TW202140490A - Pyrimido heterocyclic compounds and application thereof - Google Patents
Pyrimido heterocyclic compounds and application thereof Download PDFInfo
- Publication number
- TW202140490A TW202140490A TW110112436A TW110112436A TW202140490A TW 202140490 A TW202140490 A TW 202140490A TW 110112436 A TW110112436 A TW 110112436A TW 110112436 A TW110112436 A TW 110112436A TW 202140490 A TW202140490 A TW 202140490A
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- μmol
- add
- alkyl
- reaction
- Prior art date
Links
Images


Landscapes
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
本發明主張如下優先權: CN202010323035.4,申請日2020年04月22日; CN202010953203.8,申請日2020年09月11日; CN202011593642.9,申請日2020年12月29日; PCT/CN2021/080278,申請日2021年03月11日。The present invention claims the following priority: CN202010323035.4, application date April 22, 2020; CN202010953203.8, application date September 11, 2020; CN202011593642.9, application date December 29, 2020; PCT/CN2021/080278, application date March 11, 2021.
本發明關於一類嘧啶並雜環類化合物,具體關於式 (III) 所示化合物或其藥學上可接受的鹽。The present invention relates to a class of pyrimidoheterocyclic compounds, and specifically relates to a compound represented by formula (III) or a pharmaceutically acceptable salt thereof.
RAS癌基因突變是人類癌症中最常見的活化突變,發生在30%的人類腫瘤中。RAS基因家族包括三個亞型(KRAS、HRAS和NRAS),其中85%的RAS驅動的癌症是由KRAS亞型突變引起的。KRAS突變常見於實體腫瘤中,如:肺腺癌、胰腺導管癌和結直腸癌等。在KRAS突變腫瘤中,80%的致癌突變發生在密碼子12上,最常見的突變包括:p.G12D(41%)、p.G12V(28%)和p.G12C(14%)。RAS oncogene mutations are the most common activating mutations in human cancers, occurring in 30% of human tumors. The RAS gene family includes three subtypes (KRAS, HRAS, and NRAS), of which 85% of RAS-driven cancers are caused by mutations in the KRAS subtype. KRAS mutations are common in solid tumors, such as lung adenocarcinoma, pancreatic ductal carcinoma, and colorectal cancer. In KRAS mutant tumors, 80% of oncogenic mutations occur at codon 12. The most common mutations include: p.G12D (41%), p.G12V (28%) and p.G12C (14%).
KRAS基因的全名是Kirsten rat sarcoma viraloncogene homolog(Kristen 大鼠肉瘤病毒癌基因同源物)。KRAS在細胞生長的訊息調控中起著一個樞紐的作用,上游的EGFR(ErbB1)、HER2(ErbB2)、ErbB3和ErbB4等細胞表面受體,在接受了外界訊息之後,會通過RAS蛋白,把訊息傳遞到下游。KRAS蛋白沒有被活化的時候,與GDP(鳥嘌呤核苷酸二磷酸)緊密結合。在被SOS1等鳥嘌呤核苷酸交換因子活化後,與GTP (鳥嘌呤核苷酸三磷酸)結合,變成激酶活性的狀態。KRAS基因突變後,可以不依賴於上游生長因子受體訊息,獨立向下游通路傳輸生長和增殖的訊息,造成不受控制的細胞生長和腫瘤進展,同時KRAS基因是否有突變,也是腫瘤預後的一個重要指標。The full name of the KRAS gene is Kirsten rat sarcoma viraloncogene homolog (Kristen rat sarcoma virus oncogene homolog). KRAS plays a pivotal role in the information regulation of cell growth. The upstream cell surface receptors such as EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4, after receiving external information, will use the RAS protein to transfer the information. Pass it downstream. When KRAS protein is not activated, it binds closely to GDP (guanine nucleotide diphosphate). After being activated by guanine nucleotide exchange factors such as SOS1, it binds to GTP (guanine nucleotide triphosphate) and becomes a state of kinase activity. After KRAS gene mutation, it can independently transmit growth and proliferation messages to downstream pathways independently of upstream growth factor receptor messages, causing uncontrolled cell growth and tumor progression. At the same time, whether the KRAS gene has mutations is also one of the prognosis of tumors. Important indicators.
儘管KRAS是第一個被發現的癌基因,但長期以來被認為是不可成藥靶點。直到2019年,Amgen和Mirati Therapeutics先後公佈了的KRAS小分子抑制劑AMG510和MRTX849的臨床研究結果,首次在臨床上證實了KRAS抑制劑在臨床上治療腫瘤的有效性。AMG 510和MRTX849都屬不可逆小分抑制劑,通過與KRAS G12C突變蛋白的半胱胺酸殘基形成不可逆共價鍵,從而抑制KRAS活性。Although KRAS is the first oncogene to be discovered, it has long been regarded as an un-drugable target. Until 2019, Amgen and Mirati Therapeutics successively announced the clinical research results of KRAS small molecule inhibitors AMG510 and MRTX849, which for the first time clinically confirmed the effectiveness of KRAS inhibitors in the treatment of tumors. Both AMG 510 and MRTX849 are irreversible small-part inhibitors, which inhibit KRAS activity by forming irreversible covalent bonds with the cysteine residues of the KRAS G12C mutant protein.
統計結果顯示,肺腺癌有12-36%病人KRAS突變驅動的;27-56%結腸癌病人由KRAS驅動,此外90%的胰腺癌、21%的子宮內膜癌和12-36%的肺腺癌等都是由KRAS驅動的,病人群體巨大;在KRAS的基因突變中,97%是發生在第12位或者第13位的胺基酸殘基發生了突變,其中G12D,G12V和G13D這三種突變,但是這三種突變的成藥性較差,KRAS(G12C)突變:12位的甘胺酸被半胱胺酸替換後為共價抑制劑的開發提供了一個很好的方向。Statistics show that 12-36% of patients with lung adenocarcinoma are driven by KRAS mutations; 27-56% of colon cancer patients are driven by KRAS, in addition 90% of pancreatic cancer, 21% of endometrial cancer and 12-36% of lung Adenocarcinoma, etc. are all driven by KRAS, and the patient population is huge; 97% of KRAS gene mutations are mutations in the 12th or 13th amino acid residues, among which G12D, G12V and G13D are Three kinds of mutations, but these three kinds of mutations have poor druggability. KRAS (G12C) mutation: The replacement of glycine at position 12 by cysteine provides a good direction for the development of covalent inhibitors.
本發明提供了式(III)所示化合物或其藥學上可接受的鹽,

在本發明的一些方案中,上述各Ra
分別獨立地選自F、Cl、Br、I、OH、NH2
、CN、CH3
、CH2
CH3
、OCH3
、OCH2
CH3
、-CH=CH2
、-CH2
-CH=CH2
和
在本發明的一些方案中,上述各Ra
分別獨立地選自F、OH、NH2
、CH3
、CF3
、CH2
CH3
和
在本發明的一些方案中,上述R1 選自苯基、萘基、吲哚基和吲唑基,所述苯基、萘基、吲哚基和吲唑基任選被1、2或3個Ra 取代,其他變量如本發明所定義。In some embodiments of the present invention, the above-mentioned R 1 is selected from phenyl, naphthyl, indolyl and indazolyl, and the phenyl, naphthyl, indolyl and indazolyl are optionally selected by 1, 2 or 3 R a is substituted, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R1
選自



在本發明的一些方案中,上述R2 選自H、CH3 、CH2 CH3 和CH(CH3 )2 ,所述CH3 、CH2 CH3 和CH(CH3 )2 選任被1、2或3個Rb 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from H, CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 , and the CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 are optionally selected from 1, 2 or 3 R b substitutions, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R2 選自H和CH3 ,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from H and CH 3 , and other variables are as defined in the present invention.
在本發明的一些方案中,上述各R分別獨立地選自H、F、Cl、Br、OH、CN、CH3
、CH2
CH3
、CH2
CF3
、OCH3
、OCF3
和
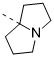
在本發明的一些方案中,上述Rc 選自四氫吡咯基和六氫-1H-吡咯里嗪基,所述四氫吡咯基和六氫-1H-吡咯里嗪基任選被1、2或3個R取代,其他變量如本發明所定義。In some embodiments of the present invention, the above-mentioned R c is selected from tetrahydropyrrolyl and hexahydro-1H-pyrrolizinyl, and the tetrahydropyrrolyl and hexahydro-1H-pyrrolizinyl are optionally selected by 1, 2 Or 3 R substitutions, and other variables are as defined in the present invention.
在本發明的一些方案中,上述Rc
選自


在本發明的一些方案中,上述Rc
選自


![]()
![]()
在本發明的一些方案中,上述R3 選自CH3 ,所述CH3 任選被1、2或3個Rc 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 3 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R c , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R3
選自



在本發明的一些方案中,上述R3
選自


在本發明的一些方案中,上述R4 選自H和CH3 ,所述CH3 任選被1、2或3個Rd 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from H and CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R d , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R4 選自H、CH3 和CH2 CN,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from H, CH 3 and CH 2 CN, and other variables are as defined in the present invention.
本發明提供了式(III)所示化合物或其藥學上可接受的鹽,
在本發明的一些方案中,上述化合物或其藥學上可接受的鹽,其選自,
在本發明的一些方案中,上述R1
選自苯基、萘基和
在本發明的一些方案中,上述R1
選自
在本發明的一些方案中,上述R2 選自H、CH3 、CH2 CH3 和CH(CH3 )2 ,所述CH3 、CH2 CH3 和CH(CH3 )2 選任被1、2或3個Rb 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from H, CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 , and the CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 are optionally selected from 1, 2 or 3 R b substitutions, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R2 選自H和CH3 ,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from H and CH 3 , and other variables are as defined in the present invention.
在本發明的一些方案中,上述Rc
選自![]()
![]()
在本發明的一些方案中,上述Rc
選自![]()
![]()
![]()
![]()
在本發明的一些方案中,上述R3 選自CH3 ,所述CH3 任選被1、2或3個Rc 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 3 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R c , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R3
選自
在本發明的一些方案中,上述R3
選自
在本發明的一些方案中,上述R4 選自H、CH3 ,所述CH3 任選被1、2或3個Rd 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from H and CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 Rd , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R4 選自H、CH3 和CH2 CN,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from H, CH 3 and CH 2 CN, and other variables are as defined in the present invention.
本發明提供了式(III)所示化合物或其藥學上可接受的鹽,
在本發明的一些方案中,上述化合物或其藥學上可接受的鹽,其選自,
在本發明的一些方案中,上述R1
選自苯基、萘基和
在本發明的一些方案中,上述R1
選自
在本發明的一些方案中,上述R2 選自H、CH3 、CH2 CH3 和CH(CH3 )2 ,所述CH3 、CH2 CH3 和CH(CH3 )2 選任被1、2或3個Rb 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from H, CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 , and the CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 are optionally selected from 1, 2 or 3 R b substitutions, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R2 選自H和CH3 ,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from H and CH 3 , and other variables are as defined in the present invention.
在本發明的一些方案中,上述Rc
選自
在本發明的一些方案中,上述R3 選自CH3 ,所述CH3 任選被1、2或3個Rc 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 3 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R c , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R3
選自
在本發明的一些方案中,上述R4 選自CH3 ,所述CH3 任選被1、2或3個Rd 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R d , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R4 選自CH2 CN,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from CH 2 CN, and other variables are as defined in the present invention.
本發明提供了式(II)所示化合物或其藥學上可接受的鹽,
在本發明的一些方案中,上述R1 選自萘基,所述萘基任選被1、2或3個Ra 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-described R 1 is selected naphthyl group, a naphthyl group optionally substituted with 1, 2 or 3 R a, the other variables are as defined in the present invention.
在本發明的一些方案中,上述R1
選自
在本發明的一些方案中,上述R2 選自CH3 、CH2 CH3 和CH(CH3 )2 ,所述CH3 、CH2 CH3 和CH(CH3 )2 選任被1、2或3個Rb 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 , and the CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 are optionally selected from 1, 2 or 3 R b substitutions, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R2 選自CH3 ,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from CH 3 , and other variables are as defined in the present invention.
在本發明的一些方案中,上述Rc
選自
在本發明的一些方案中,上述R3 選自CH3 ,所述CH3 任選被1、2或3個Rc 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 3 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R c , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R3
選自
在本發明的一些方案中,上述R4 選自CH3 ,所述CH3 任選被1、2或3個Rd 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R d , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R4 選自CH2 CN,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from CH 2 CN, and other variables are as defined in the present invention.
本發明提供了式(I)所示化合物或其藥學上可接受的鹽,
在本發明的一些方案中,上述R1 選自萘基,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 1 is selected from naphthyl, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R1
選自
在本發明的一些方案中,上述R2 選自CH3 、CH2 CH3 和CH(CH3 )2 ,所述CH3 、CH2 CH3 和CH(CH3 )2 選任被1、2或3個Rb 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 , and the CH 3 , CH 2 CH 3 and CH(CH 3 ) 2 are optionally selected from 1, 2 or 3 R b substitutions, and other variables are as defined in the present invention.
在本發明的一些方案中,上述R2 選自CH3 ,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 2 is selected from CH 3 , and other variables are as defined in the present invention.
在本發明的一些方案中,上述Rc
選自
在本發明的一些方案中,上述R3 選自CH3 ,所述CH3 任選被1、2或3個Rc 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 3 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R c , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R3
選自
在本發明的一些方案中,上述R4 選自CH3 ,所述CH3 任選被1、2或3個Rd 取代,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from CH 3 , and the CH 3 is optionally substituted with 1, 2 or 3 R d , and other variables are as defined in the present invention.
在本發明的一些方案中,上述R4 選自CH2 CN,其他變量如本發明所定義。In some aspects of the present invention, the above-mentioned R 4 is selected from CH 2 CN, and other variables are as defined in the present invention.
在本發明的一些方案中,上述化合物或其藥學上可接受的鹽,其選自,
在本發明的一些方案中,上述化合物或其藥學上可接受的鹽,其選自,



本發明還有一些方案是由上述變量任意組合而來。There are also some schemes of the present invention that come from any combination of the above variables.
本發明提供了下式化合物或其藥學上可接受的鹽,
















在本發明的一些方案中,上述化合物或其藥學上可接受的鹽,其選自,



















在本發明的一些方案中,上述化合物或其藥學上可接受的鹽,其選自,

































本發明還提供了上述的化合物或其藥學上可接受的鹽在製備治療與KRASG12C突變蛋白相關疾病的藥物中的應用。The present invention also provides the application of the above-mentioned compound or a pharmaceutically acceptable salt thereof in the preparation of a medicine for treating diseases related to KRASG12C mutant protein.
技術效果 本發明化合物對KRASG12C突變的MIA-PA-CA-2細胞株、NCI-H358細胞具有良好的細胞增殖抑制活性。有良好的肝微粒體、肝細胞、血漿、全血穩定性,有良好的PK性質,並且有顯著的抑瘤作用。Technical effect The compound of the present invention has good cell proliferation inhibitory activity on KRASG12C mutant MIA-PA-CA-2 cell line and NCI-H358 cell. It has good stability of liver microsomes, hepatocytes, plasma, and whole blood, has good PK properties, and has a significant anti-tumor effect.
相關定義 除非另有說明,本文所用的下列術語和短語旨在具有下列含義。一個特定的術語或短語在沒有特別定義的情況下不應該被認為是不確定的或不清楚的,而應該按照普通的含義去理解。當本文中出現商品名時,意在指代其對應的商品或其活性成分。Related definitions Unless otherwise stated, the following terms and phrases used herein are intended to have the following meanings. A specific term or phrase should not be considered uncertain or unclear without a special definition, but should be understood in its ordinary meaning. When a trade name appears in this article, it is meant to refer to its corresponding commodity or its active ingredient.
這裡所採用的術語“藥學上可接受的”,是針對那些化合物、材料、組合物和/或劑型而言,它們在可靠的醫學判斷的範圍之內,適用於與人類和動物的組織接觸使用,而沒有過多的毒性、刺激性、過敏性反應或其它問題或併發症,與合理的利益/風險比相稱。The term "pharmaceutically acceptable" used here refers to those compounds, materials, compositions and/or dosage forms that are within the scope of reliable medical judgment and are suitable for use in contact with human and animal tissues. , Without excessive toxicity, irritation, allergic reactions or other problems or complications, commensurate with a reasonable benefit/risk ratio.
術語“藥學上可接受的鹽”是指本發明化合物的鹽,由本發明發現的具有特定取代基的化合物與相對無毒的酸或鹼製備。當本發明的化合物中含有相對酸性的官能基時,可以通過在純的溶液或合適的惰性溶劑中用足夠量的鹼與這類化合物接觸的方式獲得鹼加成鹽。藥學上可接受的鹼加成鹽包括鈉、鉀、鈣、銨、有機胺或鎂鹽或類似的鹽。當本發明的化合物中含有相對鹼性的官能團時,可以通過在純的溶液或合適的惰性溶劑中用足夠量的酸與這類化合物接觸的方式獲得酸加成鹽。藥學上可接受的酸加成鹽的實例包括無機酸鹽,所述無機酸包括例如鹽酸、氫溴酸、硝酸、碳酸,碳酸氫根,磷酸、磷酸一氫根、磷酸二氫根、硫酸、硫酸氫根、氫碘酸、亞磷酸等;以及有機酸鹽,所述有機酸包括如乙酸、丙酸、異丁酸、馬來酸、丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、苦杏仁酸、鄰苯二甲酸、苯磺酸、對甲苯磺酸、檸檬酸、酒石酸和甲磺酸等類似的酸;還包括胺基酸(如精胺酸等)的鹽,以及如葡萄糖醛酸等有機酸的鹽。本發明的某些特定的化合物含有鹼性和酸性的官能團,從而可以被轉換成任一鹼或酸加成鹽。The term "pharmaceutically acceptable salt" refers to a salt of the compound of the present invention, which is prepared from a compound with specific substituents discovered in the present invention and a relatively non-toxic acid or base. When the compound of the present invention contains a relatively acidic functional group, a base addition salt can be obtained by contacting the compound with a sufficient amount of base in a pure solution or a suitable inert solvent. Pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amine or magnesium salt or similar salts. When the compound of the present invention contains a relatively basic functional group, the acid addition salt can be obtained by contacting the compound with a sufficient amount of acid in a pure solution or a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include inorganic acid salts including, for example, hydrochloric acid, hydrobromic acid, nitric acid, carbonic acid, hydrogen carbonate, phosphoric acid, monohydrogen phosphate, dihydrogen phosphate, sulfuric acid, Hydrogen sulfate, hydroiodic acid, phosphorous acid, etc.; and organic acid salts, the organic acid includes, for example, acetic acid, propionic acid, isobutyric acid, maleic acid, malonic acid, benzoic acid, succinic acid, suberic acid, Similar acids such as fumaric acid, lactic acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid and methanesulfonic acid; also include amino acids (such as arginine, etc.) ), as well as salts of organic acids such as glucuronic acid. Certain specific compounds of the present invention contain basic and acidic functional groups, which can be converted into any base or acid addition salt.
本發明的藥學上可接受的鹽可由含有酸根或鹼基的母體化合物通過常規化學方法合成。一般情況下,這樣的鹽的製備方法是:在水或有機溶劑或兩者的混合物中,經由游離酸或鹼形式的這些化合物與化學計量的適當的鹼或酸反應來製備。The pharmaceutically acceptable salt of the present invention can be synthesized from the parent compound containing acid or base by conventional chemical methods. In general, such salts are prepared by reacting these compounds in free acid or base form with a stoichiometric amount of an appropriate base or acid in water or an organic solvent or a mixture of both.
本發明的化合物可以存在特定的幾何或立體異構體形式。本發明設想所有的這類化合物,包括順式和反式異構體、(-)- 和 (+)-對映體、(R )- 和 (S )-對映體、非對映異構體、(D )-異構體、(L )-異構體,及其外消旋混合物和其他混合物,例如對映異構體或非對映體富集的混合物,所有這些混合物都屬本發明的範圍之內。烷基等取代基中可存在另外的不對稱碳原子。所有這些異構體以及它們的混合物,均包括在本發明的範圍之內。The compounds of the present invention may exist in specific geometric or stereoisomeric forms. The present invention contemplates all such compounds, including cis and trans isomers, (-)- and (+)-enantiomers, ( R )- and ( S )-enantiomers, diastereomers Isomers, ( D )-isomers, ( L )-isomers, and their racemic mixtures and other mixtures, such as enantiomers or diastereomer-enriched mixtures, all of these mixtures belong to the original Within the scope of the invention. Additional asymmetric carbon atoms may be present in substituents such as alkyl groups. All these isomers and their mixtures are included in the scope of the present invention.
本發明的化合物可以在一個或多個構成該化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素標記化合物,比如氚(3 H),碘-125(125 I)或C-14(14 C)。又例如,可用重氫取代氫形成氘代藥物,氘與碳構成的鍵比普通氫與碳構成的鍵更堅固,相比於未氘化藥物,氘代藥物有降低毒副作用、增加藥物穩定性、增強療效、延長藥物生物半衰期等優勢。本發明的化合物的所有同位素組成的變換,無論放射性與否,都包括在本發明的範圍之內。The compound of the present invention may contain unnatural proportions of atomic isotopes on one or more of the atoms constituting the compound. For example, compounds can be labeled with radioisotopes, such as tritium ( 3 H), iodine-125 ( 125 I), or C-14 ( 14 C). For another example, deuterium can be substituted for hydrogen to form deuterated drugs. The bond formed by deuterium and carbon is stronger than the bond formed by ordinary hydrogen and carbon. Compared with undeuterated drugs, deuterated drugs have reduced toxic side effects and increased drug stability. , Enhance the efficacy, prolong the biological half-life of drugs and other advantages. All changes in the isotopic composition of the compounds of the present invention, whether radioactive or not, are included in the scope of the present invention.
術語“任選”或“任選地”指的是隨後描述的事件或狀況可能但不是必需出現的,並且該描述包括其中所述事件或狀況發生的情況以及所述事件或狀況不發生的情況。The term "optional" or "optionally" refers to the event or condition described later that may but not necessarily occur, and the description includes the situation in which the event or condition occurs and the situation in which the event or condition does not occur .
術語“被取代的”是指特定原子上的任意一個或多個氫原子被取代基取代,取代基可以包括重氫和氫的變體,只要特定原子的價態是正常的並且取代後的化合物是穩定的。當取代基為氧(即=O)時,意味著兩個氫原子被取代。氧取代不會發生在芳香基上。術語“任選被取代的”是指可以被取代,也可以不被取代,除非另有規定,取代基的種類和數目在化學上可以實現的基礎上可以是任意的。The term "substituted" means that any one or more hydrogen atoms on a specific atom are replaced by a substituent. The substituent may include deuterium and hydrogen variants, as long as the valence of the specific atom is normal and the compound after substitution Is stable. When the substituent is oxygen (ie =O), it means that two hydrogen atoms are replaced. Oxygen substitution does not occur on aromatic groups. The term "optionally substituted" means that it can be substituted or unsubstituted. Unless otherwise specified, the type and number of substituents can be arbitrary on the basis that they can be chemically realized.
當任何變量(例如R)在化合物的組成或結構中出現一次以上時,其在每一種情況下的定義都是獨立的。因此,例如,如果一個基團被0-2個R所取代,則所述基團可以任選地至多被兩個R所取代,並且每種情況下的R都有獨立的選項。此外,取代基和/或其變體的組合只有在這樣的組合會產生穩定的化合物的情況下才是被允許的。When any variable (such as R) occurs more than once in the composition or structure of a compound, its definition in each case is independent. Thus, for example, if a group is substituted with 0-2 Rs, the group can optionally be substituted with up to two Rs, and R has independent options in each case. In addition, combinations of substituents and/or variants thereof are only permitted if such combinations result in stable compounds.
當一個連接基團的數量為0時,比如-(CRR)0 -,表示該連接基團為單鍵。When the number of a linking group is 0, such as -(CRR) 0 -, it means that the linking group is a single bond.
當其中一個變量選自單鍵時,表示其連接的兩個基團直接相連,比如A-L-Z中L代表單鍵時表示該結構實際上是A-Z。When one of the variables is selected from a single bond, it means that the two groups connected are directly connected. For example, when L in A-L-Z represents a single bond, it means that the structure is actually A-Z.
當所列舉的連接基團沒有指明其連接方向,其連接方向是任意的,例如,


除非另有規定,當某一基團具有一個或多個可連接位點時,該基團的任意一個或多個位點可以通過化學鍵與其他基團相連。當該化學鍵的連接方式是不定位的,且可連接位點存在H原子時,則連接化學鍵時,該位點的H原子的個數會隨所連接化學鍵的個數而對應減少變成相應價數的基團。所述位點與其他基團連接的化學鍵可以用直形實線鍵(









除非另有說明,用楔形實線鍵(










除非另有說明,術語“富含一種異構體”、“異構體富集”、“富含一種對映體”或者“對映體富集”指其中一種異構體或對映體的含量小於100%,並且,該異構體或對映體的含量大於等於60%,或者大於等於70%,或者大於等於80%,或者大於等於90%,或者大於等於95%,或者大於等於96%,或者大於等於97%,或者大於等於98%,或者大於等於99%,或者大於等於99.5%,或者大於等於99.6%,或者大於等於99.7%,或者大於等於99.8%,或者大於等於99.9%。Unless otherwise specified, the terms "enriched in one isomer", "enriched in isomers", "enriched in one enantiomer" or "enriched in enantiomers" refer to the content of one of the isomers or enantiomers The content is less than 100%, and the content of the isomer or enantiomer is greater than or equal to 60%, or greater than or equal to 70%, or greater than or equal to 80%, or greater than or equal to 90%, or greater than or equal to 95%, or greater than or equal to 96 %, or 97% or greater, or 98% or greater, or 99% or greater, or 99.5% or greater, or 99.6% or greater, or 99.7% or greater, or 99.8% or greater, or 99.9% or greater.
除非另有說明,術語“異構體過量”或“對映體過量”指兩種異構體或兩種對映體相對百分數之間的差值。例如,其中一種異構體或對映體的含量為90%,另一種異構體或對映體的含量為10%,則異構體或對映體過量(ee值)為80%。Unless otherwise indicated, the term "isomer excess" or "enantiomeric excess" refers to the difference between the relative percentages of two isomers or two enantiomers. For example, if the content of one isomer or enantiomer is 90%, and the content of the other isomer or enantiomer is 10%, the isomer or enantiomeric excess (ee value) is 80%.
可以通過的手性合成或手性試劑或者其他常規技術製備光學活性的(R )-和(S )-異構體以及D 和L 異構體。如果想得到本發明某化合物的一種對映體,可以通過不對稱合成或者具有手性助劑的衍生作用來製備,其中將所得非對映體混合物分離,並且輔助基團裂開以提供純的所需對映異構體。 或者,當分子中含有鹼性官能團(如胺基)或酸性官能團(如羧基)時,與適當的光學活性的酸或鹼形成非對映異構體的鹽,然後通過本領域所習知的常規方法進行非對映異構體拆分,然後回收得到純的對映體。此外,對映異構體和非對映異構體的分離通常是通過使用層析法完成的,所述層析法採用手性固定相,並任選地與化學衍生法相結合(例如由胺生成胺基甲酸鹽)。 The optically active (R )- and ( S )-isomers and D and L isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If you want to obtain an enantiomer of a compound of the present invention, it can be prepared by asymmetric synthesis or derivatization with chiral auxiliary agents, in which the resulting diastereomeric mixture is separated and the auxiliary group is cleaved to provide pure all Enantiomers are required. Alternatively, when the molecule contains a basic functional group (such as an amine group) or an acidic functional group (such as a carboxyl group), it forms a diastereomeric salt with a suitable optically active acid or base, and then passes the salt as known in the art The diastereoisomers are resolved by conventional methods, and then the pure enantiomers are recovered. In addition, the separation of enantiomers and diastereomers is usually accomplished through the use of chromatography, which employs a chiral stationary phase and is optionally combined with chemical derivatization (for example, by amine Generate carbamate).
除非另有規定,術語“C1-6 烷基”用於表示直鏈或支鏈的由1至6個碳原子組成的飽和碳氫基團。所述C1-6 烷基包括C1-5 、C1-4 、C1-3 、C1-2 、C2-6 、C2-4 、C6 和C5 烷基等;其可以是一價(如甲基)、二價(如亞甲基)或者多價(如次甲基)。C1-6 烷基的實例包括但不限於甲基 (Me)、乙基 (Et)、丙基 (包括n -丙基和異丙基)、丁基 (包括n -丁基,異丁基,s -丁基和t -丁基)、戊基 (包括n -戊基,異戊基和新戊基)、己基等。Unless otherwise specified, the term "C 1-6 alkyl" is used to indicate a linear or branched saturated hydrocarbon group composed of 1 to 6 carbon atoms. The C 1-6 alkyl group includes C 1-5 , C 1-4 , C 1-3 , C 1-2 , C 2-6 , C 2-4 , C 6 and C 5 alkyl groups, etc.; it may Is monovalent (such as methyl), divalent (such as methylene) or multivalent (such as methine). Examples of C 1-6 alkyl include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n -propyl and isopropyl), butyl (including n -butyl, isobutyl) , S -butyl and t -butyl), pentyl (including n -pentyl, isopentyl and neopentyl), hexyl, etc.
除非另有規定,術語“C1-3 烷基”用於表示直鏈或支鏈的由1至3個碳原子組成的飽和碳氫基團。所述C1-3 烷基包括C1-2 和C2-3 烷基等;其可以是一價(如甲基)、二價(如亞甲基)或者多價(如次甲基)。C1-3 烷基的實例包括但不限於甲基 (Me)、乙基 (Et)、丙基 (包括n -丙基和異丙基) 等。Unless otherwise specified, the term "C 1-3 alkyl" is used to indicate a linear or branched saturated hydrocarbon group composed of 1 to 3 carbon atoms. The C 1-3 alkyl group includes C 1-2 and C 2-3 alkyl groups, etc.; it can be monovalent (such as methyl), divalent (such as methylene) or multivalent (such as methine) . Examples of C 1-3 alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n -propyl and isopropyl), and the like.
除非另有規定,術語“C1-3 烷氧基”表示通過一個氧原子連接到分子的其餘部分的那些包含1至3個碳原子的烷基基團。所述C1-3 烷氧基包括C1-2 、C2-3 、C3 和C2 烷氧基等。C1-3 烷氧基的實例包括但不限於甲氧基、乙氧基、丙氧基 (包括正丙氧基和異丙氧基)等。Unless otherwise specified, the term "C 1-3 alkoxy" refers to those alkyl groups containing 1 to 3 carbon atoms that are attached to the rest of the molecule through an oxygen atom. The C 1-3 alkoxy group includes C 1-2 , C 2-3 , C 3 and C 2 alkoxy groups and the like. Examples of C 1-3 alkoxy include, but are not limited to, methoxy, ethoxy, propoxy (including n-propoxy and isopropoxy), and the like.
除非另有規定,術語“C1-3 烷胺基”表示通過胺基連接到分子的其餘部分的那些包含1至3個碳原子的烷基基團。所述C1-3 烷胺基包括C1-2 、C3 和C2 烷胺基等。C1-3 烷胺基的實例包括但不限於-NHCH3 、-N(CH3 )2 、-NHCH2 CH3 、-N(CH3 )CH2 CH3 、-NHCH2 CH2 CH3 、-NHCH2 (CH3 )2 等。Unless otherwise specified, the term "C 1-3 alkylamino" refers to those alkyl groups containing 1 to 3 carbon atoms that are attached to the rest of the molecule through an amine group. The C 1-3 alkylamino group includes C 1-2 , C 3 and C 2 alkylamino groups and the like. Examples of C 1-3 alkylamino groups include, but are not limited to, -NHCH 3 , -N(CH 3 ) 2 , -NHCH 2 CH 3 , -N(CH 3 )CH 2 CH 3 , -NHCH 2 CH 2 CH 3 , -NHCH 2 (CH 3 ) 2 and so on.
除非另有規定,“C2-3 烯基”用於表示直鏈或支鏈的包含至少一個碳-碳雙鍵的由2至3個碳原子組成的碳氫基團,碳-碳雙鍵可以位於該基團的任何位置上。所述C2-3 烯基包括C3 和C2 烯基;所述C2-3 烯基可以是一價、二價或者多價。C2-3 烯基的實例包括但不限於乙烯基、丙烯基等。Unless otherwise specified, "C 2-3 alkenyl" is used to mean a linear or branched hydrocarbon group consisting of 2 to 3 carbon atoms containing at least one carbon-carbon double bond, and a carbon-carbon double bond It can be located at any position of the group. The C 2-3 alkenyl group includes C 3 and C 2 alkenyl groups; the C 2-3 alkenyl group may be monovalent, divalent or multivalent. Examples of C 2-3 alkenyl include, but are not limited to, vinyl, propenyl, and the like.
除非另有規定,“C2-3 炔基”用於表示直鏈或支鏈的包含至少一個碳-碳三鍵的由2至3個碳原子組成的碳氫基團,碳-碳三鍵可以位於該基團的任何位置上。其可以是一價、二價或者多價。所述C2-3 炔基包括C3 和C2 炔基。C2-3 炔基的實例包括但不限於乙炔基、丙炔基等。Unless otherwise specified, "C 2-3 alkynyl" is used to mean a linear or branched hydrocarbon group consisting of 2 to 3 carbon atoms containing at least one carbon-carbon triple bond, and a carbon-carbon triple bond It can be located at any position of the group. It can be univalent, divalent, or multivalent. The C 2-3 alkynyl group includes C 3 and C 2 alkynyl groups. Examples of C 2-3 alkynyl include, but are not limited to, ethynyl, propynyl, and the like.
除非另有規定,本發明術語“C6-10 芳環”和“C6-10 芳基”可以互換使用,術語“C6-10 芳環”或“C6-10 芳基”表示由6至10個碳原子組成的具有共軛π電子體系的環狀碳氫基團,它可以是單環、稠合雙環或稠合三環體系,其中各個環均為芳香性的。其可以是一價、二價或者多價,C6-10 芳基包括C6-9 、C9 、C10 和C6 芳基等。C6-10 芳基的實例包括但不限於苯基、萘基 (包括1-萘基和2-萘基等)。Unless otherwise specified, the terms "C 6-10 aromatic ring" and "C 6-10 aryl" can be used interchangeably in the present invention, and the term "C 6-10 aromatic ring" or "C 6-10 aryl" means that 6 A cyclic hydrocarbon group with a conjugated π-electron system composed of up to 10 carbon atoms, which can be a monocyclic, fused bicyclic or fused tricyclic system, in which each ring is aromatic. It may be monovalent, divalent or multivalent, and C 6-10 aryl groups include C 6-9 , C 9 , C 10 and C 6 aryl groups and the like. Examples of C 6-10 aryl groups include, but are not limited to, phenyl, naphthyl (including 1-naphthyl, 2-naphthyl, etc.).
除非另有規定,本發明術語“5-10元雜芳環”和“5-10元雜芳基”可以互換使用,術語“5-10元雜芳基”是表示由5至10個環原子組成的具有共軛π電子體系的環狀基團,其1、2、3或4個環原子為獨立選自O、S和N的雜原子,其餘為碳原子。其可以是單環、稠合雙環或稠合三環體系,其中各個環均為芳香性的。其中氮原子任選地被季銨化,氮和硫雜原子可任選被氧化(即NO和S(O)p ,p是1或2)。5-10元雜芳基可通過雜原子或碳原子連接到分子的其餘部分。所述5-10元雜芳基包括5-8元、5-7元、5-6元、5元和6元雜芳基等。所述5-10元雜芳基的實例包括但不限於吡咯基 (包括N -吡咯基、2-吡咯基和3-吡咯基等)、吡唑基 (包括2-吡唑基和3-吡唑基等)、咪唑基 (包括N -咪唑基、2-咪唑基、4-咪唑基和5-咪唑基等)、噁唑基 (包括2-噁唑基、4-噁唑基和5-噁唑基等)、三唑基 (1H -1,2,3-三唑基、2H -1,2,3-三唑基、1H -1,2,4-三唑基和4H -1,2,4-三唑基等)、四唑基、異噁唑基 (3-異噁唑基、4-異噁唑基和5-異噁唑基等)、噻唑基 (包括2-噻唑基、4-噻唑基和5-噻唑基等)、呋喃基 (包括2-呋喃基和3-呋喃基等)、噻吩基 (包括2-噻吩基和3-噻吩基等)、吡啶基 (包括2-吡啶基、3-吡啶基和4-吡啶基等)、吡嗪基、嘧啶基 (包括2-嘧啶基和4-嘧啶基等)、苯並噻唑基 (包括5-苯並噻唑基等)、嘌呤基、苯並咪唑基 (包括2-苯並咪唑基等)、苯並噁唑基、吲哚基 (包括5-吲哚基等)、異喹啉基 (包括1-異喹啉基和5-異喹啉基等)、喹喔啉基 (包括2-喹喔啉基和5-喹喔啉基等)或喹啉基 (包括3-喹啉基和6-喹啉基等)。Unless otherwise specified, the terms "5-10 membered heteroaryl ring" and "5-10 membered heteroaryl group" can be used interchangeably in the present invention. The term "5-10 membered heteroaryl group" means a ring consisting of 5 to 10 ring atoms. It is composed of a cyclic group with a conjugated π-electron system, in which 1, 2, 3 or 4 ring atoms are heteroatoms independently selected from O, S and N, and the rest are carbon atoms. It can be a monocyclic, fused bicyclic or fused tricyclic system, where each ring is aromatic. Where the nitrogen atom is optionally quaternized, the nitrogen and sulfur heteroatoms may optionally be oxidized (ie NO and S(O) p , p is 1 or 2). The 5-10 membered heteroaryl group can be attached to the rest of the molecule through a heteroatom or a carbon atom. The 5-10 membered heteroaryl groups include 5-8 membered, 5-7 membered, 5-6 membered, 5 membered and 6 membered heteroaryl groups and the like. Examples of the 5-10 membered heteroaryl include, but are not limited to, pyrrolyl (including N -pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, etc.), pyrazolyl (including 2-pyrazolyl and 3-pyrrolyl, etc.) Azolyl, etc.), imidazolyl (including N -imidazolyl, 2-imidazolyl, 4-imidazolyl and 5-imidazolyl, etc.), oxazolyl (including 2-oxazolyl, 4-oxazolyl and 5- Oxazolyl, etc.), triazolyl (1 H -1,2,3-triazolyl, 2 H -1,2,3-triazolyl, 1 H -1,2,4-triazolyl and 4 H -1,2,4-triazolyl, etc.), tetrazolyl, isoxazolyl (3-isoxazolyl, 4-isoxazolyl and 5-isoxazolyl, etc.), thiazolyl (including 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, etc.), furyl (including 2-furyl and 3-furyl, etc.), thienyl (including 2-thienyl and 3-thienyl, etc.), pyridine Group (including 2-pyridyl, 3-pyridyl and 4-pyridyl, etc.), pyrazinyl, pyrimidinyl (including 2-pyrimidinyl and 4-pyrimidinyl, etc.), benzothiazolyl (including 5-benzo Thiazolyl, etc.), purinyl, benzimidazolyl (including 2-benzimidazolyl, etc.), benzoxazolyl, indolyl (including 5-indolyl, etc.), isoquinolinyl (including 1- Isoquinolinyl and 5-isoquinolinyl, etc.), quinoxalinyl (including 2-quinoxalinyl and 5-quinoxalinyl, etc.) or quinolinyl (including 3-quinolinyl and 6-quinoxalinyl, etc.) Linyl etc.).
除非另有規定,術語“4-8元雜環烷基”本身或者與其他術語聯合分別表示由4至8個環原子組成的飽和環狀基團,其1、2、3或4個環原子為獨立選自O、S和N的雜原子,其餘為碳原子,其中氮原子任選地被季銨化,氮和硫雜原子可任選被氧化 (即NO和S(O)p ,p是1或2)。其包括單環和雙環體系,其中雙環體系包括螺環、並環和橋環。此外,就該“4-8元雜環烷基”而言,雜原子可以佔據雜環烷基與分子其餘部分的連接位置。所述4-8元雜環烷基包括4-6元、5-6元、4元、5元和6元雜環烷基等。4-8元雜環烷基的實例包括但不限於氮雜環丁基、氧雜環丁基、硫雜環丁基、吡咯烷基、吡唑烷基、咪唑烷基、四氫噻吩基 (包括四氫噻吩-2-基和四氫噻吩-3-基等)、四氫呋喃基 (包括四氫呋喃-2-基等)、四氫吡喃基、哌啶基 (包括1-哌啶基、2-哌啶基和3-哌啶基等)、哌嗪基 (包括1-哌嗪基和2-哌嗪基等)、嗎福林基 (包括3-嗎福林基和4-嗎福林基等)、二噁烷基、二噻烷基、異噁唑烷基、異噻唑烷基、1,2-噁嗪基、1,2-噻嗪基、六氫噠嗪基、高哌嗪基、高哌啶基或二氧雜環庚烷基等。Unless otherwise specified, the term "4-8 membered heterocycloalkyl" by itself or in combination with other terms means a saturated cyclic group consisting of 4 to 8 ring atoms, with 1, 2, 3 or 4 ring atoms. Are heteroatoms independently selected from O, S and N, and the rest are carbon atoms, wherein nitrogen atoms are optionally quaternized, and nitrogen and sulfur heteroatoms can be optionally oxidized (ie, NO and S(O) p , p Is 1 or 2). It includes monocyclic and bicyclic ring systems, where the bicyclic ring system includes spiro, fused, and bridged rings. In addition, for the "4-8 membered heterocycloalkyl group", a heteroatom may occupy the connection position of the heterocycloalkyl group with the rest of the molecule. The 4-8 membered heterocycloalkyl group includes 4-6 membered, 5-6 membered, 4-membered, 5-membered, and 6-membered heterocycloalkyl group. Examples of 4-8 membered heterocycloalkyl include, but are not limited to, azetidinyl, oxetanyl, thietanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, tetrahydrothienyl ( Including tetrahydrothiophen-2-yl and tetrahydrothiophen-3-yl, etc.), tetrahydrofuranyl (including tetrahydrofuran-2-yl, etc.), tetrahydropyranyl, piperidinyl (including 1-piperidinyl, 2- Piperidinyl and 3-piperidinyl, etc.), piperazinyl (including 1-piperazinyl and 2-piperazinyl, etc.), moofolinyl (including 3-molofolin and 4-molofolin) Etc.), dioxazinyl, dithiazinyl, isoxazolidinyl, isothiazolidinyl, 1,2-oxazinyl, 1,2-thiazinyl, hexahydropyridazinyl, homopiperazinyl , Homopiperidinyl or dioxepanyl, etc.
除非另有規定,Cn-n+m 或Cn -Cn+m 包括n至n+m個碳的任何一種具體情況,例如C1-12 包括C1 、C2 、C3 、C4 、C5 、C6 、C7 、C8 、C9 、C10 、C11 、和C12 ,也包括n至n+m中的任何一個範圍,例如C1-12 包括C1-3 、C1-6 、C1-9 、C3-6 、C3-9 、C3-12 、C6-9 、C6-12 、和C9-12 等;同理,n元至n+m元表示環上原子數為n至n+m個,例如3-12元環包括3元環、4元環、5元環、6元環、7元環、8元環、9元環、10元環、11元環、和12元環,也包括n至n+m中的任何一個範圍,例如3-12元環包括3-6元環、3-9元環、5-6元環、5-7元環、6-7元環、6-8元環、和6-10元環等。Unless otherwise specified, C n-n+m or C n -C n+m includes any specific case of n to n+m carbons, for example, C 1-12 includes C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 , and C 12 , including any range from n to n+m, for example, C 1-12 includes C 1-3 , C 1-6 , C 1-9 , C 3-6 , C 3-9 , C 3-12 , C 6-9 , C 6-12 , and C 9-12 etc.; in the same way, n yuan to n+ The m member means that the number of atoms in the ring is from n to n+m. For example, a 3-12 membered ring includes a 3-membered ring, a 4-membered ring, a 5-membered ring, a 6-membered ring, a 7-membered ring, an 8-membered ring, a 9-membered ring, 10-membered ring, 11-membered ring, and 12-membered ring, including any range from n to n+m, for example, 3-12 membered ring includes 3-6 membered ring, 3-9 membered ring, 5-6 membered ring , 5-7 membered ring, 6-7 membered ring, 6-8 membered ring, 6-10 membered ring, etc.
術語“離去基團”是指可以被另一種官能團或原子通過取代反應(例如親核取代反應)所取代的官能團或原子。例如,代表性的離去基團包括三氟甲磺酸酯;氯、溴、碘;磺酸酯基,如甲磺酸酯、甲苯磺酸酯、對溴苯磺酸酯、對甲苯磺酸酯等;醯氧基,如乙醯氧基、三氟乙醯氧基等等。The term "leaving group" refers to a functional group or atom that can be replaced by another functional group or atom through a substitution reaction (for example, a nucleophilic substitution reaction). For example, representative leaving groups include trifluoromethanesulfonate; chlorine, bromine, and iodine; sulfonate groups such as mesylate, tosylate, p-bromobenzenesulfonate, p-toluenesulfonic acid Esters, etc.; acetoxy groups, such as acetoxy, trifluoroacetoxy and the like.
術語“保護基”包括但不限於“胺基保護基”、“羥基保護基”或“巰基保護基”。術語“胺基保護基”是指適合用於阻止胺基氮位上副反應的保護基團。代表性的胺基保護基包括但不限於:甲醯基;醯基,例如鏈烷醯基(如乙醯基、三氯乙醯基或三氟乙醯基);烷氧基羰基,如三級丁氧基羰基(Boc);芳基甲氧羰基,如苄氧羰基(Cbz)和9-芴甲氧羰基(Fmoc);芳基甲基,如苄基(Bn)、三苯甲基(Tr)、1,1-二-(4'-甲氧基苯基)甲基;甲矽烷基,如三甲基甲矽烷基(TMS)和三級丁基二甲基甲矽烷基(TBS)等等。術語“羥基保護基”是指適合用於阻止羥基副反應的保護基。代表性羥基保護基包括但不限於:烷基,如甲基、乙基和三級丁基;醯基,例如鏈烷醯基(如乙醯基);芳基甲基,如苄基(Bn),對甲氧基苄基(PMB)、9-芴基甲基(Fm)和二苯基甲基(二苯甲基,DPM);甲矽烷基,如三甲基甲矽烷基(TMS)和三級丁基二甲基甲矽烷基(TBS)等等。The term "protecting group" includes but is not limited to "amino protecting group", "hydroxy protecting group" or "sulfhydryl protecting group". The term "amino protecting group" refers to a protecting group suitable for preventing side reactions at the amino nitrogen position. Representative amine protecting groups include, but are not limited to: methanoyl; anolyte, such as alkanoyl (such as acetyl, trichloroacetyl or trifluoroacetyl); alkoxycarbonyl, such as tri Grade butoxycarbonyl (Boc); arylmethoxycarbonyl, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethyloxycarbonyl (Fmoc); arylmethyl, such as benzyl (Bn), trityl ( Tr), 1,1-bis-(4'-methoxyphenyl) methyl; silyl groups, such as trimethylsilyl (TMS) and tertiary butyldimethylsilyl (TBS) etc. The term "hydroxyl protecting group" refers to a protecting group suitable for preventing side reactions of the hydroxyl group. Representative hydroxy protecting groups include but are not limited to: alkyl groups, such as methyl, ethyl, and tertiary butyl; acyl groups, such as alkanoyl groups (such as acetyl); arylmethyl groups, such as benzyl (Bn ), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm) and diphenylmethyl (diphenylmethyl, DPM); silyl groups, such as trimethylsilyl (TMS) And tertiary butyldimethylsilyl (TBS) and so on.
本發明的化合物可以通過本領域技術人員所熟知的多種合成方法來製備,包括下面列舉的具體實施方式、其與其他化學合成方法的結合所形成的實施方式以及本領域技術上人員所熟知的等同替換方式,較佳的實施方式包括但不限於本發明的實施例。The compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments listed below, the embodiments formed by combining them with other chemical synthesis methods, and equivalents well known to those skilled in the art. Alternatively, preferred implementations include, but are not limited to, embodiments of the present invention.
本發明的化合物可以通過本領域技術人員所熟知的常規方法來確認結構,如果本發明關於化合物的絕對構型,則該絕對構型可以通過本領域常規技術手段予以確證。例如單晶X射線繞射法(SXRD),把培養出的單晶用Bruker D8 venture繞射儀收集繞射強度數據,光源為CuKα輻射,掃描方式:φ/ω 掃描,收集相關數據後,進一步採用直接法(Shelxs97)解析晶體結構,便可以確證絕對構型。The structure of the compound of the present invention can be confirmed by conventional methods well known to those skilled in the art. If the present invention relates to the absolute configuration of the compound, the absolute configuration can be confirmed by conventional technical means in the field. For example, the single crystal X-ray diffraction method (SXRD) uses the Bruker D8 venture diffractometer to collect the diffraction intensity data of the cultivated single crystal. The light source is CuKα radiation. The scanning method: φ/ω scanning. After collecting the relevant data, further Using the direct method (Shelxs97) to analyze the crystal structure, the absolute configuration can be confirmed.
本發明所使用的溶劑可經市售獲得。The solvent used in the present invention is commercially available.
化合物依據本領域常規命名原則或者使用ChemDraw®軟體命名,市售化合物採用供應商目錄名稱。Compounds are named according to conventional naming principles in the field or using ChemDraw® software, and commercially available compounds use supplier catalog names.
下面通過實施例對本發明進行詳細描述,但並不意味著對本發明任何不利限制。本文已經詳細地描述了本發明,其中也揭露了其具體實施例方式,對本領域的技術人員而言,在不脫離本發明精神和範圍的情況下針對本發明具體實施方式進行各種變化和改進將是顯而易見的。The following examples describe the present invention in detail, but they are not meant to limit the present invention in any unfavorable manner. The present invention has been described in detail herein, and its specific embodiments are also disclosed. For those skilled in the art, various changes and improvements will be made to the specific embodiments of the present invention without departing from the spirit and scope of the present invention. It is obvious.
實施例 1
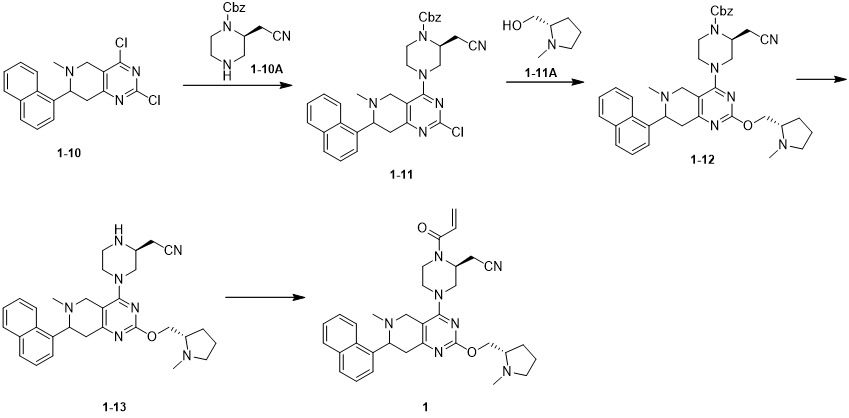
步驟2:化合物1-3的合成 將乙酸甲酯 (4.28 g, 57.83 mmol, 4.60 mL, 1.5 eq) 溶於四氫呋喃 (100 mL) 中,氮氣保護下降溫至-78℃,將六甲基二矽基胺基鋰(1 M, 59.76 mL, 1.55 eq) 緩慢滴加到反應液中。-78℃下攪拌1hr後,再將化合物1-2 (10 g, 38.56 mmol, 1 eq) 緩慢滴加到反應液中,在此溫度下繼續攪拌1hr。反應結束後,將反應液倒入飽和氯化銨水溶液(80 mL)中,乙酸乙酯(50mL × 3)萃取。合併有機相,飽和食鹽水洗滌,無水硫酸鈉乾燥,過濾,收集濾液濃縮。 粗品經管柱層析純化(石油醚/乙酸乙酯=50/1~1/1) 得到化合物1-3。1 H NMR (400 MHz, CDCl3 ) δ = 8.17 (d,J = 8.4 Hz, 1H), 7.89 (d,J = 8.0 Hz, 1H), 7.82 (d,J = 8.0 Hz, 1H), 7.57 (t,J = 6.8 Hz, 2H), 7.54 - 7.52 (m, 1H), 7.52 - 7.44 (m, 2H), 4.78 ( d,J = 2.4 Hz, 1H) , 3.69 (s, 3H), 3.09 (d,J = 6.4 Hz, 2H), 1.25 - 1.22 (s, 9H);LCMSm/z = 334.1 [M+1]+ 。Step 2: Synthesis of compound 1-3. Methyl acetate (4.28 g, 57.83 mmol, 4.60 mL, 1.5 eq) was dissolved in tetrahydrofuran (100 mL), and the temperature was reduced to -78℃ under nitrogen protection. Lithium base amide (1 M, 59.76 mL, 1.55 eq) was slowly added dropwise to the reaction solution. After stirring for 1 hr at -78°C, compound 1-2 (10 g, 38.56 mmol, 1 eq) was slowly added dropwise to the reaction solution, and stirring was continued at this temperature for 1 hr. After the reaction, the reaction solution was poured into saturated aqueous ammonium chloride solution (80 mL), and extracted with ethyl acetate (50 mL × 3). The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was collected and concentrated. The crude product is purified by column chromatography (petroleum ether/ethyl acetate=50/1~1/1) to obtain compound 1-3. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.17 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.57 ( t, J = 6.8 Hz, 2H), 7.54-7.52 (m, 1H), 7.52-7.44 (m, 2H), 4.78 (d, J = 2.4 Hz, 1H), 3.69 (s, 3H), 3.09 (d , J = 6.4 Hz, 2H), 1.25-1.22 (s, 9H); LCMS m/z = 334.1 [M+1] + .
步驟3:化合物1-4的合成 將化合物乙酸甲酯 (5.55 g, 74.98 mmol, 5.96 mL, 5 eq) 溶於四氫呋喃(50 mL) 中,氮氣保護下降溫至-78℃,將六甲基二矽基胺基鈉 (1 M, 74.98 mL, 5 eq) 加入到反應液中。在-78℃下攪拌1hr後,再將化合物1-3 (5 g,15.00 mmol, 1 eq) 緩慢滴加到反應液中,在此溫度下繼續攪拌1hr。反應完成後,將反應液倒入飽和氯化銨水溶液(50 mL)中,乙酸乙酯(50 mL × 3)萃取。合併有機相,飽和食鹽水洗滌(50 mL),無水硫酸鈉乾燥,過濾,收集濾液濃縮,得到化合物1-4,直接用於下一步反應。LCMSm/z = 376.1 [M+1]+ 。Step 3: Synthesis of compound 1-4 The compound methyl acetate (5.55 g, 74.98 mmol, 5.96 mL, 5 eq) was dissolved in tetrahydrofuran (50 mL), and the temperature was reduced to -78℃ under nitrogen protection. Sodium silylamide (1 M, 74.98 mL, 5 eq) was added to the reaction solution. After stirring at -78°C for 1 hr, compound 1-3 (5 g, 15.00 mmol, 1 eq) was slowly added dropwise to the reaction solution, and stirring was continued at this temperature for 1 hr. After the reaction was completed, the reaction solution was poured into a saturated aqueous ammonium chloride solution (50 mL), and extracted with ethyl acetate (50 mL × 3). The organic phases were combined, washed with saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was collected and concentrated to obtain compound 1-4, which was directly used in the next reaction. LCMS m/z = 376.1 [M+1] + .
步驟4:化合物1-5的合成 將化合物1-4 (5 g, 13.32 mmol, 11.92 mL, 1 eq) 溶於甲苯 (50 mL) 中,加入N,N-二甲基甲醯胺二甲基縮醛 (15.87 g, 133.16 mmol, 17.69 mL, 10 eq),在19℃下攪拌反應10hr。反應完成後,將反應液倒入飽和氯化銨水溶液(80 mL)中,乙酸乙酯(50mL ×3)萃取。合併有機相,飽和食鹽水洗滌,無水硫酸鈉乾燥,過濾,收集濾液濃縮。粗品經管柱層析(二氯甲烷/甲醇=100/1~10/1)分離純化得到化合物1-5。LCMSm/z = 431.1 [M+1]+ 。Step 4: Synthesis of compound 1-5. Dissolve compound 1-4 (5 g, 13.32 mmol, 11.92 mL, 1 eq) in toluene (50 mL) and add N,N-dimethylformamide dimethyl Acetal (15.87 g, 133.16 mmol, 17.69 mL, 10 eq), the reaction was stirred at 19° C. for 10 hr. After the reaction was completed, the reaction solution was poured into a saturated aqueous ammonium chloride solution (80 mL), and extracted with ethyl acetate (50 mL × 3). The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was collected and concentrated. The crude product was separated and purified by column chromatography (dichloromethane/methanol=100/1~10/1) to obtain compound 1-5. LCMS m/z = 431.1 [M+1] + .
步驟5:化合物1-6的合成: 將化合物1-5 (2.4 g, 5.57 mmol, 1 eq) 溶於鹽酸二氧六環(4 M, 60.00 mL) 中,在18℃下攪拌10hr。 反應完成後,反應液直接濃縮,得到化合物1-6的鹽酸鹽,直接用於下一步反應。LCMSm/z = 282.1 [M+1]+ 。Step 5: Synthesis of compound 1-6: Compound 1-5 (2.4 g, 5.57 mmol, 1 eq) was dissolved in dioxane hydrochloride (4 M, 60.00 mL), and stirred at 18° C. for 10 hr. After the completion of the reaction, the reaction solution was directly concentrated to obtain the hydrochloride salt of compound 1-6, which was directly used in the next reaction. LCMS m/z = 282.1 [M+1] + .
步驟6:化合物1-7的合成 將化合物1-6 鹽酸鹽(2 g, 6.29 mmol, 1 eq) 溶於N,N-二甲基甲醯胺 (20 mL),然後依次加入碳酸鉀(6.15 g, 18.88 mmol, 3 eq) ,碘甲烷 (1.79 g, 12.59 mmol, 783.65 µL, 2 eq),在18℃下攪拌10hr。反應完成後, 反應液倒入水中(30mL),用乙酸乙酯(30mL ×2)萃取。合併後的有機相用飽和食鹽水洗滌(50 mL),無水硫酸鈉乾燥,過濾,濃縮得到粗品。粗品經管柱層析(二氯甲烷/甲醇=50/1~10/1)分離純化得到化合物1-7。1 H NMR (400 MHz,CDCl3 ) δ = 8.47 (s, 1H), 7.96 - 7.88 (m, 2H), 7.85 (d,J = 8.4 Hz, 1H), 7.62 - 7.51 (m, 2H), 7.48 - 7.41 (m, 1H), 7.35 (d,J = 7.0 Hz, 1H), 5.52 - 5.39 (m, 1H), 3.83 (s, 3H), 3.19(s, 3H),3.23 - 3.14 (m, 1H), 2.98 - 2.87 (m, 1H)。Step 6: Synthesis of compound 1-7 Compound 1-6 hydrochloride (2 g, 6.29 mmol, 1 eq) was dissolved in N,N-dimethylformamide (20 mL), and then potassium carbonate ( 6.15 g, 18.88 mmol, 3 eq), methyl iodide (1.79 g, 12.59 mmol, 783.65 µL, 2 eq), stirred at 18°C for 10 hr. After the reaction was completed, the reaction solution was poured into water (30 mL), and extracted with ethyl acetate (30 mL × 2). The combined organic phase was washed with saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was separated and purified by column chromatography (dichloromethane/methanol=50/1~10/1) to obtain compound 1-7. 1 H NMR (400 MHz,CDCl 3 ) δ = 8.47 (s, 1H), 7.96-7.88 (m, 2H), 7.85 (d, J = 8.4 Hz, 1H), 7.62-7.51 (m, 2H), 7.48 -7.41 (m, 1H), 7.35 (d, J = 7.0 Hz, 1H), 5.52-5.39 (m, 1H), 3.83 (s, 3H), 3.19 (s, 3H), 3.23-3.14 (m, 1H) ), 2.98-2.87 (m, 1H).
步驟7:化合物1-8的合成 將化合物1-7(20 mg, 67.72 µmol, 1 eq) 溶於乙醇 (0.2 mL),1,4二氧六環 (1 mL) 中。然後加入六水合氯化鎳 (19.32 mg, 81.26 µmol, 1.2 eq) 。降溫至5~10℃後,再加入硼氫化鈉(1.28 mg, 33.86 µmol, 0.5 eq),並在10℃下反應0.5hr。反應完成後,倒入飽和氯化銨水溶液(5mL)中,用乙酸乙酯(10mL × 2)萃取。合併後的有機相用飽和食鹽水洗滌(5 mL),無水硫酸鈉乾燥,過濾,濃縮得到粗品。粗品經過薄層層析製備板分離純化(展開液:石油醚/乙酸乙酯=3/1)得到化合物1-8。1 H NMR (400 MHz, CDCl3 ) δ = 11.99 - 11.85 (m, 1H), 8.67 - 8.49 (m, 1H), 7.92 - 7.85 (m, 1H), 7.85 - 7.77 (m, 1H), 7.57 - 7.41 (m, 4H), 3.82 (s, 3H), 3.56 - 3.51 (m, 1H), 3.16 - 2.95 (m, 2H), 2.68 - 2.47 (m, 1H), 2.15 (s, 3H)。Step 7: Synthesis of compound 1-8 Compound 1-7 (20 mg, 67.72 µmol, 1 eq) was dissolved in ethanol (0.2 mL) and 1,4 dioxane (1 mL). Then add nickel chloride hexahydrate (19.32 mg, 81.26 µmol, 1.2 eq). After cooling down to 5-10°C, add sodium borohydride (1.28 mg, 33.86 µmol, 0.5 eq) and react at 10°C for 0.5hr. After the reaction is complete, pour into saturated aqueous ammonium chloride solution (5 mL), and extract with ethyl acetate (10 mL × 2). The combined organic phase was washed with saturated brine (5 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was separated and purified by thin layer chromatography preparation plate (developing solution: petroleum ether/ethyl acetate = 3/1) to obtain compound 1-8. 1 H NMR (400 MHz, CDCl 3 ) δ = 11.99-11.85 (m, 1H), 8.67-8.49 (m, 1H), 7.92-7.85 (m, 1H), 7.85-7.77 (m, 1H), 7.57- 7.41 (m, 4H), 3.82 (s, 3H), 3.56-3.51 (m, 1H), 3.16-2.95 (m, 2H), 2.68-2.47 (m, 1H), 2.15 (s, 3H).
步驟8:化合物1-9的合成 將化合物1-8 (240 mg, 807.14 µmol, 1 eq),尿素 (242.36 mg, 4.04mmol, 216.40 µL, 5 eq) 溶於乙醇 (5 mL) 中,再加入甲醇鈉(130.80 mg, 2.42 mmol, 3 eq)。85℃下反應10hr後,將反應液緩慢倒入水中,再加入乙酸乙酯(5 mL),有固體析出。過濾,收集固體得到化合物1-9。LCMSm/z = 308.1 [M+1]+ 。Step 8: Synthesis of compound 1-9. Dissolve compound 1-8 (240 mg, 807.14 µmol, 1 eq), urea (242.36 mg, 4.04 mmol, 216.40 µL, 5 eq) in ethanol (5 mL), and then add Sodium methoxide (130.80 mg, 2.42 mmol, 3 eq). After reacting at 85°C for 10 hours, the reaction solution was slowly poured into water, and ethyl acetate (5 mL) was added, and a solid precipitated out. The solid was collected by filtration to obtain compound 1-9. LCMS m/z = 308.1 [M+1] + .
步驟9:化合物1-10的合成 將化合物1-9 (400 mg, 1.30 mmol, 1 eq) 溶於三氯氧磷 (132.00 g, 860.89 mmol, 80 mL)。升溫至105℃下反應10hr後,減壓濃縮除去多餘三氯氧磷。剩餘物用乙酸乙酯(50 mL)溶解,再加入到飽和碳酸氫鈉水溶液(20 mL)中。水相用乙酸乙酯(50 mL × 3)萃取。合併後的有機相用飽和食鹽水(50 mL)洗滌,無水硫酸鈉乾燥,過濾,濃縮得到粗品。粗品用薄層層析管柱(溶析液:石油醚/乙酸乙酯=20/1~0/1)分離純化得到化合物1-10。LCMSm/z = 344.0 [M+1]+ 。Step 9: Synthesis of compound 1-10 Compound 1-9 (400 mg, 1.30 mmol, 1 eq) was dissolved in phosphorus oxychloride (132.00 g, 860.89 mmol, 80 mL). After the temperature was raised to 105°C and reacted for 10 hours, the excess phosphorus oxychloride was removed by concentration under reduced pressure. The residue was dissolved in ethyl acetate (50 mL) and added to saturated aqueous sodium bicarbonate solution (20 mL). The aqueous phase was extracted with ethyl acetate (50 mL × 3). The combined organic phase was washed with saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was separated and purified with a thin-layer chromatography column (eluent: petroleum ether/ethyl acetate=20/1~0/1) to obtain compound 1-10. LCMS m/z = 344.0 [M+1] + .
步驟10:化合物1-11的合成 將化合物1-10 (250 mg, 726.24 µmol, 1 eq),中間體1-10A鹽酸鹽(279.24 mg, 944.12 µmol, 1.3 eq) 溶於異丙醇 (2 mL),再加入N,N-二異丙基乙胺 (375.44 mg, 2.90 mmol, 505.98 µL, 4 eq) 。110℃下反應12hr後,反應液直接濃縮。剩餘物經管柱層析分離純化(溶析液:石油醚/乙酸乙酯=10/1~1/1 )得到化合物1-11。1 H NMR (400 MHz, CDCl3 ) δ = 8.60 - 8.48 (m, 1H), 7.93 - 7.87 (m, 1H), 7.86 - 7.80 (m, 1H), 7.58 - 7.34 (m, 9H), 5.21 (m, 2H), 4.77 - 4.61 (m, 1H), 4.06 (m, 2H), 3.97 - 3.75 (m, 2H), 3.62 - 3.40 (m, 3H), 3.30 - 3.00 (m, 4H), 2.78 - 2.64 (m, 1H), 2.26 (s, 1.5H), 2.21 (s, 1.5H);LCMSm/z = 567.3 [M+1]+ 。Step 10: Synthesis of compound 1-11. Compound 1-10 (250 mg, 726.24 µmol, 1 eq) and intermediate 1-10A hydrochloride (279.24 mg, 944.12 µmol, 1.3 eq) were dissolved in isopropanol (2 mL), and then add N,N-diisopropylethylamine (375.44 mg, 2.90 mmol, 505.98 µL, 4 eq). After reacting at 110°C for 12 hours, the reaction solution was directly concentrated. The residue is separated and purified by column chromatography (eluent: petroleum ether/ethyl acetate=10/1~1/1) to obtain compound 1-11. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.60-8.48 (m, 1H), 7.93-7.87 (m, 1H), 7.86-7.80 (m, 1H), 7.58-7.34 (m, 9H), 5.21 ( m, 2H), 4.77-4.61 (m, 1H), 4.06 (m, 2H), 3.97-3.75 (m, 2H), 3.62-3.40 (m, 3H), 3.30-3.00 (m, 4H), 2.78- 2.64 (m, 1H), 2.26 (s, 1.5H), 2.21 (s, 1.5H); LCMS m/z = 567.3 [M+1] + .
步驟11:化合物1-12的合成 將化合物1-11 (100 mg, 176.34 µmol, 1 eq) ,1-11A (60.93 mg, 529.03 µmol, 62.81 µL, 3 eq) 溶於1,4二氧六環 (1.5 mL) 中,再加入碳酸銫 (172.37 mg, 529.03 µmol, 3 eq),2-二環己基磷-2',6'-二異丙氧基-1,1'-聯苯 (16.46 mg, 35.27 µmol, 0.2 eq)和三(二亞苄基丙酮)二鈀 (32.30 mg, 35.27 µmol, 0.2 eq)。氮氣保護下,90℃反應24hr 。反應完成後,直接濃縮。剩餘物經管柱層析分離純化(溶析液:二氯甲烷/甲醇=100/1~10/1)得到化合物1-12。LCMSm/z = 646.4 [M+1]+ 。Step 11: Synthesis of compound 1-12. Dissolve compound 1-11 (100 mg, 176.34 µmol, 1 eq), 1-11A (60.93 mg, 529.03 µmol, 62.81 µL, 3 eq) in 1,4 dioxane (1.5 mL), add cesium carbonate (172.37 mg, 529.03 µmol, 3 eq), 2-dicyclohexylphosphorus-2',6'-diisopropoxy-1,1'-biphenyl (16.46 mg , 35.27 µmol, 0.2 eq) and tris(dibenzylideneacetone) dipalladium (32.30 mg, 35.27 µmol, 0.2 eq). Under the protection of nitrogen, react at 90°C for 24 hours. After the reaction is complete, it is directly concentrated. The residue was separated and purified by column chromatography (eluent: dichloromethane/methanol=100/1~10/1) to obtain compound 1-12. LCMS m/z = 646.4 [M+1] + .
步驟12:化合物1-13的合成 將化合物1-12(50 mg, 77.42 µmol, 1 eq) 溶於四氫呋喃 (50 mL)中,加入Pd/C (77.4 mg, 10%純度),反應體系用H2 置換三次。在15psi,20℃下,攪拌反應10hr。反應完成後,過濾,得到化合物1-13的四氫呋喃溶液(70 mL),直接用於下一步。LCMSm/z = 512.3 [M+1]+ 。Step 12: Synthesis of compound 1-13. Dissolve compound 1-12 (50 mg, 77.42 µmol, 1 eq) in tetrahydrofuran (50 mL), add Pd/C (77.4 mg, 10% purity), and use H for the reaction system 2 Replace three times. The reaction was stirred at 15 psi and 20°C for 10 hr. After the reaction is complete, filter to obtain a tetrahydrofuran solution (70 mL) of compound 1-13, which is used directly in the next step. LCMS m/z = 512.3 [M+1] + .
步驟13:化合物1的合成
往上一步反應所得到的化合物1-13四氫呋喃溶液(70 mL)中加入N,N-二異丙基乙胺 (17.18 mg, 132.90 µmol, 23.15 µL, 2 eq) , 然後降溫至-20~-30℃,加入丙烯醯氯(6.01 mg, 66.45 µmol, 5.42 µL, 1 eq) 。在此溫度下,反應30min後,反應液倒入水中(10 mL)。再用乙酸乙酯(10 mL)萃取。有機相用無水硫酸鈉乾燥,過濾,濃縮得到粗品。粗品使用高效液相層析管柱分離純化(管柱: Phenomenex Luna 80*30mm*3µm; 流動相: [10mM NH4
HCO3
水溶液-乙腈]; 乙腈%: 30%-60%, 7min)得到化合物1,化合物1經SFC鑑定由兩個非對映異構體組成(Chiralcel OD-3管柱,P1 Rt =1.93 min,P2 Rt =2.08 min,P1:P2=50.6:49.4)。1
H NMR (400 MHz, CDCl3
) δ = 8.66 - 8.53 (m, 1H), 7.93 - 7.87 (m, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.56 - 7.41 (m, 4H), 6.70 - 6.50 (m, 1H), 6.47 - 6.34 (m, 1H), 5.84 ( d, J = 7.2 Hz, 1H), 4.38 (m, 1H), 4.27 - 4.09 (m, 2H), 4.05 - 3.78 (m, 4H), 3.60 - 3.35 (m, 3H), 3.23 - 3.01 (m, 4H), 2.84 - 2.60 (m, 3H), 2.50 - 2.41 (m, 3H), 2.30 - 2.21 (m, 4H), 2.10 - 1.98 (m, 1H), 1.90 - 1.66 (m, 4H). LCMSm/z
= 566.4 [M+1]+
。Step 13: Synthesis of
實施例 2和3

步驟2:化合物2-3的合成 將鈉氫(248.01 mg, 6.20 mmol, 60% 純度, 1.2 eq) 懸浮於無水四氫呋喃(5 mL) 中,氮氣保護下冷卻到0℃,然後向其中滴加乙醯乙酸甲酯 (600 mg, 5.17 mmol, 555.56 µL, 1 eq)。攪拌10min後,滴加正丁基鋰(2.5 M, 2.27 mL, 1.1 eq),0℃繼續攪拌反應20min。再用乾冰丙酮浴將反應體系冷卻到-78℃,向其中滴加化合物2-2 (1.08 g, 5.68 mmol, 1.1 eq)的四氫呋喃(6 mL)溶液。攪拌反應30min,緩慢升至室溫攪拌30min。加水(30 mL)淬滅反應,水相用乙酸乙酯(50 mL×2)萃取。合併後的有機相用硫酸鈉乾燥、過濾除去乾燥劑、濾液減壓除去溶劑得到粗品。粗品過柱純化(乙酸乙酯/石油醚=0~20%)得到化合物2-3。1 H NMR (400 MHz, CDCl3 ) δ = 8.07 (d, J=7.6 Hz, 1H), 7.81 (d, J=8.0 Hz, 2H), 7.63 - 7.49 (m, 2H), 7.35 (t, J=8.0 Hz, 1H), 6.92 (br d, J=9.6 Hz, 1H), 3.75 (s, 3H), 3.55 (s, 2H), 3.37 (dd, J=1.6, 18.1 Hz, 1H), 3.24 (d, J=1.2 Hz, 1H), 2.86-2.77 (m, 1H)。Step 2: Synthesis of compound 2-3. Suspend sodium hydrogen (248.01 mg, 6.20 mmol, 60% purity, 1.2 eq) in anhydrous tetrahydrofuran (5 mL), cool to 0℃ under nitrogen protection, and then add ethyl acetate dropwise to it Methyl acetate (600 mg, 5.17 mmol, 555.56 µL, 1 eq). After stirring for 10 min, n-butyl lithium (2.5 M, 2.27 mL, 1.1 eq) was added dropwise, and the reaction was continued to stir for 20 min at 0°C. The reaction system was cooled to -78°C with a dry ice acetone bath, and a solution of compound 2-2 (1.08 g, 5.68 mmol, 1.1 eq) in tetrahydrofuran (6 mL) was added dropwise thereto. The reaction was stirred for 30 minutes, then slowly raised to room temperature and stirred for 30 minutes. The reaction was quenched by adding water (30 mL), and the aqueous phase was extracted with ethyl acetate (50 mL×2). The combined organic phase was dried with sodium sulfate, filtered to remove the desiccant, and the filtrate was decompressed to remove the solvent to obtain a crude product. The crude product was purified by column (ethyl acetate/petroleum ether=0-20%) to obtain compound 2-3. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.07 (d, J=7.6 Hz, 1H), 7.81 (d, J=8.0 Hz, 2H), 7.63-7.49 (m, 2H), 7.35 (t, J =8.0 Hz, 1H), 6.92 (br d, J=9.6 Hz, 1H), 3.75 (s, 3H), 3.55 (s, 2H), 3.37 (dd, J=1.6, 18.1 Hz, 1H), 3.24 ( d, J=1.2 Hz, 1H), 2.86-2.77 (m, 1H).
步驟3:化合物2-4的合成 將化合物2-3 (520 mg, 1.70 mmol, 1 eq)溶解於二氯甲烷 (5 mL) 中,然後加入N,N-二甲基甲醯胺縮二甲醇 (202.01 mg, 1.70 mmol, 225.20 µL, 1 eq)。所得反應液25℃攪拌反應1hr,然後加入三氟化硼乙醚錯合物 (240.60 mg, 1.70 mmol, 209.22 µL, 1 eq),反應液25℃攪拌反應18hr。反應液真空濃縮,殘留物中用2M鹽酸調節pH~3-4, 然後用乙酸乙酯(30 mL×3)萃取。合併後的有機相真空下濃縮得到粗品。粗品經過柱純化(乙酸乙酯/石油醚=0~35%)得到化合物2-4。1 H NMR (400 MHz, CDCl3 ) δ = 8.56 (d, J=0.8 Hz, 1H), 7.91 (t, J=8.0 Hz, 2H), 7.85 (dd, J=1.2, 8.4 Hz, 1H), 7.65 (dd, J=1.6, 7.6 Hz, 1H), 7.59 (t, J=8.0 Hz, 1H), 7.44 - 7.35 (m, 2H), 3.87 (s, 3H), 3.27 - 3.17 (m, 1H), 2.92-2.82 (m, 1H). LCMSm/z = 317.0 [M+H]+ 。Step 3: Synthesis of compound 2-4 Dissolve compound 2-3 (520 mg, 1.70 mmol, 1 eq) in dichloromethane (5 mL), and then add N,N-dimethylformamide dimethylacetal (202.01 mg, 1.70 mmol, 225.20 µL, 1 eq). The resulting reaction solution was stirred and reacted at 25°C for 1 hr, and then boron trifluoride ether complex (240.60 mg, 1.70 mmol, 209.22 µL, 1 eq) was added, and the reaction solution was stirred at 25°C for 18 hr. The reaction solution was concentrated in vacuo, and the residue was adjusted to pH~3-4 with 2M hydrochloric acid, and then extracted with ethyl acetate (30 mL×3). The combined organic phase was concentrated under vacuum to obtain a crude product. The crude product is purified by column (ethyl acetate/petroleum ether=0~35%) to obtain compound 2-4. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.56 (d, J=0.8 Hz, 1H), 7.91 (t, J=8.0 Hz, 2H), 7.85 (dd, J=1.2, 8.4 Hz, 1H), 7.65 (dd, J=1.6, 7.6 Hz, 1H), 7.59 (t, J=8.0 Hz, 1H), 7.44-7.35 (m, 2H), 3.87 (s, 3H), 3.27-3.17 (m, 1H) , 2.92-2.82 (m, 1H). LCMS m/z = 317.0 [M+H] + .
步驟4:化合物2-5的合成 將化合物2-4 (780 mg, 2.46 mmol, 1 eq) 溶解於四氫呋喃 (3 mL),在氮氣保護下冷卻到-78℃。然後向其中滴加三二級丁基硼氫化鋰 (1 M, 2.46 mL, 1 eq),-78℃攪拌反應1hr。用飽和氯化銨(5 mL)淬滅反應,然後用乙酸乙酯(50 mL×3)萃取。合併有機相,真空濃縮得到粗品。粗品過柱純化(乙酸乙酯/石油醚=0~15%)得到化合物2-5。1 H NMR (400 MHz, CDCl3 ) δ =11.81 (s, 1H), 7.99 (d, J=7.2 Hz, 1H), 7.85-7.80 (m, 2H), 7.63 - 7.53 (m, 2H), 7.36 (t, J=7.6 Hz, 1H), 6.30 (dd, J=2.8, 10.4 Hz, 1H), 4.68 - 4.62 (m, 1H), 4.56 - 4.47 (m, 1H), 3.82 (s, 3H), 3.07 - 2.98 (m, 1H), 2.57 - 2.46 (m, 1H)。Step 4: Synthesis of compound 2-5 Compound 2-4 (780 mg, 2.46 mmol, 1 eq) was dissolved in tetrahydrofuran (3 mL), and cooled to -78°C under nitrogen protection. Then, lithium tertiary butyl borohydride (1 M, 2.46 mL, 1 eq) was added dropwise, and the reaction was stirred at -78° C. for 1 hr. The reaction was quenched with saturated ammonium chloride (5 mL), and then extracted with ethyl acetate (50 mL×3). The organic phases were combined and concentrated in vacuo to obtain a crude product. The crude product was purified by column (ethyl acetate/petroleum ether=0~15%) to obtain compound 2-5. 1 H NMR (400 MHz, CDCl 3 ) δ = 11.81 (s, 1H), 7.99 (d, J=7.2 Hz, 1H), 7.85-7.80 (m, 2H), 7.63-7.53 (m, 2H), 7.36 (t, J=7.6 Hz, 1H), 6.30 (dd, J=2.8, 10.4 Hz, 1H), 4.68-4.62 (m, 1H), 4.56-4.47 (m, 1H), 3.82 (s, 3H), 3.07-2.98 (m, 1H), 2.57-2.46 (m, 1H).
步驟5:化合物2-6的合成 將化合物2-5 (497 mg, 1.56 mmol, 1 eq)溶解於甲醇(2 mL)中,然後加入2-甲基硫脲硫酸鹽 (528.27 mg, 2.81 mmol, 1.8 eq)和甲醇鈉 (421.14 mg, 7.80 mmol, 5 eq),所得反應液氮氣保護下在25℃攪拌反應18hr。減壓除去甲醇,殘留物中加入水(1 mL),然後用2M鹽酸調節pH~5-6,有大量白色固體析出來,過濾收集固體,真空乾燥得到化合物2-6。粗品直接用於下一步反應。LCMSm/z = 359.1 [M+H]+ 。Step 5: Synthesis of compound 2-6. Compound 2-5 (497 mg, 1.56 mmol, 1 eq) was dissolved in methanol (2 mL), and then 2-methylthiourea sulfate (528.27 mg, 2.81 mmol, 1.8 eq) and sodium methoxide (421.14 mg, 7.80 mmol, 5 eq), and the resulting reaction solution was stirred at 25° C. for 18 hr under the protection of nitrogen. The methanol was removed under reduced pressure, water (1 mL) was added to the residue, and the pH was adjusted to 5-6 with 2M hydrochloric acid. A large amount of white solid precipitated out. The solid was collected by filtration and dried in vacuo to obtain compound 2-6. The crude product was directly used in the next reaction. LCMS m/z = 359.1 [M+H] + .
步驟6:化合物2-7的合成 將化合物2-6 (440.00 mg, 1.23 mmol, 1 eq) 和N.N-二異丙基乙胺 (316.95 mg, 2.45 mmol, 427.15 µL, 2 eq) 加入到無水二氯甲烷(5 mL) 中,並冷卻到0℃,然後向其中加入三氟甲磺酸酐(449.74 mg, 1.59 mmol, 263.00 µL, 1.3 eq)。加完後,0℃攪拌反應60min。反應液真空下濃縮得到粗品,粗品過柱純化(乙酸乙酯/石油醚=0~6%)得到化合物2-7。1 H NMR (400 MHz, CDCl3 ) δ = 7.99 (d, J=7.2 Hz, 1H), 7.90-7.82 (m, 2H), 7.66 - 7.54 (m, 2H), 7.44 - 7.33 (m, 1H), 6.46 (dd, J=2.4, 10.4 Hz, 1H), 5.12 - 5.04 (m, 1H), 4.97 - 4.89 (m, 1H), 3.63 (dd, J=2.0, 18.0 Hz, 1H), 3.05-2.90 (m, 1H), 2.57 (s, 3H). LCMSm/z = 491.0 [M+H]+ 。Step 6: Synthesis of compound 2-7. Compound 2-6 (440.00 mg, 1.23 mmol, 1 eq) and NN-diisopropylethylamine (316.95 mg, 2.45 mmol, 427.15 µL, 2 eq) were added to the anhydrous two In methyl chloride (5 mL) and cooled to 0°C, add trifluoromethanesulfonic anhydride (449.74 mg, 1.59 mmol, 263.00 µL, 1.3 eq) to it. After the addition, the reaction was stirred at 0°C for 60 min. The reaction solution was concentrated under vacuum to obtain the crude product, and the crude product was purified by column (ethyl acetate/petroleum ether=0~6%) to obtain compound 2-7. 1 H NMR (400 MHz, CDCl 3 ) δ = 7.99 (d, J=7.2 Hz, 1H), 7.90-7.82 (m, 2H), 7.66-7.54 (m, 2H), 7.44-7.33 (m, 1H) , 6.46 (dd, J=2.4, 10.4 Hz, 1H), 5.12-5.04 (m, 1H), 4.97-4.89 (m, 1H), 3.63 (dd, J=2.0, 18.0 Hz, 1H), 3.05-2.90 (m, 1H), 2.57 (s, 3H). LCMS m/z = 491.0 [M+H] + .
步驟7:化合物2-8的合成 將化合物2-7 (121 mg, 246.48 µmol, 1 eq)和N,N-二異丙基乙胺 (95.57 mg, 739.45 µmol, 128.80 µL, 3 eq)加入到N,N-二甲基甲醯胺(1.5 mL)中,然後加入化合物1-10A鹽酸鹽(70.31 mg, 237.71 µmol, 1.1 eq),所得反應液氮氣置換後置於100℃油浴中攪拌反應1hr。反應液真空下濃縮得到粗品,粗品過柱純化(乙酸乙酯/石油醚=0~30%)得到化合物2-8。LCMSm/z = 600.2 [M+H]+ 。Step 7: Synthesis of compound 2-8. Compound 2-7 (121 mg, 246.48 µmol, 1 eq) and N,N-diisopropylethylamine (95.57 mg, 739.45 µmol, 128.80 µL, 3 eq) were added to N,N-Dimethylformamide (1.5 mL), then compound 1-10A hydrochloride (70.31 mg, 237.71 µmol, 1.1 eq) was added, the resulting reaction solution was replaced with nitrogen and placed in an oil bath at 100°C and stirred Reaction for 1hr. The reaction solution was concentrated under vacuum to obtain the crude product, and the crude product was purified by column (ethyl acetate/petroleum ether=0~30%) to obtain compound 2-8. LCMS m/z = 600.2 [M+H] + .
步驟8:化合物2-9的合成 將化合物2-8 (125 mg, 208.29 µmol, 1 eq) 溶解於二氯甲烷(1 mL) 中,然後加入間氯過氧苯甲酸(84.57 mg, 416.58 µmol, 85%純度, 2 eq),所得反應液20℃攪拌反應8hr。將反應液過濾除去不溶物,濾液真空濃縮得到粗品,粗品過柱純化(乙酸乙酯/石油醚=0~60%)得到化合物2-9。LCMSm/z = 632.3 [M+H]+ 。Step 8: Synthesis of compound 2-9. Compound 2-8 (125 mg, 208.29 µmol, 1 eq) was dissolved in dichloromethane (1 mL), and then m-chloroperoxybenzoic acid (84.57 mg, 416.58 µmol, 85% purity, 2 eq), and the resulting reaction solution was stirred and reacted at 20°C for 8 hours. The reaction solution was filtered to remove insoluble materials, and the filtrate was concentrated in vacuo to obtain a crude product. The crude product was purified by column (ethyl acetate/petroleum ether=0~60%) to obtain compound 2-9. LCMS m/z = 632.3 [M+H] + .
步驟9:化合物2-10的合成 將化合物2-9 (101 mg, 159.78 µmol, 1 eq) 和 1-11A (55.21 mg, 479.34 µmol, 56.91 µL, 3 eq) 溶解於甲苯 (0.8 mL) 中。所得溶液冷卻到-5℃,然後加入t-BuONa (30.71 mg, 319.56 µmol, 2 eq),所得反應液在-5~0℃攪拌反應1hr。將反應液用3 mL乙酸乙酯稀釋,然後用水(1 mL)和飽和食鹽水(1 mL)洗滌。有機相真空下濃縮得到粗品,粗品過柱純化(甲醇/二氯甲烷=0~8%)得到化合物2-10。LCMSm/z = 667.3 [M+H]+ 。Step 9: Synthesis of compound 2-10. Compound 2-9 (101 mg, 159.78 µmol, 1 eq) and 1-11A (55.21 mg, 479.34 µmol, 56.91 µL, 3 eq) were dissolved in toluene (0.8 mL). The resulting solution was cooled to -5°C, then t-BuONa (30.71 mg, 319.56 µmol, 2 eq) was added, and the resulting reaction solution was stirred and reacted at -5~0°C for 1 hr. The reaction solution was diluted with 3 mL of ethyl acetate, and then washed with water (1 mL) and saturated brine (1 mL). The organic phase was concentrated under vacuum to obtain the crude product, and the crude product was purified by column (methanol/dichloromethane=0~8%) to obtain compound 2-10. LCMS m/z = 667.3 [M+H] + .
步驟10:化合物2-11和3-1混合物的合成 將化合物2-10 (101 mg, 151.38 µmol, 1 eq) 溶解於二氯甲烷 (1 mL) 中,然後加入醋酸鈀 (6.80 mg, 30.28 µmol, 0.2 eq) 和三乙基矽烷 (88.01 mg, 756.90 µmol, 120.90 µL, 5 eq) ,所得反應液室溫下攪拌反應1hr。將反應液真空濃縮得到化合物2-11和3-1的混合物,該混合物不經純化直接用於下一步反應。化合物2-11:LCMSm/z = 555.3 [M+Na]+ ;化合物3-1:LCMSm/z = 521.3 [M+Na]+ 。Step 10: Synthesis of the mixture of compound 2-11 and 3-1. Dissolve compound 2-10 (101 mg, 151.38 µmol, 1 eq) in dichloromethane (1 mL), and then add palladium acetate (6.80 mg, 30.28 µmol) , 0.2 eq) and triethylsilane (88.01 mg, 756.90 µmol, 120.90 µL, 5 eq), and the resulting reaction solution was stirred for 1 hour at room temperature. The reaction solution was concentrated in vacuo to obtain a mixture of compounds 2-11 and 3-1, and the mixture was directly used in the next reaction without purification. Compound 2-11: LCMS m/z = 555.3 [M+Na] + ; Compound 3-1: LCMS m/z = 521.3 [M+Na] + .
步驟11:化合物2和3的合成 將化合物2-11和3-1的混合物溶於二氯甲烷 (1 mL)中,然後加入三乙胺 (45.95 mg, 454.14 µmol, 63.21 µL, 3 eq)。所得反應液冷卻至0℃,然後加入烯丙基醯氯 (20.55 mg, 227.07 µmol, 18.52 µL, 1.5 eq),攪拌反應30min。將反應液真空濃縮得到粗品,粗品用高效液相層析製備分離(分離條件:層析管柱: Welch Xtimate C18 150*30mm*5µm;流動相: [水(0.225%甲酸)-乙腈]; 乙腈%: 15%-55%,8min)得到化合物2和3。化合物2和3分別為一對非對映異構體。化合物2:LCMSm/z = 587.3 [M+H]+ ;化合物3:LCMSm/z = 553.3 [M+H]+ 。Step 11: Synthesis of compounds 2 and 3 The mixture of compounds 2-11 and 3-1 was dissolved in dichloromethane (1 mL), and then triethylamine (45.95 mg, 454.14 µmol, 63.21 µL, 3 eq) was added. The resulting reaction solution was cooled to 0°C, and then allyl chloride (20.55 mg, 227.07 µmol, 18.52 µL, 1.5 eq) was added, and the reaction was stirred for 30 min. The reaction solution was concentrated in vacuo to obtain the crude product. The crude product was prepared and separated by high performance liquid chromatography (Separation conditions: chromatographic column: Welch Xtimate C18 150*30mm*5µm; mobile phase: [water (0.225% formic acid)-acetonitrile]; acetonitrile %: 15%-55%, 8min) to obtain compounds 2 and 3. Compounds 2 and 3 are a pair of diastereomers, respectively. Compound 2: LCMS m/z = 587.3 [M+H] + ; Compound 3: LCMS m/z = 553.3 [M+H] + .
實施例 4



步驟2:化合物4-22的合成 將化合物4-21 (0.2 g, 887.76 µmol, 1 eq) 溶於四氫呋喃 (5 mL) 中,加入三乙胺 (269.50 mg, 2.66 mmol, 370.70 µL, 3 eq) ,氮氣保護,降溫至0℃,加入三氟乙酸酐(205.10 mg, 976.53 µmol, 135.83 µL, 1.1 eq),在0℃反應0.5 hr。倒入飽和氯化銨水溶液(10 mL),加入乙酸乙酯(5 mL * 2),飽和食鹽水(5 mL )洗滌,管柱層析純化,(石油醚/乙酸乙酯=10/1~1/1,TLC:石油醚/乙酸乙酯=3/1)得到化合物4-22。1 H NMR (400 MHz, CDCl3 ) δ = 4.86 (s, 1H), 4.51 - 4.06 (m, 2H), 3.88 (d, J = 14.0 Hz, 1H), 3.52 - 3.33 (m, 1H), 3.24 (dd, J = 4.0, 14.2 Hz, 1H), 3.12 - 2.92 (m, 1H), 2.91 - 2.73 (m, 1H), 2.67 (s, 1H), 1.50 (s, 9H);LCMS: MS m/z = 222.0 [M-100+H]+ 。Step 2: Synthesis of compound 4-22. Compound 4-21 (0.2 g, 887.76 µmol, 1 eq) was dissolved in tetrahydrofuran (5 mL), and triethylamine (269.50 mg, 2.66 mmol, 370.70 µL, 3 eq) was added , Nitrogen protection, cooling to 0°C, adding trifluoroacetic anhydride (205.10 mg, 976.53 µmol, 135.83 µL, 1.1 eq), and reacting at 0°C for 0.5 hr. Pour into saturated aqueous ammonium chloride solution (10 mL), add ethyl acetate (5 mL * 2), wash with saturated brine (5 mL), and purify by column chromatography (petroleum ether/ethyl acetate=10/1~ 1/1, TLC: petroleum ether/ethyl acetate=3/1) to obtain compound 4-22. 1 H NMR (400 MHz, CDCl 3 ) δ = 4.86 (s, 1H), 4.51-4.06 (m, 2H), 3.88 (d, J = 14.0 Hz, 1H), 3.52-3.33 (m, 1H), 3.24 (dd, J = 4.0, 14.2 Hz, 1H), 3.12-2.92 (m, 1H), 2.91-2.73 (m, 1H), 2.67 (s, 1H), 1.50 (s, 9H); LCMS: MS m/ z = 222.0 [M-100+H] + .
步驟3:化合物4-14A的合成 將化合物4-22(150 mg, 466.86 µmol, 1 eq)溶於鹽酸二氧六環(5 M, 8 mL, 85.68 eq) ,氮氣保護,18℃反應1 hr。直接旋乾,得到化合物4-14A的鹽酸鹽。LCMS: MS m/z = 222.0 [M+H]+ 。Step 3: Synthesis of compound 4-14A Compound 4-22 (150 mg, 466.86 µmol, 1 eq) was dissolved in dioxane hydrochloride (5 M, 8 mL, 85.68 eq), protected with nitrogen, and reacted at 18°C for 1 hr . Rotate directly to dryness to obtain the hydrochloride salt of compound 4-14A. LCMS: MS m/z = 222.0 [M+H] + .
實施例4的合成 步驟1:化合物4-2的合成 將水(210 mL),鹽酸(210 mL, 36~38% 質量含量)混合後,將化合物4-1(36.00 g, 176.44 mmol, 1 eq)加入其中,將溫度升至65℃,反應1 hr。然後將溫度降至0~5℃後,然後將亞硝酸鈉(14.61 g, 211.72 mmol, 1.2 eq)溶於水(70 mL)中後滴加到其中,攪拌15 min。將氯化亞銅(26.20 g, 264.65 mmol, 6.33 mL, 1.5 eq)溶於鹽酸(350 mL, 36~38% 質量含量)後,降溫至0~5℃,將上述溶液滴加到其中後,繼續反應6 hr。向反應體系中加入750 mL 二氯甲烷後攪拌20 min後,分液,有機相加入350 mL飽和食鹽水洗滌一次,加入30.00 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸,得到化合物4-2。1 H NMR (400 MHz, CDCl3 ) δ = 7.24 - 7.21 (m, 1H), 6.94 (dd, J = 2.8, 8.8 Hz, 1H), 2.43 (s, 3H)。Synthesis step 1: Synthesis of compound 4-2. After mixing water (210 mL) and hydrochloric acid (210 mL, 36~38% mass content), compound 4-1 (36.00 g, 176.44 mmol, 1 eq) ) Add to it, raise the temperature to 65°C, and react for 1 hr. Then lower the temperature to 0~5°C, then dissolve sodium nitrite (14.61 g, 211.72 mmol, 1.2 eq) in water (70 mL) and add dropwise to it, and stir for 15 min. After dissolving cuprous chloride (26.20 g, 264.65 mmol, 6.33 mL, 1.5 eq) in hydrochloric acid (350 mL, 36~38% mass content), the temperature is reduced to 0~5°C, and the above solution is added dropwise to it, Continue to react for 6 hr. After adding 750 mL of dichloromethane to the reaction system and stirring for 20 minutes, the liquid was separated, the organic phase was washed once with 350 mL of saturated brine, and 30.00 g of anhydrous sodium sulfate was added to dry it, and then filtered. The filtrate was subjected to vacuum rotation at 45°C. Steam to obtain compound 4-2. 1 H NMR (400 MHz, CDCl 3 ) δ = 7.24-7.21 (m, 1H), 6.94 (dd, J = 2.8, 8.8 Hz, 1H), 2.43 (s, 3H).
步驟2:化合物4-3的合成 將四氫呋喃(395 mL),化合物4-2(39.50 g, 176.76 mmol, 1 eq)加入預先準備好的乾淨反應瓶中,開始攪拌,將溫度降至-70~-65℃後,將二異丙基胺基鋰(2 M, 106.05 mL, 1.2 eq)滴加到其中,反應1hr。然後將N,N-二甲基甲醯胺(18.76 g, 256.70 mmol, 19.75 mL, 1.45 eq)加入其中,繼續反應1 hr。向反應體系中加入500 mL飽和氯化銨溶液後,分液,有機相用300 mL飽和食鹽水洗滌一次後加入20 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。得到化合物4-3。1 H NMR (400 MHz, CDCl3 ) δ = 10.28 (s, 1H), 7.08 (d, J = 10.8 Hz, 1H), 2.51 (s, 3H);LCMSm/z = 245.0[M+H]+ , 247.0[M+3H]+ 。Step 2: Synthesis of compound 4-3. Add tetrahydrofuran (395 mL) and compound 4-2 (39.50 g, 176.76 mmol, 1 eq) into a clean reaction flask prepared in advance, start stirring, and reduce the temperature to -70~ After -65°C, add lithium diisopropylamide (2 M, 106.05 mL, 1.2 eq) dropwise to it, and react for 1 hr. Then N,N-dimethylformamide (18.76 g, 256.70 mmol, 19.75 mL, 1.45 eq) was added to it, and the reaction was continued for 1 hr. After adding 500 mL of saturated ammonium chloride solution to the reaction system, the liquid was separated, the organic phase was washed once with 300 mL of saturated brine, and then dried by adding 20 g of anhydrous sodium sulfate, filtered, and the filtrate was subjected to reduced pressure rotary evaporation at 45°C. Compound 4-3 is obtained. 1 H NMR (400 MHz, CDCl 3 ) δ = 10.28 (s, 1H), 7.08 (d, J = 10.8 Hz, 1H), 2.51 (s, 3H); LCMS m/z = 245.0[M+H] + , 247.0[M+3H] + .
步驟3:化合物4-4的合成 將二甲亞碸(300 mL),化合物4-3(20.00 g, 79.53 mmol, 1 eq)加入預先準備好的乾淨反應瓶中,開始攪拌,然後將水合肼(48.75 g, 954.35 mmol, 47.33 mL, 98% 質量含量, 12 eq)加入其中,將溫度升至130℃,反應3 hr。與小試反應液合併後,將反應液倒入700 mL水中後,過濾,濾餅用水(100 mLx 3次)洗滌;得到的濾餅用300 mL乙酸乙酯溶解後分液,有機相加入50.00 g 無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸,得到化合物4-4。1 H NMR (400 MHz,CDCl3 ) δ = 10.38 (brs, 1H), 8.03 (s, 1H), 7.33 (s, 1H), 2.57 (s, 3H);LCMSm/z = 245.1[M+H]+ , 247.1[M+3H]+ 。Step 3: Synthesis of compound 4-4. Add dimethyl sulfoxide (300 mL) and compound 4-3 (20.00 g, 79.53 mmol, 1 eq) into a clean reaction flask prepared in advance, start stirring, and then add hydrazine hydrate (48.75 g, 954.35 mmol, 47.33 mL, 98% mass content, 12 eq) was added to it, the temperature was raised to 130°C, and the reaction was carried out for 3 hr. After combining with the small test reaction solution, the reaction solution was poured into 700 mL of water, filtered, and the filter cake was washed with water (100 mL x 3 times); the obtained filter cake was dissolved in 300 mL of ethyl acetate and then separated, and the organic phase was added 50.00 g After drying with anhydrous sodium sulfate and filtering, the filtrate was rotary evaporated under reduced pressure at 45° C. to obtain compound 4-4. 1 H NMR (400 MHz,CDCl 3 ) δ = 10.38 (brs, 1H), 8.03 (s, 1H), 7.33 (s, 1H), 2.57 (s, 3H); LCMS m/z = 245.1[M+H ] + , 247.1[M+3H] + .
步驟4:化合物4-5的合成
將二氯甲烷(200 mL),化合物4-4(20.00 g, 81.47 mmol, 1 eq)加入預先準備好的乾淨反應瓶中,開始攪拌;然後依次將對甲苯磺酸吡啶鹽(2.05 g, 8.15 mmol, 0.1 eq),2-甲羥基-3,4-二氫吡喃(20.56 g, 244.40 mmol, 22.35 mL, 3 eq)加入其中,室溫20℃,反應12 hr。向反應體系中加入200 mL水後,反應液直接分液,有機相加入20.00 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸,得到粗品化合物。粗品經管柱層析純化(石油醚/乙酸乙酯= 100/0~70/30,TLC : 石油醚/乙酸乙酯= 5/1),得到化合物4-5。1
H NMR (400 MHz, CDCl3
) δ = 7.95 (s, 1H), 7.44 (s, 1H), 5.67 (dd,J
= 2.8, 8.8 Hz, 1H), 4.02 - 3.98 (m, 1H), 3.79 - 3.71 (m, 1H), 2.57 (s, 3H), 2.54 - 2.46 (m, 1H), 2.18 - 2.05 (m, 2H), 1.80 - 1.66 (m, 3H);LCMSm/z
= 329.0[M+H]+
, 331.0[M+3H]+
。Step 4: Synthesis of compound 4-5. Add dichloromethane (200 mL) and compound 4-4 (20.00 g, 81.47 mmol, 1 eq) into a clean reaction flask prepared in advance and start stirring; then add p-toluene in sequence Sulfonic acid pyridine salt (2.05 g, 8.15 mmol, 0.1 eq), 2-methylhydroxy-3,4-dihydropyran (20.56 g, 244.40 mmol, 22.35 mL, 3 eq) were added to it, and the reaction was carried out at
步驟5:化合物4-6的合成 將四氫呋喃(160 mL),化合物4-5(16 g, 48.54 mmol, 1 eq)加入預先準備好的乾淨反應瓶中,開始攪拌。將溫度降至-70~-65℃後,將正丁基鋰(2.5 M, 21.36 mL, 1.1 eq)緩慢滴加到其中,反應1hr;然後將N,N-二甲基甲醯胺(35.48 g, 485.41 mmol, 37.35 mL, 10 eq)加入其中,繼續反應0.5hr。向反應體系中加入250 mL飽和氯化銨溶液後,分液,有機相用150 mL飽和食鹽水洗滌一次後,加入無水硫酸鈉乾燥,過濾,濾液在45℃下進行減壓旋蒸的有狀物。油狀物與7 mL乙酸乙酯混合打漿20 min後,過濾,濾餅在45℃下進行減壓旋蒸。得到化合物4-6。1 H NMR (400 MHz, CDCl3 ) δ = 10.72 (s, 1H), 8.63 (s, 1H), 7.74 (s, 1H), 5.70 (dd,J = 2.8, 8.8 Hz, 1H), 3.98 - 3.94 (m, 1H), 3.75 - 3.68 (m, 1H), 2.55 (s, 3H), 2.53 - 2.45 (m, 1H), 2.16 - 2.05 (m, 2H), 1.83 - 1.61 (m, 3H); LCMSm/z = 279.1[M+H]+ 。Step 5: Synthesis of compound 4-6 Add tetrahydrofuran (160 mL) and compound 4-5 (16 g, 48.54 mmol, 1 eq) into a clean reaction flask prepared in advance, and start stirring. After the temperature was lowered to -70~-65℃, n-butyllithium (2.5 M, 21.36 mL, 1.1 eq) was slowly added dropwise to it, and reacted for 1 hr; then N,N-dimethylformamide (35.48 g, 485.41 mmol, 37.35 mL, 10 eq) was added to it, and the reaction was continued for 0.5 hr. After adding 250 mL of saturated ammonium chloride solution to the reaction system, separate the liquids, wash the organic phase with 150 mL of saturated brine once, add anhydrous sodium sulfate to dry, filter, and perform vacuum rotary evaporation of the filtrate at 45°C. Things. After mixing the oily substance with 7 mL of ethyl acetate and beating for 20 minutes, it was filtered, and the filter cake was subjected to vacuum rotary evaporation at 45°C. Compound 4-6 is obtained. 1 H NMR (400 MHz, CDCl 3 ) δ = 10.72 (s, 1H), 8.63 (s, 1H), 7.74 (s, 1H), 5.70 (dd, J = 2.8, 8.8 Hz, 1H), 3.98-3.94 (m, 1H), 3.75-3.68 (m, 1H), 2.55 (s, 3H), 2.53-2.45 (m, 1H), 2.16-2.05 (m, 2H), 1.83-1.61 (m, 3H); LCMS m/z = 279.1[M+H] + .
步驟6:化合物7的合成
將四氫呋喃(54 mL),化合物4-6(5.4 g, 19.37 mmol, 1 eq)加入預先準備好的乾淨反應瓶中,開始攪拌;然後依次將三級丁基亞磺醯胺(2.58 g, 21.31 mmol, 232.15 µL, 1.1 eq),鈦酸四異丙酯(8.84 g, 38.75 mmol, 8.04 mL, 2 eq)加入其中,20℃反應12 hr。向反應體系中加入50 mL飽和氯化銨溶液後,分液,有機相加入3.00 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(石油醚/乙酸乙酯= 100/0~50/50,TLC : 石油醚/乙酸乙酯= 10/1),得到化合物4-7。LCMSm/z
= 382.2[M+H]+
。Step 6: Synthesis of
步驟7:化合物4-8的合成
將四氫呋喃(35 mL),鈉氫(829.50 mg, 20.74 mmol, 60%質量含量, 1.2eq
) 加入預先準備好的乾淨反應瓶中,開始攪拌,然後將溫度降至0~5℃後,將乙醯乙酸甲酯(2.41 g, 20.74 mmol, 2.23 mL, 1.2eq
)滴加到其中,反應20 min。然後將正丁基鋰(2.5 M, 7.60 mL, 1.1eq
)滴加到其中,繼續反應20 min,然後將溫度降至-70~-65℃後,將化合物4-7(6.60 g, 17.28 mmol, 1eq
)溶於四氫呋喃(35 mL)後,滴加到其中,繼續反應20 min,將溫度緩慢升至室溫20℃,繼續反應0.5 hr。將反應液倒入100 mL飽和氯化銨溶液後,與1g批次合併後,分液,有機相加入3.00 g 無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(石油醚/乙酸乙酯= 100/0~20/80,TLC:PE/EtOAc=0:1),得到化合物4-8。1
H NMR (400 MHz, CDCl3
) δ = 8.20 (s, 1H), 7.44 (d,J
= 5.6 Hz, 1H), 5.72 - 5.64 (m, 2H), 4.04 - 3.99 (m, 1H), 3.77 - 3.69 (m, 4H), 3.57 - 3.46 (m, 2H), 3.15 - 3.08 (m, 1H), 2.59 - 2.52 (m, 4H), 2.16 - 2.05 (m, 2H), 1.83 - 1.65 (m, 4H), 1.20 - 1.18 (m, 9H);LCMSm/z
= 498.2[M+H]+
。Step 7: Synthesis of compound 4-8 Add tetrahydrofuran (35 mL), sodium hydrogen (829.50 mg, 20.74 mmol, 60% mass content, 1.2 eq ) into a clean reaction flask prepared in advance, start stirring, and then lower the temperature After reaching 0~5°C, methyl acetylacetate (2.41 g, 20.74 mmol, 2.23 mL, 1.2 eq ) was added dropwise to it and reacted for 20 min. Then n-butyllithium (2.5 M, 7.60 mL, 1.1 eq ) was added dropwise to it, the reaction was continued for 20 min, and then the temperature was lowered to -70~-65℃, compound 4-7 (6.60 g, 17.28 mmol , 1 eq ) was dissolved in tetrahydrofuran (35 mL), added dropwise to it, and the reaction was continued for 20 min. The temperature was slowly raised to
步驟8:化合物4-9的合成 將甲苯(66 mL),化合物4-8(6.60 g, 13.25 mmol, 1eq )加入預先準備好的乾淨反應瓶中,開始攪拌;然後將N,N-二甲基甲醯胺二甲縮醛(4.74 g, 39.76 mmol, 5.28 mL, 3eq )加入其中,室溫20℃反應12hr。向反應體系中加入60 mL水,以及60 mL乙酸乙酯後,攪拌5 min;分液,有機相加入60 mL飽和食鹽水洗滌一次後,加入5.00 g無水硫酸鈉乾燥後,過濾,濾液在50℃下進行減壓旋蒸,得到化合物4-9,直接用於下一步。Step 8: Synthesis of compound 4-9. Add toluene (66 mL) and compound 4-8 (6.60 g, 13.25 mmol, 1 eq ) into a clean reaction flask prepared in advance, and start stirring; Methylformamide dimethyl acetal (4.74 g, 39.76 mmol, 5.28 mL, 3 eq ) was added to it, and reacted at room temperature at 20°C for 12 hours. Add 60 mL of water and 60 mL of ethyl acetate to the reaction system, stir for 5 min; separate the organic phase, wash the organic phase with 60 mL of saturated brine once, add 5.00 g of anhydrous sodium sulfate to dry, filter, and filter the filtrate at 50 Rotary evaporation under reduced pressure was carried out at °C to obtain compound 4-9, which was directly used in the next step.
步驟9:化合物4-10的合成 將化合物 4-9 (50 mg, 90.40 µmol, 1 eq) 溶於鹽酸乙酸乙酯(3 mL)中,加入到反應中,18℃攪拌20 min。直接濃縮得到粗品。得到化合物 4-10的鹽酸鹽。LCMSm/z = 320.0[M+H]+ 。Step 9: Synthesis of compound 4-10 Compound 4-9 (50 mg, 90.40 µmol, 1 eq) was dissolved in ethyl acetate hydrochloride (3 mL), added to the reaction, and stirred at 18°C for 20 min. Concentrate directly to obtain the crude product. The hydrochloride salt of compound 4-10 was obtained. LCMS m/z = 320.0 [M+H] + .
步驟10:化合物4-11的合成 將化合物4-10 (5.00 g, 14.04 mmol, 1 eq, HCl)溶於二氯甲烷 (50 mL) 中,三乙胺 (5.97 g, 58.96 mmol, 8.21 mL, 4.2 eq),二碳酸三級丁酯 (12.25 g, 56.15 mmol, 12.90 mL, 4 eq),4-二甲胺基吡啶(1.71 g, 14.04 mmol, 1 eq) 加入到反應中,18℃攪拌10 hr。與0.5g批次合併處理,飽和氯化銨水溶液淬滅(100 mL),二氯甲烷(30 mL*2次)萃取,合併有機相,無水硫酸鈉乾燥濃縮,得到粗品。粗品管柱層析(石油醚/乙酸乙酯=50/1~0/1,TLC: 石油醚/乙酸乙酯=1/1),得到化合物4-11。1 H NMR (400 MHz, CDCl3 ) δ = 9.02 (s, 1H), 8.12 (s, 1H), 7.89 (s, 1H), 6.16 (dd, J = 5.2, 8.8 Hz, 1H), 3.77 (s, 3H), 3.10 (dd, J = 8.4, 16.0 Hz, 1H), 2.82 (m, 1H), 2.48 (s, 3H), 1.63 (s, 9H), 1.18 (s, 9H)。LCMSm/z = 520.1[M+H]+ 。Step 10: Synthesis of compound 4-11. Compound 4-10 (5.00 g, 14.04 mmol, 1 eq, HCl) was dissolved in dichloromethane (50 mL), and triethylamine (5.97 g, 58.96 mmol, 8.21 mL, 4.2 eq), tertiary butyl dicarbonate (12.25 g, 56.15 mmol, 12.90 mL, 4 eq), 4-dimethylaminopyridine (1.71 g, 14.04 mmol, 1 eq) were added to the reaction and stirred at 18°C for 10 hr. Combine treatment with 0.5g batch, quench with saturated aqueous ammonium chloride solution (100 mL), extract with dichloromethane (30 mL*2 times), combine the organic phases, dry and concentrate with anhydrous sodium sulfate to obtain a crude product. Crude product column chromatography (petroleum ether/ethyl acetate=50/1-0/1, TLC: petroleum ether/ethyl acetate=1/1) to obtain compound 4-11. 1 H NMR (400 MHz, CDCl 3 ) δ = 9.02 (s, 1H), 8.12 (s, 1H), 7.89 (s, 1H), 6.16 (dd, J = 5.2, 8.8 Hz, 1H), 3.77 (s , 3H), 3.10 (dd, J = 8.4, 16.0 Hz, 1H), 2.82 (m, 1H), 2.48 (s, 3H), 1.63 (s, 9H), 1.18 (s, 9H). LCMS m/z = 520.1 [M+H] + .
步驟11:化合物4-12的合成 將化合物4-11 (3.00 g, 5.77 mmol, 1 eq) 溶於四氫呋喃 (30 mL) 中,降溫至-78℃,氮氣保護,將三二級丁基硼氫化鋰 (1 M, 5.77 mL, 1 eq) 滴加到反應液中,攪拌0.5 hr。飽和氯化銨水溶液淬滅(30mL),乙酸乙酯(20 mL x 2次)萃取,合併有機相,無水硫酸鈉乾燥濃縮,得到粗品得到化合物4-12。 LCMSm/z = 522.2[M+H]+ ,466.1[M-56+H]+ 。Step 11: Synthesis of compound 4-12. Dissolve compound 4-11 (3.00 g, 5.77 mmol, 1 eq) in tetrahydrofuran (30 mL), reduce the temperature to -78°C, protect with nitrogen, and hydrogenate the tertiary butyl borohydride Lithium (1 M, 5.77 mL, 1 eq) was added dropwise to the reaction solution and stirred for 0.5 hr. Quench with saturated aqueous ammonium chloride (30 mL), extract with ethyl acetate (20 mL x 2 times), combine the organic phases, dry and concentrate with anhydrous sodium sulfate to obtain crude product to obtain compound 4-12. LCMS m/z = 522.2 [M+H] + , 466.1 [M-56+H] + .
步驟12:化合物4-13的合成 將化合物4-12 (2.30 g, 4.41 mmol, 1 eq),2-甲基-2-硫代異尿素硫酸氫鹽 (1.66 g, 8.81 mmol, 2 eq, H2 SO4 ) 溶於甲醇 (430 mL)中,加入甲醇鈉 (476.05 mg, 8.81 mmol, 2 eq),18℃攪拌1.5 hr,再將甲醇鈉 (357.04 mg, 6.61 mmol, 1.5 eq)加入到反應液中,18℃在攪拌10 hr。旋乾,加入水(50 mL),1M 稀鹽酸調節pH=2~3,白色固體析出,過濾收集固體。粗品管柱層析純化(石油醚/乙酸乙酯=10/1~0/1,TLC:石油醚/乙酸乙酯=1/1)得到化合物4-13。 LCMSm/z = 562.1[M+H]+ 。Step 12: Synthesis of compound 4-13 Compound 4-12 (2.30 g, 4.41 mmol, 1 eq), 2-methyl-2-thioisourea hydrogen sulfate (1.66 g, 8.81 mmol, 2 eq, H 2 SO 4 ) was dissolved in methanol (430 mL), sodium methoxide (476.05 mg, 8.81 mmol, 2 eq) was added, stirred at 18°C for 1.5 hr, and then sodium methoxide (357.04 mg, 6.61 mmol, 1.5 eq) was added to the reaction In the solution, stir at 18°C for 10 hr. Rotate to dry, add water (50 mL), 1M dilute hydrochloric acid to adjust pH=2~3, a white solid will precipitate out, and the solid will be collected by filtration. The crude product was purified by column chromatography (petroleum ether/ethyl acetate=10/1-0/1, TLC: petroleum ether/ethyl acetate=1/1) to obtain compound 4-13. LCMS m/z = 562.1 [M+H] + .
步驟13:化合物4-14的合成 將化合物4-13 (0.328 g, 583.55 µmol, 1 eq) ,N,N-二異丙基乙胺 (377.09 mg, 2.92 mmol, 508.21 µL, 5 eq) 溶於二氯甲烷 (10 mL) 中,0℃加入三氟甲磺酸酐 (246.96 mg, 875.32 µmol, 144.42 µL, 1.5 eq) ,0℃攪拌1 hr。與0.56 g批次合併處理,合併倒入飽和氯化銨水溶液(50 mL)中,加入乙酸乙酯(20 mL x 3次)萃取,飽和食鹽水洗滌,無水硫酸鈉乾燥,過濾,濃縮得到粗品。粗品管柱層析(石油醚/乙酸乙酯 = 20/1~5/1)TLC(石油醚/乙酸乙酯 = 5/1),得到化合物4-14。1 H NMR (400 MHz, CDCl3) δ = 8.21 - 8.11 (m, 1H), 8.00 - 7.90 (m, 1H), 5.86 - 5.69 (m, 1H), 5.25 - 5.09 (m, 1H), 4.68 - 4.46 (m, 1H), 3.57 - 3.42 (m, 1H), 3.27 - 3.08 (m, 1H), 2.66 - 2.41 (m, 6H), 1.79 - 1.67 (m, 9H), 1.21 - 1.07 (m, 9H);LCMSm/z = 637.9[M-56+H]+ ,639.8[M-56+3H]+ 。Step 13: Synthesis of compound 4-14. Dissolve compound 4-13 (0.328 g, 583.55 µmol, 1 eq), N,N-diisopropylethylamine (377.09 mg, 2.92 mmol, 508.21 µL, 5 eq) In dichloromethane (10 mL), add trifluoromethanesulfonic anhydride (246.96 mg, 875.32 µmol, 144.42 µL, 1.5 eq) at 0°C, and stir at 0°C for 1 hr. Combine treatment with 0.56 g batches, combine and pour into saturated aqueous ammonium chloride solution (50 mL), add ethyl acetate (20 mL x 3 times) for extraction, wash with saturated brine, dry with anhydrous sodium sulfate, filter, and concentrate to obtain crude product . Crude product column chromatography (petroleum ether/ethyl acetate=20/1~5/1) TLC (petroleum ether/ethyl acetate=5/1) to obtain compound 4-14. 1 H NMR (400 MHz, CDCl3) δ = 8.21-8.11 (m, 1H), 8.00-7.90 (m, 1H), 5.86-5.69 (m, 1H), 5.25-5.09 (m, 1H), 4.68-4.46 (m, 1H), 3.57-3.42 (m, 1H), 3.27-3.08 (m, 1H), 2.66-2.41 (m, 6H), 1.79-1.67 (m, 9H), 1.21-1.07 (m, 9H) ; LCMS m/z = 637.9 [M-56+H] + , 639.8 [M-56+3H] + .
步驟14:化合物4-15的合成 將化合物4-14(630 mg, 907.60 µmol, 1 eq) ,化合物4-14A(420.90 mg, 1.63 mmol, 1.8 eq, HCl) 溶於N,N-二甲基甲醯胺 (15 mL) 中,加入N,N-二異丙基乙胺 (469.19mg, 3.63 mmol, 632.33 µL, 4 eq) ,20℃攪拌2 hr。倒入水中(30 mL),乙酸乙酯萃取(20 mL x 3),飽和食鹽水(10 mL)洗滌,無水硫酸鈉乾燥,過濾,濃縮得到粗品。管柱層析純化(石油醚/乙酸乙酯=50/1~1/1),TLC: 石油醚/乙酸乙酯=0/1得到化合物 4-15。1 H NMR (400 MHz, CDCl3) δ = 8.18 - 8.05 (m, 1H), 8.04 - 7.93 (m, 1H), 5.75 - 5.45 (m, 1H), 5.06 - 4.89 (m, 1H), 4.66 - 4.35 (m, 1H), 4.19 - 3.84 (m, 3H), 3.82 - 3.45 (m, 1H), 3.43 - 3.12 (m, 2H), 3.06 - 2.75 (m, 6H), 2.61 - 2.38 (m, 5H), 1.79 - 1.60 (m, 9H), 1.14 - 0.85 (s, 9H);LCMSm/z = 765.0[M+H]+ 。Step 14: Synthesis of compound 4-15. Compound 4-14 (630 mg, 907.60 µmol, 1 eq) and compound 4-14A (420.90 mg, 1.63 mmol, 1.8 eq, HCl) were dissolved in N,N-dimethyl To formamide (15 mL), add N,N-diisopropylethylamine (469.19 mg, 3.63 mmol, 632.33 µL, 4 eq) and stir at 20°C for 2 hr. Pour into water (30 mL), extract with ethyl acetate (20 mL x 3), wash with saturated brine (10 mL), dry with anhydrous sodium sulfate, filter, and concentrate to obtain a crude product. Purification by column chromatography (petroleum ether/ethyl acetate=50/1~1/1), TLC: petroleum ether/ethyl acetate=0/1 to obtain compound 4-15. 1 H NMR (400 MHz, CDCl3) δ = 8.18-8.05 (m, 1H), 8.04-7.93 (m, 1H), 5.75-5.45 (m, 1H), 5.06-4.89 (m, 1H), 4.66-4.35 (m, 1H), 4.19-3.84 (m, 3H), 3.82-3.45 (m, 1H), 3.43-3.12 (m, 2H), 3.06-2.75 (m, 6H), 2.61-2.38 (m, 5H) , 1.79-1.60 (m, 9H), 1.14-0.85 (s, 9H); LCMS m/z = 765.0[M+H] + .
步驟15:化合物4-16的合成 將化合物4-15(400.00 mg, 522.71 µmol,1 eq) ,溶於二氯甲烷 (8 mL) 中,加入間氯過氧苯甲酸 (200.00 mg, 985.11 µmol, 85% 質量含量, 1.88 eq) ,20℃攪拌2 hr。與200mg批次合併處理,反應液用亞硫酸鈉水溶液(20mL,10%)洗滌,無水硫酸鈉乾燥,過濾,濃縮,得到粗品。粗品管柱層析(SiO2 100目,石油醚/乙酸乙酯=50/1~1/1,TLC: 石油醚/乙酸乙酯=2/1)得到化合物4-16。LCMSm/z = 697.1[M-100+H]+ 。Step 15: Synthesis of compound 4-16 Compound 4-15 (400.00 mg, 522.71 µmol, 1 eq) was dissolved in dichloromethane (8 mL), and m-chloroperoxybenzoic acid (200.00 mg, 985.11 µmol, 85% mass content, 1.88 eq), stirring at 20°C for 2 hr. Combined with 200mg batches, the reaction solution was washed with aqueous sodium sulfite solution (20 mL, 10%), dried over anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. Crude product column chromatography (SiO 2 100 mesh, petroleum ether/ethyl acetate=50/1~1/1, TLC: petroleum ether/ethyl acetate=2/1) to obtain compound 4-16. LCMS m/z = 697.1 [M-100+H] + .
步驟16:化合物4-17的合成 將化合物1-11A (57.79 mg, 501.73 µmol, 59.57 µL, 4 eq) 溶於甲苯(1 mL)中,0℃加入三級丁醇鈉 (42.19 mg, 439.01 µmol, 3.5 eq) ,攪拌 15min,再將化合物4-16 (100.00 mg, 125.43 µmol, 1 eq) 溶於0.1mL甲苯中,緩慢加入到反應液中,0℃反應30min。水(5mL)淬滅,乙酸乙酯(5 mL x 2)萃取,合併有機相。得到化合物4-17。LCMSm/z = 636.1[M+H]+ 。Step 16: Synthesis of compound 4-17 Compound 1-11A (57.79 mg, 501.73 µmol, 59.57 µL, 4 eq) was dissolved in toluene (1 mL), and sodium tertiary butoxide (42.19 mg, 439.01 µmol) was added at 0℃ , 3.5 eq), stirred for 15 min, then compound 4-16 (100.00 mg, 125.43 µmol, 1 eq) was dissolved in 0.1 mL of toluene, slowly added to the reaction solution, and reacted at 0°C for 30 min. It was quenched with water (5 mL), extracted with ethyl acetate (5 mL x 2), and the organic phases were combined. Compound 4-17 was obtained. LCMS m/z = 636.1 [M+H] + .
步驟17:化合物4-18的合成 將化合物 4-17 (79.80 mg, 125.44 µmol, 1 eq)溶於二氯甲烷 (2 mL) 中,18℃加入N,N-二異丙基乙胺 (81.06 mg, 627.18 µmol, 109.24 µL, 5 eq) ,降溫至-78℃, 再將丙烯醯氯 (4.54 mg, 50.17 µmol, 4.09 µL, 0.4 eq) ,緩慢加入到反應液中,-78℃反應0.5hr。補加 8.00 mg 丙烯醯氯反應1hr,飽和氯化銨水溶液(5 mL)淬滅,二氯甲烷(5 mL * 2)萃取,合併有機相。粗品在碳酸鉀(1.7M,1 mL )/甲醇(1 mL)中,18℃攪拌1hr,檢測產品(time=0.943)得到化合物4-18。LCMSm/z = 690.3[M+H]+ 。Step 17: Synthesis of compound 4-18. Compound 4-17 (79.80 mg, 125.44 µmol, 1 eq) was dissolved in dichloromethane (2 mL), and N,N-diisopropylethylamine (81.06) was added at 18°C. mg, 627.18 µmol, 109.24 µL, 5 eq), cooled to -78°C, and then propylene chloride (4.54 mg, 50.17 µmol, 4.09 µL, 0.4 eq) was slowly added to the reaction solution, and reacted at -78°C for 0.5hr . Add 8.00 mg of propylene chloride to react for 1 hr, quench with saturated ammonium chloride aqueous solution (5 mL), extract with dichloromethane (5 mL * 2), and combine the organic phases. The crude product was stirred in potassium carbonate (1.7M, 1 mL)/methanol (1 mL) at 18°C for 1 hr, and the product was detected (time=0.943) to obtain compound 4-18. LCMS m/z = 690.3 [M+H] + .
步驟18:化合物4A和4B的合成 將化合物4-18 (100 mg, 144.88 µmol, 1eq ) 溶於二氯甲烷 (2 mL)、三氟乙酸 (3.08 g, 27.01 mmol, 2.00 mL, 186.45eq ) ,18℃反應1 hr。濃縮得到化合物4-19。化合物4-19使用高效液相層析管柱分離純化:層析管柱: Phenomenex luna C18 100*40mm*5 µm;流動相: [H2 O(0.1%TFA)-乙腈];乙腈%: 5%-30%,8min ,樣品在加入0.05mL稀鹽酸0.2 mL,真空下濃縮,得到化合物4A的鹽酸鹽(出峰時間:2.417min)。LCMSm/z = 590.1[M+H]+,295.9[M/2+H]+ ;和化合物4B的鹽酸鹽(出峰時間:2.388min)。LCMSm/z = 590.1[M+H]+,295.9[M/2+H]+ 。Step 18: Synthesis of compound 4A and 4B. Compound 4-18 (100 mg, 144.88 µmol, 1 eq ) was dissolved in dichloromethane (2 mL), trifluoroacetic acid (3.08 g, 27.01 mmol, 2.00 mL, 186.45 eq ) , React at 18°C for 1 hr. Concentrate to obtain compound 4-19. Compound 4-19 was separated and purified using high performance liquid chromatography column: Chromatography column: Phenomenex luna C18 100*40mm*5 µm; mobile phase: [H 2 O(0.1%TFA)-acetonitrile]; acetonitrile%: 5 %-30%, 8min, the sample was added with 0.05mL dilute hydrochloric acid 0.2mL and concentrated under vacuum to obtain the hydrochloride of compound 4A (peak time: 2.417min). LCMS m/z = 590.1 [M+H] +, 295.9 [M/2+H] + ; and the hydrochloride salt of compound 4B (peak time: 2.388 min). LCMS m/z = 590.1 [M+H]+, 295.9 [M/2+H] + .
實施例 5
步驟2:化合物5-2的合成
將二氯甲烷(25 mL),化合物5-1(1.6 g, 4.05 mmol, 1eq
)加入預先準備好的乾淨反應瓶中,開始攪拌;然後將N,N-二甲基甲醯胺二甲縮醛(724.30 mg, 6.08 mmol, 807.47 µL, 1.5eq
)加入其中,室溫20℃反應12hr。然後將溫度降至0~5℃後,將三氟化硼乙醚(575.13 mg, 4.05 mmol, 500.11 µL, 1eq
)加入其中,室溫20℃繼續反應1hr。反應液30℃進行減壓旋蒸,得到化合物5-2直接用於下一步。Step 2: Synthesis of compound 5-2. Add dichloromethane (25 mL) and compound 5-1 (1.6 g, 4.05 mmol, 1 eq ) into a clean reaction flask prepared in advance and start stirring; then add N, N -Dimethylformamide dimethyl acetal (724.30 mg, 6.08 mmol, 807.47 µL, 1.5 eq ) was added to it, and reacted at room temperature at 20°C for 12 hours. Then, after lowering the temperature to 0~5°C, add boron trifluoride ether (575.13 mg, 4.05 mmol, 500.11 µL, 1 eq ) to it, and continue the reaction at
步驟3:化合物5-3的合成 將四氫呋喃(58 mL),化合物5-2(3.9 g, 8.40 mmol, 87.233% 質量含量, 1eq )加入預先準備好的乾淨反應瓶中,開始攪拌;將溫度降至-70~-65℃後,將三二級丁基硼氫化鋰 (1 M, 9.24 mL, 1.1eq )滴加到其中,反應0.5hr。將反應液倒入50 mL飽和氯化銨溶液中後,有機相加入2.00 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(石油醚/乙酸乙酯= 100/0~70/30,TLC : 石油醚/乙酸乙酯= 5/1),得到化合物5-3。LCMS: MSm/z = 407.0[M+H]+ 。Step 3: Synthesis of compound 5-3. Add tetrahydrofuran (58 mL), compound 5-2 (3.9 g, 8.40 mmol, 87.233% mass content, 1 eq ) into a clean reaction flask prepared in advance, and start stirring; After the temperature drops to -70~-65°C, the tertiary butyl lithium borohydride (1 M, 9.24 mL, 1.1 eq ) is added dropwise to it, and the reaction is carried out for 0.5 hr. After the reaction solution was poured into 50 mL of saturated ammonium chloride solution, the organic phase was dried by adding 2.00 g of anhydrous sodium sulfate, filtered, and the filtrate was subjected to reduced pressure rotary evaporation at 45°C. The crude product was purified by column chromatography (petroleum ether/ethyl acetate=100/0~70/30, TLC: petroleum ether/ethyl acetate=5/1) to obtain compound 5-3. LCMS: MS m/z = 407.0 [M+H] + .
步驟4:化合物5-4的合成 將甲醇(4 mL),化合物5-3(0.65 g, 1.60 mmol, 1eq ), 甲基異硫脲硫酸鹽 (1.22 g, 6.39 mmol, 4eq , H2 SO4 )加入預先準備好的反應瓶中,開始攪拌。然後將甲醇鈉(172.61 mg, 3.20 mmol, 2eq )加入其中,室溫25℃反應1hr,補加甲醇鈉(172.62 mg, 3.20 mmol, 2eq )後,繼續反應15hr。反應液在45℃下進行減壓旋蒸,得到的白色固體加入10 mL水以及10 mL乙酸乙酯萃取分液,有機相用10 mL飽和食鹽水洗滌一次後,加入0.50 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(石油醚/乙酸乙酯= 100/0~40/60,TLC : 石油醚/乙酸乙酯= 1/1),得到化合物5-4。LCMS: MS m/z = 447.0[M+H]+ 。Step 4: Synthesis of compound 5-4 Methanol (4 mL), compound 5-3 (0.65 g, 1.60 mmol, 1 eq ), methyl isothiourea sulfate (1.22 g, 6.39 mmol, 4 eq , H 2 SO 4 ) Put it into the pre-prepared reaction flask and start stirring. Then sodium methoxide (172.61 mg, 3.20 mmol, 2 eq ) was added to it, and the reaction was carried out at room temperature at 25° C. for 1 hr. After additional sodium methoxide (172.62 mg, 3.20 mmol, 2 eq ) was added, the reaction was continued for 15 hr. The reaction solution was subjected to reduced pressure rotary evaporation at 45°C. The white solid obtained was extracted and separated by adding 10 mL of water and 10 mL of ethyl acetate. The organic phase was washed once with 10 mL of saturated brine, and then dried by adding 0.50 g of anhydrous sodium sulfate. , Filtration, and the filtrate was subjected to reduced pressure rotary evaporation at 45°C. The crude product was purified by column chromatography (petroleum ether/ethyl acetate = 100/0-40/60, TLC: petroleum ether/ethyl acetate = 1/1) to obtain compound 5-4. LCMS: MS m/z = 447.0 [M+H] + .
步驟5:化合物5-5的合成 將二氯甲烷(20 mL),化合物5-4(610 mg, 1.36 mmol, 1eq )加入預先準備好的乾淨反應瓶中,開始攪拌。將溫度降至0~5℃後,依次將N,N-二異丙基乙胺 (617.36 mg, 4.78 mmol, 832.02 µL, 3.5eq ),三氟甲磺酸酐(770.13 mg, 2.73 mmol, 450.37 µL, 2eq )加入其中,反應0.5hr。將反應液倒入20 mL飽和氯化銨溶液中後,分液,有機相加入10 mL飽和食鹽水洗滌一次後,加入無水硫酸鈉乾燥,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(石油醚/乙酸乙酯= 100/0~70/30,TLC : 石油醚/乙酸乙酯= 5/1),得到化合物5-5。1H NMR (400 MHz, CDCl3 ) δ = 8.26 (d,J = 5.2 Hz, 1H), 7.48 (d,J = 14.4 Hz, 1H), 5.73 - 5.67 (m, 1H), 5.53 - 5.49 (m, 1H), 5.15 (dd,J = 3.2, 15.6 Hz, 1H), 4.88 (d,J = 15.6 Hz, 1H), 4.06 - 3.99 (m, 1H), 3.80 - 3.72 (m, 1H), 3.30 - 3.25 (m, 1H), 3.12 - 3.04 (m, 1H), 2.61 - 2.49 (m, 7H), 2.19 - 2.07 (m, 2H), 1.83 - 1.68 (m, 3H)。Step 5: Synthesis of compound 5-5. Add dichloromethane (20 mL) and compound 5-4 (610 mg, 1.36 mmol, 1 eq ) into a clean reaction flask prepared in advance, and start stirring. After lowering the temperature to 0~5℃, add N,N-diisopropylethylamine (617.36 mg, 4.78 mmol, 832.02 µL, 3.5 eq ) and trifluoromethanesulfonic anhydride (770.13 mg, 2.73 mmol, 450.37 µL) in sequence , 2 eq ) was added to it and reacted for 0.5hr. After the reaction solution was poured into 20 mL of saturated ammonium chloride solution, the layers were separated, and the organic phase was washed once with 10 mL of saturated brine, dried with anhydrous sodium sulfate, filtered, and the filtrate was rotary evaporated under reduced pressure at 45°C. The crude product was purified by column chromatography (petroleum ether/ethyl acetate = 100/0~70/30, TLC: petroleum ether/ethyl acetate = 5/1) to obtain compound 5-5. 1H NMR (400 MHz, CDCl 3 ) δ = 8.26 (d, J = 5.2 Hz, 1H), 7.48 (d, J = 14.4 Hz, 1H), 5.73-5.67 (m, 1H), 5.53-5.49 (m, 1H), 5.15 (dd, J = 3.2, 15.6 Hz, 1H), 4.88 (d, J = 15.6 Hz, 1H), 4.06-3.99 (m, 1H), 3.80-3.72 (m, 1H), 3.30-3.25 (m, 1H), 3.12- 3.04 (m, 1H), 2.61-2.49 (m, 7H), 2.19-2.07 (m, 2H), 1.83-1.68 (m, 3H).
步驟6:化合物5-6的合成 將N,N-二甲基甲醯胺(5 mL),化合物5-5(0.33 g, 569.94 µmol, 1eq )加入預先準備好的乾淨反應瓶中,開始攪拌,然後依次將N,N-二異丙基乙胺(368.29 mg, 2.85 mmol, 496.35 µL, 5eq ),化合物5-5a(143 mg, 1.14 mmol, 2.00eq , 2HCl)加入其中,將溫度升至100℃,反應1hr。將反應液倒入20 mL飽和氯化胺溶液中後,加入10 mL乙酸乙酯溶液中,分液,有機相加入無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(二氯甲烷/甲醇= 100/0~85/15,TLC : 二氯甲烷/甲醇= 15/1),得到化合物5-6。1 H NMR (400 MHz, CDCl3 ) δ = 8.22 (d,J = 4.4 Hz, 1H), 7.45 (d,J = 8.8 Hz, 1H), 5.71 - 5.66 (m, 1H), 5.57 - 5.53 (m, 1H), 4.89 - 4.80 (m, 2H), 4.05 - 3.86 (m, 2H), 3.77 - 3.32 (m, 1H), 3.60 - 3.57 (m, 1H), 3.39 - 3.38 (m, 1H), 3.31 - 3.26 (m, 1H), 3.23 - 3.17 (m, 1H), 3.12 - 3.09(m, 1H), 3.02 - 2.96 (m, 3H), 2.93 - 2.83 (m, 2H), 2.57 - 2.56 (m, 1H), 2.54 - 2.52 (m, 7H), 2.16 - 2.04 (m, 2H), 1.79 - 1.71 (m, 3H)。LCMS: MS m/z = 554.0[M+H]+ 。Step 6: Synthesis of compound 5-6. Add N,N-dimethylformamide (5 mL) and compound 5-5 (0.33 g, 569.94 µmol, 1 eq ) into a clean reaction flask prepared in advance, and start Stir, then add N,N-diisopropylethylamine (368.29 mg, 2.85 mmol, 496.35 µL, 5 eq ), compound 5-5a (143 mg, 1.14 mmol, 2.00 eq , 2HCl) in sequence, and add the temperature Raise to 100°C and react for 1 hr. After the reaction solution was poured into 20 mL of saturated amine chloride solution, it was added to 10 mL of ethyl acetate solution for liquid separation, the organic phase was added with anhydrous sodium sulfate and dried, filtered, and the filtrate was subjected to reduced pressure rotary evaporation at 45°C. The crude product was purified by column chromatography (dichloromethane/methanol=100/0~85/15, TLC: dichloromethane/methanol=15/1) to obtain compound 5-6. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.22 (d, J = 4.4 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H), 5.71-5.66 (m, 1H), 5.57-5.53 (m , 1H), 4.89-4.80 (m, 2H), 4.05-3.86 (m, 2H), 3.77-3.32 (m, 1H), 3.60-3.57 (m, 1H), 3.39-3.38 (m, 1H), 3.31 -3.26 (m, 1H), 3.23-3.17 (m, 1H), 3.12-3.09 (m, 1H), 3.02-2.96 (m, 3H), 2.93-2.83 (m, 2H), 2.57-2.56 (m, 1H), 2.54-2.52 (m, 7H), 2.16-2.04 (m, 2H), 1.79-1.71 (m, 3H). LCMS: MS m/z = 554.0 [M+H] + .
步驟7:化合物5-7的合成 將化合物5-6(190 mg, 342.90 µmol, 1eq )溶於四氫呋喃(2 mL)後,開始攪拌。然後將溫度降至0~5℃後,將三氟乙酸酐(108.03 mg, 514.34 µmol, 71.54 µL, 1.5eq ),三乙胺(121.44 mg, 1.20 mmol, 167.04 µL, 3.5eq )加入其中,反應0.5hr。將反應液倒入10 mL飽和氯化銨溶液中後,加入10 mL二氯甲烷萃取,有機相用飽和食鹽水洗滌一次後,加入無水硫酸鈉乾燥,過濾,濾液在45℃下進行減壓旋蒸,得到化合物5-7。LCMS: MS m/z = 650.2[M+H]+ 。Step 7: Synthesis of compound 5-7 After dissolving compound 5-6 (190 mg, 342.90 µmol, 1 eq ) in tetrahydrofuran (2 mL), start stirring. Then, after lowering the temperature to 0~5℃, add trifluoroacetic anhydride (108.03 mg, 514.34 µmol, 71.54 µL, 1.5 eq ), triethylamine (121.44 mg, 1.20 mmol, 167.04 µL, 3.5 eq ) into it, and react 0.5hr. After the reaction solution was poured into 10 mL saturated ammonium chloride solution, 10 mL dichloromethane was added for extraction, the organic phase was washed once with saturated brine, dried with anhydrous sodium sulfate, filtered, and the filtrate was subjected to reduced pressure rotation at 45°C. Steam to obtain compound 5-7. LCMS: MS m/z = 650.2 [M+H] + .
步驟8:化合物5-8的合成 將二氯甲烷(5 mL),化合物5-7(0.2 g, 290.04 µmol, 94.281% 質量含量, 1 eq)加入預先準備好的乾淨反應瓶中,開始攪拌。然後將間氯過氧苯甲酸(143.96 mg, 667.37 µmol, 80% 質量含量, 2.30 eq)加入其中,室溫25℃反應0.5hr。將反應液倒入20 mL的硫代硫酸鈉溶液(10%)中後,加入15 mL二氯甲烷萃取,有機相加入無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸。粗品經管柱層析純化(二氯甲烷/甲醇= 100/0~85/15,TLC : 二氯甲烷/甲醇= 15/1),得到化合物5-8。LCMS: MSm/z = 682.0[M+H]+ 。Step 8: Synthesis of compound 5-8. Add dichloromethane (5 mL) and compound 5-7 (0.2 g, 290.04 µmol, 94.281% mass content, 1 eq) into a clean reaction flask prepared in advance, and start stirring. Then m-chloroperoxybenzoic acid (143.96 mg, 667.37 µmol, 80% mass content, 2.30 eq) was added to it, and reacted at room temperature at 25°C for 0.5hr. After the reaction solution was poured into 20 mL of sodium thiosulfate solution (10%), 15 mL of dichloromethane was added for extraction, and the organic phase was added with anhydrous sodium sulfate to dry, then filtered, and the filtrate was rotary evaporated under reduced pressure at 45°C. The crude product was purified by column chromatography (dichloromethane/methanol=100/0~85/15, TLC: dichloromethane/methanol=15/1) to obtain compound 5-8. LCMS: MS m/z = 682.0 [M+H] + .
步驟9:化合物5-9的合成 將甲苯(5 mL),化合物1-11A(148.59 mg, 1.29 mmol, 153.18 µL, 4eq )加入預先準備好的乾淨反應瓶中,開始攪拌。然後將溫度降至0~5℃後,將三級丁醇鈉(123.98 mg, 1.29 mmol, 4eq )加入其中,反應15 min,然後將化合物5-8(0.22 g, 322.53 µmol, 1eq )溶於0.2 mL 甲苯後,快速加入其中,反應0.5hr。將反應液倒入10 mL飽和氯化銨溶液中後,加入10 mL二氯甲烷萃取,有機相用10 mL飽和食鹽水洗滌一次後,加入0.50 g無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸,得到化合物5-9。LCMS: MSm/z = 621.4[M+H]+ 。Step 9: Synthesis of compound 5-9 Add toluene (5 mL) and compound 1-11A (148.59 mg, 1.29 mmol, 153.18 µL, 4 eq ) into a clean reaction flask prepared in advance, and start stirring. Then, after lowering the temperature to 0~5℃, add tertiary butoxide sodium (123.98 mg, 1.29 mmol, 4 eq ) to it, react for 15 min, and then compound 5-8 (0.22 g, 322.53 µmol, 1 eq ) After being dissolved in 0.2 mL of toluene, it was quickly added to it and reacted for 0.5 hr. Pour the reaction solution into 10 mL of saturated ammonium chloride solution, add 10 mL of dichloromethane for extraction, wash the organic phase with 10 mL of saturated brine once, add 0.50 g of anhydrous sodium sulfate to dry, filter, and filter the filtrate at 45°C Rotary evaporation is carried out under reduced pressure to obtain compound 5-9. LCMS: MS m/z = 621.4 [M+H] + .
步驟10:化合物5-10的合成 將二氯甲烷(5 mL),化合物5-9(98.26 mg, 125.80 µmol, 79.529% 質量含量, 1eq )加入預先準備好的反應瓶中,開始攪拌。然後將溫度降至-60℃,然後將N,N-二異丙基乙胺(162.59 mg, 1.26 mmol, 219.12 µL, 10eq )加入其中,將丙烯醯氯(17.08 mg, 188.70 µmol, 15.39 µL, 1.5eq )溶於0.3 mL二氯甲烷後,滴加到其中,反應10 min。將反應液倒入5 mL飽和氯化銨溶液中後,分液,有機相加入5 mL飽和食鹽水洗滌一次後,加入無水硫酸鈉乾燥後,過濾,濾液在35℃下進行減壓旋蒸。得到化合物5-10,直接用於下一步。LCMS: MS m/z = 675.1[M+H]+ 。Step 10: Synthesis of compound 5-10. Add dichloromethane (5 mL) and compound 5-9 (98.26 mg, 125.80 µmol, 79.529% mass content, 1 eq ) into the pre-prepared reaction flask and start stirring. Then the temperature was lowered to -60°C, and then N,N-diisopropylethylamine (162.59 mg, 1.26 mmol, 219.12 µL, 10 eq ) was added to it, and propylene chloride (17.08 mg, 188.70 µmol, 15.39 µL) , 1.5 eq ) dissolved in 0.3 mL of dichloromethane, added dropwise to it, and reacted for 10 min. The reaction solution was poured into 5 mL of saturated ammonium chloride solution, and the layers were separated. The organic phase was washed once with 5 mL of saturated brine, then dried with anhydrous sodium sulfate, filtered, and the filtrate was subjected to reduced pressure rotary evaporation at 35°C. Compound 5-10 is obtained and used directly in the next step. LCMS: MS m/z = 675.1 [M+H] + .
步驟11:化合物5A和5B的合成 將二氯甲烷/三氟乙酸(4 mL,5/3),化合物5-10(0.1 g, 148.10 µmol, 1eq )加入反應瓶中,室溫25℃反應0.5hr。將反應液緩慢滴加到15 mL飽和碳酸氫鈉溶液中後,然後加入10 mL二氯甲烷萃取,有機相用10 mL飽和食鹽水洗滌一次後,加入無水硫酸鈉乾燥後,過濾,濾液在30℃下進行減壓旋蒸,粗品使用高效液相層析管柱分離純化,方法層析管柱: Phenomenex Gemini-NX 150*30mm*5µm;流動相: [H2 O(0.1%TFA)-乙腈];乙腈%: 20%-50%,9min,得到化合物5-11。化合物5-11送SFC拆分,方法為層析管柱: DAICEL CHIRALPAK AS(250mm*30mm,10µm);流動相: [0.1%NH3 H2 O EtOH];乙醇: 50%-50%,15min。Step 11: Synthesis of compounds 5A and 5B. Add dichloromethane/trifluoroacetic acid (4 mL, 5/3) and compound 5-10 (0.1 g, 148.10 µmol, 1 eq ) into the reaction flask, and react at room temperature at 25°C 0.5hr. The reaction solution was slowly added dropwise to 15 mL of saturated sodium bicarbonate solution, and then 10 mL of dichloromethane was added for extraction. The organic phase was washed once with 10 mL of saturated brine, dried by adding anhydrous sodium sulfate, and filtered. Rotary evaporation under reduced pressure at ℃, the crude product was separated and purified by high performance liquid chromatography column, method Chromatography column: Phenomenex Gemini-NX 150*30mm*5µm; Mobile phase: [H 2 O(0.1%TFA)-acetonitrile ]; Acetonitrile%: 20%-50%, 9min, to obtain compound 5-11. Compound 5-11 is sent to SFC for resolution. The method is chromatography column: DAICEL CHIRALPAK AS (250mm*30mm, 10µm); mobile phase: [0.1%NH 3 H 2 O EtOH]; ethanol: 50%-50%, 15min .
得到5A(手性管柱出峰時間:1.516)。SFC分析方法(管柱:Chiralpak AD-3, 50×4.6 mm,I.D.,3μm;流動相:A (CO2) 和 B (異丙醇,含0.05%二乙醇胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:91.04%。1 H NMR (400 MHz, CDCl3 ) δ = 8.26 (s, 1H), 7.37 (s, 1H), 6.62 - 6.56 (m, 1H), 6.42 - 6.38 (m, 1H), 5.84 (d,J = 11.6 Hz, 1H), 5.58 (dd,J = 4.0, 11.2 Hz, 1H), 4.94 (s, 2H), 4.55 - 4.43 (m, 1H), 4.27 - 4.18 (m, 1H), 4.02 - 3.87 (m, 1H), 3.76 - 3.73 (m, 1H), 3.23 - 3.18 (m, 4H), 3.07 – 2.98 (m, 2H), 2.87 - 2.74 (m, 3H), 2.56 - 2.53 (m, 6H), 2.13 - 2.07 (m, 1H), 1.82 - 1.76 (m, 3H), 1.37 - 1.29 (m, 3H)。LCMS: MSm/z = 591.2[M+H]+ 。Get 5A (chiral column peak time: 1.516). SFC analysis method (column: Chiralpak AD-3, 50×4.6 mm, ID, 3μm; mobile phase: A (CO2) and B (isopropanol, containing 0.05% diethanolamine); gradient: B%=5~50 %, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 91.04%. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.26 (s, 1H), 7.37 (s, 1H ), 6.62-6.56 (m, 1H), 6.42-6.38 (m, 1H), 5.84 (d, J = 11.6 Hz, 1H), 5.58 (dd, J = 4.0, 11.2 Hz, 1H), 4.94 (s, 2H), 4.55-4.43 (m, 1H), 4.27-4.18 (m, 1H), 4.02-3.87 (m, 1H), 3.76-3.73 (m, 1H), 3.23-3.18 (m, 4H), 3.07- 2.98 (m, 2H), 2.87-2.74 (m, 3H), 2.56-2.53 (m, 6H), 2.13-2.07 (m, 1H), 1.82-1.76 (m, 3H), 1.37-1.29 (m, 3H) ). LCMS: MS m/z = 591.2 [M+H] + .
得到5B(手性管柱出峰時間:1.800)。SFC分析方法(管柱:Chiralpak AD-3, 50×4.6 mm,I.D.,3μm;流動相:A (CO2) 和 B (異丙醇,含0.05%二乙醇胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:99.74%。1 H NMR (400 MHz, CDCl3 ) δ = 8.31 (s, 1H), 7.36 (s, 1H), 6.63 - 6.53 (m, 1H), 6.42 - 6.37 (m, 1H), 5.83 (d,J = 11.6 Hz, 1H), 5.59 (dd,J = 4.0, 11.2 Hz, 1H), 4.98 - 4.88 (m, 2H), 4.55 - 4.80 (m, 1H), 4.24 - 4.19 (m, 1H), 4.01 - 3.97 (m, 1H), 3.93 - 3.85 (m, 1H), 3.74 - 3.69 (m, 1H), 3.56 - 3.52 (m, 1H), 3.28 - 3.05 (m, 3H), 3.03 - 2.95 (m, 1H), 2.83 - 2.69 (m, 3H), 2.58 - 2.53 (m, 6H), 2.43 - 2.33 (m, 1H), 2.12 - 2.06 (m, 1H), 1.91 - 1.86 (m, 1H), 1.81 - 1.79 (m, 2H), 1.45 - 1.30 (m, 2H)。LCMS: MSm/z = 591.2[M+H]+ 。Get 5B (chiral column peak time: 1.800). SFC analysis method (column: Chiralpak AD-3, 50×4.6 mm, ID, 3μm; mobile phase: A (CO2) and B (isopropanol, containing 0.05% diethanolamine); gradient: B%=5~50 %, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 99.74%. 1 H NMR (400 MHz, CDCl 3 ) δ = 8.31 (s, 1H), 7.36 (s, 1H ), 6.63-6.53 (m, 1H), 6.42-6.37 (m, 1H), 5.83 (d, J = 11.6 Hz, 1H), 5.59 (dd, J = 4.0, 11.2 Hz, 1H), 4.98-4.88 ( m, 2H), 4.55-4.80 (m, 1H), 4.24-4.19 (m, 1H), 4.01-3.97 (m, 1H), 3.93-3.85 (m, 1H), 3.74-3.69 (m, 1H), 3.56-3.52 (m, 1H), 3.28-3.05 (m, 3H), 3.03-2.95 (m, 1H), 2.83-2.69 (m, 3H), 2.58-2.53 (m, 6H), 2.43-2.33 (m , 1H), 2.12-2.06 (m, 1H), 1.91-1.86 (m, 1H), 1.81-1.79 (m, 2H), 1.45-1.30 (m, 2H). LCMS: MS m/z = 591.2[M +H] + .
實施例 6


步驟2:化合物6-2的合成 將化合物6-1 (200 mg, 273.15 µmol, 1eq) 溶於二氯甲烷(4 mL) 中,0℃加入三氟乙酸(3.08 g, 27.01 mmol, 2 mL, 98.89 eq) ,18℃反應0.5hr。直接旋乾,得到粗品,使用高效液相層析管柱分離純化 層析管柱: Phenomenex Gemini-NX 150*30mm*5µm;流動相: [H2 O(0.1%TFA)-乙腈];乙腈%: 30%-60%,9min,得到化合物6-2。LCMS: MSm/z = 632.3[M+H]+ 。Step 2: Synthesis of compound 6-2. Compound 6-1 (200 mg, 273.15 µmol, 1eq) was dissolved in dichloromethane (4 mL), and trifluoroacetic acid (3.08 g, 27.01 mmol, 2 mL, 98.89 eq), react at 18°C for 0.5hr. Rotate directly to dry to obtain the crude product. Use high performance liquid chromatography column to separate and purify the column: Phenomenex Gemini-NX 150*30mm*5µm; mobile phase: [H 2 O(0.1%TFA)-acetonitrile]; acetonitrile% : 30%-60%, 9min, compound 6-2 is obtained. LCMS: MS m/z = 632.3 [M+H] + .
步驟3:化合物6-3的合成 將化合物6-2 (110 mg, 174.03 µmol, 1 eq),多聚甲醛 (88.91 mg, 1.74 mmol, 10 eq) 溶於1,2-二氯乙烷 (1 mL) ,甲醇 (1 mL) 中,加入冰乙酸 (1.05 mg, 17.40 µmol, 9.95e-1 µL,0.1 eq) ,攪拌30min,在加入氰基硼氫化鈉 (21.87 mg, 348.06 µmol, 2 eq) ,25℃攪拌10 hr。倒入飽和氯化銨水溶液(10 mL),加入二氯甲烷(5 mL x 3),無水硫酸鈉乾燥,過濾,濃縮得到6-3。LCMS: MSm/z = 646.1[M+H]+ ,647.7[M+2H]+ 。Step 3: Synthesis of compound 6-3. Compound 6-2 (110 mg, 174.03 µmol, 1 eq), paraformaldehyde (88.91 mg, 1.74 mmol, 10 eq) were dissolved in 1,2-dichloroethane (1 mL), methanol (1 mL), add glacial acetic acid (1.05 mg, 17.40 µmol, 9.95e-1 µL, 0.1 eq), stir for 30 min, then add sodium cyanoborohydride (21.87 mg, 348.06 µmol, 2 eq) , Stir at 25°C for 10 hr. Pour into saturated aqueous ammonium chloride solution (10 mL), add dichloromethane (5 mL x 3), dry with anhydrous sodium sulfate, filter, and concentrate to obtain 6-3. LCMS: MS m/z = 646.1 [M+H] + , 647.7 [M+2H] + .
步驟4:化合物6-4的合成
將化合物6-3(90 mg, 139.30 µmol, 1 eq) 溶於甲醇 (3 mL) 中, 加入碳酸鉀 (1.7 M, 2.70 mL, 32.95 eq),18℃反應1 hr。飽和氯化銨水溶液(5 mL)淬滅,乙酸乙酯(5 mL x 2)萃取,合併有機相,飽和食鹽水洗滌,無水硫酸鈉乾燥,過濾,濃縮得到化合物6-4。LCMS: MSm/z
= 550.2[M+H]+
,551.8[M+2H]+
。Step 4: Synthesis of compound 6-4. Dissolve compound 6-3 (90 mg, 139.30 µmol, 1 eq) in methanol (3 mL), add potassium carbonate (1.7 M, 2.70 mL, 32.95 eq), and react at 18°
步驟5:化合物6A和6B的合成 將化合物6-4 (76 mg, 138.16 µmol, 1 eq) 溶於二氯甲烷 (20 mL) 中,加入N,N-二異丙基乙胺(267.83 mg, 2.07 mmol, 360.96 µL, 15 eq) ,-60℃加入丙烯醯氯 (12.50 mg, 138.16 µmol, 11.27 µL, 1 eq) ,-60℃反應0.5 hr。 飽和氯化銨水溶液(5 mL)淬滅,乙酸乙酯(5 mL x 2)萃取,合併有機相,濃縮得到化合物6-5,使用高效液相層析管柱分離純化:層析管柱: Phenomenex Gemini-NX C18 75*30mm*3µm;流動相: [H2 O(0.04%NH3 H2 O+10mM NH4 HCO3 )-ACN];乙腈%: 25%-55%,6min ,得到化合物6-5 ,進行SFC : 層析管柱: Phenomenex Gemini-NX C18 75*30mm*3µm;流動相: [H2 O(0.04%NH3 H2 O+10mM NH4 HCO3 )-ACN];乙腈%: 25%-55%,6min,得到化合物6A(手性管柱出峰時間=1.435min),SFC分析方法(管柱:Chiralpak AD-3, 50×4.6 mm,I.D.,3μm;流動相:A (CO2) 和 B (異丙醇,含0.05%二乙醇胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:87.38%。LCMS: MSm/z = 604.1[M+H]+,和化合物6B (手性管柱出峰時間=1.643),SFC分析方法(管柱:Chiralpak AD-3, 50×4.6 mm,I.D.,3μm;流動相:A (CO2) 和 B (異丙醇,含0.05%二乙醇胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:100%。LCMS: MSm/z = 604.1[M+H]+。Step 5: Synthesis of compounds 6A and 6B. Compound 6-4 (76 mg, 138.16 µmol, 1 eq) was dissolved in dichloromethane (20 mL), and N,N-diisopropylethylamine (267.83 mg, 2.07 mmol, 360.96 µL, 15 eq), add propylene chloride (12.50 mg, 138.16 µmol, 11.27 µL, 1 eq) at -60°C, and react at -60°C for 0.5 hr. Quench with saturated aqueous ammonium chloride (5 mL), extract with ethyl acetate (5 mL x 2), combine the organic phases, and concentrate to obtain compound 6-5, which is separated and purified using a high performance liquid chromatography column: Chromatography column: Phenomenex Gemini-NX C18 75*30mm*3µm; Mobile phase: [H 2 O(0.04%NH 3 H 2 O+10mM NH 4 HCO 3 )-ACN]; Acetonitrile%: 25%-55%, 6min, get the compound 6-5, SFC: Chromatography column: Phenomenex Gemini-NX C18 75*30mm*3µm; Mobile phase: [H 2 O(0.04%NH 3 H 2 O+10mM NH 4 HCO 3 )-ACN]; Acetonitrile %: 25%-55%, 6min, get compound 6A (chiral column peak time=1.435min), SFC analysis method (column: Chiralpak AD-3, 50×4.6 mm, ID, 3μm; mobile phase: A (CO2) and B (isopropanol, containing 0.05% diethanolamine); gradient: B%=5~50%, 3 min; flow rate: 3.4 mL/min; wavelength: 220nm; pressure: 1800psi. Optical purity: 87.38 %. LCMS: MS m/z = 604.1[M+H]+, and compound 6B (chiral column peak time = 1.643), SFC analysis method (column: Chiralpak AD-3, 50×4.6 mm, ID ,3μm; Mobile phase: A (CO2) and B (isopropanol, containing 0.05% diethanolamine); Gradient: B%=5~50%, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800 psi. Optical purity: 100%. LCMS: MS m/z = 604.1 [M+H]+.
實施例 7
步驟2:化合物7A和7B的合成 將二氯甲烷/三氟乙酸(7 mL,5/3),化合物7-1 (70 mg, 100.98 µmol, 1eq )加入反應瓶中,室溫25℃反應3hr。將反應液緩慢滴加到15 mL飽和碳酸氫鈉溶液中後,與小試反應液混合,然後加入10 mL二氯甲烷萃取,有機相用10 mL飽和食鹽水洗滌一次後,加入無水硫酸鈉乾燥後,過濾,濾液在30℃下進行減壓旋蒸得到粗品,使用高效液相層析管柱分離純化,方法為層析管柱: Phenomenex lµna C18 100*40mm*5 µm;流動相: [H2 O(0.1%TFA)-乙腈];乙腈%: 10%-35%,8min.得到化合物7-2,進行 SFC拆分純化,方法為層析管柱: DAICEL CHIRALCEL OJ(250mm*30mm,10µm);流動相: [0.1%NH3 H2 O EtOH];EtOH%: 40%-40%,15min。Step 2: Synthesis of compounds 7A and 7B. Add dichloromethane/trifluoroacetic acid (7 mL, 5/3), compound 7-1 (70 mg, 100.98 µmol, 1 eq ) into the reaction flask, and react at room temperature at 25°C 3hr. The reaction solution was slowly added dropwise to 15 mL of saturated sodium bicarbonate solution, mixed with the small test reaction solution, and then extracted with 10 mL of dichloromethane, the organic phase was washed once with 10 mL of saturated brine, and dried by adding anhydrous sodium sulfate After filtering, the filtrate was subjected to reduced-pressure rotary evaporation at 30°C to obtain the crude product, which was separated and purified by high performance liquid chromatography column. The method was chromatographic column: Phenomenex lµna C18 100*40mm*5 µm; mobile phase: [H 2 O(0.1%TFA)-Acetonitrile]; Acetonitrile%: 10%-35%, 8min. Compound 7-2 was obtained, which was separated and purified by SFC using chromatography column: DAICEL CHIRALCEL OJ(250mm*30mm, 10µm ); Mobile phase: [0.1%NH 3 H 2 O EtOH]; EtOH%: 40%-40%, 15min.
得到化合物7A(手性管柱出峰時間:1.263 min)。SFC分析方法(管柱:Chiralcel OJ-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (乙醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:91.94%。1 HNMR (400 MHz, CDCl3 ) δ = 8.29 (s, 1H), 7.35 (s, 1H), 5.61 - 5.57 (m, 1H), 5.48 - 5.32 (m, 1H), 5.28 - 5.24 (m, 1H),4.95 - 4.86 (m, 3H), 4.44 - 4.43 (m, 1H), 4.20 - 4.16 (m, 2H), 3.97 - 3.93 (m, 1H), 3.80 - 3.78 (m, 1H), 3.50 - 3.48 (m, 1H), 3.27 - 3.22 (m, 1H), 3.14 - 2.95 (m, 4H), 2.81 - 2.71 (m, 3H), 2.52 - 2.50 (m, 7H), 2.34 - 2.28 (m, 1H), 2.08 - 2.02 (m, 1H), 1.91 - 1.84 (m, 2H)。LCMS: MSm/z = 609.2[M+H]+ 。Compound 7A is obtained (peak time of chiral column: 1.263 min). SFC analysis method (column: Chiralcel OJ-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (ethanol, containing 0.05% diisopropylamine amine); gradient: B%=5~50% , 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 91.94%. 1 HNMR (400 MHz, CDCl 3 ) δ = 8.29 (s, 1H), 7.35 (s, 1H), 5.61-5.57 (m, 1H), 5.48-5.32 (m, 1H), 5.28-5.24 (m, 1H), 4.95-4.86 (m, 3H), 4.44-4.43 (m, 1H), 4.20-4.16 (m , 2H), 3.97-3.93 (m, 1H), 3.80-3.78 (m, 1H), 3.50-3.48 (m, 1H), 3.27-3.22 (m, 1H), 3.14-2.95 (m, 4H), 2.81 -2.71 (m, 3H), 2.52-2.50 (m, 7H), 2.34-2.28 (m, 1H), 2.08-2.02 (m, 1H), 1.91-1.84 (m, 2H). LCMS: MS m/z = 609.2[M+H] + .
得到化合物7B(手性管柱出峰時間:1.393 min)。SFC分析方法(管柱:Chiralcel OJ-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (乙醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:82.48%。1 HNMR (400 MHz, CDCl3) δ = 8.26 (s, 1H), 7.35 (s, 1H), 5.59 - 5.55 (m, 1H), 5.48 - 5.36 (m, 1H), 5.29 - 5.24 (m, 1H),4.93 (s, 2H), 4.42 - 4.40 (m, 1H), 4.24 - 4.20 (m, 2H), 3.73 - 3.70 (m, 1H), 3.24 - 2.98 (m, 8H), 2.90 - 2.71 (m, 3H), 2.53 - 2.48 (m, 7H), 2.33 - 2.31 (m, 1H), 2.09 - 2.04 (m, 1H), 1.89 - 1.85 (m, 2H)。LCMS: MS m/z = 609.1[M+H]+ 。Compound 7B was obtained (peak time of chiral column: 1.393 min). SFC analysis method (column: Chiralcel OJ-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (ethanol, containing 0.05% diisopropylamine amine); gradient: B%=5~50% , 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 82.48%. 1 HNMR (400 MHz, CDCl3) δ = 8.26 (s, 1H), 7.35 (s, 1H), 5.59 -5.55 (m, 1H), 5.48-5.36 (m, 1H), 5.29-5.24 (m, 1H), 4.93 (s, 2H), 4.42-4.40 (m, 1H), 4.24-4.20 (m, 2H) , 3.73-3.70 (m, 1H), 3.24-2.98 (m, 8H), 2.90-2.71 (m, 3H), 2.53-2.48 (m, 7H), 2.33-2.31 (m, 1H), 2.09-2.04 ( m, 1H), 1.89-1.85 (m, 2H). LCMS: MS m/z = 609.1 [M+H] + .
實施例8


步驟2:化合物8-3的合成 將2,2,6,6-四甲基哌啶(31.31 g, 221.65 mmol, 37.63 mL, 3eq )加入到無水四氫呋喃 (300 mL)中,降溫至-5℃,滴加正丁基鋰(2.5 M, 94.57 mL, 3.2eq ),-5~0℃反應15 min,降溫至-60℃,加入化合物8-2 (27 g, 73.88 mmol, 1eq )的四氫呋喃(60 mL)溶液,-60℃反應0.5hr,快速加入N,N-二甲基甲醯胺 (108.00 g, 1.48 mol, 113.69 mL, 20eq ),-60℃反應10 min。向反應液中加入400 mL飽和氯化銨,利用200 mLx 2的甲基三級丁基醚萃取,合併有機相,200 mL飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮得到產物粗品,粗品經石油醚:甲基三級丁基醚=5:1的混合溶劑70 mL打漿0.5 hr,過濾,濾餅旋乾,濾液拌樣後經過柱純化(石油醚:乙酸乙酯=100:0-10:1),得到的化合物8-3 。1 H NMR (400MHz, CDCl3 ) δ = 10.43 - 10.35 (m, 1H), 7.21-7.18 (m, 5H), 6.92 - 6.81 (m, 5H), 4.25 (s, 4H), 3.80 (s, 6H), 2.23 (s, 3H)。LCMS:MSm/z = 394.2[M+H]+ 。Step 2: Synthesis of compound 8-3 2,2,6,6-tetramethylpiperidine (31.31 g, 221.65 mmol, 37.63 mL, 3 eq ) was added to anhydrous tetrahydrofuran (300 mL), and the temperature was reduced to -5 ℃, add n-butyl lithium (2.5 M, 94.57 mL, 3.2 eq ) dropwise, react at -5~0℃ for 15 min, cool to -60℃, add compound 8-2 (27 g, 73.88 mmol, 1 eq ) Tetrahydrofuran (60 mL) solution was reacted at -60°C for 0.5 hr, N,N-dimethylformamide (108.00 g, 1.48 mol, 113.69 mL, 20 eq ) was quickly added, and reacted at -60°C for 10 min. 400 mL saturated ammonium chloride was added to the reaction solution, extracted with 200 mL x 2 methyl tertiary butyl ether, the organic phases were combined, washed with 200 mL saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated to obtain the crude product. Petroleum ether: Methyl tertiary butyl ether = 5:1 mixed solvent 70 mL beating for 0.5 hr, filtered, the filter cake was spin-dried, and the filtrate was mixed and purified by column (petroleum ether: ethyl acetate = 100:0-10 :1), the obtained compound 8-3. 1 H NMR (400MHz, CDCl 3 ) δ = 10.43-10.35 (m, 1H), 7.21-7.18 (m, 5H), 6.92-6.81 (m, 5H), 4.25 (s, 4H), 3.80 (s, 6H) ), 2.23 (s, 3H). LCMS: MS m/z = 394.2 [M+H] + .
步驟3:化合物8-4的合成 將化合物8-3 (17.8 g, 45.24 mmol, 1eq )加入到N,N-二甲基甲醯胺 (170 mL)中,加入溴代丁二醯亞胺 (8.05 g, 45.24 mmol, 1eq ) ,20℃反應20 min,將反應液加入到300 mL的水中,利用150 mL x 2的甲基三級丁基醚萃取,合併有機相,100 mL x 2的飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮,粗品利用乙酸乙酯:甲基三級丁基醚=1:1的混合溶劑打漿0.5 hr,過濾,將濾餅旋乾,得到化合物8-4。1 H NMR (400MHz, CDCl3 ) δ = 10.39 (s, 1H), 7.17 (d,J = 8.8 Hz, 4H), 6.89 (d,J = 8.8 Hz, 1H), 6.85-6.82 (m, 4H), 4.22 (s, 4H), 3.79 (s, 6H), 2.28 (s, 3H)。LCMS:MSm/z = 472.1[M+H]+ ,474.1[M+3H]+ 。Step 3: Synthesis of compound 8-4. Compound 8-3 (17.8 g, 45.24 mmol, 1 eq ) was added to N,N-dimethylformamide (170 mL), and bromosuccinimide was added (8.05 g, 45.24 mmol, 1 eq ), react at 20°C for 20 min, add the reaction solution to 300 mL of water, extract with 150 mL x 2 methyl tertiary butyl ether, combine the organic phases, 100 mL x 2 Washed with saturated brine, dried and filtered with anhydrous sodium sulfate and concentrated. The crude product was slurried with a mixed solvent of ethyl acetate: methyl tertiary butyl ether=1:1 for 0.5 hr, filtered, and the filter cake was spin-dried to obtain compound 8- 4. 1 H NMR (400MHz, CDCl 3 ) δ = 10.39 (s, 1H), 7.17 (d, J = 8.8 Hz, 4H), 6.89 (d, J = 8.8 Hz, 1H), 6.85-6.82 (m, 4H) , 4.22 (s, 4H), 3.79 (s, 6H), 2.28 (s, 3H). LCMS: MS m/z = 472.1 [M+H] + , 474.1 [M+3H] + .
步驟4:化合物8-5的合成 將化合物8-4 (19.3 g, 40.86 mmol, 1eq )加入到N,N-二甲基甲醯胺(190 mL)中,氮氣下加入碘化亞銅 (15.56 g, 81.72 mmol, 2eq )和氟磺醯基二氟乙酸甲酯(39.25 g, 204.30 mmol, 25.99 mL, 5eq ),100℃氮氣下反應1hr,將反應液墊矽藻土過濾,濾液加入到300 mL的水中,利用150 mL x 2的甲基三級丁基醚萃取,合併有機相,飽和食鹽水洗滌(200 mL x 2),無水硫酸鈉乾燥過濾後濃縮,粗品利用過柱純化(石油醚:乙酸乙酯=100:0-10:1,石油醚:乙酸乙酯=5:1),得到化合物8-5。1 H NMR (400MHz, CDCl3 ) δ = 10.37 (q,J = 4.0 Hz, 1H), 7.18 - 7.11 (m, 4H), 6.89 - 6.82 (m, 4H), 6.73 (d,J = 8.8 Hz, 1H), 4.36 (s, 4H), 3.81 (s, 6H), 2.37 - 2.29 (m, 3H)。LCMS: MS m/z =484.0[M+Na]+ 。Step 4: Synthesis of compound 8-5 Compound 8-4 (19.3 g, 40.86 mmol, 1 eq ) was added to N,N-dimethylformamide (190 mL), and cuprous iodide ( 15.56 g, 81.72 mmol, 2 eq ) and methyl fluorosulfonyl difluoroacetate (39.25 g, 204.30 mmol, 25.99 mL, 5 eq ), react at 100°C under nitrogen for 1 hour, filter the reaction solution with Celite, and filter the filtrate Add to 300 mL of water, extract with 150 mL x 2 methyl tertiary butyl ether, combine the organic phases, wash with saturated brine (200 mL x 2), dry and filter with anhydrous sodium sulfate, concentrate, and purify the crude product by column (Petroleum ether: ethyl acetate=100:0-10:1, petroleum ether: ethyl acetate=5:1) to obtain compound 8-5. 1 H NMR (400MHz, CDCl 3 ) δ = 10.37 (q, J = 4.0 Hz, 1H), 7.18-7.11 (m, 4H), 6.89-6.82 (m, 4H), 6.73 (d, J = 8.8 Hz, 1H), 4.36 (s, 4H), 3.81 (s, 6H), 2.37-2.29 (m, 3H). LCMS: MS m/z = 484.0 [M+Na] + .
步驟5:化合物8-6的合成 將無水四氫呋喃 (50 mL)和鈉氫(1.17 g, 29.26 mmol, 60% 質量含量, 3eq )加入到乾燥的三口燒瓶中,降溫至0℃,氮氣保護下滴加乙醯乙酸甲酯 (3.40 g, 29.26 mmol, 3.15 mL, 3eq ),0℃氮氣下反應0.5 hr,滴加正丁基鋰(2.5 M, 11.70 mL, 3eq ),0℃反應0.5 hr,降溫至-60℃,滴加化合物8-5(4.5 g, 9.75 mmol, 1eq )的四氫呋喃 (20 mL)溶液,-60℃反應0.5 hr,向反應液中加入100 mL飽和氯化銨溶液,利用30 mL的乙酸乙酯萃取,80 mL飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮得到產物粗品,合併純化,粗品利用過柱純化(石油醚:乙酸乙酯=100:0-3:1,石油醚:乙酸乙酯=3:1),得到黃色油狀的化合物8-6。1 H NMR (400MHz, CDCl3) δ = 7.18-7.15 (m, 4H), 6.90 - 6.78 (m, 4H), 6.61 (d,J = 8.8 Hz, 1H), 5.72 - 5.57 (m, 1H), 4.31 (m, 4H), 3.81(s, 6H),3.76(s, 3H), 3.56 (s, 2H), 3.50 - 3.38 (m, 1H), 2.98 - 2.93 (m, 1H), 2.38 - 2.26 (m, 3H)。LCMS: MSm/z =578.1[M+H]+ 。Step 5: Synthesis of compound 8-6. Add anhydrous tetrahydrofuran (50 mL) and sodium hydrogen (1.17 g, 29.26 mmol, 60% mass content, 3 eq ) into a dry three-necked flask, and cool to 0°C under nitrogen protection Add methyl acetylacetate (3.40 g, 29.26 mmol, 3.15 mL, 3 eq ) dropwise, react under nitrogen at 0°C for 0.5 hr, add n-butyl lithium (2.5 M, 11.70 mL, 3 eq ) dropwise, react 0.5 at 0°C hr, reduce the temperature to -60℃, add dropwise compound 8-5 (4.5 g, 9.75 mmol, 1 eq ) in tetrahydrofuran (20 mL) solution, react at -60℃ for 0.5 hr, add 100 mL saturated ammonium chloride to the reaction solution The solution was extracted with 30 mL of ethyl acetate, washed with 80 mL of saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated to obtain the crude product, which was combined and purified. The crude product was purified by column (petroleum ether: ethyl acetate=100:0-3 :1, petroleum ether: ethyl acetate=3:1) to obtain compound 8-6 as yellow oil. 1 H NMR (400MHz, CDCl3) δ = 7.18-7.15 (m, 4H), 6.90-6.78 (m, 4H), 6.61 (d, J = 8.8 Hz, 1H), 5.72-5.57 (m, 1H), 4.31 (m, 4H), 3.81(s, 6H), 3.76(s, 3H), 3.56 (s, 2H), 3.50-3.38 (m, 1H), 2.98-2.93 (m, 1H), 2.38-2.26 (m , 3H). LCMS: MS m/z = 578.1 [M+H] + .
步驟6:化合物8-7的合成 將化合物8-6 (3 g, 5.19 mmol, 1eq )加入到無水二氯甲烷 (30 mL)中,加入N,N-二甲基甲醯胺二甲基縮醛 (742.74 mg, 6.23 mmol, 828.02 µL, 1.2 eq),20℃反應16 hr,加入三氟化硼乙醚 (884.66 mg, 6.23 mmol, 769.27 µL, 1.2eq ),20℃反應1 hr。將反應液加入到20 mL飽和碳酸氫鈉溶液中,分液,水相繼續利用20 mL的二氯甲烷萃取,合併有機相,無水硫酸鈉乾燥過濾後濃縮,粗品利用過柱純化(石油醚:乙酸乙酯=100:0-3:1,石油醚:乙酸乙酯=3:1),得到化合物8-7。1 H NMR (400MHz, CDCl3 ) δ =8.43 (d,J = 0.8 Hz, 1H), 7.21 - 7.10 (m, 4H), 6.91 - 6.81 (m, 4H), 6.70 (d,J = 8.8 Hz, 1H), 5.93 (dd,J = 3.2, 14.8 Hz, 1H), 4.35 (s, 4H), 3.8(s, 3H),3.81 (s, 6H), 3.38-3.29 (m, 1H), 2.68 (dd,J = 3.6, 16.8 Hz, 1H), 2.39 - 2.24 (m, 3H)。LCMS: MSm/z =588.2[M+H]+ 。Step 6: Synthesis of compound 8-7. Compound 8-6 (3 g, 5.19 mmol, 1 eq ) was added to dry dichloromethane (30 mL), and N,N-dimethylformamide dimethyl was added Acetal (742.74 mg, 6.23 mmol, 828.02 µL, 1.2 eq) was reacted at 20°C for 16 hr. Boron trifluoride ether (884.66 mg, 6.23 mmol, 769.27 µL, 1.2 eq ) was added and reacted at 20°C for 1 hr. The reaction solution was added to 20 mL of saturated sodium bicarbonate solution and separated. The aqueous phase was extracted with 20 mL of dichloromethane. The organic phases were combined, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by column (petroleum ether: Ethyl acetate=100:0-3:1, petroleum ether: ethyl acetate=3:1) to obtain compound 8-7. 1 H NMR (400MHz, CDCl 3 ) δ =8.43 (d, J = 0.8 Hz, 1H), 7.21-7.10 (m, 4H), 6.91-6.81 (m, 4H), 6.70 (d, J = 8.8 Hz, 1H), 5.93 (dd, J = 3.2, 14.8 Hz, 1H), 4.35 (s, 4H), 3.8(s, 3H), 3.81 (s, 6H), 3.38-3.29 (m, 1H), 2.68 (dd , J = 3.6, 16.8 Hz, 1H), 2.39-2.24 (m, 3H). LCMS: MS m/z = 588.2 [M+H] + .
步驟7:化合物8-8的合成 將化合物8-7(2.1 g, 3.57 mmol, 1eq )加入到無水四氫呋喃 (21 mL)中,降溫至-60℃,氮氣保護下加入三二級丁基硼氫化鋰(1 M, 4.29 mL, 1.2eq ),-60℃反應0.5hr。將反應液加入到30 mL的飽和氯化銨中,分液萃取,有機相利用20 mL飽和食鹽水洗滌無水硫酸鈉乾燥過濾後濃縮得到產物粗品,粗品利用過柱純化(石油醚:乙酸乙酯=100:0=3:1,石油醚:乙酸乙酯=3:1),得到化合物8-8。1 H NMR (400MHz, CDCl3) δ = 7.167-7.14(m, 4H), 6.87-6.83 (m,4H), 6.63 (d,J = 8.8 Hz, 1H), 5.05-5.00 (m, 1H), 4.61-4.58 (m, 1H), 4.42 - 4.24 (m, 5H), 3.85-3.73 (m, 10H),3.13-3.05 (m, 1H), 2.47 - 2.38 (m, 1H), 2.35-2.31 (m, 3H)。LCMS: MSm/z = 600.1[M+H]+ 。Step 7: Synthesis of compound 8-8. Compound 8-7 (2.1 g, 3.57 mmol, 1 eq ) was added to anhydrous tetrahydrofuran (21 mL), the temperature was lowered to -60°C, and tertiary butyl boron was added under nitrogen protection Lithium hydride (1 M, 4.29 mL, 1.2 eq ) was reacted at -60°C for 0.5 hr. The reaction solution was added to 30 mL of saturated ammonium chloride, separated and extracted, the organic phase was washed with 20 mL of saturated brine, dried over sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by column (petroleum ether: ethyl acetate) =100:0=3:1, petroleum ether:ethyl acetate=3:1) to obtain compound 8-8. 1 H NMR (400MHz, CDCl3) δ = 7.167-7.14(m, 4H), 6.87-6.83 (m, 4H), 6.63 (d, J = 8.8 Hz, 1H), 5.05-5.00 (m, 1H), 4.61 -4.58 (m, 1H), 4.42-4.24 (m, 5H), 3.85-3.73 (m, 10H), 3.13-3.05 (m, 1H), 2.47-2.38 (m, 1H), 2.35-2.31 (m, 3H). LCMS: MS m/z = 600.1 [M+H] + .
步驟8:化合物8-9的合成 將化合物8-8 (1.27 g, 2.15 mmol, 1 eq)加入到乙醇 (15 mL)和水 (3 mL)中,加入碳酸氫鈉 (3.62 g, 43.08 mmol, 1.68 mL, 20 eq)和甲基異硫脲硫酸鹽 (4.05 g, 21.54 mmol, 10 eq),50℃反應4 hr。將反應液加入到40 mL水中,利用20 mL x 2的乙酸乙酯萃取,合併有機相,利用20 mL x 2的飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮,粗品利用過柱純化(石油醚:乙酸乙酯=100:0-1:1,石油醚:乙酸乙酯=1:1),得到化合物8-9。1 H NMR (400MHz, CDCl3) δ = 7.22 - 7.14 (m, 4H), 6.91 - 6.82 (m, 4H), 6.65 (dd,J = 8.4 Hz 1H), 5.12-5.08 (m, 1H), 4.97-4.91 (m, 1H), 4.67 - 4.57 (m, 1H), 4.45 - 4.22 (m, 4H), 3.88 - 3.74 (m, 6H), 3.43-3.35 (m, 1H), 2.77-2.72 (m, 1H), 2.59 (m, 3H), 2.40-2.31 (m, 3H)。LCMS:MSm/z =630.2[M+H]+ 。Step 8: Synthesis of compound 8-9. Compound 8-8 (1.27 g, 2.15 mmol, 1 eq) was added to ethanol (15 mL) and water (3 mL), and sodium bicarbonate (3.62 g, 43.08 mmol, 1.68 mL, 20 eq) and methyl isothiourea sulfate (4.05 g, 21.54 mmol, 10 eq), react at 50°C for 4 hr. The reaction solution was added to 40 mL of water, extracted with 20 mL x 2 ethyl acetate, combined the organic phases, washed with 20 mL x 2 saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude product was purified by column (petroleum Ether: ethyl acetate=100:0-1:1, petroleum ether: ethyl acetate=1:1) to obtain compound 8-9. 1 H NMR (400MHz, CDCl3) δ = 7.22-7.14 (m, 4H), 6.91-6.82 (m, 4H), 6.65 (dd, J = 8.4 Hz 1H), 5.12-5.08 (m, 1H), 4.97- 4.91 (m, 1H), 4.67-4.57 (m, 1H), 4.45-4.22 (m, 4H), 3.88-3.74 (m, 6H), 3.43-3.35 (m, 1H), 2.77-2.72 (m, 1H) ), 2.59 (m, 3H), 2.40-2.31 (m, 3H). LCMS: MS m/z = 630.2 [M+H] + .
步驟9:化合物8-10的合成 將化合物8-9 (0.57 g, 905.25 µmol, 1eq )加入到無水二氯甲烷 (6 mL)中,0℃加入N,N-二異丙基乙胺 (409.48 mg, 3.17 mmol, 551.86 µL, 3.5eq )和三氟甲磺酸酐 (510.81 mg, 1.81 mmol, 298.72 µL, 2eq ), 0~5℃反應0.5 hr。將反應液加入到20 mL飽和氯化銨中,利用10 mL的二氯甲烷萃取,無水硫酸鈉乾燥過濾後濃縮,粗品利用過柱純化(石油醚:乙酸乙酯=100:0-5:1,石油醚:乙酸乙酯=3:1)得到化合物8-10。1 H NMR (400MHz, CDCl3) δ = 7.21 - 7.11 (m, 4H), 6.90 - 6.80 (m, 4H), 6.66 (d,J = 8.4 Hz, 1H), 5.19-5.15 (m, 1H), 5.04 - 4.93 (m, 1H), 4.77-4.72 (m, 1H), 4.41 - 4.19 (m, 4H), 3.80 (s, 6H), 3.62-3.54 (m, 1H), 3.11 - 2.97 (m, 1H), 2.56 (s, 3H), 2.42 - 2.31 (m, 3H)。LCMS:MSm/z =762.2[M+H]+ 。Step 9: Synthesis of compound 8-10. Compound 8-9 (0.57 g, 905.25 µmol, 1 eq ) was added to dry dichloromethane (6 mL), and N,N-diisopropylethylamine ( 409.48 mg, 3.17 mmol, 551.86 µL, 3.5 eq ) and trifluoromethanesulfonic anhydride (510.81 mg, 1.81 mmol, 298.72 µL, 2 eq ), reacted at 0~5°C for 0.5 hr. The reaction solution was added to 20 mL of saturated ammonium chloride, extracted with 10 mL of dichloromethane, dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by column (petroleum ether: ethyl acetate=100:0-5:1) , Petroleum ether: ethyl acetate=3:1) to obtain compound 8-10. 1 H NMR (400MHz, CDCl3) δ = 7.21-7.11 (m, 4H), 6.90-6.80 (m, 4H), 6.66 (d, J = 8.4 Hz, 1H), 5.19-5.15 (m, 1H), 5.04 -4.93 (m, 1H), 4.77-4.72 (m, 1H), 4.41-4.19 (m, 4H), 3.80 (s, 6H), 3.62-3.54 (m, 1H), 3.11-2.97 (m, 1H) , 2.56 (s, 3H), 2.42-2.31 (m, 3H). LCMS: MS m/z = 762.2 [M+H] + .
步驟10:化合物8-11的合成 將化合物8-10 (0.45 g, 590.76 µmol, 1eq )加入到N,N-二甲基甲醯胺(5 mL)中,依次加入N,N-二異丙基乙胺(229.05 mg, 1.77 mmol, 308.69 µL, 3eq )和化合物1-10A (306.37 mg, 1.18 mmol, 2eq , HCl),50℃反應2 hr。將反應液倒入到20 mL水中,過濾,將濾餅溶於20 mL甲基三級丁基醚中,利用20 mL飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮得到化合物8-11。1 H NMR (400MHz, CDCl3) δ = 7.44 - 7.32 (m, 5H), 7.16-7.13 (m, 4H), 6.85-6.82 (m, 4H), 6.63 (d,J = 7.6 Hz, 1H), 5.21-5.15 (m, 2H), 4.80 - 4.66 (m, 3H), 4.39 - 4.22 (m, 4H), 3.93-3.88 (m, 1H), 3.80 (s, 6H), 3.71 - 3.55 (m, 1H), 3.52 - 3.29 (m, 2H), 3.25 - 3.08 (m, 3H), 3.06 - 2.96 (m, 2H), 2.91 - 2.77 (m, 1H), 2.71-2.68 (m, 1H), 2.52 (s, 3H), 2.35-2.30 (m, 3H)。LCMS: MSm/z =871.4[M+H]+ 。Step 10: Synthesis of compound 8-11. Compound 8-10 (0.45 g, 590.76 µmol, 1 eq ) was added to N,N-dimethylformamide (5 mL), followed by N,N-diiso Propylethylamine (229.05 mg, 1.77 mmol, 308.69 µL, 3 eq ) and compound 1-10A (306.37 mg, 1.18 mmol, 2 eq , HCl) were reacted at 50°C for 2 hr. The reaction solution was poured into 20 mL of water, filtered, and the filter cake was dissolved in 20 mL of methyl tertiary butyl ether, washed with 20 mL of saturated brine, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain compound 8-11. 1 H NMR (400MHz, CDCl3) δ = 7.44-7.32 (m, 5H), 7.16-7.13 (m, 4H), 6.85-6.82 (m, 4H), 6.63 (d, J = 7.6 Hz, 1H), 5.21 -5.15 (m, 2H), 4.80-4.66 (m, 3H), 4.39-4.22 (m, 4H), 3.93-3.88 (m, 1H), 3.80 (s, 6H), 3.71-3.55 (m, 1H) , 3.52-3.29 (m, 2H), 3.25-3.08 (m, 3H), 3.06-2.96 (m, 2H), 2.91-2.77 (m, 1H), 2.71-2.68 (m, 1H), 2.52 (s, 3H), 2.35-2.30 (m, 3H). LCMS: MS m/z =871.4 [M+H] + .
步驟11:化合物8-12的合成 將化合物8-11(580.00 mg, 665.94 µmol, 1eq )加入到無水二氯甲烷 (6 mL)中,加入間氯過氧苯甲酸 (359.13 mg, 1.66 mmol, 80% 質量含量, 2.5eq ),25℃反應0.5 hr,將反應液倒入20 mL的硫代硫酸鈉溶液(10%)中後,加入10 mL二氯甲烷萃取,有機相加入無水硫酸鈉乾燥後,過濾,濾液在45℃下進行減壓旋蒸,粗品利用過柱純化(石油醚:乙酸乙酯=100:0-1:1,石油醚:乙酸乙酯=1:1),得到化合物8-12。1 H NMR (400MHz, CDCl3) δ = 7.40-7.37 (m, 5H), 7.17-7.12 (m, 4H), 6.86-6.82 (m, 4H), 6.67-6.64 (d,J = 8.4 Hz, 1H), 5.19 (s, 2H), 4.86 - 4.79 (m, 2H), 4.71-4.63 (m, 1H), 4.35 - 4.24 (m, 4H), 3.82-3.81 (m, 1H), 3.80 (s, 6H), 3.64 - 3.50 (m, 2H), 3.46 - 3.33 (m, 2H), 3.30 - 3.27 (m, 4H), 3.25 - 3.11 (m, 3H), 2.71-2.65 (m, 1H), 2.52-2.45 (m, 1H), 2.38-2.30 (m, 3H)。LCMS: MSm/z =903.3[M+H]+ 。Step 11: Synthesis of compound 8-12. Compound 8-11 (580.00 mg, 665.94 µmol, 1 eq ) was added to anhydrous dichloromethane (6 mL), and m-chloroperoxybenzoic acid (359.13 mg, 1.66 mmol, 80% mass content, 2.5 eq ), react at 25°C for 0.5 hr, pour the reaction solution into 20 mL of sodium thiosulfate solution (10%), add 10 mL of dichloromethane for extraction, and dry the organic phase with anhydrous sodium sulfate After filtration, the filtrate was subjected to reduced pressure rotary evaporation at 45°C, and the crude product was purified by column (petroleum ether: ethyl acetate=100:0-1:1, petroleum ether: ethyl acetate=1:1) to obtain the compound 8-12. 1 H NMR (400MHz, CDCl3) δ = 7.40-7.37 (m, 5H), 7.17-7.12 (m, 4H), 6.86-6.82 (m, 4H), 6.67-6.64 (d, J = 8.4 Hz, 1H) , 5.19 (s, 2H), 4.86-4.79 (m, 2H), 4.71-4.63 (m, 1H), 4.35-4.24 (m, 4H), 3.82-3.81 (m, 1H), 3.80 (s, 6H) , 3.64-3.50 (m, 2H), 3.46-3.33 (m, 2H), 3.30-3.27 (m, 4H), 3.25-3.11 (m, 3H), 2.71-2.65 (m, 1H), 2.52-2.45 ( m, 1H), 2.38-2.30 (m, 3H). LCMS: MS m/z =903.3 [M+H] + .
步驟12:化合物8-13的合成 將1-11A(117.35 mg, 1.02 mmol, 120.98 µL, 4eq )加入到二氧六環(5 mL)中,降溫至0-5℃,加入三級丁醇鈉 (97.91 mg, 1.02 mmol, 4eq ),反應10 min,加入化合物8-12(230.00 mg, 254.72 µmol, 1eq )的甲苯(1 mL) 溶液,反應0.5 hr,將反應液加入到20 mL飽和氯化銨中,利用10 mL x 2的乙酸乙酯萃取,合併有機相,20 mL飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮,粗品利用過柱純化(石油醚:乙酸乙酯=1:1-0:1,二氯甲烷:甲醇=100:0-10:1,二氯甲烷:甲醇=10:1),得到化合物8-13。LCMS: MSm/z =938.2[M+H]+ 。Step 12: Synthesis of compound 8-13. Add 1-11A (117.35 mg, 1.02 mmol, 120.98 µL, 4 eq ) to dioxane (5 mL), reduce the temperature to 0-5°C, and add tertiary butanol Sodium (97.91 mg, 1.02 mmol, 4 eq ), react for 10 min, add compound 8-12 (230.00 mg, 254.72 µmol, 1 eq ) in toluene (1 mL) solution, react for 0.5 hr, add the reaction solution to 20 mL In saturated ammonium chloride, extracted with 10 mL x 2 ethyl acetate, combined the organic phases, washed with 20 mL saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by column (petroleum ether: ethyl acetate = 1 :1-0:1, dichloromethane:methanol=100:0-10:1, dichloromethane:methanol=10:1) to obtain compound 8-13. LCMS: MS m/z =938.2 [M+H] + .
步驟13:化合物8-14的合成 將化合物8-13 (0.15 g, 159.91 µmol, 1eq )加入到無水二氯甲烷 (5 mL)中,加入三氟乙酸 (0.5 mL),25℃反應2.5 hr,將反應液加入到10 mL飽和碳酸氫鈉溶液中,利用5 mL x 2的二氯甲烷萃取,合併有機相,無水硫酸鈉乾燥過濾後濃縮,得到化合物8-14。LCMS: MSm/z =698.2[M+H]+ 。Step 13: Synthesis of compound 8-14 Add compound 8-13 (0.15 g, 159.91 µmol, 1 eq ) to anhydrous dichloromethane (5 mL), add trifluoroacetic acid (0.5 mL), and react at 25°C for 2.5 hr , The reaction solution was added to 10 mL saturated sodium bicarbonate solution, extracted with 5 mL x 2 dichloromethane, the organic phases were combined, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain compound 8-14. LCMS: MS m/z = 698.2 [M+H] + .
步驟14:化合物8-15的合成 將化合物8-14(0.17 g, 243.65 µmol, 1eq )加入到無水甲醇(2 mL)和無水四氫呋喃 (2 mL)中,加入鈀碳 (0.15 g, 10% 質量含量) ,在氫氣(15 psi)25℃反應0.5 hr。反應液直接過濾,回收催化劑,濾液濃縮得到黃色固體狀的化合物8-15。 LCMS: MSm/z =564.2[M+H]+ 。Step 14: Synthesis of compound 8-15. Compound 8-14 (0.17 g, 243.65 µmol, 1 eq ) was added to anhydrous methanol (2 mL) and anhydrous tetrahydrofuran (2 mL), and palladium on carbon (0.15 g, 10% Mass content), react in hydrogen (15 psi) at 25°C for 0.5 hr. The reaction liquid was directly filtered, the catalyst was recovered, and the filtrate was concentrated to obtain compound 8-15 as a yellow solid. LCMS: MS m/z = 564.2 [M+H] + .
步驟15:化合物8A和8B的合成 將化合物8-15 (60 mg, 106.46 µmol, 1eq ) 和2-氟丙烯酸 (11.50 mg, 127.75 µmol, 1.2 eq) 和2-(7-氮雜苯並三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯 (60.72 mg, 159.69 µmol, 1.5 eq) 加入到N,N二甲基甲醯胺(1 mL)中,加入N,N-二異丙基乙胺 (41.28 mg, 319.38 µmol, 55.63 µL, 3eq ),25℃反應0.5 hr,將反應液加入到10 mL飽和氯化銨中,利用5 mL x 2的乙酸乙酯萃取,合併有機相,利用5 mL x 2的飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後濃縮得到化合物8-16,使用高效液相層析管柱分離純化(層析管柱: Phenomenex Gemini-NX 150*30mm*5µm;流動相: [H2 O(0.1%TFA)-ACN];乙腈%: 20%-50%,9min),真空下濃縮後,加入5mL去離子水和0.5mL乙腈,加入2滴1M的鹽酸溶液,真空下濃縮得到化合物8A的鹽酸鹽(出峰時間:1.379 min)。SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:80.82%。LCMS:MSm/z =636.4[M+H]+ ;得到化合物8B的鹽酸鹽(出峰時間:1.789 min)。SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:75.56%。1 H NMR (400MHz, CDCl3 ) δ = 6.59 (d,J = 8.4 Hz, 1H), 5.50 - 5.33 (m, 1H), 5.29 - 5.16 (m, 2H), 4.82 - 4.69 (m, 2H), 4.39 (dd,J =5.2, 10.8 Hz, 1H), 4.16 (dd,J =6.8, 10.4 Hz, 1H), 4.04 (s, 2H), 3.94 (d,J = 14.0 Hz, 1H), 3.68 (d,J = 11.6 Hz, 1H), 3.50 - 3.32 (m, 2H), 3.10 (br t,J = 7.2 Hz, 1H), 3.05 - 2.94 (m, 2H), 2.79 (br d,J = 7.2 Hz, 2H), 2.71-2.62 (m, 1H), 2.48 (s, 3H), 2.39 (q,J = 4.0 Hz, 3H), 2.32 - 2.22 (m, 1H), 2.11 - 1.99 (m, 1H), 1.93 - 1.66 (m, 6H)。LCMS: MSm/z =636.4[M+H]+ 。Step 15: Synthesis of Compounds 8A and 8B Compound 8-15 (60 mg, 106.46 µmol, 1 eq ) and 2-fluoroacrylic acid (11.50 mg, 127.75 µmol, 1.2 eq) and 2-(7-azabenzotriazole) Nitrozole)-N,N,N',N'-tetramethylurea hexafluorophosphate (60.72 mg, 159.69 µmol, 1.5 eq) was added to N,N dimethylformamide (1 mL) and added N,N-Diisopropylethylamine (41.28 mg, 319.38 µmol, 55.63 µL, 3 eq ), react at 25°C for 0.5 hr, add the reaction solution to 10 mL saturated ammonium chloride, and use 5 mL x 2 acetic acid Extract with ethyl ester, combine the organic phases, wash with 5 mL x 2 saturated brine, dry and filter with anhydrous sodium sulfate to obtain compound 8-16, which is separated and purified by high performance liquid chromatography column (chromatographic column: Phenomenex Gemini -NX 150*30mm*5µm; mobile phase: [H 2 O(0.1%TFA)-ACN]; acetonitrile%: 20%-50%, 9min), after concentration under vacuum, add 5mL deionized water and 0.5mL acetonitrile , Add 2 drops of 1M hydrochloric acid solution, concentrate under vacuum to obtain the hydrochloride salt of compound 8A (peak time: 1.379 min). SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (methanol, containing 0.05% diisopropylamine amine); gradient: B%=5~50% , 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 80.82%. LCMS: MS m/z =636.4[M+H] + ; Obtain the hydrochloride salt of compound 8B (peak Time: 1.789 min). SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (methanol, containing 0.05% diisopropylamine amine); gradient: B %=5~50%, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 75.56%. 1 H NMR (400MHz, CDCl 3 ) δ = 6.59 (d, J = 8.4 Hz , 1H), 5.50-5.33 (m, 1H), 5.29-5.16 (m, 2H), 4.82-4.69 (m, 2H), 4.39 (dd, J =5.2, 10.8 Hz, 1H), 4.16 (dd, J =6.8, 10.4 Hz, 1H), 4.04 (s, 2H), 3.94 (d, J = 14.0 Hz, 1H), 3.68 (d, J = 11.6 Hz, 1H), 3.50-3.32 (m, 2H), 3.10 (br t, J = 7.2 Hz, 1H), 3.05-2.94 (m, 2H), 2.79 (br d, J = 7.2 Hz, 2H), 2.71-2.62 (m, 1H), 2.48 (s, 3H), 2.39 (q, J = 4.0 Hz, 3H), 2.32-2.22 (m, 1H), 2.11-1.99 (m, 1H), 1.93-1.66 (m, 6H). LCMS: MS m/z =636.4[M+ H] + .
實施例9
步驟2:化合物9-2的合成 將N,N-二甲基甲醯胺 (6 mL)加入到乾燥的反應瓶中,再加入化合物8-10 (0.55 g, 722.04 µmol, 1 eq),N,N-二異丙基乙胺 (279.95 mg, 2.17 mmol, 377.29 uL, 3 eq) 和化合物9-1A (289.22 mg, 1.44 mmol, 2 eq),反應體系在50 °C和氮氣保護的條件下反應50分鐘,補加化合物9-1A (50 mg)繼續反應0.5小時,TLC (石油醚:乙酸乙酯=3:1)表明原料消失,出現新點。反應體系降溫至室溫後 (15 °C),將反應液加入到飽和氯化銨溶液 (30 mL)中,用甲基三級丁基醚 (10 mL x 2)萃取,合併有機相,用飽和食鹽水 (20 mL x 2)洗滌,無水硫酸鈉乾燥過濾後減壓濃縮,未純化,直接用於下一步。得到化合物9-2。LCMS m/z =812.4[M+H]+ 。Step 2: Synthesis of compound 9-2. Add N,N-dimethylformamide (6 mL) into a dry reaction flask, and then add compound 8-10 (0.55 g, 722.04 µmol, 1 eq), N ,N-Diisopropylethylamine (279.95 mg, 2.17 mmol, 377.29 uL, 3 eq) and compound 9-1A (289.22 mg, 1.44 mmol, 2 eq), the reaction system is at 50 °C under nitrogen protection After reacting for 50 minutes, compound 9-1A (50 mg) was added and the reaction was continued for 0.5 hours. TLC (petroleum ether: ethyl acetate=3:1) indicated that the raw materials disappeared and new spots appeared. After the reaction system was cooled to room temperature (15 °C), the reaction solution was added to saturated ammonium chloride solution (30 mL), extracted with methyl tertiary butyl ether (10 mL x 2), and the organic phases were combined and used Washed with saturated brine (20 mL x 2), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure without purification, and used directly in the next step. Compound 9-2 was obtained. LCMS m/z = 812.4 [M+H] + .
步驟3:化合物9-3的合成 將二氯甲烷 (10 mL)加入到乾燥的反應瓶中,再加入化合物9-2 (0.65 g, 800.57 µmol, 1 eq)和間氯過氧苯甲酸 (207.23 mg, 960.68 µmol, 80% 純度, 1.2 eq) ,反應體系在15°C的條件下反應0.5小時。將反應液倒入到水 (20 mL)中,加入硫代硫酸鈉溶液 (20 mL,10%)後,用澱粉-KI試紙檢測成陰性後,加入二氯甲烷 (20 mL)萃取,有機相加入無水硫酸鈉乾燥後過濾,濾液在40°C下進行減壓濃縮得到粗品,粗品根據TLC (石油醚:乙酸乙酯=0:1,RF=0.53)過柱純化(石油醚:乙酸乙酯=3:1到0:1),得到化合物9-3。1 H NMR (400 MHz,CDCl3 ) δ ppm 7.16 (d, J = 7.60 Hz, 4 H) 6.84 (d, J = 8.40 Hz, 4 H) 6.66 (s, 1 H) 5.26 (d, J = 10.42 Hz, 1 H), 4.85 - 4.68 (m, 2 H) 4.39 - 4.20 (m, 4 H) 4.09 - 3.90 (m, 2 H) 3.89 - 3.67 (m, 7 H) 3.66 - 3.42 (m, 2 H) 3.40 - 3.16 (m, 2 H) 3.14 - 2.75 (m, 4 H) 2.34 (d, J = 4.00 Hz, 3 H) 1.49 (s, 9 H) 1.43 - 1.37 (m, 2 H) 1.19 (m, 2 H), LCMS m/z =828.2[M+H]+ 。Step 3: Synthesis of compound 9-3. Dichloromethane (10 mL) was added to a dry reaction flask, followed by compound 9-2 (0.65 g, 800.57 µmol, 1 eq) and m-chloroperoxybenzoic acid (207.23). mg, 960.68 µmol, 80% purity, 1.2 eq), the reaction system is reacted at 15°C for 0.5 hours. Pour the reaction solution into water (20 mL), add sodium thiosulfate solution (20 mL, 10%), test negative with starch-KI test paper, add dichloromethane (20 mL) to extract, and extract the organic phase Add anhydrous sodium sulfate to dry and filter. The filtrate was concentrated under reduced pressure at 40°C to obtain a crude product. The crude product was purified by TLC (petroleum ether: ethyl acetate=0:1, RF=0.53) by column (petroleum ether: ethyl acetate) =3:1 to 0:1) to obtain compound 9-3. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.16 (d, J = 7.60 Hz, 4 H) 6.84 (d, J = 8.40 Hz, 4 H) 6.66 (s, 1 H) 5.26 (d, J = 10.42 Hz, 1 H), 4.85-4.68 (m, 2 H) 4.39-4.20 (m, 4 H) 4.09-3.90 (m, 2 H) 3.89-3.67 (m, 7 H) 3.66-3.42 (m, 2 H) ) 3.40-3.16 (m, 2 H) 3.14-2.75 (m, 4 H) 2.34 (d, J = 4.00 Hz, 3 H) 1.49 (s, 9 H) 1.43-1.37 (m, 2 H) 1.19 (m , 2 H), LCMS m/z = 828.2 [M+H] + .
步驟4:化合物9-4的合成 將甲苯 (6 mL)加入到乾燥的反應瓶中,再加入化合物9-3A (289.51 mg, 2.17 mmol, 28.68 µL, 4 eq),反應體系降溫至0°C,加入三級丁醇鈉 (208.93 mg, 2.17 mmol, 4 eq) ,反應體系在0-5°C的條件下反應10分鐘,加入含有化合物9-3 (0.45 g, 543.53 µmol, 1 eq) 的甲苯 (2 mL)溶液 ,反應體系在0-5°C的條件下反應0.5小時。反應液用的飽和氯化銨 (20 mL x 2)洗滌,再用飽和食鹽水 (20 mL)洗滌,無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步,得到化合物9-4。1 H NMR (400 MHz, CDCl3 ) δ ppm 7.15 (d, J = 7.60 Hz, 4 H) 6.84 (d, J = 8.40 Hz, 4 H) 6.62 (d, J = 7.20 Hz, 1 H) 5.29 - 5.04 (m, 3 H) 4.29 (d, J = 14.80 Hz, 4 H) 3.80 (s, 6 H) 3.44 - 3.34 (m, 2 H) 3.30 - 3.21 (m, 1 H) 3.06 - 2.79 (m, 2 H) 2.68 - 2.52 (m, 5 H) 2.47 (s, 3 H) 2.42 - 2.27 (m, 5 H) 2.25 - 2.08 (m, 5 H) 1.49 (s, 9 H) 1.38 (d, J = 6.40 Hz, 2 H) 1.14 (d, J = 6.80 Hz, 1 H)。LCMS m/z =897.3[M+H]+ 。Step 4: Synthesis of compound 9-4. Toluene (6 mL) was added to a dry reaction flask, and compound 9-3A (289.51 mg, 2.17 mmol, 28.68 µL, 4 eq) was added, and the reaction system was cooled to 0°C , Add tertiary butoxide sodium (208.93 mg, 2.17 mmol, 4 eq), react the reaction system at 0-5°C for 10 minutes, add compound 9-3 (0.45 g, 543.53 µmol, 1 eq) Toluene (2 mL) solution, the reaction system was reacted at 0-5°C for 0.5 hours. The reaction solution was washed with saturated ammonium chloride (20 mL x 2), then with saturated brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and used directly in the next step without purification to obtain compound 9-4 . 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.15 (d, J = 7.60 Hz, 4 H) 6.84 (d, J = 8.40 Hz, 4 H) 6.62 (d, J = 7.20 Hz, 1 H) 5.29- 5.04 (m, 3 H) 4.29 (d, J = 14.80 Hz, 4 H) 3.80 (s, 6 H) 3.44-3.34 (m, 2 H) 3.30-3.21 (m, 1 H) 3.06-2.79 (m, 2 H) 2.68-2.52 (m, 5 H) 2.47 (s, 3 H) 2.42-2.27 (m, 5 H) 2.25-2.08 (m, 5 H) 1.49 (s, 9 H) 1.38 (d, J = 6.40 Hz, 2 H) 1.14 (d, J = 6.80 Hz, 1 H). LCMS m/z = 897.3 [M+H] + .
步驟5:化合物9-5的合成 將二氯甲烷 (15 mL)加入到乾燥的反應瓶中,再加入化合物9-4 (0.6g, 668.91 µmol, 1 eq)和三氟乙酸 (3 mL) ,反應體系在15°C的條件下反應2.5小時,補加三氟乙酸 (0.5 mL),繼續反應1小時,繼續補加三氟乙酸 (0.5 mL),反應1小時。將反應液緩慢加入到飽和碳酸氫鈉溶液 (80 mL)中,用二氯甲烷 (30 mL x 2)萃取,合併有機相,無水硫酸鈉乾燥過濾後減壓濃縮得到粗品,粗品根據TLC(二氯甲烷:甲醇=5:1)過柱純化(二氯甲烷:甲醇=100:0-1:1),得到化合物9-5。1 H NMR (400 MHz,CDCl3 ) δ ppm 6.59 (d, J = 8.40 Hz, 1 H) 5.29 - 5.07 (m, 2 H) 4.75 - 4.67 (m, 1 H) 4.52 - 4.38 (m, 1 H) 4.32 - 4.16 (m, 1 H) 4.08 - 3.87 (m, 3 H) 3.66 - 3.26 (m, 4 H) 3.25 - 2.8 (m, 6 H) 2.72 - 2.59 (m, 1 H) 2.54 (d, J = 2.00 Hz, 3 H) 2.44 - 2.26 (m, 3H) 2.11 - 1.86 (m, 1 H) 1.52 (d, J = 6.80 Hz, 1 H) 1.26 (d, J = 6.80 Hz, 2 H)。LCMS m/z =557.3[M+H]+ 。Step 5: Synthesis of compound 9-5. Add dichloromethane (15 mL) to a dry reaction flask, then add compound 9-4 (0.6g, 668.91 µmol, 1 eq) and trifluoroacetic acid (3 mL), The reaction system was reacted at 15°C for 2.5 hours, and trifluoroacetic acid (0.5 mL) was added, and the reaction was continued for 1 hour, and trifluoroacetic acid (0.5 mL) was continued to be added, and the reaction was continued for 1 hour. The reaction solution was slowly added to saturated sodium bicarbonate solution (80 mL), extracted with dichloromethane (30 mL x 2), the organic phases were combined, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain the crude product. The crude product was obtained according to TLC (two Chloromethane: methanol = 5:1) was purified by column (dichloromethane: methanol = 100:0-1:1) to obtain compound 9-5. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 6.59 (d, J = 8.40 Hz, 1 H) 5.29-5.07 (m, 2 H) 4.75-4.67 (m, 1 H) 4.52-4.38 (m, 1 H ) 4.32-4.16 (m, 1 H) 4.08-3.87 (m, 3 H) 3.66-3.26 (m, 4 H) 3.25-2.8 (m, 6 H) 2.72-2.59 (m, 1 H) 2.54 (d, J = 2.00 Hz, 3 H) 2.44-2.26 (m, 3H) 2.11-1.86 (m, 1 H) 1.52 (d, J = 6.80 Hz, 1 H) 1.26 (d, J = 6.80 Hz, 2 H). LCMS m/z = 557.3 [M+H] + .
步驟6:化合物9A和9B的合成 將二氯甲烷 (5 mL)加入到乾燥的反應瓶中,再加入丙烯酸 (21.75 mg, 301.85 µmol, 20.72 µL, 1.2 eq) 和N,N-二異丙基乙胺 (97.53 mg, 754.62 µmol, 131.44 µL, 3 eq)。反應體系降溫至-60°C,加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽 (114.77 mg, 301.85 µmol, 1.2 eq),反應體系在-60°C的條件下反應10分鐘,再加入化合物9-5 (0.14 g, 251.54 µmol, 1 eq) ,繼續反應1小時。將反應液用二氯甲烷(10 mL)稀釋,用飽和氯化銨(10 mL x 2)溶液洗滌,用無水硫酸鈉乾燥過濾後減壓濃縮。使用高效液相層析管柱分離純化方法為{(層析管柱: Phenomenex luna C18 80*40mm*3 µm;流動相: [H2 O(0.04%HCl)-ACN];乙腈%: 20%-32%,7min] },凍乾後根據SFC進行手性分離方法為{(層析管柱: DAICEL CHIRALCEL OD(250mm*30mm,10µm);流動相: [0.1%NH3 H2 O MEOH];MeOH%: 60%-60%,9min)}。得到化合物9A(手性管柱出峰時間:1.594 min)SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:100%。1 H NMR (400 MHz, CDCl3 ) δ ppm 6.71 - 6.49 (m, 2 H) 6.42 - 6.28 (m, 1 H) 5.77 (d, J = 10.80 Hz, 1 H) 5.50 - 5.04 (m, 3 H) 4.71 (s, 3 H) 4.49 - 4.22 (m, 2 H) 4.03 (s, 3 H) 3.78 (d, J = 9.20 Hz, 1 H) 3.64 (s, 1 H) 3.51 - 3.17 (m, 4 H) 3.14 - 3.00 (m, 4 H) 2.64 - 2.48 (m, 1 H) 2.40 ( d, J = 4.00 Hz, 4 H) 1.16 (d, J = 10.40 Hz, 3 H)。LCMS m/z =611.3[M+H]+ 。Step 6: Synthesis of compounds 9A and 9B. Add dichloromethane (5 mL) to a dry reaction flask, then add acrylic acid (21.75 mg, 301.85 µmol, 20.72 µL, 1.2 eq) and N,N-diisopropyl Ethylamine (97.53 mg, 754.62 µmol, 131.44 µL, 3 eq). The reaction system was cooled to -60°C, and O-(7-azabenzotriazole-1-YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (114.77 mg, 301.85 µmol) was added , 1.2 eq), the reaction system was reacted at -60°C for 10 minutes, then compound 9-5 (0.14 g, 251.54 µmol, 1 eq) was added, and the reaction was continued for 1 hour. The reaction solution was diluted with dichloromethane (10 mL), washed with saturated ammonium chloride (10 mL x 2) solution, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The separation and purification method using high performance liquid chromatography column is {(chromatographic column: Phenomenex luna C18 80*40mm*3 µm; mobile phase: [H 2 O(0.04%HCl)-ACN]; acetonitrile%: 20% -32%,7min] }, the chiral separation method according to SFC after lyophilization is {(chromatographic column: DAICEL CHIRALCEL OD(250mm*30mm,10µm); mobile phase: [0.1%NH 3 H 2 O MEOH] ;MeOH%: 60%-60%, 9min)}. Obtain compound 9A (chiral column peak time: 1.594 min) SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3μm; flow Phase: A (CO2) and B (methanol, containing 0.05% diisopropylamine amine); Gradient: B%=5~50%, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity :100%. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 6.71-6.49 (m, 2 H) 6.42-6.28 (m, 1 H) 5.77 (d, J = 10.80 Hz, 1 H) 5.50-5.04 ( m, 3 H) 4.71 (s, 3 H) 4.49-4.22 (m, 2 H) 4.03 (s, 3 H) 3.78 (d, J = 9.20 Hz, 1 H) 3.64 (s, 1 H) 3.51-3.17 (m, 4 H) 3.14-3.00 (m, 4 H) 2.64-2.48 (m, 1 H) 2.40 (d, J = 4.00 Hz, 4 H) 1.16 (d, J = 10.40 Hz, 3 H). LCMS m/z =611.3[M+H] + .
化合物9B(手性出峰時間:1.903 min)SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:100%。1 H NMR (400 MHz, CDCl3 ) δ ppm 6.69 - 6.54 (m, 2 H) 6.42 - 6.30 (m, 1 H) 5.77 (d, J = 10.80 Hz, 1 H) 5.47 - 5.01 (m, 3 H) 4.71 (s, 3 H) 4.49 - 4.23 (m, 2 H) 4.03 (s, 3 H) 3.94 - 3.72 (m, 1 H) 3.64 (s, 1 H) 3.53 - 3.22 (m, 4 H) 3.14 - 3.01 (m, 4 H) 2.62 - 2.49 (m, 1 H) 2.40 (d, J = 4.00 Hz, 4 H) 1.16 (d, J = 10.40 Hz, 3 H)。LCMS m/z =611.3[M+H]+ 。Compound 9B (chiral peak time: 1.903 min) SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (methanol, containing 0.05% diisopropylamine) Amine); Gradient: B%=5~50%, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 100%. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 6.69 -6.54 (m, 2 H) 6.42-6.30 (m, 1 H) 5.77 (d, J = 10.80 Hz, 1 H) 5.47-5.01 (m, 3 H) 4.71 (s, 3 H) 4.49-4.23 (m , 2 H) 4.03 (s, 3 H) 3.94-3.72 (m, 1 H) 3.64 (s, 1 H) 3.53-3.22 (m, 4 H) 3.14-3.01 (m, 4 H) 2.62-2.49 (m , 1 H) 2.40 (d, J = 4.00 Hz, 4 H) 1.16 (d, J = 10.40 Hz, 3 H). LCMS m/z =611.3[M+H] + .
實施例10


步驟2:化合物10-2的合成 將化合物10-1A (2 g, 3.18 mmol, 1 eq) 溶於二氯甲烷(20 mL) 中,加入N,N-二異丙基乙胺 (1.23 g, 9.53 mmol, 1.66 mL, 3 eq),降溫至0~10°C,將三氟甲磺酸酐 (1.34 g, 4.76 mmol, 786.11 µL, 1.5 eq)緩慢加入到體系中,在此溫度下反應15min。倒入飽和氯化銨水溶液(15 mL),分液,水相用二氯甲烷(15 mL x 2)萃取,合併有機相,無水硫酸鈉乾燥,過濾,濃縮得到粗品。粗品根據TLC(PE/EtOAc=10/1 )管柱層析(PE/ETOAC=100/1~0/1)得到化合物10-2。LCMS m/z =762.2[M+H]+ 。Step 2: Synthesis of compound 10-2. Compound 10-1A (2 g, 3.18 mmol, 1 eq) was dissolved in dichloromethane (20 mL), and N,N-diisopropylethylamine (1.23 g, 9.53 mmol, 1.66 mL, 3 eq), lower the temperature to 0~10°C, slowly add trifluoromethanesulfonic anhydride (1.34 g, 4.76 mmol, 786.11 µL, 1.5 eq) into the system, and react at this temperature for 15 min. Pour into saturated aqueous ammonium chloride solution (15 mL), separate the layers, extract the aqueous phase with dichloromethane (15 mL x 2), combine the organic phases, dry with anhydrous sodium sulfate, filter, and concentrate to obtain the crude product. The crude product was subjected to TLC (PE/EtOAc=10/1) column chromatography (PE/ETOAC=100/1-0/1) to obtain compound 10-2. LCMS m/z = 762.2 [M+H] + .
步驟3:化合物10-3的合成 將N,N-二甲基甲醯胺 (2 mL)加入到一個乾燥的反應瓶中,再加入化合物10-2 (0.16 g, 210.05 µmol, 1 eq),N,N-二異丙基乙胺 (81.44 mg, 630.15 µmol, 109.76 µL, 3 eq) 和化合物10-2A (50.48 mg, 252.06 µmol, 1.2 eq),反應體系在50°C和氮氣保護的條件下反應1小時。TLC (石油醚:乙酸乙酯=3:1)表明原料消失,出現新點。向反應液中加入甲基三級丁基醚 (10 mL),用飽和氯化銨溶液 (20 mL x 2)洗滌後,再用飽和食鹽水(10 mL)洗滌,用無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步,得到化合物10-3。1 H NMR (400 MHz, CDCl3 ) δ ppm 7.12 (d, J = 8.80 Hz, 4 H) 6.81 (d, J = 8.80 Hz, 4 H) 6.60 (d, J = 8.80 Hz, 1 H) 5.19 (d, J = 8.00 Hz, 1 H) 4.72 (s, 2 H) 4.37 - 4.22 (m, 4 H) 3.88 (d, J = 13.2 Hz, 1 H) 3.77 (s, 6 H) 3.71 - 3.56 (dd, J = 12.80, 13.20 Hz, 2 H) 3.40 (dd, J = 12.40, 12.40 Hz, 1 H) 3.31 (m, 2 H) 3.20 (s, 1 H) 3.03 - 2.91 (m, 2 H) 2.50 (s, 3 H) 2.35 - 2.25 (m, 3 H) 1.46 (s, 9 H) 1.13 (d, J = 6.80 Hz, 3 H)。Step 3: Synthesis of compound 10-3. Add N,N-dimethylformamide (2 mL) into a dry reaction flask, and then add compound 10-2 (0.16 g, 210.05 µmol, 1 eq), N,N-Diisopropylethylamine (81.44 mg, 630.15 µmol, 109.76 µL, 3 eq) and compound 10-2A (50.48 mg, 252.06 µmol, 1.2 eq), the reaction system is at 50°C under nitrogen protection React for 1 hour. TLC (petroleum ether: ethyl acetate=3:1) indicates that the raw material disappeared and new spots appeared. Methyl tertiary butyl ether (10 mL) was added to the reaction solution, washed with saturated ammonium chloride solution (20 mL x 2), washed with saturated brine (10 mL), dried with anhydrous sodium sulfate and filtered It was concentrated under reduced pressure and used directly in the next step without purification to obtain compound 10-3. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.12 (d, J = 8.80 Hz, 4 H) 6.81 (d, J = 8.80 Hz, 4 H) 6.60 (d, J = 8.80 Hz, 1 H) 5.19 ( d, J = 8.00 Hz, 1 H) 4.72 (s, 2 H) 4.37-4.22 (m, 4 H) 3.88 (d, J = 13.2 Hz, 1 H) 3.77 (s, 6 H) 3.71-3.56 (dd , J = 12.80, 13.20 Hz, 2 H) 3.40 (dd, J = 12.40, 12.40 Hz, 1 H) 3.31 (m, 2 H) 3.20 (s, 1 H) 3.03-2.91 (m, 2 H) 2.50 ( s, 3 H) 2.35-2.25 (m, 3 H) 1.46 (s, 9 H) 1.13 (d, J = 6.80 Hz, 3 H).
步驟4:化合物10-4的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物10-3 (0.21 g, 258.64 µmol, 1 eq)和間氯過氧苯甲酸 (66.95 mg, 310.37 µmol, 80%純度, 1.2 eq) ,反應體系在15°C的條件下反應0.5小時。向反應液中加入硫代硫酸鈉 (15 mL,10%)溶液,澱粉- KI試紙檢測為陰性後,用二氯甲烷 (15 mL)萃取,無水硫酸鈉乾燥過濾後減壓濃縮得到粗品,粗品根據TLC (石油醚:乙酸乙酯=0:1)過柱純化(石油醚:乙酸乙酯=10:1到0:1)得到化合物10-4和10-4A。1 H NMR (400 MHz, CDCl3 ) δ ppm 7.20 (s, 4 H), 6.84 (d, J = 8.80 Hz, 4 H), 6.79~6.62(s,1 H), 5.24 (d, J = 10.80 Hz, 1 H), 4.87 - 4.74 (m, 2 H), 4.36 (s, 4 H), 3.99 - 3.83 (m, 3 H), 3.83 - 3.71 (m, 7 H), 3.62 - 3.43 (m, 2 H), 3.40 - 3.27 (m, 3 H), 3.22 - 3.06 (m, 2 H), 2.92 (d, J = 5.20 Hz, 1 H), 2.34 (s, 3 H) ,1.49 (s, 9 H), 1.16 (d, J = 6.80 Hz, 3 H)。LCMS m/z =828.2M+H]+ 。Step 4: Synthesis of compound 10-4 Dichloromethane (5 mL) was added to a dry reaction flask, then compound 10-3 (0.21 g, 258.64 µmol, 1 eq) and m-chloroperoxybenzoic acid ( 66.95 mg, 310.37 µmol, 80% purity, 1.2 eq), the reaction system was reacted at 15°C for 0.5 hours. Add sodium thiosulfate (15 mL, 10%) solution to the reaction solution. After the starch-KI test paper is negative, it is extracted with dichloromethane (15 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a crude product. According to TLC (petroleum ether: ethyl acetate=0:1), column purification (petroleum ether: ethyl acetate=10:1 to 0:1) was used to obtain compounds 10-4 and 10-4A. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.20 (s, 4 H), 6.84 (d, J = 8.80 Hz, 4 H), 6.79~6.62(s,1 H), 5.24 (d, J = 10.80 Hz, 1 H), 4.87-4.74 (m, 2 H), 4.36 (s, 4 H), 3.99-3.83 (m, 3 H), 3.83-3.71 (m, 7 H), 3.62-3.43 (m, 2 H), 3.40-3.27 (m, 3 H), 3.22-3.06 (m, 2 H), 2.92 (d, J = 5.20 Hz, 1 H), 2.34 (s, 3 H) ,1.49 (s, 9 H), 1.16 (d, J = 6.80 Hz, 3 H). LCMS m/z = 828.2M+H] + .
步驟3:化合物10-5的合成 將甲苯 (1 mL)加入到一個乾燥的反應瓶中,再加入化合物1-11A (38.95 mg, 338.19 µmol, 4 eq),反應體系降溫至0°C加入三級丁醇鈉 (32.50 mg, 338.19 µmol, 4 eq),反應10分鐘後,加入含有化合物10-4 (0.07 g, 84.55 µmol, 1 eq)和10-4A (71.35 mg, 84.55 µmol, 1 eq)混合物的甲苯混合溶液(1 mL),反應0.5小時。向反應液中加入10mL乙酸乙酯,然後分別用10mL的飽和氯化銨溶液和飽和食鹽水洗滌,有機相用無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步。得到化合物10-5。LCMS m/z =879.3[M+H]+ 。Step 3: Synthesis of compound 10-5. Toluene (1 mL) was added to a dry reaction flask, and compound 1-11A (38.95 mg, 338.19 µmol, 4 eq) was added. The reaction system was cooled to 0°C and three Grade sodium butoxide (32.50 mg, 338.19 µmol, 4 eq), after 10 minutes of reaction, add 10-4 (0.07 g, 84.55 µmol, 1 eq) and 10-4A (71.35 mg, 84.55 µmol, 1 eq) The toluene mixed solution (1 mL) of the mixture was reacted for 0.5 hours. 10 mL of ethyl acetate was added to the reaction solution, and then washed with 10 mL of saturated ammonium chloride solution and saturated brine, the organic phase was dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and used directly in the next step without purification. Compound 10-5 was obtained. LCMS m/z = 879.3 [M+H] + .
步驟4:化合物10-6的合成 將二氯甲烷 (5 mL)加入到乾燥的反應瓶中,再加入化合物10-5 (0.16 g,182.03 µmol, 1 eq)和三氟乙酸 (1.25 mL) ,反應體系在18°C的條件下攪拌1.5小時。補加三氟乙酸 (0.25 mL),繼續反應1.5小時,向反應液中加入水 (5 mL),收集水相,用飽和碳酸氫鈉溶液調節pH為8,用二氯甲烷 (20 mL x 2)萃取,合併有機相,用無水硫酸鈉乾燥過濾後減壓濃縮,未純化,直接用於下一步。得到化合物10-6。LCMS m/z =539.2[M+H]+ 。Step 4: Synthesis of compound 10-6. Add dichloromethane (5 mL) to a dry reaction flask, then add compound 10-5 (0.16 g, 182.03 µmol, 1 eq) and trifluoroacetic acid (1.25 mL), The reaction system was stirred at 18°C for 1.5 hours. Add additional trifluoroacetic acid (0.25 mL), continue the reaction for 1.5 hours, add water (5 mL) to the reaction solution, collect the aqueous phase, adjust the pH to 8 with saturated sodium bicarbonate solution, and use dichloromethane (20 mL x 2 ) Extract, combine the organic phases, dry with anhydrous sodium sulfate, filter, concentrate under reduced pressure, without purification, and directly use in the next step. Compound 10-6 was obtained. LCMS m/z = 539.2 [M+H] + .
步驟5:化合物10的合成
將二氯甲烷 (5 mL)加入到乾燥的反應瓶中,再加入化合物丙烯酸 (5.54 mg, 76.87 µmol, 5.28 µL, 2 eq),化合物10-6 (23 mg, 38.43 µmol,90% 純度, 1 eq)和N,N-二異丙基乙胺 (14.90 mg, 115.30 µmol, 20.08 µL, 3 eq),反應體系降溫至-60°C,再加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(17.54 mg,46.12 µmol, 1.2 eq) ,然後攪拌0.5小時。該反應與化合物10-6(19.49 mg)批次合併處理,向反應液中加入水 (5 ml),分液,有機相減壓濃縮,高效液相層析管柱分離,純化方法為{層析管柱: Phenomenex luna C18 80*40mm*3 µm;: [H2
O(0.04%HCl)-ACN]; ;乙腈%: 20%-40%,7min}得到化合物10。LCMS m/z =593.4[M+H]+
。Step 5: Synthesis of
實施例11

步驟2:化合物11-3的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物11-2 (0.20 g, 242.14 µmol, 1 eq)和間氯過氧苯甲酸 (62.68 mg, 290.57 µmol, 80% 純度, 1.2 eq),反應體系在15 °C的條件下反應0.5小時,與化合物11-2(50 mg)批次合併處理。向反應液中加入硫代硫酸鈉 (15 mL, 10%)溶液,澱粉-KI試紙檢測為陰性後,用二氯甲烷 (10 mL)萃取,無水硫酸鈉乾燥過濾後減壓濃縮粗品,粗品根據TLC (石油醚:乙酸乙酯=0:1)過柱純化(石油醚:乙酸乙酯=10:1到0:1)得到化合物11-3和化合物11-3A的混合物。1 H NMR (400 MHz, CDCl3 ) δ ppm 7.17 (d, J = 6.40 Hz, 4 H) 6.84 (d, J = 8.40 Hz, 4 H) 6.67 (s, 1 H) 5.23 (d, J = 11.20 Hz, 1 H) 4.90 - 4.74 (m, 2 H) 4.32 (d, J = 12.00 Hz, 4 H) 3.84 - 3.75 (m, 9 H) 3.68 - 3.39 (m, 3 H) 3.31 - 3.10 (m, 3 H) 2.89 (d, J=10.40 Hz, 2 H) 2.34 (d, J=3.60 Hz, 3 H) 1.49 (s, 9 H) 1.42 - 1.29 (m, 6 H)。LCMS m/z =842.2[M+H]+ 。Step 2: Synthesis of compound 11-3. Dichloromethane (5 mL) was added to a dry reaction flask, followed by compound 11-2 (0.20 g, 242.14 µmol, 1 eq) and m-chloroperoxybenzoic acid ( 62.68 mg, 290.57 µmol, 80% purity, 1.2 eq), the reaction system was reacted at 15 °C for 0.5 hours, and then combined with compound 11-2 (50 mg). Add sodium thiosulfate (15 mL, 10%) solution to the reaction solution. After the starch-KI test paper is negative, it is extracted with dichloromethane (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. TLC (petroleum ether: ethyl acetate=0:1) column purification (petroleum ether: ethyl acetate=10:1 to 0:1) to obtain a mixture of compound 11-3 and compound 11-3A. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.17 (d, J = 6.40 Hz, 4 H) 6.84 (d, J = 8.40 Hz, 4 H) 6.67 (s, 1 H) 5.23 (d, J = 11.20 Hz, 1 H) 4.90-4.74 (m, 2 H) 4.32 (d, J = 12.00 Hz, 4 H) 3.84-3.75 (m, 9 H) 3.68-3.39 (m, 3 H) 3.31-3.10 (m, 3 H) 2.89 (d, J=10.40 Hz, 2 H) 2.34 (d, J=3.60 Hz, 3 H) 1.49 (s, 9 H) 1.42-1.29 (m, 6 H). LCMS m/z = 842.2 [M+H] + .
步驟3:化合物11-4的合成 將甲苯 (1 mL)加入到一個乾燥的反應瓶中,再加入化合物1-11A (46.51 mg, 403.82 µmol, 4 eq),反應體系降溫至0°C加入三級丁醇鈉 (38.81 mg, 403.82 µmol, 4 eq),反應10分鐘後,加入含有化合物11-3 (0.085 g, 100.96 µmol, 1 eq)和11-3A(86.62 mg, 100.96 µmol, 1 eq)混合物的甲苯混合溶液 (1 mL),反應體系攪拌0.5小時。與化合物11-3(10 mg)批次合併處理。向反應液中加入10mL乙酸乙酯,然後分別用10mL的飽和氯化銨溶液和飽和食鹽水洗滌,用無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步,得到化合物11-4。LCMS m/z =893.4[M+H]+ 。Step 3: Synthesis of compound 11-4. Toluene (1 mL) was added to a dry reaction flask, and compound 1-11A (46.51 mg, 403.82 µmol, 4 eq) was added. The reaction system was cooled to 0°C and three Grade sodium butoxide (38.81 mg, 403.82 µmol, 4 eq), after 10 minutes of reaction, add compounds 11-3 (0.085 g, 100.96 µmol, 1 eq) and 11-3A (86.62 mg, 100.96 µmol, 1 eq) The mixture was a toluene mixed solution (1 mL), and the reaction system was stirred for 0.5 hours. Combined with compound 11-3 (10 mg) batches for processing. Add 10 mL of ethyl acetate to the reaction solution, then wash with 10 mL of saturated ammonium chloride solution and saturated brine, dry with anhydrous sodium sulfate, filter, concentrate under reduced pressure, and use it directly in the next step without purification to obtain compound 11-4 . LCMS m/z = 893.4 [M+H] + .
步驟4:化合物11-5的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物11-4 (0.13 g,145.57 µmol, 1 eq)和三氟乙酸 (1.25 mL) 中,反應體系在18°C的條件下反應1.5小時,補加三氟乙酸 (0.25 mL),繼續反應1.5小時,與11-4 (15mg)批次合併處理,向反應液中加入水 (5 mL),收集水相,用飽和碳酸氫鈉溶液調節pH為8,再用的二氯甲烷 (20 mL x 2)萃取,合併有機相,用無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步,得到化合物11-5。LCMS m/z =553.2[M+H]+ 。Step 4: Synthesis of compound 11-5. Dichloromethane (5 mL) was added to a dry reaction flask, followed by compound 11-4 (0.13 g, 145.57 µmol, 1 eq) and trifluoroacetic acid (1.25 mL) In the reaction system, the reaction system was reacted at 18°C for 1.5 hours, and trifluoroacetic acid (0.25 mL) was added, and the reaction was continued for 1.5 hours. The reaction was combined with 11-4 (15mg) batches, and water (5 mg) was added to the reaction solution. mL), collect the aqueous phase, adjust the pH to 8 with saturated sodium bicarbonate solution, and extract with dichloromethane (20 mL x 2). Combine the organic phases, dry with anhydrous sodium sulfate, filter, and concentrate under reduced pressure. Used in the next step to obtain compound 11-5. LCMS m/z = 553.2 [M+H] + .
步驟5:化合物11的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物丙烯酸 (14.08 mg, 195.44 µmol, 13.41 µL, 2 eq),化合物11-5 (60.00 mg, 97.72µmol, 90% 純度, 1 eq)和N,N-二異丙基乙胺 (37.89 mg, 293.16 µmol, 51.06 µL, 3 eq) ,反應體系降溫至-60°C。加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽 (44.59 mg, 117.26 µmol, 1.2 eq),然後攪拌0.5小時。該反應與化合物11-5(20 mg)批次合併處理,向反應液中加入水 (5 ml),分液,有機相減壓濃縮,高效液相層析管柱分離純化方法為{層析管柱: Phenomenex luna C18 80*40mm *3 µm;流動相: [H2 O (0.04%HCl)-ACN];乙腈%: 15%-40%,7min}得化合物11。1 H NMR (400 MHz, CD3 OD) δ = 6.92 - 6.75 (m, 1H), 6.74 - 6.70 (m, 1H), 6.33 - 6.24 (m, 1H), 5.88 - 5.77 (m, 1H), 5.28 - 5.18 (m, 1H), 4.82 - 4.63 (m, 2H), 4.56 - 4.43 (m, 1H), 4.42 - 4.23 (m, 1H), 4.01 - 3.81 (m, 2H), 3.79 - 3.59 (m, 2H), 3.57 - 3.41 (m, 1H), 3.32 - 3.20 (m, 5H), 3.17 - 3.02 (m, 3H), 2.84 (m, 2H), 2.51 - 2.35 (m, 3H), 2.31 - 1.99 (m, 3H), 1.49 - 1.29 (m, 6H)。LCMS m/z =607.5[M+H]+ 。Step 5: Synthesis of Compound 11 Dichloromethane (5 mL) was added to a dry reaction flask, followed by compound acrylic acid (14.08 mg, 195.44 µmol, 13.41 µL, 2 eq), compound 11-5 (60.00 mg, 97.72 µmol, 90% purity, 1 eq) and N,N-diisopropylethylamine (37.89 mg, 293.16 µmol, 51.06 µL, 3 eq), the reaction system was cooled to -60°C. Add O-(7-azabenzotriazole-1-YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (44.59 mg, 117.26 µmol, 1.2 eq), and then stir for 0.5 hours . The reaction was combined with compound 11-5 (20 mg) in batches. Water (5 ml) was added to the reaction solution, separated, and the organic phase was concentrated under reduced pressure. The high performance liquid chromatography column separation and purification method was {chromatography Column: Phenomenex luna C18 80*40mm *3 µm; mobile phase: [H 2 O (0.04%HCl)-ACN]; acetonitrile%: 15%-40%, 7min} to obtain compound 11. 1 H NMR (400 MHz, CD 3 OD) δ = 6.92-6.75 (m, 1H), 6.74-6.70 (m, 1H), 6.33-6.24 (m, 1H), 5.88-5.77 (m, 1H), 5.28 -5.18 (m, 1H), 4.82-4.63 (m, 2H), 4.56-4.43 (m, 1H), 4.42-4.23 (m, 1H), 4.01-3.81 (m, 2H), 3.79-3.59 (m, 2H), 3.57-3.41 (m, 1H), 3.32-3.20 (m, 5H), 3.17-3.02 (m, 3H), 2.84 (m, 2H), 2.51-2.35 (m, 3H), 2.31-1.99 ( m, 3H), 1.49-1.29 (m, 6H). LCMS m/z = 607.5 [M+H] + .
實施例12
步驟2:化合物12-3的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物12-2 (0.18 g, 221.70 µmol, 1 eq)和間氯過氧苯甲酸 (57.39 mg, 266.03 µmol, 80% 純度, 1.2 eq) ,反應體系在15°C的條件下反應0.5小時。與化合物12-2(30 mg)批次合併處理。向反應液中加入硫代硫酸鈉 (10 mL,10%)溶液,澱粉-KI試紙檢測為陰性後,用二氯甲烷 (3 mL)萃取,無水硫酸鈉乾燥過濾後減壓濃縮得到粗品,粗品根據TLC (石油醚:乙酸乙酯=0:1),過柱純化(石油醚:乙酸乙酯=10:1到0:1)得到化合物12-3。1 H NMR (400 MHz, CDCl3 ) δ ppm 7.17 (br s, 4 H) 6.84 (br d,J = 8.40 Hz, 4 H) 6.67 (br s, 1 H) 5.25 (br d,J = 9.60 Hz, 1 H) 4.89 - 4.64 (m, 2 H) 4.32 (br d,J = 10.40 Hz, 4 H) 4.09 - 3.86 (m, 3 H) 3.84 - 3.68 (m, 7 H) 3.64 - 3.51 (m, 1 H) 3.37 - 2.97 (m, 4 H) 2.95 - 2.81 (m, 3 H) 2.34 (br d,J = 3.60 Hz, 3 H) 1.49 (s, 9 H) 1.41 (br s, 3 H)。LCMS m/z =828.2[M+H]+ 。Step 2: Synthesis of compound 12-3. Dichloromethane (5 mL) was added to a dry reaction flask, followed by compound 12-2 (0.18 g, 221.70 µmol, 1 eq) and m-chloroperoxybenzoic acid ( 57.39 mg, 266.03 µmol, 80% purity, 1.2 eq), the reaction system is reacted at 15°C for 0.5 hours. Combined with compound 12-2 (30 mg) batches for processing. Add sodium thiosulfate (10 mL, 10%) solution to the reaction solution. After the starch-KI test paper is negative, it is extracted with dichloromethane (3 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a crude product. According to TLC (petroleum ether: ethyl acetate=0:1) and column purification (petroleum ether: ethyl acetate=10:1 to 0:1), compound 12-3 was obtained. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.17 (br s, 4 H) 6.84 (br d, J = 8.40 Hz, 4 H) 6.67 (br s, 1 H) 5.25 (br d, J = 9.60 Hz , 1 H) 4.89-4.64 (m, 2 H) 4.32 (br d, J = 10.40 Hz, 4 H) 4.09-3.86 (m, 3 H) 3.84-3.68 (m, 7 H) 3.64-3.51 (m, 1 H) 3.37-2.97 (m, 4 H) 2.95-2.81 (m, 3 H) 2.34 (br d, J = 3.60 Hz, 3 H) 1.49 (s, 9 H) 1.41 (br s, 3 H). LCMS m/z = 828.2 [M+H] + .
步驟3:化合物12-4的合成 將甲苯 (1 mL)加入到乾燥的反應瓶中,再加入化合物12-3A (66.52 mg, 471.06 µmol, 3 eq),反應體系降溫至0°C,再加入加入三級丁醇鈉 (45.27 mg, 471.06 µmol, 3 eq),攪拌10分鐘後,加入含有化合物12-3 (0.13 g, 157.02 µmol, 1 eq)的甲苯溶液(0.5 mL),攪拌0.5小時。與化合物12-3(20 mg)批次合併處理,向反應液中加入10mL乙酸乙酯,然後分別用10mL的飽和氯化銨溶液和飽和食鹽水洗滌,無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步,得到化合物12-4。LCMS m/z =905.3[M+H]+ 。Step 3: Synthesis of compound 12-4. Toluene (1 mL) was added to a dry reaction flask, compound 12-3A (66.52 mg, 471.06 µmol, 3 eq) was added, the reaction system was cooled to 0°C, and then added Sodium tertiary butoxide (45.27 mg, 471.06 µmol, 3 eq) was added, and after stirring for 10 minutes, a toluene solution (0.5 mL) containing compound 12-3 (0.13 g, 157.02 µmol, 1 eq) was added, and the mixture was stirred for 0.5 hours. Combine it with compound 12-3 (20 mg) in batches, add 10 mL of ethyl acetate to the reaction solution, wash with 10 mL of saturated ammonium chloride solution and saturated brine, dry and filter with anhydrous sodium sulfate and concentrate under reduced pressure. It was used directly in the next step without purification to obtain compound 12-4. LCMS m/z =905.3 [M+H] + .
步驟4:化合物12-5的合成 將二氯甲烷 (6 mL)加入到乾燥的反應瓶中,再加入化合物12-4 (0.18 g,198.89 µmol, 1 eq)和三氟乙酸 (1.5 mL) ,反應體系在18°C的條件下攪拌3.5小時。向反應液中加入水 (5 mL),收集水相,用飽和碳酸氫鈉溶液調節反應液的pH為8,用二氯甲烷 (20 mL x 2)萃取,合併有機相,無水硫酸鈉乾燥過濾後減壓濃縮,未純化直接用於下一步,得到化合物12-5。LCMS m/z =565.2[M+H]+ 。Step 4: Synthesis of compound 12-5. Add dichloromethane (6 mL) to a dry reaction flask, then add compound 12-4 (0.18 g, 198.89 µmol, 1 eq) and trifluoroacetic acid (1.5 mL), The reaction system was stirred at 18°C for 3.5 hours. Add water (5 mL) to the reaction solution, collect the aqueous phase, adjust the pH of the reaction solution to 8 with saturated sodium bicarbonate solution, extract with dichloromethane (20 mL x 2), combine the organic phases, dry and filter with anhydrous sodium sulfate Then it was concentrated under reduced pressure and used directly in the next step without purification to obtain compound 12-5. LCMS m/z = 565.2 [M+H] + .
步驟5:化合物12的合成 將二氯甲烷 (5 mL)加入到乾燥的反應瓶中開始攪拌,再加入化合物丙烯酸 (11.49 mg, 159.40 µmol, 10.94 µL, 2 eq),化合物12-5 (50 mg, 79.70 µmol,90% 純度, 1 eq)和N,N-二異丙基乙胺 (30.90 mg, 239.10 µmol, 41.65 µL, 3 eq),反應體系降溫至-60°C,加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(36.37 mg,95.64 µmol, 1.2 eq) ,然後攪拌0.5小時。該反應與化合物12-5(20 mg)批次合併處理。向反應液中加入水 (5 ml),分液,有機相用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品高效液相層析管柱分離純化{層析管柱: Phenomenex luna C18 80*40mm *3 µm;流動相: [H2 O(0.04%HCl)-ACN];乙腈%: 15%-40%,7min},得化合物12(出峰時間:1.509)。 SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi。光學純度:99.4%。1 H NMR (400 MHz, CD3 OD) δ = 6.89 - 6.71 (m, 1H), 6.71 - 6.65 (d,J = 8.8 Hz , 1H), 6.32 - 6.19 (m, 1H), 5.84 - 5.75 (m, 1H), 5.26 - 5.15 (m, 1H), 4.68 - 4.60 (m, 1H), 4.52 (s, 2H), 4.50 - 4.45 (m, 1H), 4.39 - 4.32 (m, 1H), 4.27 - 4.16 (m, 1H), 4.15 - 3.89 (m, 2H), 3.76 - 3.61 (m, 2H), 3.60 - 3.32 (m, 2H), 3.28 - 3.13 (m, 3H), 3.09 - 3.00 (m, 1H), 2.96 - 2.85 (m, 1H), 2.38 - 2.32 (m, 3H), 2.32 - 2.24 (m, 2H), 2.24 - 2.12 (m, 4H), 2.12 - 2.03 (m, 2H), 1.42 - 1.31 (m, 3H)。LCMS m/z =619.3[M+H]+ 。Step 5: Synthesis of compound 12 Dichloromethane (5 mL) was added to the dry reaction flask and stirred, then compound acrylic acid (11.49 mg, 159.40 µmol, 10.94 µL, 2 eq) was added, compound 12-5 (50 mg , 79.70 µmol, 90% purity, 1 eq) and N,N-diisopropylethylamine (30.90 mg, 239.10 µmol, 41.65 µL, 3 eq), the reaction system is cooled to -60°C, and O-(7 -Azabenzotriazole-1-YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (36.37 mg, 95.64 µmol, 1.2 eq), and then stirred for 0.5 hours. The reaction was combined with batches of compound 12-5 (20 mg). Add water (5 ml) to the reaction solution, separate the layers, dry and filter the organic phase with anhydrous sodium sulfate, and concentrate the filtrate under reduced pressure to obtain the crude product. The crude product is separated and purified by HPLC column {chromatography column: Phenomenex luna C18 80 *40mm *3 µm; mobile phase: [H 2 O(0.04%HCl)-ACN]; acetonitrile%: 15%-40%, 7min}, to obtain compound 12 (peak time: 1.509). SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (methanol, containing 0.05% diisopropylamine amine); gradient: B%=5~50% , 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi. Optical purity: 99.4%. 1 H NMR (400 MHz, CD 3 OD) δ = 6.89-6.71 (m, 1H), 6.71-6.65 (d, J = 8.8 Hz , 1H), 6.32-6.19 (m, 1H), 5.84-5.75 (m, 1H), 5.26-5.15 (m, 1H), 4.68-4.60 (m, 1H), 4.52 (s , 2H), 4.50-4.45 (m, 1H), 4.39-4.32 (m, 1H), 4.27-4.16 (m, 1H), 4.15-3.89 (m, 2H), 3.76-3.61 (m, 2H), 3.60 -3.32 (m, 2H), 3.28-3.13 (m, 3H), 3.09-3.00 (m, 1H), 2.96-2.85 (m, 1H), 2.38-2.32 (m, 3H), 2.32-2.24 (m, 2H), 2.24-2.12 (m, 4H), 2.12-2.03 (m, 2H), 1.42-1.31 (m, 3H). LCMS m/z =619.3[M+H] + .
實施例13
步驟2:化合物13-3的合成 將二氯甲烷 (10 mL)加入到一個乾燥的反應瓶中,再加入化合物13-2 (200.00 mg, 246.33 µmol, 1eq)開始攪拌,然後加入間氯過氧苯甲酸(60.01 mg, 295.59 µmol, 85% 純度, 1.2 eq),反應體系在25°C的條件下攪拌1小時。將反應液用二氯甲烷 (20 mL)稀釋,然後5%硫代硫酸鈉 (10 mL)溶液和飽和食鹽水 (10 mL)各洗滌一次,用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品TLC (石油醚 : 乙酸乙酯= 1:1),過柱純化(石油醚: 乙酸乙酯 =90:10到50:50)得到產物,得化合物13-3。LCMS m/z =828.3[M+H]+ 。Step 2: Synthesis of compound 13-3. Add dichloromethane (10 mL) to a dry reaction flask, then add compound 13-2 (200.00 mg, 246.33 µmol, 1eq) to start stirring, then add m-chloroperoxy Benzoic acid (60.01 mg, 295.59 µmol, 85% purity, 1.2 eq), the reaction system was stirred at 25°C for 1 hour. The reaction solution was diluted with dichloromethane (20 mL), and then washed once with 5% sodium thiosulfate (10 mL) solution and saturated brine (10 mL), dried with anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product , Crude product TLC (petroleum ether: ethyl acetate = 1:1), column purification (petroleum ether: ethyl acetate = 90:10 to 50:50) to obtain the product, compound 13-3. LCMS m/z = 828.3 [M+H] + .
步驟3:化合物13-4的合成 將甲苯 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物1-11A (112.68 mg, 978.35 µmol, 4.5 eq)開始攪拌,然後加入三級丁醇鈉 (94.02 mg, 978.35 µmol, 4.5 eq),反應體系降溫至0°C攪拌10分鐘,然後加入化合物13-3 (180 mg, 217.41 µmol, 1 eq),反應體系在0°C的條件下攪拌1小時。該反應與化合物13-3(60 mg)批次合併處理。向反應液中加入乙酸乙酯 (30 mL)萃取,有機相溶液用飽和氯化銨 (10mL)溶液和飽和食鹽水(10mL)各洗滌一次,用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步,得化合物13-4。LCMS m/z =879.3[M+H]+ 。Step 3: Synthesis of compound 13-4. Toluene (5 mL) was added to a dry reaction flask, and compound 1-11A (112.68 mg, 978.35 µmol, 4.5 eq) was added to start stirring, and then sodium tertiary butoxide was added (94.02 mg, 978.35 µmol, 4.5 eq), the reaction system was cooled to 0°C and stirred for 10 minutes, then compound 13-3 (180 mg, 217.41 µmol, 1 eq) was added, and the reaction system was stirred at 0°C for 1 Hour. The reaction was combined with a batch of compound 13-3 (60 mg). Ethyl acetate (30 mL) was added to the reaction solution for extraction, the organic phase solution was washed once with saturated ammonium chloride (10 mL) solution and saturated brine (10 mL), dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product , The crude product was directly used in the next step without purification to obtain compound 13-4. LCMS m/z = 879.3 [M+H] + .
步驟4:化合物13-5的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物13-4 (260 mg, 295.79 µmol, 1 eq)開始攪拌,然後加入乙酸鉀 (2.82 g, 24.70 mmol, 1.83 mL, 83.50 eq),反應體系在20°C的條件下攪拌2小時。向反應液中加入水 (30 mL)分液,水相用飽和碳酸氫鈉調節pH= 9,然後用乙酸乙酯(15 mL x 2) 萃取,合併有機相,用飽和食鹽水 (10 mL)洗滌,用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步,得到化合物13-5。LCMS m/z =539.2[M+H]+ 。Step 4: Synthesis of compound 13-5. Dichloromethane (5 mL) was added to a dry reaction flask, and compound 13-4 (260 mg, 295.79 µmol, 1 eq) was added to start stirring, and then potassium acetate ( 2.82 g, 24.70 mmol, 1.83 mL, 83.50 eq), the reaction system was stirred at 20°C for 2 hours. Water (30 mL) was added to the reaction solution for liquid separation. The aqueous phase was adjusted to pH = 9 with saturated sodium bicarbonate, and then extracted with ethyl acetate (15 mL x 2). The organic phases were combined and saturated brine (10 mL) It was washed, dried with anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product. The crude product was directly used in the next step without purification to obtain compound 13-5. LCMS m/z = 539.2 [M+H] + .
步驟5:化合物13的合成
將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合丙烯酸 (10.84 mg, 150.40 µmol, 10.32 µL, 1 eq),化合物13-5 (90 mg, 150.40 µmol,90% 純度, 1 eq)和N,N-二異丙基乙胺 (58.31 mg, 451.19 µmol, 78.59 µL, 3 eq)開始攪拌,反應體系降溫至-60°C,加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(68.62 mg,180.47 µmol, 1.2 eq),然後攪拌0.5小時。該反應與化合物13-5(30 mg)批次合併處理,向反應液中加入水 (5 mL)分液,有機相溶液直接減壓濃縮得到粗品,粗品高效液相層析管柱分離純化{ 層析管柱: Welch Xtimate C18 100*25mm*3µm;流動相: [H2
O(0.05%HCl)-ACN];乙腈%: 15%-45%,8min}得化合物13(出峰時間:1.683)。SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi,光學純度:95.48%。1
H NMR (400 MHz, CD3
OD) δ = 6.87 - 6.73 (m, 1H), 6.68 (d,J
= 8.4 Hz, 1H), 6.25 (dd,J
= 3.8, 16.6 Hz, 1H), 5.78 (d,J
= 11.6 Hz, 1H), 5.20 (dd,J
= 4.0, 11.2 Hz, 1H), 4.83 - 4.75 (m, 2H), 4.74 - 4.61 (m, 2H), 4.60 - 4.45 (m, 2H), 4.32 (d,J
= 13.0 Hz, 1H), 4.17 - 3.92 (m, 1H), 3.90 - 3.80 (m, 1H), 3.71 - 3.57 (m, 2H), 3.55 - 3.42 (m, 1H), 3.39 - 3.32 (m, 1H), 3.27 - 3.18 (m, 2H), 3.04 (s, 3H), 2.99 - 2.85 (m, 2H), 2.41 - 2.30 (m, 4H), 2.22 - 1.95 (m, 3H), 1.11 (d,J
= 6.6 Hz, 3H), LCMS m/z =593.3[M+H]+
。Step 5: Synthesis of
實施例14
步驟2:化合物14-3的合成 將二氯甲烷 (10 mL)加入到一個乾燥的反應瓶中,再加入化合物14-2 (350.18 mg, 423.97 µmol,1 eq),開始攪拌,然後加入間氯過氧苯甲酸 (109.75 mg, 508.77 µmol, 80% 純度, 1.2 eq),反應體系在25°C的條件下攪拌1小時。檢測到一個極性變大的主點形成。將反應液用二氯甲烷 (10 mL)稀釋,然後5%硫代硫酸鈉 (10 mL)溶液和飽和食鹽水 (10 mL)各洗滌一次,用無水硫酸鈉乾燥過濾後濾液減壓濃縮得到粗品。粗品TLC (石油醚 : 乙酸乙酯= 0:1),過柱純化(石油醚 : 乙酸乙酯 = 90:10到50:50)得到化合物14-3。LCMS m/z =842.3[M+H]+ 。Step 2: Synthesis of compound 14-3. Add dichloromethane (10 mL) to a dry reaction flask, then add compound 14-2 (350.18 mg, 423.97 µmol, 1 eq), start stirring, and then add m-chlorine Peroxybenzoic acid (109.75 mg, 508.77 µmol, 80% purity, 1.2 eq), the reaction system was stirred at 25°C for 1 hour. The formation of a major dot with increased polarity is detected. The reaction solution was diluted with dichloromethane (10 mL), and then washed once with 5% sodium thiosulfate (10 mL) solution and saturated brine (10 mL), dried with anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product . Crude TLC (petroleum ether: ethyl acetate = 0:1), column purification (petroleum ether: ethyl acetate = 90:10 to 50:50) to obtain compound 14-3. LCMS m/z =842.3 [M+H] + .
步驟3:化合物14-4的合成 將甲苯 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物1-11A (98.73 mg, 857.24 µmol, 4.5 eq),開始攪拌,然後加入三級丁醇鈉 (82.38 mg,857.24 µmol, 4.5 eq),反應體系降溫至0°C攪拌10分鐘,然後加入含有化合物14-3 (160.39 mg, 190.50 µmol, 1 eq)的甲苯(2 mL)溶液,反應體系在0°C的條件下攪拌1小時。該反應與化合物14-3(50 mg)批次合併處理。向反應液中加入乙酸乙酯 (30 mL)萃取,收集有機相溶液用飽和氯化銨(10 mL)溶液和飽和食鹽水(10 mL)各洗滌一次,用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步,得化合物14-4。LCMS m/z =893.4[M+H]+ 。Step 3: Synthesis of compound 14-4. Add toluene (5 mL) to a dry reaction flask, then add compound 1-11A (98.73 mg, 857.24 µmol, 4.5 eq), start stirring, and then add tertiary butanol Sodium (82.38 mg, 857.24 µmol, 4.5 eq), the reaction system was cooled to 0°C and stirred for 10 minutes, and then a toluene (2 mL) solution containing compound 14-3 (160.39 mg, 190.50 µmol, 1 eq) was added to the reaction system Stir at 0°C for 1 hour. This reaction was combined with a batch of compound 14-3 (50 mg). Ethyl acetate (30 mL) was added to the reaction solution for extraction, the organic phase solution was collected and washed with saturated ammonium chloride (10 mL) solution and saturated brine (10 mL) each, dried over anhydrous sodium sulfate and filtered, and the filtrate was decompressed Concentrate to obtain the crude product, which is directly used in the next step without purification to obtain compound 14-4. LCMS m/z = 893.4 [M+H] + .
步驟4:化合物14-5的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物14-4 (260 mg,291.15 µmol, 1 eq) 開始攪拌,然後加入三氟乙酸 (2.77 g, 24.31 mmol, 1.8 mL, 83.50 eq),反應體系在20°C的條件下攪拌2小時。向反應液中加入二氯甲烷 (20 mL)溶液稀釋反應液,然後加入水 (20 mL)分液,水相用飽和碳酸氫鈉調節pH =8,然後用乙酸乙酯 (20 mL x 2)溶液萃取,合併有機相,用飽和食鹽水 (10 mL)洗滌,用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步,得化合物14-5。LCMS m/z =553.2[M+H]+ 。Step 4: Synthesis of compound 14-5 Dichloromethane (5 mL) was added to a dry reaction flask, and compound 14-4 (260 mg, 291.15 µmol, 1 eq) was added to start stirring, and then trifluoroacetic acid was added (2.77 g, 24.31 mmol, 1.8 mL, 83.50 eq), the reaction system was stirred at 20°C for 2 hours. Dichloromethane (20 mL) was added to the reaction solution to dilute the reaction solution, and then water (20 mL) was added for separation. The aqueous phase was adjusted to pH = 8 with saturated sodium bicarbonate, and then ethyl acetate (20 mL x 2) The solution was extracted, the organic phases were combined, washed with saturated brine (10 mL), dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product. The crude product was directly used in the next step without purification to obtain compound 14-5. LCMS m/z = 553.2 [M+H] + .
步驟5:化合物14的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,再加入化合物丙烯酸 (10.56 mg, 146.58 µmol, 10.06 µL, 1eq ),化合物14-5 (90 mg, 146.58 µmol, 90%純度, 1eq )和N,N-二異丙基乙胺 (56.83 mg, 439.73 µmol, 76.59 µL, 3eq )開始攪拌,反應體系降溫至-60°C,加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(66.88 mg,175.89 µmol, 1.2eq ) ,然後攪拌0.5小時。向反應液中加入水 (5 mL)淬滅反應後分液, 有機相用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品。粗品進行高效液相層析管柱分離純化方法為{層析管柱: Welch Xtimate C18 100*25mm*3µm;流動相: [H2 O(0.05%HCl)-ACN];乙腈%: 15%-45%, 8min},得化合物14。1 H NMR (400 MHz, CDCl3 )6.69 - 6.62 (m, 1H), 6.62 - 6.48 (m, 1H), 6.47 - 6.34 (m, 1H), 5.91 - 5.76 (m, 1H), 5.34 (s, 1H), 5.25 - 5.14 (m, 1H), 5.10 - 4.99 (m, 1H), 4.86 - 4.64 (m, 2H), 4.62 - 4.30 (m, 2H), 4.21 - 4.11 (m, 1H), 4.06 - 3.93 (m, 2H), 3.90 - 3.73 (m, 2H), 3.71 - 3.59 (m, 1H), 3.57 - 3.49 (m, 3H), 3.47 - 3.33 (m, 1H), 3.28 - 3.15 (m, 2H), 3.09 - 2.92 (m, 1H), 2.50 - 2.35 (m, 4H), 2.26 - 2.09 (m, 2H), 1.43 - 1.32 (m, 4H), 1.31 - 1.25 (m, 2H), LCMS m/z =607.4[M+H]+ 。Step 5: Synthesis of compound 14 Dichloromethane (5 mL) was added to a dry reaction flask, followed by compound acrylic acid (10.56 mg, 146.58 µmol, 10.06 µL, 1 eq ), compound 14-5 (90 mg, 146.58 µmol, 90% purity, 1 eq ) and N,N-diisopropylethylamine (56.83 mg, 439.73 µmol, 76.59 µL, 3 eq ) began to stir, the reaction system was cooled to -60°C, and O-( 7-Azabenzotriazole-1-YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (66.88 mg, 175.89 µmol, 1.2 eq ), and then stirred for 0.5 hours. Water (5 mL) was added to the reaction solution to quench the reaction and liquid separation, the organic phase was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product. The crude product is separated and purified by high performance liquid chromatography column as {chromatographic column: Welch Xtimate C18 100*25mm*3µm; mobile phase: [H 2 O(0.05%HCl)-ACN]; acetonitrile%: 15%- 45%, 8min} to obtain compound 14. 1 H NMR (400 MHz, CDCl 3 ) 6.69-6.62 (m, 1H), 6.62-6.48 (m, 1H), 6.47-6.34 (m, 1H), 5.91-5.76 (m, 1H), 5.34 (s, 1H), 5.25-5.14 (m, 1H), 5.10-4.99 (m, 1H), 4.86-4.64 (m, 2H), 4.62-4.30 (m, 2H), 4.21-4.11 (m, 1H), 4.06- 3.93 (m, 2H), 3.90-3.73 (m, 2H), 3.71-3.59 (m, 1H), 3.57-3.49 (m, 3H), 3.47-3.33 (m, 1H), 3.28-3.15 (m, 2H) ), 3.09-2.92 (m, 1H), 2.50-2.35 (m, 4H), 2.26-2.09 (m, 2H), 1.43-1.32 (m, 4H), 1.31-1.25 (m, 2H), LCMS m/ z =607.4[M+H] + .
實施例15

步驟2:化合物15-3的合成 將二氯甲烷 (3 mL)加入到一個乾燥的反應瓶中,加入化合物15-2 (0.24 g, 295.59 µmol, 1 eq)開始攪拌,再加入間氯過氧苯甲酸 (66.95 mg, 310.37 µmol, 80% 純度 1.05 eq),反應體系在25°C的條件下反應30分鐘。向反應液中加入亞硫酸鈉水溶液 (5 mL 5%)淬滅,反應液分液後,水相用二氯甲烷 (5 mL x 2)萃取,合併有機相,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品。水相用澱粉碘化鉀試紙檢測,無氧化性後,水相遺棄。粗品根據TLC (石油醚:乙酸乙酯=1:1,產品Rf=0.51)經管柱層析純化(石油醚:乙酸乙酯=50:1到0:1)得到產物。得到化合物15-3。1 H NMR (400 MHz, CDCl3 ) δ = 7.17 - 7.06 (m, 4H), 6.83 - 6.72 (m, 4H), 6.71 - 6.60 (m, 1H), 5.24 - 5.10 (m, 1H), 4.87 - 4.64 (m, 2H), 4.44 - 4.17 (m, 4H), 3.98 - 3.81 (m, 2H), 3.78 - 3.69 (m, 6H), 3.67 - 3.43 (m, 2H), 3.20 - 2.97 (m, 4H), 2.88 - 2.78 (m, 3H), 2.32 - 2.20 (m, 3H), 1.47 - 1.37 (m, 9H), 1.32 - 1.23 (m, 3H)。LCMS m/z =828.3 M+H]+ 。Step 2: Synthesis of compound 15-3. Add dichloromethane (3 mL) to a dry reaction flask, add compound 15-2 (0.24 g, 295.59 µmol, 1 eq) and start stirring, then add m-chloroperoxy Benzoic acid (66.95 mg, 310.37 µmol, 80% purity 1.05 eq), the reaction system was reacted at 25°C for 30 minutes. The reaction solution was quenched by adding sodium sulfite aqueous solution (5 mL 5%). After the reaction solution was separated, the aqueous phase was extracted with dichloromethane (5 mL x 2), the organic phases were combined, anhydrous sodium sulfate was added to dry and filtered, and the filtrate was depressurized Concentrate to obtain crude product. The water phase is tested with starch potassium iodide test paper. After it is non-oxidizing, the water phase is discarded. The crude product was purified by column chromatography (petroleum ether: ethyl acetate = 50:1 to 0:1) according to TLC (petroleum ether: ethyl acetate = 1:1, product Rf = 0.51) to obtain the product. Compound 15-3 was obtained. 1 H NMR (400 MHz, CDCl 3 ) δ = 7.17-7.06 (m, 4H), 6.83-6.72 (m, 4H), 6.71-6.60 (m, 1H), 5.24-5.10 (m, 1H), 4.87- 4.64 (m, 2H), 4.44-4.17 (m, 4H), 3.98-3.81 (m, 2H), 3.78-3.69 (m, 6H), 3.67-3.43 (m, 2H), 3.20-2.97 (m, 4H) ), 2.88-2.78 (m, 3H), 2.32-2.20 (m, 3H), 1.47-1.37 (m, 9H), 1.32-1.23 (m, 3H). LCMS m/z = 828.3 M+H] + .
步驟3:化合物15-4的合成 將甲苯 (1 mL)加入到一個乾燥的反應瓶中,加入化合物15-3 (180 mg, 217.41 µmol, 1 eq)開始攪拌,反應體系降溫至0-5°C,加入三級丁醇鈉 (2.68 mg, 652.23 µmol, 3 eq),攪拌10分鐘,將含有化合物1-11A (75.12 mg, 652.23 µmol, 77.44 µL, 3 eq)的甲苯(0.3 mL)加入到上述反應液中,反應體系在0-5°C的條件下反應30分鐘。將反應液倒入飽和氯化銨水溶液 (5 mL)中,用乙酸乙酯 (5 mL x 3)萃取,合併有機相,用飽和食鹽水 (5 mL)洗滌,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步。得到化合物15-4。LCMS m/z =879.4[M+H]+ 。Step 3: Synthesis of compound 15-4. Toluene (1 mL) was added to a dry reaction flask, compound 15-3 (180 mg, 217.41 µmol, 1 eq) was added to start stirring, and the reaction system was cooled to 0-5° C, add tertiary butoxide sodium (2.68 mg, 652.23 µmol, 3 eq), stir for 10 minutes, add toluene (0.3 mL) containing compound 1-11A (75.12 mg, 652.23 µmol, 77.44 µL, 3 eq) to In the above reaction solution, the reaction system is reacted at 0-5°C for 30 minutes. The reaction solution was poured into saturated aqueous ammonium chloride solution (5 mL), extracted with ethyl acetate (5 mL x 3), combined the organic phases, washed with saturated brine (5 mL), added anhydrous sodium sulfate, dried and filtered, and the filtrate Concentrate under reduced pressure to obtain the crude product, which was used directly in the next step without purification. Compound 15-4 was obtained. LCMS m/z =879.4 [M+H] + .
步驟4:化合物15-5的合成 將二氯甲烷 (4 mL)加入到一個乾燥的反應瓶中,加入化合物15-4 (160 mg,182.03 µmol, 1 eq)開始攪拌,反應體系降溫至0-5°C,加入三氟乙酸 (1.23 g, 10.81 mmol, 800.00 µL, 59.36 eq),攪拌4小時。反應液加入到飽和碳酸氫鈉水溶液 (10 mL)中,分液,用二氯甲烷 (5 mL x 2)萃取,合併有機相,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步,得到化合物15-5。LCMS m/z =539.2[M+H]+ 。Step 4: Synthesis of compound 15-5. Dichloromethane (4 mL) was added to a dry reaction flask, compound 15-4 (160 mg, 182.03 µmol, 1 eq) was added to start stirring, and the reaction system was cooled to 0- At 5°C, add trifluoroacetic acid (1.23 g, 10.81 mmol, 800.00 µL, 59.36 eq) and stir for 4 hours. The reaction solution was added to a saturated aqueous sodium bicarbonate solution (10 mL), separated, extracted with dichloromethane (5 mL x 2), combined the organic phases, added anhydrous sodium sulfate, dried and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product. Purification was used directly in the next step to obtain compound 15-5. LCMS m/z = 539.2 [M+H] + .
步驟5:化合物15的合成 將二氯甲烷 (10 mL)加入到一個乾燥的反應瓶中,加入化合物15-5 (0.06 g, 111.40 µmol, 1 eq) 和化合物丙烯酸(16.06 mg, 222.81 µmol, 15.29 µL, 2 eq)開始攪拌,再加入N,N-二異丙基乙胺 (28.80 mg, 222.81 µmol, 38.81 µL, 2 eq),反應體系降溫至-60°C,加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(63.54 mg,167.11 µmol, 1.5 eq),反應體系在-60°C的條件下反應0.5小時。與化合物15-5(20mg)批次合併處理,向反應液中加入二氯甲烷 (5 mL),用飽和氯化銨溶液 (5 mL x 2)洗滌,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品。進行高效液相層析管柱分離純化,方法為{層析管柱: Phenomenex Luna C18 200*40mm*10µm;流動相: [H2 O(0.04%HCl)-ACN];乙腈%: 1%-50%,8min},機分液中加入1滴氨水,溶液顯示為鹼性,濃縮除去有機溶劑後凍乾,得到化合物15。1 H NMR (400 MHz, CD3 OD) δ = 7.24 - 7.07 (m, 2H), 6.80 (dd,J = 10.8, 16.8 Hz, 1H), 6.71 (d,J = 8.6 Hz, 1H), 6.24 (d,J = 16.8 Hz, 1H), 5.79 (d,J = 11.7 Hz, 1H), 5.24 - 5.16 (m, 1H), 4.76 (d,J = 13.8 Hz, 3H), 4.57 (dd,J = 7.2, 12.5 Hz, 2H), 4.07 - 3.86 (m, 3H), 3.73 (s, 1H), 3.17 (d,J = 11.4 Hz, 3H), 3.07 (s, 3H), 2.90 (d,J = 14.8 Hz, 1H), 2.46 - 2.34 (m, 4H), 2.33 - 2.30 (m, 1H), 2.26 - 1.95 (m, 3H), 1.41 (s, 3H)。LCMS m/z =593.2[M+H]+ 。Step 5: Synthesis of compound 15 Dichloromethane (10 mL) was added to a dry reaction flask, compound 15-5 (0.06 g, 111.40 µmol, 1 eq) and compound acrylic acid (16.06 mg, 222.81 µmol, 15.29) were added µL, 2 eq) start stirring, then add N,N-diisopropylethylamine (28.80 mg, 222.81 µmol, 38.81 µL, 2 eq), the reaction system is cooled to -60°C, and O-(7-nitrogen Heterobenzotriazole-1-YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (63.54 mg, 167.11 µmol, 1.5 eq), the reaction system is reacted at -60°C 0.5 hours. Combine it with compound 15-5 (20mg) in batches, add dichloromethane (5 mL) to the reaction solution, wash with saturated ammonium chloride solution (5 mL x 2), add anhydrous sodium sulfate, dry and filter, and reduce the pressure of the filtrate Concentrate to obtain crude product. Carry out high performance liquid chromatography column separation and purification, the method is {chromatographic column: Phenomenex Luna C18 200*40mm*10µm; mobile phase: [H 2 O(0.04%HCl)-ACN]; acetonitrile%: 1%- 50%, 8min}, 1 drop of ammonia was added to the mechanical separation solution, the solution was alkaline, concentrated to remove the organic solvent and lyophilized to obtain compound 15. 1 H NMR (400 MHz, CD 3 OD) δ = 7.24-7.07 (m, 2H), 6.80 (dd, J = 10.8, 16.8 Hz, 1H), 6.71 (d, J = 8.6 Hz, 1H), 6.24 ( d, J = 16.8 Hz, 1H), 5.79 (d, J = 11.7 Hz, 1H), 5.24-5.16 (m, 1H), 4.76 (d, J = 13.8 Hz, 3H), 4.57 (dd, J = 7.2 , 12.5 Hz, 2H), 4.07-3.86 (m, 3H), 3.73 (s, 1H), 3.17 (d, J = 11.4 Hz, 3H), 3.07 (s, 3H), 2.90 (d, J = 14.8 Hz , 1H), 2.46-2.34 (m, 4H), 2.33-2.30 (m, 1H), 2.26-1.95 (m, 3H), 1.41 (s, 3H). LCMS m/z =593.2 [M+H] + .
實施例16
步驟2:化合物16-3的合成 將N,N-二甲基甲醯胺 (3 mL)加入到一個乾燥的反應瓶中,加入化合物16-2(230 mg, 312.15 µmol, 1 eq)開始攪拌,再加入N,N-二異丙基乙胺 (121.03 mg, 936.46 µmol, 163.11 µL, 3 eq)和二三級丁基二碳酸酯(74.94 mg, 343.37 µmol, 78.88 µL, 1.1 eq),反應體系在20°C的條件下反應10小時。反應液倒入飽和氯化銨水溶液 (15 mL)中,用乙酸乙酯 (10 mL x 2)萃取,合併有機相,用飽和食鹽水(5 mL)洗滌,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品。粗品根據TLC (石油醚:乙酸乙酯=3:1 )經管柱層析純化(石油醚:乙酸乙酯=100:1-0:1)得到化合物16-3。1 H NMR (400 MHz, CDCl3 ) δ = 7.16 (d,J = 8.4 Hz, 4H), 6.85 (d,J = 8.6 Hz, 4H), 6.64 (d,J = 8.0 Hz, 1H), 5.22 ( d,J = 7.2 Hz, 1H), 4.90 - 4.68 (m, 2H), 4.61 (s, 1H), 4.41 - 4.21 (m, 4H), 4.04 (s, 1H), 3.80 (s, 6H), 3.71 (s, 1H), 3.50 (d,J = 11.0 Hz, 2H), 3.30 (s, 1H), 3.24 - 3.02 (m, 2H), 2.90 (d,J = 2.0 Hz, 1H), 2.78 - 2.58 (m, 2H), 2.55 (s, 3H), 2.34 (d,J = 4.0 Hz, 3H), 1.51 (s,9H)。LCMS m/z =837.2[M+H]+ 。Step 2: Synthesis of compound 16-3. Add N,N-dimethylformamide (3 mL) to a dry reaction flask, add compound 16-2 (230 mg, 312.15 µmol, 1 eq) and start stirring , Then add N,N-diisopropylethylamine (121.03 mg, 936.46 µmol, 163.11 µL, 3 eq) and di-tertiary butyl dicarbonate (74.94 mg, 343.37 µmol, 78.88 µL, 1.1 eq), and react The system reacts for 10 hours at 20°C. The reaction solution was poured into a saturated aqueous ammonium chloride solution (15 mL), extracted with ethyl acetate (10 mL x 2), combined the organic phases, washed with saturated brine (5 mL), added anhydrous sodium sulfate, dried and filtered, and the filtrate was reduced Concentrate by pressure to obtain a crude product. The crude product was purified by column chromatography (petroleum ether: ethyl acetate=100:1-0:1) according to TLC (petroleum ether: ethyl acetate=3:1) to obtain compound 16-3. 1 H NMR (400 MHz, CDCl 3 ) δ = 7.16 (d, J = 8.4 Hz, 4H), 6.85 (d, J = 8.6 Hz, 4H), 6.64 (d, J = 8.0 Hz, 1H), 5.22 ( d, J = 7.2 Hz, 1H), 4.90-4.68 (m, 2H), 4.61 (s, 1H), 4.41-4.21 (m, 4H), 4.04 (s, 1H), 3.80 (s, 6H), 3.71 (s, 1H), 3.50 (d, J = 11.0 Hz, 2H), 3.30 (s, 1H), 3.24-3.02 (m, 2H), 2.90 (d, J = 2.0 Hz, 1H), 2.78-2.58 ( m, 2H), 2.55 (s, 3H), 2.34 (d, J = 4.0 Hz, 3H), 1.51 (s, 9H). LCMS m/z = 837.2 [M+H] + .
步驟3:化合物16-4的合成 將二氯甲烷 (0.3 mL)加入到一個乾燥的反應瓶中,加入化合物16-3(230 mg, 274.81 µmol,1 eq)開始攪拌,再加入間氯過氧苯甲酸 (61.37 mg, 302.29 µmol, 85% 純度, 1.1 eq),反應體系在20°C的條件下反應1小時。將反應液倒入5%亞硫酸鈉水溶液 (5 mL)中,分液,水相用二氯甲烷 (5 mL x 2)萃取,合併有機相,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,水相用澱粉碘化鉀試紙檢測,沒有氧化性後遺棄水相。粗品根據TLC (石油醚:乙酸乙酯=1:1)經管柱層析純化(石油醚:乙酸乙酯=100:1-0:1)得到化合物16-4。 LCMS m/z =853.2[M+H]+ 。Step 3: Synthesis of compound 16-4. Add dichloromethane (0.3 mL) to a dry reaction flask, add compound 16-3 (230 mg, 274.81 µmol, 1 eq) and start stirring, then add m-chloroperoxy Benzoic acid (61.37 mg, 302.29 µmol, 85% purity, 1.1 eq), the reaction system was reacted at 20°C for 1 hour. Pour the reaction solution into 5% sodium sulfite aqueous solution (5 mL), separate the layers, extract the aqueous phase with dichloromethane (5 mL x 2), combine the organic phases, add anhydrous sodium sulfate, dry and filter, and concentrate the filtrate under reduced pressure to obtain a crude product. The water phase is tested with starch potassium iodide test paper, and the water phase is discarded after there is no oxidation. The crude product was purified by column chromatography (petroleum ether: ethyl acetate = 100:1-0:1) according to TLC (petroleum ether: ethyl acetate=1:1) to obtain compound 16-4. LCMS m/z = 853.2 [M+H] + .
步驟4:化合物16-5的合成 將甲苯 (2 mL)加入到一個乾燥的反應瓶中,加入化合物16-4(158 mg, 185.24 µmol, 1 eq)開始攪拌,反應體系降溫至0°C,再加入三級丁醇鈉 (35.60 mg, 370.49 µmol, 2 eq),攪拌15分鐘,加入化合物12-3A(65.40 mg, 463.11 µmol, 2.5 eq),反應體系在0°C的條件下反應30分鐘。反應液倒入飽和氯化銨水溶液 (5 mL)中,用乙酸乙酯 (5 mL x 3)萃取,合併有機相,用飽和食鹽水 (3 mL)洗滌,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,未純化直接用於下一步,得到化合物16-5。LCMS m/z =930.4[M+H]+ 。Step 4: Synthesis of compound 16-5. Toluene (2 mL) was added to a dry reaction flask, compound 16-4 (158 mg, 185.24 µmol, 1 eq) was added to start stirring, and the reaction system was cooled to 0°C. Then add tertiary sodium butoxide (35.60 mg, 370.49 µmol, 2 eq), stir for 15 minutes, add compound 12-3A (65.40 mg, 463.11 µmol, 2.5 eq), and react the reaction system at 0°C for 30 minutes . The reaction solution was poured into saturated aqueous ammonium chloride (5 mL), extracted with ethyl acetate (5 mL x 3), combined the organic phases, washed with saturated brine (3 mL), added anhydrous sodium sulfate, dried and filtered, and the filtrate was reduced The crude product was obtained by pressure concentration, which was directly used in the next step without purification to obtain compound 16-5. LCMS m/z =930.4 [M+H] + .
步驟5:化合物16-6的合成 將二氯甲烷 (5 mL)加入到一個乾燥的反應瓶中,加入化合物16-5(0.18 g, 193.54 µmol, 1 eq)開始攪拌,再加入三氟乙酸 (1 mL),反應體系在18°C的條件下反應3小時。向反應液中加入水 (10 mL)萃取分液,收集水相,用飽和碳酸氫鈉溶液調節pH為8,用二氯甲烷 (20 mL x 2)萃取,合併有機相,加入無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品未純化直接用於下一步,得到化合物16-6。LCMS m/z =590.2[M+H]+ 。Step 5: Synthesis of compound 16-6. Add dichloromethane (5 mL) to a dry reaction flask, add compound 16-5 (0.18 g, 193.54 µmol, 1 eq) and start stirring, then add trifluoroacetic acid ( 1 mL), the reaction system was reacted at 18°C for 3 hours. Water (10 mL) was added to the reaction solution for extraction and liquid separation. The aqueous phase was collected, adjusted to pH 8 with saturated sodium bicarbonate solution, and extracted with dichloromethane (20 mL x 2). The organic phases were combined and dried by adding anhydrous sodium sulfate. After filtration, the filtrate was concentrated under reduced pressure to obtain a crude product. The crude product was directly used in the next step without purification to obtain compound 16-6. LCMS m/z =590.2 [M+H] + .
步驟6:化合物16的合成
將化合物16-6(62.77 mg, 106.46 µmol, 1 eq),2-氟丙烯酸(19.17 mg, 212.92 µmol, 2 eq), N,N-二異丙基乙胺 (41.28 mg, 319.38 µmol, 55.63 µL, 3 eq) ,溶於DCM (5 mL) ,降溫至-60°C。加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(48.58 mg, 127.75 µmol, 1.2 eq) ,然後攪拌0.5hr。與化合物16-6(20.92 mg)批次合併處理。向反應液中加入5mL水,分液,有機相濃縮,高效液相層析管柱分離純機分純化,純化方法{層析管柱: Phenomenex luna C18 80*40mm*3 µm;流動相: [H2
O(0.04%HCl)-ACN];乙腈%: 20%-40%,7min}得化合物16。1
H NMR (400MHz, CD3
OD) δ = 6.73 (d, J=8.6 Hz, 1H), 5.45 - 5.21 (m, 3H), 4.87 - 4.80 (m, 2H), 4.57 (s, 2H), 4.19 (br d, J=13.7 Hz, 1H), 3.98 (br d, J=13.1 Hz, 1H), 3.75 - 3.66 (m, 2H), 3.56 - 3.49 (m, 1H), 3.37 (s, 3H), 3.32 - 3.27 (m, 2H), 3.26 - 3.11 (m, 1H), 3.06 - 2.87 (m, 1H), 3.06 - 2.87 (m, 1H), 3.06 - 2.87 (m, 1H), 2.44 - 2.03 (m, 12H)。LCMS m/z =662.4[M+H]+
。Step 6: Synthesis of
實施例17
步驟2:化合物17-3的合成 將二氯甲烷 (25 mL)加入到乾燥的反應瓶中,再加入化合物17-2 (2.3 g, 3.12 mmol, 1 eq)開始攪拌,反應體系降溫至0 °C,加入三乙胺 (789.67 mg, 7.80 mmol, 1.09 mL, 2.5 eq)和三氟乙酸酐 (983.42 mg, 4.68 mmol, 651.27 µL, 1.5 eq),反應體系在0-5°C的條件下反應0.5小時。與化合物17-2(0.3g)批次合併處理,向反應液中加入的飽和氯化銨溶液 (20 mL x 2)洗滌後,用無水硫酸鈉乾燥過濾後,濾液減壓濃縮得到粗品,粗品未純化,直接用於下一步。得到化合物17-3。1 H NMR (400 MHz, CDCl3 ) δ ppm 7.15 (d,J = 8.40 Hz, 4 H), 6.84 (d,J = 8.40 Hz, 4 H), 6.65 (br d,J = 8.40 Hz, 1 H), 5.29 - 5.20 (m, 1 H), 4.77 (s, 2 H), 4.38 - 4.24 (m, 4 H), 4.05 - 3.88 (m, 2 H), 3.80 (s, 6 H), 3.78 - 3.60 (m, 2 H), 3.59 - 3.37 (m, 2 H), 3.14 - 2.99 (m, 2 H), 2.98 - 2.93 (m, 1 H), 2.91 - 2.86 (m, 1 H), 2.78 (t,J = 6.80 Hz, 1 H), 2.53 (s, 3 H), 2.39 - 2.30 (m, 3 H),LCMS m/z =833.1[M+H]+ 。Step 2: Synthesis of compound 17-3. Dichloromethane (25 mL) was added to a dry reaction flask, and compound 17-2 (2.3 g, 3.12 mmol, 1 eq) was added to start stirring, and the reaction system was cooled to 0 ° C, add triethylamine (789.67 mg, 7.80 mmol, 1.09 mL, 2.5 eq) and trifluoroacetic anhydride (983.42 mg, 4.68 mmol, 651.27 µL, 1.5 eq), and the reaction system is reacted at 0-5°C 0.5 hours. Combined with compound 17-2 (0.3g) in batches, the reaction solution was washed with saturated ammonium chloride solution (20 mL x 2), dried with anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure to obtain a crude product. It was not purified and used directly in the next step. Compound 17-3 was obtained. 1 H NMR (400 MHz, CDCl 3 ) δ ppm 7.15 (d, J = 8.40 Hz, 4 H), 6.84 (d, J = 8.40 Hz, 4 H), 6.65 (br d, J = 8.40 Hz, 1 H ), 5.29-5.20 (m, 1 H), 4.77 (s, 2 H), 4.38-4.24 (m, 4 H), 4.05-3.88 (m, 2 H), 3.80 (s, 6 H), 3.78- 3.60 (m, 2 H), 3.59-3.37 (m, 2 H), 3.14-2.99 (m, 2 H), 2.98-2.93 (m, 1 H), 2.91-2.86 (m, 1 H), 2.78 ( t, J = 6.80 Hz, 1 H), 2.53 (s, 3 H), 2.39-2.30 (m, 3 H), LCMS m/z =833.1[M+H] + .
步驟3:化合物17-4的合成
將二氯甲烷 (30 mL)加入到乾燥的反應瓶中,再加入化合物17-3 (2.6 g, 3.12 mmol, 1eq
)開始攪拌,再加入間氯過氧苯甲酸 (697.20 mg, 3.43 mmol, 85% 純度, 1.1eq
),反應體系在18°C的條件下反應0.5小時。與化合物17-3(0.2 g)批次合併處理,向反應液中加入硫代硫酸鈉溶液 (20 mL 10%),用澱粉-KI試紙檢測為陰性後,用二氯甲烷 (20 mL x 2)萃取,加入無水硫酸鈉乾燥過濾後,濾液減壓濃縮得到粗品,粗品根據TLC (石油醚:乙酸乙酯=0:1)過柱純化(石油醚:乙酸乙酯=10:1-0:1),得到化合物17-4。1
H NMR (400 MHz, CDCl3
) δ ppm 7.15 (d,J
= 8.40 Hz, 4 H), 6.84 (d,J
= 8.40 Hz, 4 H), 6.66 (d,J
= 8.40 Hz, 1 H), 5.27 (d,J
= 9.20 Hz, 1 H), 4.93 - 4.982 (m, 2 H), 4.38 - 4.24 (m, 4 H), 4.10 - 3.99 (m, 2 H), 3.98 - 3.88 (m, 1 H), 3.87 - 3.68 (m, 8 H), 3.67 - 3.54 (m, 1 H), 3.54 - 2.98 (m, 3 H), 2.93 - 2.79 (m, 4 H), 2.78 - 2.65 (m, 1 H), 2.35 (d,J
= 3.60 Hz, 3 H), LCMS m/z =849.1[M+H]+
。Step 3: Synthesis of compound 17-4. Add dichloromethane (30 mL) to a dry reaction flask, then add compound 17-3 (2.6 g, 3.12 mmol, 1 eq ) and start stirring, then add m-chloroperoxy Benzoic acid (697.20 mg, 3.43 mmol, 85% purity, 1.1 eq ), the reaction system was reacted at 18°C for 0.5 hours. Combine the batch with compound 17-3 (0.2 g). Add sodium thiosulfate solution (20
步驟4:化合物17-5的合成 將甲苯 (1 mL)加入到乾燥的反應瓶中,再加入化合物17-4A(78.77 mg, 494.80 µmol, 3 eq)開始攪拌,反應體系降溫至0°C,加入三級丁醇鈉 (47.55 mg, 494.80 µmol, 3 eq),攪拌10分鐘後,加入含有化合物17-4 (0.14 g, 164.93 µmol, 1 eq)的甲苯溶液 (0.5 mL),繼續反應0.5小時。與化合物17-4(20 mg)批次合併處理,向反應液加入乙酸乙酯 (5 mL)稀釋,依次用飽和氯化銨 (10 mL x 2)溶液和飽和食鹽水 (10 mL)洗滌,加入無水硫酸鈉乾燥過濾後,濾液減壓濃縮得到粗品,粗品未純化,直接用於下一步,得到化合物17-5。LCMS m/z =848.3[M+H]+ 。Step 4: Synthesis of compound 17-5. Toluene (1 mL) was added to a dry reaction flask, and compound 17-4A (78.77 mg, 494.80 µmol, 3 eq) was added to start stirring. The reaction system was cooled to 0°C. Add tertiary butoxide sodium (47.55 mg, 494.80 µmol, 3 eq), stir for 10 minutes, add compound 17-4 (0.14 g, 164.93 µmol, 1 eq) in toluene solution (0.5 mL), continue the reaction for 0.5 hours . Combine it with compound 17-4 (20 mg) in batches, add ethyl acetate (5 mL) to the reaction solution to dilute, and wash with saturated ammonium chloride (10 mL x 2) solution and saturated brine (10 mL) successively, After adding anhydrous sodium sulfate, drying and filtering, the filtrate was concentrated under reduced pressure to obtain a crude product. The crude product was not purified and was directly used in the next step to obtain compound 17-5. LCMS m/z = 848.3 [M+H] + .
步驟5:化合物17-6的合成 將二氯甲烷 (12 mL)加入到乾燥的反應瓶中,再加入化合物17-5 (160.00 mg, 188.70 µmol, 1eq )開始攪拌,再加入三氟乙酸 (2 mL),反應體系在18°C的條件下反應2小時。與化合物17-5(20 mg)批次合併處理,向反應液中加入水 (10 mL),分液萃取,水相用飽和碳酸氫鈉溶液至pH=8,用的二氯甲烷 (10 mL x 2)萃取,合併有機相,用無水硫酸鈉乾燥過濾後,濾液減壓濃縮得到粗品,粗品未純化,直接用於下一步,得到化合物17-6。LCMS m/z =608.3[M+H]+ 。Step 5: Synthesis of compound 17-6. Add dichloromethane (12 mL) to a dry reaction flask, then add compound 17-5 (160.00 mg, 188.70 µmol, 1 eq ) and start stirring, then add trifluoroacetic acid ( 2 mL), the reaction system was reacted at 18°C for 2 hours. Combine treatment with compound 17-5 (20 mg) in batches, add water (10 mL) to the reaction solution, separate and extract the aqueous phase with saturated sodium bicarbonate solution to pH=8, use dichloromethane (10 mL) x 2) Extract, combine the organic phases, dry and filter with anhydrous sodium sulfate, and concentrate the filtrate under reduced pressure to obtain a crude product. The crude product is not purified and used directly in the next step to obtain compound 17-6. LCMS m/z = 608.3 [M+H] + .
步驟6:化合物17的合成 將二氯甲烷 (5 mL)加入到乾燥的反應瓶中,再加入化合物17-6 (50 mg, 82.29 µmol, 1 eq)和2-氟丙烯酸 (14.82 mg, 164.58 µmol, 2 eq)以及N,N-二異丙基乙胺 (31.90 mg, 246.87 µmol, 43.00 µL, 3 eq)開始攪拌,反應體系降溫至-60°C,再加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽 (37.55 mg, 98.75 µmol, 1.2 eq),然後攪拌0.5小時。合併處理,向反應液中加入水 (5 mL)淬滅反應,分液,有機相用無水硫酸鈉乾燥過濾,濾液減壓濃縮得到粗品,粗品高效液相層析管柱分離純化 {層析管柱: Welch Xtimate C18 100*25mm*3µm; 流動相:[H2 O(0.05%HCl) -ACN];乙腈%: 20%-50%,8min},得到化合物17。SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3μm;流動相:A (CO2) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi,光學純度:99.21%,出峰時間:1.840。1 H NMR (400 MHz, CD3 OD) δ = 6.80 - 6.68 (m, 1H), 5.73 - 5.51 (m, 1H), 5.46 - 5.19 (m, 3H), 5.05 - 4.90 (m, 3H), 4.74 - 4.58 (m, 2H), 4.37 - 4.26 (m, 1H), 4.20 - 4.06 (m, 2H), 4.05 - 3.84 (m, 3H), 3.79 - 3.59 (m, 2H), 3.54 - 3.43 (m, 1H), 3.42 - 3.35 (m, 1H), 3.31 - 3.24 (m, 1H), 3.13 - 2.89 (m, 3H), 2.82 - 2.52 (m, 2H), 2.50 - 2.42 (m, 1H), 2.41 - 2.30 (m, 5H), 2.29 - 2.18 (m, 1H)。Step 6: Synthesis of Compound 17 Dichloromethane (5 mL) was added to a dry reaction flask, followed by compound 17-6 (50 mg, 82.29 µmol, 1 eq) and 2-fluoroacrylic acid (14.82 mg, 164.58 µmol) , 2 eq) and N,N-diisopropylethylamine (31.90 mg, 246.87 µmol, 43.00 µL, 3 eq) start stirring, the reaction system is cooled to -60°C, and then O-(7-azabenzene And triazole-1-YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (37.55 mg, 98.75 µmol, 1.2 eq), and then stirred for 0.5 hours. Combine the treatments, add water (5 mL) to the reaction solution to quench the reaction, separate the layers, dry the organic phase with anhydrous sodium sulfate and filter, and concentrate the filtrate under reduced pressure to obtain the crude product. The crude product is separated and purified by HPLC column {chromatography tube Column: Welch Xtimate C18 100*25mm*3µm; mobile phase: [H 2 O(0.05%HCl) -ACN]; acetonitrile%: 20%-50%, 8min}, to obtain compound 17. SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3μm; mobile phase: A (CO2) and B (methanol, containing 0.05% diisopropylamine amine); gradient: B%=5~50% , 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi, optical purity: 99.21%, peak time: 1.840. 1 H NMR (400 MHz, CD 3 OD) δ = 6.80-6.68 (m, 1H), 5.73-5.51 (m, 1H), 5.46-5.19 (m, 3H), 5.05-4.90 (m, 3H), 4.74-4.58 (m, 2H), 4.37-4.26 (m, 1H), 4.20- 4.06 (m, 2H), 4.05-3.84 (m, 3H), 3.79-3.59 (m, 2H), 3.54-3.43 (m, 1H), 3.42-3.35 (m, 1H), 3.31-3.24 (m, 1H) ), 3.13-2.89 (m, 3H), 2.82-2.52 (m, 2H), 2.50-2.42 (m, 1H), 2.41-2.30 (m, 5H), 2.29-2.18 (m, 1H).
實施例18
步驟2:化合物18-2的合成 將化合物18-1(0.4 g, 455.07 µmol, 1 eq)加入到無水二氯甲烷 (12 mL)中,加入三氟乙酸 (2.4 mL) ,25°C反應1.5小時,與化合物18-1(50 mg)批次合併處理,向反應液緩慢加入飽和碳酸氫鈉至pH為7-8,利用20 mL的二氯甲烷萃取,無水硫酸鈉乾燥過濾後濃縮旋乾得到化合物18-2。LCMS m/z =539.1[M+H]+ 。Step 2: Synthesis of compound 18-2. Compound 18-1 (0.4 g, 455.07 µmol, 1 eq) was added to anhydrous dichloromethane (12 mL), trifluoroacetic acid (2.4 mL) was added, and the reaction was carried out at 25°C for 1.5 20 hours, combined with compound 18-1 (50 mg) batches, slowly add saturated sodium bicarbonate to the reaction solution to a pH of 7-8, extract with 20 mL of dichloromethane, dry and filter with anhydrous sodium sulfate, concentrate and spin dry Compound 18-2 was obtained. LCMS m/z = 539.1 [M+H] + .
步驟5:化合物18A和18B的合成 將化合物18-2 (36.80 mg, 510.60 µmol, 35.04 µL, 1.1 eq),丙烯酸(36.80 mg, 510.60 µmol, 35.04 µL, 1.1 eq)和N,N-二異丙基乙胺 (179.97 mg, 1.39 mmol, 242.55 µL, 3 eq)加入到無水二氯甲烷(5 mL)中,降溫至-60°C,加入O-(7-氮雜苯並三氮唑-1-YL)-N,N,N,N-四甲基脲六氟膦鹽(176.50 mg, 464.18 µmol, 1 eq),-60°C反應30分鐘,將反應液利用10 mL二氯甲烷稀釋,用10 mL x 2的飽和氯化銨洗滌,無水硫酸鈉乾燥過濾後濃縮, 進行高效液相層析管柱分離純化(管柱: Phenomenex luna C18 100*40mm*5 µm;流動相: [H2 O (0.1%TFA)-ACN];乙腈%: 10%-40%,8min)後凍乾,SFC進行手性分離(管柱: DAICEL CHIRALCEL OD(250mm*30mm,10µm);流動相: [0.1%NH3 H2 O ETOH];乙醇%: 50%-50%,15min),得到化合物18A (手性出峰時間:1.479)。SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3µm;流動相:A (CO2 ) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi,光學純度100%。MS m/z =593.3[M+H]+ ,1 H NMR (400MHz,CDCl3 ) δ = 6.66 - 6.50 (m, 2H), 6.36 (d, J = 16.8 Hz, 1H), 5.76 (d, J=10.0 Hz, 1H), 5.20 (d, J = 7.6 Hz, 1H), 4.58-4.53 (m, 1H), 4.45 - 4.20 (m, 2H), 4.01 (s, 3H), 3.85 - 3.23 (m, 6H), 3.04 - 2.86 (m, 2H), 2.67 (s, 2H), 2.47 - 2.33 (m, 3H), 2.22 - 1.53 (m, 8H), 1.23 - 1.04 (m, 3H)。Step 5: Synthesis of Compound 18A and 18B Compound 18-2 (36.80 mg, 510.60 µmol, 35.04 µL, 1.1 eq), acrylic acid (36.80 mg, 510.60 µmol, 35.04 µL, 1.1 eq) and N,N-diisopropyl Ethylethylamine (179.97 mg, 1.39 mmol, 242.55 µL, 3 eq) was added to anhydrous dichloromethane (5 mL), the temperature was reduced to -60°C, and O-(7-azabenzotriazole-1 -YL)-N,N,N,N-tetramethylurea hexafluorophosphonium salt (176.50 mg, 464.18 µmol, 1 eq), react at -60°C for 30 minutes, dilute the reaction solution with 10 mL of dichloromethane, Wash with 10 mL x 2 saturated ammonium chloride, dry and filter with anhydrous sodium sulfate, concentrate, and perform high performance liquid chromatography column separation and purification (column: Phenomenex luna C18 100*40mm*5 µm; mobile phase: [H 2 O (0.1%TFA)-ACN]; Acetonitrile%: 10%-40%, 8min) and then freeze-dried, SFC for chiral separation (column: DAICEL CHIRALCEL OD (250mm*30mm, 10µm); mobile phase: [0.1 %NH 3 H 2 O ETOH]; ethanol %: 50%-50%, 15 min), to obtain compound 18A (chiral peak time: 1.479). SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3µm; mobile phase: A (CO 2 ) and B (methanol, containing 0.05% diisopropylamine amine); gradient: B%=5~50 %, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi, optical purity 100%. MS m/z =593.3[M+H] + , 1 H NMR (400MHz,CDCl 3 ) δ = 6.66 -6.50 (m, 2H), 6.36 (d, J = 16.8 Hz, 1H), 5.76 (d, J=10.0 Hz, 1H), 5.20 (d, J = 7.6 Hz, 1H), 4.58-4.53 (m, 1H), 4.45-4.20 (m, 2H), 4.01 (s, 3H), 3.85-3.23 (m, 6H), 3.04-2.86 (m, 2H), 2.67 (s, 2H), 2.47-2.33 (m, 3H), 2.22-1.53 (m, 8H), 1.23-1.04 (m, 3H).
得到化合物18B (手性出峰時間:1.642) SFC分析方法(管柱:Chiralcel OD-3, 50×4.6mm I.D., 3µm;流動相:A (CO2 ) 和 B (甲醇,含0.05%二異丙胺胺);梯度:B%=5~50%,3 min;流速: 3.4 mL/min;波長: 220nm;壓力: 1800psi,光學純度97.8%。LCMS m/z =593.3[M+H]+ 。1 H NMR (400MHz,CDCl3 ) δ = 6.72 - 6.48 (m, 2H), 6.35 (dd, J = 1.6, 16.8 Hz, 1H), 5.76 (d, J = 10.4 Hz, 1H), 5.20 (d, J = 7.6 Hz, 1H), 4.83 - 4.57 (m, 3H), 4.18 - 3.99 (m, 3H), 3.94 - 3.51 (m, 2H), 3.50 - 3.20 (m, 2H), 3.11 - 2.75 (m, 6H), 2.43 - 2.35 (m, 3H), 2.35 – 1.80 (m, 8H), 1.38 (d, J = 6.4 Hz, 3H)。Obtained compound 18B (chiral peak time: 1.642) SFC analysis method (column: Chiralcel OD-3, 50×4.6mm ID, 3µm; mobile phase: A (CO 2 ) and B (methanol, containing 0.05% diisocyanate) Propylamine amine); Gradient: B%=5-50%, 3 min; Flow rate: 3.4 mL/min; Wavelength: 220nm; Pressure: 1800psi, optical purity 97.8%. LCMS m/z =593.3[M+H] + . 1 H NMR (400MHz,CDCl 3 ) δ = 6.72-6.48 (m, 2H), 6.35 (dd, J = 1.6, 16.8 Hz, 1H), 5.76 (d, J = 10.4 Hz, 1H), 5.20 (d, J = 7.6 Hz, 1H), 4.83-4.57 (m, 3H), 4.18-3.99 (m, 3H), 3.94-3.51 (m, 2H), 3.50-3.20 (m, 2H), 3.11-2.75 (m, 6H), 2.43-2.35 (m, 3H), 2.35-1.80 (m, 8H), 1.38 (d, J = 6.4 Hz, 3H).
實施例19
實施例20
步驟2: 中間體20-2的製備 20℃條件下,將化合物17-4A(12.26 mg, 77.02 µmol)溶於無水四氫呋喃(2 mL)中,加入三級丁醇鈉(7.40 mg, 77.02 µmol),反應液繼續攪拌30分鐘,加入化合物20-1(50 mg, 59.25 µmol)的四氫呋喃(0.5 mL)溶液,反應液在該溫度下繼續攪拌0.5小時。減壓除去有機溶劑,所得粗產物經薄層層析製備板(展開劑:二氯甲烷:甲醇=10:1)分離純化,得到化合物20-2。MS m/z =923.6 [M+H]+ 。Step 2: Preparation of Intermediate 20-2 At 20℃, dissolve compound 17-4A (12.26 mg, 77.02 µmol) in anhydrous tetrahydrofuran (2 mL), and add tertiary butoxide sodium (7.40 mg, 77.02 µmol) , The reaction solution was stirred for 30 minutes, compound 20-1 (50 mg, 59.25 µmol) in tetrahydrofuran (0.5 mL) was added, and the reaction solution was stirred at this temperature for 0.5 hours. The organic solvent was removed under reduced pressure, and the obtained crude product was separated and purified by thin layer chromatography preparation plate (developing solvent: dichloromethane: methanol = 10:1) to obtain compound 20-2. MS m/z = 923.6 [M+H] + .
步驟3: 化合物20-3的製備 將化合物20-2(45 mg, 48.75 µmol)溶於無水二氯甲烷(2 mL)中,加入三氟乙酸(1.5 mL),反應液在20℃下繼續攪拌2小時。減壓除去溶劑,所得粗產物溶於20 mL二氯甲烷中,加入固體碳酸氫鈉3g,室溫下繼續攪拌1小時,過濾,減壓除去有機溶劑,得到粗產物20-3,該化合物不經進一步純化直接用於下一步反應。Step 3: Preparation of compound 20-3 Compound 20-2 (45 mg, 48.75 µmol) was dissolved in dry dichloromethane (2 mL), trifluoroacetic acid (1.5 mL) was added, and the reaction solution was stirred at 20°C for 2 hours. The solvent was removed under reduced pressure, and the obtained crude product was dissolved in 20 mL of dichloromethane, and 3 g of solid sodium bicarbonate was added. The mixture was stirred for 1 hour at room temperature, filtered and the organic solvent was removed under reduced pressure to obtain the crude product 20-3. After further purification, it was directly used in the next reaction.
步驟5: 化合物20的製備
20°C條件下,將化合物20-3(20 mg, 34.33 µmol)溶於無水二氯甲烷(2 mL)中,加入二異丙基乙胺(13.31 mg, 102.99 µmol, 17.94 µL)和烯丙醯氯(4.66 mg, 51.49 µmol, 4.20 µL),反應液在該溫度下繼續攪拌16小時。減壓除去有機溶劑,所得粗產物通過高效液相層析法(管柱: Welch Xtimate C18 100*40mm*3µm;流動相: [水(0.075%三氟乙酸)-乙腈];乙腈: 22%-52%, 8分鐘)分離純化,得到化合物20的三氟乙酸鹽。MS m/z =637.4 [M+H]+
。Step 5: Preparation of
生物測試數據: 實驗例1:化合物對KRASG12C 突變的MIA-PA-CA-2細胞增殖抑制作用的測試 1.1實驗目的 測試化合物對KRASG12C 突變的MIA-PA-CA-2細胞增殖抑制的IC50 。Biological test data: Experimental example 1: Test of the compound's inhibitory effect on the proliferation of KRAS G12C mutant MIA-PA-CA-2 cells 1.1 Experimental purpose The IC 50 of the compound to inhibit the proliferation of KRAS G12C mutant MIA-PA-CA-2 cells .
1.2試劑 本研究使用的主要試劑包括CellTiter-Glo(Promega,貨號:G7573)。1.2 Reagents The main reagents used in this study include CellTiter-Glo (Promega, catalog number: G7573).
1.3儀器 本研究所使用主要儀器為PerkinElmer EnVision多功能微量盤分光光譜儀。1.3 Apparatus The main instrument used in this research is the PerkinElmer EnVision Multi-Function Micro Disk Spectrometer.
1.4實驗方法
1)貼壁細胞經胰酶消化處理成細胞懸液,並對細胞懸液進行計數備用。
2)取適量細胞至離心管並用細胞培養液補足至需要體積,鋪至96孔盤,終密度為2000 細胞/孔,100 μL培養液。
3)培養24hr後,將化合物用DMSO配置成10 mM,並用DPBS(杜比可磷酸緩衝液)以3倍梯度稀釋9個點,每孔加入10 μL,兩複孔。實驗對照孔(Con)每孔加入10 μL DPBS。
4)同天,取一塊未加藥處理的細胞培養盤,加入50 μL CellTiter Glo,利用EnVision進行螢光讀值,標記為Day0讀值。
5)加藥處理後的細胞培養72 h後,去除培養盤,向細胞盤中加入50 μL CellTiter Glo,利用EnVision進行螢光讀值。
6) 數據分析:按下列公式計算各孔對細胞的抑制率:
1.5 實驗結果
表1. 本發明化合物對KRASG12C突變的MIA-PA-CA-2細胞增殖抑制的測試結果
實驗例2:H358細胞實驗 2.1實驗目的 測試化合物對KRASG12C 突變的H358細胞增殖抑制的IC50 。IC 50 H358 cells 2.1 Experimental test object of the test compound KRAS G12C mutant H358 cell proliferation: Experimental Example 2.
2.2試劑 本研究使用的主要試劑包括RPMI-1640培養基,盤尼西林/鏈黴素抗生素購自維森特,胎牛血清購自Biosera。CellTiter-Glo(細胞活率化學發光檢測試劑)試劑購自Promega。NCI-H358細胞株購自中國科學院細胞庫。2.2 Reagents The main reagents used in this study included RPMI-1640 medium, penicillin/streptomycin antibiotics were purchased from Vicente, and fetal bovine serum was purchased from Biosera. CellTiter-Glo (cell viability chemiluminescence detection reagent) reagent was purchased from Promega. The NCI-H358 cell line was purchased from the Cell Bank of the Chinese Academy of Sciences.
2.3儀器 本研究所使用主要儀器為Nivo多標記分析儀(PerkinElmer)。2.3 Apparatus The main instrument used in this research is the Nivo multi-label analyzer (PerkinElmer).
2.4實驗方法: 1)將NCI-H358細胞種於白色96孔盤中,80μL細胞懸液每孔,其中包含4000個NCI-H358細胞。細胞盤置於二氧化碳培養箱中過夜培養。 2)將待測化合物用微量吸管進行5倍稀釋至第9個濃度,即從2 mM稀釋至5.12nM,設置雙複孔實驗。向中間盤中加入78 μL培養基,再按照對應位置,轉移2 μL每孔的梯度稀釋化合物至中間盤,混勻後轉移20μL每孔到細胞盤中。轉移到細胞盤中的化合物濃度範圍是10μM至0.0256nM。細胞盤置於二氧化碳培養箱中培養5天。另準備一塊細胞盤,在加藥當天讀取訊號值作為最大值(下面方程式中Max值)參與數據分析。向此細胞盤每孔加入25 μL細胞活率化學發光檢測試劑,室溫孵育10分鐘使發光訊號穩定。採用多標記分析儀讀數。 3)向細胞盤中加入每孔25 μL的細胞活率化學發光檢測試劑,室溫孵育10分鐘使發光訊號穩定。採用多標記分析儀讀數。2.4 Experimental method: 1) Plant NCI-H358 cells in a white 96-well plate, 80 μL of cell suspension per well, which contains 4000 NCI-H358 cells. The cell dish was placed in a carbon dioxide incubator for overnight culture. 2) Dilute the test compound 5-fold with a micropipette to the 9th concentration, that is, from 2 mM to 5.12 nM, and set up a double-well experiment. Add 78 μL of culture medium to the middle plate, and then transfer 2 μL of the serially diluted compound per well to the middle plate according to the corresponding position. After mixing, transfer 20 μL of each well to the cell plate. The concentration of the compound transferred to the cell dish ranges from 10 μM to 0.0256 nM. The cell dish was placed in a carbon dioxide incubator for 5 days. In addition, prepare a cell plate, and read the signal value as the maximum value (Max value in the following equation) on the day of dosing to participate in data analysis. Add 25 μL of cell viability chemiluminescence detection reagent to each well of this cell dish, and incubate at room temperature for 10 minutes to stabilize the luminescence signal. Use multi-marker analyzer to read. 3) Add 25 μL of cell viability chemiluminescence detection reagent per well to the cell dish, and incubate at room temperature for 10 minutes to stabilize the luminescence signal. Use multi-marker analyzer to read.
數據分析: 利用方程式(Sample-Min)/(Max-Min)*100%將原始數據換算成抑制率,IC50 的值即可通過四參數進行曲線擬合得出(GraphPad Prism中"log(inhibitor) vs. response -- Variable slope" 模式得出)。表2提供了本發明的化合物對NCI-H358細胞增殖的抑制活性。Data analysis: Using the equation (Sample-Min)/(Max-Min)*100% to convert the original data into the inhibition rate, the value of IC 50 can be obtained by four-parameter curve fitting ("log(inhibitor in GraphPad Prism) ) vs. response - Variable slope" mode). Table 2 provides the inhibitory activity of the compounds of the present invention on the proliferation of NCI-H358 cells.
表2. 本發明化合物對KRASG12C突變的H358細胞增殖抑制的測試結果
實驗例3:肝細胞代謝穩定性 實驗目的:評定受試化合物分別在CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴、人肝細胞中的代謝穩定性。Experimental example 3: Metabolic stability of liver cells Experimental purpose: to evaluate the metabolic stability of the test compounds in CD-1 mice, SD rats, Migru dogs, stone crab macaques, and human hepatocytes.
實驗操作:準備若干96孔樣品沉澱盤,分別命名為T0、T15、T30、T60、T90、T120、T0-MC、T120-MC和空白基質。提前取出復甦培養液和孵育培養液,放置在37˚C水浴鍋中預熱。從液氮罐中取出凍存的肝細胞,立即浸沒到37˚C水浴中(約90秒)。待凍存部分溶化鬆動後,分別倒入含有40 mL復甦培養液的離心管中,輕柔的顛倒讓細胞在復甦培養液中重懸。室溫條件下,100 ×g離心5分鐘,移除上清液,用適當體積的孵育培養液重懸肝細胞,用台盼藍染色法計算細胞活率。將198 μL的肝細胞混懸液 (0.51×106 cells/mL)加入到已預熱的孵育盤中,培養液對照組加入198 μL不含肝細胞的孵育培養液至T0-MC和T120-MC孵育盤中,所有孵育盤在37℃培養箱中預孵育10分鐘。然後加入2 μL供試品和對照化合物工作液,混勻,立即將孵育盤放入培養箱內的搖盤機中,啟動計時器開始反應。每個化合物的每個時間點準備2個重複樣本。孵育條件為37˚C、飽和濕度、含5%CO2 。測試體系中,供試品的終濃度為1 µM,對照品的終濃度為3 µM,肝細胞的終濃度為0.5×106 cells /mL,總有機溶劑的終濃度為0.96%,其中DMSO的終濃度為0.1%。相應時間點孵育結束時,取出孵育盤,取出25 μL化合物和對照化合物與細胞的混合液加入到含有125 μL終止液(含有200 ng/mL甲苯磺丁脲和拉貝諾爾的乙腈溶液)的樣品盤中。對於Blank樣品盤,直接加入25 μL不含肝細胞的孵育培養液。所有樣品盤封膜後在搖盤機上以600 rpm搖10分鐘後,3220×g離心20分鐘。供試品和對照品上清液用超純水以1:3的比例稀釋。所有樣品混勻後用LC/MS/MS的方法進行分析。 實驗結果如表3所示。Experimental operation: Prepare a number of 96-well sample precipitation trays, named T0, T15, T30, T60, T90, T120, T0-MC, T120-MC and blank matrix respectively. Take out the resuscitation medium and incubation medium in advance, and place them in a 37˚C water bath to preheat. Take out the frozen liver cells from the liquid nitrogen tank and immediately immerse them in a 37˚C water bath (about 90 seconds). After the frozen parts are melted and loosened, pour them into centrifuge tubes containing 40 mL of resuscitation medium, gently invert them to resuspend the cells in the resuscitation medium. Centrifuge at 100 × g for 5 minutes at room temperature, remove the supernatant, resuspend the hepatocytes in an appropriate volume of incubation medium, and calculate the cell viability by trypan blue staining. Add 198 μL of hepatocyte suspension (0.51×106 cells/mL) to the pre-heated incubation plate, and add 198 μL of incubation medium without hepatocytes to T0-MC and T120-MC for the control group. In the incubation dish, all the incubation dishes are pre-incubated for 10 minutes in a 37°C incubator. Then add 2 μL of the working solution of the test substance and the reference compound, mix them, and immediately put the incubation plate into the shaker in the incubator, and start the timer to start the reaction. Prepare 2 replicate samples for each time point for each compound. The incubation conditions are 37˚C, saturated humidity, and 5% CO 2 . In the test system, the final concentration of the test substance is 1 µM, the final concentration of the reference substance is 3 µM, the final concentration of hepatocytes is 0.5×106 cells/mL, and the final concentration of total organic solvents is 0.96%. The concentration is 0.1%. At the end of the incubation at the corresponding time point, take out the incubation plate, take out 25 μL of the mixture of compound and control compound and cells and add to the sample containing 125 μL of stop solution (containing 200 ng/mL tolbutamide and Rabenol in acetonitrile) Intraday. For the Blank sample plate, add 25 μL of incubation medium without hepatocytes directly. After sealing all the sample pans, they were shaken on a plate shaker at 600 rpm for 10 minutes, and then centrifuged at 3220×g for 20 minutes. The supernatant of the test substance and the reference substance was diluted with ultrapure water in a ratio of 1:3. All samples were mixed and analyzed by LC/MS/MS method. The experimental results are shown in Table 3.
表3. 受試化合物CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴、人肝細胞中的代謝穩定性
實驗例4: 體外肝微粒體穩定性研究 實驗目的:評定受試化合物分別在CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴、人肝微粒體中的代謝穩定性。Experimental example 4: In vitro liver microsome stability study Experimental purpose: to evaluate the metabolic stability of the test compounds in CD-1 mice, SD rats, Migru dogs, stone crab macaques, and human liver microsomes.
實驗操作:準備2塊96孔孵育盤,分別命名為T60孵育盤和NCF60孵育盤。 在T60孵育盤和NCF60孵育盤上分別加入445μL微粒體工作液(肝微粒體蛋白濃度為0.56mg/mL),然後將上述孵育盤放置於37°C水浴鍋中預孵育大約10分鐘。Experimental operation: Prepare two 96-well incubation plates, named T60 incubation plate and NCF60 incubation plate, respectively. Add 445μL of microsomal working solution (liver microsomal protein concentration is 0.56mg/mL) on the T60 incubation plate and NCF60 incubation plate respectively, and then place the above incubation plate in a 37°C water bath for pre-incubation for about 10 minutes.
預孵育結束後,在T60孵育盤和NCF60孵育盤上分別加入5μL供試品或對照化合物工作液,混勻。在NCF60孵育盤上每孔添加50μL磷酸鉀鹽緩衝液啟動反應;在T0終止盤中加入180μL的終止液(含200ng/mL甲苯磺丁脲和200ng/mL拉貝洛爾的乙腈溶液)和6 uL的NADPH再生體系工作液,從T60孵育盤中取出54μL樣品至T0終止盤(T0樣品產生)。在T60孵育盤上每孔添加44μLNADPH再生體系工作液啟動反應。在Blank盤中只添加54μL微粒體工作液、6 uL的NADPH再生體系工作液和180μL的終止液。因此,在供試品或對照化合物的樣品中,化合物、睪固酮、雙氯芬酸和普羅帕酮的反應終濃度為1μM,肝微粒體的濃度為0.5mg/mL,DMSO和乙腈在反應體系中的終濃度分別為0.01%(v/v)和0.99%(v/v)。After the pre-incubation is over, add 5 μL of the test product or control compound working solution to the T60 incubation plate and the NCF60 incubation plate, respectively, and mix well. Add 50μL of potassium phosphate buffer to each well on the NCF60 incubation plate to start the reaction; add 180μL of stop solution (containing 200ng/mL tolbutamide and 200ng/mL labetalol in acetonitrile) and 6 uL NADPH regeneration system working solution, take 54μL sample from T60 incubation plate to T0 stop plate (T0 sample generation). Add 44μL of NADPH regeneration system working solution to each well on the T60 incubation plate to start the reaction. Add only 54μL of microsomal working solution, 6 uL of NADPH regeneration system working solution and 180μL of stop solution to the Blank disk. Therefore, in the test product or control compound sample, the final reaction concentration of compound, testosterone, diclofenac and propafenone is 1μM, the concentration of liver microsomes is 0.5mg/mL, and the final concentration of DMSO and acetonitrile in the reaction system They are 0.01% (v/v) and 0.99% (v/v).
孵育適當時間(如5、15、30、45和60分鐘)後,分別在每個終止盤的樣品孔中加入180μL的終止液(含200ng/mL 甲苯磺丁脲和200ng/mL拉貝洛爾的乙腈溶液),之後從T60孵育盤中取出60μL樣品以終止反應。所有樣品盤搖勻並在3220 ×g離心20分鐘,然後每孔取80μL上清液稀釋到240μL純水中用於液相層析串聯質譜分析,液相層析串聯質譜分析所有樣品進樣分析。After incubating for an appropriate time (such as 5, 15, 30, 45, and 60 minutes), add 180 μL of stop solution (containing 200 ng/mL tolbutamide and 200 ng/mL labetalol to the sample wells of each stop plate). Acetonitrile solution), and then take out 60μL sample from the T60 incubation plate to stop the reaction. Shake all sample plates and centrifuge at 3220 × g for 20 minutes, and then take 80 μL of supernatant from each well and dilute to 240 μL of pure water for liquid chromatography tandem mass spectrometry analysis, and liquid chromatography tandem mass spectrometry analysis for all samples injection analysis .
表4. 受試化合物CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴、人肝微粒體的代謝穩定性
實驗例5: 血漿穩定研究 實驗目的:評定受試化合物分別在CD-1小鼠、人的血漿穩定性。Experimental Example 5: Study on Plasma Stability Experimental purpose: to evaluate the plasma stability of the test compounds in CD-1 mice and humans.
實驗操作:將凍存的血漿解凍10~20分鐘,待血漿完全解凍後,將其置於離心機中以3220×g離心5分鐘,去除其中存在的懸浮物和沉澱物。準備96孔孵育盤,分別命名為T0、T10、T30、T60、T120。將孵育盤中對應地加入98 μL的小鼠、大鼠、犬、猴和人空白血漿,然後加入2 μL化合物或對照化合物的工作液加入到對應的孵育盤中,每個樣品準備兩個平行孔。所有的樣品在37˚C水浴鍋中進行孵育。化合物和對照化合物比沙可啶、馬來酸依那普利、普魯卡因和普魯本辛的最終孵育濃度為2 μM,最終有機相含量為2.0%。每一個孵育時間點結束時,取出相應的孵育盤,向每個對應的樣品孔中加入400 μL含有200 ng/mL甲苯磺丁脲和拉貝諾爾的乙腈溶液沉澱蛋白。所有樣品盤封膜並搖勻後,3220×g離心20 分鐘。取50 µL上清液加入100 µL超純水稀釋,所有樣品混勻後用LC/MS/MS的方法分析。Experimental operation: Thaw the frozen plasma for 10-20 minutes. After the plasma is completely thawed, place it in a centrifuge at 3220×g for 5 minutes to remove suspended solids and sediments. Prepare 96-well incubation plates, named T0, T10, T30, T60, T120. Correspondingly add 98 μL of mouse, rat, dog, monkey and human blank plasma to the incubation plate, and then add 2 μL of the compound or the working solution of the control compound to the corresponding incubation plate. Prepare two parallels for each sample hole. All samples were incubated in a 37˚C water bath. The final incubation concentration of the compound and the control compound bisacodyl, enalapril maleate, procaine and probensine was 2 μM, and the final organic phase content was 2.0%. At the end of each incubation time point, take out the corresponding incubation plate, and add 400 μL of acetonitrile solution containing 200 ng/mL tolbutamide and Rabenol to each corresponding sample well to precipitate the protein. After sealing and shaking all sample pans, centrifuge at 3220×g for 20 minutes. Dilute 50 µL of the supernatant with 100 µL of ultrapure water, mix all samples and analyze them by LC/MS/MS.
表5. 受試化合物CD-1小鼠、人的血漿穩定性
實驗例6: 全血穩定研究 實驗目的:評定受試化合物分別在評定受試化合物分別在CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴的全血穩定性。Experimental Example 6: Whole blood stability study The purpose of the experiment: to evaluate the whole blood stability of the test compounds in CD-1 mice, SD rats, Migru dogs, and stone crab macaques.
實驗操作:在實驗當天或實驗的前一天,採用抗凝劑EDTA-K2採集新鮮的CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴全血。在實驗開始前,將全血與PBS進行1:1(v:v)混合,放置於37℃水浴鍋中預熱10~20分鐘。準備96孔孵育盤,分別命名為T0、T30、T60、T240。在對應的孵育盤中,包括T0、T30、T60和T240孵育盤,將2 μL化合物或對照化合物的工作液與98 μL的小鼠、大鼠、犬、猴和人空白全血混合,每個樣品準備兩個平行孔。所有的樣品在37˚C水浴鍋中進行孵育。化合物的最終孵育濃度為5 μM,對照化合物的最終孵育濃度為2 μM。每一個時間點孵育結束時,取出相應的孵育盤,立即向對應的樣品孔中加入100 μL超純水,混勻,然後加入800 μL含有200 ng/mL甲苯磺丁脲和拉貝諾爾的乙腈溶液沉澱蛋白。樣品盤封膜並搖勻後,3220×g離心20 分鐘。取150 µL上清液用LC/MS/MS的方法分析。Experimental operation: On the day of the experiment or the day before the experiment, the anticoagulant EDTA-K2 was used to collect fresh whole blood of CD-1 mice, SD rats, MiGru dogs, and stone crab macaques. Before the start of the experiment, mix the whole blood with PBS 1:1 (v:v), and place it in a 37°C water bath for preheating for 10-20 minutes. Prepare 96-well incubation plates, named T0, T30, T60, T240. In the corresponding incubation plates, including T0, T30, T60 and T240 incubation plates, mix 2 μL of the working solution of the compound or control compound with 98 μL of blank whole blood from mice, rats, dogs, monkeys and humans, each Prepare two parallel holes for the sample. All samples were incubated in a 37˚C water bath. The final incubation concentration of the compound was 5 μM, and the final incubation concentration of the control compound was 2 μM. At the end of each time point of incubation, take out the corresponding incubation plate, immediately add 100 μL of ultrapure water to the corresponding sample well, mix well, and then add 800 μL of acetonitrile containing 200 ng/mL tolbutamide and Rabenol The solution precipitates the protein. After sealing and shaking the sample pan, centrifuge at 3220×g for 20 minutes. Take 150 µL of the supernatant and analyze it by LC/MS/MS.
表6. 受試化合物CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴的全血穩定性
實驗例7: 蛋白結合率研究 實驗目的:採用平衡透析法測定受試化合物在CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴和人血漿中的蛋白結合率。Experimental Example 7: Study on Protein Binding Rate Experimental purpose: The balance dialysis method was used to determine the protein binding rate of the test compound in the plasma of CD-1 mice, SD rats, Migru dogs, stone crab macaques and humans.
實驗操作:採用上述五個物種的血漿分別配製化合物濃度為2µM的血漿樣品,置於96孔平衡透析裝置中,在37±1°C下用磷酸鹽緩衝溶液透析4 h。本實驗採用華法林作為對照化合物。血漿和透析緩衝液中待測物的濃度用LC-MS/MS法進行測定。Experimental operation: The plasma samples of the above five species were used to prepare plasma samples with a compound concentration of 2 µM, placed in a 96-well balanced dialysis device, and dialyzed with phosphate buffer solution at 37±1°C for 4 h. This experiment uses warfarin as the control compound. The concentration of the analyte in plasma and dialysis buffer was determined by LC-MS/MS method.
表7. 受試化合物CD-1小鼠、SD大鼠、米格魯犬、石蟹獼猴、人的蛋白結合率
實驗例8: 體內藥物動力學研究 1)SD大鼠口服及靜脈注射受試化合物的藥物動力學研究 受試化合物與5%二甲基亞碸/95%(10%羥丙基-β-環糊精)溶液混合,渦旋並超聲,製備得到1 mg/mL澄清溶液,微孔濾膜過濾後備用。選取7至10週齡的雄性SD大鼠,靜脈注射、口服給予候選化合物溶液。收集一定時間的全血,製備得到血漿,以LC-MS/MS方法分析藥物濃度,並用Phoenix WinNonlin 軟體(美國Pharsight公司)計算藥動參數。實驗結果如表8所示:Experimental Example 8: In vivo pharmacokinetic study 1) Study on the pharmacokinetics of the test compound in SD rats by oral and intravenous injection The test compound was mixed with 5% dimethyl sulfoxide/95% (10% hydroxypropyl-β-cyclodextrin) solution, vortexed and sonicated to prepare a clear solution of 1 mg/mL, which was filtered through a microporous membrane. use. Male SD rats aged 7 to 10 weeks were selected, and the candidate compound solution was administered intravenously or orally. Collect whole blood for a certain period of time, prepare plasma, analyze drug concentration by LC-MS/MS method, and calculate pharmacokinetic parameters with Phoenix WinNonlin software (Pharsight, USA). The experimental results are shown in Table 8:
表8. 受試化合物的藥物動力學結果
2) CD小鼠口服及靜脈注射受試化合物的藥物動力學研究 受試化合物與5%二甲基亞碸/95%(10%羥丙基-β-環糊精)溶液混合,渦旋並超聲,製備得到1 mg/mL澄清溶液,微孔濾膜過濾後備用。選取7至10週齡的雄性CD小鼠,靜脈注射、口服給予候選化合物溶液。收集一定時間的全血,製備得到血漿,以LC-MS/MS方法分析藥物濃度,並用Phoenix WinNonlin 軟體(美國Pharsight公司)計算藥動參數。實驗結果如表9所示:2) Studies on the pharmacokinetics of oral and intravenous injection of test compounds in CD mice The test compound was mixed with 5% dimethyl sulfoxide/95% (10% hydroxypropyl-β-cyclodextrin) solution, vortexed and sonicated to prepare a clear solution of 1 mg/mL, which was filtered through a microporous membrane. use. Male CD mice aged 7 to 10 weeks were selected, and the candidate compound solution was administered intravenously or orally. Collect whole blood for a certain period of time, prepare plasma, analyze drug concentration by LC-MS/MS method, and calculate pharmacokinetic parameters with Phoenix WinNonlin software (Pharsight, USA). The experimental results are shown in Table 9:
表9. 受試化合物的藥物動力學結果
實驗例9:體內藥效學研究 人胰腺癌Mia PaCa-2細胞裸小鼠皮下移植腫瘤Balb/c Nude小鼠模型的體內藥效學研究。Experimental Example 9: In vivo pharmacodynamic study In vivo pharmacodynamics study of human pancreatic cancer Mia PaCa-2 cells transplanted subcutaneously in nude mice Balb/c Nude mouse model.
1.細胞培養和腫瘤組織準備 細胞培養:人胰腺癌Mia PaCa-2細胞(ATCC-CRL-1420)體外單層培養,培養條件為DMEM培養基中加10%胎牛血清,2.5%馬血清,37℃5%二氧化碳孵箱培養。一週兩次用胰酶-EDTA進行常規消化處理繼代。當細胞飽和度為80%-90%,數量到達要求時,收取細胞,計數,重懸於適量PBS中,1:1加入基質膠,獲取細胞密度為25 x 106 cells/mL的細胞懸液。1. Cell culture and tumor tissue preparation Cell culture: Human pancreatic cancer Mia PaCa-2 cells (ATCC-CRL-1420) are cultured in a monolayer in vitro. The culture conditions are DMEM medium with 10% fetal bovine serum, 2.5% horse serum, and 37℃5% carbon dioxide incubator. Use pancreatin-EDTA for routine digestion and subculture twice a week. When the cell saturation is 80%-90% and the number reaches the requirement, the cells are collected, counted, and resuspended in an appropriate amount of PBS. Matrigel is added 1:1 to obtain a cell suspension with a cell density of 25 x 106 cells/mL.
細胞接種:將0.2 mL(5×106 cells/mouse個)Mia PaCa-2細胞(加基質膠,體積比為1:1)皮下接種於每隻小鼠的右後背,腫瘤平均體積達到190 mm3時,根據腫瘤體積進行隨機分組,按照表10中的方案開始給藥。Cell inoculation: 0.2 mL (5×106 cells/mouse) of Mia PaCa-2 cells (with matrigel, volume ratio 1:1) subcutaneously inoculated on the right back of each mouse, when the average tumor volume reaches 190 mm3 , Randomly grouped according to tumor volume, and started administration according to the schedule in Table 10.
表10. 實驗動物分組及給藥方案
2.腫瘤測量和實驗指標 每週兩次用游標卡尺測量腫瘤直徑。腫瘤體積的計算公式為:V = 0.5a × b2 ,a和b分別表示腫瘤的長徑和短徑。2. Tumor measurement and experimental indicators Use vernier calipers to measure tumor diameter twice a week. The calculation formula for tumor volume is: V = 0.5a × b 2 , where a and b represent the long diameter and short diameter of the tumor, respectively.
化合物的抑瘤療效用TGI(%)或相對腫瘤增殖率T/C(%)評價。 相對腫瘤增殖率T/C(%) = TRTV / CRTV × 100 %(TRTV:治療組RTV;CRTV:陰性對照組RTV)。根據腫瘤測量的結果計算出相對腫瘤體積(relative tμmor volμme,RTV),計算公式為 RTV = Vt / V0,其中V0是分組給藥時(即D0)測量所得平均腫瘤體積,Vt為某一次測量時的平均腫瘤體積,TRTV與CRTV取同一天數據。The anti-tumor efficacy of the compound was evaluated by TGI (%) or the relative tumor proliferation rate T/C (%). Relative tumor proliferation rate T/C (%) = TRTV / CRTV × 100% (TRTV: treatment group RTV; CRTV: negative control group RTV). Calculate the relative tumor volume (relative tμmor volμme, RTV) based on the results of the tumor measurement. The calculation formula is RTV = Vt / V0, where V0 is the average tumor volume measured during group administration (ie D0), and Vt is the time of a certain measurement. The average tumor volume, TRTV and CRTV data on the same day.
TGI (%),反映腫瘤生長抑制率。TGI(%)=[(1-(某處理組給藥結束時平均瘤體積-該處理組開始給藥時平均瘤體積))/(溶劑對照組治療結束時平均瘤體積-溶劑對照組開始治療時平均瘤體積)]×100%。TGI (%), reflects the tumor growth inhibition rate. TGI(%)=[(1-(Average tumor volume at the end of a certain treatment group-average tumor volume at the beginning of the treatment group))/(Average tumor volume at the end of treatment in the solvent control group-start treatment in the solvent control group Time average tumor volume)]×100%.
3.實驗結果 實驗結果如圖1、2所示。 給藥22天時結果如表11所示。3. Experimental results The experimental results are shown in Figures 1 and 2. The results at 22 days of administration are shown in Table 11.
表11. 給藥第22天下的T/C及TGI
無。none.
圖1. 不同給藥量隨著時間延長的腫瘤體積變化。 圖2. 不同給藥量隨著時間延長的動物體重變化。Figure 1. Tumor volume changes with different doses over time. Figure 2. Changes in animal body weight with different doses over time.

Claims (22)

































































































Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010323035.4 | 2020-04-22 | ||
| CN202010323035 | 2020-04-22 | ||
| CN202010953203 | 2020-09-11 | ||
| CN202010953203.8 | 2020-09-11 | ||
| CN202011593642.9 | 2020-12-29 | ||
| CN202011593642 | 2020-12-29 | ||
| WOPCT/CN2021/080278 | 2021-03-11 | ||
| PCT/CN2021/080278 WO2021180181A1 (en) | 2020-03-12 | 2021-03-11 | Pyrimidoheterocyclic compounds and application thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202140490A true TW202140490A (en) | 2021-11-01 |
| TWI811656B TWI811656B (en) | 2023-08-11 |
Family
ID=80783163
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW110112436A TWI811656B (en) | 2020-04-22 | 2021-04-06 | Pyrimido heterocyclic compounds and application thereof |
Country Status (1)
| Country | Link |
|---|---|
| TW (1) | TWI811656B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI820901B (en) * | 2021-09-10 | 2023-11-01 | 大陸商德昇濟醫藥(無錫)有限公司 | Crystal form of pyrimido-heterocyclic compounds and preparation method thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017201161A1 (en) * | 2016-05-18 | 2017-11-23 | Mirati Therapeutics, Inc. | Kras g12c inhibitors |
| TW201942116A (en) * | 2018-02-09 | 2019-11-01 | 美商輝瑞股份有限公司 | Tetrahydroquinazoline derivatives useful as anticancer agents |
-
2021
- 2021-04-06 TW TW110112436A patent/TWI811656B/en active
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI820901B (en) * | 2021-09-10 | 2023-11-01 | 大陸商德昇濟醫藥(無錫)有限公司 | Crystal form of pyrimido-heterocyclic compounds and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI811656B (en) | 2023-08-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN115298174B (en) | Pyrimidine heterocyclic compound and application thereof | |
| CN115380026B (en) | Protein degradation modulators and methods of use thereof | |
| CN112689638B (en) | Cyclopropylamine compound serving as LSD1 inhibitor and application thereof | |
| CN116848112A (en) | Pyridine [4,3-d ] pyrimidine compounds | |
| TW202120506A (en) | Pyrimidopyrrole compound as dna-pk inhibitor | |
| TW202233630A (en) | Pyrimidinopyrane compounds | |
| JP2022550427A (en) | Compounds and uses thereof as PD-1/PD-L1 small molecule inhibitors | |
| CN111344290A (en) | Macrocyclic compound as Wee1 inhibitor and application thereof | |
| EP4269403A1 (en) | Aromatic heterocyclic compound, and pharmaceutical composition and application thereof | |
| TW202138372A (en) | Aminopyrimidine compound and derivative thereof as dna-pk inhibitor | |
| TW202346306A (en) | Bridged ring substituted heteroaryl fused pyran derivatives and applications thereof | |
| TWI811656B (en) | Pyrimido heterocyclic compounds and application thereof | |
| EP4349828A1 (en) | Nitrogen-containing heterocyclic compound, preparation method therefor and application thereof | |
| CN115667247A (en) | Pyridine derivatives and application thereof | |
| BR122023026304B1 (en) | PYRIMIDOHETEROCYCLIC COMPOUNDS AND THEIR USE FOR THE MANUFACTURING OF A MEDICINE TO TREAT RELATED DISEASES | |
| TWI795129B (en) | Pyridopyrimidinone compounds | |
| EA047880B1 (en) | PYRIMIDO-HETERACYCLIC COMPOUNDS AND THEIR APPLICATIONS | |
| TW202225162A (en) | Aromatic heterocyclic compound, and pharmaceutical composition and application thereof | |
| CN116981660A (en) | Fluoro vinyl biphenyl derivative and application thereof |