KR20080034993A - 선택적 hm74a 작동제로서의 크산틴 유도체 - Google Patents
선택적 hm74a 작동제로서의 크산틴 유도체 Download PDFInfo
- Publication number
- KR20080034993A KR20080034993A KR1020087005717A KR20087005717A KR20080034993A KR 20080034993 A KR20080034993 A KR 20080034993A KR 1020087005717 A KR1020087005717 A KR 1020087005717A KR 20087005717 A KR20087005717 A KR 20087005717A KR 20080034993 A KR20080034993 A KR 20080034993A
- Authority
- KR
- South Korea
- Prior art keywords
- mmol
- chloro
- butyl
- mixture
- purine
- Prior art date
Links
- 0 CCCCCN(c1c(C(N2CCCC3=C*(Cc(c(F)ccc4)c4Cl)N=C3)=O)[n]c(*)n1)C2=O Chemical compound CCCCCN(c1c(C(N2CCCC3=C*(Cc(c(F)ccc4)c4Cl)N=C3)=O)[n]c(*)n1)C2=O 0.000 description 7
- WWPCFNUYRIATBU-UHFFFAOYSA-N CCCCCN(c1c(C(N2CCCCc3n[o]c(-c4c[s]cc4)n3)=O)[nH]c(Cl)n1)C2=O Chemical compound CCCCCN(c1c(C(N2CCCCc3n[o]c(-c4c[s]cc4)n3)=O)[nH]c(Cl)n1)C2=O WWPCFNUYRIATBU-UHFFFAOYSA-N 0.000 description 1
- DWAUCXWIUDXLIP-UHFFFAOYSA-M CCCCCN(c1c(C(N2CCCc3ccc[o]3)=O)[n-]c(Cl)n1)C2=O Chemical compound CCCCCN(c1c(C(N2CCCc3ccc[o]3)=O)[n-]c(Cl)n1)C2=O DWAUCXWIUDXLIP-UHFFFAOYSA-M 0.000 description 1
- LCFDYOGBKPHLRY-UHFFFAOYSA-N CCCCN(c1c(C(N2CCCCC(O)=O)=O)[nH]c(Cl)n1)C2=O Chemical compound CCCCN(c1c(C(N2CCCCC(O)=O)=O)[nH]c(Cl)n1)C2=O LCFDYOGBKPHLRY-UHFFFAOYSA-N 0.000 description 1
- LXUSRVAAVXLQDE-UHFFFAOYSA-N CCCCN(c1c(C(N2CCCCc3n[o]c(-c(cc4)ccc4O)n3)=O)[nH]c(Cl)n1)C2=O Chemical compound CCCCN(c1c(C(N2CCCCc3n[o]c(-c(cc4)ccc4O)n3)=O)[nH]c(Cl)n1)C2=O LXUSRVAAVXLQDE-UHFFFAOYSA-N 0.000 description 1
- DITNVHWVFHZLDT-UHFFFAOYSA-N O=C1N(CCCCBr)CCC1Cc1ccccc1 Chemical compound O=C1N(CCCCBr)CCC1Cc1ccccc1 DITNVHWVFHZLDT-UHFFFAOYSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/04—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/04—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two oxygen atoms
- C07D473/06—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two oxygen atoms with radicals containing only hydrogen and carbon atoms, attached in position 1 or 3
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Emergency Medicine (AREA)
- Nutrition Science (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
본 발명은 크산틴 유도체인 화학식 I의 화합물, 이러한 유도체의 제조 방법, 이러한 화합물을 함유하는 제약학적 제제, 질병의 치료법, 예를 들어 HM74A 수용체의 활성화 부족이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병을 치료함에 있어서 화합물의 용도에 관한 것이다.
<화학식 I>

크산틴 유도체, HM74A 수용체, 지질대사의 이상, 제약학적 제제.
Description
본 발명은 크산틴 유도체, 상기 유도체의 제조 방법, 이러한 화합물을 함유하는 제약학적 제제, 및 HM74A 수용체의 활성화 부족(under-activation)이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병의 치료 요법에서 화합물의 용도에 관한 것이다.
이상지혈증은 정도를 벗어난 지방단백질 프로파일을 가진 개인을 설명하기 위해 사용되는 일반적인 용어이다. 임상적으로, 이상지혈증을 갖고 따라서 심혈관 질병의 위험이 있는 환자의 치료를 위해 사용되는 화합물의 주요 부류는 스타틴, 피브레이트, 담즙산 결합 수지 및 니코틴산이다. 다양한 형태의 이상지혈증을 가진 환자에서 40년 이상에 걸쳐 니코틴산 (니아신, 비타민 B)이 임상적으로 사용되었다. 니코틴산의 주요 작용 방식은 호르몬-민감성 트리글리세리드 리파제(HSL)의 억제를 통한 것이고, 그 결과 혈장 비-에스테르화 지방산(NEFA)을 저하시키며 이것은 다시 LDL 및 VLDL (저 밀도 및 초 저 밀도 지방단백질)의 생성을 감소시키기 위해 간 지방 대사를 변경시킨다. 감소된 VLDL 수준은 콜레스테롤 에스테르 전달 단백질(CETP) 활성을 저하시키는 것으로 생각되고 그 결과 HDL (고 밀도 지방단백질) 수준을 증가시키며 관찰된 심혈관계 이점의 원인이 될 수 있다. 따라서, 니코틴산 은 지방단백질 프로파일에서 매우 바람직한 변화를 일으키고; HDL을 증가시키면서 VLDL 및 LDL의 수준을 감소시킨다. 니코틴산은 질병을 변형시키는 이점을 갖고 죽상경화증 병변의 진행을 감소시키고 퇴보를 증가시키며, 여러 번의 시도에서 심혈관 사건의 수를 감소시킨다는 것이 증명되었다.
니코틴산 처리에 의한 HSL의 억제 관찰은 아데닐릴 시클라제의 G-단백질-매개 억제에 의해 유발되는 세포 고리형 아데노신 모노포스페이트 (cAMP)의 감소에 의해 매개된다. 최근들어, G-단백질 결합 수용체 HM74 및 HM74A가 니코틴산의 수용체로서 확인되었다 [PCT 특허출원 WO 02/84298; Wise 등, J Biol Chem., 2003, 278 (11), 9869-9874]. 인간 HM74A의 DNA 서열은 진뱅크(Genbank)에서 수탁번호 AY 148884로 찾을 수 있다. 2개의 추가의 보고서는 이러한 발견을 뒷받침하지만 ([Tunaru 등, Nature Medicine, 2003, 9(3), 352-255] 및 [Soga 등, Biochem Biophys Res Commun., 2003, 303 (1) 364-369], 명명법은 약간 다르다. Tunaru의 보고서에서 그들은 인간 HM74이라 일컫는 것을 본 발명자들은 HM74A로 일컬으며, Soga 보고서에서 HM74b가 HM74A와 동일하다. HM74A 및/또는 HM74를 발현하기 위해 형질감염된 세포는 니코틴산에 노출된 후에 Gi G-단백질 매개 반응을 유도하는 능력을 얻는다. HM74A의 상동체 (m-PUMA-G)가 부족한 생쥐에서 니코틴산은 혈장 NEFA 수준을 감소시키지 못한다.
선행 기술에서 특정한 크산틴 유도체가 합성되고 개시되어 있다. 예를 들어 EP 0389282호는 뇌혈관 질환의 잠재적인 매개인자로서 크산틴 유도체를 개시하고 있다. 크산틴 유도체의 범위는 문헌 [Jacobson 등, J.Med.Chem., 1993, 36, 2639-2644]에 의하여 아데노신 수용체 길항제로서 확인되었다.
본 발명자들은, 니코틴산 수용체 HM74A의 선택적 작동제이고 따라서 이러한 수용체의 활성화 부족이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병의 치료, 예방 및 억제에서 잠재적인 이점을 갖는 크산틴 유도체의 군을 제시한다.
발명의 요약
본 발명은 크산틴 유도체 및, 예를 들어 HM74A 수용체의 활성화 부족이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병, 예를 들어, 이상지혈증 또는 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 지질 대사의 이상, 및 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심혈관 질병의 치료를 위한 치료법에서 이러한 유도체의 용도를 제공한다. 따라서, 화합물들은 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 및 뇌졸중뿐만 아니라 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진, 비만과 관련된 심혈관 증상을 위한 치료제로서 유리한 것으로 밝혀질 수 있다. 화합물들은 하기 기재된 바와 같이 염증성 질병 또는 상태의 치료에서 사용될 수 있다.
여기에 기재된 중간체, 제제, 방법 및 공정은 본 발명의 추가의 구현양태를 형성한다.
본 발명의 하나의 측면에 따르면, 화학식 I의 화합물로부터 선택된 하나 이상의 화학 물질 및 제약학적으로 허용가능한 그의 유도체가 제공된다.

상기 식에서,
R1은 -(알킬렌)m-X-(알킬렌)n-Y를 나타내고;
여기에서, m 및 n은 알킬렌 사슬에 있는 탄소 원자의 수를 나타내고;
X는 헤테로아릴 및 헤테로시클릴로부터 선택된 기를 나타내고;
Y는 아릴, 헤테로아릴 및 O-아릴로부터 선택된 기를 나타내고; 이것은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, 할로겐, -(CH2)qNR5R7, -(CH2)q-(O)p-(CH2)q-N(R5)C(O)OR8, -(CH2)q-N(R5)C(O)R8, -(CH2)q-(O)p-(CH2)q-C(O)NR5R6, -(CH2)q-N(R5)C(O)NR5R6, -(CH2)q-C(O)N((CH2)mOH)R5, -(CH2)q-N(R5)-S(O)2R8, -CH2-S(O)2NR5R6, -C1-6 할로알킬, -OCF3, -OCH(F)2, -OCH2F, -C(O)OR5, -OR5, -R8CN, CN, -SO2R9, -(CH2)n헤테로아릴 -(CH2)n헤테로시클릴, -(CH2)n시클로알킬, -(CH2)n시클로알케닐 및 -(CH2)n아릴로부터 독립적으로 선택된 하나 이상의 기로 임의로 치환될 수 있고;
R2는 시클로알킬, C1 -6 할로알킬, 할로겐, -CN 및 -OR4로부터 독립적으로 선택된 하나 이상의 기로 임의로 치환될 수 있는 C1 -6 알킬을 나타내고;
R3은 할로겐을 나타내고;
R4는 수소, C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)n시클로알킬, -(CH2)n시클로알케닐, -(CH2)n헤테로시클릴, -(CH2)n 아릴 및 -(CH2)n 헤테로아릴로부터 선택된 기를 나타내고;
R5 및 R6은 수소 및 C1 -4 알킬로부터 독립적으로 선택되고;
R7은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)t시클로알킬, -(CH2)t시클로알케닐, -(CH2)t헤테로시클릴, -(CH2)t 아릴 및 -(CH2)t 헤테로아릴로부터 선택된 기를 나타내고;
R8은 C1 -4 알킬을 나타내고;
R9은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)n시클로알킬, -(CH2)n시클로알케닐, -(CH2)n헤테로시클릴, -(CH2)n 아릴, -(CH2)n 헤테로아릴 및 CN으로부터 선택된 기를 나타내고;
m은 3 및 4로부터 선택된 정수를 나타내고;
n은 0 및 1로부터 선택된 정수를 나타내고;
p는 0 및 1로부터 선택된 정수를 나타내고;
q는 0, 1 및 2로부터 선택된 정수를 나타내고;
t는 1 및 2로부터 선택된 정수를 나타낸다.
화합물(들)은 HM74A 수용체의 활성화 부족이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병의 치료에서, 예를 들어, 이상지혈증 또는 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 지질 대사의 이상, 및 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심혈관 질병의 치료에서 유용한 것으로 생각된다. 따라서, 화합물들은 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 및 뇌졸중뿐만 아니라 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진, 비만과 관련된 심혈관 증상을 위한 치료제로서 유리한 것으로 밝혀질 수 있다. 따라서, 본 발명의 화합물은 HM74A의 작동제 또는 부분 작동제로서 용도를 가질 수 있다. 화합물은 또한 하기 기재된 것과 같은 염증성 질병 또는 상태의 치료에서 사용될 수 있다.
본 발명의 하나의 구현양태에서, X는 헤테로아릴을 나타낸다. 다른 구현양태에서, X는 질소 헤테로원자를 포함하는 헤테로아릴, 예를 들어 트리아졸릴, 푸라자닐, 옥사디아졸릴, 테트라졸릴, 이미다졸릴 또는 피라졸릴을 나타낸다. 추가의 구현양태에서, X는 옥사디아졸릴 및 테트라졸릴로부터 선택된 기를 나타낸다.
다른 구현양태에서, Y는 아릴로부터 선택된 임의로 치환된 기, 예를 들어 페닐 또는 나프틸, 헤테로아릴, 예를 들어 피리디닐, 티아졸릴, 티에닐, 벤조푸라닐 또는 인돌릴 및 O-아릴, 예를 들어 O-페닐을 나타낸다. 추가의 구현양태에서, Y는 아릴 및 헤테로아릴로부터 선택된 임의로 치환된 기를 나타낸다. 하나의 구현양태에서, Y는 아릴로부터 선택된다.
본 발명의 하나의 구현양태에서, Y는 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, 할로겐, -NH2, -(CH2)q-(O)p-(CH2)q-N(R5)C(O)OR8, -(CH2)q-N(R5)C(O)R8, -(CH2)q-(O)p-(CH2)q-C(O)NR5R6, -(CH2)q-N(R5)C(O)N(R5)R6, -(CH2)q-C(O)N((CH2)mOH)R5, -(CH2)q-N(R5)-S(O)2R8, -CH2-S(O)2N(R5)R6, -C1 -6 할로알킬, -OCF3, -OCH(F)2, -OCH2F, -C(O)OR5, -OR5, -R8CN, CN, -SO2R9, -(CH2)n헤테로아릴, -(CH2)n헤테로시클릴, -(CH2)n시클로알킬, -(CH2)n시클로알케닐 및 -(CH2)n아릴의 하나 이상의 기로 임의로 치환될 수 있다.
추가의 구현양태에서, Y는 OR5, 예를 들어 OH 또는 OCH3, 할로겐, 예를 들어 F 또는 Cl, 아릴, 예를 들어 페닐, C1 -6 할로알킬, 예를 들어 CF3 또는 CH2CF3, OCF3, R8CN, CN, (CH2)q-N(R5)-S(O)2R8, 예를 들어 NHSO2CH3 및 SO2R9, 예를 들어 SO2CH3으로부터 하나 이상의 기에 의해 치환된다.
추가의 구현양태에서, Y는 OR5, 할로겐, C1 -6 할로알킬 및 -(CH2)q-N(R5)C(O)R8로부터 선택된 하나 이상의 기로 치환된다.
다른 구현양태에서, Y는 할로겐 및 C1 -6 할로알킬로부터 선택된 하나 이상의 기로 치환된다.
또 다른 구현양태에서, Y는 더욱 치환되지 않는다.
본 발명의 하나의 구현양태에서, X 및 Y는 각각 독립적으로 질소 헤테로원자를 포함한 헤테로아릴을 나타낸다. 추가의 구현양태에서, X는 옥사디아졸릴을 나타내고 Y는 피리디닐을 나타낸다. 다른 구현양태에서, X는 테트라졸릴을 나타내고 Y는 페닐을 나타낸다. 본 발명의 다른 구현양태에서, X는 옥사디아졸릴을 나타내고 Y는 페닐을 나타낸다.
본 발명의 하나의 구현양태에서, m은 4이고 n은 0이다. 추가의 구현양태에서, m은 3이고 n은 1이다.
본 발명의 하나의 구현양태에서, R2는 C3 -6 알킬로부터 선택되고, 예를 들어 부틸 또는 펜틸, 예를 들어 n-부틸 또는 n-펜틸이다.
본 발명의 추가의 구현양태에서, R3은 염소 및 브롬으로부터 선택된다. 다 른 구현양태에서, R3은 염소를 나타낸다.
본 발명의 하나의 구현양태에서, R7은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)t시클로알킬, -(CH2)n시클로알케닐, -(CH2)t헤테로시클릴, -(CH2)t아릴 및 -(CH2)t헤테로아릴로부터 선택된 기를 나타낸다.
본 발명의 하나의 구현양태에서, X는 옥사디아졸릴을 나타내고, Y는 페닐을 나타내고, R2는 부틸이고, R3은 염소를 나타내고, m은 4이고 n은 0이다.
입체이성질체에 관하여, 화학식 I의 화합물은 하나 이상의 비대칭 탄소 원자를 가질 수 있고, 라세미체, 라세미 혼합물로서 그리고 각각의 거울상이성질체 또는 부분입체이성질체로서 발생할 수 있다. 그들의 혼합물을 포함하여 이러한 모든 이성질체 형태가 본 발명에 포함된다.
화학식 I의 화합물이 알케닐 또는 알케닐렌 기를 함유하는 경우에, 시스(E) 및 트랜스(Z) 이성질체가 발생할 수 있다. 본 발명은 본 발명의 화합물의 각각의 입체이성질체, 및 적절하다면 이들의 혼합물과 함께 개개의 호변이성질체 형태를 포함한다.
부분입체이성질체 또는 시스 및 트랜스 이성질체의 분리는 통상적인 기술에 의해, 예를 들어 상응하는 광학적 순수 중간체로부터 제조될 수 있는 약제의 입체이성질체 혼합물의 분별 결정화, 크로마토그래피 또는 HPLC에 의해, 또는 적절한 키랄 지지체를 사용하여 상응하는 라세미체의 HPLC와 같은 분할에 의해, 또는 적절 하다면 적절한 광학 활성 산 또는 염기와의 상응하는 라세미체의 반응에 의해 형성된 부분입체이성질체 염의 분별 결정화에 의해 달성될 수 있다.
또한, 화학식 I의 화합물의 일부 결정 형태는 다형태로서 존재할 수 있고 본 발명에 포함된다. 하나의 형태는 다른 형태에 비해 장점을 가질 수 있고, 예를 들어 하나의 형태가 다른 형태에 비해 개선된 안정성을 가질 수 있다.
본 발명은 특별한 구현양태의 조합을 포함하고 상기 기재된 특정한 치환기의 모든 조합을 포함하는 것으로 이해되어야 한다.
본 명세서 및 첨부된 청구의 범위에 있어서, 용어 "포함한다(comprise)" 및 "포함한다(include)" 및 "포함한다", "포함하는", "포함한다" 및 "포함하는"과 같은 변형은 포괄적인 것으로 해석되어야 한다. 다시 말해서, 이러한 용어들은 구체적으로 열거되지 않은 다른 요소 또는 정수들의 가능한 포함을 전달하는 것으로 해석되어야 한다.
여기에서 사용된 용어 "알킬" (기로서 또는 기의 일부로서 사용될 때)은 달리 규정되지 않는 한 특정한 수의 탄소 원자를 함유하는 직쇄 또는 분지쇄 탄화수소 사슬을 가리킨다. 예를 들어, C3-C6 알킬은 적어도 3개 및 많아야 6개 탄소 원자를 함유하는 직쇄 또는 분지쇄 탄화수소 사슬을 의미한다. 여기에서 사용된 알킬의 예는 이에 한정되지 않지만 메틸 (Me), 에틸 (Et), n-프로필 및 i-프로필을 포함한다.
여기에서 사용된 용어 "알킬렌"은 직쇄 및 분지쇄 포화 또는 불포화 사슬 또 는 고리형 포화 탄화수소 링커 기를 의미한다. 알킬렌 기의 예는 메틸렌 (-CH2-), 에틸렌 (-CH2CH2-), 에텐 (-CH=CH-), 또는 시클로프로필렌 등을 포함한다. 예를 들어, 여기에서 사용된 -(알킬렌)m- (여기에서, m은 3이다)은 -(CH2)3-, -C(CH3)2-, -CH2CH=CH- 또는 -시클로프로필렌- 등을 나타낸다. 예를 들어, 여기에서 사용된 바와 같이 -(alk)m- (여기에서, m은 4이다)는 -(CH2)4-, -CH2C(CH3)2-, -CH2CH=CHCH2- 또는 -CH2시클로프로필렌 등을 나타낸다. 예를 들어 여기에서 사용된 바와 같이 -(알킬렌)n (여기에서 n=1)은 -CH2-를 의미한다. 여기에서 사용된 -(알킬렌)n (여기에서 n=0)은 이 위치에 알킬렌 링커가 없음을 의미한다.
여기에서 사용된 용어 "알케닐"은 하나 이상의 이중 결합을 함유하는 특정한 수의 탄소 원자를 함유하는 직쇄 또는 분지쇄 탄화수소 사슬을 가리킨다.
여기에서 사용된 용어 "알키닐"은 하나 이상의 삼중 결합을 함유하는 특정한 수의 탄소 원자를 함유하는 직쇄 또는 분지쇄 탄화수소 사슬을 가리킨다.
여기에서 사용된 용어 "시클로알킬"은 3 내지 8개 탄소 원자의 포화 단일고리형 탄화수소 고리를 가리킨다. 이러한 기의 예는 시클로프로필, 시클로부틸, 시클로펜틸, 시클로헥실, 시클로헵틸 및 시클로옥틸을 포함한다.
여기에서 사용된 용어 "시클로알케닐"은 하나 이상의 탄소-탄소 이중 결합을 함유하는 3 내지 8개 탄소 원자의 불포화 비-방향족 단일고리형 탄화수소 고리를 가리킨다. 이러한 기의 예는 시클로프로페닐, 시클로부테닐, 시클로펜테닐, 시클 로헥세닐, 시클로헵테닐, 시클로옥테닐 등을 포함한다.
여기에서 사용된 용어 "아릴"은 적어도 하나의 고리가 방향족인 C6 -12 단일고리형, 이고리형 또는 삼고리형 탄화수소 고리를 가리킨다. 이러한 기의 예는 페닐, 나프틸 또는 테트라히드로나프탈레닐 등을 포함한다.
여기에서 사용된 용어 "헤테로아릴"은 1 내지 4개 헤테로원자를 함유하는 5-6 원 단일고리형 방향족 고리 또는 융합된 8-10 원 이고리형 방향족 고리를 가리키고, 이들은 독립적으로 산소, 질소 및 황으로부터 선택된다. 고리 탄소 원자 상에 하나 이상의 임의의 옥소 치환기가 존재할 수 있다. 이러한 단일고리형 방향족 고리의 예는 티에닐, 푸릴, 푸라자닐, 피롤릴, 트리아졸릴, 테트라졸릴, 이미다졸릴, 옥사졸릴, 티아졸릴, 옥사디아졸릴, 이소티아졸릴, 이속사졸릴, 티아디아졸릴, 피라닐, 피라졸릴, 피리미딜, 피리다지닐, 피라지닐, 피리딜, 트리아지닐, 테트라지닐 등을 포함한다. 이러한 융합된 방향족 고리의 예는 퀴놀리닐, 이소퀴놀리닐, 퀴나졸리닐, 퀴녹살리닐, 프테리디닐, 신놀리닐, 프탈라지닐, 나프티리디닐, 인돌릴, 이소인돌릴, 아자인돌릴, 인돌리지닐, 인다졸릴, 퓨리닐, 피롤로피리디닐, 푸로피리디닐, 벤조푸라닐, 이소벤조푸라닐, 벤조티에닐, 벤조이미다졸릴, 벤족사졸릴, 벤조이속사졸릴, 벤조티아졸릴, 벤조이소티아졸릴, 벤족사디아졸릴, 벤조티아디아졸릴 등을 포함한다.
여기에서 사용된 용어 "헤테로시클릴"은 각각 산소, 질소 또는 황으로부터 독립적으로 선택된 1 내지 4개 헤테로원자를 함유하는 포화 또는 부분적 불포화될 수 있는 4-7 원 단일고리형 고리 또는 융합된 8-12 원 이고리형 고리를 가리킨다. 고리 탄소 원자 위에 하나 이상의 임의의 옥소 치환기가 존재할 수 있다. 이러한 단일고리형 고리의 예는 피롤리디닐, 아제티디닐, 피라졸리디닐, 옥사졸리디닐, 피페리디닐, 피페라지닐, 모르폴리닐, 티오모르폴리닐, 티아졸리디닐, 히단토이닐, 발레로락타밀, 옥시라닐, 옥세타닐, 디옥소라닐, 디옥사닐, 옥사티올라닐, 옥사티아닐, 디티아닐, 디히드로푸라닐, 테트라히드로푸라닐, 디히드로피라닐, 테트라히드로피라닐, 테트라히드로피리디닐, 테트라히드로피리미디닐, 테트라히드로티오페닐, 테트라히드로티오피라닐, 디아제파닐, 아제파닐 등을 포함한다. 이러한 이고리형 고리의 예는 인돌리닐, 이소인돌리닐, 벤조피라닐, 퀴누클리디닐, 2,3,4,5-테트라히드로-1H-3-벤즈아제핀, 테트라히드로이소퀴놀리닐 등을 포함한다.
여기에서 사용된 용어 "할로겐" 또는 "할로"는 예를 들어 불소, 염소, 브롬 또는 요오드 원자를 가리킨다.
여기에서 사용된 용어 "C1 -6 할로알킬"은 여기에 정의된 C1 -6 알킬 기를 가리키고, 여기에서 적어도 하나의 수소 원자가 할로겐으로 대체된다. 이러한 기의 예는 플루오로에틸, 트리플루오로메틸, 트리플루오로에틸 등을 포함한다.
여기에서 사용된 바와 같이, 기가 다른 기에 의해 "치환"되거나 "하나 이상의 치환기"를 갖는 것으로 언급된다면, 이러한 치환을 위해 특별한 위치가 규정되지 않는 한, 치환은 기의 어느 위치에 존재할 수 있는 것으로 이해된다.
여기에서 사용된 용어 "제약학적으로 허용가능한 유도체"는 본 발명의 화합 물의 제약학적으로 허용가능한 유도체, 예를 들어 염, 용매화물 또는 에스테르를 가리키고, 인간과 같은 포유동물에 투여될 때, 이러한 화합물 또는 그의 활성 대사물을 (직접적 또는 간접적으로) 제공할 수 있다. 이러한 유도체는 과다한 실험 없이도 당업자에게 명백하고, 참고문헌으로 포함되어 있는 문헌 [Burger's Medicinal Chemistry And Drug Discovery, 제5판, Vol. 1: Principles And Practice]의 교시내용을 참조한다.
환자에 투여하기 위한 제약학적 제제에 포함될 수 있는 성분 (활성 성분, 희석제, 부형제 또는 담체)에 관련하여 여기에서 사용된 용어 "제약학적으로 허용가능한"은 제약학적 제제에 존재하는 다른 성분들과 상용성이고 그의 수용자에게 해가 없다는 의미에서 허용될 수 있는 성분을 가리킨다.
여기에서 사용된 용어 "용매화물"은 용질 (본 발명에서 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체) 및 용매에 의해 형성된 가변 화학양론의 착물을 가리킨다. 본 발명의 목적을 위한 이러한 용매는 용질의 생물학적 활성을 방해하지 않을 수 있다. 사용된 용매는 제약학적으로 허용가능한 용매일 수 있다. 적절한 제약학적으로 허용가능한 용매의 예는 물, 에탄올 및 아세트산을 포함한다. 사용될 수 있는 용매의 예는 물이고, 이 경우에 용매화물은 당해 용질의 수화물로 일컬어질 수 있다.
제약학적 용도를 위하여, 상기 언급된 "염 또는 용매화물"은 제약학적으로 허용가능한 염 또는 용매화물일 것으로 이해된다. 그러나, 기타 염 또는 용매화물은 예를 들어 화학식 I의 화합물의 제조에서 또는 제약학적으로 허용가능한 염 또 는 용매화물의 제조에서 사용될 수 있다.
제약학적으로 허용가능한 염은 문헌 [Berge, Bighley and Monkhouse, J. Pharm. Sci. 1977, 66, 1-19]에 기재된 것을 포함한다. 적절한 제약학적으로 허용가능한 염은 알칼리 금속 수산화물과 같은 알칼리 금속 염기의 첨가로부터 형성된 알칼리 금속 염을 포함한다. 적절한 알칼리 금속 염의 예는 나트륨 염 및 포타슘 염을 포함한다. 제약학적으로 허용가능한 적절한 다른 염은 알칼리 토금속 염, 예컨대 칼슘 염 및 마그네슘 염, 암모늄 염; 또는 에탄올아민, 트리에탄올아민, 에틸렌 디아민, 트리에틸아민, 콜린 및 메글루민과 같은 유기 염기와의 염; 또는 아르기닌, 리신 및 히스티딘과 같은 아미노산과의 염을 포함한다.
에스테르는 당연히 활성일 수 있고/있거나 인체에서 생체내 조건 하에 가수분해될 수 있다. 적절한 제약학적으로 허용가능한 생체내 가수분해가능한 에스테르 기는 인체에서 모 산 또는 그의 염을 남기기 위해 쉽게 파괴되는 것을 포함한다. 에스테르는 상응하는 알콜과의 반응과 관련하여 당 기술분야에서 공지된 방법에 의하여 카르복실 산 (-C(O)OH) 기에서 형성될 수 있다. 예를 들어, 에스테르는 C1 -6 알킬 에스테르, 예를 들어 메틸 에스테르, 에틸 에스테르 등 일 수 있다.
여기에서 사용된 용어 "본 발명의 화합물"은 화학식 I에 따른 화합물 및 제약학적으로 허용가능한 그의 유도체를 의미한다. 용어 "본 발명의 화합물"은 상기 정의된 것과 같이 본 발명의 화합물의 어느 것을 의미한다.
여기에서 사용된 용어 "하나 이상의 화학 물질"은 화학식 I의 화합물 및 제 약학적으로 허용가능한 그의 유도체로 구성된 화합물의 군에서 선택된 하나 이상의 화학 물질을 의미한다.
본 발명의 하나의 측면에서, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1이 제공된다. 본 발명의 다른 측면에서, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 2가 제공된다.
실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1 및 2의 샘플에 관한 열 분석을 수행하였다. 따라서, 각각 160℃ 이상 및 147℃ 이상의 DSC (±0.5 ℃)에 의해 측정된 융점 개시를 갖는, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (형태 1 또는 2)이 제공된다.
이하 기재된 바와 같이 제조된, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1 및 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 2의 샘플은 도 1 내지 2의 X-선 분말 회절 패턴을 제공하였다. X-선 회절 패턴은 결정질 형태에 대해 특유하다. 실질적으로 결정질인 형태는 2 세타 각(°)으로 표현될 수 있는 특유한 일련의 회절 피크를 가진 회절 패턴을 나타낸다.
2 세타 회절 각은 X-선 회절 패턴에서 다양한 피크의 위치를 설명한다. 관찰된 2 세타 각에서의 약간의 변동은 사용된 특정한 회절계 및 분석자의 샘플 제조 기술을 근거로 하여 예측된다.
3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온의 실질적으로 결정질인 형태는 특징적인 2 세타 각 피크의 존재에 의해 또는 특정한 결정질 형태의 특징인 여러 개의 2 세타 각에 의해 확인될 수 있다. 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (형태 1)을 확인하기 위하여, 2 세타 각 (±0.1도)로 표현되는 하기 위치: 5.4, 6.7, 9.7, 11.1, 12.9, 14.0, 15.6, 16.3, 16.7, 23.1도에서 이러한 피크들이 일어난다. 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (형태 2)을 확인하기 위하여, 2 세타 각 (±0.1도)으로 표현되는 하기 위치: 5.2, 6.6, 10.4, 11.2, 13.4, 15.6, 18.1, 19.5, 20.9 도에서 이러한 피크들이 일어난다. 하나의 구현양태에서, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1 및 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 2를 확인하기 위해 2 세타 각의 적어도 하나가 사용된다. 다른 구현양태에서, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1, 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5- (2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 2를 확인하기 위하여, 상기 2 세타 각의 적어도 2, 3, 4 또는 5개 (적용가능하다면)가 사용된다.
2 세타 각 지정의 각각에 일부 오차 한계가 존재한다. 상기 2 세타 각에서의 오차 한계는 상기 각각의 피크 지정에 대해 대략 ±0.1도이다.
일부 오차 한계가 2 세타 각의 지정에서 가능하기 때문에, 특정한 결정질 형태를 확인하기 위하여 X-선 분말 회절 패턴을 비교하는 바람직한 방법은, 공지된 형태의 X-선 분말 회절 패턴 위에 알려지지 않은 형태의 X-선 분말 회절 패턴을 겹쳐 놓는 것이다. 예를 들어, 당업자라면 여기에 기재된 방법을 사용하여 수득된 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온의 확인되지 않은 형태의 X-선 분말 회절 패턴을 겹칠 수 있고 (예를 들어 도 3 참조), 확인되지 않은 형태의 X-선 회절 패턴이 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1 또는 2의 X-선 분말 회절 패턴과 실질적으로 동일한지의 여부를 쉽게 결정할 수 있다. X-선 분말 회절 패턴이 도 1 내지 2에 나타낸 것과 실질적으로 동일하다면, 이전의 형태를 쉽고 정확하게 확인할 수 있다.
여기에서 사용된 용어 "실질적으로 결정질인 형태"는, 비결정질 형태 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온을 실질적으로 갖지 않는 것을 의미한다. "실질적으로 갖지 않는"이란, 50% 미만의 비결정질 형태를 함유하고, 하나의 측면에서 20% 미만의 비결정 질 형태를 함유하고 다른 측면에서 10% 미만의 비결정질 형태를 함유하고, 다른 측면에서 5% 미만의 비결정질 형태를 함유하고, 다른 측면에서 2% 미만의 비결정질 형태를 함유하고, 다른 측면에서 1% 미만의 비결정질 형태를 함유함을 의미한다.
본 발명은 여기에 기재된 것과 같은 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태의 제조 방법을 제공한다.
화학식 I의 화합물은, 이상지혈증 및 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 많은 지질 대사 질병, 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심장혈관 질병, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진, 비만의 증상을 치료하고 개선하는데 잠재적인 치료적 장점을 갖는다. 따라서, 화합물들은 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 및 뇌졸중의 치료제로서 유리한 것으로 밝혀질 수 있다.
HM74 및 HM74A 수용체는 염증 (WO 02084298)에 연관된 것으로 보고되었다. 염증은 외상에 대한 혈관, 세포 및 신경학적 반응의 군을 나타낸다. 염증은 단핵구, 호중구 및 과립구와 같은 염증성 세포가 조직으로 이동하는 것으로 특징화될 수 있다. 이것은 보통 내피 장벽 기능의 감소 및 조직 내로의 부종과 관련된다. 질병에 관한 염증은 전형적으로 만성 염증이라 일컬어진다. 이러한 만성 염증은 질병 증상을 통해 그 자체로 명백히 나타날 수 있다. 따라서, 항-염증 요법의 목 적은 이러한 만성 염증을 감소시키는 것이고 치유 및 조직 회복의 생리학적 과정이 진행될 수 있도록 하는 것이다.
본 발명의 화합물이 유용한 것으로 증명될 수 있는 염증 질병 또는 상태의 예는 관절, 예를 들어 관절염 (예, 류마티스성 관절염, 골관절염, 인공 관절 기능상실) 또는 위장관 (예, 궤양 대장염, 크론병 및 기타 염증성 장 및 위장 병, 위염 및 감염으로부터 생긴 점막 염증, 비-스테로이드성 항-염증 약물에 의해 유발된 창자병증), 폐 (예, 성인 호흡곤란증후군, 천식, 낭성섬유증, 또는 만성 폐쇄성 폐 질환), 심장 (예, 심근염), 신경 조직 (예, 다발경화증), 췌장 (예, 진성 당뇨병 및 그의 합병증과 관련된 염증), 신장 (예, 사구체신염), 피부 (예, 피부염, 건선, 습진, 두드러기, 화상), 눈 (녹내장) 뿐만 아니라 이식된 기관 (예, 거부) 및 다-기관 질환 (예, 전신홍반루푸스, 패혈증) 및 바이러스 또는 세균 감염의 염증 후유증 및 죽상경화증과 관련된 염증 상태 및 예를 들어 뇌에서의 저산소 또는 허혈성 발작 (재관류와 함께 또는 재관류 없이) 또는 허혈성 심장병의 질환을 포함한다.
하나의 구현양태에서, 본 발명의 화합물은 죽상경화증, 동맥경화증, 고트리글리세리드혈증 및 혼합된 이상지혈증을 포함하여 염증, 당뇨병 및 심혈관 질병 또는 상태의 치료 및 예방에서 유용하다.
니코틴산은 가능하다면 높은 수준 (일일 그램 양)으로 투여되기 때문에 상당한 부작용 프로파일을 갖는다. 가장 일반적인 부작용은 강한 피부 홍조이다. 본 발명의 특정한 구현양태에서 화합물은 니코틴산에 비해 감소된 부작용을 나타낼 수 있다. HM74A는 니코틴산에 대해 높은 친화력 수용체로서 확인되는 반면 HM74는 낮 은 친화력 수용체이다. 본 발명의 화합물은 HM74에 대해서보다 HM74A에 대해 높은 친화력을 나타내고 따라서 선택적 HM74A 작동제 또는 부분 작동제로서 유용할 수 있다.
HM74A를 활성화하기 위한 화학식 I의 화합물의 잠재력은 예를 들어 하기 시험관내 전 세포 분석을 사용하여 증명될 수 있다.
시험관내
시험
일시적 형질감염을 위하여, HEK293T 세포 (SV40 큰 T-항원을 안정하게 발현하는 HEK293 세포)를 10% 태아 소 혈청 및 2mM 글루타민을 함유하는 DMEM에 유지하였다. 세포를 90mm 배양 접시에 접종하고 형질감염에 앞서서 60-80% 전면생장(confluence) (18-24시간)까지 생육시켰다. 인간 HM74A (진뱅크TM 수탁 번호 AY-148884)를 포유동물 발현 벡터 (pcDNA3; 인비트로겐)에 서브클로닝하고 리포펙타민(Lipofectamine)TM 시약을 사용하여 형질감염시켰다. 형질감염을 위하여, 9 ㎍의 DNA를 0.6 ml의 옵티-MEM (라이프테크놀로지스 인코포레이티드) 중에서 30 ㎕ 리포펙타민과 함께 혼합하고 1.6 ml의 옵티-MEM의 첨가에 앞서서 실온에서 30분 동안 배양하였다. 세포를 5시간 동안 리포펙타민/DNA 혼합물에 노출시키고, DMEM 중의 20% (v/v) 태아 소 혈청 6 ml를 첨가하였다. 세포를 형질감염 후 48시간에 수집하였다. 16시간 동안 배지에 50 ngml-1로 보충함으로써 백일해 독소 처리를 수행하였다. 모든 일시적 형질감염 연구는 Gi /o G 단백질, Go1 α와 함께 수용체의 공동-형 질감염과 연관되었다.
안정한 세포 주의 발생을 위하여 상기 방법을 사용하여 6웰 접시에 접종된 CHO-K1 세포를 형질감염시키고 30% 전면생장까지 생육시켰다. 세포를 10% 태아 소 혈청 및 2 mM 글루타민을 함유한 DMEM-Ham's F-12 배지 (인비트로겐으로부터 입수가능함)에 유지하였다. 형질감염 후 48시간에 항생물질 내성 세포의 선택을 위하여 배지를 400 ㎍/ml 제네티신 (G418, 기브코)로 보충하였다. FM74A를 안정하게 발현하는 클론성 CHO-K1 세포 주는, 니코틴산의 첨가 후에 [35S]-GTPγS 결합 측정에 의해 입증되었다.
P2 막 제조 - 수집 후에 -80 ℃에서 동결된 세포 페이스트로부터 혈장 막-함유 P2 입자 분획을 제조하였다. 4 ℃에서 모든 절차를 수행하였다. 세포 펠릿을 1ml의 10mM 트리스-HCl 및 0.1mM EDTA, pH 7.5 (완충액 A)에 재현탁시키고, 울트라 투랙스(Ultra Turrax)와 함께 20초 동안 균질화한 다음 25-게이지 니들을 통해 통과시켰다 (5 회). 핵을 펠릿화하기 위하여 세포 용해물을 마이크로원심분리에서 1,000 g에서 10분 동안 원심분리하고, 16,000 g에서 30분 동안 마이크로원심분리에 의해 비파괴 세포 및 P2 입상 분획을 회수하였다. P2 입상 분획을 완충액 A에 재현탁시키고 필요할 때까지 -80 ℃에서 저장하였다.
[35S]-GTPγS 결합 - 앞서 기재된 방법을 근거로 하여 384-웰 방식으로 실온에서 분석을 수행하였다 [Wieland, T. 및 Jakobs,K.H. (1994) Methods Enzymol. 237, 3-13]. 간략하게, 표준 또는 시험 화합물의 희석을 제조하고 10 ㎕의 부피로 384-웰 플레이트에 첨가하였다. 막 (HM74A 또는 HM74)을 사포닌 (60 ㎍/ml), 리드시커(Leedseeker) WGA 비드 (아머샴; 250 ㎍/웰) 및 10 μM GDP으로 보충된 분석 완충액 (20mM HEPES, 100mM NaCl, 10mM MgCl2, pH 7.4)에 희석하였으며, 그 결과 각각의 웰에 첨가된 20㎕ 부피는 5 ㎍의 막을 함유한다. [35S]-GTPγS (1170 Ci/밀리몰, 아머샴)을 분석 완충액에서 희석하고 (1:1500) 20 ㎕를 각각의 웰에 첨가하였다. 방사능표지물질의 첨가 후에, 플레이트를 밀봉하고 펄스 회전시키고 실온에서 4시간 동안 배양하였다. 배양 기간의 마지막에 플레이트를 리드시커 기계 (뷰룩스 플러스; 퍼킨-엘머(VIEWLUX PLUS; Perkin-Elmer))위에서 판독하여 특이적 결합 수준을 결정하였다.
최종 분석 부피를 10 ㎕로 감소시킴으로써 이러한 분석을 정련하였다. 이러한 10㎕ 분석을 위하여, 재검사 프로토콜을 사용하였다. 이것은 단지 384-웰 플레이트의 웰 당 10 nl의 표준 또는 시험 화합물, 및 1.5 ㎍ 막 및 100 ㎍ 리드시커 WGA 비드의 사용과 관련되었다. 저 부피 프로토콜을 위하여, 막, 비드 및 [35S]-GTPγS를 함께 혼합한 다음 이러한 혼합물의 10 ㎕를 각각의 웰에 분배하였다. 배양 및 플레이트 판독은 10 ㎕ 및 50 ㎕ 분석에 대해 동일하였다.
모든 예시된 화합물들을 상기 기재된 [35S]-GTPγS 결합 분석의 한쪽 또는 양쪽에서 시험하였다 (즉, 10 ㎕ 및 50 ㎕ 분석).
XC 50 소프트웨어 패키지를 사용하여 4 매개변수 기호논리학 방정식을 사용 하여 수행되는 곡선 접합에 의해 데이터를 분석하였다 (어느 하나의 곡선으로부터 결실된 최대 2개 점). 특이적 결합을 니코틴산 결합의 최대 반응에 비교하여 pEC50 및 % 효율로서 환산하였다.
생체내
시험
연구에 앞서 적어도 12시간 동안 금식시킨 수컷 스파그-돌리(Spague-Dawley) 쥐 (200-250 g)에서 본 발명의 화합물을 시험할 수 있다. 화합물을 1 또는 3 mg/kg (5 ml/kg)으로 정맥내 투여하거나 또는 1 내지 30 mg/kg (10 ml/kg) 범위의 투여량으로 경구 가비지에 의하여 투여하였다. 투여 전 및 투여 후 3회 (투여 후 15분 내지 6시간의 범위의 시간)에 혈액 샘플 (0.3 ml 꼬리 정맥 출혈)을 취할 수 있다. 각각의 혈액 샘플을 헤파린 관 (벡톤 딕킨슨 마이크로테이너, PST LH)에 옮기고 원심분리 (5 분 동안 10,000 g)하여 혈장 샘플을 생성하였다. 통상적으로 입수가능한 키트 (랜독스(Randox)를 사용하여, 비-에스테르화 지방산 (NEFA)의 수준에 대해 혈장 샘플을 분석하였다. 투여-전 수준에 비하여, 혈장 NEFA 수준의 억제를 HM74A 작동제 활성에 대한 대용으로서 사용한다.
본 발명의 화합물이 니코틴산과 관련된 세정 반응을 나타내는지를 결정하기 위하여, 이들을 의식이 있는 기니 피그에 투여할 수 있다. 수컷 던킨 하틀리(Dunkin Hartley) 기니 피그 (300-600 g; 1군 당 n=10-20)를 적어도 12시간 동안 금식시키지만, 실험 전 24시간을 초과하여 금식시키지 않았다. 각각의 동물로부터 회복 마취 하 (추가의 O2를 가진 이소플루란 3.5% (1L/분))에서 심장 천자에 의해 연구-전 혈액 샘플 (0.5 ml)을 취한다. 적외선 온도 탐침 위에 각 동물의 왼쪽 귀를 놓음으로써 귀 온도 측정을 행한다. 투여 전 5분으로부터 투여 후 30분까지 1분 간격으로 측정을 행한다. 투여 후 2시간까지 15분 간격으로 온도 측정을 행한다. 동물은 경구 가비지 (5 ml/kg)에 의해 시험 화합물을 수용한다. 말단 마취 하에서 심장 천자에 의해 혈액 샘플 (0.5 ml)을 취한다. 투여 후 0.5, 1, 2, 3 및 4시간에서 데이터를 제공하기 위해 각각의 동물로부터 혈액 샘플을 취한다. 모든 혈액 샘플을 5분 동안 혈액 롤러 위에 놓은 다음, 연구 마지막까지 얼음 위에 보관한다. 원심분리 후에 (5분 동안 12000 g) 혈장을 새로운 관으로 옮기고 NEFA 농도를 위해 검사할 때까지 -20 ℃에서 보관한다.
화학식 I에 따른 일부 화합물을 합성하고 (하기 합성예 참조) 상기 언급된 [35S]-GTPγS 결합 분석에서 시험한다.
8-클로로-3-(3,3-디메틸부틸)-1-[2-(에틸옥시)에틸]-3,7-디히드로-1H-퓨린-2,6-디온을 포함하여 화학식 I에 따른 일부 화합물은 화학식 I에 따른 다른 화합물의 제조에서 중간체로서 사용된다.
예시된 화합물 (실시예 1- 512)는, 이들이 시험된 상기 기재된 [35S]-GTPγS 결합 분석에서 4.3 (± 0.3 로그 단위) 이상의 pEC50 및 30% 이상 (니코틴산에 대해)의 효능을 갖는다.
일반적 정제 및 분석 방법:
LC/MS: 방법
하기 용출 구배 0-0.7분 0% B, 0.7-4.2 분 0→100% B, 4.2-4.6분 100% B, 4.6-4.8분 100→0% B를 사용하여, 3 ml/분의 유량에서 0.1% HCO2H 및 물 중의 0.01M 아세트산암모늄 (용매 A), 및 95% MeCN 및 5% 물 (0.5% HCO2H 함유) (용매 B)로 용출시키면서 수펠코실(Supelcosil)TM ABZ+PLUS 컬럼 (수펠코) (3 ㎛, 3.3cm×4.6 mm ID) 상에서 분석적 HPLC를 수행하였다. 215 내지 330 nm의 범위에서 다이오드 어레이 UV 검출을 수행하였다. 전자분무 포지티브 이온화 [(MH+ 및 M(NH4)+ 분자 이온을 수득하기 위해 ES+ve] 또는 전자분무 네가티브 이온화 [(M-H)- 분자 이온을 수득하기 위해 ES-ve] 모드를 사용하여 워터스 ZQ 질량 분광계 상에서 질량 스펙트럼(MS)을 기록하였다. 단지 주요 동위원소의 모 이온(parent ion) 만이 예시된다.
테트라메틸실란을 표준으로서 사용하여 브루커(Bruker) DPX 400MHz 분광계를 사용하여 1H NMR 스펙트럼을 기록하였다.
바이오태그(Biotage)TM 크로마토그래피는, KPSil (실리카)로 예비-충진된 카트릿지를 사용하여, 바이오태그 AB에 의해 시판되는 플래시 40i 또는 플래시 150i 정제 체계를 사용하여 수행되는 정제를 가리킨다.
컴패니온 (Companion)TM 시스템이란 텔레다인 이스코(Teledyne Isco) 콤비플 래시 컴패니온TM 정제 시스템을 가리킨다. 이것은 UV 역치에 의한 자동화 분획 수집을 유발하는 능력과 함께 통합적인 가변 파장 UV 검출을 가진 구배 제어 정제 시스템이다.
질량 지정 오토프레프(MDAP)란, 수펠코실(Supelcosil)TM ABZ + 5㎛ 컬럼 (10 cm × 20 mm i.d.) 또는 수펠코실TM ABZ + 10㎛ 컬럼 (15 cm × 30 mm i.d.)를 사용하여 용매 A: 물 중의 0.1% HCO2H 및 용매 B: 95% MeCN, 5% 물 (0.5% HCO2H 함유)의 적절한 구배에 의하여 고 성능 액체 크로마토그래피에 의해 물질이 정제되는 방법을 가리킨다. 워터스 2767 주입/수집장치는 주요 질량을 검출하면서 마이크로매스 ZQ 질량 분광계에 의해 유발되었다 (마이크로매스 매스링스(Mircomass MassLynx) 소프트웨어 사용).
조제 HPLC (오토프레프 HPLC 또는 오토프레프)란, 물 중의 0.1% HCO2H 및 MeCN (0.5% HCO2H를 가짐)의 적절한 구배를 사용하여 수펠코실(Supelcosil)TM ABZ+5㎛ 컬럼 (10cm × 21.2 mm i.d.) 상에서 고 성능 액체 크로마토그래피에 의해 물질이 정제되는 방법을 가리킨다. 길슨(Gilson) 233 분획 수집장치는 UV 검출에 의해 유발되었다.
SPE (고체 상 추출)는 정제를 위해 사용되는 흡수제로 사전-충진된 폴리에틸렌 카트릿지의 사용을 가리킨다. 이러한 카트릿지에 함유된 흡수제는 규정될 것이 다. 사용된 예는 하기 상세히 기재된다:
C18 SPE란 40 μM C18 작용화 실리카 흡수제 (배리언 인코포레이티드에 의해 시판됨)으로 예비-충진된 카트릿지의 사용을 가리킨다. 화합물을 전형적으로 50:50 DMSO/MeOH 중에서 MeCN으로 미리 조절된 카트릿지에 부하하고 물 중의 5% MeCN으로 평형상태화한다. 생성물을 물 중의 0.1% HCO2H 및 MeCN의 적절한 구배로 용출시켰다 (0.5% HCO2H).
아미노프로필 SPE 또는 컬럼은 40 ㎛ - 120 ㎛ 아미노프로필 작용화 실리카 (배리언 인코포레이티드에 의해 시판됨)으로 예비-충진된 카트릿지의 사용을 가리킨다. 조 생성물을 전형적으로 DCM/MeOH 혼합물 중에서 MeOH로 미리 상태조절된 카트릿지에 부하한다. 중성 성분을 MeOH 및/또는 DCM (3 또는 4개 컬럼 부피)로 용출하고 산성 성분을 AcOH의 일부를 함유하는 용출제로 용출시켰다 (2-20%).
오아시스(Oasis)TM 카트릿지/오아시스TM SPE란 워터스 코포레이션에 의해 제조된 중합체 흡수제로 충진된 SPE 카트릿지를 가리킨다. 이들은 전형적으로 3 컬럼 부피의 MeOH로 상태조절되고 샘플이 부하되기 전에 물로 평형화된다. 염 및 무기물을 물로 용출시키고 생성물을 전형적으로 MeOH 또는 MeCN으로 용출시킨다.
그린하우스(GreenHouse)TM 란 RDT Ltd.UK으로부터 입수가능한 24개 반응 병렬 합성장치 단을 가리킨다.
상기 나타낸 바와 같이, 화학식 I의 화합물은 인간 또는 동물용 의약에서 예 를 들어 이상지혈증 및 고지방단백혈증의 관리에서 HM74A의 활성화제로서 용도를 가질 수 있다.
따라서, 본 발명의 다른 구현양태로서, 이상지혈증 또는 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 지질 대사의 이상 및 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심혈관 질병, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진 및 비만의 치료에서, 인간 또는 동물용 의약에서 사용하기 위한 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체가 제공된다. 따라서, 화합물들은 또한 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 및 뇌졸중의 치료에서의 용도를 제공한다.
본 발명의 추가의 구현양태로서, 이상지혈증 또는 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 지질 대사의 이상 및 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심혈관 질병, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진 및 비만의 치료를 위한 약제의 제조에서 사용하기 위한, 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체가 제공된다. 따라서, 화합물들은 또한 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 및 뇌졸중의 치료에서의 용도를 제공한다.
여기에서 치료라는 언급은 증상의 예방, 재발 방지 또는 억제뿐만 아니라 확립된 상태의 치료까지 확장된다는 것을 이해해야 할 것이다.
본 발명의 하나의 구현양태에서, 이상지혈증 및 고지방단백혈증을 포함한 지질 대사의 이상의 치료에서 사용하기 위한 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체가 제공된다. 예를 들어, 당뇨병성 이상지혈증, 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진, 비만, 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 뇌졸중 및 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심혈관 질병의 치료에서 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체의 용도가 제공된다.
본 발명의 구현양태는 특정한 구현양태의 조합을 포함하고 화학식 I의 화합물에 대해 여기에 기재된 특정한 치환기의 모든 조합을 포함하는 것으로 이해된다.
추가로, 본 발명은 관절, 예를 들어 관절염 (예, 류마티스성 관절염, 골관절염, 인공 관절 기능상실) 또는 위장관 (예, 궤양 대장염, 크론병 및 기타 염증성 장 및 위장 질환, 위염 및 감염으로부터 생긴 점막 염증, 비-스테로이드성 항-염증 약물에 의해 유발된 창자병증), 폐 (예, 성인 호흡곤란증후군, 천식, 낭성섬유증, 또는 만성 폐쇄성 폐 질환), 심장 (예, 심근염), 신경 조직 (예, 다발경화증), 췌장 (예, 진성 당뇨병 및 그의 합병증과 관련된 염증), 신장 (예, 사구체신염), 피부 (예, 피부염, 건선, 습진, 두드러기, 화상), 눈 (녹내장) 뿐만 아니라 이식된 기관 (예, 거부) 및 다-기관 질환 (예, 전신홍반루푸스, 패혈증)의 염증성 질병 또는 상태 및 바이러스 또는 세균 감염의 염증 후유증 및 죽상경화증과 관련된 염증 상태 및 예를 들어 뇌에서의 저산소 또는 허혈성 발작 (재관류와 함께 또는 재관류 없이) 또는 허혈성 심장병의 치료를 위한 약제의 제조에서, 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체의 용도를 제공한다.
추가의 또는 대안적인 구현양태에서, 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체의 유효량을 인간 또는 동물 피험자에게 투여하는 것을 포함하는, HM74A 수용체의 활성화 부족이 상태의 원인이 되거나 또는 수용체의 활성화가 유익하게 될 상태를 가진 인간 또는 동물 피험자의 치료 방법이 제공된다.
또한, 본 발명의 구현양태는 특정한 구현양태의 조합을 포함하고 화학식 I의 화합물에 대해 상기 기재된 특정한 치환기의 모든 조합을 포함하는 것으로 이해된다.
하나의 구현양태에서, 본 발명은 인간 또는 동물 피험자에게 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 염의 유효량을 투여하는 것을 포함하는, 이상지혈증 또는 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 지질 대사의 이상 및 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심혈관 질병, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진 및 비만의 치료 방법을 제공한다. 따라서, 이러한 화합물은 인간 또는 동물 피험자에게 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체의 유효량을 투여하는 것을 포함하는, 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 및 뇌졸중의 치료 방법에서 유리할 것이다.
바람직한 생물학적 효과를 달성하기 위해 필요한 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체의 양은 물론 여러 요인, 예를 들어 투여 방식 및 수용자의 정확한 임상 조건에 의존될 것이다. 일반적으로, 1일 투여량은 0.1 mg 내지 1 g/kg, 전형적으로 0.1 내지 100 mg/kg의 범위일 것이다. 정맥내 투여량은 일반적으로 0.01 mg 내지 0.1 g/kg, 전형적으로 0.01 mg 내지 10 mg/kg의 범위일 수 있고, 이것은 1 분 당 0.1 ㎍ 내지 1 mg의 주입으로서 편리하게 투여될 수 있다. 이러한 목적을 위해 적절한 주입 유체는 예를 들어 밀리리터 당 0.01 ㎍ 내지 0.1 mg를 함유할 수 있다. 단위 투여량은 예를 들어 0.01 ㎍ 내지 1 g의 본 발명의 화합물을 함유할 수 있다. 주사용 앰풀은 예를 들어 0.01 ㎍ 내지 0.1 g을 함유할 수 있고, 경구 투여가능한 단위 투여 제제, 예컨대 정제 또는 캡슐은 예를 들어 0.1 mg 내지 1 g, 예를 들어 5 mg 내지 50 mg을 함유할 수 있다.
화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체는 HM74A 수용체의 활성화 부족이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병의 치료에서 그 자체로서 화합물로서 사용될 수 있고, 그의 예는 본 발명의 화합물이 제약학적 제제의 형태로 허용가능한 담체와 함께 존재하는 것이다. 물론, 담체는 제제의 다른 성분과 상용성이라는 의미에서 허용가능해야 하고 수용자에게 해로워서는 안된다. 담체는 고체 또는 액체 또는 양쪽 모두일 수 있고 단위-투여 제제, 예를 들어 0.05 내지 95 중량%의 본 발명의 화합물을 함유할 수 있는 정제로서 본 발명의 화합물과 함께 제형될 수 있다.
제제는 경구, 직장, 국소, 협측 (예, 설하) 및 비경구 (예, 피하, 근육내, 피내 또는 정맥내) 투여를 위해 적절한 것을 포함한다.
본 발명에 따르면, 성분들을 혼합하는 것을 포함하여, 이러한 제약학적 조성물의 제조 방법이 제공된다.
경구 투여를 위해 적절한 제제는 별개의 단위, 예컨대 각각 소정 량의 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 함유하는 캡슐, 카세제, 함당정제 또는 정제로서; 분말 또는 과립으로서; 수성 또는 비-수성 액체 중의 용액 또는 현탁액으로서; 또는 수-중-유 또는 유-중-수 에멀젼으로서 존재할 수 있다. 일반적으로, 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 액체 또는 미세하게 분리된 고체 담체 또는 양쪽 모두와 균일하고 긴밀하게 혼합한 다음 필요하다면 생성물을 성형함으로써 제제가 제조된다. 예를 들어 정제는 화학식 I의 화합물 또는 그의 제약학적으로 허용가능한 유도체의 분말 또는 과립을 임의로 하나 이상의 보조 성분과 함께 압축 또는 성형함으로써 제조될 수 있다. 압축된 정제들은, 적절한 기계에서 임의로 결합제, 윤활제, 불활성 희석제 및/또는 표면 활성/분산제와 혼합된 자유-유동 형태, 예컨대 분말 또는 과립의 화합물을 압축함으로써 제조될 수 있다. 성형된 정제는 적절한 기계에서 불활성 액체 희석제와 습윤된 분말화 화합물을 성형함으로써 제조될 수 있다.
경구 투여를 위한 정제 및 캡슐은 결합제, 예를 들어 시럽, 아라비아고무, 젤라틴, 소르비톨, 트라가칸트, 전분 또는 폴리비닐 피롤리돈의 점액; 충진제, 예를 들어 락토스, 미세결정질 셀룰로스, 당, 옥수수-전분, 인산칼슘 또는 소르비톨; 윤활제, 예를 들어 마그네슘 스테아레이트, 스테아르산, 탈크, 폴리에틸렌 글리콜 또는 실리카; 붕해제, 예를 들어 감자 전분, 크로스카르멜로스 나트륨 또는 나트륨 전분 글리콜레이트; 또는 습윤제, 예컨대 나트륨 라우릴 설페이트와 같은 통상적인 부형제를 함유할 수 있다. 정제를 당 기술분야에 공지된 방법에 따라 코팅할 수 있다. 경구 액체 제제는 예를 들어 수성 또는 유성 현탁액, 용액, 에멀젼, 시럽 또는 엘릭시르의 형태일 수 있거나, 또는 사용 전에 물 또는 기타 적절한 부형제와의 구성을 위한 건조한 제품으로서 존재할 수 있다. 이러한 액체 제제는 통상적인 첨가제, 예컨대 현탁제, 예를 들어 소르비톨 시럽, 메틸 셀룰로스, 글루코스/당 시럽, 젤라틴, 히드록시메틸 셀룰로스, 히드록시프로필메틸 셀룰로스, 카르복시메틸 셀룰로스, 알루미늄 스테아레이트 겔 또는 수소첨가된 식용 유지; 유화제, 예를 들어 레시틴, 소르비탄 모노-올레에이트 또는 아라비아고무; 비-수성 부형제 (식용 오일을 포함할 수 있음), 예를 들어 아몬드유, 분별된 코코넛 유, 유성 에스테르, 프로필렌 글리콜 또는 에틸 알콜; 또는 보존제, 예를 들어 메틸 또는 프로필 p-히드록시벤조에이트 또는 소르빈산을 함유할 수 있다. 제제는 적절하다면 완충제 염, 향미제, 착색제, 및/또는 감미제 (예, 만니톨)을 함유할 수 있다.
협측 (설하) 투여를 위해 적절한 제제는 본 발명의 화합물을 향미 기재, 통상 슈크로스 및 아라비아고무 또는 트라가간트에 포함하는 함당정제, 및 본 발명의 화합물을 불활성 기재, 예컨대 젤라틴 및 글리세린 또는 슈크로스 및 아라비아고무에 포함하는 향정을 포함한다.
비경구 투여를 위해 적절한 본 발명의 제제는 편리하게는 화학식 I의 화합물의 무균 수성 제제 또는 제약학적으로 허용가능한 그의 유도체를 포함하고, 제제는 수용자의 혈액과 등장성일 수 있다. 투여가 피하, 근육내 또는 피내 주사에 의해 실행될 수 있긴 하지만 이러한 제제는 정맥내 투여될 수 있다. 이러한 제제는 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 물과 혼합하고, 얻어진 용액을 무균이 되게 하고 혈액과 등장성이 되게 함으로써 편리하게 제조될 수 있다. 본 발명에 따른 주사용 조성물은 일반적으로 0.1 내지 5% w/w의 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 함유할 것이다.
따라서, 일시 주사 또는 연속 주입에 의한 비경구 투여를 위하여 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 포함하는 비경구 투여를 위해 적절한 본 발명의 제제를 제형할 수 있고, 단위 투여 형태, 예를 들어 앰풀, 바이알, 작은 부피의 주입 또는 미리 충진된 주사기로 제공할 수 있거나 첨가된 보존제를 가진 다-용량 용기에 존재할 수 있다. 조성물은 수성 또는 비-수성 부형제 중의 용액, 현탁액 또는 에멀젼으로서 형태를 취할 수 있고, 산화방지제, 완충제, 항균제 및/또는 독성 조절제와 같은 제형화제를 함유할 수 있다. 경구 투여를 위해 적절한 제제의 예는 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 살균 염수 중의 10% DMSO 및 90% 탄산수소나트륨 중에 포함하는 제제를 포함한다. 정맥내 투여를 위해 적절한 제제의 예는 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 살균 수 중의 5% 또는 10% DMSO 및 95% 또는 90% 탄산수소나트륨 중에 포함하는 제제를 포함한다. 대안적으로, 치료적 활성 약제는 사용 전에 적절한 부형제, 예를 들어 살균, 비병원성 물과의 구성을 위한 분말 형태일 수 있다. 건조 고체는 살균 분말을 무균적으로 각각의 살균 용기 내에 충진하거나 살균 용액을 무균적으로 각각의 용기 내에 충진하고 동결 건조함으로써 제조될 수 있다.
직장 투여를 위해 적절한 제제는 단위-투여 좌약으로서 존재할 수 있다. 이들은 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 하나 이상의 통상적인 고체 담체, 예를 들어 코코넛 버터 또는 글리세리드와 혼합한 다음 얻어진 혼합물을 성형함으로써 제조될 수 있다.
피부에 국소 적용을 위해 적절한 제제는 연고, 크림, 로션, 페이스트, 겔, 스프레이, 에어로졸 또는 오일의 형태를 취할 수 있다. 사용될 수 있는 담체는 바셀린, 라놀린, 폴리에틸렌 글리콜, 알콜 및 2 이상의 조합을 포함한다. 화학식 I의 화합물 또는 제약학적으로 허용가능한 그의 유도체는 일반적으로 조성물의 0.1 내지 15% w/w, 예를 들어 0.5 내지 2%의 농도로 존재한다.
여기에서 사용된 국소 투여에 의하여, 본 발명자들은 통기 및 흡입에 의한 투여를 포함한다. 국소 투여를 위해 다양한 유형의 제제의 예는 연고, 크림, 로션, 분말, 페서리, 스프레이, 에어로졸, 캡슐, 또는 흡입기 또는 취입기에서 사용하기 위한 카트릿지, 또는 점적제 (예, 점안제 또는 점비제)를 포함한다.
연고 및 크림은 예를 들어 적절한 증점 및/또는 겔화제 및/또는 용매의 첨가와 함께 수성 또는 유성 기재와 함께 제형될 수 있다. 이러한 기재는 예를 들어 물 및/또는 오일, 예컨대 액체 파라핀 또는 식물성 오일, 예컨대 낙화생 유 또는 피마자 유 또는 폴리에틸렌 글리콜과 같은 용매를 포함할 수 있다.
사용될 수 있는 증점제는 연질 파라핀, 알루미늄 스테아레이트, 세토스테아 릴 알콜, 폴리에틸렌 글리콜, 미세결정질 왁스 및 밀랍을 포함한다.
로션은 수성 또는 유성 기재와 함께 제형될 수 있고 일반적으로 하나 이상의 유화제, 안정화제, 분산제, 현탁제 또는 증점제를 함유할 것이다
외부 적용을 위한 분말은 적절한 분말 기재, 예를 들어 탈크, 락토스 또는 전분과 함께 형성될 수 있다. 점적제는 하나 이상의 분산제, 가용화제 또는 현탁제를 포함하는 수성 또는 비-수성 기재와 함께 제형될 수 있다.
스프레이 조성물은 예를 들어 수용액 또는 현탁액으로서 또는 적절한 추진제, 예를 들어 디클로로디플루오로메탄, 트리클로로플루오로메탄, 디클로로테트라플루오로에탄, 1,1,1,2,3,3,3-헵타플루오로프로판, 1,1,1,2-테트라플루오로에탄, 이산화탄소 또는 기타 적절한 기체를 사용하여 가압 팩으로부터 전달되는 에어로졸로서 제형될 수 있다.
예를 들어 젤라틴의 흡입기 또는 취입기에서 사용하기 위한 캡슐 및 카트릿지는 본 발명의 화합물의 분말 혼합물 및 락토스 또는 전분과 같은 적절한 분말 기재를 함유하도록 제형될 수 있다.
본 발명에 따른 제약학적 조성물은 다른 치료제와 함께, 예를 들어 다른 부류의 이상지혈증 약물 (예, 스타틴, 피브레이트, 담즙산 결합 수지 또는 니코틴산)과 조합하여 사용될 수 있다.
본 발명의 화합물은 하나 이상의 다른 치료적 활성 약제와 조합하여, 예를 들어 다른 부류의 이상지혈증 약물, 예를 들어 3-히드록시-3-메틸글루타릴-조효소 A 환원효소 억제제 (스타틴) 또는 피브레이트 또는 담즙산 결합 수지 또는 니코틴 산과 조합하여 사용될 수 있다. 추가의 구현양태에서, 본 발명은 HM74A 수용체의 활성화 부족이 질병의 원인이 되거나 수용체의 활성화가 유익하게 될 질병의 치료에서 이러한 조합물의 용도, 및 이상지혈증 및 고지방단백혈증, 예컨대 당뇨병성 이상지혈증 및 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증을 포함한 많은 지질 대사의 이상, 죽상경화증, 동맥경화증 및 고트리글리세리드혈증을 포함한 심장혈관 질병, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진 및 비만의 조합 요법을 위한 약제의 제조에서 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체의 용도를 제공한다.
본 발명의 화합물이 다른 치료적 활성 약제와 조합하여 사용될 때, 화합물은 통상적인 경로에 의해 함께 또는 별도로, 연속적으로 또는 동시에 투여될 수 있다.
상기 언급된 조합물은 제약학적 제제의 형태에서 사용하기 위해 편리하게 제공할 수 있고, 따라서 상기 정의된 조합물을 제약학적으로 허용가능한 담체 또는 부형제와 함께 최적으로 포함하는 제약학적 제제는 본 발명의 추가의 구현양태를 포함한다. 이러한 조합물의 각각의 성분은 별개의 또는 조합된 제약학적 제형에서 함께 또는 별도로, 연속적으로 또는 동시에 투여될 수 있다.
동일한 제제에서 조합될 때, 2개의 성분들이 안정해야 하고 상호 간에 및 제제의 다른 성분과 함께 상용가능해야 하며 투여를 위해 제형될 수 있는 것으로 이해된다. 별개로 제형될 때, 이들은 당 기술분야에서 이러한 화합물을 위해 공지된 것과 같은 방식으로 편리하게 편리한 제형으로 제공될 수 있다.
동일한 질병에 대해 활성인 두 번째 치료제와 조합될 때, 각각의 성분의 투 여량은 화합물이 단독으로 사용될 때와 상이할 수 있다. 적절한 투여량은 당업자에 의해 쉽게 이해될 것이다.
따라서, 본 발명은 추가의 구현양태에서 화학식 I의 적어도 하나의 화합물 또는 제약학적으로 허용가능한 그의 유도체를 다른 치료적 활성 약제와 함께 포함하는 조합물을 제공한다.
상기 언급된 조합물은 제약학적 제제의 형태에서 사용하기 위해 편리하게 제공될 수 있고, 따라서 상기 정의된 조합물을 제약학적으로 허용가능한 담체와 함께 포함하는 제약학적 제제가 본 발명의 추가의 구현양태를 나타낸다.
본 발명의 화합물 및 제약학적으로 허용가능한 그의 유도체는 본 발명의 추가의 구현양태를 구성하는 이하 기재된 방법에 의하여 제조될 수 있다.
하나의 구현양태에서, 본 발명은
적절한 출발 물질, 예를 들어 하기 화학식 II의 화합물(들)로부터
(i) N7 보호된 크산틴의 N1에서의 알킬화;
(ii) N7 보호된 크산틴의 N3에서의 알킬화;
(iii) C8에서의 할로겐화; 및
(iv) N7의 탈보호를 임의의 순서로 수행하는 것을 포함하되,
단, 탈보호는 알킬화 후에 수행하는 것인, 화학식 I의 화합물(들)의 제조 방법을 제공한다.

상기 식에서, PG = 보호기
방법 1:
R1이 헤테로시클릴, 헤테로아릴 또는 아릴을 포함하고, R1이 Cl을 나타내는 화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법.

i) 알릴 브로마이드에 의한 구아닌의 알킬화
ii) 아질산나트륨과의 디아조화에 이어서 가수분해에 의해 크산틴을 형성
iii) 염소화
iv) N3에서의 알킬화 (적절한 염기의 예는 Na2CO3, Cs2CO3, K2CO3를 포함한다)
v) N1에서의 알킬화 (적절한 염기의 예는 Na2CO3, Cs2CO3, K2CO3를 포함한다)
vi) 알릴 기의 팔라듐 촉매화 제거.
상기 식에서, L은 이탈기, 예를 들어 할로겐을 나타낸다.
방법 2:
화학식 I의 화합물(들)의 제조를 위해 유용할 수 있는, R1이 아미드, 카르바메이트 또는 우레아를 포함하는 중간체를 제조하기 위한 본 발명에 따른 방법.

상기 식에서, L은 이탈기, 예를 들어 할로겐을 나타내고, d는 (m-1)을 나타내고 (즉 d는 이전의 메틸렌 = m을 가짐), R은 -(알킬렌)n-Y를 나타내고, Q는 존재하거나 존재하지 않을 수 있고 존재한다면 O 또는 NR5를 나타낸다.
방법 3:
화학식 I의 화합물(들)의 제조를 위해 유용할 수 있는, R1이 "역" 카르바메이트 또는 에스테르를 포함하는 중간체의 제조 방법.

상기 식에서, L은 이탈기, 예를 들어 할로겐을 나타내고, d는 (m-1)을 나타내고, R은 -(alk)n-Y를 나타낸다.
방법 4:
화학식 I의 화합물(들)의 제조를 위해 유용할 수 있는, R1이 에스테르 또는 아미드를 포함하는 중간체를 제조하기 위한 본 발명에 따른 방법.

상기 식에서, L은 이탈기, 예를 들어 할로겐을 나타내고, d는 (m-1)을 나타 내고, R은 -NR5R7또는 -OR5를 나타낸다.
방법 5:
X가 피라졸, 이미다졸 또는 테트라졸을 포함하는, 화학식 I의 화합물을 제조하기 위한 본 발명에 따른 방법.

상기 식에서, L은 이탈기, 예를 들어 할로겐을 나타내고, d는 (m-1)을 나타내고, R은 -(alk)n-Y를 나타낸다.
방법 6:
X가 옥사디아졸을 포함하는 화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법.

상기 식에서, L은 이탈기, 예를 들어 할로겐을 나타내고, d는 (m-1)을 나타내고, R은 알킬기를 나타내고 R'는 -(alk)n-Y를 나타낸다.
방법 7:
X가 옥사디아졸을 포함하는 화학식 I의 화합물을 제조하기 위한 본 발명에 따른 방법.

상기 식에서,
L은 이탈기, 예를 들어 할로겐을 나타내고, d는 (m-1)을 나타내고, R은 알킬 기를 나타내고 R'는 (alk)n-Y를 나타낸다.
방법 8:
화학식 I의 화합물(들)의 제조를 위해 유용할 수 있는, R3이 CN인 중간체를 제조하기 위한 본 발명에 따른 방법.
이것은 방법 1의 단계 (i) 및 (ii)에 이어서 다음 단계들을 포함한다.

iii) N3에서의 알킬화
iv) N1에서의 알킬화
v) LiHMDS에 의한 리튬화 및 DMF 반응정지에 의하여 C8에서의 알데히드의 형성
vi) 알데히드의 니트릴로의 전환
vii) 알릴 기의 팔라듐 촉매화 제거
상기 식에서, L은 이탈기를 나타낸다.
방법 9:
R3가 Cl 또는 Br인 화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법은 방법 8의 단계 (i) 내지 (iv)에 이어서 다음 단계를 포함한다:

i) NCS 또는 NBS를 사용하여 C8에서의 할로겐화
ii) 알릴 기의 팔라듐 촉매화 제거
방법 10:
R3이 Cl인 화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법은 다음 단계를 포함한다.

i) N3에서의 알킬화
ii) N1에서의 알킬화
iii) 탈벤질화
iv) C8에서의 염소화
상기 식에서, L은 이탈기를 나타낸다.
방법 11:
R1이 R2와 상이하고 R3이 Cl인 화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법은 방법 1의 단계 (i) 내지 (v) (식에서, 방법 1로부터의 R2는 특별하게는 SEM 또는 MEM이다)에 이어서 다음 단계들을 포함한다.

vi) MEM 또는 SEM 보호 기의 절단
vii) N3의 알킬화에 이어서 알릴 기의 팔라듐 촉매화 제거
상기 식에서, L은 이탈기를 나타낸다.
방법 12:
R3이 Cl, Br 또는 I인 화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법은 방법 8의 단계 (i) 내지 (iv)에 이어서 하기 단계들을 포함한다.

v) 알릴 기의 팔라듐 촉매화 제거
vi) NCS, NBS 또는 NIS를 사용하여 C8에서의 할로겐화
방법 13:
화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법은 다음을 포함 한다.

i) 피리미딘디온 형성
ii) N1에서의 알킬화
iii) 니트로화
iv) Na2S2O4 또는 유사한 환원제를 사용한 환원
v) 크산틴 형성
vi) NCS를 사용하여 C8에서의 할로겐화
상기 식에서, L은 이탈기를 나타낸다.
방법 14:
화학식 I의 화합물(들)을 제조하기 위한 본 발명에 따른 방법.

상기 식에서, L은 이탈기를 나타낸다.
상기 기재된 일반적 방법에 대한 추가의 단계로서, 특히 하기 실시예의 제조에서 사용하기 위하여, 화합물을 정제하기 위한 몇 가지 방법이 존재하고 그의 하나 이상은 본 발명에서 사용될 수 있고, 예를 들어 MDAP를 사용하거나, 에틸 아세테이트, 무수 에탄올, 아세토니트릴 또는 메탄올과 같은 하나 이상의 적절한 용매로부터 재결정화하거나, 또는 실리카 레디셉(Silica Redisep)TM 카트릿지와 같은 정제 컬럼을 사용하고 이어서 에틸 아세테이트를 함유하는 디클로로메탄과 같은 적절한 용매로 용출하는 것이다.
원하거나 필요하다면, 상기 합성 방법의 어느 하나의 최종 단계로서, 화학식 I의 화합물을 제약학적으로 허용가능한 유도체로 전환될 수 있고, 예를 들어 화학식 I의 화합물을 염 형태로 전환할 수 있거나 또는 그 역으로 전환할 수 있고, 또는 하나의 염을 다른 제약학적으로 허용가능한 염 형태로 전환할 수 있다. 이러한 방법이 당업자에게 공지될 것이다.
약어
AcOH 아세트산
atm 대기압
br 브로드(broad) (NMR)
CDI 카르보닐디이미다졸
d 더블렛(doublet) (NMR)
DBAD 디-t-부틸아조디카르복실레이트
DCM 디클로로메탄
DIPEA 디이소프로필에틸아민
DMSO 디메틸술폭시드
DMF N,N-디메틸포름아미드
EtOAc 에틸 아세테이트
EtOH 에탄올
h 시간(들)
IPA 이소프로필 알콜
m 멀티플렛 (NMR)
MDAP 질량 지정 오토프레프(mass directed autoprep)
MeCN 아세토니트릴
MeOH 메탄올
min 분(들)
NCS N-클로로숙신이미드
NBS N-브로모숙신이미드
NIS N-요오도숙신이미드
q 쿼테트 (NMR)
rt 실온
RT 체류 시간
s 싱글렛 (NMR)
SPE 고체상 추출 카트릿지
t 트리플렛 (NMR)
TFA 트리플루오로아세트산
THF 테트라히드로푸란
DMEM 둘베코 변형 이글 배지
HEPES 4-(2-히드록시에틸)피페라진-1-에탄술폰산
LiHMDS 리튬 헥사메틸디실릴아미드
△ 열
SEM 2-(트리메틸실릴)에톡시메틸
MEM 2-메톡시에톡시메틸
Boc t-부톡시카르보닐
THP 테트라히드로피란
도 1: 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥 사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1의 XRPD 데이터
도 2: 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 2의 XRPD 데이터
도 3: 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1 및 형태 2의 XRPD 데이터의 겹침
하기 비-제한적 실시예들은 본 발명을 예증한다:
합성 예
하기 예시된 화합물에 표시된 (Z)-입체화학의 지정은 실험 데이터에 의해 확인되지 않는다는 것을 주목해야 한다. 당업자라면 E & Z 이성질체 간의 상호전환이 존재할 수 있음을 인지할 것이다 [Dondoni, Alessandro; Lunazzi, Lodovico; Giorgianni, Patrizia; Macciantelli, Dante. Carbon-nitrogen rotational barrier as a stereochemical probe of benzamidoximes. Journal of Organic Chemistry (1975), 40(20), 2979-80].
실시예
1: 8-
클로로
-1-(3-{1-[(2-
클로로
-6-
플루오로페닐
)
메틸
]-1H-
피라졸
-4-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-퓨
린
-2,6-
디온
a) 8-클로로-1-(3-{1-[(2-클로로-6-플루오로페닐)메틸]-1H-피라졸-4-일}프로필)-3-펜틸-3,7-디히드로-1H-퓨린-2,6-디온

건조 DMF (2 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-1-[3-(1H-피라졸-4-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온 (61 mg, 0.15 밀리몰)을 탄산나트륨 (64 mg, 0.6 밀리몰)과 함께 교반하고, 2-클로로-6-플루오로벤질 브로마이드 (134 mg, 0.6 밀리몰)을 질소 하에 45 ℃에서 18 시간 동안 가열하였다. 실온으로 냉각한 후에, 공기를 빼내고 질소를 다시 넣음으로써 혼합물을 탈기시키고, 테트라키스(트리페닐포스핀)팔라듐(0) (35 mg, 0.303 밀리몰) 및 모르폴린 (0.13 ml)과 함게 5.5 시간 동안 교반하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 분리하고 증발시키고 잔류물을 아미노프로필 SPE (5 g, THF-MeOH (1:1)로 세척한 다음 순수한 MeOH로 세척하고 마지막으로 5% AcOH를 함유하는 DCM-MeOH (1:1)로 용출하였다)에 의해 정제하여 표제 화합물 (57 mg)을 고체로서 수득하였다.
LC/MS: m/z 507 [MH]+, RT 3.64 분
b) 8-클로로-3-펜틸-7-(2-프로펜-1-일)-1-[3-(1H-피라졸-4-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온

3-펜틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (5 g, 16.86 밀리몰) 및 3-(1H-피라졸-4-일)프로판-1-올 (2.12 g, 16.8 밀리몰)을 건조 THF (150 ml) 중에서 3 ℃에서 교반하였다. 디벤질 아조디카르복실레이트 (10.05 g, 33.7 밀리몰)를 첨가한 다음 건조 THF (70 ml) 중의 트리페닐포스핀 (8.83 g, 33.7 밀리몰)을 적가하였다. 혼합물을 실온으로 가온하고 18시간 동안 교반하였다. 물 (1 ml)을 첨가하고, 용매를 증발시켰다. 잔류물을 Et2O (200 ml) 중에 취하고, 그로 부터 대부분 트리페닐포스핀 옥사이드인 백색 고체가 결정화되고 이것을 여과해 내었다. 여액을 농축하고 에테르-시클로헥산으로부터 추가의 부산물을 결정화하였다. 나머지 여액을 농축하고 (19.2 g), EtOAc-시클로헥산 (2:1)로 용출되는 바이오태그(Biotage)TM 시스템 (400 g) 위에서 정제하여 표제 화합물을 백색 고체 (2.89 g)로서 수득하였다.
LC/MS: m/z 405 [MH]+, RT 3.19 분
상응하는 벤질 할라이드로부터 실시예 1에 대한 것과 유사한 방법을 사용하여 하기 화합물 (표 1)을 제조하였다.

표 1로부터의 선택된 실시예에 대한 NMR 세부사항
실시예 2: 8-클로로-3-펜틸-1-(3-{1-[(2,4,6-트리플루오로페닐)메틸]-1H-피라졸-4-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온

실시예
6: 8-
클로로
-1-(3-{1-[(2,4-
디플루오로페닐
)
메틸
]-1H-
피라졸
-4-일}프로필)-3-펜틸-3,7-
디히드로
-1H-퓨린-2,6-
디온

8-클로로-3-펜틸-7-(2-프로펜-1-일)-1-[3-(1H-피라졸-4-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온 (81 mg, 0.2 밀리몰) 및 탄산나트륨 (85 mg, 0.8 밀리몰)을 무수 DMF (2 ml) 중에서 2,4-디플루오로벤질 브로마이드 (166 mg, 0.8 밀리몰)과 함께 45 ℃에서 18시간 동안 교반하였다. 혼합물을 탈기시키고, 테트라키스(트리페닐포스핀)팔라듐(0) (46 mg, 0.04 밀리몰) 및 모르폴린 (176 mg, 2 밀리몰)과 함께 실온에서 6 시간 동안 교반하였다. 반응을 진행시키고 아미노프로필 SPE (5 g, THF-MeOH (1:1)로 세척한 다음 순수한 MeOH로 세척하고 마지막으로 5% AcOH를 함유하는 DCM-MeOH (1:1)로 용출한다)에 의해 정제하여 표제 화합물 (37.7 mg)을 고체로서 수득하였다.
LC/MS: m/z 491 [MH]+, RT 3.42 분
실시예 6에 대한 것과 유사한 방법을 사용하여 상응하는 벤질 할라이드로부터 하기 화합물 (표 1)을 제조하였다.

표 2로부터의 선택된 실시예에 대한 NMR 세부사항
실시예 6:
실시예 7:
8-클로로-3-펜틸-1-{3-[1-(페닐메틸)-1H-피라졸-4-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온:

실시예
11: 8-
클로로
-1-(3-{1-[(2-
클로로페닐
)
메틸
]-1H-
피라졸
-4-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-퓨린-2,6-
디온

2-클로로벤질 브로마이드 (164 mg, 0.8 밀리몰)로부터 제조되는 것 이외에는, 8-클로로-1-(3-{1-[(2,4-디플루오로페닐)메틸]-1H-피라졸-4-일}프로필)-3-펜틸-3,7-디히드로-1H-퓨린-2,6-디온, 실시예 6에 대한 방법에 의해 제조하였다. 그러나, 탈보호 단계를 완결하기 위하여, 추가의 테트라키스(트리페닐포스핀)팔라듐(0) (40 mg) 및 모르폴린 (0.15 ml)을 첨가하고, 추가의 5.5 시간 동안 교반을 계속하였다. 상기와 같은 아미노프로필 SPE에 의한 정제는 고체로서 표제 화합물을 제공하였다 (42 mg).
LC/MS: m/z 489 [MH]+, RT 3.67 분
실시예
12: 3-부틸-8-
클로로
-1-{3-[1-(
페닐메틸
)-1H-
이미다졸
-4-일}프로필)-3,7-
디히드로
-1H-퓨린-2,6-
디온
a) 3-부틸-8-클로로-1-{3-[1-(페닐메틸)-1H-이미다졸-4-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

무수 THF (5 ml) 중의 3-부틸-8-클로로-1-[4-(1H-이미다졸-4-일)부틸]-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (300 mg, 0.77 밀리몰)의 용액을 벤젤 브로마이드 (144 mg, 0.84 밀리몰) 및 DIPEA (147 ㎕, 0.84 밀리몰)로 처리하였다. 혼합물을 질소 하에 실온에서 4일 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. 원료를 0.5-5% MeOH/DCM 구배를 사용하여 실리카 SPE 컬럼에 의해 정제하였다. 생성물 분획을 합하고 고 진공 하에서 농축하였다. 생성물을 THF (5 ml)에 용해시키고 이어서 Pd(PPh3)4 (88 mg, 0.077 밀리몰) 및 모르폴린 (670㎕, 7.67 밀리몰)을 첨가하고, 혼합물을 질소 하에 실온에서 3 시간 동안 교반하였다. 88 mg의 Pd(PPh3)4 (0.077 밀리몰)을 첨가하고, 혼합물을 질소 하에 16 시간 동안 실온에서 교반하였다. 혼합물을 EtOAc와 H2O 사이에 분배하였다. 유기 층을 분리하고, 수성 층을 EtOAc (×2)로 추출하였다. 유기 층을 합하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하였다. 조 생성물을 MDAP에 의해 정제하여 백색 고체로서 표제 화합물을 수득하였다 (9 mg, 2%).

b) 3-부틸-8-클로로-1-[3-(1H-이미다졸-4-일)프로필]-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온
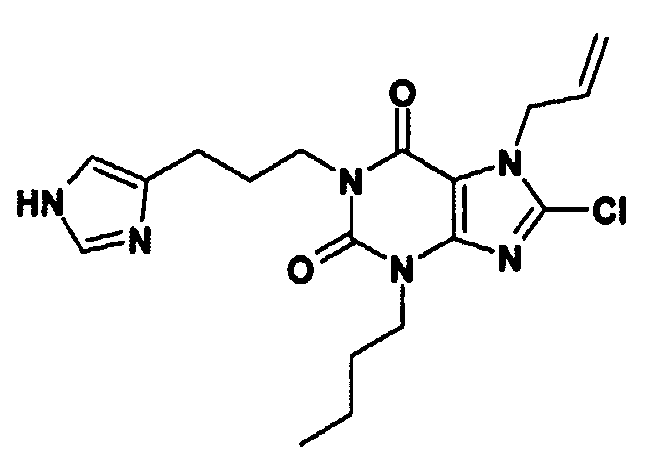
무수 THF (60 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (2.8 g, 9.9 밀리몰)의 용액을 무수 THF (10 ml) 및 PPh3 (3.4 g, 13 밀리몰)중에서 3-(1H-이미다졸-4-일)-1-프로판올 (1.5 g, 12 밀리몰)로 처리하였다. DBAD (2.9 g, 13 밀리몰)을 한번 분량으로 첨가하고 혼합물을 질소 하에 18시간 동안 실온에서 교반하였다. 혼합물을 EtOAc와 H2O 사이에 분배하였다. 수성 층을 추출하고 EtOAc로 세척하였다. 유기 층을 합하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. 조 생성물을 MeOH/EtOAc 구배 (0.5%-7% MeOH)를 사용하여 실리카 SPE 카트릿지에 의해 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (2.16g, 55%).
LC/MS: m/z 391 [MH]+, RT 2.40분
c) 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온

무수 DMF (19 ml) 중의 3-부틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (3.34g, 13.4 밀리몰)의 용액에 NCS (1.97 g, 14.8 밀리몰)를 첨가하고, 실온에서 질소 하에 22 시간 동안 교반하였다. 혼합물을 진공 하에 농축하여 황색 고체를 수득하고, 이것을 여과하고 MeOH로 세척하여 첫 번째 수확물을 제공하였다. 여액을 고체로 농축하고 MeOH로 세척하여 두 번째 수확물을 제공하고, 2가지 경우를 더욱 반복하여 표제 화합물을 제공하였다. 최종 세척 시에, 여액을 EtOAc:시클로헥산(1:1)으로 용출되는 SPE (Si, 20 g) 카트릿지에 의해 더욱 정제하였다. 합한 고체를 진공 하에 건조시켜 표제 화합물 (2.42g, 64%)을 수득하였다.
LC/MS: m/z 283 [MH]+
실시예
13: 3-부틸-8-
클로로
-1-(3-{1-[2,3-
디플루오로페닐
)
메틸
]-1H-
이미다졸
-4-일}프로필)-3,7-
디히드로
-1H-퓨린-2,6-
디온

무수 DMF (3 ml) 중의 3-부틸-8-클로로-1-[4-(1H-이미다졸-4-일)부틸]-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (150 mg, 0.38 밀리몰)의 용액을 1-(브로모메틸)-2,4-디플루오로벤젠 (54 ㎕, 0.42 밀리몰) 및 DIPEA (73 ㎕, 0.42 밀리몰)로 처리하였다. 혼합물을 질소 하에서 3일 동안 실온에서 교반하였다. 혼합물을 EtOAc 및 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하였다. DCM을 사용하여 물질을 컬럼에 부하하고, 불순물을 씻어낸 다음 0-5% MeOH/DCM 구배에 의해 화합물을 용출함으로써 실리카 SPE 컬럼 상에서 조 생성물을 정제하였다. 생성물 분획을 합하고 농축하고 잔류물을 무수 DMF (3 ml)에 용해시켰다. 용액을 탈기시키고, Pd(PPh3)4 (39 mg, 0.034 밀리몰) 및 모르폴린 (200 ㎕, 2.3 밀리몰)을 첨가하고, 혼합물을 실온에서 질소 하에서 3 시간 동안 교반하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고 불순물을 씻어내고 이어서 0-5% AcOH/MeOH 구배에 의해 생성물을 용출함으로써 아미노프로필 SPE에 의해 조 생성물을 정제하였다. 생성물 분획을 합하고 고 진공에 의해 농축하여 백색 고체로서 표제 화합물을 수득하였다 (14 mg, 7%).
LC/MS: m/z 477 [MH]+, RT 2.54mm
실시예
14: 3-부틸-8-
클로로
-1-[3-(1-{[2-(
트리플루오로메틸
)
페닐
]
메틸
}-1H-
이미다졸
-4-일)프로필]-3,7-
디히드로
-1H-퓨린-2,6-
디온

무수 DMF (3 ml) 중의 3-부틸-8-클로로-1-[4-(1H-이미다졸-4-일)부틸]-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (150 mg, 0.38 밀리몰)의 용액을 1-(클로로메틸)-2-(트리플루오로메틸)벤젠 (61 ㎕, 0.42 밀리몰) 및 DIPEA (73 ㎕, 0.42 밀리몰)로 처리하였다. 혼합물을 실온에서 질소 하에 3 일 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. DCM을 사용하여 물질을 컬럼에 부하하고 불순물을 씻어낸 다음 0-5% MeOH/DCM 구배를 사용하여 화합물을 용출시킴으로써 실리카 SPE 컬럼 상에서 조 생성물을 정제하였다. 생성물 분획을 합하고 농축하고 잔류물을 무수 DMF (3 ml)에 용해시켰다. 용액을 탈기시킨 다음 Pd(PPh3)4 (35 mg, 0.030 밀리몰) 및 모르폴린 (174 ㎕, 2.0 밀리몰)을 첨가하고, 혼합물을 질소 하에 실온에서 3 시간 동안 교반하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고 불순물을 씻어낸 다음 0-5% AcOH/MeOH 구배에 의해 생성물을 용출함으로써 아미노프로필 SPE에 의해 조 생성물을 정제하였다. 생성물 분획을 합하고 고 진공에 의해 농축하여 백색 고체로서 표제 화합물을 얻었다 (50 mg, 26%).
LC/MS: m/z 509 [MH]+, RT 2.64 분
실시예
15: 3-부틸-8-
클로로
-1-[3-(1-{[3-(
트리플루오로메틸
)
페닐
]
메틸
}-1H-
이미다졸
-4-일)프로필]-3,7-
디히드로
-1H-퓨린-2,6-
디온

무수 DMF (3 ml) 중의 3-부틸-8-클로로-1-[4-(1H-이미다졸-4-일)부틸]-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (150 mg, 0.38 밀리몰)의 용액을 1-(클로로메틸)-3-(트리플루오로메틸)벤젠 (65 ㎕, 0.42 밀리몰) 및 DIPEA (73 ㎕, 0.42 밀리몰)로 처리하였다. 혼합물을 실온에서 질소 하에 3일 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. DCM을 사용하여 물질을 컬럼에 부하하고 불순물을 씻어낸 다음 0-5% MeOH/DCM 구배를 사용하여 화합물을 용출시킴으로써 실리카 SPE 컬럼 상에서 조 생성물을 정제하였다. 생성물 분획을 합하고 농축하고 잔류물을 무수 DMF (3 ml)에 용해시켰다. 용액을 탈기시킨 다음 Pd(PPh3)4 (30 mg, 0.027 밀리몰) 및 모르폴린 (156 ㎕, 1.8 밀리몰)을 첨가하고, 혼합물을 질소 하에 실온에서 3 시간 동안 교반하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고 불순물을 씻어낸 다음 0-5% AcOH/MeOH 구배에 의해 생성물을 용출함으로써 아미노프로필 SPE에 의해 조 생성물을 정제하였다. 생성물 분획을 합하고 고 진공에 의해 농축하여 백색 고체로서 표제 화합물을 얻었다 (18 mg, 9%).
LC/MS: m/z 509 [MH]+, RT 2.78 분
실시예
16: 3-부틸-8-
클로로
-1-{3-[3-(
페닐메틸
)-1H-1,2,4-
트리아졸
-1-일]프로필}-3,7-디
히
드로-1H-퓨린-2,6-
디온
a) 3-부틸-8-클로로-1-{3-[3-(페닐메틸)-1H-1,2,4-트리아졸-1-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

진공을 가함으로써 THF (7 ml) 중의 3-부틸-8-클로로-1-{3-[3-(페닐메틸)-1H-1,2,4-트리아졸-1-일]프로필}-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (669 mg 1.39 밀리몰)의 용액을 탈기시키고 질소를 도입하였다. Pd(PPh3)4 (160 mg, 0.14 밀리몰)을 첨가하고, 혼합물을 다시 한번 탈기시켰다. 모르폴린 (1.2 ml, 13.9 밀리몰)을 첨가하고, 혼합물을 질소 하에 18시간 동안 교반한 다음, 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 수성 층을 EtOAc로 다시 추출하였다. 합한 추출물을 염수로 세척하고, 건조시키고 (MgSO4) 농축하여 황색 잔류물을 수득하였다. MeOH를 첨가하고, 2-3% AcOH/MeOH로 용출하면서 생성물을 아미노프로필 SPE를 통과시켰다. 생성물 분획을 합하고 농축하여 담황색 고체 (380 mg)를 수득하였다. 대략 물질의 사분의 일을 오토프레프 HPLC에 의해 정제하고, 나머지를 MeOH:DMSO (1:1)로부터 결정화하여 표제 화합물을 백색 고체로서 수득하였다 (125 mg, 31%).

b) 3-부틸-8-클로로-1-{3-[3-(페닐메틸)-1H-1,2,4-트리아졸-1-일]프로필}-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온

MeOH (40 ml) 중의 3-(페닐메틸)-1H-1,2,4-트리아졸 (2.1g, 13.2 밀리몰)의 용액을 MeOH (29 ml) 중의 0.5M NaOMe로 처리한 다음 1,3-디브로모프로판 (1.7 ml) 로 처리하였다. 50℃에서 5 시간 동안 교반한 후에, 혼합물을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하여 오일 잔류물을 수득하였다 (1.0 g). 이것에 부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (917 mg, 3.2 밀리몰) 및 Cs2CO3 (1.2 g, 3.6 밀리몰)을 첨가하였다. DMF (15 ml)를 첨가하고, 혼합물을 50 ℃에서 20시간 동안 교반한 다음 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. 얻어진 오일 (1.52g)을 EtOAc/시클로헥산 혼합물로 용출시키는 실리카 SPE (50 g) 컬럼을 통과시켰다. 트리아졸의 2개의 이성질체 생성물을 2:1 혼합물로서 표제 화합물을 위해 황색 페이스트로서 수득하였다 (697 mg, 존재하는 이성질체의 비율을 기초로 하여 67%).
LC/MS: m/z 482 [MH]+, RT 3.3 분
실시예
17: 8-
클로로
-3-
펜틸
-1-{3-[5-(
페닐메틸
)-2H-
테트라졸
-2-일]프로필}-3,7-
디히드로
-1H-퓨린-2,6-
디온

MeOH (5 ml) 중의 5-벤질-1H-테트라졸 (1.0g, 6.24 밀리몰)의 용액을 1-클로로-3-요오도프로판 (1.0 ml, 9.36 밀리몰) 및 MeOH 중의 0.5M NaOMe 용액 (4.7 ml, 9.36 밀리몰)으로 처리하였다. 반응을 환류 하에 18시간 동안 가열시킨 다음 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 다시 한번 추출하였다. 합한 추출물을 염수로 세척하고, 건조시키고 (MgSO4), 농축하여 황색 고체를 수득하였다 (796 mg). 700 mg의 이 물질을, 75 ℃에서 24시간 동안 DMF (20 ml) 중에서, 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2.6-디온 (732 mg, 2.47 밀리몰) 및 Cs2CO3 (967 mg, 3.0 밀리몰)와 반응시켰다. 반응을 실온으로 냉각하고 진공을 가하여 혼합물을 탈기시킨 다음 질소를 도입하였다. Pd(PPh3)4 (428 mg, 0.37 밀리몰)을 첨가하고, 혼합물을 다시 한번 탈기시켰다. 모르폴린 (2.1 ml, 24.7 밀리몰)을 첨가하고 혼합물을 질소 하에 3 시간 동안 교반한 다음 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 다시 한번 추출하였다. 합한 추출물을 농축하고 황색 잔류물을 수득하였다. MeOH를 첨가한 다음 생성물을 2-3% AcOH/MeOH로 용출하면서 아미노프로필 SPE를 통과시켰다. 생성물 분획을 합하고, 농축한 다음 MDAP에 의해 정제하여 백색 고체로서 표제 화합물을 수득하였다 (35 mg, 3%).

실시예
18: 3-부틸-8-
클로로
-1-{3-[5-(
페닐메틸
)-2H-
테트라졸
-2-일]프로필}-3,7-
디히드로
-1H-퓨린-2.6-
디온

MeOH (30 ml) 중의 5-벤질-1H-테트라졸 (1.8 g, 11.2 밀리몰)의 용액을 1,3-디브로모프로판 (5.7 ml, 56.2 밀리몰) 및 MeOH 중의 0.5M NaOMe (31.5 ml)으로 처리하고, 질소 하에 40 ℃에서 60시간 동안 교반하였다. 혼합물을 2M HCl(수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. SPE (20 g 실리카, 시클로헥산/EtOAc 혼합물)에 의해 그리고 컴패니온(Companion)TM 시스템 (실리카 SPE, 시클로헥산/EtOAc 혼합물)에 의해 부분 정제하여 오일을 수득하고 (1.98g, 이성질체의 혼합물의 62%, 2-(3-브로모프로필)-5-(페닐메틸)-2H-테트라졸을 위하여 2:1), 이것을 이후의 단계에서 원료로 사용하였다. 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (1.74g, 6.1 밀리몰), 조 2-(3-브로모프로필)-5-(페닐메틸)-2H-테트라졸 (1.9g, 6.8 밀리몰), Cs2CO3 (2.2g, 6.8 밀리몰) 및 DMF (60 ml)의 혼합물을 질소 하에 45 ℃에서 24시간 동안 교반하였다. 진공을 가하여 혼합물을 탈기시키고 질소를 도입하였다. Pd(PPh3)4 (705 mg, 0.61 밀리몰)을 첨가하고, 혼합물을 다시 한번 탈기시켰다. 모르폴린 (5.4 ml, 61.4 밀리몰)을 첨가하고, 혼합물을 질소 하에 4 시간 동안 교반한 다음 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고 건조시키고 (MgSO4) 농축하여, 황색 잔류물을 수득하였다. MeOH를 첨가하고 2% AcOH/MeOH로 용출하면서 생성물을 아미노프로필 컬럼에 통과시켰다. 생성물을 EtOAc/시클로헥산 혼합물을 사용하여 컴패니온TM 시스템에 의해 더욱 정제하였다. 얻어진 고체를 끓는 Et2O와 함께 교반하고, 실온으로 냉각한 후 여과하였다. 표제 화합물을 백색 고체 (1.01g, 37%)로서 수집하여 50 ℃에서 진공하에 건조시켰다.

실시예
19: 8-
클로로
-1-(3-{5-[(4-
플루오로페닐
)
메틸
]-2H-
테트라졸
-2-일}프로필)-3-(4,4,4-트
리플루
오로부틸)-3,7-
디히드로
-1H-퓨린-2,6-
디온
a) 8-클로로-1-(3-{5-[(4-플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3-(4,4,4-트리플루오로부틸)-3,7-디히드로-1H-퓨린-2,6-디온

5-[(4-플루오로페닐)메틸]-1H-테트라졸 (75 mg, 0.4 밀리몰)을 탄산칼륨 (100 mg, 0.7 밀리몰) 및 DMF (3 ml)로 처리하였다. 혼합물을 DMF (0.5 ml) 중의 3-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-3-(4,4,4-트리플루오로부틸)-2,3,6,7-테트라히드로-1H-퓨린-1-일]프로필 메탄술포네이트 (100 mg, 0.2 밀리몰)의 용액으로 처리하였다. 혼합물을 교반하고 60 ℃에서 3시간 동안 가열하고, 이어서 냉각하고 증발시켰다. 잔류물을 클로로포름 (4 ml)과 물 (2 cm3) 사이에 분배하였다. 1cm3의 포화 수성 중탄산나트륨 (3 ml)를 각각에 첨가하였다. 혼합물을 분리하고 유기 상을 증발시켰다. 잔류물을 무수 THF (3 ml)에 용해시키고 진공과 질소 압력의 조심스러운 연속 적용에 의해 혼합물을 탈기시켰다. 혼합물을 테트라키스 (트리페닐포스핀)팔라듐(0) (10 mg, 0.008 밀리몰) 및 모르폴린 (0.2 ml, 2.3 밀리몰)으로 처리하고 질소 대기 하에 2 시간동안 교반하였다. 혼합물을 증발시키고 클로로포름 (4 ml)과 포화 수성 염화암모늄 (3 ml) 사이에 분배하였다. 혼합물을 분리하고, 수성 상을 클로로포름으로 재추출하였다. 유기 상을 증발시키고, 잔류물을 MeOH (3 ml) 에 용해시켰다. 용액을 2 g 아미노프로필 SPE 위에 첨가하고 MeOH로 세척하였다 (15 ml). MeOH (20 ml) 중의 AcOH의 3% v/v 용액을 가진 카트릿지로부터 원하는 생성물을 용출하였다. 분획을 함유하는 생성물을 합하고, 증발시키고 잔류물을 플래시 컬럼 크로마토그래피에 의해 정제하였다 (구배 용출: 10:1 시클로헥산/EtOAc 내지 EtOAc). 생성물 함유 분획을 합하고 증발시켜 무색 오일로서 생성물을 수득하였다. 최소의 디에틸 에테르 중에서 분쇄하여 생성물을 고화시키고, 완전히 가열시켜 표제 화합물을 백색 고체 (18.7 mg, 18%)로 수득하였다.

b) 3-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-3-(4,4,4-트리플루오로부틸)-2,3,6,7-테트라히드로-1H-퓨린-1-일]프로필 메탄술포네이트
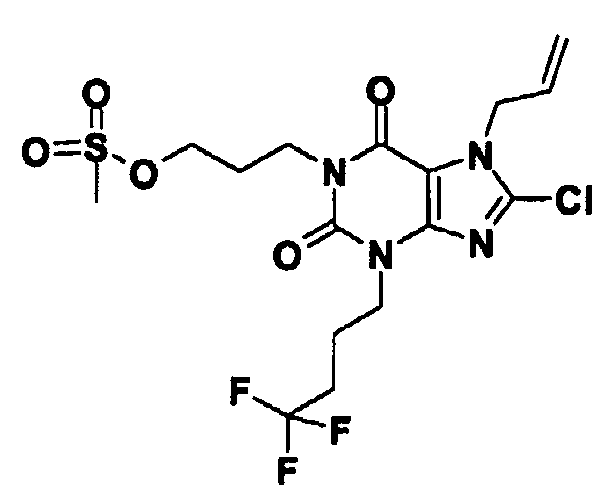
DCM (20 ml) 중의 8-클로로-1-(3-히드록시프로필)-7-(2-프로펜-1-일)-3-(4,4,4-트리플루오로부틸)-3,7-디히드로-1H-퓨린-2,6-디온 (0.82g, 2.1 밀리몰)의 용액을 트리에틸아민 (0.42 ml, 3.1 밀리몰) 및 메탄술폰 안히드라이드 (0.40 g, 2.3 밀리몰)로 처리하였다. 1 시간 후에, 혼합물을 포화 수성 중탄산나트륨 (20 ml)으로 처리하였다. 혼합물을 분리하고 유기 상을 건조시키고 (MgSO4), 여과하고 증발시켜 표제 화합물 (0.91g)을 수득하고, 추가의 정제 없이 이것을 사용하였다.
LC/MS: m/z 473 [MH]+, RT 3.17 분.
c) 8-클로로-1-(3-히드록시프로필)-7-(2-프로펜-1-일)-3-(4,4,4-트리플루오로부틸)-3,7-디히드로-1H-퓨린-2,6-디온

DMF (15 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3-(4,4,4-트리플루오로부틸)-3,7-디히드로-1H-퓨린-2,6-디온 (1.0g, 3.0 밀리몰)의 용액을 탄산세슘 (1.16g, 3.6 밀리몰) 및 3-브로모-1-프로판올 (0.3 ml, 3.3 밀리몰)로 처리하였다. 혼합물을 60 ℃에서 4시간 동안 가열한 다음 냉각하고 증발시켰다. 잔류물을 EtOAc (50 ml)와 물 (50 ml) 사이에 분배하였다. 유기 상을 건조시키고 (MgSO4), 여과하고 증발시켰다. 시클로헥산으로부터 EtOAc로의 구배 용출을 사용하여 플래시 크로마토그래피에 의해 생성물을 정제하였다. 생성물-함유 분획을 합하고 증발시켜 무색 오일로서 표제 화합물을 수득하였다 (0.82 g, 75%).
LC/MS: m/z 395 [MH]+, RT 2.90 분
d) 8-클로로-7-(2-프로펜-1-일)-3-(4,4,4-트리플루오로부틸)-3,7-디히드로-1H-퓨린-2,6-디온

DMF (20 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (2.0 g, 8.8 밀리몰)의 용액을 탄산나트륨 (1.15g, 10.8 밀리몰) 및 4-브로모-1,1,1-트리플루오로부탄 (1.86g, 9.7밀리몰)로 처리하였다. 혼합물을 50 ℃에서 18시간 동안 교반한 다음 냉각하고 증발시켰다. 잔류물을 EtOAc (100 ml)와 포화된 수성 중탄산나트륨 (50 ml) 사이에 분배하였다. 유기 층을 건조시키고 (MgSO4) 여과하고 증발시켰다. 잔류물을 디에틸 에테르와 시클로헥산의 혼합물에서 분쇄한 다음, 생성물을 여과해 내고 건조시켜 표제 화합물을 백색 고체로서 수득하였다 (1.18g, 40%).
LC/MS: m/z 337 [MH]+, RT 2.83 분
e) 5-[(4-플루오로페닐)메틸]-1H-테트라졸

트리에틸암모늄 클로라이드 (4.14g, 30밀리몰) 및 아지드화 나트륨 (1.95g, 30밀리몰)의 혼합물을 톨루엔 (14 ml) 중의 (4-플루오로페닐)아세토니트릴 (1.35g, 10밀리몰)의 용액으로 처리하고, 혼합물을 교반하고 100 ℃에서 5시간 동안 가열하였다. 냉각된 혼합물을 물 (10 ml)로 처리하고 혼합물을 분리하였다. 수성 상을 교반하고 생성물이 용액으로부터 침전될 때까지 진한 염산으로 적가 처리하였다. 침전된 생성물을 여과 제거하고, 물로 세척하고 건조시켜 백색 고형물로서 표제 화합물을 수득하였다 (1.27 g, 72%).
LC/MS: m/z 179 [MH]+, RT 2.24 분
적절한 메탄술포네이트 및 테트라졸을 사용하여 실시예 19: 8-클로로-1-(3-{5-[(4-플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3-(4,4,4-트리플루오로부틸)-3,7-디히드로-1H-퓨린-2,6-디온에 대한 것과 유사한 방법을 사용하여 표 3의 화합물을 제조하였다. 표준 상 크로마토그래피 후에 불충분하게 순수한 화합물을 더욱 정제하기 위해 MDAP를 사용하였다. 하기 절차에 따라서 메탄술포네이트 중간체 및 그들의 전구체 알콜을 제조하였다.
3-[8-
클로로
-2,6-
디옥소
-7-(2-
프로펜
-1-일)-3-프로필-2,3,6,7-
테트라히드로
-1H-퓨린-1-일]프로필
메탄술포네이트

DCM (50 ml) 중의 8-클로로-1-(3-히드록시프로필)-7-(2-프로펜-1-일)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온 (1.99 g, 6.1 밀리몰)의 용액을 트리에틸아민 (1.2 ml, 8.6 밀리몰) 및 메탄술폰 안히드라이드 (1.2 g, 6.9 밀리몰)로 처리하였다. 1.5 시간 후에 혼합물을 물 (50 ml)로 처리하였다. 혼합물을 분리하고 수성 상을 DCM (25 ml)로 추출하고, 합한 유기 상을 건조시키고 (MgSO4), 여과하고 증발시켜 담황색 오일로서 표제 화합물을 수득하고 (2.38 g), 이것을 추가의 정제 없이 사용하였다.
LC/MS: m/z 405 [MH]+, RT 2.93 분
3-[3-부틸-8-
클로로
-2,6-
디옥소
-7-(2-
프로펜
-1-일)-2,3,6,7-
테트라히드로
-1H-퓨린-1-일]프로필
메탄술포네이트

3-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-3-프로필-2,3,6,7-테트라히드로-1H-퓨린-1-일]프로필 메탄술포네이트를 위해 사용된 방법에 따라 제조하여 표제 화합물을 담황색 오일로서 수득하였다 (2.44g).
LC/MS: m/z 419 [MH]+, RT 3.14 분
8-
클로로
-1-(3-히드록시프로필)-7-(2-
프로펜
-1-일)-3-프로필-3,7-
디히드로
-1H-퓨린-2,6-디온

DMF (20 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온 (3.0g, 11.1 밀리몰)의 용액을 탄산세슘 (3.7g, 11.4 밀리몰) 및 3-브로모-1-프로판올 (1.6 g, 11.5 밀리몰)로 처리하였다. 혼합물을 60 ℃에서 4시간 동안 가열한 다음 냉각시키고 증발시켰다. 잔류물을 EtOAc (60 ml)와 포화 수성 중탄산나트륨 (50 ml) 사이에 분배하였다. 수성 상을 EtOAc (60 ml)로 추출하고, 합한 유기 상을 건조시키고 (MgSO4), 여과하고 증발시켰다. 컴패니온TM 시스템 및 시클로헥산으로부터 EtOAc로의 구배 용출을 사용하여 생성물을 정제하였다. 생성물 함유 분획을 합하고 증발시켜 무색 오일로서 표제 화합물 (2.6 g)을 수득하였다.
LC/MS: m/z 327 [MH]+, RT 2.62 분
3-부틸-8-
클로로
-1-(3-히드록시프로필)-7-(2-
프로펜
-1-일)-3,7-
디히드로
-1H-퓨린-2,6-디온

8-클로로-1-(3-히드록시프로필)-7-(2-프로펜-1-일)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온을 위해 사용된 방법에 따라 제조하여 표제 화합물을 무색 오일로서 수득하였다 (2.3g).
LC/MS: m/z 341 [MH]+, RT 2.85 분
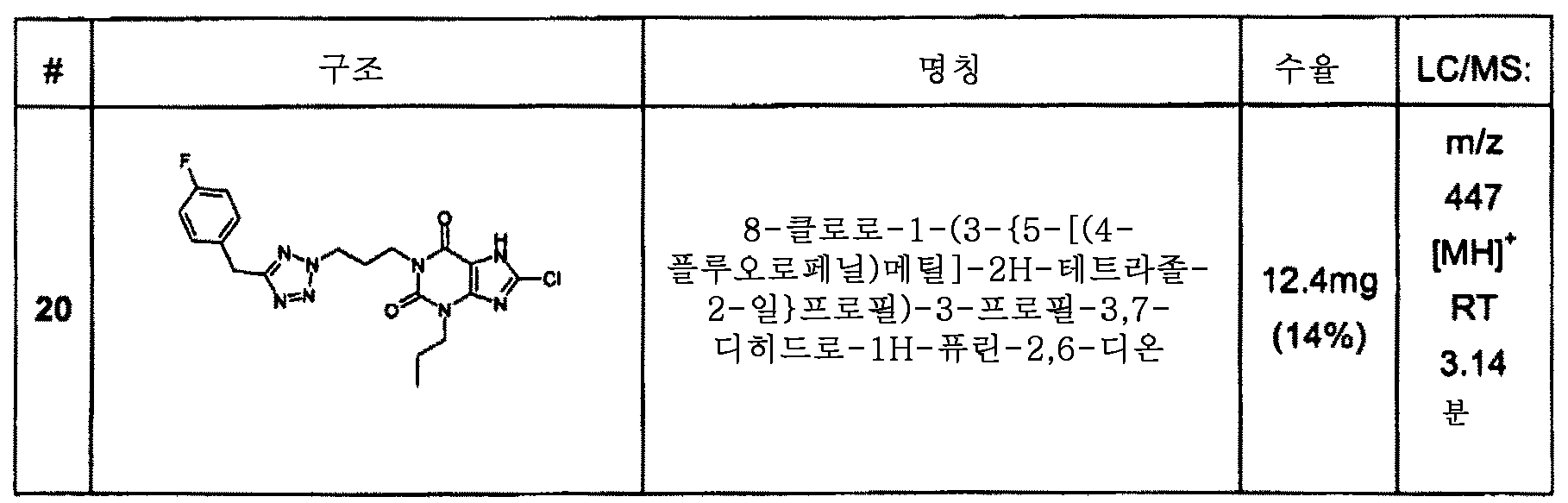








표 3으로부터 선택된
실시예에
대한
NMR
세부사항
실시예 20 : 8-클로로-1-(3-{5-[(4-플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온

실시예 23 : 8-클로로-3-(4,4,4-트리플루오로부틸)-1-(3-{5-[(2,4,6-트리플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온

실시예 24

실시예 27
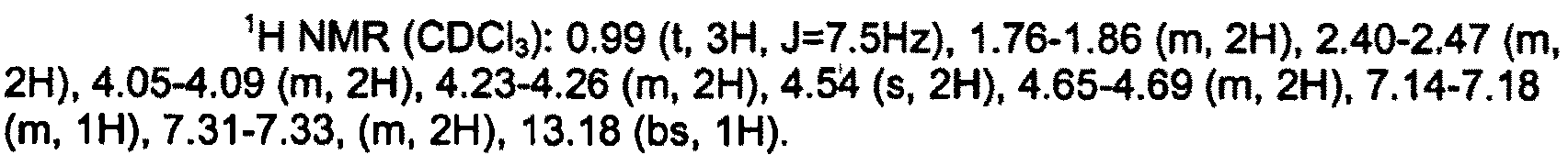
실시예 28
실시예 29

실시예 30 : 3-부틸-8-클로로-1-(3-{5-[(3-플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온
실시예 31
실시예 32 : 3-부틸-8-클로로-1-(3-{5-[(4-플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온
실시예 33 : 3-부틸-8-클로로-1-(3-{5-[(4-클로로페닐)메틸]-2H-테트라졸-2-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온
실시예 48 : 8-클로로-1-(3-{5-[(2,4-디플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온
실시예 51 : 8-클로로-3-프로필-1-(3-{5-[(2,4,6-트리플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온
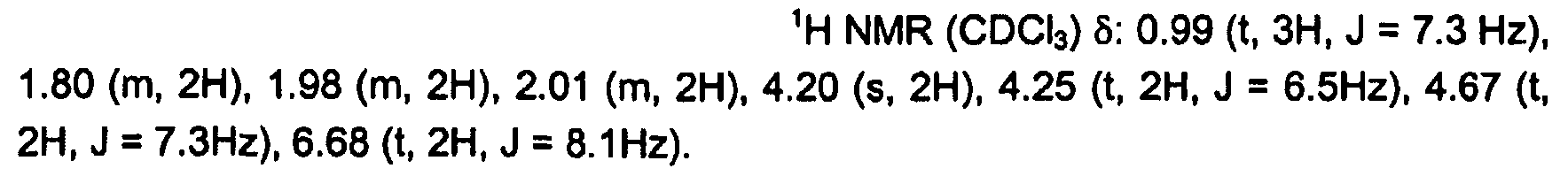
실시예 52
실시예 59
실시예 61
실시예 64 : 3-부틸-8-클로로-1-(3-{5-[(2,4,6-트리플루오로페닐)메틸]-2H-테트라졸-2-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온

실시예
68: 8-
클로로
-3-
펜틸
-1-{3-[3-(
페닐메틸
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-디
히
드로-1H-퓨린-2,6-
디온
a) 8-클로로-3-펜틸-1-{3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]프로필}-디히드로-1H-퓨린-2,6-디온

THF (4 ml) 중의 3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]-1-프로판올} (88 mg, 0.4 밀리몰)의 용액을 질소 하에서 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3.7-디히드로-1H-퓨린-2,6-디온 (100 mg, 0.34 밀리몰) 및 PPh3 (115 mg, 0.44 밀리몰)으로 처리하였다. DBAD (101 mg, 0.44 밀리몰)를 한번 분량으로 첨가하고, 반응을 5 시간 동안 반응시켰다. 진공을 가함으로써 혼합물을 탈기시키고 질소를 도입하였다. Pd(PPh3)4 (39 mg, 0.034 밀리몰)을 첨가하고, 혼합물을 한번 더 탈기시켰다. 모르폴린 (294 ㎕, 3.4 밀리몰)을 첨가하고, 혼합물을 질소 하에 3 시간 동안 교반하였다. 혼합물을 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하였다. MDAP에 의해 정제 후에 표제 화합물을 백색 고체로서 수득하였다 (64 mg, 42%).

b) 3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]-1-프로판올

γ-부티로락톤 (223 ㎕, 2.9 밀리몰), 벤즈아미딘 옥심 (480 mg, 3.2 밀리몰), EtOH (1.3 ml) 중의 NaOEt의 21% 용액 및 EtOH (3 ml)의 혼합물을 140 ℃에서 10 분 동안 마이크로파에서 가열하였다. 혼합물을 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하였다. 컴패니온TM 시스템을 사용하여 실리카 위에서 표제 화합물을 정제하여 담황색 오일을 수득하였다 (143 mg, 23%).
LC/MS: m/z 219 [MH]+, RT 2.4 분
실시예
69: 8-
클로로
-1-(3-{3-[(4-
클로로페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-퓨린-2,6-
디온
a) 8-클로로-1-(3-{3-[(4-클로로페닐)메틸]-1,2,4-옥사디아졸-5-일}프로필)-3-펜틸-3,7-디히드로-1H-퓨린-2,6-디온

플라스크에서 연속하여 공기를 빼내고 질소를 재도입하여 (3회), DMF (5 ml) 중의 8-클로로-1-(3-{3-[(4-클로로페닐)메틸]-1,2,4-옥사디아졸-5-일}프로필)-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.18 g, 0.34 밀리몰)의 용액을 탈기시키고, 모르폴린 (0.5 ml, 5.8 밀리몰) 및 Pd(PPh3)4 (80 mg, 0.068 밀리몰)를 첨가하였다. 용액을 72 시간 동안 교반한 다음 농축하고 잔류물을 아미노프로필 SPE (10 g) 위에 MeOH와 함께 부하하였다. MeOH로의 용출에 이어서 5% AcOH/MeOH에 의해 용출하여 담황색 고체로서 표제 화합물을 제공하고, 이것을 에테르로 세척하여 백색 고체를 수득하였다 (0.053 g, 32%).
LC/MS: m/z 491 [MH]+, RT 3.69 분
b) 8-클로로-1-(3-{3-[(4-클로로페닐)메틸]-1,2,4-옥사디아졸-5-일}프로필)-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온

i) γ-부티로락톤 (8 ml, 104 밀리몰), 4-클로로벤즈아미드 옥심 (3.0 g, 16.25 밀리몰), MeOH 중의 NaOMe의 30% 용액 (5 ml) 및 MeOH (80 ml)의 혼합물을 30시간 동안 환류시키고 냉각하고 농축하였다. DCM/EtOH/0.88 수성 암모니아 (200:8:1)로 용출시키면서 실리카 위에서 플래시 크로마토그래피에 의해 잔류물을 정제하여 황색 오일 (13 g)을 제공하였다. 이 물질을 DCM (150 ml)에 용해시키고 2M 수산화나트륨 (100 ml)으로 세척하고 유기물을 분리시키고, 건조시키고 농축하여 점성 오일으로서 3-{3-[(4-클로로페닐)메틸]-1,2,4-옥사디아졸-5-일}-1-프로판올을 점성 오일 (3.95 g, 96%)으로서 수득하고 이것을 이후의 단계에서 사용하였다.
ii) THF (5 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.10 g, 0.34 밀리몰), 3-{3-[(4-클로로페닐)메틸]-1,2,4-옥사디아졸-5-일}-1-프로판올 (0.086 g, 0.34 밀리몰) 및 트리페닐포스핀 (0.186g, 0.69 밀리몰)의 용액에 디벤질아조디카르복실레이트 (0.204g, 0.68 밀리몰)을 첨가하고 18 시간 동안 용액을 교반하였다. 용액을 농축하고 잔류물을 처음에 DCM으로 용출시키고 이어서 DCM/Et2O 혼합물로 용출시키면서 실리카 (20 g, SPE) 위에서 크로마토그래피 하여, 디벤질아조디카르복실레이트 부산물 (0.18 g)으로 오염된 표제 화합물을 수득하였다. 탈보호 단계에서 그대로 물질을 사용하였다.
LC/MS: m/z 531 [MH]+, RT 3.83 분
실시예
70: 8-
클로로
-1-{3-[3-(
페닐메틸
)-1,2,4-
옥사디아졸
-5-일]프로필}-3-프로필-3,7-디히드로-1H-퓨린-2,6-
디온
a) 8-클로로-1-{3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]프로필}-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온

THF (4 ml)중의 8-클로로-7-(2-프로펜-1-일)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온 (200 mg, 0.74 밀리몰)의 용액을 3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]-1-프로판올 (195 mg, 0.89 밀리몰) 및 PPh3 (254 mg, 0.96 밀리몰)로 처리하였다. DBAD (223 mg, 0.96 밀리몰)을 한번 분량으로 첨가하고, 혼합물을 실온에서 18 시간 동안 질소 하에서 교반하였다. 혼합물을 EtOAc와 2M HCl (수성)에서 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하였다. 0-70% 시클로헥산/EtOAc 구배를 사용하여 실리카 SPE 컬럼에 의하여 조 생성물을 정제하였다. 생성물 분획을 조합하고 농축하고 0-60% 시클로헥산/EtOAc 구배를 사용하여 실리카 SPE 컬럼에 의해 정제하였다. 생성물 분획을 합하고 농축한 다음 무수 THF (4 ml)에 용해시켰다. 용액을 고 진공에 의해 탈기시킨 다음 Pd(PPh3)4 (86 mg, 0.074 밀리몰) 및 모르폴린 (644 ㎕, 7.4 밀리몰)을 첨가하고, 혼합물을 실온에서 질소 하에 1일 동안 교반하였다. 혼합물을 EtOAc와 HCl (수성) 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 고 진공에 의해 농축하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고 불순물을 씻어낸 다음 2-4% AcOH/MeOH 구배에 의해 생성물을 용출함으로써 아미노프로필 SPE에 의해 조 생성물을 정제하였다. 생성물 분획을 합하고, 고 진공에 의해 농축하여 표제 화합물을 백색 고체로서 수득하였다 (74 mg, 23%).

b) 8-클로로-7-(2-프로펜-1-일)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온

DMF (40 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (1.5 g, 6.6 밀리몰), 1-요오도프로판 (1.2g, 6.9 밀리몰) 및 탄산나트륨 (0.9 g, 8.5 밀리몰)의 혼합물을 50 ℃에서 18 시간 동안 가열하였다. 반응 혼합물을 진공 하에 농축하고, 잔류물을 물 (60 ml)으로 처리하고, EtOAc (3×80 ml)으로 추출하였다. 합한 유기 추출물을 건조시키고 (MgSO4) 여과하고 증발시켰다. 잔류물을 에테르/시클로헥산으로 분쇄하고, 고체를 여과해 내고 건조시켜 표제 화합물 (0.82 g, 46%)을 수득하였다.
LC/MS: m/z 269 [MH]+
실시예
71: 8-
클로로
-3-
펜틸
-1-{3-[3-(3-
티에닐메틸
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-
디히드로
-1H-퓨린-2,6-
디온
a) 8-클로로-3-펜틸-1-{3-[3-(3-티에닐메틸)-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

에틸 4-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (70 mg, 0.19 밀리몰), N-히드록시-2-(3-티에닐)에탄이미다미드 (36 mg, 0.23 밀리몰), EtOH 중의 NaOEt의 21% 용액 (78 ㎕, 0.21 밀리몰) 및 EtOH (1.5 ml)의 혼합물을 마이크로파에서 140 ℃에서 10분 동안 가열하였다. 냉각 후에, 반응을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 다시 추출하였다. 합한 추출물을 농축하고 MDAP에 의해 정제하였다. 표제 화합물을 1.4-디옥산으로부터 동결 건조하여 백색 고체를 수득하였다 (27 mg, 31%).
LC/MS: m/z 463 [MH]+, RT 3.4 분
b) 에틸 4-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트

무수 DMF (35 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (3.0 g, 10.1 밀리몰)의 용액을 Cs2CO3 (3.6 g, 11.1 밀리몰) 및 에틸 4-브로모부티레이트 (1.6 ml, 11.1 밀리몰)로 처리하였다. 혼합물을 실온에서 18 시간 동안 교반한 다음 약한 진공 하에서 탈기시킨 다음 질소를 도입하였다. 이것을 2회 반복하였다. Pd(PPh3)4 (1.17 g, 1.0 밀리몰)를 첨가하고, 혼합물을 다시 한번 탈기시켰다. 모르폴린 (8.8 ml, 101 밀리몰)을 첨가하고, 실온에서 3 시간 동안 교반하였다. 혼합물을 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4), 농축하여 황색 고체 (5.16 g)을 수득하였다. 잔류물을 MeOH에 취하고 2개의 동일 분량으로 나눈 다음, 각각 MeOH에 이어서 5% AcOH/MeOH로 용출시키면서 아미노프로필 SPE (20 g)을 통과시켰다. 생성물 분획을 합하고 농축하여 거의 백색 고체로서 표제 화합물을 수득하였다 (3.01 g, 80%).
LC/MS: m/z 371 [MH]+, RT 3.2 분
실시예
72: 3-부틸-8-
클로로
-1-{3-[3-(2,3-
디플루오로벤질
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-
디히드로
-1H-퓨린-2,6-
디온
a) 3-부틸-8-클로로-1-{3-[3-(2,3-디플루오로벤질)-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

2,3-디플루오로페닐아세토니트릴 (23 mg, 0.15 밀리몰)을 EtOH (1 ml)에 용해시켰다. 히드록실아민 히드로클로라이드 (14 mg, 0.20 밀리몰)을 첨가한 다음 물 (0.5 ml) 및 탄산칼륨 (41 mg, 0.3 밀리몰)을 첨가하였다. 혼합물을 환류 하에 밤새 가열한 다음 냉각하고 EtOAc와 염수 사이에 분배하였다. 유기 상을 증발시키고 이렇게 수득된 조 아미독심을 EtOH (1 ml)에 용해시켰다. 에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (43 mg, 0.12 밀리몰) 및 21 중량% 에탄올성 에톡시화 나트륨 (0.067 ml, 0.18 밀리몰)을 첨가하고, 혼합물을 마이크로파 반응기에서 140 ℃에서 10 분 동안 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 증발시키고 생성물을 MDAP에 의해 정제하여 표제 화합물을 고체로서 제공하였다 (13 mg).

b) 에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트

건조 DMF (100 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (6.0 g, 21.24 밀리몰)에 Cs2CO3 (7.62 g, 23.36 밀리몰)에 이어서 에틸 4-브로모부티레이트 (4.556 g, 23.36 밀리몰)을 첨가하였다. 혼합물을 55 에서 18 시간 동안 가열한 다음 냉각하고 이어서 반복하여 공기를 빼내고 질소를 다시 넣음으로써 탈기시켰다. 모르폴린 (14.9 g, 171 밀리몰)을 첨가한 다음 테트라키스(트리페닐포스핀)팔라듐(0) (4.0 g, 3.46 밀리몰)을 첨가하고, 혼합물을 4 시간 동안 교반하였다. EtOAc (300 ml) 및 2M HCl (150 ml) 및 물(100 ml)을 첨가하고, 유기 상을 분리하고 염수 (3×100 ml)로 세척하고 여과하였다. 여액을 건조시키고 (Na2SO4) 증발시켰다. THF/MeOH (1:1)에 부하하고 THF/MeOH (1:1) 및 순수한 MeOH로 세척하고 5% 첨가된 AcOH를 함유하는 DCM/MeOH (1:1)로 생성물을 용출시켜 조 생성물 (10 g)을 아미노프로필 SPE (3×20g)에 의해 정제하여, 표제 화합물을 수득하였다 (5.08 g).

실시예
73: 3-부틸-8-
클로로
-1-{3-[3-(2-
클로로벤질
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-
디히드로
-1H-퓨린-2,6-
디온

에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (53 mg, 0.15 밀리몰) 및 (1Z)-2-(2-클로로페닐)-N-히드록시에탄이미다미드 (30 mg, 0.18 밀리몰; 번호 1, 표 7)를 EtOH (0.75 ml)에서 21% 에탄올성 에톡시화 나트륨 (0.083 ml, 0.22 밀리몰)로 140 ℃에서 10분 동안 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고 유기 상을 증발시켰다. 생성물을 MDAP에 의해 정제하여 고체로서 표제 화합물을 수득하였다 (34.8 mg).

실시예
74: 3-부틸-8-
클로로
-1-{3-[3-(4-
플루오로벤질
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-
디히드로
-1H-퓨린-2,6-
디온

(1Z)-2-(4-플루오로페닐)-N-히드록시에탄이미다미드 (28 mg, 0.18 밀리몰; 번호 2, 표 7)로부터 출발하여 고체로서 표제 화합물을 유사하게 수득하였다 (10.0 mg).
LC/MS: m/z 461 [MH]+, RT 3.49 분
실시예
75: 3-부틸-8-
클로로
-1-{3-[3-(2,3-
디클로로벤질
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (53 mg, 0.15 밀리몰) 및 (1Z)-2-(2,3-디클로로페닐)-N-히드록시에탄이미다미드 (36 mg, 0.165 밀리몰; 번호 3, 표 7) 및 21% 에탄올성 에톡시화 나트륨 (0.083 ml, 0.22 밀리몰)을, 마이크로파 반응기에서 140 ℃에서 10분 동안 EtOH (0.75 ml) 중에서 함께 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 분리하고 증발시키고 생성물을 MDAP에 의해 정제하여 고체 (42.1 mg)로서 표제 화합물을 수득하였다.
LC/MS: m/z 511, 513, 515 (동위원소) [MH]+, RT 3.66 분
(실시예 87 (표 4)의 경우에, 처리 동안에 EtOAc로의 추출에 앞서서 pH를 5로 조절하고; 실시예 88 (표 4)의 경우에 조 생성물을 EtOH (1 ml) 및 2M NaOH (0.5 ml)와 함께 18 시간 동안 교반하고, 실시예 89 (표 4)의 경우에 처리를 반복하고 MDAP에 의해 정제하기에 앞서서 EtOH (0.75 ml) 및 2M NaOH (0.5 ml)와 함께 18 시간 동안 교반하는 것을 제외하고는) 적절한 아미독심을 사용하여 실시예 75에 대해서와 유사한 방법을 사용하여 하기 화합물들 (표 4)을 제조하였다.



표 4로부터 선택된 실시예에 대한 NMR 세부사항
실시예 76: 3-부틸-8-클로로-1-{3-[3-(3-플루오로벤질)-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

실시예 77: 3-부틸-8-클로로-1-{3-[3-(3,4-디플루오로벤질)-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온
실시예 79: 1-{3-[3-(1,3-벤조디옥솔-5-일메틸)-1,2,4-옥사디아졸-5-일]프로필}-3-부틸-8-클로로-3,7-디히드로-1H-퓨린-2,6-디온

실시예 87: 3-부틸-8-클로로-1-{3-[3-(1H-인돌-3-일메틸)-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

실시예 88: 3-부틸-8-클로로-1-(3-{3-[3-(히드록시페닐)메틸]-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

실시예 89: N-[3-{(5-[3-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)프로필]-1,2,4-옥사디아졸-3-일}메틸)페닐]메탄술폰아미드

실시예
90: 3-부틸-8-
클로로-1-(3-{3-[(3,4-디클로로페닐)메틸]-1,2,4-옥사
디아졸-5-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (71 mg, 0.2 밀리몰), (1Z)-2-(3,4-디클로로페닐)-N-히드록시에탄이미다미드 (48 mg, 0.22 밀리몰) 및 21 중량% 에탄올성 에톡시화 나트륨 (0.111 ml, 0.3 밀리몰)을 마이크로파 반응기에서 140 ℃에서 10분 동안 EtOH (1 ml) 중에서 함께 가열하였다. 혼합물을 EtOAc 와 2M HCl 사이에 분배하고, 유기 상을 분리하고 증발시키고, 조 생성물을 MDAP에 의해 정제하여 고체 (48.8 mg)으로서 표제 화합물을 수득하였다.
LC/MS: m/z 511, 513 [MH]+, RT 3.65 분
(실시예 91을 위해, 아미독심이 히드로클로라이드 염이 되도록 하기 위하여 0.185 ml (0.5 밀리몰)의 21% 에톡시화 나트륨를 첨가하는 것 이외에는) 실시예 90에서와 유사한 방법을 사용하여 적절한 아미독심을 사용하여 하기 화합물 (표 5)을 제조하였다.


표 5로부터 선택된 실시예에 대한 NMR 세부사항
실시예 91
실시예 94: 3-부틸-8-클로로-1-(3-{3-[(4-클로로페닐)메틸]-1,2,4-옥사디아졸-5-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온

실시예 96: 3-부틸-8-클로로-1-(3-{3-[(4-히드록시페닐)메틸]-1,2,4-옥사디아졸-5-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온

실시예 97: 3-부틸-8-클로로-1-[3-(3-{[3-(트리플루오로메틸)-1H-피라졸-1-일]메틸}-1,2,4-옥사디아졸-5-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온

실시예
99: 3-부틸-8-
클로로
-1-[3-(3-{[3-(
에틸옥시
)-4-
히드록시페닐
]
메틸
}-1,2,4-옥
사
디아졸-5-일)프로필]-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (53 mg, 0.15 밀리몰) 및 (1Z)-2-[3-(에틸옥시)-4-히드록시페닐]-N-히드록시에탄이미다미드 (35 mg, 0.165 밀리몰; 번호 11, 표 7)를 EtOH (0.75 ml) 중에서 혼합하였다. 에탄올성 에톡시화 나트륨 (21 중량%, 0.083 ml, 0.22 밀리몰)를 첨가하고, 혼합물을 마이크로파에서 140 ℃에서 10분 동안 가열하였다. 추가의 0.055 ml (0.15 밀리몰)의 NaOEt 용액을 첨가하고 혼합물을 추가로 10 분 동안 140 ℃에서 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 증발시키고 MDAP에 의해 정제하여 표제 화합물을 고체로서 수득하였다 (29.6 mg).
LC/MS: m/z 503 [MH]+, RT 3.15 분
(HCl 처리 및 MDAP에 의한 정제를 반복하기 전에 잔류 출발 에스테르를 가수분해하기 위하여, 실시예 100 (표 6)에 대하여 처리 후의 조 생성물을 EtOH (1 ml) 및 2M NaOH (0.5 ml)와 함께 밤새 교반하는 것 이외에는) 적절한 아미독심을 사용하여 실시예 99에 대해서와 유사한 방법을 사용하여 하기 화합물들 (표 6)을 제조하였다.

표 6으로부터 선택된 실시예를 위한 NMR 세부사항
실시예 101: N-[3-({5-[3-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)프로필]-1,2,4-옥사디아졸-3-일}메틸)페닐]아세트아미드

실시예
102: 3-부틸-8-
클로로
-1-(3-{3-[(2-
클로로
-4-
플루오로페닐
)
메틸
]-1,2,4-옥
사
디아졸-5-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (100 mg, 0.28 밀리몰) 및 (1Z)-2-(2-클로로-4-플루오로페닐)-N-히드록시에탄이미다미드 (62.4 mg, 0.308 밀리몰) 및 21 중량% 에탄올성 에톡시화 나트륨 (0.157 ml, 0.42 밀리몰)을 마이크로파 반응기에서 EtOH (1.5 ml)에서 140 ℃에서 10분 동안 함께 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배함으로써 처리하였다. 유기 상을 증발시키고 MDAP에 의해 정제하여 표제 화합물을 고체로서 수득하였다 (73 mg).
LC/MS: m/z 495 [MH]+, RT 3.55 분
실시예
103: 8-
클로로
-3-에틸-1-{3-[3-(
페닐메틸
)-1,2,4-옥사디아졸-5-일]프로필}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온
a) 8-클로로-3-에틸-1-{3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

무수 THF (4 ml) 중에서 8-클로로-3-에틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (150 mg, 0.59 밀리몰)의 용액을 3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]-1-프로판올 (154 mg, 0.71 밀리몰) 및 트리페닐포스핀 (200 mg, 0.76 밀리몰)으로 처리하였다. PBAD (162 mg, 0.71 밀리몰)을 한번 분량으로 첨가하고, 혼합물을 18시간 동안 질소 하에 실온에서 교반하였다. 혼합물을 고 진공에 의해 탈기시키고, 이어서 Pd(PPh3)4 (68 mg, 0.059 밀리몰) 및 모르폴린 (515 ㎕, 5.9 밀리몰)을 첨가하였다. 혼합물을 실온에서 질소 하에 3시간 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 고 진공에 의해 농축하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고 불순물을 씻어낸 다음 2% AcOH/MeOH를 사용하여 화합물을 용출시킴으로써 아미노프로필 SPE에 의해 조 물질을 정제하였다. UV 활성 분획을 합하고 고 진공에 의해 농축하였다. 생성물을 MDAP에 의해 더욱 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (61 mg, 25%).

b) 8-클로로-3-에틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온

무수 DMF (100 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (10 g, 0.044 몰)의 용액을 요오도에탄 (5.4 ml, 0.068 몰) 및 Na2CO3 (4.9 g, 0.046 몰)로 처리하였다. 반응 혼합물을 실온에서 질소 하에 2일 동안 교반하였다. 요오도에탄 (0.35 ml, 0.0044몰)을 첨가하고, 혼합물을 실온에서 1일 동안 교반하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하였다. 유기 층을 분리하고, 포화 아황산나트륨 용액 및 염수로 연속하여 세척하고, 건조시키고 (MgSO4) 농축하였다. 조 고형물을 Et2O로 세척하여 백색 고체로서 표제 화합물을 수득하였다 (8.37 g, 75%).
LC/MS: m/z 255 [MH]+, RT 2.35 분
실시예
104: 8-
클로로
-1-(3-{3-[(3-
클로로페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (70 mg, 0.19 밀리몰)을 EtOH에 용해시켰다. 용액을 EtOH 중의 NaOEt의 21% 용액 (78 ㎕, 0.21 밀리몰) 및 (1Z)-2-(3-클로로페닐)-N-히드록시에탄이미다미드 (38 mg, 0.21 밀리몰)로 처리하였다. 반응을 140 ℃에서 마이크로파에서 10분 동안 가열하였다. 혼합물을 EtOH와 2M HCl (수성) 사이에 분배하였다. 유기 층을 경사분리하고 농축하였다. 조 생성물을 MDAP에서 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (46 mg, 49%).

실시예
105: 8-
클로로
-1-(3-{3-[(3,4-
디클로로페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (70 mg, 0.19 밀리몰)을 EtOH에 용해시켰다. 용액을 EtOH 중의 NaOEt의 21% 용액 (78 ㎕, 0.21 밀리몰) 및 (1Z)-2-(3,4-디클로로페닐)-N-히드록시에탄이미다미드 (46 mg, 0.21 밀리몰)로 처리하였다. 반응을 140 ℃에서 마이크로파에서 10분 동안 가열하였다. 혼합물을 EtOH와 2M HCl (수성) 사이에 분배하였다. 유기 층을 경사분리하고 농축하였다. 조 생성물을 MDAP에서 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (66 mg, 66%).
LC/MS: m/z 527 [MH]+, RT 3.80 분
실시예
106: 8-
클로로
-1-(3-{3-[(2,6-
디클로로페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (70 mg, 0.19 밀리몰)을 EtOH에 용해시켰다. 용액을 EtOH 중의 NaOEt의 21% 용액 (78 ㎕, 0.21 밀리몰) 및 (1Z)-2-(2,6-디클로로페닐)-N-히드록시에탄이미다미드 (46 mg, 0.21 밀리몰)로 처리하였다. 반응을 140 ℃에서 마이크로파에서 10분 동안 가열하였다. 혼합물을 EtOH와 2M HCl (수성) 사이에 분배하였다. 유기 층을 경사분리하고 질소 송풍에 의해 농축하였다. 조 생성물을 MDAP에서 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (80 mg, 80%).
LC/MS: m/z 526 [MH]+, RT 3.6 분
실시예
107: 8-
클로로
-1-(3-{3-[(2-
클로로
-4-
플로오로페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (70 mg, 0.19 밀리몰)을 EtOH에 용해시켰다. 용액을 EtOH 중의 NaOEt의 21% 용액 (78 ㎕, 0.21 밀리몰) 및 (1Z)-2-(2-클로로-4-플루오로페닐)-N-히드록시에탄이미다미드 (42 mg, 0.21 밀리몰)로 처리하였다. 반응을 140 ℃에서 마이크로파에서 10분 동안 가열하였다. 혼합물을 EtOH와 2M HCl (수성) 사이에 분배하였다. 유기 층을 경사분리하고 농축하였다. 조 생성물을 MDAP에서 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (65 mg, 67%).
LC/MS: m/z 509 [MH]+, RT 3.63 분
실시예
108: 3-부틸-8-
클로로
-1-{3-[3-(
페닐메틸
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온

무수 THF (4 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (205 mg, 0.73 밀리몰)의 용액을 3-[3-(페닐메틸)-1,2,4-옥사디아졸-5-일]-1-프로판올 (190 mg, 0.87 밀리몰) 및 PPh3 (247 mg, 0.94 밀리몰)로 처리하였다. DBAD (217 mg, 0.94 밀리몰)을 한번 분량으로 첨가하고 혼합물을 실온에서 질소 하에 18시간 동안 교반하였다. 고 진공에 의해 혼합물을 탈기한 다음, Pd(PPh3)4 (84 mg, 0.073 밀리몰) 및 모르폴린 (636 ㎕, 7.3 밀리몰)을 첨가하였다. 혼합물을 실온에서 질소 하에 3시간 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하고, 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 농축하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고 불순물을 씻어낸 다음 2-4% AcOH/MeOH 구배를 사용하여 컬럼으로부터 화합물을 제거함으로써 아미노프로필 컬럼에 의해 조 물질을 정제하였다. MDAP에 의해 추가의 정제를 실행하여 표제 화합물을 백색 고체로서 수득하였다 (75 mg, 23%).

실시예
109: 8-
클로로
-1-(3-{3-[(4-
히드록시페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (29 mg, 0.078 밀리몰) 및 (1Z)-N-히드록시-2-(4-히드록시페닐)에탄이미다미드 (14 mg, 0.084 밀리몰)를 EtOH (1 ml)중에서 21% 에탄올성 에톡시화 나트륨 (0.043 ml, 0.117 밀리몰)와 함께 마이크로파 조사 하에서 140 ℃에서 10분 동안 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 증발시켰다. 이 물질을 EtOAc와 2M HCl 사이에 분배함으로써 다시 처리하기 전에 EtOH (1 ml)와 2M NaOH (0.5 ml)와 함께 18시간 동안 교반하였다. MDAP에 의해 정제하여 표제 화합물을 수득하였다 (6.5 mg).

실시예
110: 3-부틸-8-
클로로
-1-(3-{3-[(페닐옥시)메틸]-1,2,4-옥사디아졸-5-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

EtOH (1 ml) 중의 에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (26 mg, 0.073 밀리몰) 및 (1Z)-N-히드록시-2-(페닐옥시)에탄이미다미드 히드로클로라이드 (16 mg, 0.079 밀리몰)에 21중량% 에탄올성 에톡시화 나트륨 용액 (0.068 ml, 0.183 밀리몰)을 첨가하고, 혼합물을 마이크로파 조사 하에서 140 ℃에서 10분 동안 가열하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 건조시키고 (Na2SO4) 증발시키고 MDAP에 의해 정제하여, 에테르로 분쇄 시에 고화된 고무로서 표제 화합물을 수득하였다 (5.9 mg).
LC/MS: m/z 459 [MH]+, RT 3.39 분

실시예
111: 3-부틸-8-
클로로
-1-(3-{3-[(3,5-
디클로로페닐
)
메틸
]-1,2,4-
옥사
디아졸-5-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

건조 EtOH (2 ml) 중의 에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (185 mg, 0.52 밀리몰) 및 (1Z)-2-(3,5-디클로로페닐)-N-히드록시에탄이미다미드 (126 mg, 0.58 밀리몰; 번호 23, 표 7)에 21중량% 에탄올성 에톡시화 나트륨 용액 (0.29 ml, 0.78 밀리몰)을 첨가하고, 혼합물을 마이크로파에 의해 140 ℃에서 10분 동안 가열하였다. EtOAc와 2M HCl 사이에 분배함으로써 반응을 처리하고 유기 상을 증발시켰다. MDAP에 의해 정제하여 표제 화합물을 고체로서 수득하였다 (135 mg).
LC/MS: m/z 511 [MH]+, RT 3.71 분
실시예
112: 3-부틸-8-
클로로
-1-(3-{3-[(2,4,6-
트리플루오로페닐
)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

(1Z)-N-히드록시-2-(2,4,6-트리플루오로페닐)에탄이미다미드 (119 mg, 0.58 밀리몰; 번호 24, 표 7)로부터 출발하여 135 mg의 수율로 유사하게 제조하였다.

아미독심
:
하기 상세히 기재되고 표 7에서 유사물로 예시된 방법에 의해 이들을 이용할 수 있다.



여기에서 사용된 것과 같은  는 한정되지 않은 기하구조의 이중 결합을 나타낸다는 것을 주목해야 한다.
는 한정되지 않은 기하구조의 이중 결합을 나타낸다는 것을 주목해야 한다.
방법 A
상응하는 니트릴 (0.5 밀리몰)을 EtOH (1.5 ml)중에서 50% 히드록실아민 수용액 (0.08 ml, 1.3 밀리몰)과 함께 가열하고 65 ℃에서 4.5시간 동안 가열하였다. 냉각 후에, 조 반응 혼합물을 SCX SPE 카트릿지 (2 g)에 부하하고 MeOH로 세척한 다음 아미독심 생성물을 MeOH 중에서 2M 암모늄으로 용출하였다.
방법 B
생성물을 조 반응 혼합물로부터 결정화하고 SCX 대신에 여과에 의해 단리하는 것을 제외하고는 방법 A와 유사하다.
방법 C
5 g SCX 카트릿지 위에서 생성물을 정제하는 것 이외에는 방법 A와 유사하다.
방법 D
가열 기간이 18시간인 것 이외에는 방법 C와 유사하다.
방법 E
규모가 0.753 밀리몰의 니트릴인 것 이외에는 방법 C와 유사하다.
방법 F
규모가 1.5 밀리몰의 니트릴이고 정제가 10g SCX 카트릿지인 것 이외에는 방법 A와 유사하다.
방법 G
가열 시간이 2.75시간이고 규모가 0.25밀리몰의 니트릴인 것 이외에는 방법 A와 유사하다.
실시예
113: 3-부틸-8-
클로로
-1-(3-{5-[(3-
클로로페닐
)
메틸
]-1,2,4-
옥사디아졸
-3-일}프로필)-3,7-
디히드로
-1H-퓨린-2,6-
디온
a) 3-부틸-8-클로로-1-(3-{5-[(3-클로로페닐)메틸]-1,2,4-옥사디아졸-3-일}프로필)-3,7-디히드로-1H-퓨린-2,6-디온

(3-클로로페닐)아세트산 (0.1 밀리몰), N-[3-(디메틸아미노)프로필]-N'-에틸카르보디이미드 히드로클로라이드 (21 mg, 0.11 밀리몰) 및 1H-1,2,3-벤조트리아졸-1-올 (15 mg, 0.11 밀리몰)을 1-메틸-2-피롤리디논 (1 ml)에서 교반하였다. 여기에 (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드 (34 mg, 0.1 밀리몰)을 첨가하고 혼합물을 실온에서 17시간 동안 교반한 다음 80 ℃에서 24시간 동안 교반하였다. 조제 HPLC (오토 프레프)에 의하여 반응 혼합물을 추가의 변형 없이 정제하여 표제 화합물을 수득하였다 (13 mg, 27%).

b) (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드

4-(3-부틸-8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일)부탄니트릴 (1 g, 0.0032 몰)을 EtOH (3.5 ml) 및 물 (1.8 ml) 중에서 교반하였다. 히드록실아민 히드로클로라이드 (344 mg, 0.0049 몰) 및 탄산칼륨 (652 mg, 0.0049 몰)을 첨가하고, 혼합물을 80 ℃에서 3일 동안 가열하였다. 냉각 후에, 조 반응 혼합물을 증발시켰다. 조 생성물을 물에 용해시키고 HCl로 pH7로 중화시키고, 오아시스(Oasis)TM 카트릿지(2 g)에 부하하였다. 이것을 물로 용출하여 염을 제거하고 MeOH로 용출하여 표제 화합물을 수득하였다 (957 mg, 86%).
LC/MS: m/z 343, 345 [MH]+, RT 2.04 분
c) 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일]부탄니트릴

4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일]부탄니트릴 (2.1 g, 6 밀리몰)을 질소 탈기된 DCM (20 ml) 및 AcOH (2 ml)의 혼합물 중에서 교반하였다. 테트라키스(트리페닐포스핀)팔라듐 (675 mg, 0.6 밀리몰) 및 페닐실란 (7.4 ml, 60 밀리몰)을 첨가하고, 혼합물을 실온에서 2일 동안 교반하였다. 이것을 증발시키고, 잔류물을 디에틸에테르:시클로헥산 (1:1)의 혼합물로 분쇄하여 표제 화합물 (1.47 g, 60%)을 백색 고체로서 수득하였다.
LC/MS: m/z 310 [MH]+, RT 2.66 분
d) 4-[3-부틸-8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]부탄니트릴

건조 MeCN (20 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (2.0g, 0.0072 몰)에 Cs2CO3 (4.68 g, 0.0144몰)을 첨가한 다음 브로모부티로니트릴 (1.38 g, 0.0094 몰)을 첨가하였다. 혼합물을 80 ℃에서 18시간 동안 가열한 다음 냉각하였다. 반응 혼합물을 증발시키고 조 생성물을 EtOAc와 HCl (2N) 사이에 분배하였다. 유기 상을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 증발시켜 조 생성물을 수득하였다. 이것을 시클로헥산:에틸아세테이트 (2:1 내지 1:1)로 용출하면서 실리카 SPE (50 g)에 의해 정제하여 투명한 오일로서 표제 화합물을 수득하였다 (2.1 g, 85%).
LC/MS: m/z 350 [MH]+, RT 3.10 분
실시예 113에 대한 것과 유사한 방법을 사용하여 상응하는 산 및 (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드로부터 하기 화합물 (표 8)을 제조하였다.

실시예에서 *가 사용된 경우에, 이것은 크산틴 코어에 대한 R기의 부착 지점을 나타낸다.


표 8로부터 선택된 실시예에 대한 NMR 세부사항
실시예 115:

실시예
121: 8-
클로로
-3-
펜틸
-1-{3-[5-(
페닐메틸
)-1,2,4-
옥사디아졸
-3-일]프로필}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온
a) 8-클로로-3-펜틸-1-{3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

THF (5 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.20, 0.67 밀리몰)의 교반된 용액에 3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]-1-프로판올 (0.162 g, 0.74 밀리몰), DBAD (0.186 g, 0.81 밀리몰) 및 트리페닐포스핀 (0.212 g, 0.81 밀리몰)을 첨가하고, 용액을 18시간 동안 교반하였다. 용액에 Pd(PPh3)4 (75 mg, 0.067 밀리몰) 및 모르폴린 (600 ㎕, 6.7 밀리몰)을 첨가하고 질소 하에 실온에서 추가로 3시간 동안 교반하였다. 75mg의 Pd(PPh3)4를 첨가하고, 혼합물을 추가로 3시간 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4), 농축하였다. MeOH를 사용하여 화합물을 컬럼에 부하하고, 불순물을 씻어낸 다음 2-4% AcOH/MeOH를 사용하여 화합물을 용출함으로써 조 물질을 아미노프로필 SPE에 의해 정제하였다. 생성물 분획을 합하고 농축하여 MDAP에 의해 더욱 정제하였다. 생성물 분획을 합하고 농축하여 표제 화합물을 백색 고체 (51 mg, 20%)로서 수득하였다.
LC/MS: m/z 457 [MH]+, RT 3.54 분
b) 3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]-1-프로판올

(1E)-4,4-비스(에틸옥시)-N-히드록시부탄이미다미드 (3.2 g, 16.8 밀리몰), 에틸 페닐아세테이트 (2.3 ml, 14.4 밀리몰) 및 에톡시화 나트륨 (EtOH 중의 21% 용액, 6.4 ml)의 혼합물을 마이크로파에서 140 ℃에서 10분 동안 가열하였다. 물질을 두 번째 반응으로부터의 것과 조합하고 (1.2 g의 (1E)-4,4-비스(에틸옥시)-N-히드록시부탄이미다미드를 사용하여, 상기 기재된 바와 같이 수행됨) 1M HCl 용액과 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고 건조시키고 농축하여, 5-[3,3-비스(에틸옥시)프로필]-5-(페닐메틸)-1,2,4-옥사디아졸을 제공하고 이것을 이후의 단계에서 정제없이 사용하였다.
EtOH (75 ml) 중의 조 3-[3,3-비스(에틸옥시)페닐]-5-(페닐메틸)-1,2,4-옥사디아졸 (5.63 g, 19.4 밀리몰)을 p-톨루엔술폰산 (0.738g, 3.9 밀리몰)과 21시간 동안 교반하고, 혼합물을 EtOAc와 물 사이에 분배하였다. 유기물을 단리하고 물 및 염수로 세척하고 건조시키고 적색 오일로 농축하였다. 이 물질은 상당한 양의 아세탈을 함유하고, 따라서 오일을 THF (15 ml)중에 용해시키고 2M HCl 용액으로 2시간 동안 처리한 다음 EtOAc와 물 사이에 분배하였다. 유기물을 단리하고 염수로 세척하고 건조시키고 농축하여 3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]프로판올을 적색/갈색 오일 (3.77g)로서 수득하였으며, 이것을 이후의 단계에서 원료로 사용하였다.
MeOH (60 ml) 중의 조 3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]프로판올 (3.76 g, 17.4 밀리몰)의 용액을 0 ℃로 냉각하고, 붕수소화나트륨 (0.724g, 19.1 밀리몰)을 30분에 걸쳐 적가하였다. 냉각 배쓰를 제거하고 용액을 추가로 1시간 동안 교반한 다음 1M HCl과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 추출하였다. 합한 추출물을 염수로 세척하고, 건조시키고 오렌지색 액체로 농축하였다. 이것을 시클로헥산/EtOAc (20% 내지 80% 구배 용출)로 용출하면서 50 g 실리카 SPE 위에서 정제하여 표제 화합물을 황색 오일 (2.24g)로서 수득하였다.
LC/MS: m/z 210 [MH]+
c) (1E)-4,4-비스(에틸옥시)-N-히드록시부탄이미다미드

물 (20 ml) 및 EtOH (40 ml) 중의 3-시아노프로피온알데히드 디에틸아세탈 (6.12g, 39 밀리몰), 히드록실아민 히드로클로라이드 (4.06g, 58.4 밀리몰), 탄산칼륨 (10.76g, 77.9 밀리몰)의 혼합물을 24시간 동안 환류시켰다. 혼합물을 냉각한 다음 물과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 추출하였다. 합한 유기 분획을 염수로 세척하고 건조시키고 농축하여 표제 화합물을 ~20% 출발 니트릴로 오염된 무색 오일로서 수득하였다 (6.03 g, 81%).
LC/MS: m/z 191 [MH]+
실시예
122: 8-
클로로
-1-{3-[5-(
페닐메틸
)-1,2,4-
옥사디아졸
-3-일]프로필}-3-프로필-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

THF (4 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3-프로필-3,7-디히드로-1H-퓨린-2,6-디온 (200 mg, 0.74 밀리몰)의 용액을 3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]-1-프로판올 (195 mg, 0.89 밀리몰) 및 PPh3 (254 mg, 0.96 밀리몰)로 처리하였다. DBAD (223 mg, 0.96 밀리몰)를 한번 분량으로 첨가하고 혼합물을 실온에서 질소 하에서 18시간 동안에 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 고 진공에 의해 농축하였다. 조 생성물을 0-70% 시클로헥산/EtOAc 구배를 사용하여 실리카 SPE 컬럼 위에서 정제하였다. 생성물 분획을 합하고 고 진공에 의해 농축하고 0-60% 시클로헥산/EtOAc 구배를 사용하여 실리카 SPE 컬럼 위에서 정제하였다. 생성물 분획을 합하고 농축한 다음 무수 THF (4 ml) 중에 용해시켰다. 용액을 고 진공에 의해 탈기시키고 Pd(PPh3)4 (61 mg, 0.053 밀리몰) 및 모르폴린 (460 ㎕, 5.3 밀리몰)을 첨가하고 혼합물을 실온에서 질소 하에 1일 동안 교반하였다. 혼합물을 EtOAc와 2M HCl (수성) 사이에 분배하였다. 유기 층을 분리하고, 염수로 세척하고, 건조시키고 (MgSO4) 고 진공에 의해 농축하였다. MeOH를 사용하여 화합물을 컬럼 상에 부하하고 불순물을 씻어낸 다음 2-4% AcOH/MeOH 구배를 사용하여 생성물을 용출함으로써 아미노프로필 SPE에 의해 조 생성물을 정제하였다. 생성물 분획을 합하고 농축하여 표제 화합물을 백색 고체로서 수득하였다 (36 mg, 11%).

실시예
123: 3-부틸-8-클로로-1-{3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]프로필}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

THF (25 ml) 중의 3-[5-(페닐메틸)-1,2,4-옥사디아졸-3-일]-1-프로판올 (594 mg, 2.7 밀리몰)의 용액을 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (700 mg, 2.48 밀리몰) 및 PPh3 (779 mg, 2.97 밀리몰)로 질소 하에 처리하였다. DBAD (684 mg, 2.97 밀리몰)를 한번 분량으로 첨가하고, 반응을 60시간 동안 반응시켰다. 혼합물을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. MeOH를 잔류물에 첨가한 다음 생성물을 2-4% AcOH/MeOH로 용출하면서 아미노프로필 컬럼을 통과시켰다. 생성물 분획을 조합하고 농축하였다. 회백색 잔류물을 EtOAc:시클로헥산 (1:1)로부터 재결정하여 표제 화합물을 백색 고체로서 수득하였다 (696 mg, 63%).

실시예
124: 3-부틸-8-
클로로
-1-(3-{5-[(3-
클로로
-4-
히드록시페닐
)
메틸
]-1,2,4-
옥사디아졸
-3-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

DMSO (1 ml) 중의 3-클로로-4-히드록시페닐아세트산 (24 mg, 0.13 밀리몰)의 용액을 CDI (21 mg, 0.13 밀리몰)로 처리하고 30분 동안 반응시켰다. (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드 (50 mg, 0.15 밀리몰)을 첨가하고, 혼합물을 마이크로파에서 120 ℃에서 15분 동안 가열하였다. 용액을 MDAP에 의해 직접 정제하여 표제 화합물을 백색 고체로서 수득하였다 (12 mg, 17%).
LC/MS: m/z 493 [MH]+, RT 3.2 분
실시예
125: 3-부틸-8-
클로로
-1-(3-{5-{[3-
클로로
-2-(
메틸옥시
)
페닐
]
메틸
}-1,2,4-옥사디아졸-3-일)프로필]-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

DMF (1.5 ml) 중의 [3-클로로-2-(메틸옥시)페닐]아세트산 (32 mg, 0.16 밀리몰)의 혼합물을 CDI (26 mg, 0.16 밀리몰)로 처리하고 45분 동안 반응시켰다. (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드 (60 mg, 0.18 밀리몰)을 첨가하고, 혼합물을 마이크로파에서 140 ℃에서 15분 동안 가열하였다. 냉각 후에, 반응을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리한 다음 농축하고 MDAP에 의해 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (25 mg, 28%).
LC/MS: m/z 507 [MH]+, RT 3.5 분
실시예
126: 3-부틸-8-
클로로
-1-(3-{5-[(3-
플루오로
-4-
히드록시페닐
)
메틸
]-1,2,4-옥사디아졸-3-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

DMF (1.5 ml) 중의 (3-플루오로-4-히드록시페닐)아세트산 (27 mg, 0.16 밀리몰)의 혼합물을 CDI (26 mg, 0.16 밀리몰)로 처리하고 45분 동안 반응시켰다. (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드 (60 mg, 0.18 밀리몰)을 첨가하고, 혼합물을 마이크로파에서 140 ℃에서 15분 동안 가열하였다. 냉각 후에, 반응을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리한 다음 농축하고 MDAP에 의해 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (10 mg, 12%).
LC/MS: m/z 477 [MH]+, RT 3.2 분
실시예
127: 8-
클로로
-3-
펜틸
-1-[4-(5-
페닐
-1,2,4-
옥사디아졸
-3-일)부틸]-3,7-디
히
드로-1H-
퓨린
-2,6-
디온
a) 8-클로로-3-펜틸-1-[4-(5-페닐-1,2,4-옥사디아졸-3-일)부틸]-3,7-디히드로-1H-퓨린-2,6-디온의 제조

벤조산 (18 mg, 0.15 밀리몰)을 DMSO (0.3 ml) 중의 1H-1,2,3-벤조트리아졸-1-올 수화물 (25 mg, 0.19 밀리몰)의 용액으로 처리하였다. 여기에 DMSO (0.3 ml) 중의 N-[3-(디메틸아미노)프로필]-N'-에틸카르보디이미드 히드로클로라이드 (29 mg, 0.15 밀리몰)의 용액/현탁액을 첨가한 다음 DMSO (0.3 ml) 중의 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (55 mg, 0.15 밀리몰)의 용액을 첨가하였다. 혼합물을 40 ℃에서 1시간 동안 가열한 다음 80 ℃에서 5시간 동안 가열하고 냉각하였다. 혼합물을 MDAP에 의해 정제하였다. 생성물-함유 분획을 질소 기류에 의해 송풍 건조시켜 백색 고체로서 표제 화합물을 수득하였다 (17.2 mg, 25%).

b) 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드

EtOH (30 ml) 중의 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄니트릴 (3.0g, 8.9 밀리몰)의 용액을 물 (15 ml), 탄산칼륨 (1.48 g, 10.7 밀리몰) 및 히드록실아민 히드로클로라이드 (0.74 g, 10.7 밀리몰)로 처리한 다음, 70 ℃에서 밤새 가열하였다. 추가의 탄산칼륨 (1.5 g, 10.9 밀리몰) 및 히드록실아민 히드로클로라이드 (1.0 g, 14.5 밀리몰)를 조심스럽게 혼합물에 첨가하고, 이어서 90 ℃로 24시간 동안 가열하였다. 혼합물을 냉각하고 진공 하에서 농축하여 대부분의 EtOH를 제거하였다. 잔류 혼합물을 물 (30 ml)로 처리하고 2M 수성 염산의 조심스러운 첨가에 의해 pH 7로 산성화하였다. 침전된 고체를 여과 제거하고 물로 세척한 다음 디에틸 에테르로 세척하고 완전히 건조시켜 백색 고체로서 표제 화합물을 수득하였다 (2.80 g, 85%).
LC/MS: m/z 371 [MH]+, RT 2.27 분
c) 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄니트릴

DMF (100 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (4.0 g, 13.5 밀리몰)의 용액을 탄산세슘 (4.83 g, 14.8 밀리몰) 및 5-브로모펜탄니트릴 (1.73 ml, 1.48 밀리몰)로 처리하였다. 혼합물을 50 ℃에서 질소 대기 하에 19시간 동안 가열한 다음 냉각하였다. 혼합물을 반복적인 연속 진공 적용에 이어서 질소 압력에 의해 탈기시켰다. 이어서, 혼합물을 테트라키스(트리페닐포스핀)팔라듐(0) (1.1g, 0.94 밀리몰) 및 모르폴린 (11.8 ml, 136 밀리몰)으로 첨가하였다. 혼합물을 질소 대기 중에서 3시간 동안 교반한 다음 EtOAc와 2M 수성 염산 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하여 황색 유성 잔류물을 수득하였다. 이것을 MeOH에 용해시키고, 4개 분량으로 동일하게 분리하고 각각의 분량을 20 g 아미노프로필 SPE에 적용한 다음 MeOH로 씻어내었다. 원하는 생성물을 MeOH 중의 AcOH의 5% v/v 용액으로 카트릿지로부터 용출시켰다. 생성물-함유 분획을 합하고 농축하여 담황색 고체로서 표제 화합물을 수득하였다 (4.03 g, 88%).
LC/MS: m/z 338 [MH]+, RT 3.05 분
상응하는 산으로부터 실시예 127 (8-클로로-3-펜틸-1-[4-(5-펜틸-1,2,4-옥사디아졸-3-일)부틸]-3,7-디히드로-1H)퓨린-2,6-디온에 대해서와 유사한 방법을 사용하여 하기 화합물들을 제조하였다.

또한, 실시예 128, 8-클로로-3-펜틸-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온은 하기 스펙트럼 데이터를 갖는다:

실시예
132: 8-
클로로
-1-{4-[5-(4-
히드록시페닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3-프로필-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

4-히드록시벤조산 (18 mg, 0.13 밀리몰) 및 CDI (24 mg, 0.15 밀리몰)을 무수 DMSO (0.9 ml) 중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(8-클로로-2,6-디옥소-3-프로필-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (50 mg, 0.15 밀리몰; 실시예 128 (b)에 기재된 바와 같이 (1Z)-5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드와 유사한 방법으로 제조됨)을 첨가하고 혼합물을 90 ℃에서 2시간 동안 교반하였다. 반응 혼합물을 MDAP에 의해 정제하였다. 생성물 분획을 합하고 고 진공 하에 농축하여 백색 고체로서 표제 화합물을 수득하였다 (7 mg, 11%).
LC/MS: m/z 443 [MH]+, RT 3.28 분
실시예
133: 3-부틸-8-
클로로
-1-{4-[5-(2,6-
디플루오로페닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

2,6-디플루오로벤조산 (40 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)을 무수 DMSO (0.9 ml) 중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고 혼합물을 90 ℃에서 16시간 동안 교반하였다. 반응 혼합물을 MDAP에 의해 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (18 mg, 15%).
LC/MS: m/z 479 [MH]+, RT 3.40 분
실시예
134: 3-부틸-8-
클로로
-1-{4-[5-(2-
플루오로페닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

2-플루오로벤조산 (36 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)을 무수 DMSO (0.9 ml) 중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고 혼합물을 90 ℃에서 16시간 동안 교반하였다. 반응 혼합물을 MDAP에 의해 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (33 mg, 29%).
LC/MS: m/z 461 [MH]+, RT 3.44 분
실시예
135: 3-부틸-8-
클로로
-1-{4-[5-(4-
클로로
-2-
피리디닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

4-클로로-2-피리딘카르복실산 (40 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)을 무수 DMSO (0.9 ml) 중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고 혼합물을 90 ℃에서 16시간 동안 교반하였다. 반응 혼합물을 MDAP에 의해 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (13 mg, 11%).

실시예
136: 3-부틸-8-
클로로
-1-{4-[5-(3-
메틸
-2-
피리디닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

3-메틸-2-피리딘카르복실산 (35 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)을 무수 DMSO (0.9 ml) 중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고 혼합물을 90 ℃에서 16시간 동안 교반하였다. 반응 혼합물을 MDAP에 의해 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (14 mg, 12%).
LC/MS: m/z 458 [MH]+, RT 3.13 분
실시예
137: 3-부틸-8-
클로로
-1-{4-[5-(2-
피리디닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온
방법 A

2-피리딘카르복실산 (31 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)을 무수 DMSO (0.5 ml) 중에서 실온에서 1시간 동안 교반하였다. DMSO (0.4 ml) 중의 (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)의 용액을 첨가하고 혼합물을 90 ℃에서 16시간 동안 교반하였다. 반응 혼합물을 MDAP에 의해 직접 정제하였다. 생성물 분획을 합하고 농축하여 백색 고체로서 표제 화합물을 수득하였다 (14 mg, 12%).

방법 B

2-피리딘카르복실산 (675 mg, 5.3 밀리몰) 및 CDI (909 mg, 5.6 밀리몰)을 무수 DMF (30 ml) 중에서 실온에서 질소 하에 90분 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (2.0 g, 5.6 밀리몰) 및 DMF (10 ml)를 첨가하고 혼합물을 100 ℃에서 20시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각하고, 포화 NH4Cl(수성) 용액과 EtOAc 사이에 분배하였다. 유기 층을 분리하고, 수성 용액을 EtOAc으로 추출하였다. 합한 추출물을 염수로 세척하고, MgSO4로 건조시키고 농축하여 오렌지색 액체를 수득하였다. 이것을 컴패니온(Companion)TM 시스템을 사용하여 정제하여 2개의 동일한 백색 고체를 수득하였다 (649 mg; 240 mg).
LC/MS: m/z 444 [MH]+, RT 3.04 분
방법 C

12L 둥근 바닥 플라스크에 오버헤드, 기계적 교반기, J-KEM 온도 조절장치를 가진 온도 탐침, 응축기 및 질소 유입 어댑터를 장착하였다. 플라스크에 피콜린산 (0.180 kg, 1.46 몰), MIBK (4.0 L), 1,1'-카르보닐디이미다졸 (0.23kg, 1.42 몰) 및 더 많은 MIBK (0.66 L)을 넣었다. 혼합물을 교반하고 대략 1시간에 걸쳐 50 ℃로 가온하고, 온도를 56 ℃로 올렸다. 50 ℃로 가열하는 동안에 고체를 용해시키고 이산화탄소가 발생되었다. 50 ℃에서 1시간 후에, (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (0.467 kg, 1.31 몰)를 반응에 첨가하였다. 혼합물을 1시간에 걸쳐 90 ℃로 가온하였다. 90 ℃에서 5.5시간 가열한 후에, 반응의 HPLC 분석은 반응이 완결되었음을 나타내었다. 가열을 끄고, 1.0N 염산 용액 (2.33L)을 첨가하였다. 온도를 61 ℃로 떨어뜨렸다. 밤새 교반 후에, 생성물을 침전시키고 여과하였다. 필터케이크를 물 (1× 2.23L, 1 × 2.43L)로 세척하고 헵탄 (1.40L)으로 세척하였다. 습윤 케이크를 진공 오븐에서 50 ℃에서 22시간 동안 건조시켜 396g의 생성물 (68%)을 수득하였다. HPLC 분석 97.7% (AUC) tR= 18.6분
실시예 137의 방법 A, B 및 C는 실질적으로 결정질인 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 2를 생성한다.
방법 D
3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 형태 1
반응 용기에 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (1 중량), 아세톤 (20 부피) 및 물 (0.6 부피)을 넣었다. 혼합물을 교반하고 50 내지 60 ℃로 가온하고, 최대 1 시간 동안 교반하였다. 용액을 형성하고 이 온도에서 1마이크론 필터를 통해 두 번째 반응 용기에 여과함으로써 정화시켰다. 용액을 대략 3시간 동안 33 내지 38 ℃로 냉각하고, 이 온도에서 3-부틸-8-클로로-1-{4-[5-(2-피리디닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (형태 1, 0.01 중량%)로 접종하였다. 묽은 현탁액을 이 온도에서 최소 1시간 동안 교반한 다음 20 내지 25 ℃로 냉각하고, 이 온도에서 최소 12 시간 동안 유지하였다. 이렇게 형성된 현탁액을 13 내지 17 ℃로 냉각하고, 이 온도에서 최소 1시간 동안 유지하였다. 이어서, 현탁액을 샘플채취하고, 고체를 실험실에서 여과에 의해 수집하였다. 고체를 건조시키고, xrpd/DSC에 의해 분석하여 형태를 검사하였다. 형태가 요구된다면 (형태 1), 회분을 여과하고, 세척하고 (2×3 vol 아세톤) 진공 오븐에서 50 ℃에서 건조시켰다. 일단 회분을 열고 나서, 분석은 용매 수준 (아세톤, 물)이 허용가능한 수준임을 나타낸다.
기대 수율 (75 내지 80% w/w)
*에서 취해진 샘플의 형태가 순수한 형태 1과 다른 것으로 밝혀진다면, 회분을 35 내지 45 ℃에서 재가열하고 이 온도에서 최소 1시간 동안 교반하였다. 묽은 현탁액을 20 내지 25 ℃로 냉각하고 이 온도에서 최소 12시간 동안 유지하였다. 이렇게 형성된 현탁액을 13 내지 17 ℃로 냉각하고 이 온도에서 최소 1시간 동안 유지하였다. 현탁액을 샘플채취하고, 실험에서 여과에 의해 고체를 수집하였다. 고체를 건조시키고 형태를 점검하기 위해 xrpd/DSC에 의해 분석하였다. 형태가 요구된다면 (형태 1), 회분을 여과시키고 세척하고 앞서 기재된 바와 같이 건조시켰다. 형태가 순수한 형태 1이 아니라면, 만족스런 결과가 수득될 때까지 **로부터의 순환을 반복하였다.
X-선 분말
회절
(
XRPD
)
X-선 분말 회절 (XRPD) 데이터를 도 1 내지 3에 나타낸다. 데이터는 패널리티컬 (PANalytical) X'Pert Pro 분말 회절계, 모델 PW 3040/60, 일련번호 DY 1850 위에서 X'셀러레이터 검출장치를 사용하여 획득되었다. 획득 조건은 복사선: CuKα, 발생장치 장력: 40kV, 발생장치 전류: 45 mA, 출발 각: 2.0°2θ, 말단 각: 40.0°2θ, 단계 크기: 0.0167°2θ, 단계 당 시간: 31.75 초였다. 몇몇 밀리그램의 샘플을 Si 웨이퍼 (제로 배경) 평판 위에 설치함으로써 샘플을 제조하여, 분말의 얇은 층을 얻었다.
실시예
138: 3-부틸-8-
클로로
-1-{4-[5-(4-
히드록시페닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

4-히드록시벤조산 (35 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)를 무수 DMSO (0.9 ml)중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고, 혼합물을 90 ℃에서 16시간 동안 교반하였다. 혼합물을 MDAP에 의해 정제하여 표제 화합물을 백색 고체로서 수득하였다 (5 mg, 4%).
LC/MS: m/z 459 [MH]+, RT 3.24 분
실시예
139: 8-
클로로
-1-[4-(5-
페닐
-1,2,4-
옥사디아졸
-3-일)부틸]--3-프로필-3,7-디히드로-1H-
퓨린
-2,6-
디온

벤조산 (9 mg, 0.074 밀리몰) 및 CDI (13 mg, 0.081 밀리몰)를 무수 DMSO (0.9 ml)중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(8-클로로-2,6-디옥소-3-프로필-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (28 mg, 0.081 밀리몰)을 첨가하고, 혼합물을 80 ℃에서 4시간 동안 교반하였다. 혼합물을 MDAP에 의해 정제하여 표제 화합물을 백색 고체로서 수득하였다 (0.6 mg, 2%).

실시예
140: 3-부틸-8-
클로로
-1-{4-[5-(2-
클로로
-6-
플루오로페닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

2-클로로-6-플루오로벤조산 (44 mg, 0.25 밀리몰) 및 CDI (45 mg, 0.28 밀리몰)를 무수 DMSO (0.9 ml)중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고, 혼합물을 90 ℃에서 16시간 동안 교반하였다. 혼합물을 MDAP에 의해 정제하였다. 생성물 분획을 합하고 농축하여 표제 화합물을 백색 고체로서 수득하였다 (6.4 mg, 5%).
LC/MS: m/z 495 [MH]+, RT 3.58 분
실시예
141: 3-부틸-8-
클로로
-1-{4-[5-(5-히드록시-2-
피리디닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-퓨린-2,6-
디온

5-히드록시-2-피리딘카르복실산 (24 mg, 0.17 밀리몰) 및 CDI (31 mg, 0.19 밀리몰)를 무수 DMSO (0.9 ml)중에서 실온에서 1시간 동안 교반하였다. (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (68 mg, 0.19 밀리몰)을 첨가하고, 혼합물을 90 ℃에서 16시간 동안 교반하였다. 혼합물을 MDAP에 의해 정제하고 생성물 분획을 농축하여 표제 화합물을 백색 고체로서 수득하였다 (19 mg, 24%).
LC/MS: m/z 459 [MH]+, RT 3.03 분
실시예
142: 8-
클로로
-3-
펜틸
-1-{4-[5-(3-
티에닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온

EtOH (1 ml)중의 (1Z)-5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (50 mg, 0.13 밀리몰)의 용액을 EtOH 중의 NaOEt의 21% 용액 (55 ㎕, 0.21 밀리몰) 및 에틸 3-티오펜카르복실레이트 (18 ㎕, 0.13 밀리몰)로 처리하였다. 혼합물을 150 ℃에서 10분 동안 마이크로파에서 가열하였다. 냉각 후에, 반응을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 다시 추출하였다. 합한 추출물을 농축하고 MDAP에 의해 정제하였다. 표제 화합물을 회백색 고체로서 수득하였다 (20 mg, 32%).
LC/MS: m/z 463 [MH]+, RT 3.6 분
실시예
143: 8-
클로로
-3-
펜틸
-1-{4-[5-(2-
티에닐
)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온

2-티오펜카르복실산 (14 mg, 0.11 밀리몰)을 NMP (0.9 ml)에 용해시키고 CDI (18 mg, 0.11 밀리몰)로 처리하였다. 1시간 후에, (1Z)-5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (50 mg, 0.13 밀리몰)를 첨가하고 혼합물을 마이크로파에서 150 ℃에서 15분 동안 가열하였다. 용액을 MDAP에 의해 직접적으로 정제하여 표제 화합물을 수득한 다음 1,4-디옥산으로부터 동결 건조하여 표제 화합물을 백색 고형물을 수득하였다 (19 mg, 31%).
LC/MS: m/z 463 [MH]+, RT 3.5 분
실시예
144: 8-
클로로
-3-
펜틸
-1-{4-[5-(1,3-티아졸-2-일)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

EtOH (1.5 ml)중의 (1Z)-5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (50 mg, 0.13 밀리몰)의 용액을 EtOH 중의 NaOEt의 21% 용액 (50 ㎕, 0.13 밀리몰) 및 에틸 1,3-티아졸-2-카르복실레이트 (18 mg, 0.11 밀리몰)로 처리하였다. 혼합물을 170 ℃에서 10분 동안 마이크로파에서 가열하였다. 냉각 후에, 반응을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 농축하여 MDAP에 의해 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (13 mg, 21%).

실시예
145: 3-부틸-8-
클로로
-1-{4-[3-(2-
피리디닐
)-1,2,4-
옥사디아졸
-5-일]부틸}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온
a) 3-부틸-8-클로로-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온의 제조

EtOH (2 ml) 중의 에틸 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (120 mg, 0.32 밀리몰) 및 N-히드록시-2-피리딘카르복시미다미드 (50 mg, 0.36 밀리몰)의 혼합물에 EtOH 중의 에톡시화 나트륨의 21% (w/v) 용액 (0.225 ml, 0.62 밀리몰) 을 첨가한 다음 마이크로파 오븐에서 140 ℃에서 밀봉된 바이알 내에서 10분 동안 가열하였다. 냉각된 혼합물을 증발 건조시키고, 잔류물을 클로로포름 (5 ml)과 포화된 수성 염화암모늄 (5 ml) 사이에 분배하였다. 유기 상을 증발 건조시키고, 조 생성물을 MDAP에 의해 정제하였다. 생성물 함유 분획을 합하고 증발 건조시켰다. 생성물을 소량의 디에틸 에테르 중에 고체로 분쇄한 다음 건조시켜 백색 고체로서 표제 화합물을 수득하였다 (44 mg, 31%).

b) N-히드록시-2-피리딘카르복시미다미드의 제조

EtOH (30 ml) 중의 2-피리딘카르보니트릴 (3 g, 29 밀리몰) 및 탄산칼륨 (4.1 g, 30 밀리몰)의 혼합물에 물 (15 ml)을 첨가하고, 조심스럽게 히드록실아민 히드로클로라이드 (2.9 g, 42 밀리몰)를 첨가한 다음 6시간 동안 가열환류시키고, 냉각하고 증발 건조시켰다. 잔류물을 물(100 ml)로 처리하고, 현탁된 고체 생성물을 여과해 내고 물로 세척하고 건조시켜 표제 화합물을 백색 고체로서 수득하였다 (2.28 g, 57%).
실시예
146: 3-부틸-8-클로로-1-[4-(3-페닐-1,2,4-옥사디아졸-5-일)부틸]-3,7-디
히
드로-1H-
퓨린
-2,6-
디온
방법 A
a) 3-부틸-8-클로로-1-[4-(3-페닐-1,2,4-옥사디아졸-5-일)부틸]-3,7-디히드로-1H-퓨린-2,6-디온

에틸 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (74 mg, 0.2 밀리몰) 및 벤즈아미독심 (30 mg, 0.22 밀리몰)을 건조 EtOH (1 ml)에 현탁시키고 에탄올성 에톡시화 나트륨 (21 중량%, 0.111 ml, 0.3 밀리몰)를 첨가하였다. 고체가 용해될 때까지 혼합물을 서서히 가온한 다음 마이크로파 반응기에서 140 ℃에서 10분 동안 가열하였다. 이어서, 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 건조시키고 (Na2SO4) 증발시켰다. MDAP는 순수한 표제 화합물을 제공하였다 (40.7 mg).

b) 에틸 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트

건조 DMF (25 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (1.5g, 5.31 밀리몰)에 Cs2CO3 (1.905 g, 5.84 밀리몰)을 첨가한 다음 에틸 5-브로모발레레이트 (1.46 g, 6.99 밀리몰)을 첨가하였다. 혼합물을 55 ℃에서 18시간 동안 가열한 다음 냉각하였다. 반복적으로 공기를 빼내고 질소를 다시 넣음으로써 탈기시키고, 이어서 모르폴린 (3.70 ml, 42.5 밀리몰) 및 테트라키스(트리페닐포스핀)팔라듐(0) (1.0g, 0.865 밀리몰)을 첨가하고 혼합물을 5시간 동안 교반하였다. EtOAc (75 ml), 2M HCl (40 ml) 및 물 (20 ml)을 첨가하고 유기 상을 분리하고 염수로 세척하고 (3 × 25 ml) 여과하여 일부 불용성 황색 고체를 제거하고 건조시키고 (Na2SO4) 증발시켰다. 잔류물 (2.5 g)을 아미노프로필 SPE (20 g)에 의해 정제하고, THF-MeOH (1:1)중에 부하하고, MeOH로 세척하고 5% 첨가된 AcOH를 함유하는 DCM-MeOH (1:1)로 생성물을 용출하여 표제 화합물을 수득하였다 (1.53 g).
LC/MS: m/z 371 [MH]+, RT 3.18 분
방법 B
a) 3-부틸-8-클로로-1-[4-(3-페닐-1,2,4-옥사디아졸-5-일)부틸]-3,7-디히드로-1H-퓨린-2,6-디온

DMF (15 ml) 중의 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산 (1.89 g, 5.5 밀리몰)의 용액에 CDI (0.98 g, 6.1 밀리몰)을 첨가하고 질소 하에 1.5시간 동안 교반하였다. 벤즈아미독심 (0.91 g, 6.1 밀리몰)을 첨가하고 혼합물을 110 ℃에서 밤새 교반하였다. 반응 혼합물을 EtOAc와 2M HCl 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 증발시켰다. 조 생성물을 메탄올로부터 결정화한 다음 컴패니온TM 시스템 및 시클로헥산으로부터 EtOAc까지의 구배 용출을 사용하여 더욱 정제하였다. 생성물 함유 분획을 합하고 증발시켜 백색 고체로서 표제 화합물을 수득하였다 (850 mg).

b) 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산

에틸 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (2.8g, 7.55 밀리몰), LiOH (542 mg, 22.7밀리몰), 물 (2.5ml) 및 메탄올 (50ml)의 혼합물을 실온에서 60시간 동안 교반하였다. 혼합물을 물과 EtOAc 사이에 분배하고 수성 상의 pH를 pH 4 내지 5로 조절하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 증발시켜 백색 고체로서 표제 화합물을 수득하였다 (2.18g).
LC/MS: m/z 343 [MH]+, RT 2.69 분
실시예
147: 8-
클로로
-3-
펜틸
-1-[4-(3-
페닐
-1,2,4-
옥사디아졸
-5-일)부틸]-3,7-디
히
드로-1H-
퓨린
-2,6-
디온

EtOH (1.5ml) 중의 메틸 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (50 mg, 0.13 밀리몰), 벤즈아미딘 옥심 (20mg, 0.15 밀리몰) 및 EtOH 중의 NaOEt의 21% 용액 (76 ㎕, 0.20 밀리몰)의 혼합물을 마이크로파에서 140℃에서 10분 동안 가열하였다. 냉각 후에 반응을 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고 건조시키고 (MgSO4) 농축하였다. MDAP에 의한 정제는 백색 고체로서 표제 화합물을 제공하였다 (25 mg, 41%).

실시예
148: 3-부틸-8-클로로-1-{4-[3-(3-히드록시페닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

에틸 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (50 mg, 0.13 밀리몰), N,3-디히드록시벤젠카르복시미다미드 (25mg, 0.16 밀리몰), EtOH 중의 NaOEt의 21% 용액 (55 ㎕, 0.15 밀리몰) 및 EtOH (1.5 ml)의 혼합물을 마이크로파에서 180℃에서 10분 동안 가열하였다. EtOH 중의 NaOEt의 21% 용액의 다른 분취량 (55 ㎕, 0.21밀리몰)을 첨가하고, 혼합물을 마이크로파에서 175 ℃에서 30분 동안 가열하였다. 냉각 후에 반응을 2M HCl (수성) 및 EtOAc 사이에 분배하였다. 유기 층을 분리하고 농축하고 MDAP에 의해 정제하였다. 표제 화합물을 회백색 고체로서 수득하였다 (20 mg, 32%).
LC/MS: m/z 459 [MH]+, RT 3.3 분
실시예
149: 8-
클로로
-1-{4-[3-(4-
히드록시페닐
)-1,2,4-
옥사디아졸
-5-일]부틸}-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

DMF (2 ml) 중의 5-(8-클로로-2,6-디옥소-3-펜틸-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산 (50 mg, 0.14 밀리몰)의 용액을 CDI (23 mg, 0.14 밀리몰)로 처리하고 실온에서 30분 동안 교반하였다. N,4-디히드록시벤젠카르복시미다미드 (26 mg, 0.17밀리몰)를 첨가하고 혼합물을 마이크로파에서 120 ℃에서 15분 동안 가열하였다. 냉각 후에 반응을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 농축하고 MDAP에 의해 정제하였다. 표제 화합물을 회백색 고체로서 수득하였다 (17 mg, 26%).
LC/MS: m/z 473 [MH]+, RT 3.5 분
실시예
150: 3-부틸-8-
클로로
-1-[4-(5-
페닐
-2H-
테트라졸
-2-일)부틸]-3,7-디히드로-1H-
퓨린
-2,6-
디온

4-[3-부틸-8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]부틸 메탄술포네이트 (50 mg, 0.12 밀리몰), Cs2CO3 (45 mg, 0.14 밀리몰) 및 DMF (3 ml)의 혼합물을 5-페닐-1H-테트라졸 (20 mg, 0.14 밀리몰)로 처리하고 50 ℃에서 60시간 동안 교반하였다. 냉각 후에, 진공을 가한 다음 질소를 도입함으로써 혼합물을 탈기시켰다. Pd(PPh3)4 (20 mg, 0.017 밀리몰)을 첨가하고, 혼합물을 한번 더 탈기시켰다. 모르폴린 (150 ㎕, 1.7밀리몰)을 첨가하고 혼합물을 질소 하에 18시간 동안 교반한 다음 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 다시 추출하였다. 합한 추출물을 농축하고 황색 잔류물을 수득하였다. MeOH를 첨가한 다음 생성물을 2% AcOH/MeOH로 용출하면서 NH2-프로필 컬럼을 통과시켰다. MDAP에 의한 추가의 정제는 회백색 고체로서 표제 화합물을 제공하였다 (15 mg, 29%).

실시예
151: 3-부틸-8-
클로로
-1-[4-(5-옥소-4-
페닐
-4,5-
디히드로
-1H-
테트라
졸-1-일)부틸]-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

4-[3-부틸-8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]부틸 메탄술포네이트 (50 mg, 0.12 밀리몰), Cs2SO3 (45 mg, 0.14 밀리몰) 및 DMF (3 ml)의 혼합물을 1-페닐-1,2-디히드로-5H-테트라졸-5-온 (23 mg, 0.14 밀리몰)로 처리하고 50 ℃에서 60시간 동안 교반하였다. 냉각 후에, 진공을 가하고 질소를 도입함으로써 혼합물을 탈기시켰다. Pd(PPh3)4 (20 mg, 0.017 밀리몰)을 첨가하고 혼합물을 한번 더 탈기시켰다. 모르폴린 (150 ㎕, 1.7밀리몰)을 첨가하고, 혼합물을 질소 하에 18시간 동안 교반한 다음 2M HCl(수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 수성 층을 EtOAc로 다시 추출하였다. 합한 추출물을 농축하고 황색 잔류물을 수득하였다. MeOH를 첨가한 다음 생성물을 2% AcOH/MeOH로 용출하면서 아미노프로필 컬럼을 통과시켰다. MDAP에 의한 추가의 정제는 회백색 고체로서 표제 화합물을 제공하였다 (27 mg, 51%). NB. 약 10% O-알킬화 물질이 존재한다.

실시예
152: 3-부틸-8-
클로로
-1-{4-[3-(4-
히드록시페닐
)-1,2,4-
옥사디아졸
-5-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

DMF (4 ml) 중의 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산 (100 mg, 0.29 밀리몰)의 교반된 용액을 CDI (52 mg, 0.32밀리몰)로 처리하였다. 1시간 후에, N,4-디히드록시벤젠카르복시미다미드를 첨가하고 혼합물을 100 ℃에서 6시간 동안 가열하였다. 냉각 시에, 반응 혼합물을 2M HCl (수성)과 EtOAc 사이에 분배하였다. 유기 층을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. MDAP에 의한 정제는 연한 회색 고체로서 표제 화합물을 제공하였다 (72 mg).
LC/MS: m/z 459 [MH]+, RT 3.27 분
적절한 아미독심을 사용하여 실시예 146에 대해서와 유사한 방법에 의해 하기 화합물들 (표 10)을 제조하였다.

비교예
A: 3-부틸-8-
클로로
-1-{3-[3-(1-
페닐시클로펜틸
)-1,2,4-
옥사디아졸
-5-일]프로필}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

건조 MeOH (0.75 ml) 중의 에틸 4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)부타노에이트 (53 mg, 0.15 밀리몰), N-히드록시-1-페닐시클로펜탄카르복시미다미드 (34 mg, 0.165 밀리몰) 및 메톡시화 나트륨 (20 mg, 0.37 밀리몰)을 마이크로파 반응기에서 140 ℃에서 10분 동안 가열하였다. 이어서, 혼합물을 에틸 아세테이트와 2M HCl 사이에 분배하고, 유기 상을 증발시키고 생성물을 MDAP에 의해 정제하여 고체로서 표제 화합물을 수득하였다 (29.1 mg).
LC/MS: m/z 497 [MH]+, RT 3.76 분
실시예
156: 1-[3-(3-
비시클로
[4.2.0]옥타-1,3,5-트리엔-7-일-1,2,4-옥사디아졸-5-일)프로필]-3-부틸-8-
클로로
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

적절한 아미독심을 사용하여 비교예 A에 대해서와 유사한 방법을 사용하여 화합물을 제조하였다.
수율 (mg): 28.8
LC/MS: m/z 455 [MH]+, RT 3.43 분
실시예
157: 3-부틸-8-
클로로
-1-[3-(3-{[4-(
메틸옥시
)
페닐
]
메틸
}-1,2,4-
옥사디아졸
-5-일)프로필]-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

HPLC를 사용한 추가의 최종 정제 단계를 사용하는 것 이외에는, 실시예 93를 위해서와 유사한 방법을 사용하여 제조하였다. 수율 6.0 mg
LC/MS: m/z 473 [MH]+, RT 3.27 분
[실시예 162를 위하여, 일반적인 EtOAc/HCl 처리 및 MDAP 이전에 2M NaOH (0.5 ml)와 함께 밤새 EtOH (0.75 ml) 중에서 조 생성물을 교반하고; 실시예 165의 제조로부터 실시예 164를 불순물로 단리하고 HPLC에 의해 그것으로부터 분리하고; 실시예 166 및 167를 위하여 수성 처리 동안에 pH를 추출 이전에 대략 5로 조절하고; 추가로 실시예 167을 MDAP 후에 실리카 SPE (2 g, DCM-MeOH 40:1 이어서 20:1)에 의해 정제하는 것 이외에는] 적절한 아미독심을 사용하여 실시예 75에 대한 것과 유사한 방법에 의해 하기 화합물들 (표 11)을 제조하였다.



[실시예 170을 실시예 90의 규모의 반으로 수행하고 처리 동안에 수성상을 추출 전에 중화시키고; 실시예 171을 실시예 90의 규모의 반으로 수행하고 조 생성물을 처리 및 MDAP 이전에 5시간 동안 EtOH (1 ml) 중의 2M NaOH (0.5 ml)와 함께 교반하고; 실시예 175를 위해 0.185 ml (0.5 밀리몰)의 21% NaOEt를 사용하는 것 이외에는] 적절한 아미독심을 사용하여 실시예 90에 대한 것과 유사한 방법에 의해 하기 화합물들 (표 12)을 제조하였다.



실시예
180: 3-부틸-8-
클로로
-1-(3-{3-[(2-옥소-2,3-
디히드로
-1H-
벤즈이미다
졸-5-일)
메틸
]-1,2,4-
옥사디아졸
-5-일}프로필)-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

21% 에톡시화 나트륨 (0.11ml)의 2당량을 사용하고, 과다한 가열 시간이 20분인 것 이외에는 실시예 99를 위해 유사한 방법에 의해 합성하고, 생성물을 여과에 의해 단리한 다음 고온 MeOH로 분쇄하였다. 수율 14.5 mg
LC/MS: m/z 499 [MH]+, RT 2.78 분
[실시예 181-186을 50 mg의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-1-[3-(1H-피라졸-4-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온으로부터 출발하여 모두 합성하고; 실시예 184, 186, 188, 189 및 190을 아미노프로필 SPE 후에 MDAP에 의해서 추가로 정제하고; 실시예 185를 아미노프로필 SPE 후에 MeOH로부터의 재결정화에 의해서 추가로 정제하고; 실시예 191을 위하여 128 mg (1.2 밀리몰)의 탄산나트륨을 사용하고; 처리 동안에 추출에 앞서서 수성 상을 pH6으로 조절하고; 생성물을 MDAP에 이어서 HPLC에 의해 정제하고; 실시예 184를 위하여, 처리 동안에 침전된 고체를 SPE 이전에 EtOAc 추출물과 조합하는 것 이외에는] 실시예 1을 위한 것과 유사한 방법에 의해 하기 화합물들 (표 13)을 제조하였다.
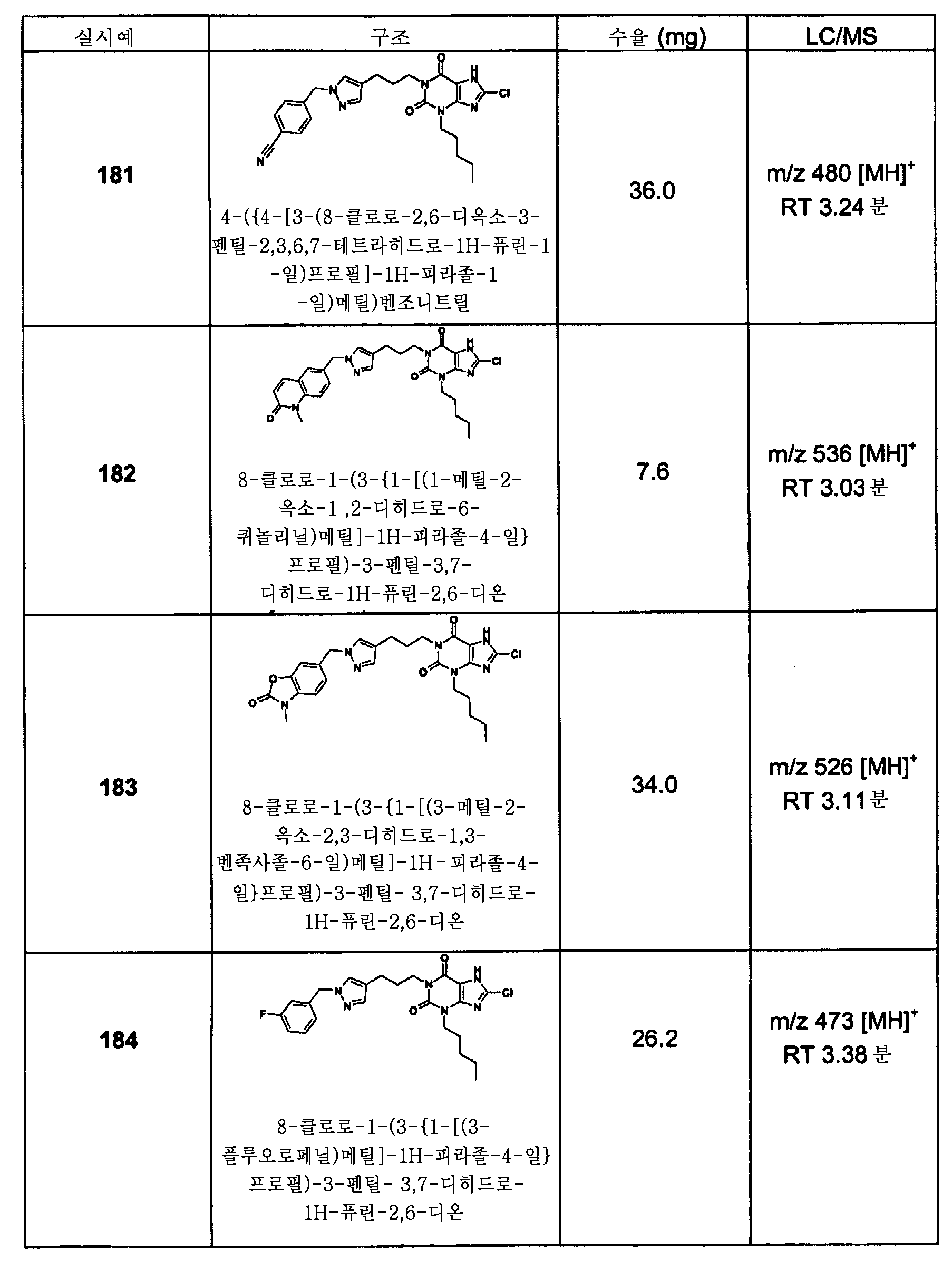


실시예
192: 8-
클로로
-1-(3-{1-[(4-
클로로페닐
)
메틸
]-1H-
피라졸
-4-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-퓨린-2,6-
디온

건조 DMF (1.5 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-1-[3-(1H-피라졸-4-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온 (50 mg, 0.123 밀리몰)을 탄산나트륨 (75 mg, 0.708 밀리몰) 및 4-클로로벤질 브로마이드 (150 mg, 0.73 밀리몰)와 함께 40 ℃에서 18시간 동안 교반하였다. 혼합물을 EtOAc와 물 사이에 분배하고, 유기 상을 염수로 세척하고 건조하고 증발시켰다. 생성물을 실리카 위에 표준 상 크로마토그래피에 의해 정제하여 (컴패니온 시스템, EtOAc-시클로헥산 구배) 오일을 수득하였다 (44 mg). 이것을 질소 하에서 테트라키스(트리페닐포스핀)팔라듐(0) (19 mg) 및 모르폴린 (0.072 ml)과 함께 탈기된 건조 DMF (1 ml)에서 6시간 동안 교반하였다. 혼합물을 EtOAc와 2M HCl 사이에 분배하고, 유기 상을 증발시키고 일반적인 아미노프로필 SPE 절차에 의해 정제하였다. 수율 21.0 mg
LC/MS: m/z 489 [MH]+, RT 3.59 분
[실시예 193을 위하여, 두 번째 분량의 Pd(PPh3)4를 5시간 후에 첨가하고, 교반을 밤새 계속하고, HPLC에 의해 최종 정제를 달성하며; 실시예 195를 위하여 MDAP에 의한 추가의 정제가 요구되고; 실시예 200을 위하여 MeOH로부터의 재결정화에 의한 추가의 정제가 요구되며; 실시예 201을 위하여 MeOH로의 분쇄에 의한 추가의 정제가 요구되는 것 이외에는] 실시예 6에서와 유사한 방법에 의해 하기 화합물들 (표 14)을 제조하였다.



실시예
202: 8-
클로로
-1-(3-{1-[(5-
클로로
-2-티에닐]메틸]-1H-피라졸-4-일}프로필)-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

건조 THF (1 ml) 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-1-[3-(1H-피라졸-4-일)프로필]-3,7-디히드로-1H-퓨린-2,6-디온 (61 mg, 0.15 밀리몰)에 -78 ℃에서 질소 하에 t-부톡시화칼륨 (THF 중의 1M, 0.15 ml)을 첨가한 다음 2-클로로-5-(클로로메틸)티오펜 (25 mg, 0.15 밀리몰)을 첨가하였다. 교반을 -78 ℃에서 15분 동안, 이어서 실온에서 1시간 동안, 최종적으로 60 ℃에서 18시간 동안 계속하였다. 용액을 탈기시키고 모르폴린 (0.13 ml) 및 테트라키스(트리페닐포스핀)팔라듐(0) (35 mg)을 첨가하고 6시간 동안 교반을 계속하였다. 추가 량 (0.2 ml 모르폴린, 50 mg Pd(PPh3)4)을 첨가하고, 교반을 밤새 계속하였다. EtOAc와 2M HCl 사이의 분배에 의해 처리하여, 유기 상이 증발되고 표준 아미노프로필 SPE 절차에 의해 정제한 다음 MDAP에 의해 정제하여 표제 화합물을 백색 고체로서 수득하였다 (5.1 mg).
LC/MS: m/z 495 [MH]+, RT 3.68분
실시예
203: 3-부틸-8-
클로로
-1-{3-[5-(
페닐메틸
)-1,3,4-
옥사디아졸
-2-일]프로필}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

건조 DMF (2 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (99 mg, 0.35 밀리몰)에 탄산세슘 (137 mg, 0.42 밀리몰)을 첨가한 다음 건조 DMF (1 ml)중의 2-(3-클로로프로필)-5-(페닐메틸)-1,3,4-옥사디아졸 (99 mg, 0.42 밀리몰)의 용액을 첨가하였다. 혼합물을 질소 하에서 교반하고, 55 ℃에서 2.5시간 동안 가열한 다음 실온에서 밤새 교반하였다. 반복적으로 공기를 빼내고 질소를 넣음으로써 혼합물을 탈기시킨 다음, 테트라키스 (트리페닐포스핀)팔라듐(0) (81mg, 0.07 밀리몰) 및 모르폴린 (0.305 ml, 3.5 밀리몰)을 첨가하였고, 교반을 5시간 동안 계속하였다. EtOAc 및 2M HCl을 첨가하고, 혼합물을 20분 동안 교반한 다음 여과하였다. 유기 상을 분리하고 증발시키고, THF-MeOH (1:1)에 이어서 MeOH로 세척하고, 산성 생성물을 5% 첨가 AcOH를 함유하는 DCM-MeOH (1:1)로 용출함으로써 아미노프로필 SPE (5 g)에 의해 생성물을 정제하였다. 이렇게 수득된 생성물을 MDAP에 의해 더욱 정제하여 표제 화합물 (92 mg)을 수득하였다.
LC/MS: m/z 443 [MH]+, RT 3.18분
실시예 211이 HPLC에 의해 더욱 정제되는 것 이외에는 실시예 203을 위한 것과 유사한 방법에 의해 하기 화합물들 (표 15)을 제조하였다.

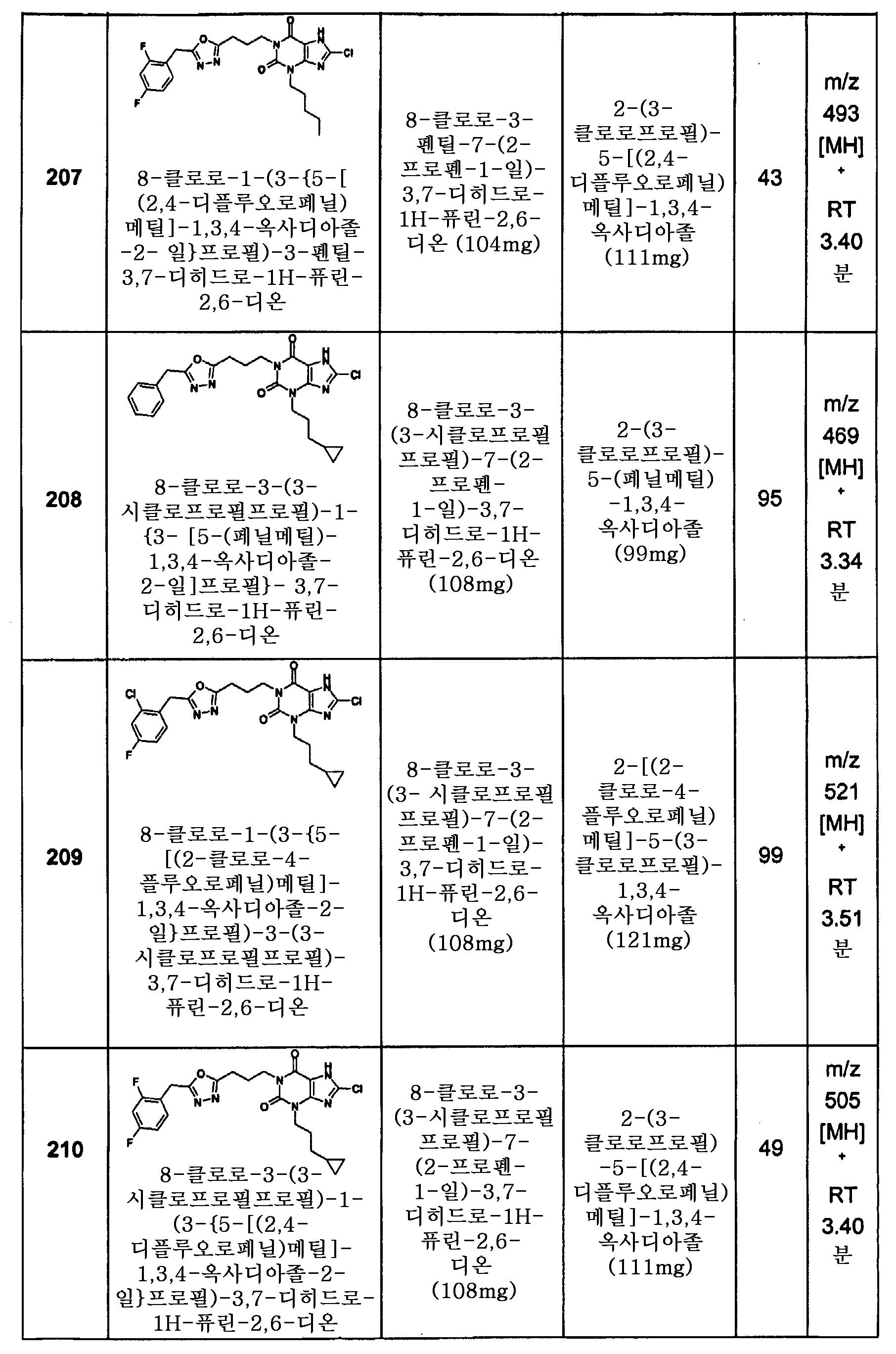

표 15로부터
클로로프로필
1,3,4-
옥사디아졸
중간체의 합성:
2-[(2-클로로-4-플루오로페닐)메틸]-5-(3-클로로프로필)-1,3,4-옥사디아졸
2-(3-클로로프로필)-5-[(2,4-디플루오로페닐)메틸]-1,3,4-옥사디아졸
2-(3-클로로프로필)-5-(페닐메틸)-1,3,4-옥사디아졸
디아실 히드라진 (500 mg, 하기 합성)을 건조 톨루엔 (4 ml)중에서 교반하고 옥시염화인 (4 ml)을 첨가하였다. 혼합물을 90 ℃에서 2시간 동안 가열한 다음 냉각하고 용매를 증발시켰다. 잔류물을 건조 톨루엔에 용해시키고 증발시킨 다음 EtOAc와 수성 NaHCO3 사이에 분배하였다. 유기 상을 염수로 세척하고 Na2SO4 상에서 건조시키고 증발시켜 무색 오일로서 필요한 옥사디아졸을 수득하였다. 이것을 더 이상 정제시키지 않았으며, 상기 기재된 바와 같이 크산틴과 직접적으로 반응시켰다.

4-클로로-N'-(페닐아세틸)부타노히드라지드의 제조

건조 DCM (10 ml) 중의 4-클로로부티릴 클로라이드 (1.12ml, 10밀리몰)에 40분에 걸쳐 건조 DCM (40 ml)중의 페닐아세트 히드라지드 (1.5g, 10밀리몰) 및 DIPEA (1.77 ml, 10.2 밀리몰)의 혼합물을 실온에서 적가하였다. 조밀한 백색 침전물이 형성되었다. 추가의 20분 후에, 2M HCl (30 ml)를 첨가하고 표제 화합물 (백색 고체)를 여과 제거하고 물로 세척하고 건조하였다 (2.24 g).
LC/MS: m/z 255 [MH]+, RT 2.20분
4-클로로-N'-[(2-클로로-4-플루오로페닐)아세틸]부타노히드라지드의 제조

(i) 건조 DCM (15 ml) 중의 2-클로로-4-플루오로페닐아세틸 클로라이드 (10밀리몰)의 용액을 20분에 걸쳐 건조 DCM (20 ml) 중의 t-부틸 카르바제이트 (1.32 g, 10 밀리몰) 및 DIPEA (1.77 ml, 10.2 밀리몰)의 혼합물에 첨가하였다. 추가로 2시간 동안 교반한 후에, 혼합물을 1M HCl로 세척한 다음 수성 NaHCO3로 세척하였다. 이 시점에서 백색 고체가 침전되고 이것을 여과해 내고 물 및 DCM으로 세척한 다음 건조시켜 1,1-디메틸에틸 2-[(2-클로로-4-플루오로페닐)아세틸]히드라진카르복실레이트 (1.94 g)를 수득하였다.
(ii) 이 화합물 (1.92 g, 6.34 밀리몰)을 디옥산 (2 ml)에 현탁시키고, 디옥산 중의 4M HCl(5 ml)을 첨가하였다. 조밀한 백색 침전물이 형성되었다. 1시간 후에 혼합물을 EtOAc와 포화 수성 NaHCO3 사이에 분배하고 유기 상을 염수로 세척하고, 건조시키고 (Na2SO4) 증발시켜 2-(2-클로로-4-플루오로페닐)아세토히드라지드를 백색 고체 (1.07g)로서 수득하였다.
(iii) 건조 DCM (65 ml) 중의 2-(2-클로로-4-플루오로페닐)아세토히드라지드 (909 mg, 4.5 밀리몰) 및 DIPEA (0.817 ml, 4.7 밀리몰)의 혼합물을 20분에 걸쳐 건조 DCM (5 ml) 중의 4-클로로부티릴 클로라이드 (0.505 ml, 4.5 밀리몰)에 첨가하였다. 1.5 시간 후에, 2M HCl을 첨가하고 침전된 4-클로로-N'-[(2-클로로-4-플루오로페닐)아세틸]부타노히드라지드를 여과해 내고 물로 세척하고 건조시켰다 (1.24g).
LC/MS: m/z 307 [MH]+, RT 2.61분
2-(3-클로로프로필)-5-[(2,4-디플루오로페닐)메틸]-1,3,4-옥사디아졸의 제조

(i) 건조 DCM (15 ml) 중의 2,4-디플루오로페닐아세틸 클로라이드 (10 밀리몰)의 용액을 10분에 걸쳐 건조 DCM (20 ml) 중의 t-부틸 카르바제이트 (1.32g, 10밀리몰) 및 DIPEA (1.77 ml, 10.2 밀리몰)의 혼합물에 첨가하였다. 1.5시간 동안 교반 후에, 혼합물을 1M HCl로 세척하고 이어서 수성 NaHCO3로 세척하였다. 유기 상을 증발시켜 1,1-디메틸에틸-2-[(2,4-디플루오로페닐)아세틸]히드라진카르복실레이트를 백색 고체로서 수득하였다.
(ii) 디옥산 (5 ml) 중의 1,1-디메틸에틸 2-[(2,4-디플루오로페닐)아세틸]히드라진카르복실레이트 (10밀리몰)을 디옥산 중의 4M HCl (8 ml)과 함께 1.5시간 동안 교반하였다. 혼합물을 EtOAc와 포화 수성 NaHCO3 사이에 분배하고, 유기 상을 염수로 세척하고, 건조시키고 (Na2SO4) 증발시켰다. 반응이 불완전하였으며, 따라서 잔류물을 디옥산 중의 4M HCl (10 ml)과 함께 2.5시간 동안 다시 교반하였다. 이전과 같은 처리는 2-(2,4-디플루오로페닐)아세토히드라지드를 고체로서 제공하였다 (570 mg).
(iii) 건조 DCM (30 ml) 중의 2-(2,4-디플루오로페닐)아세토히드라지드 (570 mg, 3.06 밀리몰) 및 DIPEA (0.553 ml, 3.2 밀리몰)의 혼합물을 15분에 걸쳐 건조 DCM (5 ml) 중의 4-클로로부티릴 클로라이드 (0.343 ml, 3.06 밀리몰)에 첨가하였다. 즉시 백색 침전물이 형성되었다. 1시간 동안 교반한 후에, 2M HCl (20 ml)를 첨가하고 고체 2-(3-클로로프로필)-5-[(2,4-디플루오로페닐)메틸]-1,3,4-옥사디아졸을 여과해 내고 물로 세척하고 건조시켰다 (726 mg).
LC/MS: m/z 291 [MH]+, RT 2.45분
실시예
212: 3-부틸-8-
클로로
-1-[4-(3-
페닐
-5-
이속사졸릴
)부틸]-3,7-
디히드
로-1H-
퓨린
-2,6-
디온
a) 3-부틸-8-클로로-1-[4-(3-페닐-5-이속사졸릴)부틸]-3,7-디히드로-1H-퓨린-2,6-디온

3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (100 mg, 0.354 밀리몰) 및 4-(3-페닐-5-이속사졸릴)-1-부탄올 (77 mg, 0.355 밀리몰)을 질소 하에서 건조 THF (4 ml) 중에서 용해시켰다. 건조 THF (2 ml) 중의 디벤질 아조디카르복실레이트 (94%, 224 mg, 0.708 밀리몰)의 용액을 첨가하였다. 혼합물을 0 ℃로 냉각하고, 건조 THF (1 ml) 중의 트리페닐포스핀의 용액 (185 mg, 0.708 밀리몰)을 첨가하였다. 혼합물을 0 ℃에서 20분 동안 교반한 다음 실온에서 밤새 교반하였다. 혼합물을 탈기한 다음 모르폴린 (0.308 ml) 및 테트라키스(트리페닐포스핀)팔라듐(0) (82 mg)과 함께 4.5시간 동안 교반하였다. 추가의 60mg의 테트라키스(트리페닐포스핀)팔라듐(0)을 첨가하고 밤새 교반을 계속하였다. EtOAc와 2M HCl 사이에서 분배시킴으로써 반응을 처리하고, 유기 상을 증발시키고, THF-MeOH (1:1), MeOH로 세척하고 5% AcOH를 함유하는 DCM-MeOH (1:1)로 용출시키면서 아미노프로필 SPE (5 g)에 의해 정제하였다. MDAP에 의한 추가의 정제는 표제 화합물 (56 mg)을 제공하였다.
LC/MS: m/z 442 [MH]+, RT 3.59분
b) 4-(3-페닐-5-이속사졸릴)-1-부탄올

건조 DCM (6 ml) 중의 N-히드록시벤젠카르복시미도일 (622 mg, 4 밀리몰)에 5-헥신-1-올 (431 mg, 4.4 밀리몰)을 첨가하였다. 트리에틸아민 (0.612 ml, 4.4 밀리몰)을 10분에 걸쳐 적가할 때 혼합물을 질소 하에 0 ℃로 냉각하였다. 0 ℃에서 추가로 20분 동안, 이어서 실온에서 밤새 교반하였다. 혼합물을 물로 세척하고 유기 상을 증발시켰다. 생성물을 EtOAc-시클로헥산 (1:2에 이어서 3:1)로 용출하면서 실리카 SPE (20 g)에 의해 정제하여 백색 왁스질 고체 (443 mg)를 수득하였다.
LC/MS: m/z 218 [MH]+, RT 2.74분
실시예
213: 3-부틸-8-
클로로
-1-{3-[3-(페닐메틸)-5-이속사졸릴]프로필}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온
a) 3-부틸-8-클로로-1-{3-[3-(페닐메틸)-5-이속사졸릴]프로필}-3,7-디히드로-1H-퓨린-2,6-디온

3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (50 mg, 0.177 밀리몰) 및 3-[3-(페닐메틸)-5-이속사졸릴]-1-프로판올 (38.4 mg, 0.177 밀리몰)로부터 출발하여 절반의 몰 량을 사용하여 3-부틸-8-클로로-1-[4-(3-페닐-5-이속사졸릴)부틸]-3,7-디히드로-1H-퓨린-2,6-디온 (실시예 213)과 유사하게 제조하였다. 수율 24.2 mg, LC/MS: m/z 442 [MH]+, RT 3.43분
b) 3-[3-(페닐메틸)-5-이속사졸릴]-1-프로판올

N-히드록시-2-페닐에탄이미도일 클로라이드 (253 mg, 1.5 밀리몰) 및 4-펜틴-1-올 (139 mg, 1.65 밀리몰)을 사용하여 4-(3-페닐-5-이속사졸릴)-1-부탄올에서와 같이 합성하였다. 수율 61 mg의 담황색 오일
LC/MS: m/z 218 [MH]+, RT 2.62분
비교예
B: 8-
클로로
-1-[3-(2-
푸라닐
)프로필]-3-
펜틸
-3,7-
디히드로
-1H-
퓨린
-2,6-디온, 나트륨 염

교반기가 장착된 그린하우스(GreenHouse)TM 관에 THF 중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.13 밀리몰)의 0.54M 용액의 0.25 ml 분취량을 넣었다. 혼합물에 THF (0.25ml) 중의 3-(2-푸라닐)-1-프로판올 (21 mg, 0.16 밀리몰, 1.2 당량)을 첨가한 다음, THF 중의 비스(1,1-디메틸에틸) (E)-1,2-디아젠디카르복실레이트 (0.18 밀리몰)의 0.71M 용액의 0.25 ml 분취량에 이어서 THF 중의 트리페닐포스핀 (0.18 밀리몰)의 0.71M 용액의 0.25 ml 분취량을 첨가하였다. 용액을 질소 하에 그린하우스TM에서 16시간 동안에 교반하였다. 혼합물에 THF 중의 비스(1,1-디메틸에틸) (E)-1,2-디아젠디카르복실레이트 (0.36 밀리몰)의 1.4M 용액의 0.25 ml 분취량을 첨가한 다음 THF 중의 트리페닐포스핀 (0.36 밀리몰)의 1.4M 용액 0.25 ml 분취량을 첨가하였다. 혼합물을 질소 기류 하에 16시간 동안 교반하였다.
테트라키스(트리페닐포스핀)팔라듐(0) (16 mg, 0.014 밀리몰) 및 모르폴린 (0.12 ml, 1.35 밀리몰)을 혼합물에 첨가하고, 이것을 질소 기류 하에 16시간 동안 교반하였다. 반응 혼합물을 질소 하에 농축하고 조 물질을 수성 NaOH 용액 (0.5 ml, 2M)에 용해시켰다. 얻어진 용액을 아미노프로필 SPE (DCM 및 MeOH 중의 AcOH로 용출)을 사용하여 정제하였다. C18 SPE를 사용한 추가의 정제 (물, 암모니아 및 MeCN 혼합물로 용출됨)는 표제 화합물을 투명한 고무로서 제공하였다 (22 mg, 45%).

비교예 B에 대해서와 유사한 방법에 의해 하기 화합물들 (표 16)을 제조하였다.

상응하는 산 및 (1Z)-4-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시부탄이미다미드로부터 실시예 113에 대해서와 유사한 방법에 의해 하기 화합물들 (표 17)을 제조하였다.


실시예
221: 8-클로로-3-프로필-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온
a) 8-클로로-3-프로필-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온

DMF (3 ml) 중의 8-클로로-7-(2-프로펜-1-일)-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (40 mg, 0.09 밀리몰)의 용액을 탄산칼륨 (15 mg, 0.11 밀리몰) 및 1-요오도프로판 (19 mg, 0.11 밀리몰)로 처리하였다. 혼합물을 40 ℃에서 3시간 동안 가열한 다음 70 ℃에서 추가로 3시간 동안 가열하였다. 혼합물을 냉각하고 연속적으로 진공 및 질소 기체를 가함으로써 탈기시켰다. 이어서, 혼합물을 테트라키스(트리페닐포스핀)팔라듐(0) (10 mg, 0.009 밀리몰) 및 모르폴린 (0.1 ml, 1.2 밀리몰)의 용액으로 처리하고 이어서 밤새 교반하였다. 혼합물을 증발시키고 클로로포름 (2 ml)과 물 (2 ml) 사이에 분배하였다. 수성 상을 추가의 클로로포름 (2 ml)으로 추출하고, 합한 유기물을 증발시키고 잔류물을 메탄올 (2 ml)에 용해시켰다. 용액을 1 g 아미노프로필 SPE에 적용하고, 메탄올로 용출한 다음 메탄올 중의 5% 아세트산으로 용출하였다. 생성물-함유 분획을 모으고, 증발시키고 생성물을 MDAP에 의해 정제하여 8-클로로-3-프로필-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (1.4 mg)을 백색 고체로서 수득하였다.
LC/MS: m/z 430 [MH]+, RT 2.84분
b) 8-클로로-7-(2-프로펜-1-일)-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온

무수 THF (20 ml) 중의 N-히드록시-2-피리딘카르복시미다미드 (1.15 g, 8.4 밀리몰)의 현탁액을 메톡시화 나트륨 (0.38 g, 7.0 밀리몰)로 처리하고 혼합물을 5분 동안 교반하였다. 혼합물을 에틸 5-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]펜타노에이트 (2 g, 5.6 밀리몰)로 처리하고 모든 물질이 용해될 때까지 약 5분 동안 교반하였다. 혼합물을 밀봉하고 마이크로파에서 120 ℃에서 15분 동안 가열한 다음 냉각하고 에틸 아세테이트(100 ml)와 포화 수성 중탄산나트륨 (50 ml) 사이에 분배하였다. 수성 상을 추가의 에틸 아세테이트 (50 ml)로 추출하고, 합한 유기물을 건조시키고 (MgSO4), 여과하고 증발시켰다. 1:9 에틸 아세테이트/시클로헥산으로부터 에틸 아세테이트까지의 구배 용출을 사용하여 플래시 크로마토그래피에 의해 생성물을 정제하여, 8-클로로-7-(2-프로펜-1-일)-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 (1.49g)을 백색 고체로서 수득하였다.
LC/MS: m/z 428 [MH]+, RT 2.70분
에틸 4-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]부타노에이트를 사용하여, 8-클로로-1-(3-{3-[(2,4-디플루오로페닐)메틸]-1,2,4-옥사디아졸-5-일}프로필)-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온을 유사하게 제조하였다.
LC/MS: m/z 463 [MH]+, RT 3.09분
c) 에틸 5-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]펜타노에이트

DMF (10 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3-({[2-(트리메틸실릴)에틸]옥시}메틸)-3,7-디히드로-1H-퓨린-2,6-디온 (10g, 28 밀리몰)의 용액을 탄산칼륨 (4.8g, 35 밀리몰) 및 에틸 5-브로모펜타노에이트 (6.5g, 31 밀리몰)로 처리하고, 이어서 70 ℃로 3시간 동안 가열하고 냉각하고 증발시켰다. 잔류물을 에틸 아세테이트 (100 ml)와 물 (50 ml) 사이에 분배하였다. 유기 상을 건조시키고 (MgSO4), 여과하고 증발시키고 조 중간체를 디클로로메탄 (90 ml)에 용해시키고 트리플루오로아세트산 (17ml)으로 처리하고 혼합물을 주위 온도에서 밤새 교반하였다. 톨루엔 (50 ml)을 첨가하고 혼합물을 증발 건조시켰다. 시클로헥산으로부터 에틸 아세테이트까지의 구배 용출을 사용하는 플래시 크로마토그래피에 의해 생성물을 정제하여 8.65 g의 에틸 5-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]펜타노에이트를 백색 고체로서 수득하였다.
LC/MS: m/z 355 [MH]+, RT 2.75분
유사하게, 에틸 4-[8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]부타노에이트를 제조하였다.
LC/MS: m/z 341 [MH]+, RT 2.61분
d) 8-클로로-7-(2-프로펜-1-일)-3-({[2-(트리메틸실릴)에틸]옥시}메틸)-3,7-디히드로-1H-퓨린-2,6-디온

DMF (80 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (5 g, 22.1 밀리몰)의 용액에 2,2-(트리메틸실릴)에톡시메틸 클로라이드 (4.3 ml, 24.2 밀리몰) 및 탄산나트륨 (2.6 g, 24.2 밀리몰)을 첨가하였다. 실온에서 밤새 교반 후에, 추가의 2,2-(트리메틸실릴)에톡시메틸 클로라이드 (4.3 ml, 24.2 밀리몰) 및 탄산나트륨 (1.3g, 12.1 밀리몰)을 첨가하고 2시간 동안 교반을 계속하였다. 반응 혼합물을 5% LiCl 수성과 에틸 아세테이트 사이에 분배하였다. 유기 추출물을 분리하고 염수로 세척하고 건조시키고 (MgSO4) 농축하였다. 1:4-1:2 에틸 아세테이트/시클로헥산으로 용출되는 실리카 카트릿지를 사용하는 바이오태그(Biotage)TM 크로마토그래피에 의해 정제하여 표제 화합물을 수득하였다 (3.14 g, 40%). m/z 374 [MNH4 +]
8-클로로-7-(2-프로펜-1-일)-1-{4-[3-(2-피리디닐)-1,2,4-옥사디아졸-5-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온 및 적절한 알킬화제로부터, 실시예 221에 대해서와 유사한 방법에 의해 하기 화합물 (표 18)을 제조하였다.


8-클로로-1-(3-{3-[(2,4-디플루오로페닐)메틸]-1,2,4-옥사디아졸-5-일]프로필)-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 및 적절한 알킬화제로부터, 실시예 221에 대해서와 유사한 방법에 의해 하기 화합물 (표 19)을 제조하였다.




실시예
246: 8-
클로로
-1-{4-[2-옥소-3-(
페닐메틸
)-1-
피롤리디닐
]부틸}-3-
펜틸
-3,7-디히드로-1H-
퓨린
-2,6-
디온
a) 8-클로로-1-{4-[2-옥소-3-(페닐메틸)-1-피롤리디닐]부틸}-3-펜틸-3,7-디히드로-1H-퓨린-2,6-디온

THF (10 ml) 중의 8-클로로-1-{4-[2-옥소-3-(페닐메틸)-1-피롤리디닐]부틸}-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.35g, 0.67밀리몰), Pd(PPh3)4 (0.082g, 0.07밀리몰) 및 모르폴린 (0.6 ml, 6.7 밀리몰)의 용액을 탈기시키고 (연속 탈기에 이어서 질소 첨가 3회) 이어서 4시간 동안 교반하였다. 이어서, 용액을 아미노프로필 SPE (5 g)에 부하하고 MeOH로 용출시킨 다음 5% AcOH/MeOH로 용출하여 소량의 불순물을 함유하는 표제 화합물을 제공하였다. 실리카 (10 g SPE, 구배 용출 에테르/에틸 아세테이트 1:0 내지 0:1) 위에서의 정제는 투명한 오일로서 표제 화합물을 제공하였다 (0.10 g, 31%).
LC/MS: m/z 486 [MH]+
b) 8-클로로-1-{4-[2-옥소-3-(페닐메틸)-1-피롤리디닐]부틸}-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온

1-(4-브로모부틸)-3-(페닐메틸)-2-피롤리디논을 알킬화제로서 사용하고 염기로서 탄산칼륨을 사용하고 50 ℃에서 18시간 동안 가열하여 8-클로로-1-(2-히드록시-6-페닐헥실)-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온에서와 같이 제조하였다.
수율 86%
LC/MS: m/z 526 [MH]+
c) 1-(4-브로모부틸)-3-(페닐메틸)-2-피롤리디논

DMF (5 ml) 중의 3-(페닐메틸)-2-피롤리디논 (0.23g, 1.3 밀리몰) 및 1,4-디브로모부탄 (0.57 g, 4.2 밀리몰)의 용액에 t-부톡시화 나트륨 (0.151 g, 1.6 밀리몰)을 첨가하고 용액을 18시간 동안 교반하였다. 용액을 농축하고 잔류물을 실리카 위에서 크로마토그래피하여 (20 g SPE, 시클로헥산으로 용출한 다음 DCM으로 용출) 미량의 DMF를 함유하는 무색 오일로서 표제 화합물을 수득하였다 (0.25 g, 61%).
LC/MS: m/z 311 [MH]+
실시예
247: 8-
클로로
-1-{4-[2-옥소-1-(
페닐메틸
)-3-
피롤리디닐
]부틸}-3-
펜틸
-3,7-디히드로-1H-
퓨린
-2,6-
디온
a) 8-클로로-1-{4-[2-옥소-1-(페닐메틸)-3-피롤리디닐]부틸}-3-펜틸-3,7-디히드로-1H-퓨린-2,6-디온

THF (5 ml)중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.086 g, 0.29 밀리몰) 및 3-(4-브로모부틸)-1-(페닐메틸)-2-피롤리디논 (0.17 g, 0.55 밀리몰, 2-(페닐메틸)-2-아자스피로[4.4]노난-1-온과의 1:1 혼합물)의 용액에 탄산칼륨 (0.08 g, 0.58 밀리몰)을 첨가하고 혼합물을 가열하고 50 ℃에서 18시간 동안 교반하였다. 용액을 냉각한 다음 탈기시키고 (연속 탈기에 이어서 질소 첨가 3회), Pd(PPh3)4 (0.09 g, 0.077 밀리몰)에 이어서 모르폴린 (0.2 ml, 2.2 밀리몰)을 첨가하고 용액을 주변 온도에서 18시간 동안 교반하였다. 용액을 에틸 아세테이트와 묽은 염산 사이에 분리하고 유기물을 염수로 세척하고 건조시키고 농축하였다. MeOH에 이어서 5% AcOH/MeOH로 용출되는 아미노프로필 SPE (5 g)를 사용하여 잔류물을 정제하여 황색 오일로서 표제 화합물을 수득하였으며 이것은 에테르 하에 정치할 때 결정화되었다 (0.031g, 22%).
LC/MS: m/z 486 [MH]+
b) 3-(4-브로모부틸)-1-(페닐메틸)-2-피롤리디논

-78 ℃에서 THF (10 ml) 중의 1-(페닐메틸)-2-피롤리디논 (0.47 g, 2.7밀리몰)의 용액에 리튬 헥사메틸디실릴라진 (2.8 ml, 2.7 밀리몰, 1M 용액)을 5분에 걸쳐 첨가하였다. 15분 후에, 1,4-디브로모부탄 (0.32 ml, 2.7 밀리몰)을 첨가하고 용액을 주변 온도에서 2시간 동안 달성한 다음 추가의 18시간 동안 교반하였다. 용액을 에틸 아세테이트와 물 사이에 분리하고 유기물을 단리하고 건조시키고 농축하였다. 시클로헥산에 이어서 DCM 및 마지막으로 에테르로 용출되는 실리카 (20 g, SPE) 위에서의 크로마토그래피는 표제 화합물과 2-(페닐메틸)-2-아자스피로 [4.4]노난-1-온 (0.17g)의 1:1 혼합물인 투명 오일을 제공하였다. 이것을 추가의 정제없이 이후 단계에서 사용하였다.
LC/MS: m/z 310, 312 [MH]+
비교예
C: 8-
클로로
-1-(5-{5-[(3,4-
디클로로페닐
)
메틸
]-2H-
테트라졸
-2-일}
펜틸
)-3-펜틸-3,7-
디히드로
-1H-퓨린-2,6-
디온

THF (5 ml)중의 8-클로로-3-펜틸-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (0.18g, 0.61 밀리몰)의 용액에 5-{5-[(3,4-디클로로페닐)메틸]-2H-테트라졸-2-일}-1-펜탄올 (0.191 g, 0.61 밀리몰; 실시예 35에서와 유사한 방식으로 제조됨), 트리페닐포스핀 (0.36g, 1.3 밀리몰) 및 최종적으로 디벤질아조디카르복실레이트 (0.40 g, 1.3밀리몰)을 첨가하였다. 용액을 18시간 동안 교반하고 그 후에 Pd(PPh3)4 (0.16g, 0.137 밀리몰)에 이어서 모르폴린 (0.75 ml, 8.3 밀리몰)을 첨가하고, 용액을 주변 온도에서 6시간 동안 교반하였다. 용액을 아미노프로필 SPE (5 g) 위에 부하하고 MeOH에 이어서 5% AcOH/MeOH를 사용하여 용출하여, 미량의 불순물을 함유하는 표제 화합물을 수득하였다. 또한, 에테르로 용출되는 크로마토그래피 (실리카 SPE, 20 g)은 백색 고체로서 표제 화합물을 수득하였다 (0.061 g, 18%)
LC/MS: m/z 553 [MH]+
실시예
248: 8-
클로로
-1-{3-[5-(4-
클로로페닐
)-1H-
피라졸
-3-일]프로필}-3-
펜
틸-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

3-[5-(4-클로로페닐)-1H-피라졸-3-일]-1-프로판올을 사용하여 8-클로로-1-(5-{5-[(3,4-디클로로페닐)메틸]-2H-테트라졸-2-일}펜틸)-3-펜틸-3,7-디히드로-1H-퓨린-2,6-디온 (비교예 C)에서와 같이 제조하였다. 최종 생성물 물질을 에테르로 세척하여 표제 화합물을 크림색 고체로서 수득하였다 (30%).
LC/MS: m/z 475 [MH]+
실시예
249: 3-부틸-8-
클로로
-1-{4-[5-(1-
메틸
-1H-1,2,3-
트리아졸
-4-일)-1,2,4-
옥사디아졸
-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온

DMF (0.5 ml) 중의 1-메틸-1H-1,2,3-트리아졸-4-카르복실산 (18 mg, 0.14 밀리몰)의 용액을 CDI (23 mg, 0.14 밀리몰)로 실온에서 1시간 동안 처리하였다. DMSO (0.4 ml)중의 (1Z)-5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (50 mg, 0.14 밀리몰)의 용액을 혼합물에 첨가하고 이어서 100 ℃로 18시간 동안 가열하였다. 반응 혼합물을 MDAP에 의해 정제하고 표제 화합물을 백색 고체로서 수득하였다 (18 mg)
LC/MS: m/z 448 [MH]+, RT 2.86 분
적절한 카르복실산을 사용하여 실시예 249에서와 유사한 방법에 의해 하기 화합물들 (표 20)을 제조하였다.







실시예
291: 8-
클로로
-1-{4-[3-(2,4-
디플루오로페닐
)-1,2,4-
옥사디아졸
-5-일]부틸}-3-에틸-3,7-
디히드로
-1H-
퓨린
-2,6-
디온
a) 8-클로로-1-{4-[3-(2,4-디플루오로페닐)-1,2,4-옥사디아졸-5-일]부틸}-3-에틸-3,7-디히드로-1H-퓨린-2,6-디온

DMSO (1 ml) 중의 5-(8-클로로-3-에틸-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산 (0.05g, 0.16 밀리몰)의 용액을 CDI (0.029g, 0.18 밀리몰)로 처리하고, 혼합물을 실온에서 1시간 동안 교반하였다. 이어서, 혼합물을 2,4-디플루오로벤즈아미독심 (0.03g, 0.18밀리몰)로 처리한 다음 120 ℃로 30분 동안 가열하였다. MDAP를 사용하여 조 혼합물로부터 생성물을 정제하였다. 질소 기류를 사용하여 생성물-함유 분획을 증발시키고, 얻어진 무색 고무를 에테르 중에서 분쇄시키고 건조시켜 백색 고체로서 표제 화합물을 수득하였다 (50 mg, 70%).
LC/MS: m/z 451 [MH]+, RT 2.23 분
b) 5-(8-클로로-3-에틸-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산

메탄올 (75 ml) 중의 에틸 5-(8-클로로-3-에틸-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 (2.3g, 6.7 밀리몰)의 용액을 물(3 ml) 및 수산화리튬(0.481g, 20.1밀리몰)로 처리하고, 혼합물을 40 ℃에서 17시간 동안 교반하였다. 혼합물을 증발 건조시키고, 잔류물을 50 ml의 에틸 아세테이트 및 50 ml의 물로 처리하였다. 2개 상을 분리하고 수성 상을 2M 수성 염산을 사용하여 pH 5로 조절하였다. 침전된 생성물을 여과해 내고 건조시켜 백색 고체로서 5-(8-클로로-3-에틸-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄산을 수득하였다 (1.99 g, 95%).
LC/MS: m/z 315 [MH]+, RT 2.34 분
c) 에틸 5-(8-클로로-3-에틸-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트

DMF (40 ml) 중의 8-클로로-7-(2-프로펜-1-일)-3-에틸-3,7-디히드로-1H-퓨린-2,6-디온 (3 g, 11.8 밀리몰)의 용액을 탄산칼륨 (1.9 g, 14.1 밀리몰) 및 에틸 5-브로모펜타노에이트 (2.24 ml, 14.1 밀리몰)로 처리하고 혼합물을 질소 대기 중에서 70 ℃에서 5시간 동안 가열한 다음 냉각하였다. 반복적으로 진공을 가하여 혼합물을 탈기시킨 다음 질소 기체로 다시 충진하여 테트라키스(트리페닐포스핀)팔라듐(0) (1.36 g, 1.1 밀리몰) 및 모르폴린 (10.3 ml, 118 밀리몰)로 처리하였다. 혼합물을 질소 대기 중에서 4시간 동안 교반한 다음 증발 건조시켰다. 잔류물을 100 ml의 에틸 아세테이트 및 100 ml의 물 사이에 분배하였다. 수성 상을 100 ml의 에틸 아세테이트로 재-추출하고 합한 유기물을 황산 마그네슘 상에서 건조하고 여과하고 증발시켰다. 잔류물을 디에틸 에테르에 분쇄하고 여과하고 건조시켜 에틸 5-(8-클로로-3-에틸-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜타노에이트 화합물을 백색 고체로서 수득하였다 (2.3 g, 57%).
LC/MS: m/z 343 [MH]+, RT 2.73 분
실시예 291에 대해서와 유사한 방법에 의해 하기 화합물 (표 21)을 제조하였다.




적절한 산을 사용하여 실시예 127에 대해서와 유사한 방법을 사용하여 하기 화합물들 (표 22)을 제조하였다.



적절한 테트라졸 및 3-[3-알킬-8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]프로필 메탄술포네이트를 사용하여, 실시예 19에 대해서와 유사한 방법에 의해 하기 화합물들 (표 23)을 제조하였다.
표준 상 크로마토그래피 후에 불충분하게 순수한 화합물을 더욱 정제하기 위하여 MDAP를 사용하였다.



















실시예
408: 3-부틸-8-
클로로
-1-{3-[4-(
페닐메틸
)-1-
피페라지닐
]프로필}-3,7-디
히
드로-1H-
퓨린
-2,6-
디온

DMF (5 ml) 중의 3-[3-부틸-8-클로로-2,6-디옥소-7-(2-프로펜-1-일)-2,3,6,7-테트라히드로-1H-퓨린-1-일]프로필 메탄술포네이트 (0.08 g, 0.19 밀리몰)의 용액을 탄산칼륨 (0.08 g, 0.6 밀리몰) 및 1-벤질피페라진 (0.04 g, 0.23 밀리몰)으로 처리하고 70 ℃에서 2시간 동안 가열하였다. 혼합물을 냉각하고 증발 건조시키고 10 ml의 DCM 및 10 ml의 물 사이에 분배하였다. 유기 상을 증발 건조시키고 잔류물을 무수 THF (5 ml)에 용해시켰다. 반응 혼합물에 반복적으로 진공을 가함으로써 용액을 조심스럽게 탈기시키고 질소 기체로 다시 충진한 다음 테트라키스(트리페닐포스핀)팔라듐(0) (0.010 g, 0.009 밀리몰) 및 모르폴린 (0.200 ml, 2.3 밀리몰)으로 처리하고 혼합물을 질소 대기에서 2시간 동안 교반하였다. 혼합물을 증발시켜 잔류물을 5 ml의 메탄올에 취하고 2 g 아미노프로필 SPE 카트릿지에 첨가한 다음 메탄올로 세척하고 메탄올 중의 아세트산의 3% 용액을 사용하여 생성물을 용출시켰다. 생성물-함유 분획을 모으고 증발 건조시켰다. 이어서 DCM/2% 아세트산으로부터 DCM/20% MeOH/2% 아세트산까지의 구배 용출을 사용하여 생성물을 플래시 크로마토그래피에 의해 정제하고, 최종 생성물을 1,4-디옥산으로부터 동결 건조시켜 백색 고체로서 표제 화합물을 수득하였다 (0.021 g, 24%).
LC/MS: m/z 459 [MH]+, RT 2.37 분
상기 기재된 적절한 일반적 방법에 의하여 하기 화합물들 (표 24)을 제조하였다.








실시예
463: 3-부틸-8-
클로로
-1-{4-[5-(2-
플루오로
-4-
히드록시페닐
)-1,2,4-옥사디아졸-3-일]부틸}-3,7-
디히드로
-1H-
퓨린
-2,6-
디온
a) 3-부틸-8-클로로-1-{4-[5-(2-플루오로-4-히드록시페닐)-1,2,4-옥사디아졸-3-일]부틸}-3,7-디히드로-1H-퓨린-2,6-디온

무수 DMSO (0.5 ml) 중의 CDI (45 mg, 0.28 밀리몰)을 2-플루오로-4-히드록시벤조산 (40 mg, 0.25 밀리몰)에 첨가하고 실온에서 2시간 동안 교반하였다. DMSO (0.4 ml) 중의 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드 (100 mg, 0.28 밀리몰)을 첨가하고 얻어진 혼합물을 90 ℃에서 18시간 동안 가열하였다. MDAP에 의한 정제는 고체로서 표제 화합물을 제공하였다 (38 mg, 28%).

b) 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)-N-히드록시펜탄이미다미드

5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄니트릴 (8.5g, 26 밀리몰)을 EtOH (100 ml)에 용해시켰다. 히드록실아민 (물 중의 50%; 2.6 ml, 39 밀리몰)을 첨가하고 혼합물을 80 ℃에서 48시간 동안 질소 하에 가열하였다. 반응 혼합물을 진공 하에 농축하고 얻어진 고체를 메탄올로 세척하고 건조시켜 고체로서 표제 화합물을 수득하였다 (5.9g, 47%).
LC/MS: m/z 357 [MH]+, RT 2.17 분
c) 5-(3-부틸-8-클로로-2,6-디옥소-2,3,6,7-테트라히드로-1H-퓨린-1-일)펜탄니트릴

5-브로모펜탄니트릴 (4.54ml, 39 밀리몰) 및 탄산세슘 (12.7g)을 DMF (100 ml) 중의 3-부틸-8-클로로-7-(2-프로펜-1-일)-3,7-디히드로-1H-퓨린-2,6-디온 (10g, 35 밀리몰)의 용액에 첨가하고, 혼합물을 40 ℃에서 밤새 질소 하에 교반하고 냉각하였다.
이어서, 반복적으로 진공을 가하고 질소 압력을 가함으로써 혼합물을 탈기시켰다. 이어서, 혼합물을 테트라키스(트리페닐포스핀)팔라듐(0) (2.86g, 2.5 밀리몰) 및 모르폴린 (30.8 ml, 350 밀리몰)로 처리하였다. 혼합물을 질소 대기 하에 3시간 동안 교반한 다음 EtOAc와 2M 염산 사이에 분배하였다. 수성 층을 분리하고 EtOAc (×2)로 추출하였다. 합한 유기 상을 진공 하에 농축하여 고체를 수득하고 이것을 에테르로 세척하고 여과하고 건조시켰다. 여액을 농축하고 MeOH에 이어서 3% AcOH/MeOH로 용출하면서 아미노프로필 컬럼 위에서 정제하였다. 생성물-함유 분획을 합하고 농축하여 고체를 수득하고 이것을 여과된 생성물과 조합하였다. 표제 화합물을 고체로서 수득하였다 (10.5g, 93%).
LC/MS: m/z 324 [MH]+, RT 2.75 분
적절한 카르복실산을 사용하여 실시예 463에 대해서와 유사한 방법에 의해 하기 화합물들 (표 25)을 제조하였다.






상기 출발 물질은 상업적으로 구입될 수 있고/있거나 문헌에서 입수가능한 방법에 따라 만들 수 있다.
이에 한정되지 않지만 본 명세서에 인용된 특허 및 특허 출원을 포함한 모든 공보들은, 각각의 공보가 참고문헌으로서 마치 완전히 기재된 것처럼 구체적 및 개별적으로 표시된 바와 같이, 본 명세서에서 참고문헌으로 포함된다.
Claims (21)
- 하기 화학식 I의 화합물로부터 선택된 하나 이상의 화학 물질 및 제약학적으로 허용가능한 그의 유도체.<화학식 I>
 상기 식에서,R1은 -(알킬렌)m-X-(알킬렌)n-Y를 나타내고;여기에서, m 및 n은 알킬렌 사슬에 있는 탄소 원자의 수를 나타내고;X는 헤테로아릴 및 헤테로시클릴로부터 선택된 기를 나타내고;Y는 아릴, 헤테로아릴 및 O-아릴로부터 선택된 기를 나타내고; 이것은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, 할로겐, -(CH2)qNR5R7, -(CH2)q-(O)p-(CH2)q-N(R5)C(O)OR8, -(CH2)q-N(R5)C(O)R8, -(CH2)q-(O)p-(CH2)q-C(O)NR5R6, -(CH2)q-N(R5)C(O)NR5R6, -(CH2)q-C(O)N((CH2)mOH)R5, -(CH2)q-N(R5)-S(O)2R8, -CH2-S(O)2NR5R6, -C1-6 할로알킬, -OCF3, -OCH(F)2, -OCH2F, -C(O)OR5, -OR5, -R8CN, CN, -SO2R9, -(CH2)n헤테로아릴, -(CH2)n헤테로시클릴, -(CH2)n시클로알킬, -(CH2)n시클로알케닐 및 -(CH2)n아릴로부터 독립적으로 선택된 하나 이상의 기로 임의로 치환될 수 있고;R2는 시클로알킬, C1 -6 할로알킬, 할로겐, -CN 및 -OR4로부터 독립적으로 선택된 하나 이상의 기로 임의로 치환될 수 있는 C1 -6 알킬을 나타내고;R3은 할로겐을 나타내고;R4는 수소, C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)n시클로알킬, -(CH2)n시클로알케닐, -(CH2)n헤테로시클릴, -(CH2)n 아릴 및 -(CH2)n 헤테로아릴로부터 선택된 기를 나타내고;R5 및 R6은 수소 및 C1 -4 알킬로부터 독립적으로 선택되고;R7은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)t시클로알킬, -(CH2)n시클로알케닐, -(CH2)t헤테로시클릴, -(CH2)t 아릴 및 -(CH2)t 헤테로아릴로부터 선택된 기를 나타내고;R8은 C1 -4 알킬을 나타내고;R9은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)n시클로알킬, -(CH2)n시클로알케닐, -(CH2)n헤테로시클릴, -(CH2)n 아릴, -(CH2)n 헤테로아릴 및 CN으로부터 선택된 기를 나타내고;m은 3 및 4로부터 선택된 정수를 나타내고;n은 0 및 1로부터 선택된 정수를 나타내고;p는 0 및 1로부터 선택된 정수를 나타내고;q는 0, 1 및 2로부터 선택된 정수를 나타내고;t는 1 및 2로부터 선택된 정수를 나타낸다.
상기 식에서,R1은 -(알킬렌)m-X-(알킬렌)n-Y를 나타내고;여기에서, m 및 n은 알킬렌 사슬에 있는 탄소 원자의 수를 나타내고;X는 헤테로아릴 및 헤테로시클릴로부터 선택된 기를 나타내고;Y는 아릴, 헤테로아릴 및 O-아릴로부터 선택된 기를 나타내고; 이것은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, 할로겐, -(CH2)qNR5R7, -(CH2)q-(O)p-(CH2)q-N(R5)C(O)OR8, -(CH2)q-N(R5)C(O)R8, -(CH2)q-(O)p-(CH2)q-C(O)NR5R6, -(CH2)q-N(R5)C(O)NR5R6, -(CH2)q-C(O)N((CH2)mOH)R5, -(CH2)q-N(R5)-S(O)2R8, -CH2-S(O)2NR5R6, -C1-6 할로알킬, -OCF3, -OCH(F)2, -OCH2F, -C(O)OR5, -OR5, -R8CN, CN, -SO2R9, -(CH2)n헤테로아릴, -(CH2)n헤테로시클릴, -(CH2)n시클로알킬, -(CH2)n시클로알케닐 및 -(CH2)n아릴로부터 독립적으로 선택된 하나 이상의 기로 임의로 치환될 수 있고;R2는 시클로알킬, C1 -6 할로알킬, 할로겐, -CN 및 -OR4로부터 독립적으로 선택된 하나 이상의 기로 임의로 치환될 수 있는 C1 -6 알킬을 나타내고;R3은 할로겐을 나타내고;R4는 수소, C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)n시클로알킬, -(CH2)n시클로알케닐, -(CH2)n헤테로시클릴, -(CH2)n 아릴 및 -(CH2)n 헤테로아릴로부터 선택된 기를 나타내고;R5 및 R6은 수소 및 C1 -4 알킬로부터 독립적으로 선택되고;R7은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)t시클로알킬, -(CH2)n시클로알케닐, -(CH2)t헤테로시클릴, -(CH2)t 아릴 및 -(CH2)t 헤테로아릴로부터 선택된 기를 나타내고;R8은 C1 -4 알킬을 나타내고;R9은 C1 -6 알킬, C2 -6 알케닐, C2 -6 알키닐, -(CH2)n시클로알킬, -(CH2)n시클로알케닐, -(CH2)n헤테로시클릴, -(CH2)n 아릴, -(CH2)n 헤테로아릴 및 CN으로부터 선택된 기를 나타내고;m은 3 및 4로부터 선택된 정수를 나타내고;n은 0 및 1로부터 선택된 정수를 나타내고;p는 0 및 1로부터 선택된 정수를 나타내고;q는 0, 1 및 2로부터 선택된 정수를 나타내고;t는 1 및 2로부터 선택된 정수를 나타낸다. - 제1항에 있어서, X가 헤테로아릴로부터 선택된 기를 나타내는 것인 하나 이상의 화학 물질.
- 제1항 또는 제2항에 있어서, R2가 C3 -6 알킬로부터 선택되는 것인 하나 이상의 화학 물질.
- 제2항 또는 제3항에 있어서, X가 질소 헤테로원자를 포함한 헤테로아릴로부터 선택된 기를 나타내는 것인 하나 이상의 화학 물질.
- 제4항에 있어서, X가 옥사디아졸릴 또는 테트라졸을 나타내는 것인 하나 이상의 화학 물질.
- 제1항 내지 제5항 중 어느 한 항에 있어서, Y가 아릴 및 헤테로아릴로부터 선택된 기를 나타내는 것인 하나 이상의 화학 물질.
- 제1항 내지 제6항 중 어느 한 항에 있어서, Y가 하나 이상의 할로겐 및 C1 -6 할로알킬로 임의로 치환된 것인 하나 이상의 화학 물질.
- 제1항 내지 제7항 중 어느 한 항에 있어서, R3이 염소를 나타내는 것인 하나 이상의 화학 물질.
- 제1항 내지 제8항 중 어느 한 항에 있어서, Y가 페닐을 나타내고, m이 3이고, n이 1인 하나 이상의 화학 물질.
- 제9항에 있어서, X가 테트라졸릴을 나타내고, R2가 부틸을 나타내고, R3가 염소를 나타내는 것인 하나 이상의 화학 물질.
- 제8항에 있어서, X가 옥사디아졸릴을 나타내고, Y가 피리디닐을 나타내고, R2가 부틸을 나타내고, R3가 염소를 나타내고, m이 4이고, n이 0인 하나 이상의 화학 물질.
- 제1항 내지 제11항 중 어느 한 항에 있어서, 인간 또는 동물용 의약에서 사용하기 위한 하나 이상의 화학 물질.
- 제1항 내지 제11항 중 어느 한 항에 있어서, 지질 대사의 이상, 예컨대 이상지혈증 및 고지방단백혈증 및/또는 염증성 질환 또는 상태의 치료에서 사용하기 위한 하나 이상의 화학 물질.
- 제1항 내지 제11항 중 어느 한 항에 있어서, 당뇨병성 이상지혈증, 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증, 심혈관 질병, 예컨대 죽상경화증, 동맥경화증 및 고트리글리세리드혈증, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진, 비만, 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 말초 혈관 질병 또는 뇌졸중의 치료에서 사용하기 위한 하나 이상의 화학 물질.
- 제1항 내지 제11항 중 어느 한 항에 있어서, 당뇨병성 이상지혈증, 혼합성 이상지혈증, 심부전, 고콜레스테롤혈증, 심혈관 질병, 예컨대 죽상경화증, 동맥경 화증 및 고트리글리세리드혈증, 유형 II 진성 당뇨병, 유형 I 당뇨병, 인슐린 내성, 고지질혈증, 신경성 식욕부진, 비만, 관상 동맥 질병, 혈전증, 협심증, 만성 신부전, 또는 뇌졸중의 치료를 위한 약제의 제조에서 사용하기 위한 하나 이상의 화학 물질.
- 제1항 내지 제7항 중 어느 한 항에 따른 하나 이상의 화학 물질의 유효량을 인간 또는 동물 피험자에게 투여하는 것을 포함하는, HM74A 수용체의 활성화 부족이 상태의 원인이 되거나 수용체의 활성화가 유익하게 될 상태를 가진 인간 또는 동물 피험자의 치료 방법.
- 제16항에 있어서, 인간 또는 동물 피험자가 지질 대사의 이상, 예컨대 이상지혈증 또는 고지방단백혈증, 또는 염증성 질환 또는 상태를 가진 것인 방법.
- 제1항 내지 제11항 중 어느 한 항에 따른 하나 이상의 화학 물질 및 하나 이상의 제약학적으로 허용가능한 희석제, 부형제 또는 담체를 포함하는 제약학적 제제.
- 제1항 내지 제11항 중 어느 한 항에 따른 하나 이상의 화학 물질을 하나 이상의 치료적 활성 약제와 함께 포함하는, 별개의 또는 조합된 제약학적 제제에서 함께 또는 별도로, 연속적으로 또는 동시에 투여하기 위한 조합물.
- (i) 제1항 내지 제11항 중 어느 한 항에 따른 하나 이상의 화학 물질;(ii) 스타틴, 피브레이트, 담즙산 결합 수지 및 니코틴산으로부터 선택된 하나 이상의 치료적 활성 약제; 및(iii) 하나 이상의 제약학적으로 허용가능한 희석제, 부형제 또는 담체를 포함하는 제약학적 제제.
- 적절한 출발 물질, 예를 들어 하기 화학식 II의 화합물(들)로부터(i) N7 보호된 크산틴의 N1에서의 알킬화;(ii) N7 보호된 크산틴의 N3에서의 알킬화;(iii) C8에서의 할로겐화; 및(iv) N7의 탈보호를 임의의 순서로 수행하는 것을 포함하되,단, 탈보호는 알킬화 후에 수행하는 것인,제1항 내지 제11항 중 어느 한 항에 따른 하나 이상의 화학 물질의 제조 방법.<화학식 II>
 상기 식에서, PG는 보호기이다.
상기 식에서, PG는 보호기이다.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0516464A GB0516464D0 (en) | 2005-08-10 | 2005-08-10 | Novel compounds |
| GB0516464.5 | 2005-08-10 | ||
| GB0607736.6 | 2006-04-19 | ||
| GB0607736A GB0607736D0 (en) | 2006-04-19 | 2006-04-19 | Novel compounds |
| GB0614569A GB0614569D0 (en) | 2006-07-21 | 2006-07-21 | Novel compounds |
| GB0614569.2 | 2006-07-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20080034993A true KR20080034993A (ko) | 2008-04-22 |
Family
ID=37189397
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020087005724A KR20080038396A (ko) | 2005-08-10 | 2006-08-08 | 선택적 hm74a 작동제로서의 크산틴 유도체 |
| KR1020087005717A KR20080034993A (ko) | 2005-08-10 | 2006-08-08 | 선택적 hm74a 작동제로서의 크산틴 유도체 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020087005724A KR20080038396A (ko) | 2005-08-10 | 2006-08-08 | 선택적 hm74a 작동제로서의 크산틴 유도체 |
Country Status (29)
| Country | Link |
|---|---|
| US (3) | US20100179128A1 (ko) |
| EP (3) | EP2272848B1 (ko) |
| JP (2) | JP5112316B2 (ko) |
| KR (2) | KR20080038396A (ko) |
| AR (1) | AR055369A1 (ko) |
| AT (1) | ATE487719T1 (ko) |
| AU (2) | AU2006278216A1 (ko) |
| BR (2) | BRPI0615145A2 (ko) |
| CA (2) | CA2618963A1 (ko) |
| CR (2) | CR9748A (ko) |
| CY (1) | CY1111757T1 (ko) |
| DE (1) | DE602006018151D1 (ko) |
| DK (1) | DK1912991T3 (ko) |
| EA (2) | EA200800564A1 (ko) |
| ES (1) | ES2401128T3 (ko) |
| HK (1) | HK1116779A1 (ko) |
| HR (1) | HRP20100725T1 (ko) |
| IL (2) | IL189083A0 (ko) |
| MA (2) | MA29692B1 (ko) |
| MX (2) | MX2008001929A (ko) |
| MY (1) | MY142067A (ko) |
| NO (2) | NO20081211L (ko) |
| NZ (1) | NZ565494A (ko) |
| PE (1) | PE20070405A1 (ko) |
| PL (1) | PL1912991T3 (ko) |
| PT (1) | PT1912991E (ko) |
| SI (1) | SI1912991T1 (ko) |
| TW (1) | TW200800217A (ko) |
| WO (2) | WO2007017262A1 (ko) |
Families Citing this family (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7495005B2 (en) * | 2002-08-22 | 2009-02-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Xanthine derivatives, their preparation and their use in pharmaceutical compositions |
| AU2005212816C1 (en) * | 2004-02-14 | 2010-03-04 | Glaxosmithkline Llc | Novel compounds |
| DE102004030502A1 (de) | 2004-06-24 | 2006-01-12 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue Imidazole und Triazole, deren Herstellung und Verwendung als Arzneimittel |
| DE602006018151D1 (de) * | 2005-08-10 | 2010-12-23 | Glaxosmithkline Llc | Xanthinderivate als selektive hm74a-agonisten |
| MX2009000170A (es) | 2006-06-23 | 2009-01-23 | Incyte Corp | Derivados de purinona como agonistas de hm74a. |
| WO2007150026A2 (en) | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
| AU2007283113A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| EP2025674A1 (de) | 2007-08-15 | 2009-02-18 | sanofi-aventis | Substituierte Tetrahydronaphthaline, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| DE102007054497B3 (de) | 2007-11-13 | 2009-07-23 | Sanofi-Aventis Deutschland Gmbh | Neue kristalline Diphenylazetidinonhydrate und Verfahren zu deren Herstellung |
| UY31968A (es) | 2008-07-09 | 2010-01-29 | Sanofi Aventis | Nuevos derivados heterocíclicos, sus procesos para su preparación, y sus usos terapéuticos |
| BRPI0922134A2 (pt) | 2008-12-08 | 2015-08-18 | Glaxosmithkline Llc | Compostos |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| ES2443016T3 (es) | 2009-08-26 | 2014-02-17 | Sanofi | Nuevos hidratos cristalinos de fluoroglicósidos heteroaromáticos, productos farmacéuticos que comprenden estos compuestos, y su empleo |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| KR101799429B1 (ko) * | 2010-05-03 | 2017-11-21 | 에스케이바이오팜 주식회사 | 신경 세포 사멸 또는 신경 퇴화를 억제하기 위한 약학적 조성물 |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| US8530413B2 (en) | 2010-06-21 | 2013-09-10 | Sanofi | Heterocyclically substituted methoxyphenyl derivatives with an oxo group, processes for preparation thereof and use thereof as medicaments |
| TW201215387A (en) | 2010-07-05 | 2012-04-16 | Sanofi Aventis | Spirocyclically substituted 1,3-propane dioxide derivatives, processes for preparation thereof and use thereof as a medicament |
| TW201221505A (en) | 2010-07-05 | 2012-06-01 | Sanofi Sa | Aryloxyalkylene-substituted hydroxyphenylhexynoic acids, process for preparation thereof and use thereof as a medicament |
| TW201215388A (en) | 2010-07-05 | 2012-04-16 | Sanofi Sa | (2-aryloxyacetylamino)phenylpropionic acid derivatives, processes for preparation thereof and use thereof as medicaments |
| EP2606045B1 (en) * | 2010-08-16 | 2016-01-06 | Boehringer Ingelheim International GmbH | Oxadiazole inhibitors of leukotriene production |
| WO2012040137A1 (en) * | 2010-09-23 | 2012-03-29 | Boehringer Ingelheim International Gmbh | Oxadiazole inhibitors of leukotriene production |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| EP2683705B1 (de) | 2011-03-08 | 2015-04-22 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| EP2683699B1 (de) | 2011-03-08 | 2015-06-24 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| EP2683700B1 (de) | 2011-03-08 | 2015-02-18 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP2760862B1 (en) | 2011-09-27 | 2015-10-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| JP5992096B2 (ja) * | 2012-05-30 | 2016-09-14 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | Pde10阻害剤としてのトリアゾロ化合物 |
| SG11201506479TA (en) * | 2013-03-15 | 2015-09-29 | Hydra Biosciences Inc | Substituted xanthines and methods of use thereof |
| BR112015028173A2 (pt) | 2013-05-10 | 2017-07-25 | Nimbus Apollo Inc | inibidores de acc e usos dos mesmos |
| BR112015028152A2 (pt) | 2013-05-10 | 2017-07-25 | Nimbus Apollo Inc | inibidores de acc e usos dos mesmos |
| WO2014182951A1 (en) | 2013-05-10 | 2014-11-13 | Nimbus Apollo, Inc. | Acc inhibitors and uses thereof |
| BR112015028171A8 (pt) * | 2013-05-10 | 2017-08-22 | Nimbus Apollo Inc | Inibidores de acc e seu uso |
| CN104211702B (zh) * | 2013-05-29 | 2018-08-31 | 中国医学科学院药物研究所 | 取代黄嘌呤类化合物及其制备方法和用途 |
| CN105646492B (zh) * | 2014-11-14 | 2019-04-09 | 中国医学科学院药物研究所 | 含五元芳杂环的取代黄嘌呤类化合物及其制备方法和用途 |
| DE102017000359A1 (de) | 2016-08-26 | 2018-03-01 | Bihl+Wiedemann Gmbh | Diagnose-Repeater für AS-Interface Netze |
| CN110461838B (zh) * | 2017-03-07 | 2022-05-06 | 豪夫迈·罗氏有限公司 | 噁二唑瞬时受体电位通道抑制剂 |
| CN112724141A (zh) * | 2021-01-21 | 2021-04-30 | 南京艾美斐生物医药科技有限公司 | 一种gpr109a蛋白受体抑制剂及其制备和应用 |
| AU2023216305A1 (en) * | 2022-02-03 | 2024-08-08 | D.E. Shaw Research, Llc | N3-substituted uracil compounds as trpa1 inhibitors |
| WO2024163333A1 (en) * | 2023-01-30 | 2024-08-08 | D. E. Shaw Research, Llc | Bicyclic imide compounds as trpa1 inhibitors |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2797220A (en) * | 1954-12-02 | 1957-06-25 | Univ Michigan | Substituted paraxanthines |
| GB8906792D0 (en) | 1989-03-23 | 1989-05-10 | Beecham Wuelfing Gmbh & Co Kg | Treatment and compounds |
| IL100088A (en) | 1990-11-21 | 1995-07-31 | Smithkline Beecham Corp | FNT inhibitor preparations containing histogenic transducts converted at positions 1, 3, and 8 |
| AU3725893A (en) | 1992-02-21 | 1993-09-13 | Smithkline Beecham Corporation | TNF inhibitors |
| JP2753395B2 (ja) | 1992-03-04 | 1998-05-20 | セル・セラピューティックス・インコーポレーテッド | 鏡像異性体ヒドロキシル化キサンチン化合物 |
| US6469017B1 (en) * | 1998-01-16 | 2002-10-22 | Cell Therapeutics, Inc. | Method of inhibiting interleukin-12 signaling |
| US6693105B1 (en) * | 1992-11-16 | 2004-02-17 | Cell Therapeutics, Inc. | Hydroxyl-containing compounds |
| US5804584A (en) * | 1992-11-16 | 1998-09-08 | Cell Therapeutics, Inc. | Therapeutic compounds containing a monocyclic five- to six- membered ring structure having one to two nitrogen atoms |
| US5473070A (en) | 1992-11-16 | 1995-12-05 | Cell Therapeutics, Inc. | Substituted long chain alcohol xanthine compounds |
| IL109161A0 (en) | 1993-03-31 | 1994-06-24 | Cell Therapeutics Inc | Amino alcohol derivatives, methods for the preparation thereof, and pharmaceutical compositions containing the same |
| US5670506A (en) | 1993-04-05 | 1997-09-23 | Cell Therapeutics, Inc. | Halogen, isothiocyanate or azide substituted xanthines |
| WO1994024133A1 (en) | 1993-04-09 | 1994-10-27 | Cell Therapeutics, Inc. | Ring-substituted cell signaling inhibitors |
| AU1868195A (en) | 1994-01-28 | 1995-08-15 | Cell Therapeutics, Inc. | Cell signaling inhibitors |
| US6780865B1 (en) * | 1994-02-18 | 2004-08-24 | Cell Therapeutics, Inc. | Compounds having selective hydrolytic potentials |
| US6878715B1 (en) * | 1994-02-18 | 2005-04-12 | Cell Therapeutics, Inc. | Therapeutic compounds for inhibiting interleukin-12 signals and method for using same |
| JPH09511496A (ja) * | 1994-02-18 | 1997-11-18 | セル・セラピューティックス・インコーポレーテッド | 細胞内メッセンジャー |
| US6103730A (en) * | 1994-03-24 | 2000-08-15 | Cell Therapeutics, Inc. | Amine substituted compounds |
| US5807861A (en) * | 1994-03-24 | 1998-09-15 | Cell Therapeutics, Inc. | Amine substituted xanthinyl compounds |
| US6323201B1 (en) * | 1994-12-29 | 2001-11-27 | The Regents Of The University Of California | Compounds for inhibition of ceramide-mediated signal transduction |
| AR015966A1 (es) | 1997-10-17 | 2001-05-30 | Smithkline Beecham Corp | Uso de un compuesto inhibidor de pde4 para la preparacion de un medicamento util para el tratamiento de prurito |
| ES2254163T3 (es) | 1999-04-09 | 2006-06-16 | Cell Therapeutics, Inc. | Compuestos terapeuticos para inhibir la señalizacion de la interleucina-12 y metodos para el uso de la misma. |
| US20030207901A1 (en) * | 1999-07-27 | 2003-11-06 | Cell Therapeutics, Inc. | Hydroxyl-containing compounds |
| US6821978B2 (en) * | 2000-09-19 | 2004-11-23 | Schering Corporation | Xanthine phosphodiesterase V inhibitors |
| US6586429B2 (en) * | 2000-11-29 | 2003-07-01 | Cell Therapeutics, Inc. | Tricyclic fused xanthine compounds and their uses |
| DK1757606T3 (da) * | 2001-02-24 | 2009-09-07 | Boehringer Ingelheim Pharma | Anvendelse af xanthinderivater som l gemidler samt fremgangsm de til deres fremstilling |
| AU2002242910A1 (en) | 2001-04-11 | 2002-10-28 | Glaxo Group Limited | Medicaments which are modulators of hm74 and/or hm74a activity |
| JP2005502624A (ja) * | 2001-07-03 | 2005-01-27 | ノボ ノルディスク アクティーゼルスカブ | 糖尿病を治療するための、dpp−ivを阻害するプリン誘導体 |
| DE60304911D1 (de) * | 2002-02-25 | 2006-06-08 | Eisai Co Ltd | Xanthin-Derivate als DPP-IV-Inhibitoren |
| US7482337B2 (en) * | 2002-11-08 | 2009-01-27 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Xanthine derivatives, the preparation thereof and their use as pharmaceutical compositions |
| CN1227492C (zh) * | 2003-03-11 | 2005-11-16 | 张跃 | 一种吸收式空调主体 |
| GB0319124D0 (en) | 2003-08-14 | 2003-09-17 | Smithkline Beecham Corp | Chemical compounds |
| AU2005212816C1 (en) * | 2004-02-14 | 2010-03-04 | Glaxosmithkline Llc | Novel compounds |
| WO2006045564A1 (en) | 2004-10-22 | 2006-05-04 | Smithkline Beecham Corporation | Xanthine derivatives with hm74a receptor activity |
| US20090209561A1 (en) * | 2004-10-22 | 2009-08-20 | Richard Jonathan Daniel Hatley | Xanthine Derivatives with HM74A Receptor Activity |
| RU2007118932A (ru) * | 2004-10-22 | 2008-11-27 | Смитклайн Бичам Корпорейшн (US) | Производные ксантина с активностью нм74а |
| JP2008530074A (ja) * | 2005-02-14 | 2008-08-07 | スミスクライン・ビーチャム・コーポレイション | Hm74a受容体アゴニストとしてのアントラニル酸誘導体 |
| GB0516462D0 (en) * | 2005-08-10 | 2005-09-14 | Smithkline Beecham Corp | Novel compounds |
| DE602006018151D1 (de) * | 2005-08-10 | 2010-12-23 | Glaxosmithkline Llc | Xanthinderivate als selektive hm74a-agonisten |
| DE102008009758A1 (de) * | 2008-02-18 | 2009-08-20 | Beiersdorf Ag | Verwendung von 3-(4-Hydroxy-3-methoxyphenyl)-1-(4-hydroxyphenyl)-propan-1-on zur verbesserten Hautkonturierung bzw. gegen Cellulite |
| BRPI0922134A2 (pt) | 2008-12-08 | 2015-08-18 | Glaxosmithkline Llc | Compostos |
-
2006
- 2006-08-08 DE DE602006018151T patent/DE602006018151D1/de active Active
- 2006-08-08 AR ARP060103459A patent/AR055369A1/es unknown
- 2006-08-08 MX MX2008001929A patent/MX2008001929A/es not_active Application Discontinuation
- 2006-08-08 AU AU2006278216A patent/AU2006278216A1/en not_active Abandoned
- 2006-08-08 CA CA002618963A patent/CA2618963A1/en not_active Abandoned
- 2006-08-08 US US12/063,432 patent/US20100179128A1/en not_active Abandoned
- 2006-08-08 SI SI200630916T patent/SI1912991T1/sl unknown
- 2006-08-08 PE PE2006000963A patent/PE20070405A1/es not_active Application Discontinuation
- 2006-08-08 NZ NZ565494A patent/NZ565494A/en not_active IP Right Cessation
- 2006-08-08 EA EA200800564A patent/EA200800564A1/ru unknown
- 2006-08-08 EP EP10172674A patent/EP2272848B1/en active Active
- 2006-08-08 AT AT06763016T patent/ATE487719T1/de active
- 2006-08-08 CA CA002626723A patent/CA2626723A1/en not_active Abandoned
- 2006-08-08 BR BRPI0615145-0A patent/BRPI0615145A2/pt not_active Application Discontinuation
- 2006-08-08 EP EP06763016A patent/EP1912991B1/en active Active
- 2006-08-08 WO PCT/EP2006/007869 patent/WO2007017262A1/en active Application Filing
- 2006-08-08 MY MYPI20063833A patent/MY142067A/en unknown
- 2006-08-08 JP JP2008525474A patent/JP5112316B2/ja not_active Expired - Fee Related
- 2006-08-08 JP JP2008525475A patent/JP2009504592A/ja not_active Withdrawn
- 2006-08-08 KR KR1020087005724A patent/KR20080038396A/ko not_active Application Discontinuation
- 2006-08-08 WO PCT/EP2006/007865 patent/WO2007017261A1/en active Application Filing
- 2006-08-08 DK DK06763016.0T patent/DK1912991T3/da active
- 2006-08-08 AU AU2006278215A patent/AU2006278215A1/en not_active Abandoned
- 2006-08-08 MX MX2008001931A patent/MX2008001931A/es active IP Right Grant
- 2006-08-08 EP EP06776699A patent/EP1912992A1/en not_active Withdrawn
- 2006-08-08 KR KR1020087005717A patent/KR20080034993A/ko not_active Application Discontinuation
- 2006-08-08 EA EA200800555A patent/EA014556B1/ru not_active IP Right Cessation
- 2006-08-08 BR BRPI0614270-2A patent/BRPI0614270A2/pt not_active IP Right Cessation
- 2006-08-08 US US12/063,434 patent/US8143264B2/en not_active Expired - Fee Related
- 2006-08-08 PT PT06763016T patent/PT1912991E/pt unknown
- 2006-08-08 ES ES10172674T patent/ES2401128T3/es active Active
- 2006-08-08 TW TW095128952A patent/TW200800217A/zh unknown
- 2006-08-08 PL PL06763016T patent/PL1912991T3/pl unknown
-
2008
- 2008-01-28 IL IL189083A patent/IL189083A0/en unknown
- 2008-01-28 IL IL189081A patent/IL189081A0/en unknown
- 2008-02-08 MA MA30628A patent/MA29692B1/fr unknown
- 2008-02-08 MA MA30629A patent/MA29693B1/fr unknown
- 2008-02-21 CR CR9748A patent/CR9748A/es not_active Application Discontinuation
- 2008-02-21 CR CR9749A patent/CR9749A/es not_active Application Discontinuation
- 2008-03-07 NO NO20081211A patent/NO20081211L/no not_active Application Discontinuation
- 2008-03-07 NO NO20081212A patent/NO20081212L/no not_active Application Discontinuation
- 2008-07-08 HK HK08107505.5A patent/HK1116779A1/xx not_active IP Right Cessation
-
2010
- 2010-12-27 HR HR20100725T patent/HRP20100725T1/hr unknown
-
2011
- 2011-01-12 CY CY20111100033T patent/CY1111757T1/el unknown
- 2011-04-18 US US13/088,962 patent/US20110257205A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20080034993A (ko) | 선택적 hm74a 작동제로서의 크산틴 유도체 | |
| JP5020848B2 (ja) | Hm74a受容体活性を有する薬剤 | |
| KR20070070231A (ko) | Hm74a 수용체 활성을 가지는 크산틴 유도체 | |
| JP2009504593A (ja) | 選択的hm74aアゴニストとしてのキサンチン誘導体 | |
| JP2018515559A (ja) | ヒドロキシプリン類化合物及びその応用 | |
| CN101282977B (zh) | 作为选择性hm74a激动剂的黄嘌呤衍生物 | |
| US20090209561A1 (en) | Xanthine Derivatives with HM74A Receptor Activity | |
| ES2354723T3 (es) | Derivados de xantina como agonistas selectivos de hm74a. | |
| MXPA06009269A (en) | Novel compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E601 | Decision to refuse application |