保持在細胞儲槽/攪拌器中。施加至細胞之所有溶液(包括胞內溶液)將維持在室溫下(19℃至30℃)下。 在密封製程期間,將使用上文所描述之標準電解液
。施加至細胞之所有溶液(包括移液管溶液)將維持在室溫下(19℃至30℃)下。在膜片電極與經轉染個別HEK293細胞之間形成千兆歐姆密封之後,將僅灌注不含 Mg 的電解液
且細胞膜將會破裂以確保電氣進入細胞內部(全細胞膜片組態)。將在向膜片鉗位細胞施加300 µM NMDA (及8.0 µM甘胺酸)後量測內向電流5秒。在整個實驗期間,細胞將在-80 mV之保持電位下經電壓鉗位。 為了分析測試化合物,NMDA受體將由下文所描述之300 µM NMDA及8.0 µM甘胺酸及測試化合物組合刺激。將在施加之間進行利用測試化合物之三十二個預洗滌步驟。 表A:施加協議;測試化合物之使用依賴性

 1.在0℃下向TBAF (3.04 mL,1 M於THF中,3.04 mmol,Aldrich)於THF (100 mL)中之溶液,接著向S-200-INT-2
(19 g,60.8 mmol)於THF (100 mL)中之溶液中逐滴添加TMSCF3
(25.8 g,182 mmol)。在0℃下攪拌混合物30 min。在0℃下向混合物添加TBAF (200 mL,1 M於THF中,200 mmol,國內的)。在0℃下再攪拌混合物30 min。向混合物添加NH4
Cl (100 mL,飽和水溶液)。真空濃縮混合物。向殘餘物添加PE/EtOAc (400 mL,1:1),分離有機層,其與其他兩個批次(2×10 gS200-INT-2
)合併。合併之有機層用水(300 mL)、鹽水(300 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到油狀物。使殘餘物溶解於DCM (150 mL)中且用PE (750 mL)稀釋。將溶液倒入矽膠管柱(500 g,100至200目)且用PE:DCM:EtOAc=5:1:0.05至5:1:0.1溶離,得到呈油狀物之S200-CF3_1B
(12 g,70%純度,17%產量)及不純S200-CF3_1A
。該不純自MeCN (250 mL)再結晶,得到呈固體之S200-CF3_1A
(6.5 g)。自MeCN過濾藉由矽膠管柱(PE:DCM:EtOAc=50:1:1至20:1:1)純化得到粗產物,該粗產物自MeCN (20 mL)再結晶,得到呈固體之S-200-CF3_1A
(1 g,16%總產量)。注意 :
自 3 JH,CF3
(FDCS)鑑別200-CF3_1A
及200-CF3_1B
。(J. Org. Chem. 2015, 80, 1754
.S-200-CF3_1A: 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.33 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H); 2.49 (s, 2H); 2.11-1.97 (m, 4H), 1.95-1.32 (m, 14H), 1.30-0.98 (m, 7H), 0.59 (s, 3H)。S-200-CF3_1B: 1 H NMR
(400 MHz, CDCl3
)δ 5.54-5.41 (m, 1H), 4.86 (s, 1H), 4.72 (s, 1H); 2.78-2.65 (m, 1H); 2.18-1.97 (m, 3H), 1.95-1.35 (m, 16H), 1.32-0.98 (m, 7H), 0.59 (s, 3H)。 2.向S-200-CF3_1A
(8 g,20.9 mmol)於THF (80 mL)中之溶液中添加9-BBN二聚體(5.85 g,24 mmol)。在40℃下攪拌混合物1 h。將混合物冷卻至0℃。向該混合物逐滴添加EtOH (12 mL)、NaOH (41.8 mL,5 M水溶液)及H2
O2
(20.9 mL,10 M水溶液)。在50℃下攪拌混合物1 h。在冷卻後向混合物添加Na2
SO3
(100 mL,25%,水溶液)。用EtOAc (300 mL)萃取混合物。有機層經分離且藉由矽膠管柱(PE:EtOAc=10:1至5:1)純化,得到呈固體之S-200-CF3_2A
(7.1 g,85%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.42-5.32 (m, 1H), 3.64 (dd,J
= 3.2, 10.4 Hz, 1H), 3.37 (dd,J
= 6.8, 10.4 Hz, 1H), 2.49 (s, 2H), 2.32-1.92 (m, 4H), 1.92-1.70 (m, 4H), 1.70-1.29 (m, 8H), 1.29-0.91 (m, 11H), 0.71 (s, 3H)。 3.在25℃下向S-200-CF3_5A
(3g,7.49 mmol)於DCM (50 mL)中之溶液中添加DMP (6.31 g,14.9 mmol),在25℃下攪拌30 min之後,反應混合物用飽和NaHCO3
(100 mL)淬滅且添加DCM (100 mL)且攪拌10 min。DCM相經分離且用Na2
S2
O3
飽和水溶液(2×100 mL)洗滌。合併之有機層用飽和鹽水(2×100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(5%至20% EtOAc/PE)純化,得到呈固體之N-004-027_1
(1.5 g,50%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.58-9.55 (m, 1H), 5.38-5.36 (m, 1H), 2.49 (s, 1H), 2.40-2.25 (m, 1H), 2.23-1.60 (m, 10H), 1.53-1.20 (m, 9H), 1.15-1.00 (m, 7H), 0.78-0.64 (m, 3H)。 4.在0℃下向N-004-027_1
(1.5 g,3.76 mmol)於無水THF (40 mL)中之溶液中添加CsF (1.42 g,9.40 mmol)。在0℃下攪拌20 min之後,在0℃下添加TMSCF3
(1.33 g,9.40 mmol)且攪拌30 min。顏色變成淡黃色。添加TBAF.3H2
O (4.74 g,15.0 mmol)且在50℃下攪拌30 min。將反應混合物倒入冰水(100 mL)中。用EtOAc (2×100 mL)萃取水相。合併之有機相用飽和鹽水(2×100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈黃色固體之異構體混合物(1.45 g,粗產物),其藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之53
(340 mg,24%)及呈白色固體之1
(200 mg,14%)。1: 1 H NMR
(400 MHz, CDCl3
) δ 5.38-5.36 (m, 1H), 4.10-4.00 (m, 1H), 2.49 (s, 2H), 2.19-2.12 (m, 1H), 2.06-1.61 (m, 10H), 1.53-1.29 (m, 6H), 1.27-0.98 (m, 10H), 0.71 (s, 3H)。 在2 min層析中,LCMS
Rt=1.121 min,30-90AB_2MIN_E,純度100%,MS
50-100_1_4min.m,C24
H33
F6
O[M+H-H2
O]+
451,實驗值451。1: 1 H NMR
(400 MHz, CDCl3
) δ 5.38-5.36 (m, 1H), 4.10-4.00 (m, 1H), 2.49 (s, 2H), 2.19-2.12 (m, 1H), 2.06-1.61 (m, 10H), 1.53-1.29 (m, 6H), 1.27-0.98 (m, 10H), 0.71 (s, 3H)。 在2 min層析中,LCMS
Rt=1.121 min,30-90AB_2MIN_E,純度100%,MS
50-100_1_4min.m,C24
H33
F6
O[M+H-H2
O]+
451,實驗值451。實例 2 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (2) 之合成
1.在0℃下向TBAF (3.04 mL,1 M於THF中,3.04 mmol,Aldrich)於THF (100 mL)中之溶液,接著向S-200-INT-2
(19 g,60.8 mmol)於THF (100 mL)中之溶液中逐滴添加TMSCF3
(25.8 g,182 mmol)。在0℃下攪拌混合物30 min。在0℃下向混合物添加TBAF (200 mL,1 M於THF中,200 mmol,國內的)。在0℃下再攪拌混合物30 min。向混合物添加NH4
Cl (100 mL,飽和水溶液)。真空濃縮混合物。向殘餘物添加PE/EtOAc (400 mL,1:1),分離有機層,其與其他兩個批次(2×10 gS200-INT-2
)合併。合併之有機層用水(300 mL)、鹽水(300 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到油狀物。使殘餘物溶解於DCM (150 mL)中且用PE (750 mL)稀釋。將溶液倒入矽膠管柱(500 g,100至200目)且用PE:DCM:EtOAc=5:1:0.05至5:1:0.1溶離,得到呈油狀物之S200-CF3_1B
(12 g,70%純度,17%產量)及不純S200-CF3_1A
。該不純自MeCN (250 mL)再結晶,得到呈固體之S200-CF3_1A
(6.5 g)。自MeCN過濾藉由矽膠管柱(PE:DCM:EtOAc=50:1:1至20:1:1)純化得到粗產物,該粗產物自MeCN (20 mL)再結晶,得到呈固體之S-200-CF3_1A
(1 g,16%總產量)。注意 :
自 3 JH,CF3
(FDCS)鑑別200-CF3_1A
及200-CF3_1B
。(J. Org. Chem. 2015, 80, 1754
.S-200-CF3_1A: 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.33 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H); 2.49 (s, 2H); 2.11-1.97 (m, 4H), 1.95-1.32 (m, 14H), 1.30-0.98 (m, 7H), 0.59 (s, 3H)。S-200-CF3_1B: 1 H NMR
(400 MHz, CDCl3
)δ 5.54-5.41 (m, 1H), 4.86 (s, 1H), 4.72 (s, 1H); 2.78-2.65 (m, 1H); 2.18-1.97 (m, 3H), 1.95-1.35 (m, 16H), 1.32-0.98 (m, 7H), 0.59 (s, 3H)。 2.向S-200-CF3_1A
(8 g,20.9 mmol)於THF (80 mL)中之溶液中添加9-BBN二聚體(5.85 g,24 mmol)。在40℃下攪拌混合物1 h。將混合物冷卻至0℃。向該混合物逐滴添加EtOH (12 mL)、NaOH (41.8 mL,5 M水溶液)及H2
O2
(20.9 mL,10 M水溶液)。在50℃下攪拌混合物1 h。在冷卻後向混合物添加Na2
SO3
(100 mL,25%,水溶液)。用EtOAc (300 mL)萃取混合物。有機層經分離且藉由矽膠管柱(PE:EtOAc=10:1至5:1)純化,得到呈固體之S-200-CF3_2A
(7.1 g,85%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.42-5.32 (m, 1H), 3.64 (dd,J
= 3.2, 10.4 Hz, 1H), 3.37 (dd,J
= 6.8, 10.4 Hz, 1H), 2.49 (s, 2H), 2.32-1.92 (m, 4H), 1.92-1.70 (m, 4H), 1.70-1.29 (m, 8H), 1.29-0.91 (m, 11H), 0.71 (s, 3H)。 3.在25℃下向S-200-CF3_5A
(3g,7.49 mmol)於DCM (50 mL)中之溶液中添加DMP (6.31 g,14.9 mmol),在25℃下攪拌30 min之後,反應混合物用飽和NaHCO3
(100 mL)淬滅且添加DCM (100 mL)且攪拌10 min。DCM相經分離且用Na2
S2
O3
飽和水溶液(2×100 mL)洗滌。合併之有機層用飽和鹽水(2×100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(5%至20% EtOAc/PE)純化,得到呈固體之N-004-027_1
(1.5 g,50%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.58-9.55 (m, 1H), 5.38-5.36 (m, 1H), 2.49 (s, 1H), 2.40-2.25 (m, 1H), 2.23-1.60 (m, 10H), 1.53-1.20 (m, 9H), 1.15-1.00 (m, 7H), 0.78-0.64 (m, 3H)。 4.在0℃下向N-004-027_1
(1.5 g,3.76 mmol)於無水THF (40 mL)中之溶液中添加CsF (1.42 g,9.40 mmol)。在0℃下攪拌20 min之後,在0℃下添加TMSCF3
(1.33 g,9.40 mmol)且攪拌30 min。顏色變成淡黃色。添加TBAF.3H2
O (4.74 g,15.0 mmol)且在50℃下攪拌30 min。將反應混合物倒入冰水(100 mL)中。用EtOAc (2×100 mL)萃取水相。合併之有機相用飽和鹽水(2×100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈黃色固體之異構體混合物(1.45 g,粗產物),其藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之53
(340 mg,24%)及呈白色固體之1
(200 mg,14%)。1: 1 H NMR
(400 MHz, CDCl3
) δ 5.38-5.36 (m, 1H), 4.10-4.00 (m, 1H), 2.49 (s, 2H), 2.19-2.12 (m, 1H), 2.06-1.61 (m, 10H), 1.53-1.29 (m, 6H), 1.27-0.98 (m, 10H), 0.71 (s, 3H)。 在2 min層析中,LCMS
Rt=1.121 min,30-90AB_2MIN_E,純度100%,MS
50-100_1_4min.m,C24
H33
F6
O[M+H-H2
O]+
451,實驗值451。1: 1 H NMR
(400 MHz, CDCl3
) δ 5.38-5.36 (m, 1H), 4.10-4.00 (m, 1H), 2.49 (s, 2H), 2.19-2.12 (m, 1H), 2.06-1.61 (m, 10H), 1.53-1.29 (m, 6H), 1.27-0.98 (m, 10H), 0.71 (s, 3H)。 在2 min層析中,LCMS
Rt=1.121 min,30-90AB_2MIN_E,純度100%,MS
50-100_1_4min.m,C24
H33
F6
O[M+H-H2
O]+
451,實驗值451。實例 2 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (2) 之合成 
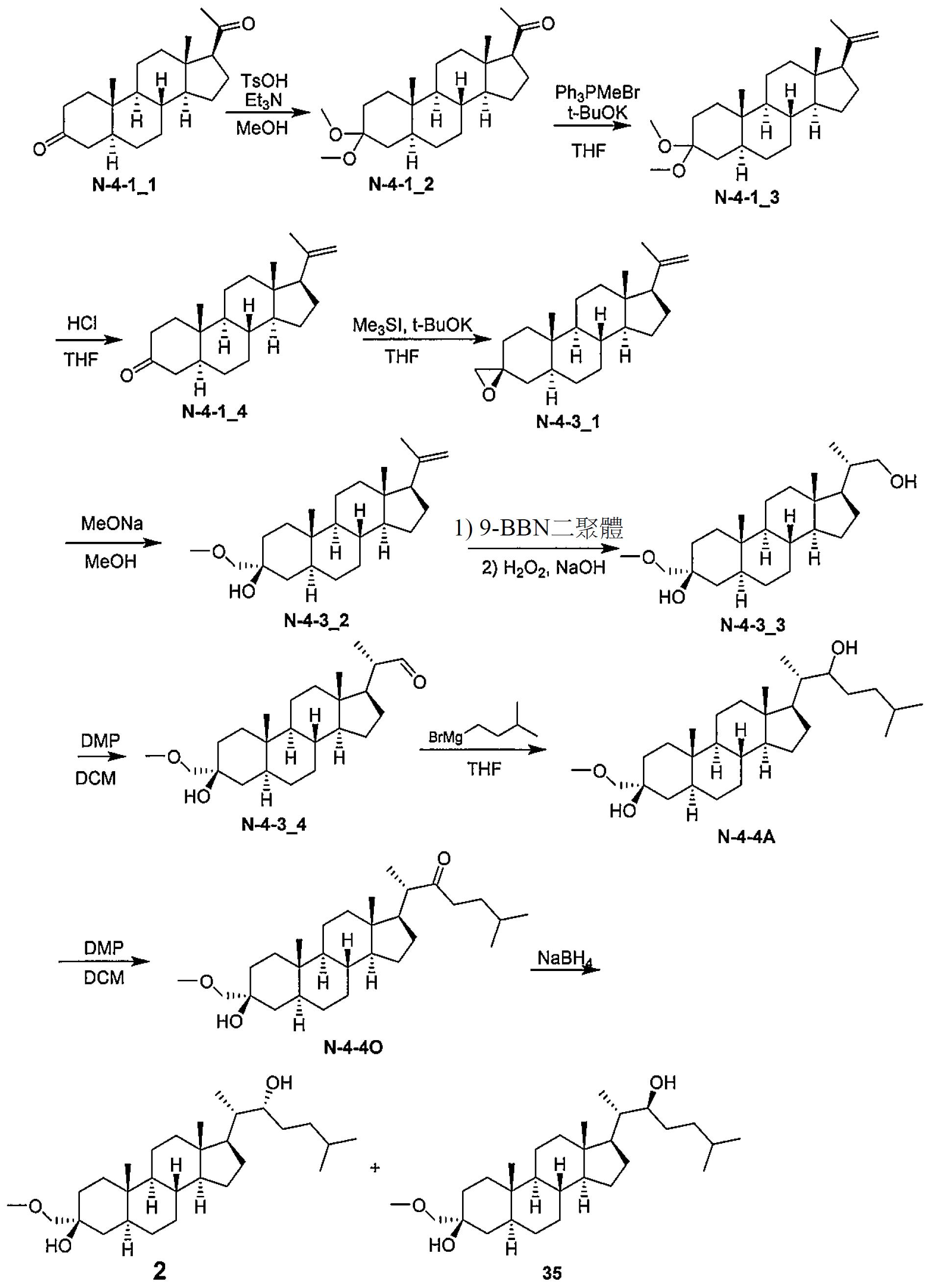 1.在20℃下向N-4-4_1
(50 g,157 mmol)於無水甲醇(500 mL)中之懸浮液一次性添加無水TsOH (2.84 g,15.7 mmol)。使混合物升溫直至60℃且攪拌1 h。反應混合物用Et3
N (1.58 g,15.7 mmol)淬滅且攪拌另外30 min。濾出沈澱之固體,用甲醇(250 mL)洗滌且於空氣中乾燥,得到呈固體之N-4-1_2
(51 g,90%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.18 (s, 3H), 3.14 (s, 3H); 2.54-2.48 (m, 1H); 2.10-2.00 (m, 4H); 1.95-1.75 (m, 2H), 1.65-1.50 (m, 7H), 1.48-0.80 (m, 11H), 0.78-0.75 (m, 4H), 0.59 (s, 3H)。 2.在20℃下於N2
下向Ph3
PMeBr (75 g,210 mmol)於無水THF (500 mL)中之懸浮液逐份添加t-BuOK (23.5 g,210 mmol)。混合物變成深橙色且在20℃下攪拌30 min。隨後添加N-4-1_2
(51 g,140 mmol)。使混合物升溫至40℃且攪拌1 h。將反應混合物冷卻且逐份倒入NH4
Cl水溶液(冰) (400mL)中。分離所得混合物;用THF (200 mL)萃取含水層。合併之有機層直接用作N-4-1_3
之溶液而無需進一步純化。 3.在20℃下向N-4-1_3
(50.4 g,139 mmol)於THF (700 mL)中之溶液中添加HCl水溶液(1 M,208 mL,208 mmol)。在20℃下攪拌混合物1小時,且固體沈澱。將水(200 mL)添加至混合物中,且濾出沈澱之固體,用水洗滌且乾燥,得到呈固體之N-4-1_4
(41 g,94%)。 1 H NMR
(400 MHz, CDCl3
)δ 4.85 (s, 1H), 4.70 (s, 1H); 2.38-2.25 (m, 3H); 2.10-1.98 (m, 3H), 1.88-1.49 (m, 10H), 1.40-1.08 (m, 11H), 0.97-0.72 (m, 2H), 0.58 (s, 3H)。 4.在25℃下於N2
下向Me3
SI (101 g,496 mmol)於無水THF (400 mL)中之溶液中逐份添加t-BuOK (58.3 g,520 mmol)且攪拌30 min。添加N-4-1_4
(39 g,124 mmol)於無水THF (300 mL)中之溶液。使反應混合物升溫至50℃且攪拌2小時。將反應混合物冷卻至25℃且用NH4
Cl水溶液(500 mL)處理。用EtOAc (2×500 mL))萃取水相。合併之有機相用鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至10/1)純化,得到呈固體之N-4-3_1
(35 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 4.84 (s, 1H), 4.70 (s, 1H); 2.65-2.55 (m, 2H); 2.10-1.98 (m, 2H), 1.92-1.49 (m, 13H), 1.40-1.13 (m, 8H), 0.99-0.69 (m, 6H), 0.57 (s, 3H)。 5.在25℃下向N-4-3_1
(35 g,647 mmol)於無水MeOH (500 mL)中之溶液中添加MeONa (57.2 g,1.06 mol)且在N2
下攪拌混合物30 min。使反應混合物升溫至70℃且於N2
下攪拌回流3小時。將反應混合物冷卻至25℃且用水(500 mL)處理。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc=10/1至6/1)純化,得到呈固體之N-4-3_2
(25 g,不純)。粗產物在25℃下自PE (250 mL)濕磨1 h。過濾懸浮液且真空乾燥濾餅以獲得呈固體之N-4-3_2
(15 g,25%)。 1 H NMR
(400 MHz, CDCl3
)δ 4.86 (s, 1H), 4.72 (s, 1H), 3.46-3.37 (m, 5H), 2.54 (s, 1H), 2.07-1.99 (m, 1H), 1.89-1.52 (m, 15H), 1.41-1.06 (m, 10H), 0.86 (s, 3H); 0.58 (s, 3H) 6.在0℃下向N-4-3_2
(15 g,41.6 mmol)於無水THF (200 mL)中之溶液中添加9-BBN二聚體(27.7 g,124 mmol)且於N2
下攪拌30 min。使反應混合物升溫至50℃且攪拌1 h。將反應混合物冷卻至0℃且添加EtOH (50 mL),隨後在0℃下極緩慢地添加NaOH (41.6 mL,5M,208 mmol)。緩慢地添加H2
O2
(23.5 g,208 mmol,30%於水中)同時使內部溫度保持在10℃以下。使混合物升溫至50℃且再攪拌1小時。將反應混合物冷卻,逐份倒入冰水(500 mL)中且過濾。真空濃縮濾液以提供呈油狀物之N-4-3_3
(14 g,粗產物)。粗殘餘物直接用於下一步驟。 7.在25℃下將DMP (3.35 g,7.92 mmol)添加至N-4_3
(1 g,2.64 mmol)於DCM (20 mL)中之混合物中。使反應混合物升溫至40℃且攪拌1 h。在pH 7至8及10℃以下用NaHCO3
飽和水溶液淬滅反應混合物。過濾懸浮液。濾液中之DCM相經分離且用NaHCO3
/Na2
S2
O3
飽和水溶液(1:1,2×50 mL)、鹽水(2×50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮以獲得固體。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之N-4-3_4
(0.6 g,60%)。 1 H NMR
(400 MHz, CDCl3
)δ 9.57 (s, 1H), 3.40-3.34 (m, 5H); 2.38-2.28 (m, 1H); 1.94-1.76 (m, 2H), 1.74-1.35 (m, 16H), 1.06-0.82 (m, 10H), 0.73-0.64 (m, 5H)。 8.在0℃下於N2
下向N-4-3_4
(0.6 g,1.59 mmol)於無水THF (10 mL)中之溶液中添加異戊基溴化鎂(4.37 mL,8.74 mmol,2 M於二乙醚中)。使反應混合物升溫至25℃且攪拌1小時。向反應混合物中添加NH4
Cl 飽和水溶液(50 mL)。用EtOAc (3×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-4-4A
(0.5g,粗產物)。 1 H NMR
(400 MHz, CDCl3
)δ 3.64-3.60 (m, 1H), 3.40-3.37 (m, 5H); 2.02-1.79 (m, 3H); 1.75-1.50 (m, 11H), 1.25-1.10 (m, 14H), 0.99-0.75 (m, 14H), 0.70-0.64 (m, 4H)。 9.在25℃下向N-4-4A
(0.5 g,粗產物)於DCM (20 mL)中之溶液中添加DMP (1.88 g,4.44 mmol)。使反應混合物升溫至40℃且攪拌1小時。在pH 7至8及10℃以下用NaHCO3
飽和水溶液淬滅反應混合物。過濾懸浮液。DCM相經分離且用NaHCO3
/Na2
S2
O3
飽和水溶液(1:1,2×50 mL)、鹽水(2×50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-4-4O
(0.4 g,粗產物),其直接用於下一步驟。 在25℃下向N-4-4O
(0.4 g,0.895 mmol)於MeOH (4 mL)中之溶液中緩慢地添加NaBH4
(0.340 g,8.95 mmol)且攪拌2小時。用DCM (2 × 20 mL)萃取水相。合併之有機相用飽和鹽水(2×20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc=8/1至5/1)純化,得到呈固體之35
(150 mg,不純)及2
(130 mg,不純)。在82o
C下回流1小時自MeCN (3 mL)再結晶2
(130 mg,不純)。攪拌混合物且冷卻至25℃。過濾懸浮液且真空濃縮濾液以提供呈固體之2
(50 mg,12%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.63-3.61 (m, 1H), 3.41-3.38 (m, 5H); 2.51 (s, 1H); 1.97-1.81 (m, 2H), 1.71-1.31 (m, 15H), 1.26-1.03 (m, 10H), 0.97-0.78 (m, 14H), 0.71-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.350 min,30-90 AB,純度99%,C29
H48
O [M+H-2H2
O]+
之MS ESI計算值413,實驗值413。實例 3 : (3S,8R,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (3) 之合成
1.在20℃下向N-4-4_1
(50 g,157 mmol)於無水甲醇(500 mL)中之懸浮液一次性添加無水TsOH (2.84 g,15.7 mmol)。使混合物升溫直至60℃且攪拌1 h。反應混合物用Et3
N (1.58 g,15.7 mmol)淬滅且攪拌另外30 min。濾出沈澱之固體,用甲醇(250 mL)洗滌且於空氣中乾燥,得到呈固體之N-4-1_2
(51 g,90%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.18 (s, 3H), 3.14 (s, 3H); 2.54-2.48 (m, 1H); 2.10-2.00 (m, 4H); 1.95-1.75 (m, 2H), 1.65-1.50 (m, 7H), 1.48-0.80 (m, 11H), 0.78-0.75 (m, 4H), 0.59 (s, 3H)。 2.在20℃下於N2
下向Ph3
PMeBr (75 g,210 mmol)於無水THF (500 mL)中之懸浮液逐份添加t-BuOK (23.5 g,210 mmol)。混合物變成深橙色且在20℃下攪拌30 min。隨後添加N-4-1_2
(51 g,140 mmol)。使混合物升溫至40℃且攪拌1 h。將反應混合物冷卻且逐份倒入NH4
Cl水溶液(冰) (400mL)中。分離所得混合物;用THF (200 mL)萃取含水層。合併之有機層直接用作N-4-1_3
之溶液而無需進一步純化。 3.在20℃下向N-4-1_3
(50.4 g,139 mmol)於THF (700 mL)中之溶液中添加HCl水溶液(1 M,208 mL,208 mmol)。在20℃下攪拌混合物1小時,且固體沈澱。將水(200 mL)添加至混合物中,且濾出沈澱之固體,用水洗滌且乾燥,得到呈固體之N-4-1_4
(41 g,94%)。 1 H NMR
(400 MHz, CDCl3
)δ 4.85 (s, 1H), 4.70 (s, 1H); 2.38-2.25 (m, 3H); 2.10-1.98 (m, 3H), 1.88-1.49 (m, 10H), 1.40-1.08 (m, 11H), 0.97-0.72 (m, 2H), 0.58 (s, 3H)。 4.在25℃下於N2
下向Me3
SI (101 g,496 mmol)於無水THF (400 mL)中之溶液中逐份添加t-BuOK (58.3 g,520 mmol)且攪拌30 min。添加N-4-1_4
(39 g,124 mmol)於無水THF (300 mL)中之溶液。使反應混合物升溫至50℃且攪拌2小時。將反應混合物冷卻至25℃且用NH4
Cl水溶液(500 mL)處理。用EtOAc (2×500 mL))萃取水相。合併之有機相用鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至10/1)純化,得到呈固體之N-4-3_1
(35 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 4.84 (s, 1H), 4.70 (s, 1H); 2.65-2.55 (m, 2H); 2.10-1.98 (m, 2H), 1.92-1.49 (m, 13H), 1.40-1.13 (m, 8H), 0.99-0.69 (m, 6H), 0.57 (s, 3H)。 5.在25℃下向N-4-3_1
(35 g,647 mmol)於無水MeOH (500 mL)中之溶液中添加MeONa (57.2 g,1.06 mol)且在N2
下攪拌混合物30 min。使反應混合物升溫至70℃且於N2
下攪拌回流3小時。將反應混合物冷卻至25℃且用水(500 mL)處理。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc=10/1至6/1)純化,得到呈固體之N-4-3_2
(25 g,不純)。粗產物在25℃下自PE (250 mL)濕磨1 h。過濾懸浮液且真空乾燥濾餅以獲得呈固體之N-4-3_2
(15 g,25%)。 1 H NMR
(400 MHz, CDCl3
)δ 4.86 (s, 1H), 4.72 (s, 1H), 3.46-3.37 (m, 5H), 2.54 (s, 1H), 2.07-1.99 (m, 1H), 1.89-1.52 (m, 15H), 1.41-1.06 (m, 10H), 0.86 (s, 3H); 0.58 (s, 3H) 6.在0℃下向N-4-3_2
(15 g,41.6 mmol)於無水THF (200 mL)中之溶液中添加9-BBN二聚體(27.7 g,124 mmol)且於N2
下攪拌30 min。使反應混合物升溫至50℃且攪拌1 h。將反應混合物冷卻至0℃且添加EtOH (50 mL),隨後在0℃下極緩慢地添加NaOH (41.6 mL,5M,208 mmol)。緩慢地添加H2
O2
(23.5 g,208 mmol,30%於水中)同時使內部溫度保持在10℃以下。使混合物升溫至50℃且再攪拌1小時。將反應混合物冷卻,逐份倒入冰水(500 mL)中且過濾。真空濃縮濾液以提供呈油狀物之N-4-3_3
(14 g,粗產物)。粗殘餘物直接用於下一步驟。 7.在25℃下將DMP (3.35 g,7.92 mmol)添加至N-4_3
(1 g,2.64 mmol)於DCM (20 mL)中之混合物中。使反應混合物升溫至40℃且攪拌1 h。在pH 7至8及10℃以下用NaHCO3
飽和水溶液淬滅反應混合物。過濾懸浮液。濾液中之DCM相經分離且用NaHCO3
/Na2
S2
O3
飽和水溶液(1:1,2×50 mL)、鹽水(2×50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮以獲得固體。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之N-4-3_4
(0.6 g,60%)。 1 H NMR
(400 MHz, CDCl3
)δ 9.57 (s, 1H), 3.40-3.34 (m, 5H); 2.38-2.28 (m, 1H); 1.94-1.76 (m, 2H), 1.74-1.35 (m, 16H), 1.06-0.82 (m, 10H), 0.73-0.64 (m, 5H)。 8.在0℃下於N2
下向N-4-3_4
(0.6 g,1.59 mmol)於無水THF (10 mL)中之溶液中添加異戊基溴化鎂(4.37 mL,8.74 mmol,2 M於二乙醚中)。使反應混合物升溫至25℃且攪拌1小時。向反應混合物中添加NH4
Cl 飽和水溶液(50 mL)。用EtOAc (3×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-4-4A
(0.5g,粗產物)。 1 H NMR
(400 MHz, CDCl3
)δ 3.64-3.60 (m, 1H), 3.40-3.37 (m, 5H); 2.02-1.79 (m, 3H); 1.75-1.50 (m, 11H), 1.25-1.10 (m, 14H), 0.99-0.75 (m, 14H), 0.70-0.64 (m, 4H)。 9.在25℃下向N-4-4A
(0.5 g,粗產物)於DCM (20 mL)中之溶液中添加DMP (1.88 g,4.44 mmol)。使反應混合物升溫至40℃且攪拌1小時。在pH 7至8及10℃以下用NaHCO3
飽和水溶液淬滅反應混合物。過濾懸浮液。DCM相經分離且用NaHCO3
/Na2
S2
O3
飽和水溶液(1:1,2×50 mL)、鹽水(2×50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-4-4O
(0.4 g,粗產物),其直接用於下一步驟。 在25℃下向N-4-4O
(0.4 g,0.895 mmol)於MeOH (4 mL)中之溶液中緩慢地添加NaBH4
(0.340 g,8.95 mmol)且攪拌2小時。用DCM (2 × 20 mL)萃取水相。合併之有機相用飽和鹽水(2×20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc=8/1至5/1)純化,得到呈固體之35
(150 mg,不純)及2
(130 mg,不純)。在82o
C下回流1小時自MeCN (3 mL)再結晶2
(130 mg,不純)。攪拌混合物且冷卻至25℃。過濾懸浮液且真空濃縮濾液以提供呈固體之2
(50 mg,12%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.63-3.61 (m, 1H), 3.41-3.38 (m, 5H); 2.51 (s, 1H); 1.97-1.81 (m, 2H), 1.71-1.31 (m, 15H), 1.26-1.03 (m, 10H), 0.97-0.78 (m, 14H), 0.71-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.350 min,30-90 AB,純度99%,C29
H48
O [M+H-2H2
O]+
之MS ESI計算值413,實驗值413。實例 3 : (3S,8R,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (3) 之合成  1.
在35℃下於氮氣下將t-BuOH (350 mL)裝入三頸圓底燒瓶且於氮氣下攪拌10 min。將t-BuOK (90.5 g,807 mmol)添加至混合物中且於氮氣下攪拌15 min。將S-310-B9_1
(20 g, 73.4 mmol)添加至以上混合物且在35℃下於氮氣下攪拌1.5小時。將反應混合物倒入至10%乙酸水溶液(500 mL)中且在35℃以下攪拌15 min。添加水(500 mL)且攪拌混合物30 min。用碳酸氫鈉(500 mL)將混合物之pH調節為7至8且攪拌30 min。用PE (2×500 mL)萃取混合物。有機層經分離,用鹽水(500 mL)洗滌,經無水硫酸鈉乾燥,過濾且在35℃以下濃縮,得到呈油狀之S-200-N19-3_1
(17 g,粗產物)。粗殘餘物直接用於下一步驟。2.
在0℃下向2,6-二第三丁基-4-甲苯酚(100 g,453 mmol)於甲苯(300 ml)中之溶液中逐滴添加AlMe3
(113 mL,226 mmol,2 M於甲苯中)。在25℃下攪拌混合物1小時以產生MAD。在-70℃下將S-200-N19-3_1
(10 g,36.7 mmol)於甲苯(50 mL)中之溶液逐滴添加至MAD溶液。在-70℃下攪拌1小時之後,在-70℃下逐滴添加MeMgBr (36.6 mL,110 mmol,3M於乙基醚中)。在-70℃下攪拌所得溶液1小時。在-70℃下用飽和檸檬酸(400 ml)淬滅反應混合物。在25℃下攪拌10 min後,過濾所得混合物且用EtOAc (2×200 ml)洗滌。合併之有機層經分離,用鹽水(2×200 ml)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(PE/EtOAc=10/1至5/1)純化以產生呈固體之S-200-N19-3_2
(7.6 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 5.45-5.40 (m, 1H), 2.51-2.38 (m, 1H), 2.49-2.21 (m, 1H), 2.14-1.88 (m, 5H), 1.86-1.77 (m, 2H), 1.73-1.38 (m, 8H), 1.34-1.22 (m, 4H), 0.95-0.81 (m, 8H)。 3.在40℃下於N2
下向PPh3
EtBr (37.1 g,100 mmol)於THF (200 mL)中之懸浮液中添加t-BuOK (11.2 g,100 mmol)。在20℃下攪拌10 min後,添加S-200-N19-3_2
(7.6 g, 25.1 mmol)。在40℃下攪拌反應混合物1小時。反應物在0℃下用NH4
Cl水溶液(200 mL)淬滅,用EtOAc (3×200 mL)萃取。合併之有機相用鹽水(200 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由Combi-flash (0%至30% EtOAc/PE)純化,得到呈固體之S-200-N19-3_3
(5 g,63%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.45-5.35 (m, 1H), 5.20-5.00 (m, 1H), 2.41-2.30 (m, 1H), 2.29-2.12 (m, 3H), 2.09-1.76 (m, 6H), 1.69-1.38 (m, 15H), 1.35-0.94 (m, 7H)。4.
在0℃下於N2
下向S-200-N19-3_3
(2 g,6.35 mmol)於THF (20 mL)中之溶液中添加9-BNN二聚體(3.09 g,12.7 mmol)。在60℃下攪拌溶液1小時。冷卻至0℃後,極緩慢地添加EtOH (20 ml)及NaOH (12.7 ml,5M,63.5 mmol)之溶液。添加後,緩慢地添加H2
O2
(2.15 mg,6.35 mmol,30%於水中),且內部溫度維持在10℃以下。在60℃下於N2
下攪拌混合物1小時。將混合物重新冷卻至30℃。將水(100 mL)添加至溶液且用EtOAc (100 mL)萃取含水層。有機層用(2×100 mL)鹽水洗滌。合併之有機層經無水Na2
SO4
乾燥,且藉由矽膠層析(PE/EtOAc=2/1)純化,得到呈固體之S-200-N19-4_1
(1.6 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.45-5.35 (m, 1H), 3.75-3.62 (m, 1H), 2.28-2.19 (m, 1H), 2.10-1.75 (m, 7H), 1.71-0.97 (m, 19H), 0.92-0.75 (m, 4H), 0.68 (s, 3H)。5.
向S-200-N19-4_1
(1.6 g,4.81 mmol)於DCM (20 mL)中之溶液中添加矽膠(2 g)及PCC (2.07 g,9.62 mmol)。在25℃下攪拌混合物3小時。向混合物添加PE (50 mL)。混合物經由矽膠墊過濾,且固體用PE/DCM (30 mL/30 mL)洗滌。過濾混合物且真空濃縮濾液。殘餘物藉由矽膠層析(PE/EtOAc=10/1至5/1)純化,得到呈固體之S-200-N19-4_2
(1.2 g,不純),其在回流下自MeCN (10 mL)再結晶以提供呈固體之S-200-N19-4_2
(1.0 g,84.0%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.40-5.35 (m, 1H), 2.61-2.45 (m, 1H), 2.30-2.10 (m, 5H), 2.00-1.75 (m, 6H), 1.70-1.10 (m, 14H), 0.90-0.75 (m, 4H); 0.633 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.058 min,30-90 AB,純度100%,C22
H34
[M+H-H2
O]+
之MS ESI計算值313,實驗值313。 6.在40℃下於N2
下向Ph3
PMeBr (11.1 g,31.4 mmol)於THF (50 mL)中之懸浮液中添加t-BuOK (3.51 g,31.4 mmol)。在25℃下攪拌10 min之後,添加S-200-N19-4_2
(2.6 g,7.86 mmol)。在40℃下攪拌反應混合物1 h。反應物在0℃下用NH4
Cl水溶液(100 mL)淬滅,其用EtOAc (2×100 mL)萃取。合併之有機相用鹽水(2 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由Combi-flash (0%至30% EtOAc/PE)純化,得到呈固體之S-200-N19-4_3
(2.4 g,93%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.45-5.35 (m, 1H), 4.86-4.83 (m, 1H), 8.70-4.65 (m, 1H), 2.27-2.20 (m, 1H), 2.10-1.90 (m, 4H), 1.89-1.50 (m, 11H), 1.49-1.30 (m, 3H), 1.28-1.00 (m, 6H), 0.80-0.60 (m, 5H), 0.59 (s, 3H)。 7.在0℃下於N2
下將9-BBN二聚體(9.27 g,38.0 mmol)添加至S-200-N19-4_3
(5 g,15.2 mmol)於THF (60 mL)中之溶液中。在60℃下攪拌溶液1 h。冷卻至0℃後,極緩慢地添加EtOH (60 ml)及NaOH (30.4 ml,5M,152 mmol)之溶液。添加後,緩慢地添加H2
O2
(15.2 ml,152 mmol,30%於水中),且內部溫度維持在10℃以下。在60℃下於N2
下攪拌混合物1小時。將混合物重新冷卻至30℃。將水(100 mL)與EtOH (100 ml)添加至溶液。獲得懸浮液,其經過濾且真空濃縮以產生呈固體之S-200-N19-4_4
(5 g,粗產物)。 1 H NMR
(400MHz, CDCl3) δ 5.44-5.32 (m, 1H), 3.68-3.59 (m, 1H), 3.39-3.35 (m, 1H), 2.29-2.19 (m, 1H), 2.08-1.89 (m, 4H), 1.88-1.75 (m, 3H), 1.62-1.60 (m, 2H), 1.56-1.39 (m, 6H), 1.36-1.24 (m, 3H), 1.23-1.11 (m, 4H), 1.08-0.98 (m, 4H), 0.92-0.75 (m, 5H), 0.70 (s, 3H)。 8.在25℃下將戴斯-馬丁高碘烷(2.44 g,5.76 mmol)添加至S-200-N19-4_4
(1 g,2.88 mmol)於DCM (150 mL)中之溶液中。在25℃下攪拌反應物1小時。在25℃下攪拌反應物30 min。在0℃下將混合物倒入飽和Na2
S2
O3
(100 ml)中,其用DCM (3×100 ml)萃取。合併之有機層用飽和NaHCO3
(100 mL×2)、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,藉由矽膠管柱(PE/EtOAc=10:1)將粗產物純化得到呈固體之S-500-15-2_1
(800 mg,80%)。 1 H NMR
(400MHz, CDCl3) δ 9.58-9.57 (m, 1H), 5.40-5.38 (m, 1H), 2.37-2.35 (m, 1H), 2.25-2.23 (m, 1H), 2.08-1.76 (m, 7H), 1.65-1.63 (m, 2H), 1.53-1.37 (m, 5H), 1.31-1.21 (m, 4H), 1.19-1.00 (m, 6H), 0.90-0.80 (m, 5H), 0.73 (s, 3H)。 9.在60℃下將1-溴-3-甲基丁烷(4 g,26.4 mmol)於THF (27 mL)中之溶液逐滴添加至Mg (947 mg,39.5 mmol)及I2
(33.5 mg,0.132 mmol)於THF (3 mL)中之懸浮液。在60℃下攪拌混合物1小時。在0℃下於N2
下將新鮮製備的異戊基溴化鎂(30 mL,0.88 M於THF中,26.4 mmol)添加至S-500-15-2_1
(800 mg,2.32 mmol)於THF (2 mL)中之溶液中。在0℃下攪拌混合物1小時。向混合物添加NH4
Cl (50 mL,飽和水溶液)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,藉由矽膠層析(PE/EtOAc=10/1至5/1)將粗產物純化得到呈固體之44
(720 mg,75%)。 1 H NMR
(400MHz, CDCl3) δ 5.40-5.38 (m, 1H), 3.63-3.61 (m, 1H), 2.23-2.21 (m, 1H), 2.10-1.74 (m, 7H), 1.69-1.58 (m, 2H), 1.54-1.34 (m, 8H), 1.33-1.00 (m, 11H), 0.95-0.75 (m, 14H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.289 min,30-90 AB,純度100%,C28
H45
[M+H-2H2
O]+之MS ESI計算值381,實驗值381。 10a.在25℃下於N2
下向44
(300 mg,0.720 mmol)於THF (14 mL)中之溶液中添加苯甲酸(348 mg,2.85 mmol)及三苯基膦(1.11 g,4.27 mmol)。在25℃下攪拌20 min之後,在0℃下於N2
下添加DIAD (780 mg,3.86 mmol)。在0℃下攪拌混合物20 min隨後升溫至25℃且在25℃下攪拌17小時。添加水(100 mL)且用EtOAc (2×100 mL)萃取混合物。有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到待純化之粗產物(1.5 g,粗產物)。 10b.在25℃下於N2
下向44
(1.9 g,4.55 mmol)於THF (70 mL)中之溶液中添加苯甲酸(2.19 g,18.0 mmol)及三苯膦(7.07 g,27.0 mmol)。在25℃下攪拌20 min之後,在0℃下於N2
下添加DIAD (4.93 g,24.4 mmol)。在0℃下攪拌混合物20 min隨後升溫至25℃且在25℃下攪拌17小時。添加水(250 mL)且用EtOAc (2×250 mL)萃取混合物。有機相用鹽水(2×300 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物。與另一批次之300 mg之44
合併,粗產物藉由矽膠管柱(PE/EtOAc=8/1)純化,得到呈油狀物之S-500-15-1_1
(1.2 g,不純),其直接用於下一步驟。 11.在25℃下向S-500-15-1_1
(1.2 g,不純)於THF/MeOH (2 mL/2 mL)中之溶液中添加NaOH (400 mg)及H2
O (2 mL)。在50℃下攪拌反應物16 h。冷卻後,反應混合物用H2
O (20 mL)稀釋且用EtOAc (2×30 mL)萃取。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮。粗產物藉由矽膠管柱(PE/EtOAc=4/1)純化,得到產物3
(150 mg,不純),其藉由在25℃下用MeCN (5 mL)濕磨純化,得到呈固體之3
(30 mg,純,及100 mg,不純)。 1 H NMR
(400MHz, CDCl3) δ 5.39-5.37 (m, 1H), 3.63-3.59 (m, 1H), 2.26-2.21 (m, 1H), 2.09-1.88 (m, 4H), 1.86-1.76 (m, 2H), 1.75-1.61 (m, 3H), 1.54-1.32 (m, 7H), 1.32-1.08 (m, 10H), 1.07-0.96 (m, 1H), 0.95-0.74 (m, 14H), 0.95-0.74 (m, 1H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.281 min,30-90 AB,純度98%,C28
H47
O [M+H-H2
O]+之MS ESI計算值399,實驗值399。實例 4 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己 基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (4) 之合成
1.
在35℃下於氮氣下將t-BuOH (350 mL)裝入三頸圓底燒瓶且於氮氣下攪拌10 min。將t-BuOK (90.5 g,807 mmol)添加至混合物中且於氮氣下攪拌15 min。將S-310-B9_1
(20 g, 73.4 mmol)添加至以上混合物且在35℃下於氮氣下攪拌1.5小時。將反應混合物倒入至10%乙酸水溶液(500 mL)中且在35℃以下攪拌15 min。添加水(500 mL)且攪拌混合物30 min。用碳酸氫鈉(500 mL)將混合物之pH調節為7至8且攪拌30 min。用PE (2×500 mL)萃取混合物。有機層經分離,用鹽水(500 mL)洗滌,經無水硫酸鈉乾燥,過濾且在35℃以下濃縮,得到呈油狀之S-200-N19-3_1
(17 g,粗產物)。粗殘餘物直接用於下一步驟。2.
在0℃下向2,6-二第三丁基-4-甲苯酚(100 g,453 mmol)於甲苯(300 ml)中之溶液中逐滴添加AlMe3
(113 mL,226 mmol,2 M於甲苯中)。在25℃下攪拌混合物1小時以產生MAD。在-70℃下將S-200-N19-3_1
(10 g,36.7 mmol)於甲苯(50 mL)中之溶液逐滴添加至MAD溶液。在-70℃下攪拌1小時之後,在-70℃下逐滴添加MeMgBr (36.6 mL,110 mmol,3M於乙基醚中)。在-70℃下攪拌所得溶液1小時。在-70℃下用飽和檸檬酸(400 ml)淬滅反應混合物。在25℃下攪拌10 min後,過濾所得混合物且用EtOAc (2×200 ml)洗滌。合併之有機層經分離,用鹽水(2×200 ml)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(PE/EtOAc=10/1至5/1)純化以產生呈固體之S-200-N19-3_2
(7.6 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 5.45-5.40 (m, 1H), 2.51-2.38 (m, 1H), 2.49-2.21 (m, 1H), 2.14-1.88 (m, 5H), 1.86-1.77 (m, 2H), 1.73-1.38 (m, 8H), 1.34-1.22 (m, 4H), 0.95-0.81 (m, 8H)。 3.在40℃下於N2
下向PPh3
EtBr (37.1 g,100 mmol)於THF (200 mL)中之懸浮液中添加t-BuOK (11.2 g,100 mmol)。在20℃下攪拌10 min後,添加S-200-N19-3_2
(7.6 g, 25.1 mmol)。在40℃下攪拌反應混合物1小時。反應物在0℃下用NH4
Cl水溶液(200 mL)淬滅,用EtOAc (3×200 mL)萃取。合併之有機相用鹽水(200 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由Combi-flash (0%至30% EtOAc/PE)純化,得到呈固體之S-200-N19-3_3
(5 g,63%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.45-5.35 (m, 1H), 5.20-5.00 (m, 1H), 2.41-2.30 (m, 1H), 2.29-2.12 (m, 3H), 2.09-1.76 (m, 6H), 1.69-1.38 (m, 15H), 1.35-0.94 (m, 7H)。4.
在0℃下於N2
下向S-200-N19-3_3
(2 g,6.35 mmol)於THF (20 mL)中之溶液中添加9-BNN二聚體(3.09 g,12.7 mmol)。在60℃下攪拌溶液1小時。冷卻至0℃後,極緩慢地添加EtOH (20 ml)及NaOH (12.7 ml,5M,63.5 mmol)之溶液。添加後,緩慢地添加H2
O2
(2.15 mg,6.35 mmol,30%於水中),且內部溫度維持在10℃以下。在60℃下於N2
下攪拌混合物1小時。將混合物重新冷卻至30℃。將水(100 mL)添加至溶液且用EtOAc (100 mL)萃取含水層。有機層用(2×100 mL)鹽水洗滌。合併之有機層經無水Na2
SO4
乾燥,且藉由矽膠層析(PE/EtOAc=2/1)純化,得到呈固體之S-200-N19-4_1
(1.6 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.45-5.35 (m, 1H), 3.75-3.62 (m, 1H), 2.28-2.19 (m, 1H), 2.10-1.75 (m, 7H), 1.71-0.97 (m, 19H), 0.92-0.75 (m, 4H), 0.68 (s, 3H)。5.
向S-200-N19-4_1
(1.6 g,4.81 mmol)於DCM (20 mL)中之溶液中添加矽膠(2 g)及PCC (2.07 g,9.62 mmol)。在25℃下攪拌混合物3小時。向混合物添加PE (50 mL)。混合物經由矽膠墊過濾,且固體用PE/DCM (30 mL/30 mL)洗滌。過濾混合物且真空濃縮濾液。殘餘物藉由矽膠層析(PE/EtOAc=10/1至5/1)純化,得到呈固體之S-200-N19-4_2
(1.2 g,不純),其在回流下自MeCN (10 mL)再結晶以提供呈固體之S-200-N19-4_2
(1.0 g,84.0%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.40-5.35 (m, 1H), 2.61-2.45 (m, 1H), 2.30-2.10 (m, 5H), 2.00-1.75 (m, 6H), 1.70-1.10 (m, 14H), 0.90-0.75 (m, 4H); 0.633 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.058 min,30-90 AB,純度100%,C22
H34
[M+H-H2
O]+
之MS ESI計算值313,實驗值313。 6.在40℃下於N2
下向Ph3
PMeBr (11.1 g,31.4 mmol)於THF (50 mL)中之懸浮液中添加t-BuOK (3.51 g,31.4 mmol)。在25℃下攪拌10 min之後,添加S-200-N19-4_2
(2.6 g,7.86 mmol)。在40℃下攪拌反應混合物1 h。反應物在0℃下用NH4
Cl水溶液(100 mL)淬滅,其用EtOAc (2×100 mL)萃取。合併之有機相用鹽水(2 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由Combi-flash (0%至30% EtOAc/PE)純化,得到呈固體之S-200-N19-4_3
(2.4 g,93%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.45-5.35 (m, 1H), 4.86-4.83 (m, 1H), 8.70-4.65 (m, 1H), 2.27-2.20 (m, 1H), 2.10-1.90 (m, 4H), 1.89-1.50 (m, 11H), 1.49-1.30 (m, 3H), 1.28-1.00 (m, 6H), 0.80-0.60 (m, 5H), 0.59 (s, 3H)。 7.在0℃下於N2
下將9-BBN二聚體(9.27 g,38.0 mmol)添加至S-200-N19-4_3
(5 g,15.2 mmol)於THF (60 mL)中之溶液中。在60℃下攪拌溶液1 h。冷卻至0℃後,極緩慢地添加EtOH (60 ml)及NaOH (30.4 ml,5M,152 mmol)之溶液。添加後,緩慢地添加H2
O2
(15.2 ml,152 mmol,30%於水中),且內部溫度維持在10℃以下。在60℃下於N2
下攪拌混合物1小時。將混合物重新冷卻至30℃。將水(100 mL)與EtOH (100 ml)添加至溶液。獲得懸浮液,其經過濾且真空濃縮以產生呈固體之S-200-N19-4_4
(5 g,粗產物)。 1 H NMR
(400MHz, CDCl3) δ 5.44-5.32 (m, 1H), 3.68-3.59 (m, 1H), 3.39-3.35 (m, 1H), 2.29-2.19 (m, 1H), 2.08-1.89 (m, 4H), 1.88-1.75 (m, 3H), 1.62-1.60 (m, 2H), 1.56-1.39 (m, 6H), 1.36-1.24 (m, 3H), 1.23-1.11 (m, 4H), 1.08-0.98 (m, 4H), 0.92-0.75 (m, 5H), 0.70 (s, 3H)。 8.在25℃下將戴斯-馬丁高碘烷(2.44 g,5.76 mmol)添加至S-200-N19-4_4
(1 g,2.88 mmol)於DCM (150 mL)中之溶液中。在25℃下攪拌反應物1小時。在25℃下攪拌反應物30 min。在0℃下將混合物倒入飽和Na2
S2
O3
(100 ml)中,其用DCM (3×100 ml)萃取。合併之有機層用飽和NaHCO3
(100 mL×2)、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,藉由矽膠管柱(PE/EtOAc=10:1)將粗產物純化得到呈固體之S-500-15-2_1
(800 mg,80%)。 1 H NMR
(400MHz, CDCl3) δ 9.58-9.57 (m, 1H), 5.40-5.38 (m, 1H), 2.37-2.35 (m, 1H), 2.25-2.23 (m, 1H), 2.08-1.76 (m, 7H), 1.65-1.63 (m, 2H), 1.53-1.37 (m, 5H), 1.31-1.21 (m, 4H), 1.19-1.00 (m, 6H), 0.90-0.80 (m, 5H), 0.73 (s, 3H)。 9.在60℃下將1-溴-3-甲基丁烷(4 g,26.4 mmol)於THF (27 mL)中之溶液逐滴添加至Mg (947 mg,39.5 mmol)及I2
(33.5 mg,0.132 mmol)於THF (3 mL)中之懸浮液。在60℃下攪拌混合物1小時。在0℃下於N2
下將新鮮製備的異戊基溴化鎂(30 mL,0.88 M於THF中,26.4 mmol)添加至S-500-15-2_1
(800 mg,2.32 mmol)於THF (2 mL)中之溶液中。在0℃下攪拌混合物1小時。向混合物添加NH4
Cl (50 mL,飽和水溶液)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,藉由矽膠層析(PE/EtOAc=10/1至5/1)將粗產物純化得到呈固體之44
(720 mg,75%)。 1 H NMR
(400MHz, CDCl3) δ 5.40-5.38 (m, 1H), 3.63-3.61 (m, 1H), 2.23-2.21 (m, 1H), 2.10-1.74 (m, 7H), 1.69-1.58 (m, 2H), 1.54-1.34 (m, 8H), 1.33-1.00 (m, 11H), 0.95-0.75 (m, 14H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.289 min,30-90 AB,純度100%,C28
H45
[M+H-2H2
O]+之MS ESI計算值381,實驗值381。 10a.在25℃下於N2
下向44
(300 mg,0.720 mmol)於THF (14 mL)中之溶液中添加苯甲酸(348 mg,2.85 mmol)及三苯基膦(1.11 g,4.27 mmol)。在25℃下攪拌20 min之後,在0℃下於N2
下添加DIAD (780 mg,3.86 mmol)。在0℃下攪拌混合物20 min隨後升溫至25℃且在25℃下攪拌17小時。添加水(100 mL)且用EtOAc (2×100 mL)萃取混合物。有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到待純化之粗產物(1.5 g,粗產物)。 10b.在25℃下於N2
下向44
(1.9 g,4.55 mmol)於THF (70 mL)中之溶液中添加苯甲酸(2.19 g,18.0 mmol)及三苯膦(7.07 g,27.0 mmol)。在25℃下攪拌20 min之後,在0℃下於N2
下添加DIAD (4.93 g,24.4 mmol)。在0℃下攪拌混合物20 min隨後升溫至25℃且在25℃下攪拌17小時。添加水(250 mL)且用EtOAc (2×250 mL)萃取混合物。有機相用鹽水(2×300 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物。與另一批次之300 mg之44
合併,粗產物藉由矽膠管柱(PE/EtOAc=8/1)純化,得到呈油狀物之S-500-15-1_1
(1.2 g,不純),其直接用於下一步驟。 11.在25℃下向S-500-15-1_1
(1.2 g,不純)於THF/MeOH (2 mL/2 mL)中之溶液中添加NaOH (400 mg)及H2
O (2 mL)。在50℃下攪拌反應物16 h。冷卻後,反應混合物用H2
O (20 mL)稀釋且用EtOAc (2×30 mL)萃取。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮。粗產物藉由矽膠管柱(PE/EtOAc=4/1)純化,得到產物3
(150 mg,不純),其藉由在25℃下用MeCN (5 mL)濕磨純化,得到呈固體之3
(30 mg,純,及100 mg,不純)。 1 H NMR
(400MHz, CDCl3) δ 5.39-5.37 (m, 1H), 3.63-3.59 (m, 1H), 2.26-2.21 (m, 1H), 2.09-1.88 (m, 4H), 1.86-1.76 (m, 2H), 1.75-1.61 (m, 3H), 1.54-1.32 (m, 7H), 1.32-1.08 (m, 10H), 1.07-0.96 (m, 1H), 0.95-0.74 (m, 14H), 0.95-0.74 (m, 1H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.281 min,30-90 AB,純度98%,C28
H47
O [M+H-H2
O]+之MS ESI計算值399,實驗值399。實例 4 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己 基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (4) 之合成 
 1.在0℃下將t-BuLi (90.7 mL,118 mmol,1.3 M於正己烷中,3.0當量)添加至氯基(甲氧基甲基)三苯基磷烷(40.4 g,118 mmol,3.0當量)於THF (200 mL)之溶液中。添加之後,在0℃下攪拌反應混合物1小時。在0℃下將混合物添加至S-500-6-29_2A
(5 g,39.6 mmol,1.0當量)於THF (50 mL)中之溶液中且在15℃下攪拌反應混合物2 h。混合物用NH4
Cl(100 mL,10%)處理且用EtOAc (2×200 mL)萃取。有機相經分離且真空濃縮,得到S-500-6-29_2B
(18.0 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.74 (s, 1H), 3.52 (s, 3H), 2.20-2.15 (m, 2H), 1.95-1.90 (m, 2H), 1.26-1.16 (m, 4H), 0.90 (s, 6H)。 2.在15℃下將TFA (21.4 mL,290 mmol)添加至S-500-6-29_2B
(5.6 g,不純)於DCM (25 mL)中之攪拌溶液中且在15℃下攪拌1.5 h。反應混合物用NaHCO3
飽和水溶液(10 mL)淬滅且用EtOAc (2×20 mL)萃取。合併之有機層經無水Na2
SO4
乾燥且在減壓下濃縮,得到呈油狀物之S-500-6-29_2C
(5.0 g,粗產物),其用於下一步驟中而無需純化。 1 H NMR
(400 MHz, CDCl3
) δ 9.64 (s, 1H), 2.15-2.05 (m, 1H), 1.80-1.60 (m, 2H), 1.70-1.35 (m, 4H), 1.25-1.15 (m, 2H), 0.91 (s, 3H), 0.87 (s, 3H)。3.
在15℃下於N2
下將NaBH4
(1.61 g,42.7 mmol)添加至S-500-6-29_2C
(5.0 g,35.6 mmol)於MeOH (50 mL)中之溶液中。在15℃下攪拌混合物1小時。將混合物倒入水(50 mL)中且攪拌20分鐘。用EtOAc (3×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈油狀物之S-500-6-29_2D
(5.6 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.47-3.42 (m, 2H), 1.60-1.50 (m, 2H), 1.42-1.30 (m, 4H), 1.25-1.0 (m, 4H), 0.91 (s, 3H), 0.87 (s, 3H)。 4.在15℃下於N2
下將TsCl (8.23 g,43.2 mmol)添加至S-500-6-29_2D
(5.6 g,39.3 mmol)於吡啶(50 mL)中之溶液中。在15℃下攪拌混合物16小時。將混合物倒入水(50 mL)中且攪拌20分鐘。用DCM (3×40 mL)萃取水相。合併之有機相用飽和鹽水(2×200 mL)、HCl (0.5 M,50 ml)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到油狀物,其在68o
C下自己烷(50 mL)再結晶得到呈固體之S-500-6-29_2E
(4.2 g,61%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.80-7.76 (m, 2H), 7.35-7.25 (m, 2H), 3.86-3.80 (m, 2H), 2.45 (s, 3H), 1.60-1.45 (m, 3H), 1.40-1.30 (m, 2H), 1.20-1.05 (m, 4H), 0.88 (s, 3H), 0.82 (s, 3H)。5.
將LiBr (2.33 g,26.9 mmol)添加至S-500-6-29_2E
(2 g,6.74 mmol)於丙酮(50 mL)中之溶液中。在65℃下攪拌混合物12小時。混合物用水(50 mL)淬滅且用MTBE (3×20 mL)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈液體之S-500-6-29_2
(1.3 g,粗產物)。與另一批次之2.2 g之S-500-6-29_2E
合併,合併之粗產物經由小矽膠過濾且用PE (100 mL)洗滌且濃縮,得到呈油狀物之S-500-6-29_2
(2.6 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.34-3.28 (m, 2H), 1.72-1.64 (m, 2H), 1.60-1.48 (m, 1H), 1.42-1.35 (m, 2H), 1.28-1.18 (m, 4H), 0.91 (s, 3H), 0.87 (s, 3H)。 6.在25℃下向Ph3
PMeBr (167 g,470 mmol)於THF (900 mL)中之溶液中添加t
-BuOK (52.7 g,470 mmol)。將反應混合物加熱至60℃且攪拌1小時。添加孕烯醇酮
(50 g,157 mmol)。在60℃下攪拌反應混合物1小時。添加飽和NH4
Cl (900 mL)。用EtOAc (2×1000 mL)萃取混合物。合併之有機層用鹽水(2×2000 mL)洗滌,經Na2
SO4
乾燥且真空濃縮,得到呈油狀物之粗產物,其藉由矽膠管柱層析(PE:EtOAc=20:1至5:1)純化,得到呈固體之S-200-INT_1
(45 g,91.2%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.40-5.30 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 3.60-3.40 (m, 1H), 2.40-2.20 (m, 2H), 2.05-1.90 (m, 2H), 1.85-1.60 (m, 9H), 1.53-1.40 (m, 5H), 1.25-0.90 (m, 9H), 0.59 (s, 3H)。 7.在20℃下向S-200-INT_1
(45 g,143 mmol)於DCM (1500 mL)中之溶液中添加DMP (108 g,257 mmol)。在20℃下攪拌混合物2小時。添加水(800 mL)且添加NaHCO3
(200 g固體)。過濾混合物。濾液用飽和Na2
S2
O3
(2×2000 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到S-200-INT_2
於DCM (100 mL)中之溶液,其直接用於下一步驟。 8.在10℃下向BHT (191 g,866 mmol)於甲苯(500 mL)中之溶液中添加AlMe3
(2 M於甲苯中,216 mL,433 mmol)且攪拌1小時。在-78℃下向混合物添加S-200-INT_2
(理論質量:44.6 g)於DCM (100 mL)中之溶液。在-78℃下攪拌混合物1小時。在-78℃下添加EtMgBr (141 mL,426 mmol)。在-78℃下攪拌混合物20 min。添加飽和檸檬酸(1 L)。有機相經分離,用鹽水(600 mL)洗滌,經Na2
SO4
乾燥且真空濃縮,得到粗產物,其藉由矽膠管柱層析(PE:EtOAc=50:1至30:1)純化,得到呈固體之S-200-INT_3a
(27 g,55%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.35-5.25 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.40-2.30 (m, 1H), 2.10-1.60 (m, 14H), 1.50-0.75 (m, 17H), 0.58 (s, 3H)。 9.將9-BBN二聚體(17.6 g,72.5 mmol)添加至S-200-INT_3E
(5 g,14.5 mmol)於THF (40 mL)中之溶液中。在60℃下於N2
下攪拌混合物3小時,且形成固體。向反應混合物添加乙醇(8.33 mL,145 mmol)及NaOH (28.9 mL,5 M,145 mmol)。混合物變得澄清。在25℃下逐滴添加H2
O2
(14.4 mL,10 M,145 mmol)且使內部溫度升至回流(75℃)。混合物冷卻,在添加且攪拌1小時之後,形成固體。在25℃下向混合物添加Na2
SO3
(20 mL,20%水溶液)。用EtOAc (2×100 mL)萃取混合物。合併之有機相用鹽水(2×200 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,且藉由矽膠管柱(PE/EtOAc=10/1至3/1)純化,以提供呈固體之S-200-INT_4E
(3.5 g,67%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.68-3.60 (m, 1H), 3.41-3.32 (m, 1H), 2.40-2.32 (m, 1H), 2.03-1.93 (m, 2H), 1.92-1.65 (m, 4H), 1.58-1.16 (m, 13H), 1.16-0.90 (m, 11H), 0.90-0.81 (m, 3H), 0.73-0.62 (s, 3H)。 10.在25℃下將DMP (4.66 g,11.0 mmol)添加至S-200-INT_4E
(2 g,5.54 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌反應混合物10 min。在25℃下用NaHCO3
飽和水溶液(30 mL)淬滅反應混合物。分離DCM層且用DCM (30 mL)萃取水相。合併之有機相用Na2
SO3
飽和水溶液(3×50 mL)、鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-200-INT_5E
(2.0 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.59-9.56 (m, 1H), 5.31-5.26 (m, 1H), 2.42-2.10 (m, 2H), 2.10-1.80 (m, 4H), 1.79-1.54 (m, 7H), 1.54-1.31 (m, 7H), 1.28-0.90 (m, 9H), 0.90-0.81 (m, 4H), 0.73 (s, 3H)。 11.在75℃下將S-500-6-29_2
(2.56 g,12.5 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (600 mg,25.0 mmol)及I2
(63.4 mg,0.25 mmol)於THF (3 mL)中之懸浮液中。在75℃下攪拌混合物1小時。冷卻後,在15℃下緩慢地添加S-500-6-1_1
(1 g,2.78 mmol)於THF (30 mL)中之溶液。添加之後,在15℃下攪拌混合物2小時,用飽和NH4
Cl (40 mL)及飽和檸檬酸(20 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之S-500-6-29_1
(800 mg,60%)之混合物。 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.19 (m, 1H), 3.88-3.71 (m, 1H), 2.42-2.29 (m, 1H), 2.07-1.86 (m, 4H), 1.78-1.59 (m, 4H), 1.54-1.31 (m, 13H), 1.29-1.13 (m, 8H), 1.12-0.99 (m, 8H), 0.94-0.79 (m, 13H), 0.68 (s, 3H)。 12.將DMP (1.39 g,3.30 mmol)添加至S-500-6-29_1
(800 mg,1.65 mmol)於DCM (30 mL)中之溶液中。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之粗產物S-500-6-29_2
(800 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.25 (m, 1H), 2.54-2.43 (m, 1H), 2.40-2.21 (m, 3H), 2.07-1.87 (m, 3H),1.81-1.57 (m, 7H), 1.53-1.39 (m, 7H), 1.38-1.29 (m, 3H), 1.27-1.16 (m, 4H), 1.15-1.04 (m, 8H), 1.03 (s, 3H), 1.00-0.92 (m, 2H), 0.91-0.80 (m, 9H), 0.69 (s, 3H)。 13.將NaBH4
(2.80 g,82.5 mmol)每五分鐘添加五次至S-500-6-29_2
(800 mg,1.65 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (50 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到49
(290 mg,36%)及呈固體之12
(120 mg,45%)。49: 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.07-1.87 (m, 4H), 1.76-1.69 (m, 1H), 1.66-1.55 (m, 5H), 1.53-1.42 (m, 7H), 1.41-1.31 (m, 5H), 1.30-1.12 (m, 8H), 1.11-1.05 (m, 3H), 1.03 (s, 3H), 1.01-0.92 (m, 2H), 0.91-0.82 (m, 12H), 0.68 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.718 min,30-90AB_E,純度98%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。12: 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.06-1.95 (m, 3H), 1.77-1.58 (m, 7H), 1.54-1.28 (m, 12H), 1.27-1.06 (m, 11H), 1.03 (s, 3H), 1.00-0.95 (m, 2H), 0.93-0.82 (m, 12H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.708 min,30-90AB_E,純度100%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。 14.將Pd(OH)2
(200 mg,無水)添加至49
(140 mg,0.288 mmol)於MeOH (30 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到59
(27 mg,19%)及呈固體之4
(42 mg,30%)。4: 1 H NMR
(400 MHz, CDCl3
) δ 3.84-3.76 (m, 1H), 1.98-1.85 (m, 2H), 1.69-1.54 (m, 9H), 1.53-1.46 (m, 3H), 1.45-1.28 (m, 9H), 1.27-1.20 (m, 4H), 1.19-1.13 (m, 5H), 1.12-1.02 (m, 4H), 1.01-0.92 (m, 2H), 0.91-0.85 (m, 12H), 0.82 (s, 3H), 0.70-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.799 min,30-90AB_E,純度100%,C33
H55
[M+H-H2
O]+
之MS ESI計算值451,實驗值451。實例 5 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (5) 之合成
1.在0℃下將t-BuLi (90.7 mL,118 mmol,1.3 M於正己烷中,3.0當量)添加至氯基(甲氧基甲基)三苯基磷烷(40.4 g,118 mmol,3.0當量)於THF (200 mL)之溶液中。添加之後,在0℃下攪拌反應混合物1小時。在0℃下將混合物添加至S-500-6-29_2A
(5 g,39.6 mmol,1.0當量)於THF (50 mL)中之溶液中且在15℃下攪拌反應混合物2 h。混合物用NH4
Cl(100 mL,10%)處理且用EtOAc (2×200 mL)萃取。有機相經分離且真空濃縮,得到S-500-6-29_2B
(18.0 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.74 (s, 1H), 3.52 (s, 3H), 2.20-2.15 (m, 2H), 1.95-1.90 (m, 2H), 1.26-1.16 (m, 4H), 0.90 (s, 6H)。 2.在15℃下將TFA (21.4 mL,290 mmol)添加至S-500-6-29_2B
(5.6 g,不純)於DCM (25 mL)中之攪拌溶液中且在15℃下攪拌1.5 h。反應混合物用NaHCO3
飽和水溶液(10 mL)淬滅且用EtOAc (2×20 mL)萃取。合併之有機層經無水Na2
SO4
乾燥且在減壓下濃縮,得到呈油狀物之S-500-6-29_2C
(5.0 g,粗產物),其用於下一步驟中而無需純化。 1 H NMR
(400 MHz, CDCl3
) δ 9.64 (s, 1H), 2.15-2.05 (m, 1H), 1.80-1.60 (m, 2H), 1.70-1.35 (m, 4H), 1.25-1.15 (m, 2H), 0.91 (s, 3H), 0.87 (s, 3H)。3.
在15℃下於N2
下將NaBH4
(1.61 g,42.7 mmol)添加至S-500-6-29_2C
(5.0 g,35.6 mmol)於MeOH (50 mL)中之溶液中。在15℃下攪拌混合物1小時。將混合物倒入水(50 mL)中且攪拌20分鐘。用EtOAc (3×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈油狀物之S-500-6-29_2D
(5.6 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.47-3.42 (m, 2H), 1.60-1.50 (m, 2H), 1.42-1.30 (m, 4H), 1.25-1.0 (m, 4H), 0.91 (s, 3H), 0.87 (s, 3H)。 4.在15℃下於N2
下將TsCl (8.23 g,43.2 mmol)添加至S-500-6-29_2D
(5.6 g,39.3 mmol)於吡啶(50 mL)中之溶液中。在15℃下攪拌混合物16小時。將混合物倒入水(50 mL)中且攪拌20分鐘。用DCM (3×40 mL)萃取水相。合併之有機相用飽和鹽水(2×200 mL)、HCl (0.5 M,50 ml)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到油狀物,其在68o
C下自己烷(50 mL)再結晶得到呈固體之S-500-6-29_2E
(4.2 g,61%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.80-7.76 (m, 2H), 7.35-7.25 (m, 2H), 3.86-3.80 (m, 2H), 2.45 (s, 3H), 1.60-1.45 (m, 3H), 1.40-1.30 (m, 2H), 1.20-1.05 (m, 4H), 0.88 (s, 3H), 0.82 (s, 3H)。5.
將LiBr (2.33 g,26.9 mmol)添加至S-500-6-29_2E
(2 g,6.74 mmol)於丙酮(50 mL)中之溶液中。在65℃下攪拌混合物12小時。混合物用水(50 mL)淬滅且用MTBE (3×20 mL)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈液體之S-500-6-29_2
(1.3 g,粗產物)。與另一批次之2.2 g之S-500-6-29_2E
合併,合併之粗產物經由小矽膠過濾且用PE (100 mL)洗滌且濃縮,得到呈油狀物之S-500-6-29_2
(2.6 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.34-3.28 (m, 2H), 1.72-1.64 (m, 2H), 1.60-1.48 (m, 1H), 1.42-1.35 (m, 2H), 1.28-1.18 (m, 4H), 0.91 (s, 3H), 0.87 (s, 3H)。 6.在25℃下向Ph3
PMeBr (167 g,470 mmol)於THF (900 mL)中之溶液中添加t
-BuOK (52.7 g,470 mmol)。將反應混合物加熱至60℃且攪拌1小時。添加孕烯醇酮
(50 g,157 mmol)。在60℃下攪拌反應混合物1小時。添加飽和NH4
Cl (900 mL)。用EtOAc (2×1000 mL)萃取混合物。合併之有機層用鹽水(2×2000 mL)洗滌,經Na2
SO4
乾燥且真空濃縮,得到呈油狀物之粗產物,其藉由矽膠管柱層析(PE:EtOAc=20:1至5:1)純化,得到呈固體之S-200-INT_1
(45 g,91.2%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.40-5.30 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 3.60-3.40 (m, 1H), 2.40-2.20 (m, 2H), 2.05-1.90 (m, 2H), 1.85-1.60 (m, 9H), 1.53-1.40 (m, 5H), 1.25-0.90 (m, 9H), 0.59 (s, 3H)。 7.在20℃下向S-200-INT_1
(45 g,143 mmol)於DCM (1500 mL)中之溶液中添加DMP (108 g,257 mmol)。在20℃下攪拌混合物2小時。添加水(800 mL)且添加NaHCO3
(200 g固體)。過濾混合物。濾液用飽和Na2
S2
O3
(2×2000 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到S-200-INT_2
於DCM (100 mL)中之溶液,其直接用於下一步驟。 8.在10℃下向BHT (191 g,866 mmol)於甲苯(500 mL)中之溶液中添加AlMe3
(2 M於甲苯中,216 mL,433 mmol)且攪拌1小時。在-78℃下向混合物添加S-200-INT_2
(理論質量:44.6 g)於DCM (100 mL)中之溶液。在-78℃下攪拌混合物1小時。在-78℃下添加EtMgBr (141 mL,426 mmol)。在-78℃下攪拌混合物20 min。添加飽和檸檬酸(1 L)。有機相經分離,用鹽水(600 mL)洗滌,經Na2
SO4
乾燥且真空濃縮,得到粗產物,其藉由矽膠管柱層析(PE:EtOAc=50:1至30:1)純化,得到呈固體之S-200-INT_3a
(27 g,55%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.35-5.25 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.40-2.30 (m, 1H), 2.10-1.60 (m, 14H), 1.50-0.75 (m, 17H), 0.58 (s, 3H)。 9.將9-BBN二聚體(17.6 g,72.5 mmol)添加至S-200-INT_3E
(5 g,14.5 mmol)於THF (40 mL)中之溶液中。在60℃下於N2
下攪拌混合物3小時,且形成固體。向反應混合物添加乙醇(8.33 mL,145 mmol)及NaOH (28.9 mL,5 M,145 mmol)。混合物變得澄清。在25℃下逐滴添加H2
O2
(14.4 mL,10 M,145 mmol)且使內部溫度升至回流(75℃)。混合物冷卻,在添加且攪拌1小時之後,形成固體。在25℃下向混合物添加Na2
SO3
(20 mL,20%水溶液)。用EtOAc (2×100 mL)萃取混合物。合併之有機相用鹽水(2×200 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,且藉由矽膠管柱(PE/EtOAc=10/1至3/1)純化,以提供呈固體之S-200-INT_4E
(3.5 g,67%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.68-3.60 (m, 1H), 3.41-3.32 (m, 1H), 2.40-2.32 (m, 1H), 2.03-1.93 (m, 2H), 1.92-1.65 (m, 4H), 1.58-1.16 (m, 13H), 1.16-0.90 (m, 11H), 0.90-0.81 (m, 3H), 0.73-0.62 (s, 3H)。 10.在25℃下將DMP (4.66 g,11.0 mmol)添加至S-200-INT_4E
(2 g,5.54 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌反應混合物10 min。在25℃下用NaHCO3
飽和水溶液(30 mL)淬滅反應混合物。分離DCM層且用DCM (30 mL)萃取水相。合併之有機相用Na2
SO3
飽和水溶液(3×50 mL)、鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-200-INT_5E
(2.0 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.59-9.56 (m, 1H), 5.31-5.26 (m, 1H), 2.42-2.10 (m, 2H), 2.10-1.80 (m, 4H), 1.79-1.54 (m, 7H), 1.54-1.31 (m, 7H), 1.28-0.90 (m, 9H), 0.90-0.81 (m, 4H), 0.73 (s, 3H)。 11.在75℃下將S-500-6-29_2
(2.56 g,12.5 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (600 mg,25.0 mmol)及I2
(63.4 mg,0.25 mmol)於THF (3 mL)中之懸浮液中。在75℃下攪拌混合物1小時。冷卻後,在15℃下緩慢地添加S-500-6-1_1
(1 g,2.78 mmol)於THF (30 mL)中之溶液。添加之後,在15℃下攪拌混合物2小時,用飽和NH4
Cl (40 mL)及飽和檸檬酸(20 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之S-500-6-29_1
(800 mg,60%)之混合物。 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.19 (m, 1H), 3.88-3.71 (m, 1H), 2.42-2.29 (m, 1H), 2.07-1.86 (m, 4H), 1.78-1.59 (m, 4H), 1.54-1.31 (m, 13H), 1.29-1.13 (m, 8H), 1.12-0.99 (m, 8H), 0.94-0.79 (m, 13H), 0.68 (s, 3H)。 12.將DMP (1.39 g,3.30 mmol)添加至S-500-6-29_1
(800 mg,1.65 mmol)於DCM (30 mL)中之溶液中。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之粗產物S-500-6-29_2
(800 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.25 (m, 1H), 2.54-2.43 (m, 1H), 2.40-2.21 (m, 3H), 2.07-1.87 (m, 3H),1.81-1.57 (m, 7H), 1.53-1.39 (m, 7H), 1.38-1.29 (m, 3H), 1.27-1.16 (m, 4H), 1.15-1.04 (m, 8H), 1.03 (s, 3H), 1.00-0.92 (m, 2H), 0.91-0.80 (m, 9H), 0.69 (s, 3H)。 13.將NaBH4
(2.80 g,82.5 mmol)每五分鐘添加五次至S-500-6-29_2
(800 mg,1.65 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (50 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到49
(290 mg,36%)及呈固體之12
(120 mg,45%)。49: 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.07-1.87 (m, 4H), 1.76-1.69 (m, 1H), 1.66-1.55 (m, 5H), 1.53-1.42 (m, 7H), 1.41-1.31 (m, 5H), 1.30-1.12 (m, 8H), 1.11-1.05 (m, 3H), 1.03 (s, 3H), 1.01-0.92 (m, 2H), 0.91-0.82 (m, 12H), 0.68 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.718 min,30-90AB_E,純度98%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。12: 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.06-1.95 (m, 3H), 1.77-1.58 (m, 7H), 1.54-1.28 (m, 12H), 1.27-1.06 (m, 11H), 1.03 (s, 3H), 1.00-0.95 (m, 2H), 0.93-0.82 (m, 12H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.708 min,30-90AB_E,純度100%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。 14.將Pd(OH)2
(200 mg,無水)添加至49
(140 mg,0.288 mmol)於MeOH (30 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到59
(27 mg,19%)及呈固體之4
(42 mg,30%)。4: 1 H NMR
(400 MHz, CDCl3
) δ 3.84-3.76 (m, 1H), 1.98-1.85 (m, 2H), 1.69-1.54 (m, 9H), 1.53-1.46 (m, 3H), 1.45-1.28 (m, 9H), 1.27-1.20 (m, 4H), 1.19-1.13 (m, 5H), 1.12-1.02 (m, 4H), 1.01-0.92 (m, 2H), 0.91-0.85 (m, 12H), 0.82 (s, 3H), 0.70-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.799 min,30-90AB_E,純度100%,C33
H55
[M+H-H2
O]+
之MS ESI計算值451,實驗值451。實例 5 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (5) 之合成 
 1.在50℃至55℃下於N2
下將1-溴-3,3-二甲基丁烷(3.68 g,22.3 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (1.08 g,44.6 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液中。在55℃下攪拌混合物1小時。隨後在0℃下將S-500-6-1_1
(0.8 g,2.23 mmol)於THF (5 mL)中之溶液添加至新鮮製備的(3,3-二甲基丁基)溴化鎂(22.3 mmol於10 mL之THF中)。在15℃下攪拌混合物2小時。向混合物添加檸檬酸(20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到5
(580 mg,P1,58%)及54
(50 mg,5%,不純)。5: 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.65-3.54 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.84 (m, 4H), 1.76-1.38 (m, 15H), 1.38-1.00 (m, 12H), 0.93-0.80 (m, 15H), 0.70 (s, 3H)。54: 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.62-3.52 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.90 (m, 3H), 1.75-1.00 (m, 28H), 1.00-0.75 (m, 18H), 0.70 (s, 3H)。 2.在20℃下將DMP (1.1 g,2.6 mmol)及水(1滴)添加至5
(580 mg,1.3 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌混合物2 h。將NaHCO3
飽和溶液(20 mL)及Na2
S2
O3
(20 mL,飽和)添加至混合物中。用EtOAc (50 mL)萃取混合物。有機層用NaHCO3
/Na2
S2
O3
(20+20 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾,真空濃縮,得到呈固體之S-500-6-1_3
(520 mg,90%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.38-5.18 (m, 1H), 2.62-2.22 (m, 4H), 2.11-1.85 (m, 3H), 1.78-1.57 (m, 7H), 1.57-1.32 (m, 8H), 1.32-1.21 (m, 2H), 1.19-1.09 (m, 5H), 1.08-1.01 (m, 4H), 1.00-0.91 (m, 1H), 0.90-0.80 (m, 12H), 0.70 (s, 3H)。 3.在15℃下將NaBH4
(1.77 g,46.8 mmol)逐份添加至S-500-6-1_3
(520 mg,1.17 mmol)於THF (5 mL)及MeOH (10 mL)中之溶液中。在15℃下攪拌混合物20 min。混合物用NH4
Cl (20 mL,飽和水溶液)淬滅且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到5
(300 mg,不純)及54
(170 mg,不純)。 4.不純5
(300 mg,不純)藉由閃蒸塔(0%至12% EtOAc/PE)純化,得到固體。在60℃下使固體溶解於MeCN (50 mL)中且真空濃縮,得到呈固體之5
(270 mg,52%)。5: 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.67-3.54 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.84 (m, 4H), 1.78-1.57 (m, 5H), 1.55-1.38 (m, 12H), 1.38-1.07 (m, 7H), 1.03 (s, 3H), 0.93-0.89 (m, 12H), 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H)。 在7.0 min層析中,LCMS
Rt=5.587 min,30-90_AB_E,純度96.5%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 6 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-5- 環丙基 -3- 羥基戊 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (6) 之合成
1.在50℃至55℃下於N2
下將1-溴-3,3-二甲基丁烷(3.68 g,22.3 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (1.08 g,44.6 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液中。在55℃下攪拌混合物1小時。隨後在0℃下將S-500-6-1_1
(0.8 g,2.23 mmol)於THF (5 mL)中之溶液添加至新鮮製備的(3,3-二甲基丁基)溴化鎂(22.3 mmol於10 mL之THF中)。在15℃下攪拌混合物2小時。向混合物添加檸檬酸(20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到5
(580 mg,P1,58%)及54
(50 mg,5%,不純)。5: 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.65-3.54 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.84 (m, 4H), 1.76-1.38 (m, 15H), 1.38-1.00 (m, 12H), 0.93-0.80 (m, 15H), 0.70 (s, 3H)。54: 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.62-3.52 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.90 (m, 3H), 1.75-1.00 (m, 28H), 1.00-0.75 (m, 18H), 0.70 (s, 3H)。 2.在20℃下將DMP (1.1 g,2.6 mmol)及水(1滴)添加至5
(580 mg,1.3 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌混合物2 h。將NaHCO3
飽和溶液(20 mL)及Na2
S2
O3
(20 mL,飽和)添加至混合物中。用EtOAc (50 mL)萃取混合物。有機層用NaHCO3
/Na2
S2
O3
(20+20 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾,真空濃縮,得到呈固體之S-500-6-1_3
(520 mg,90%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.38-5.18 (m, 1H), 2.62-2.22 (m, 4H), 2.11-1.85 (m, 3H), 1.78-1.57 (m, 7H), 1.57-1.32 (m, 8H), 1.32-1.21 (m, 2H), 1.19-1.09 (m, 5H), 1.08-1.01 (m, 4H), 1.00-0.91 (m, 1H), 0.90-0.80 (m, 12H), 0.70 (s, 3H)。 3.在15℃下將NaBH4
(1.77 g,46.8 mmol)逐份添加至S-500-6-1_3
(520 mg,1.17 mmol)於THF (5 mL)及MeOH (10 mL)中之溶液中。在15℃下攪拌混合物20 min。混合物用NH4
Cl (20 mL,飽和水溶液)淬滅且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到5
(300 mg,不純)及54
(170 mg,不純)。 4.不純5
(300 mg,不純)藉由閃蒸塔(0%至12% EtOAc/PE)純化,得到固體。在60℃下使固體溶解於MeCN (50 mL)中且真空濃縮,得到呈固體之5
(270 mg,52%)。5: 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.67-3.54 (m, 1H), 2.41-2.31 (m, 1H), 2.11-1.84 (m, 4H), 1.78-1.57 (m, 5H), 1.55-1.38 (m, 12H), 1.38-1.07 (m, 7H), 1.03 (s, 3H), 0.93-0.89 (m, 12H), 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H)。 在7.0 min層析中,LCMS
Rt=5.587 min,30-90_AB_E,純度96.5%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 6 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-5- 環丙基 -3- 羥基戊 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (6) 之合成  1.在50℃至55℃下於N2
下將(2-溴乙基)環丙烷(1.8 g,12 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (641 mg,26.4 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液中。在55℃下攪拌1小時之後,用THF (10 mL)稀釋混合物。在0℃下將格林納溶液添加至S-500-6-1_1
(0.8 g,2.23 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌4小時之後,反應物用NH4
Cl (20 mL,10%水溶液)淬滅且用EtOAc (30 mL)萃取。有機層經分離且真空濃縮,得到呈固體之混合物(1g,粗產物),該混合物藉由閃蒸塔(0%至25% DCM/EtOAc (1/1)/PE)分離,得到S-500-6-20
(700 mg,73%,不純)及呈固體之S-500-6-19
(70 mg,7%,不純)。2.
在20℃下將DMP (1.38 g,3.26 mmol)及水(1滴)添加至S-500-6-20
(700 mg,1.63 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌2 h之後,混合物用NaHCO3
(20 mL,飽和)及Na2
S2
O3
(20 mL,飽和)處理且用EtOAc (50 mL)萃取。有機層用飽和NaHCO3
/Na2
S2
O3
(2× (20 mL/20 mL))洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-500-6-19_3
(700 mg,100%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.35-5.20 (m, 1H), 2.72-2.26 (m, 4H), 2.17-1.87 (m, 3H), 1.82-1.35 (m, 13H), 1.35-1.20 (m, 2H), 1.20-0.91 (m, 12H), 0.85 (t,J
= 7.2 Hz, 3H), 0.80-0.62 (m, 4H), 0.53-0.33 (m, 2H), 0.12-0.00 (m, 2H)。 3.在15℃下將NaBH4
(2.46 g,65.1 mmol)逐份添加至S-500-6-1_3
(700 mg,1.63 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌20 min之後,用NH4
Cl (20 mL,飽和水溶液)淬滅混合物且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到760 mg呈固體之混合物,其藉由閃蒸塔(0%至35% DCM/EtOAc (1/1)/PE)分離,得到69
(330 mg,47%)及呈固體之6
(250 mg,35%,不純)。不純6
(250 mg)進一步藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到呈固體之6
(170 mg,23%)。6: 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.24 (m, 1H), 3.77-3.66 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.91 (m, 3H), 1.79-1.59 (m, 6H), 1.55-1.21 (m, 14H), 1.21-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.95 (m, 1H), 0.93 (d,J
= 6.8 Hz, 3H) 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H), 0.68-0.62 (m, 1H), 0.49-0.38 (m, 2H), 0.11-0.02 (m, 2H)。 在2.0 min層析中,LCMS
Rt=1.380 min,30-90_AB_E,純度100%,C29
H47
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。實例 7 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1R,2S)-1- 羥基 -1-( 吡啶 -3- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (7) 之合成
1.在50℃至55℃下於N2
下將(2-溴乙基)環丙烷(1.8 g,12 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (641 mg,26.4 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液中。在55℃下攪拌1小時之後,用THF (10 mL)稀釋混合物。在0℃下將格林納溶液添加至S-500-6-1_1
(0.8 g,2.23 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌4小時之後,反應物用NH4
Cl (20 mL,10%水溶液)淬滅且用EtOAc (30 mL)萃取。有機層經分離且真空濃縮,得到呈固體之混合物(1g,粗產物),該混合物藉由閃蒸塔(0%至25% DCM/EtOAc (1/1)/PE)分離,得到S-500-6-20
(700 mg,73%,不純)及呈固體之S-500-6-19
(70 mg,7%,不純)。2.
在20℃下將DMP (1.38 g,3.26 mmol)及水(1滴)添加至S-500-6-20
(700 mg,1.63 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌2 h之後,混合物用NaHCO3
(20 mL,飽和)及Na2
S2
O3
(20 mL,飽和)處理且用EtOAc (50 mL)萃取。有機層用飽和NaHCO3
/Na2
S2
O3
(2× (20 mL/20 mL))洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-500-6-19_3
(700 mg,100%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.35-5.20 (m, 1H), 2.72-2.26 (m, 4H), 2.17-1.87 (m, 3H), 1.82-1.35 (m, 13H), 1.35-1.20 (m, 2H), 1.20-0.91 (m, 12H), 0.85 (t,J
= 7.2 Hz, 3H), 0.80-0.62 (m, 4H), 0.53-0.33 (m, 2H), 0.12-0.00 (m, 2H)。 3.在15℃下將NaBH4
(2.46 g,65.1 mmol)逐份添加至S-500-6-1_3
(700 mg,1.63 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌20 min之後,用NH4
Cl (20 mL,飽和水溶液)淬滅混合物且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到760 mg呈固體之混合物,其藉由閃蒸塔(0%至35% DCM/EtOAc (1/1)/PE)分離,得到69
(330 mg,47%)及呈固體之6
(250 mg,35%,不純)。不純6
(250 mg)進一步藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到呈固體之6
(170 mg,23%)。6: 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.24 (m, 1H), 3.77-3.66 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.91 (m, 3H), 1.79-1.59 (m, 6H), 1.55-1.21 (m, 14H), 1.21-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.95 (m, 1H), 0.93 (d,J
= 6.8 Hz, 3H) 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H), 0.68-0.62 (m, 1H), 0.49-0.38 (m, 2H), 0.11-0.02 (m, 2H)。 在2.0 min層析中,LCMS
Rt=1.380 min,30-90_AB_E,純度100%,C29
H47
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。實例 7 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1R,2S)-1- 羥基 -1-( 吡啶 -3- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (7) 之合成 
 1.在0℃下於N2
下向BHT (416 g,1.88 mol)於甲苯(1500 mL)中之溶液逐滴添加三甲基鋁(2 M於甲苯中,469 mL,939 mmol)。在0℃下攪拌混合物30 min且直接用作MAD
(0.47 M於甲苯中)之溶液而無需進一步純化。在-70℃下於N2
下向MAD
(0.47 M於甲苯中,2.01 L,945 mmol)之溶液逐滴添加N-005 _1
(100 g,315 mmol)於甲苯(800 mL)中之溶液。在-70℃下攪拌混合物30 min。向以上混合物逐滴添加EtMgBr (3 M於乙基醚中,315 mL,945 mmol)。在-70℃下攪拌所得混合物1小時。將反應混合物倒入冰冷卻之檸檬酸水溶液中(1000 mL),用EtOAc (2×600 mL)萃取。合併之有機層用鹽水(500 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠層析(0%至20% EtOAc/PE)純化,得到85 g呈固體之N-005_2
(78%產量)。 1 H NMR
(400 MHz, CDCl3
) δ 2.55-2.46 (m, 1H), 2.19-2.12 (m, 1H), 2.11-2.09 (m, 3H), 2.08-1.96 (m, 1H), 1.71-1.48 (m, 10H), 1.47-1.31 (m, 5H), 1.30-1.09 (m, 7H), 1.06-0.94 (m, 2H), 0.92-0.87 (m, 3H), 0.86-0.79 (m, 3H), 0.75-0.64 (m, 1H), 0.60(s, 3H)。2.
在15℃下於N2
下向MePPh3
Br (174 g,0.49 mol)於THF (1000 mL)中之懸浮液中添加t-BuOK (54.9 g,0.49 mol)。在50℃下攪拌30 min之後,在65℃以下逐份添加N-005_2
(85 g,245 mmol)於THF (800 mL)中之溶液。在50℃下攪拌混合物1小時,用NH4
Cl (1000 mL)淬滅,用EtOAc (2×900 mL)萃取。有機層經分離,真空濃縮,得到粗產物,該粗產物在50℃下自MeOH/水(1.5 L,1:1)濕磨。冷卻之後過濾混合物且濾餅用MeOH/水(2×500 mL,1:1)洗滌,真空濃縮,得到呈固體之N-005_3
(75 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.85-4.82 (m, 1H), 4.71-4.68 (m, 1H), 2.06-1.94 (m, 1H), 1.86-1.78 (m, 1H), 1.76-1.71 (m, 4H), 1.70-1.62 (m, 4H), 1.61-1.48 (m, 6H), 1.47-1.30 (m, 3H), 1.29-1.05 (m, 8H), 1.04-0.92 (m, 1H), 0.91-0.82 (m, 6H), 0.76-0.63 (m, 1H), 0.56 (s, 3H)。 3.於N2下向N-005_3
(75 g,217 mmol)於THF (1800 mL)中之溶液中添加9-BBN二聚體(105 g,434 mmol)。在60℃下攪拌混合物3小時。向反應混合物逐份添加乙醇(124 mL,2.17 mol)及NaOH水溶液(434 mL,5 M,2.17 mmol)。隨後在0℃下逐滴添加H2
O2
(217 mL,10 M,2.17 mol)。使混合物升溫至65℃且攪拌1小時且用水(1.5 L)稀釋。用EtOAc (2×800 mL)萃取反應混合物。合併之有機層添加飽和水溶液Na2
S2
O3
(600 mL)且攪拌1小時。反應物藉由碘化鉀澱粉測試紙檢查以確認過量的H2
O2
經破壞。隨後有機相用飽和鹽水(2×500 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-005_4
(78 g,粗產物)。在15℃下粗產物N-005_4
(78 g,不純)自MeOH/H2O=10/1濕磨,得到呈固體之N-005_4
(70 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.60 (m, 1H), 3.41-3.32 (m, 1H), 1.99-1.92 (m, 1H), 1.88-1.75 (m, 1H), 1.69-1.45 (m, 10H), 1.44-1.29 (m, 4H), 1.28-1.15 (m, 6H), 1.14-0.91 (m, 8H), 0.90-0.79 (m, 7H), 0.67 (s, 3H)。 4.向N-005_4
(70 g,193 mmol)於DCM (800 mL)中之溶液中添加DMP (122 g,289 mmol)。此後,在15℃下攪拌反應物30 min。反應混合物添加NaHCO3
飽和水溶液(500 ml)且在15℃下攪拌20 min。添加Na2
S2
O3
飽和水溶液(600 mL)且在15℃下再攪拌混合物1小時。反應物藉由碘化鉀澱粉測試紙檢查以確認過量的DMP經破壞。用DCM (2×400 mL)萃取水相。合併之有機層用NaHCO3
飽和水溶液(400 mL)及鹽水(400 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-005_5
(70 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 9.58-9.55 (m, 1H), 2.39-2.30 (m, 1H), 1.95-1.78 (m, 2H), 1.69-1.42 (m, 10H), 1.41-1.30 (m, 4H), 1.29-1.14 (m, 5H), 1.13-0.95 (m, 6H), 0.94-0.86 (m, 4H), 0.85-0.81 (m, 3H), 0.69 (m, 4H)。5.
將i-PrMgCl (2.49 mL,4.98 mmol,2 M於乙醚中)逐滴添加至3-溴吡啶(875 mg,5.54 mmol)於THF (5 mL)中之溶液中。在25℃下攪拌1 h之後,添加N-8-7_1
(200 mg,0.554 mmol)於THF (5 mL)中之溶液。在25℃下攪拌16小時之後,反應混合物用NH4
Cl (50 mL,10%水溶液)淬滅且用EtOAc (2×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由閃蒸塔(0%至50% EtOAc/DCM)純化,得到呈固體之N-8-19_1
(100 mg,41%)。6.N-8-19_1
(100 mg, 0.227 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:50-50% B (A= 0.05% NH3
/H2
O,B= MeOH),流速:80 mL/min)分離,得到7
(峰值1,57 mg,57%)及呈固體之89
(峰值2,8 mg,8%)。 在3 min層析中,SFC
峰值1:Rt=1.798 min且峰值2 Rt=1.985 min,AD-H_3UM_4_5_40_4ML (「Chiralpak AD-3 50×4.6mm I.D.,3um 移動相:A:CO2
,B:異丙醇(0.05% DEA),梯度:在1.4 min內B為5%至40%且保持40% 1.05 min,隨後B為5%持續0.35 min,流速:4 mL/min,管柱溫度:40℃」)。7: 1 H NMR
(400 MHz, CDCl3
) δ 8.56-8.52 (m,1H), 8.49-8.45 (m, 1H), 7.68-7.62 (m, 1H), 7.29-7.24 (m, 1H), 5.01-4.95 (m,1H), 2.11-2.01 (m, 1H), 1.96-1.89 (m, 1H), 1.83-1.76 (m, 1H), 1.73-1.63 (m, 4H), 1.59-1.47 (m, 6H),1.43-1.29 (m, 4H), 1.27-1.20 (m, 4H), 1.19-1.06 (m, 4H), 1.03-0.92 (m, 1H), 0.91-0.85 (m, 4H), 0.83 (s, 3H), 0.77-0.73 (m, 3H), 0.70-0.64 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.017 min,10-80AB_E,純度100%,C29
H46
NO2
[M+H]+
之MS ESI計算值440,實驗值440。 在3 min層析中,SFC
Rt=1.780 min,AD-H_3UM_4_5_40_4ML,100%de。實例 8 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (8) 之合成
1.在0℃下於N2
下向BHT (416 g,1.88 mol)於甲苯(1500 mL)中之溶液逐滴添加三甲基鋁(2 M於甲苯中,469 mL,939 mmol)。在0℃下攪拌混合物30 min且直接用作MAD
(0.47 M於甲苯中)之溶液而無需進一步純化。在-70℃下於N2
下向MAD
(0.47 M於甲苯中,2.01 L,945 mmol)之溶液逐滴添加N-005 _1
(100 g,315 mmol)於甲苯(800 mL)中之溶液。在-70℃下攪拌混合物30 min。向以上混合物逐滴添加EtMgBr (3 M於乙基醚中,315 mL,945 mmol)。在-70℃下攪拌所得混合物1小時。將反應混合物倒入冰冷卻之檸檬酸水溶液中(1000 mL),用EtOAc (2×600 mL)萃取。合併之有機層用鹽水(500 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠層析(0%至20% EtOAc/PE)純化,得到85 g呈固體之N-005_2
(78%產量)。 1 H NMR
(400 MHz, CDCl3
) δ 2.55-2.46 (m, 1H), 2.19-2.12 (m, 1H), 2.11-2.09 (m, 3H), 2.08-1.96 (m, 1H), 1.71-1.48 (m, 10H), 1.47-1.31 (m, 5H), 1.30-1.09 (m, 7H), 1.06-0.94 (m, 2H), 0.92-0.87 (m, 3H), 0.86-0.79 (m, 3H), 0.75-0.64 (m, 1H), 0.60(s, 3H)。2.
在15℃下於N2
下向MePPh3
Br (174 g,0.49 mol)於THF (1000 mL)中之懸浮液中添加t-BuOK (54.9 g,0.49 mol)。在50℃下攪拌30 min之後,在65℃以下逐份添加N-005_2
(85 g,245 mmol)於THF (800 mL)中之溶液。在50℃下攪拌混合物1小時,用NH4
Cl (1000 mL)淬滅,用EtOAc (2×900 mL)萃取。有機層經分離,真空濃縮,得到粗產物,該粗產物在50℃下自MeOH/水(1.5 L,1:1)濕磨。冷卻之後過濾混合物且濾餅用MeOH/水(2×500 mL,1:1)洗滌,真空濃縮,得到呈固體之N-005_3
(75 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.85-4.82 (m, 1H), 4.71-4.68 (m, 1H), 2.06-1.94 (m, 1H), 1.86-1.78 (m, 1H), 1.76-1.71 (m, 4H), 1.70-1.62 (m, 4H), 1.61-1.48 (m, 6H), 1.47-1.30 (m, 3H), 1.29-1.05 (m, 8H), 1.04-0.92 (m, 1H), 0.91-0.82 (m, 6H), 0.76-0.63 (m, 1H), 0.56 (s, 3H)。 3.於N2下向N-005_3
(75 g,217 mmol)於THF (1800 mL)中之溶液中添加9-BBN二聚體(105 g,434 mmol)。在60℃下攪拌混合物3小時。向反應混合物逐份添加乙醇(124 mL,2.17 mol)及NaOH水溶液(434 mL,5 M,2.17 mmol)。隨後在0℃下逐滴添加H2
O2
(217 mL,10 M,2.17 mol)。使混合物升溫至65℃且攪拌1小時且用水(1.5 L)稀釋。用EtOAc (2×800 mL)萃取反應混合物。合併之有機層添加飽和水溶液Na2
S2
O3
(600 mL)且攪拌1小時。反應物藉由碘化鉀澱粉測試紙檢查以確認過量的H2
O2
經破壞。隨後有機相用飽和鹽水(2×500 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-005_4
(78 g,粗產物)。在15℃下粗產物N-005_4
(78 g,不純)自MeOH/H2O=10/1濕磨,得到呈固體之N-005_4
(70 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.60 (m, 1H), 3.41-3.32 (m, 1H), 1.99-1.92 (m, 1H), 1.88-1.75 (m, 1H), 1.69-1.45 (m, 10H), 1.44-1.29 (m, 4H), 1.28-1.15 (m, 6H), 1.14-0.91 (m, 8H), 0.90-0.79 (m, 7H), 0.67 (s, 3H)。 4.向N-005_4
(70 g,193 mmol)於DCM (800 mL)中之溶液中添加DMP (122 g,289 mmol)。此後,在15℃下攪拌反應物30 min。反應混合物添加NaHCO3
飽和水溶液(500 ml)且在15℃下攪拌20 min。添加Na2
S2
O3
飽和水溶液(600 mL)且在15℃下再攪拌混合物1小時。反應物藉由碘化鉀澱粉測試紙檢查以確認過量的DMP經破壞。用DCM (2×400 mL)萃取水相。合併之有機層用NaHCO3
飽和水溶液(400 mL)及鹽水(400 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-005_5
(70 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 9.58-9.55 (m, 1H), 2.39-2.30 (m, 1H), 1.95-1.78 (m, 2H), 1.69-1.42 (m, 10H), 1.41-1.30 (m, 4H), 1.29-1.14 (m, 5H), 1.13-0.95 (m, 6H), 0.94-0.86 (m, 4H), 0.85-0.81 (m, 3H), 0.69 (m, 4H)。5.
將i-PrMgCl (2.49 mL,4.98 mmol,2 M於乙醚中)逐滴添加至3-溴吡啶(875 mg,5.54 mmol)於THF (5 mL)中之溶液中。在25℃下攪拌1 h之後,添加N-8-7_1
(200 mg,0.554 mmol)於THF (5 mL)中之溶液。在25℃下攪拌16小時之後,反應混合物用NH4
Cl (50 mL,10%水溶液)淬滅且用EtOAc (2×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由閃蒸塔(0%至50% EtOAc/DCM)純化,得到呈固體之N-8-19_1
(100 mg,41%)。6.N-8-19_1
(100 mg, 0.227 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:50-50% B (A= 0.05% NH3
/H2
O,B= MeOH),流速:80 mL/min)分離,得到7
(峰值1,57 mg,57%)及呈固體之89
(峰值2,8 mg,8%)。 在3 min層析中,SFC
峰值1:Rt=1.798 min且峰值2 Rt=1.985 min,AD-H_3UM_4_5_40_4ML (「Chiralpak AD-3 50×4.6mm I.D.,3um 移動相:A:CO2
,B:異丙醇(0.05% DEA),梯度:在1.4 min內B為5%至40%且保持40% 1.05 min,隨後B為5%持續0.35 min,流速:4 mL/min,管柱溫度:40℃」)。7: 1 H NMR
(400 MHz, CDCl3
) δ 8.56-8.52 (m,1H), 8.49-8.45 (m, 1H), 7.68-7.62 (m, 1H), 7.29-7.24 (m, 1H), 5.01-4.95 (m,1H), 2.11-2.01 (m, 1H), 1.96-1.89 (m, 1H), 1.83-1.76 (m, 1H), 1.73-1.63 (m, 4H), 1.59-1.47 (m, 6H),1.43-1.29 (m, 4H), 1.27-1.20 (m, 4H), 1.19-1.06 (m, 4H), 1.03-0.92 (m, 1H), 0.91-0.85 (m, 4H), 0.83 (s, 3H), 0.77-0.73 (m, 3H), 0.70-0.64 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.017 min,10-80AB_E,純度100%,C29
H46
NO2
[M+H]+
之MS ESI計算值440,實驗值440。 在3 min層析中,SFC
Rt=1.780 min,AD-H_3UM_4_5_40_4ML,100%de。實例 8 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (8) 之合成 
 1.
在25℃以下於N2
氛圍下向BHT (1.97 kg,8.94 mol)於甲苯(1 L)中之溶液中逐滴添加AlMe3
(2.14 L,2.0 M於甲苯中,4.28 mol)。在25℃下攪拌所得混合物1小時。在-70℃下添加於DCM (3 L)中之S-200-INT_2
(794 g,85%重量百分比,2.16 mol)。在-70℃下攪拌混合物1小時。在-70℃下添加MeMgBr (862 mL,3.0 M於二乙醚中,2.59 mol)。在-70℃下攪拌反應混合物10 min。混合物用飽和檸檬酸(3 L)淬滅,用EtOAc (2×2 L)萃取。合併之有機相用鹽水(2 L)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到殘餘物,其在25℃下自MeCN (3 L)濕磨得到呈固體之S-200-INT_3
(340 g,43%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.34-
5.26 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.50-2.35 (m, 1H), 2.07-
1.94 (m, 3H), 1.91-
1.84 (m, 1H), 1.83-
1.63 (m, 8H), 1.58-
1.33 (m, 6H), 1.27-
1.13 (m, 3H), 1.12 (s, 3H), 1.10-
1.05 (m, 1H), 1.02 (s, 3H), 1.00-
0.92 (m, 1H), 0.58 (s, 3H)。2.
在15℃下於N2
下向S-200-INT_3
(149 g,453 mmol)及9-BBN二聚體(127 g,520 mmol)之混合物添加THF (1 L)。在60℃下攪拌反應混合物1小時。將混合物冷卻至15℃。在15℃下添加EtOH (208 g,4.53 mol)。在15℃下逐滴添加NaOH水溶液(906 mL,5 M,4.53 mol)。在15℃下逐滴添加H2
O2
(514 g,30%,4.53 mol)。在60℃下攪拌所得混合物1小時。生產一種固體。用乙醇(200 mL)洗滌該固體,得到固體,其在80℃下在回流下用EtOH (2.3 L)及水(2.5 L)連續濕磨,得到呈固體之15-3b
(131 g,84%)。自乙醇之濾液真空濃縮,得到呈固體之15-
3b
(30 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
)δ5.35-
5.24 (m, 1H), 3.67-
3.61 (m, 1H), 3.42-
3.33 (m, 1H), 2.50-2.35 (m, 1H), 2.07-
1.92 (m, 3H), 1.88-
1.65 (m, 3H), 1.60-
1.38 (m, 9H), 1.37-
1.26 (m, 1H), 1.26-
1.12 (m, 4H), 1.11 (s, 3H), 1.08 (s, 1H), 1.05 (d,J
= 6.8 Hz, 3H), 1.01 (s, 3H), 1.00-
0.91 (m, 1H), 0.70 (s, 3H)。 3.將DMP (2.44 g,5.76 mmol)添加至15-3b
(1 g,2.88 mmol)於DCM (10 mL)中之溶液中。此後,在25℃下攪拌反應物10 min。藉由添加NaHCO3
飽和水溶液(20 mL)及Na2
S2
O3
飽和水溶液(20 mL)來淬滅反應混合物,用DCM (2×50 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3×50 mL)及鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-500-2-9_1
(1 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.57 (brs, 1H), 5.35-5.25 (m, 1H), 2.50-2.30 (m, 2H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.75-1.65 (m, 1H), 1.65-1.60 (m, 3H), 1.55-1.50 (m, 2H), 1.50-1.40 (m, 2H), 1.40-1.30 (m, 1H), 1.25-1.20 (m, 2H), 1.20-1.15 (m, 2H), 1.15-1.10 (m, 6H), 1.05-0.95 (m, 5H), 0.90-0.70 (m, 1H), 0.68 (s, 3H)。 4.在60℃下攪拌鎂(641 mg,26.4 mmol)及I2
(33.5 mg,0.132 mmol)之混合物,且於N2
下逐滴添加異戊基溴化鎂(2 g,13.2 mmol)於THF (20 mL)中之溶液。此後,在60℃下攪拌反應混合物1小時。反應混合物直接用作異戊基溴化鎂溶液而無需任何純化。在0℃下於N2
下向S-500-2-9_1
(1 g,2.90 mmol)於THF (10 mL)中之溶液添加格林納溶液。此後,在25℃下攪拌反應混合物1小時。反應混合物添加NH4
Cl飽和水溶液(50 mL),用EtOAc (2 × 50 mL)萃取,用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(EtOAc/ PE=1/4)純化,得到呈固體之不純S-500-2-9-1A
(560 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 5.28-5.25 (m, 1H), 3.90-3.80 (m, 0.25H), 3.68-3.58 (m, 0.75H), 2.48-2.36 (m, 1H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.80-1.75 (m, 1H), 1.75-1.52 (m, 6H) 1.52-1.42 (m, 6H), 1.42-1.32 (m, 3H), 1.32-1.22 (m, 3H), 1.22-1.12 (m, 3H), 1.12-1.02 (m, 2H), 1.01 (s, 3H), 1.00-0.92 (m, 1H), 0.92-0.85 (m, 9H), 0.85-0.77 (m, 1H), 0.69 (s, 3H)。 5.S-500-2-9-1A
(560 mg)藉由SFC (管柱:Chiralcel OD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃)純化,得到呈固體之不純30
(160 mg)及呈固體之75
(265 mg,47%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.30 (m, 1H), 3.70-3.60 (m, 1H), 2.50-2.40 (m, 1H), 2.05-1.90 (m, 4H), 1.85-1.75 (m, 2H), 1.75-1.60 (m, 1H), 1.55-1.45 (m, 8H), 1.45-1.25 (m, 8H), 1.25-1.10 (m, 4H), 1.10-1.05 (m, 2H), 1.02 (s, 3H), 0.99-0.91 (m, 3H), 0.91-0.89 (m, 4H), 0.88 (s, 3H), 0.69 (s, 3H)。 在1.5 min層析中,LCMS
Rt=1.162 min,5-95 AB,純度99%,C28
H45
[M+H-2H2
O]+
之MS ESI計算值381,實驗值381。 6.將無水Pd(OH)2
/C (100 mg)添加至75
(230 mg,0.551 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液中。在50℃下於H2
及50 Psi下攪拌反應混合物24 h。此後,HNMR顯示反應完成。反應混合物用濾紙過濾且真空濃縮,得到不純產物。不純產物用MeCN (3 mL)再結晶得到呈灰白色固體之8
(68 mg,30%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.65-3.55 (m, 1H), 2.00-1.80 (m, 2H), 1.76-1.60 (m, 3H), 1.55-1.48 (m, 3H), 1.48-1.38 (m, 4H), 1.38-1.26 (m, 7H), 1.26-1.23 (m, 4H), 1.23-1.06 (m, 5H), 1.06-1.02 (m, 3H), 1.02-095 (m, 1H), 0.95-0.85 (m, 10H), 0.81 (s, 3H), 0.70-0.60 (m, 4H)。 在1.5 min層析中,LCMS
Rt=1.171 min,5-95 AB,純度100%。 C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 9 : (3S,5R,8R,9S,10S,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 - 17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (9) 之合成
1.
在25℃以下於N2
氛圍下向BHT (1.97 kg,8.94 mol)於甲苯(1 L)中之溶液中逐滴添加AlMe3
(2.14 L,2.0 M於甲苯中,4.28 mol)。在25℃下攪拌所得混合物1小時。在-70℃下添加於DCM (3 L)中之S-200-INT_2
(794 g,85%重量百分比,2.16 mol)。在-70℃下攪拌混合物1小時。在-70℃下添加MeMgBr (862 mL,3.0 M於二乙醚中,2.59 mol)。在-70℃下攪拌反應混合物10 min。混合物用飽和檸檬酸(3 L)淬滅,用EtOAc (2×2 L)萃取。合併之有機相用鹽水(2 L)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到殘餘物,其在25℃下自MeCN (3 L)濕磨得到呈固體之S-200-INT_3
(340 g,43%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.34-
5.26 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.50-2.35 (m, 1H), 2.07-
1.94 (m, 3H), 1.91-
1.84 (m, 1H), 1.83-
1.63 (m, 8H), 1.58-
1.33 (m, 6H), 1.27-
1.13 (m, 3H), 1.12 (s, 3H), 1.10-
1.05 (m, 1H), 1.02 (s, 3H), 1.00-
0.92 (m, 1H), 0.58 (s, 3H)。2.
在15℃下於N2
下向S-200-INT_3
(149 g,453 mmol)及9-BBN二聚體(127 g,520 mmol)之混合物添加THF (1 L)。在60℃下攪拌反應混合物1小時。將混合物冷卻至15℃。在15℃下添加EtOH (208 g,4.53 mol)。在15℃下逐滴添加NaOH水溶液(906 mL,5 M,4.53 mol)。在15℃下逐滴添加H2
O2
(514 g,30%,4.53 mol)。在60℃下攪拌所得混合物1小時。生產一種固體。用乙醇(200 mL)洗滌該固體,得到固體,其在80℃下在回流下用EtOH (2.3 L)及水(2.5 L)連續濕磨,得到呈固體之15-3b
(131 g,84%)。自乙醇之濾液真空濃縮,得到呈固體之15-
3b
(30 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
)δ5.35-
5.24 (m, 1H), 3.67-
3.61 (m, 1H), 3.42-
3.33 (m, 1H), 2.50-2.35 (m, 1H), 2.07-
1.92 (m, 3H), 1.88-
1.65 (m, 3H), 1.60-
1.38 (m, 9H), 1.37-
1.26 (m, 1H), 1.26-
1.12 (m, 4H), 1.11 (s, 3H), 1.08 (s, 1H), 1.05 (d,J
= 6.8 Hz, 3H), 1.01 (s, 3H), 1.00-
0.91 (m, 1H), 0.70 (s, 3H)。 3.將DMP (2.44 g,5.76 mmol)添加至15-3b
(1 g,2.88 mmol)於DCM (10 mL)中之溶液中。此後,在25℃下攪拌反應物10 min。藉由添加NaHCO3
飽和水溶液(20 mL)及Na2
S2
O3
飽和水溶液(20 mL)來淬滅反應混合物,用DCM (2×50 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3×50 mL)及鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-500-2-9_1
(1 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.57 (brs, 1H), 5.35-5.25 (m, 1H), 2.50-2.30 (m, 2H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.75-1.65 (m, 1H), 1.65-1.60 (m, 3H), 1.55-1.50 (m, 2H), 1.50-1.40 (m, 2H), 1.40-1.30 (m, 1H), 1.25-1.20 (m, 2H), 1.20-1.15 (m, 2H), 1.15-1.10 (m, 6H), 1.05-0.95 (m, 5H), 0.90-0.70 (m, 1H), 0.68 (s, 3H)。 4.在60℃下攪拌鎂(641 mg,26.4 mmol)及I2
(33.5 mg,0.132 mmol)之混合物,且於N2
下逐滴添加異戊基溴化鎂(2 g,13.2 mmol)於THF (20 mL)中之溶液。此後,在60℃下攪拌反應混合物1小時。反應混合物直接用作異戊基溴化鎂溶液而無需任何純化。在0℃下於N2
下向S-500-2-9_1
(1 g,2.90 mmol)於THF (10 mL)中之溶液添加格林納溶液。此後,在25℃下攪拌反應混合物1小時。反應混合物添加NH4
Cl飽和水溶液(50 mL),用EtOAc (2 × 50 mL)萃取,用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(EtOAc/ PE=1/4)純化,得到呈固體之不純S-500-2-9-1A
(560 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 5.28-5.25 (m, 1H), 3.90-3.80 (m, 0.25H), 3.68-3.58 (m, 0.75H), 2.48-2.36 (m, 1H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.80-1.75 (m, 1H), 1.75-1.52 (m, 6H) 1.52-1.42 (m, 6H), 1.42-1.32 (m, 3H), 1.32-1.22 (m, 3H), 1.22-1.12 (m, 3H), 1.12-1.02 (m, 2H), 1.01 (s, 3H), 1.00-0.92 (m, 1H), 0.92-0.85 (m, 9H), 0.85-0.77 (m, 1H), 0.69 (s, 3H)。 5.S-500-2-9-1A
(560 mg)藉由SFC (管柱:Chiralcel OD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃)純化,得到呈固體之不純30
(160 mg)及呈固體之75
(265 mg,47%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.30 (m, 1H), 3.70-3.60 (m, 1H), 2.50-2.40 (m, 1H), 2.05-1.90 (m, 4H), 1.85-1.75 (m, 2H), 1.75-1.60 (m, 1H), 1.55-1.45 (m, 8H), 1.45-1.25 (m, 8H), 1.25-1.10 (m, 4H), 1.10-1.05 (m, 2H), 1.02 (s, 3H), 0.99-0.91 (m, 3H), 0.91-0.89 (m, 4H), 0.88 (s, 3H), 0.69 (s, 3H)。 在1.5 min層析中,LCMS
Rt=1.162 min,5-95 AB,純度99%,C28
H45
[M+H-2H2
O]+
之MS ESI計算值381,實驗值381。 6.將無水Pd(OH)2
/C (100 mg)添加至75
(230 mg,0.551 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液中。在50℃下於H2
及50 Psi下攪拌反應混合物24 h。此後,HNMR顯示反應完成。反應混合物用濾紙過濾且真空濃縮,得到不純產物。不純產物用MeCN (3 mL)再結晶得到呈灰白色固體之8
(68 mg,30%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.65-3.55 (m, 1H), 2.00-1.80 (m, 2H), 1.76-1.60 (m, 3H), 1.55-1.48 (m, 3H), 1.48-1.38 (m, 4H), 1.38-1.26 (m, 7H), 1.26-1.23 (m, 4H), 1.23-1.06 (m, 5H), 1.06-1.02 (m, 3H), 1.02-095 (m, 1H), 0.95-0.85 (m, 10H), 0.81 (s, 3H), 0.70-0.60 (m, 4H)。 在1.5 min層析中,LCMS
Rt=1.171 min,5-95 AB,純度100%。 C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 9 : (3S,5R,8R,9S,10S,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 - 17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (9) 之合成 
 1.將Pd/C (5 g,<1%水)添加至N-004-022_1
(50 g,151 mmol)於MeOH (100 mL)及THF (100 mL)中之溶液中。隨後在25℃下於30 psi氫氣下氫化溶液48小時。混合物經由矽藻土墊過濾且真空濃縮濾液,得到呈固體之N-004-022_2
(50 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.65-3.55 (m, 1H), 3.40-3.3 (m, 1H), 2.80-2.60 (m, 1H), 2.40-2.30 (m, 1H), 2.25-2.10 (m, 1H), 2.10-1.95 (m, 3H), 1.80-1.65 (m, 3H), 1.65-1.53 (m, 1H), 1.53-1.40 (m, 4H), 1.40-1.01 (m, 17H), 0.70 (s, 3H)。 2.在60℃下於N2
下加熱三甲基氧化鋶碘(19.8 g,90.2 mmol)及t-BuOK (10.1 g,90.2 mmol)於DMSO (200 mL)中之經攪拌溶液1小時。將N-004-022_2
(15 g,45.1 mmol)添加至反應混合物中且在60℃下攪拌10 min。用水(1000 mL)處理反應物且用EtOAc (2×500 mL)萃取。合併之有機相用水(2×500 mL)、鹽水(300 mL)洗滌,經無水Na2
SO4
乾燥,過濾,真空濃縮,得到呈固體之N-004-022_3
(15.5 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.18-4.08 (m, 1H), 3.67-3.60 (m, 1H), 3.40-3.30 (m, 1H), 2.70-2.50 (m, 3H), 2.40-2.30 (m, 1H), 2.01-1.50 (m, 14H), 1.40-0.65 (m, 14H), 0.68 (s, 3H)。 3.在25℃下於N2
下將MeONa (12.0 g,223 mmol)添加至N-004-022_3
(15.5 g,44.7 mmol)於MeOH (500 mL)中之溶液中。在70℃回流下於N2
下攪拌混合物16小時。用水(500 mL)處理反應物。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-004-022_4
(15 g,粗產物)。粗產物N-004-022_4
(15 g)藉由矽膠層析(PE/EtOAc=10/1至5/1)純化,得到呈固體之N-004-022_4
(7.4 g,50%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.76-3.73 (m, 1H), 3.64-3.60 (m, 1H), 3.40-3.33 (m, 4H), 3.22-3.16 (m, 2H), 2.01-1.69 (m, 6H), 1.62-1.51 (m, 4H), 1.44-1.31 (m, 13H), 1.10-0.99 (m, 5H), 0.97 (s, 3H), 0.67 (s, 3H)。 4.將DMP (1.56 g,3.69 mmol)添加至N-004-022_4
(1.4 g,3.69 mmol)於DCM (15 mL)中之溶液中。此後,在25℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(20 mL)淬滅直至pH=9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×10 mL)、NaHCO3
飽和溶液(10 mL)、鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物用MeCN (10 mL)濕磨,得到呈固體之N-004-022_5
(700,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 9.56-9.58 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.40-2.31 (m, 1H), 2.01-1.50 (m, 11H), 1.47-1.01 (m, 16H), 0.97 (s, 3H), 0.70 (s, 3H)。
1.將Pd/C (5 g,<1%水)添加至N-004-022_1
(50 g,151 mmol)於MeOH (100 mL)及THF (100 mL)中之溶液中。隨後在25℃下於30 psi氫氣下氫化溶液48小時。混合物經由矽藻土墊過濾且真空濃縮濾液,得到呈固體之N-004-022_2
(50 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.65-3.55 (m, 1H), 3.40-3.3 (m, 1H), 2.80-2.60 (m, 1H), 2.40-2.30 (m, 1H), 2.25-2.10 (m, 1H), 2.10-1.95 (m, 3H), 1.80-1.65 (m, 3H), 1.65-1.53 (m, 1H), 1.53-1.40 (m, 4H), 1.40-1.01 (m, 17H), 0.70 (s, 3H)。 2.在60℃下於N2
下加熱三甲基氧化鋶碘(19.8 g,90.2 mmol)及t-BuOK (10.1 g,90.2 mmol)於DMSO (200 mL)中之經攪拌溶液1小時。將N-004-022_2
(15 g,45.1 mmol)添加至反應混合物中且在60℃下攪拌10 min。用水(1000 mL)處理反應物且用EtOAc (2×500 mL)萃取。合併之有機相用水(2×500 mL)、鹽水(300 mL)洗滌,經無水Na2
SO4
乾燥,過濾,真空濃縮,得到呈固體之N-004-022_3
(15.5 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.18-4.08 (m, 1H), 3.67-3.60 (m, 1H), 3.40-3.30 (m, 1H), 2.70-2.50 (m, 3H), 2.40-2.30 (m, 1H), 2.01-1.50 (m, 14H), 1.40-0.65 (m, 14H), 0.68 (s, 3H)。 3.在25℃下於N2
下將MeONa (12.0 g,223 mmol)添加至N-004-022_3
(15.5 g,44.7 mmol)於MeOH (500 mL)中之溶液中。在70℃回流下於N2
下攪拌混合物16小時。用水(500 mL)處理反應物。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-004-022_4
(15 g,粗產物)。粗產物N-004-022_4
(15 g)藉由矽膠層析(PE/EtOAc=10/1至5/1)純化,得到呈固體之N-004-022_4
(7.4 g,50%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.76-3.73 (m, 1H), 3.64-3.60 (m, 1H), 3.40-3.33 (m, 4H), 3.22-3.16 (m, 2H), 2.01-1.69 (m, 6H), 1.62-1.51 (m, 4H), 1.44-1.31 (m, 13H), 1.10-0.99 (m, 5H), 0.97 (s, 3H), 0.67 (s, 3H)。 4.將DMP (1.56 g,3.69 mmol)添加至N-004-022_4
(1.4 g,3.69 mmol)於DCM (15 mL)中之溶液中。此後,在25℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(20 mL)淬滅直至pH=9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×10 mL)、NaHCO3
飽和溶液(10 mL)、鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物用MeCN (10 mL)濕磨,得到呈固體之N-004-022_5
(700,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 9.56-9.58 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.40-2.31 (m, 1H), 2.01-1.50 (m, 11H), 1.47-1.01 (m, 16H), 0.97 (s, 3H), 0.70 (s, 3H)。 5.在0℃下於N2
下向N-004-022_5
(200 mg,0.531 mmol)、CsF (40.2 mg,0.265 mmol)於THF (5 mL))於中之溶液添加TMSCF3
(187 mg,1.32 mmol)。在25℃下攪拌混合物1小時。向混合物添加TBAF.3H2O (836 mg,2.65 mmol)。在25℃下攪拌2小時之後,用50% NH4
Cl (20 mL)淬滅混合物且用EtOAc (2 × 10 mL)萃取。合併之有機相用鹽水(20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA=10/1)純化,得到9
(56 mg,24%)及呈白色固體之71
(30 mg,不純)。9: 1 H NMR
(400 MHz, CDCl3
) δ 4.05-3.95 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.00-1.83 (m, 5H), 1.77-1.68 (m, 2H), 1.64-1.47 (m, 8H), 1.43-1.35 (m, 5H), 1.31-1.08 (m, 6H), 1.06-1.00 (m, 3H), 0.97 (s, 3H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.156 min,30-90AB_2min.lcm,純度100%,C25
H41
F3
O3
[M+Na]+
之MS ESI計算值469,實驗值469。實例 10 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1S,2S)-1- 環丙基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (10) 之合成
5.在0℃下於N2
下向N-004-022_5
(200 mg,0.531 mmol)、CsF (40.2 mg,0.265 mmol)於THF (5 mL))於中之溶液添加TMSCF3
(187 mg,1.32 mmol)。在25℃下攪拌混合物1小時。向混合物添加TBAF.3H2O (836 mg,2.65 mmol)。在25℃下攪拌2小時之後,用50% NH4
Cl (20 mL)淬滅混合物且用EtOAc (2 × 10 mL)萃取。合併之有機相用鹽水(20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA=10/1)純化,得到9
(56 mg,24%)及呈白色固體之71
(30 mg,不純)。9: 1 H NMR
(400 MHz, CDCl3
) δ 4.05-3.95 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.00-1.83 (m, 5H), 1.77-1.68 (m, 2H), 1.64-1.47 (m, 8H), 1.43-1.35 (m, 5H), 1.31-1.08 (m, 6H), 1.06-1.00 (m, 3H), 0.97 (s, 3H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.156 min,30-90AB_2min.lcm,純度100%,C25
H41
F3
O3
[M+Na]+
之MS ESI計算值469,實驗值469。實例 10 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1S,2S)-1- 環丙基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (10) 之合成 
 1.
在0℃下將於THF (5 mL)中之N-8-7_1
(500 mg,1.38 mmol)添加至環丙基溴化鎂(1 g,13.7 mL,0.5M於THF中)於THF (5 mL)中之溶液中且在25℃下攪拌4小時。混合物添加NH4
Cl (20 mL,10%水溶液)且用EtOAc (2×30 mL)萃取。有機層經分離且真空濃縮,得到殘餘物。殘餘物藉由矽膠層析純化用PE/EtOAc=1/1溶離,得到呈固體之N-8-13_1
(140 mg,25%)。 在2.0 min層析中,LCMS
Rt=1.192 min,30-90AB_2MIN_E,純度99%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 2.將DMP (294 mg,0.694 mmol)添加至N-8-13_1
(140 mg,0.347 mmol)於DCM (5 mL)中之溶液中。此後,在25℃下攪拌反應混合物1 h。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH變成約9為止。過濾混合物。分離DCM層且用DCM (100 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×100 mL)、飽和NaHCO3
(100 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-8-13_2
(140 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 2.65-2.55 (m, 1H), 1.95-1.90 (m, 2H), 1.50-1.15 (m, 19H), 1.14-0.95 (m, 7H), 0.94-0.80 (m, 12H), 0.69 (s, 3H)。 3.向N-8-13_2 (140 mg,0.347 mmol)於MeOH (1 mL)及THF (1 mL)中之溶液每五分鐘添加五次NaBH4 (1.18 g,17.4 mmol)。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (50 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之10
(26 mg,19%)及呈固體之90
(12 mg,9%)。10: 1 H NMR
(400 MHz, CDCl3
) δ 2.85-2.80 (m, 1H), 2.00-1.95 (m, 1H), 1.90-1.80 (m, 1H), 1.55-1.10 (m, 16H), 1.09-0.80 (m, 17H), 0.70-0.60 (m, 5H), 0.58-0.43 (m, 3H), 0.32-0.34 (m, 1H), 0.13-0.06 (m, 1H)。 在7.0 min層析中,LCMS
Rt=3.840 min,30-90AB_7MIN_E,純度97%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在30 min層析中,HPLC
Rt=13.470 min,70-90AB_1_30MIN.M,純度97%。實例 11 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (11) 之合成
1.
在0℃下將於THF (5 mL)中之N-8-7_1
(500 mg,1.38 mmol)添加至環丙基溴化鎂(1 g,13.7 mL,0.5M於THF中)於THF (5 mL)中之溶液中且在25℃下攪拌4小時。混合物添加NH4
Cl (20 mL,10%水溶液)且用EtOAc (2×30 mL)萃取。有機層經分離且真空濃縮,得到殘餘物。殘餘物藉由矽膠層析純化用PE/EtOAc=1/1溶離,得到呈固體之N-8-13_1
(140 mg,25%)。 在2.0 min層析中,LCMS
Rt=1.192 min,30-90AB_2MIN_E,純度99%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 2.將DMP (294 mg,0.694 mmol)添加至N-8-13_1
(140 mg,0.347 mmol)於DCM (5 mL)中之溶液中。此後,在25℃下攪拌反應混合物1 h。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH變成約9為止。過濾混合物。分離DCM層且用DCM (100 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×100 mL)、飽和NaHCO3
(100 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-8-13_2
(140 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 2.65-2.55 (m, 1H), 1.95-1.90 (m, 2H), 1.50-1.15 (m, 19H), 1.14-0.95 (m, 7H), 0.94-0.80 (m, 12H), 0.69 (s, 3H)。 3.向N-8-13_2 (140 mg,0.347 mmol)於MeOH (1 mL)及THF (1 mL)中之溶液每五分鐘添加五次NaBH4 (1.18 g,17.4 mmol)。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (50 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之10
(26 mg,19%)及呈固體之90
(12 mg,9%)。10: 1 H NMR
(400 MHz, CDCl3
) δ 2.85-2.80 (m, 1H), 2.00-1.95 (m, 1H), 1.90-1.80 (m, 1H), 1.55-1.10 (m, 16H), 1.09-0.80 (m, 17H), 0.70-0.60 (m, 5H), 0.58-0.43 (m, 3H), 0.32-0.34 (m, 1H), 0.13-0.06 (m, 1H)。 在7.0 min層析中,LCMS
Rt=3.840 min,30-90AB_7MIN_E,純度97%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在30 min層析中,HPLC
Rt=13.470 min,70-90AB_1_30MIN.M,純度97%。實例 11 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (11) 之合成  1.在0℃下將TMSCF3
(493 mg,3.47mmol)添加至S-500-6-1_1
(500 mg,1.39 mmol)及CsF (105 mg,695 μmol)於THF (5 mL)中之溶液中。在25℃下攪拌混合物1小時且用TBAF.3H2
O (1.09 g,3.47 mmol)處理。在25℃下攪拌混合物2小時,用水(100 mL)淬滅且用EtOAc (2 × 50 mL)萃取。合併之有機相用鹽水(100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA=10/1)純化,得到呈固體之S-500-6-1_2
(400 mg,67%)。 1 H NMR
(400 MHz, CDCl3) δ 5.33-5.24 (m, 1H), 4.06-4.00 (m, 1H), 2.38-2.35 (m, 1H), 2.08-1.82 (m, 6H), 1.77-1.69 (m, 1H), 1.62-1.20 (m, 13H), 1.16-1.00 (m, 8H), 0.99-0.92 (m, 1H), 0.87-0.83 (m, 4H), 0.74-0.64 (m, 3H)。2.S-500-6-1_2
(350 mg)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
.H2
O ETOH,梯度:35%至35%,流速(ml/min):60 mL/min,25℃)純化,得到81
(峰值1,130 mg,37%)及呈固體之62
(峰值2,180 mg,52%)。81: 1 H NMR
(400 MHz, CDCl3) δ 5.34-5.24 (m, 1H), 4.09-4.00 (m, 1H), 2.43-2.33 (m, 1H), 2.14 (d,J
= 4Hz, 1H), 2.07-1.80 (m, 5H), 1.77-1.55 (m, 5H), 1.53-1.30 (m, 7H), 1.28-1.00 (m, 11H), 1.00-0.91 (m, 1H), 0.85 (t,J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.220 min,30-90 AB,純度100%,C25
H38
F3
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,SFC
峰值1:Rt=4.561 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),100%de。3.
將Pd(OH)2
(0.2 g,<1%水)添加至81
(110 mg,0.256 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中。在50℃下於50 psi氫氣下氫化溶液48小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到56
(38 mg,35%)及呈固體之11
(42 mg,38%)。11: 1 H NMR
(400 MHz, CDCl3) δ 4.09-3.99 (m, 1H), 2.11 (d,J
= 6.0 Hz, 1H), 1.99-1.80 (m, 3H), 1.70-1.55 (m, 6H), 1.53-1.30 (m, 8H), 1.27-0.96 (m, 11H), 0.95-0.81 (m, 7H), 0.70-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.247 min,30-90 AB,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 12 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (12) 之合成
1.在0℃下將TMSCF3
(493 mg,3.47mmol)添加至S-500-6-1_1
(500 mg,1.39 mmol)及CsF (105 mg,695 μmol)於THF (5 mL)中之溶液中。在25℃下攪拌混合物1小時且用TBAF.3H2
O (1.09 g,3.47 mmol)處理。在25℃下攪拌混合物2小時,用水(100 mL)淬滅且用EtOAc (2 × 50 mL)萃取。合併之有機相用鹽水(100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA=10/1)純化,得到呈固體之S-500-6-1_2
(400 mg,67%)。 1 H NMR
(400 MHz, CDCl3) δ 5.33-5.24 (m, 1H), 4.06-4.00 (m, 1H), 2.38-2.35 (m, 1H), 2.08-1.82 (m, 6H), 1.77-1.69 (m, 1H), 1.62-1.20 (m, 13H), 1.16-1.00 (m, 8H), 0.99-0.92 (m, 1H), 0.87-0.83 (m, 4H), 0.74-0.64 (m, 3H)。2.S-500-6-1_2
(350 mg)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
.H2
O ETOH,梯度:35%至35%,流速(ml/min):60 mL/min,25℃)純化,得到81
(峰值1,130 mg,37%)及呈固體之62
(峰值2,180 mg,52%)。81: 1 H NMR
(400 MHz, CDCl3) δ 5.34-5.24 (m, 1H), 4.09-4.00 (m, 1H), 2.43-2.33 (m, 1H), 2.14 (d,J
= 4Hz, 1H), 2.07-1.80 (m, 5H), 1.77-1.55 (m, 5H), 1.53-1.30 (m, 7H), 1.28-1.00 (m, 11H), 1.00-0.91 (m, 1H), 0.85 (t,J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.220 min,30-90 AB,純度100%,C25
H38
F3
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,SFC
峰值1:Rt=4.561 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),100%de。3.
將Pd(OH)2
(0.2 g,<1%水)添加至81
(110 mg,0.256 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中。在50℃下於50 psi氫氣下氫化溶液48小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到56
(38 mg,35%)及呈固體之11
(42 mg,38%)。11: 1 H NMR
(400 MHz, CDCl3) δ 4.09-3.99 (m, 1H), 2.11 (d,J
= 6.0 Hz, 1H), 1.99-1.80 (m, 3H), 1.70-1.55 (m, 6H), 1.53-1.30 (m, 8H), 1.27-0.96 (m, 11H), 0.95-0.81 (m, 7H), 0.70-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.247 min,30-90 AB,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 12 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (12) 之合成  1.將NaBH4
(2.80 g,82.5 mmol)每五分鐘添加五次至S-500-6-29_2
(800 mg,1.65 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (50 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到S-500-6-30
(290 mg,36%)及呈固體之12
(120 mg,45%)。12: 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.06-1.95 (m, 3H), 1.77-1.58 (m, 7H), 1.54-1.28 (m, 12H), 1.27-1.06 (m, 11H), 1.03 (s, 3H), 1.00-0.95 (m, 2H), 0.93-0.82 (m, 12H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.708 min,30-90AB_E,純度100%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。實例 13 : (1S,3R,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (13) 之合成
1.將NaBH4
(2.80 g,82.5 mmol)每五分鐘添加五次至S-500-6-29_2
(800 mg,1.65 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (50 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到S-500-6-30
(290 mg,36%)及呈固體之12
(120 mg,45%)。12: 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.06-1.95 (m, 3H), 1.77-1.58 (m, 7H), 1.54-1.28 (m, 12H), 1.27-1.06 (m, 11H), 1.03 (s, 3H), 1.00-0.95 (m, 2H), 0.93-0.82 (m, 12H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.708 min,30-90AB_E,純度100%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。實例 13 : (1S,3R,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (13) 之合成 
 1.向N-4-1_4
(27 g,85.8 mmol)於THF (200 mL)中之溶液中添加CsF (25.9 g,171 mmol)及TMSCF3
(24.3 g,171 mmol)。在10℃下攪拌混合物1小時。向混合物中添加水(10 mL)及TBAF.3H2
O (30 g)。在30℃下再攪拌混合物2小時。真空濃縮混合物。使殘餘物溶解於EtOAc (500 ml)中,用水(2×500 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮且藉由閃蒸塔(DCM/EtOAc(1:1)於PE中,0%至10%)純化,得到N-4-1_5
(27 g,82%)及呈固體之N-4-1_5A
(3.5 g,11%)。N-4-1_5: 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.12-1.94 (m, 3H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.60 (m, 5H), 1.58-1.48 (m, 2H), 1.45-1.09 (m, 10H), 1.01-0.89 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.56 (s, 3H)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.09-1.99 (m, 1H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.52 (m, 9H), 1.45-1.06 (m, 10H), 1.00-1.81 (m, 2H), 0.79 (s, 3H), 0.56 (s, 3H)。 2.向N-4-1_5
(23 g,59.8 mmol)於THF (250 mL)中之溶液中添加9-BBN二聚體(29 g,119 mmol),在40℃下於N2
下攪拌16小時。向反應混合物添加乙醇(34.3 mL,598 mmol)及NaOH (119 mL,5 M,598 mmol)。混合物變得澄清。在25℃下逐滴添加H2
O2
(59.8 mL,10 M,598 mmol)且使內部溫度升至回流(70℃)。添加之後將混合物冷卻至30℃。向該混合物添加Na2
SO3
(100 mL,20%水溶液)。有機層經分離且倒入水(800 mL)中。形成固體。過濾混合物且固體用水洗滌,真空乾燥且用MeCN (250 mL)濕磨,得到固體。該固體在60℃下自MeOH/水(250 mL/12.5 mL)濕磨且在冷卻至15℃之後過濾。真空乾燥固體,得到呈固體之N-4-1_6
(16.4 g,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.69-3.60 (m, 1H), 3.39-3.29 (m, 1H), 2.09-2.01 (m, 1H), 1.99-1.92 (m, 1H), 1.87-1.75 (m, 2H), 1.72-1.43 (m, 7H), 1.42-1.07 (m, 11H), 1.03 (d,J
= 6.8 Hz, 3H), 1.01-0.86 (m, 3H), 0.85 (s, 3H), 0.73-0.69 (m, 1H), 0.67 (s, 3H)。3.
向N-4-1_6
(5 g,12.4 mmol)於DCM (200 mL)中之懸浮液中添加水(223 mg,12.4 mmol)及DMP (10.5 g,24.8 mmol)。在15℃下攪拌混合物15 min。混合物用NaHCO3
/Na2
S2
O3
(200 mL/200 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-4-1_7
(4.5 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.60-9.51 (m, 1H), 2.40-2.30 (m, 1H), 2.12-1.78 (m, 5H), 1.75-1.59 (m, 4H), 1.57-1.15 (m, 11H), 1.14-0.84 (m, 8H), 0.78-0.63 (m, 5H)。 4.在0℃下將MeLi (7.75 mL,1.6 M,12.4 mmol)添加至N-4-1_7
(1 g,2.49 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌混合物1 h。向混合物添加NH4
Cl (10%,20 mL)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,得到呈膠狀之混合物(1 g)。混合物N-3-2_1
(1 g)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到91
(450 mg)及22
(460 mg)及130 mg混合物。91
(450 mg)自MeCN (10 mL)再結晶得到呈固體之91
(50 mg),22
(460 mg)自MeCN (10 mL)再結晶兩次得到呈固體之22
(50 mg)。91: 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.88 (m, 1H), 2.11-2.02 (m, 1H), 2.00 (s, 1H), 1.98-1.88 (m, 2H), 1.85-1.79 (m, 1H), 1.73-1.58 (m, 4H), 1.52-1.20 (m, 11H), 1.19-1.11 (m, 4H), 1.10-1.00 (m, 3H), 0.97-0.89 (m, 4H), 0.85 (s, 3H), 0.75-0.68 (m, 1H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.155 min,30-90_AB_E,純度100%,C24
H38
F3
O [M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt=5.23 min,30-90_AB_E,純度98.88%,d.e. 100%。22: 1 H NMR
(400 MHz, CDCl3
) δ 3.97-3.82 (m, 1H), 2.10-1.92 (m, 3H), 1.85-1.78 (m, 1H), 1.77-1.60 (m, 5H), 1.59-1.06 (m, 13H), 1.05-0.81 (m, 12H), 0.74-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.136 min,30-90_AB_E,純度100%,C24
H38
F3
O [M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt=5.05 min,30-90_AB_E,純度100%,d.e. 100%。5.
向N-3-2_1
(0.88 g,2.11 mmol)於DCM (20 mL)中之溶液中添加水(2滴)及DMP (1.78 g,4.22 mmol)。在25℃下攪拌混合物30 min。混合物用NaHCO3
/Na2
S2
O3
(30 mL/30 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-3-2_2
(0.85 g,97%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.53-2.42 (m, 1H), 2.13-2.00 (m, 4H), 1.97-1.78 (m, 2H), 1.75-1.45 (m, 9H), 1.43-1.13 (m, 9H), 1.11 (d,J
= 8.4 Hz, 3H), 1.07-1.00 (m, 1H), 0.98-0.88 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.67 (s, 3H)。 6.在0℃下向N-3-2_2
(0.85 g,2.05 mmol)於DCM (5 mL)中之溶液中添加咪唑(279 mg,4.10 mmol)及TMSCl (333 mg,3.07 mmol)。在0℃下攪拌混合物0.5 h。添加混合物,藉由NaHCO3
(20 mL,飽和)淬滅且用PE (15 mL)萃取。有機層經分離,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-3-2_2A
(0.98 g,98%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.53-2.42 (m, 1H), 2.13-2.03 (m, 4H), 1.97-1.78 (m, 2H), 1.75-1.31 (m, 11H), 1.31-1.00 (m, 10H), 1.00-0.88 (m, 1H), 0.83 (s, 3H), 0.75-0.61 (m, 4H), 0.15 (s, 9H)。 7.在-70℃下將BuLi (0.384 mL,2.5 M,0.615 mmol)添加至於THF (0.5 mL)中之i-Pr2
NH (62.2 mg,0.615 mmol)中且在0℃下攪拌10 min。在-70℃下添加於THF (1 mL)中之N-3-2_2A
(0.2 g,0.41 mmol)且在-70℃下攪拌30 min。在-70℃下添加苯甲醛(91.3 mg,0.861 mmol)於THF (0.5 mL)中之溶液且在-70℃下攪拌15 min。將NH4
Cl (1 mL,飽和水溶液)添加至混合物且用EtOAc (10 mL)萃取。有機層經分離,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈油狀物之N-3-2_3C
(250 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 7.46-7.30 (m, 5H), 5.70-5.55 (m, 1H), 3.60-3.25 (m, 1H), 2.90-2.70 (m, 2H), 2.55-2.41 (m, 1H), 2.16-2.00 (m, 2H), 1.96-1.75 (m, 3H), 1.50-1.15 (m, 9H), 1.13-1.05 (m, 4H), 1.05-0.88 (m, 4H), 0.87-0.80 (m, 5H), 0.73-0.62 (m, 5H), 0.15 (s, 9H)。8.
在0℃下將LiAlH4
(159 mg,4.20 mmol)添加至N-3-2_3C
(250 mg,0.421 mmol)於THF (10 mL)中之溶液中。在0℃下攪拌混合物5 min。將水(0.16 mL)、NaOH (0.16 mL,15%水溶液)及水(0.48 mL)以本文中所撰寫之順序添加至混合物。過濾混合物且用THF (30 mL)洗滌固體。真空濃縮合併之濾液,得到呈固體之N-3-2_4C
(250 mg,100%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.42-7.28 (m, 5H), 5.70-5.30 (m, 1H), 4.15-3.65 (m, 1H), 2.18-1.55 (m, 9H), 1.53-1.00 (m, 15H), 1.00-0.75 (m, 9H), 0.75-0.50 (m, 4H), 0.15 (s, 9H)。 9.將TBAF (219 mg,0.84 mmol)添加至N-3-2_4C
(250 mg,0.42 mmol)於THF (2mL)中之溶液中。在25℃下攪拌混合物3 h。真空濃縮混合物。使殘餘物溶解於EtOAc (10 mL)中且用水(3×10 mL)洗滌,藉由閃蒸塔(10%至25% EtOAc/PE)純化,得到呈固體之N-3-2_5C
(150 mg,68%)。10.
混合物N-3-2_5C
(150 mg)藉由SFC (儀器:MG-II;管柱:IC (250mm×30mm,10 um);條件:0.1%NH3
H2
O MeOH;開始B:30%;結束B:30%;流速(mL/min):60;注射:300)分離,得到不純79
(35 mg,不純)、31
與25
之混合物(55 mg)及13
(28 mg,不純)。 不純79
(35 mg)藉由閃蒸塔(10%至30% EtOAc/PE)純化,真空濃縮溶離劑。使殘餘物溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到呈固體之79
(12 mg)。25
及13
(55 mg)藉由SFC(儀器:MG-II;管柱:AD (250mm×30mm,5um);條件:0.1%NH3
H2
O MeOH;開始B:35%;結束B:35%;流速(mL/min):60;注射70)分離。溶離劑中之每一者分別真空濃縮,溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到均呈固體之25
(28 mg)及13
(7 mg)。 不純31
(28 mg)藉由SFC (儀器:SFC 17;管柱:AS (250mm×30mm,5um),條件:0.1%NH3
H2
O EtOH;開始B:30%;結束B:30%;流速(mL/min):50;注射:60)純化,得到固體。使殘餘物溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到呈固體之31
(9 mg)。四種異構體之 SFC :在
10 min層析中,峰值1:Rt=1.501 min,峰值2:Rt=1.730 min及峰值3:Rt=1.943 min,IC-3_MeOH(DEA)_40_2.5ML (「管柱:ChiralPak IC-3 150×4.6mm I.D.,3um;梯度:40%甲醇(0.05% DEA)/CO2
;流速:2.5mL/min;管柱溫度:40℃」)。25 及 13 之 SFC :在
8 min層析中,峰值1:Rt=4.411 min及峰值2:Rt=4.920 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN (「管柱:Chiralpak AD-3 100×4.6mm I.D.,3um;移動相:A:CO2
,B:甲醇(0.05% DEA);梯度:在4.5min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續1 min;流速:2.8mL/min;管柱溫度:40℃」)。79: 1 H NMR
(400 MHz, CDCl3
) δ 7.43-7.28 (m, 5H), 5.05-4.94 (m, 1H), 4.04-3.91 (m, 1H), 2.51 (brs, 1H), 2.07-1.78 (m, 6H), 1.70-1.61 (m, 4H), 1.51-1.41 (m, 3H), 1.39-1.12 (m, 11H), 1.05-0.98 (m, 2H), 0.91-0.81 (m, 7H), 0.71-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.298 min,10-80AB_2MIN_E,純度96.7%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.483 min,IC-3_MeOH(DEA)_40_2.5ML,100%de。25: 1 H NMR
(400 MHz, CDCl3
) δ 7.42-7.28 (m, 5H), 4.97-4.81 (m, 1H), 4.12-3.92 (m, 1H), 3.23 (brs, 1H), 2.69 (brs, 1H), 2.10-1.88 (m, 3H), 1.82-1.62 (m, 7H), 1.48-1.18 (m, 10H), 1.10-0.88 (m, 8H), 0.87-0.78 (m, 4H), 0.70-0.58 (m, 4H)。 在2 min層析中,LCMS
Rt=1.319 min,10-80AB_2MIN_E,純度97.0%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.718 min,IC-3_MeOH(DEA)_40_2.5ML,98.26%de。 在8 min層析中,SFC
Rt=4.367 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,100%de。31: 1 H NMR
(400 MHz, CDCl3
) δ 7.45-7.28 (m, 5H), 5.02-4.81 (m, 1H), 4.18-3.98 (m, 1H), 3.35 (brs, 1H), 2.47 (brs, 1H), 2.15-1.72 (m, 8H), 1.53-1.31 (m, 8H), 1.30-1.03 (m, 8H), 0.99-0.89 (m, 4H), 0.89-0.78 (m, 4H), 0.75-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.327 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.929 min,IC-3_MeOH(DEA)_40_2.5ML,98.4%de。13: 1 H NMR
(400 MHz, CDCl3
) δ 7.40-7.28 (m, 5H), 5.12-5.07 (m, 1H), 3.95-3.88 (m, 1H), 2.76 (brs, 1H), 2.08-1.78 (m, 6H), 1.75-1.60 (m, 5H), 1.51-1.38 (m, 4H), 1.36-1.09 (m, 9H), 1.00-0.89 (m, 6H), 0.83 (s, 3H), 0.71-0.64 (m, 1H), 0.63 (s, 3H)。 在2 min層析中,LCMS
Rt=1.309 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.683 min,IC-3_MeOH(DEA)_40_2.5ML,98.94%de。 在8 min層析中,SFC
Rt=4.785 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,94.03%de。實例 14 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R,E)-3- 羥基 -5- 苯基戊 -4- 烯 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (14) 之合成
1.向N-4-1_4
(27 g,85.8 mmol)於THF (200 mL)中之溶液中添加CsF (25.9 g,171 mmol)及TMSCF3
(24.3 g,171 mmol)。在10℃下攪拌混合物1小時。向混合物中添加水(10 mL)及TBAF.3H2
O (30 g)。在30℃下再攪拌混合物2小時。真空濃縮混合物。使殘餘物溶解於EtOAc (500 ml)中,用水(2×500 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮且藉由閃蒸塔(DCM/EtOAc(1:1)於PE中,0%至10%)純化,得到N-4-1_5
(27 g,82%)及呈固體之N-4-1_5A
(3.5 g,11%)。N-4-1_5: 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.12-1.94 (m, 3H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.60 (m, 5H), 1.58-1.48 (m, 2H), 1.45-1.09 (m, 10H), 1.01-0.89 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.56 (s, 3H)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.09-1.99 (m, 1H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.52 (m, 9H), 1.45-1.06 (m, 10H), 1.00-1.81 (m, 2H), 0.79 (s, 3H), 0.56 (s, 3H)。 2.向N-4-1_5
(23 g,59.8 mmol)於THF (250 mL)中之溶液中添加9-BBN二聚體(29 g,119 mmol),在40℃下於N2
下攪拌16小時。向反應混合物添加乙醇(34.3 mL,598 mmol)及NaOH (119 mL,5 M,598 mmol)。混合物變得澄清。在25℃下逐滴添加H2
O2
(59.8 mL,10 M,598 mmol)且使內部溫度升至回流(70℃)。添加之後將混合物冷卻至30℃。向該混合物添加Na2
SO3
(100 mL,20%水溶液)。有機層經分離且倒入水(800 mL)中。形成固體。過濾混合物且固體用水洗滌,真空乾燥且用MeCN (250 mL)濕磨,得到固體。該固體在60℃下自MeOH/水(250 mL/12.5 mL)濕磨且在冷卻至15℃之後過濾。真空乾燥固體,得到呈固體之N-4-1_6
(16.4 g,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.69-3.60 (m, 1H), 3.39-3.29 (m, 1H), 2.09-2.01 (m, 1H), 1.99-1.92 (m, 1H), 1.87-1.75 (m, 2H), 1.72-1.43 (m, 7H), 1.42-1.07 (m, 11H), 1.03 (d,J
= 6.8 Hz, 3H), 1.01-0.86 (m, 3H), 0.85 (s, 3H), 0.73-0.69 (m, 1H), 0.67 (s, 3H)。3.
向N-4-1_6
(5 g,12.4 mmol)於DCM (200 mL)中之懸浮液中添加水(223 mg,12.4 mmol)及DMP (10.5 g,24.8 mmol)。在15℃下攪拌混合物15 min。混合物用NaHCO3
/Na2
S2
O3
(200 mL/200 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-4-1_7
(4.5 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.60-9.51 (m, 1H), 2.40-2.30 (m, 1H), 2.12-1.78 (m, 5H), 1.75-1.59 (m, 4H), 1.57-1.15 (m, 11H), 1.14-0.84 (m, 8H), 0.78-0.63 (m, 5H)。 4.在0℃下將MeLi (7.75 mL,1.6 M,12.4 mmol)添加至N-4-1_7
(1 g,2.49 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌混合物1 h。向混合物添加NH4
Cl (10%,20 mL)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,得到呈膠狀之混合物(1 g)。混合物N-3-2_1
(1 g)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到91
(450 mg)及22
(460 mg)及130 mg混合物。91
(450 mg)自MeCN (10 mL)再結晶得到呈固體之91
(50 mg),22
(460 mg)自MeCN (10 mL)再結晶兩次得到呈固體之22
(50 mg)。91: 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.88 (m, 1H), 2.11-2.02 (m, 1H), 2.00 (s, 1H), 1.98-1.88 (m, 2H), 1.85-1.79 (m, 1H), 1.73-1.58 (m, 4H), 1.52-1.20 (m, 11H), 1.19-1.11 (m, 4H), 1.10-1.00 (m, 3H), 0.97-0.89 (m, 4H), 0.85 (s, 3H), 0.75-0.68 (m, 1H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.155 min,30-90_AB_E,純度100%,C24
H38
F3
O [M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt=5.23 min,30-90_AB_E,純度98.88%,d.e. 100%。22: 1 H NMR
(400 MHz, CDCl3
) δ 3.97-3.82 (m, 1H), 2.10-1.92 (m, 3H), 1.85-1.78 (m, 1H), 1.77-1.60 (m, 5H), 1.59-1.06 (m, 13H), 1.05-0.81 (m, 12H), 0.74-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.136 min,30-90_AB_E,純度100%,C24
H38
F3
O [M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt=5.05 min,30-90_AB_E,純度100%,d.e. 100%。5.
向N-3-2_1
(0.88 g,2.11 mmol)於DCM (20 mL)中之溶液中添加水(2滴)及DMP (1.78 g,4.22 mmol)。在25℃下攪拌混合物30 min。混合物用NaHCO3
/Na2
S2
O3
(30 mL/30 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-3-2_2
(0.85 g,97%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.53-2.42 (m, 1H), 2.13-2.00 (m, 4H), 1.97-1.78 (m, 2H), 1.75-1.45 (m, 9H), 1.43-1.13 (m, 9H), 1.11 (d,J
= 8.4 Hz, 3H), 1.07-1.00 (m, 1H), 0.98-0.88 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.67 (s, 3H)。 6.在0℃下向N-3-2_2
(0.85 g,2.05 mmol)於DCM (5 mL)中之溶液中添加咪唑(279 mg,4.10 mmol)及TMSCl (333 mg,3.07 mmol)。在0℃下攪拌混合物0.5 h。添加混合物,藉由NaHCO3
(20 mL,飽和)淬滅且用PE (15 mL)萃取。有機層經分離,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-3-2_2A
(0.98 g,98%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.53-2.42 (m, 1H), 2.13-2.03 (m, 4H), 1.97-1.78 (m, 2H), 1.75-1.31 (m, 11H), 1.31-1.00 (m, 10H), 1.00-0.88 (m, 1H), 0.83 (s, 3H), 0.75-0.61 (m, 4H), 0.15 (s, 9H)。 7.在-70℃下將BuLi (0.384 mL,2.5 M,0.615 mmol)添加至於THF (0.5 mL)中之i-Pr2
NH (62.2 mg,0.615 mmol)中且在0℃下攪拌10 min。在-70℃下添加於THF (1 mL)中之N-3-2_2A
(0.2 g,0.41 mmol)且在-70℃下攪拌30 min。在-70℃下添加苯甲醛(91.3 mg,0.861 mmol)於THF (0.5 mL)中之溶液且在-70℃下攪拌15 min。將NH4
Cl (1 mL,飽和水溶液)添加至混合物且用EtOAc (10 mL)萃取。有機層經分離,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈油狀物之N-3-2_3C
(250 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 7.46-7.30 (m, 5H), 5.70-5.55 (m, 1H), 3.60-3.25 (m, 1H), 2.90-2.70 (m, 2H), 2.55-2.41 (m, 1H), 2.16-2.00 (m, 2H), 1.96-1.75 (m, 3H), 1.50-1.15 (m, 9H), 1.13-1.05 (m, 4H), 1.05-0.88 (m, 4H), 0.87-0.80 (m, 5H), 0.73-0.62 (m, 5H), 0.15 (s, 9H)。8.
在0℃下將LiAlH4
(159 mg,4.20 mmol)添加至N-3-2_3C
(250 mg,0.421 mmol)於THF (10 mL)中之溶液中。在0℃下攪拌混合物5 min。將水(0.16 mL)、NaOH (0.16 mL,15%水溶液)及水(0.48 mL)以本文中所撰寫之順序添加至混合物。過濾混合物且用THF (30 mL)洗滌固體。真空濃縮合併之濾液,得到呈固體之N-3-2_4C
(250 mg,100%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.42-7.28 (m, 5H), 5.70-5.30 (m, 1H), 4.15-3.65 (m, 1H), 2.18-1.55 (m, 9H), 1.53-1.00 (m, 15H), 1.00-0.75 (m, 9H), 0.75-0.50 (m, 4H), 0.15 (s, 9H)。 9.將TBAF (219 mg,0.84 mmol)添加至N-3-2_4C
(250 mg,0.42 mmol)於THF (2mL)中之溶液中。在25℃下攪拌混合物3 h。真空濃縮混合物。使殘餘物溶解於EtOAc (10 mL)中且用水(3×10 mL)洗滌,藉由閃蒸塔(10%至25% EtOAc/PE)純化,得到呈固體之N-3-2_5C
(150 mg,68%)。10.
混合物N-3-2_5C
(150 mg)藉由SFC (儀器:MG-II;管柱:IC (250mm×30mm,10 um);條件:0.1%NH3
H2
O MeOH;開始B:30%;結束B:30%;流速(mL/min):60;注射:300)分離,得到不純79
(35 mg,不純)、31
與25
之混合物(55 mg)及13
(28 mg,不純)。 不純79
(35 mg)藉由閃蒸塔(10%至30% EtOAc/PE)純化,真空濃縮溶離劑。使殘餘物溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到呈固體之79
(12 mg)。25
及13
(55 mg)藉由SFC(儀器:MG-II;管柱:AD (250mm×30mm,5um);條件:0.1%NH3
H2
O MeOH;開始B:35%;結束B:35%;流速(mL/min):60;注射70)分離。溶離劑中之每一者分別真空濃縮,溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到均呈固體之25
(28 mg)及13
(7 mg)。 不純31
(28 mg)藉由SFC (儀器:SFC 17;管柱:AS (250mm×30mm,5um),條件:0.1%NH3
H2
O EtOH;開始B:30%;結束B:30%;流速(mL/min):50;注射:60)純化,得到固體。使殘餘物溶解於MeCN/水(20 mL,4:1)中且真空濃縮,得到呈固體之31
(9 mg)。四種異構體之 SFC :在
10 min層析中,峰值1:Rt=1.501 min,峰值2:Rt=1.730 min及峰值3:Rt=1.943 min,IC-3_MeOH(DEA)_40_2.5ML (「管柱:ChiralPak IC-3 150×4.6mm I.D.,3um;梯度:40%甲醇(0.05% DEA)/CO2
;流速:2.5mL/min;管柱溫度:40℃」)。25 及 13 之 SFC :在
8 min層析中,峰值1:Rt=4.411 min及峰值2:Rt=4.920 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN (「管柱:Chiralpak AD-3 100×4.6mm I.D.,3um;移動相:A:CO2
,B:甲醇(0.05% DEA);梯度:在4.5min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續1 min;流速:2.8mL/min;管柱溫度:40℃」)。79: 1 H NMR
(400 MHz, CDCl3
) δ 7.43-7.28 (m, 5H), 5.05-4.94 (m, 1H), 4.04-3.91 (m, 1H), 2.51 (brs, 1H), 2.07-1.78 (m, 6H), 1.70-1.61 (m, 4H), 1.51-1.41 (m, 3H), 1.39-1.12 (m, 11H), 1.05-0.98 (m, 2H), 0.91-0.81 (m, 7H), 0.71-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.298 min,10-80AB_2MIN_E,純度96.7%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.483 min,IC-3_MeOH(DEA)_40_2.5ML,100%de。25: 1 H NMR
(400 MHz, CDCl3
) δ 7.42-7.28 (m, 5H), 4.97-4.81 (m, 1H), 4.12-3.92 (m, 1H), 3.23 (brs, 1H), 2.69 (brs, 1H), 2.10-1.88 (m, 3H), 1.82-1.62 (m, 7H), 1.48-1.18 (m, 10H), 1.10-0.88 (m, 8H), 0.87-0.78 (m, 4H), 0.70-0.58 (m, 4H)。 在2 min層析中,LCMS
Rt=1.319 min,10-80AB_2MIN_E,純度97.0%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.718 min,IC-3_MeOH(DEA)_40_2.5ML,98.26%de。 在8 min層析中,SFC
Rt=4.367 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,100%de。31: 1 H NMR
(400 MHz, CDCl3
) δ 7.45-7.28 (m, 5H), 5.02-4.81 (m, 1H), 4.18-3.98 (m, 1H), 3.35 (brs, 1H), 2.47 (brs, 1H), 2.15-1.72 (m, 8H), 1.53-1.31 (m, 8H), 1.30-1.03 (m, 8H), 0.99-0.89 (m, 4H), 0.89-0.78 (m, 4H), 0.75-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.327 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.929 min,IC-3_MeOH(DEA)_40_2.5ML,98.4%de。13: 1 H NMR
(400 MHz, CDCl3
) δ 7.40-7.28 (m, 5H), 5.12-5.07 (m, 1H), 3.95-3.88 (m, 1H), 2.76 (brs, 1H), 2.08-1.78 (m, 6H), 1.75-1.60 (m, 5H), 1.51-1.38 (m, 4H), 1.36-1.09 (m, 9H), 1.00-0.89 (m, 6H), 0.83 (s, 3H), 0.71-0.64 (m, 1H), 0.63 (s, 3H)。 在2 min層析中,LCMS
Rt=1.309 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.683 min,IC-3_MeOH(DEA)_40_2.5ML,98.94%de。 在8 min層析中,SFC
Rt=4.785 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,94.03%de。實例 14 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R,E)-3- 羥基 -5- 苯基戊 -4- 烯 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (14) 之合成 
 1.在-70℃下將BuLi (0.384 mL,2.5 M,0.615 mmol)添加至於THF (0.5 mL)中之i-Pr2
NH (62.2 mg,0.615 mmol)中且在0℃下攪拌10 min。在-70℃下添加於THF (1 mL)中之N-3-2_2A (0.2 g,0.41 mmol)且在-70℃下攪拌1 h。在-70℃下添加苯甲醛(91.3 mg,0.861 mmol)於THF (0.5 mL)中之溶液且在20℃下攪拌4 h。將NH4
Cl (1 mL,飽和水溶液)添加至混合物且用EtOAc (10 mL)萃取。有機層經分離,經Na2
SO4
乾燥,過濾且真空濃縮,藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之N-3-2_3D
(150 mg,64%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.64-7.55 (m, 3H), 7.43-7.39 (m, 3H), 6.79 (d,J
= 16.0 Hz, 1H), 2.86-2.73 (m, 1H), 2.15-2.08 (m, 1H), 2.00-1.90 (m, 1H), 1.88-1.80 (m, 1H), 1.72-1.59 (m, 5H), 1.53-1.22 (m, 9H), 1.21-1.03 (m, 7H), 0.99-0.88 (m, 1H), 0.84 (s, 3H), 0.75-0.61 (m, 4H), 0.15 (s, 9H)。2.
將TBAF (135 mg,0.52 mmol)添加至N-3-2_3D
(150 mg,0.26 mmol)於THF (1 mL)中之溶液中。在20℃下攪拌混合物20 h。向混合物添加EtOAc (5 mL)。混合物用水(2×5 mL)、鹽水(5 mL)洗滌,真空濃縮,得到呈固體之N-003-005_1
(140 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 7.64-7.53 (m, 3H), 7.43-7.35 (m, 3H), 6.79 (d,J
= 16.0 Hz, 1H), 2.88-2.73 (m, 1H), 2.13-1.90 (m, 3H), 1.88-1.78 (m, 1H), 1.77-1.90 (m, 5H), 1.60-1.22 (m, 8H), 1.21-0.88 (m, 9H), 0.86 (s, 3H), 0.75-0.61 (m, 4H)。3.
在20℃下向N-003-005_1
(140 mg,0.278 mmol)於THF (2 mL)及MeOH (1 mL)中之溶液逐份添加NaBH4
(419 mg,11.1 mmol)。在20℃下攪拌混合物10 min。反應物用水(20 mL)及NH4
Cl (20mL,飽和)淬滅。用EtOAc (50 mL)萃取混合物。真空濃縮有機層且藉由prep-TLC (PE/EtOAc=4/1)純化,得到均呈固體之N-003-005
(50 mg,不純)及N-003-006
(50 mg)。 使14 (50 mg)溶解於MeCN (20 mL)中且真空濃縮,得到呈固體之14 (29 mg)。14: 1 H NMR
(400 MHz, CDCl3
) δ 7.44-7.38 (m, 2H), 7.37-7.29 (m, 2H), 7.25-7.18 (m, 1H), 6.59 (d,J
= 16.0 Hz, 1H), 6.24 (dd,J
= 7.2, 16.0 Hz, 1H), 4.40-4.30 (m, 1H), 2.08-1.92 (m, 3H), 1.89-1.77 (m, 3H), 1.68-1.60 (m, 3H), 1.50-1.08 (m, 13H), 1.03-0.82 (m, 9H), 0.72-0.62 (m, 4H)。 在2 min層析中,LCMS
Rt=1.236 min,30-90AB_2MIN_E,純度99.0%,C31
H42
F3
O [M+H-H2
O]+
之MS ESI計算值487,實驗值487。 在8 min層析中,HPLC
Rt=5.89 min,30-90_AB_1.2ml,98.1% d.e. (220 nm)實例 15 : 合成 (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (15)
1.在-70℃下將BuLi (0.384 mL,2.5 M,0.615 mmol)添加至於THF (0.5 mL)中之i-Pr2
NH (62.2 mg,0.615 mmol)中且在0℃下攪拌10 min。在-70℃下添加於THF (1 mL)中之N-3-2_2A (0.2 g,0.41 mmol)且在-70℃下攪拌1 h。在-70℃下添加苯甲醛(91.3 mg,0.861 mmol)於THF (0.5 mL)中之溶液且在20℃下攪拌4 h。將NH4
Cl (1 mL,飽和水溶液)添加至混合物且用EtOAc (10 mL)萃取。有機層經分離,經Na2
SO4
乾燥,過濾且真空濃縮,藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之N-3-2_3D
(150 mg,64%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.64-7.55 (m, 3H), 7.43-7.39 (m, 3H), 6.79 (d,J
= 16.0 Hz, 1H), 2.86-2.73 (m, 1H), 2.15-2.08 (m, 1H), 2.00-1.90 (m, 1H), 1.88-1.80 (m, 1H), 1.72-1.59 (m, 5H), 1.53-1.22 (m, 9H), 1.21-1.03 (m, 7H), 0.99-0.88 (m, 1H), 0.84 (s, 3H), 0.75-0.61 (m, 4H), 0.15 (s, 9H)。2.
將TBAF (135 mg,0.52 mmol)添加至N-3-2_3D
(150 mg,0.26 mmol)於THF (1 mL)中之溶液中。在20℃下攪拌混合物20 h。向混合物添加EtOAc (5 mL)。混合物用水(2×5 mL)、鹽水(5 mL)洗滌,真空濃縮,得到呈固體之N-003-005_1
(140 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 7.64-7.53 (m, 3H), 7.43-7.35 (m, 3H), 6.79 (d,J
= 16.0 Hz, 1H), 2.88-2.73 (m, 1H), 2.13-1.90 (m, 3H), 1.88-1.78 (m, 1H), 1.77-1.90 (m, 5H), 1.60-1.22 (m, 8H), 1.21-0.88 (m, 9H), 0.86 (s, 3H), 0.75-0.61 (m, 4H)。3.
在20℃下向N-003-005_1
(140 mg,0.278 mmol)於THF (2 mL)及MeOH (1 mL)中之溶液逐份添加NaBH4
(419 mg,11.1 mmol)。在20℃下攪拌混合物10 min。反應物用水(20 mL)及NH4
Cl (20mL,飽和)淬滅。用EtOAc (50 mL)萃取混合物。真空濃縮有機層且藉由prep-TLC (PE/EtOAc=4/1)純化,得到均呈固體之N-003-005
(50 mg,不純)及N-003-006
(50 mg)。 使14 (50 mg)溶解於MeCN (20 mL)中且真空濃縮,得到呈固體之14 (29 mg)。14: 1 H NMR
(400 MHz, CDCl3
) δ 7.44-7.38 (m, 2H), 7.37-7.29 (m, 2H), 7.25-7.18 (m, 1H), 6.59 (d,J
= 16.0 Hz, 1H), 6.24 (dd,J
= 7.2, 16.0 Hz, 1H), 4.40-4.30 (m, 1H), 2.08-1.92 (m, 3H), 1.89-1.77 (m, 3H), 1.68-1.60 (m, 3H), 1.50-1.08 (m, 13H), 1.03-0.82 (m, 9H), 0.72-0.62 (m, 4H)。 在2 min層析中,LCMS
Rt=1.236 min,30-90AB_2MIN_E,純度99.0%,C31
H42
F3
O [M+H-H2
O]+
之MS ESI計算值487,實驗值487。 在8 min層析中,HPLC
Rt=5.89 min,30-90_AB_1.2ml,98.1% d.e. (220 nm)實例 15 : 合成 (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (15)  1.將Pd(OH)2
/C (100 mg)添加至44
(100 mg)於MeOH/THF (2 mL/2 mL)中之溶液中。在50℃下於H2
(50 psi)下攪拌混合物20 h。過濾混合物。濃縮濾液得到100 mg固體。NMR顯示9%54
保留。在相同條件下再氫化不純樣本3次。過濾混合物。濃縮濾液且藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到呈固體之5
(7 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.48 (m, 1H), 2.00-1.55 (m, 9H), 1.50-1.22 (m, 15H), 1.19-1.03 (m, 8H), 0.96 (s, 3H), 0.91-0.81 (m, 15H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.492 min,30-90_AB_E,純度100%,C30
H51
[M+H-2H2
O]+
之MS ESI計算值411,實驗值411。實例 16 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (16) 之合成
1.將Pd(OH)2
/C (100 mg)添加至44
(100 mg)於MeOH/THF (2 mL/2 mL)中之溶液中。在50℃下於H2
(50 psi)下攪拌混合物20 h。過濾混合物。濃縮濾液得到100 mg固體。NMR顯示9%54
保留。在相同條件下再氫化不純樣本3次。過濾混合物。濃縮濾液且藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到呈固體之5
(7 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.48 (m, 1H), 2.00-1.55 (m, 9H), 1.50-1.22 (m, 15H), 1.19-1.03 (m, 8H), 0.96 (s, 3H), 0.91-0.81 (m, 15H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.492 min,30-90_AB_E,純度100%,C30
H51
[M+H-2H2
O]+
之MS ESI計算值411,實驗值411。實例 16 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (16) 之合成  1.將Pd(OH)2
(0.2 g,<1%水)添加至62
(160 mg,0.373 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中。在50℃下於50 psi氫氣下氫化溶液16小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到呈固體之44
(27 mg,17%)及16
(117 mg,73%)。16: 1 H NMR
(400 MHz, CDCl3) δ 4.04-3.96 (m, 1H), 1.98-1.83 (m, 4H), 1.69-1.59 (m, 3H), 1.56-1.20 (m, 13H), 1.17-0.95 (m, 8H), 0.91-0.83 (m, 8H), 0.70-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt = 1.240 min,30-90 AB,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 17 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (17) 之合成
1.將Pd(OH)2
(0.2 g,<1%水)添加至62
(160 mg,0.373 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中。在50℃下於50 psi氫氣下氫化溶液16小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到呈固體之44
(27 mg,17%)及16
(117 mg,73%)。16: 1 H NMR
(400 MHz, CDCl3) δ 4.04-3.96 (m, 1H), 1.98-1.83 (m, 4H), 1.69-1.59 (m, 3H), 1.56-1.20 (m, 13H), 1.17-0.95 (m, 8H), 0.91-0.83 (m, 8H), 0.70-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt = 1.240 min,30-90 AB,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 17 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (17) 之合成  1.將Pd(OH)2
/C (100 mg)添加至5
(250 mg)於MeOH/THF (2 mL/2 mL)中之溶液中。在50℃下於H2
(50 psi)下攪拌混合物20 小時。過濾混合物。濃縮濾液得到250 mg固體。NMR顯示70%5
保留。在相同條件下再氫化不純樣本3次。過濾混合物。濃縮濾液且藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到呈固體之17
(3 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 3.63-3.50 (m, 1H), 1.98-1.55 (m, 8H), 1.49-1.37 (m, 8H), 1.35-1.21 (m, 8H), 1.19-1.01 (m, 8H), 0.97 (s, 3H), 0.91-0.82 (m, 15H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.529 min,30-90_AB_E,純度95.6%,C30
H51
[M+H-2H2
O]+
之MS ESI計算值411,實驗值411。實例 18 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4-( 四氫 -2H- 哌喃 -4- 基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (18) 之合成
1.將Pd(OH)2
/C (100 mg)添加至5
(250 mg)於MeOH/THF (2 mL/2 mL)中之溶液中。在50℃下於H2
(50 psi)下攪拌混合物20 小時。過濾混合物。濃縮濾液得到250 mg固體。NMR顯示70%5
保留。在相同條件下再氫化不純樣本3次。過濾混合物。濃縮濾液且藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到呈固體之17
(3 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 3.63-3.50 (m, 1H), 1.98-1.55 (m, 8H), 1.49-1.37 (m, 8H), 1.35-1.21 (m, 8H), 1.19-1.01 (m, 8H), 0.97 (s, 3H), 0.91-0.82 (m, 15H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.529 min,30-90_AB_E,純度95.6%,C30
H51
[M+H-2H2
O]+
之MS ESI計算值411,實驗值411。實例 18 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4-( 四氫 -2H- 哌喃 -4- 基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (18) 之合成  1.
在20℃下於氮氣下向Me3
SI (8.44 g,41.4 mmol)於無水THF (50 mL)中之懸浮液中添加t-BuOK (4.64 g,41.4 mmol)。在20℃下攪拌混合物1小時,且添加N-005_5
(5 g,13.8 mmol)。使所得混合物升溫至45℃且攪拌4小時。將反應混合物冷卻至室溫,用水(50 mL)淬滅且用EtOAc (2×50 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-005_001
(2.7 g,52%)。 在2.0 min層析中,LCMS
Rt=1.324 min,30-90AB_E,純度92%,C25
H41
O [M+H-H2
O]+
之MS ESI計算值357,實驗值357。 2.在60℃下於N2
下將4-氯四氫-2H-哌喃(1 g,8.29 mmol)於無水THF (8 mL)中之溶液逐滴添加至Mg (401 mg,16.5 mmol)及I2
(105 mg,0.414 mmol)於無水THF (2 mL)中之混合物。在60℃下攪拌混合物10 min。溫度升至66℃。再攪拌反應混合物30 min,冷卻至室溫,其直接用作(四氫-2H-哌喃-4-基)氯化鎂(0.83 M於THF中)之溶液。 3.在20℃下於氮氣下將(四氫-2H-哌喃-4-基)-氯化鎂(0.83 M於THF中,6.38 mL,5.30 mmol)之溶液逐滴添加至N-005_001
(400 mg,1.06 mmol)及CuI (20.1 mg,0.106 mmol)於無水THF (10 mL)中之懸浮液中。在20℃下攪拌混合物18小時。反應混合物用水(10 mL)及飽和NH4
Cl (10 mL)淬滅,用EtOAc (2×15 mL)萃取。合併之有機層用鹽水(15 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮,得到呈固體之N-6-9/10
(760 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.01-3.92 (m, 3H), 3.83-3.62 (m, 1H), 3.42-3.32 (m, 3H), 1.97-1.87 (m, 1H), 1.68-1.58 (m, 7H), 1.57-1.45 (m, 6H), 1.43-1.29 (m, 8H), 1.24-0.95 (m, 10H), 0.91-0.79 (m, 9H), 0.73-0.56 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.332 min,10-80AB,純度93%,C30
H52
NaO3
[M+Na]+
之MS ESI計算值483,實驗值483。 4.將BzCl (691 mg,4.92 mmol)及DMAP (20 mg,0.164 mmol)添加至N-6-9/10
(760 mg,1.64 mmol)於吡啶(10 mL)中之溶液中。在20℃下攪拌混合物2小時。反應混合物用水(15 mL)淬滅,用EtOAc (2×20 mL)萃取。合併之有機層用10% HCl水溶液(2×20 mL)、飽和NaHCO3
(40 mL)及鹽水(20 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-6-9_1
(400 mg,43%)。 1 H NMR
(400 MHz, CDCl3
) δ 8.08-8.01 (m, 1H), 8.08-8.01 (m, 1H), 7.61-7.42 (m, 3H), 5.42-5.29 (m, 1H), 3.99-3.85 (m, 2H), 3.41-3.24 (m, 2H), 2.06-1.65 (m, 5H), 1.65-1.57 (m, 5H), 1.54-1.42 (m, 6H), 1.42-1.14 (m, 11H), 1.14-0.90 (m, 8H), 0.89-0.77 (m, 7H), 0.69-0.51 (m, 4H)。N-6-9_1
(400 mg,0.708 mmol)經分離且藉由SFC (管柱:C2 250mm×30mm,10 um,梯度:45-45% B (A= 0.1%NH3
/H2
O,B= MeOH ),流速:60 mL/min)純化,得到呈固體之N-6-9_2
(峰值1,Rt=3.926 min,80 mg,20%)及呈固體之N-6-10_1
(峰值2,Rt=4.893 min,180 mg,45%)。N-6-9_2: 1 H NMR
(400 MHz, CDCl3
) δ 8.06-8.00 (m, 2H), 7.60-7.53 (m, 1H), 7.49-7.42 (m, 2H), 5.35-5.28 (m, 1H), 3.98-3.91 (m, 2H), 3.41-3.31 (m, 2H), 1.88-1.67 (m, 5H), 1.66-1.57 (m, 4H), 1.54-1.36 (m, 10H), 1.35-1.16 (m, 8H), 1.08-0.88 (m, 8H), 0.88-0.82 (m, 4H), 0.80 (s, 3H), 0.64 (s,3H), 0.61-0.54 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.540 min,30-90AB,純度96%,C30
H49
O [M-BzOH-H2
O+H]+
之MS ESI計算值425,實驗值425。 在8 min層析中,SFC
Rt=3.789 min,管柱:Lux Cellulose-2 150×4.6mm I.D.,3 μm;移動相:40% 甲醇(0.05% DEA)/CO2;流速:2.5mL/min;管柱溫度:40℃,97%de。N-6-10_1: 1 H NMR
(400 MHz, CDCl3
) δ 8.06-8.01 (m, 2H), 7.61-7.53 (m, 1H), 7.49-7.42 (m, 2H), 5.41-5.33 (m, 1H), 3.98-3.86 (m, 2H), 3.40-3.27 (m, 2H), 2.05-1.91 (m, 2H), 1.84-1.72 (m, 2H), 1.66-1.59 (m, 3H), 1.55-1.38 (m, 9H), 1.37-1.16 (m, 11H), 1.13-1.00 (m, 6H), 1.00-0.90 (m, 2H), 0.89-0.79 (m, 7H), 0.67 (s, 3H), 0.63-0.54 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.507 min,30-90AB,純度97%,C30
H49
O [M-BzOH-H2
O+H]+
之MS ESI計算值425,實驗值425。 在8 min層析中,SFC
Rt=4.699 min,管柱:Lux Cellulose-2 150×4.6mm I.D.,3 μm;移動相:40% 甲醇(0.05% DEA)/CO2;流速:2.5mL/min;管柱溫度:40℃,97%de。 5.將水(1 mL)及KOH (78.5 mg,1.40 mmol)添加至N-6-9_2
(80 mg,0.141 mmol)於THF (2 mL)及甲醇(1 mL)中之溶液中。在50℃下攪拌混合物18小時。將反應混合物冷卻,用水(5 mL)稀釋,用10% HCl (0.2 mL)酸化且用EtOAc (3 × 5 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(10%至30% EtOAc/PE)純化,得到呈固體之18
(13 mg,20%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.05-3.89 (m, 3H), 3.45-3.34 (m, 2H), 1.94-1.87 (m, 1H), 1.86-1.76 (m, 1H), 1.72-1.59 (m, 6H), 1.54-1.45 (m, 4H), 1.44-1.28 (m, 9H), 1.28-1.15 (m, 7H), 1.13-0.92 (m, 5H), 0.91-0.85 (m, 4H), 0.84-0.79 (m, 6H), 0.70-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.213 min,30-90AB,純度100%。 C30
H49
O [M-2H2
O+H]+
之MS ESI計算值425,實驗值425。實例 19 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1- 苯基丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (19) 之合成
1.
在20℃下於氮氣下向Me3
SI (8.44 g,41.4 mmol)於無水THF (50 mL)中之懸浮液中添加t-BuOK (4.64 g,41.4 mmol)。在20℃下攪拌混合物1小時,且添加N-005_5
(5 g,13.8 mmol)。使所得混合物升溫至45℃且攪拌4小時。將反應混合物冷卻至室溫,用水(50 mL)淬滅且用EtOAc (2×50 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-005_001
(2.7 g,52%)。 在2.0 min層析中,LCMS
Rt=1.324 min,30-90AB_E,純度92%,C25
H41
O [M+H-H2
O]+
之MS ESI計算值357,實驗值357。 2.在60℃下於N2
下將4-氯四氫-2H-哌喃(1 g,8.29 mmol)於無水THF (8 mL)中之溶液逐滴添加至Mg (401 mg,16.5 mmol)及I2
(105 mg,0.414 mmol)於無水THF (2 mL)中之混合物。在60℃下攪拌混合物10 min。溫度升至66℃。再攪拌反應混合物30 min,冷卻至室溫,其直接用作(四氫-2H-哌喃-4-基)氯化鎂(0.83 M於THF中)之溶液。 3.在20℃下於氮氣下將(四氫-2H-哌喃-4-基)-氯化鎂(0.83 M於THF中,6.38 mL,5.30 mmol)之溶液逐滴添加至N-005_001
(400 mg,1.06 mmol)及CuI (20.1 mg,0.106 mmol)於無水THF (10 mL)中之懸浮液中。在20℃下攪拌混合物18小時。反應混合物用水(10 mL)及飽和NH4
Cl (10 mL)淬滅,用EtOAc (2×15 mL)萃取。合併之有機層用鹽水(15 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮,得到呈固體之N-6-9/10
(760 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.01-3.92 (m, 3H), 3.83-3.62 (m, 1H), 3.42-3.32 (m, 3H), 1.97-1.87 (m, 1H), 1.68-1.58 (m, 7H), 1.57-1.45 (m, 6H), 1.43-1.29 (m, 8H), 1.24-0.95 (m, 10H), 0.91-0.79 (m, 9H), 0.73-0.56 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.332 min,10-80AB,純度93%,C30
H52
NaO3
[M+Na]+
之MS ESI計算值483,實驗值483。 4.將BzCl (691 mg,4.92 mmol)及DMAP (20 mg,0.164 mmol)添加至N-6-9/10
(760 mg,1.64 mmol)於吡啶(10 mL)中之溶液中。在20℃下攪拌混合物2小時。反應混合物用水(15 mL)淬滅,用EtOAc (2×20 mL)萃取。合併之有機層用10% HCl水溶液(2×20 mL)、飽和NaHCO3
(40 mL)及鹽水(20 mL)洗滌,經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-6-9_1
(400 mg,43%)。 1 H NMR
(400 MHz, CDCl3
) δ 8.08-8.01 (m, 1H), 8.08-8.01 (m, 1H), 7.61-7.42 (m, 3H), 5.42-5.29 (m, 1H), 3.99-3.85 (m, 2H), 3.41-3.24 (m, 2H), 2.06-1.65 (m, 5H), 1.65-1.57 (m, 5H), 1.54-1.42 (m, 6H), 1.42-1.14 (m, 11H), 1.14-0.90 (m, 8H), 0.89-0.77 (m, 7H), 0.69-0.51 (m, 4H)。N-6-9_1
(400 mg,0.708 mmol)經分離且藉由SFC (管柱:C2 250mm×30mm,10 um,梯度:45-45% B (A= 0.1%NH3
/H2
O,B= MeOH ),流速:60 mL/min)純化,得到呈固體之N-6-9_2
(峰值1,Rt=3.926 min,80 mg,20%)及呈固體之N-6-10_1
(峰值2,Rt=4.893 min,180 mg,45%)。N-6-9_2: 1 H NMR
(400 MHz, CDCl3
) δ 8.06-8.00 (m, 2H), 7.60-7.53 (m, 1H), 7.49-7.42 (m, 2H), 5.35-5.28 (m, 1H), 3.98-3.91 (m, 2H), 3.41-3.31 (m, 2H), 1.88-1.67 (m, 5H), 1.66-1.57 (m, 4H), 1.54-1.36 (m, 10H), 1.35-1.16 (m, 8H), 1.08-0.88 (m, 8H), 0.88-0.82 (m, 4H), 0.80 (s, 3H), 0.64 (s,3H), 0.61-0.54 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.540 min,30-90AB,純度96%,C30
H49
O [M-BzOH-H2
O+H]+
之MS ESI計算值425,實驗值425。 在8 min層析中,SFC
Rt=3.789 min,管柱:Lux Cellulose-2 150×4.6mm I.D.,3 μm;移動相:40% 甲醇(0.05% DEA)/CO2;流速:2.5mL/min;管柱溫度:40℃,97%de。N-6-10_1: 1 H NMR
(400 MHz, CDCl3
) δ 8.06-8.01 (m, 2H), 7.61-7.53 (m, 1H), 7.49-7.42 (m, 2H), 5.41-5.33 (m, 1H), 3.98-3.86 (m, 2H), 3.40-3.27 (m, 2H), 2.05-1.91 (m, 2H), 1.84-1.72 (m, 2H), 1.66-1.59 (m, 3H), 1.55-1.38 (m, 9H), 1.37-1.16 (m, 11H), 1.13-1.00 (m, 6H), 1.00-0.90 (m, 2H), 0.89-0.79 (m, 7H), 0.67 (s, 3H), 0.63-0.54 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.507 min,30-90AB,純度97%,C30
H49
O [M-BzOH-H2
O+H]+
之MS ESI計算值425,實驗值425。 在8 min層析中,SFC
Rt=4.699 min,管柱:Lux Cellulose-2 150×4.6mm I.D.,3 μm;移動相:40% 甲醇(0.05% DEA)/CO2;流速:2.5mL/min;管柱溫度:40℃,97%de。 5.將水(1 mL)及KOH (78.5 mg,1.40 mmol)添加至N-6-9_2
(80 mg,0.141 mmol)於THF (2 mL)及甲醇(1 mL)中之溶液中。在50℃下攪拌混合物18小時。將反應混合物冷卻,用水(5 mL)稀釋,用10% HCl (0.2 mL)酸化且用EtOAc (3 × 5 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(10%至30% EtOAc/PE)純化,得到呈固體之18
(13 mg,20%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.05-3.89 (m, 3H), 3.45-3.34 (m, 2H), 1.94-1.87 (m, 1H), 1.86-1.76 (m, 1H), 1.72-1.59 (m, 6H), 1.54-1.45 (m, 4H), 1.44-1.28 (m, 9H), 1.28-1.15 (m, 7H), 1.13-0.92 (m, 5H), 0.91-0.85 (m, 4H), 0.84-0.79 (m, 6H), 0.70-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.213 min,30-90AB,純度100%。 C30
H49
O [M-2H2
O+H]+
之MS ESI計算值425,實驗值425。實例 19 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1- 苯基丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (19) 之合成  1.在0℃下將N-8-7_1
(300 mg,0.832 mmol)於THF (5 mL)中之溶液添加至PhMgBr (1.38 mL,3 M於乙醚中,4.15 mmol)於THF (10 mL)中之溶液中,隨後在0℃下攪拌反應混合物3小時。接下來,在25℃下攪拌反應混合物5小時。在0℃下用水(10 mL)淬滅反應混合物。過濾溶液且用EtOAc (10 mL)洗滌濾餅。用EtOAc (3×15 mL)萃取水相。合併之有機相用飽和鹽水(2×10 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至1/1)純化,得到呈固體之粗產物(200 mg)。粗產物藉由SFC (管柱:AD (250mm×30mm,5um),梯度:25-25% B (A= 0.1%NH3/H2O,B= EtOH),流速:60 mL/min)純化,得到55
(峰值2,55 mg, 15%)及呈固體之19
(峰值1,21 mg,6%)。19: 1 H NMR
(400MHz, CDCl3
) δ 7.40-7.20 (m, 5H), 4.85- 4.80 (m, 1H), 2.10-1.60 (m, 5H), 1.55-1.05 (m, 17H), 0.95-0.75 (m, 14H), 0.71 (s, 3H), 0.60-0.50 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.208 min,30-90AB_2 min.,純度100%,C30
H43
[M-2H2
O+H]+
之MS ESI計算值403,實驗值403。 在3 min層析中,SFC
Rt=1.047 min,OJ_3_EtOH_DEA_5_40_25ML,100%de。實例 20 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (20) 之合成
1.在0℃下將N-8-7_1
(300 mg,0.832 mmol)於THF (5 mL)中之溶液添加至PhMgBr (1.38 mL,3 M於乙醚中,4.15 mmol)於THF (10 mL)中之溶液中,隨後在0℃下攪拌反應混合物3小時。接下來,在25℃下攪拌反應混合物5小時。在0℃下用水(10 mL)淬滅反應混合物。過濾溶液且用EtOAc (10 mL)洗滌濾餅。用EtOAc (3×15 mL)萃取水相。合併之有機相用飽和鹽水(2×10 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至1/1)純化,得到呈固體之粗產物(200 mg)。粗產物藉由SFC (管柱:AD (250mm×30mm,5um),梯度:25-25% B (A= 0.1%NH3/H2O,B= EtOH),流速:60 mL/min)純化,得到55
(峰值2,55 mg, 15%)及呈固體之19
(峰值1,21 mg,6%)。19: 1 H NMR
(400MHz, CDCl3
) δ 7.40-7.20 (m, 5H), 4.85- 4.80 (m, 1H), 2.10-1.60 (m, 5H), 1.55-1.05 (m, 17H), 0.95-0.75 (m, 14H), 0.71 (s, 3H), 0.60-0.50 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.208 min,30-90AB_2 min.,純度100%,C30
H43
[M-2H2
O+H]+
之MS ESI計算值403,實驗值403。 在3 min層析中,SFC
Rt=1.047 min,OJ_3_EtOH_DEA_5_40_25ML,100%de。實例 20 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (20) 之合成 
 1.
向12
(100 mg,0.206 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(150 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之87
(12 mg,12%)及20
(11 mg,11%)。20: 1 H NMR
(400 MHz, CDCl3
) δ 3.83-3.75 (m, 1H), 1.99-1.92 (m, 1H), 1.71-1.57 (m, 8H), 1.52-1.43 (m, 3H), 1.41-1.29 (m, 8H), 1.27-1.13 (m, 11H), 1.12-1.04 (m, 4H), 1.03-0.94 (m, 3H), 0.91-0.86 (m, 12H), 0.82 (s, 3H), 0.68-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.748 min,30-90AB_E,純度100%,C33
H55
[M+H-H2
O]+
之MS ESI計算值451,實驗值451。實例 21 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (21) 之合成
1.
向12
(100 mg,0.206 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(150 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之87
(12 mg,12%)及20
(11 mg,11%)。20: 1 H NMR
(400 MHz, CDCl3
) δ 3.83-3.75 (m, 1H), 1.99-1.92 (m, 1H), 1.71-1.57 (m, 8H), 1.52-1.43 (m, 3H), 1.41-1.29 (m, 8H), 1.27-1.13 (m, 11H), 1.12-1.04 (m, 4H), 1.03-0.94 (m, 3H), 0.91-0.86 (m, 12H), 0.82 (s, 3H), 0.68-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.748 min,30-90AB_E,純度100%,C33
H55
[M+H-H2
O]+
之MS ESI計算值451,實驗值451。實例 21 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (21) 之合成  1.在25℃下於N2
下將EtMgBr (0.553 mL,3 M於乙醚中,1.66 mmol)逐滴添加至N-8_1
(250 mg,0.8320 mmol)於THF (3 mL)中之溶液中。在25℃下攪拌混合物1小時,用飽和NH4
Cl (10 mL)淬滅且用EtOAc (3×15 mL)萃取。有機層用鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物N-8-24_1
,其藉由閃蒸塔(0%至15% EtOAc/PE)純化得到呈固體之21
(130 mg,不純)。不純N-8-24
(130 mg,0.3327 mmol)在85℃下自MeCN (3 mL)再結晶,得到呈固體之純21
(111 mg,86%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.62-3.50 (m, 1H), 2.02-1.81 (m, 2H), 1.72-1.59 (m, 3H), 1.56-1.46 (m, 4H), 1.45-1.17 (m, 12H), 1.16-1.00 (m, 5H), 0.99-0.85 (m, 11H), 0.84-0.78 (m, 4H), 0.66 (s, 4H)。 在10 min層析中,HPLC
Rt=5.73 min,30-90_AB_1.2 mL_E,純度100%。 C26
H43
[M+H-2H2
O]+
之MS ESI計算值355,實驗值355。實例 22 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥丁 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (22) 之合成
1.在25℃下於N2
下將EtMgBr (0.553 mL,3 M於乙醚中,1.66 mmol)逐滴添加至N-8_1
(250 mg,0.8320 mmol)於THF (3 mL)中之溶液中。在25℃下攪拌混合物1小時,用飽和NH4
Cl (10 mL)淬滅且用EtOAc (3×15 mL)萃取。有機層用鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物N-8-24_1
,其藉由閃蒸塔(0%至15% EtOAc/PE)純化得到呈固體之21
(130 mg,不純)。不純N-8-24
(130 mg,0.3327 mmol)在85℃下自MeCN (3 mL)再結晶,得到呈固體之純21
(111 mg,86%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.62-3.50 (m, 1H), 2.02-1.81 (m, 2H), 1.72-1.59 (m, 3H), 1.56-1.46 (m, 4H), 1.45-1.17 (m, 12H), 1.16-1.00 (m, 5H), 0.99-0.85 (m, 11H), 0.84-0.78 (m, 4H), 0.66 (s, 4H)。 在10 min層析中,HPLC
Rt=5.73 min,30-90_AB_1.2 mL_E,純度100%。 C26
H43
[M+H-2H2
O]+
之MS ESI計算值355,實驗值355。實例 22 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥丁 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (22) 之合成  1.在0℃下將MeLi (7.75 mL,1.6 M,12.4 mmol)添加至N-4-1_7
(1 g,2.49 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌混合物1小時。向混合物添加NH4
Cl (10%,20 mL)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,得到呈膠狀之混合物(1 g)。混合物(1 g)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到91
(450 mg)及22
(460 mg)及130 mg之混合物。91
(450 mg)自MeCN (10 mL)再結晶得到呈固體之91
(50 mg)。22
(460 mg)自MeCN (10 mL)再結晶兩次得到呈固體之22
(50 mg)。22: 1 H NMR
(400 MHz, CDCl3
) δ 3.97-3.82 (m, 1H), 2.10-1.92 (m, 3H), 1.85-1.78 (m, 1H), 1.77-1.60 (m, 5H), 1.59-1.06 (m, 13H), 1.05-0.81 (m, 12H), 0.74-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.136 min,30-90_AB_E,純度100%,C24
H38
F3
O [M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt=5.05 min,30-90_AB_E,純度100%,d.e. 100%。實例 23 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (23) 之合成
1.在0℃下將MeLi (7.75 mL,1.6 M,12.4 mmol)添加至N-4-1_7
(1 g,2.49 mmol)於THF (10 mL)中之溶液中。在15℃下攪拌混合物1小時。向混合物添加NH4
Cl (10%,20 mL)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,得到呈膠狀之混合物(1 g)。混合物(1 g)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到91
(450 mg)及22
(460 mg)及130 mg之混合物。91
(450 mg)自MeCN (10 mL)再結晶得到呈固體之91
(50 mg)。22
(460 mg)自MeCN (10 mL)再結晶兩次得到呈固體之22
(50 mg)。22: 1 H NMR
(400 MHz, CDCl3
) δ 3.97-3.82 (m, 1H), 2.10-1.92 (m, 3H), 1.85-1.78 (m, 1H), 1.77-1.60 (m, 5H), 1.59-1.06 (m, 13H), 1.05-0.81 (m, 12H), 0.74-0.62 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.136 min,30-90_AB_E,純度100%,C24
H38
F3
O [M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt=5.05 min,30-90_AB_E,純度100%,d.e. 100%。實例 23 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (23) 之合成  1.
在0℃下向S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液緩慢地添加丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)。添加之後,在15℃下攪拌混合物1小時。用飽和NH4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之72
(500 mg,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 2.向72
(150 mg,0.372 mmol)於MeOH (20 mL)中之溶液中添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之23
(9 mg,6%)及38
(43 mg,29%)。23: 1 H NMR
(400 MHz, CDCl3
) δ 3.71-3.62 (m, 1H), 2.01-1.83 (m, 3H), 1.82-1.72 (m, 1H), 1.69-1.57 (m, 3H), 1.51-1.37 (m, 9H), 1.36-1.22 (m, 9H), 1.20-1.00 (m, 8H), 0.97 (s, 3H), 0.94-0.87 (m, 8H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.440 min,30-90AB_E,純度98.8%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 24 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (24) 之合成
1.
在0℃下向S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液緩慢地添加丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)。添加之後,在15℃下攪拌混合物1小時。用飽和NH4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之72
(500 mg,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 2.向72
(150 mg,0.372 mmol)於MeOH (20 mL)中之溶液中添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之23
(9 mg,6%)及38
(43 mg,29%)。23: 1 H NMR
(400 MHz, CDCl3
) δ 3.71-3.62 (m, 1H), 2.01-1.83 (m, 3H), 1.82-1.72 (m, 1H), 1.69-1.57 (m, 3H), 1.51-1.37 (m, 9H), 1.36-1.22 (m, 9H), 1.20-1.00 (m, 8H), 0.97 (s, 3H), 0.94-0.87 (m, 8H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.440 min,30-90AB_E,純度98.8%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 24 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (24) 之合成  1.在60℃下向Mg (4.35 g,179 mmol)及I2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。混合物用THF (10 mL)稀釋並直接使用。在0℃下於N2
下向S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。將NH4
Cl (20 mL,飽和水溶液)添加至混合物。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH3
CN (10 mL)再結晶,得到呈固體之77
(255 mg,21%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.306 min,30-90 AB,純度100%,C29
H49
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。 2.在25℃下於N2
下將苯甲酸(508 mg,4.16 mmol)及三苯基膦(1.63 g,6.24 mmol)添加至77
(900 mg,2.08 mmol)於THF (30 mL)中之溶液中。在25℃下攪拌混合物20 min。在0℃下於N2
下添加DIAD (1.26 g,6.24 mmol)。在0℃下攪拌混合物20 min,升溫至25℃且在25℃下攪拌16小時。反應混合物用水(60 mL)淬滅且用MTBE (3×30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物,該粗產物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈油狀物之不純產物S-500-2-15_1
(900 mg),其直接用於下一步驟。3.
將NaOH溶液(974 mg於6 mL H2
O中,16.8 mmol)添加至S-500-2-15_1
(900 mg,1.68 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在50℃下加熱混合物16小時。反應混合物用飽和NH4
Cl (60 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾濃縮,且藉由combi-flash (0%至15% EtOAc/PE)純化,得到210 mg固體,其藉由SFC (管柱:AD (250mm×30mm,5um),梯度:35-35% B (A= 0.1%NH3
/H2
O,B= MeOH),流速:80 mL/min)純化,得到呈固體之52
(150 mg,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 3.64-3.58 (m, 1H), 2.40-2.30 (m, 1H), 2.02-1.92 (m, 3H), 1.80-1.58 (m, 7H), 1.56-1.31 (m, 9H), 1.30-1.05 (m, 8H), 1.03 (s, 3H), 1.02-0.96 (m, 2H), 0.95-0.86 (m, 9H), 0.85-0.80 (m, 3H), 0.69 (s, 3H)。 在2 min層析中,LCMS
tR
=1.335 min,30-90AB_ELSD,純度100.0%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。 4.向52
(50 mg,0.116 mmol)於MeOH (10 mL)中之溶液添加Pd(OH)2
(200 mg)。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之24
(15 mg,30%)及呈固體之96
(1.2 mg,3%)。24: 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.52 (m, 1H), 2.02-1.91 (m, 1H), 1.74-1.57(m, 7H), 1.52-1.44 (m, 2H), 1.43-1.29 (m, 7H), 1.28-1.04 (m, 11H), 1.03-0.94 (m, 3H), 0.94-0.85 (m, 13H), 0.82 (s, 3H), 0.71-0.60 (m, 4H)。 在2 min層析中,LCMS
tR
=1.342 min,30-90AB_ELSD,純度100.0%,C29
H49
[M+H-2H2
O]+
之MS ESI計算值397,實驗值397。實例 25 : (1S,3S,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (25) 之合成
1.在60℃下向Mg (4.35 g,179 mmol)及I2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。混合物用THF (10 mL)稀釋並直接使用。在0℃下於N2
下向S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。將NH4
Cl (20 mL,飽和水溶液)添加至混合物。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH3
CN (10 mL)再結晶,得到呈固體之77
(255 mg,21%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.306 min,30-90 AB,純度100%,C29
H49
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。 2.在25℃下於N2
下將苯甲酸(508 mg,4.16 mmol)及三苯基膦(1.63 g,6.24 mmol)添加至77
(900 mg,2.08 mmol)於THF (30 mL)中之溶液中。在25℃下攪拌混合物20 min。在0℃下於N2
下添加DIAD (1.26 g,6.24 mmol)。在0℃下攪拌混合物20 min,升溫至25℃且在25℃下攪拌16小時。反應混合物用水(60 mL)淬滅且用MTBE (3×30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物,該粗產物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈油狀物之不純產物S-500-2-15_1
(900 mg),其直接用於下一步驟。3.
將NaOH溶液(974 mg於6 mL H2
O中,16.8 mmol)添加至S-500-2-15_1
(900 mg,1.68 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在50℃下加熱混合物16小時。反應混合物用飽和NH4
Cl (60 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾濃縮,且藉由combi-flash (0%至15% EtOAc/PE)純化,得到210 mg固體,其藉由SFC (管柱:AD (250mm×30mm,5um),梯度:35-35% B (A= 0.1%NH3
/H2
O,B= MeOH),流速:80 mL/min)純化,得到呈固體之52
(150 mg,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 3.64-3.58 (m, 1H), 2.40-2.30 (m, 1H), 2.02-1.92 (m, 3H), 1.80-1.58 (m, 7H), 1.56-1.31 (m, 9H), 1.30-1.05 (m, 8H), 1.03 (s, 3H), 1.02-0.96 (m, 2H), 0.95-0.86 (m, 9H), 0.85-0.80 (m, 3H), 0.69 (s, 3H)。 在2 min層析中,LCMS
tR
=1.335 min,30-90AB_ELSD,純度100.0%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。 4.向52
(50 mg,0.116 mmol)於MeOH (10 mL)中之溶液添加Pd(OH)2
(200 mg)。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之24
(15 mg,30%)及呈固體之96
(1.2 mg,3%)。24: 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.52 (m, 1H), 2.02-1.91 (m, 1H), 1.74-1.57(m, 7H), 1.52-1.44 (m, 2H), 1.43-1.29 (m, 7H), 1.28-1.04 (m, 11H), 1.03-0.94 (m, 3H), 0.94-0.85 (m, 13H), 0.82 (s, 3H), 0.71-0.60 (m, 4H)。 在2 min層析中,LCMS
tR
=1.342 min,30-90AB_ELSD,純度100.0%,C29
H49
[M+H-2H2
O]+
之MS ESI計算值397,實驗值397。實例 25 : (1S,3S,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (25) 之合成  25
之製備可見於實例13中。25: 1 H NMR
(400 MHz, CDCl3
) δ 7.42-7.28 (m, 5H), 4.97-4.81 (m, 1H), 4.12-3.92 (m, 1H), 3.23 (brs, 1H), 2.69 (brs, 1H), 2.10-1.88 (m, 3H), 1.82-1.62 (m, 7H), 1.48-1.18 (m, 10H), 1.10-0.88 (m, 8H), 0.87-0.78 (m, 4H), 0.70-0.58 (m, 4H)。 在2 min層析中,LCMS
Rt=1.319 min,10-80AB_2MIN_E,純度97.0%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.718 min,IC-3_MeOH(DEA)_40_2.5ML,98.26%de。 在8 min層析中,SFC
Rt=4.367 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,100%de。實例 26 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((S)-3- 羥基 -3- 甲基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (26) 之合成
25
之製備可見於實例13中。25: 1 H NMR
(400 MHz, CDCl3
) δ 7.42-7.28 (m, 5H), 4.97-4.81 (m, 1H), 4.12-3.92 (m, 1H), 3.23 (brs, 1H), 2.69 (brs, 1H), 2.10-1.88 (m, 3H), 1.82-1.62 (m, 7H), 1.48-1.18 (m, 10H), 1.10-0.88 (m, 8H), 0.87-0.78 (m, 4H), 0.70-0.58 (m, 4H)。 在2 min層析中,LCMS
Rt=1.319 min,10-80AB_2MIN_E,純度97.0%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.718 min,IC-3_MeOH(DEA)_40_2.5ML,98.26%de。 在8 min層析中,SFC
Rt=4.367 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,100%de。實例 26 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((S)-3- 羥基 -3- 甲基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (26) 之合成 
 1.在0℃下於N2
下將MeMgBr (0.83 mL,2.49 mmol,3 M於乙醚中)逐滴添加至於N-8-7_1
(300 mg,0.832 mmol)於THF (20 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應混合物用飽和NH4
Cl (50 mL)淬滅且用EtOAc (2×10 mL)萃取。合併層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到殘餘物,其藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之N-8-22_1
(100 mg,31%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.99-3.88 (m, 1H), 1.98-1.84 (m, 2H), 1.69-1.57 (m, 6H), 1.52-1.45 (m, 2H), 1.44-1.28 (m, 3H), 1.26-1.17 (m, 5H), 1.16-1.11 (m, 5H), 1.10-0.95 (m, 5H), 0.93-0.86 (m, 7H), 0.84-0.80 (m, 4H), 0.69-0.62 (m, 4H)。2.
將DMP (224 mg,0.53 mmol)添加至N-8-22_1
(100 mg,0.265 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌10 min之後,用NaHCO3
飽和溶液(30 mL)淬滅反應混合物直至含水層之pH為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至20% EtOAc/DCM)純化,得到呈固體之N-8-22_2
(80mg,80%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.54-2.42 (m, 1H), 2.09 (s, 3H), 1.94-1.87 (m, 1H), 1.71-1.59 (m, 4H), 1.54-1.45 (m, 3H), 1.44-1.30 (m, 4H), 1.29-1.16 (m, 6H), 1.15-1.07 (m, 5H), 1.06-0.92 (m, 4H), 0.91-0.79 (m, 7H), 0.74-0.61 (m, 4H)。 3.於N2
下將MeMgBr (0.353 mL,1.06 mmol,3 M於乙醚中)添加至N-8-22_2
(80 mg,0.213 mmol)於THF (5 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應混合物用NH4
Cl飽和水溶液(30 mL)淬滅且用EtOAc (20 mL×3)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,該粗產物藉由矽膠管柱(0%至10% EtOAc/PE)純化,得到呈固體之26
(7 mg,8%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.08-2.01 (m, 1H), 1.97-1.86 (m, 1H), 1.69-1.57 (m, 6H), 1.53-1.45 (m, 3H), 1.40-1.27 (m, 5H), 1.26-1.17 (m, 8H), 1.14 (s, 3H), 1.13-1.01 (m, 3H), 0.99-0.92 (m, 5H), 0.91-0.85 (m, 4H), 0.82 (s, 3H), 0.70 (s, 3H), 0.67-0.60 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.240 min,30-90AB_E,純度100%,C26
H43
[M+H-2H2
O]+
之MS ESI計算值355,實驗值355。實例 27 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (27) 之合成
1.在0℃下於N2
下將MeMgBr (0.83 mL,2.49 mmol,3 M於乙醚中)逐滴添加至於N-8-7_1
(300 mg,0.832 mmol)於THF (20 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應混合物用飽和NH4
Cl (50 mL)淬滅且用EtOAc (2×10 mL)萃取。合併層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到殘餘物,其藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之N-8-22_1
(100 mg,31%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.99-3.88 (m, 1H), 1.98-1.84 (m, 2H), 1.69-1.57 (m, 6H), 1.52-1.45 (m, 2H), 1.44-1.28 (m, 3H), 1.26-1.17 (m, 5H), 1.16-1.11 (m, 5H), 1.10-0.95 (m, 5H), 0.93-0.86 (m, 7H), 0.84-0.80 (m, 4H), 0.69-0.62 (m, 4H)。2.
將DMP (224 mg,0.53 mmol)添加至N-8-22_1
(100 mg,0.265 mmol)於DCM (10 mL)中之溶液中。在20℃下攪拌10 min之後,用NaHCO3
飽和溶液(30 mL)淬滅反應混合物直至含水層之pH為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至20% EtOAc/DCM)純化,得到呈固體之N-8-22_2
(80mg,80%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.54-2.42 (m, 1H), 2.09 (s, 3H), 1.94-1.87 (m, 1H), 1.71-1.59 (m, 4H), 1.54-1.45 (m, 3H), 1.44-1.30 (m, 4H), 1.29-1.16 (m, 6H), 1.15-1.07 (m, 5H), 1.06-0.92 (m, 4H), 0.91-0.79 (m, 7H), 0.74-0.61 (m, 4H)。 3.於N2
下將MeMgBr (0.353 mL,1.06 mmol,3 M於乙醚中)添加至N-8-22_2
(80 mg,0.213 mmol)於THF (5 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應混合物用NH4
Cl飽和水溶液(30 mL)淬滅且用EtOAc (20 mL×3)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,該粗產物藉由矽膠管柱(0%至10% EtOAc/PE)純化,得到呈固體之26
(7 mg,8%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.08-2.01 (m, 1H), 1.97-1.86 (m, 1H), 1.69-1.57 (m, 6H), 1.53-1.45 (m, 3H), 1.40-1.27 (m, 5H), 1.26-1.17 (m, 8H), 1.14 (s, 3H), 1.13-1.01 (m, 3H), 0.99-0.92 (m, 5H), 0.91-0.85 (m, 4H), 0.82 (s, 3H), 0.70 (s, 3H), 0.67-0.60 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.240 min,30-90AB_E,純度100%,C26
H43
[M+H-2H2
O]+
之MS ESI計算值355,實驗值355。實例 27 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (27) 之合成 
 1.在0℃下於N2
下經30 min之時段向N-014-012_4
(300 mg,0.8366 mmol)於THF (20 mL)中之溶液逐滴添加異丙基氯化鎂(1.25 mL,2.50 mmol,2 M)之溶液,在此期間將溫度維持在0℃以下。在20℃下再攪拌反應混合物2小時,得到懸浮液。反應混合物添加NH4
Cl飽和水溶液(15 mL)且攪拌20 min,隨後用EtOAc (3×10mL)萃取混合物。合併之有機相用鹽水(2×10 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,以獲得呈固體之N-014-001_1
(360 mg,粗產物),1
H NMR顯示所需產物,且直接用於下一步驟。 2.X1
(150 mg,0.37 mmol)藉由SFC (管柱:Chiralpak AS-H 250×30 5u;條件:0.1% NH3
H2
O EtOH;開始B:20%;結束B:20%;流速(ml/min):65)純化,以獲得37
(峰值2,46 mg,31%)及呈固體之27
(峰值1,27 mg,18%)。 37: 27: 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.42-3.34 (m, 1H), 2.43-2.34 (m, 1H), 2.06-1.91 (m, 3H), 1.90-1.75 (m, 2H), 1.74-1.66 (m, 2H), 1.63-1.58 (m, 3H), 1.54-1.26 (m, 11H), 1.22-1.04 (m, 3H), 1.03-0.99 (m, 3H), 0.97-0.93 (m, 7H), 0.92-0.87 (m, 3H), 0.86-0.77 (m, 3H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.228 min,30-90AB_2MIN_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。 在10 min層析中,SFC
Rt=2.440 min,OJ_3_EtOH_DEA_5_40_25ML (「管柱:Chiralcel OJ-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),97.38%de。實例 28 : (3S,5S,8R,9R,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 ) -13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (28) 之合成
1.在0℃下於N2
下經30 min之時段向N-014-012_4
(300 mg,0.8366 mmol)於THF (20 mL)中之溶液逐滴添加異丙基氯化鎂(1.25 mL,2.50 mmol,2 M)之溶液,在此期間將溫度維持在0℃以下。在20℃下再攪拌反應混合物2小時,得到懸浮液。反應混合物添加NH4
Cl飽和水溶液(15 mL)且攪拌20 min,隨後用EtOAc (3×10mL)萃取混合物。合併之有機相用鹽水(2×10 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,以獲得呈固體之N-014-001_1
(360 mg,粗產物),1
H NMR顯示所需產物,且直接用於下一步驟。 2.X1
(150 mg,0.37 mmol)藉由SFC (管柱:Chiralpak AS-H 250×30 5u;條件:0.1% NH3
H2
O EtOH;開始B:20%;結束B:20%;流速(ml/min):65)純化,以獲得37
(峰值2,46 mg,31%)及呈固體之27
(峰值1,27 mg,18%)。 37: 27: 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.42-3.34 (m, 1H), 2.43-2.34 (m, 1H), 2.06-1.91 (m, 3H), 1.90-1.75 (m, 2H), 1.74-1.66 (m, 2H), 1.63-1.58 (m, 3H), 1.54-1.26 (m, 11H), 1.22-1.04 (m, 3H), 1.03-0.99 (m, 3H), 0.97-0.93 (m, 7H), 0.92-0.87 (m, 3H), 0.86-0.77 (m, 3H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.228 min,30-90AB_2MIN_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。 在10 min層析中,SFC
Rt=2.440 min,OJ_3_EtOH_DEA_5_40_25ML (「管柱:Chiralcel OJ-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),97.38%de。實例 28 : (3S,5S,8R,9R,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 ) -13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (28) 之合成 
 1.
在Ar下向44
(200 mg,0.480 mmol)於MeOH/THF (10 mL/10 mL)中之溶液添加Pd/C (無水,200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物48小時,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用THF (100 mL)洗滌。濃縮濾液,得到呈固體之28
(30 mg,15%)及呈固體之82
(30 mg,15%)。28: 1 H NMR
(400MHz, CDCl3) δ 3.63-3.61 (m, 1H), 1.98-1.76 (m, 4H), 1.72-1.55 (m, 7H), 1.55-1.47 (m, 4H), 1.46-1.23 (m, 6H), 1.22-0.97 (m, 11H), 0.92-0.78 (m, 12H), 0.76-0.54 (m, 5H)。 在2 min層析中,LCMS
Rt=1.298 min,30-90AB,純度100%,C28
H47
[M+H-2H2
O]+之MS ESI計算值383,實驗值383。實例 29 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (29) 之合成
1.
在Ar下向44
(200 mg,0.480 mmol)於MeOH/THF (10 mL/10 mL)中之溶液添加Pd/C (無水,200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物48小時,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用THF (100 mL)洗滌。濃縮濾液,得到呈固體之28
(30 mg,15%)及呈固體之82
(30 mg,15%)。28: 1 H NMR
(400MHz, CDCl3) δ 3.63-3.61 (m, 1H), 1.98-1.76 (m, 4H), 1.72-1.55 (m, 7H), 1.55-1.47 (m, 4H), 1.46-1.23 (m, 6H), 1.22-0.97 (m, 11H), 0.92-0.78 (m, 12H), 0.76-0.54 (m, 5H)。 在2 min層析中,LCMS
Rt=1.298 min,30-90AB,純度100%,C28
H47
[M+H-2H2
O]+之MS ESI計算值383,實驗值383。實例 29 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (29) 之合成  1.在60℃下向Mg (4.35 g,179 mmol)及I2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。混合物用THF (10 mL)稀釋並直接使用。在0℃下於N2
下向S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。將NH4
Cl (20 mL,飽和水溶液)添加至混合物。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH3
CN (10 mL)再結晶,得到呈固體之77
(255 mg,21%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.306 min,30-90 AB,純度100%,C29
H49
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。2.
於Ar下將Pd(OH)2
(無水,20%,50.0 mg)添加至77
(100 mg,232 umol)於THF(10 mL)及MeOH (10 mL)中之溶液中。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物16 小時。反應混合物經由矽藻土墊過濾且用THF (3×10 mL)洗滌。濃縮濾液。殘餘物藉由矽膠層析(PE/EtOAc=20/1)純化,得到呈固體之29
(7.00 mg,7%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.60 (m, 1H), 1.96-1.88 (m, 2H), 1.68-1.60 (m, 3H), 1.53-1.47 (m, 7H), 1.39-1.23 (m, 13H), 1.16-0.95 (m, 7H), 0.90-0.86 (m, 12H), 0.83 (s, 3H), 0.66-0.63 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.603 min,30-90 AB_ELSD,純度97%,C29
H49
[M+H-H2
O]+
之MS ESI計算值397,實驗值397。實例 30 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (30) 之合成
1.在60℃下向Mg (4.35 g,179 mmol)及I2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。混合物用THF (10 mL)稀釋並直接使用。在0℃下於N2
下向S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。將NH4
Cl (20 mL,飽和水溶液)添加至混合物。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH3
CN (10 mL)再結晶,得到呈固體之77
(255 mg,21%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.306 min,30-90 AB,純度100%,C29
H49
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。2.
於Ar下將Pd(OH)2
(無水,20%,50.0 mg)添加至77
(100 mg,232 umol)於THF(10 mL)及MeOH (10 mL)中之溶液中。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物16 小時。反應混合物經由矽藻土墊過濾且用THF (3×10 mL)洗滌。濃縮濾液。殘餘物藉由矽膠層析(PE/EtOAc=20/1)純化,得到呈固體之29
(7.00 mg,7%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.60 (m, 1H), 1.96-1.88 (m, 2H), 1.68-1.60 (m, 3H), 1.53-1.47 (m, 7H), 1.39-1.23 (m, 13H), 1.16-0.95 (m, 7H), 0.90-0.86 (m, 12H), 0.83 (s, 3H), 0.66-0.63 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.603 min,30-90 AB_ELSD,純度97%,C29
H49
[M+H-H2
O]+
之MS ESI計算值397,實驗值397。實例 30 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (30) 之合成 
 1.
在25℃下於N2
下將苯甲酸(2.03 g,16.7 mmol)及三苯基膦(6.57 g,25.1 mmol)添加至S-500-2-10
(3.5 g,8.39 mmol)於THF (30 mL)中之溶液中。在25℃下攪拌混合物20 min。在0℃下於N2
下添加DIAD (5.07 g,25.1 mmol)。在0℃下攪拌混合物20 min,隨後升溫至25℃且攪拌1 h。反應混合物用水(100 mL)淬滅且用MTBE (3×30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物,該粗產物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到300 mg呈油狀物之粗產物S-500-2-9_1
,其直接用於下一步驟。 2.將NaOH (1.14 g於3 mL H2
O中,28.7 mmol)添加至S-500-2-9_1
(300 mg,0.576 mmol)於THF (5 mL)及MeOH (3 mL)中之溶液中。在50℃下攪拌混合物16小時。混合物用飽和NH4
Cl (20 mL)淬滅,且用EtOAc (3×10 mL)萃取。合併之有機相用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮,且藉由combi-flash (0%至10% EtOAc/PE)純化,得到呈固體之30
(12 mg,5%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.28 (m, 1H), 3.63-3.59 (m, 1H), 2.44-2.40 (m, 1H), 2.05-1.90 (m, 3H), 1.80-1.62 (m, 4H), 1.61-1.58 (m, 3H), 1.56-1.30 (m, 9H), 1.28-1.03 (m, 10H), 1.01 (s, 3H), 0.99-0.85 (m, 10H), 0.69 (s, 3H)。 在2 min層析中,LCMS
tR
=1.260 min,30-90AB_ELSD,純度100.0%,C28
H45
[M+H-2H2
O]+
之MS ESI計算值381,實驗值381。實例 31 : (1R,3R,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (31) 之合成
1.
在25℃下於N2
下將苯甲酸(2.03 g,16.7 mmol)及三苯基膦(6.57 g,25.1 mmol)添加至S-500-2-10
(3.5 g,8.39 mmol)於THF (30 mL)中之溶液中。在25℃下攪拌混合物20 min。在0℃下於N2
下添加DIAD (5.07 g,25.1 mmol)。在0℃下攪拌混合物20 min,隨後升溫至25℃且攪拌1 h。反應混合物用水(100 mL)淬滅且用MTBE (3×30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,真空濃縮,得到粗產物,該粗產物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到300 mg呈油狀物之粗產物S-500-2-9_1
,其直接用於下一步驟。 2.將NaOH (1.14 g於3 mL H2
O中,28.7 mmol)添加至S-500-2-9_1
(300 mg,0.576 mmol)於THF (5 mL)及MeOH (3 mL)中之溶液中。在50℃下攪拌混合物16小時。混合物用飽和NH4
Cl (20 mL)淬滅,且用EtOAc (3×10 mL)萃取。合併之有機相用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮,且藉由combi-flash (0%至10% EtOAc/PE)純化,得到呈固體之30
(12 mg,5%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.28 (m, 1H), 3.63-3.59 (m, 1H), 2.44-2.40 (m, 1H), 2.05-1.90 (m, 3H), 1.80-1.62 (m, 4H), 1.61-1.58 (m, 3H), 1.56-1.30 (m, 9H), 1.28-1.03 (m, 10H), 1.01 (s, 3H), 0.99-0.85 (m, 10H), 0.69 (s, 3H)。 在2 min層析中,LCMS
tR
=1.260 min,30-90AB_ELSD,純度100.0%,C28
H45
[M+H-2H2
O]+
之MS ESI計算值381,實驗值381。實例 31 : (1R,3R,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (31) 之合成  31
之合成可見於實例13中。31: 1 H NMR
(400 MHz, CDCl3
) δ 7.45-7.28 (m, 5H), 5.02-4.81 (m, 1H), 4.18-3.98 (m, 1H), 3.35 (brs, 1H), 2.47 (brs, 1H), 2.15-1.72 (m, 8H), 1.53-1.31 (m, 8H), 1.30-1.03 (m, 8H), 0.99-0.89 (m, 4H), 0.89-0.78 (m, 4H), 0.75-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.327 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.929 min,IC-3_MeOH(DEA)_40_2.5ML,98.4%de。實例 32 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 苯基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (32) 之合成
31
之合成可見於實例13中。31: 1 H NMR
(400 MHz, CDCl3
) δ 7.45-7.28 (m, 5H), 5.02-4.81 (m, 1H), 4.18-3.98 (m, 1H), 3.35 (brs, 1H), 2.47 (brs, 1H), 2.15-1.72 (m, 8H), 1.53-1.31 (m, 8H), 1.30-1.03 (m, 8H), 0.99-0.89 (m, 4H), 0.89-0.78 (m, 4H), 0.75-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.327 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.929 min,IC-3_MeOH(DEA)_40_2.5ML,98.4%de。實例 32 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 苯基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (32) 之合成  1.
在20℃下於氮氣下將t-BuOK (4.64 g,41.4 mmol)添加至Me3
SI (8.44 g,41.4 mmol)於無水THF (50 mL)中之懸浮液中。在20℃下攪拌混合物1小時,且添加N-005_5
(5 g,13.8 mmol)。使所得混合物升溫至45℃且攪拌4小時。使反應混合物冷卻至室溫,用水(50 mL)淬滅,用EtOAc (2×50 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-005_001
(2.7 g,52%)。 在2.0 min層析中,LCMS
Rt=1.324 min,30-90AB_E,純度92%,C25
H41
O [M+H-H2
O]+
之MS ESI計算值357,實驗值357。 2.在0℃下於氮氣下將CuI (10.1 mg,0.0534 mmol)及PhMgBr (1 M於THF中,2.66 mL,2.66 mmol)添加至N-005_001
(200 mg,0.534 mmol)於無水THF (20 mL)中之溶液中。使混合物逐漸升溫至15℃且攪拌16小時。反應混合物用NH4
Cl水溶液(20 mL)淬滅,用EtOAc (2×10 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0-5% EtOAc/PE)純化,得到呈固體之NA-6-5/6
(190 mg,79%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.35-7.27 (m, 2H), 7.25-7.18 (m, 3H), 3.95-3.81 (m, 1H), 2.87-2.39 (m, 2H), 2.04-1.92 (m, 1H), 1.89-1.80 (m, 1H), 1.71-1.58 (m, 4H), 1.56-1.43 (m, 6H), 1.41-1.27 (m, 5H), 1.26-1.18 (m, 4H), 1.18-1.08 (m, 2H), 1.06-0.96 (m, 5H), 0.92-0.79 (m, 8H), 0.73-0.55 (m, 4H)。3.N-6-5/6
(190 mg,0.420 mmol)藉由製備型HPLC (管柱:YMC-Actus Triart C18 100×30mm×5um;條件:水(0.05%HCl)-ACN;梯度:90-100%B;流速:25 mL/min)分離,得到呈固體之93
(56 mg,30%)及呈固體之32
(12 mg,6%)。32: 1 H NMR
(400 MHz, CDCl3
) δ 7.36-7.29 (m, 2H), 7.25-7.19 (m, 3H), 3.89-3.83 (m, 1H), 2.79-2.72 (m, 1H), 2.49-2.40 (m, 1H), 2.05-1.98 (m, 1H), 1.92-1.79 (m, 2H), 1.72-1.51 (m, 9H), 1.44-1.31 (m, 5H), 1.30-1.09 (m, 7H), 1.08-0.96 (m, 5H), 0.92-0.81 (m, 7H), 0.74-0.63 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.343 min,30-90AB,純度100%,C31
H47
O [M+H-H2
O]+
之MS ESI計算值435,實驗值435。實例 33 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4-(3- 甲基氧雜環丁 -3- 基 ) 丁 -2- 基 )-10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (33) 之合成
1.
在20℃下於氮氣下將t-BuOK (4.64 g,41.4 mmol)添加至Me3
SI (8.44 g,41.4 mmol)於無水THF (50 mL)中之懸浮液中。在20℃下攪拌混合物1小時,且添加N-005_5
(5 g,13.8 mmol)。使所得混合物升溫至45℃且攪拌4小時。使反應混合物冷卻至室溫,用水(50 mL)淬滅,用EtOAc (2×50 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-005_001
(2.7 g,52%)。 在2.0 min層析中,LCMS
Rt=1.324 min,30-90AB_E,純度92%,C25
H41
O [M+H-H2
O]+
之MS ESI計算值357,實驗值357。 2.在0℃下於氮氣下將CuI (10.1 mg,0.0534 mmol)及PhMgBr (1 M於THF中,2.66 mL,2.66 mmol)添加至N-005_001
(200 mg,0.534 mmol)於無水THF (20 mL)中之溶液中。使混合物逐漸升溫至15℃且攪拌16小時。反應混合物用NH4
Cl水溶液(20 mL)淬滅,用EtOAc (2×10 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮。殘餘物藉由矽膠管柱層析(0-5% EtOAc/PE)純化,得到呈固體之NA-6-5/6
(190 mg,79%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.35-7.27 (m, 2H), 7.25-7.18 (m, 3H), 3.95-3.81 (m, 1H), 2.87-2.39 (m, 2H), 2.04-1.92 (m, 1H), 1.89-1.80 (m, 1H), 1.71-1.58 (m, 4H), 1.56-1.43 (m, 6H), 1.41-1.27 (m, 5H), 1.26-1.18 (m, 4H), 1.18-1.08 (m, 2H), 1.06-0.96 (m, 5H), 0.92-0.79 (m, 8H), 0.73-0.55 (m, 4H)。3.N-6-5/6
(190 mg,0.420 mmol)藉由製備型HPLC (管柱:YMC-Actus Triart C18 100×30mm×5um;條件:水(0.05%HCl)-ACN;梯度:90-100%B;流速:25 mL/min)分離,得到呈固體之93
(56 mg,30%)及呈固體之32
(12 mg,6%)。32: 1 H NMR
(400 MHz, CDCl3
) δ 7.36-7.29 (m, 2H), 7.25-7.19 (m, 3H), 3.89-3.83 (m, 1H), 2.79-2.72 (m, 1H), 2.49-2.40 (m, 1H), 2.05-1.98 (m, 1H), 1.92-1.79 (m, 2H), 1.72-1.51 (m, 9H), 1.44-1.31 (m, 5H), 1.30-1.09 (m, 7H), 1.08-0.96 (m, 5H), 0.92-0.81 (m, 7H), 0.74-0.63 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.343 min,30-90AB,純度100%,C31
H47
O [M+H-H2
O]+
之MS ESI計算值435,實驗值435。實例 33 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4-(3- 甲基氧雜環丁 -3- 基 ) 丁 -2- 基 )-10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (33) 之合成  1.在25℃下向N-014-005_1
(10 g,97.9 mmol)於DCM (100 mL)中之溶液中添加1-甲基-1H-咪唑(16.0 g,195 mmol)及TEA (19.7 g,195 mmol)。將TsCl (37.1 g,195 mmol)添加至溶液中。在25℃下攪拌反應混合物2小時。混合物用水(2×100 mL)、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈淡黃色固體之N-014-005_2
(25 g,粗產物),其藉由矽膠管柱層析(0%至15% EtOAc/PE)純化,得到呈白色固體之N-014-005_2
(23.6 g,95%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.80-7.68 (m, 2H), 7.41-7.26 (m, 2H), 3.40-3.29 (m, 4H), 4.12-4.00 (s, 2H), 2.44 (s, 3H), 1.28 (s, 3H)。 2.向N-014-005_2
(10 g,39.0 mmol)於丙酮(100 mL)中之溶液中添加LiBr (13.5 g,156 mmol)。在65℃下攪拌混合物1小時。混合物在0℃下用水(200 mL)淬滅且用己烷(3×200 mL)萃取。合併之有機相用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈黃色液體之N-014-005_3
(2.54 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.50-4.30 (m, 4H), 3.64 (s, 2H), 1.58 (s, 1H), 1.43 (s, 3H)。
1.在25℃下向N-014-005_1
(10 g,97.9 mmol)於DCM (100 mL)中之溶液中添加1-甲基-1H-咪唑(16.0 g,195 mmol)及TEA (19.7 g,195 mmol)。將TsCl (37.1 g,195 mmol)添加至溶液中。在25℃下攪拌反應混合物2小時。混合物用水(2×100 mL)、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈淡黃色固體之N-014-005_2
(25 g,粗產物),其藉由矽膠管柱層析(0%至15% EtOAc/PE)純化,得到呈白色固體之N-014-005_2
(23.6 g,95%)。 1 H NMR
(400 MHz, CDCl3
) δ 7.80-7.68 (m, 2H), 7.41-7.26 (m, 2H), 3.40-3.29 (m, 4H), 4.12-4.00 (s, 2H), 2.44 (s, 3H), 1.28 (s, 3H)。 2.向N-014-005_2
(10 g,39.0 mmol)於丙酮(100 mL)中之溶液中添加LiBr (13.5 g,156 mmol)。在65℃下攪拌混合物1小時。混合物在0℃下用水(200 mL)淬滅且用己烷(3×200 mL)萃取。合併之有機相用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈黃色液體之N-014-005_3
(2.54 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 4.50-4.30 (m, 4H), 3.64 (s, 2H), 1.58 (s, 1H), 1.43 (s, 3H)。 3.在50℃至55℃下於N2
下向Mg (807 mg,33.2 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液逐滴添加N-014-005_3
(2.5 g,15.1 mmol)於THF (8 mL)中之溶液。在55℃下攪拌混合物1 h。用THF (10 mL)稀釋混合物,且直接用於下一步驟中而無需監測。在0℃下向N-14-12_4
(1.01 g,2.83 mmol)於THF (10 mL)中之溶液添加新鮮製備的3-[(溴鎂)甲基]-3-甲基環氧丙烷(15 mmol於20 mL THF中)。在15℃下攪拌混合物4 h。向混合物添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。分離有機層且真空濃縮。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈白色固體之混合物(190 mg,15%),其藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3H2O ETOH,梯度:50%至50%,流速(ml/min):60mL/min,25℃)純化,得到33
(峰值1,110 mg,9%)及呈白色固體之70
(峰值2,30mg,不純)。不純70
(30 mg,不純)藉由矽膠管柱層析(15% EtOAc/PE)純化,得到呈白色固體之70
(10 mg,5%)。33: 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 4.59-4.70 (m, 1H), 4.50-4.48 (m, 1H),4.36-4.33 (m, 1H), 3.83 (s, 1H), 2.40-2.33 (m, 1H), 2.10-1.50 (m, 17H), 1.49-1.35 (m, 9H), 1.30-0.80 (m, 13H), 0.68 (s, 3H)。 在3 min層析中,LCMS
Rt=1.069 min,30-90AB_2MIN_E.M,純度100%,C29
H49
O3
[M+H]+
之MS ESI計算值445,實驗值445。實例 34 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (34) 之合成
3.在50℃至55℃下於N2
下向Mg (807 mg,33.2 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液逐滴添加N-014-005_3
(2.5 g,15.1 mmol)於THF (8 mL)中之溶液。在55℃下攪拌混合物1 h。用THF (10 mL)稀釋混合物,且直接用於下一步驟中而無需監測。在0℃下向N-14-12_4
(1.01 g,2.83 mmol)於THF (10 mL)中之溶液添加新鮮製備的3-[(溴鎂)甲基]-3-甲基環氧丙烷(15 mmol於20 mL THF中)。在15℃下攪拌混合物4 h。向混合物添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。分離有機層且真空濃縮。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈白色固體之混合物(190 mg,15%),其藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3H2O ETOH,梯度:50%至50%,流速(ml/min):60mL/min,25℃)純化,得到33
(峰值1,110 mg,9%)及呈白色固體之70
(峰值2,30mg,不純)。不純70
(30 mg,不純)藉由矽膠管柱層析(15% EtOAc/PE)純化,得到呈白色固體之70
(10 mg,5%)。33: 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 4.59-4.70 (m, 1H), 4.50-4.48 (m, 1H),4.36-4.33 (m, 1H), 3.83 (s, 1H), 2.40-2.33 (m, 1H), 2.10-1.50 (m, 17H), 1.49-1.35 (m, 9H), 1.30-0.80 (m, 13H), 0.68 (s, 3H)。 在3 min層析中,LCMS
Rt=1.069 min,30-90AB_2MIN_E.M,純度100%,C29
H49
O3
[M+H]+
之MS ESI計算值445,實驗值445。實例 34 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (34) 之合成  1.
在0℃下將於THF (5 mL)中之N-8-7_1
(500 mg,1.38 mmol)添加至異丙基氯化鎂(708 mg,3.44 mL,2 M於THF中)於THF (5 mL)中之溶液中。在25℃下攪拌混合物4小時。向混合物添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (2 × 30 mL)萃取混合物。有機層經分離且真空濃縮,得到殘餘物。殘餘物藉由矽膠層析純化用PE/EtOAc=3/1溶離,得到呈固體之N-8-11_1
(170 mg,30%)。該不純產物(120 mg)藉由矽膠層析進一步純化用PE/EtOAc=3/1溶離,得到呈固體之N-8-11_1
(50 mg,42%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.20-3.10 (m, 1H), 2.00-1.80 (m, 2H), 1.70-1.60 (m, 2H), 1.55-1.40 (m, 4H), 1.39-0.95 (m, 19H), 0.90-0.80 (m, 15H), 0.70-0.60 (m, 5H)。 2.將DMP (457 mg,1.08 mmol)添加至N-8-11_1
(220 mg,0.543 mmol)於DCM (5 mL)中之溶液中。在25℃下攪拌10 min之後,反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH變成約9為止。過濾混合物。分離DCM層,且用DCM (100 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×100 mL)、飽和NaHCO3
(100 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-8-11_2
(140 mg,64%)。 在2.0 min層析中,LCMS
Rt=1.300 min,30-90AB_2MIN_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。 3.將NaBH4
(1.17 g,17.3 mmol)每五分鐘添加五次至N-8-11_2
(140 mg,0.347 mmol)於MeOH (2 mL)及THF (2 mL)中之溶液中。在15℃下攪拌混合物30分鐘。用飽和NH4
Cl (50 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到N-8-11_1
(140 mg,不純)。N-8-11_1
藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之34
(50 mg,不純)及呈固體之65
(10 mg,不純)。4.N-8-11_1
(50 mg,0.123 mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之34
(30 mg,不純)。N-8-11_1
(30 mg,0.0741mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之34
(9 mg,30%)。34: 1 H NMR
(400 MHz, CDCl3
) δ 3.18-3.07 (m, 1H), 1.98-1.81 (m, 2H), 1.71-1.58 (m, 6H), 1.53-1.31 (m, 7H), 1.30-0.98 (m, 14H), 0.97-0.78 (m, 14H), 0.70-0.60 (m, 4H)。 在7.0 min層析中,LCMS
Rt=4.387 min,30-90AB_7MIN_E,純度97.6%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 35 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (35) 之合成
1.
在0℃下將於THF (5 mL)中之N-8-7_1
(500 mg,1.38 mmol)添加至異丙基氯化鎂(708 mg,3.44 mL,2 M於THF中)於THF (5 mL)中之溶液中。在25℃下攪拌混合物4小時。向混合物添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (2 × 30 mL)萃取混合物。有機層經分離且真空濃縮,得到殘餘物。殘餘物藉由矽膠層析純化用PE/EtOAc=3/1溶離,得到呈固體之N-8-11_1
(170 mg,30%)。該不純產物(120 mg)藉由矽膠層析進一步純化用PE/EtOAc=3/1溶離,得到呈固體之N-8-11_1
(50 mg,42%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.20-3.10 (m, 1H), 2.00-1.80 (m, 2H), 1.70-1.60 (m, 2H), 1.55-1.40 (m, 4H), 1.39-0.95 (m, 19H), 0.90-0.80 (m, 15H), 0.70-0.60 (m, 5H)。 2.將DMP (457 mg,1.08 mmol)添加至N-8-11_1
(220 mg,0.543 mmol)於DCM (5 mL)中之溶液中。在25℃下攪拌10 min之後,反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH變成約9為止。過濾混合物。分離DCM層,且用DCM (100 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×100 mL)、飽和NaHCO3
(100 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之N-8-11_2
(140 mg,64%)。 在2.0 min層析中,LCMS
Rt=1.300 min,30-90AB_2MIN_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。 3.將NaBH4
(1.17 g,17.3 mmol)每五分鐘添加五次至N-8-11_2
(140 mg,0.347 mmol)於MeOH (2 mL)及THF (2 mL)中之溶液中。在15℃下攪拌混合物30分鐘。用飽和NH4
Cl (50 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到N-8-11_1
(140 mg,不純)。N-8-11_1
藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之34
(50 mg,不純)及呈固體之65
(10 mg,不純)。4.N-8-11_1
(50 mg,0.123 mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之34
(30 mg,不純)。N-8-11_1
(30 mg,0.0741mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之34
(9 mg,30%)。34: 1 H NMR
(400 MHz, CDCl3
) δ 3.18-3.07 (m, 1H), 1.98-1.81 (m, 2H), 1.71-1.58 (m, 6H), 1.53-1.31 (m, 7H), 1.30-0.98 (m, 14H), 0.97-0.78 (m, 14H), 0.70-0.60 (m, 4H)。 在7.0 min層析中,LCMS
Rt=4.387 min,30-90AB_7MIN_E,純度97.6%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 35 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (35) 之合成  1.在25℃下將NaOH (71.9 mg,180 mmol)添加至N-4-4B
(20 mg,0.0361 mmol)於THF/MeOH (2 mL)中之溶液中。使反應混合物升溫至50℃且攪拌1 h。使反應混合物冷卻且添加水(20 mg)。用EtOAc (3 × 10 mL)萃取水相。合併之有機相用飽和鹽水(2×20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化以提供呈白色固體之35
(8 mg,50%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.63-3.61 (m, 1H), 3.41-3.38 (m, 5H); 2.51 (s, 1H); 1.97-1.81 (m, 1H), 1.71-1.54 (m, 8H), 1.51-1.48 (m, 4H), 1.25-1.10 (m, 15H), 0.99-0.80 (m, 9H), 0.78-0.75 (m, 4H),0.71-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.301 min,30-90 AB,純度96%,C29
H48
O [M+H-2H2
O]+
之MS ESI計算值413,實驗值413。實例 36 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (36) 之合成
1.在25℃下將NaOH (71.9 mg,180 mmol)添加至N-4-4B
(20 mg,0.0361 mmol)於THF/MeOH (2 mL)中之溶液中。使反應混合物升溫至50℃且攪拌1 h。使反應混合物冷卻且添加水(20 mg)。用EtOAc (3 × 10 mL)萃取水相。合併之有機相用飽和鹽水(2×20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化以提供呈白色固體之35
(8 mg,50%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.63-3.61 (m, 1H), 3.41-3.38 (m, 5H); 2.51 (s, 1H); 1.97-1.81 (m, 1H), 1.71-1.54 (m, 8H), 1.51-1.48 (m, 4H), 1.25-1.10 (m, 15H), 0.99-0.80 (m, 9H), 0.78-0.75 (m, 4H),0.71-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.301 min,30-90 AB,純度96%,C29
H48
O [M+H-2H2
O]+
之MS ESI計算值413,實驗值413。實例 36 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (36) 之合成  1.將Pd(OH)2
(200 mg)添加至30
(100 mg,0.239 mmol)於MeOH (10 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0%至10% EtOAc/PE)純化,得到47
(21 mg,21%)及呈固體之36
(1 mg,1%)。37: 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.55 (m, 1H), 2.05-1.77 (m, 3H), 1.72-1.63 (m, 3H), 1.55-1.48 (m, 3H), 1.47-1.31 (m, 9H), 1.29-1.12 (m, 13H), 1.11-1.00 (m, 3H), 0.96 (s, 3H), 0.93-0.87 (m, 9H), 0.67 (s, 3H)。 在2 min層析中,LCMS
tR
=1.296 min,30-90AB_ELSD,純度100.0%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 37 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (37) 之合成
1.將Pd(OH)2
(200 mg)添加至30
(100 mg,0.239 mmol)於MeOH (10 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0%至10% EtOAc/PE)純化,得到47
(21 mg,21%)及呈固體之36
(1 mg,1%)。37: 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.55 (m, 1H), 2.05-1.77 (m, 3H), 1.72-1.63 (m, 3H), 1.55-1.48 (m, 3H), 1.47-1.31 (m, 9H), 1.29-1.12 (m, 13H), 1.11-1.00 (m, 3H), 0.96 (s, 3H), 0.93-0.87 (m, 9H), 0.67 (s, 3H)。 在2 min層析中,LCMS
tR
=1.296 min,30-90AB_ELSD,純度100.0%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 37 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (37) 之合成  1.N-014-001_1
(150 mg,0.37 mmol)藉由SFC (管柱:Chiralpak AS-H 250×30 5u;條件:0.1% NH3
H2
O EtOH;開始B:20%;結束B:20%;流速(ml/min):65)純化,以獲得37
(峰值2,46 mg,31%)及呈固體之27
(峰值1,27 mg,18%)。 014-001A: 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.28 (m, 1H), 3.18-3.09 (m, 1H), 2.39-2.35 (m, 1H), 2.06-1.81 (m, 4H), 1.73-1.57 (m, 6H), 1.54-1.41 (m, 8H), 1.40-1.26 (m, 3H), 1.24-1.11 (m, 3H),1.10-0.97 (m, 6H), 0.96-0.92 (m, 1H), 0.90-0.85 (m, 5H), 0.84-0.76 (m, 4H), 0.69 (s, 3H)。 在2 min層析中,LCMS
Rt=1.207 min,30-90AB_2MIN_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。 在10 min層析中,SFC
Rt=2.635 min,OJ_3_EtOH_DEA_5_40_25ML (「管柱:Chiralcel OJ-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),98.66%de。實例 38 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (38) 之合成
1.N-014-001_1
(150 mg,0.37 mmol)藉由SFC (管柱:Chiralpak AS-H 250×30 5u;條件:0.1% NH3
H2
O EtOH;開始B:20%;結束B:20%;流速(ml/min):65)純化,以獲得37
(峰值2,46 mg,31%)及呈固體之27
(峰值1,27 mg,18%)。 014-001A: 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.28 (m, 1H), 3.18-3.09 (m, 1H), 2.39-2.35 (m, 1H), 2.06-1.81 (m, 4H), 1.73-1.57 (m, 6H), 1.54-1.41 (m, 8H), 1.40-1.26 (m, 3H), 1.24-1.11 (m, 3H),1.10-0.97 (m, 6H), 0.96-0.92 (m, 1H), 0.90-0.85 (m, 5H), 0.84-0.76 (m, 4H), 0.69 (s, 3H)。 在2 min層析中,LCMS
Rt=1.207 min,30-90AB_2MIN_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。 在10 min層析中,SFC
Rt=2.635 min,OJ_3_EtOH_DEA_5_40_25ML (「管柱:Chiralcel OJ-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),98.66%de。實例 38 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (38) 之合成 
 1.在0℃下將丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)緩慢地添加至S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液中。添加之後,在15℃下攪拌混合物1小時。混合物用飽和NH4
Cl (40 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之72
(500 mg,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 2.向72
(150 mg,0.372 mmol)於MeOH (20 mL)中之溶液中添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到23
(9 mg,6%)及呈固體之38
(43 mg,29%)。38: 1 H NMR
(400 MHz, CDCl3
) δ 3.71-3.62 (m, 1H), 1.99-1.82 (m, 2H), 1.70-1.56 (m, 6H), 1.54-1.45 (m, 3H), 1.44-1.38 (m, 3H), 1.37-1.17 (m, 10H), 1.16-1.01 (m, 5H), 1.00-0.85 (m, 11H), 0.82 (s, 3H), 0.70-0.60 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.397 min,30-90AB_E,純度100%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 39 : (3S,8S,9S,10R,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (39) 之合成
1.在0℃下將丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)緩慢地添加至S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液中。添加之後,在15℃下攪拌混合物1小時。混合物用飽和NH4
Cl (40 mL)淬滅,且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之72
(500 mg,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 2.向72
(150 mg,0.372 mmol)於MeOH (20 mL)中之溶液中添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到23
(9 mg,6%)及呈固體之38
(43 mg,29%)。38: 1 H NMR
(400 MHz, CDCl3
) δ 3.71-3.62 (m, 1H), 1.99-1.82 (m, 2H), 1.70-1.56 (m, 6H), 1.54-1.45 (m, 3H), 1.44-1.38 (m, 3H), 1.37-1.17 (m, 10H), 1.16-1.01 (m, 5H), 1.00-0.85 (m, 11H), 0.82 (s, 3H), 0.70-0.60 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.397 min,30-90AB_E,純度100%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 39 : (3S,8S,9S,10R,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (39) 之合成  1. 在35℃下於N2
下將t-BuOH (600 mL)饋入至三頸圓底燒瓶中,隨後添加t-BuOK (101 g,905 mmol)。在35℃下攪拌30分鐘之後,將N-004-029_1
(50 g,151 mmol)添加至上述混合物且在35℃下攪拌1小時。將反應混合物倒入至5%乙酸水溶液(2 L)中,在此期間將溫度維持在低於10℃。添加冰水(1 L)。用NaHCO3
將混合物之pH調節至約7至8且過濾。將濾餅溶解於DCM (1.5 L)中。合併之有機相用水(2×500 ml)、鹽水(2×500 ml)洗滌,經Na2
SO4
乾燥,過濾且在低於35℃下於真空中濃縮,得到呈固體之N-004-029_2
(45 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.32 (m, 1H), 3.71-3.58 (m, 1H), 3.38-3.25 (m, 2H), 2.90-2.78 (m, 1H), 2.55-2.20 (m, 2H), 2.13-1.92 (m, 3H), 1.90-1.59 (m, 5H), 1.46-1.14 (m, 10H), 1.12-0.96 (m, 6 H), 0.72 (s, 3H)。2.
在0℃下於N2
下將n-BuLi (108 mL,272 mmol,2.5 M於己烷中)逐滴添加至Me3
SI (73.8 g,362 mmol)於無水THF (300 mL)中之混合物中。在0℃下攪拌30分鐘之後,在-40℃下添加N-004-029_2
(30 g,90.7 mmol)於無水THF (600 mL)中之溶液。在-40℃下攪拌混合物2小時且在25℃下攪拌16小時。將反應混合物倒入至冰水(1 L)中。用EtOAc (2×500 mL))萃取水相。合併之有機相用鹽水(2×500 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之N-004-029_3
(1.8 g,6%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.25 (m, 1H), 3.66-3.61 (m, 1H), 3.39-3.31 (m, 1H), 2.93-2.86 (m, 1H), 2.59-2.53 (m, 1H), 2.20-1.93 (m, 4H), 1.89-1.14 (m, 15H), 1.12-0.90 (m, 9H), 0.71 (s, 3H)。 3. 在25℃下於N2
下將MeONa (5.61 g,104 mmol)添加至N-004-029_3
(1.8 g,5.22 mmol)於MeOH (20 mL)中之溶液中。在50℃下攪拌12小時之後,將水(100 mL)添加至混合物中且攪拌10分鐘。用EtOAc (2×80 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之N-004-029_4
(1.5 g,76%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.28 (m, 1H), 3.70-3.54 (m, 1H), 3.40-3.35 (m, 6H), 3.28-3.16 (m, 2H), 2.40-2.35 (m, 1H), 2.09-1.90 (m, 5H), 1.87-1.57 (m, 11H), 1.34-1.06 (m, 10H), 0.70 (s, 3H)。 4. 在25℃下將DMP (2.53 g,5.97 mmol)添加至N-004-029_4
(1.5 g,3.98 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌30 min之後,在25℃下用飽和NaHCO3
(50 mL)淬滅反應混合物。將DCM (50 mL)添加至混合物中且攪拌10 min。分離DCM相且用飽和Na2
S2
O3
水溶液(2×50 mL)、鹽水(2×50 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(5%至25% EtOAc/PE)純化,得到呈固體之N-004-029_5
(0.6 g,40%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.59-9.57 (m, 1H), 5.32-5.29 (m, 1H), 3.37 (s, 3H), 3.30-3.15 (m, 2H), 2.44-2.31 (m, 2H), 2.13-1.40 (m, 16H), 1.27-1.02 (m, 10H), 0.73 (s, 3H)。 5. 在0℃下將CsF (607 mg,4.00 mmol)添加至N-004-029_5
(0.6 g,1.60 mmol)於無水THF (20 mL)中之溶液中。在攪拌20 min之後,在0℃下添加TMSCF3
(568 mg,4.00 mmol)且攪拌混合物1小時。將TBAF.3H2
O (2.02 g,6.40 mmol)添加至混合物中,在50℃下攪拌該混合物1小時。將反應混合物倒入至冰水(50 mL)中。用EtOAc (2×80 mL)萃取水相。將合併之有機相用飽和鹽水(2×80 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之N-004-029A
(450 mg,63%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.29 (m, 1H), 4.11-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.43-2.36 (m, 1H), 2.26-1.82 (m, 7H), 1.78-1.61 (m, 5H), 1.34-0.80 (m, 15H), 0.75-0.67 (m, 3H)。 6.N-004-029A
(0.45 g,1.01 mmol)藉由SFC (管柱:AD (250 mm×30 mm,5 um),條件:0.1% NH3
H2
O ETOH,開始B:30%,結束B:30%)純化,得到呈白色固體之39
(PK1:120 mg,26.7%)及呈白色固體之95
(PK2:200 mg,44.5%)。 藉由NOE確認39 之
結構。39 : 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.29 (m, 1H), 4.06-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.43-2.36 (m, 1H), 2.25-2.19 (m, 1H), 2.15-2.07 (m, 1H), 2.04-1.60 (m, 9H), 1.55-1.34 (m, 5H), 1.25-0.88 (m, 11H), 0.70 (s, 3H)。LCMS
Rt=2 min層析中1.078 min,30-90AB_2MIN_E,純度100%,C24
H34
F3
O[M-CH5
O2
]+
之MS ESI計算值395,實驗值395。實例 40 : (3S,5S,8R,9R,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (40) 之合成
1. 在35℃下於N2
下將t-BuOH (600 mL)饋入至三頸圓底燒瓶中,隨後添加t-BuOK (101 g,905 mmol)。在35℃下攪拌30分鐘之後,將N-004-029_1
(50 g,151 mmol)添加至上述混合物且在35℃下攪拌1小時。將反應混合物倒入至5%乙酸水溶液(2 L)中,在此期間將溫度維持在低於10℃。添加冰水(1 L)。用NaHCO3
將混合物之pH調節至約7至8且過濾。將濾餅溶解於DCM (1.5 L)中。合併之有機相用水(2×500 ml)、鹽水(2×500 ml)洗滌,經Na2
SO4
乾燥,過濾且在低於35℃下於真空中濃縮,得到呈固體之N-004-029_2
(45 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.32 (m, 1H), 3.71-3.58 (m, 1H), 3.38-3.25 (m, 2H), 2.90-2.78 (m, 1H), 2.55-2.20 (m, 2H), 2.13-1.92 (m, 3H), 1.90-1.59 (m, 5H), 1.46-1.14 (m, 10H), 1.12-0.96 (m, 6 H), 0.72 (s, 3H)。2.
在0℃下於N2
下將n-BuLi (108 mL,272 mmol,2.5 M於己烷中)逐滴添加至Me3
SI (73.8 g,362 mmol)於無水THF (300 mL)中之混合物中。在0℃下攪拌30分鐘之後,在-40℃下添加N-004-029_2
(30 g,90.7 mmol)於無水THF (600 mL)中之溶液。在-40℃下攪拌混合物2小時且在25℃下攪拌16小時。將反應混合物倒入至冰水(1 L)中。用EtOAc (2×500 mL))萃取水相。合併之有機相用鹽水(2×500 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之N-004-029_3
(1.8 g,6%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.25 (m, 1H), 3.66-3.61 (m, 1H), 3.39-3.31 (m, 1H), 2.93-2.86 (m, 1H), 2.59-2.53 (m, 1H), 2.20-1.93 (m, 4H), 1.89-1.14 (m, 15H), 1.12-0.90 (m, 9H), 0.71 (s, 3H)。 3. 在25℃下於N2
下將MeONa (5.61 g,104 mmol)添加至N-004-029_3
(1.8 g,5.22 mmol)於MeOH (20 mL)中之溶液中。在50℃下攪拌12小時之後,將水(100 mL)添加至混合物中且攪拌10分鐘。用EtOAc (2×80 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之N-004-029_4
(1.5 g,76%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.28 (m, 1H), 3.70-3.54 (m, 1H), 3.40-3.35 (m, 6H), 3.28-3.16 (m, 2H), 2.40-2.35 (m, 1H), 2.09-1.90 (m, 5H), 1.87-1.57 (m, 11H), 1.34-1.06 (m, 10H), 0.70 (s, 3H)。 4. 在25℃下將DMP (2.53 g,5.97 mmol)添加至N-004-029_4
(1.5 g,3.98 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌30 min之後,在25℃下用飽和NaHCO3
(50 mL)淬滅反應混合物。將DCM (50 mL)添加至混合物中且攪拌10 min。分離DCM相且用飽和Na2
S2
O3
水溶液(2×50 mL)、鹽水(2×50 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(5%至25% EtOAc/PE)純化,得到呈固體之N-004-029_5
(0.6 g,40%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.59-9.57 (m, 1H), 5.32-5.29 (m, 1H), 3.37 (s, 3H), 3.30-3.15 (m, 2H), 2.44-2.31 (m, 2H), 2.13-1.40 (m, 16H), 1.27-1.02 (m, 10H), 0.73 (s, 3H)。 5. 在0℃下將CsF (607 mg,4.00 mmol)添加至N-004-029_5
(0.6 g,1.60 mmol)於無水THF (20 mL)中之溶液中。在攪拌20 min之後,在0℃下添加TMSCF3
(568 mg,4.00 mmol)且攪拌混合物1小時。將TBAF.3H2
O (2.02 g,6.40 mmol)添加至混合物中,在50℃下攪拌該混合物1小時。將反應混合物倒入至冰水(50 mL)中。用EtOAc (2×80 mL)萃取水相。將合併之有機相用飽和鹽水(2×80 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之N-004-029A
(450 mg,63%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.29 (m, 1H), 4.11-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.43-2.36 (m, 1H), 2.26-1.82 (m, 7H), 1.78-1.61 (m, 5H), 1.34-0.80 (m, 15H), 0.75-0.67 (m, 3H)。 6.N-004-029A
(0.45 g,1.01 mmol)藉由SFC (管柱:AD (250 mm×30 mm,5 um),條件:0.1% NH3
H2
O ETOH,開始B:30%,結束B:30%)純化,得到呈白色固體之39
(PK1:120 mg,26.7%)及呈白色固體之95
(PK2:200 mg,44.5%)。 藉由NOE確認39 之
結構。39 : 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.29 (m, 1H), 4.06-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.43-2.36 (m, 1H), 2.25-2.19 (m, 1H), 2.15-2.07 (m, 1H), 2.04-1.60 (m, 9H), 1.55-1.34 (m, 5H), 1.25-0.88 (m, 11H), 0.70 (s, 3H)。LCMS
Rt=2 min層析中1.078 min,30-90AB_2MIN_E,純度100%,C24
H34
F3
O[M-CH5
O2
]+
之MS ESI計算值395,實驗值395。實例 40 : (3S,5S,8R,9R,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (40) 之合成  1. 在25℃下於Ar下,將Pd(OH)2
(100 mg,無水)添加至3
(30 mg,0.072 mmol)於MeOH/THF (5 mL/5mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌反應物48 h。過濾混合物且在真空中濃縮濾液,得到粗產物,其藉由矽膠管柱(PE/EtOAc=10/1至5/1)純化,得到呈固體之41
(10 mg,33%)。 1 H NMR
(400MHz, CDCl3) δ 3.62-3.59 (m, 1H), 1.99-1.91 (m, 1H), 1.88-1.76 (m, 2H), 1.74-1.61 (m, 6H), 1.46-1.29 (m, 6H), 1.26-1.09 (m, 9H), 1.08-0.99 (m, 6H), 0.95-0.78 (m, 14H), 0.74-0.58 (m, 5H)。 在2 min層析中,LCMS
Rt=1.491 min,30-90 AB,純度99%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 41 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基庚 -5- 炔 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (41) 之合成
1. 在25℃下於Ar下,將Pd(OH)2
(100 mg,無水)添加至3
(30 mg,0.072 mmol)於MeOH/THF (5 mL/5mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌反應物48 h。過濾混合物且在真空中濃縮濾液,得到粗產物,其藉由矽膠管柱(PE/EtOAc=10/1至5/1)純化,得到呈固體之41
(10 mg,33%)。 1 H NMR
(400MHz, CDCl3) δ 3.62-3.59 (m, 1H), 1.99-1.91 (m, 1H), 1.88-1.76 (m, 2H), 1.74-1.61 (m, 6H), 1.46-1.29 (m, 6H), 1.26-1.09 (m, 9H), 1.08-0.99 (m, 6H), 0.95-0.78 (m, 14H), 0.74-0.58 (m, 5H)。 在2 min層析中,LCMS
Rt=1.491 min,30-90 AB,純度99%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 41 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基庚 -5- 炔 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (41) 之合成 
 1. 在-20℃下將n-BuLi (13.8 mL,34.5 mmol,2.5 M於己烷中)逐滴添加至丁-2-炔(1.86 g,34.5 mmol)於於THF (100 mL)中之溶液中。-20℃下攪拌溶液2.5小時,且接著將其冷卻至-78℃,添加含N-8-7_1
(5.0 g,13.8 mmol)之THF (100 mL)。在此溫度下攪拌溶液30分鐘,接著在-20℃下攪拌1小時,且隨後20℃下攪拌18小時。所得凝膠藉由倒入至飽和NH4
Cl (100 mL)中淬滅,隨後用EtOAc (2×150 mL)萃取。合併之有機層用水(40 mL)及鹽水(40 mL)洗滌,經Na2
SO4
乾燥,經過濾且藉由閃蒸塔純化用含0至20%EtOAc之PE溶離,得到呈油狀物之N-8-7_2B
(1 g,粗產物)及N-8-7_2B
(1.7 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.90-3.75 (m, 1H), 2.45-2.35 (m, 1H), 2.25-2.05 (m, 3H), 1.95-1.85 (m, 2H), 1.85-1.40 (m, 8H), 1.40-1.20 (m, 9H), 1.20-0.75 (m, 18H), 0.65 (s, 4H)。 在10 min層析中,SFC 峰值1:Rt = 3.008 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6 mm I.D.,3 um行動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。 2. 粗產物N-8-7_2B
(250 mg,0.868 mmol)藉由SFC (管柱:AD (250 mm×30 mm,10 um),梯度:35%至35% B (A= 0.1% NH3
/H2
O,B = EtOH),流速:60 mL/min)進一步純化,得到呈固體之41
(峰值2,81 mg,33%)及呈固體之68
(峰值1,78 mg,31%)。41 : 1 H NMR
(400 MHz, CDCl3
) δ 3.82-3.70 (m, 1H), 2.79-2.08 (m, 2H), 2.00-1.90 (m, 1H), 1.80 (s, 4H), 1.78-1.69 (m, 1H), 1.69-1.42 (m, 10H), 1.40-1.31 (m, 4H), 1.31-1.18 (m, 4H), 1.18-0.92 (m, 6H), 0.92-0.85 (m, 7H),0.82 (s, 3H), 0.66 (s, 3H)。LCMS
Rt=2 min層析中之1.206 min,30-90AB_2MIN_E,純度100%,C28
H45
O [M+H-H2
O]+
之MS ESI計算值397,實驗值397。 在10 min層析中,SFC
Rt=5.823 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 42 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (42) 之合成
1. 在-20℃下將n-BuLi (13.8 mL,34.5 mmol,2.5 M於己烷中)逐滴添加至丁-2-炔(1.86 g,34.5 mmol)於於THF (100 mL)中之溶液中。-20℃下攪拌溶液2.5小時,且接著將其冷卻至-78℃,添加含N-8-7_1
(5.0 g,13.8 mmol)之THF (100 mL)。在此溫度下攪拌溶液30分鐘,接著在-20℃下攪拌1小時,且隨後20℃下攪拌18小時。所得凝膠藉由倒入至飽和NH4
Cl (100 mL)中淬滅,隨後用EtOAc (2×150 mL)萃取。合併之有機層用水(40 mL)及鹽水(40 mL)洗滌,經Na2
SO4
乾燥,經過濾且藉由閃蒸塔純化用含0至20%EtOAc之PE溶離,得到呈油狀物之N-8-7_2B
(1 g,粗產物)及N-8-7_2B
(1.7 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.90-3.75 (m, 1H), 2.45-2.35 (m, 1H), 2.25-2.05 (m, 3H), 1.95-1.85 (m, 2H), 1.85-1.40 (m, 8H), 1.40-1.20 (m, 9H), 1.20-0.75 (m, 18H), 0.65 (s, 4H)。 在10 min層析中,SFC 峰值1:Rt = 3.008 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6 mm I.D.,3 um行動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。 2. 粗產物N-8-7_2B
(250 mg,0.868 mmol)藉由SFC (管柱:AD (250 mm×30 mm,10 um),梯度:35%至35% B (A= 0.1% NH3
/H2
O,B = EtOH),流速:60 mL/min)進一步純化,得到呈固體之41
(峰值2,81 mg,33%)及呈固體之68
(峰值1,78 mg,31%)。41 : 1 H NMR
(400 MHz, CDCl3
) δ 3.82-3.70 (m, 1H), 2.79-2.08 (m, 2H), 2.00-1.90 (m, 1H), 1.80 (s, 4H), 1.78-1.69 (m, 1H), 1.69-1.42 (m, 10H), 1.40-1.31 (m, 4H), 1.31-1.18 (m, 4H), 1.18-0.92 (m, 6H), 0.92-0.85 (m, 7H),0.82 (s, 3H), 0.66 (s, 3H)。LCMS
Rt=2 min層析中之1.206 min,30-90AB_2MIN_E,純度100%,C28
H45
O [M+H-H2
O]+
之MS ESI計算值397,實驗值397。 在10 min層析中,SFC
Rt=5.823 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 42 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (42) 之合成  :
: 1. 在25℃下將4-甲基苯磺酸(2.70 g,15.7 mmol)添加至N-4-1_1
(50 g,157 mmol)於MeOH (500 mL)中之溶液中。在65℃下攪拌混合物1小時。使反應混合物冷卻至25℃且添加TEA (2.16 mL,15.7 mmol)。攪拌混合物0.5 h。沈澱物藉由過濾採集且用甲醇(2×100 mL)洗滌,得到呈固體之N-4-1_2
(50 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.25-3.05 (m, 6H), 2.60-2.40 (m, 1H), 2.20-2.05 (m, 4H), 2.00-1.95 (m, 1H), 1.90-1.80 (m, 1H), 1.75-1.50 (m, 6H), 1.49-1.05 (m, 12H), 1.04-0.95 (m, 1H), 0.78 (s, 3H), 0.59 (s, 3H)。 2. 在25℃下將t-BuOK (23.0 g,205 mmol)添加至溴基(甲基)三苯基磷烷(73.2 g,205 mmol)於THF (500 mL)中之溶液中。將混合物加熱至45℃且攪拌1小時。添加N-4-1_2
(50 g,137 mmol)。在45℃下攪拌混合物2小時。混合物用NH4
Cl (200 mL)淬滅且用THF (3×100 mL)萃取。有機層經鹽水(200 mL)洗滌,經Na2
SO4
乾燥且過濾,得到混合物(50 g,500 mL),該混合物不經進一步純化即用於下一步驟中。3.
將HCl水溶液(207 mL,1 M於水中)添加至N-4-1_3
(50 g,138 mmol)於THF (500 mL)中之溶液中。在25℃下攪拌混合物0.5 h。過濾混合物且將濾餅溶解於DCM (200 mL)中並用鹽水(100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈固體之N-4-1_4
(39 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.45-2.20 (m, 3H), 2.15-2.00 (m, 3H), 1.90-1.65 (m, 8H), 1.60-1.50 (m, 2H), 1.45-1.05 (m, 8H), 1.00 (s, 3H) 0.90-0.85 (m, 1H), 0.80-0.75 (m, 1H), 0.58 (s, 3H)。 4. 將CsF (25.9 g,171 mmol)及TMSCF3
(24.3 g,171 mmol)添加至N-4-1_4
(27 g,85.8 mmol)於THF (200 mL)中之溶液中。在10℃下攪拌混合物1小時。將水(10 mL)及TBAF.3H2
O (30 g)添加至混合物中。在30℃下再攪拌混合物2小時。真空濃縮混合物。將殘餘物溶解於EtOAc (500 mL)中,用水(2×500 mL)洗滌,經Na2
SO4
乾燥,經過濾,在真空中濃縮且藉由閃蒸塔(DCM/EtOAc (1:1)/PE,0%至10%)純化,得到呈固體之N-4-1_5
(27 g,82%)及N-4-1_5A
(3.5 g,11%) 。N-4-1_5 : 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.12-1.94 (m, 3H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.60 (m, 5H), 1.58-1.48 (m, 2H), 1.45-1.09 (m, 10H), 1.01-0.89 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.56 (s, 3H)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.09-1.99 (m, 1H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.52 (m, 9H), 1.45-1.06 (m, 10H), 1.00-1.81 (m, 2H), 0.79 (s, 3H), 0.56 (s, 3H)。 5. 將9-BBN二聚體(29 g,119 mmol)添加至N-4-1_5
(23 g, 59.8 mmol)於THF (250 mL)中之溶液中且在40℃下於N2
下攪拌混合物16小時。將乙醇(34.3 mL,598 mmol)及NaOH (119 mL,5 M,598 mmol)添加至反應混合物。使混合物變得澄清。在25℃下逐滴添加H2
O2
(59.8 mL,10 M,598 mmol)且使內部溫度升至回流(70℃)。在添加之後,使混合物冷卻至30℃。向混合物添加Na2
SO3
(100 mL,20%水溶液)。有機層經分離且倒入水(800 mL)中。形成固體。過濾混合物且該固體用水洗滌,在真空下乾燥且用MeCN (250 mL)濕磨,得到固體。該固體在60℃下自MeOH/水(250 mL/12.5 mL)濕磨且在冷卻至15℃之後過濾。在真空下乾燥固體,得到呈固體之N-4-1_6
(16.4 g,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.69-3.60 (m, 1H), 3.39-3.29 (m, 1H), 2.09-2.01 (m, 1H), 1.99-1.92 (m, 1H), 1.87-1.75 (m, 2H), 1.72-1.43 (m, 7H), 1.42-1.07 (m, 11H), 1.03 (d,J
= 6.8 Hz, 3H), 1.01-0.86 (m, 3H), 0.85 (s, 3H), 0.73-0.69 (m, 1H), 0.67 (s, 3H)。6.
將水(223 mg,12.4 mmol)及DMP (10.5 g,24.8 mmol)添加至N-4-1_6
(5 g,12.4 mmol)於DCM (200 mL)中之懸浮液中。在15℃下攪拌混合物15 min。混合物用NaHCO3
/Na2
S2
O3
(200 mL/200 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-4-1_7
(4.5 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.60-9.51 (m, 1H), 2.40-2.30 (m, 1H), 2.12-1.78 (m, 5H), 1.75-1.59 (m, 4H), 1.57-1.15 (m, 11H), 1.14-0.84 (m, 8H), 0.78-0.63 (m, 5H)。 7. 在50℃至55℃下於N2
下將1-溴-3-甲基丁烷(2.79 g,18.5 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (899 mg,37 mmol)及I2
(1 mg)於THF (2 mL) 於中之懸浮液中。在55℃下攪拌混合物1小時,得到異戊基溴化鎂溶液。在0℃下將新鮮製備的異戊基溴化鎂(18.5 mmol於10 mL THF中)添加至N-4-1_7
(0.5 g,1.24 mmol)於THF (5 mL)之溶液中。在15℃下攪拌混合物2小時。向混合物中添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈固體之N-4-2
(0.6 g,粗產物)。 8. 在15℃下將水(1滴)及DMP (1.06 g,2.52 mmol)添加至N-4-2 (0.6 g,1.26 mmol)於DCM (20 mL)中之溶液中。在15℃下攪拌混合物1 h。混合物用NaHCO3
/Na2
S2
O3
(20 mL/20 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈固體之N-4-1_8
(0.6 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 2.59-2.30 (m, 3H), 2.11-1.78 (m, 4H), 1.75-1.36 (m, 13H), 1.35-0.98 (m, 11H), 0.91-0.82 (m, 10H), 0.78-0.70 (m, 1H), 0.67 (s, 3H)。 9. 在15℃下將NaBH4
(0.96 g,25.4 mmol)逐份添加至N-4-1_8
(0.6 g,1.27 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌混合物30 min。向混合物中添加NH4
Cl (50 mL,10%)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到不純42
及85
。N42
在15℃下自MeCN (10 mL)濕磨且在真空中乾燥,得到呈固體之42
(153 mg,25%)。85
藉由閃蒸塔(0%至15% EtOAc於PE中)純化,得到油狀物,用MeCN (5 mL)及水(5 mL)處理該油狀物,且真空濃縮,得到呈固體之85
(70 mg,12%)。42 : 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.55 (m, 1H), 2.01-1.78 (m, 6H), 1.71-1.59 (m, 4H), 1.51-1.15 (m, 16H), 1.09-1.02 (m, 3H), 0.92-0.81 (m, 13H), 0.72-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.378 min,30-90_AB_E,純度100%,C28
H46
F3
O [M+H-H2
O]+
之MS ESI計算值 455,實驗值455。 在10.0 min層析中,HPLC
Rt=5.38 min,50-100_AB_E,純度99.58%。實例 43 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (43) 之合成
1. 在25℃下將4-甲基苯磺酸(2.70 g,15.7 mmol)添加至N-4-1_1
(50 g,157 mmol)於MeOH (500 mL)中之溶液中。在65℃下攪拌混合物1小時。使反應混合物冷卻至25℃且添加TEA (2.16 mL,15.7 mmol)。攪拌混合物0.5 h。沈澱物藉由過濾採集且用甲醇(2×100 mL)洗滌,得到呈固體之N-4-1_2
(50 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.25-3.05 (m, 6H), 2.60-2.40 (m, 1H), 2.20-2.05 (m, 4H), 2.00-1.95 (m, 1H), 1.90-1.80 (m, 1H), 1.75-1.50 (m, 6H), 1.49-1.05 (m, 12H), 1.04-0.95 (m, 1H), 0.78 (s, 3H), 0.59 (s, 3H)。 2. 在25℃下將t-BuOK (23.0 g,205 mmol)添加至溴基(甲基)三苯基磷烷(73.2 g,205 mmol)於THF (500 mL)中之溶液中。將混合物加熱至45℃且攪拌1小時。添加N-4-1_2
(50 g,137 mmol)。在45℃下攪拌混合物2小時。混合物用NH4
Cl (200 mL)淬滅且用THF (3×100 mL)萃取。有機層經鹽水(200 mL)洗滌,經Na2
SO4
乾燥且過濾,得到混合物(50 g,500 mL),該混合物不經進一步純化即用於下一步驟中。3.
將HCl水溶液(207 mL,1 M於水中)添加至N-4-1_3
(50 g,138 mmol)於THF (500 mL)中之溶液中。在25℃下攪拌混合物0.5 h。過濾混合物且將濾餅溶解於DCM (200 mL)中並用鹽水(100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈固體之N-4-1_4
(39 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.45-2.20 (m, 3H), 2.15-2.00 (m, 3H), 1.90-1.65 (m, 8H), 1.60-1.50 (m, 2H), 1.45-1.05 (m, 8H), 1.00 (s, 3H) 0.90-0.85 (m, 1H), 0.80-0.75 (m, 1H), 0.58 (s, 3H)。 4. 將CsF (25.9 g,171 mmol)及TMSCF3
(24.3 g,171 mmol)添加至N-4-1_4
(27 g,85.8 mmol)於THF (200 mL)中之溶液中。在10℃下攪拌混合物1小時。將水(10 mL)及TBAF.3H2
O (30 g)添加至混合物中。在30℃下再攪拌混合物2小時。真空濃縮混合物。將殘餘物溶解於EtOAc (500 mL)中,用水(2×500 mL)洗滌,經Na2
SO4
乾燥,經過濾,在真空中濃縮且藉由閃蒸塔(DCM/EtOAc (1:1)/PE,0%至10%)純化,得到呈固體之N-4-1_5
(27 g,82%)及N-4-1_5A
(3.5 g,11%) 。N-4-1_5 : 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.12-1.94 (m, 3H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.60 (m, 5H), 1.58-1.48 (m, 2H), 1.45-1.09 (m, 10H), 1.01-0.89 (m, 1H), 0.85 (s, 3H), 0.78-0.68 (m, 1H), 0.56 (s, 3H)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.09-1.99 (m, 1H), 1.89-1.78 (m, 2H), 1.75 (s, 3H), 1.72-1.52 (m, 9H), 1.45-1.06 (m, 10H), 1.00-1.81 (m, 2H), 0.79 (s, 3H), 0.56 (s, 3H)。 5. 將9-BBN二聚體(29 g,119 mmol)添加至N-4-1_5
(23 g, 59.8 mmol)於THF (250 mL)中之溶液中且在40℃下於N2
下攪拌混合物16小時。將乙醇(34.3 mL,598 mmol)及NaOH (119 mL,5 M,598 mmol)添加至反應混合物。使混合物變得澄清。在25℃下逐滴添加H2
O2
(59.8 mL,10 M,598 mmol)且使內部溫度升至回流(70℃)。在添加之後,使混合物冷卻至30℃。向混合物添加Na2
SO3
(100 mL,20%水溶液)。有機層經分離且倒入水(800 mL)中。形成固體。過濾混合物且該固體用水洗滌,在真空下乾燥且用MeCN (250 mL)濕磨,得到固體。該固體在60℃下自MeOH/水(250 mL/12.5 mL)濕磨且在冷卻至15℃之後過濾。在真空下乾燥固體,得到呈固體之N-4-1_6
(16.4 g,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.69-3.60 (m, 1H), 3.39-3.29 (m, 1H), 2.09-2.01 (m, 1H), 1.99-1.92 (m, 1H), 1.87-1.75 (m, 2H), 1.72-1.43 (m, 7H), 1.42-1.07 (m, 11H), 1.03 (d,J
= 6.8 Hz, 3H), 1.01-0.86 (m, 3H), 0.85 (s, 3H), 0.73-0.69 (m, 1H), 0.67 (s, 3H)。6.
將水(223 mg,12.4 mmol)及DMP (10.5 g,24.8 mmol)添加至N-4-1_6
(5 g,12.4 mmol)於DCM (200 mL)中之懸浮液中。在15℃下攪拌混合物15 min。混合物用NaHCO3
/Na2
S2
O3
(200 mL/200 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-4-1_7
(4.5 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.60-9.51 (m, 1H), 2.40-2.30 (m, 1H), 2.12-1.78 (m, 5H), 1.75-1.59 (m, 4H), 1.57-1.15 (m, 11H), 1.14-0.84 (m, 8H), 0.78-0.63 (m, 5H)。 7. 在50℃至55℃下於N2
下將1-溴-3-甲基丁烷(2.79 g,18.5 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (899 mg,37 mmol)及I2
(1 mg)於THF (2 mL) 於中之懸浮液中。在55℃下攪拌混合物1小時,得到異戊基溴化鎂溶液。在0℃下將新鮮製備的異戊基溴化鎂(18.5 mmol於10 mL THF中)添加至N-4-1_7
(0.5 g,1.24 mmol)於THF (5 mL)之溶液中。在15℃下攪拌混合物2小時。向混合物中添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈固體之N-4-2
(0.6 g,粗產物)。 8. 在15℃下將水(1滴)及DMP (1.06 g,2.52 mmol)添加至N-4-2 (0.6 g,1.26 mmol)於DCM (20 mL)中之溶液中。在15℃下攪拌混合物1 h。混合物用NaHCO3
/Na2
S2
O3
(20 mL/20 mL,飽和)洗滌兩次,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈固體之N-4-1_8
(0.6 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 2.59-2.30 (m, 3H), 2.11-1.78 (m, 4H), 1.75-1.36 (m, 13H), 1.35-0.98 (m, 11H), 0.91-0.82 (m, 10H), 0.78-0.70 (m, 1H), 0.67 (s, 3H)。 9. 在15℃下將NaBH4
(0.96 g,25.4 mmol)逐份添加至N-4-1_8
(0.6 g,1.27 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌混合物30 min。向混合物中添加NH4
Cl (50 mL,10%)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到不純42
及85
。N42
在15℃下自MeCN (10 mL)濕磨且在真空中乾燥,得到呈固體之42
(153 mg,25%)。85
藉由閃蒸塔(0%至15% EtOAc於PE中)純化,得到油狀物,用MeCN (5 mL)及水(5 mL)處理該油狀物,且真空濃縮,得到呈固體之85
(70 mg,12%)。42 : 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.55 (m, 1H), 2.01-1.78 (m, 6H), 1.71-1.59 (m, 4H), 1.51-1.15 (m, 16H), 1.09-1.02 (m, 3H), 0.92-0.81 (m, 13H), 0.72-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.378 min,30-90_AB_E,純度100%,C28
H46
F3
O [M+H-H2
O]+
之MS ESI計算值 455,實驗值455。 在10.0 min層析中,HPLC
Rt=5.38 min,50-100_AB_E,純度99.58%。實例 43 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (43) 之合成  1. 向62
(160 mg,0.373 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中添加Pd(OH)2
(0.2 g,<1%水)。在50℃下於50 psi氫氣下氫化溶液16小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到43
(27 mg,17%)及呈白色固體之16
(117 mg,73%)。43 : 1 H NMR
(400 MHz, CDCl3) δ.4.05-3.99 (m, 1H), 1.99-1.81 (m, 5H), 1.79-1.72 (m, 1H), 1.70-1.56 (m, 3H), 1.53-1.35 (m, 7H), 1.35-1.07 (m, 12H), 1.04-1.02 (m, 3H), 0.97 (s, 3H), 0.92 (t,J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt = 1.271 min,30-90 AB,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 44 : (3S,8R,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (44) 之合成
1. 向62
(160 mg,0.373 mmol)於MeOH (2 mL)及THF (1 mL)中之溶液中添加Pd(OH)2
(0.2 g,<1%水)。在50℃下於50 psi氫氣下氫化溶液16小時。隨後混合物經由矽藻土墊過濾且真空濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到43
(27 mg,17%)及呈白色固體之16
(117 mg,73%)。43 : 1 H NMR
(400 MHz, CDCl3) δ.4.05-3.99 (m, 1H), 1.99-1.81 (m, 5H), 1.79-1.72 (m, 1H), 1.70-1.56 (m, 3H), 1.53-1.35 (m, 7H), 1.35-1.07 (m, 12H), 1.04-1.02 (m, 3H), 0.97 (s, 3H), 0.92 (t,J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt = 1.271 min,30-90 AB,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 44 : (3S,8R,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (44) 之合成  1.
在60℃下將1-溴-3-甲基丁烷(4 g,26.4 mmol)於THF (27 mL)中之溶液逐滴添加至Mg (947 mg,39.5 mmol)及I2
(33.5 mg,0.132 mmol)於THF (3 mL)中之懸浮液中。在60℃下攪拌混合物1小時。在0℃下於N2
下將新鮮製備的異戊基溴化鎂(30 mL,0.88 M於THF中,26.4 mmol)添加至S-500-15-2_1
(800 mg,2.32 mmol)於THF (2 mL)中之溶液中。在0℃下攪拌混合物1 h。將NH4
Cl (50 mL,飽和水溶液)添加至混合物,用EtOAc (2 × 50 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到粗產物,其藉由矽膠(PE/EtOAc=10/1至5/1)純化,得到呈白色固體之44
(720 mg,75%)。 1H NMR (400MHz, CDCl3) δ 5.40-5.38 (m, 1H), 3.63-3.61 (m, 1H), 2.23-2.21 (m, 1H), 2.10-1.74 (m, 7H), 1.69-1.58 (m, 2H), 1.54-1.34 (m, 8H), 1.33-1.00 (m, 11H), 0.95-0.75 (m, 14H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.289 min,30-90 AB,純度100%,C28H45 [M+H-2H2O]+
之MS ESI計算值381,實驗值381。實例 45 : (3S,5R,8R,9R,10S,13S,14S,17R)-3-( 甲氧基甲基 )-13- 甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (45) 之合成
1.
在60℃下將1-溴-3-甲基丁烷(4 g,26.4 mmol)於THF (27 mL)中之溶液逐滴添加至Mg (947 mg,39.5 mmol)及I2
(33.5 mg,0.132 mmol)於THF (3 mL)中之懸浮液中。在60℃下攪拌混合物1小時。在0℃下於N2
下將新鮮製備的異戊基溴化鎂(30 mL,0.88 M於THF中,26.4 mmol)添加至S-500-15-2_1
(800 mg,2.32 mmol)於THF (2 mL)中之溶液中。在0℃下攪拌混合物1 h。將NH4
Cl (50 mL,飽和水溶液)添加至混合物,用EtOAc (2 × 50 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到粗產物,其藉由矽膠(PE/EtOAc=10/1至5/1)純化,得到呈白色固體之44
(720 mg,75%)。 1H NMR (400MHz, CDCl3) δ 5.40-5.38 (m, 1H), 3.63-3.61 (m, 1H), 2.23-2.21 (m, 1H), 2.10-1.74 (m, 7H), 1.69-1.58 (m, 2H), 1.54-1.34 (m, 8H), 1.33-1.00 (m, 11H), 0.95-0.75 (m, 14H), 0.70 (s, 3H)。 在2 min層析中,LCMS
Rt=1.289 min,30-90 AB,純度100%,C28H45 [M+H-2H2O]+
之MS ESI計算值381,實驗值381。實例 45 : (3S,5R,8R,9R,10S,13S,14S,17R)-3-( 甲氧基甲基 )-13- 甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (45) 之合成 
 1.
向G-020-004_1
(100 g,364 mmol)於無水甲醇(1 L)中之溶液中添加TsOH (6.26 g,36.4 mmol)。在60℃下攪拌混合物18小時。反應混合物經濃縮以移除大部分溶劑,用Et3
N (3.7 g)中和,用EtOAc (600 mL)稀釋,用水(500 mL)及鹽水(500 mL)洗滌。有機層經濃縮,得到呈油狀物之G-020-004_2
(133 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.20 (s, 3H), 3.14 (s, 3H), 2.63-2.39 (m, 2H), 2.14-2.03 (m, 2H), 1.97-1.89 (m, 2H), 1.86-1.77 (m, 3H), 1.64-1.60 (m, 2H), 1.56-1.49 (m, 3H), 1.47-1.42 (m, 2H), 1.40-1.32 (m, 2H), 1.29-1.23 (m, 3H), 1.16-1.06 (m, 2H), 0.87 (s, 3H)。2.
向Ph3
PEtBr (308 g,830 mmol)於20℃下氮氣下之無水THF (700 mL)中之懸浮液中添加t-BuOK (93.1 g,830 mmol)。在20℃下攪拌1小時之後,將G-020-004_2
(133 g,415 mmol)於無水THF (300 mL)中之溶液添加至混合物。使所得混合物升溫至50℃且攪拌4小時。反應混合物經冷卻,用水(400 mL)及飽和NH4
Cl (300 mL)淬滅,攪拌30 min。分離有機層,且用THF (300 mL)萃取水相。合併之有機層直接用於下一步驟中。 3. 向G-020-004_3
(137 g,412 mmol,理論上)於THF (1.3 L)中之溶液中添加HCl水溶液(1 M,618 mL,618 mmol)。在20℃下攪拌1小時之後,反應混合物用飽和NaHCO3
(800 mL)淬滅且用EtOAc (2×500 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮,得到固體(300 g)。自石油醚(800 mL)濕磨固體18小時。過濾出固體,且用石油醚(400 mL)洗滌濾餅。濃縮濾液,得到殘餘物(117 g)。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-004-023_5
(70 g)。 1 H NMR
(400 MHz, CDCl3
) δ 5.17-5.09 (m, 1H), 2.65-2.55 (m, 1H), 2.43-2.34 (m, 1H), 2.33-2.15 (m, 6H), 2.11-2.05 (m, 1H), 1.83-1.70 (m, 2H), 1.68-1.64 (m, 4H), 1.63-1.59 (m, 2H), 1.58-1.46 (m, 3H), 1.42-1.25 (m, 3H), 1.25-1.14 (m, 3H), 0.91 (s, 3H)。4.
在60℃下於N2
下加熱三甲基氧化鋶碘(30.5 g,139 mmol)及t-BuOK (15.5 g,139 mmol)於DMSO (200 mL)中之經攪拌溶液1.0 h;將N-004-023_5
(20 g,69.8 mmol)添加至反應混合物且在60℃下攪拌10分鐘。用水(1000 mL)處理反應物。用EtOAc (2×500 mL))萃取水相。合併之有機相用水(2×500 mL)、鹽水(300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-004-023_6
(20.5 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.13-5.10 (m, 1H), 2.62-2.60 (m, 2H), 2.25-2.20 (m, 5H), 2.00-1.48 (m, 12H), 1.46-1.00 (m, 8H), 0.98-0.89 (m, 4H)。 5. 在25℃下於N2
下將MeONa (18.4 g,341 mmol)添加至N-004-023_6
(20.5 g,68.2 mmol)於MeOH (500 mL)中之溶液中。在70℃回流下於N2
下攪拌混合物16 h。用水(500 mL)處理反應物。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,經過濾且濃縮物藉由矽膠層析(PE/EtOAc=10/1至6/1)純化,得到呈固體之N-004-023_7
(20 g,88%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.12-5.06 (m, 1H), 3.38 (s, 3H), 3.19 (s, 2H), 2.25-2.22 (m, 1H), 2.20-2.09 (m, 3H), 1.66-1.63 (m, 3H), 1.60-1.24 (m, 14H), 1.22-1.00 (m, 6H), 0.87 (s, 3H)。 6. 在0℃下於N2
下將9-BBN二聚體(29.2 g,120 mmol)添加至N-004-023_7
(20 g,60.1 mmol)於THF (100 mL)中之溶液中。在65℃下攪拌溶液2小時。冷卻至0℃後,添加EtOH (34.9 mL,601 mmol)。接著極緩慢地添加NaOH (120 mL,5 M,601 mmol)之溶液。添加後,緩慢添加H2
O2
(68.0 g,601 mmol,30%於水中),且將內部溫度維持在低於10℃。在75℃下於N2
下攪拌混合物1小時。將混合物再冷卻至25℃。將混合物添加至H2
O (2 L)。攪拌混合物30分鐘。沈澱物藉由過濾採集且用H2
O (2×500 mL)洗滌,得到呈固體之N-004-023_8
(17.8 g,85%) 。 1 H NMR
(400 MHz, CDCl3
) δ 3.70-3.55 (m, 1H), 3.38 (s, 3H), 3.19 (m, 2H), 2.11-1.86 (m, 4H), 1.80-1.25 (m, 13H), 1.23-0.88 (m, 12H), 0.68 (s, 3H)。
1.
向G-020-004_1
(100 g,364 mmol)於無水甲醇(1 L)中之溶液中添加TsOH (6.26 g,36.4 mmol)。在60℃下攪拌混合物18小時。反應混合物經濃縮以移除大部分溶劑,用Et3
N (3.7 g)中和,用EtOAc (600 mL)稀釋,用水(500 mL)及鹽水(500 mL)洗滌。有機層經濃縮,得到呈油狀物之G-020-004_2
(133 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.20 (s, 3H), 3.14 (s, 3H), 2.63-2.39 (m, 2H), 2.14-2.03 (m, 2H), 1.97-1.89 (m, 2H), 1.86-1.77 (m, 3H), 1.64-1.60 (m, 2H), 1.56-1.49 (m, 3H), 1.47-1.42 (m, 2H), 1.40-1.32 (m, 2H), 1.29-1.23 (m, 3H), 1.16-1.06 (m, 2H), 0.87 (s, 3H)。2.
向Ph3
PEtBr (308 g,830 mmol)於20℃下氮氣下之無水THF (700 mL)中之懸浮液中添加t-BuOK (93.1 g,830 mmol)。在20℃下攪拌1小時之後,將G-020-004_2
(133 g,415 mmol)於無水THF (300 mL)中之溶液添加至混合物。使所得混合物升溫至50℃且攪拌4小時。反應混合物經冷卻,用水(400 mL)及飽和NH4
Cl (300 mL)淬滅,攪拌30 min。分離有機層,且用THF (300 mL)萃取水相。合併之有機層直接用於下一步驟中。 3. 向G-020-004_3
(137 g,412 mmol,理論上)於THF (1.3 L)中之溶液中添加HCl水溶液(1 M,618 mL,618 mmol)。在20℃下攪拌1小時之後,反應混合物用飽和NaHCO3
(800 mL)淬滅且用EtOAc (2×500 mL)萃取。合併之有機層經無水硫酸鈉乾燥,過濾且濃縮,得到固體(300 g)。自石油醚(800 mL)濕磨固體18小時。過濾出固體,且用石油醚(400 mL)洗滌濾餅。濃縮濾液,得到殘餘物(117 g)。殘餘物藉由矽膠管柱層析(0%至10% EtOAc/PE)純化,得到呈固體之N-004-023_5
(70 g)。 1 H NMR
(400 MHz, CDCl3
) δ 5.17-5.09 (m, 1H), 2.65-2.55 (m, 1H), 2.43-2.34 (m, 1H), 2.33-2.15 (m, 6H), 2.11-2.05 (m, 1H), 1.83-1.70 (m, 2H), 1.68-1.64 (m, 4H), 1.63-1.59 (m, 2H), 1.58-1.46 (m, 3H), 1.42-1.25 (m, 3H), 1.25-1.14 (m, 3H), 0.91 (s, 3H)。4.
在60℃下於N2
下加熱三甲基氧化鋶碘(30.5 g,139 mmol)及t-BuOK (15.5 g,139 mmol)於DMSO (200 mL)中之經攪拌溶液1.0 h;將N-004-023_5
(20 g,69.8 mmol)添加至反應混合物且在60℃下攪拌10分鐘。用水(1000 mL)處理反應物。用EtOAc (2×500 mL))萃取水相。合併之有機相用水(2×500 mL)、鹽水(300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之N-004-023_6
(20.5 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.13-5.10 (m, 1H), 2.62-2.60 (m, 2H), 2.25-2.20 (m, 5H), 2.00-1.48 (m, 12H), 1.46-1.00 (m, 8H), 0.98-0.89 (m, 4H)。 5. 在25℃下於N2
下將MeONa (18.4 g,341 mmol)添加至N-004-023_6
(20.5 g,68.2 mmol)於MeOH (500 mL)中之溶液中。在70℃回流下於N2
下攪拌混合物16 h。用水(500 mL)處理反應物。用DCM (2×300 mL)萃取水相。合併之有機相用飽和鹽水(2×300 mL)洗滌,經無水Na2
SO4
乾燥,經過濾且濃縮物藉由矽膠層析(PE/EtOAc=10/1至6/1)純化,得到呈固體之N-004-023_7
(20 g,88%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.12-5.06 (m, 1H), 3.38 (s, 3H), 3.19 (s, 2H), 2.25-2.22 (m, 1H), 2.20-2.09 (m, 3H), 1.66-1.63 (m, 3H), 1.60-1.24 (m, 14H), 1.22-1.00 (m, 6H), 0.87 (s, 3H)。 6. 在0℃下於N2
下將9-BBN二聚體(29.2 g,120 mmol)添加至N-004-023_7
(20 g,60.1 mmol)於THF (100 mL)中之溶液中。在65℃下攪拌溶液2小時。冷卻至0℃後,添加EtOH (34.9 mL,601 mmol)。接著極緩慢地添加NaOH (120 mL,5 M,601 mmol)之溶液。添加後,緩慢添加H2
O2
(68.0 g,601 mmol,30%於水中),且將內部溫度維持在低於10℃。在75℃下於N2
下攪拌混合物1小時。將混合物再冷卻至25℃。將混合物添加至H2
O (2 L)。攪拌混合物30分鐘。沈澱物藉由過濾採集且用H2
O (2×500 mL)洗滌,得到呈固體之N-004-023_8
(17.8 g,85%) 。 1 H NMR
(400 MHz, CDCl3
) δ 3.70-3.55 (m, 1H), 3.38 (s, 3H), 3.19 (m, 2H), 2.11-1.86 (m, 4H), 1.80-1.25 (m, 13H), 1.23-0.88 (m, 12H), 0.68 (s, 3H)。 7. 在25℃下將矽膠(24 g)及PCC (24.5 g,114 mmol)添加至N-004-023_8
(20 g,57.0 mmol)於DCM (500 mL)中之懸浮液中。在25℃下攪拌混合物2小時。過濾混合物且用DCM (2×100 mL)洗滌濾餅。真空濃縮合併之濾液。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之NA-004-023_9
(19 g,95%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.39 (s, 3H), 3.20 (s, 2H), 2.60-2.52 (m, 1H), 2.20-2.10 (m, 5H), 1.99-1.80 (m, 3H), 1.75-1.40 (m, 12H), 1.30-1.04 (m, 7H), 0.61 (s, 3H)。 8. 在25℃下將t-BuOK (12.2 g,54.5 mmol)添加至MePPh3
Br (38.9 g,109 mmol)於THF (300 mL)中之懸浮液中。在添加之後,將反應混合物加熱至45℃且攪拌1小時。接著添加N-004-023_9 (19 g,35.9 mmol)且在45℃下攪拌反應混合物16小時。用NH4
Cl (100 mL,飽和水溶液)處理混合物。分離有機層。用EtOAc (2×300 mL)萃取水相。將合併之有機相用飽和鹽水(2×200 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。在25℃下自MeOH/H2
O (100 mL/100 mL)濕磨殘餘物,得到呈固體之N-004-023_10
(17 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.68 (s, 1H), 3.39 (s, 3H), 3.19 (s, 2H), 2.10-2.04 (m, 1H), 2.03-1.90 (m, 3H), 1.75-1.56 (m, 12H), 1.49-1.25 (m, 4H), 1.22-0.89 (m, 8H), 0.57 (s, 3H)。 9. 將9-BBN二聚體(29.5 g,122 mmol)添加至N-004-023_10
(17 g,49.0 mmol)於無水THF (300 mL)中之溶液中且在0℃下於N2
下攪拌30 min。使反應混合物升溫至25℃ (室溫)且攪拌2小時。冷卻反應混合物。在0℃下藉由EtOH (100 mL)淬滅混合物。極緩慢地添加NaOH (98.0 mL,490 mol,5 M於水中)。在添加之後,添加緩慢H2
O2
(44.5 mL,490 mmol,11 M)直至內部溫度不再上升且在此期間將溫度維持在低於30℃。在50℃下再攪拌混合物1小時。接著混合物經冷卻,用水(2 L)處理且攪拌30 min。真空中過濾懸浮液,得到呈固體之N-004-023_11
(17 g, 粗產物)。在25℃下自MeOH/H2
O (100/100 mL)濕磨N-004-023_11
(17 g,46.6 mmol)且攪拌1小時。在真空下過濾懸浮液,以獲得呈固體之N-004-023_11
(14 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 3.79-3.66 (m, 1H), 3.50-3.37 (m, 4H), 3.28 (s, 2H), 2.43 (s, 1H), 2.26-1.98 (m, 3H), 2.18-2.12 (m, 1H), 1.95-1.60 (m, 11H), 1.34-1.04 (m, 14H), 0.76 (s, 3H)。 10. 在25℃下將DMP (9.24 g,21.8 mmol)添加至N-004-023_11
(4 g,10.9 mmol)於DCM (80 ml)中之溶液中。將一滴水添加至混合物且攪拌30分鐘。在低於10℃下用NaHCO3
飽和水溶液(pH=7至8)淬滅反應混合物。分離濾液中之DCM相位且用飽和NaHCO3
/Na2S2
O3
水溶液(1:1,2×50 mL)洗滌。有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈油狀物之N-004-023_12
(1.8 g,46%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.57-9.55 (m, 1H), 3.38 (s, 3H), 3.20 (s, 2H), 2.39-2.26 (m, 1H), 2.17-2.06 (m, 1H), 2.05-1.75 (m, 4H), 1.74-1.53 (m, 8H), 1.85-1.00 (m, 15H), 0.74 (s, 3H)。 11. 在0℃下將CsF (1.86 g,12.3 mmol)添加至N-004-023_12
(1.8 g,4.96 mmol)於無水THF (20 mL)中之溶液中。在0℃下攪拌20 min之後,在0℃下添加TMSCF3
(1.74 g,12.3 mmol)且攪拌1小時,接著添加TBAF.3H2
O (6.25 g,19.8 mmol) 。使混合反應物升溫至50℃且再攪拌1小時。將反應混合物倒入至冰水(50 mL)中且攪拌10 min。用EtOAc (2×80 mL)萃取水相。將合併之有機相用飽和鹽水(2×80 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之N-004-023_13
(1.2 g,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.03-3.98 (m, 1H), 3.39 (s, 3H), 3.20 (s, 2H), 2.17-1.80 (m, 7H), 1.73-1.41 (m, 10H), 1.28-0.95 (m, 13H), 0.71 (s, 3H)。 12. 在25℃下將DMP (2.34 g,5.54 mmol)添加至N-004-023_13
(1.2 g,2.77 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌30分鐘之後,在低於10℃下用飽和NaHCO3
水溶液(30 mL) (pH=7至8)淬滅反應混合物。接著添加DCM (30 mL)且攪拌混合物10 min。過濾懸浮液。分離濾液中之DCM相位且用飽和NaHCO3
/Na2
S2
O3
水溶液(1:1,2×50 mL)洗滌。有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈油狀物之N-004-023_13A
(1.2 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.39 (s, 3H), 3.20 (s, 2H), 3.05-2.95 (m, 1H), 1.91-1.51 (m, 10H), 1.46-1.20 (m, 10H), 1.17-0.96 (m, 8H), 0.72 (s, 3H)。13.
在0℃將NaBH4
(210 mg,5.56 mmol)添加至N-004-023_13A
(1.2 g,2.78 mmol)於MeOH (5 mL)之溶液中且攪拌30 min。在用MeOH/H2
O (20/20 mL)處理之後,攪拌混合物10 min。用EtOAc (2×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之48
(57 mg,5% )及呈固體之45
(200 mg,不純)。45
(200 mg,0.462 mmol)藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之45
(120 mg,10% )。45
: 1 H NMR
(400 MHz, CDCl3
) δ 4.01-3.98 (m, 1H), 3.38 (s, 3H), 3.19 (s, 2H), 2.15-2.10 (m, 1H), 2.05-1.80 (m, 5H), 1.72-1.55 (m, 5H), 1.54-1.34 (m, 6H), 1.31-1.20 (m, 4H), 1.16-0.95 (m, 9H), 0.71 (s, 3H)。 在2 min層析中,LCMS
Rt=1.129 min,30-90AB_2MIN_E,純度100%,C24
H38
F3
[M -HO3
]+
之MS ESI計算值383,實驗值383。實例 46 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (46) 之合成
7. 在25℃下將矽膠(24 g)及PCC (24.5 g,114 mmol)添加至N-004-023_8
(20 g,57.0 mmol)於DCM (500 mL)中之懸浮液中。在25℃下攪拌混合物2小時。過濾混合物且用DCM (2×100 mL)洗滌濾餅。真空濃縮合併之濾液。殘餘物藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之NA-004-023_9
(19 g,95%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.39 (s, 3H), 3.20 (s, 2H), 2.60-2.52 (m, 1H), 2.20-2.10 (m, 5H), 1.99-1.80 (m, 3H), 1.75-1.40 (m, 12H), 1.30-1.04 (m, 7H), 0.61 (s, 3H)。 8. 在25℃下將t-BuOK (12.2 g,54.5 mmol)添加至MePPh3
Br (38.9 g,109 mmol)於THF (300 mL)中之懸浮液中。在添加之後,將反應混合物加熱至45℃且攪拌1小時。接著添加N-004-023_9 (19 g,35.9 mmol)且在45℃下攪拌反應混合物16小時。用NH4
Cl (100 mL,飽和水溶液)處理混合物。分離有機層。用EtOAc (2×300 mL)萃取水相。將合併之有機相用飽和鹽水(2×200 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。在25℃下自MeOH/H2
O (100 mL/100 mL)濕磨殘餘物,得到呈固體之N-004-023_10
(17 g,90%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.68 (s, 1H), 3.39 (s, 3H), 3.19 (s, 2H), 2.10-2.04 (m, 1H), 2.03-1.90 (m, 3H), 1.75-1.56 (m, 12H), 1.49-1.25 (m, 4H), 1.22-0.89 (m, 8H), 0.57 (s, 3H)。 9. 將9-BBN二聚體(29.5 g,122 mmol)添加至N-004-023_10
(17 g,49.0 mmol)於無水THF (300 mL)中之溶液中且在0℃下於N2
下攪拌30 min。使反應混合物升溫至25℃ (室溫)且攪拌2小時。冷卻反應混合物。在0℃下藉由EtOH (100 mL)淬滅混合物。極緩慢地添加NaOH (98.0 mL,490 mol,5 M於水中)。在添加之後,添加緩慢H2
O2
(44.5 mL,490 mmol,11 M)直至內部溫度不再上升且在此期間將溫度維持在低於30℃。在50℃下再攪拌混合物1小時。接著混合物經冷卻,用水(2 L)處理且攪拌30 min。真空中過濾懸浮液,得到呈固體之N-004-023_11
(17 g, 粗產物)。在25℃下自MeOH/H2
O (100/100 mL)濕磨N-004-023_11
(17 g,46.6 mmol)且攪拌1小時。在真空下過濾懸浮液,以獲得呈固體之N-004-023_11
(14 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 3.79-3.66 (m, 1H), 3.50-3.37 (m, 4H), 3.28 (s, 2H), 2.43 (s, 1H), 2.26-1.98 (m, 3H), 2.18-2.12 (m, 1H), 1.95-1.60 (m, 11H), 1.34-1.04 (m, 14H), 0.76 (s, 3H)。 10. 在25℃下將DMP (9.24 g,21.8 mmol)添加至N-004-023_11
(4 g,10.9 mmol)於DCM (80 ml)中之溶液中。將一滴水添加至混合物且攪拌30分鐘。在低於10℃下用NaHCO3
飽和水溶液(pH=7至8)淬滅反應混合物。分離濾液中之DCM相位且用飽和NaHCO3
/Na2S2
O3
水溶液(1:1,2×50 mL)洗滌。有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈油狀物之N-004-023_12
(1.8 g,46%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.57-9.55 (m, 1H), 3.38 (s, 3H), 3.20 (s, 2H), 2.39-2.26 (m, 1H), 2.17-2.06 (m, 1H), 2.05-1.75 (m, 4H), 1.74-1.53 (m, 8H), 1.85-1.00 (m, 15H), 0.74 (s, 3H)。 11. 在0℃下將CsF (1.86 g,12.3 mmol)添加至N-004-023_12
(1.8 g,4.96 mmol)於無水THF (20 mL)中之溶液中。在0℃下攪拌20 min之後,在0℃下添加TMSCF3
(1.74 g,12.3 mmol)且攪拌1小時,接著添加TBAF.3H2
O (6.25 g,19.8 mmol) 。使混合反應物升溫至50℃且再攪拌1小時。將反應混合物倒入至冰水(50 mL)中且攪拌10 min。用EtOAc (2×80 mL)萃取水相。將合併之有機相用飽和鹽水(2×80 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之N-004-023_13
(1.2 g,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.03-3.98 (m, 1H), 3.39 (s, 3H), 3.20 (s, 2H), 2.17-1.80 (m, 7H), 1.73-1.41 (m, 10H), 1.28-0.95 (m, 13H), 0.71 (s, 3H)。 12. 在25℃下將DMP (2.34 g,5.54 mmol)添加至N-004-023_13
(1.2 g,2.77 mmol)於DCM (30 mL)中之溶液中。在25℃下攪拌30分鐘之後,在低於10℃下用飽和NaHCO3
水溶液(30 mL) (pH=7至8)淬滅反應混合物。接著添加DCM (30 mL)且攪拌混合物10 min。過濾懸浮液。分離濾液中之DCM相位且用飽和NaHCO3
/Na2
S2
O3
水溶液(1:1,2×50 mL)洗滌。有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮,得到呈油狀物之N-004-023_13A
(1.2 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 3.39 (s, 3H), 3.20 (s, 2H), 3.05-2.95 (m, 1H), 1.91-1.51 (m, 10H), 1.46-1.20 (m, 10H), 1.17-0.96 (m, 8H), 0.72 (s, 3H)。13.
在0℃將NaBH4
(210 mg,5.56 mmol)添加至N-004-023_13A
(1.2 g,2.78 mmol)於MeOH (5 mL)之溶液中且攪拌30 min。在用MeOH/H2
O (20/20 mL)處理之後,攪拌混合物10 min。用EtOAc (2×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之48
(57 mg,5% )及呈固體之45
(200 mg,不純)。45
(200 mg,0.462 mmol)藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之45
(120 mg,10% )。45
: 1 H NMR
(400 MHz, CDCl3
) δ 4.01-3.98 (m, 1H), 3.38 (s, 3H), 3.19 (s, 2H), 2.15-2.10 (m, 1H), 2.05-1.80 (m, 5H), 1.72-1.55 (m, 5H), 1.54-1.34 (m, 6H), 1.31-1.20 (m, 4H), 1.16-0.95 (m, 9H), 0.71 (s, 3H)。 在2 min層析中,LCMS
Rt=1.129 min,30-90AB_2MIN_E,純度100%,C24
H38
F3
[M -HO3
]+
之MS ESI計算值383,實驗值383。實例 46 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (46) 之合成 
 1.
在75℃下將(溴甲基)環戊烷(2.25 g,13.8 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (662 mg,27.6 mmol)及I2
(70 mg,0.276 mmol)於THF (3 mL)中之懸浮液中。在75℃下攪拌混合物1小時。冷卻後,在15℃下緩慢地添加S-500-6-1_1
(1 g,2.78 mmol)於THF (30 mL)中之溶液。在添加之後,在15℃下攪拌混合物2小時,用飽和NH4
Cl (40 mL)及飽和檸檬酸(20 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之S-500-6-13_1
及22位置處之異構體的混合物(900 mg,73%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.23 (m, 1H), 3.77-3.67 (m, 1H), 2.40-2.30 (m, 1H), 2.07-1.89 (m, 4H), 1.88-1.69 (m, 4H), 1.67-1.59 (m, 4H), 1.55-1.26 (m, 15H), 1.16-1.05 (m, 5H), 1.05-1.00 (m, 4H), 0.99-0.81 (m, 8H), 0.68 (s, 3H)。 2.將DMP (1.72 g,4.06 mmol)添加至S-500-6-13_1
(900 mg,2.03 mmol)於DCM (30 mL)中之溶液中。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之粗產物S-500-6-13_
2 (900 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.25 (m, 1H), 2.54-2.42 (m, 2H), 2.41-2.33 (m, 1H), 2.30-2.17 (m, 1H), 2.06-1.90 (m, 3H), 1.87-1.78 (m, 2H), 1.73-1.66 (m, 2H), 1.65-1.35 (m, 15H), 1.33-1.21 (m, 2H), 1.17-0.92 (m, 13H), 0.88-0.82 (m, 3H), 0.69 (s, 3H)。 3. 將NaBH4
(3.46 g,102 mmol)每五分鐘添加五次至S-500-6-13_2
(900 mg,2.04 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘,用飽和NH4
Cl (50 mL) 淬滅且用EtOAc (3×20 mL)萃取。將合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之不純46
(120 mg),其再次藉由SFC (管柱:AD (250 mm×30 mm,5um),梯度:45%至45% B (A= 0.05% NH3
/H2
O,B= MeOH),流速:60 mL/min)純化,得到呈固體之純46
(100 mg,84%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.76-3.67 (m, 1H), 2.40-2.33 (m, 1H), 2.08-1.91 (m, 4H), 1.90-1.78 (m, 2H), 1.77-1.55 (m, 10H), 1.54-1.31 (m, 9H), 1.26-1.22 (m, 2H), 1.22-1.05 (m, 6H), 1.03 (s, 3H), 1.01-0.89 (m, 5H), 0.89-0.82 (m, 3H), 0.69 (s, 3H)。LCMS
Rt=2.0 min層析中1.474 min,30-90AB_E,純度99%,C30
H49
O [M+H-H2
O]+
之MS ESI計算值425,實驗值425。實例 47 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (47) 之合成
1.
在75℃下將(溴甲基)環戊烷(2.25 g,13.8 mmol)於THF (8 mL)中之溶液逐滴添加至Mg (662 mg,27.6 mmol)及I2
(70 mg,0.276 mmol)於THF (3 mL)中之懸浮液中。在75℃下攪拌混合物1小時。冷卻後,在15℃下緩慢地添加S-500-6-1_1
(1 g,2.78 mmol)於THF (30 mL)中之溶液。在添加之後,在15℃下攪拌混合物2小時,用飽和NH4
Cl (40 mL)及飽和檸檬酸(20 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(2×30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之S-500-6-13_1
及22位置處之異構體的混合物(900 mg,73%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.23 (m, 1H), 3.77-3.67 (m, 1H), 2.40-2.30 (m, 1H), 2.07-1.89 (m, 4H), 1.88-1.69 (m, 4H), 1.67-1.59 (m, 4H), 1.55-1.26 (m, 15H), 1.16-1.05 (m, 5H), 1.05-1.00 (m, 4H), 0.99-0.81 (m, 8H), 0.68 (s, 3H)。 2.將DMP (1.72 g,4.06 mmol)添加至S-500-6-13_1
(900 mg,2.03 mmol)於DCM (30 mL)中之溶液中。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3×40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈固體之粗產物S-500-6-13_
2 (900 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.25 (m, 1H), 2.54-2.42 (m, 2H), 2.41-2.33 (m, 1H), 2.30-2.17 (m, 1H), 2.06-1.90 (m, 3H), 1.87-1.78 (m, 2H), 1.73-1.66 (m, 2H), 1.65-1.35 (m, 15H), 1.33-1.21 (m, 2H), 1.17-0.92 (m, 13H), 0.88-0.82 (m, 3H), 0.69 (s, 3H)。 3. 將NaBH4
(3.46 g,102 mmol)每五分鐘添加五次至S-500-6-13_2
(900 mg,2.04 mmol)於MeOH (5 mL)及THF (5 mL)中之溶液中。在15℃下攪拌混合物30分鐘,用飽和NH4
Cl (50 mL) 淬滅且用EtOAc (3×20 mL)萃取。將合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到呈固體之不純46
(120 mg),其再次藉由SFC (管柱:AD (250 mm×30 mm,5um),梯度:45%至45% B (A= 0.05% NH3
/H2
O,B= MeOH),流速:60 mL/min)純化,得到呈固體之純46
(100 mg,84%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.76-3.67 (m, 1H), 2.40-2.33 (m, 1H), 2.08-1.91 (m, 4H), 1.90-1.78 (m, 2H), 1.77-1.55 (m, 10H), 1.54-1.31 (m, 9H), 1.26-1.22 (m, 2H), 1.22-1.05 (m, 6H), 1.03 (s, 3H), 1.01-0.89 (m, 5H), 0.89-0.82 (m, 3H), 0.69 (s, 3H)。LCMS
Rt=2.0 min層析中1.474 min,30-90AB_E,純度99%,C30
H49
O [M+H-H2
O]+
之MS ESI計算值425,實驗值425。實例 47 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (47) 之合成  1.將Pd(OH)2
(200 mg)添加至30
(100 mg,0.239 mmol)於MeOH (10 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾,濃縮且藉由combi-flash (0%至10% EtOAc/PE)純化,得到47
(21 mg,21%)及呈白色固體之36
(1 mg,1%)。47 : 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.54 (m, 1H), 2.02-1.90 (m, 1H), 1.76-1.57 (m, 6H), 1.54-1.27 (m, 10H), 1.26-1.21 (m, 7H), 1.20-1.08 (m, 5H), 1.07-0.95 (m, 3H), 0.94-0.83 (m, 10H), 0.81 (s, 3H), 0.72-0.60 (m, 4H)。LCMS
tR
=2 min層析中1.290 min,30-90AB_ELSD,純度100.0%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 48 : (3S,5R,8R,9R,10S,13S,14S,17R)-3-( 甲氧基甲基 )-13- 甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (48) 之合成
1.將Pd(OH)2
(200 mg)添加至30
(100 mg,0.239 mmol)於MeOH (10 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾,濃縮且藉由combi-flash (0%至10% EtOAc/PE)純化,得到47
(21 mg,21%)及呈白色固體之36
(1 mg,1%)。47 : 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.54 (m, 1H), 2.02-1.90 (m, 1H), 1.76-1.57 (m, 6H), 1.54-1.27 (m, 10H), 1.26-1.21 (m, 7H), 1.20-1.08 (m, 5H), 1.07-0.95 (m, 3H), 0.94-0.83 (m, 10H), 0.81 (s, 3H), 0.72-0.60 (m, 4H)。LCMS
tR
=2 min層析中1.290 min,30-90AB_ELSD,純度100.0%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 48 : (3S,5R,8R,9R,10S,13S,14S,17R)-3-( 甲氧基甲基 )-13- 甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (48) 之合成  在0℃下將NaBH4
(210 mg,5.56 mmol)添加至N-004-023_13A
(1.2 g,2.78 mmol)於1. MeOH (5 mL)中之溶液中攪拌且30 min。在用MeOH/H2
O (20/20 mL)處理之後,攪拌混合物10 min。用EtOAc (2×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之48
(57 mg,5% )及呈固體之45
(200 mg,不純)。45
(200 mg,0.462 mmol)藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之45
(120 mg,10%)。48 : 1 H NMR
(400 MHz, CDCl3
) δ 4.07-4.01 (m, 1H), 3.39 (s, 3H), 3.19 (s, 2H), 2.30-2.22 (m, 1H), 2.14-2.05 (m, 1H), 2.00-1.80 (m, 4H), 1.72-1.57 (m, 6H), 1.49-1.20 (m, 9H), 1.18-0.95 (m, 9H), 0.68 (s, 3H)。 在2 min層析中,LCMS
Rt=1.085 min,30-90AB_2MIN_E,純度100%,C24
H38
F3
[M -HO3
]+
之MS ESI計算值383,實驗值383。實例 49 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (49) 之合成
在0℃下將NaBH4
(210 mg,5.56 mmol)添加至N-004-023_13A
(1.2 g,2.78 mmol)於1. MeOH (5 mL)中之溶液中攪拌且30 min。在用MeOH/H2
O (20/20 mL)處理之後,攪拌混合物10 min。用EtOAc (2×50 mL)萃取水相。合併之有機相用飽和鹽水(2×50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至20% EtOAc/PE)純化,得到呈固體之48
(57 mg,5% )及呈固體之45
(200 mg,不純)。45
(200 mg,0.462 mmol)藉由閃蒸塔(0%至30% EtOAc/PE)純化,得到呈固體之45
(120 mg,10%)。48 : 1 H NMR
(400 MHz, CDCl3
) δ 4.07-4.01 (m, 1H), 3.39 (s, 3H), 3.19 (s, 2H), 2.30-2.22 (m, 1H), 2.14-2.05 (m, 1H), 2.00-1.80 (m, 4H), 1.72-1.57 (m, 6H), 1.49-1.20 (m, 9H), 1.18-0.95 (m, 9H), 0.68 (s, 3H)。 在2 min層析中,LCMS
Rt=1.085 min,30-90AB_2MIN_E,純度100%,C24
H38
F3
[M -HO3
]+
之MS ESI計算值383,實驗值383。實例 49 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (49) 之合成  49
之合成描述於實例4中。49 : 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.07-1.87 (m, 4H), 1.76-1.69 (m, 1H), 1.66-1.55 (m, 5H), 1.53-1.42 (m, 7H), 1.41-1.31 (m, 5H), 1.30-1.12 (m, 8H), 1.11-1.05 (m, 3H), 1.03 (s, 3H), 1.01-0.92 (m, 2H), 0.91-0.82 (m, 12H), 0.68 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.718 min,30-90AB_E,純度98%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。實例 50 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (50) 之合成
49
之合成描述於實例4中。49 : 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.85-3.77 (m, 1H), 2.40-2.32 (m, 1H), 2.07-1.87 (m, 4H), 1.76-1.69 (m, 1H), 1.66-1.55 (m, 5H), 1.53-1.42 (m, 7H), 1.41-1.31 (m, 5H), 1.30-1.12 (m, 8H), 1.11-1.05 (m, 3H), 1.03 (s, 3H), 1.01-0.92 (m, 2H), 0.91-0.82 (m, 12H), 0.68 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.718 min,30-90AB_E,純度98%,C33
H53
[M+H-2H2
O]+
之MS ESI計算值449,實驗值449。實例 50 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (50) 之合成 
 1.
在0℃下於N2
下將MeMgBr (0.83 mL,2.49 mmol,3 M於乙醚中)逐滴添加至N-8-7_1
(300 mg,0.832 mmol)於THF (20 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應物用飽和NH4
Cl (50 mL)淬滅且用EtOAc (2×10 mL)萃取。合併之相位用鹽水洗滌(10 mL),經Na2
SO4
乾燥,經過濾,濃縮且藉由閃蒸塔(步驟2) (0%至10% EtOAc/PE)純化,得到呈固體之50
(40 mg,29%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.89 (m, 1H), 1.99-1.84 (m, 2H), 1.69-1.56 (m, 6H), 1.54-1.45 (m, 2H), 1.43-1.29 (m, 6H), 1.28-1.17 (m, 4H), 1.17-1.12 (m, 4H), 1.12-0.94 (m, 5H), 0.92-0.84 (m, 7H), 0.82 (s, 3H), 0.68-0.61 (m, 4H)。 在7.0 min層析中,LCMS
Rt = 3.428 min,30-90AB_E,純度100%,C25
H41
[M+H-2H2
O]+
之MS ESI計算值341,實驗值341。實例 51 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (51) 之合成
1.
在0℃下於N2
下將MeMgBr (0.83 mL,2.49 mmol,3 M於乙醚中)逐滴添加至N-8-7_1
(300 mg,0.832 mmol)於THF (20 mL)中之溶液中。在20℃下攪拌30分鐘之後,反應物用飽和NH4
Cl (50 mL)淬滅且用EtOAc (2×10 mL)萃取。合併之相位用鹽水洗滌(10 mL),經Na2
SO4
乾燥,經過濾,濃縮且藉由閃蒸塔(步驟2) (0%至10% EtOAc/PE)純化,得到呈固體之50
(40 mg,29%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.89 (m, 1H), 1.99-1.84 (m, 2H), 1.69-1.56 (m, 6H), 1.54-1.45 (m, 2H), 1.43-1.29 (m, 6H), 1.28-1.17 (m, 4H), 1.17-1.12 (m, 4H), 1.12-0.94 (m, 5H), 0.92-0.84 (m, 7H), 0.82 (s, 3H), 0.68-0.61 (m, 4H)。 在7.0 min層析中,LCMS
Rt = 3.428 min,30-90AB_E,純度100%,C25
H41
[M+H-2H2
O]+
之MS ESI計算值341,實驗值341。實例 51 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3-( 甲氧基甲基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (51) 之合成  1.
在25℃下於N2
下將S-500-6-29_2
(999 mg,1.22 M於THF中,4.87 mmol)逐滴添加至N-8-1_1
(210 mg,05576 mmol)於THF (2 mL)中之溶液中。在25℃下攪拌16小時之後,反應混合物用飽和NH4
Cl (10 mL)淬滅且用乙酸乙酯(3×10 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到粗產物,其藉由閃蒸塔(0%至15% EtOAc/PE)純化2次,得到不純產物(30 mg)。不純產物藉由ELSD製備型HPLC (管柱:Phenomenex Synergi C18 150×30 mm×4 um,梯度:90%至95% B (A=水(0.05% HCl),B=MeCN),流速:25 mL/min)進一步純化,得到呈固體之純51
(4 mg,1.4%產率)。 1 H NMR
(400 MHz, CDCl3
) δ 3.84-3.76 (m, 1H), 3.45-3.32 (m, 5H), 2.62-2.39 (m, 1H), 1.99-1.85 (m, 2H), 1.73-1.62 (m, 4H), 1.53-1.40 (m, 7H), 1.39-1.31 (m, 5H), 1.30-1.21 (m, 7H), 1.20-1.13 (m, 4H), 1.12-1.10 (m, 5H), 0.99-0.93 (m, 1H), 0.89-0.86 (m, 6H), 0.85 (s, 3H), 0.83 (s, 3H), 0.68-0.61 (m, 4H)。 在7.0 min層析中,LCMS
Rt = 5.669 min,30-90AB_E,純度100%,C33
H55
O [M+H-2H2
O]+
之MS ESI計算值467,實驗值467。實例 52 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (52) 之合成
1.
在25℃下於N2
下將S-500-6-29_2
(999 mg,1.22 M於THF中,4.87 mmol)逐滴添加至N-8-1_1
(210 mg,05576 mmol)於THF (2 mL)中之溶液中。在25℃下攪拌16小時之後,反應混合物用飽和NH4
Cl (10 mL)淬滅且用乙酸乙酯(3×10 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到粗產物,其藉由閃蒸塔(0%至15% EtOAc/PE)純化2次,得到不純產物(30 mg)。不純產物藉由ELSD製備型HPLC (管柱:Phenomenex Synergi C18 150×30 mm×4 um,梯度:90%至95% B (A=水(0.05% HCl),B=MeCN),流速:25 mL/min)進一步純化,得到呈固體之純51
(4 mg,1.4%產率)。 1 H NMR
(400 MHz, CDCl3
) δ 3.84-3.76 (m, 1H), 3.45-3.32 (m, 5H), 2.62-2.39 (m, 1H), 1.99-1.85 (m, 2H), 1.73-1.62 (m, 4H), 1.53-1.40 (m, 7H), 1.39-1.31 (m, 5H), 1.30-1.21 (m, 7H), 1.20-1.13 (m, 4H), 1.12-1.10 (m, 5H), 0.99-0.93 (m, 1H), 0.89-0.86 (m, 6H), 0.85 (s, 3H), 0.83 (s, 3H), 0.68-0.61 (m, 4H)。 在7.0 min層析中,LCMS
Rt = 5.669 min,30-90AB_E,純度100%,C33
H55
O [M+H-2H2
O]+
之MS ESI計算值467,實驗值467。實例 52 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (52) 之合成  1. 將NaOH溶液(974 mg於6 mL H2
O中,16.8 mmol)添加至S-500-2-15_1
(900 mg,1.68 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在50℃下加熱混合物16小時。反應混合物用飽和NH4
Cl (60 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到210 mg固體,其藉由SFC (管柱:AD (250 mm×30 mm,5 um),梯度:35%至35% B (A= 0.1% NH3
/H2
O,B= MeOH),流速:80 mL/min)純化,得到呈固體之52
(150 mg,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 3.64-3.58 (m, 1H), 2.40-2.30 (m, 1H), 2.02-1.92 (m, 3H), 1.80-1.58 (m, 7H), 1.56-1.31 (m, 9H), 1.30-1.05 (m, 8H), 1.03 (s, 3H), 1.02-0.96 (m, 2H), 0.95-0.86 (m, 9H), 0.85-0.80 (m, 3H), 0.69 (s, 3H). 在2 min層析中,LCMS
tR
=1.335 min,30-90AB_ELSD,純度100.0%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。實例 53 : (3S,8S,9S,10R,13S,14S,17R)-10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-3-( 三氟甲基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (53) 之合成
1. 將NaOH溶液(974 mg於6 mL H2
O中,16.8 mmol)添加至S-500-2-15_1
(900 mg,1.68 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在50℃下加熱混合物16小時。反應混合物用飽和NH4
Cl (60 mL)淬滅且用EtOAc (3×20 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到210 mg固體,其藉由SFC (管柱:AD (250 mm×30 mm,5 um),梯度:35%至35% B (A= 0.1% NH3
/H2
O,B= MeOH),流速:80 mL/min)純化,得到呈固體之52
(150 mg,68%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 3.64-3.58 (m, 1H), 2.40-2.30 (m, 1H), 2.02-1.92 (m, 3H), 1.80-1.58 (m, 7H), 1.56-1.31 (m, 9H), 1.30-1.05 (m, 8H), 1.03 (s, 3H), 1.02-0.96 (m, 2H), 0.95-0.86 (m, 9H), 0.85-0.80 (m, 3H), 0.69 (s, 3H). 在2 min層析中,LCMS
tR
=1.335 min,30-90AB_ELSD,純度100.0%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。實例 53 : (3S,8S,9S,10R,13S,14S,17R)-10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-3-( 三氟甲基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (53) 之合成  1. 在0℃下向N-004-027_1
(1.5 g,3.76 mmol)於無水THF (40 mL)中之溶液中添加CsF (1.42 g,9.40 mmol)。在0℃下攪拌20 min之後,在0℃下添加TMSCF3
(1.33 g,9.40 mmol)且攪拌30 min。顏色變為淡黃色。添加TBAF.3H2
O (4.74 g,15.0 mmol)且在50℃下攪拌30 min。將反應混合物倒入至冰水(100 mL)中。用EtOAc (2×100 mL)萃取水相。合併之有機相用飽和鹽水(2×100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈黃色固體之異構體混合物(1.45 g,粗產物),其藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之53
(340 mg,24%)及呈白色固體之1
(200 mg,14%)。53 : 1 H NMR
(400 MHz, CDCl3
) δ 5.38-5.36 (m, 1H), 4.06-3.94 (m, 1H), 2.49 (s, 2H), 2.09-1.58 (m, 13H), 1.48-0.85 (m, 14H), 0.73 (s, 3H)。 在2 min層析中,LCMS
Rt=1.134 min,30-90AB_2MIN_E,純度99%。MS
50-100_1_4min.m,C24
H33
F6
O[M+H-H2
O]+
之MS ESI計算值451,實驗值451。實例 54 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (54) 之合成
1. 在0℃下向N-004-027_1
(1.5 g,3.76 mmol)於無水THF (40 mL)中之溶液中添加CsF (1.42 g,9.40 mmol)。在0℃下攪拌20 min之後,在0℃下添加TMSCF3
(1.33 g,9.40 mmol)且攪拌30 min。顏色變為淡黃色。添加TBAF.3H2
O (4.74 g,15.0 mmol)且在50℃下攪拌30 min。將反應混合物倒入至冰水(100 mL)中。用EtOAc (2×100 mL)萃取水相。合併之有機相用飽和鹽水(2×100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮,得到呈黃色固體之異構體混合物(1.45 g,粗產物),其藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之53
(340 mg,24%)及呈白色固體之1
(200 mg,14%)。53 : 1 H NMR
(400 MHz, CDCl3
) δ 5.38-5.36 (m, 1H), 4.06-3.94 (m, 1H), 2.49 (s, 2H), 2.09-1.58 (m, 13H), 1.48-0.85 (m, 14H), 0.73 (s, 3H)。 在2 min層析中,LCMS
Rt=1.134 min,30-90AB_2MIN_E,純度99%。MS
50-100_1_4min.m,C24
H33
F6
O[M+H-H2
O]+
之MS ESI計算值451,實驗值451。實例 54 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6,6- 二甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (54) 之合成  1. 在15℃下將NaBH4
(1.77 g,46.8 mmol)逐份添加至S-500-6-1_3
(520 mg,1.17 mmol)於THF (5 mL)及MeOH (10 mL)中之溶液中。在15℃下攪拌混合物20 min。混合物用NH4
Cl (20 mL,飽和水溶液)淬滅且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到S-500-6-1
(300 mg,不純)及54
(170 mg,不純)。 不純54
(220 mg,不純)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到固體。在60℃下將固體溶解於MeCN (50 mL)中且真空濃縮,得到呈固體之54
(120 mg,23%)。54 : 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.62-3.52 (m, 1H), 2.42-2.31 (m, 1H), 2.11-1.90 (m, 3H), 1.72-1.35 (m, 15H), 1.29-1.08 (m, 8H), 1.03 (s, 3H), 1.01-0.96 (m, 2H), 0.93 (d,J
= 6.8 Hz, 3H), 0.90 (s, 9H), 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H)。 在7.0 min層析中,LCMS
Rt = 5.463 min,30-90_AB_E,純度100%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 55 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1R,2S)-1- 羥基 -1- 苯基丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇
(55)之合成
1. 在15℃下將NaBH4
(1.77 g,46.8 mmol)逐份添加至S-500-6-1_3
(520 mg,1.17 mmol)於THF (5 mL)及MeOH (10 mL)中之溶液中。在15℃下攪拌混合物20 min。混合物用NH4
Cl (20 mL,飽和水溶液)淬滅且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到混合物,該混合物藉由閃蒸塔(0%至15% EtOAc/PE)分離,得到S-500-6-1
(300 mg,不純)及54
(170 mg,不純)。 不純54
(220 mg,不純)藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到固體。在60℃下將固體溶解於MeCN (50 mL)中且真空濃縮,得到呈固體之54
(120 mg,23%)。54 : 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.24 (m, 1H), 3.62-3.52 (m, 1H), 2.42-2.31 (m, 1H), 2.11-1.90 (m, 3H), 1.72-1.35 (m, 15H), 1.29-1.08 (m, 8H), 1.03 (s, 3H), 1.01-0.96 (m, 2H), 0.93 (d,J
= 6.8 Hz, 3H), 0.90 (s, 9H), 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H)。 在7.0 min層析中,LCMS
Rt = 5.463 min,30-90_AB_E,純度100%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 55 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1R,2S)-1- 羥基 -1- 苯基丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇
(55)之合成 
 1. 在0℃下將N-8-7_1
(300 mg,0.832 mmol)於THF (5 mL)中之溶液添加至PhMgBr (1.38 mL,3 M於乙醚中,4.15 mmol)於THF (10 mL)中之溶液中,隨後在0℃下攪拌反應混合物3小時。接著,在25℃下攪拌反應混合物5小時。在0℃下用水(10 mL)淬滅反應混合物。過濾溶液且用EtOAc (10 mL)洗滌濾餅。用EtOAc (3×15 mL)萃取水相。合併之有機相用飽和鹽水(2×10 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至1/1)純化,得到呈固體之59
及19
(200 mg,粗產物)。粗產物藉由SFC (管柱:AD (250 mm×30 mm,5um),梯度:25%至25% B (A= 0.1% NH3
/H2
O,B= EtOH ),流速:60 mL/min)純化,得到55
(峰值2,55 mg, 15%)及呈固體之19
(峰值1,21 mg,6%)。55 : 1 H NMR
(400MHz, CDCl3
) δ 7.38-7.28 (m, 4H), 7.25-7.20 (m, 1H), δ4.95- 4.90 (m, 1H), 2.13-2.01 (m, 1H), 1.98-1.88 (m, 1H), 1.77-1.59 (m, 6H) , 1.57-1.43 (m, 6H), 1.43-0.93 (m, 13H), 0.91-0.85 (m, 3H), 0.83 (s, 3H), 0.76-0.72 (m, 3H), 0.68 (s, 4H)。 在2.0層析層析中,LCMS
Rt=1.239 min,30-90AB_2 min.,純度100%,C30
H43
[M-2H2
O+H]+
之MS ESI計算值403,實驗值403。 在3 min層析中,SFC Rt=1.192 min,OJ_3_EtOH_DEA_5_40_25ML,99%de。實例 56 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (56) 之合成
1. 在0℃下將N-8-7_1
(300 mg,0.832 mmol)於THF (5 mL)中之溶液添加至PhMgBr (1.38 mL,3 M於乙醚中,4.15 mmol)於THF (10 mL)中之溶液中,隨後在0℃下攪拌反應混合物3小時。接著,在25℃下攪拌反應混合物5小時。在0℃下用水(10 mL)淬滅反應混合物。過濾溶液且用EtOAc (10 mL)洗滌濾餅。用EtOAc (3×15 mL)萃取水相。合併之有機相用飽和鹽水(2×10 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc=20/1至1/1)純化,得到呈固體之59
及19
(200 mg,粗產物)。粗產物藉由SFC (管柱:AD (250 mm×30 mm,5um),梯度:25%至25% B (A= 0.1% NH3
/H2
O,B= EtOH ),流速:60 mL/min)純化,得到55
(峰值2,55 mg, 15%)及呈固體之19
(峰值1,21 mg,6%)。55 : 1 H NMR
(400MHz, CDCl3
) δ 7.38-7.28 (m, 4H), 7.25-7.20 (m, 1H), δ4.95- 4.90 (m, 1H), 2.13-2.01 (m, 1H), 1.98-1.88 (m, 1H), 1.77-1.59 (m, 6H) , 1.57-1.43 (m, 6H), 1.43-0.93 (m, 13H), 0.91-0.85 (m, 3H), 0.83 (s, 3H), 0.76-0.72 (m, 3H), 0.68 (s, 4H)。 在2.0層析層析中,LCMS
Rt=1.239 min,30-90AB_2 min.,純度100%,C30
H43
[M-2H2
O+H]+
之MS ESI計算值403,實驗值403。 在3 min層析中,SFC Rt=1.192 min,OJ_3_EtOH_DEA_5_40_25ML,99%de。實例 56 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (56) 之合成  1. 向81
(1 g,3.26 mmol)於MeOH (30 mL)及THF (10 mL)中之溶液中添加Pd(OH)2
(1 g,<1%水)。接著在50℃下於50 psi下氫化混合物48小時。在無監測之情況下經由矽藻土墊過濾混合物且在真空中濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到呈白色固體之56
(331 mg,33%)。 1 H NMR
(400 MHz, CDCl3) δ 4.09-3.99 (m, 1H), 2.18-2.13 (m, 1H), 1.99-1.78 (m, 4H), 1.75-1.59 (m, 3H), 1.50-1.3 (m, 7H), 1.34-1.22 (m, 6H), 1.21-1.00 (m, 10H), 0.96 (s, 3H), 0.94-0.89 (m, 3H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.184 min,30-90AB_E,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 57 : (3S,5R,8R,9S,10S,13S,14S,17R)-3,10,13- 三甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (57) 之合成
1. 向81
(1 g,3.26 mmol)於MeOH (30 mL)及THF (10 mL)中之溶液中添加Pd(OH)2
(1 g,<1%水)。接著在50℃下於50 psi下氫化混合物48小時。在無監測之情況下經由矽藻土墊過濾混合物且在真空中濃縮濾液。殘餘物藉由閃蒸塔(PE/EtOAc=10/1至5/1)純化,得到呈白色固體之56
(331 mg,33%)。 1 H NMR
(400 MHz, CDCl3) δ 4.09-3.99 (m, 1H), 2.18-2.13 (m, 1H), 1.99-1.78 (m, 4H), 1.75-1.59 (m, 3H), 1.50-1.3 (m, 7H), 1.34-1.22 (m, 6H), 1.21-1.00 (m, 10H), 0.96 (s, 3H), 0.94-0.89 (m, 3H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.184 min,30-90AB_E,純度100%,C25
H40
F3
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 57 : (3S,5R,8R,9S,10S,13S,14S,17R)-3,10,13- 三甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (57) 之合成  1. 在10℃下於N2
下攪拌LiCl (13.9 g,329 mmol,無水)於THF (500 mL,無水)中之懸浮液30分鐘。在10℃下添加FeCl3
(27.8 g,172 mmol,無水)。使混合物冷卻至-30℃。在-30℃下向混合物中滴落添加MeMgBr (209 mL,3M於二乙醚中)。在-30℃下攪拌10分鐘之後,在-30℃下添加S-500-2-12_1
(50 g,157 mmol)。在-15℃下攪拌混合物2小時且用檸檬酸(500 mL,10%水溶液)淬滅。用EtOAc (3×800 mL)萃取混合物。合併之有機相用飽和鹽水(300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,其藉由矽膠管柱(PE/DCM/EtOAc=1/1/1)純化,得到呈固體之S-500-2-12_2
(50 g,86%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.57-2.48 (m, 1H), 2.23-2.13 (m, 1H), 2.06-1.78 (m, 3H), 1.64-1.25 (m, 14H), 1.24-1.01 (m, 10H), 0.96 (s, 3H), 0.74 (s, 1H), 0.60 (s, 3H)。 2. 在20℃下向PPh3
MeBr (79.7 g,244 mmol)於THF (400 mL)中之懸浮液中添加t-BuOK (25.1 g,224 mmol)。在40℃下攪拌30 min之後,在40℃下添加S-500-2-12_2
(50 g,150 mmol)於THF (100 mL)中之溶液且在40℃下攪拌反應混合物1小時。將反應混合物倒入至50 g冰中且攪拌15分鐘。分離有機層且用用EtOAc (3×50 mL)萃取水相。在真空中濃縮合併之有機相,得到油狀物。粗產物在MeOH/H2
O (200 mL/200 mL)中經濕磨且過濾,得到呈固體之S-500-2-12_3
(55 g,88%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.69 (s, 1H), 2.06-1.79 (m, 4H), 1.75 (s, 3H), 1.73-1.58 (m, 4H), 1.56-1.25 (m, 9H), 1.22 (s, 3H), 1.21-1.02 (m, 6H), 1.01-0.94 (s, 3H), 0.93-0.74 (m, 1H), 0.55 (s, 3H)。3.
向S-500-2-12_3
(55 g,166 mmol)於THF (500 mL)中之溶液中添加9-BBN二聚體(60.7 g,249 mmol)且在25℃下於N2
下攪拌1小時,形成固體。向反應混合物添加乙醇(95.3 mL,1.66 mmol)及NaOH (166 mL,5 M,830 mmol)。使混合物變得澄清。在25℃下逐滴添加H2
O2
(132 mL,10 M,1.32 mmol)且使內部溫度升至回流(75℃)。添加且攪拌16小時之後使混合物冷卻,形成固體。在25℃下向混合物中添加Na2
S2O3
(500 mL,20%水溶液)及水(1 L)。攪拌混合物1小時。在關閉攪拌器之後,形成澄清底層及上懸浮液層。丟棄澄清底層。向上懸浮液層添加水(2 L)。攪拌混合物15分鐘。過濾混合物,得到呈固體之S-500-2-12_4
(50 g,不純)。在100℃下在EtOH/H2
O (90 mL/10 mL)中濕磨S-500-2-12_4
(50 g,143 mmol,不純)2小時,接著使其冷卻至15℃且過濾,得到呈固體之S-500-2-12_4
(38 g,不純)。在100℃下在EtOH/H2
O (45 mL/5 mL)中濕磨S-500-2-12_4
(38 g,109 mmol,不純)2小時,接著使其冷卻至15℃且過濾,得到呈固體之S-500-2-12_4
(28 g,43%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.67-3.59 (m, 1H), 3.39-3.32 (m, 1H), 2.01-1.75 (m, 4H), 1.69-1.58 (m, 3H), 1.54-1.24 (m, 10H), 1.23-1.14 (m, 9H), 1.09-1.02 (m, 5H), 0.96 (s, 3H), 0.74 (m, 1H), 0.67 (s, 3H)。 4. 向N-004-016_1
(10.0 g,28.6 mmol)於DCM (100 mL)中之溶液中添加DMP (24.2 g,57.2mmol)。接著將H2
O (0.2 mL)添加至混合物。此後,在25℃下攪拌反應物1小時。向反應混合物中添加飽和NaHCO3
水溶液 (100 mL)。過濾混合物且用DCM (2×100 mL)洗滌濾餅。分離混合物,且用DCM (2×100 mL)萃取水相。合併之有機層用飽和NaHCO3
/Na2
S2O3
水溶液(100 mL/100 mL)及鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到白色固體。殘餘物藉由矽膠層析(PE/EtOAc=0%至20%)純化,得到呈白色固體之N-004-016_2
(3.5 g,35%)。 1 H NMR
(400 MHz, CDCl3) δ 9.58-9.54 (m, 1H), 2.39-2.32 (m, 1H), 1.96-1.77 (m, 4H), 1.69-1.31 (m, 14H), 1.23-1.16 (m, 6H), 1.14-1.02 (m, 5H), 0.96 (s, 3H), 0.76-0.59 (m, 4H)。 5. 在0℃下向N-004-016_2
(1.5 g,4.32 mmol)、CsF (328 mg,2.16 mmol)於THF (10 mL)中之溶液中添加TMSCF3
(1.53 g,10.8 mmol) 。在25℃下攪拌混合物1小時。向混合物中添加TBAF.3
H2
O (3.4 g,10.8 mmol)。在25℃下攪拌混合物2小時。混合物用水(20 mL)淬滅且用EtOAc (2×30 mL)萃取。合併之有機相用鹽水(50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之N-004-017_1
(700 mg,39%)。 1 H NMR
(400 MHz, CDCl3) δ 4.07-3.96 (m, 1H), 2.21-2.09 (m, 1H), 1.99-1.77 (m, 5H), 1.72-1.29 (m, 15H), 1.24-1.20 (m, 4H), 1.13-1.01 (m, 5H), 0.96 (s, 3H), 0.89-0.84 (m, 1H), 0.76-0.64 (m, 3H), 0.60 (s, 1H)。 6. 在25℃下向N-004-017_1
(700 mg,1.68 mmol)於吡啶(5 mL)中之溶液中添加苯甲醯氯(354 mg,2.52 mmol)及DMAP (102 mg,0.84 mmol)。將混合物加熱至60℃且攪拌10小時。反應混合物用EtOAc (10 mL)稀釋,接著用水(10 mL)淬滅。用EtOAc (2×20 mL)萃取水溶液。合併之有機層用鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到白色固體。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之N-004-017_2
(600 mg,68%)。 在2 min層析中,LCMS
Rt
=1.464 min,30-90AB_2MIN_E.M,純度92%。 SFC條件:峰值 1
:Rt=2.434 min且在8 min層析中峰值 2
:Rt=2.541 min,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M (管柱:Chiralcel OD -3 100×4.6 mm I.D.,3um行動相:A:CO2 B:甲醇(0.05% DEA),梯度:在4.5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續1 min,流速:2.8 mL/min,min管柱溫度:40℃)。7. N-004-017_2
(600 mg,1.15 mmol) 藉由SFC (管柱:Chiralcel OD (250 mm×30 mm,5 um),梯度:20%至20% B(A=0.1% NH3
/H2
O,B=MeOH),流速:60 mL/min)純化,得到N-004-017_3
(峰值2,190 mg,不純,31%)、呈白色固體之N-004-018_1
(峰值1,180 mg,30%)。不純N-004-017_3
(190 mg,0.36 mmol) 藉由SFC (管柱:OD (250 mm×30 mm,5 um),梯度:20%至20% B (A=0.1% NH3
/H2
O,B=MeOH),流速:60 mL/min)純化,得到呈白色固體之N-004-017_3
(100 mg,53%)。N-004-018_1 : 1 H NMR
(400 MHz, CDCl3) δ 8.12-8.06 (m, 2H), 7.65-7.59 (m, 1H), 7.53-7.46 (m, 2H), 5.68-5.58 (m, 1H), 2.15-2.03 (m, 2H), 1.97-1.69 (m, 3H), 1.67-1.57 (m, 3H), 1.44-1.24 (m, 9H), 1.23-1.13 (m, 11H), 1.12-1.97 (m, 3H), 0.94 (s, 3H), 0.72 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.525 min,30-90AB_2min_220&254.lcm,純度100%。 在8 min層析中,SFC _D1
Rt = 2.450 min,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M,100%de。N-004-017_3 : 1 H NMR
(400 MHz, CDCl3) δ 8.14-8.06 (m, 2H), 7.64-7.57 (m, 1H), 7.52-7.44 (m, 2H), 5.63-5.52 (m, 1H), 2.11 (s, 1H), 2.06-1.77 (m, 6H), 1.72-1.62 (m, 3H), 1.44-1.32 (m, 8H), 1.28-1.18 (m, 12H), 0.99-0.93 (m, 4H), 0.65 (s, 3H)。 在2 min層析中,LCMS
Rt = 1.529 min,30-90AB_2min_220&254.lcm,純度100%。 在8 min層析中,SFC
Rt = 2.544 mi,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M,98%de。 8. 向N-004-018_1
(180 mg,0.34 mmol)於THF (3 mL)及MeOH (1.5 mL)及水(1.5 mL)中之溶液中添加KOH (96.5 mg,1.72 mmol)。在60℃下攪拌混合物16小時。將混合物倒入水(20 mL)中且用EtOAc (2×40 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之57
(114 mg,79%)。 1 H NMR
(400 MHz, CDCl3) δ 4.00 (brs, 1H), 2.03-1.77 (m, 8H), 1.69-1.61 (m, 2H), 1.54-1.49 (m, 1H), 1.47-1.24 (m, 10H), 1.22 (s, 3H), 1.21-1.10 (m, 4H), 1.08-1.01 (m, 4H), 0.96 (s, 3H), 0.69 (s, 3H)。 在2 min層析中,LCMS
Rt = 1.169 min,30-90AB_2MIN_E.M,純度95%,C24
H38
F3
O[M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10 min最終C18 3×50 mm 3 um中,HPLC
Rt = 5.44 min,30-90_AB_1.2ML_E.MET,純度100%。實例 58 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1R,2S)-1- 環戊基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (58) 之合成
1. 在10℃下於N2
下攪拌LiCl (13.9 g,329 mmol,無水)於THF (500 mL,無水)中之懸浮液30分鐘。在10℃下添加FeCl3
(27.8 g,172 mmol,無水)。使混合物冷卻至-30℃。在-30℃下向混合物中滴落添加MeMgBr (209 mL,3M於二乙醚中)。在-30℃下攪拌10分鐘之後,在-30℃下添加S-500-2-12_1
(50 g,157 mmol)。在-15℃下攪拌混合物2小時且用檸檬酸(500 mL,10%水溶液)淬滅。用EtOAc (3×800 mL)萃取混合物。合併之有機相用飽和鹽水(300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,其藉由矽膠管柱(PE/DCM/EtOAc=1/1/1)純化,得到呈固體之S-500-2-12_2
(50 g,86%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.57-2.48 (m, 1H), 2.23-2.13 (m, 1H), 2.06-1.78 (m, 3H), 1.64-1.25 (m, 14H), 1.24-1.01 (m, 10H), 0.96 (s, 3H), 0.74 (s, 1H), 0.60 (s, 3H)。 2. 在20℃下向PPh3
MeBr (79.7 g,244 mmol)於THF (400 mL)中之懸浮液中添加t-BuOK (25.1 g,224 mmol)。在40℃下攪拌30 min之後,在40℃下添加S-500-2-12_2
(50 g,150 mmol)於THF (100 mL)中之溶液且在40℃下攪拌反應混合物1小時。將反應混合物倒入至50 g冰中且攪拌15分鐘。分離有機層且用用EtOAc (3×50 mL)萃取水相。在真空中濃縮合併之有機相,得到油狀物。粗產物在MeOH/H2
O (200 mL/200 mL)中經濕磨且過濾,得到呈固體之S-500-2-12_3
(55 g,88%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.69 (s, 1H), 2.06-1.79 (m, 4H), 1.75 (s, 3H), 1.73-1.58 (m, 4H), 1.56-1.25 (m, 9H), 1.22 (s, 3H), 1.21-1.02 (m, 6H), 1.01-0.94 (s, 3H), 0.93-0.74 (m, 1H), 0.55 (s, 3H)。3.
向S-500-2-12_3
(55 g,166 mmol)於THF (500 mL)中之溶液中添加9-BBN二聚體(60.7 g,249 mmol)且在25℃下於N2
下攪拌1小時,形成固體。向反應混合物添加乙醇(95.3 mL,1.66 mmol)及NaOH (166 mL,5 M,830 mmol)。使混合物變得澄清。在25℃下逐滴添加H2
O2
(132 mL,10 M,1.32 mmol)且使內部溫度升至回流(75℃)。添加且攪拌16小時之後使混合物冷卻,形成固體。在25℃下向混合物中添加Na2
S2O3
(500 mL,20%水溶液)及水(1 L)。攪拌混合物1小時。在關閉攪拌器之後,形成澄清底層及上懸浮液層。丟棄澄清底層。向上懸浮液層添加水(2 L)。攪拌混合物15分鐘。過濾混合物,得到呈固體之S-500-2-12_4
(50 g,不純)。在100℃下在EtOH/H2
O (90 mL/10 mL)中濕磨S-500-2-12_4
(50 g,143 mmol,不純)2小時,接著使其冷卻至15℃且過濾,得到呈固體之S-500-2-12_4
(38 g,不純)。在100℃下在EtOH/H2
O (45 mL/5 mL)中濕磨S-500-2-12_4
(38 g,109 mmol,不純)2小時,接著使其冷卻至15℃且過濾,得到呈固體之S-500-2-12_4
(28 g,43%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.67-3.59 (m, 1H), 3.39-3.32 (m, 1H), 2.01-1.75 (m, 4H), 1.69-1.58 (m, 3H), 1.54-1.24 (m, 10H), 1.23-1.14 (m, 9H), 1.09-1.02 (m, 5H), 0.96 (s, 3H), 0.74 (m, 1H), 0.67 (s, 3H)。 4. 向N-004-016_1
(10.0 g,28.6 mmol)於DCM (100 mL)中之溶液中添加DMP (24.2 g,57.2mmol)。接著將H2
O (0.2 mL)添加至混合物。此後,在25℃下攪拌反應物1小時。向反應混合物中添加飽和NaHCO3
水溶液 (100 mL)。過濾混合物且用DCM (2×100 mL)洗滌濾餅。分離混合物,且用DCM (2×100 mL)萃取水相。合併之有機層用飽和NaHCO3
/Na2
S2O3
水溶液(100 mL/100 mL)及鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到白色固體。殘餘物藉由矽膠層析(PE/EtOAc=0%至20%)純化,得到呈白色固體之N-004-016_2
(3.5 g,35%)。 1 H NMR
(400 MHz, CDCl3) δ 9.58-9.54 (m, 1H), 2.39-2.32 (m, 1H), 1.96-1.77 (m, 4H), 1.69-1.31 (m, 14H), 1.23-1.16 (m, 6H), 1.14-1.02 (m, 5H), 0.96 (s, 3H), 0.76-0.59 (m, 4H)。 5. 在0℃下向N-004-016_2
(1.5 g,4.32 mmol)、CsF (328 mg,2.16 mmol)於THF (10 mL)中之溶液中添加TMSCF3
(1.53 g,10.8 mmol) 。在25℃下攪拌混合物1小時。向混合物中添加TBAF.3
H2
O (3.4 g,10.8 mmol)。在25℃下攪拌混合物2小時。混合物用水(20 mL)淬滅且用EtOAc (2×30 mL)萃取。合併之有機相用鹽水(50 mL)洗滌,經無水Na2
SO4
乾燥,過濾且在真空中濃縮。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之N-004-017_1
(700 mg,39%)。 1 H NMR
(400 MHz, CDCl3) δ 4.07-3.96 (m, 1H), 2.21-2.09 (m, 1H), 1.99-1.77 (m, 5H), 1.72-1.29 (m, 15H), 1.24-1.20 (m, 4H), 1.13-1.01 (m, 5H), 0.96 (s, 3H), 0.89-0.84 (m, 1H), 0.76-0.64 (m, 3H), 0.60 (s, 1H)。 6. 在25℃下向N-004-017_1
(700 mg,1.68 mmol)於吡啶(5 mL)中之溶液中添加苯甲醯氯(354 mg,2.52 mmol)及DMAP (102 mg,0.84 mmol)。將混合物加熱至60℃且攪拌10小時。反應混合物用EtOAc (10 mL)稀釋,接著用水(10 mL)淬滅。用EtOAc (2×20 mL)萃取水溶液。合併之有機層用鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到白色固體。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之N-004-017_2
(600 mg,68%)。 在2 min層析中,LCMS
Rt
=1.464 min,30-90AB_2MIN_E.M,純度92%。 SFC條件:峰值 1
:Rt=2.434 min且在8 min層析中峰值 2
:Rt=2.541 min,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M (管柱:Chiralcel OD -3 100×4.6 mm I.D.,3um行動相:A:CO2 B:甲醇(0.05% DEA),梯度:在4.5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續1 min,流速:2.8 mL/min,min管柱溫度:40℃)。7. N-004-017_2
(600 mg,1.15 mmol) 藉由SFC (管柱:Chiralcel OD (250 mm×30 mm,5 um),梯度:20%至20% B(A=0.1% NH3
/H2
O,B=MeOH),流速:60 mL/min)純化,得到N-004-017_3
(峰值2,190 mg,不純,31%)、呈白色固體之N-004-018_1
(峰值1,180 mg,30%)。不純N-004-017_3
(190 mg,0.36 mmol) 藉由SFC (管柱:OD (250 mm×30 mm,5 um),梯度:20%至20% B (A=0.1% NH3
/H2
O,B=MeOH),流速:60 mL/min)純化,得到呈白色固體之N-004-017_3
(100 mg,53%)。N-004-018_1 : 1 H NMR
(400 MHz, CDCl3) δ 8.12-8.06 (m, 2H), 7.65-7.59 (m, 1H), 7.53-7.46 (m, 2H), 5.68-5.58 (m, 1H), 2.15-2.03 (m, 2H), 1.97-1.69 (m, 3H), 1.67-1.57 (m, 3H), 1.44-1.24 (m, 9H), 1.23-1.13 (m, 11H), 1.12-1.97 (m, 3H), 0.94 (s, 3H), 0.72 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.525 min,30-90AB_2min_220&254.lcm,純度100%。 在8 min層析中,SFC _D1
Rt = 2.450 min,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M,100%de。N-004-017_3 : 1 H NMR
(400 MHz, CDCl3) δ 8.14-8.06 (m, 2H), 7.64-7.57 (m, 1H), 7.52-7.44 (m, 2H), 5.63-5.52 (m, 1H), 2.11 (s, 1H), 2.06-1.77 (m, 6H), 1.72-1.62 (m, 3H), 1.44-1.32 (m, 8H), 1.28-1.18 (m, 12H), 0.99-0.93 (m, 4H), 0.65 (s, 3H)。 在2 min層析中,LCMS
Rt = 1.529 min,30-90AB_2min_220&254.lcm,純度100%。 在8 min層析中,SFC
Rt = 2.544 mi,OD_MEOH(DEA)_5_40_2,8ML_8MIN.M,98%de。 8. 向N-004-018_1
(180 mg,0.34 mmol)於THF (3 mL)及MeOH (1.5 mL)及水(1.5 mL)中之溶液中添加KOH (96.5 mg,1.72 mmol)。在60℃下攪拌混合物16小時。將混合物倒入水(20 mL)中且用EtOAc (2×40 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至15% EtOAc/PE)純化,得到呈白色固體之57
(114 mg,79%)。 1 H NMR
(400 MHz, CDCl3) δ 4.00 (brs, 1H), 2.03-1.77 (m, 8H), 1.69-1.61 (m, 2H), 1.54-1.49 (m, 1H), 1.47-1.24 (m, 10H), 1.22 (s, 3H), 1.21-1.10 (m, 4H), 1.08-1.01 (m, 4H), 0.96 (s, 3H), 0.69 (s, 3H)。 在2 min層析中,LCMS
Rt = 1.169 min,30-90AB_2MIN_E.M,純度95%,C24
H38
F3
O[M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10 min最終C18 3×50 mm 3 um中,HPLC
Rt = 5.44 min,30-90_AB_1.2ML_E.MET,純度100%。實例 58 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1R,2S)-1- 環戊基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (58) 之合成  1.
在0℃下於N2
下將含N-8-7_1
(500 mg, 1.38 mmol)之THF (5 mL)添加至環戊基溴化鎂(1.38 mL,3 M於THF中)。在15℃下攪拌18小時之後,反應混合物用飽和NH4
Cl (10 mL)淬滅,且用EtOAc (2×10 mL)萃取。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之N-8-15_1
(170 mg,29%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.39-3.22 (m, 1H), 2.00-1.81 (m, 4H), 1.70-1.41 (m, 12H), 1.41-1.13 (m, 13H), 1.13-0.95 (m, 6H), 0.95-0.79 (m, 11H), 0.65 (s, 3H)。 2. 將DMP (0.881 g,2.08 mmol)添加至N-8-15_1
(300 mg,0.696 mmol)於DCM (20 mL)中之溶液中。在15℃下攪拌10 min之後,反應混合物用飽和NaHCO3
(10 mL)淬滅。混合物用DCM (3×20 mL)萃取。合併之有機相用飽和Na2
S2
O3
(3×20 mL)及鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至20% EtOAc/PE)純化,得到呈固體之N-8-15_2
(240 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 2.98-2.89 (m, 1H), 2.62-2.55 (m, 1H), 1.98-1.87 (m, 1H), 1.81-1.72 (m, 4H), 1.71-1.49 (m, 10H), 1.41-1.29 (m, 4H), 1.29-1.19 (m, 6H), 1.14-0.98 (m, 9H), 0.94-0.87 (m, 4H), 0.82 (s, 3H), 0.67 (m, 5H)。3.
向N-8-15_2
(240 mg,0.559 mmol)於MeOH (3 mL)及THF (2 mL)中之混合物添加NaBH4 (550 mg,14.5 mmol)。在15℃下攪拌混合物0.5 h。添加另一批NaBH4
(550 mg,14.5 mmol)。再攪拌反應混合物1 h。向反應混合物中添加水(5 mL)。用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之58
(7 mg,5%),且78
(50 mg,不純)藉由閃蒸塔(0-5% EtOAc/PE)進一步純化,得到呈固體之78
(17 mg,12%)。58 : 1 H NMR
(400 MHz, CDCl3
) δ 3.64-3.59 (m, 1H), 2.09-1.90 (m, 2H), 1.89-1.70 (m, 4H), 1.70-1.45 (m, 11H), 1.45-1.32 (m, 5H), 1.32-1.19 (m, 9H), 1.19-1.08 (m, 3H),1.08-0.98 (m, 5 H), 0.98-0.89 (m, 4 H), 0.84 (s, 3H), 0.68 (s, 3H)。 在7 min層析中,LCMS
Rt=4.832 min,30-90AB_7MIN_E,純度100%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。 在10 min層析中,HPLC
Rt=6.338 min,50-100AB_10MIN.M,純度98%。實例 59 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (59) 之合成
1.
在0℃下於N2
下將含N-8-7_1
(500 mg, 1.38 mmol)之THF (5 mL)添加至環戊基溴化鎂(1.38 mL,3 M於THF中)。在15℃下攪拌18小時之後,反應混合物用飽和NH4
Cl (10 mL)淬滅,且用EtOAc (2×10 mL)萃取。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0%至10% EtOAc/PE)純化,得到呈固體之N-8-15_1
(170 mg,29%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.39-3.22 (m, 1H), 2.00-1.81 (m, 4H), 1.70-1.41 (m, 12H), 1.41-1.13 (m, 13H), 1.13-0.95 (m, 6H), 0.95-0.79 (m, 11H), 0.65 (s, 3H)。 2. 將DMP (0.881 g,2.08 mmol)添加至N-8-15_1
(300 mg,0.696 mmol)於DCM (20 mL)中之溶液中。在15℃下攪拌10 min之後,反應混合物用飽和NaHCO3
(10 mL)淬滅。混合物用DCM (3×20 mL)萃取。合併之有機相用飽和Na2
S2
O3
(3×20 mL)及鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0%至20% EtOAc/PE)純化,得到呈固體之N-8-15_2
(240 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 2.98-2.89 (m, 1H), 2.62-2.55 (m, 1H), 1.98-1.87 (m, 1H), 1.81-1.72 (m, 4H), 1.71-1.49 (m, 10H), 1.41-1.29 (m, 4H), 1.29-1.19 (m, 6H), 1.14-0.98 (m, 9H), 0.94-0.87 (m, 4H), 0.82 (s, 3H), 0.67 (m, 5H)。3.
向N-8-15_2
(240 mg,0.559 mmol)於MeOH (3 mL)及THF (2 mL)中之混合物添加NaBH4 (550 mg,14.5 mmol)。在15℃下攪拌混合物0.5 h。添加另一批NaBH4
(550 mg,14.5 mmol)。再攪拌反應混合物1 h。向反應混合物中添加水(5 mL)。用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之58
(7 mg,5%),且78
(50 mg,不純)藉由閃蒸塔(0-5% EtOAc/PE)進一步純化,得到呈固體之78
(17 mg,12%)。58 : 1 H NMR
(400 MHz, CDCl3
) δ 3.64-3.59 (m, 1H), 2.09-1.90 (m, 2H), 1.89-1.70 (m, 4H), 1.70-1.45 (m, 11H), 1.45-1.32 (m, 5H), 1.32-1.19 (m, 9H), 1.19-1.08 (m, 3H),1.08-0.98 (m, 5 H), 0.98-0.89 (m, 4 H), 0.84 (s, 3H), 0.68 (s, 3H)。 在7 min層析中,LCMS
Rt=4.832 min,30-90AB_7MIN_E,純度100%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。 在10 min層析中,HPLC
Rt=6.338 min,50-100AB_10MIN.M,純度98%。實例 59 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (59) 之合成  1. 將Pd(OH)2
(200 mg,無水)添加至S-500-6-30
(140 mg,0.288 mmol)於MeOH (30 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到S-500-6-25
(27 mg,19%)及呈固體之S-500-6-26
(42 mg,30%)。S-500-6-25 : 1 H NMR
(400 MHz, CDCl3
) δ 3.84-3.77 (m, 1H), 1.99-1.84 (m, 2H), 1.81-1.72 (m, 1H), 1.68-1.56 (m, 4H), 1.53-1.43 (m, 5H), 1.42-1.32 (m, 9H), 1.31-1.23 (m, 5H), 1.22-.12 (m, 7H), 1.12-1.00 (m, 5H), 0.99-0.95 (m, 4H), 0.94-0.85 (m, 12H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.797 min,30-90AB_E,純度100%,C33
H55
[M+H-2H2
O]+
之MS ESI計算值451,實驗值451。實例 60 : (3S,5S,8R,9S,10S,13S,14S,17R)-3 乙基 -17-((2S,3R)-3- 羥基 -4-((1R,2S)-2- 甲基環丙基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (60) 之合成
1. 將Pd(OH)2
(200 mg,無水)添加至S-500-6-30
(140 mg,0.288 mmol)於MeOH (30 mL)中之溶液中。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0%至15% EtOAc/PE)純化,得到S-500-6-25
(27 mg,19%)及呈固體之S-500-6-26
(42 mg,30%)。S-500-6-25 : 1 H NMR
(400 MHz, CDCl3
) δ 3.84-3.77 (m, 1H), 1.99-1.84 (m, 2H), 1.81-1.72 (m, 1H), 1.68-1.56 (m, 4H), 1.53-1.43 (m, 5H), 1.42-1.32 (m, 9H), 1.31-1.23 (m, 5H), 1.22-.12 (m, 7H), 1.12-1.00 (m, 5H), 0.99-0.95 (m, 4H), 0.94-0.85 (m, 12H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.797 min,30-90AB_E,純度100%,C33
H55
[M+H-2H2
O]+
之MS ESI計算值451,實驗值451。實例 60 : (3S,5S,8R,9S,10S,13S,14S,17R)-3 乙基 -17-((2S,3R)-3- 羥基 -4-((1R,2S)-2- 甲基環丙基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (60) 之合成 
 1.
在0℃下向N-8-7_2
(1 g,2.41 mmol)於THF (50 mL)中之溶液添加LiAlH4
(914 mg,24.1 mmol)。在66℃下加熱灰色懸浮液18小時。將反應混合物冷卻至0℃,藉由冰水(914 mg)、接著15% w/w NaOH水溶液(914 mg)及水(2.74 g)淬滅。過濾混合物且濾餅用DCM (3 × 50 mL)洗滌。濃縮濾液,得到殘餘物,其藉由急驟層析(乙酸乙酯,10%於PE中)純化兩次,得到N-8-7_3
(192 mg,19%)及呈油狀物之N-8-7_3A
(397 mg,39%)。N-8-7_3 : 1 H NMR
(400 MHz, CDCl3) δ 5.59 - 5.36 (m, 2H), 3.69 - 3.61 (m, 1H), 2.25 - 2.12 (m, 1H), 2.08 - 1.81 (m, 3H), 1.68 (d, J = 10.0 Hz, 3H), 1.64 - 1.54 (m, 9H), 1.53 - 1.15 (m, 11H), 1.14 - 0.92 (m, 5H), 0.92 - 0.85 (m, 5H), 0.83 (s, 4H), 0.69 - 0.60 (m, 4H)。N-8-7_3A : 1 H NMR
(400 MHz, CDCl3
) δ 5.62 - 5.37 (m, 2H), 3.62 (br d,J
=10.0 Hz, 1H), 2.20 - 2.06 (m, 1H), 1.99 - 1.61 (m, 6H), 1.61 - 1.44 (m, 11H), 1.43 - 1.18 (m, 5H), 1.16 - 0.94 (m, 6H), 0.94 - 0.85 (m, 5H), 0.82 (s, 5H), 0.70 - 0.58 (m, 6H)。2.
在0℃下經15 min之時段向二乙基鋅(1 M於甲苯中,4.31 mL,4.31 mmol)於0℃下之DCM (15 ml )中之溶液添加CH2
I2
(2.31 g,8.63 mmol)。在0℃下攪拌乳白色懸浮液10 min,且經由注射器快速添加Charette配位體((4R,5R)-2-(第三丁基)-N4,N4,N5,N5-四甲基-1,3,2-二氧硼戊環-4,5-二甲醯胺) (233 mg,0.8638 mmol)及N-8-7_3A
(300 mg,0.7199 mmol)於DCM (20 ml)中之預製溶液,隨即反應混合物變為澄清。使溶液達至25℃且在此溫度下攪拌16 h。接著藉由添加飽和NH4
Cl水溶液(150 ml)淬滅反應,分離各相且用DCM (3×100 ml)萃取水相。合併之有機相用飽和NaHCO3
水溶液(150 mL)、飽和Na2
S2
O3
水溶液(150 mL)及鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在減壓下濃縮,且殘餘物藉由閃蒸塔層析(11%乙酸乙酯/PE)純化,得到呈固體之N-8-7_4A
(140 mg,45%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.81-3.72 (m, 1H), 1.97-1.90 (m, 1H), 1.76-1.58 (m, 4H), 1.53-1.44 (m, 4H), 1.43 - 1.15 (m, 10H), 1.13-0.91 (m, 8H), 0.90-0.78 (m, 12H), 0.66 (s, 3H), 0.54-0.34 (m, 4H), 0.32-0.22 (m, 2H), 0.19-0.12 (m, 1H)。 3. 在25℃下向N-8-7_4A
(140 mg,0.325 mmol)於吡啶(5 mL)中之溶液中添加苯甲醯氯(91.3 mg,0.65 mmol),隨後添加DMAP (15.8 mg,0.13 mmol)。在60℃下攪拌反應混合物16小時。用DCM (80 mL)稀釋反應混合物。DCM相位用水(100 mL)、1.0 M HCl水溶液(2×100 mL)、10% NaHCO3
(2×100 mL)水溶液、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到油狀物,其藉由閃蒸塔(1%乙酸乙酯/PE)純化,得到呈油狀物之N-8-7_5A
(180 mg,不純),其藉由閃蒸塔(PE)進一步純化,得到呈固體之N-8-7_5A
(110 mg,61%)。 在2 min層析中,LCMS
Rt=1.439 min,5-95AB_220&254,純度93%,C36
H53
O2
[M-H2
O+H]+
之MS ESI計算值517.8,實驗值517.8。SFC
峰值1:Rt=4.079 min,且在10 min層析中峰值2:Rt=4.345 min,AD_3_EtOH_DEA_5_40_25ML (「Chiralpak AD-3 150×4.6 mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。4. N-8-7A_5
(110 mg,0.206 mmol)藉由SFC (管柱:AD (250 mm×30 mm,5 um),梯度:30%至30% B (A=0.1% NH3
/H2
O,B=EtOH),流速:60 mL/min)純化,得到呈固體之不純N-8-7A_6
(峰值1,54 mg,50%)及呈固體之不純N-8-8A_1
(峰值2,23 mg,不純)。 在10 min層析中,SFC
Rt=4.088 min,AD_3_EtOH_DEA_5_40_25ML,100%de。5.
向N-8-7A_6
(54 mg,0.101 mmol)於THF/MeOH (1.5 mL/1.5 mL)中之溶液添加含KOH (45.2 mg,0.807 mmol)之水(0.5 mL)。在50℃下攪拌反應混合物16小時。向混合物中添加HCl (0.2 M,50 mL)。用DCM (2 × 60 mL)萃取懸浮液。合併之有機相用3% NaHCO3
水溶液(80 mL)、鹽水(80 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到固體,其藉由快速層析(15%乙酸乙酯/PE)純化,得到呈固體之60
(21 mg,48%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.80-3.75 (m, 1H), 1.97-1.91 (m, 1H), 1.76-1.58 (m, 7H), 1.54-1.27 (m, 7H), 1.26-1.06 (m, 7H), 1.04 (d,J
= 6.0 Hz, 4H), 0.95 (s, 3H), 0.90-0.84 (m, 8H), 0.82 (s, 4H), 0.66 (s, 3H), 0.64-0.59 (m, 1H), 0.52-0.37 (m, 2H), 0.32-0.22 (m, 2H)。 在2 min層析中,LCMS
Rt=1.327 min,30-90AB_2MIN_E,純度100%,C29
H49
O [M-H2
O+H]+
之MS ESI計算值413.4,實驗值413.4。實例 61 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (61) 之合成
1.
在0℃下向N-8-7_2
(1 g,2.41 mmol)於THF (50 mL)中之溶液添加LiAlH4
(914 mg,24.1 mmol)。在66℃下加熱灰色懸浮液18小時。將反應混合物冷卻至0℃,藉由冰水(914 mg)、接著15% w/w NaOH水溶液(914 mg)及水(2.74 g)淬滅。過濾混合物且濾餅用DCM (3 × 50 mL)洗滌。濃縮濾液,得到殘餘物,其藉由急驟層析(乙酸乙酯,10%於PE中)純化兩次,得到N-8-7_3
(192 mg,19%)及呈油狀物之N-8-7_3A
(397 mg,39%)。N-8-7_3 : 1 H NMR
(400 MHz, CDCl3) δ 5.59 - 5.36 (m, 2H), 3.69 - 3.61 (m, 1H), 2.25 - 2.12 (m, 1H), 2.08 - 1.81 (m, 3H), 1.68 (d, J = 10.0 Hz, 3H), 1.64 - 1.54 (m, 9H), 1.53 - 1.15 (m, 11H), 1.14 - 0.92 (m, 5H), 0.92 - 0.85 (m, 5H), 0.83 (s, 4H), 0.69 - 0.60 (m, 4H)。N-8-7_3A : 1 H NMR
(400 MHz, CDCl3
) δ 5.62 - 5.37 (m, 2H), 3.62 (br d,J
=10.0 Hz, 1H), 2.20 - 2.06 (m, 1H), 1.99 - 1.61 (m, 6H), 1.61 - 1.44 (m, 11H), 1.43 - 1.18 (m, 5H), 1.16 - 0.94 (m, 6H), 0.94 - 0.85 (m, 5H), 0.82 (s, 5H), 0.70 - 0.58 (m, 6H)。2.
在0℃下經15 min之時段向二乙基鋅(1 M於甲苯中,4.31 mL,4.31 mmol)於0℃下之DCM (15 ml )中之溶液添加CH2
I2
(2.31 g,8.63 mmol)。在0℃下攪拌乳白色懸浮液10 min,且經由注射器快速添加Charette配位體((4R,5R)-2-(第三丁基)-N4,N4,N5,N5-四甲基-1,3,2-二氧硼戊環-4,5-二甲醯胺) (233 mg,0.8638 mmol)及N-8-7_3A
(300 mg,0.7199 mmol)於DCM (20 ml)中之預製溶液,隨即反應混合物變為澄清。使溶液達至25℃且在此溫度下攪拌16 h。接著藉由添加飽和NH4
Cl水溶液(150 ml)淬滅反應,分離各相且用DCM (3×100 ml)萃取水相。合併之有機相用飽和NaHCO3
水溶液(150 mL)、飽和Na2
S2
O3
水溶液(150 mL)及鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在減壓下濃縮,且殘餘物藉由閃蒸塔層析(11%乙酸乙酯/PE)純化,得到呈固體之N-8-7_4A
(140 mg,45%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.81-3.72 (m, 1H), 1.97-1.90 (m, 1H), 1.76-1.58 (m, 4H), 1.53-1.44 (m, 4H), 1.43 - 1.15 (m, 10H), 1.13-0.91 (m, 8H), 0.90-0.78 (m, 12H), 0.66 (s, 3H), 0.54-0.34 (m, 4H), 0.32-0.22 (m, 2H), 0.19-0.12 (m, 1H)。 3. 在25℃下向N-8-7_4A
(140 mg,0.325 mmol)於吡啶(5 mL)中之溶液中添加苯甲醯氯(91.3 mg,0.65 mmol),隨後添加DMAP (15.8 mg,0.13 mmol)。在60℃下攪拌反應混合物16小時。用DCM (80 mL)稀釋反應混合物。DCM相位用水(100 mL)、1.0 M HCl水溶液(2×100 mL)、10% NaHCO3
(2×100 mL)水溶液、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且在真空中濃縮,得到油狀物,其藉由閃蒸塔(1%乙酸乙酯/PE)純化,得到呈油狀物之N-8-7_5A
(180 mg,不純),其藉由閃蒸塔(PE)進一步純化,得到呈固體之N-8-7_5A
(110 mg,61%)。 在2 min層析中,LCMS
Rt=1.439 min,5-95AB_220&254,純度93%,C36
H53
O2
[M-H2
O+H]+
之MS ESI計算值517.8,實驗值517.8。SFC
峰值1:Rt=4.079 min,且在10 min層析中峰值2:Rt=4.345 min,AD_3_EtOH_DEA_5_40_25ML (「Chiralpak AD-3 150×4.6 mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,接著B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。4. N-8-7A_5
(110 mg,0.206 mmol)藉由SFC (管柱:AD (250 mm×30 mm,5 um),梯度:30%至30% B (A=0.1% NH3
/H2
O,B=EtOH),流速:60 mL/min)純化,得到呈固體之不純N-8-7A_6
(峰值1,54 mg,50%)及呈固體之不純N-8-8A_1
(峰值2,23 mg,不純)。 在10 min層析中,SFC
Rt=4.088 min,AD_3_EtOH_DEA_5_40_25ML,100%de。5.
向N-8-7A_6
(54 mg,0.101 mmol)於THF/MeOH (1.5 mL/1.5 mL)中之溶液添加含KOH (45.2 mg,0.807 mmol)之水(0.5 mL)。在50℃下攪拌反應混合物16小時。向混合物中添加HCl (0.2 M,50 mL)。用DCM (2 × 60 mL)萃取懸浮液。合併之有機相用3% NaHCO3
水溶液(80 mL)、鹽水(80 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到固體,其藉由快速層析(15%乙酸乙酯/PE)純化,得到呈固體之60
(21 mg,48%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.80-3.75 (m, 1H), 1.97-1.91 (m, 1H), 1.76-1.58 (m, 7H), 1.54-1.27 (m, 7H), 1.26-1.06 (m, 7H), 1.04 (d,J
= 6.0 Hz, 4H), 0.95 (s, 3H), 0.90-0.84 (m, 8H), 0.82 (s, 4H), 0.66 (s, 3H), 0.64-0.59 (m, 1H), 0.52-0.37 (m, 2H), 0.32-0.22 (m, 2H)。 在2 min層析中,LCMS
Rt=1.327 min,30-90AB_2MIN_E,純度100%,C29
H49
O [M-H2
O+H]+
之MS ESI計算值413.4,實驗值413.4。實例 61 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (61) 之合成  1.向S-500-6-11_1
(580 mg,1.44 mmol)於DCM (30 mL)中之溶液添加DMP (1.22 g,2.88 mmol)。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈白色固體之粗產物S-500-6-11_1A
(550 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.34-5.25 (m, 1H), 2.58-2.29 (m, 4H), 2.08-1.90 (m, 3H), 1.78-1.56 (m, 9H), 1.54-1.35 (m, 6H), 1.31-1.21 (m, 2H), 1.19-1.08 (m, 5H), 1.06-0.99 (m, 5H), 0.93-0.82 (m, 6H), 0.69 (s, 3H)。 2.以五個兩分鐘間隔向S-500-6-11A
(550 mg,1.37 mmol)於THF (4 mL)及MeOH (2 mL)中之溶液添加NaBH4
(1.39 g,41.1 mmol)。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (20 mL)淬滅,且用EtOAc (3 × 6 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈白色固體之不純61
(120 mg),其再藉由combi-flash (0-15% EtOAc/PE)進一步純化,得到呈白色固體之純61
(150 mg,75%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.25 (m, 1H), 3.73-3.59 (m, 1H), 2.44-2.29 (m, 1H), 2.08-1.92 (m, 3H),1.76-1.57 (m, 6H), 1.54-1.26 (m, 10H), 1.25-1.18 (m, 3H), 1.17-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.88 (m, 8H), 0.87-0.82 (m, 3H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.345 min,30-90AB_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。實例 62 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (62) 之合成
1.向S-500-6-11_1
(580 mg,1.44 mmol)於DCM (30 mL)中之溶液添加DMP (1.22 g,2.88 mmol)。此後,在15℃下攪拌反應混合物10 min。反應混合物用NaHCO3
飽和水溶液(50 mL)淬滅直至含水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 40 mL)、飽和NaHCO3
(40 mL)、鹽水(40 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到呈白色固體之粗產物S-500-6-11_1A
(550 mg,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 5.34-5.25 (m, 1H), 2.58-2.29 (m, 4H), 2.08-1.90 (m, 3H), 1.78-1.56 (m, 9H), 1.54-1.35 (m, 6H), 1.31-1.21 (m, 2H), 1.19-1.08 (m, 5H), 1.06-0.99 (m, 5H), 0.93-0.82 (m, 6H), 0.69 (s, 3H)。 2.以五個兩分鐘間隔向S-500-6-11A
(550 mg,1.37 mmol)於THF (4 mL)及MeOH (2 mL)中之溶液添加NaBH4
(1.39 g,41.1 mmol)。在15℃下攪拌混合物30分鐘。混合物用飽和NH4
Cl (20 mL)淬滅,且用EtOAc (3 × 6 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈白色固體之不純61
(120 mg),其再藉由combi-flash (0-15% EtOAc/PE)進一步純化,得到呈白色固體之純61
(150 mg,75%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.25 (m, 1H), 3.73-3.59 (m, 1H), 2.44-2.29 (m, 1H), 2.08-1.92 (m, 3H),1.76-1.57 (m, 6H), 1.54-1.26 (m, 10H), 1.25-1.18 (m, 3H), 1.17-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.88 (m, 8H), 0.87-0.82 (m, 3H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.345 min,30-90AB_E,純度100%,C27
H45
O [M+H-H2
O]+
之MS ESI計算值385,實驗值385。實例 62 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (62) 之合成 
 1.在0℃下向S-500-6-1_1
(500 mg,1.39 mmol)及CsF (105 mg,695 umol)於THF (5 mL)中之溶液添加TMSCF3
(493 mg,3.47mmol)。在25℃下攪拌混合物1小時且用TBAF.3H2
O (1.09 g,3.47 mmol)處理。在25℃下攪拌混合物2小時,用水(100 mL)淬滅且用EtOAc (2 × 50 mL)萃取。合併之有機相用鹽水(100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE:EA=10:1)純化,得到呈白色固體之S-500-6-1_2
(400 mg,67%)。 1 H NMR
(400 MHz, CDCl3) δ 5.33-5.24 (m, 1H), 4.06-4.00 (m, 1H), 2.38-2.35 (m, 1H), 2.08-1.82 (m, 6H), 1.77-1.69 (m, 1H), 1.62-1.20 (m, 13H), 1.16-1.00 (m, 8H), 0.99-0.92 (m, 1H), 0.87-0.83 (m, 4H), 0.74-0.64 (m, 3H)。 2. 3.5 gS-500-6-1_2
藉由SFC (管柱:AD (250mm×30mm,5um),梯度:40-40% B (A=0.05% NH3
/H2
O,B=MeOH),流速:200 mL/min)分離,得到純81
(1 g,28%,峰值1)及呈白色固體之62
(1871 mg,53%,峰值2)。62 : 1 H NMR
(400 MHz, CDCl3) δ 5.30-5.28 (m, 1H), 4.03-3.99 (m, 1H), 2.38-2.34 (m, 1H), 2.10-1.83 (m, 6H), 1.78-1.55 (m, 5H), 1.52-1.32 (m, 6H), 1.31-1.01 (m, 12H), 0.98-0.92 (s, 1H), 0.85 (t,J
= 8 Hz, 3H), 0.73 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.219 min,30-90 AB,純度100%,C25
H38
F3
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,SFC
峰值2:Rt=5.262 min,AD_3_EtOH_DEA_5_40_25ML,99%de。實例 63 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4-( 四氫 -2H- 哌喃 -4- 基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (63) 之合成
1.在0℃下向S-500-6-1_1
(500 mg,1.39 mmol)及CsF (105 mg,695 umol)於THF (5 mL)中之溶液添加TMSCF3
(493 mg,3.47mmol)。在25℃下攪拌混合物1小時且用TBAF.3H2
O (1.09 g,3.47 mmol)處理。在25℃下攪拌混合物2小時,用水(100 mL)淬滅且用EtOAc (2 × 50 mL)萃取。合併之有機相用鹽水(100 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE:EA=10:1)純化,得到呈白色固體之S-500-6-1_2
(400 mg,67%)。 1 H NMR
(400 MHz, CDCl3) δ 5.33-5.24 (m, 1H), 4.06-4.00 (m, 1H), 2.38-2.35 (m, 1H), 2.08-1.82 (m, 6H), 1.77-1.69 (m, 1H), 1.62-1.20 (m, 13H), 1.16-1.00 (m, 8H), 0.99-0.92 (m, 1H), 0.87-0.83 (m, 4H), 0.74-0.64 (m, 3H)。 2. 3.5 gS-500-6-1_2
藉由SFC (管柱:AD (250mm×30mm,5um),梯度:40-40% B (A=0.05% NH3
/H2
O,B=MeOH),流速:200 mL/min)分離,得到純81
(1 g,28%,峰值1)及呈白色固體之62
(1871 mg,53%,峰值2)。62 : 1 H NMR
(400 MHz, CDCl3) δ 5.30-5.28 (m, 1H), 4.03-3.99 (m, 1H), 2.38-2.34 (m, 1H), 2.10-1.83 (m, 6H), 1.78-1.55 (m, 5H), 1.52-1.32 (m, 6H), 1.31-1.01 (m, 12H), 0.98-0.92 (s, 1H), 0.85 (t,J
= 8 Hz, 3H), 0.73 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.219 min,30-90 AB,純度100%,C25
H38
F3
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,SFC
峰值2:Rt=5.262 min,AD_3_EtOH_DEA_5_40_25ML,99%de。實例 63 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4-( 四氫 -2H- 哌喃 -4- 基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (63) 之合成  1.向N-6-10_1
(180 mg,0.318 mmol)於THF (2 mL)及甲醇(1 mL)中之溶液添加水(1 mL)及KOH (177 mg,3.17 mmol)。在50℃下攪拌混合物18小時。將反應混合物冷卻,用水(5 mL)稀釋,用10% HCl (0.2 mL)酸化且用EtOAc (3 × 5 mL)萃取。合併之有機層經無水硫酸鈉乾燥、過濾且濃縮。殘餘物藉由矽膠管柱層析(10-30% EtOAc/PE)純化,得到呈固體之63
(108 mg,74%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.90 (m, 2H), 3.85-3.77 (m, 1H), 3.44-3.33 (m, 2H), 1.99-1.82 (m, 2H), 1.69-1.58 (m, 6H), 1.57-1.45 (m, 6H), 1.43-1.29 (m, 7H), 1.28-1.14 (m, 7H), 1.13-0.95 (m, 5H), 0.93-0.84 (m, 7H), 0.83 (s, 3H), 0.69-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.167 min,30-90AB,純度100%。 C30
H49
O [M-2H2
O+H]+
之MS
ESI計算值425,實驗值425。實例 64 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (64) 之合成
1.向N-6-10_1
(180 mg,0.318 mmol)於THF (2 mL)及甲醇(1 mL)中之溶液添加水(1 mL)及KOH (177 mg,3.17 mmol)。在50℃下攪拌混合物18小時。將反應混合物冷卻,用水(5 mL)稀釋,用10% HCl (0.2 mL)酸化且用EtOAc (3 × 5 mL)萃取。合併之有機層經無水硫酸鈉乾燥、過濾且濃縮。殘餘物藉由矽膠管柱層析(10-30% EtOAc/PE)純化,得到呈固體之63
(108 mg,74%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.90 (m, 2H), 3.85-3.77 (m, 1H), 3.44-3.33 (m, 2H), 1.99-1.82 (m, 2H), 1.69-1.58 (m, 6H), 1.57-1.45 (m, 6H), 1.43-1.29 (m, 7H), 1.28-1.14 (m, 7H), 1.13-0.95 (m, 5H), 0.93-0.84 (m, 7H), 0.83 (s, 3H), 0.69-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.167 min,30-90AB,純度100%。 C30
H49
O [M-2H2
O+H]+
之MS
ESI計算值425,實驗值425。實例 64 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (64) 之合成 
 1.在-70℃下於N2
下向E-3_1
(20.0 g,63.1 mmol)於THF (300 mL)中之溶液緩慢添加EtMgBr (42 mL,126 mmol,3 M於醚中)。添加之後,在-70℃下攪拌混合物2小時。混合物用飽和NH4
Cl (500 mL)淬滅,且用EtOAc (3 ×500 mL)萃取。合併之有機相用鹽水(500 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之E-3_2
(6.50 g,30%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.57-2.49 (m, 1H), 2.23-1.80 (m, 6H), 1.78-1.52 (m, 4H), 1.50-1.02 (m, 17H), 0.97 (s, 3H), 0.95-0.80 (m, 4H), 0.60 (s, 3H)。 2.在15℃下於N2
下向MePPh3
Br (13.3 g,37.4 mmol)於THF (200 mL)中之懸浮液添加t-BuOK(4.19 g,37.4 mmol)。在50℃下攪拌混合物30 min。在50℃以下向混合物逐份添加E-3_2
(6.50 g,18.7 mmol)。在50℃下攪拌混合物1小時。向混合物添加NH4
Cl (400 mL)。有機層經分離且真空濃縮,得到粗產物,其在50℃下自MeOH/水(200 mL,1:1)濕磨而得。冷卻之後過濾混合物,且固體用MeOH/水(2 × 30 mL,1:1)洗滌並真空濃縮,得到呈固體之E-3_3
(5.8 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.06-1.97 (m, 1H), 1.94-1.55 (m, 12H), 1.53-1.05 (m, 16H), 0.97 (s, 3H), 0.95-0.85 (m, 3H), 0.55 (s, 3H)。 3.在15℃下於N2
下向E-3_3
(5.80 g,16.8 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(8.19 g,33.6 mmol)。在60℃下攪拌反應混合物1小時。將混合物冷卻至15℃。在15℃下添加乙醇(7.72 g,168 mmol)。在15℃下逐滴添加NaOH水溶液(33.6 mL,5 M,168 mmol)。在15℃下逐滴添加H2
O2
(16.8 mL,10.0 M,168 mmol)。在60℃下攪拌所得混合物1小時。用EtOAc (3 × 100 mL)萃取水相。合併之有機相用鹽水(2 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。在65℃下自CH3
OH/H2
O = 1/1 (150 mL)濕磨殘餘物,得到呈固體之E-3_4
(2.80 g,46%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.61 (m, 1H), 3.38-3.32 (m, 1H), 2.03-1.56 (m, 4H), 1.56-1.51 (m, 5H), 1.51-1.10 (m, 16H), 1.10-1.02 (m, 6H), 0.97 (s, 3H), 0.96-0.88 (m, 3H), 0.67 (s, 3H)。 4.向E-3_4
(2.50 g,6.89 mmol)於DCM (50 mL)中之溶液添加DMP (5.80 g,13.7 mmol)。此後,在20℃下攪拌反應物30 min。向反應混合物添加NaHCO3
飽和水溶液(50 mL)、Na2
S2
O3
飽和水溶液(30 mL),用DCM (2 × 20mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 10 mL)及鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之E-3_5
(2.45 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.60-9.55 (m, 1H), 2.26 (s, 1H), 1.98-1.70 (m, 6H), 1.70-1.51 (m, 6H), 1.51-1.00 (m, 12H), 1.00-0.89 (m, 10H), 1.75-0.65 (m, 4H)。 5.在0℃下於N2
下向E-3_5
(2.45 g,6.79 mmol)於THF (10 mL)中之溶液添加異丁基溴化鎂(33.9 mL,2 M於THF中,67.9 mmol)。在20℃下攪拌混合物16小時。向混合物添加NH4
Cl (20 mL,飽和水溶液),用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(20 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,且藉由閃蒸塔(0-20% EtOAc/PE)純化,得到呈固體之E-3_6
(1.6 g,55%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.65-3.55 (m, 1H), 2.01-1.85 (m, 3H), 1.85-1.49 (m, 3H), 1.49-1.36 (m, 12H), 1.36-1.22 (m, 10H), 1.22-1.02 (m, 9H), 1.02-0.98 (m, 4H), 0.98-0.80 (m, 7H), 0.66 (s, 3H)。6.
向E-3_6
(1.6 g,3.69 mmol)於DCM (30 mL)中之溶液添加DMP (3.12 g,7.38 mmol)。此後,在20℃下攪拌反應物30 min。向反應混合物添加NaHCO3
飽和水溶液(20 mL)、Na2
S2
O3
飽和水溶液(20 mL),接著用DCM (2 × 20 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 10 mL)及鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之E-3_7
(1.5 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 2.55-2.80 (m, 3H), 2.80-2.23 (m, 1H), 1.98-1.81 (m, 2H), 1.81-1.1.69 (m, 1H), 1.69-1.25 (m, 16H), 1.25-1.01 (m, 10H), 1.01-0.81 (m, 14H), 0.67 (s, 3H)。 7.在0℃下向E-3_7
(1.45 g,3.36 mmol)於MeOH (20 mL)中之溶液一次性添加NaBH4
(255 mg,6.72 mmol)。添加之後,在20℃下攪拌混合物1小時且用NH4
Cl (20 mL,飽和水溶液)淬滅。用DCM (2 × 20 mL)萃取混合物。合併之有機相用(2 × 10 mL)鹽水洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由閃蒸塔(0-20% EtOAc/PE)純化,得到呈固體之E-3_8
(1.2 g,83%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.80-3.70 (m, 1H), 3.70-3.55 (m, 1H), 2.05-1.82 (m, 3H), 1.82-1.55 (m, 4H), 1.55-1.35 (m, 5H), 1.35-1.00 (m, 18H), 1.00-0.79 (m, 17H), 0.66 (s, 3H)。 8.向E-3_8
(1.15 g,2.65 mmol)於吡啶(20 mL)中之溶液添加苯甲醯氯(1.85 g,13.2 mmol)。在20℃下攪拌反應混合物4小時。將反應混合物倒入水(20 mL)中。用EtOAc (2 × 20 mL)萃取混合物。合併之有機相用飽和鹽水(2 × 10 mL)洗滌,經無水Na2
SO4
乾燥,過濾濃縮且藉由閃蒸塔(0-10% EtOAc/PE)純化,得到呈固體之混合物產物(1.45 g,不純)。混合物產物(1.45 g,不純)藉由SFC (管柱:AD (250mm×50mm,10um),梯度:30-30% B (A=0.1% NH3/H2O,B=EtOH),流速:200 mL/min)純化,得到呈固體之E-3_9
(峰值2,470 mg,33%,DE%=100%)及呈固體之E-3_9A
(峰值1,600 mg,42%,DE%=99.1%)。E-3_9A : 1 H NMR
(400 MHz, CDCl3
) δ 8.10-7.99 (m, 2H), 7.60-7.50 (m, 1H), 7.50-7.38 (m, 2H), 5.15-5.05 (m, 1H), 2.05-1.70 (m, 6H), 1.70-1.35 (m, 5H), 1.35-1.05 (m, 19H), 1.05-0.82 (m, 17H), 0.65 (s, 3H)。 在10 min層析中,SFC
Rt=3.344 min,AD_3_EtOH_DEA_5_40_25ML,100%de。E-3_9 : 1 H NMR
(400 MHz, CDCl3
) δ 8.10-7.99 (m, 2H), 7.60-7.50 (m, 1H), 7.50-7.38 (m, 2H), 5.25-5.15 (m, 1H), 2.05-1.80 (m, 3H), 1.80-1.45 (m, 15H), 1.45-1.09 (m, 13H), 1.09-0.85 (m, 16H), 0.68 (s, 3H)。 在10 min層析中,SFC
Rt=3.851 min,AD_3_EtOH_DEA_5_40_25ML,99.1%de。 9.在25℃下向E-3_9A
(600 mg,1.11 mmol)於THF (2 mL)及MeOH (2 mL)中之溶液添加NaOH(531 mg,13.3 mmol)及H2
O (0.5 mL)。在50℃下攪拌溶液48小時。添加水(10 mL)。用EtOAc (2 × 10 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,其用MeCN (10 mL)濕磨,得到呈固體之所需產物69
(473 mg,99%) 。 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.55 (m, 1H), 2.01-1.85 (m, 3H), 1.85-1.70 (m, 1H), 1.70-1.45 (m, 8H), 1.45-1.22 (m, 13H), 1.22-1.05 (m, 8H), 1.05-1.86 (m, 15H), 0.66 (s, 3H)。 在2 min層析中,LCMS
tR=1.403 min,30-90AB_ELSD,純度100.0%,C29
H49
[M+H-2H2
O]+
之MS ESI計算值397,實驗值397。實例 65 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (65) 之合成
1.在-70℃下於N2
下向E-3_1
(20.0 g,63.1 mmol)於THF (300 mL)中之溶液緩慢添加EtMgBr (42 mL,126 mmol,3 M於醚中)。添加之後,在-70℃下攪拌混合物2小時。混合物用飽和NH4
Cl (500 mL)淬滅,且用EtOAc (3 ×500 mL)萃取。合併之有機相用鹽水(500 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之E-3_2
(6.50 g,30%)。 1 H NMR
(400 MHz, CDCl3
) δ 2.57-2.49 (m, 1H), 2.23-1.80 (m, 6H), 1.78-1.52 (m, 4H), 1.50-1.02 (m, 17H), 0.97 (s, 3H), 0.95-0.80 (m, 4H), 0.60 (s, 3H)。 2.在15℃下於N2
下向MePPh3
Br (13.3 g,37.4 mmol)於THF (200 mL)中之懸浮液添加t-BuOK(4.19 g,37.4 mmol)。在50℃下攪拌混合物30 min。在50℃以下向混合物逐份添加E-3_2
(6.50 g,18.7 mmol)。在50℃下攪拌混合物1小時。向混合物添加NH4
Cl (400 mL)。有機層經分離且真空濃縮,得到粗產物,其在50℃下自MeOH/水(200 mL,1:1)濕磨而得。冷卻之後過濾混合物,且固體用MeOH/水(2 × 30 mL,1:1)洗滌並真空濃縮,得到呈固體之E-3_3
(5.8 g,不純)。 1 H NMR
(400 MHz, CDCl3
) δ 4.84 (s, 1H), 4.70 (s, 1H), 2.06-1.97 (m, 1H), 1.94-1.55 (m, 12H), 1.53-1.05 (m, 16H), 0.97 (s, 3H), 0.95-0.85 (m, 3H), 0.55 (s, 3H)。 3.在15℃下於N2
下向E-3_3
(5.80 g,16.8 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(8.19 g,33.6 mmol)。在60℃下攪拌反應混合物1小時。將混合物冷卻至15℃。在15℃下添加乙醇(7.72 g,168 mmol)。在15℃下逐滴添加NaOH水溶液(33.6 mL,5 M,168 mmol)。在15℃下逐滴添加H2
O2
(16.8 mL,10.0 M,168 mmol)。在60℃下攪拌所得混合物1小時。用EtOAc (3 × 100 mL)萃取水相。合併之有機相用鹽水(2 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。在65℃下自CH3
OH/H2
O = 1/1 (150 mL)濕磨殘餘物,得到呈固體之E-3_4
(2.80 g,46%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.61 (m, 1H), 3.38-3.32 (m, 1H), 2.03-1.56 (m, 4H), 1.56-1.51 (m, 5H), 1.51-1.10 (m, 16H), 1.10-1.02 (m, 6H), 0.97 (s, 3H), 0.96-0.88 (m, 3H), 0.67 (s, 3H)。 4.向E-3_4
(2.50 g,6.89 mmol)於DCM (50 mL)中之溶液添加DMP (5.80 g,13.7 mmol)。此後,在20℃下攪拌反應物30 min。向反應混合物添加NaHCO3
飽和水溶液(50 mL)、Na2
S2
O3
飽和水溶液(30 mL),用DCM (2 × 20mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 10 mL)及鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之E-3_5
(2.45 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.60-9.55 (m, 1H), 2.26 (s, 1H), 1.98-1.70 (m, 6H), 1.70-1.51 (m, 6H), 1.51-1.00 (m, 12H), 1.00-0.89 (m, 10H), 1.75-0.65 (m, 4H)。 5.在0℃下於N2
下向E-3_5
(2.45 g,6.79 mmol)於THF (10 mL)中之溶液添加異丁基溴化鎂(33.9 mL,2 M於THF中,67.9 mmol)。在20℃下攪拌混合物16小時。向混合物添加NH4
Cl (20 mL,飽和水溶液),用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(20 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,且藉由閃蒸塔(0-20% EtOAc/PE)純化,得到呈固體之E-3_6
(1.6 g,55%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.65-3.55 (m, 1H), 2.01-1.85 (m, 3H), 1.85-1.49 (m, 3H), 1.49-1.36 (m, 12H), 1.36-1.22 (m, 10H), 1.22-1.02 (m, 9H), 1.02-0.98 (m, 4H), 0.98-0.80 (m, 7H), 0.66 (s, 3H)。6.
向E-3_6
(1.6 g,3.69 mmol)於DCM (30 mL)中之溶液添加DMP (3.12 g,7.38 mmol)。此後,在20℃下攪拌反應物30 min。向反應混合物添加NaHCO3
飽和水溶液(20 mL)、Na2
S2
O3
飽和水溶液(20 mL),接著用DCM (2 × 20 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 10 mL)及鹽水(20 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之E-3_7
(1.5 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 2.55-2.80 (m, 3H), 2.80-2.23 (m, 1H), 1.98-1.81 (m, 2H), 1.81-1.1.69 (m, 1H), 1.69-1.25 (m, 16H), 1.25-1.01 (m, 10H), 1.01-0.81 (m, 14H), 0.67 (s, 3H)。 7.在0℃下向E-3_7
(1.45 g,3.36 mmol)於MeOH (20 mL)中之溶液一次性添加NaBH4
(255 mg,6.72 mmol)。添加之後,在20℃下攪拌混合物1小時且用NH4
Cl (20 mL,飽和水溶液)淬滅。用DCM (2 × 20 mL)萃取混合物。合併之有機相用(2 × 10 mL)鹽水洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由閃蒸塔(0-20% EtOAc/PE)純化,得到呈固體之E-3_8
(1.2 g,83%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.80-3.70 (m, 1H), 3.70-3.55 (m, 1H), 2.05-1.82 (m, 3H), 1.82-1.55 (m, 4H), 1.55-1.35 (m, 5H), 1.35-1.00 (m, 18H), 1.00-0.79 (m, 17H), 0.66 (s, 3H)。 8.向E-3_8
(1.15 g,2.65 mmol)於吡啶(20 mL)中之溶液添加苯甲醯氯(1.85 g,13.2 mmol)。在20℃下攪拌反應混合物4小時。將反應混合物倒入水(20 mL)中。用EtOAc (2 × 20 mL)萃取混合物。合併之有機相用飽和鹽水(2 × 10 mL)洗滌,經無水Na2
SO4
乾燥,過濾濃縮且藉由閃蒸塔(0-10% EtOAc/PE)純化,得到呈固體之混合物產物(1.45 g,不純)。混合物產物(1.45 g,不純)藉由SFC (管柱:AD (250mm×50mm,10um),梯度:30-30% B (A=0.1% NH3/H2O,B=EtOH),流速:200 mL/min)純化,得到呈固體之E-3_9
(峰值2,470 mg,33%,DE%=100%)及呈固體之E-3_9A
(峰值1,600 mg,42%,DE%=99.1%)。E-3_9A : 1 H NMR
(400 MHz, CDCl3
) δ 8.10-7.99 (m, 2H), 7.60-7.50 (m, 1H), 7.50-7.38 (m, 2H), 5.15-5.05 (m, 1H), 2.05-1.70 (m, 6H), 1.70-1.35 (m, 5H), 1.35-1.05 (m, 19H), 1.05-0.82 (m, 17H), 0.65 (s, 3H)。 在10 min層析中,SFC
Rt=3.344 min,AD_3_EtOH_DEA_5_40_25ML,100%de。E-3_9 : 1 H NMR
(400 MHz, CDCl3
) δ 8.10-7.99 (m, 2H), 7.60-7.50 (m, 1H), 7.50-7.38 (m, 2H), 5.25-5.15 (m, 1H), 2.05-1.80 (m, 3H), 1.80-1.45 (m, 15H), 1.45-1.09 (m, 13H), 1.09-0.85 (m, 16H), 0.68 (s, 3H)。 在10 min層析中,SFC
Rt=3.851 min,AD_3_EtOH_DEA_5_40_25ML,99.1%de。 9.在25℃下向E-3_9A
(600 mg,1.11 mmol)於THF (2 mL)及MeOH (2 mL)中之溶液添加NaOH(531 mg,13.3 mmol)及H2
O (0.5 mL)。在50℃下攪拌溶液48小時。添加水(10 mL)。用EtOAc (2 × 10 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物,其用MeCN (10 mL)濕磨,得到呈固體之所需產物69
(473 mg,99%) 。 1 H NMR
(400 MHz, CDCl3
) δ 3.68-3.55 (m, 1H), 2.01-1.85 (m, 3H), 1.85-1.70 (m, 1H), 1.70-1.45 (m, 8H), 1.45-1.22 (m, 13H), 1.22-1.05 (m, 8H), 1.05-1.86 (m, 15H), 0.66 (s, 3H)。 在2 min層析中,LCMS
tR=1.403 min,30-90AB_ELSD,純度100.0%,C29
H49
[M+H-2H2
O]+
之MS ESI計算值397,實驗值397。實例 65 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- 甲基戊 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (65) 之合成 
 N-8-11_1
(30 mg,0.0741 mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之65
(9 mg,30%)。65 : 1 H NMR
(400 MHz, CDCl3
) δ 3.18-3.07 (m, 1H), 1.98-1.81 (m, 2H), 1.71-1.58 (m, 6H), 1.53-1.31 (m, 7H), 1.30-0.98 (m, 14H), 0.97-0.78 (m, 14H), 0.70-0.60 (m, 4H)。 在7.0 min層析中,LCMS
Rt=4.387 min,30-90AB_7MIN_E,純度97.6%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 66 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- (2- 甲基環丙基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (66) 之合成
N-8-11_1
(30 mg,0.0741 mmol,不純)藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之65
(9 mg,30%)。65 : 1 H NMR
(400 MHz, CDCl3
) δ 3.18-3.07 (m, 1H), 1.98-1.81 (m, 2H), 1.71-1.58 (m, 6H), 1.53-1.31 (m, 7H), 1.30-0.98 (m, 14H), 0.97-0.78 (m, 14H), 0.70-0.60 (m, 4H)。 在7.0 min層析中,LCMS
Rt=4.387 min,30-90AB_7MIN_E,純度97.6%,C27
H45
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 66 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4- (2- 甲基環丙基 ) 丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (66) 之合成  1.
在0℃下向N-8-7_2
(1 g,2.41 mmol)於THF (50 mL)中之溶液添加LiAlH4
(914 mg,24.1 mmol)。在66℃下加熱灰色懸浮液18小時。將反應混合物冷卻至0℃,藉由冰水(914 mg)、接著15% w/w NaOH水溶液(914 mg)、水(2.74 g)淬滅。過濾混合物且濾餅用DCM (3 × 50 mL)洗滌。濃縮濾液,得到殘餘物,其藉由急驟層析(乙酸乙酯,10%於PE中)純化兩次,得到N-8-7_3
(192 mg,19%)及呈油狀物之N-8-7_3A
(397 mg,39%)。N-8-7_3 :
1H NMR (400 MHz, CDCl3) δ 5.59 - 5.36 (m, 2H), 3.69 - 3.61 (m, 1H), 2.25 - 2.12 (m, 1H), 2.08 - 1.81 (m, 3H), 1.68 (d, J = 10.0 Hz, 3H), 1.64 - 1.54 (m, 9H), 1.53 - 1.15 (m, 11H), 1.14 - 0.92 (m, 5H), 0.92 - 0.85 (m, 5H), 0.83 (s, 4H), 0.69 - 0.60 (m, 4H)。N-8-7_3A : 1 H NMR
(400 MHz, CDCl3
) δ 5.62 - 5.37 (m, 2H), 3.62 (br d,J
=10.0 Hz, 1H), 2.20 - 2.06 (m, 1H), 1.99 - 1.61 (m, 6H), 1.61 - 1.44 (m, 11H), 1.43 - 1.18 (m, 5H), 1.16 - 0.94 (m, 6H), 0.94 - 0.85 (m, 5H), 0.82 (s, 5H), 0.70 - 0.58 (m, 6H)。 2.在0℃下經15 min之時段向二乙基鋅(1 M於甲苯中,2.59 mL,2.59 mmol)於DCM (10 ml )中之溶液添加CH2
I2
(1.38 g,5.18 mmol)。在0℃下攪拌乳白色懸浮液10 min,且經由注射器快速添加Charette配位體((4R,5R)-2-(第三丁基)-N4,N4,N5,N5-四甲基-1,3,2-二氧硼戊環-4,5-二甲醯胺) (139 mg,0.5182 mmol)及N-8-7_3
(180 mg,0.4319 mmol)於DCM (15 mL)中之預製溶液,隨即反應混合物變為澄清。將溶液升溫至25℃且在25℃下攪拌16 h。接著藉由添加NH4
Cl飽和水溶液(150 ml)來淬滅反應物。分離各相且用DCM (3 × 60 ml)萃取水相。合併之有機相用NaHCO3
飽和水溶液(150 mL)、Na2
S2
O3
飽和水溶液(150 mL)及鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且減壓濃縮,且殘餘物藉由閃蒸塔層析(11% 乙酸乙酯/PE)純化,得到呈固體之N-8-7_4
(60 mg,32%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.77 (br s, 1H), 1.99-1.87 (m, 2H), 1.70-1.56 (m, 6H), 1.54-1.34 (m, 6H), 1.31-1.16 (m, 5H), 1.15-1.05 (m, 1H), 1.15-0.97 (m, 7H), 0.99-0.94 (m, 1H), 0.92-0.77 (m, 10H), 0.69-0.58 (m, 6H), 0.52-0.13 (m, 5H)。 3.在25℃下向N-8-7_4A
(60 mg,0.1393 mmol)於吡啶(3 mL)中之溶液添加苯甲醯氯(39.1 mg,0.2786 mmol)隨後添加DMAP (6.79 mg,0.05572 mmol)。在60℃下攪拌反應混合物16小時。用DCM (80 mL)稀釋反應混合物。DCM相用水(100 mL)、1.0 M HCl水溶液(2 × 100 mL)、10% NaHCO3
水溶液(2 × 100 mL)、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到油狀物,其藉由閃蒸塔(1% 乙酸乙酯/PE)純化,得到呈油狀物之N-8-7_5
(24 mg,32%)。 在2 min層析中,LCMS
Rt=1.431 min,5-95AB_220&254,純度90%,C36
H53
O2
[M-H2
O+H]+
之MS ESI計算值517.3,實驗值517.3。 在10 min層析中,SFC
峰值1:Rt = 5.703 min,AD_3_EtOH_DEA_5_40_25ML (「Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。4. N-8-7_5
(24 mg,0.04487 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:40-40% B (A=0.1% NH3
/H2
O,B=EtOH),流速:50 mL/min)純化,得到呈固體之不純N-8-7_6
(RT:5.732,19 mg,不純)。未獲得異構體。 在2 min層析中,LCMS
Rt=1.435 min,5-95AB_220&254,純度98%,C36
H53
O2
[M-H2
O+H]+
之MS ESI計算值517.3,實驗值517.3。 在10 min層析中,SFC
Rt=5.732 min,AD_3_EtOH_DEA_5_40_25ML,97.76%de。 5.向N-8-7_6
(19 mg,0.0355 mmol)於THF/MeOH (0.5 ml/0.5 mL)中之溶液添加含KOH (19.8 mg,0.3552 mmol)之水(0.2 mL)。在55℃下攪拌反應混合物16小時。向混合物添加HCl (0.2 M,50 mL)。用DCM (2 × 60 mL)萃取懸浮液。合併之有機相用3% NaHCO3
水溶液(80 mL)及鹽水(80 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到固體,其藉由急驟層析(乙酸乙酯/PE,15%)純化,得到呈固體之66
(2 mg,13%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.77 (br s, 1H), 1.98-1.87 (m, 2H), 1.70-1.57 (m, 7H), 1.54 - 1.28 (m, 11H), 1.26 - 0.94 (m, 12H), 0.91 - 0.84 (m, 7H), 0.83 (s, 3H), 0.72 - 0.60 (m, 4H), 0.51 - 0.15 (m, 3H)。 在2 min層析中,LCMS
Rt=1.315 min,30-90AB_2MIN_E,純度100%,C29
H49
O [M-H2
O+H]+
之MS ESI計算值395.3,實驗值395.3。實例 67 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (67) 之合成
1.
在0℃下向N-8-7_2
(1 g,2.41 mmol)於THF (50 mL)中之溶液添加LiAlH4
(914 mg,24.1 mmol)。在66℃下加熱灰色懸浮液18小時。將反應混合物冷卻至0℃,藉由冰水(914 mg)、接著15% w/w NaOH水溶液(914 mg)、水(2.74 g)淬滅。過濾混合物且濾餅用DCM (3 × 50 mL)洗滌。濃縮濾液,得到殘餘物,其藉由急驟層析(乙酸乙酯,10%於PE中)純化兩次,得到N-8-7_3
(192 mg,19%)及呈油狀物之N-8-7_3A
(397 mg,39%)。N-8-7_3 :
1H NMR (400 MHz, CDCl3) δ 5.59 - 5.36 (m, 2H), 3.69 - 3.61 (m, 1H), 2.25 - 2.12 (m, 1H), 2.08 - 1.81 (m, 3H), 1.68 (d, J = 10.0 Hz, 3H), 1.64 - 1.54 (m, 9H), 1.53 - 1.15 (m, 11H), 1.14 - 0.92 (m, 5H), 0.92 - 0.85 (m, 5H), 0.83 (s, 4H), 0.69 - 0.60 (m, 4H)。N-8-7_3A : 1 H NMR
(400 MHz, CDCl3
) δ 5.62 - 5.37 (m, 2H), 3.62 (br d,J
=10.0 Hz, 1H), 2.20 - 2.06 (m, 1H), 1.99 - 1.61 (m, 6H), 1.61 - 1.44 (m, 11H), 1.43 - 1.18 (m, 5H), 1.16 - 0.94 (m, 6H), 0.94 - 0.85 (m, 5H), 0.82 (s, 5H), 0.70 - 0.58 (m, 6H)。 2.在0℃下經15 min之時段向二乙基鋅(1 M於甲苯中,2.59 mL,2.59 mmol)於DCM (10 ml )中之溶液添加CH2
I2
(1.38 g,5.18 mmol)。在0℃下攪拌乳白色懸浮液10 min,且經由注射器快速添加Charette配位體((4R,5R)-2-(第三丁基)-N4,N4,N5,N5-四甲基-1,3,2-二氧硼戊環-4,5-二甲醯胺) (139 mg,0.5182 mmol)及N-8-7_3
(180 mg,0.4319 mmol)於DCM (15 mL)中之預製溶液,隨即反應混合物變為澄清。將溶液升溫至25℃且在25℃下攪拌16 h。接著藉由添加NH4
Cl飽和水溶液(150 ml)來淬滅反應物。分離各相且用DCM (3 × 60 ml)萃取水相。合併之有機相用NaHCO3
飽和水溶液(150 mL)、Na2
S2
O3
飽和水溶液(150 mL)及鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且減壓濃縮,且殘餘物藉由閃蒸塔層析(11% 乙酸乙酯/PE)純化,得到呈固體之N-8-7_4
(60 mg,32%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.77 (br s, 1H), 1.99-1.87 (m, 2H), 1.70-1.56 (m, 6H), 1.54-1.34 (m, 6H), 1.31-1.16 (m, 5H), 1.15-1.05 (m, 1H), 1.15-0.97 (m, 7H), 0.99-0.94 (m, 1H), 0.92-0.77 (m, 10H), 0.69-0.58 (m, 6H), 0.52-0.13 (m, 5H)。 3.在25℃下向N-8-7_4A
(60 mg,0.1393 mmol)於吡啶(3 mL)中之溶液添加苯甲醯氯(39.1 mg,0.2786 mmol)隨後添加DMAP (6.79 mg,0.05572 mmol)。在60℃下攪拌反應混合物16小時。用DCM (80 mL)稀釋反應混合物。DCM相用水(100 mL)、1.0 M HCl水溶液(2 × 100 mL)、10% NaHCO3
水溶液(2 × 100 mL)、鹽水(100 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到油狀物,其藉由閃蒸塔(1% 乙酸乙酯/PE)純化,得到呈油狀物之N-8-7_5
(24 mg,32%)。 在2 min層析中,LCMS
Rt=1.431 min,5-95AB_220&254,純度90%,C36
H53
O2
[M-H2
O+H]+
之MS ESI計算值517.3,實驗值517.3。 在10 min層析中,SFC
峰值1:Rt = 5.703 min,AD_3_EtOH_DEA_5_40_25ML (「Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。4. N-8-7_5
(24 mg,0.04487 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:40-40% B (A=0.1% NH3
/H2
O,B=EtOH),流速:50 mL/min)純化,得到呈固體之不純N-8-7_6
(RT:5.732,19 mg,不純)。未獲得異構體。 在2 min層析中,LCMS
Rt=1.435 min,5-95AB_220&254,純度98%,C36
H53
O2
[M-H2
O+H]+
之MS ESI計算值517.3,實驗值517.3。 在10 min層析中,SFC
Rt=5.732 min,AD_3_EtOH_DEA_5_40_25ML,97.76%de。 5.向N-8-7_6
(19 mg,0.0355 mmol)於THF/MeOH (0.5 ml/0.5 mL)中之溶液添加含KOH (19.8 mg,0.3552 mmol)之水(0.2 mL)。在55℃下攪拌反應混合物16小時。向混合物添加HCl (0.2 M,50 mL)。用DCM (2 × 60 mL)萃取懸浮液。合併之有機相用3% NaHCO3
水溶液(80 mL)及鹽水(80 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到固體,其藉由急驟層析(乙酸乙酯/PE,15%)純化,得到呈固體之66
(2 mg,13%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.77 (br s, 1H), 1.98-1.87 (m, 2H), 1.70-1.57 (m, 7H), 1.54 - 1.28 (m, 11H), 1.26 - 0.94 (m, 12H), 0.91 - 0.84 (m, 7H), 0.83 (s, 3H), 0.72 - 0.60 (m, 4H), 0.51 - 0.15 (m, 3H)。 在2 min層析中,LCMS
Rt=1.315 min,30-90AB_2MIN_E,純度100%,C29
H49
O [M-H2
O+H]+
之MS ESI計算值395.3,實驗值395.3。實例 67 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (67) 之合成  1.向32
(80 mg,0.18 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(160 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到67
(10 mg,12%)及呈固體之84
(30 mg,37%)。67 : 1 H NMR
(400 MHz, CDCl3
) δ 3.76-3.66 (m, 1H), 2.01-1.78 (m, 5H), 1.76-1.58 (m, 7H), 1.52-1.31 (m, 13H), 1.28-1.10 (m, 10H), 1.09-0.99 (m, 4H), 0.96 (s, 3H), 0.93-0.86 (m, 6H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.508 min,30-90AB_E,純度100%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 68 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基庚 -5- 炔 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (68) 之合成
1.向32
(80 mg,0.18 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(160 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到67
(10 mg,12%)及呈固體之84
(30 mg,37%)。67 : 1 H NMR
(400 MHz, CDCl3
) δ 3.76-3.66 (m, 1H), 2.01-1.78 (m, 5H), 1.76-1.58 (m, 7H), 1.52-1.31 (m, 13H), 1.28-1.10 (m, 10H), 1.09-0.99 (m, 4H), 0.96 (s, 3H), 0.93-0.86 (m, 6H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.508 min,30-90AB_E,純度100%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 68 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基庚 -5- 炔 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (68) 之合成  1.粗產物N-8-7_2B
(250 mg,0.868 mmol)藉由SFC (管柱:AD (250 mm×30 mm,10 um),梯度:35-35% B (A=0.1% NH3
/H2
O,B=EtOH),流速:60 mL/min)進一步純化,得到呈固體之41
(峰值2,81 mg,33%)及呈固體之68
(峰值1,78 mg,31%)。68 : 1 H NMR
(400 MHz, CDCl3
) δ 3.87-3.78 (m, 1H), 2.21-2.12 (m, 1H), 1.99-1.86 (m, 2H), 1.80 (s, 3H), 1.73-1.51 (m, 8H), 1.51-1.42 (m, 4H), 1.42-1.20 (m, 8H), 1.20-0.95 (m,7H), 0.95-0.79 (m, 8H), 0.95 (s, 4H)。 在2 min層析中,LCMS
Rt=1.188 min,30-90AB_2MIN_E,純度100%,C28
H45
O [M+H-H2
O]+
之MS ESI計算值397,實驗值397。 在10 min層析中,SFC
Rt=6.465 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 69 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-5- 環丙基 -3- 羥基戊 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (69) 之合成
1.粗產物N-8-7_2B
(250 mg,0.868 mmol)藉由SFC (管柱:AD (250 mm×30 mm,10 um),梯度:35-35% B (A=0.1% NH3
/H2
O,B=EtOH),流速:60 mL/min)進一步純化,得到呈固體之41
(峰值2,81 mg,33%)及呈固體之68
(峰值1,78 mg,31%)。68 : 1 H NMR
(400 MHz, CDCl3
) δ 3.87-3.78 (m, 1H), 2.21-2.12 (m, 1H), 1.99-1.86 (m, 2H), 1.80 (s, 3H), 1.73-1.51 (m, 8H), 1.51-1.42 (m, 4H), 1.42-1.20 (m, 8H), 1.20-0.95 (m,7H), 0.95-0.79 (m, 8H), 0.95 (s, 4H)。 在2 min層析中,LCMS
Rt=1.188 min,30-90AB_2MIN_E,純度100%,C28
H45
O [M+H-H2
O]+
之MS ESI計算值397,實驗值397。 在10 min層析中,SFC
Rt=6.465 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 69 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-5- 環丙基 -3- 羥基戊 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (69) 之合成  1.在15℃下向S-500-6-19_3
(700 mg,1.63 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液逐份添加NaBH4
(2.46 g,65.1 mmol)。在15℃下攪拌20 min之後,用NH4
Cl (20 mL,飽和水溶液)淬滅混合物且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到呈固體之760 mg混合物,其藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到69
(330 mg,47%)及呈固體之6
(250 mg,35%,不純)。不純6
(250 mg)進一步藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到呈固體之6
(170 mg,23%)。69 : 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.23 (m, 1H), 3.75-3.63 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.85 (m, 4H), 1.78-1.59 (m, 5H), 1.53-1.38 (m, 9H), 1.38-1.05 (m, 9H), 1.03 (s, 3H), 1.00-0.91 (m, 1H), 0.91 (d,J
= 6.4 Hz, 3H) 0.85 (t,J
= 7.6 Hz, 3H), 0.69 (s, 3H), 0.68-0.60 (m, 1H), 0.45-0.36 (m, 2H), 0.09--0.08 (m, 2H)。 在2.0 min層析中,LCMS
Rt=1.387 min,30-90_AB_E,純度98.1%,C29
H47
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。S-500-6-19 : 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.24 (m, 1H), 3.77-3.66 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.91 (m, 3H), 1.79-1.59 (m, 6H), 1.55-1.21 (m, 14H), 1.21-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.95 (m, 1H), 0.93 (d,J
= 6.8 Hz, 3H) 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H), 0.68-0.62 (m, 1H), 0.49-0.38 (m, 2H), 0.11-0.02 (m, 2H)。 在2.0 min層析中,LCMS
Rt=1.380 min,30-90_AB_E,純度100%,C29
H47
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。實例 70 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4-(3- 甲基氧雜環丁 -3- 基 ) 丁 -2- 基 )-10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (70) 之合成
1.在15℃下向S-500-6-19_3
(700 mg,1.63 mmol)於THF (5 mL)及MeOH (5 mL)中之溶液逐份添加NaBH4
(2.46 g,65.1 mmol)。在15℃下攪拌20 min之後,用NH4
Cl (20 mL,飽和水溶液)淬滅混合物且用EtOAc (50 mL)萃取。有機層經分離且真空濃縮,得到呈固體之760 mg混合物,其藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到69
(330 mg,47%)及呈固體之6
(250 mg,35%,不純)。不純6
(250 mg)進一步藉由閃蒸塔(0-35% DCM/EtOAc (1/1)/PE)分離,得到呈固體之6
(170 mg,23%)。69 : 1 H NMR
(400 MHz, CDCl3
) δ 5.33-5.23 (m, 1H), 3.75-3.63 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.85 (m, 4H), 1.78-1.59 (m, 5H), 1.53-1.38 (m, 9H), 1.38-1.05 (m, 9H), 1.03 (s, 3H), 1.00-0.91 (m, 1H), 0.91 (d,J
= 6.4 Hz, 3H) 0.85 (t,J
= 7.6 Hz, 3H), 0.69 (s, 3H), 0.68-0.60 (m, 1H), 0.45-0.36 (m, 2H), 0.09--0.08 (m, 2H)。 在2.0 min層析中,LCMS
Rt=1.387 min,30-90_AB_E,純度98.1%,C29
H47
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。S-500-6-19 : 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.24 (m, 1H), 3.77-3.66 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.91 (m, 3H), 1.79-1.59 (m, 6H), 1.55-1.21 (m, 14H), 1.21-1.06 (m, 4H), 1.03 (s, 3H), 1.00-0.95 (m, 1H), 0.93 (d,J
= 6.8 Hz, 3H) 0.85 (t,J
= 7.6 Hz, 3H), 0.70 (s, 3H), 0.68-0.62 (m, 1H), 0.49-0.38 (m, 2H), 0.11-0.02 (m, 2H)。 在2.0 min層析中,LCMS
Rt=1.380 min,30-90_AB_E,純度100%,C29
H47
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。實例 70 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -4-(3- 甲基氧雜環丁 -3- 基 ) 丁 -2- 基 )-10,13- 二甲基 - 2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (70) 之合成 
 1.在50℃至55℃下於N2
下向Mg (807 mg,33.2 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液逐滴添加N-014-005_3
(2.5 g,15.1 mmol)於THF (8 mL)中之溶液。在55℃下攪拌混合物1 h。用THF (10 mL)稀釋混合物,且在不經監測的情況下直接用於下一步驟中。在0℃下向N-14-12_4
(1.01 g,2.83 mmol)於THF (10 mL)中之溶液添加新鮮製備的3-[(溴鎂)甲基]-3-甲基環氧丙烷(15 mmol於20 ml THF中)。在15℃下攪拌混合物4 h。向混合物添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。分離有機層且真空濃縮。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈白色固體之混合物
(190 mg,15%),其藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3H2O ETOH,梯度:50%至50%,流速(ml/min):60mL/min,25℃)純化,得到33
(峰值1,110 mg,9%)及呈白色固體之70
(峰值2,30mg,不純)。不純70
(30 mg,不純)藉由矽膠管柱層析(15% EtOAc/PE)純化,得到呈白色固體之70
(10 mg,5%)。33 : 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 4.59-4.70 (m, 1H), 4.50-4.48 (m, 1H),4.36-4.33 (m, 1H), 3.83 (s, 1H), 2.40-2.33 (m, 1H), 2.10-1.50 (m, 17H), 1.49-1.35 (m, 9H), 1.30-0.80 (m, 13H), 0.68 (s, 3H)。 在3 min層析中,LCMS
Rt=1.069 min,30-90AB_2MIN_E.M,純度100%,C29
H49
O3
[M+H]+
之MS ESI計算值445,實驗值445。70 : 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 4.10-4.02 (m, 1H), 3.81-3.70 (m, 1H), 3.50-3.41 (m, 2H), 3.34-3.30 (m, 1H), 2.35-2.31 (m, 1H), 2.10-1.50 (m, 18H), 1.49-1.05 (m, 13H), 1.05-0.90 (m, 4H), 0.90-0.80 (m, 3H), 0.67 (s, 3H)。 在3 min層析中,LCMS
Rt=1.115 min,30-90AB_2MIN_E.M,純度100%,C29
H49
O3
[M+H]+
之MS ESI計算值445,實驗值445。實例 71 : (3S,5R,8R,9S,10S,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 - 17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (71) 之合成
1.在50℃至55℃下於N2
下向Mg (807 mg,33.2 mmol)及I2
(1 mg)於THF (2 mL)中之懸浮液逐滴添加N-014-005_3
(2.5 g,15.1 mmol)於THF (8 mL)中之溶液。在55℃下攪拌混合物1 h。用THF (10 mL)稀釋混合物,且在不經監測的情況下直接用於下一步驟中。在0℃下向N-14-12_4
(1.01 g,2.83 mmol)於THF (10 mL)中之溶液添加新鮮製備的3-[(溴鎂)甲基]-3-甲基環氧丙烷(15 mmol於20 ml THF中)。在15℃下攪拌混合物4 h。向混合物添加NH4
Cl (20 mL,10%水溶液)。用EtOAc (30 mL)萃取混合物。分離有機層且真空濃縮。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈白色固體之混合物
(190 mg,15%),其藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3H2O ETOH,梯度:50%至50%,流速(ml/min):60mL/min,25℃)純化,得到33
(峰值1,110 mg,9%)及呈白色固體之70
(峰值2,30mg,不純)。不純70
(30 mg,不純)藉由矽膠管柱層析(15% EtOAc/PE)純化,得到呈白色固體之70
(10 mg,5%)。33 : 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 4.59-4.70 (m, 1H), 4.50-4.48 (m, 1H),4.36-4.33 (m, 1H), 3.83 (s, 1H), 2.40-2.33 (m, 1H), 2.10-1.50 (m, 17H), 1.49-1.35 (m, 9H), 1.30-0.80 (m, 13H), 0.68 (s, 3H)。 在3 min層析中,LCMS
Rt=1.069 min,30-90AB_2MIN_E.M,純度100%,C29
H49
O3
[M+H]+
之MS ESI計算值445,實驗值445。70 : 1 H NMR
(400 MHz, CDCl3
) δ 5.30-5.26 (m, 1H), 4.10-4.02 (m, 1H), 3.81-3.70 (m, 1H), 3.50-3.41 (m, 2H), 3.34-3.30 (m, 1H), 2.35-2.31 (m, 1H), 2.10-1.50 (m, 18H), 1.49-1.05 (m, 13H), 1.05-0.90 (m, 4H), 0.90-0.80 (m, 3H), 0.67 (s, 3H)。 在3 min層析中,LCMS
Rt=1.115 min,30-90AB_2MIN_E.M,純度100%,C29
H49
O3
[M+H]+
之MS ESI計算值445,實驗值445。實例 71 : (3S,5R,8R,9S,10S,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 - 17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (71) 之合成  1.在0℃下於N2
下向N-004-022_5
(200 mg,0.531 mmol)、CsF (40.2 mg,0.265 mmol)於THF (5 mL)中之溶液添加TMSCF3
(187 mg,1.32 mmol)。在25℃下攪拌混合物1小時。向混合物添加TBAF.3H2O (836 mg,2.65 mmol)。在25℃下攪拌2小時之後,用50% NH4
Cl (20 mL)淬滅混合物且用EtOAc (2 × 10 mL)萃取。合併之有機相用鹽水(20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA = 10/1)純化,得到9
(56 mg,24%)及呈白色固體之71
(30 mg,不純)。 在25℃下自正己烷(2 mL)再結晶71
(30 mg,0.067 mmol),得到呈白色固體之71
(24 mg,10%)。71 : 1 H NMR
(400 MHz, CDCl3
) δ 4.08-4.00 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.22-2.15 (m, 1H), 2.02-1.77 (m, 5H), 1.75-1.68 (m, 2H), 1.64-1.52 (m, 5H), 1.47-1.31 (m, 6H), 1.28-1.01 (m, 10H), 0.97 (s, 3H), 0.67 (s, 3H)。 在2 min層析中,LCMS
Rt = 1.105 min,30-90AB_POS_E.M,純度100%,C25
H41
F3
O3
[M+Na]+
之MS ESI計算值469,實驗值469。實例 72 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (72) 之合成
1.在0℃下於N2
下向N-004-022_5
(200 mg,0.531 mmol)、CsF (40.2 mg,0.265 mmol)於THF (5 mL)中之溶液添加TMSCF3
(187 mg,1.32 mmol)。在25℃下攪拌混合物1小時。向混合物添加TBAF.3H2O (836 mg,2.65 mmol)。在25℃下攪拌2小時之後,用50% NH4
Cl (20 mL)淬滅混合物且用EtOAc (2 × 10 mL)萃取。合併之有機相用鹽水(20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮。殘餘物藉由矽膠層析(100至200目矽膠,PE/EA = 10/1)純化,得到9
(56 mg,24%)及呈白色固體之71
(30 mg,不純)。 在25℃下自正己烷(2 mL)再結晶71
(30 mg,0.067 mmol),得到呈白色固體之71
(24 mg,10%)。71 : 1 H NMR
(400 MHz, CDCl3
) δ 4.08-4.00 (m, 1H), 3.39 (s, 3H), 3.24-3.18 (m, 2H), 2.22-2.15 (m, 1H), 2.02-1.77 (m, 5H), 1.75-1.68 (m, 2H), 1.64-1.52 (m, 5H), 1.47-1.31 (m, 6H), 1.28-1.01 (m, 10H), 0.97 (s, 3H), 0.67 (s, 3H)。 在2 min層析中,LCMS
Rt = 1.105 min,30-90AB_POS_E.M,純度100%,C25
H41
F3
O3
[M+Na]+
之MS ESI計算值469,實驗值469。實例 72 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基己 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (72) 之合成  1.在0℃下向S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液緩慢地添加丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)。添加之後,在15℃下攪拌混合物1小時。用飽和NH4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2 × 30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之72
(500 mg,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。實例 73 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (73) 之合成。
1.在0℃下向S-500-6-1_1
(800 mg,2.23 mmol)於THF (30 mL)中之溶液緩慢地添加丙基溴化鎂(3.34 mL,6.69 mmol,2 M於THF中)。添加之後,在15℃下攪拌混合物1小時。用飽和NH4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2 × 30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之72
(500 mg,56%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.31-5.26 (m, 1H), 3.72-3.64 (m, 1H), 2.41-2.31 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.69 (m, 1H), 1.62-1.54 (m, 3H), 1.52-1.38 (m, 9H), 1.37-1.16 (m, 6H), 1.15-1.01 (m, 7H), 0.99-0.88 (m, 7H), 0.87-0.82 (m, 3H), 0.68 (s, 3H)。 在7.0 min層析中,LCMS
Rt=4.979 min,30-90AB_E,純度98.8%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。實例 73 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (73) 之合成。 
 1.在N2
下向G-21-1
(40 g,110 mmol)於無水二噁烷(1 L)中之溶液添加甲醇鈉(29.7 g,550 mmol)。在110℃下攪拌混合物16小時。TLC顯示起始物質幾乎完全耗盡。移除溶劑至1/3體積,且用2 M HCl酸化混合物至pH=5-6,用DCM (500 mL × 3)萃取,用碳酸氫鈉水溶液(500 mL)及鹽水(500 mL)洗滌,經硫酸鈉乾燥且濃縮。殘餘物藉由矽膠管柱層析(PE:EA:MeOH = 3:1:0.1)純化,得到呈白色固體之G-21-4A
(11 g,33.1%)。 2.在-70℃下向液態銨(1000 mL,製備於13-601中歷時1.5小時)逐份添加Li (2.54 g,363 mmol)之溶液。在-70℃下攪拌混合物30 min直至所有Li溶解為止。逐滴添加G-21-4A
(11 g,36.3 mmol)及第三-BuOH (5.38 g,72.6 mmol)於400 ml無水四氫呋喃中之溶液,且攪拌90 min直至反應混合物變成淺黃色為止。TLC (PE:EA= 1:1,PMA)顯示大部分STM經消耗。添加氯化銨(15 g)且使過量氨蒸發。用0.5N HCl (500 mL)及二氯甲烷(500 mL × 2)萃取殘餘物。合併之有機層用NaHCO3
飽和溶液洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到G-21-5A
及G-21-5B
(10 g,不純)之混合物,其未經進一步純化即直接用於下一步驟中。 3.向G-21-5A
及G-21-5B
(10 g,27.9 mmol)於100 mL無水二氯甲烷中之溶液添加PCC (16.6 g,65.6 mmol)及矽膠(16.6 g)。在25℃下攪拌2 h之後,TLC (PE:EA = 1:1,PMA)顯示STM經消耗。濃縮所得溶液且藉由矽膠層析(石油醚/乙酸乙酯= 5:1至2:1)純化,得到呈白色固體之G-21-6A
(4.6 g,46.4%)。 4.在0℃下於氮氣下向BHT (34.8 g,158 mmol)於甲苯(120 mL)中之溶液逐滴添加三甲基鋁(2 M於甲苯中,39.5 mL,79.1 mmol)。在20℃下攪拌30分鐘之後,在-70℃下於氮氣下逐滴添加G-21-6A
(8 g,26.4 mmol)於甲苯(80 mL)中之溶液。在-70℃下攪拌30 min之後,逐滴添加MeMgBr (3 M於乙醚中,26.3 mL,79.1 mmol,3 M於醚中)。在-70℃下攪拌所得混合物1小時,緩慢地倒入冰冷的檸檬酸水溶液(300 mL)且用EtOAc (3 × 100 mL)萃取。合併之有機層用鹽水(300 mL)洗滌,經無水硫酸鈉乾燥,過濾,濃縮且藉由combi-flash (0-40% EtOAc/PE)純化,得到呈白色固體之N-4-10_1
(6.5 g,77%)。 1 H NMR
(400 MHz, CDCl3
)δ 2.60-2.49 (m, 1H), 2.47-2.37 (m, 2H), 2.34-2.19 (m, 3H), 2.14-2.03 (m, 1H), 2.00-1.82 (m, 3H), 1.73-1.58 (m, 3H), 1.56-1.46 (m, 2H), 1.36-1.26 (m, 3H), 1.24 (s, 3H), 1.21-1.07 (m, 2H), 1.04 (s, 3H), 1.00-0.84 (m, 1H), 0.82 (s, 3H)。 5.在20℃下於N2
下向溴(乙基)三苯基磷烷(22.6 g,61.1 mmol)於無水THF (200 mL)中之懸浮液添加2-甲基丙-2-醇化鉀(6.84 g,61.1 mmol)。在40℃下攪拌30分鐘之後,緩慢添加N-4-10_1
(6.5 g,20.4 mmol)於無水THF (50 mL)中之溶液。在40℃下攪拌所得混合物10分鐘,接著用NH4
Cl水溶液(400 mL)淬滅且用EtOAc (2 × 150 mL)萃取。合併之有機相經硫酸鈉乾燥,過濾,濃縮且藉由矽膠管柱層析(0-25% EtOAc/PE)純化,得到呈白色固體之N-4-10_2
(5.5 g,82%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.22-5.13 (m, 1H), 2.91-2.81 (m, 1H), 2.62-2.51 (m, 1H), 2.50-2.39 (m, 2H), 2.38-2.24 (m, 1H), 1.91-1.81(m, 1H), 1.80-1.70 (m, 4H), 1.55-1.41 (m, 4H), 1.36-1.25 (m, 5H), 1.23 (s, 3H), 1.21-1.04 (m, 3H), 1.01 (s, 3H), 0.98-0.84 (m, 2H), 0.81 (s, 3H)。 6.在15℃下於N2
下向N-4-10_2
(5.5 g,16.6 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(8.10 g,33.2 mmol)。在50℃下攪拌1小時之後,將混合物冷卻至15℃。在15℃以下逐滴添加NaOH水溶液(33.2 mL,5 M,166 mmol),接著在15℃以下逐滴添加H2
O2
(18.8 g,30%,166 mmol)。用EtOAc (3 × 100 mL)萃取混合物。合併之有機相用飽和Na2
S2
O3
(5 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到7 g粗產物,其直接用於下一步驟中。 7.向N-4-10_3
(7 g,19.9 mmol)於DCM (300 mL)中之溶液添加DMP (25.2 g,59.6 mmol)。在20℃下攪拌10 min之後,用NaHCO3
飽和溶液(500 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層,且用DCM (200 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 400 mL)、飽和NaHCO3
(400 mL)、鹽水(400 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-20% EtOAc/DCM)純化,得到呈白色固體之N-4-10_4
(4 g,58%)。 1 H NMR
(400 MHz, CDCl3
)δ 2.77-2.67 (m, 1H), 2.65-2.38 (m, 3H), 2.32-2.17 (m, 1H), 2.09 (s, 3H), 1.88-1.63 (m, 7H), 1.59-1.49 (m, 3H), 1.35-1.21 (m, 7H), 1.19-1.09 (m, 2H), 1.01 (s, 3H), 0.96-0.84 (m, 1H), 0.57 (s, 3H)。 8.向MePh3
PBr (8.18 g,23.0 mmol)於THF (100 mL)中之懸浮液添加t-BuOK (2.57 g,23.0 mmol)。在40℃下攪拌10分鐘之後,在20℃下向N-4-10_4
(4 g,11.5 mmol)於THF (50 mL)中之溶液緩慢地逐滴添加混合物。添加之後,用NH4
Cl (200 mL)淬滅混合物且用EtOAc (3 × 80 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-25% EtOAc/PE)純化,得到呈白色固體之N-4-10_5
(3.2 g,80%)。 1 H NMR
(400 MHz, CDCl3
)δ 4.88 (s, 1H), 4.70 (s, 1H), 2.48-2.37 (m, 2H), 2.31-2.22 (m, 2H), 1.89-1.77 (m, 4H), 1.75-1.61 (m, 7H), 1.54-1.45 (m, 2H), 1.34-1.29 (m, 2H), 1.28-1.24 (m, 3H), 1.23 (s, 3H), 1.17-1.05 (m, 2H), 1.01 (s, 3H), 0.93-0.83 (m, 1H), 0.51 (s, 3H)。 9.在15℃下於N2
下向N-4-10_5
(3.2 g,9.28 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(4.51 g,18.5 mmol)。在50℃下攪拌1小時之後,將混合物冷卻至15℃。在15℃以下逐滴添加NaOH水溶液(18.5 mL,5 M,92.8 mmol),接著在15℃以下逐滴添加H2
O2
(10.5 g,30%,92.8 mmol)。用EtOAc (3 × 100 mL)萃取混合物。合併之有機相用飽和Na2
S2
O3
(5 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到5 g粗產物,其直接用於下一步驟中。 10.向N-4-10_5A
(5 g,13.7 mmol)於DCM (300 mL)中之溶液添加DMP (11.6 g,27.4 mmol)。在20℃下攪拌10 min之後,用NaHCO3
飽和溶液 (300 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (100 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 300 mL)、飽和NaHCO3
(300 mL)、鹽水(300 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-10% 丙酮/DCM)純化,得到呈白色固體之N-4-10_7
(1 g,20%)。 1 H NMR
(400 MHz, CDCl3
)δ 9.58-9.55 (m, 1H), 2.52-2.27 (m, 4H), 2.08-1.96 (m, 1H), 1.84-1.62 (m, 8H), 1.51-1.39 (m, 3H), 1.32-1.21 (m, 7H), 1.17-1.06 (m, 5H), 1.01 (s, 3H), 0.94-0.83 (m, 1H), 0.66 (s, 3H)。 11.在0℃下於N2
下向N-4-10_6
(400 mg,0.832 mmol)於THF (20 mL)中之溶液逐滴添加異戊基溴化鎂(1.65 mL,3.30 mmol,2 M於醚中)。在0℃下攪拌10分鐘之後,用飽和NH4
Cl (60 mL)淬滅混合物且用EtOAc (2 × 30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-10% 丙酮/DCM)純化,得到呈白色固體之73
(200 mg,42%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.65-3.56 (m, 1H), 2.55-2.49 (m, 1H), 2.46-2.38 (m, 1H), 2.32-2.25 (m, 1H), 2.10-1.98 (m, 1H), 1.83-1.62 (m, 7H), 1.57-1.44 (m, 4H), 1.42-1.25 (m, 7H), 1.24-1.20 (m, 4H), 1.19-1.04 (m, 5H), 1.01 (s, 3H), 0.94-0.82 (m, 10H), .0.63 (s, 3H)。 在7.0 min層析中,LCMS
Rt=3.381 min,30-90AB_E,純度100%,C28
H47
O2
[M+H-H2
O]+
之MS ESI計算值415,實驗值415。實例 74 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (74) 之合成
1.在N2
下向G-21-1
(40 g,110 mmol)於無水二噁烷(1 L)中之溶液添加甲醇鈉(29.7 g,550 mmol)。在110℃下攪拌混合物16小時。TLC顯示起始物質幾乎完全耗盡。移除溶劑至1/3體積,且用2 M HCl酸化混合物至pH=5-6,用DCM (500 mL × 3)萃取,用碳酸氫鈉水溶液(500 mL)及鹽水(500 mL)洗滌,經硫酸鈉乾燥且濃縮。殘餘物藉由矽膠管柱層析(PE:EA:MeOH = 3:1:0.1)純化,得到呈白色固體之G-21-4A
(11 g,33.1%)。 2.在-70℃下向液態銨(1000 mL,製備於13-601中歷時1.5小時)逐份添加Li (2.54 g,363 mmol)之溶液。在-70℃下攪拌混合物30 min直至所有Li溶解為止。逐滴添加G-21-4A
(11 g,36.3 mmol)及第三-BuOH (5.38 g,72.6 mmol)於400 ml無水四氫呋喃中之溶液,且攪拌90 min直至反應混合物變成淺黃色為止。TLC (PE:EA= 1:1,PMA)顯示大部分STM經消耗。添加氯化銨(15 g)且使過量氨蒸發。用0.5N HCl (500 mL)及二氯甲烷(500 mL × 2)萃取殘餘物。合併之有機層用NaHCO3
飽和溶液洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到G-21-5A
及G-21-5B
(10 g,不純)之混合物,其未經進一步純化即直接用於下一步驟中。 3.向G-21-5A
及G-21-5B
(10 g,27.9 mmol)於100 mL無水二氯甲烷中之溶液添加PCC (16.6 g,65.6 mmol)及矽膠(16.6 g)。在25℃下攪拌2 h之後,TLC (PE:EA = 1:1,PMA)顯示STM經消耗。濃縮所得溶液且藉由矽膠層析(石油醚/乙酸乙酯= 5:1至2:1)純化,得到呈白色固體之G-21-6A
(4.6 g,46.4%)。 4.在0℃下於氮氣下向BHT (34.8 g,158 mmol)於甲苯(120 mL)中之溶液逐滴添加三甲基鋁(2 M於甲苯中,39.5 mL,79.1 mmol)。在20℃下攪拌30分鐘之後,在-70℃下於氮氣下逐滴添加G-21-6A
(8 g,26.4 mmol)於甲苯(80 mL)中之溶液。在-70℃下攪拌30 min之後,逐滴添加MeMgBr (3 M於乙醚中,26.3 mL,79.1 mmol,3 M於醚中)。在-70℃下攪拌所得混合物1小時,緩慢地倒入冰冷的檸檬酸水溶液(300 mL)且用EtOAc (3 × 100 mL)萃取。合併之有機層用鹽水(300 mL)洗滌,經無水硫酸鈉乾燥,過濾,濃縮且藉由combi-flash (0-40% EtOAc/PE)純化,得到呈白色固體之N-4-10_1
(6.5 g,77%)。 1 H NMR
(400 MHz, CDCl3
)δ 2.60-2.49 (m, 1H), 2.47-2.37 (m, 2H), 2.34-2.19 (m, 3H), 2.14-2.03 (m, 1H), 2.00-1.82 (m, 3H), 1.73-1.58 (m, 3H), 1.56-1.46 (m, 2H), 1.36-1.26 (m, 3H), 1.24 (s, 3H), 1.21-1.07 (m, 2H), 1.04 (s, 3H), 1.00-0.84 (m, 1H), 0.82 (s, 3H)。 5.在20℃下於N2
下向溴(乙基)三苯基磷烷(22.6 g,61.1 mmol)於無水THF (200 mL)中之懸浮液添加2-甲基丙-2-醇化鉀(6.84 g,61.1 mmol)。在40℃下攪拌30分鐘之後,緩慢添加N-4-10_1
(6.5 g,20.4 mmol)於無水THF (50 mL)中之溶液。在40℃下攪拌所得混合物10分鐘,接著用NH4
Cl水溶液(400 mL)淬滅且用EtOAc (2 × 150 mL)萃取。合併之有機相經硫酸鈉乾燥,過濾,濃縮且藉由矽膠管柱層析(0-25% EtOAc/PE)純化,得到呈白色固體之N-4-10_2
(5.5 g,82%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.22-5.13 (m, 1H), 2.91-2.81 (m, 1H), 2.62-2.51 (m, 1H), 2.50-2.39 (m, 2H), 2.38-2.24 (m, 1H), 1.91-1.81(m, 1H), 1.80-1.70 (m, 4H), 1.55-1.41 (m, 4H), 1.36-1.25 (m, 5H), 1.23 (s, 3H), 1.21-1.04 (m, 3H), 1.01 (s, 3H), 0.98-0.84 (m, 2H), 0.81 (s, 3H)。 6.在15℃下於N2
下向N-4-10_2
(5.5 g,16.6 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(8.10 g,33.2 mmol)。在50℃下攪拌1小時之後,將混合物冷卻至15℃。在15℃以下逐滴添加NaOH水溶液(33.2 mL,5 M,166 mmol),接著在15℃以下逐滴添加H2
O2
(18.8 g,30%,166 mmol)。用EtOAc (3 × 100 mL)萃取混合物。合併之有機相用飽和Na2
S2
O3
(5 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到7 g粗產物,其直接用於下一步驟中。 7.向N-4-10_3
(7 g,19.9 mmol)於DCM (300 mL)中之溶液添加DMP (25.2 g,59.6 mmol)。在20℃下攪拌10 min之後,用NaHCO3
飽和溶液(500 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層,且用DCM (200 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 400 mL)、飽和NaHCO3
(400 mL)、鹽水(400 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-20% EtOAc/DCM)純化,得到呈白色固體之N-4-10_4
(4 g,58%)。 1 H NMR
(400 MHz, CDCl3
)δ 2.77-2.67 (m, 1H), 2.65-2.38 (m, 3H), 2.32-2.17 (m, 1H), 2.09 (s, 3H), 1.88-1.63 (m, 7H), 1.59-1.49 (m, 3H), 1.35-1.21 (m, 7H), 1.19-1.09 (m, 2H), 1.01 (s, 3H), 0.96-0.84 (m, 1H), 0.57 (s, 3H)。 8.向MePh3
PBr (8.18 g,23.0 mmol)於THF (100 mL)中之懸浮液添加t-BuOK (2.57 g,23.0 mmol)。在40℃下攪拌10分鐘之後,在20℃下向N-4-10_4
(4 g,11.5 mmol)於THF (50 mL)中之溶液緩慢地逐滴添加混合物。添加之後,用NH4
Cl (200 mL)淬滅混合物且用EtOAc (3 × 80 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-25% EtOAc/PE)純化,得到呈白色固體之N-4-10_5
(3.2 g,80%)。 1 H NMR
(400 MHz, CDCl3
)δ 4.88 (s, 1H), 4.70 (s, 1H), 2.48-2.37 (m, 2H), 2.31-2.22 (m, 2H), 1.89-1.77 (m, 4H), 1.75-1.61 (m, 7H), 1.54-1.45 (m, 2H), 1.34-1.29 (m, 2H), 1.28-1.24 (m, 3H), 1.23 (s, 3H), 1.17-1.05 (m, 2H), 1.01 (s, 3H), 0.93-0.83 (m, 1H), 0.51 (s, 3H)。 9.在15℃下於N2
下向N-4-10_5
(3.2 g,9.28 mmol)於THF (100 mL)中之混合物添加9-BBN二聚體(4.51 g,18.5 mmol)。在50℃下攪拌1小時之後,將混合物冷卻至15℃。在15℃以下逐滴添加NaOH水溶液(18.5 mL,5 M,92.8 mmol),接著在15℃以下逐滴添加H2
O2
(10.5 g,30%,92.8 mmol)。用EtOAc (3 × 100 mL)萃取混合物。合併之有機相用飽和Na2
S2
O3
(5 × 100 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到5 g粗產物,其直接用於下一步驟中。 10.向N-4-10_5A
(5 g,13.7 mmol)於DCM (300 mL)中之溶液添加DMP (11.6 g,27.4 mmol)。在20℃下攪拌10 min之後,用NaHCO3
飽和溶液 (300 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層且用DCM (100 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 300 mL)、飽和NaHCO3
(300 mL)、鹽水(300 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-10% 丙酮/DCM)純化,得到呈白色固體之N-4-10_7
(1 g,20%)。 1 H NMR
(400 MHz, CDCl3
)δ 9.58-9.55 (m, 1H), 2.52-2.27 (m, 4H), 2.08-1.96 (m, 1H), 1.84-1.62 (m, 8H), 1.51-1.39 (m, 3H), 1.32-1.21 (m, 7H), 1.17-1.06 (m, 5H), 1.01 (s, 3H), 0.94-0.83 (m, 1H), 0.66 (s, 3H)。 11.在0℃下於N2
下向N-4-10_6
(400 mg,0.832 mmol)於THF (20 mL)中之溶液逐滴添加異戊基溴化鎂(1.65 mL,3.30 mmol,2 M於醚中)。在0℃下攪拌10分鐘之後,用飽和NH4
Cl (60 mL)淬滅混合物且用EtOAc (2 × 30 mL)萃取。合併之有機相用鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-10% 丙酮/DCM)純化,得到呈白色固體之73
(200 mg,42%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.65-3.56 (m, 1H), 2.55-2.49 (m, 1H), 2.46-2.38 (m, 1H), 2.32-2.25 (m, 1H), 2.10-1.98 (m, 1H), 1.83-1.62 (m, 7H), 1.57-1.44 (m, 4H), 1.42-1.25 (m, 7H), 1.24-1.20 (m, 4H), 1.19-1.04 (m, 5H), 1.01 (s, 3H), 0.94-0.82 (m, 10H), .0.63 (s, 3H)。 在7.0 min層析中,LCMS
Rt=3.381 min,30-90AB_E,純度100%,C28
H47
O2
[M+H-H2
O]+
之MS ESI計算值415,實驗值415。實例 74 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (74) 之合成 
 1.在35℃下於N2
下將t-BuOH (1.7 L)裝入三頸圓底燒瓶且攪拌10 min。向混合物添加t-BuOK (292 g,2.61 mol)且攪拌直至反應物變得澄清。此後,向以上混合物添加S-310-B9_1
(65 g, 238 mmol)且在35℃下於N2
下攪拌1.5 h。將反應混合物倒入10%乙酸水溶液(2 L)中且攪拌30 min,在此期間將溫度維持在10℃以下。接著混合物用水(1.5 L)處理且用NaHCO3
將pH值調整至7至8,且攪拌混合物30 min。水相用MTBE (3 L)萃取。分離有機層,用鹽水(3 × 1 L)洗滌,經無水Na2
SO4
乾燥,過濾並在35℃以下濃縮,得到呈油狀物之S-200-N19-3_1
(65 g,粗產物)。粗殘餘物直接用於下一步驟。 2.在0℃下向2,6-二第三丁基-4-甲苯酚(340 g,1.54 mol)於甲苯(700 mL)中之溶液逐滴添加AlMe3
(385 mL, 770 mmol, 2 M於甲苯中)。在25℃下攪拌混合物1小時且直接用作MAD溶液。在-70℃下於N2
下經30 min之時段向MAD溶液添加200-N19-3_1
(60 g,220 mmol)於無水甲苯(200 mL)及無水DCM (200 mL)中之溶液。在-70℃下攪拌反應混合物1 h。接著在-70℃下逐滴添加MeMgBr (220 mL, 660 mmol, 3 M於乙基醚中)且攪拌1 h。在0℃下將反應物倒入檸檬酸飽和水溶液(2 L)中且攪拌30 min,用EtOAc萃取(2 × 1 L)。合併之有機相用飽和鹽水(2×1 L)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc = 10/1至5/1)純化,得到呈固體之200-N19-M22_1
(33 g,52%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.46-5.42 (m, 1H), 2.25-2.40 (m, 1H), 2.21-1.60 (m, 13H),1.35-1.21 (m, 4H), 1.13 (s, 3H), 0.98-0.83 (m, 6H)。 3.在25℃下於N2
下向Ph3
PEtBr (102 g,277 mmol)於無水THF (500 mL)中之懸浮液一次性添加t-BuOK (31.0 g,277 mmol)。在25℃下攪拌30 min之後,添加200-N19-M22_1
(20 g, 69.3 mmol)且在25℃下攪拌2 h。在0℃下用NH4
Cl水溶液(800 mL)淬滅反應物,用EtOAc (2 × 500 mL)萃取。合併之有機相用鹽水(2 × 500 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc = 10/1至5/1)純化,得到呈固體之N-4-14_1
(15 g,72%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.40 (m, 1H), 5.16-5.10 (m, 1H), 2.41-2.33 (m, 1H), 2.28-1.86 (m, 8H), 1.78-1.71 (m, 1H), 1.69-1.50 (m, 11H), 1.41-1.10 (m, 6H), 0.94-0.81 (s, 3H)。4.
向N-4-14_1
(30 g, 99.8 mmol)於無水THF (500 mL)中之溶液添加9-BNN二聚體(66.9 g, 299 mmol)且在0℃下於N2
下攪拌30 min。使反應混合物升溫至50℃且攪拌1小時。冷卻至0℃之後,添加EtOH (100 mL)。極緩慢地添加NaOH水溶液(99.8 mL, 5 M, 499 mmol)。緩慢添加H2
O2
(53.0 g,499 mmol, 30%於水中)且將內部溫度維持在30℃以下。將混合物升溫至50℃且攪拌1小時。冷卻反應混合物且添加冰水(1 L)並攪拌30 min。過濾且真空濃縮,得到呈固體之N-4-11_2
(30 g,粗產物)。粗殘餘物直接用於下一步驟。 5.向N-4-14_2
(30 g,粗產物)於DCM (500 mL)中之溶液添加矽膠(150 g)及PCC (81.0 g,376 mmol)。使反應混合物升溫至40℃且攪拌1小時。冷卻反應混合物,用PE (500 mL)處理,經由矽膠墊過濾,且固體用PE/DCM (500/500 mL)洗滌。母液經過濾且真空濃縮,得到粗產物。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之N-4-14_3
(20 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.40 (m, 1H), 2.57-2.50 (m, 1H), 2.21-2.08 (m, 6H), 1.77-1.43 (m, 10H), 1.37-1.12 (m, 9H), 1.00-0.82 (m, 2H), 0.64 (s, 3H)。 6.在0℃下於N2
下向Ph3
PMeBr (44.8 g,126 mmol)於無水THF (300 mL)中之懸浮液一次性添加t-BuOK (14.1 g,126 mmol)。在25℃下攪拌反應混合物30 min。添加N-4-14_3
(20 g,63.1 mmol)。使反應混合物升溫至40℃且攪拌1小時。在0℃下將反應物倒入冰水(500 mL)中。用EtOAc (2 × 400 mL)萃取水相。合併之有機相用鹽水(2 × 300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠管柱(PE/EtOAc = 8/1-5/1)純化,得到呈固體之N-4-14_4
(19 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.40 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.23-2.13 (m, 2H), 1.87-1.64 (m, 11H), 1.42-1.40 (m, 2H), 1.29-1.08 (m, 8H), 0.97-0.80 (m, 3H), 0.59 (s, 3H)。 7.在0℃下於N2
下向N-4-14_4
(19 g,60.4 mmol)於無水THF (300 mL)中之溶液一次性添加9-BNN二聚體 (40.5 g,181 mmol)。將混合物升溫至50℃且攪拌1 h。冷卻反應混合物且添加EtOH (100 mL)。極緩慢添加NaOH水溶液(60.3 mL,5 M,302 mmol)。緩慢添加H2
O2
(34.0 g,302 mmol, 30%於水中) 且將內部溫度維持在10℃以下。將混合物升溫至50℃且攪拌1小時。冷卻之後,添加冰水(1 L)且攪拌30 min。濾出沈澱固體。濾餅在空氣中乾燥,得到呈固體之N-4-11_5
(17 g,粗產物),其直接用於下一步驟。 8.在25℃下向N-4-14_5
(17 g,粗產物)於DCM (300 mL)中之溶液一次性添加矽膠(60 g)及PCC (43.9 g,204 mmol)。使反應混合物升溫至40℃且攪拌1小時。冷卻反應混合物,且添加PE (200 mL)。混合物經由矽膠墊過濾,且固體用PE/DCM (200/200 mL)洗滌。過濾且真空濃縮,得到固體。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之N-4-14_6
(5.5 g,不純)。在82℃下自MeCN (50 mL)再結晶殘餘物,得到呈固體之N-4-14_6
(5 g,91%)。 1 H NMR
(400 MHz, CDCl3
)δ 9.60-9.56 (m, 1H), 5.42-5.40 (m, 1H), 2.58-2.51 (m, 1H),2.40-1.85 (m, 9H), 1.44-1.04 (m, 16H), 1.00-0.80 (m, 3H), 0.75-0.71 (m, 3H)。 9.在25℃下於N2
下向Mg (3.96 g,165 mmol)及I2
(1 mg)於無水THF (20 mL)中之懸浮液逐滴添加1
(12.5 g,82.7 mmol)於無水THF (63 mL)中之溶液,且將內部溫度升高至65℃且攪拌2小時。混合物直接用於下一步驟中。在0℃下於N2
下向N-4-14_6
(5 g,15.1 mmol)於無水THF (50 mL)中之溶液一次性添加異戊基溴化鎂(83.0 mL,1 M於THF中)。使反應混合物升溫至15℃且攪拌1小時。向反應混合物添加NH4
Cl 飽和水溶液(100 mL)。用EtOAc (3 × 100 mL)萃取水相。合併之有機相用飽和鹽水(2 × 200 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(0-20% EtOAc/PE)純化,得到呈固體之N-4-14_7
(2.5 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.40-5.37 (m, 1H), 3.67-3.61 (m, 1H), 2.19-1.71 (m, 9H), 1.64-1.28 (m, 13H), 1.18-1.03 (m, 7H), 0.95-0.78 (m, 12H), 0.69 (s, 3H)。 10.在25℃下向DMP (10.5 g,24.8 mmol)添加含N-4-14_7
(2.5 g,6.20 mmol)之DCM (40 mL)。使反應混合物升溫至40℃且攪拌1 h。在pH 7至8且低於10℃下用NaHCO3
飽和水溶液淬滅反應混合物。過濾懸浮液。分離濾液中之DCM相且用NaHCO3
/Na2
S2
O3
飽和水溶液(1:1,2 × 30 mL)洗滌,合併之有機相用飽和鹽水(2 × 30 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到固體。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈固體之N-4- 14_7O
(1.5 g,60%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 2.56-2.31 (m, 3H), 2.19-1.83 (m, 5H), 1.84-1.42 (m, 12H), 1.30-0.97 (m, 12H), 0.96-0.77 (m, 8H), 0.74-0.70 (m, 3H)。 11.在25℃下向N-4-14_7O
(1.5 g,3.74)於MeOH (10 mL)中之溶液緩慢添加NaBH4
(1.42 g,37.4 mmol),且攪拌2小時。添加冰水(100 mL)且攪拌混合物30 min。用DCM (2 × 20 mL)萃取水相。合併之有機相用飽和鹽水(2 × 20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之N-4-14_7
(1 g, 67%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.65-3.63 (m, 1H), 2.20-2.16 (m, 1H), 2.11-1.88 (m, 5H), 1.86-1.54 (m, 10H), 1.33-0.99 (m, 14H), 0.95-0.79 (m, 11H), 0.70 (s, 3H)。SFC
峰值1:Rt=4.644 min,且在10 min層析中峰值2 Rt=5.240 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。 12.N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始:B:40%,結束B:40%)純化,得到80
(峰值2,300 mg,不純)及呈固體之83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之80
(150 mg,15%)。在82℃下回流1 小時,自MeCN (3 mL)再結晶83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之83
(150 mg,15%)。83 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.62-3.60 (m, 1H), 2.22-1.89 (m, 6H), 1.64-1.49 (m, 9H), 1.46-1.11 (m, 16H), 0.98-0.86 (m, 10H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.268 min,30-90 AB,純度100%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=4.609 min,AD_3_EtOH_DEA_5_40_25ML,100%de。80 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.63-3.62 (m, 1H), 2.22-1.67 (m, 10H), 1.64-1.36 (m, 12H), 1.16-1.03 (m, 8H), 0.98-0.80 (m, 11H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.276 min,30-90 AB,純度99%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=5.236 min,AD_3_EtOH_DEA_5_40_25ML,100%de。 13. 在Ar下向80
(150 mg,0.372 mmol)於THF (3 mL)及MeOH (3 mL)中之溶液添加無水Pd(OH)2
(200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物12 h,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用DCM (3 × 50 mL)洗滌。真空濃縮濾液以提供油狀物。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之74
(20 mg,13%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.63-3.61 (m, 1H), 1.96-1.85 (m, 2H), 1.78-1.50 (m, 8H), 1.45-1.19 (s, 12H), 1.17-1.00 (m, 11H), 0.98-0.83 (m, 11H), 0.72-0.59 (m, 3H)。 在2.0 min層析中,LCMS
Rt=1.333 min,30-90 AB,純度99%,C27
H44
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 75 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (75) 之合成
1.在35℃下於N2
下將t-BuOH (1.7 L)裝入三頸圓底燒瓶且攪拌10 min。向混合物添加t-BuOK (292 g,2.61 mol)且攪拌直至反應物變得澄清。此後,向以上混合物添加S-310-B9_1
(65 g, 238 mmol)且在35℃下於N2
下攪拌1.5 h。將反應混合物倒入10%乙酸水溶液(2 L)中且攪拌30 min,在此期間將溫度維持在10℃以下。接著混合物用水(1.5 L)處理且用NaHCO3
將pH值調整至7至8,且攪拌混合物30 min。水相用MTBE (3 L)萃取。分離有機層,用鹽水(3 × 1 L)洗滌,經無水Na2
SO4
乾燥,過濾並在35℃以下濃縮,得到呈油狀物之S-200-N19-3_1
(65 g,粗產物)。粗殘餘物直接用於下一步驟。 2.在0℃下向2,6-二第三丁基-4-甲苯酚(340 g,1.54 mol)於甲苯(700 mL)中之溶液逐滴添加AlMe3
(385 mL, 770 mmol, 2 M於甲苯中)。在25℃下攪拌混合物1小時且直接用作MAD溶液。在-70℃下於N2
下經30 min之時段向MAD溶液添加200-N19-3_1
(60 g,220 mmol)於無水甲苯(200 mL)及無水DCM (200 mL)中之溶液。在-70℃下攪拌反應混合物1 h。接著在-70℃下逐滴添加MeMgBr (220 mL, 660 mmol, 3 M於乙基醚中)且攪拌1 h。在0℃下將反應物倒入檸檬酸飽和水溶液(2 L)中且攪拌30 min,用EtOAc萃取(2 × 1 L)。合併之有機相用飽和鹽水(2×1 L)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc = 10/1至5/1)純化,得到呈固體之200-N19-M22_1
(33 g,52%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.46-5.42 (m, 1H), 2.25-2.40 (m, 1H), 2.21-1.60 (m, 13H),1.35-1.21 (m, 4H), 1.13 (s, 3H), 0.98-0.83 (m, 6H)。 3.在25℃下於N2
下向Ph3
PEtBr (102 g,277 mmol)於無水THF (500 mL)中之懸浮液一次性添加t-BuOK (31.0 g,277 mmol)。在25℃下攪拌30 min之後,添加200-N19-M22_1
(20 g, 69.3 mmol)且在25℃下攪拌2 h。在0℃下用NH4
Cl水溶液(800 mL)淬滅反應物,用EtOAc (2 × 500 mL)萃取。合併之有機相用鹽水(2 × 500 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠層析(PE/EtOAc = 10/1至5/1)純化,得到呈固體之N-4-14_1
(15 g,72%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.40 (m, 1H), 5.16-5.10 (m, 1H), 2.41-2.33 (m, 1H), 2.28-1.86 (m, 8H), 1.78-1.71 (m, 1H), 1.69-1.50 (m, 11H), 1.41-1.10 (m, 6H), 0.94-0.81 (s, 3H)。4.
向N-4-14_1
(30 g, 99.8 mmol)於無水THF (500 mL)中之溶液添加9-BNN二聚體(66.9 g, 299 mmol)且在0℃下於N2
下攪拌30 min。使反應混合物升溫至50℃且攪拌1小時。冷卻至0℃之後,添加EtOH (100 mL)。極緩慢地添加NaOH水溶液(99.8 mL, 5 M, 499 mmol)。緩慢添加H2
O2
(53.0 g,499 mmol, 30%於水中)且將內部溫度維持在30℃以下。將混合物升溫至50℃且攪拌1小時。冷卻反應混合物且添加冰水(1 L)並攪拌30 min。過濾且真空濃縮,得到呈固體之N-4-11_2
(30 g,粗產物)。粗殘餘物直接用於下一步驟。 5.向N-4-14_2
(30 g,粗產物)於DCM (500 mL)中之溶液添加矽膠(150 g)及PCC (81.0 g,376 mmol)。使反應混合物升溫至40℃且攪拌1小時。冷卻反應混合物,用PE (500 mL)處理,經由矽膠墊過濾,且固體用PE/DCM (500/500 mL)洗滌。母液經過濾且真空濃縮,得到粗產物。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之N-4-14_3
(20 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.40 (m, 1H), 2.57-2.50 (m, 1H), 2.21-2.08 (m, 6H), 1.77-1.43 (m, 10H), 1.37-1.12 (m, 9H), 1.00-0.82 (m, 2H), 0.64 (s, 3H)。 6.在0℃下於N2
下向Ph3
PMeBr (44.8 g,126 mmol)於無水THF (300 mL)中之懸浮液一次性添加t-BuOK (14.1 g,126 mmol)。在25℃下攪拌反應混合物30 min。添加N-4-14_3
(20 g,63.1 mmol)。使反應混合物升溫至40℃且攪拌1小時。在0℃下將反應物倒入冰水(500 mL)中。用EtOAc (2 × 400 mL)萃取水相。合併之有機相用鹽水(2 × 300 mL)洗滌,經無水Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由矽膠管柱(PE/EtOAc = 8/1-5/1)純化,得到呈固體之N-4-14_4
(19 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.43-5.40 (m, 1H), 4.85 (s, 1H), 4.71 (s, 1H), 2.23-2.13 (m, 2H), 1.87-1.64 (m, 11H), 1.42-1.40 (m, 2H), 1.29-1.08 (m, 8H), 0.97-0.80 (m, 3H), 0.59 (s, 3H)。 7.在0℃下於N2
下向N-4-14_4
(19 g,60.4 mmol)於無水THF (300 mL)中之溶液一次性添加9-BNN二聚體 (40.5 g,181 mmol)。將混合物升溫至50℃且攪拌1 h。冷卻反應混合物且添加EtOH (100 mL)。極緩慢添加NaOH水溶液(60.3 mL,5 M,302 mmol)。緩慢添加H2
O2
(34.0 g,302 mmol, 30%於水中) 且將內部溫度維持在10℃以下。將混合物升溫至50℃且攪拌1小時。冷卻之後,添加冰水(1 L)且攪拌30 min。濾出沈澱固體。濾餅在空氣中乾燥,得到呈固體之N-4-11_5
(17 g,粗產物),其直接用於下一步驟。 8.在25℃下向N-4-14_5
(17 g,粗產物)於DCM (300 mL)中之溶液一次性添加矽膠(60 g)及PCC (43.9 g,204 mmol)。使反應混合物升溫至40℃且攪拌1小時。冷卻反應混合物,且添加PE (200 mL)。混合物經由矽膠墊過濾,且固體用PE/DCM (200/200 mL)洗滌。過濾且真空濃縮,得到固體。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之N-4-14_6
(5.5 g,不純)。在82℃下自MeCN (50 mL)再結晶殘餘物,得到呈固體之N-4-14_6
(5 g,91%)。 1 H NMR
(400 MHz, CDCl3
)δ 9.60-9.56 (m, 1H), 5.42-5.40 (m, 1H), 2.58-2.51 (m, 1H),2.40-1.85 (m, 9H), 1.44-1.04 (m, 16H), 1.00-0.80 (m, 3H), 0.75-0.71 (m, 3H)。 9.在25℃下於N2
下向Mg (3.96 g,165 mmol)及I2
(1 mg)於無水THF (20 mL)中之懸浮液逐滴添加1
(12.5 g,82.7 mmol)於無水THF (63 mL)中之溶液,且將內部溫度升高至65℃且攪拌2小時。混合物直接用於下一步驟中。在0℃下於N2
下向N-4-14_6
(5 g,15.1 mmol)於無水THF (50 mL)中之溶液一次性添加異戊基溴化鎂(83.0 mL,1 M於THF中)。使反應混合物升溫至15℃且攪拌1小時。向反應混合物添加NH4
Cl 飽和水溶液(100 mL)。用EtOAc (3 × 100 mL)萃取水相。合併之有機相用飽和鹽水(2 × 200 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(0-20% EtOAc/PE)純化,得到呈固體之N-4-14_7
(2.5 g,不純)。 1 H NMR
(400 MHz, CDCl3
)δ 5.40-5.37 (m, 1H), 3.67-3.61 (m, 1H), 2.19-1.71 (m, 9H), 1.64-1.28 (m, 13H), 1.18-1.03 (m, 7H), 0.95-0.78 (m, 12H), 0.69 (s, 3H)。 10.在25℃下向DMP (10.5 g,24.8 mmol)添加含N-4-14_7
(2.5 g,6.20 mmol)之DCM (40 mL)。使反應混合物升溫至40℃且攪拌1 h。在pH 7至8且低於10℃下用NaHCO3
飽和水溶液淬滅反應混合物。過濾懸浮液。分離濾液中之DCM相且用NaHCO3
/Na2
S2
O3
飽和水溶液(1:1,2 × 30 mL)洗滌,合併之有機相用飽和鹽水(2 × 30 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到固體。殘餘物藉由閃蒸塔(0-30% EtOAc/PE)純化,得到呈固體之N-4- 14_7O
(1.5 g,60%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 2.56-2.31 (m, 3H), 2.19-1.83 (m, 5H), 1.84-1.42 (m, 12H), 1.30-0.97 (m, 12H), 0.96-0.77 (m, 8H), 0.74-0.70 (m, 3H)。 11.在25℃下向N-4-14_7O
(1.5 g,3.74)於MeOH (10 mL)中之溶液緩慢添加NaBH4
(1.42 g,37.4 mmol),且攪拌2小時。添加冰水(100 mL)且攪拌混合物30 min。用DCM (2 × 20 mL)萃取水相。合併之有機相用飽和鹽水(2 × 20 mL)洗滌,經無水Na2
SO4
乾燥,過濾且真空濃縮以獲得固體。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之N-4-14_7
(1 g, 67%)。 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.65-3.63 (m, 1H), 2.20-2.16 (m, 1H), 2.11-1.88 (m, 5H), 1.86-1.54 (m, 10H), 1.33-0.99 (m, 14H), 0.95-0.79 (m, 11H), 0.70 (s, 3H)。SFC
峰值1:Rt=4.644 min,且在10 min層析中峰值2 Rt=5.240 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」)。 12.N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始:B:40%,結束B:40%)純化,得到80
(峰值2,300 mg,不純)及呈固體之83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之80
(150 mg,15%)。在82℃下回流1 小時,自MeCN (3 mL)再結晶83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之83
(150 mg,15%)。83 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.62-3.60 (m, 1H), 2.22-1.89 (m, 6H), 1.64-1.49 (m, 9H), 1.46-1.11 (m, 16H), 0.98-0.86 (m, 10H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.268 min,30-90 AB,純度100%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=4.609 min,AD_3_EtOH_DEA_5_40_25ML,100%de。80 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.63-3.62 (m, 1H), 2.22-1.67 (m, 10H), 1.64-1.36 (m, 12H), 1.16-1.03 (m, 8H), 0.98-0.80 (m, 11H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.276 min,30-90 AB,純度99%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=5.236 min,AD_3_EtOH_DEA_5_40_25ML,100%de。 13. 在Ar下向80
(150 mg,0.372 mmol)於THF (3 mL)及MeOH (3 mL)中之溶液添加無水Pd(OH)2
(200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物12 h,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用DCM (3 × 50 mL)洗滌。真空濃縮濾液以提供油狀物。殘餘物藉由矽膠層析(PE/EtOAc = 8/1至5/1)純化,得到呈固體之74
(20 mg,13%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.63-3.61 (m, 1H), 1.96-1.85 (m, 2H), 1.78-1.50 (m, 8H), 1.45-1.19 (s, 12H), 1.17-1.00 (m, 11H), 0.98-0.83 (m, 11H), 0.72-0.59 (m, 3H)。 在2.0 min層析中,LCMS
Rt=1.333 min,30-90 AB,純度99%,C27
H44
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 75 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (75) 之合成  1.
向15_3a
(1 g,2.88 mmol)於DCM (10 mL)中之溶液添加DMP (2.44 g,5.76 mmol)。此後,在25℃下攪拌反應物10 min。反應混合物藉由添加NaHCO3
飽和水溶液(20 mL)及Na2
S2
O3
飽和水溶液(20 mL)淬滅,用DCM (2×50 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 50 mL)及鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-500-2-9_1
(1 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.57 (br. s, 1H), 5.35-5.25 (m, 1H), 2.50-2.30 (m, 2H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.75-1.65 (m, 1H), 1.65-1.60 (m, 3H), 1.55-1.50 (m, 2H), 1.50-1.40 (m, 2H), 1.40-1.30 (m, 1H), 1.25-1.20 (m, 2H), 1.20-1.15 (m, 2H), 1.15-1.10 (m, 6H), 1.05-0.95 (m, 5H), 0.90-0.70 (m, 1H), 0.68 (s, 3H)。 2.在60℃下攪拌鎂(641 mg,26.4 mmol)及I2
(33.5 mg,0.132 mmol)之混合物,且於N2
下逐滴添加異戊基溴化鎂(2 g,13.2 mmol)於THF (20 mL)中之溶液。此後,在60℃下攪拌反應混合物1 h。反應混合物直接用作異戊基溴化鎂溶液而無需任何純化。在0℃下於N2
下向S-500-2-9_1
(1 g,2.90 mmol)於THF (10 mL)中之溶液添加格林納溶液。此後,在25℃下攪拌反應混合物1 h。反應混合物添加NH4
Cl飽和水溶液(50 mL),用EtOAc (2 × 50 mL)萃取,用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(EtOAc/ PE = 1/4)純化,得到呈固體之不純S-500-2- 9_2
(560 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 5.28-5.25 (m, 1H), 3.90-3.80 (m, 0.25H), 3.68-3.58 (m, 0.75H), 2.48-2.36 (m, 1H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.80-1.75 (m, 1H), 1.75-1.52 (m, 6H) 1.52-1.42 (m, 6H), 1.42-1.32 (m, 3H), 1.32-1.22 (m, 3H), 1.22-1.12 (m, 3H), 1.12-1.02 (m, 2H), 1.01 (s, 3H), 1.00-0.92 (m, 1H), 0.92-0.85 (m, 9H), 0.85-0.77 (m, 1H), 0.69 (s, 3H)。 3.S-500-2-9_2
(560 mg)藉由SFC (管柱:Chiralcel OD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃)純化,得到呈固體之不純30
(160 mg)及呈固體之75
(265 mg,47%)。75 : 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.30 (m, 1H), 3.70-3.60 (m, 1H), 2.50-2.40 (m, 1H), 2.05-1.90 (m, 4H), 1.85-1.75 (m, 2H), 1.75-1.60 (m, 1H), 1.55-1.45 (m, 8H), 1.45-1.25 (m, 8H), 1.25-1.10 (m, 4H), 1.10-1.05 (m, 2H), 1.02 (s, 3H), 0.99-0.91 (m, 3H), 0.91-0.89 (m, 4H), 0.88 (s, 3H), 0.69 (s, 3H)。 在1.5 min層析中,LCMS
Rt=1.162 min,5-95 AB,純度99%,C28
H45
[M+H-2H2
O]+
之MS ESI計算值381,實驗值381。實例 76 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((S)-1-(1- 羥基環丙基 ) 乙基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (76) 之合成
1.
向15_3a
(1 g,2.88 mmol)於DCM (10 mL)中之溶液添加DMP (2.44 g,5.76 mmol)。此後,在25℃下攪拌反應物10 min。反應混合物藉由添加NaHCO3
飽和水溶液(20 mL)及Na2
S2
O3
飽和水溶液(20 mL)淬滅,用DCM (2×50 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 50 mL)及鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到呈固體之S-500-2-9_1
(1 g,粗產物)。 1 H NMR
(400 MHz, CDCl3
) δ 9.57 (br. s, 1H), 5.35-5.25 (m, 1H), 2.50-2.30 (m, 2H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.75-1.65 (m, 1H), 1.65-1.60 (m, 3H), 1.55-1.50 (m, 2H), 1.50-1.40 (m, 2H), 1.40-1.30 (m, 1H), 1.25-1.20 (m, 2H), 1.20-1.15 (m, 2H), 1.15-1.10 (m, 6H), 1.05-0.95 (m, 5H), 0.90-0.70 (m, 1H), 0.68 (s, 3H)。 2.在60℃下攪拌鎂(641 mg,26.4 mmol)及I2
(33.5 mg,0.132 mmol)之混合物,且於N2
下逐滴添加異戊基溴化鎂(2 g,13.2 mmol)於THF (20 mL)中之溶液。此後,在60℃下攪拌反應混合物1 h。反應混合物直接用作異戊基溴化鎂溶液而無需任何純化。在0℃下於N2
下向S-500-2-9_1
(1 g,2.90 mmol)於THF (10 mL)中之溶液添加格林納溶液。此後,在25℃下攪拌反應混合物1 h。反應混合物添加NH4
Cl飽和水溶液(50 mL),用EtOAc (2 × 50 mL)萃取,用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮,得到粗產物。粗產物藉由矽膠管柱(EtOAc/ PE = 1/4)純化,得到呈固體之不純S-500-2- 9_2
(560 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 5.28-5.25 (m, 1H), 3.90-3.80 (m, 0.25H), 3.68-3.58 (m, 0.75H), 2.48-2.36 (m, 1H), 2.05-1.95 (m, 3H),1.95-1.80 (m, 1H), 1.80-1.75 (m, 1H), 1.75-1.52 (m, 6H) 1.52-1.42 (m, 6H), 1.42-1.32 (m, 3H), 1.32-1.22 (m, 3H), 1.22-1.12 (m, 3H), 1.12-1.02 (m, 2H), 1.01 (s, 3H), 1.00-0.92 (m, 1H), 0.92-0.85 (m, 9H), 0.85-0.77 (m, 1H), 0.69 (s, 3H)。 3.S-500-2-9_2
(560 mg)藉由SFC (管柱:Chiralcel OD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃)純化,得到呈固體之不純30
(160 mg)及呈固體之75
(265 mg,47%)。75 : 1 H NMR
(400 MHz, CDCl3
) δ 5.35-5.30 (m, 1H), 3.70-3.60 (m, 1H), 2.50-2.40 (m, 1H), 2.05-1.90 (m, 4H), 1.85-1.75 (m, 2H), 1.75-1.60 (m, 1H), 1.55-1.45 (m, 8H), 1.45-1.25 (m, 8H), 1.25-1.10 (m, 4H), 1.10-1.05 (m, 2H), 1.02 (s, 3H), 0.99-0.91 (m, 3H), 0.91-0.89 (m, 4H), 0.88 (s, 3H), 0.69 (s, 3H)。 在1.5 min層析中,LCMS
Rt=1.162 min,5-95 AB,純度99%,C28
H45
[M+H-2H2
O]+
之MS ESI計算值381,實驗值381。實例 76 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((S)-1-(1- 羥基環丙基 ) 乙基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (76) 之合成  1.向N-8-7_1
(500 mg,1.38 mmol)於MeCN (30 mL)中之溶液添加NaClO2
(374 mg,4.14 mmol),TEMPO (645 mg,4.14 mmol)及NaClO (10 mL,10%於水中)。在50℃下攪拌混合物48小時之後,出現固體。藉由過濾收集固體,且用DCM (5 mL)濕磨,得到呈固體之N-8-26_1
(180 mg,34%)。 1 H NMR
(400 MHz, MeOD) δ 2.30-2.20 (m, 1H), 1.98-1.91 (m,1H), 1.86-1.72 (m, 1H), 1.71-1.63 (m, 1H), 1.62-1.49 (m, 8H), 1.43-1.31 (m, 4H), 1.30-1.20 (m, 4H), 1.19-1.07 (m, 8H), 1.06-0.86 (m, 8H), 0.75-0.65 (m, 4H)。 2.向N-8-26_1
(180 mg,0.477 mmol)於DMF (5 mL)中之溶液添加K2
CO3
(328 mg,2.38 mmol)及MeI (686 mg,4.77 mmol)。在20℃下攪拌16小時之後,用50% NH4
Cl (20 mL)淬滅混合物且用EtOAc (3 × 10 mL)萃取。合併之有機相用LiCl (3%於水中,30 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之N-8-26_2
(160 mg,86%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.63 (s, 3H), 2.45-2.36 (m, 1H), 1.92-1.85 (m, 1H), 1.74-1.58 (m, 6H), 1.56-1.46 (m, 4H), 1.42-1.19 (m, 9H), 1.18-1.15 (m, 3H), 1.13-0.93 (m, 4H), 0.91-0.84 (m, 4H), 0.82 (s, 3H), 0.70-0.61 (m, 4H)。 3.在20℃下向N-8-26_2
(80 mg,0.204 mmol)於THF (2 mL)中之溶液添加Ti(i-PrO)4
(57.9 mg,0.204 mmol)及EtMgBr (0.204 mL,3 M於Et2
O,0.612 mmol)。在20℃下攪拌30分鐘之後,用NH4
Cl (30 mL)飽和溶液淬滅反應混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮得到粗產物,其藉由矽膠管柱(0-10% EtOAc/ PE)純化,得到呈固體之76
(16 mg,20%)。 1 H NMR
(400 MHz, CDCl3
) δ 1.98-1.86 (m, 2H), 1.69-1.58 (m, 6H), 1.54-1.44 (m, 3H), 1.44-1.29 (m, 4H), 1.28-1.18 (m, 4H), 1.18-1.10 (m, 5H), 1.09-0.93 (m, 4H), 0.91-0.81 (m, 9H), 0.71-0.57 (m, 6H), 0.31-0.24 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.184 min,30-90AB_E,純度100%,C26
H41
[M+H]+
之MS ESI計算值353,實驗值353。實例 77 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (77) 之合成
1.向N-8-7_1
(500 mg,1.38 mmol)於MeCN (30 mL)中之溶液添加NaClO2
(374 mg,4.14 mmol),TEMPO (645 mg,4.14 mmol)及NaClO (10 mL,10%於水中)。在50℃下攪拌混合物48小時之後,出現固體。藉由過濾收集固體,且用DCM (5 mL)濕磨,得到呈固體之N-8-26_1
(180 mg,34%)。 1 H NMR
(400 MHz, MeOD) δ 2.30-2.20 (m, 1H), 1.98-1.91 (m,1H), 1.86-1.72 (m, 1H), 1.71-1.63 (m, 1H), 1.62-1.49 (m, 8H), 1.43-1.31 (m, 4H), 1.30-1.20 (m, 4H), 1.19-1.07 (m, 8H), 1.06-0.86 (m, 8H), 0.75-0.65 (m, 4H)。 2.向N-8-26_1
(180 mg,0.477 mmol)於DMF (5 mL)中之溶液添加K2
CO3
(328 mg,2.38 mmol)及MeI (686 mg,4.77 mmol)。在20℃下攪拌16小時之後,用50% NH4
Cl (20 mL)淬滅混合物且用EtOAc (3 × 10 mL)萃取。合併之有機相用LiCl (3%於水中,30 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之N-8-26_2
(160 mg,86%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.63 (s, 3H), 2.45-2.36 (m, 1H), 1.92-1.85 (m, 1H), 1.74-1.58 (m, 6H), 1.56-1.46 (m, 4H), 1.42-1.19 (m, 9H), 1.18-1.15 (m, 3H), 1.13-0.93 (m, 4H), 0.91-0.84 (m, 4H), 0.82 (s, 3H), 0.70-0.61 (m, 4H)。 3.在20℃下向N-8-26_2
(80 mg,0.204 mmol)於THF (2 mL)中之溶液添加Ti(i-PrO)4
(57.9 mg,0.204 mmol)及EtMgBr (0.204 mL,3 M於Et2
O,0.612 mmol)。在20℃下攪拌30分鐘之後,用NH4
Cl (30 mL)飽和溶液淬滅反應混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機層用鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾且真空濃縮得到粗產物,其藉由矽膠管柱(0-10% EtOAc/ PE)純化,得到呈固體之76
(16 mg,20%)。 1 H NMR
(400 MHz, CDCl3
) δ 1.98-1.86 (m, 2H), 1.69-1.58 (m, 6H), 1.54-1.44 (m, 3H), 1.44-1.29 (m, 4H), 1.28-1.18 (m, 4H), 1.18-1.10 (m, 5H), 1.09-0.93 (m, 4H), 0.91-0.81 (m, 9H), 0.71-0.57 (m, 6H), 0.31-0.24 (m, 1H)。 在2.0 min層析中,LCMS
Rt=1.184 min,30-90AB_E,純度100%,C26
H41
[M+H]+
之MS ESI計算值353,實驗值353。實例 77 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (77) 之合成  1.在60℃下向Mg (4.35 g,179 mmol)及I2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。用THF (10 mL)稀釋混合物且直接使用。在0℃下於N2
下向S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。向混合物添加NH4
Cl (20 mL,飽和水溶液)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH3
CN (10 mL)再結晶,得到呈固體之77
(255 mg,21%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.306 min,30-90 AB,純度100%,C29
H49
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 78 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1S,2S)-1- 環戊基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (78) 之合成
1.在60℃下向Mg (4.35 g,179 mmol)及I2
(20 mg)於THF (2 mL)中之懸浮液逐滴添加1-溴-3-甲基丁烷(11.7 g,78 mmol)於THF (8 mL)中之溶液。在60℃下攪拌混合物1小時。用THF (10 mL)稀釋混合物且直接使用。在0℃下於N2
下向S-200-INT_5E
(1.0 g,2.78 mmol)於THF (5 mL)中之溶液添加新鮮製備的異戊基溴化鎂(19.5 mL,3.9 M於THF中,76 mmol)。在0℃下攪拌混合物1小時。向混合物添加NH4
Cl (20 mL,飽和水溶液)。用EtOAc (2 × 30 mL)萃取混合物。合併之有機相用鹽水(100 mL)洗滌,經Na2
SO4
乾燥,真空濃縮,藉由矽膠(PE/EtOAc=20/1至10/1)純化,且自CH3
CN (10 mL)再結晶,得到呈固體之77
(255 mg,21%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.66-3.59 (m, 1H), 2.42-2.32 (m, 1H), 2.07-1.85 (m, 4H), 1.77-1.58 (m, 4H), 1.55-1.38 (m, 10H), 1.38-1.19 (m, 5 H), 1.19-1.00 (m, 8H), 1.00-0.81 (m, 13H), 0.69 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.306 min,30-90 AB,純度100%,C29
H49
O [M+H-H2
O]+
之MS ESI計算值413,實驗值413。實例 78 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1S,2S)-1- 環戊基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (78) 之合成  1. 向N-8-15_2
(240 mg,0.559 mmol)於MeOH (3 mL)及THF (2 mL)中之混合物添加NaBH4
(550 mg,14.5 mmol)。在15℃下攪拌混合物0.5 h。添加另一批次之NaBH4
(550 mg,14.5 mmol)。再攪拌反應混合物1小時。向反應混合物添加水(5 mL)。用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之58
(7 mg,5%),且78
(50 mg,不純)藉由閃蒸塔(0-5% EtOAc/PE)進一步純化,得到呈固體之78
(17 mg,12%)。78 : 1 H NMR
(400 MHz, CDCl3
) δ 3.38-3.47 (m, 1H), 2.01-1.82 (m, 4H), 1.71-1.53 (m, 11H), 1.53-1.48 (m, 4H), 1.48-1.30 (m, 5H), 1.30-1.11 (m, 7H), 1.11-0.98 (m, 5H),0.98-0.85 (m, 7 H), 0.85-0.80 (m, 3 H), 0.65 (s, 3H)。 在2 min層析中,LCMS
Rt=1.358 min,30-90AB_7MIN_E,純度100%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。 在10 min層析中,HPLC
Rt=6.093 min,50-100AB_10 MIN.M,純度98%。實例 79 : (1R,3S,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (79) 之合成
1. 向N-8-15_2
(240 mg,0.559 mmol)於MeOH (3 mL)及THF (2 mL)中之混合物添加NaBH4
(550 mg,14.5 mmol)。在15℃下攪拌混合物0.5 h。添加另一批次之NaBH4
(550 mg,14.5 mmol)。再攪拌反應混合物1小時。向反應混合物添加水(5 mL)。用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之58
(7 mg,5%),且78
(50 mg,不純)藉由閃蒸塔(0-5% EtOAc/PE)進一步純化,得到呈固體之78
(17 mg,12%)。78 : 1 H NMR
(400 MHz, CDCl3
) δ 3.38-3.47 (m, 1H), 2.01-1.82 (m, 4H), 1.71-1.53 (m, 11H), 1.53-1.48 (m, 4H), 1.48-1.30 (m, 5H), 1.30-1.11 (m, 7H), 1.11-0.98 (m, 5H),0.98-0.85 (m, 7 H), 0.85-0.80 (m, 3 H), 0.65 (s, 3H)。 在2 min層析中,LCMS
Rt=1.358 min,30-90AB_7MIN_E,純度100%,C29
H47
[M+H-2H2
O]+
之MS ESI計算值395,實驗值395。 在10 min層析中,HPLC
Rt=6.093 min,50-100AB_10 MIN.M,純度98%。實例 79 : (1R,3S,4S)-4-((3S,5S,8R,9S,10S,13S,14S,17R)-3- 羥基 -10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -17- 基 )-1- 苯基戊烷 -1,3- 二醇 (79) 之合成  79
之合成描述於實例13中。79 : 1 H NMR
(400 MHz, CDCl3
) δ 7.43-7.28 (m, 5H), 5.05-4.94 (m, 1H), 4.04-3.91 (m, 1H), 2.51 (brs, 1H), 2.07-1.78 (m, 6H), 1.70-1.61 (m, 4H), 1.51-1.41 (m, 3H), 1.39-1.12 (m, 11H), 1.05-0.98 (m, 2H), 0.91-0.81 (m, 7H), 0.71-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.298 min,10-80AB_2MIN_E,純度96.7%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.483 min,IC-3_MeOH(DEA)_40_2.5ML,100%de。13 : 1 H NMR
(400 MHz, CDCl3
) δ 7.40-7.28 (m, 5H), 5.12-5.07 (m, 1H), 3.95-3.88 (m, 1H), 2.76 (brs, 1H), 2.08-1.78 (m, 6H), 1.75-1.60 (m, 5H), 1.51-1.38 (m, 4H), 1.36-1.09 (m, 9H), 1.00-0.89 (m, 6H), 0.83 (s, 3H), 0.71-0.64 (m, 1H), 0.63 (s, 3H)。 在2 min層析中,LCMS
Rt=1.309 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.683 min,IC-3_MeOH(DEA)_40_2.5ML,98.94%de。 在8 min層析中,SFC
Rt=4.785 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,94.03%de。實例 80 : (3S,8R,9S,10R,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (80) 之合成
79
之合成描述於實例13中。79 : 1 H NMR
(400 MHz, CDCl3
) δ 7.43-7.28 (m, 5H), 5.05-4.94 (m, 1H), 4.04-3.91 (m, 1H), 2.51 (brs, 1H), 2.07-1.78 (m, 6H), 1.70-1.61 (m, 4H), 1.51-1.41 (m, 3H), 1.39-1.12 (m, 11H), 1.05-0.98 (m, 2H), 0.91-0.81 (m, 7H), 0.71-0.60 (m, 4H)。 在2 min層析中,LCMS
Rt=1.298 min,10-80AB_2MIN_E,純度96.7%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在10 min層析中,SFC
Rt=1.483 min,IC-3_MeOH(DEA)_40_2.5ML,100%de。13 : 1 H NMR
(400 MHz, CDCl3
) δ 7.40-7.28 (m, 5H), 5.12-5.07 (m, 1H), 3.95-3.88 (m, 1H), 2.76 (brs, 1H), 2.08-1.78 (m, 6H), 1.75-1.60 (m, 5H), 1.51-1.38 (m, 4H), 1.36-1.09 (m, 9H), 1.00-0.89 (m, 6H), 0.83 (s, 3H), 0.71-0.64 (m, 1H), 0.63 (s, 3H)。 在2 min層析中,LCMS
Rt=1.309 min,10-80AB_2MIN_E,純度100%,C31
H45
F3
O3
Na [M+Na]+
之MS ESI計算值545,實驗值545。 在5 min層析中,SFC
Rt=1.683 min,IC-3_MeOH(DEA)_40_2.5ML,98.94%de。 在8 min層析中,SFC
Rt=4.785 min,AD_MEOH(DEA)_5_40_2,8ML_8MIN,94.03%de。實例 80 : (3S,8R,9S,10R,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (80) 之合成 
 1.N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始:B:40%,結束B:40%)純化,得到80
(峰值2,300 mg,不純)及呈固體之83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空中過濾懸浮液,得到呈固體之80
(150 mg,15%)。在82℃下回流1 小時,自MeCN (3 mL)再結晶83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空中過濾懸浮液,得到呈固體之83
(150 mg,15%)。80 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.63-3.62 (m, 1H), 2.22-1.67 (m, 10H), 1.64-1.36 (m, 12H), 1.16-1.03 (m, 8H), 0.98-0.80 (m, 11H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.276 min,30-90 AB,純度99%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=5.236 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 81 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (81) 之合成
1.N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始:B:40%,結束B:40%)純化,得到80
(峰值2,300 mg,不純)及呈固體之83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空中過濾懸浮液,得到呈固體之80
(150 mg,15%)。在82℃下回流1 小時,自MeCN (3 mL)再結晶83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空中過濾懸浮液,得到呈固體之83
(150 mg,15%)。80 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.63-3.62 (m, 1H), 2.22-1.67 (m, 10H), 1.64-1.36 (m, 12H), 1.16-1.03 (m, 8H), 0.98-0.80 (m, 11H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.276 min,30-90 AB,純度99%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=5.236 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 81 : (3S,8S,9S,10R,13S,14S,17R)-3- 乙基 -10,13- 二甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (81) 之合成  1. S-500-6-1_2
(350 mg)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
.H2
O EtOH,梯度:35%至35%,流速 (mL/min):60 mL/min,25℃)純化,得到81
(峰值1,130 mg,37%)及呈白色固體之65
(峰值2,180 mg,52%)。81 : 1 H NMR
(400 MHz, CDCl3) δ 5.34-5.24 (m, 1H), 4.09-4.00 (m, 1H), 2.43-2.33 (m, 1H), 2.14 (d,J
= 4Hz, 1H), 2.07-1.80 (m, 5H), 1.77-1.55 (m, 5H), 1.53-1.30 (m, 7H), 1.28-1.00 (m, 11H), 1.00-0.91 (m, 1H), 0.85 (t,J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.220 min,30-90 AB,純度100%,C25
H38
F3
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,SFC
__E1峰值1:Rt=4.561 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),100%de。實例 82 : (3S,5R,8R,9R , 10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (82) 之合成
1. S-500-6-1_2
(350 mg)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
.H2
O EtOH,梯度:35%至35%,流速 (mL/min):60 mL/min,25℃)純化,得到81
(峰值1,130 mg,37%)及呈白色固體之65
(峰值2,180 mg,52%)。81 : 1 H NMR
(400 MHz, CDCl3) δ 5.34-5.24 (m, 1H), 4.09-4.00 (m, 1H), 2.43-2.33 (m, 1H), 2.14 (d,J
= 4Hz, 1H), 2.07-1.80 (m, 5H), 1.77-1.55 (m, 5H), 1.53-1.30 (m, 7H), 1.28-1.00 (m, 11H), 1.00-0.91 (m, 1H), 0.85 (t,J
= 8 Hz, 3H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.220 min,30-90 AB,純度100%,C25
H38
F3
O [M+H-H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,SFC
__E1峰值1:Rt=4.561 min,AD_3_EtOH_DEA_5_40_25ML (「管柱:Chiralpak AD-3 150×4.6mm I.D.,3um 移動相:A:CO2 B:乙醇(0.05% DEA),梯度:在5 min內B為5%至40%且保持40% 2.5 min,隨後B為5%持續2.5 min,流速:2.5 mL/min,管柱溫度:35℃」),100%de。實例 82 : (3S,5R,8R,9R , 10S,13S,14S,17R)-3- 乙基 -17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-13- 甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (82) 之合成  1.在Ar下向44
(200 mg,0.480 mmol)於MeOH/THF (10 mL/10 mL)中之溶液添加Pd/C (無水,200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物48小時,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用THF (100 mL)洗滌。濃縮濾液,得到呈固體之28
(30 mg,15%)及呈固體之82
(30 mg,15%)。82 : 1 H NMR
(400MHz, CDCl3) δ 3.63-3.61 (m, 1H), 2.13-2.00 (m, 1H), 1.99-1.81 (m, 2H), 1.72-1.57 (m, 6H), 1.54-1.34 (m, 11H), 1.33-1.16 (m, 7H), 1.15-0.96 (m, 5H), 0.92-0.85 (m, 13H), 0.81-0.69 (m, 1H), 0.67 (s, 3H)。 在2 min層析中,LCMS
Rt=1.348 min,30-90AB,純度100%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 83 : (3S,8R,9S,10R,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (83) 之合成
1.在Ar下向44
(200 mg,0.480 mmol)於MeOH/THF (10 mL/10 mL)中之溶液添加Pd/C (無水,200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物48小時,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用THF (100 mL)洗滌。濃縮濾液,得到呈固體之28
(30 mg,15%)及呈固體之82
(30 mg,15%)。82 : 1 H NMR
(400MHz, CDCl3) δ 3.63-3.61 (m, 1H), 2.13-2.00 (m, 1H), 1.99-1.81 (m, 2H), 1.72-1.57 (m, 6H), 1.54-1.34 (m, 11H), 1.33-1.16 (m, 7H), 1.15-0.96 (m, 5H), 0.92-0.85 (m, 13H), 0.81-0.69 (m, 1H), 0.67 (s, 3H)。 在2 min層析中,LCMS
Rt=1.348 min,30-90AB,純度100%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 83 : (3S,8R,9S,10R,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (83) 之合成  1.N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始:B:40%,結束B:40%)純化,得到80
(峰值2,300 mg,不純)及呈固體之83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之80
(150 mg,15%)。在82℃下回流1 h,自MeCN (3 mL)再結晶83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之83
(150 mg,15%)。83 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.62-3.60 (m, 1H), 2.22-1.89 (m, 6H), 1.64-1.49 (m, 9H), 1.46-1.11 (m, 16H), 0.98-0.86 (m, 10H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.268 min,30-90 AB,純度100%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=4.609 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 84 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (84) 之合成
1.N-4-14_7
(1 g,2.48 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始:B:40%,結束B:40%)純化,得到80
(峰值2,300 mg,不純)及呈固體之83
(峰值1,250 mg,不純)。在82℃下回流1小時,自MeCN (4 mL)再結晶80
(300 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之80
(150 mg,15%)。在82℃下回流1 h,自MeCN (3 mL)再結晶83
(250 mg,不純)。將攪拌的混合物冷卻至25℃。在真空下過濾懸浮液,得到呈固體之83
(150 mg,15%)。83 : 1 H NMR
(400 MHz, CDCl3
)δ 5.41-5.39 (m, 1H), 3.62-3.60 (m, 1H), 2.22-1.89 (m, 6H), 1.64-1.49 (m, 9H), 1.46-1.11 (m, 16H), 0.98-0.86 (m, 10H), 0.70 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.268 min,30-90 AB,純度100%,C27
H42
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在10 min層析中,SFC
Rt=4.609 min,AD_3_EtOH_DEA_5_40_25ML,100%de。實例 84 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (84) 之合成 
 1.向32
(80 mg,0.18 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(160 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到67
(10 mg,12%)及呈固體之84
(30 mg,37%)。84 : 1 H NMR
(400 MHz, CDCl3
) δ 3.75-3.66 (m, 1H), 2.00-1.90 (m, 2H), 1.86-1.75 (m, 2H), 1.73-1.55 (m, 11H), 1.53-1.26 (m, 9H), 1.25-1.15 (m, 6H), 1.14-1.03 (m, 5H), 1.02-0.92 (m, 3H), 0.91-0.85 (m, 6H), 0.82 (s, 3H), 0.72-0.58 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.518 min,30-90AB_E,純度100%,C30
H49
[M+H-H2
O]+
之MS ESI計算值409,實驗值409。實例 85 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (85) 之合成
1.向32
(80 mg,0.18 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(160 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到67
(10 mg,12%)及呈固體之84
(30 mg,37%)。84 : 1 H NMR
(400 MHz, CDCl3
) δ 3.75-3.66 (m, 1H), 2.00-1.90 (m, 2H), 1.86-1.75 (m, 2H), 1.73-1.55 (m, 11H), 1.53-1.26 (m, 9H), 1.25-1.15 (m, 6H), 1.14-1.03 (m, 5H), 1.02-0.92 (m, 3H), 0.91-0.85 (m, 6H), 0.82 (s, 3H), 0.72-0.58 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.518 min,30-90AB_E,純度100%,C30
H49
[M+H-H2
O]+
之MS ESI計算值409,實驗值409。實例 85 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (85) 之合成  1.在15℃下將NaBH4
(0.96 g,25.4 mmol)逐份添加至N-4-1_8
(0.6 g,1.27 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌混合物30 min。向混合物添加NH4
Cl (50 ml,10%)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到不純42
及85
。在15℃下自MeCN (10 mL)濕磨42
且在真空中乾燥,得到呈固體之42
(153 mg,25%)。85
藉由閃蒸塔(0-15% EtOAc/PE)純化以提供油狀物,用MeCN (5 mL)及水(5 mL)處理該油狀物,真空濃縮,得到呈固體之85
(70 mg,12%)。85 : 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.55 (m, 1H), 2.10-1.91 (m, 3H), 1.88-1.78 (m, 1H), 1.72-1.55 (m, 6H), 1.50-1.38 (m, 9H), 1.37-0.95 (m, 10H), 0.94-0.79 (m, 13H), 0.75-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.343 min,30-90_AB_E,純度100%,C28
H46
F3
O [M+H-H2
O]+
之MS ESI計算值 455,實驗值455。 在10.0 min層析中,HPLC
Rt=5.14 min,50-100_AB_E,純度98.56%。實例 86 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1-( 四氫 -2H- 哌喃 -4- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (86) 之合成
1.在15℃下將NaBH4
(0.96 g,25.4 mmol)逐份添加至N-4-1_8
(0.6 g,1.27 mmol)於THF (10 mL)及MeOH (5 mL)中之溶液中。在15℃下攪拌混合物30 min。向混合物添加NH4
Cl (50 ml,10%)。用EtOAc (2 × 50 mL)萃取混合物。合併之有機層經Na2
SO4
乾燥,過濾且真空濃縮,且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到不純42
及85
。在15℃下自MeCN (10 mL)濕磨42
且在真空中乾燥,得到呈固體之42
(153 mg,25%)。85
藉由閃蒸塔(0-15% EtOAc/PE)純化以提供油狀物,用MeCN (5 mL)及水(5 mL)處理該油狀物,真空濃縮,得到呈固體之85
(70 mg,12%)。85 : 1 H NMR
(400 MHz, CDCl3
) δ 3.66-3.55 (m, 1H), 2.10-1.91 (m, 3H), 1.88-1.78 (m, 1H), 1.72-1.55 (m, 6H), 1.50-1.38 (m, 9H), 1.37-0.95 (m, 10H), 0.94-0.79 (m, 13H), 0.75-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt=1.343 min,30-90_AB_E,純度100%,C28
H46
F3
O [M+H-H2
O]+
之MS ESI計算值 455,實驗值455。 在10.0 min層析中,HPLC
Rt=5.14 min,50-100_AB_E,純度98.56%。實例 86 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1-( 四氫 -2H- 哌喃 -4- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (86) 之合成  1.在70℃下向Mg (486 mg,20 mmol)及I2
(1 mg)之混合物逐滴添加4-氯四氫-2H-哌喃(1.2 g,10 mmol)於THF (5 mL)中之溶液。在50℃下攪拌混合物0.5 h,用THF (5 mL)稀釋且直接使用。在0℃下於N2
下向(四氫-2H-哌喃-4-基)氯化鎂(4.14 mL,1 M於THF中)之溶液添加含N-8-7_1
(500 mg,1.38 mmol)之THF (5 mL)。在15℃下再攪拌混合物18小時。用飽和NH4
Cl (5 mL)淬滅反應混合物,且用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(5 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈固體之N-8-17_1
(350mg,57%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.08-3.91 (m, 2H), 3.43-3.31 (m, 2H), 3.31-3.25 (m, 1H), 1.98-1.91 (m, 2H), 1.91-1.80 (m, 1H),1.70-1.50 (m, 10H), 1.50-1.41 (m, 2H), 1.41-1.32 (m, 5H), 1.32-1.15 (m, 9H), 1.15-0.92 (m, 6H), 0.92-0.83 (m, 7H), 0.65 (s, 3H)。2.
向N-8-17_1
(300 mg,0.671 mmol)於DCM (5 mL)中之溶液添加DMP (0.852 g,2.01 mmol)。在15℃下攪拌10 min之後,用飽和NaHCO3 (20 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層,且用DCM (2 × 20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 20 mL)、飽和NaHCO3 (20 mL)、鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-30% EtOAc/PE)純化,得到呈固體之N-8-17_3
(200 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 4.08-3.91 (m, 2H), 3.50-3.31 (m, 2H), 2.73-2.51 (m, 2H), 1.98-1.79 (m, 1H), 1.79-1.42 (m, 16H), 1.42-1.18 (m, 7H), 1.18-0.93 (m, 8H), 0.93-0.79 (m, 6H), 0.68 (s, 4H)。 3.在0℃下向N-8-17_3
(200 mg,0.449 mmol)於THF (5 mL)中之混合物添加LiAlH4 (50.9 mg,1.34 mmol)。在15℃下攪拌0.5 h之後,用水(3 mL)淬滅反應混合物,且用EtOAc (2 × 10 mL)萃取。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之86
(23mg,11%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.08-3.91 (m, 2H), 3.41-3.31 (m, 2H), 3.31-3.22(m, 1H), 2.01-1.79 (m, 3H), 1.70-1.61 (m, 1H), 1.61-1.53 (m, 8H), 1.53-1.51 (m, 1H), 1.51-1.39 (m, 5H), 1.39-1.13 (m, 8H), 1.13-0.92 (m, 5H), 0.92-0.85 (m, 7H), 0.82 (s, 3H), 0.66 (s, 4H)。 在7 min層析中,LCMS
Rt=4.832 min,30-90AB_7MIN_E,純度100%,C29
H47
O [M+H-2H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,HPLC
Rt=6.338 min,30-90AB_1.2 mL e. Met,100%純度。實例 87 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (87) 之合成
1.在70℃下向Mg (486 mg,20 mmol)及I2
(1 mg)之混合物逐滴添加4-氯四氫-2H-哌喃(1.2 g,10 mmol)於THF (5 mL)中之溶液。在50℃下攪拌混合物0.5 h,用THF (5 mL)稀釋且直接使用。在0℃下於N2
下向(四氫-2H-哌喃-4-基)氯化鎂(4.14 mL,1 M於THF中)之溶液添加含N-8-7_1
(500 mg,1.38 mmol)之THF (5 mL)。在15℃下再攪拌混合物18小時。用飽和NH4
Cl (5 mL)淬滅反應混合物,且用EtOAc (2 × 10 mL)萃取所得混合物。合併之有機層用鹽水(5 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈固體之N-8-17_1
(350mg,57%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.08-3.91 (m, 2H), 3.43-3.31 (m, 2H), 3.31-3.25 (m, 1H), 1.98-1.91 (m, 2H), 1.91-1.80 (m, 1H),1.70-1.50 (m, 10H), 1.50-1.41 (m, 2H), 1.41-1.32 (m, 5H), 1.32-1.15 (m, 9H), 1.15-0.92 (m, 6H), 0.92-0.83 (m, 7H), 0.65 (s, 3H)。2.
向N-8-17_1
(300 mg,0.671 mmol)於DCM (5 mL)中之溶液添加DMP (0.852 g,2.01 mmol)。在15℃下攪拌10 min之後,用飽和NaHCO3 (20 mL)淬滅反應混合物直至含水層之pH值變為約9為止。過濾混合物。分離DCM層,且用DCM (2 × 20 mL)萃取水相。合併之有機相用Na2
S2
O3
飽和水溶液(3 × 20 mL)、飽和NaHCO3 (20 mL)、鹽水(50 mL)洗滌,經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (0-30% EtOAc/PE)純化,得到呈固體之N-8-17_3
(200 mg)。 1 H NMR
(400 MHz, CDCl3
) δ 4.08-3.91 (m, 2H), 3.50-3.31 (m, 2H), 2.73-2.51 (m, 2H), 1.98-1.79 (m, 1H), 1.79-1.42 (m, 16H), 1.42-1.18 (m, 7H), 1.18-0.93 (m, 8H), 0.93-0.79 (m, 6H), 0.68 (s, 4H)。 3.在0℃下向N-8-17_3
(200 mg,0.449 mmol)於THF (5 mL)中之混合物添加LiAlH4 (50.9 mg,1.34 mmol)。在15℃下攪拌0.5 h之後,用水(3 mL)淬滅反應混合物,且用EtOAc (2 × 10 mL)萃取。合併之有機層用鹽水(10 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-5% EtOAc/PE)純化,得到呈固體之86
(23mg,11%)。 1 H NMR
(400 MHz, CDCl3
) δ 4.08-3.91 (m, 2H), 3.41-3.31 (m, 2H), 3.31-3.22(m, 1H), 2.01-1.79 (m, 3H), 1.70-1.61 (m, 1H), 1.61-1.53 (m, 8H), 1.53-1.51 (m, 1H), 1.51-1.39 (m, 5H), 1.39-1.13 (m, 8H), 1.13-0.92 (m, 5H), 0.92-0.85 (m, 7H), 0.82 (s, 3H), 0.66 (s, 4H)。 在7 min層析中,LCMS
Rt=4.832 min,30-90AB_7MIN_E,純度100%,C29
H47
O [M+H-2H2
O]+
之MS ESI計算值411,實驗值411。 在10 min層析中,HPLC
Rt=6.338 min,30-90AB_1.2 mL e. Met,100%純度。實例 87 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3R)-4-(4,4- 二甲基環己基 )-3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (87) 之合成  1.向12
(100 mg,0.206 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(150 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之87
(12 mg,12%)及20
(11 mg,11%)。87 : 1 H NMR
(400 MHz, CDCl3
) δ 3.82-3.75 (m, 1H), 2.00-1.83 (m, 2H), 1.80-1.58 (m, 7H), 1.52-1.42 (m,4H), 1.40-1.27 (m, 10H), 1.25-1.14 (m, 10H), 1.13-0.98 (m, 6H), 0.96 (s, 3H), 0.94-0.82 (m, 12H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.734 min,30-90AB_E,純度100%,C33
H55
[M+H-2H2
O]+
之MS ESI計算值451,實驗值451。實例 88 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (88) 之合成
1.向12
(100 mg,0.206 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(150 mg,無水)。在50℃下於H2
(50 Psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之87
(12 mg,12%)及20
(11 mg,11%)。87 : 1 H NMR
(400 MHz, CDCl3
) δ 3.82-3.75 (m, 1H), 2.00-1.83 (m, 2H), 1.80-1.58 (m, 7H), 1.52-1.42 (m,4H), 1.40-1.27 (m, 10H), 1.25-1.14 (m, 10H), 1.13-0.98 (m, 6H), 0.96 (s, 3H), 0.94-0.82 (m, 12H), 0.67 (s, 3H)。 在2.0 min層析中,LCMS
Rt=1.734 min,30-90AB_E,純度100%,C33
H55
[M+H-2H2
O]+
之MS ESI計算值451,實驗值451。實例 88 : (3S,5S,8R,9R,10S,13S,14S,17R)-17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-3,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (88) 之合成  1.在Ar下向83
(150 mg,0.372 mmol)於MeOH (3 mL)及THF (3 mL)中之溶液添加Pd(OH)2
(200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物12 h,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用DCM (3 × 50 mL)洗滌。真空濃縮濾液以提供油狀物。殘餘物藉由矽膠層析(PE/EtOAc=8/1至5/1)純化,得到呈固體之88
(18 mg,12%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.62-3.59 (m, 1H), 1.97-1.81 (m, 2H), 1.76-1.50 (m, 12H), 1.46-1.28 (m, 5H), 1.24-1.03 (m, 12H), 0.95-0.83 (m, 11H), 0.74-0.57 (m, 5H)。 在1.317 min層析中,LCMS
Rt=2.0 min,30-90 AB,純度99%,C27
H44
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 89 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1-( 吡啶 -3- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (89) 之合成
1.在Ar下向83
(150 mg,0.372 mmol)於MeOH (3 mL)及THF (3 mL)中之溶液添加Pd(OH)2
(200 mg)。將懸浮液真空脫氣且用H2
淨化三次。在50℃下於H2
(50 psi)下攪拌混合物12 h,得到黑色懸浮液。反應混合物經由矽藻土墊過濾且用DCM (3 × 50 mL)洗滌。真空濃縮濾液以提供油狀物。殘餘物藉由矽膠層析(PE/EtOAc=8/1至5/1)純化,得到呈固體之88
(18 mg,12%)。 1 H NMR
(400 MHz, CDCl3
)δ 3.62-3.59 (m, 1H), 1.97-1.81 (m, 2H), 1.76-1.50 (m, 12H), 1.46-1.28 (m, 5H), 1.24-1.03 (m, 12H), 0.95-0.83 (m, 11H), 0.74-0.57 (m, 5H)。 在1.317 min層析中,LCMS
Rt=2.0 min,30-90 AB,純度99%,C27
H44
[M+H-2H2
O]+
之MS ESI計算值369,實驗值369。實例 89 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((1S,2S)-1- 羥基 -1-( 吡啶 -3- 基 ) 丙 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (89) 之合成  1.N-8-19_1 (3340)
(100 mg,0.227 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:50-50% B (A=0.05% NH3/H2O,B=MeOH),流速:80 mL/min)分離,得到7
(峰值1,57 mg,57%)及呈固體之89
(峰值2,8 mg,8%)。 在3 min層析中,SFC
峰值1:Rt=1.798 min且峰值2 Rt=1.985 min,AD-H_3UM_4_5_40_4ML (「Chiralpak AD-3 50×4.6mm I.D.,3um 移動相:A:CO2
,B:異丙醇(0.05% DEA),梯度:在1.4 min內B為5%至40%且保持40% 1.05 min,隨後B為5%持續0.35 min,流速:4 mL/min,管柱溫度:40℃」)。8
之結構藉由x射線晶體分析法確認。實例 90 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1R,2S)-1- 環丙基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (90) 之合成
1.N-8-19_1 (3340)
(100 mg,0.227 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),梯度:50-50% B (A=0.05% NH3/H2O,B=MeOH),流速:80 mL/min)分離,得到7
(峰值1,57 mg,57%)及呈固體之89
(峰值2,8 mg,8%)。 在3 min層析中,SFC
峰值1:Rt=1.798 min且峰值2 Rt=1.985 min,AD-H_3UM_4_5_40_4ML (「Chiralpak AD-3 50×4.6mm I.D.,3um 移動相:A:CO2
,B:異丙醇(0.05% DEA),梯度:在1.4 min內B為5%至40%且保持40% 1.05 min,隨後B為5%持續0.35 min,流速:4 mL/min,管柱溫度:40℃」)。8
之結構藉由x射線晶體分析法確認。實例 90 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((1R,2S)-1- 環丙基 -1- 羥基丙 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (90) 之合成  1.向N-8-13_2
(140 mg,0.347 mmol)於MeOH (1 mL)及THF (1 mL)中之溶液每五分鐘添加五次NaBH4
(1.18 g,17.4 mmol)。在15℃下攪拌混合物30分鐘。用飽和NH4
Cl (50 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之10
(26 mg,19%)及呈固體之90
(12 mg,9%)。90 : 1 H NMR
(400 MHz, CDCl3
) δ 3.00-2.80 (m, 1H), 2.01-1.95 (m, 1H), 1.75-1.60 (m, 5H), 1.47-1.18 (m, 13H), 1.15-0.79 (m, 18H), 0.70-0.60 (m, 4H), 0.58-0.50 (m, 1H), 0.48-0.40 (m, 1H), 0.38-0.30 (m, 1H), 0.24-0.16 (m, 1H)。 在7.0 min層析中,LCMS
Rt=3.796 min,30-90AB_7MIN_E,純度100%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在30 min層析中,HPLC
Rt= 13.689 min,70-90AB_1_30MIN.M,純度98%。實例 91 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥丁 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (91) 之合成
1.向N-8-13_2
(140 mg,0.347 mmol)於MeOH (1 mL)及THF (1 mL)中之溶液每五分鐘添加五次NaBH4
(1.18 g,17.4 mmol)。在15℃下攪拌混合物30分鐘。用飽和NH4
Cl (50 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相經Na2
SO4
乾燥,過濾,濃縮且藉由combi-flash (25% EtOAc/PE)純化,得到呈固體之10
(26 mg,19%)及呈固體之90
(12 mg,9%)。90 : 1 H NMR
(400 MHz, CDCl3
) δ 3.00-2.80 (m, 1H), 2.01-1.95 (m, 1H), 1.75-1.60 (m, 5H), 1.47-1.18 (m, 13H), 1.15-0.79 (m, 18H), 0.70-0.60 (m, 4H), 0.58-0.50 (m, 1H), 0.48-0.40 (m, 1H), 0.38-0.30 (m, 1H), 0.24-0.16 (m, 1H)。 在7.0 min層析中,LCMS
Rt=3.796 min,30-90AB_7MIN_E,純度100%,C27
H43
[M+H-2H2
O]+
之MS ESI計算值367,實驗值367。 在30 min層析中,HPLC
Rt= 13.689 min,70-90AB_1_30MIN.M,純度98%。實例 91 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥丁 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (91) 之合成  91
之合成描述於實例13中。91 : 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.88 (m, 1H), 2.11-2.02 (m, 1H), 2.00 (s, 1H), 1.98-1.88 (m, 2H), 1.85-1.79 (m, 1H), 1.73-1.58 (m, 4H), 1.52-1.20 (m, 11H), 1.19-1.11 (m, 4H), 1.10-1.00 (m, 3H), 0.97-0.89 (m, 4H), 0.85 (s, 3H), 0.75-0.68 (m, 1H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt= 1.155 min,30-90_AB_E,純度100%,C24
H38
F3
O[M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt= 5.23 min,30-90_AB_E,純度98.88%,d.e. 100%。實例 92 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (92) 之合成
91
之合成描述於實例13中。91 : 1 H NMR
(400 MHz, CDCl3
) δ 3.98-3.88 (m, 1H), 2.11-2.02 (m, 1H), 2.00 (s, 1H), 1.98-1.88 (m, 2H), 1.85-1.79 (m, 1H), 1.73-1.58 (m, 4H), 1.52-1.20 (m, 11H), 1.19-1.11 (m, 4H), 1.10-1.00 (m, 3H), 0.97-0.89 (m, 4H), 0.85 (s, 3H), 0.75-0.68 (m, 1H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt= 1.155 min,30-90_AB_E,純度100%,C24
H38
F3
O[M+H-H2
O]+
之MS ESI計算值399,實驗值399。 在10.0 min層析中,HPLC
Rt= 5.23 min,30-90_AB_E,純度98.88%,d.e. 100%。實例 92 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-3- 羥基 -6- 甲基庚 -2- 基 )-3,10,13- 三甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (92) 之合成  1.向N-4-16_1
(1.00 g,2.86 mmol)於DCM (20 mL)中之溶液添加DMP (2.42 g,5.72 mmol)。此後,在15℃下攪拌反應物10 min。向反應混合物添加NaHCO3飽和水溶液(20 mL)及Na2
S2
O3
飽和水溶液(20 mL),接著用DCM (2 × 20 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 60 mL)及鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾且真空下濃縮,得到固體。殘餘物藉由矽膠層析(PE/EtOAc=0%至30%)純化,得到呈固體之N-4-16_2
(800 mg,81%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.58-9.53 (m, 1H), 2.41-2.31 (m, 1H), 1.96-1.81 (m, 4H), 1.89-1.34 (m, 10H), 1.32-1.21 (m, 10H), 1.16-1.09 (m, 5H), 0.97 (s, 3H), 0.89-0.84 (m, 1H), 0.69 (s, 3H)。2.
在25℃下於N2
下向異戊基溴化鎂(4.32 mL,2 M於醚中,8.65 mmol)之溶液中添加含N-4-16_2
(300 mg,0.86 mmol)之THF (10 mL)。在25℃下攪拌混合物30分鐘,藉由飽和NH4
Cl (10 mL)來淬滅且用(3 × 10 mL)乙酸乙酯萃取。有機層用鹽水(20 mL)洗滌,經Na2
SO4
乾燥且過濾,真空濃縮且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈固體之N-4-16
(200 mg,不純),其在25℃下於MeCN (10 mL)中濕磨,得到呈固體之92
(141 mg,70%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.67-3.57 (m, 1H), 2.01-1.77 (m, 4H), 1.67-1.57 (m, 4H), 1.55-1.26 (m, 14H), 1.25-1.21 (m, 5H), 1.19-0.99 (m, 7H), 0.96 (s, 3H), 0.93-0.84 (m, 9H), 0.66 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.367 min,30-90AB_2MIN_E,純度100%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 93 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 苯基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (93) 之合成
1.向N-4-16_1
(1.00 g,2.86 mmol)於DCM (20 mL)中之溶液添加DMP (2.42 g,5.72 mmol)。此後,在15℃下攪拌反應物10 min。向反應混合物添加NaHCO3飽和水溶液(20 mL)及Na2
S2
O3
飽和水溶液(20 mL),接著用DCM (2 × 20 mL)萃取。合併之有機層用NaHCO3
飽和水溶液(3 × 60 mL)及鹽水(60 mL)洗滌,經Na2
SO4
乾燥,過濾且真空下濃縮,得到固體。殘餘物藉由矽膠層析(PE/EtOAc=0%至30%)純化,得到呈固體之N-4-16_2
(800 mg,81%)。 1 H NMR
(400 MHz, CDCl3
) δ 9.58-9.53 (m, 1H), 2.41-2.31 (m, 1H), 1.96-1.81 (m, 4H), 1.89-1.34 (m, 10H), 1.32-1.21 (m, 10H), 1.16-1.09 (m, 5H), 0.97 (s, 3H), 0.89-0.84 (m, 1H), 0.69 (s, 3H)。2.
在25℃下於N2
下向異戊基溴化鎂(4.32 mL,2 M於醚中,8.65 mmol)之溶液中添加含N-4-16_2
(300 mg,0.86 mmol)之THF (10 mL)。在25℃下攪拌混合物30分鐘,藉由飽和NH4
Cl (10 mL)來淬滅且用(3 × 10 mL)乙酸乙酯萃取。有機層用鹽水(20 mL)洗滌,經Na2
SO4
乾燥且過濾,真空濃縮且藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈固體之N-4-16
(200 mg,不純),其在25℃下於MeCN (10 mL)中濕磨,得到呈固體之92
(141 mg,70%)。 1 H NMR
(400 MHz, CDCl3
) δ 3.67-3.57 (m, 1H), 2.01-1.77 (m, 4H), 1.67-1.57 (m, 4H), 1.55-1.26 (m, 14H), 1.25-1.21 (m, 5H), 1.19-0.99 (m, 7H), 0.96 (s, 3H), 0.93-0.84 (m, 9H), 0.66 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.367 min,30-90AB_2MIN_E,純度100%,C28
H47
[M+H-2H2
O]+
之MS ESI計算值383,實驗值383。實例 93 : (3S,5S,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -4- 苯基丁 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (93) 之合成  1.N-6-5 及 N-6-6
(190 mg,0.420 mmol)之混合物藉由製備型HPLC (管柱:YMC-Actus Triart C18 100×30mm×5um;條件:水(0.05% HCl)-ACN;梯度:90-100% B;流速:25 mL/min)分離,得到呈固體之93
(56 mg,30%)及呈固體之32
(12 mg,6%)。93 : 1 H NMR
(400 MHz, CDCl3
) δ 7.34-7.28 (m, 2H), 7.25-7.18 (m, 3H), 3.95-3.86 (m, 1H), 2.87-2.75 (m, 1H), 2.69-2.58 (m, 1H), 2.01-1.91 (m, 1H), 1.90-1.79 (m, 1H), 1.69-1.58 (m, 4H), 1.55-1.41 (m, 6H), 1.40-1.11 (m, 11H), 1.07-0.95 (m, 6H), 0.91-0.80 (m, 7H), 0.69-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt= 1.334 min,30-90AB,純度100%,C31
H48
O2
Na[M+Na]+
之MS ESI計算值475,實驗值475。實例 94 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S
,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (94) 之合成
1.N-6-5 及 N-6-6
(190 mg,0.420 mmol)之混合物藉由製備型HPLC (管柱:YMC-Actus Triart C18 100×30mm×5um;條件:水(0.05% HCl)-ACN;梯度:90-100% B;流速:25 mL/min)分離,得到呈固體之93
(56 mg,30%)及呈固體之32
(12 mg,6%)。93 : 1 H NMR
(400 MHz, CDCl3
) δ 7.34-7.28 (m, 2H), 7.25-7.18 (m, 3H), 3.95-3.86 (m, 1H), 2.87-2.75 (m, 1H), 2.69-2.58 (m, 1H), 2.01-1.91 (m, 1H), 1.90-1.79 (m, 1H), 1.69-1.58 (m, 4H), 1.55-1.41 (m, 6H), 1.40-1.11 (m, 11H), 1.07-0.95 (m, 6H), 0.91-0.80 (m, 7H), 0.69-0.59 (m, 4H)。 在2.0 min層析中,LCMS
Rt= 1.334 min,30-90AB,純度100%,C31
H48
O2
Na[M+Na]+
之MS ESI計算值475,實驗值475。實例 94 : (3S,5R,8R,9S,10S,13S,14S,17R)-17-((2S
,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (94) 之合成 
 1.向100
(150 mg,0.338 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到94
(6 mg,4%)及呈固體之98
(46 mg,30%)。94 : 1 H NMR
(400 MHz, CDCl3
) δ 3.78-3.66 (m, 1H), 1.98-1.72 (m, 7H), 1.69-1.59 (m, 4H), 1.48-1.32 (m, 12H), 1.27-1.07 (m, 12H), 1.06-1.00 (m, 3H), 0.97 (s, 3H), 0.94-0.85 (m, 7H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt= 1.639 min,30-90AB_E,純度98.8%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 95 : (3S,8S,9S,10R,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (95) 之合成
1.向100
(150 mg,0.338 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到94
(6 mg,4%)及呈固體之98
(46 mg,30%)。94 : 1 H NMR
(400 MHz, CDCl3
) δ 3.78-3.66 (m, 1H), 1.98-1.72 (m, 7H), 1.69-1.59 (m, 4H), 1.48-1.32 (m, 12H), 1.27-1.07 (m, 12H), 1.06-1.00 (m, 3H), 0.97 (s, 3H), 0.94-0.85 (m, 7H), 0.66 (s, 3H)。 在2.0 min層析中,LCMS
Rt= 1.639 min,30-90AB_E,純度98.8%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 95 : (3S,8S,9S,10R,13S,14S,17R)-3-( 甲氧基甲基 )-10,13- 二甲基 -17-((2S,3R)-4,4,4- 三氟 -3- 羥丁 -2- 基 )-2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (95) 之合成  1.N-004-029A
(0.45 g,1.01 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始B:30%,結束B:30%)純化,得到呈白色固體之39
(PK1
:120 mg,26.7%)及呈白色固體之95
(PK2
:200 mg,44.5%)。95 : 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.29 (m, 1H), 4.06-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.42-2.33 (m, 1H), 2.17-2.07 (m, 1H), 2.20-1.85 (m, 5H), 1.77-1.63 (m, 4H), 1.51-0.83 (m, 17H), 0.73 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.103 min,30-90AB_2MIN_E,純度100%,C24
H34
F3
O[M-CH5
O2
]+
之MS ESI計算值395,實驗值395。實例 96 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (96) 之合成
1.N-004-029A
(0.45 g,1.01 mmol)藉由SFC (管柱:AD (250mm×30mm,5um),條件:0.1% NH3
H2
O ETOH,開始B:30%,結束B:30%)純化,得到呈白色固體之39
(PK1
:120 mg,26.7%)及呈白色固體之95
(PK2
:200 mg,44.5%)。95 : 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.29 (m, 1H), 4.06-3.99 (m, 1H), 3.37 (s, 3H), 3.30-3.19 (m, 2H), 2.54 (s, 1H), 2.42-2.33 (m, 1H), 2.17-2.07 (m, 1H), 2.20-1.85 (m, 5H), 1.77-1.63 (m, 4H), 1.51-0.83 (m, 17H), 0.73 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.103 min,30-90AB_2MIN_E,純度100%,C24
H34
F3
O[M-CH5
O2
]+
之MS ESI計算值395,實驗值395。實例 96 : (3S,5R,8R,9S,10S,13S,14S,17R)-3- 乙基 -17-((2S,3R)-3- 羥基 -6- 甲基庚 -2- 基 )-10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (96) 之合成  1.向52
(50 mg,0.116 mmol)於MeOH (10 mL)中之溶液添加Pd(OH)2
(200 mg)。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之24
(15 mg,30%)及呈固體之96
(1.2 mg,3%)。96 : 1 H NMR
(400 MHz, CDCl3
) δ 3.67-3.55 (m, 1H), 2.01-1.83 (m, 2H), 1.80-1.62 (m, 4H), 1.61-1.56 (m, 2H), 1.55-1.50 (m, 1H), 1.49-1.31 (m, 10H), 1.30-1.10 (m, 11H), 1.09-1.00 (m, 3H), 0.96 (s, 3H), 0.94-0.86 (m, 12H), 0.67(s, 3H)。 在2 min層析中,LCMS
tR= 1.326 min,30-90AB_ELSD,純度100.0%,C29
H49
[M+H-2H2
O]+
之MS ESI計算值397,實驗值397。實例 97 : (3S,5R,8R,9S,10S,13S,14S,17R)-3,10,13- 三甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (97) 之合成
1.向52
(50 mg,0.116 mmol)於MeOH (10 mL)中之溶液添加Pd(OH)2
(200 mg)。在50℃下於H2
(50 Psi)下攪拌混合物。混合物經過濾、濃縮且藉由combi-flash (0-10% EtOAc/PE)純化,得到呈固體之24
(15 mg,30%)及呈固體之96
(1.2 mg,3%)。96 : 1 H NMR
(400 MHz, CDCl3
) δ 3.67-3.55 (m, 1H), 2.01-1.83 (m, 2H), 1.80-1.62 (m, 4H), 1.61-1.56 (m, 2H), 1.55-1.50 (m, 1H), 1.49-1.31 (m, 10H), 1.30-1.10 (m, 11H), 1.09-1.00 (m, 3H), 0.96 (s, 3H), 0.94-0.86 (m, 12H), 0.67(s, 3H)。 在2 min層析中,LCMS
tR= 1.326 min,30-90AB_ELSD,純度100.0%,C29
H49
[M+H-2H2
O]+
之MS ESI計算值397,實驗值397。實例 97 : (3S,5R,8R,9S,10S,13S,14S,17R)-3,10,13- 三甲基 -17-((2S,3S)-4,4,4- 三氟 -3- 羥丁 -2- 基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (97) 之合成  1.向N-004-017_3
(100 mg,0.19 mmol)於THF (2 mL)及MeOH (1 mL)以及水(1 mL)中之溶液添加KOH (53.8 mg,0.96 mmol)。在60℃下攪拌混合物16小時。將混合物倒入水(20 mL)中且用EtOAc (2 × 40 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈白色固體之97
(80 mg,不純)。97
(80 mg,0.19 mmol)藉由閃蒸塔(0-10% EtOAc/PE)純化,得到97
(60 mg,不純)。在0℃下於N2
下向97
(40 mg,0.096 mmol)於甲醇(3 mL)中之溶液一次性添加NaBH4
(10.8 mg,0.28 mmol)。在0℃下攪拌混合物30 min。將混合物倒入水(10 mL)中並攪拌20 min。用EtOAc (3 × 10 mL)萃取水相。合併之有機相用鹽水(2 × 20 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到粗產物,該粗產物與另一批次的20 mg不純97
合併,殘餘物藉由閃蒸塔(0-10% EtOAc/PE)純化,得到呈白色固體之97
(31 mg,31%)。 1 H NMR
(400 MHz, CDCl3) δ 4.11-3.96 (m, 1H), 2.18-2.11 (d,J ab
= 6.4 Hz, 1H), 2.02-1.77 (m, 5H), 1.68-1.57 (m, 3H), 1.49-1.24 (m, 11H), 1.23-1.19 (m, 5H), 1.18-1.01 (m, 7H), 0.96 (s, 3H), 0.67 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.124 min,30-90AB_2MIN_E.M,純度100%,C24
H38
F3
O[M+H-H2
O]+
之MS ESI計算值399,實驗值399。實例 98 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (98) 之合成
1.向N-004-017_3
(100 mg,0.19 mmol)於THF (2 mL)及MeOH (1 mL)以及水(1 mL)中之溶液添加KOH (53.8 mg,0.96 mmol)。在60℃下攪拌混合物16小時。將混合物倒入水(20 mL)中且用EtOAc (2 × 40 mL)萃取。合併之有機層用鹽水(30 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮。殘餘物藉由閃蒸塔(0-15% EtOAc/PE)純化,得到呈白色固體之97
(80 mg,不純)。97
(80 mg,0.19 mmol)藉由閃蒸塔(0-10% EtOAc/PE)純化,得到97
(60 mg,不純)。在0℃下於N2
下向97
(40 mg,0.096 mmol)於甲醇(3 mL)中之溶液一次性添加NaBH4
(10.8 mg,0.28 mmol)。在0℃下攪拌混合物30 min。將混合物倒入水(10 mL)中並攪拌20 min。用EtOAc (3 × 10 mL)萃取水相。合併之有機相用鹽水(2 × 20 mL)洗滌,經Na2
SO4
乾燥,過濾且濃縮,得到粗產物,該粗產物與另一批次的20 mg不純97
合併,殘餘物藉由閃蒸塔(0-10% EtOAc/PE)純化,得到呈白色固體之97
(31 mg,31%)。 1 H NMR
(400 MHz, CDCl3) δ 4.11-3.96 (m, 1H), 2.18-2.11 (d,J ab
= 6.4 Hz, 1H), 2.02-1.77 (m, 5H), 1.68-1.57 (m, 3H), 1.49-1.24 (m, 11H), 1.23-1.19 (m, 5H), 1.18-1.01 (m, 7H), 0.96 (s, 3H), 0.67 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.124 min,30-90AB_2MIN_E.M,純度100%,C24
H38
F3
O[M+H-H2
O]+
之MS ESI計算值399,實驗值399。實例 98 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基十六氫 -1H- 環戊 [a] 菲 -3- 醇 (98) 之合成  1.向100
(150 mg,0.338 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到94
(6 mg,4%)及呈固體之98
(46 mg,30%)。98 : 1 H NMR
(400 MHz, CDCl3
) δ 3.78-3.67 (m, 1H), 1.97-1.74 (m, 6H), 1.68-1.56 (m, 8H), 1.53-1.45 (m, 4H), 1.44-1.31 (m, 10H), 1.28-1.21 (m, 1H), 1.16-0.96 (m, 9H), 0.91-0.85 (m, 6H), 0.82 (s, 3H), 0.69-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt= 1.582 min,30-90AB_E,純度100%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 99 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S,E)-3- 羥基 -5- 苯基戊 -4- 烯 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (99) 之合成
1.向100
(150 mg,0.338 mmol)於MeOH (20 mL)中之溶液添加Pd(OH)2
(300 mg,無水)。在50℃下於H2
(50 psi)下攪拌混合物48小時。混合物經過濾、濃縮且藉由combi-flash (0-15% EtOAc/PE)純化,得到94
(6 mg,4%)及呈固體之98
(46 mg,30%)。98 : 1 H NMR
(400 MHz, CDCl3
) δ 3.78-3.67 (m, 1H), 1.97-1.74 (m, 6H), 1.68-1.56 (m, 8H), 1.53-1.45 (m, 4H), 1.44-1.31 (m, 10H), 1.28-1.21 (m, 1H), 1.16-0.96 (m, 9H), 0.91-0.85 (m, 6H), 0.82 (s, 3H), 0.69-0.61 (m, 4H)。 在2.0 min層析中,LCMS
Rt= 1.582 min,30-90AB_E,純度100%,C30
H49
[M+H-2H2
O]+
之MS ESI計算值409,實驗值409。實例 99 : (3S,5S,8R,9S,10S,13S,14S,17R)-17-((2S,3S,E)-3- 羥基 -5- 苯基戊 -4- 烯 -2- 基 )-10,13- 二甲基 -3-( 三氟甲基 ) 十六氫 -1H- 環戊 [a] 菲 -3- 醇 (99) 之合成 
 1.在20℃下向N-003-005_1
(140 mg,0.278 mmol)於THF (2 mL)及MeOH (1 mL)中之溶液逐份添加NaBH4
(419 mg,11.1 mmol)。在20℃下攪拌混合物10 min。用水(20 mL)及NH4
Cl (20 mL,飽和)淬滅反應物。用EtOAc (50 mL)萃取混合物。真空濃縮有機層且藉由prep-TLC (PE/EtOAc=4/1)純化,得到均呈固體之N-003-005
(50 mg,不純)及14
(50 mg)。 不純99
(50 mg)藉由SFC (儀器:SFC 1;管柱:OD (250mm×30mm,5um);條件:0.1% NH3
H2
O EtOH;開始B:40%;結束B:40%;流速(mL/min):50;注入:60)純化以提供溶解於MeCN (20 mL)中之固體且真空濃縮,得到呈固體之99
(17 mg)。99 : 1 H NMR
(400 MHz, CDCl3
) δ 7.44-7.36 (m, 2H), 7.36-7.28 (m, 2H), 7.25-7.20 (m, 1H), 6.58 (d,J
= 16.0 Hz, 1H), 6.24 (dd,J
= 4.8, 16.0 Hz, 1H), 4.49-4.40 (m, 1H), 2.09-1.91 (m, 4H), 1.86-1.75 (m, 1H), 1.72-1.58 (m, 5H), 1.52-1.04 (m, 14H), 0.94 (d,J
= 6.4 Hz, 3H), 0.91-0.87 (m, 1H), 0.86 (s, 3H), 0.75-0.70 (m, 1H), 0.69 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.280 min,30-90AB_2MIN_E,純度98.5%,C31
H42
F3
O [M+H-H2
O]+
之MS ESI計算值487,實驗值487。 在8 min層析中,HPLC
Rt= 6.29 min,30-90_AB_1.2ml,100% d.e.。實例 100 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (100) 之合成
1.在20℃下向N-003-005_1
(140 mg,0.278 mmol)於THF (2 mL)及MeOH (1 mL)中之溶液逐份添加NaBH4
(419 mg,11.1 mmol)。在20℃下攪拌混合物10 min。用水(20 mL)及NH4
Cl (20 mL,飽和)淬滅反應物。用EtOAc (50 mL)萃取混合物。真空濃縮有機層且藉由prep-TLC (PE/EtOAc=4/1)純化,得到均呈固體之N-003-005
(50 mg,不純)及14
(50 mg)。 不純99
(50 mg)藉由SFC (儀器:SFC 1;管柱:OD (250mm×30mm,5um);條件:0.1% NH3
H2
O EtOH;開始B:40%;結束B:40%;流速(mL/min):50;注入:60)純化以提供溶解於MeCN (20 mL)中之固體且真空濃縮,得到呈固體之99
(17 mg)。99 : 1 H NMR
(400 MHz, CDCl3
) δ 7.44-7.36 (m, 2H), 7.36-7.28 (m, 2H), 7.25-7.20 (m, 1H), 6.58 (d,J
= 16.0 Hz, 1H), 6.24 (dd,J
= 4.8, 16.0 Hz, 1H), 4.49-4.40 (m, 1H), 2.09-1.91 (m, 4H), 1.86-1.75 (m, 1H), 1.72-1.58 (m, 5H), 1.52-1.04 (m, 14H), 0.94 (d,J
= 6.4 Hz, 3H), 0.91-0.87 (m, 1H), 0.86 (s, 3H), 0.75-0.70 (m, 1H), 0.69 (s, 3H)。 在2 min層析中,LCMS
Rt= 1.280 min,30-90AB_2MIN_E,純度98.5%,C31
H42
F3
O [M+H-H2
O]+
之MS ESI計算值487,實驗值487。 在8 min層析中,HPLC
Rt= 6.29 min,30-90_AB_1.2ml,100% d.e.。實例 100 : (3S,8S,9S,10R,13S,14S,17R)-17-((2S,3S)-4- 環戊基 -3- 羥丁 -2- 基 )-3- 乙基 -10,13- 二甲基 -2,3,4,7,8,9,10,11,12,13,14,15,16,17- 十四氫 -1H- 環戊 [a] 菲 -3- 醇 (100) 之合成  1. 在75℃下向Mg (528 mg,22.0 mmol)及I2
(55.8 mg,0.22 mmol)於THF (3 mL)中之懸浮液逐滴添加(溴甲基)環戊烷(1.8 g,11.0 mmol)於THF (11 mL)中之溶液。在75℃下攪拌混合物1小時。在15℃下向於S-500-6-1_1
(800mg,2.23mmol)於THF (30 mL)中之溶液緩慢添加(環戊基甲基)溴化鎂(11.1mL,11.1mmol,1 M於THF中)之混合物。添加之後,在15℃下攪拌混合物1小時。用飽和NH4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2 × 30 mL)洗滌,經Na2
SO4
乾燥,過濾及濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之100
(350 mg,35%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.77-3.69 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.89 (m, 4H), 1.88-1.69 (m, 4H), 1.68-1.55 (m, 6H), 1.54-1.27 (m, 12H), 1.26-1.15 (m, 2H), 1.14-1.05 (m, 4H), 1.04-0.99 (m, 5H), 0.98-0.88 (m, 4H), 0.87-0.81 (m, 3H), 0.69 (s, 3H)。 在7.0 min層析中,LCMS
Rt= 5.661 min,30-90AB_E,純度100%,C30
H47
[M+H-2H2
O]+
之MS ESI計算值407,實驗值407。表 1. 例示性化合物之資料。
1. 在75℃下向Mg (528 mg,22.0 mmol)及I2
(55.8 mg,0.22 mmol)於THF (3 mL)中之懸浮液逐滴添加(溴甲基)環戊烷(1.8 g,11.0 mmol)於THF (11 mL)中之溶液。在75℃下攪拌混合物1小時。在15℃下向於S-500-6-1_1
(800mg,2.23mmol)於THF (30 mL)中之溶液緩慢添加(環戊基甲基)溴化鎂(11.1mL,11.1mmol,1 M於THF中)之混合物。添加之後,在15℃下攪拌混合物1小時。用飽和NH4
Cl (40 mL)淬滅混合物,且用EtOAc (3 × 20 mL)萃取。合併之有機相用鹽水(2 × 30 mL)洗滌,經Na2
SO4
乾燥,過濾及濃縮,且藉由combi-flash (0-15% EtOAc/PE)純化,得到呈固體之100
(350 mg,35%)。 1 H NMR
(400 MHz, CDCl3
) δ 5.32-5.26 (m, 1H), 3.77-3.69 (m, 1H), 2.41-2.31 (m, 1H), 2.09-1.89 (m, 4H), 1.88-1.69 (m, 4H), 1.68-1.55 (m, 6H), 1.54-1.27 (m, 12H), 1.26-1.15 (m, 2H), 1.14-1.05 (m, 4H), 1.04-0.99 (m, 5H), 0.98-0.88 (m, 4H), 0.87-0.81 (m, 3H), 0.69 (s, 3H)。 在7.0 min層析中,LCMS
Rt= 5.661 min,30-90AB_E,純度100%,C30
H47
[M+H-2H2
O]+
之MS ESI計算值407,實驗值407。表 1. 例示性化合物之資料。 

 表示單鍵或雙鍵,其中當
表示單鍵或雙鍵,其中當 中之一者為雙鍵時,另一
中之一者為雙鍵時,另一 為單鍵且R6不存在;且其中當
為單鍵且R6不存在;且其中當 兩者均為單鍵時,R6為氫。
兩者均為單鍵時,R6為氫。





 或
或 ;或 g.該式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物:
;或 g.該式(I-63)之化合物選自式(I-E-i63)或(I-E-ii63)之化合物: 或
或

 表示單鍵或雙鍵,其中當
表示單鍵或雙鍵,其中當 中之一者為雙鍵時,另一
中之一者為雙鍵時,另一 為單鍵且R6不存在;且其中當
為單鍵且R6不存在;且其中當 兩者均為單鍵時,R6為氫。
兩者均為單鍵時,R6為氫。











 及
及










 及
及
