KR101480530B1 - 8-아자비시클로[3.2.1]옥탄 화합물의 결정형 - Google Patents
8-아자비시클로[3.2.1]옥탄 화합물의 결정형 Download PDFInfo
- Publication number
- KR101480530B1 KR101480530B1 KR1020097020094A KR20097020094A KR101480530B1 KR 101480530 B1 KR101480530 B1 KR 101480530B1 KR 1020097020094 A KR1020097020094 A KR 1020097020094A KR 20097020094 A KR20097020094 A KR 20097020094A KR 101480530 B1 KR101480530 B1 KR 101480530B1
- Authority
- KR
- South Korea
- Prior art keywords
- oct
- crystalline
- cyclohexylmethyl
- ethyl
- crystalline salt
- Prior art date
Links
Images





Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/46—8-Azabicyclo [3.2.1] octane; Derivatives thereof, e.g. atropine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
본 발명은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염 또는 그의 용매화물인 결정형 염 형태에 관한 것이다. 본 발명은 또한 이러한 결정형 염 형태를 포함하는 약제학적 조성물, 이러한 결정형 염 형태를 사용하여 뮤 오피오이드 수용체 활성과 관련되는 질병을 치료하는 방법, 및 이러한 결정형 염 형태를 제조하는데 유용한 방법을 제공한다.
오피오이드, 결정
Description
본 발명은 뮤(mu) 오피오이드(opioid) 수용체 안타고니스트(antagonist)로 유용한 8-아자비시클로[3.2.1]옥탄 화합물의 결정형 염 형태(crystalline salt form)에 관한 것이다. 본 발명은 또한 상기 결정형 화합물을 포함하는 약제학적 조성물, 뮤 오피오이드 수용체 활성에 의해 매개되는 의학적 상태를 치료하거나 또는 개선하기 위하여 상기 화합물을 사용하는 방법, 및 상기 화합물을 제조하는데 유용한 방법에 관한 것이다.
일반적으로 양수된, 2006년 3월 1일에 출원된 미국 가출원 No. 60/777,962호, 2006년 8월 30일에 출원된 60/841,028호, 및 미국출원 No. 11/711,961호는 뮤 오피오이드 수용체 활성에 의해 매개되는 의학적 상태를 치료하거나 또는 개선하는데 유용하다고 기대되는 뮤 오피오이드 수용체 안타고니스트인 8-아자비시클로[3.2.1]옥탄 화합물을 개시한다. 구체적으로, 화합물 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈 아미드 황산염은 이러한 출원에서 뮤 오피오이드 수용체 안타고니스트 활성을 입증하면서 구체적으로 개시되어 있다.
3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈 아미드(이하에서 화합물 1)의 화학 구조는 하기에서 보여진다:

이 화합물을 치료제로 효과적으로 사용하기 위해서는, 쉽게 제조될 수 있고 허용가능한 화학적 및 물리적 안정성을 갖는 고체 상태의 염을 갖는 것이 바람직할 것이다. 예를 들면, 열에 안정하고 예를 들면 약 175℃ 또는 약 180℃를 초과하는 온도에서 안정하고 흡습성이 없거나 또는 조해성이 없어서, 물질의 처리 및 보관을 용이하게 하도록 하는, 염 형태를 갖는 것은 매우 바람직할 것이다. 결정형 고체는 제조된 제품의 순도와 안정성을 높이기 위해서, 무정형에 비해 일반적으로 바람직하다.
화합물 1의 결정형 염 형태(crystalline salt form)는 이전에 보고되지 않았다.
따라서, 흡습성이 없고 조해성이 없으며, 탁월한 열적 안정성을 나타내는 화합물 1의 안정한 결정형 염 형태(crystalline salt form)에 대한 필요성이 존재한다.
발명의 요약
본 발명은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염(crystalline sulfate salt) 또는 그의 용매화물을 제공한다. 일 구체예에서, 본 발명의 결정형 염 형태는 화합물 1의 결정형 황산염이다. 또 다른 구체예에서, 본 발명의 결정형 염은 화합물 1의 황산염의 결정형 수화물이다.
놀랍게도, 본 발명의 결정형 황산염은 약 190 내지 약 205℃ 범위의 녹는점 미만에서 유의성 있는 과열(thermal event)을 나타내지 않았고 실온에서 약 2% 내지 약 90%의 상대 습도 범위에 노출되었을 때 약 0.3% 미만의 중량 변화를 나타내는 것으로 밝혀졌다. 게다가, 본 발명의 결정형 황산염 또는 그의 수화물 둘 다 실온에서 약 90% 이하의 상대 습도에 노출되었을 때 조해성이 없었다.
본 발명의 결정형 염은 다른 용도 중에서도, 뮤 오피오이드 수용체 활성에 의해 매개되는 의학적 상태를 치료하거나 또는 개선하기 위한 약제학적 조성물을 제조하는데 유용할 것으로 기대된다. 따라서, 본 발명 조성물의 또 다른 구체예에서, 본 발명은 약제학적으로 허용가능한 담체, 및 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염 또는 그의 용매화물을 포함하는 약제학적 조성물을 제공한다.
본 발명은 또한 뮤 오피오이드 수용체 안타고니스트에 의한 치료에 의해 개선되는 질병 또는 상태 예를 들면 위장관의 감소된 운동성으로 인한 질환을 치료하거나 또는 개선하는 방법을 제공하고, 상기 방법은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염 또는 그의 용매화물의 치료적 유효량을 포유 동물에게 투여하는 단계를 포함한다.
본 발명은 오피오이드에 의해 유도된 장 기능 장애(bowel dysfunction) 또는 수술후 장폐색(post-operative ileus)을 치료하는 방법을 제공하고, 상기 방법은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염 또는 그의 용매화물의 치료적 유효량을 포유 동물에게 투여하는 단계를 포함한다.
또 다른 방법 구체예에서, 본 발명은 본 발명의 결정형 황산염의 제조 방법을 제공하고, 상기 방법은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드를 황산과 접촉시켜 반응 혼합물을 생성하고, 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함한다.
본 발명은 본 발명의 결정형 황산염을 제조하기 위한 추가적인 방법을 제공하고, 상기 방법은 메탄올을 포함하는 희석제에서 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 결정형 수화물을 분산시켜 반응 혼합물을 생성하고, 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함한다.
또 다른 방법 구체예에서, 본 발명은 화합물 1의 결정형 황산염의 제조 방법을 제공하고, 상기 방법은 (a) 히드록시기가 보호된 화합물 1의 보호된 전구체와 황산을 접촉시켜, 제1 반응 혼합물을 생성하는 단계; (b) 제1 반응 혼합물로부터 화합물 1의 중간 등급의(intermediate grade) 고체형 황산염을 분리하는 단계; (c) 중간 등급의 고체형 황산염을 메탄올을 포함하는 희석제에서 분산시켜, 제2 반응 혼합물을 생성하는 단계; 및 (d) 제2 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함한다.
관련 조성물 구체예에서, 본 발명은 화합물 1의 상기 보호된 전구체의 제조를 위해 유용한, N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르 중아황산염(bisulfite) 부가물(adduct)을 제공한다.
본 발명은 또한 치료 또는 의약으로 사용하기 위하여 본 명세서에서 기술된 본 발명의 결정형 황산염뿐만 아니라, 의약 제조 특히 포유 동물에서 뮤 오피오이드 수용체 활성과 관련되는 질환을 치료하기 위한 의약의 제조에서 본 발명의 결정형 황산염의 용도를 제공한다.
발명의 상세한 설명
본 발명은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염 또는 그의 용매화물을 제공한다.
정의
본 발명의 화합물, 조성물 및 방법을 기술할 때, 하기의 용어들은 달리 지시되지 않는다면 하기의 의미를 갖는다.
용어 "치료적 유효량"은 치료를 필요로 하는 환자에게 투여될 때 치료를 달성하는데 충분한 함량을 의미한다.
용어 "치료"는 본 명세서에서 사용될 때 포유 동물(특히, 인간)과 같은 환자에서 질병, 질환, 또는 의학적 상태의 치료를 의미하고, (a) 질병, 질환, 또는 의학적 상태가 발생하는 것을 예방하는 것, 즉 환자의 예방적 치료;
(b) 질병, 질환, 또는 의학적 상태를 개선하는 것, 즉 다른 치료제의 효과를 중화시키는 것을 포함하여 환자에서 질병, 질환, 또는 의학적 상태를 제거하거나 또는 이들의 퇴행을 야기시키는 것;
(c) 질병, 질환, 또는 의학적 상태를 억제하는 것, 즉 환자에서 질병, 질환, 또는 의학적 상태의 진전을 늦추거나 억제하는 것; 또는
(d) 환자에서 질병, 질환, 또는 의학적 상태의 증상을 경감시키는 것을 포함한다.
용어 "용매화물"은 용질 즉, 본 발명의 화합물 또는 그의 약제학적으로 허용가능한 염의 하나 이상의 분자, 및 용매 하나 이상의 분자에 의해 형성된 복합체 또는 응집체를 의미한다. 이런 용매화물은 통상적으로는 용질과 용매의 실질적으로 고정된 몰 비율을 갖는 결정형 고체이다. 대표적인 용매는 예를 들면, 물, 메탄올, 에탄올, 이소프로판올, 아세트산, 및 그 등가물을 포함한다. 용매가 물일 때, 형성된 용매화물은 특히 수화물로 명명된다.
용어 "결정형 황산염", 또는 택일적으로는 "결정형 황산염(무수 형태)" 또는 "무수 황산염"이 본 명세서에서 사용될 때, 이것은 결정 격자에서 용매 분자의 실질적으로 고정된 몰 분율을 포함하지 않는 결정형 고체, 즉 용매화물 또는 수화물이 아닌 결정형 고체를 의미한다. 본 발명의 용매화물, 또는 특히 수화물은 명시적으로 확인된다.
단수 형태 "하나", "하나", "하나", 및 "상기"가 본 명세서 및 첨부된 청구범위에서 사용될 때, 이것은 내용이 명백하게 다른 것을 나타내지 않는다면, 복수에 대한 언급을 포함할 수 있다는 것이 주지되어야 한다.
활성제
본 발명에서 활성제, 즉 화합물 1은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드로 나타낸다. 택일적으로는, AutoNom 소프트웨어(MDL Information Systems, GmbH, Frankfurt, 독일)에서 실시되는 바와 같은 IUPAC 협정을 사용한다면, 화합물은 3-((1R,3R,5S)-8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자비시클로[3.2.1]옥트-3-일)벤즈아미드로 나타내어진다. 따라서, 본 명세서에서 사용되는 이름은 명시적으로 표시된 8-아자비시클로[3.2.1]옥탄 기에 대하여 치환된 페닐기의 엔도 배열을 갖는 IUPAC 표시와 일치한다. 또 다른 일반적인 명명법에서, 분자의 "((S)-2,3-디히드록시-프로피오닐)아미노" 부분은 ((S)-2,3-디히드록시-1-옥소프로필)아미노 또는 ((S)-2,3-디히드록시프로판아미도)로 다양하게 표시된다.
본 발명의 염 형태
일 구체예에서, 본 발명은 결정형 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염을 제공한다.
일 구체예에서, 본 발명의 결정형 염은 6.58±0.20, 7.52±0.20, 9.35±0.20, 14.69±0.20, 16.01±0.20, 17.45±0.20, 17.99±0.20, 18.62±0.20, 19.76±0.20, 21.11±0.20, 22.07±0.20, 23.18±0.20, 23.74±0.20, 24.56±0.20, 25.63±0.20, 26.45±0.20, 27.86±0.20, 28.31±0.20, 29.54±0.20, 30.59±0.20, 31.58±0.20, 33.89±0.20, 및 36.02±0.20로부터 선택되는 2θ 값에서 3개 이상 및 4개 이상의 회절 피크를 포함하는, 2개 이상의 회절 피크를 갖는 X-선 분말 회절(x-ray powder diffraction XRPD) 패턴을 특징으로 한다. 구체적으로, 이러한 구체예에서, 결정형은 14.69±0.20, 16.01±0.20, 21.11±0.20, 22.07±0.20, 및23.18±0.20로부터 선택되는 2θ 값에서 3개 이상 및 4개 이상의 회절 피크를 포함하는, 2개 이상의 회절 피크를 갖는 X-선 분말 회절 패턴을 특징으로 한다.
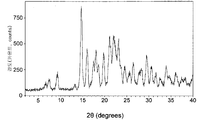
분말 X-선 회절 분야에서 잘 알려져 있는 바와 같이, XRPD 스펙트럼의 피크 위치는 상대적인 피크의 높이 보다는, 시료 제조 및 장치 구조의 상세부와 같은 실험상의 상세부에 비교적 덜 민감하다. 따라서, 일 구체예에서, 화합물 1의 결정형 황산염은 피크 위치가 도 1에서 도시되는 위치와 실질적으로 일치하는 x-선 분말 회절 패턴을 특징으로 한다.
결정형 황산염 구조는 하기의 격자 파라미터를 제공하는, 단일 결정 x-선 회절 분석을 추가적인 특징으로 한다: 단위 격자(unit cell)는 a = 6.8239 Å, b = 16.2275 Å, c = 24.2021 Å, α= β= γ= 90°의 규모를 갖는 사방정계이고; 2680.0 Å3의 격자 부피(cell volume, V); 계산된 밀도는 1.38 g/cm3이고; 공간군(space group)은 P212121(#19)이다. 그 결과 생성되는 분자 구조는 화학 조성물이 황산염 반대 이온(counterion) 대 화합물의 1:1 몰 비율로 된 화합물 1의 황산염의 구조이고 비대칭형 단위 격자는 물 또는 다른 용매 분자를 포함하지 않음을 확인시켜 준다. 유래된 원자 위치로부터 예상되는 X-선 분말 회전 피크는 관찰된 XRPD 패턴과 상당히 일치한다.
또 다른 실시 형태에서, 본 발명의 결정형 황산염은 고온에 노출되었을 때 황산염의 반응(behavior)을 특징으로 한다. 도 2에서 입증되는 바와 같이, 매우 결정형인 시료의 시차 주사 열량계(differential scanning calorimetry DSC) 자취(trace)는 약 190℃ 내지 약 205℃ 범위에서, 용융 전이(melt transition)로 확인되는, 흡열성 열 흐름의 피크를 나타낸다. 열 중량 분석(thermal gravimetric analysis TGA) 자취는 녹는점 미만의 온도에서 유의성 있는 중량 손실을 보여주지 않는다. 열 분해는 대략 녹은 후에 발생한다.
또 다른 실시 형태에서, 결정형 황산염은 약 430, 590, 639, 705, 867, 1036, 1053, 1105, 1171, 1231, 1277, 1375, 1391, 1452, 1476, 1553, 1596, 1639, 1664, 2852, 2907, 2928, 2967, 3168, 및 3357 cm-1에서 유의성 있는 흡수 밴드를 보여주는 적외선 흡수 스펙트럼을 특징으로 한다.
화합물 1의 결정형 황산염은 도 3에서 보여지는 바와 같이, 예외적으로 낮은 수준의 흡습성을 가지면서(즉, 실온에서 2% 상대 습도 내지 90% 상대 습도의 습도 범위에서 약 0.3% 미만의 중량 증가) 가역적인 흡착/탈착 프로파일(sorption/desorption profile)을 갖는 것으로 입증되었다.
추가적으로, 화합물 1의 결정형 황산염은 높아지는 온도와 습도에 노출된 후 안정한 것으로 밝혀졌다. 40℃ 및 75% 상대 습도에서 4주 동안 보관한 후, HPLC에 의한 분석은 화학적 분해를 보여주지 않았고, DSC, TGA, 또는 XRPD 결과에서 검출가능한 변화가 없었다.
또 다른 실시 형태에서, 본 발명은 화합물 1의 황산염의 결정형 수화물을 제공한다.
하나의 실시 형태에서, 본 발명의 황산염의 결정형 수화물은 9.41±0.20, 9.98±0.20, 15.17±0.20, 16.70±0.20, 18.59±0.20, 19.46±0.20, 19.91±0.20, 20.63±0.20, 21.35±0.20, 21.89±0.20, 23.00±0.20, 24.20±0.20, 25.40±0.20, 26.03±0.20, 27.44±0.20, 28.46±0.20, 29.45±0.20, 31.22±0.20, 31.82±0.20, 33.17±0.20, 33.56±0.20 및 36.89±0.20으로부터 선택되는 2θ 값에서 3개 이상 및 4개 이상의 피크를 포함하는, 두 개 이상의 회절 피크를 갖는 X-선 분말 회절(XRPD) 패턴을 특징으로 한다. 구체적으로, 이 실시 형태에서, 결정형은 16.70±0.20, 18.59±0.20, 19.46±0.20, 19.91±0.20, 23.00±0.20, 및 24.20±0.20으로부터 선택되는 2θ 값에서 3개 이상 및 4개 이상의 회절 피크를 포함하는, 2개 이상의 회절 피크를 갖는 X-선 분말 회절 패턴을 특징으로 한다.
또 다른 실시 형태에서, 화합물 1의 황산염의 결정형 수화물은 피크 위치가 도 4에 표시된 것과 실질적으로 일치하는 X-선 분말 회절 패턴을 특징으로 한다.
본 발명의 황산염의 결정형 수화물은 도 5에서 나타나는 바와 같이, 1분 당 10℃의 가열 속도에서 분석되었을 때, 약 125 내지 약 133℃ 범위에서 흡열성 열 흐름의 첫 번째 피크와 약 178 내지 약 183℃ 범위에서 두 번째 피크인 두 개의 흡열 발생(endothermic event)을 나타내는 시차 주사 열량계(DSC) 자취를 특징으로 한다. 열 중량 분석(TGA) 자취는 약 60 내지 약 140℃에서 첫 번째 과열(thermal event)과 약 140 내지 약 190℃에서 두 번째 과열(thermal event)을 보여준다. 첫 번째 과열(thermal event)에서 증발된 물질의 IR과 연결된 TGA에 의한 분석은 화합물 1 염산염 1 몰당 물 약 1 몰을 갖는 수화물 조성물과 일치한다.
놀랍게도, 화합물 1의 황산염의 결정형 수화물은 흡습성이 낮은 것으로 입증되었다. 도 6에서 나타나는 바와 같이, 결정형 수화물은 상대 습도의 전 범위에 걸쳐 약 0.3% 미만의 중량 증가가 있고 약 2% 내지 약 90% 상대 습도의 전 범위 및 실온에서 가역적인 흡착/탈착 프로파일을 나타낸다.
본 발명의 염 형태의 이러한 특징은 하기 실시예에서 추가로 설명된다.
합성 방법
활성제 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드는 하기 실시예에서 기술된 방법을 사용하거나 또는 이 출원의 종래 기술 부분에서 열거된 일반적으로-양수된 미국 출원에서 기술된 방법을 사용하고, 쉽게 입수가능한 출발 물질로부터 제조될 수 있다.
하나의 제조 방법에서, 본 발명의 결정형 황산염은 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드와 황산 약 1 몰의 등가물을 포함하는 약 0.5 내지 약 1.5 몰 등가물을 접촉시킴으로써 제조된다. 일반적으로, 이 반응은 약 60 내지 약 65℃를 포함하는 약 0℃ 내지 약 65℃ 범위의 온도에서 비활성 희석제에서 수행된다. 적절한 비활성 희석제는 예를 들면, 약 10% 물을 포함하는 메탄올과 물의 배합물 뿐만 아니라, 메탄올, 톨루엔, 디클로로메탄, 및 톨루엔과 아세토니트릴, 디클로로메탄과 아세토니트릴, 톨루엔, 아세토니트릴, 및 물과 같은 배합물을 포함한다. 이러한 희석제를 사용하여, 약 50 내지 약 100 mg/mL를 포함하는, 약 5 내지 약 400 mg/mL의 농도를 갖는 반응 혼합물이 제조되고, 선택적으로는 교반을 가하면서, 약 2시간 내지 약 24시간 동안 유지된다. 얻은 혼합물을 유지 기간 동안 약 5 내지 약 20℃까지 냉각될 수 있다.
반응이 완료된 후, 본 발명의 결정형 염은 여과, 농축, 원심 분리, 및 그 등가물과 같은 임의의 통상적인 방법에 의해 반응 혼합물로부터 분리된다.
택일적으로는, 본 발명의 결정형 황산염은 수화물 형태의 재결정화에 의해 제조된다. 결정형 수화물은 상기에서 기술된 바와 같은 비활성 희석제에서 약 5 내지 약 400 mg/ml의 농도에서 분산된다. 메탄올, 또는 약 3:1 내지 약 9:1의 비율로 된 메탄올 : 물 배합물이 이러한 반응을 위해 특히 유용한 희석제이다. 반응 혼합물은 통상적으로는 교반을 가하면서, 약 0 내지 약 65℃ 범위의 온도에서, 약 1시간 내지 약 6시간을 포함하는 약 1시간 내지 약 24시간 동안 유지된다. 통상적으로, 반응 혼합물은 유지 기간 동안 약 65℃부터 약 5 내지 약 20℃까지 냉각된다. 수득율을 높이기 위해서는, 약 5 내지 약 20℃의 온도에서, 약 1시간 내지 약 6 시간을 포함하는 약 1시간 내지 약 24시간의 시간 기간 동안 반응 혼합물을 유지하기 전에, 반응 혼합물 용액의 부피를 약 50%까지 줄일 수 있다. 그 결과 생성된 결정은 통상적으로 회수된다.
화합물 1의 결정형 황산염과 화합물 1의 황산염의 결정형 수화물 둘 다 화합물 1의 보호된 전구체로부터 편리하게 제조된다. 하기 실시예에서 기술되는 바와 같이, 활성제를 제조하기 위해서는, 중아황산염 부가물 3으로부터 재생된 N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르인 보호된 알데히드 2가 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)-벤즈아미드 히드로클로라이드 4와 커플링되어 보호된 전구체 5를 제공하고, 이것은 탈보호되어 3-엔도-{8-[2-(시클로헥실메틸아미노)에틸]-8-아자-비시클로[3.2.1]옥트-3-일}벤즈아미드 6을 제공한다.

중간체 6과 리튬 (4S)-2,2-디메틸-1,3-디옥솔란-4-카르복실레이트 7의 반응은 보호된 중간체 (S)-2,2-디메틸-[1,3]디옥솔란-4-카르복시산 {2-[3-(3-카르바모일-페닐)-8-아자비시클로[3.2.1]옥트-8-일]에틸}시클로헥실메틸-아미드 8을 제공한다.

보호된 중간체 8은 약 20 내지 약 30℃의 온도에서, 에틸 아세테이트 또는 이소프로필 아세테이트와 같은 비활성 희석제에서, 황산 수용액 약 0.8 내지 약 1.3 당량, 통상적으로는 약 1 내지 약 1.2 당량과 접촉된다. 얻은 반응 혼합물과 섞일 수 있고 얻은 생성물이 덜 용해될 수 있는 제2 희석제는 반응 혼합물에 통상적으로 포함된다. 아세토니트릴이 제2 희석제로 유용하다. 얻은 반응 혼합물은 통상적으로는 약 2 내지 약 72시간 동안 교반되어, 화합물 8의 탈보호와 화합물 1의 중간 등급의 고체형 황산염을 생성하고, 이것은 통상적으로는 주로 화합물 1의 황산염의 결정형 수화물이 된다. 중간 등급의 생성물은 통상적으로 예를 들면 여과에 의해 분리될 수 있다.
하기 실시예 2에서 기술된 바와 같이, 중간 등급의 황산염 생성물의 재결정화에 의해서, 예를 들면 중간 등급의 생성물을 아세토니트릴에서 가열과 함께 현탁시키고, 물을 첨가하여 용해를 촉진하고, 실온까지 냉각시키고, 결정화된 수화물 형태를 분리함으로써, 수화물 형태가 얻어질 수 있다.
화합물 1의 결정형 황산염은 상기에서 기술된 탈보호 단계의 중간 등급의 고체형 생성물로부터 얻어질 수 있다. 중간 등급의 생성물은 약 50 내지 약 200 mg/mL을 포함하는 약 5 내지 약 400 mg/mL의 농도에서 메탄올을 포함하는 비활성 희석제에 분산된다. 놀랍게도, 약 0 내지 약 15% 물, 및 약 5 내지 약 15% 물을 포함하는 25% 이하의 물을 갖는, 메탄올과 물의 배합물이 무수 결정형 염을 제조하는데 유용한 희석제인 것으로 결정되었다. 특히, 약 10% 물을 포함하는 메탄올과 물의 배합물은 중간 등급의 생성물을 본 발명에 따른 무수 황산염으로 재결정화하는데 유용하다.
통상의 재결정화 방법에서, 완전한 용해가 얻어질 때까지 반응 혼합물은 가열되고, 예를 들면 반응 혼합물은 약 65℃까지 가열되고, 그런 다음 약 2시간 내지 약 24시간의 기간에 걸쳐 약 5 내지 약 22℃까지 냉각된다. 선택적으로는, 반응 혼합물이 용해 온도 미만에 있을 때에는, 무수 황산염의 종자 결정(seed crystal)이 첨가될 수 있다. 그 결과 얻어지는 결정은 통상적으로, 예를 들면 여과에 의해 회수된다.
또 다른 방법에 따르면, 수화물 형태는 결정형 황산염(무수)으로부터 제조될 수 있다. 통상적으로, 결정형 황산염은 예를 들면 결정형 황산염으로부터 제조된 용액의 동결 건조 또는 급속 용매 증발(rapid evaporation)에 의해, 더 가용성이고 무정형인 형태로 먼저 전환된다. 그런 다음, 무정형인 고체형 황산 물질을 수성 용매 시스템, 예를 들면 25% 물과 75% 아세토니트릴의 수성 용매 시스템에서 분산되고, 선택적으로는 약 0 내지 약 65℃ 범위의 온도에서, 약 12시간보다 많은 또는 약 24시간보다 많은 시간 기간 동안 선택적으로 흔든다. 통상적으로, 온도는 먼저 약 65℃까지 높여지고, 그런 다음 약 5 내지 약 20℃까지 낮추어진다. 그 결과 얻어지는 결정형 수화물 형태는 통상적으로 회수된다.
따라서, 방법 실시 형태에서, 다른 방법들 중에서도, 본 발명은 화합물 1의 결정형 황산염을 제조하는 방법을 제공하고, 상기 방법은 (a) 히드록시기가 보호된 화합물 1의 보호된 전구체를 황산과 접촉시켜, 제1 반응 혼합물을 생성하는 단계; (b) 제1 반응 혼합물로부터 화합물 1의 중간 등급의 고체형 황산염을 분리하는 단계; (c) 중간 등급의 고체형 황산염을 메탄올을 포함하는 희석제에서 분산시켜, 제2 반응 혼합물을 생성하는 단계; 및 (d) 제2 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함한다.
추가적으로, 조성물 실시 형태에서, 본 발명은 화합물 1을 제조하는데 유용한, N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르 중아황산염 부가물 3을 제공한다. 제조예 1에서 기술된 바와 같이, 중아황산염 부가물 3은 소듐 트리아세톡시보로히드라이드를 사용하는 2,2-디에톡시에틸아민에 의한 시클로헥산카르복스알데히드의 환원성 아민화(reductive amination), 그 다음에 아미노-보호기의 첨가, 알데히드 작용기의 탈보호, 및 아황산나트륨과의 반응에 의한 중아황산염으로 전환에 의해 제조될 수 있다. 택일적으로는, 최초의 환원성 아민화는 촉매에 의한 수소화반응에 의해 수행될 수 있다. 적절한 수소화반응 촉매는 팔라듐, 백금, 및 라니 니켈(Raney nickel) 촉매를 포함하지만, 이들에 제한되는 것은 아니다.
약제학적 조성물
본 발명의 결정형 황산염 형태는 통상적으로는 약제학적 조성물 또는 제제의 형태로 환자에게 투여된다. 이러한 약제학적 조성물은 제한되지는 않지만 경구, 직장, 질내, 코, 흡입, 국소(경피를 포함) 및 비경구 투여 방법을 포함하는 임의의 허용가능한 투여 경로에 의해 환자에게 투여될 수 있다.
따라서, 본 발명 조성물 실시 형태 중 하나에서, 본 발명은 약제학적으로 허용가능한 담체 또는 부형제와, 화합물 1의 결정형 황산염 또는 그의 용매화물의 치료적 유효량을 포함하는 약제학적 조성물에 관한 것이다. 선택적으로는, 이러한 약제학적 조성물은 필요하다면, 다른 치료제 및/또는 제제화제(formulating agent)를 포함할 수 있다. 조성물을 논의할 때, 용어 "본 발명의 염"은 화합물 1의 결정형 황산염뿐만 아니라 그의 용매화물 특히 수화물을 포함한다고 이해된다.
본 발명의 약제학적 조성물은 통상적으로는 본 발명의 염 형태로 존재하는, 활성제의 치료적 유효량을 포함한다. 통상적으로, 이러한 약제학적 조성물은 활성제 약 0.1 내지 약 95 중량%; 바람직하게는, 활성제 약 5 내지 약 70 중량%; 및 더 바람직하게는 활성제 약 10 내지 약 60 중량%를 포함할 것이다.
임의의 통상적인 담체 또는 부형제가 본 발명의 약제학적 조성물에 사용될 수 있다. 특정 담체 또는 부형제, 또는 담체 또는 부형제의 배합에 대한 선택은 특정 환자를 치료하는데 사용되는 투여 경로 또는 의학적 상태 또는 질병 상태의 형태에 의존할 것이다. 이와 관련하여, 특정 투여 경로를 위한 적절한 약제학적 조성물의 제조는 약제학 분야에서 당업자들의 범위 내에 있다. 추가적으로, 본 발명의 약제학적 조성물에 사용되는 담체 또는 부형제는 상업적으로 입수가능하다. 추가적인 예로서, 통상의 제제화 방법은 Remington: The Science and Practice of Pharmacy, 제20판, Lippincott Williams & White, Baltimore, Maryland (2000); 및 H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 제7판, Lippincott Williams & White, Baltimore, Maryland (1999)에서 기술되어 있다.
약제학적으로 허용가능한 담체로 도움이 될 수 있는 물질의 대표적인 예는 하기의 물질을 포함하지만, 이들에 제한되는 것은 아니다: 락토오스, 글루코스 및 수크로스와 같은 당류; 옥수수 전분 및 감자 전분과 같은 전분; 미세결정형 셀룰로오스와 같은 셀룰로오스, 및 소듐 카르복시메틸 셀룰로오스, 에틸 셀룰로오스 및 셀룰로오스 아세테이트와 같은 셀룰로오스 유도체; 분말형 트래거컨스; 맥아; 젤라틴; 탈크; 코코아 버터 및 좌제용 왁스와 같은 부형제; 땅콩유, 면실유, 홍화씨유, 참기름, 올리브유, 옥수수유 및 콩기름과 같은 오일; 프로필렌 글리콜과 같은 글리콜; 글리세린, 소르비톨, 만니톨 및 폴리에틸렌 글리콜과 같은 폴리올; 에틸 올레이트 및 에틸 라우레이트와 같은 에스테르; 아가; 마그네슘 수산화물과 알루미늄 수산화물과 같은 완충제; 알긴산; 발열 물질이 없는 물(pyrogen-free water); 등상성 식염수; 링거 용액; 에틸 알코올; 포스페이트 완충 용액; 및 약제학적 조성물에서 사용되는 다른 무독성인 양립가능한 물질.
약제학적 조성물은 통상적으로는 활성제를 약제학적으로 허용가능한 담체와 하나 이상의 선택적인 성분을 완전히 및 직접적으로 혼합하거나 또는 블렌딩(blending)함으로써 제조된다. 그 결과 얻은 균일하게 혼합된 혼합물은 통상의 방법과 장치를 사용하여, 정제, 캡슐, 환약 및 그 등가물로 형상화되거나 또는 충진될 수 있다.
본 발명의 약제학적 조성물은 바람직하게는 단위 투여 제형(unit dosage form)으로 포장된다. 용어 "단위 투여 제형"은 환자에게 투여하는데 적절한 물리적으로 분리된 단위, 즉 단독으로 또는 하나 이상의 추가적인 단위와 배합되어 목표로 하는 치료 효과를 만들어내도록 계산된 미리 정해진 활성제의 함량을 포함하는 각각의 단위를 나타낸다. 예를 들면, 그러한 단위 투여 제형은 캡슐, 정제, 환약 및 그 등가물, 또는 비경구 투여를 위해 적절한 단위 포장형(unit package)이 될 수 있다.
하나의 구체예에서, 본 발명의 약제학적 조성물은 경구 투여에 적절하다. 경구 투여에 적절한 약제학적 조성물은 캡슐, 정제, 환약, 로젠지(lozenge), 샤셋(cachet), 당의정(dragee), 분말, 과립의 형태; 수성 또는 비-수성 액체 중 용액 또는 현탁액; 또는 수중유형(oil-in-water) 또는 유중수형(water-in-oil) 액체 에멀젼; 또는 엘릭시르(elixir) 또는 시럽; 및 그 등가물이 될 수 있고; 각각은 본 발명 화합물의 미리 정해진 함량을 활성 성분으로 포함한다.
고체 투여 제형(예를 들면, 캡슐, 정제, 환약 및 그 등가물)으로 경구 투여를 목적으로 할 때, 본 발명의 약제학적 조성물은 통상적으로는 활성제와, 소듐 시트레이트 또는 디칼슘 포스페이트와 같은 하나 이상의 약제학적으로 허용가능한 담체를 포함할 것이다. 선택적으로 또는 택일적으로, 이러한 고체 투여 제형은 또한 전분, 미세결정형 셀룰로오스, 락토오스, 수크로스, 글루코스, 만니톨, 및/또는 규산과 같은 충진제 또는 증량제(extender); 카르복시메틸셀룰로오스, 알기네이트, 젤라틴, 폴리비닐피롤리돈, 수크로스 및/또는 아카시아와 같은 결합제; 글리세롤과 같은 습윤제(humectant); 아가-아가, 칼슘 카보네이트, 감자 또는 타피오카 전분, 알긴산, 임의의 실리케이트, 및/또는 소듐 카보네이트와 같은 붕해제(disintegrating agent); 파라핀과 같은 용해 지연제; 4차 암모늄 화합물과 같은 흡수 촉진제; 세틸 알코올 및/또는 글리세롤 모노스테아레이트와 같은 보습제(wetting agent); 카올린 및/또는 벤토나이트 클레이와 같은 흡수제; 탈크, 칼슘 스테아레이트, 마그네슘 스테아레이트, 고체형 폴리에틸렌 글리콜, 소듐 라우릴 술페이트, 및/또는 이들의 혼합물과 같은 윤활제; 착색제; 및 완충제를 포함할 수 있다.
방출제, 보습제, 코팅제, 감미제, 향미제 및 향료, 방부제와 항산화제가 또한 본 발명 약제학적 조성물에 존재할 수 있다. 약제학적으로 허용가능한 항산화제의 예는 아스코르브산, 시스테인 히드로클로라이드, 소듐 비술페이트, 소듐 메타비술페이트, 소듐 술파이트 및 그 등가물과 같은 수-가용성 항산화제; 아스코르빌 팔미테이트, 부틸화된 히드록시아니솔, 부틸화된 히드록시톨루엔, 레시친, 프로필 갈레이트, 알파-토코페롤, 및 그 등가물과 같은 오일-가용성 항산화제; 및 시트르산, 에틸렌디아민 테트라아세트산, 소르비톨, 타르타르산, 인산, 및 그 등가물과 같은 금속-킬레이팅제를 포함한다. 정제, 캡슐, 환약 및 그 등가물을 위한 코팅제는 셀룰로오스 아세테이트 프탈레이트, 폴리비닐 아세테이트 프탈레이트, 히드록시프로필 메틸셀룰로오스 프탈레이트, 메타크릴산-메타크릴산 에스테르 공중합체, 셀룰로오스 아세테이트 트리멜러테이트(trimellitate), 카르복시메틸 에틸 셀룰로오스, 히드록시프로필 메틸 셀룰로오스 아세테이트 숙시네이트, 및 그 등가물과 같은 장용 코팅(enteric coating)을 위해 사용되는 코팅제를 포함한다.
본 발명의 약제학적 조성물은 예를 들면 다양한 비율의 히드록시프로필 메틸 셀룰로오스; 또는 다른 중합체 매트릭스, 리포좀 및/또는 미소 구체(microsphere)를 사용함으로써, 활성제의 서 방출 또는 제어 방출을 제공하도록 제제화될 수 있다. 추가적으로, 본 발명의 약제학적 조성물은 불투명화제를 선택적으로 포함할 수 있고, 위장관의 일부 부분에서만, 또는 우선적으로, 선택적으로는 지연된 방식으로 활성제를 방출하도록 제제화될 수 있다. 사용될 수 있는 임베딩 조성물(embedding composition)의 예는 중합체 물질과 왁스를 포함한다. 활성제는 또한 필요하다면, 하나 이상의 상기 기술된 부형제와 함께 마이크로캡슐화될 수 있다.
경구 투여를 위한 적절한 액체 투여 제형은 예를 들면, 약제학적으로 허용가능한 에멀젼, 마이크로에멀젼, 용액, 현탁액, 시럽 및 엘릭시르를 포함한다. 액체 투여 제형은 통상적으로는 활성제와, 예를 들면, 물 또는 다른 용매와 같은 비활성 희석제, 에틸 알코올, 이소프로필 알코올, 에틸 카보네이트, 에틸 아세테이트, 벤질 알코올, 벤질 벤조에이트, 프로필렌 글리콜, 1,3-부틸렌 글리콜, 오일(예를 들면, 면실유, 땅콩유, 옥수수유, 발아유, 올리브유, 피마자유 및 참기름), 글리세롤, 테트라히드로푸릴 알코올, 폴리에틸렌 글리콜 및 소르비탄의 지방산 에스테르와 같은 가용화제 및 유화제, 및 이들의 혼합물과 같은 비활성 희석제를 포함한다. 활성 성분에 추가적으로, 현탁액은 예를 들면, 에톡시화된 이소스테아릴 알코올, 폴리옥시에틸렌 소르비톨 및 소르비탄 에스테르, 미세결정형 셀룰로오스, 알루미늄 메타히드록시드, 벤토나이트, 아가-아가 및 트래거컨스와 같은 현탁제, 및 이들의 혼합물을 포함할 수 있다.
본 발명의 염은 또한 비경구로(예를 들면, 정맥내, 피하, 근육내, 또는 복강내 주입) 투여될 수 있다. 비경구 투여를 위해서, 활성제는 예를 들면, 멸균된 수용액, 식염수, 프로필렌 글리콜과 같은 저분자량 알코올, 폴리에틸렌 글리콜, 식물성 오일, 젤라틴, 에틸 올레이트와 같은 지방산 에스테르, 및 그 등가물을 포함하는 비경구 투여를 위한 적절한 매체(vehicle)와 혼합된다. 비경구 제제는 또한 하나 이상의 항산화제, 가용화제, 안정화제, 방부제, 보습제, 유화제, 또는 분산제를 포함할 수 있다. 이러한 제제는 멸균한 주입가능한 매질, 멸균제, 여과, 조사, 또는 열을 사용하여 멸균될 수 있다.
택일적으로는, 본 발명의 약제학적 조성물은 흡입 투여를 위해 제제화된다. 흡입 투여를 위한 적절한 약제학적 조성물은 통상적으로 에어로졸 또는 분말의 제형으로 될 것이다. 이러한 조성물은 정량 투여 흡입기(metered-dose inhaler), 건조 분말 흡입기, 네뷸라이저 또는 유사한 전달 장치와 같은 잘 알려진 전달 장치를 사용하여 일반적으로 투여된다.
본 발명의 약제학적 조성물이 가압된 용기를 사용하여 흡입에 의해 투여될 때, 본 발명의 조성물은 통상적으로는 활성 성분과, 디클로로디플루오로메탄, 트리클로로플루오로메탄, 디클로로테트라플루오로에탄, 이산화탄소 또는 다른 적절한 가스와 같은 적절한 추진체를 포함할 것이다. 추가적으로, 약제학적 조성물은 본 발명의 화합물과 분말 흡입기에서 사용하는데 적절한 분말을 포함하는 캡슐 또는 카트리지(예를 들면, 젤라틴으로 만들어짐)의 형태로 될 수 있다. 적절한 분말 기제는 예를 들면 락토오스 또는 전분을 포함한다.
본 발명의 염은 또한 공지된 경피 전달 시스템과 부형제를 사용하여 경피로 투여될 수 있다. 예를 들면, 활성제는 프로필렌 글리콜, 폴리에틸렌 글리콜 모노라우레이트, 아자시클로알칸-2-온 및 그 등가물과 같은 투과 증진제와 혼합되어, 패치 또는 유사한 전달 시스템 안에 통합(incorporate)될 수 있다. 필요하다면, 겔화제, 유화제 및 완충제를 포함하는 추가적인 부형제가 이러한 경피용 조성물에 사용될 수 있다.
필요하다면, 본 발명의 염은 하나 이상의 다른 치료제와 배합되어 투여될 수 있다. 이러한 실시 형태에서, 본 발명의 염은 다른 치료제와 물리적으로 혼합되어 두 개의 제제를 포함하는 하나의 조성물을 생성하거나; 또는 각각의 제제가 분리되고 별개인 조성물에 존재하여 환자에게 동시에 또는 연속적으로 투여된다.
예를 들면, 본 발명의 염은 통상의 방법과 장비를 사용하여 제2 치료제와 배합되어, 화합물 1과 제2 치료제를 포함하는 조성물을 생성할 수 있다. 추가적으로, 치료제는 약제학적으로 허용가능한 담체와 배합되어, 본 발명의 염, 제2 치료제 및 약제학적으로 허용가능한 담체를 포함하는 약제학적 조성물을 생성할 수 있다. 이 구체예에서, 조성물의 구성 성분은 통상적으로는 혼합되거나 블렌딩되어 물리적 혼합물을 만들어낸다. 그런 다음, 물리적 혼합물은 본 명세서에서 기술된 임의의 경로를 사용하여 치료적 유효량으로 투여된다. 택일적으로는, 치료제는 환자에게 투여 전에 분리되고 별개로 존재할 수 있다. 이 구체예에서, 활성제는 투여 전에는 물리적으로 함께 혼합되어 있지는 않지만, 별개의 조성물로서 동시에 또는 별개의 시간에 투여된다. 이러한 조성물은 별개로 포장되거나 또는 키트로서 함께 포장될 수 있다. 키트에 있는 두 개의 치료제는 동일한 투여 경로에 의해 또는 서로 다른 투여 경로에 의해 투여될 수 있다.
본 발명의 활성제와 양립가능한 임의의 치료제가 제2 치료제로 사용될 수 있다. 특히, 뮤 오피오이드 수용체 길항작용을 제외한 기작을 통하여 작용하는 위장운동촉진제(prokinetic agent)가 본 발명의 화합물과 배합되어 사용될 수 있다. 예를 들면, 테가세로드(tegaserod), 렌자프리드(renzapride), 모사프리드(mosapride), 프루칼로프리드(prucalopride), 1-이소프로필-1H-인다졸-3-카르복시산{(1S,3R,5R)-8-[2-(4-아세틸피페라진-1-일)에틸]-8-아자비시클로[3.2.1]옥트-3-일}아미드, 1-이소프로필-2-옥소-1,2-디히드로퀴놀린-3-카르복시산 {(1S,3R,5R)-8-[(R)-2-히드록시-3-(메탄술포닐-메틸-아미노)프로필]-8-아자비시클로[3.2.1]옥트-3-일}아미드, 또는 4-(4-{[(2-이소프로필-1H-벤조이미다졸-4-카르보닐)아미노]메틸}-피페리딘-1-일메틸)피페리딘-1-카르복시산 메틸 에스테르와 같은 5-HT4 수용체 아고니스트(agonist)가 제2 치료제로 사용될 수 있다.
추가적인 유용한 위장운동촉진제는 5-HT3 수용체 아고니스트(예를 들면, 푸모세트랙(pumosetrag)), 5-HT1A 수용체 안타고니스트(antagonist)(예를 들면, AGI 001), 알파-2-델타 리간드(예를 들면, PD-217014), 염소 채널 개방자(chloride channel opener)(예를 들면, 루비프로스톤(lubiprostone)), 도파민 안타고니스트(예를 들면, 이토프리드(itopride), 메타클로프라미드(metaclopramide), 돔페리돈(domperidone)), GABA-B 아고니스트(예를 들면, 바클로펜(baclofen), AGI 006), 카파(kappa) 오피오이드 아고니스트(예를 들면, 아시마돌린(asimadoline)), 무스카린 M1 및 M2 안타고니스트(예를 들면, 아코티아미드(acotiamide)), 모틸린(motilin) 아고니스트(예를 들면, 미템시날(mitemcinal)), 구아닐레이트 시클라제 활성화제(예를 들면, MD-1100 및 그렐린(ghrelin) 아고니스트(예를 들면, Tzp 101, RC 1139)를 포함하지만, 이들에 제한되는 것은 아니다.
추가적으로, 본 발명의 염은 오피오이드 치료제와 배합될 수 있다. 이러한 오피오이드 제는 모르핀(morphine), 페시딘(pethidine), 코데인(codeine), 디히드로코데인(dihydrocodeine), 옥시콘틴(oxycontin), 옥시코돈(oxycodone), 히드로코돈(hydrocodone), 수펜타닐(sufentanil), 펜타닐(fentanyl), 레미펜타닐(remifentanil), 부프레노르핀(buprenorphine), 메싸돈(methadone), 및 헤로인을 포함하지만, 이들에 제한되는 것은 아니다.
이러한 치료제의 수 개의 추가적인 예들은 당해 분야에서 알려져 있고, 이러한 공지된 치료제는 본 발명의 화합물과 배합되어 사용될 수 있다. 제2 제제(들)가 포함될 때, 제2 제제는 치료적 유효량으로 존재하고, 즉 본 발명의 화합물과 동시 투여될 때 치료적 유효 효과를 생성하는 임의의 함량으로 존재한다. 본 발명의 화합물과 배합되어 투여되는 다른 치료제를 위한 적절한 투여량은 통상적으로는 약 0.05 ㎍/일 내지 약 100 mg/일 범위가 될 것이다.
따라서, 본 발명의 약제학적 조성물은 하기에서 기술된 바와 같은 제2 치료제를 선택적으로 포함한다.
하기의 실시예는 본 발명의 대표적인 약제학적 조성물을 설명한다.
제제 실시예 A: 경구 투여를 위한 경질 젤라틴 캡슐
본 발명의 염(50 g), 분무-건조된 락토오스(200 g), 및 마그네슘 스테아레이트(10 g)를 완전히 혼합한다. 그 결과 얻은 조성물을 경질 젤라텐 캡슐 안에 넣는다(캡슐 1개당 조성물 260 mg).
제제 실시예 B: 경구 투여를 위한 경질 젤라틴 캡슐
본 발명의 염(20 mg), 전분(89 mg), 미세결정형 셀룰로오스(89 mg), 및 마그네슘 스테아레이트(2 mg)를 완전히 혼합하고, 그런 다음 No. 45 메쉬 U.S. 체를 통과시킨다. 그 결과 얻은 조성물을 경질 젤라텐 캡슐에 넣는다(캡슐 1개당 조성물 200 mg).
제제 실시예 C: 경구 투여를 위한 젤라틴 캡슐
본 발명의 염(10 mg), 폴리옥시에틸렌 소르비탄 모노올레이트(50 mg), 및 전분 분말(250 mg)을 완전히 혼합하고, 그런 다음 젤라틴 캡슐에 넣는다(캡슐 1개당 조성물 310 mg).
제제 실시예 D: 경구 투여를 위한 정제
본 발명의 염(5 mg), 전분(50 mg), 및 미세결정형 셀룰로오스(35 mg)를 No. 45 메쉬 U.S. 체를 통과시키고, 완전히 혼합한다. 그 결과 얻은 분말을 폴리비닐피롤리돈 용액(물에서 10 wt%, 4 mg)과 혼합하고, 얻은 혼합물을 No. 14 메쉬 U.S. 체를 통과시킨다. 이렇게 얻은 과립을 50-60℃에서 건조시키고 No. 18 메쉬 U.S. 체를 통과시킨다. 얻은 과립에, No. 60 메쉬 U.S. 체를 미리 통과시킨 소듐 카르복시메틸 전분(4.5 mg), 마그네슘 스테아레이트(0.5 mg), 및 탈크(1 mg)를 첨가한다. 혼합한 후에, 얻은 혼합물을 타정기에서 압착하여, 100 mg 무게의 정제를 얻는다.
제제 실시예 E: 경구 투여를 위한 정제
본 발명의 염(25 mg), 미세결정형 셀룰로오스(400 mg), 흄드 이산화규소(fumed silicon dioxide)(10 mg), 및 스테아르산(5 mg)을 완전히 혼합하고, 그런 다음 압착하여, 정제(정제 1개당 조성물 440 mg)를 형성한다.
제제 실시예 F: 경구 투여를 위한 단일-층의 정제(single-scored Tablet)
본 발명의 염(15 mg), 옥수수 전분(50 mg), 크로스카르멜로스 소듐(25 mg), 락토오스(120 mg), 및 마그네슘 스테아레이트(5 mg)를 완전히 혼합하고, 그런 다음 압착하여, 단일-층의 정제(정제 1개당 조성물 215 mg)를 형성한다.
제제 실시예 G: 경구 투여를 위한 현탁액
하기의 성분들을 완전히 혼합하여 현탁액 10 mL 당 활성 성분 100 mg을 포함하는 경구 투여를 위한 현탁액을 형성한다.
| 성분 | 함량 |
| 본 발명의 염 | 0.1 g |
| 푸마르산 | 0.5 g |
| 염화 나트륨 | 2.0 g |
| 메틸 파라벤 | 0.15 g |
| 프로필 파라벤 | 0.05 g |
| 과립화된 당 | 25.5 g |
| 소르비톨(70% 용액) | 12.85 g |
| Veegum k(Vanderbilt Co.) | 1.0 g |
| 향미제 | 0.035 ml |
| 착색제 | 0.5 mg |
| 증류수 | 100 ml까지 채움 |
제제 실시예 H: 건조 분말 조성물
본 발명의 미분화된 염(1 mg)을 락토오스(25 mg)와 혼합하고, 그런 다음 젤라틴 흡입 카트리지에 넣는다. 카트리지의 내용물은 분말 흡입기를 사용하여 투여된다.
제제 실시예 J: 주사가능한 제제
본 발명의 염(0.1 g)을 0.1 M 소듐 시트레이트 완충 용액(15 mL)과 혼합한다. 그 결과 용액의 pH를 1 N 염산 수용액 또는 1 N 수산화나트륨 수용액을 사용하여 pH 6으로 맞춘다. 시트레이트 완충액 중 멸균된 보통의 식염수를 첨가하여 전체 부피로 20 mL를 제공한다.
특정한 투여 경로에 적절한 본 발명 염의 임의의 형태(즉, 결정형 염 또는 용매화물)는 상기 논의된 약제학적 조성물에 사용될 수 있음은 이해될 것이다.
용도
본 발명의 활성제인 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염은 뮤 오피오이드 수용체에서 안타고니스트이므로, 본 발명의 염은 뮤 오피오이드 수용체에 의해 매개되거나 또는 뮤 오피오이드 수용체 활성과 관련되는 의학적 상태 즉 뮤 오피오이드 수용체 안타고니스트에 의한 치료에 의해 개선되는 의학적 상태를 치료하는데 유용할 것으로 기대된다. 구체적으로, 본 발명의 염은 오피오이드 진통제의 사용과 관련되는 부작용, 즉 총체적으로는 오피오이드에 의해 유도된 장 기능장애로 칭해지는, 변비, 감소된 위 배출증(decreased gastric emptying), 복통(abdominal pain), 고창증(bloating), 구역질, 및 위 식도 역류증(gastroesophageal reflux)을 치료하는데 유용할 것으로 기대된다. 본 발명의 염 형태는 또한 복부 또는 다른 수술 이후에 발생하는 위장관의 감소된 운동성으로 인한 질환인 수술후 장폐색을 치료하는데 유용할 것으로 기대된다. 추가적으로, 화합물 1과 같은 뮤 오피오이드 수용체 안타고니스트 화합물은 오피오이드-유도된 구역질과 구토를 없앨 수 있다고 제안되고 있다.
화합물 1은 동물 모델에서 위장(gastrointestinal GI) 관의 운동성을 증가시키는 것으로 나타났으므로, 본 발명의 염은 인간을 포함하는 포유 동물에서 감소된 운동성에 의해 야기되는 GI 관의 질환을 치료하는데 유용할 것으로 기대된다. 이러한 GI 운동성 질환은 예를 들면, 만성 변비, 변비-우세형 과민성 대장 증후군(constipation-predominant irritable bowel syndrome C-IBS), 당뇨병성 및 특발성 위마비(diabetic and idiopathic gastroparesis), 및 기능성 소화 불량을 포함한다.
그러므로, 하나의 실시 형태에서, 본 발명은 포유 동물에서 위장관의 운동성을 증가시키는 방법을 제공하고, 상기 방법은 약제학적으로 허용가능한 담체와 본 발명의 염을 포함하는 약제학적 조성물의 치료적 유효량을 포유 동물에게 투여하는 단계를 포함한다.
본 발명의 염이 GI 관의 감소된 운동성으로 인한 질환 또는 뮤 오피오이드 수용체에 의해 매개되는 다른 상태를 치료하는데 사용될 때, 본 발명의 염은 다른 투여 제형이 사용될 수 있지만, 통상적으로는 하루에 1회 투여 또는 하루에 복수 회 투여로 경구로 투여될 것이다. 예를 들면, 특히 수술후 장폐색을 치료하는데 사용될 때, 본 발명의 화합물은 비경구로 투여될 수 있다. 1회 투여 당 투여되는 활성 성분의 함량 또는 하루에 투여되는 전체 함량은 통상적으로는 치료될 상태, 선택된 투여 경로, 투여되는 실제 화합물 및 그의 상대적 활성, 개개 환자의 나이, 체중 및 반응, 환자 증상의 심각성, 및 그 등가물을 포함하는 관련 상황의 관점에서, 의사에 의해 결정될 것이다.
GI 관의 감소된 운동성으로 인한 질환 또는 뮤 오피오이드 수용체에 의해 매개되는 다른 질환을 치료하기 위한 적절한 투여량은 활성제 약 0.0007 내지 약 1.4 mg/kg/일을 포함하는 활성제 약 0.0007 내지 약 20 mg/kg/일의 범위가 될 것이다. 평균 70 kg인 사람의 경우, 이것은 하루에 활성제 약 0.05 내지 약 100 mg의 함량이 될 것이다.
본 발명의 하나의 실시 형태에서, 본 발명의 화합물은 뮤 오피오이드에 의해 유도된 장 기능 장애를 치료하는데 사용된다. 본 발명의 화합물이 오피오이드에 의해 유도된 장 기능 장애를 치료하는데 사용될 때, 본 발명의 화합물은 통상적으로는 하루에 1회 투여 또는 하루에 복수회 투여로 경구로 투여될 것이다. 바람직하게는 오피오이드에 의해 유도된 장 기능 장애를 치료하기 위한 투여량은 하루에 약 0.05 내지 약 100 mg 범위에 있을 것이다.
본 발명의 또 다른 실시 형태에서, 본 발명의 화합물은 수술후 장폐색을 치료하는데 사용된다. 본 발명의 화합물이 수술후 장폐색을 치료하는데 사용될 때, 본 발명의 화합물은 통상적으로는 하루에 1회 투여 또는 하루에 복수 회 투여로 경구로 또는 정맥 내로 투여될 것이다. 바람직하게는 수술후 장폐색을 치료하기 위한 투여량은 하루에 약 0.05 내지 약 100 mg 범위에 있을 것이다.
본 발명은 또한 뮤 오피오이드 수용체 활성과 관련되는 질병 또는 상태를 갖는 포유 동물을 치료하는 방법을 제공하고, 상기 방법은 본 발명의 화합물 또는 본 발명의 화합물을 포함하는 약제학적 조성물의 치료적 유효량을 포유 동물에게 투여하는 단계를 포함한다.
본 발명의 활성제는 선택적으로는 또 다른 치료제 또는 치료제들과 배합되어, 특히 뮤 오피오이드 기작이 아닌 기작(non-mu opioid mechanism)을 통한 위장운동촉진제와 배합되어 투여된다. 따라서, 또 다른 실시 형태에서, 본 발명의 방법과 조성물은 또 다른 위장운동촉진제의 치료적 유효량을 추가적으로 포함한다.
상기에서 기술된 바와 같이, 본 발명의 염은 뮤 오피오이드 수용체 안타고니스트이다. 따라서, 본 발명은 포유 동물에서 뮤 오피오이드 수용체를 길항시키는(antagonizing) 방법을 제공하고, 상기 방법은 포유 동물에게 본 발명의 염을 투여하는 단계를 포함한다.
다른 특징들 중에서도, 유리 염기 및 황산염 형태로 된 본 발명의 활성제는 뮤 오피오이드 수용체에 대한 강력한 결합을 나타내고 뮤 오피오이드 기능성 분석에서 작용이 거의 없거나 완전히 없는 것으로 밝혀졌다. 따라서, 본 발명의 염은 강력한 뮤 오피오이드 수용체 안타고니스트이다. 추가적으로, 활성제는 동물 모델에서 중추 신경계와 비교할 때 주로 말초 활성을 나타내었다. 따라서, 본 발명의 염은 무통증의 좋은 중추적인 효과와 저촉되지 않고 GI 운동성에서 오피오이드-유도된 감소를 역전시킬 것으로 기대된다. 본 발명 화합물의 이러한 특징뿐만 아니라 용도는 당해 분야의 당업자들에게 잘 알려져 있는 다양한 인 비트로 및 인 비보 분석을 사용하여 입증될 수 있다. 대표적인 분석을 하기 실시예에서 추가적으로 상세하게 기술한다.
하기의 합성 실시예와 생물학적 실시예는 본 발명을 설명하기 위해 제공되었고, 어떠한 방식으로든지 본 발명의 범위를 제한하도록 해석되어서는 안 된다. 하기의 실시예에서, 하기의 줄임말은 달리 지시되지 않는다면 하기의 의미를 갖는다. 하기에서 정의되지 않은 줄임말은 이들의 일반적으로 허용되는 의미를 갖는다.
DIPEA = N,N-디이소프로필에틸아민
DMF = N,N-디메틸포름아미드
EtOAc = 에틸 아세테이트
EtOH = 에탄올
MeTHF = 2-메틸테트라히드로푸란
MTBE = 터트-부틸 메틸 에테르
NaHMDS = 소듐 비스(트리메틸실릴)아미드
PyBop = 벤조트리아졸-1-일옥시트리피롤리디노-포스포늄 헥사플루오로포스페이트
psi = pounds per square inch
Rt = 잔류 시간(retention time)
THF = 테트라히드로푸란
반응물과 용매를 상업적 공급원(Aldrich, Fluka, Sigma, 등)으로부터 구입하였고, 추가로 정제하지 않고 사용하였다. 반응은 달리 표시되지 않는다면, 질소 분위기 하에서 수행하였다. 반응 혼합물의 진전을 얇은 막 크로마토그래피(TLC), 분석용 고 성능 액체 크로마토그래피(anal. HPLC), 및 질량 분석기로 모니터하였다. 생성물의 엔도/엑소 비율을 하기에서 기술된 프로토콜을 사용하여 HPLC 분석법으로 결정하였다. 반응 혼합물을 각각의 반응에서 구체적으로 기술한 바에 따라 워크 업(work up) 하였고; 통상적으로 이들을 추출 및 온도- 및 용매-의존성 결정화, 및 침전과 같은 다른 정제 방법에 의해 정제하였다. 반응 생성물의 규명은 질량 및 1H-NMR 분광기로 일반적으로 수행하였다. NMR 측정을 위해서는, 시료를 중수소화된 용매(CD3OD, CDCl3, 또는 DMSO-d6)에 용해시켰고, 표준 관찰 조건 하에서 Varian Gemini 2000 기구(300 MHz)를 사용하여 1H-NMR 스펙트럼을 얻었다. 화합물의 질량 분석에 의한 확인은 Applied Biosystems(Foster City, CA) 모델 API 150 EX 장치 또는 Agilent (Palo Alto, CA) 모델 1100 LC/MSD 장치에 의한 전자분무 이온화 방 법(electrospray ionization method ESMS)으로 수행하였다.
일반적인 HPLC 조건
컬럼: Zorbax SB-Aq, 5 ㎛ 4.6 x 250 mm
컬럼 온도: 40℃
유속: 1.0 mL/분
이동상: A = 물/ACN (98:2) + 0.1 % TFA
B = 물/ACN (10:90) + 0.1 % TFA,
주입 부피: 10 ㎕
검출기 파장: 214 nm
HPLC 방법 1
미정제된 화합물을 약 1 mg/mL로 물/ACN (50:50)에서 용해시켰고, 20분에 걸쳐 하기의 농도 구배 (시간(분)/ % B): 0/10, 2.5/20, 9/75, 15/90, 17/90, 18/10, 20/10을 사용하여 분석하였다.
HPLC 방법 2
화합물을 약 1 mg/mL로 물/ACN (90:10)에서 용해시켰고, 30분에 걸쳐 하기의 농도 구배 (시간 (분)/ % B): 0/10, 13/10, 23/65, 28/90, 29/90, 30/10를 사용하여 분석하였다.
HPLC 방법 3
화합물을 약 1 mg/mL로 물/ACN (90:10)에서 용해시켰고, 55분에 걸쳐 학의 농도 구배 (시간 (분)/ % B): 0/10, 10/20, 46/75, 47/90, 50/10, 55/10을 사용하 여 분석하였다.
제조예 1: N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르 중아황산염 부가물의 합성
a. N-시클로헥실메틸-(2,2-디에톡시에틸)아민의 제조
2,2-디에톡시에틸아민(209 mL, 1.43 mol)과 MeTHF(1050 L)의 혼합물에, 시클로헥산카르브알데히드(107 mL, 0.89 mol)를 첨가하였다. 얻은 반응 혼합물을 실온에서 30분 동안 교반하였고, 0℃까지 냉각시켰다. 소듐 트리아세톡시보로히드라이드(378 g, 1.79 mol)를 40분 후에 첨가하였고, 얻은 반응 혼합물을 2시간 동안 교반하였고 0℃까지 냉각시켰다. 1M NaOH (1 L)를 첨가하였다. 유기층을 염화나트륨 수용액(1:1, 2 x 1 L)으로 세척하였고 부피를 ~ 20%까지 줄였다. MeTHF (1 L)를 첨가하였고, 부피를 ~ 20%까지 줄였다. 얻은 미정제된 중간체 용액을 다음 단계에서 바로 사용하였다.
b. N-시클로헥실메틸-(2,2-디에톡시에틸)카르밤산 벤질 에스테르의 제조
이전 단계의 생성물(-213 g, -0.9 mol)에, MeTHF (2 L)와 DIPEA (233 mL, 1.34 mol)를 첨가하였다. 얻은 반응 혼합물을 0℃까지 냉각시켰고, 벤질클로로포르메이트(140 mL, 0.98 mol)를 적가하였다. 얻은 반응 혼합물을 0℃에서 30분 동안, 0℃에서 실온까지 2시간 동안, 그런 다음 실온에서 1시간 동안 교반하였다. 물(1.6 L)을 첨가하였고, 얻은 반응 혼합물을 10분 동안 교반하였다. 층 분리를 하였고, 얻은 유기층을 탄산수소나트륨 수용액(1.6 L)과 물(1.6 L)로 세척하였다. 층 분리를 하였고, 얻은 유기층을 약 20%까지 줄였다. MeTHF (1 L)를 첨가하였고 부피를 ~ 20%까지 줄였다. 얻은 미정제된 표제 중간체 용액을 다음 단계에서 바로 사용하였다.
c. N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르 중아황산염 부가물의 합성
이전 단계의 생성물(-302 g, -0.62 mol)과 아세토니트릴(2 L)에, 1M HCl (2 L)을 첨가하였고, 얻은 반응 혼합물을 30℃에서 7시간 동안 교반하였다. 에틸 아세테이트(2 L)를 첨가하였고, 얻은 반응 혼합물을 10분 동안 교반하였다. 층 분리를 하였고, 유기층을 1 M HCl (1.5 L)로 세척하였고, 다시 층 분리를 하였고, 유기 층을 0.5 M HCl (1 L)로 세척하였다. 아황산나트륨(71.4 g, 0.69 mol)을 첨가하였고, 얻은 반응 혼합물을 밤새 교반하였고, 그런 다음 여과하였다. 반응기와 여과 고체(filter cake)를 에틸 아세테이트(1 L)로 세척하였다. 그 결과 얻은 용액을 진공에서 2시간 동안 감압 하에서 밤새 건조시켜, 표제 화합물(199 g, HPLC에 의할 때>99 % 면적 순도)을 흰색 고체로 얻었다. 얻은 여과액을 동일한 방법으로 처리하여 표제 화합물(30 g)의 두 번째 분량을 얻었다.
제조예 2: 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)-벤즈아미드의 합성
a. 8-벤질-3-엑소-(3-메톡시페닐)-8-아자비시클로[3.2.1]옥탄-3-올의 제조
3L 플라스크에 세슘 클로라이드(cerous chloride) 분말(194 g, 0.79 mol)을 첨가하였다. 플라스크에 질소를 충진하였고, THF (800 mL)를 첨가하였다. 얻은 반응 혼합물을 25℃에서 1시간 동안 교반하였다. 얻은 혼합물에, THF 중 ~ 1M 3-메톡시페닐 마그네슘 브로마이드(800 mL, 0.87 mol)를 적가하였다. 그 결과 얻은 슬러 리를 3℃에서 1.5시간 동안 교반하였다. 내부 온도를 -5℃를 유지하면서, THF (200 mL) 중 8-벤질-8-아자-비시클로[3.2.1]옥탄-3-온(120.4 g, 0.56 mol)을 적가하였다. 그 결과 얻은 용액을 15분 동안 교반하였다. 얻은 반응 혼합물을 10℃에서 온도를 유지하고 있는 6 N HCl (800 mL)을 포함하는 플라스크에 첨가하였다. 용매를 회전식 증발기에 의해 제거한 후에, 얻은 반응 혼합물을 실온에서 밤새 교반하였다. 얻은 고체를 여과에 의해 분리하였고, 6N HCl (70 mL)과 아세토니트릴 (3 x 70 mL)로 세척하였고, 건조시켜 표제 중간체의 HCl 염을 회백색 고체(161 g)로 얻었다.
b. 8-벤질-3-(3-메톡시페닐)-8-아자비시클로[3.2.1]옥트-2-엔의 제조
3 L 플라스크에, 8-벤질-3-엑소-(3-메톡시페닐)-8-아자비시클로[3.2.1]옥탄-3-올 히드로클로라이드(383.9 g, 1.06 mol), 6 M HCl (800 mL), 및 MeTHF (200 mL)를 첨가하였다. 그 결과 얻은 슬러리를 질소 하에서 70℃에서 2.5시간 동안 가열하였다. 얻은 반응 혼합물을 12 L 반응기로 옮겼고 10℃까지 냉각시켰다. 반응 플라스크를 MeTHF (1 L)로 세척하였고, 상기 12 L 반응기에 첨가하였다. NaOH (물에서 50 wt %, 200 mL)를 첨가하였고, pH -13이 될 때까지 추가량의 NaOH (50 wt %, 150 mL)를 부분으로 나누어 첨가하였다. 층 분리를 하였고, 물 층을 MeTHF (1 L)로 추출하였고, 합한 MeTHF 층을 염화나트륨 수용액(brine 1 L)으로 세척하였다. 용매를 회전식 증발기로 30 내지 40℃에서 줄여, 표제 중간체(360 g)를 끈적한 오일로 얻었다. EtOH (1.5 L)를 첨가하였고, 부피를 ~ 500 mL까지 줄였고, 그런 다음 1.8 L로 맞추었다.
c. 3-엔도-(3-메톡시페닐)-8-아자비시클로[3.2.1]옥탄의 제조
이전 단계에서 제조한 8-벤질-3-(3-메톡시페닐)-8-아자비시클로[3.2.1]옥트-2-엔(EtOH 중 95 %, 400 mL, 0.20 mol)에, 6 M HCl (45 mL)를 첨가하였고, 그런 다음 MeTHF (50 mL)를 첨가하였다. 얻은 반응 혼합물에 질소를 충전하였고, 40℃까지 가열하였고, 탄소상 팔라듐(10 중량%, 8 g)을 첨가하였다. 반응기에 수소를 가압하였고(3 x 20 psi), 그런 다음 20 psi에서 40℃에서 18시간 동안 수소화반응시켰다. 얻은 반응 혼합물을 셀라이트(Celite)를 통해 여과하였고, 농축하였고, MeTHF (2 x 100 mL)로 세척하였고, 조립질의 유리 필터를 통해 여과하였고, MeTHF (10 mL)로 세척하였고, 필터 상에서 건조시켜, 표제 중간체(31 g, 단일 이성질체, HPLC에 의할 때 엑소 이성질체 검출되지 않음))의 HCl 염을 흰색 고체로 얻었다. 추가적으로 생성물 5.2 g을 모액으로부터 회수하였다.
d. 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)-페놀의 제조
500 mL 플라스크에, 3-엔도-(3-메톡시페닐)-8-아자비시클로[3.2.1]옥탄 히드로클로라이드(115 g, 0.45 mol)와 브롬화수소산(물에서 48 중량%, 100 mL, 0.88 mol)을 첨가하였다. 얻은 혼합물을 120℃까지 가열하였고, 그 온도에서 교반하면서 24시간 동안 유지하였다. 추가량의 브롬화수소산 용액(25 mL)을 첨가하였고, 얻은 반응 혼합물을 교반하면서 6시간 동안 가열하였고, 그런 다음 70℃까지 냉각시켰다. 아세토니트릴(200 mL)을 첨가하였고, 그 결과 얻은 슬러리를 10℃까지 냉각시켰고, 그런 다음 여과하였고, 얻은 여과 고체를 아세토니트릴(50 mL)로 세척하여, 표제 중간체의 HBr 염(99 g, > 99 % 순도)을 흰색의 과립형 고체로 얻었다.
e. 2,2,2-트리플루오로-1-[3-엔도-(3-히드록시페닐)-8-아자비시클로[3.2.1]옥트-8-일]에타논의 제조
3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)-페놀 히드로브로마이드(54.4 g, 0.19 mol), 톨루엔(210 mL), 및 트리에틸아민(40 mL, 0.29 mol)의 용액에, 트리플루오로아세트산 무수물(54 mL, 0.38 mol)을 20분에 걸쳐 첨가하였다. 얻은 반응 혼합물을 40℃에서 2시간 동안 교반하였다. 에틸 아세테이트(370 mL)와 염화나트륨 수용액(1:1, 265 mL)을 첨가하였다. 얻은 반응 혼합물을 15분 동안 교반하였고, 층 분리를 하였다. 얻은 유기층에 탄산수소나트륨 포화 수용액(300 mL)을 첨가하였고, 얻은 혼합물을 격렬하게 밤새 교반하였다. 층 분리를 하였고, 얻은 유기층을 염화나트륨 수용액(1:1, 265 mL)으로 세척하였고, 황산 나트륨으로 물을 제거하였고, 대부분의 용매를 회전식 증발기에 의해 제거하였다. 톨루엔(100 mL)을 첨가하였고, 회전식 증발기에 의해 용매를 제거하여, 미정제된 표제 중간체를 얻었다.
f. 트리플루오로메탄술폰산 3-엔도-[8-(2,2,2-트리플루오로-아세틸)-8-아자비시클로[3.2.1]옥트-3-일]페닐 에스테르의 제조
500 mL 플라스크에, 이전 단계의 중간체(32.8 g, 0.11 mol)의 에틸 아세테이트 용액(220 mL)을 첨가하였고, 트리에틸아민(23 mL. 0.17 mol)을 첨가하였다. 그 결과 얻은 용액을 5℃까지 냉각시켰고, 트리플루오로메탄 술포닐 클로라이드(14 mL, 0.13 mol)를 적가하였다. 얻은 혼합물을 25℃까지 가온시켰고, 그 온도에서 1시간 동안 교반하였다. 탄산수소나트륨 포화 수용액(200 mL)을 첨가하였고, 층 분리를 하였고, 얻은 유기층에 염화나트륨 수용액(150 mL)을 첨가하였고, 다시 층 분 리를 하였고, 얻은 유기층으로부터 용매를 제거하여, 미정제된 표제 중간체를 얻었다.
g. 3-엔도-[8-(2,2,2-트리플루오로아세틸)-8-아자비시클로[3.2.1]옥트-3-일]-벤조니트릴의 제조
100 mL 플라스크에, 트리플루오로메탄술폰산 3-엔도-[8-(2,2,2-트리플루오로-아세틸)-8-아자비시클로[3.2.1]옥트-3-일]페닐 에스테르(25.3 g, 58.7 mmol), 트리스(디벤질리덴아세톤)디팔라듐(0)(0.81 g, 0.9 mmol), 1,1'-비스(디페닐포스피노)페로센(1.01 g, 1.8 mmol), 및 시안화 아연(4.2 g, 35.8 mmol)을 첨가하였다. 플라스크에 질소를 5분 동안 3회 충전하였고, 그런 다음 집중 진공 하에서 5분 동안 두었다. 플라스크에 DMF (150 mL)와 증류수 (2.5 mL)를 첨가하였다. 얻은 용액을 교반하면서 10분 동안 질소를 충전하였고, 120℃까지 가열하였고, 120℃에서 질소 하에서 4시간 동안 교반하였다. 반응이 완료되었을 때, 동일한 방법으로 제조한 이전 단계의 생성물 20 g을 첨가하였고, 20분 동안 교반하였다.
대부분의 용매를 증류에 의해 제거하였고, 용액을 22℃까지 냉각시켰다. 얻은 용액에 에틸 아세테이트(445 mL)를 첨가하였고, 그 결과 얻은 용액을 셀라이트를 통해 여과하였다. 탄산수소나트륨 수용액(450 mL)을 첨가하였고, 그 결과 얻은 용액을 15분 동안 교반하였다. 층 분리를 하였고, 얻은 유기층을 희석시킨 염화나트륨 수용액(2 x 95 mL)으로 세척하였고, 황산 나트륨을 통해 여과하였다. 에틸 아세테이트를 제거하여, 부피를 약 50 mL로 줄였다. 이소프로필 알코올(150 mL)을 첨가하였고, 얻은 용액을 22℃에서 1시간 동안 교반하였다. 고체를 여과에 의해 분리 하였고, 이소프로필 알코올(2 x 25 mL)로 세척하여, 표제 중간체(33.5 g, HPLC에 의한 순도 100%)를 회백색/밝은 갈색 고체로 얻었다. 생성물의 두 번째 수확물(6.3 g, HPLC 순도 > 98 %)을 여과액으로부터 얻었다.
h. 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)-벤즈아미드의 합성
황산(96 %, 12 mL) 중 3-엔도-[8-(2,2,2-트리플루오로아세틸)-8-아자비시클로[3.2.1]옥트-3-일]-벤조니트릴(10 g, 32 mmol)의 용액을 교반하면서 50℃까지 가열하였고, 이 온도에서 교반하면서 2시간 동안 유지하였다. 얻은 반응 혼합물을 22℃까지 냉각시켰고, 5 N NaOH (90 mL)와 메탄올 (100 mL)을 포함하고 10℃까지 냉각시킨 500 mL 플라스크에 천천히 첨가하였다. 염 침전물을 여과하였고, 얻은 여과액을 22℃에서 1시간 동안 교반하였다. 얻은 반응 혼합물을 감압 하에서 농축시켰다. 얻은 잔여물에 MeTHF (150 mL)를 첨가하였고, 얻은 반응 혼합물을 22℃에서 5분 동안 교반하였다. 층 분리를 하였고, 얻은 수용액 층에 MeTHF (100 mL)를 첨가하였다. 층 분리를 하였고, 합한 유기층에 염화나트륨 수용액(150 mL)을 첨가하였다. 층 분리를 하였고, 얻은 유기층에서 탄산 칼륨으로 물을 제거하였고, 여과하였고, 용매를 제거하였다. 얻은 잔여물에 EtOH (25 mL)와 농축된 HCl (2.6 mL)의 혼합물을 첨가하였고, MTBE (25 mL)를 첨가하였고, 얻은 용액을 22℃에서 교반하였다. 침전된 고체를 여과하였고, 공기 중에서 건조시켜, 표제 화합물의 HCl 염(8 g, HPLC 순도 97%)을 흰색 고체로 얻었다.
제조예 3: 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)벤즈아미드의 합성
a. 트리플루오로-메탄술폰산 8-벤질-8-아자비시클로[3.2.1]옥트-2-엔-3-일 에스테르의 제조
500 mL 플라스크에, 8-벤질-8-아자비시클로[3.2.1]옥탄-3-온 히드로클로라이드(50.4 g, 200 mmol), EtOAc(160 mL), 및 4 N NaOH(50 mL)를 첨가하였다. 얻은 반응 혼합물을 30℃까지 가열하였고, 이 온도에서 1시간 동안 교반하였다. 층 분리를 하였고, 수용액 층을 버렸다. 유기층의 부피를 회전식 증발에 의해 ~40 mL까지 줄였고, THF (270 mL)를 첨가하였다.
그 결과 얻은 용액을 1L 플라스크에 첨가하였고, -20℃까지 냉각시켰다. 상기 플라스크에 NaHMDS 용액(THF에서 1 M, 230 mL, 230 mmol)을 15분에 걸쳐 첨가하였다. 그 결과 얻은 반응 혼합물을 -20±5℃에서 1시간 동안 교반하였다. 얻은 반응 혼합물에 N-페닐-비스(트리플루오로메탄술폰이미드)(82.2 g, 230 mmol)를 5분에 걸쳐 분획으로 나누어 첨가하였고, 얻은 혼합물을 -20℃ 내지 -10℃에서 1시간 동안 교반하였다. 그 결과 얻은 반응 혼합물에 1 N NaOH (200 mL)를 첨가하였고, 그 결과 얻은 혼합물을 교반하면서 22℃까지 가온시켰다. 용매를 회전식 증발기에서 30℃에서 일 부분 제거하여 450 mL 부피로 만들었다. 남아있는 반응 혼합물에 EtOAc(300 mL)와 헵탄(150 mL)을 첨가하였다. 그 결과 얻은 혼합물을 22℃에서 5분 동안 교반하였다. 층 분리를 하였고 수용액 층을 버렸다. 유기층을 1N NaOH (3 x 450 mL)로 세척하였다. 수용액 층을 버렸다. 유기층을 회전식 증발기에 의해 농축시켜, 표제 중간체를 얻었다(77 g, HPLC 방법 1에 의한 순도 >96 %).
1H NMR (d6-DMSO, 400 MHz): δ (ppm) 7.25-7.35 (m, 5H), 6.05 (d, J=5.2, 1H), 3.64 (q, J=13.2, 2H), 3.40-3.44 (m, 2H), 2.77 (d, J=16.4, 1H), 1.79-2.09 (m, 5H), 1.52- 1.59 (m, 1H).
b. 3-(8-벤질-8-아자비시클로[3.2.1]옥트-2-엔-3-일)벤즈아미드의 제조
이전 단계의 미정제된 생성물에, THF (420 mL)를 첨가하였고, 그 결과 얻은 용액에 질소를 5분 동안 충전시켰다. 2 L 플라스크에, 3-카르바모일페닐 보론산(98 %, 33.0 g, 200 mmol), 팔라듐 (II) 아세테이트(98 %. 0.46 g, 2 mmol), 1,1'-비스(디페닐포스피노)페로센(97 %, 1.1 g, 2 mmol) 및 포타슘 플루오라이드(34.9 g, 600 mmol)를 첨가하였고, 뒤이어 상기 트리플루오로-메탄술폰산 8-벤질-8-아자비시클로[3.2.1]옥트-2-엔-3-일 에스테르의 THF 용액을 첨가하였다. 그 결과 얻은 혼합물을 질소로 5분 동안 충전시켰고, 질소 하에서 환류 상태(67℃)로 가열하였고, 2시간 동안 교반하였다. 얻은 반응 혼합물을 30℃까지 냉각시켰고, 그런 다음 EtOAc (500 mL)와 1N NaOH (500 mL)를 첨가하였고, 얻은 혼합물을 22℃에서 10분 동안 교반하였다. 층 분리를 하였고, 수용액 층을 버렸다. 유기층을 브라인(250 mL)과 물(250 mL)의 혼합물로 세척하였고, 5분 동안 교반하였다. 층 분리를 하였고, 수용액 층을 버렸다. 유기층에서 짧게 Na2SO4로 물을 제거하였고, 여과하였고, 용매를 일부분 제거하였다. 용매 제거 동안에 생성물을 연한 노란색 고체로 침전시켰다. 그 결과 얻은 슬러리(약 200 mL)를 여과하였고, 얻은 고체를 차가운 EtOAc(0℃, 100 mL)로 세척하였고, 고 진공 하에서 25℃에서 건조시켜, 표제 중간체(42.5 g)를 연한 노란색 고체로 얻었다.
얻은 모액과 상기 세척액을 합하였고 농축시켰으며, 그 결과 얻은 슬러리(약 100 mL)를 5℃에서 30분 동안 교반시켰고 여과하였다. 여과된 고체를 차가운 EtOAc (0℃, 30 mL)로 세척하였고, 고 진공 하에서 물을 제거하여, 표제 중간체의 두 번째 수확물(7 g, 합한 수득율 78 %, HPLC 방법 1에 의한 순도> 98.5 %)을 얻었다.
(m/z): [M+H]+ C21H22N2O, 계산값 319.18; 측정값 319.4. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.9 (s, 1H), 7.63 (d, J=6.4, 1H), 7.57 (d, J=6.4, 1H), 7.21-7.42 (m, 6H), 6.38 (d, J=4.4, 1H), 6.13 (s, br, 1H), 5.83 (s, br, 1H), 3.68-3.76 (m, 2H), 3.46-3.51 (m, 2H), 2.92 (d, J=17.2, 1H), 2.18-2.26 (m, 1H), 2.04-2.12 (m, 2H), 1.86-1.92 (m, 1H), 1.58-1.65 (m, 1H).
c. 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)벤즈아미드의 합성
1L 수소화반응 용기에, 3-(8-벤질-8-아자비시클로[3.2.1]옥트-2-엔-3-일)벤즈아미드(40 g, 125 mmol), EtOH (800 mL), 6 M HCl (42 mL) 및 물 (80 mL)을 첨가하였고, 얻은 혼합물을 완전한 용해가 관찰될 때까지 22℃에서 교반하였다. 얻은 반응 혼합물에 질소를 5분 동안 충전시키면서 5분 동안 30℃까지 가열하였다. 얻은 혼합물에 10 wt % Pd/C (물에서 50 %, 4 g)를 첨가하였다. 얻은 혼합물을 가열하면서 대기압 하에서 5-10분 동안 수소로 충전하였다. 그 결과 얻은 혼합물을 < 5 psi (<0.34 atm)의 수소 흐름 하에서 5시간 동안 50℃에서 교반하였고, 그 결과 HPLC 분석에 따르면, 반응물의 >99 %가 변환되었다. 그 결과 얻은 용액을 30℃까지 가열하였고, 셀라이트를 통해 여과하여, 엔도의 Rt=10.97, 엑소의 Rt=12.67인 HPLC 방법 2에 의할 때, 엔도:엑소의 비율이 ~ 93:7인 표제 화합물의 미정제된 HCl 염의 용액을 얻었다. (m/z): [M+H]+ C14H18N2O, 계산값 231.15; 측정값 231.2.
EtOH (-80 mL)에서 30℃에서 공비 증류에 의하여 미정제된 생성물로부터 물을 제거하여, 슬러리를 얻었고, 얻은 슬러리를 완전히 용해될 때까지 60℃까지 가열하였다. 얻은 용액을 35℃까지 냉각시켰고, 생성물의 시드 결정(seed crystal)(0.05 g)을 첨가하였다(시드 결정은 제조예 2에서 기술된 방법에 따라 제조하였다). 그 결과 얻은 슬러리를 22℃에서 30분 동안 교반하였고, MTBE (120 mL)를 천천히 첨가하였고, 얻은 슬러리를 22℃에서 4시간 동안 교반하였고, 0℃에서 1시간 동안 교반하였다. 그 결과 얻은 고체를 여과하였고, 차가운 EtOH로 세척하였고, 고 진공 하에서 건조시켜 흰색 분말로서 표제 화합물의 HCl 염(75 % 수득율, 엔도 Rt=8.67, 엑소 Rt=9.43 HPLC 방법 3에 의할 때 >98.5% 순도 < 0.4% 엑소 이성질체)을 얻었다.
(m/z): [M+H]+ C14H18N2O 계산값 231.15; 측정값 231.2. 1H NMR (d6-DMSO, 400 MHz): δ (ppm) 9.13 (s, br, 1H), 9.03 (s, br, 1H), 8.05 (s, 1H), 7.93 (s, 1H), 7.73 (d, J=I.6, 1H), 7.58 (d, J=7.6, 1H), 7.40 (t, J=I.6, 2H), 3.97 (s, 2H), 3.17-3.23 (m, 1H), 2.39- 2.46 (m, 2H), 2.19-2.24 (m, 2H), 1.86-1.89 (m, 2H), 1.59-1.63 (m, 2H).
실시예 1A: 결정형 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로 피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 합성
a. N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르의 제조
100 mL 플라스크에, N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르 중아황산염 부가물(3.94 g, 1 mmol)과 MeTHF(35 mL)를 첨가하였고, 뒤이어 물(25 mL)을 첨가하였다. 그 결과 얻은 슬러리를 실온에서 5분 동안 교반하였고, 1 M NaOH (8 mL)를 첨가하였다. 그 결과 얻은 반응 혼합물을 실온에서 45분 동안 교반하였다. 층 분리를 하였고, 유기층의 부피를 -8 mL까지 줄여, 미정제된 표제 중간체를 얻었다.
b. 2-[3-엔도-(3-카르바모일페닐)-8-아자비시클로[3.2.1]옥트-8-일]-에틸}시클로헥실메틸-카르밤산 벤질 에스테르의 제조
이전 단계의 생성물에, DMF (15 mL)를 첨가하였고, 뒤이어 3-엔도-(8-아자비시클로[3.2.1]옥트-3-일)-벤즈아미드 히드로클로라이드(2.67 g, 1 mmol)를 첨가하였고 그 다음에 DMF (10 mL)를 첨가하였다. 얻은 혼합물을 실온에서 30분 동안 교반하였고, 10℃까지 냉각시켰고, 그런 다음 소듐 트리아세톡시보로히드라이드(4.25 g, 2 mmol)를 첨가하였다. 그 결과 얻은 반응 혼합물을 실온에서 90분 동안 교반하였고 그런 다음 10℃까지 냉각시켰다. 이소프로필 아세테이트(100 mL)를 첨가하였고, 그 다음에 1 M NaOH (50 mL)를 첨가하였다. 얻은 혼합물을 15분 동안 교반하였고, 층 분리를 하였다. 얻은 유기층을 물에서의 브라인(1:1, 2 x 50 mL)으로 세척하였고, 얻은 유기층의 부피를 ~ 10 mL로 줄여, 미정제된 표제 중간체를 얻었다.
c. 3-엔도-{8-[2-(시클로헥실메틸아미노)에틸]-8-아자-비시클로[3.2.1]옥트- 2-일}벤즈아미드의 제조
이전 단계의 생성물에, EtOH (30 mL)와 농축된 HCl (1.5 mL)을 첨가하였다. 얻은 용액에 질소를 충전하였고, 탄소상 10% 팔라듐(470 mg)을 첨가하였고, 얻은 혼합물에 질소를 5분 동안 충전하였고, 그런 다음 30 psi에서 밤새 수소화반응시켰다. 2분 동안 질소를 충전한 후에, 얻은 용액을 셀라이트를 통해 여과하였고, 용매를 ~10 mL로 줄였다. 이소프로필 아세테이트(40 mL)와 1 M NaOH (20 mL)를 첨가하였다. 층 분리를 하였고, 얻은 유기층을 브라인(20 mL)으로 세척하였고, 층 분리를 하였고, 유기 용매를 5-10 mL로 줄였다. 이소프로필 아세테이트(20 mL)를 첨가하였고, 부피를 ~ 8 mL로 줄였고, 이소프로필 아세테이트(20 mL)를 첨가하였다. 그 결과 얻은 슬러리를 실온에서 2시간 동안 교반하였다. 생성물을 여과로 분리하였고, 반응 플라스크와 여과하여 얻은 고체(filter cake)를 이소프로필 아세테이트(10 mL)로 세척하여, 회백색 중간체로 표제 중간체(2.4 g, 98% 순도)를 얻었다.
d. 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염(수화물 형태)의 제조
500 mL 플라스크에, 3-엔도-{8-[2-(시클로헥실메틸아미노)에틸]-8-아자-비시클로[3.2.1]옥트-3-일}벤즈아미드(31 g, 83.9 mmol) 및 DMF(150 mL)를 첨가하였다. 얻은 혼합물을 10분 동안 교반하였고, 그런 다음 벤조트리아졸-1-일옥시트리스(피롤리디노)-포스포늄 헥사플루오로-포스페이트(56.8 g, 109 mmol)와 리튬 (4S)-2,2-디메틸-1,3-디옥솔란-4-카르복실레이트(15.6 g, 92.3 mmol)를 첨가하였고, 얻은 혼합물을 실온에서 2시간 동안 교반하였다. 에틸 아세테이트(600 mL)와 0.5 M NaOH (300 mL)를 첨가하였고, 층 분리를 하였다. 얻은 유기층은 미정제된 분리되지 않은 (S)-2,2-디메틸-[1,3]디옥솔란-4-카르복시산 {2-[3-(3-카르바모일-페닐)-8-아자비시클로[3.2.1]옥트-8-일]에틸}시클로헥실메틸-아미드(~84 mmol)를 포함하였다.
얻은 유기층을 물에서의 브라인(1:1, 2 x 300 mL)으로 세척하였고 층 분리를 하였다. 얻은 유기층에 2 M H2SO4 (42 mL)를 첨가하였고, 얻은 반응 혼합물을 실온에서 밤새 교반하였다. 아세토니트릴(300 mL)을 첨가하였고, 그 결과 얻은 슬러리를 2시간 동안 교반하였다. 생성물을 여과에 의해 분리하였고, 얻은 여과된 고체를 아세토니트릴(200 mL)로 세척하였고, 공기 중에서 2시간 동안 건조시켰고 그런 다음 진공 하에서 실온에서 20시간 동안 건조시켜, 표제 화합물(40 g, HPLC에 의할 때 97 %)을 흰색 분말로 얻었다.
e. 결정형 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 합성
100 mL 플라스크에, 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염 수화물 형태(2 g)와 MeOH (40 mL)를 첨가하였다. 그 결과 얻은 슬러리를 질소 하에서 20분 동안 65℃까지 가열하여, 완전히 용해되도록 하였다. 얻은 용액을 교반하면서 실온까진 냉각시켰다. 용매 약 20 mL를 약한 감압 하에서 제거하였고, 그 결과 얻은 슬러리를 실온에서 밤새 교반하였다. 생성물을 여과에 의해 분리하였고, 플라스크와 여과하여 얻은 교체를 아세토니트릴(2 x 5 mL)로 세척하였다. 여과하여 얻은 고체를 공기 중에서 2시간 동안 건조시켰고, 그런 다음 진공 하에서 실온에서 밤새 건조시켜, 표제 화합물(1.71 g, HPLC에 의할 때 > 99% 순도, ~85% 수득율)을 흰색 분말로 얻었다.
상기 방법에 따라 제조한 시료는 1H NMR (400 MHz, DMSO d6): δ (ppm) 9.08 & 8.94 (br 2세트, IH), 7.99-8.04 (m, 2H), 7.74-7.76 (m, 1H), 7.68-7.70 (m, 1H), 7.41-7.45 (m, 2H), 4.81, 5.00 및 5.30 (br 3세트, 2H), 4.34 (deformed m, 1H), 4.00 & 4.05 (deformed m, 2H), 3.01-3.25 및 3.47-3.55 및 3.75-3.82 (m 3세트, 10H), 2.50-2.55 (m, 2H), 1.99 (deformed m, 2H), 1.56-1.70(m, 8H), 1.15-1.19 (m, 3H), 0.89-0.99 (m, 2H)를 특징으로 한다.
실시예 1B: 결정형 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 합성
a. 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 제조
3-엔도-{8-[2-(시클로헥실메틸아미노)에틸]-8-아자-비시클로[3.2.1]옥트-3-일}벤즈아미드(100 g, 270.6 mmol)와 DMF (480 mL)의 혼합물을 10분 동안 교반하였고, 0℃에서 냉각시켰다. 벤조트리아졸-1-일옥시트리스(피롤리디노)포스포늄 헥사플루오로포스페이트(183 g, 352 mmol)와 리튬(4S)-2,2-디메틸-1,3-디옥솔란-4-카르복실레이트(49.3 g, 324 mmol)를 0℃에서 한 번에 첨가하였다. 얻은 반응 혼합물을 실온에서 6시간 동안 교반하였다. 이소프로필 아세테이트(2.0 L)와 1 M NaOH(1.0 L)를 첨가하였고, 얻은 반응 혼합물을 15분 동안 교반하였고, 층 분리를 하였다. 얻은 유기층을 물에서의 브라인(1:1, 2 x 1.0 L)으로 세척하였고, 층 분리를 하였다. 얻은 유기층에서 부피를 1/4로 줄였고(-500 mL), 아세토니트릴을 첨가하였고, 얻은 반응 혼합물을 균일하게 될 때까지 교반하여, 이소프로필 아세테이트와 아세토니트릴 중 중간체 (S)-2,2-디메틸-1,3-디옥솔란-4-카르복시산 2-[3-엔도-(3-카르바모일-페닐)-8-아자비시클로[3.2.1]옥트-8-일]-에틸-시클로헥실메틸-아미드의 용액을 얻었다.
이소프로필 아세테이트/아세토니트릴(22.5 mL) 중 중간체(3.03 g, 6.09 mmol)의 상기 용액 중 일 분획을 물에서의 2.0 M 황산과 합하였고, 25℃에서 20시간 동안 유지시켰고, 그런 다음 교반하면서 10℃에서 5시간 동안 유지시켰다. 얻은 반응 용액을 여과하였고, 여과하여 얻은 고체를 아세토니트릴(25 mL)로 세척하였고, 건조시켜, 결정형 수화물 형태가 우세한, 표제 화합물의 중간 등급의 황산염(2.91 g, HPLC에 의한 순도 99.4%)을 흰색 고체로 얻었다.
b. 결정형 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 합성
이전 단계에서 제조한 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염 중간 등급(154.0 g, 277.1 mmol)과 메탄올/10% 물(616 mL)의 혼합물을 교반하면서 45분 동안 65℃에서 가열하였다. 그 결과 얻은 반응 혼합물을 55℃까지 냉각시켰고, 표제 생성물의 시드 결정(120 mg)을 첨가하였고, 얻은 반응 혼합물을 55℃에서 1시 간 동안 교반하였고, 온도를 10℃/h의 속도로 20℃까지 낮추었고, 그런 다음 8시간 동안 유지하였다. 얻은 반응 혼합물을 5℃까지 냉각시켰고, 30분 동안 유지하였고, 여과하였다. 여과하여 얻은 고체를 메탄올(2 x 25 mL)로 세척하였고, 고 진공 하에서 밤새 건조시켜, 표제 화합물(126.3 g, 99.9 % 순도)을 얻었다.
실시예 2: 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염(수화물 형태)의 재결정
화합물 1 황산염(수화물 형태)(920 mg)을 아세토니트릴(5 mL)에서 현탁시켰고, 65℃까지 가열하였다. 그런 다음, 물(2.4 mL)을 적가하여, 완전히 용해가 되도록 하였다. 그 결과 얻은 용액을 20분에 걸쳐 실온까지 냉각시켰다. 약 35℃에서 결정핵 생성(nucleation)이 관찰되었다. 얻은 고체를 진공 여과에 의해 분리하였고, 아세토니트릴(5 mL)로 세척하였고, 건조시켜 표제 화합물을 얻었다.
실시예 3: 3-엔도-(8-{2-시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 결정화
화합물 1 황산염(수화물 형태)(50 mg)을 물(10 %)과 메탄올(90 %) 용매 혼합물(0.83 mL)에서 분산시켰고, 교반하면서 60℃까지 가열하였다. 그 결과 얻은 용액을 2시간에 걸쳐 실온까지 냉각시켰다. 그 결과 얻은 고체를 진공 여과에 의해 분리하여, 표제 화합물(8 mg)을 얻었다.
실시예 4: 3-엔도-(8-{2-시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 결정화
화합물 1 황산염(수화물 형태)(42 mg)을 물(25 %)과 메탄올(75 %) 용매 혼합물(0.42 mL)에 분산시켰고, 교반하면서 60℃까지 가열하였다. 그 결과 얻은 용액을 실온까지 냉각시켰다. 회전식 증발에 의해 부피를 50%까지 줄였고, 얻은 용액을 실온에서 밤새 유지하였다. 그 결과 얻은 고체를 진공 여과에 의해 분리하여, 표제 화합물(8 mg)을 얻었다.
실시예 5: 3-엔도-(8-{2-시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 결정화
화합물 1(11 mg)을 톨루엔(22 %)과 아세토니트릴(78 %) 용매 혼합물(0.2 mL)에 용해시켰다. 아세토니트릴(0.15 mL)을 첨가하였고, 그 다음에 아세토니트릴 중 0.04 M 황산(0.59 mL)을 첨가하였다. 산을 첨가한 후에, 고체 침전물이 형성되었다. 얻은 반응 혼합물을 실온에서 12시간 동안 방치하였다. 그 결과 얻은 고체를 여과에 의해 분리하여, 표제 화합물을 얻었다.
실시예 6:
3-엔도-(8-{2-시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 결정화
화합물 1(38 mg)을 디클로로메탄(0.5 mL)에 용해시켰다. 얻은 용액에, 아세토니트릴 중 0.04 M 황산(1.91 mL)을 첨가하였다. 산을 첨가한 후에, 고체 침전물이 형성되었다. 얻은 반응 혼합물을 실온에서 12시간 동안 방치하였다. 그 결과 얻은 고체를 여과에 의해 분리하여, 표제 화합물을 얻었다.
실시예 7: 3-엔도-(8-{2-시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염의 결정화
화합물 1(22 mg)을 톨루엔(23 %)과 아세토니트릴(77 %) 용매 혼합물(0.41 mL)에 용해시켰다. 얻은 용액에, 아세토니트릴 중 0.04 M 황산(1.20 mL)을 첨가하였다. 산을 첨가한 후에, 고체 침전물이 형성되었다. 얻은 반응 혼합물에 물(0.16 mL)을 첨가하여, 침전물을 용해시켰다. 2시간 후에 결정핵 생성이 관찰되었다. 그 결과 얻은 고체를 진공 여과에 의해 분리하여, 표제 화합물을 얻었다.
실시예 8:
3-엔도-(8-{2-시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드 황산염(수화물 형태)의 결정화
결정형 화합물 1 황산염(7.1 g)을 물(42 mL)과 아세토니트릴(25 mL)의 용매 혼합물에 용해시켰다. 얻은 용액을 동결 건조시켜, 무정형의 황산염을 생성하였다. 얻은 무정형 염(6.6 g)을 아세토니트릴(75 %)과 물(25 %)의 용매 혼합물(34.6 mL)에 분산시켰고, 교반하면서 10분 동안 65℃까지 가열하였고, 주변 온도에 도달할 때까지 교반하면서 냉각시켰다. 12시간 후에, 그 결과 얻은 고체를 진공 여과에 의해 분리하여, 표제 화합물(5.4 g)을 얻었다.
실시예 9-17: 본 발명의 염 형태의 특징
실시예 1A에서 제조한 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염(화합물 1) 시료, 및 실시예 2에서 제조한 화합물 1의 황산염의 결정형 수화물 시료를 X-선 분말 회절(XRPD), 시차 주사 열량계(DSC), 열중량 분석(thermogravimetric analysis TGA), 적외선 분광(infrared spectroscopy IR) 및 이온 크로마토그래피로 분석하였다.
실시예 10: X-선 분말 회절
Cu Ka (30.0 kV, 15.0 mA) 조사(radiation)를 사용하는 리가쿠 회절계(Rigaku diffractometer)로, 도 1과 4의 X-선 분말 회절 패턴을 얻었다. 분석은 2 내지 40°의 범위에서 0.03°의 단계별 크기(step size)로 1분에 3°의 연속-스캔 모드에서 실행하는 고니오미터(goniometer)로 수행하였다. 시료는 분말로 된 물질의 엷은 층으로서 석영 시료 고정기(quartz specimen holder)에서 제조하였다. 장치를 실리콘 표준 물질로 조정하였다.
실시예 11: 열 분석
TA Instruments Model Q-100 모듈(module)을 사용하여, 시차 주사 열량 분석(DSC)을 수행하였다. Q SeriesTM 소프트웨어를 위한 TA Instruments Thermal Advantage를 사용하여 데이타를 모았고 분석하였다. 뚜껑이 있는 알루미늄 팬에서 시료 약 1-10 mg을 정확하게 무게를 달았다. 5℃부터 통상적으로는 265℃까지의 10℃/분의 선형 가열 램프를 사용하여, 시료를 평가하였다. 사용하는 동안 DSC 셀(cell)을 건조한 질소로 충전시켰다. 화합물 1의 결정형 황산염 시료와 화합물 1의 황산염의 결정형 수화물의 대표적인 DSC 자취를 각각 도 2와 5에 나타내었다.
TA Instruments Model Q-500 모듈(module)을 사용하여, 열중량 분석(TGA)을 수행하였다. Q SeriesTM 소프트웨어를 위한 TA Instruments Thermal Advantage를 사용하여 데이타를 모았고 분석하였다. 백금 받침대 상의 알루미늄 팬에 약 1-5 mg의 무게가 나가는 시료를 놓았고, 실온부터 300℃까지 10℃/분의 선형 가열 속도로 스캐닝(scan)하였다. 사용하는 동안 저울과 노 챔버(furnace chamber)를 질소로 충전하였다. 화합물 2의 결정형 황산염 시료와 화합물 1의 황산염의 결정형 수화물의 대표적인 TGA 자취를 각각 도 2와 5에 또한 나타내었다.
실시예 12: 동적 수분 흡수 평가(Dynamic Moisture Sorption Assessment)
VTI 대기압 하의 미량 천칭(atmospheric microbalance), SGA-100 시스템(VTI Corp., Hialeah, FL 33016)를 사용하여, 25℃에서 동적 수분 흡수(Dynamic moisture sorption DMS) 평가를 수행하였다. 대략 5-10 mg의 시료 크기를 사용하였고, 습도는 분석 시점에서 주변 값으로 맞추었다. 통상의 DMS 분석은 3개의 스캔으로 구성되어 있다: 5 % RH/단계의 스캔 속도로 주변 습도에서 2% 상대 습도(relative humidity RH), 2% RH에서 90% RH, 90% RH에서 5% RH. 질량은 2분마다 측정하였고, 시료의 질량이 5개의 연속적인 지점 동안 0.02% 이내로 안정하였을 때 RH를 다음 값(± 5 %RH)으로 바꾸었다. 화합물 1의 결정형 황산염 시료와 화합물 1의 황산염의 결정형 수화물의 대표적인 DMS 자취를 각각 도 3과 6에서 나타내었다.
실시예 13: 적외선 분석
확산 반사율 적외선 푸리에 변환 분광기(diffuse reflectance infrared fourier transform spectroscopy DRIFTS) 모듈이 장착된 Avatar 360 FT-IR 분광계를 ±사용하고 4000 내지 400 cm-1의 진동수 범위에 걸쳐 적외선(IR) 흡수 스펙트럼을 결정하였다. 본 발명의 결정형 황산염 시료의 대표적인 IR 흡수 스펙트럼은 430 ±1, 590±1, 639±1, 705±1, 867±1, 1036±1, 1053±1, 1105±1, 1171±1, 1231±1, 1277±1, 1375±1, 1391±1, 1452±1, 1476±1, 1553±1, 1596±1, 1639±1, 1664±1, 2852±1, 2907±1, 2928±1, 2967±1, 3168±1, 및 3357±1 cm-1에서 유효한 흡수 밴드를 가졌다.
실시예 14: X-선 회절 결정 구조 분석
0.43 x 0.05 x 0.031 mm 크기를 갖는 화합물 1의 황산염의 덩어리 결정을 유리 섬유 위에 놓았다. Cu Ka 조사를 사용하는 SMART 버젼 5.630 소프트웨어 (Bruker, 2003)에 의해 제어되고, 13.5 cm의 창 직경(window diameter)을 갖는 Bruker SMART 6K CCD x-선 면적 검출기를 사용하여, X-선 회절 결정 구조 데이타를 얻었다. 시료에서 검출기 거리는 5.039 cm이었다. -153±1℃의 온도에서 데이타를 모았고, SHELXS 버젼 6.14 (Bruker, 2003) 소프트웨어를 사용하여 분석하였다. 하기의 격자 파라미터를 도출하였다: 단위 격자는 a = 6.8239 Å, b = 16.2275 Å, c = 24.2021 Å, α= β= γ= 90°의 규모를 갖는 사방정계이고; 2680.0 Å3의 격자 부피(V); 계산된 밀도는 1.38 g/cm3이고; 공간군(space group)은 P212121(#19)이다. Mercury 1.4 소프트웨어에 따라 유도된 원자 위치로부터 예상한 분말 X-선 회절 피크는 시각적 검사에 의할 때 도 1의 실험 결과와 상당히 일치하는 것으로 판단되었다.
실시예 15: 고체 상태 안정성 평가
본 발명의 황산염 시료를 복수 개의 개발된 유리 바이알에서 20℃ 및 60% 상대 습도에서, 40℃ 및 75% 상대 습도에서 보관하였다. 특정한 간격으로, 대표적인 바이알의 내용물을 제거하였고, DSC, TGA, PXRD, 및 화학적 순도를 위해 HPLC로 분석하였다. 4주 보관한 후에, 두 개의 조건 중 어느 하나에서도 보관된 시료의 XRPD 패턴뿐만 아니라 DSC 또는 TGA 온도 기록도에서 검출가능한 변화가 없었다. 보관된 시료의 화학적 순도는 HPLC에 의할 때 99.7%에서 변하지 않았다.
실시예 16: 반대 이온 함유량(counterion content)의 결정
본 발명 황산염 시료를 음이온 자체-재생 억제제(anion self-regenerating suppressor), 전도도 검출기, IonPac AS11-HC 분석용 음이온-교환 컬럼, 및 IonPac AG11-HC 보조 컬럼이 장착된 Dionex ICS-2000 이온 크로마토그래피 시스템을 사용하는 황산염 이온 크로마토그래피에 의해 분석하였다. 시료 중 황산염 함량은 모 화합물(parent compound) 1 몰당 황산염 이온의 1 몰당 당량에 대한 17.6%의 이론적인 황산염 함량과 견줄 수 있는 17.1%인 것으로 결정되었다.
실시예 17: 수화물의 물 함량의 결정
최초의 중량 손실 동안에 증발되는 물질의 IR 분석과 연결된 TGA에 의해 본 발명 수화물 시료를 분석하였다. TGA 자취는 화합물 1 황산염의 일수화물에 대한 3.1%의 이론적인 중량 손실과 견줄 수 있는 100℃ 미만에서 3.2%의 중량 손실을 보여준다. 증발된 물질의 IR 스펙트럼은 물의 표준 IR 스펙트럼과 일치하였다.
분석 1: 인간 뮤(Mu), 인간 델타(Delta) 및 기니아 피그 카파(Kappa) 오피오이드 수용체에서 방사성리간드 결합 분석
a. 막 제조
인간 뮤 오피오이드 또는 기니아 피그 카파 수용체 cDNA로 안정하게 트랜스펙션된 CHO-K1(중국 햄스터 난소, Chinese Hamster Ovary) 세포를 10% FBS, 100 유닛/ml 페니실린 - 100 ㎍/mL 스트렙토마이신 및 800 ㎍/mL 제네티신(Geneticin)을보충한 Ham's-F12 배지로 구성되는 배지에서 5% CO2, 습한 배양기에서 37℃에서 길렀다. 수용체 발현 수준(Bmax ~2.0 및 -0.414 pmol/단백질 mg, 각각)은 막 방사성리간드 결합 분석에서 [3H]-디프레노르핀(Diprenorphine)(특이적 활성(specific activity) ~ 50-55 Ci/mmol)을 사용하여 결정하였다.
세포를 80-95% 컨플루언시(confluency) 상태로 길렀다(<25 계대 배양). 세포주 계대를 위해서, 세포 단일층을 실온에서 5분 동안 배양하였고, 5 mM EDTA를 보충한 PBS 10 mL에서 기계 교반으로 수확하였다. 재현탁한 다음, 세포를 40 mL 신선한 성장 배지로 옮겼고 1000 rpm에서 5분 동안 원심분리하였고, 적절한 분리 비율로 신선한 성장 배지에서 재현탁하였다.
막 제조를 위해서는, 세포를 PBS 중 5 mM EDTA로 약한 기계 교반으로 수확하였고 그 다음에 원심분리(5분 동안 2500 g)하였다. 얻은 펠렛을 pH 7.4의 분석용 버퍼(Assay Buffer)(50 mM 4-(2-히드록시에틸)피페라진-1-에탄술폰산 N-(2-히드록시에틸)피페라진-N-(2-에탄술폰산)(HEPES))에서 재현탁하였고, 얼음 위에서 폴리트론 세포 분쇄기(polytron disrupter)로 균질화시켰다. 그 결과 얻은 균질 현탁액을 원심분리(1200 g에서 5분)하였고, 얻은 펠렛을 버렸고,얻은 상층액을 원심분 리(40000 g에서 5분)하였다. 얻은 펠렛을 분석용 버퍼에서 재현탁에 의해 1회 세척하였고, 그 다음에 추가로 원심분리(40000 g에서 20분)하였다. 마지막으로 얻은 펠렛을 분석용 버퍼(등가물 1 T-225 플라스크/분석용 버퍼 1 mL)에서 재현탁하였다. 단백질 농도를 Bio-Rad Bradford Protein Assay 키트를 사용하여 결정하였고, 막을 필요할 때까지 동결시킨 분획 상태로 -80℃에서 보관하였다.
인간 델타 오피오이드 수용체(Human delta opioid receptor hDOP) 막을 Perkin Elmer로부터 구입하였다. [3H]-나트린돌(Natrindole) 방사성리간드 결합 분석에서 포화 분석에 의해 결정된 막의 기록된 KD 및 Bmax는 각각 0.14 nM (pKD= 9.85) 및 2.2 pmol/단백질 mg이었다. 단백질 농도는 Bio-Rad Bradford Protein Assay 키트를 사용하여 결정하였다. 막은 필요할 때까지 동결시킨 분획 상태로 -80℃에서 보관하였다.
b. 방사성리간드 결합 분석
방사성리간드 결합 분석은 0.025% 소혈청 알부민(bovine serum albumin BSA)을 보충한 분석용 버퍼에서 적절한 함량의 막 단백질(뮤, 델타 및 카파 각각 ~3, ~2 및 ~20㎍)을 포함하는 전체 분석 부피 200㎕에서 Axygen 1.1 mL 딥 웰(deep well) 96-웰(96-well) 폴리프로필렌 분석 플레이트에서 수행하였다. 방사성리간드의 KD 값을 결정하기 위한 포화 결합 연구는 0.001 nM - 5 nM 범위의 8-12개의 서로 다른 농도에서 [3H]-디프레노르핀을 사용하여 수행하였다. 화합물의 pKi 값을 결정하기 위한 치환 분석(displacement assay)은 뮤, 델타, 및 카파 각각 0.5, 1.2, 및 0.7 nM과 10 pM - 100 μM의 11개 농도의 화합물에서 [3H]-디프레노르핀으로 수행하였다.
결합 데이타는 1-부위 경쟁에 대한 3-파라미터 모델을 사용하는 GraphPad Prism Software 패키지(package)(GraphPad Software, Inc., San Diego, CA)를 갖는 비선형 회귀 분석법으로 분석하였다. 곡선의 최소값은 10 μM 날옥손(naloxone)의 존재 하에서 결정된 바와 같이, 비특이적 결합에 대한 값으로 고정하였다. 테스트 화합물의 Ki 값은 [L]이 [3H]-디프레노르핀의 농도인 쳉-프루소프 식(Cheng-Prusoff equation)(Ki = IC50/(l+([L]/Kd))을 사용하고, 최적의(best fit) IC50 값, 및 방사성리간드의 Kd 값으로부터 Prism에서 계산하였다. 결과는 Ki 값의 음의 십진 로그값인 pKi로 표현한다.
이 분석법에서 더 높은 pKi 값을 갖는 테스트 화합물은 뮤, 델타, 또는 카파 오피오이드 수용체에 대해 더 높은 결합 친화도를 갖는다. 화합물 1의 황산염은 인간 뮤 오피오이드 수용체에서 pKi 값 9.9를 나타내었다.
분석 2: 인간 뮤-오피오이드 수용체를 발현하는 CHO-K1 세포로부터 제조된 막에서 뮤-오피오이드 수용체의 아고니스트 매개된 활성화
이 분석법에서는, 테스트 화합물의 역가 및 고유 활성(intrinsic activity) 값을 인간 뮤 오피오이드 수용체를 발현하는 CHO-K1 세포로부터 제조된 막에서 수 용체 활성화 다음에, 존재하는 결합된 GTP-Eu의 함량을 측정함으로써 결정하였다.
a. 뮤 오피오이드 수용체 막 제조:
인간 뮤 오피오이드 수용체(Human mu opioid receptor hMOP) 막은 상기에서 기술한 바와 같이 제조하거나 또는 Perkin Elmer로부터 구입하였다. [3H]-디프레노르핀 방사성리간드 결합 분석에서 포화 분석법에 의해 결정된 구입된 막의 기록된 pKd 및 Bmax는 각각 10.06 및 2.4 pmol/단백질 mg이었다. 단백질 농도는 Bio-Rad Bradford Protein Assay 키트를 사용하여 결정하였다. 막은 필요할 때까지 동결시킨 분획 상태로 -80℃에서 보관하였다. 동결 건조된 GTP-Eu와 GDP를 이중 증류시킨 H2O에서 각각 10 μM 및 2 mM로 희석하였고, 그런 다음 혼합하였고, 실온에서 30분 동안 방치시켰고, -20℃에서 보관하기 위한 개개의 분획 시료로 옮겼다.
b. 인간 뮤 GTP-Eu 뉴클레오티드 교환 분석(Human mu GTP-Eu nucleotide exchange assay)
GTP-Eu 뉴클레오티드 교환 분석은 제조자의 설명서에 따라 AcroWell 96 웰 필터 플레이트에서 DELPHIA GTP-결합 키트(Perkin/Elmer)를 사용하여 수행하였다. 막을 상기에서 기술한 바와 같이 제조하였고, 분석을 시작하기 전에 분획을 분석용 버퍼(5OmM HEPES, 25℃에서 pH 7.4)에서 200 ㎍/mL의 농도로 희석하였고, 그런 다음 폴리트론 균질화기(Polytron homogenizer)를 사용하여 10초 동안 균질화시켰다. 테스트 화합물은 DMSO 중 10 mM 스톡 용액으로 수용되었고, 0.1 % BSA를 포함하는 분석용 버퍼에서 400 μM로 희석되었고, 및 40 pM - 80 μM 범위의 화합물의 10개 농도를 만들기 위한 계대 희석(serial dilution, 1:5)을 하였고, GDP 및 GTP-Eu를 분석용 버퍼에서 각각 4 μM 및 40 nM로 희석시켰다. 분석은 10 mM MgCl2, 50 mM NaCl, 및 0.0125% BSA에서 희석된 막 단백질 5㎍, 10 pM - 20μM 범위의 테스트 화합물, 1 μM GDP, 및 10 nM GTP-Eu를 포함하는 전체 100㎕의 부피에서 수행하였다(최종 분석 농도). DAMGO(Tyr-D-Ala-Gly-(메틸)Phe-Gly-올) 농도-반응성 곡선(12.8 pM - 1 μM의 범위)을 모든 플레이트에 포함시켰다.
분석 플레이트를 분석하기 전에 즉시 제조한 다음, 분석용 버퍼 25㎕, 테스트 화합물 25㎕, 및 GDP 및 GTP-Eu 25㎕를 첨가하였다. 막 단백질 25㎕를 첨가함으로써 분석을 개시하였고 30분 동안 배양시켰다. 그런 다음, 분석 플레이트를 10-12 in. Hg로 조절되는 중앙 집중식 진공(house vacuum)에 연결된 Waters 진공 매니폴드(vacuum manifold)로 여과하였고, 실온의 GTP Wash Solution(2 x 300 mL)으로 세척하였다. 플레이트의 아래 부분을 빨아들여, 초과량의 액체를 제거하였다. 그런 다음, 플레이트를 즉시 읽어, Packard Fusion Plate Reader Vehicle 상에서 Time Resolved Fluorescence(TRF)를 측정함으로써, 결합된 GTP-Eu의 함량을 결정하였다: DMSO는 최종 분석 농도 1%를 초과하지 않는다.
결합된 GTP-Eu의 함량은 테스트 화합물에 의한 뮤 오피오이드 수용체의 활성화 정도에 비례한다. 퍼센트로 표시되는 고유 활성(intrinsic activity IA)은 테스트 화합물에 의한 활성화에 대해 관찰된 결합된 GTP-Eu의 함량에 대하여 완전한 아고니스트(IA=1OO)로 추정되는 DAMG에 의한 활성화에 대해 관찰된 함량의 비율로 결정하였다. 화합물 1의 황산염은 이 분석에서 -5의 고유 활성을 나타내었다. 따라서, 본 발명의 황산염은 안타고니스트인 것으로 나타났다.
분석 3: 인 비보 효능 랫트 모델
이 분석에서는, 테스트 화합물의 효능을 위장관 통과(gastrointestinal transit) 모델에서 평가하였고, 이것은 말초 활성을 평가한다. 이 연구는 Theravance, Inc.의 Institutional Animal Care and Use Committee에서 승인을 받았고, 미국 국립 과학원(National Academy of Sciences)(ⓒ1996)에 의해 발행된 실험 동물의 관리와 사용에 관한 지침(Guide for the Care and Use of Laboratory Animals)와 일치한다.
a. 랫트 위 비움 분석 (Rat Gastric Emptying Assay)
테스트 화합물을 랫트 위 비움 분석법에서 평가하여, 이들 화합물의 로페라마이드-유도된 지연된 위 비움을 역행시키는 능력을 결정하였다. 랫트에게 0.001 내지 약 30 밀리그램/킬로그램(mg/kg) 범위의 투여량으로 정맥내, 피하, 근육내 또는 경구 투여 경로에 의해 테스트 화합물 또는 비히클(vehicle)을 투여하기 전에, 랫트를 밤새 절식시켰다. 테스트 화합물의 투여 다음에 1 mg/kg의 투여량으로 로페라마이드 또는 비히클을 피하 투여하였다. 로페라마이드 또는 비히클 투여하고 분 후에, 영양을 제공하지 않고, 흡수할 수 없는 활성탄 식이(charcoal meal)를 경구 위관 영양법으로 투여하였고, 동물들을 실험하는 60분 동안 물에 자유롭게 접근할 수 있도록 하였다. 그런 다음, 동물들을 이산화탄소 질식 상태로 안락사시켰고, 개흉술을 하였고, 위를 조심스럽게 절개하였다. 위를 아래쪽의 식도 괄약근과 유문 괄약근에서 연결시켜, 조직 제거 동안 추가적인 비움의 발생을 막았다. 봉합사를 제거한 후에 위 중량을 결정하였다.
b. 데이타 분석과 결과
데이타는 GraphPad Prism Software 패키지(package)(GraphPad Software, Inc., San Diego, CA)를 사용하여 분석하였다. S자 모양의 투여량 반응(가변 기울기) 모델을 사용하는 비-선형 회귀 분석에 의해서 퍼센트 역전 곡선(percent reversal curve)을 해석하였고, 최적의 ID50 값을 계산하였다. 곡선의 최소값과 최대값은 각각 로페라마이드 대조군 값(0% 역전을 나타냄)과 비히클 대조군 값(100% 역전을 나타냄)으로 고정하였다. 결과는 킬로그램 당 밀리그램으로서, 로페라마이드 효과의 50% 역전을 위해 필요한 투여량인 ID5O으로 표현하였다. 화합물 1의 황산염은 경구로 투여될 때, 위 비움 모델(gastric emptying model)에서 0.26 mg/kg의 ID5O값을 나타내었다.
본 발명이 특정 구체예 또는 그의 실시 형태를 참고로 하여 기술되어 있지만, 본 발명의 본래의 정신과 범위로부터 벗어나지 않는다면 다양하게 변화시킬 수 있거나 또는 등가물로 치환될 수 있다고 당해 분야의 당업자들은 이해할 것이다. 추가적으로, 적용가능한 특허 상태 및 규정에 의해 허용되는 정도로, 본 명세서에서 인용된 모든 간행물, 특허 및 특허 출원은 각각의 문서가 본 명세서에서 참조로서 개별적으로 통합되어 있는 것과 동일한 정도로 전문으로 본 명세서에서 참조로 통합되어 있다.
본 발명의 다양한 구체예는 첨부된 도면을 참조로 하여 설명된다.
도 1은 본 발명의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염의 x-선 분말 회절(XRPD) 패턴을 보여준다.
도 2는 본 발명의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염의 시차 주사 열량법(DSC) 자취(오른쪽의 수직 축)과 열 중량 분석(TGA) 자취(왼쪽의 수직 축)를 보여준다.
도 3은 본 발명의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염의 동적 수분 흡수(DMS) 자취를 보여준다.
도 4는 본 발명의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 결정형 수화물의 X-선 분말 회절(XRPD) 패턴을 보여준다.
도 5는 본 발명의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 결정형 수화물의 시차 주사 열량법(DSC) 자취(오른쪽 수직 축)와 열 중량 분석(TGA) 자취(왼쪽 수직 축)를 보여준다.
도 6은 본 발명의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로 피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 결정형 수화물의 동적 수분 흡수(DMS) 자취를 보여준다.
Claims (28)
- 6.58±0.20, 7.52±0.20, 9.35±0.20, 14.69±0.20, 16.01±0.20, 17.45±0.20, 17.99±0.20, 18.62±0.20, 19.76±0.20, 21.11±0.20, 22.07±0.20, 23.18±0.20, 23.74±0.20, 24.56±0.20, 25.63±0.20, 26.45±0.20, 27.86±0.20, 28.31±0.20, 29.54±0.20, 30.59±0.20, 31.58±0.20, 33.89±0.20, 및 36.02±0.20으로부터 선택되는 2θ 값에서 2개 이상의 회절 피크를 갖는 x-선 분말 회절 패턴을 특징으로 하는, 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염, 또는9.41±0.20, 9.98±0.20, 15.17±0.20, 16.70±0.20, 18.59±0.20, 19.46±0.20, 19.91±0.20, 20.63±0.20, 21.35±0.20, 21.89±0.20, 23.00±0.20, 24.20±0.20, 25.40±0.20, 26.03±0.20, 27.44±0.20, 28.46±0.20, 29.45±0.20, 31.22±0.20, 31.82±0.20, 33.17±0.20, 33.56±0.20, 및 36.89±0.20으로부터 선택되는 2θ 값에서 2개 이상의 회절 피크를 갖는 x-선 분말 회절 패턴을 특징으로 하는 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 수화물인 결정형 염 형태.
- 제1항에 있어서, 상기 결정형 염 형태는 결정형 황산염인 것인 결정형 염 형태.
- 삭제
- 제2항에 있어서, 상기 x-선 분말 회절 패턴은 14.69±0.20, 16.01±0.20, 21.11±0.20, 22.07±0.20, 및 23.18±0.20으로부터 선택되는 2θ 값에서 2개 이상의 회절 피크를 포함하는 것인 결정형 염 형태.
- 제2항에 있어서, 상기 결정형 염 형태는 피크 위치가 하기에 표시된 패턴의 피크 위치와 일치하는 x-선 분말 회절 패턴을 특징으로 하는 것인 결정형 염 형태:

- 제2항에 있어서, 상기 결정형 염 형태는 190℃ 내지 205℃의 온도에서 흡열성 열 흐름의 최대값을 보여주는, 1분당 10℃의 가열 속도로 기록된 시차 주사 열량 분석 자취(differential scanning calorimetry trace)를 특징으로 하는 것인 결정형 염 형태.
- 제2항에 있어서, 상기 결정형 염 형태는 하기에 표시된 것과 일치하는 시차 주사 열량 분석 자취를 특징으로 하는 것인 결정형 염 형태:

- 제1항에 있어서, 상기 결정형 염 형태는 황산염의 수화물인 것인 결정형 염 형태.
- 삭제
- 제8항에 있어서, 상기 x-선 분말 회절 패턴은 16.70±0.20, 18.59±0.20, 19.46±0.20, 19.91±0.20, 23.00±0.20, 및 24.20±0.20으로부터 선택되는 2θ 값에서 2개 이상의 회절 피크를 포함하는 것인 결정형 염 형태.
- 제8항에 있어서, 상기 결정형 염 형태는 피크 위치가 하기에 표시된 패턴의 피크 위치와 일치하는 x-선 분말 회절 패턴을 특징으로 하는 것인 결정형 염 형태:

- 제8항에 있어서, 상기 결정형 염 형태는 하기에 표시된 것과 일치하는 시차 주사 열량 분석 자취를 특징으로 하는 것인 결정형 염 형태:

- 약제학적으로 허용가능한 담체와 제1항, 제2항, 제4항 내지 제8항, 및 제10항 내지 제12항 중 어느 한 항의 결정형 염 형태를 포함하는, 오피오이드에 의해 유도된 장 기능 장애 또는 수술후 장폐색의 치료용 약제학적 조성물.
- 제1항, 제2항, 및 제4항 내지 제7항 중 어느 한 항의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염의 제조 방법으로서, 상기 방법은(a) 히드록시기가 보호된 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 보호된 전구체와 황산을 접촉시켜, 제1 반응 혼합물을 생성하는 단계;(b) 상기 제1 반응 혼합물로부터 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 중간 등급의 고체형 황산염을 분리하는 단계;(c) 상기 중간 등급의 고체형 황산염을 메탄올을 포함하는 희석제에 분산시켜, 제2 반응 혼합물을 생성하는 단계; 및(d) 상기 제2 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함하는 것인 방법.
- 제14항에 있어서, 상기 보호된 전구체는 (S)-2,2-디메틸-[1,3]디옥솔란-4-카르복시산 {2-[3-(3-카르바모일-페닐)-8-아자비시클로[3.2.1]옥트-8-일]에틸}시클로헥실메틸-아미드인 것인 방법.
- 제15항에 있어서, 상기 메탄올을 포함하는 희석제는 25% 이하의 물을 추가로 포함하는 것인 방법.
- 제15항에 있어서, 상기 메탄올을 포함하는 희석제는 5% 내지 15%의 물을 포함하는 것인 방법.
- 제1항, 제2항, 및 제4항 내지 제7항 중 어느 한 항의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염의 제조 방법으로서, 상기 방법은(a) 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드를 황산과 접촉시켜, 반응 혼합물을 생성하는 단계; 및(b) 상기 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함하는 것인 방법.
- 제1항, 제2항, 및 제4항 내지 제7항 중 어느 한 항의 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 결정형 황산염의 제조 방법으로서, 상기 방법은(a) 3-엔도-(8-{2-[시클로헥실메틸-((S)-2,3-디히드록시-프로피오닐)아미노]에틸}-8-아자-비시클로[3.2.1]옥트-3-일)벤즈아미드의 황산염의 결정형 수화물을 메탄올을 포함하는 희석제에서 분산시켜, 반응 혼합물을 생성하는 단계; 및(b) 상기 반응 혼합물로부터 결정형 황산염을 분리하는 단계를 포함하는 것인 방법.
- 제19항에 있어서, 상기 메탄올을 포함하는 희석제는 25% 이하의 물을 추가로 포함하는 것인 방법.
- N-시클로헥실메틸-(2-옥소에틸)-카르밤산 벤질 에스테르의 중아황산염 부가물.
- 치료에 사용하기 위한 제1항, 제2항, 제4항 내지 제8항, 및 제10항 내지 제12항 중 어느 한 항의 결정형 염 형태.
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90409007P | 2007-02-28 | 2007-02-28 | |
| US60/904,090 | 2007-02-28 | ||
| PCT/US2008/002583 WO2008106159A1 (en) | 2007-02-28 | 2008-02-27 | Crystalline forms of an 8-azabicyclo[3.2.1]octane compound |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20090115767A KR20090115767A (ko) | 2009-11-05 |
| KR101480530B1 true KR101480530B1 (ko) | 2015-01-08 |
Family
ID=39620376
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020097020094A KR101480530B1 (ko) | 2007-02-28 | 2008-02-27 | 8-아자비시클로[3.2.1]옥탄 화합물의 결정형 |
Country Status (27)
| Country | Link |
|---|---|
| US (7) | US7943772B2 (ko) |
| EP (1) | EP2132204B1 (ko) |
| JP (2) | JP5512289B2 (ko) |
| KR (1) | KR101480530B1 (ko) |
| CN (1) | CN101616916B (ko) |
| AR (2) | AR065554A1 (ko) |
| AU (1) | AU2008219615B2 (ko) |
| BR (1) | BRPI0807974B8 (ko) |
| CA (1) | CA2678073C (ko) |
| CL (1) | CL2008000573A1 (ko) |
| CO (1) | CO6220856A2 (ko) |
| CY (1) | CY1115077T1 (ko) |
| DK (1) | DK2132204T3 (ko) |
| ES (1) | ES2402031T3 (ko) |
| HK (1) | HK1139399A1 (ko) |
| HR (1) | HRP20130193T1 (ko) |
| IL (1) | IL200131A (ko) |
| MX (1) | MX2009009141A (ko) |
| MY (1) | MY148318A (ko) |
| NZ (1) | NZ578871A (ko) |
| PL (1) | PL2132204T3 (ko) |
| PT (1) | PT2132204E (ko) |
| RU (1) | RU2458061C2 (ko) |
| SI (1) | SI2132204T1 (ko) |
| TW (1) | TWI409067B (ko) |
| WO (1) | WO2008106159A1 (ko) |
| ZA (1) | ZA200905832B (ko) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY145633A (en) | 2006-03-01 | 2012-03-15 | Theravance Inc | 8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
| TWI409067B (zh) | 2007-02-28 | 2013-09-21 | Theravance Inc | 8-氮雜雙環〔3.2.1〕辛烷化合物之結晶型 |
| TWI415850B (zh) * | 2007-07-20 | 2013-11-21 | Theravance Inc | 製備mu類鴉片受體拮抗劑之中間物的方法 |
| TWI423801B (zh) | 2007-08-27 | 2014-01-21 | Theravance Inc | 作為μ類鴉片受體拮抗劑之8-氮雜雙環〔3.2.1〕辛基-2-羥基苯甲醯胺化合物 |
| EP2185553B1 (en) * | 2007-08-27 | 2012-06-27 | Theravance, Inc. | Amidoalkyl-8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
| EP2195313B1 (en) * | 2007-08-27 | 2014-04-02 | Theravance, Inc. | Heteroarylalkyl-8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
| EP2195314B1 (en) * | 2007-08-27 | 2011-03-23 | Theravance, Inc. | Disubstituted alkyl-8-azabicyclo [3.2.1.]octane compounds as mu opioid receptor antagonists |
| RU2561873C2 (ru) * | 2009-09-18 | 2015-09-10 | Адолор Корпорейшн | Применение антагонистов опиоидных рецепторов при заболеваниях желудочно-кишечного тракта |
| LT3277278T (lt) | 2015-04-02 | 2020-03-25 | Theravance Biopharma R&D Ip, Llc | Kombinuota vaisto forma mu opioidinio receptoriaus antagonisto ir opioidinio agento |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007103187A2 (en) * | 2006-03-01 | 2007-09-13 | Theravance, Inc. | 8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5159081A (en) | 1991-03-29 | 1992-10-27 | Eli Lilly And Company | Intermediates of peripherally selective n-carbonyl-3,4,4-trisubstituted piperidine opioid antagonists |
| US6900228B1 (en) | 1998-03-10 | 2005-05-31 | Research Triangle Institute | Opiate compounds, methods of making and methods of use |
| GB9810671D0 (en) | 1998-05-18 | 1998-07-15 | Pfizer Ltd | Anti-pruritic agents |
| TWI244481B (en) | 1998-12-23 | 2005-12-01 | Pfizer | 3-azabicyclo[3.1.0]hexane derivatives useful in therapy |
| GB9912410D0 (en) | 1999-05-28 | 1999-07-28 | Pfizer Ltd | Compounds useful in therapy |
| US6469030B2 (en) | 1999-11-29 | 2002-10-22 | Adolor Corporation | Methods for the treatment and prevention of ileus |
| US6467084B1 (en) * | 1999-12-16 | 2002-10-15 | Emware, Inc. | Systems and methods for reprogramming an embedded device with program code using relocatable program code |
| AU2001241676A1 (en) | 2000-03-03 | 2001-09-17 | Ortho-Mcneil Pharmaceutical, Inc. | 3-(diarylmethylene)-8-azabicyclo(3.2.1)octane derivatives |
| US20020025948A1 (en) | 2000-06-23 | 2002-02-28 | Banks Bernard Joseph | 3-azabicyclo[3.1.0]hexane derivatives useful in therapy |
| EP1440059B1 (en) | 2001-10-22 | 2008-04-16 | Pfizer Products Inc. | 3-azabicyclo(3.1.0)hexane derivatives as opioid receptor antagonists |
| US7381721B2 (en) | 2003-03-17 | 2008-06-03 | Adolor Corporation | Substituted piperidine compounds |
| CA2522323C (en) | 2003-04-14 | 2009-09-15 | Pfizer Products Inc. | 3-azabicyclo[3.2.1]octane derivatives as opioid receptor ligands |
| US20040204453A1 (en) | 2003-04-14 | 2004-10-14 | Pfizer Inc | 4-Phenyl-piperidine compounds and their use as modulators of opioid receptors |
| US7056930B2 (en) * | 2003-04-14 | 2006-06-06 | Pfizer Inc. | 2-Azabicyclo[3.3.1]nonane derivatives |
| CA2522199A1 (en) | 2003-04-15 | 2004-10-28 | Pfizer Products Inc. | 3-benzhydrylidene-8-aza-bicyclo¬3.2.1|octane derivatives with opioid receptor activity |
| US6992090B2 (en) | 2003-06-16 | 2006-01-31 | Adolor Corporation | Substituted piperidine compounds and methods of their use |
| US7435822B2 (en) | 2004-02-03 | 2008-10-14 | Janssen Pharmaceutica N.V. | 3-(diheteroarylmethylene)-8-azabicyclo[3.2.1]octane and 3-((aryl)(heteroaryl)methylene)-8-azabicyclo[3.2.1]octane derivatives |
| US7087749B2 (en) | 2004-03-11 | 2006-08-08 | Adolor Corporation | Substituted piperidine compounds and methods of their use |
| US20100284960A1 (en) | 2006-11-07 | 2010-11-11 | Nektar Therapeutics Al, Corporation | Dosage Forms and Co-Administration of Opioid Agonist and Opioid Antagonist |
| TWI409067B (zh) | 2007-02-28 | 2013-09-21 | Theravance Inc | 8-氮雜雙環〔3.2.1〕辛烷化合物之結晶型 |
-
2008
- 2008-02-15 TW TW097105472A patent/TWI409067B/zh active
- 2008-02-25 CL CL200800573A patent/CL2008000573A1/es unknown
- 2008-02-27 KR KR1020097020094A patent/KR101480530B1/ko active IP Right Grant
- 2008-02-27 US US12/072,534 patent/US7943772B2/en active Active
- 2008-02-27 JP JP2009551709A patent/JP5512289B2/ja active Active
- 2008-02-27 ES ES08726162T patent/ES2402031T3/es active Active
- 2008-02-27 CN CN2008800059690A patent/CN101616916B/zh active Active
- 2008-02-27 PT PT87261624T patent/PT2132204E/pt unknown
- 2008-02-27 MY MYPI20093401A patent/MY148318A/en unknown
- 2008-02-27 EP EP08726162A patent/EP2132204B1/en active Active
- 2008-02-27 CA CA2678073A patent/CA2678073C/en active Active
- 2008-02-27 RU RU2009135834/04A patent/RU2458061C2/ru active
- 2008-02-27 NZ NZ578871A patent/NZ578871A/en not_active IP Right Cessation
- 2008-02-27 SI SI200830928T patent/SI2132204T1/sl unknown
- 2008-02-27 MX MX2009009141A patent/MX2009009141A/es active IP Right Grant
- 2008-02-27 WO PCT/US2008/002583 patent/WO2008106159A1/en active Application Filing
- 2008-02-27 PL PL08726162T patent/PL2132204T3/pl unknown
- 2008-02-27 DK DK08726162.4T patent/DK2132204T3/da active
- 2008-02-27 AU AU2008219615A patent/AU2008219615B2/en not_active Ceased
- 2008-02-27 BR BRPI0807974A patent/BRPI0807974B8/pt active IP Right Grant
- 2008-02-28 AR ARP080100850A patent/AR065554A1/es not_active Application Discontinuation
-
2009
- 2009-07-29 IL IL200131A patent/IL200131A/en active IP Right Grant
- 2009-08-21 ZA ZA200905832A patent/ZA200905832B/xx unknown
- 2009-08-26 CO CO09089674A patent/CO6220856A2/es active IP Right Grant
-
2010
- 2010-06-15 HK HK10106008.5A patent/HK1139399A1/xx unknown
-
2011
- 2011-04-06 US US13/081,162 patent/US8247555B2/en active Active
-
2012
- 2012-07-17 US US13/550,781 patent/US8536335B2/en active Active
-
2013
- 2013-03-05 HR HRP20130193AT patent/HRP20130193T1/hr unknown
- 2013-04-08 CY CY20131100293T patent/CY1115077T1/el unknown
- 2013-05-10 JP JP2013099851A patent/JP2013151564A/ja not_active Withdrawn
- 2013-08-13 US US13/965,691 patent/US8816091B2/en active Active
-
2014
- 2014-07-16 US US14/333,003 patent/US9290491B2/en active Active
-
2016
- 2016-02-10 US US15/040,191 patent/US9949963B2/en active Active
-
2017
- 2017-07-31 AR ARP170102183A patent/AR109224A2/es unknown
-
2018
- 2018-03-13 US US15/919,678 patent/US10426766B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007103187A2 (en) * | 2006-03-01 | 2007-09-13 | Theravance, Inc. | 8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10426766B2 (en) | Crystalline forms of an 8-azabicyclo[3.2.1]octane compound | |
| US20210024515A1 (en) | 8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists | |
| EP2195314B1 (en) | Disubstituted alkyl-8-azabicyclo [3.2.1.]octane compounds as mu opioid receptor antagonists | |
| JP2010520198A5 (ko) | ||
| EP2185553B1 (en) | Amidoalkyl-8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists | |
| ES2465621T3 (es) | Compuestos de heteroarilalquil-8-azabiciclo[3.2.1]octano, como antagonistas de los receptores opiodes mu | |
| US8969367B2 (en) | Crystalline forms of a 3-carboxypropyl-aminotetralin compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| N231 | Notification of change of applicant | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| FPAY | Annual fee payment |
Payment date: 20171226 Year of fee payment: 4 |
|
| FPAY | Annual fee payment |
Payment date: 20181220 Year of fee payment: 5 |
|
| FPAY | Annual fee payment |
Payment date: 20191219 Year of fee payment: 6 |