TW202317117A - 一種三環化合物的用途 - Google Patents
一種三環化合物的用途 Download PDFInfo
- Publication number
- TW202317117A TW202317117A TW111138872A TW111138872A TW202317117A TW 202317117 A TW202317117 A TW 202317117A TW 111138872 A TW111138872 A TW 111138872A TW 111138872 A TW111138872 A TW 111138872A TW 202317117 A TW202317117 A TW 202317117A
- Authority
- TW
- Taiwan
- Prior art keywords
- mutation
- egfr
- compound
- acceptable salt
- pharmaceutically acceptable
- Prior art date
Links
Images











Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
本發明提供一種式(A)化合物或其可藥用的鹽在製備治療EGFR、FGFR2、KIT、ALK和/或ROS1突變介導的癌症的藥物中的用途。該化合物對該些突變類型介導的癌症具有明顯的抑制活性。
Description
本申請要求申請日為2021年10月14日的中國專利申請202111200131.0、申請日為2022年3月31日的中國專利申請202210330403.7的優先權,2022年10月8日遞交的中國專利申請202211221824.2的優先權,本申請引用上述中國專利申請的全文。
本發明提供了一種三環化合物或其可藥用的鹽在治療EGFR、FGFR2、KIT、ALK和/或ROS1突變介導的癌症中的用途。
EGFR,即表皮生長因數受體(epidermal growth factor receptor),廣泛分佈於哺乳動物上皮細胞、成纖維細胞、膠質細胞等細胞表面。EGFR訊號通路對細胞的生長、增殖和分化等生理過程發揮著重要的作用。EGFR突變也是NSCLC患者中最常見的一種突變類型,尤其是在亞洲人群中可以占到40%~50%。因此EGFR一直是藥物研發領域的最熱門靶點之一。
目前,上市的EGFR抑制劑分為第一、二、三代。第一代為可逆的靶向藥物,針對L858R突變以及Del19突變,例如吉非替尼、厄洛替尼、埃克替尼。第二代為不可逆的靶向藥物,例如阿法替尼以及達克替尼。第一、二代靶向藥物雖然療效顯著,但多數患者都會在使用藥物1-2年後出現耐藥性。EGFR抑制劑耐藥的患者中,有50%的耐藥與T790M突變有關。第三代EGFR靶向藥物奧希替尼能克服由於T790M突變引起的腫瘤耐藥,給更多的肺癌患者帶來了更好的生存獲益。然而第三代靶向藥也不可避免的產生耐藥,其耐藥原因包括進一步的C797S突變,G724S突變、L792H突變、E709K突變以及EGFR擴增等。目前臨床上對於奧希替尼耐藥的應對尚無成熟的治療手段,臨床需求迫在眉睫。
成纖維細胞生長因數及其受體(FGFR)驅動影響細胞增殖、遷移和存活的重要發育訊號傳導途徑。異常的FGF訊號傳導在許多癌症中起作用。FGFR家族由FGFR1、FGFR2、FGFR3和FGFR4組成。FGFR是在一部分腫瘤中透過基因擴增、突變或染色體易位或重排啟動的酪氨酸激酶。FGFR1的擴增發生在鱗狀細胞肺癌和雌激素受體陽性乳腺癌中。FGFR2還在胃癌和乳腺癌中擴增。在子宮內膜癌中觀察到FGFR突變,並在膀胱癌中觀察到FGFR3突變。
c-KIT的編碼產物是一種分子量為145千道爾頓的具有酪氨酸激酶活性的跨膜受體蛋白,它在細胞外區域有5個免疫球蛋白G樣結構域,因此,它屬於III型酪氨酸激酶超家族成員。生理情況下,c-KIT少量表達於肥大細胞、幹細胞、精細胞及腸Cajal細胞等。生理情況下,當幹細胞因數(c-KIT的配體)結合c-KIT的免疫球蛋白G樣結構域時,c-KIT分子發生同源二聚化,使得臨膜域的Y568和Y570酪氨酸殘基發生自動磷酸化,進而導致細胞內許多底物蛋白的酪氨酸殘基發生磷酸化,引起細胞增殖有關的多條訊號轉導通路啟動,包括Jak-Stat3/Stat5通路、Src激酶、Ras-MEK-Erk1/2及PI3K-AKT通路,從而使細胞增殖。c-KIT酪氨酸激酶域的功能獲得性點突變可引起配體非依賴性持續啟動,進而導致細胞的失控性生長和對凋亡的抵抗。已經明確c-KIT突變是造成胃腸間質瘤(GIST)、系統性肥大細胞增多症的原因,與小細胞肺癌有密切關係。
基因融合是由兩個或多個基因的編碼區首尾相連,置於同一套調控序列(包括啟動子、增強子、核糖體結合序列、終止子等)控制之下,構成的嵌合基因。在NSCLC中已發現棘皮動物微管結合蛋白4(EML4)基因和間變性淋巴瘤激酶(ALK)基因的融合。EML4-ALK融合基因是發生於非小細胞肺癌中的促癌的基因突變,占非小細胞肺癌發生率的4-5%。EML4-ALK導致酪氨酸激酶異常表達,引起細胞的惡性轉化。SLC34A2-ROS1融合基因在NSCLC中的發生率約為1.0%-3.4%,在EGFR/KRAS/ALK均陰性的人群中的發生率則可至5.7%,病理類型主要是腺癌。SLC34A2-ROS1基因發生融合時丟失細胞外區域,保留跨膜和細胞內酪氨酸激酶區域,融合位元點主要發生在ROS1基因的第32、34、35、36號外顯子。ROS1受體酪氨酸激酶參與啟動多條下游訊號轉導通路,包括RAS-MAPK/ERK、PI3K/AKT/mTOR、JAK/STAT3、PLC/IP3和SHP2/VAV3途徑,進而調控腫瘤細胞的生長增殖、細胞週期、分化、轉移和遷移。ROS1基因和ALK基因在酪氨酸激酶區域序列存在49%同源性,而在激酶催化區的ATP結合位點二者同源性高達77%,ROS1融合基因為肺癌的個體化治療提供新的方案,明確ROS1融合基因在肺腺癌中的陽性率,對臨床實踐具有重要的意義。
本申請人在專利PCT/CN2021/086941中公開了一種針對C797S突變的小分子EGFR抑制劑,其結構如式(A)所示,該小分子抑制劑針對EGFR L858R/T790M/C797S突變以及EGFR Del19/T790M/C797S突變具有良好的激酶抑制活性和細胞抗增殖活性,同時該分子在小鼠模型上體現了較好的抗腫瘤活性及耐受性。為了提高該化合物的臨床價值,開發其更多的用途具有重要的意義。

本發明提供了一種三環化合物或其可藥用的鹽在製備治療EGFR、FGFR2、KIT、ALK和/或ROS1突變介導的癌症的藥物中的用途。
具體的,本發明提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,所述的EGFR突變類型為Del19突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,所述的EGFR突變類型為L858R突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,所述的EGFR突變類型為不伴隨C797S突變的T790M突變。
在本發明的一些方案中,上述用途中所述的不伴隨C797S突變的T790M突變選自以下的一種或組合:L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,所述的EGFR突變類型為Del19/C797S雙突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,所述的EGFR突變類型為L858R/C797S雙突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR擴增介導的癌症的藥物中的用途。
在本發明的一些方案中,上述EGFR擴增為Del19/T790M/C797S三突變、L858R/T790M/D537H三突變以及V674L/E746_A750del/T790M三突變EGFR的擴增。
在本發明的一些方案中,上述EGFR擴增為伴隨著Del19/T790M/C797S三突變的EGFR擴增、伴隨著L858R/T790M/D537H三突變的EGFR擴增或伴隨著V674L/E746_A750del/T790M三突變的EGFR擴增。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,所述的EGFR突變類型為20外顯子插入突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變或擴增介導的癌症的藥物中的用途,所述的EGFR突變類型選自以下的一種或任意組合:Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變;EGFR擴增選自Del19/T790M/C797S三突變、L858R/T790M/D537H三突變、V674L/E746_A750del/T790M三突變EGFR的擴增。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EGFR突變或擴增介導的癌症的藥物中的用途,其中所述的EGFR突變類型選自以下的一種或任意組合:Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變;所述的EGFR擴增選自以下的一種或任意組合:伴隨著Del19/T790M/C797S三突變、L858R/T790M/D537H三突變或V674L/E746_A750del/T790M三突變的EGFR擴增。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療FGFR2高表達的癌症的藥物中的用途。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療C-KIT突變的癌症的藥物中的用途,所述的C-KIT突變類型為V560G突變和/或D816Y突變和/或D816H突變和/或V559和V560氨基酸缺失突變和/或D816V突變。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療EML4-ALK融合蛋白介導的癌症的藥物中的用途。
在本發明還提供式(A)化合物或其可藥用的鹽在製備治療EML4-ALK融合蛋白 L1196M突變和/或F1174L突變和/或L1196M/L1198F雙突變介導的癌症的藥物中的用途。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療SLC34A2-ROS1融合蛋白介導的癌症的藥物中的用途。
本發明還提供式(A)化合物或其可藥用的鹽在製備治療SLC34A2-ROS1融合蛋白 D2033N突變介導的癌症的藥物中的用途。
在本發明的一些方案中,上述任一用途中的(A)化合物的可藥用鹽為鹽酸鹽。
在本發明的一些方案中,上述任一用途中的(A)化合物的可藥用鹽為一鹽酸鹽。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變介導的癌症中的用途,所述的EGFR突變類型為Del19突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變介導的癌症中的用途,所述的EGFR突變類型為L858R突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變介導的癌症中的用途,所述的EGFR突變類型為不伴隨C797S突變的T790M突變。
在本發明的一些方案中,上述用途中所述的不伴隨C797S突變的T790M突變選自以下的一種或組合:L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變介導的癌症中的用途,所述的EGFR突變類型為Del19/C797S雙突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變介導的癌症中的用途,所述的EGFR突變類型為L858R/C797S雙突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR擴增介導的癌症中的用途。
在本發明的一些方案中,上述用途中所述的EGFR擴增為Del19/T790M/C797S三突變、L858R/T790M/D537H三突變以及V674L/E746_A750del/T790M三突變EGFR的擴增。
在本發明的一些方案中,上述用途中的EGFR擴增為伴隨著Del19/T790M/C797S三突變、L858R/T790M/D537H三突變或V674L/E746_A750del/T790M三突變的EGFR擴增。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變介導的癌症中的用途,所述的EGFR突變類型為20外顯子插入突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變或擴增介導的癌症中的用途,所述的EGFR突變類型選自以下的一種或任意組合:Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變;EGFR擴增選自Del19/T790M/C797S三突變、L858R/T790M/D537H三突變、V674L/E746_A750del/T790M三突變EGFR的擴增。
本發明還提供式(A)化合物或其可藥用的鹽在治療EGFR突變或擴增介導的癌症中的用途,其中所述的EGFR突變類型選自以下的一種或任意組合:Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變;所述的EGFR擴增選自以下的一種或任意組合:伴隨著Del19/T790M/C797S三突變、L858R/T790M/D537H三突變或V674L/E746_A750del/T790M三突變的EGFR擴增。
本發明還提供了治療EGFR突變或擴增介導的癌症的方法,其包括向患者施用式(A)化合物或其可藥用的鹽, 所述的EGFR突變類型選自以下的一種或任意組合:Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變;所述的EGFR擴增選自以下的一種或任意組合:伴隨著Del19/T790M/C797S三突變、L858R/T790M/D537H三突變或V674L/E746_A750del/T790M三突變的EGFR擴增。
本發明還提供了式(A)化合物在製備上述Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變的EGFR突變調節劑中的用途。
在本發明的一些方案中,上述EGFR突變調節劑為EGFR上述突變的抑制劑中的用途。
本發明還提供了式(A)化合物在製備上述Del19/T790M/C797S三突變、上述L858R/T790M/D537H三突變或上述V674L/E746_A750del/T790M三突變的EGFR擴增的調節劑中的用途。
在本發明的一些方案中,上述任一用途中的癌症為肺癌。
在本發明的一些方案中,上述任一用途中的癌症為非小細胞肺癌。
在本發明的一些方案中,上述任一用途中的癌症為未曾接受過治療的非小細胞肺癌。
在本發明的一些方案中,上述任一用途中的癌症為既往接受過EGFR抑制劑治療後,產生耐藥的非小細胞肺癌。
在本發明的一些方案中,上述的EGFR抑制劑包括第一代EGFR抑制劑、第二代或第三代EGFR抑制劑。
在本發明的一些方案中,上述第一代EGFR抑制劑包括吉非替尼、埃克替尼、厄洛替尼。
在本發明的一些方案中,上述第二代EGFR抑制劑包括阿法替尼、達克替尼。
在本發明的一些方案中,上述第三代EGFR抑制劑包括奧希替尼。
本發明還提供式(A)化合物或其可藥用的鹽在治療FGFR2高表達的癌症中的用途。
本發明還提供式(A)化合物或其可藥用的鹽在治療C-KIT突變的癌症中的用途,所述的C-KIT突變類型為V560G突變和/或D816Y突變和/或D816H突變和/或559和560氨基酸缺失突變和/或D816V突變。
本發明還提供式(A)化合物或其可藥用的鹽在治療EML-ALK融合蛋白介導的癌症中的用途。
在本發明還提供式(A)化合物或其可藥用的鹽在治療EML4-ALK融合蛋白 L1196M突變和/或F1174L突變和/或L1196M/L1198F雙突變介導的癌症中的用途。
本發明還提供式(A)化合物或其可藥用的鹽在治療SLC34A2-ROS1融合蛋白介導的癌症中的用途。
本發明還提供式(A)化合物或其可藥用的鹽在治療SLC34A2-ROS1融合蛋白 D2033N突變介導的癌症中的用途。
本發明還提供了治療上述FGFR2高表達的癌症、上述C-KIT突變的癌症、上述EML-ALK融合蛋白介導的癌症、上述SLC34A2-ROS1融合蛋白介導的癌症的方法,其包括向患者施用式(A)化合物或其可藥用的鹽。
本發明還提供了式(A)化合物在製備上述FGFR2、上述C-KIT突變、上述EML-ALK融合蛋白、上述SLC34A2-ROS1的調節劑中的用途。
在本發明的一些方案中,上述調節劑為抑制劑。
在本發明的一些方案中,上述任一用途中的式(A)化合物的可藥用鹽為鹽酸鹽。
在本發明的一些方案中,上述任一用途中的式(A)化合物的可藥用鹽為一鹽酸鹽。
技術效果
本發明式(A)化合物不僅對L858R/T790M/C797S三突變以及Del19/T790M/C797S三突變具有較好的活性,同時對L858R或者Del19的單突變、20外顯子插入突變、L858R/T790M或L858R/C797S或Del19/C797S的雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變以及伴隨EGFR擴增的Del19/T790M/C797S三突變、L858R/T790M/D537H三突變、V674L/E746_A750del/T790M三突變也有著較好的體外激酶或細胞抗增殖活性,並且該化合物在Del19單突變、L858R單突變以及Del19/C797S雙突變的小鼠模型體現出了較好的抗腫瘤活性,且小鼠耐受性良好。
除此之外,本發明式(A)化合物對FGFR2高表達、C-KIT V560G突變、C-KIT D816Y突變、C-KIT D816H突變、C-KIT V559和V560氨基酸缺失突變、C-KIT D816V突變、EML4-ALK融合蛋白突變、EML4-ALK融合蛋白L1196M或F1174L突變或L1196M/L1198F雙突變、SLC34A2-ROS1融合蛋白突變、SLC34A2-ROS1融合蛋白D2033N突變的細胞系均有著較好的抗增殖活性。
定義和說明
除非另有說明,本文所用的下列術語和短語旨在具有下列含義。一個特定的術語或短語在沒有特別定義的情況下不應該被認為是不確定的或不清楚的,而應該按照普通的含義去理解。
術語“藥學上可接受的鹽”是指本發明化合物與相對無毒的酸或堿製備得到的衍生物。這些鹽可以在化合物合成、分離、純化期間就被製備,或者單獨使用經過純化的化合物的游離形式與適合的酸或堿反應。當化合物中含有相對酸性的官能團時,與鹼金屬、鹼土金屬氫氧化物或有機胺反應得到堿加成鹽,包括基於鹼金屬與鹼土金屬的陽離子以及無毒的銨、季銨和胺陽離子,還涵蓋氨基酸的鹽等。當化合物中含有相對鹼性的官能團時,與有機酸或無機酸反應得到酸加成鹽。在本發明中,所述EGFR突變介導的腫瘤或癌症指的是在這些腫瘤或癌症患者中可檢測出EGFR的癌症驅動突變(driver mutation),包括但不限於Del19突變、L858R突變、T790M突變,20外顯子插入突變(Exon 20 ins),C797S等突變。其中,Del19突變是指第19號外顯子內部分堿基的缺失造成了非移碼性部分氨基酸缺失;L858R指的是由於堿基的錯義突變造成了858號氨基酸由L變成了R;T790M指的是由於基因中堿基的錯義突變造成了790號氨基酸由T變成了M;20外顯子插入(Exon 20 ins)突變是指發生於EGFR的20外顯子的框內重複/插入的突變;C797S突變是指797位的半胱氨酸殘基突變成絲氨酸。在本發明中,所述EGFR突變不但包括上述EGFR的單突變型,更包括T790M、Del19、L858R、Exon 20 ins、C797S以及其他位元點自由組合的複合突變型,包括但不限於L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變等。
在本發明中,所述EGFR擴增是指EGFR基因拷貝數的增加或蛋白的高水準表達。它可以發生在突變細胞上,也可以發生在沒有突變(野生型)的EGFR受體細胞上。
下面透過實施例對本發明進行詳細描述,但並不意味著對本發明任何不利限制。本發明的化合物可以透過所屬技術領域中具有通常知識者所熟知的多種合成方法來製備,包括下面列舉的具體實施方式、其與其他化學合成方法的結合所形成的實施方式以及本領域技術上人員所熟知的等同替換方式,優選的實施方式包括但不限於本發明的實施例。對所屬技術領域中具有通常知識者而言,在不脫離本發明精神和範圍的情況下針對本發明具體實施方式進行各種變化和改進將是顯而易見的。
實施例
1
:式(
A
)化合物的製備
1.1
中間體
6A
的製備

化合物
6A-1:
將化合物
1C-4(3.5 g, 15.5 mmol)溶於乙腈(40 mL),在0 °C條件下,加入N-碘代丁二醯亞胺(4.9 g, 21.7 mmol)。室溫反應攪拌5小時,LCMS監控顯示原料消失後,減壓濃縮,加入水(30 mL),用二氯甲烷(45 mL×3次)萃取,合併有機相,有機相先用飽和食鹽水(60 mL × 2次)洗滌,然後用無水硫酸鈉乾燥,過濾,減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:乙酸乙酯/石油醚 = 1 / 1)得到3.57 g化合物
6A-1。
MS (ESI, m/z): 352.0 [M + H]
+。
化合物
6A-2:
將化合物6A-1(3.4 g, 9.7 mmol)和1A(3.7 g, 12.5 mmol)溶於1,4-二氧六環(30 mL)和水(6 mL)中,向上述反應液中加入碳酸鉀(2.7 g, 19.4 mmol)和[1,1'-雙(二苯基膦)二茂鐵]二氯化鈀二氯甲烷絡合物(790 mg, 1.0 mmol)。在氮氣保護下,將反應體系加熱至80 °C並繼續攪拌2小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫,減壓濃縮,所得殘餘物用矽膠柱層析純化(洗脫劑:乙酸乙酯/石油醚 = 2 / 1)得到2.8 g化合物6A-2。
MS (ESI, m/z): 395.3 [M + H]
+。
化合物
6A-3:
將化合物6A-2(2.7 g, 6.8 mmol)溶於N,N-二甲基甲醯胺(28 mL)中。隨後,向上述反應液中加入碳酸鉀(1.9 g,13.5 mmol)。將反應體系加熱至100 °C並繼續攪拌24小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫並加入水(50 mL)淬滅。混合液用乙酸乙酯(60 mL×4次)萃取,合併有機相,有機相先用飽和食鹽水(50 mL×3次)洗滌,然後用無水硫酸鈉乾燥,過濾,最後減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/乙酸乙酯 = 15 / 1)得到1.2 g化合物6A-3。
MS (ESI, m/z): 375.2 [M + H]
+。
化合物
6A-4:
將化合物6A-3(1.2 g, 3.3 mmol)溶於氯化氫的1,4-二氧六環溶液(4 M, 15 mL)中。在30 °C條件下攪拌6小時,LCMS監控顯示原料消失後,將反應液濃縮,加入水(40 mL),用飽和碳酸氫鈉水溶液調節其pH到9。混合液用氯仿/異丙醇 = 3 / 1(50 mL×3次)萃取,合併有機相,然後用無水硫酸鈉乾燥,過濾,最後減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/甲醇 = 30 / 1)得到842 mg化合物6A-4。
MS (ESI, m/z): 275.0 [M + H]
+。
化合物
6A-5:
將化合物6A-4(300 mg,1.1 mmol)和碳酸銫(1.07 g,3.3 mmol)溶於N, N-二甲基甲醯胺(6 mL)中。隨後,向上述反應液中加入碘代異丙烷(1.86 g,10.9 mmol)。將反應體系加熱至80 °C並繼續攪拌16小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫並加入水(30 mL)淬滅。混合液用乙酸乙酯(50 mL×3次)萃取,合併有機相,有機相先用飽和食鹽水(50 mL×3次)洗滌,然後用無水硫酸鈉乾燥,過濾,最後減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/甲醇 = 10 / 1)得到75 mg化合物6A-5。
MS (ESI, m/z): 317.2 [M + H]
+。
中間體
6A:
將化合物6A-5(75 mg,0.2 mmol)溶於乙醇(8 mL)和水(1.6 mL)中。隨後,向上述反應液中加入氯化銨(50.7 mg,0.9 mmol)和還原鐵粉(132.4 mg,2.4 mmol)。將反應體系加熱至80 °C並繼續攪拌5小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫並減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/甲醇 = 10 / 1)得到48 mg化合物6A。
MS (ESI, m/z): 287.2 [M + H]
+。
中間體
35A
的製備

化合物
35A-1
:
將6-氨基喹喔啉(10 g, 68.89 mmol)溶於濃硫酸(20 mL)中。在0 °C條件下,向反應液中分批加入硝酸鉀(9.054 g, 89.55 mmol)並在該溫度下繼續攪拌30分鐘。LCMS監控顯示原料消失後,將反應液倒入冰水(100 g)中。用1 M氫氧化鈉水溶液調節其pH到8。混合液用乙酸乙酯(200 mL × 2次)萃取,合併有機相,有機相先用飽和食鹽水(100 mL × 3次)洗滌,然後用無水硫酸鈉乾燥,過濾,減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/甲醇 = 10 / 1)得到2 g化合物35A-1。
MS (ESI) M/Z: 191.2 [M + H]
+。
1.2
中間體
35A
:
將化合物35A-1(2 g, 10.5 mmol)溶於N, N-二甲基甲醯胺(20 mL)中。將反應液降至0 °C,氮氣保護下,分批加入氫化鈉(60wt, 1.3 g, 31.5 mmol)並繼續攪拌20分鐘。隨後,向上述反應液中加入2,4-二氯-5-溴嘧啶(4.8 g, 21.0 mmol),將反應升至室溫並繼續攪拌1小時。LCMS監控顯示原料消失後,將反應液降溫至0°C並加入飽和氯化銨水溶液(80 mL)淬滅。混合液用乙酸乙酯(100 mL × 3次)萃取,合併有機相,有機相先用飽和食鹽水(80 mL × 3次)洗滌,然後用無水硫酸鈉乾燥,過濾,減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:乙酸乙酯/石油醚 = 1 / 2)得到2.7 g化合物35A。
MS (ESI, m/z): 381.0, 383.0 [M + H]
+。

化合物
53A
:
將化合物6A(2.7 g, 9.43 mmol)和35A(3.6 g, 9.43 mmol)溶於N-甲基吡咯烷酮(30 mL)中。隨後,向上述反應液中加入甲烷磺酸(2.72 g, 28.28 mmol)。將反應體系加熱至95 °C並繼續攪拌3小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫並經反相C18柱純化。純化條件:色譜柱330 g C18反相柱;流動相水(含0.1%甲酸)和乙腈;流速70 mL/分鐘;梯度在20分鐘內,乙腈從10%升到50%;檢測波長254 nm。收集產品,減壓濃縮,得到3.4 g化合物53A。
MS (ESI, m/z): 631.2, 633.2 [M + H]
+。
化合物
53B
:
將化合物53A(3.4 g, 5.38 mmol)溶於乙醇(40 mL)和水(8 mL)的混合溶劑中。隨後,向上述反應液中加入鐵粉(1.50 g, 26.92 mmol)和氯化銨(0.86 g, 16.15 mmol)並將反應體系加熱至80 °C並繼續攪拌2小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫並減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/甲醇 = 10 / 1),得到2.8 g化合物53B。
MS (ESI, m/z): 601.2, 603.2 [M + H]
+。
1.3
化合物
A
:
將化合物53B(5g, 8.31 mmol)溶於吡啶(50 mL)中。隨後向反應液中滴加甲基磺醯氯(1.9 g, 16.62 mmol)。將反應體系升溫至50°C並繼續攪拌2小時。LCMS監控顯示原料消失後,將反應液冷卻至室溫並減壓濃縮。殘餘物溶於甲醇/四氫呋喃(1/1,50 mL)的混合溶劑中,在0°C條件下,向反應液中加入氫氧化鈉(2 M,5 mL)的水溶液。將反應體系升溫至室溫並繼續攪拌1小時後,減壓濃縮。所得殘餘物用矽膠柱層析純化(洗脫劑:二氯甲烷/甲醇 = 10/1),粗產品經過二氯甲烷/甲醇(20/1,30 mL)打漿後,用乙腈/水(50 mL)凍乾,得到3 g化合物A。
MS (ESI, m/z): 679.0, 681.0 [M + H]
+。
1H NMR (400 MHz, DMSO-d
6) δ 9.88 (br s, 1H), 8.94 (d, J = 2.0 Hz, 1H), 8.85 (d, J = 2.0 Hz, 1H), 8.76 (s, 1H), 8.67 (br s, 1H), 8.35 (s, 1H), 8.27 (s, 1H), 7.73 (s, 1H), 7.49 (s, 1H), 7.38 (s, 1H), 6.58 (s, 1H), 3.99-3.91 (m, 1H), 3.76 (s, 3H), 3.71 (s, 3H), 3.21 (t, J = 5.6 Hz, 2H), 3.00 (s, 3H), 2.94 (t, J = 5.6 Hz, 2H), 1.29 (d, J = 6.4 Hz, 6H)。
1.4
化合物
A
鹽酸鹽
:
將化合物A(67 g, 98.59 mmol)溶於二氯甲烷(880 mL)和甲醇(440 mL)的混合溶劑中並在室溫下繼續攪拌1小時。隨後在室溫條件下,向反應液中滴加氯化氫的甲醇溶液(4 M, 24.65 mL, 98.59 mmol)。該反應體系繼續在室溫攪拌4小時後,將反應液減壓濃縮至70 mL。向上述混合物中加入甲基叔丁基醚(880 mL)並繼續在室溫攪拌2小時。析出的固體經過過濾,乙腈/水(500 mL)凍乾,得到60.2 g化合物A鹽酸鹽。
MS (ESI) M/Z: 679.0, 681.0 [M + H]
+。
1H NMR (300 MHz, DMSO-d
6) δ 9.97 (s, 1H), 9.30-9.16 (m, 2H), 8.99 (s, 1H), 8.91 (s, 1H), 8.43 (s, 2H), 7.75-7.30 (m, 3H), 6.57 (s, 1H), 3.95-3.86 (m, 1H), 3.79 (s, 3H), 3.69 (s, 3H), 3.28-3.14 (m, 2H), 3.03 (s, 3H), 2.98-2.88 (m, 2H), 1.27 (brs, 6H)。
實施例
2
:生物學測試評價:
(一)體外酶學實驗
本實驗採用螢光共振能量轉移 (TR-FRET) 的方法測試化合物A對 EGFR WT 、EGFR Del19、EGFR L858R、 EGFR L858R/T790M、EGFR L858R/C797S和 EGFR ex19del/C797S 激酶活性的抑制作用,並得出化合物A對 EGFR 激酶活性的半數抑制濃度 IC
50。
2.
實驗材料
EGFR、EGFR Del19、EGFR L858R 、EGFR L858R/T790M、EGFR L858R/C797S、EGFR ex19del/C797S 重組酶購自 Signalchem 公司。
HTRF KinEASE-TK kit 試劑盒,購自 Cisbio 公司。
DTT,MnCl
2,MgCl
2購自Sigma公司。
ATP 購自 Promega 公司。
3.
實驗方法
1)準備 1×工作液: 5mM MgCl
2;1mM DTT;1mM MnCl
2和 1× 激酶緩衝液(試劑盒中),其中 EGFR L858R/T790M 的緩衝液中加入了SEB。
2)使用 Echo 550 (Labcyte) 轉移 10nL(或1μL) 梯度稀釋的化合物到 384 孔實驗板中。
3)加入 5 μL (或2μL) 2× 重組酶溶液到 384 孔實驗板中,室溫孵育 10 分鐘。
4)加入 5 μL(或2μL) 2× TK-substrate-biotin 底物溶液(包含 ATP)到 384 孔實驗板中,室溫孵育 40 分鐘(或1h)。
5)加入 5 μL 含有Sa-XL 665 HTRF 檢測液,以及 5 μL TK-antibody-Cryptate,室溫孵育 1 小時。
6)酶標儀檢測各孔的 615nm 和 665nm 螢光訊號值。
7)計算每孔螢光訊號 665nm/615nm 的比值。
8)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
激酶活性抑制結果見表1。
4.
實驗結果及結論
從表1中我們可以看出,化合物A對Del19單突變、L858R單突變激酶均有很好的抑制作用。
表
1.
酶學抑制結果
| 激酶 | 化合物 A IC 50(nM) |
| EGFR (WT) | 1.01 |
| EGFR Del19 | 0.48 |
| EGFR L858R | 0.36 |
| EGFR ex19del/C797S | 0.59 |
| EGFR L858R/C797S | 0.32 |
| EGFR L858R/T790M | 2.55 |
(二)細胞增殖抑制實驗
I. A431
細胞增殖抑制實驗
本實驗採用 CellTiter-Glo 的方法測試化合物A對A431細胞增殖的抑制作用,並得出化合物抑制細胞生長半數的濃度 IC
50。
1.實驗材料
A431 細胞購自 ATCC。
DMEM 培養基,胎牛血清(FBS),Penicillin-Streptomycin購自GIBCO。
Brigatinib 購自 Selleck 公司。
CellTiter-Glo 試劑,購自 Promega 公司。
2.實驗方法
1)按照每孔 800 個細胞的密度將 A431 細胞接種於 384 孔培養板,每孔 30μl,置於細胞培養箱中培養 24 小時 (37 ℃,5%CO
2)。
2)Day 0:使用 Echo 向培養板細胞中加入 30 nL 梯度稀釋的待測化合物,DMSO終濃度為 0.1%,將培養板置於細胞培養箱中孵育 72 小時(37 ℃,5%CO
2)。空白對照加入每孔 30 nL的 DMSO。
3)Day 3:每孔加入 30 μL Cell Titer-Glo 試劑,室溫避光 30 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
II. NCI-H3255
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對 NCI-H3255(EGFR L858R 突變)細胞增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
NCI-H3255 細胞,購自南京科佰生物科技有限公司。
1640 培養基,胎牛血清(FBS),Penicillin-Streptomycin,購自 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)將 NCI-H3255 細胞接種於 384 孔培養板,每孔 30μL。
2)Day0:使用 Echo 向培養板細胞中加入 30nL 梯度稀釋的待測化合物,DMSO終濃度為 0.1%,將培養板置於細胞培養箱中孵育 72 小時 (37℃,5%CO
2)。空白對照加入每孔 30nL 的 DMSO。
3)Day3:每孔加入 30μL Cell Titer-Glo 試劑,室溫避光 30 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
細胞活性抑制結果見表 2。
III. PC-9
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對 PC-9 (EGFR Del19 突變)細胞增殖的抑制作用,並得出化合物抑制細胞生長半數的濃度 IC
50。
2.實驗材料
PC-9 細胞,購自European Collection of Authenticated Cell Cultures。
1640 培養基,胎牛血清(FBS),Penicillin-Streptomycin,購自 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)將 PC-9 細胞接種於 384 孔培養板,每孔 30μL。
2)Day0:使用 Echo 向培養板細胞中加入 30nL 梯度稀釋的待測化合物,DMSO終濃度為 0.1%,將培養板置於細胞培養箱中孵育 72 小時(37℃,5%CO
2)。空白對照加入每孔 30nL 的 DMSO。
3)Day3:每孔加入 30μL Cell Titer-Glo 試劑,室溫避光 30 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
細胞活性抑制結果見表 2。
IV. NCI-H1975
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對 NCI-H1975 (EGFR L858R/T790M突變)細胞增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
NCI-H1975 細胞來自ATCC。
1640 培養基,胎牛血清(FBS),Penicillin-Streptomycin購自 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)將 NCI-H1975 細胞接種於 384 孔培養板,每孔 30μL。
2)Day0:使用 Echo 向培養板細胞中加入 30nL 梯度稀釋的待測化合物,DMSO終濃度為 0.1%,將培養板置於細胞培養箱中孵育 72 小時(37℃,5%CO
2)。空白對照加入每孔 30nL 的 DMSO。
3)Day3:每孔加入 30μL Cell Titer-Glo 試劑,室溫避光 30 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的IC
50。
細胞活性抑制結果見表 2。
V. HCC827
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對 HCC827(EGFR Del突變) 細胞增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
HCC827 細胞,購自ATCC。
1640 培養基,胎牛血清(FBS),Penicillin-Streptomycin,購自 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)將 HCC827 細胞接種於 384 孔培養板,每孔 30μL。
2)Day0:使用 Echo 向培養板細胞中加入 30nL 梯度稀釋的待測化合物,DMSO終濃度為 0.1%,將培養板置於細胞培養箱中孵育 72 小時(37℃,5%CO
2)。空白對照加入每孔 30nL 的 DMSO。
3)Day3:每孔加入 30μL Cell Titer-Glo 試劑,室溫避光 30 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
細胞活性抑制結果見表 2。
VI. Ba/F3 EGFR-Del19/G724S/T790M
、
Ba/F3 EGFR-E709K/T790M/L858R
、
Ba/F3 EGFR-L858R/T790M/L792H
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對Ba/F3 EGFR-Del19/G724S/T790M和 Ba/F3 EGFR-E709K/T790M/L858R 和 Ba/F3 EGFR-L858R/T790M/L792H 細胞增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
Ba/F3 EGFR-Del19/G724S/T790M 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 EGFR-E709K/T790M/L858R 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 EGFR-L858R/T790M/L792H 細胞來自康源博創生物科技(北京)有限公司。
1640 培養基,胎牛血清(FBS)購自 Hyclone 和 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)收穫處於對數生長期的細胞進行細胞計數。用台盼藍排斥法檢測細胞活力,確保細胞活力在 90% 以上。
2)使用完全培養基調整細胞密度,隨後接種於 96 孔細胞培養板,每孔接種90 μL,共 3000 個細胞。
3)將 96 孔板中的細胞置於 37℃、5% CO
2條件下培養。
4)配製 10 倍藥物溶液,轉移連續稀釋化合物各 10 μL 至 96 孔細胞板的相應實驗孔中,使得化合物檢測濃度為 1 µM 起,9 個濃度,3 倍稀釋,然後,每個藥物濃度設置三個複孔。
5)將已加藥的 96 孔板置於 37℃、5% CO
2條件下繼續培養 72 小時,之後進行 CTG 分析。
6)融化 CTG 試劑並平衡細胞板至室溫 30 分鐘。
7)每孔加入等體積的 CTG 溶液。
8)在定軌搖床上振動 5 分鐘使細胞裂解。
9)將細胞板放置於室溫 20 分鐘以穩定冷光訊號。
10)讀取冷光值,收集資料。
11)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
細胞活性抑制結果見表 2。
VII. Ba/F3 (EGFR-Del19/C797S)
、
Ba/F3 (EGFR-L858R/C797S)
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對Ba/F3 (EGFR-Del19/C797S)和 Ba/F3 (EGFR-L858R/C797S) 細胞增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
Ba/F3 EGFR-Del19/C797S 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 EGFR-L858R/C797S細胞來自康源博創生物科技(北京)有限公司。
1640 培養基,胎牛血清(FBS)購自 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)按照每孔 3000 個細胞的密度將 Ba/F3 (EGFR Del19/C797S) 和 Ba/F3 (EGFR L858R/C797S) 細胞分別接種於 96 孔培養板,每孔 90 μL。
2)Day 0:向培養板細胞中加入 10 μL 梯度稀釋的待測化合物,DMSO 終濃度為 0.2%,將培養板置於細胞培養箱中孵育 72 小時(37℃,5%CO
2)。空白對照加入每孔 10 μL 的 DMSO。
3)Day3:每孔加入 100 μL CellTiter-Glo 試劑,室溫避光振盪 10 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5) 使用GraphPad Prism 軟體進行資料分析,得出化合物的 IC
50。
細胞活性抑制結果見表 2。
VIII. NCI-H716
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對 NCI-H716 (FGFR2高表達)細胞增殖的抑制作用,並得出化合物抑制細胞生長半數的濃度 IC
50。
2.實驗材料
NCI-H716 細胞,購自ATCC 。
1640 培養基,胎牛血清(FBS),Penicillin-Streptomycin,購自 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)將 NCI-H716 細胞接種於 96 孔培養板,每孔 100 μL。
2)Day0:向培養板細胞中加入92 μL培養基和 8 μL 梯度稀釋的待測化合物,DMSO終濃度為 0.1%,將培養板置於細胞培養箱中孵育 72 小時(37℃,5%CO
2)。空白對照加入每孔 92 μL培養基和 8 μL 的 DMSO。
3)Day3:每孔加入 100 μL Cell Titer-Glo 試劑,室溫避光 30 分鐘。
4)Envision 酶標儀(PerkinElmer)檢測化學發光訊號。
5)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
細胞活性抑制結果見表 2。
IX. Ba/F3 C-KIT-V560G
、
Ba/F3 C-KIT-D816Y
、
Ba/F3 C-KIT-D816H
、
Ba/F3 C-KIT-Del(V559V560)
、
Ba/F3 C-KIT-D816V
、
NCI-H3122
(
EML4-ALK
)、
Ba/F3-EML4-ALK-L1196M
、
Ba/F3 EML4-ALK-F1174L
、
Ba/F3-EML4-ALK-L1196M/L1198F
、
Ba/F3 SLC34A2/ROS1
、
BaF3 SLC34A2-ROS1-D2033N
細胞增殖抑制實驗
1.實驗目的
本實驗採用CellTiter-Glo 的方法測試化合物A對Ba/F3 C-KIT-V560G、Ba/F3 C-KIT-D816Y、Ba/F3 C-KIT-D816H、Ba/F3 C-KIT-Del(V559-V560) 、Ba/F3 C-KIT-D816V、NCI-H3122(EML4-ALK)、Ba/F3-EML4-ALK-L1196M、Ba/F3 EML4-ALK-F1174L、Ba/F3-EML4-ALK-L1196M/L1198F、Ba/F3 SLC34A2/ROS1、 Ba/F3 SLC34A2-ROS1-D2033N細胞增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
Ba/F3 C-KIT-V560G 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 C-KIT-D816Y 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 C-KIT-D816H 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 C-KIT-Del(V559V560) 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 C-KIT-D816V 細胞來自康源博創生物科技(北京)有限公司。
NCI-H3122(EML4-ALK)細胞來自康源博創生物科技(北京)有限公司。
Ba/F3-EML4-ALK-L1196M細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 EML4-ALK-F1174L 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3-EML4-ALK-L1196M/L1198F 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 SLC34A2/ROS1 細胞來自康源博創生物科技(北京)有限公司。
Ba/F3 SLC34A2-ROS1-D2033N細胞來自康源博創生物科技(北京)有限公司。
1640 培養基,胎牛血清(FBS)購自 Hyclone 和 GIBCO。
CellTiter-Glo 試劑,購自 Promega 公司。
3.實驗方法
1)收穫處於對數生長期的細胞進行細胞計數。用台盼藍排斥法檢測細胞活力,確保細胞活力在 90% 以上。
2)使用完全培養基調整細胞密度,隨後接種於 96 孔細胞培養板,每孔接種90 μL,共 3000 個細胞。
3)將 96 孔板中的細胞置於 37℃、5% CO
2條件下培養。
4)配製 10 倍藥物溶液,轉移連續稀釋化合物各 10 μL 至 96 孔細胞板的相應實驗孔中,每個藥物濃度設置三個複孔。
5)將已加藥的 96 孔板置於 37℃、5% CO
2條件下繼續培養 72 小時,之後進行 CTG 分析。
6)融化 CTG 試劑並平衡細胞板至室溫 30 分鐘。
7)每孔加入等體積的 CTG 溶液。
8)在定軌搖床上振動 5 分鐘使細胞裂解。
9)將細胞板放置於室溫 20 分鐘以穩定冷光訊號。
10)讀取冷光值,收集資料。
11)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
細胞活性抑制結果見表 2。
4.實驗結果及結論
從表 2 中的實驗結果可以看出,本發明化合物A對 NCI-H3255 L858R EGFR突變 、PC9 Del19 EGFR突變 、HCC827 Del19 EGFR突變 、NCI-H1975 L858R/T790M EGFR突變、 Ba/F3 (Del19/G724S/T790M) EGFR 三突變細胞株、 Ba/F3 (L858R/T790M/L792H) EGFR 三突變細胞株、Ba/F3 (E709K/T790M/L858R) EGFR 三突變細胞株、Ba/F3 (Del19/C797S) EGFR 雙突變細胞株和 Ba/F3 ( L858R/C797S) EGFR 雙突變細胞株、NCI-H716(FGFR2)、Ba/F3 C-KIT-V560G、Ba/F3 C-KIT-D816Y、Ba/F3 C-KIT-D816H、Ba/F3 C-KIT-Del(V559V560) 、Ba/F3 C-KIT-D816V、NCI-H3122(EML4-ALK)、Ba/F3-EML4-ALK-L1196M、Ba/F3 EML4-ALK-F1174L、Ba/F3-EML4-ALK-L1196M/L1198F、Ba/F3 SLC34A2/ROS1、 Ba/F3 SLC34A2-ROS1-D2033N的細胞增殖有較好的抑制作用;對 EGFR 野生型細胞系 A431的抑制作用較弱,有較好的選擇性。
表
2.
細胞增殖抑制試驗資料結果
| 細胞及突變類型 | 化合物 A IC 50(nM) |
| A431 EGFR WT | 439.7 |
| NCI-H3255(L858R) | 21.24 |
| PC-9(Del19) | 85.65 |
| NCI-H1975(L858R/T790M) | 50.42 |
| HCC827(Del19) | 11.43 |
| Ba/F3 (Del19/G724S/T790M) | 32.15 |
| Ba/F3 (L858R/T790M/L792H) | 13.79 |
| Ba/F3 (E709K/T790M/L858R) | 1.44 |
| Ba/F3 (EGFR Del19/C797S) | 1.915 |
| Ba/F3 (EGFR L858R/C797S) | 5.456 |
| NCI-H716(FGFR2) | 17.29 |
| Ba/F3 C-KIT-V560G | 122.66 |
| Ba/F3 C-KIT-D816Y | 8.09 |
| Ba/F3 C-KIT-D816H | 6.54 |
| Ba/F3 C-KIT-Del(V559V560) | 19.04 |
| Ba/F3 C-KIT-D816V | 17.48 |
| Ba/F3 SLC34A2/ROS1 | 1.89 |
| BaF3 SLC34A2-ROS1-D2033N | 1.69 |
| NCI-H3122(EML4-ALK) | 34.38 |
| Ba/F3-EML4-ALK-L1196M | 14.73 |
| Ba/F3 EML4-ALK-F1174L | 30.53 |
| Ba/F3-EML4-ALK-L1196M/L1198F | 64.56 |
(三)
PDO
增殖抑制試驗
1.實驗目的
本實驗採用 CellTiter-Glo 的方法測試化合物A對奧西替尼耐藥的 PDO(Patient Derived Tumor Organoids)增殖的抑制作用,並得出化合物A抑制細胞生長半數的濃度 IC
50。
2.實驗材料
PDO,來自北京科途醫學科技有限公司。
Accutase,購自Sigma。
K2 Oncology,來自北京科途醫學科技有限公司。
CellTiter-Glo 試劑,購自 Promega 公司。
3.
實驗方法
1)PDO 在 6 孔細胞培養板中生長到直徑 200 μm後,用細胞消化液在 37℃消化 10-30 分鐘,用頭部灼燒鈍化的巴適管吹打幾次,直到細胞呈單細胞或者寡細胞團的狀態。
2)細胞計數後用預冷的完全培養基調整濃度到 100-160 個細胞/μL,加入等體積的 20% 的基質膠,完全混合後,按照 50 μL /孔,加入低吸附細胞培養板底部。
3)在 37 ℃培養箱孵育 30 分鐘後,上層加入 50 μL /孔完全培養基,在37℃,5% 二氧化碳培養箱中繼續培養 2 天。
4)向培養板細胞中加入梯度稀釋的待測化合物,DMSO 終濃度為 0.2%,將培養板置於細胞培養箱中孵育 5 天(37℃,5%CO
2)。
5)加入70 μL化學發光細胞裂解液,震盪5分鐘後,輕輕吹打至細胞完全裂解,從細胞培養板中轉移100 μL到白色低透酶標檢測板中。
6)在化學發光酶標儀(FLUOstar Omega)中檢測化學發光訊號。
7)使用 GraphPad Prism 軟體進行資料分析,得出化合物A的 IC
50。
4.實驗結果及結論
從表 3 結果來看,化合物A對奧西替尼耐藥的 PDO 增殖有較好的抑制作用。
表
3.PDO
增殖抑制試驗資料結果
| PDO 細胞 | 致病突變 | 化合物 A IC 50(nM) |
| KOLU-1121X | EGFR Del19/T790M/C797S/, EGFR 擴增 | 171.2 |
| KOLU-1216X | EGFR V674L/E746_A750del/T790M,EGFR 擴增 | 135.2 |
| KOLU-1102X | EGFR L858R/T790M/D537H,EGFR 擴增 | 164.8 |
| KOLU-1249X | EGFR 20ins L747_P753 | 134.4 |
(四)
EGFR Del19/C797S
突變的體內藥效研究
1.實驗目的
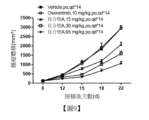
評價化合物A(本實驗使用的為化合物A的鹽酸鹽)連續14天口服給藥,對Ba/F3 EGFR Del19/C797S的抗腫瘤活性及毒副作用。
2.實驗材料
NU/NU小鼠,雌性,SPF級,購自北京維通利華實驗動物技術有限公司。
Ba/F3 EGFR Del19/C797S細胞,購自康源博創生物科技(北京)有限公司。
3.實驗步驟
3.1 細胞培養
Ba/F3 EGFR Del19/C797S細胞用含有10%的胎牛血清的RPMI1640培養基在37℃,5%二氧化碳培養箱中培養,收集指數生長期的細胞進行接種。
3.2 細胞接種
在無菌條件下,取體外培養的Ba/F3 EGFR Del19/C797S細胞懸液,離心後調整細胞濃度至3×10
7個/mL,接種於小鼠右側腋窩皮下(0.1 mL/只),接種當天設為第0天。
3.3 腫瘤分組、給藥及測量
a,當平均腫瘤體積100-200mm
3時,挑選32只腫瘤體積適中小鼠入組,按照腫瘤體積大小隨機分為4組:G1:溶媒對照組、G2:化合物A(15 mg/kg)、G3:化合物A(30 mg/kg)和G4:化合物A(65 mg/kg),8只/組。
b,動物分組後開始給藥,給藥體積均為10 mL/kg,口服給藥(po);每天稱重給藥1次,連續給藥14天;每週測量瘤徑2次。
c,腫瘤體積(Tumor volume,TV):每週測量2次腫瘤體積,以觀察瘤塊體積變化和生長速度。腫瘤體積V=1/2×a×b
2,其中a、b分別表示腫瘤長徑和短徑。化合物對腫瘤組織的生長抑制作用採用腫瘤生長抑制率TGI(%)評價。TGI(%)=[1-(某給藥組的平均腫瘤體積-該給藥組分組當天的平均腫瘤體積)/(陰性對照組的平均腫瘤體積-陰性對照組分組當天的平均腫瘤體積)]×100%。給藥組和陰性對照組取同一天數據。
d,在試驗過程中密切觀察小鼠生活狀態,包括外觀體征、一般行為活動、精神狀態、攝食情況、呼吸狀態、糞便和尿液性狀、注射局部及其它毒性表現。
e,試驗達到終點後,將小鼠實施安樂死,動物屍體凍存至冰櫃,移交至有資質的醫療廢棄物處理單位進行處置。
4.
實驗結果
表
4.
實驗資料
| 分組 | 劑量 ( mg/kg ) | 腫瘤體積 a , b ( mm 3 ) | TGI (%) | 體重改變( % ) |
| G1:溶媒對照組 | - | 1855±141.9 | - | 14.9 |
| G2:化合物A | 15 | 1030±114.7** | 47.4 | 11.1 |
| G3:化合物A | 30 | 847.3±66.6** | 57.9 | 8.9 |
| G4:化合物A | 65 | 318.8±49.2** | 88.3 | 3.5 |
a,平均值±標準誤;
b,P值腫瘤體積進行統計分析,與G1組比較,*P﹤0.05;**P﹤0.01。
5.實驗結論
從上數結果可以看出,化合物A給藥劑量15、30、65mg/kg均能顯著抑制腫瘤生長(圖1),且呈明顯的量效關係,小鼠耐受性良好(圖2)。
(五)
L858R
突變的體內藥效研究
1.實驗目的
評價化合物A連續22天口服給藥,對NCI-H3255(L858R)的抗腫瘤活性及毒副作用。
2.實驗材料
NOD SCID小鼠,雌性,SPF級,購自北京華阜康生物科技股份有限公司。
NCI-H3255(L858R)細胞,購自南京科佰生物科技有限公司。
3.實驗步驟
3.1 細胞培養
用含有滅活的10% 胎牛血清的RPMI-1640培養基在37℃、5% CO
2的培養箱中培養NCI-H3255腫瘤細胞,待細胞長滿後分瓶傳代。將處於對數生長期的腫瘤細胞用於體內腫瘤的接種。
3.2 細胞接種
將無血清RPMI-1640培養液重懸的NCI-H3255腫瘤細胞1 × 10
7/100 μL 濃度接種於實驗動物的右側脅肋部皮下,接種當天設為第0天。
3.3 腫瘤分組、給藥及測量
a,當平均腫瘤體積約236mm
3時,挑選24只腫瘤體積適中小鼠入組,按照腫瘤體積大小隨機分為3組:G1:溶媒對照組、G2:化合物A(15 mg/kg)和G3:化合物A(60 mg/kg),8只/組。
b,動物分組後開始給藥,給藥體積均為10 mL/kg,口服給藥(po);每天稱重給藥1次,連續給藥22天;每週測量瘤徑2次。
c,腫瘤體積(Tumor volume,TV):每週測量2次腫瘤體積,以觀察瘤塊體積變化和生長速度。腫瘤體積V=1/2×a×b
2,其中a、b分別表示腫瘤長徑和短徑。化合物對腫瘤組織的生長抑制作用採用腫瘤生長抑制率TGI(%)評價。TGI(%)=[1-(某給藥組的平均腫瘤體積-該給藥組分組當天的平均腫瘤體積)/(陰性對照組的平均腫瘤體積-陰性對照組分組當天的平均腫瘤體積)]×100%。給藥組和陰性對照組取同一天數據。
d,在試驗過程中密切觀察小鼠生活狀態,包括外觀體征、一般行為活動、精神狀態、攝食情況、呼吸狀態、糞便和尿液性狀、注射局部及其它毒性表現。
e,試驗達到終點後,將小鼠實施安樂死,動物屍體凍存至冰櫃,移交至有資質的醫療廢棄物處理單位進行處置。
4.實驗結果
表
5.
實驗資料
| 分組 | 劑量 ( mg/kg ) | 腫瘤體積 a , b ( mm 3 ) | TGI (%) | 體重改變( % ) |
| G1:溶媒對照組 | - | 924 ± 71 | - | 3.7 |
| G2:化合物A | 15 | 64±6** | 125 | 1.9 |
| G3:化合物A | 60 | 23±2** | 131 | -9.7 |
a,平均值±標準誤;
b,P值腫瘤體積進行統計分析,與G1組比較,*P﹤0.05;**P﹤0.01。
5.實驗結論
從上數結果可以看出,化合物A給藥劑量15、60mg/kg均能顯著抑制腫瘤生長(圖3),且呈一定的量效關係。小鼠耐受性良好(圖4)。
(六)
Del19
突變的體內藥效研究
1.實驗目的
評價化合物A連續21天口服給藥,對PC-9(Del19)的抗腫瘤活性及毒副作用。
2.實驗材料
CB-17 SCID小鼠,雌性,SPF級,購自北京華阜康生物科技股份有限公司。
PC-9(Del19)細胞,購自European Collection of Authenticated Cell Cultures。
3.實驗步驟
3.1 細胞培養
用含有滅活的10% 胎牛血清,100 U/mL 的青黴素和100 µg/mL 的鏈黴素的RPMI-1640培養基在37℃、5% CO
2的培養箱中培養PC-9(Del19)腫瘤細胞,待細胞長滿後分瓶傳代。將處於對數生長期的腫瘤細胞用於體內腫瘤的接種。
3.2 細胞接種
將無血清RPMI-1640培養液重懸的PC-9(Del19)腫瘤細胞5 × 106/100 uL濃度接種於實驗動物的右側脅肋部皮下,接種當天設為第0天。
3.3 腫瘤分組、給藥及測量
a,當平均腫瘤體積約181mm
3時,挑選20只腫瘤體積適中小鼠入組,按照腫瘤體積大小隨機分為4組:G1:溶媒對照組、G2:Gefitinib(吉非替尼,100 mg/kg)、G3:化合物A(15 mg/kg)和G4:化合物A(45/60 mg/kg),5只/組。
b,動物分組後開始給藥,給藥體積均為10 mL/kg,口服給藥(po);每天稱重給藥1次,連續給藥21天;每週測量瘤徑2次。
c,腫瘤體積(Tumor volume,TV):每週測量2次腫瘤體積,以觀察瘤塊體積變化和生長速度。腫瘤體積V=1/2×a×b
2,其中a、b分別表示腫瘤長徑和短徑。化合物對腫瘤組織的生長抑制作用採用腫瘤生長抑制率TGI(%)評價。TGI(%)=[1-(某給藥組的平均腫瘤體積-該給藥組分組當天的平均腫瘤體積)/(陰性對照組的平均腫瘤體積-陰性對照組分組當天的平均腫瘤體積)]×100%。給藥組和陰性對照組取同一天數據。
d,在試驗過程中密切觀察小鼠生活狀態,包括外觀體征、一般行為活動、精神狀態、攝食情況、呼吸狀態、糞便和尿液性狀、注射局部及其它毒性表現。
e,試驗達到終點後,將小鼠實施安樂死,動物屍體凍存至冰櫃,移交至有資質的醫療廢棄物處理單位進行處置。
4.實驗結果
表6.實驗資料
| 分組 | 劑量 (mg/kg) | 腫瘤體積 a , b(mm 3) | TGI (%) | 體重改變(%) |
| G1:溶媒對照組 | - | 2085±372 | - | 6.3 |
| G2:Gefitinib | 100 | 15±1** | 109 | -0.1 |
| G3:化合物A | 15 | 1043±82** | 55 | 10.2 |
| G4:化合物A | 45/60 | 190±35** | 100 | -12.0 |
a,平均值±標準誤;
b,P值腫瘤體積進行統計分析,與G1組比較,*P﹤0.05;**P﹤0.01。
5.實驗結論
從上數結果可以看出,化合物A劑量15、45/60mg/kg和Gefitinib劑量100mg/kg均能顯著抑制腫瘤生長,且化合物A呈明顯的量效關係(圖5)。化合物A 45/60mg/kg與Gefitinib 100mg/kg療效相當無統計差異。小鼠耐受性良好(圖6)。
(七)Osimertinib(奧希替尼)耐藥人源肺癌PDX模型研究
1.實驗目的
評價化合物A連續21天口服給藥,對Osimertinib耐藥人源肺癌PDX模型LD1-0025-200717的抗腫瘤活性及毒副作用。
2.實驗材料
NU/NU小鼠,雌性,SPF級,購自浙江維通利華實驗動物技術有限公司。
LD1-0025-200717,人源肺癌腫瘤組織,54歲男性患者,臨床診斷:左上肺原發性支氣管肺癌,腺癌;EGFR三突變,19del & T790M & C797S;Osimertinib耐藥;PDX病理診斷:低-中分化腺癌。傳至FP2+5代用於本次藥效試驗。
3.實驗步驟
3.1 人源肺癌移植瘤體內接種
將LD1-0025-200717腫瘤組織均勻切成大小約為3mm×3mm×3mm(約50~90mg)腫瘤塊並接種於NU/NU小鼠右側皮下。隨後觀察接種後小鼠並監測腫瘤的生長。
3.2 腫瘤分組、給藥及測量
a,當腫瘤平均體積達到143.53 mm
3時,根據瘤體積大小隨機分為3組:G1:溶媒對照組、G2:化合物A(15 mg/kg)和G3:化合物A(60 mg/kg),每組8只。分組當天為第0天。
b,動物分組當天開始給藥,給藥體積均為10 mL/kg,口服給藥(po);每天稱重給藥1次,連續給藥21天;每週測量瘤徑2次。
c,腫瘤體積(Tumor volume,TV):每週測量2次腫瘤體積,以觀察瘤塊體積變化和生長速度。腫瘤體積V=1/2×a×b
2,其中a、b分別表示腫瘤長徑和短徑。化合物對腫瘤組織的生長抑制作用採用腫瘤生長抑制率TGI(%)評價。TGI(%)=[1-(某給藥組的平均腫瘤體積-該給藥組分組當天的平均腫瘤體積)/(陰性對照組的平均腫瘤體積-陰性對照組分組當天的平均腫瘤體積)]×100%。給藥組和陰性對照組取同一天數據。
d,在試驗過程中密切觀察小鼠生活狀態,包括外觀體征、一般行為活動、精神狀態、攝食情況、呼吸狀態、糞便和尿液性狀、注射局部及其它毒性表現。
e,試驗達到終點後,將小鼠實施安樂死,動物屍體凍存至冰櫃,移交至有資質的醫療廢棄物處理單位進行處置。
4.實驗結果
表7.實驗資料
| 分組 | 劑量 (mg/kg) | 腫瘤體積 a , b(mm 3) | TGI(%) | 體重改變(%) |
| G1:Vehicle | - | 606.5±77.2 | - | 10.1 |
| G2:化合物A | 15 | 333.6±49.5** | 59.2 | 5.9 |
| G3:化合物A | 60 | 121.8±21.3** | 115 | 5.2 |
a,平均值±標準誤;
b,P值腫瘤體積進行統計分析,與G1組比較,*P﹤0.05;**P﹤0.01。
5.實驗結論
從以上結果可以看出,在Osimertinib耐藥人源肺癌PDX模型LD1-0025-200717中,化合物A給藥劑量15、60mg/kg均能顯著抑制腫瘤生長,且呈明顯的量效關係(圖7)。小鼠耐受性良好(圖8)。
(八)L858R/C797S突變的體內藥效研究
1.實驗目的
評價化合物A連續14天口服給藥,對Ba/F3 EGFR L858R/C797S的抗腫瘤活性及毒副作用。
2.實驗材料
NU/NU小鼠,雌性,SPF級,購自北京維通利華實驗動物技術有限公司。
Ba/F3 EGFR L858R/C797S細胞,購自康源博創生物科技(北京)有限公司。
3.實驗步驟
3.1 細胞培養
用含有10%的胎牛血清的RPMI1640培養基在37℃,5%二氧化碳培養箱中培養。細胞培養起始濃度5×10
6個,隔2-3天待細胞長滿後分瓶傳代。將處於對數生長期的腫瘤細胞用於體內腫瘤的接種。
3.2 細胞接種
將Ba/F3 EGFR L858R/C797S細胞以2×10
6個/0.1mL接種於NU/NU小鼠的右側腋窩皮下,接種當天設為第0天。
3.3 腫瘤分組、給藥及測量
a,當平均腫瘤體積約120mm
3時,按照腫瘤體積大小隨機分為5組:G1:Vehicle、G2:Osimertinib(10 mg/kg)、G3:化合物A(15 mg/kg)、G4:化合物A(30 mg/kg)和G5:化合物A(65 mg/kg),7只/組。
b,動物分組後開始給藥,給藥體積均為10 mL/kg,口服給藥(po);每天稱重給藥1次,連續給藥14天;每週測量瘤徑2次。
c,腫瘤體積(Tumor volume,TV):每週測量2次腫瘤體積,以觀察瘤塊體積變化和生長速度。腫瘤體積V=1/2×a×b
2,其中a、b分別表示腫瘤長徑和短徑。化合物對腫瘤組織的生長抑制作用採用腫瘤生長抑制率TGI(%)評價。TGI(%)=[1-(某給藥組的平均腫瘤體積-該給藥組分組當天的平均腫瘤體積)/(陰性對照組的平均腫瘤體積-陰性對照組分組當天的平均腫瘤體積)]×100%。給藥組和陰性對照組取同一天數據。
d,在試驗過程中密切觀察小鼠生活狀態,包括外觀體征、一般行為活動、精神狀態、攝食情況、呼吸狀態、糞便和尿液性狀、注射局部及其它毒性表現。
e,試驗達到終點後,將小鼠實施安樂死,動物屍體凍存至冰櫃,移交至有資質的醫療廢棄物處理單位進行處置。
4.實驗結果
表8.實驗資料
| 分組 | 劑量 (mg/kg) | 腫瘤體積 a , b(mm 3) | TGI(%) | 體重改變(%) |
| G1:溶媒對照組 | - | 2944±118.6 | - | 25.6 |
| G2:Osimertinib | 10 | 2949±134.0 | -0.2 | 24.3 |
| G3:化合物A | 15 | 2079±109.9** | 30.7 | 22.3 |
| G4:化合物A | 30 | 1581±139.2** | 48.3 | 15.5 |
| G5:化合物A | 65 | 1059±99.9** | 66.7 | 13.7 |
a,平均值±標準誤;
b,P值腫瘤體積進行統計分析,與G1組比較,*P﹤0.05;**P﹤0.01。
5.實驗結論
從上數結果可以看出,化合物A給藥劑量15、30、65mg/kg均能顯著抑制腫瘤生長,且呈明顯的量效關係,Osimertinib對L858R/C797S雙突變模型幾乎無抑制作用(圖9),證實其耐藥性。小鼠耐受性良好(圖10)。
(九)L858R/T790M突變的體內藥效研究
1.實驗目的
評價化合物A連續21天口服給藥,對H1975(L858R/T790M)的抗腫瘤活性及毒副作用。
2.實驗材料
BALB/c裸小鼠,雌性,SPF級,江蘇集萃藥康生物科技股份有限公司。
H1975細胞,購自ATCC。
3.實驗步驟
3.1 細胞培養
用含有RPMI1640培養基中加10%胎牛血清,1%雙抗(青黴素/鏈黴素溶液),37℃ 5% CO
2培養。一周兩次0.25%胰酶常規離心處理傳代。當細胞飽和度為80%-90%,數量到達要求時,收取細胞,計數,接種。
3.2 細胞接種
將含有5×10
6個H1975細胞的PBS(終體積為100 uL)皮下接種於每只小鼠的右前肢腋窩皮下,入組動物腫瘤平均體積達到127 mm
3時開始分組給藥。
3.3 腫瘤分組、給藥及測量
a,當平均腫瘤體積達到127mm
3時,按照腫瘤體積大小隨機分為4組:G1:溶媒對照組、G2:化合物A(15 mg/kg)、G3:化合物A(30 mg/kg)和G4:化合物A(65 mg/kg),8只/組。分組當天為第0天。
b,動物分組後開始給藥,給藥體積均為10 mL/kg,口服給藥(po);每天稱重給藥1次,連續給藥21天;每週測量瘤徑2次。
c,腫瘤體積(Tumor volume,TV):每週測量2次腫瘤體積,以觀察瘤塊體積變化和生長速度。腫瘤體積V=1/2×a×b
2,其中a、b分別表示腫瘤長徑和短徑。化合物對腫瘤組織的生長抑制作用採用腫瘤生長抑制率TGI(%)評價。TGI(%)=[1-(某給藥組的平均腫瘤體積-該給藥組分組當天的平均腫瘤體積)/(陰性對照組的平均腫瘤體積-陰性對照組分組當天的平均腫瘤體積)]×100%。給藥組和陰性對照組取同一天數據。
d,在試驗過程中密切觀察小鼠生活狀態,包括外觀體征、一般行為活動、精神狀態、攝食情況、呼吸狀態、糞便和尿液性狀、注射局部及其它毒性表現。
e,試驗達到終點後,將小鼠實施安樂死,動物屍體凍存至冰櫃,移交至有資質的醫療廢棄物處理單位進行處置。
4.實驗結果
表9.實驗資料
| 分組 | 劑量 (mg/kg) | 腫瘤體積 a , b(mm 3) | TGI (%) | 體重改變(%) |
| G1:溶媒對照組 | - | 2,609 ± 265 | - | 16.9 |
| G2:化合物A | 15 | 333 ± 97* | 91.7 | 6.7 |
| G3:化合物A | 30 | 178 ± 35** | 97.9 | 6.3 |
| G4:化合物A | 65 | 25 ± 4** | 104 | 5.6 |
a,平均值±標準誤;
b,P值腫瘤體積進行統計分析,與G1組比較,*P﹤0.05;**P﹤0.01。
5.實驗結論
從上數結果可以看出,化合物A劑量15、30、65mg/kg均能顯著抑制腫瘤生長,且化合物A呈一定的量效關係(圖11)。小鼠耐受性良好(圖12)。
無
圖1為EGFR Del19/C797S突變的體內藥效研究中的動物腫瘤生長曲線圖。
圖2為EGFR Del19/C797S突變的體內藥效研究中的動物體重曲線圖。
圖3為EGFR L858R突變的體內藥效研究中的動物腫瘤生長曲線圖。
圖4為EGFR L858R突變的體內藥效研究中的動物體重曲線圖。
圖5為EGFR Del19突變的體內藥效研究中的動物腫瘤生長曲線圖。
圖6為EGFR Del19突變的體內藥效研究中的動物體重曲線圖。
圖7為Osimertinib耐藥人源肺癌PDX模型研究中的動物腫瘤生長曲線圖。
圖8為Osimertinib耐藥人源肺癌PDX模型研究中的動物體重曲線圖。
圖9為EGFR L858R/C797S突變的體內藥效研究中的動物腫瘤生長曲線圖。
圖10為EGFR L858R/C797S突變的體內藥效研究中的動物體重曲線圖。
圖11為EGFR L858R/T790M突變的體內藥效研究中的動物腫瘤生長曲線圖。
圖12為EGFR L858R/T790M突變的體內藥效研究中的動物體重曲線圖。
Claims (17)
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,其中所述的EGFR突變類型為Del19突變。

- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,其中所述的EGFR突變類型為L858R突變。
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,其中所述的EGFR突變類型為不伴隨C797S突變的T790M突變; 優選地,所述的不伴隨C797S突變的T790M突變選自以下的一種L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變。
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,其中所述的EGFR突變類型為Del19/C797S雙突變。
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,其中所述的EGFR突變類型為L858R/C797S雙突變。
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR擴增介導的癌症的藥物中的用途,優選地,所述的EGFR擴增為伴隨著Del19/T790M/C797S三突變、L858R/T790M/D537H三突變或V674L/E746_A750del/T790M三突變的EGFR擴增。
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變介導的癌症的藥物中的用途,其中所述的EGFR突變類型為20外顯子插入突變; 優選地,所述的20外顯子插入突變為伴隨L747_P753突變的插入突變。
- 一種式(A)化合物或其可藥用的鹽在製備治療EGFR突變或擴增介導的癌症的藥物中的用途,其中所述的EGFR突變類型選自以下的一種或任意組合:Del19突變、L858R突變、L858R/T790M雙突變、Del19/G724S/T790M三突變、L858R/T790M/L792H三突變、E709K/T790M/L858R三突變、Del19/C797S雙突變、L858R /C797S雙突變、20外顯子突變;EGFR擴增可選自以下一種或任意組合:伴隨著Del19/T790M/C797S三突變、L858R/T790M/D537H三突變或V674L/E746_A750del/T790M三突變的EGFR擴增。
- 如請求項1至請求項8中任一項所述的用途,其中所述癌症為肺癌; 優選地,所述癌症為非小細胞肺癌; 優選地,所述癌症為未曾接受過治療的非小細胞肺癌; 優選地,所述癌症為既往接受過EGFR抑制劑治療後,產生耐藥的非小細胞肺癌。
- 如請求項9所述的用途,其中所述的EGFR抑制劑包括第一代EGFR抑制劑、第二代EGFR抑制劑或第三代EGFR抑制劑; 優選地,所述的第一代EGFR抑制劑包括吉非替尼、埃克替尼、厄洛替尼; 優選地,所述的第二代EGFR抑制劑包括阿法替尼、達克替尼; 優選地,所述的第三代EGFR抑制劑包括奧希替尼、阿美替尼、伏美替尼、貝福替尼。
- 一種式(A)化合物或其可藥用的鹽在製備治療FGFR2高表達的癌症的藥物中的用途,。
- 一種式(A)化合物或其可藥用的鹽在製備治療C-KIT突變的癌症的藥物中的用途,其中所述的C-KIT突變類型為V560G突變和/或D816Y突變和/或D816H突變和/或V559和V560氨基酸缺失突變和/或D816V突變。
- 一種式(A)化合物或其可藥用的鹽在製備治療EML4-ALK融合蛋白介導的癌症的藥物中的用途,。
- 一種式(A)化合物或其可藥用的鹽在製備治療EML4-ALK融合蛋白 L1196M突變和/或F1174L突變和/或L1196M/L1198F雙突變介導的癌症的藥物中的用途,。
- 一種式(A)化合物或其可藥用的鹽在製備治療SLC34A2-ROS1融合蛋白介導的癌症的藥物中的用途,。
- 一種式(A)化合物或其可藥用的鹽在製備治療SLC34A2-ROS1融合蛋白 D2033N突變介導的癌症的藥物中的用途,。
- 如請求項1至請求項16中任一項所述的用途,其中(A)化合物的可藥用鹽為鹽酸鹽。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111200131.0 | 2021-10-14 | ||
| CN202111200131 | 2021-10-14 | ||
| CN202210330403 | 2022-03-31 | ||
| CN202210330403.7 | 2022-03-31 | ||
| CN202211221824.2 | 2022-10-08 | ||
| CN202211221824 | 2022-10-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| TW202317117A true TW202317117A (zh) | 2023-05-01 |
Family
ID=85987313
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW111138872A TW202317117A (zh) | 2021-10-14 | 2022-10-13 | 一種三環化合物的用途 |
Country Status (3)
| Country | Link |
|---|---|
| CN (1) | CN118043329A (zh) |
| TW (1) | TW202317117A (zh) |
| WO (1) | WO2023061434A1 (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024046405A1 (zh) * | 2022-09-01 | 2024-03-07 | 齐鲁制药有限公司 | 一种egfr激酶抑制剂的用途 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2555803T3 (es) * | 2006-10-23 | 2016-01-08 | Cephalon, Inc. | Fusión de derivados bicíclicos 2,4-diaminopirimidina como utilizar inhibidores ALK y c-Met |
| KR101364277B1 (ko) * | 2006-12-08 | 2014-02-21 | 아이알엠 엘엘씨 | 단백질 키나제 억제제로서의 화합물 |
| WO2020216371A1 (zh) * | 2019-04-26 | 2020-10-29 | 江苏先声药业有限公司 | Egfr抑制剂及其应用 |
| BR112022019888A2 (pt) * | 2020-04-14 | 2022-11-22 | Qilu Pharmaceutical Co Ltd | Compostos tricíclicos como inibidores de egfr |
-
2022
- 2022-10-13 TW TW111138872A patent/TW202317117A/zh unknown
- 2022-10-13 CN CN202280066992.0A patent/CN118043329A/zh active Pending
- 2022-10-13 WO PCT/CN2022/125057 patent/WO2023061434A1/zh active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023061434A1 (zh) | 2023-04-20 |
| CN118043329A (zh) | 2024-05-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2747260C2 (ru) | Ингибитор рфрф4, способ его получения и его фармацевтическое применение | |
| JP6898868B2 (ja) | Hpk1阻害剤およびそれを用いる方法 | |
| US6608056B1 (en) | Fused heteroaryl derivatives | |
| US9956222B2 (en) | Quinazoline derivative as tyrosine-kinase inhibitor, preparation method therefor and application thereof | |
| US8258152B2 (en) | N-substituted azaindoles and methods of use | |
| JP5689069B2 (ja) | ピラゾロピリジンpi3k阻害剤化合物及び使用方法 | |
| WO2017080338A1 (zh) | 丙烯酸类衍生物、其制备方法及其在医药上的用途 | |
| TW200902531A (en) | Imidazolopyrimidine analogs and their use as PI3 kinase and mTOR inhibitors | |
| US20110130406A1 (en) | Pyrazolo-pyridines as tyrosine kinase inhibitors | |
| AU2006322187A1 (en) | Pyrazolo[1,5-a]pyridine-3-carboxylic acids as EphB and VEGFR2 kinase inhibitors | |
| AU2006319401A1 (en) | 3-(substituted amino)-pyrazolo[3,4-d]pyrimidines as EphB and VEGFR2 kinase inhibitors | |
| AU2005303965A1 (en) | 1,4 substituted pyrazolopyrimidines as kinase inhibitors | |
| BRPI0710943A2 (pt) | compostos farmacêuticos | |
| TW200407131A (en) | Novel benzoimidazole derivatives useful as antiproliferative agents | |
| CN109745321B (zh) | 包含fgfr4抑制剂的药物组合物 | |
| CN111566100A (zh) | 嘧啶类化合物、其制备方法及其医药用途 | |
| WO2013071698A1 (zh) | 三环类PI3K和/或mTOR抑制剂 | |
| CN114644627A (zh) | AhR抑制剂及其用途 | |
| WO2023061434A1 (zh) | 一种三环化合物的用途 | |
| CN110520416A (zh) | 多取代吡啶酮类衍生物、其制备方法及其医药用途 | |
| TWI546304B (zh) | Protein tyrosine kinase inhibitors and their use | |
| WO2020221209A1 (zh) | 一种cd73抑制剂,其制备方法和应用 | |
| WO2022197913A1 (en) | Bicyclic derivatives which can be used to treat cancer | |
| WO2017211216A1 (zh) | 稠合嘧啶哌啶环衍生物及其制备方法和应用 | |
| WO2014154026A1 (zh) | PI3K和/或mTOR抑制剂的前药 |