JP7545622B2 - 5,6-ジドヒロチエノ[3,4-h]キナゾリン系化合物 - Google Patents
5,6-ジドヒロチエノ[3,4-h]キナゾリン系化合物 Download PDFInfo
- Publication number
- JP7545622B2 JP7545622B2 JP2023547663A JP2023547663A JP7545622B2 JP 7545622 B2 JP7545622 B2 JP 7545622B2 JP 2023547663 A JP2023547663 A JP 2023547663A JP 2023547663 A JP2023547663 A JP 2023547663A JP 7545622 B2 JP7545622 B2 JP 7545622B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- added
- μmol
- crude product
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 125000002294 quinazolinyl group Chemical class N1=C(N=CC2=CC=CC=C12)* 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 459
- 125000000217 alkyl group Chemical group 0.000 claims description 59
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 34
- 150000003839 salts Chemical class 0.000 claims description 34
- 206010028980 Neoplasm Diseases 0.000 claims description 29
- 125000003545 alkoxy group Chemical group 0.000 claims description 26
- 229910052740 iodine Inorganic materials 0.000 claims description 20
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 18
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 18
- 108010056274 polo-like kinase 1 Proteins 0.000 claims description 18
- 102100031463 Serine/threonine-protein kinase PLK1 Human genes 0.000 claims description 17
- 125000004455 (C1-C3) alkylthio group Chemical group 0.000 claims description 15
- 101100054666 Streptomyces halstedii sch3 gene Proteins 0.000 claims description 14
- 125000003282 alkyl amino group Chemical group 0.000 claims description 14
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 14
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims description 11
- 125000000304 alkynyl group Chemical group 0.000 claims description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 10
- 239000003112 inhibitor Substances 0.000 claims description 10
- 125000004193 piperazinyl group Chemical group 0.000 claims description 10
- 206010009944 Colon cancer Diseases 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 9
- 125000005842 heteroatom Chemical group 0.000 claims description 9
- 229910052717 sulfur Inorganic materials 0.000 claims description 9
- 125000004429 atom Chemical group 0.000 claims description 8
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 7
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 239000012634 fragment Substances 0.000 claims description 5
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 5
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 claims description 4
- 229940079593 drug Drugs 0.000 claims description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 273
- 238000006243 chemical reaction Methods 0.000 description 177
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 156
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 147
- 239000012043 crude product Substances 0.000 description 136
- 239000000243 solution Substances 0.000 description 133
- 230000002829 reductive effect Effects 0.000 description 119
- 230000015572 biosynthetic process Effects 0.000 description 109
- 238000003786 synthesis reaction Methods 0.000 description 109
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 103
- 239000000203 mixture Substances 0.000 description 95
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 94
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 90
- 239000012074 organic phase Substances 0.000 description 82
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 77
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 74
- 238000005481 NMR spectroscopy Methods 0.000 description 72
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 63
- -1 CH2OCH3 Chemical group 0.000 description 61
- 239000002904 solvent Substances 0.000 description 58
- 239000012071 phase Substances 0.000 description 56
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 52
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 47
- 239000000706 filtrate Substances 0.000 description 45
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 43
- 238000010898 silica gel chromatography Methods 0.000 description 42
- 229910001873 dinitrogen Inorganic materials 0.000 description 39
- 238000010828 elution Methods 0.000 description 39
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 36
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 34
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 30
- 238000012360 testing method Methods 0.000 description 30
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 29
- 239000007864 aqueous solution Substances 0.000 description 28
- 238000002953 preparative HPLC Methods 0.000 description 28
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 27
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 27
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 27
- 239000001099 ammonium carbonate Substances 0.000 description 27
- 239000003208 petroleum Substances 0.000 description 26
- 238000002360 preparation method Methods 0.000 description 26
- 238000004128 high performance liquid chromatography Methods 0.000 description 24
- 239000003643 water by type Substances 0.000 description 24
- 238000005160 1H NMR spectroscopy Methods 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 235000019270 ammonium chloride Nutrition 0.000 description 18
- 238000000926 separation method Methods 0.000 description 17
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 13
- 235000011114 ammonium hydroxide Nutrition 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 238000004809 thin layer chromatography Methods 0.000 description 12
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 11
- 241001465754 Metazoa Species 0.000 description 11
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- HXRAMSFGUAOAJR-UHFFFAOYSA-N n,n,n',n'-tetramethyl-1-[(2-methylpropan-2-yl)oxy]methanediamine Chemical compound CN(C)C(N(C)C)OC(C)(C)C HXRAMSFGUAOAJR-UHFFFAOYSA-N 0.000 description 11
- 238000010791 quenching Methods 0.000 description 11
- 229920006395 saturated elastomer Polymers 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 10
- ZEEBGORNQSEQBE-UHFFFAOYSA-N [2-(3-phenylphenoxy)-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound C1(=CC(=CC=C1)OC1=NC(=CC(=C1)CN)C(F)(F)F)C1=CC=CC=C1 ZEEBGORNQSEQBE-UHFFFAOYSA-N 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 8
- 229910000024 caesium carbonate Inorganic materials 0.000 description 8
- 238000000605 extraction Methods 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- 238000000034 method Methods 0.000 description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 8
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 8
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 8
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- QHLVBNKYJGBCQJ-UHFFFAOYSA-N 1-(2-hydroxyethyl)-8-[5-(4-methylpiperazin-1-yl)-2-(trifluoromethoxy)anilino]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CN(C)CCN1C1=CC=C(OC(F)(F)F)C(NC=2N=C3C=4N(CCO)N=C(C=4CCC3=CN=2)C(N)=O)=C1 QHLVBNKYJGBCQJ-UHFFFAOYSA-N 0.000 description 7
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 7
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 7
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 7
- BYRPTKZOXNFFDB-UHFFFAOYSA-N lithium;bis(trimethylsilyl)azanide;oxolane Chemical compound [Li+].C1CCOC1.C[Si](C)(C)[N-][Si](C)(C)C BYRPTKZOXNFFDB-UHFFFAOYSA-N 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- 108091000080 Phosphotransferase Proteins 0.000 description 6
- 102000020233 phosphotransferase Human genes 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- 101000729945 Homo sapiens Serine/threonine-protein kinase PLK2 Proteins 0.000 description 5
- 101000691614 Homo sapiens Serine/threonine-protein kinase PLK3 Proteins 0.000 description 5
- 101000582914 Homo sapiens Serine/threonine-protein kinase PLK4 Proteins 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 102100031462 Serine/threonine-protein kinase PLK2 Human genes 0.000 description 5
- 102100026209 Serine/threonine-protein kinase PLK3 Human genes 0.000 description 5
- 102100030267 Serine/threonine-protein kinase PLK4 Human genes 0.000 description 5
- SAHIZENKTPRYSN-UHFFFAOYSA-N [2-[3-(phenoxymethyl)phenoxy]-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound O(C1=CC=CC=C1)CC=1C=C(OC2=NC(=CC(=C2)CN)C(F)(F)F)C=CC=1 SAHIZENKTPRYSN-UHFFFAOYSA-N 0.000 description 5
- 125000005219 aminonitrile group Chemical group 0.000 description 5
- 208000029742 colonic neoplasm Diseases 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 239000011259 mixed solution Substances 0.000 description 5
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 4
- 101000582926 Dictyostelium discoideum Probable serine/threonine-protein kinase PLK Proteins 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 239000005909 Kieselgur Substances 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 229910002092 carbon dioxide Inorganic materials 0.000 description 4
- 229940125758 compound 15 Drugs 0.000 description 4
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 229940015915 onvansertib Drugs 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 4
- 230000003285 pharmacodynamic effect Effects 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 229930195734 saturated hydrocarbon Natural products 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 3
- 238000011729 BALB/c nude mouse Methods 0.000 description 3
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- 206010052358 Colorectal cancer metastatic Diseases 0.000 description 3
- 229940126657 Compound 17 Drugs 0.000 description 3
- 206010052804 Drug tolerance Diseases 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- 229940125797 compound 12 Drugs 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229940125810 compound 20 Drugs 0.000 description 3
- 229940126086 compound 21 Drugs 0.000 description 3
- 229940126208 compound 22 Drugs 0.000 description 3
- 229940125833 compound 23 Drugs 0.000 description 3
- 238000007405 data analysis Methods 0.000 description 3
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical group CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000010253 intravenous injection Methods 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 238000004237 preparative chromatography Methods 0.000 description 3
- ROSDSFDQCJNGOL-UHFFFAOYSA-N protonated dimethyl amine Natural products CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 3
- 125000003003 spiro group Chemical group 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- HAKVNWBUOCSHTR-UHFFFAOYSA-N (9,9-dimethylxanthen-1-yl)-diphenylphosphane Chemical compound C=12C(C)(C)C3=CC=CC=C3OC2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 HAKVNWBUOCSHTR-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 2
- KVIZTDNKHOCNAM-UHFFFAOYSA-N 4-methylpiperazin-2-one Chemical compound CN1CCNC(=O)C1 KVIZTDNKHOCNAM-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000031648 Body Weight Changes Diseases 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- 229910020700 Na3VO4 Inorganic materials 0.000 description 2
- 102000014434 POLO box domains Human genes 0.000 description 2
- 108050003399 POLO box domains Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 2
- 229910000564 Raney nickel Inorganic materials 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 230000004579 body weight change Effects 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 239000005018 casein Substances 0.000 description 2
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 2
- 235000021240 caseins Nutrition 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 2
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 229940071870 hydroiodic acid Drugs 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 238000003674 kinase activity assay Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 2
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 239000010413 mother solution Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 238000004467 single crystal X-ray diffraction Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- VVKMHTWFAUCCOD-UHFFFAOYSA-N 1-(3-aminopropyl)-8-[3-[2-(dimethylamino)-2-oxoethyl]anilino]-n-[(2-methylpyridin-4-yl)methyl]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical group CN(C)C(=O)CC1=CC=CC(NC=2N=C3C=4N(CCCN)N=C(C=4CCC3=CN=2)C(=O)NCC=2C=C(C)N=CC=2)=C1 VVKMHTWFAUCCOD-UHFFFAOYSA-N 0.000 description 1
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- MEBFFOKESLAUSJ-UHFFFAOYSA-N 2-[tert-butyl(dimethyl)silyl]oxyacetaldehyde Chemical compound CC(C)(C)[Si](C)(C)OCC=O MEBFFOKESLAUSJ-UHFFFAOYSA-N 0.000 description 1
- LSTRKXWIZZZYAS-UHFFFAOYSA-N 2-bromoacetyl bromide Chemical compound BrCC(Br)=O LSTRKXWIZZZYAS-UHFFFAOYSA-N 0.000 description 1
- HNNUBQWDWJNURV-UHFFFAOYSA-N 2-bromoethynyl-tri(propan-2-yl)silane Chemical compound CC(C)[Si](C(C)C)(C(C)C)C#CBr HNNUBQWDWJNURV-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- ROADCYAOHVSOLQ-UHFFFAOYSA-N 3-oxetanone Chemical compound O=C1COC1 ROADCYAOHVSOLQ-UHFFFAOYSA-N 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- 206010000117 Abnormal behaviour Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 101100408813 Drosophila melanogaster polo gene Proteins 0.000 description 1
- 102100030011 Endoribonuclease Human genes 0.000 description 1
- 101710199605 Endoribonuclease Proteins 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 101710113029 Serine/threonine-protein kinase Proteins 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ABRVLXLNVJHDRQ-UHFFFAOYSA-N [2-pyridin-3-yl-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound FC(C1=CC(=CC(=N1)C=1C=NC=CC=1)CN)(F)F ABRVLXLNVJHDRQ-UHFFFAOYSA-N 0.000 description 1
- WDZVWBWAUSUTTO-UHFFFAOYSA-N [bromo(difluoro)methyl]-trimethylsilane Chemical compound C[Si](C)(C)C(F)(F)Br WDZVWBWAUSUTTO-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- RBYGDVHOECIAFC-UHFFFAOYSA-L acetonitrile;palladium(2+);dichloride Chemical compound [Cl-].[Cl-].[Pd+2].CC#N.CC#N RBYGDVHOECIAFC-UHFFFAOYSA-L 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- XQPRBTXUXXVTKB-UHFFFAOYSA-M caesium iodide Chemical compound [I-].[Cs+] XQPRBTXUXXVTKB-UHFFFAOYSA-M 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- STIAPHVBRDNOAJ-UHFFFAOYSA-N carbamimidoylazanium;carbonate Chemical compound NC(N)=N.NC(N)=N.OC(O)=O STIAPHVBRDNOAJ-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 238000002038 chemiluminescence detection Methods 0.000 description 1
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- YOXHCYXIAVIFCZ-UHFFFAOYSA-N cyclopropanol Chemical compound OC1CC1 YOXHCYXIAVIFCZ-UHFFFAOYSA-N 0.000 description 1
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 1
- 230000002380 cytological effect Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- XGZNHFPFJRZBBT-UHFFFAOYSA-N ethanol;titanium Chemical compound [Ti].CCO.CCO.CCO.CCO XGZNHFPFJRZBBT-UHFFFAOYSA-N 0.000 description 1
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000005917 in vivo anti-tumor Effects 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- JCIVHYBIFRUGKO-UHFFFAOYSA-N lithium;2,2,6,6-tetramethylpiperidine Chemical compound [Li].CC1(C)CCCC(C)(C)N1 JCIVHYBIFRUGKO-UHFFFAOYSA-N 0.000 description 1
- YTJXGDYAEOTOCG-UHFFFAOYSA-N lithium;di(propan-2-yl)azanide;oxolane Chemical compound [Li+].C1CCOC1.CC(C)[N-]C(C)C YTJXGDYAEOTOCG-UHFFFAOYSA-N 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 150000004701 malic acid derivatives Chemical class 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 239000012982 microporous membrane Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 1
- JKEMAHLSSQQCDX-UHFFFAOYSA-N n,n-bis(methylamino)formamide Chemical compound CNN(NC)C=O JKEMAHLSSQQCDX-UHFFFAOYSA-N 0.000 description 1
- DQZSVGBAVRIWMP-UHFFFAOYSA-N n,n-bis(trimethylsilyl)hexan-1-amine Chemical compound CCCCCCN([Si](C)(C)C)[Si](C)(C)C DQZSVGBAVRIWMP-UHFFFAOYSA-N 0.000 description 1
- ILBIXZPOMJFOJP-UHFFFAOYSA-N n,n-dimethylprop-2-yn-1-amine Chemical compound CN(C)CC#C ILBIXZPOMJFOJP-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003506 n-propoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- YWWARDMVSMPOLR-UHFFFAOYSA-M oxolane;tetrabutylazanium;fluoride Chemical compound [F-].C1CCOC1.CCCC[N+](CCCC)(CCCC)CCCC YWWARDMVSMPOLR-UHFFFAOYSA-M 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003246 quinazolines Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- CQLFBEKRDQMJLZ-UHFFFAOYSA-M silver acetate Chemical compound [Ag+].CC([O-])=O CQLFBEKRDQMJLZ-UHFFFAOYSA-M 0.000 description 1
- 229940071536 silver acetate Drugs 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- SANWDQJIWZEKOD-UHFFFAOYSA-N tributyl(furan-2-yl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CC=CO1 SANWDQJIWZEKOD-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description
CN202110172946.6、出願日は:2021年02月08日であり、
CN202110655630.2、出願日は:2021年06月11日であり、
CN202110864387.5、出願日は:2021年07月29日であり、
CN202111137961.3、出願日は:2021年09月27日であり、
CN202111473661.2、出願日は2021年11月29日である。

T1は、CR1及びNから選択され、
T2は、CH及びNから選択され、
R1は、Hから選択され、
R2は、H、CN、C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ、S(=O)2C1-3アルキル、C2-3アルキニル、C3-5シクロアルキル、-O-C3-5シクロアルキル及び5員ヘテロアリールから選択され、前記C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ、S(=O)2C1-3アルキル、C2-3アルキニル、C3-5シクロアルキル、-O-C3-5シクロアルキル及び5員ヘテロアリールは、任意選択で1、2又は3つのRbにより置換され、
R3は、C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルから選択され、前記C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルは、任意選択で1、2又は3つのRcにより置換され、
R4は、C1-3アルキル及びC1-3アルコキシから選択され、前記C1-3アルキル及びC1-3アルコキシは、任意選択で1、2又は3つのRdにより置換され、
R5は、H及びOHから選択され、
或いは、R1及びR3はそれらに連結された原子と環を形成し、構造フラグメント

各Rbは、それぞれ独立してF、Cl、Br、I、OH及びOCH3から選択され、
各Rcは、それぞれ独立して=O、C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルから選択され、前記C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルは、任意選択で1、2又は3つのRにより置換され、
各Rdは、それぞれ独立してF、Cl、Br及びIから選択され、
各Rは、それぞれ独立してF、Cl、Br、I及びOHから選択され、
前記「ヘテロシクロブチル」及び「7~9員ヘテロシクロアルキル」のヘテロ原子は、N、O及びSから選択される。









T1は、CR1及びNから選択され、
T2は、CH及びNから選択され、
R1は、Hから選択され、
R2は、H、CN、C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ、S(=O)2C1-3アルキル、C2-3アルキニル、C3-5シクロアルキル、-O-C3-5シクロアルキル及び5員ヘテロアリールから選択され、前記C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ、S(=O)2C1-3アルキル、C2-3アルキニル、C3-5シクロアルキル、-O-C3-5シクロアルキル及び5員ヘテロアリールは、任意選択で1、2又は3つのRbにより置換され、
R3は、C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルから選択され、前記C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルは、任意選択で1、2又は3つのRcにより置換され、
R4は、C1-3アルキル及びC1-3アルコキシから選択され、前記C1-3アルキル及びC1-3アルコキシは、任意選択で1、2又は3つのRdにより置換され、
或いは、R1及びR3はそれらに連結された原子と環を形成し、構造フラグメント

各Rbは、それぞれ独立してF、Cl、Br、I、OH及びOCH3から選択され、
各Rcは、それぞれ独立して=O、C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルから選択され、前記C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルは、任意選択で1、2又は3つのRにより置換され、
各Rdは、それぞれ独立してF、Cl、Br及びIから選択され、
各Rは、それぞれ独立してF、Cl、Br、I及びOHから選択され、
前記「ヘテロシクロブチル」及び「7~9員ヘテロシクロアルキル」のヘテロ原子は、N、O及びSから選択される。


本発明の一部の形態において、前記R2は、H、CN、SCH3、SCH2CH2OH、S(=O)2CH3、OCH2CH3、OCH2CH2OH、OCH2CH2OCH3、CH3、CH2CH2OH、CH2OCH3、シクロプロピル、







T1は、CR1及びNから選択され、
T2は、CH及びNから選択され、
R1は、Hから選択され、
R2は、H、CN、C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ及びC3-5シクロアルキルから選択され、前記C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ及びC3-5シクロアルキルは、任意選択で1、2又は3つのRbにより置換され、
R3は、C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルから選択され、前記C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルは、任意選択で1、2又は3つのRcにより置換され、
R4は、C1-3アルキル及びC1-3アルコキシから選択され、前記C1-3アルキル及びC1-3アルコキシは、任意選択で1、2又は3つのRdにより置換され、
或いは、R1及びR3はそれらに連結された原子と環を形成し、構造フラグメント

各Raは、それぞれ独立してF、Cl、Br、I、CH3及びCF3から選択され、
各Rbは、それぞれ独立してF、Cl、Br、I、OH及びOCH3から選択され、
各Rcは、それぞれ独立して=O、C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルから選択され、前記C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルは、任意選択で1、2又は3つのRにより置換され、
各Rdは、それぞれ独立してF、Cl、Br及びIから選択され、
各Rは、それぞれ独立してF、Cl、Br、I及びOHから選択され、
前記「ヘテロシクロブチル」及び「7~9員ヘテロシクロアルキル」のヘテロ原子は、N、O及びSから選択される。






T1は、CR1及びNから選択され、
T2は、CH及びNから選択され、
R1は、Hから選択され、
R2は、C1-3アルキル、C1-3アルコキシ及びC1-3アルキルチオから選択され、前記C1-3アルキル、C1-3アルコキシ及びC1-3アルキルチオは、任意選択で1、2又は3つのRbにより置換され、
R3は、C1-4アルキル及びピペラジニルから選択され、前記C1-4アルキル及びピペラジニルは、任意選択で1、2又は3つのRcにより置換され、
R4は、C1-3アルキル及びC1-3アルコキシから選択され、前記C1-3アルキル及びC1-3アルコキシは、任意選択で1、2又は3つのRdにより置換され、
或いは、R1及びR3はそれらに連結された原子とピロリジニルを形成し、前記ピロリジニルは、任意選択で

各Raは、それぞれ独立してF、Cl、Br、I、CH3及びCF3から選択され、
各Rbは、それぞれ独立してF、Cl、Br、I及びOHから選択され、
各Rcは、それぞれ独立してC1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルから選択され、前記C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルは、任意選択で1、2又は3つのRにより置換され、
各Rdは、それぞれ独立してF、Cl、Br及びIから選択され、
各Rは、それぞれ独立してF、Cl、Br、I及びOHから選択され、
前記ヘテロシクロブチルのヘテロ原子は、N、O及びSから選択される。
本発明の一部の形態において、前記R2は、SCH3、SCH2CH3、SCH(CH3)2、OCH3、OCH2CH3、OCH(CH3)2、CH3、CH2CH3及びCH(CH3)2から選択され、前記SCH3、SCH2CH3、SCH(CH3)2、OCH3、OCH2CH3、OCH(CH3)2、CH3、CH2CH3及びCH(CH3)2は、任意選択で1、2又は3つのRbにより置換され、他の変量は本発明で定義された通りである。





ニルを形成し、構造フラグメント





Rcは、本発明で定義された通りであり、
L1は、O及びSから選択され、
R6は、C1-3アルキル及びC3-5シクロアルキルから選択される。


別途に説明しない限り、本明細書で用いられる以下の用語及び連語は以下の意味を含む
。一つの特定の用語又は連語は、特別に定義されない場合、不確定又は不明瞭ではなく、普通の定義として理解されるべきである。本明細書で商品名が出た場合、相応の商品又はその活性成分を指す。
そのうち一つの変量が単結合の場合、それで連結する2つの基が直接連結し、例えばA-L-ZにおけるLが単結合を表す場合、この構造は実際にA-Zになる。










個の環原子は独立してO、S及びNのヘテロ原子から選ばれ、残りは炭素原子である。窒素原子が任意に四級化されており、窒素及び硫黄ヘテロ原子は任意に酸化される(即ち、NO及びS(O)P、pは1又は2である。)。それは、単環式及び二環式環系を含み、ここで、二環式環系にはスピロ環、縮合環及び架橋環が含まれる。更に、「7~9員ヘテロシクロアルキル」に関して、ヘテロ原子はヘテロシクロアルキルと分子他の部分の連結位置を占めることができる。前記7~9員ヘテロシクロアルキルは、7員、8員及び9員ヘテロシクロアルキルを含む。7~9員ヘテロシクロアルキルの実例には、

本発明に使用されたすべての溶媒は市販品から得ることができる。
本発明の化合物は、PLK1に対してより優れた阻害効果を有し、且つPLK1を選択的に阻害することができ、細胞増殖に対してより優れた阻害活性を示し、in vivo薬効研究において有意な抗腫瘍効果を示し、動物に良好な耐薬性を示している。本発明の
化合物は、良好な薬物動態学的特性を有している。

化合物1-1(500mg、1.85mmol、1eq)をテトラヒドロフラン(5mL)に溶解させ、20℃でtert-ブトキシビス(ジメチルアミノ)メタン(966.93mg、5.55mol、3eq)を加え、添加完了後、90℃に昇温させ12時間反応させ、LCMSで原料の消失を検出した後、20℃に冷却させ、オイルポンプで減圧濃縮して溶媒を除去して化合物1-2を得、当該粗生成物を直接に次のステップの反応に使用した。
1H NMR (400 MHz, CDCl3) δ: 1.30 (t, J = 7.04 Hz, 3 H), 2.51 (s, 3 H), 2.78 - 2.82 (m, 2 H), 3.05 (s, 6 H), 3.07 - 3.11 (m, 2 H), 4.25 (q, J = 7.04 Hz, 2 H), 7.52 (s, 1 H)。
化合物1-2(180mg、553.09μmol、1eq)及び化合物1-3(193.05mg、608.40μmol、1.1eq)をN,N-ジメチルホルムアミド(4mL)に順次に溶解させ、110℃に昇温させて20時間反応させ、LCMSで原料1-2の消失を検出した後、水(20mL)を加えて反応系を希釈し、酢酸エチル(20mL×3)で抽出し、分離し、有機相を合わせ、飽和食塩水(50mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾液を濾過し、減圧濃縮して粗生成物を得、粗生成物を薄層ク
ロマトグラフィー(展開剤:ジクロロメタン:メタノール=20:1)で分離して化合物1-4を得た。
LCMS: m/z (ESI) = 580.17 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.46 - 1.50 (m, 3 H), 2.30 (s, 3 H), 2.53 (s, 4 H), 2.58 - 2.62 (m, 3 H), 2.66 - 2.70 (m, 2 H), 3.09 - 3.32 (m, 6 H), 4.25 - 4.29 (m,
2 H), 6.35 - 6.45 (m, 1 H), 7.03 - 7.07
(m, 1 H), 7.24 (s, 1 H), 8.21 (s, 1 H),
8.32 - 8.36 (m, 1 H)。
化合物1-4(90mg、155.26μmol、1eq)をテトラヒドロフラン(1mL)に溶解させ、0℃に冷却させ、塩化アンモニウム(49.83mg、931.59μmol、6.0eq)を加え、1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(1.55mL、10eq)を加え、添加完了後、25℃に昇温させ、当該温度で2時間反応させた。エタノール(2mL)を加えて反応をクエンチングさせ、減圧濃縮して溶媒を除去して粗生成物を得、粗生成物を分取HPLC(HPLC製造方法:Phenomenex分取クロマトグラフ;カラム:C18 80×40mm×3μm;移動相A:0.05%のアンモニア水を含む水溶液、移動相B:アセトニトリル;実行勾配:B%:38%~68%、8分間実行。)で分離・精製して化合物1を得た。
LCMS: m/z (ESI) = 551.15 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 2.22 (s, 3 H), 2.43 - 2.46 (m, 4 H), 2.54 (s, 3 H), 2.72 - 2.76 (m, 2 H), 3.08 - 3.18 (m, 6 H), 6.68 - 6.73 (m, 1 H), 7.15 - 7.21 (m,
1 H), 7.44 (s, 2 H), 7.48 - 7.54 (m, 1 H), 8.33 (s, 1 H), 8.48 (s, 1 H)。

ナトリウムtert-ブトキシド(127.13mg、1.32mmol、2eq)をテトラヒドロフラン(1.6mL)に溶解させ、20℃でエタノール(154μL、1.32mmol、1.9eq)を加え、30分間撹拌した後反応系を0℃に冷却させ、化合物2-1(200mg、661.45μmol、1eq)を加え、添加完了後、1時間反応を続け、反応完了後、水(20mL)を加えて反応をクエンチングさせ、酢酸エチル(30mL×3)で抽出し、有機相を合わせ、有機相を飽和食塩水(50mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液:石油エーテル:酢酸エチル=4:1)で精製して化合物2-2を得た。
1H NMR (400 MHz, CDCl3) δ: 1.38 - 1.42 (m, 3 H), 1.62 - 1.67 (m, 3 H), 2.02 - 2.14 (m, 2 H), 2.49 - 2.60 (m, 2 H), 3.20 - 3.27 (m, 2 H), 4.34 - 4.38 (m, 4 H)。
化合物2-2(80mg、298.14μmol、1eq)をテトラヒドロフラン(0.5mL)に溶解させ、tert-ブトキシビス(ジメチルアミノ)メタン(156.4mg、894.10μmol、3eq)を加え、90℃に昇温させて12時間反応させた。反応完了後20℃に冷却させ、オイルポンプで減圧濃縮して溶媒を除去して化合物2-3を得、当該粗生成物を直接に次のステップの反応に使用した。
1H NMR (400 MHz, CDCl3) δ: 1.26 - 1.32 (m, 3 H), 1.45 - 1.52 (m, 3 H), 2.73 - 2.76 (m, 2 H), 3.00 - 3.07 (m, 8 H), 4.19 - 4.25 (m, 4 H), 7.51 (s, 1 H)。
化合物2-3(10mg、30.92μmol、1eq)及び化合物1-3(8.33mg、26.26μmol、0.849eq)をN,N-ジメチルホルムアミド(0.5mL)に順次に溶解させ、110℃に昇温させて12時間反応させ、反応完了後、水(2mL)を加えて反応系を希釈し、酢酸エチル(2mL×3)で抽出し、有機相を合わせ、飽和食塩水(3mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、粗生成物を薄層クロマトグラフィー(展開剤:ジクロロメタン:メタノール=20:1)で分離して化合物2-4を得た。
LCMS: m/z (ESI) = 578.20 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.30 - 1.33 (m, 3 H), 1.45 - 1.50 (m, 3 H), 2.30 (s, 3 H), 2.53 (s, 4 H), 2.68 - 2.72 (m, 2 H), 3.15 - 3.25 (m, 6 H), 4.25 - 4.29 (m,
4 H), 4.36 - 4.43 (m , 1 H), 7.01 - 7.08 (m, 1 H), 7.24 (s, 1 H), 8.21 (s, 1 H), 8.30 - 8.37 (m, 1 H)。
化合物2-4(80mg、138.50μmol、1eq)をテトラヒドロフラン(0.5mL)に溶解させ、塩化アンモニウム(45mg、831.00μmol、6eq)を加えて反応系を0℃に冷却させ、1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(1.39mL、1.39mmol、10eq)をゆっくりと滴下し、添加完了後、20℃にゆっくりと昇温させ、当該温度で2時間反応させ、エタノール(3mL)を加えて反応をクエンチングさせ、減圧濃縮して溶媒を除去して粗生成物を得、粗生成物を分取HPLC(高速液体製造方法:Phenomenex分取クロマトグラフ;カラム:C18 80×40mm×3μm;移動相A:0.05%のアンモニア水を含む水溶液、移動相B:アセトニトリル;実行勾配:B%:36%~66%、8分間実行。)で分離・精製して化合物2を得た。
LCMS: m/z (ESI) = 549.19 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.27 - 1.37 (m, 3 H), 2.22 (s, 3 H), 2.34 (s, 3 H), 2.68 (s, 2 H), 3.04 - 3.20 (m, 7 H), 4.19
- 4.26 (m, 2 H), 6.67 (s, 1 H), 7.17 (s, 1 H), 7.35 (s, 2 H), 7.84 (s, 1 H), 8.16 - 8.24 (m, 1 H), 8.32 (s, 1 H)。

化合物2-1(450mg、1.49mmol、1eq)をエタノール(5mL)に溶
解させ、2-メルカプトエタノール(151.17mg、1.93mmol、134.97μL、1.3eq)及びトリエチルアミン(301.19mg、2.98mmol、414.29μL、2eq)を加え、20℃で2時間撹拌した。反応系に水(10mL)を加えて希釈し、更に酢酸エチル(10mL×3)で抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮し、乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~70:30)で精製して化合物3-2を得た。
LCMS: m/z (ESI) = 301.0 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 4.30 - 4.39 (m, 2 H), 3.99 - 4.05 (m, 2 H), 3.26 - 3.34 (m, 2 H), 3.17 - 3.24 (m, 2 H), 2.53 - 2.61 (m, 2 H), 2.06 - 2.11 (m, 2 H), 1.34 - 1.42 (m, 3 H)。
化合物3-2(260mg、865.53μmol、1eq)をテトラヒドロフラン(5mL)に溶解させ、tert-ブトキシビス(ジメチルアミノ)メタン(452.54mg、2.60mmol、536.18μL、3eq)を加え、80℃で16時間撹拌した。反応系を20℃に冷却させ、更に反応系に水(20mL)を加えて希釈し、次に、酢酸エチル(10mL)を加えて撹拌し、濾過し、ケーキをエタノール(10mL)で0.5時間スラリー化させ、濾過し、ケーキをオイルポンプで減圧濃縮して残留物を除去して化合物3-3を得た。
1H NMR (400 MHz, CDCl3) δ: 7.65 (s, 1 H), 4.28 - 4.37 (m, 2 H), 3.88 - 3.98 (m, 2 H), 3.21 - 3.28 (m, 2 H), 3.15 - 3.20 (m, 2 H), 3.14 (s, 6 H), 2.82 - 2.91 (m,
2 H), 1.32 - 1.43 (m, 3 H)。
化合物3-3(300mg、843.95μmol、1eq)をN,N-ジメチルアミノホルムアミド(5mL)に溶解させ、化合物1-3(267.79mg、843.95μmol、1eq)を加え、110℃で16時間撹拌した。反応系に飽和食塩水(15mL)を加え、酢酸エチル(5mL×3)で抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物3-4を得た。
LCMS: m/z (ESI) = 610.20 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 8.29 (s, 1 H), 7.88 - 7.93 (m, 1 H), 7.15 - 7.22 (m, 1 H), 6.66 - 6.76 (m, 1 H), 4.29 - 4.36 (m, 2 H), 3.79 - 3.89 (m, 2 H), 3.26 - 3.30 (m, 6 H), 3.21 - 3.25 (m, 4 H), 2.78
- 2.85 (m, 2 H), 2.69 (s, 3 H), 2.39 - 2.44 (m, 2 H), 1.33 - 1.39 (m, 3 H)。
化合物3-4(100mg、164.02μmol、1eq)をジクロロメタン(2mL)に溶解させ、トリエチルアミン(24.90mg、246.03μmol、34.24μL、1.5eq)及び4-ジメチルアミノピリジン(2.00mg、16.40μmol、0.1eq)を加え、次にtert-ブチルジメチルシリルクロリド(29.67mg、196.82μmol、24.12μL、1.2eq)を加え、20℃で20時間撹拌した。反応溶液を減圧濃縮して乾燥させて粗生成物を得、粗生成物を薄層クロマトグラフィー(展開剤:ジクロロメタン/メタノール=10:1)で分離・精製して化合物3-5を得た。
LCMS: m/z (ESI) = 724.30 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 8.29 (s, 1 H), 7.94 - 8.00 (m, 1 H), 7.12 - 7.20 (m, 1 H), 6.65 - 6.73 (m, 1 H), 4.27 - 4.36 (m, 2 H), 3.91 - 3.99 (m, 2 H), 3.25 - 3.29 (m, 6 H), 3.21 - 3.25 (m, 2 H), 2.78
- 2.87 (m, 2 H), 2.60 - 2.70 (m, 4 H), 2.37 (s, 3 H), 1.32 - 1.40 (m, 3 H), 0.84 (s, 9 H), -0.01 (s, 6 H)。
化合物3-5(70mg、96.69μmol、1eq)をテトラヒドロフラン(1mL)に溶解させ、塩化アンモニウム(31.03mg、580.16μmol、6eq)を加え、次に0℃に冷却させ、1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(966.93μL、10eq)を加え、20℃に昇温させ4時間撹拌した。反応系に水(5mL)を加え、次に、酢酸エチル(5mL×3)を加えて抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を除去して化合物3-6を得た。
LCMS: m/z (ESI) = 695.20 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 8.29 (s, 1 H), 7.99 - 8.03 (m, 1 H), 7.14 - 7.21 (m, 1 H), 6.64 - 6.71 (m, 1 H), 3.90 - 3.96 (m, 2 H), 3.24 - 3.29 (m, 6 H), 3.21 - 3.24 (m, 2 H), 2.79 - 2.86 (m, 2 H), 2.58
- 2.65 (m, 4 H), 2.37 (s, 3 H), 0.84 (s, 9 H), -0.02 (s, 6 H)。
化合物3-6(60mg、86.34μmol、1eq)をテトラヒドロフラン(0.5mL)溶液に溶解させ、次に、1Mのフッ化テトラブチルアンモニウムのテトラヒドロフラン溶液(172.69μL、2eq)を加え、得られた反応溶液を20℃で2時間撹拌した。反応系に水(10mL)を加えて洗浄し、次に、酢酸エチル(10mL×3)を加えて抽出し、分離し、有機相を合わせた後無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して粗生成物を得た。粗生成物を分取HPLC(高速液体製造方法:Waters
Xbridge BEH分取クロマトグラフ;カラム:C18 100×30mm×10μm;移動相A:10mMの炭酸水素アンモニウム水溶液(0.05%のアンモニア水
を含む)、移動相B:アセトニトリル;実行勾配:B%:25%~55%、8分間実行。)で分離・精製して化合物3を得た。
LCMS: m/z (ESI) = 581.30 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 8.43 (s, 1 H), 8.32 (s, 1 H), 7.40 - 7.48 (m, 3 H),
7.13 - 7.20 (m, 1 H), 6.68 - 6.75 (m, 1
H), 5.02 - 5.09 (m, 1 H), 3.63 - 3.71 (m, 2 H), 3.12 - 3.17 (m, 4 H), 3.04 - 3.11 (m, 4 H), 2.71 - 2.76 (m, 2 H), 2.42 - 2.46 (m, 4 H), 2.07 (s, 3 H)。

化合物4-1(10g、45.87mmol、1eq)をジクロロメタン(300mL)に溶解させ、N,N-ジイソプロピルエチルアミン(8.90g、68.89mmol、12.00mL、1.50eq)及びクロロメチルメチルエーテル(4.47g、55.52mmol、4.22mL、1.21eq)を加え、20℃で4時間撹拌した。反応溶液を減圧濃縮して乾燥させた後粗生成物に水(150mL)及びジクロロメタン(150mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で分離・精製して化合物4-2を得た。
1H NMR (400 MHz, CDCl3) δ: 3.53 (s, 3 H), 5.29 (s, 2 H), 7.20 - 7.28 (m, 1 H), 7
.61 (s, 1 H), 7.92 - 7.99 (m, 1 H)。
化合物4-2(2g、7.63mmol、1eq)をトルエン(12mL)及びジメチルスルホキシド(4mL)に溶解させ、更にN-メチルモルホリン(1.15g、11.45mmol、1.27mL、1.5eq)、炭酸セシウム(7.46g、22.90mmol、3eq)、トリス(ジベンジリデンアセトン)ジパラジウム(139.77mg、152.64μmol、0.02eq)、(S)-(-)-2,2-ビス(ジ-p-トリルホスフィノ)-1,1-ビナフチル(207.22mg、305.28μmol、0.04eq)を順次に加え、窒素ガスで保護し、90℃で16時間撹拌した。反応溶液を珪藻土を敷いた漏斗で濾過し、酢酸エチル(150mL)でケーキを濯ぎ、濾液を減圧濃縮して溶媒を乾燥させた後、粗生成物に酢酸エチル(50mL)及び飽和食塩水(50mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~98:2)で分離・精製して化合物4-3を得た。
LCMS: m/z (ESI) =282.0 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 2.37 (s, 3 H), 2.55 - 2.61 (m, 4 H), 3.14 - 3.24 (m, 4 H), 3.53 (s, 3 H), 5.20 (s, 2 H), 7.09
(s, 1 H), 7.22 - 7.26 (m, 1 H), 7.30 -7.36 (m, 1 H)。
化合物4-3(950mg、3.38mmol、1eq)をジクロロメタン(10mL)に溶解させ、次に、メタノール(8.4mL)及び12Mの濃塩酸(1.6mL、5.69eq)を加え、15℃で16時間撹拌した。35℃に昇温させ、8時間撹拌した。減圧濃縮して溶媒を乾燥させて化合物4-4の塩酸塩を得た。
LCMS: m/z (ESI) =238.1 [M+H] +。
化合物4-4の塩酸塩(400mg、1.46mmol、1eq)をジクロロメタン(8mL)に溶解させ、氷浴で0℃に冷却させた後、水酸化カリウム(491.95mg、8.77mmol、6eq)及び水(2.4mL)の混合溶液を加え、0℃で(ブロモジフルオロメチル)トリメチルシラン(605.72mg、2.92mmol、2eq)を加え、20℃で16時間撹拌した。反応溶液にジクロロメタン(5mL)及び水(5mL)を加えて抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を薄層クロマトグラフィー(展開剤:ジクロロメタン/メタノール=10:1)で分離して化合物4-5を得た。
1H NMR (400 MHz, CDCl3) δ: 2.44 - 2.54 (m, 3 H), 2.67 - 2.84 (m, 4 H), 3.37 (s, 4 H), 6.26 - 6.75 (m, 1 H), 7.06 - 7.10 (m, ,1 H), 7.26 (s, 1 H), 7.30- 7.40 (m,
1 H)。
マイクロアルゴンガスの環境で10%の純度の湿式パラジウム炭素(50mg)、メタノール(5mL)及び化合物4-5(30mg、104.43μmol、1eq)を順次に加え、反応溶液を水素ガス(15psi)条件で15℃の温度で2時間反応させた。反応溶液を直接に濾過し、濾液を減圧濃縮して溶媒を乾燥させて化合物4-6を得た。
1H NMR (400 MHz, CD3OD) δ: 2.92 (s, 3 H), 3.05 - 3.71 (m, 8 H), 6.26 -6.32 (m, 1
H), 6.36 - 6.78 (m, 2 H), 6.91 -6.95 (m, 1 H)。
化合物4-6(18mg、69.96μmol、1eq)を6Mの塩酸水溶液(180.00μL、15.44eq)に溶解させ、次に、アミノニトリル(61.92mg、1.40mmol、61.92μL、20eq)を加え、60℃で1時間撹拌した。反応溶液に水(5mL)及びジクロロメタン(5mL)を加え、分離し、水相に更に水酸化ナトリウム固体を加えて水相のpHを12を超えるように調節し、酢酸エチル(5mL×2)を加えて抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて化合物4-7を得た。
LCMS: m/z (ESI) =300.1 [M+H] +。
化合物1-2(20mg、44.68μmol、2.4eq)をN,N-ジメチルホルムアミド(0.5mL)に溶解させ、次に、化合物4-7(6.26mg、18.62μmol、1eq)を加え、反応溶液を110℃の温度で12時間撹拌して反応させた。反応溶液に飽和食塩水(5mL)及び水(5mL)を加え、更に酢酸エチル(5mL×6)で抽出し、分離し、有機相を合わせ、有機相を飽和食塩水(5mL×6)で洗浄し、無水硫酸ナトリウムで乾燥させて濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(高速液体製造方法:Phenomenex分取クロマトグラフ;カラム:C18 75×30mm×3μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:55%~85%、12分間実行。)で分離・精製して化合物4-8を得た。
LCMS: m/z (ESI) =562.2 [M+H] +。
1H NMR (400 MHz, CD3OD) δ: 1.20- 1.28 (m, 3 H), 2.36 (s, 3 H), 2.58 - 2.65 (m, 7
H), 2.80 -2.86 (m, 2 H), 3.19 - 3.26 (m, 4 H), 3.25 -3.30 (m, 2 H), 4.30 -4.36 (m, 2 H), 6.69 -6.73 (m, 2 H), 7.04 - 7.13 (m, 1 H), 8.00 -8.05 (m, 1 H), 8.28 (s, 1 H)。
化合物4-8(2mg、3.56μmol、1eq)を無水テトラヒドロフラン(1mL)に溶解させ、塩化アンモニウム(38.09mg、712.17μmol、200e
q)及び1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(1.42mL、400eq)を加え、20℃で3時間撹拌した。反応溶液にメタノール(5mL)を加えてクエンチングさせ、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(高速液体製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:C18 100×25mm×5μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:20%~55%、10分間実行。)で分離・精製して化合物4を得た。
LCMS: m/z (ESI) =533.2 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 2.36 (s, 3 H), 2.59 - 2.69 (m, 5 H), 2.82 -2.86 (m, 2
H), 3.11 - 3.27 (m, 8 H), 6.50 - 6.93 (m, 2 H), 7.05 - 7.25 (m, 1 H), 8.03 (d, J=2.86 Hz, 1 H), 8.29 (s, 1 H)。

化合物5-1(2g、8.26mmol、1eq)をトルエン(20mL)に溶解させ、N-メチルピペラジン(827.81mg、8.26mmol、916.73μL、1.00eq)、ナトリウムtert-ブトキシド(1.19g、12.40mmol、1.5eq)、トリス(ジベンジリデンアセトン)ジパラジウム(378.41mg、413.23μmol、0.05eq)及び(R)-(+)-2,2-ビス(ジフェニルホスフィノ)-1,1-ビナフタレン(257.31mg、413.23μmol、0.05eq)を加え、窒素ガスで保護し、80℃で16時間撹拌した。反応溶液に水(5mL)を加えてクエンチングさせ、酢酸エチル(5mL×3)で抽出し、分離し、有機相を合わ
せ、有機相を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~95:5)で精製して化合物5-2を得た。
LCMS: m/z (ESI) = 262.30 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 8.07 - 8.14 (m, 1 H), 7.32 - 7.39 (dd, J = 9.2 Hz, 1 H), 6.57 - 6.67 (d, J = 9.2 Hz, 1 H), 3.52 - 3.61 (t, J = 5.2 Hz, 4 H), 2.46 - 2.58 (t, J = 5.2 Hz, 4 H), 2.36 (s, 3 H)。
-78℃で窒素ガスの保護下で、化合物5-2(500mg、1.91mmol、1eq)をテトラヒドロフラン(5mL)溶液に溶解させ、2Mのジイソプロピルアミドリチウムテトラヒドロフラン溶液(1.44mL、1.5eq)を加え、得られた反応溶液を-78℃で2時間撹拌し、次に、ヨード(728.65mg、2.87mmol、1.5eq)の無水テトラヒドロフラン(2mL)溶液を加え、反応溶液を-78℃で2時間撹拌して反応させ、次に、反応系を80℃に昇温させ16時間撹拌した。反応系に水(10mL)を加えて反応をクエンチングさせ、酢酸エチル(15mL×3)で抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~95:5)で精製して化合物5-3を得た。
LCMS: m/z (ESI) = 388.00 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 7.99 - 8.06 (m, 1 H), 7.09 (s, 1 H), 3.55 - 3.67 (m, 4 H), 2.48 - 2.65 (m, 4 H), 2.40 (s, 3 H)。
化合物1-2(300mg、921.81μmol、1eq)をN,N-ジメチルホルムアミド(5mL)に溶解させ、炭酸グアニジン(415.20mg、2.30mmol、2.5eq)を加え、110℃で3時間撹拌した。反応溶液に水(10mL)を加え、0.5時間撹拌し、濾過し、ケーキをメタノール(10mL)で濯ぎ、次に、オイルポンプで残留物を減圧除去して化合物5-4を得た。
LCMS: m/z (ESI) =321.90 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 8.13 (s, 1 H), 6.31 - 6.39 (m, 2 H), 4.22 - 4.31 (m, 2 H), 3.14 - 3.21 (m, 2 H), 2.65 - 2.72 (m, 2 H), 2.61 (s, 3 H), 1.25 - 1.33 (m, 3 H)。
化合物5-4(49.81mg、154.98μmol、1eq)を1,4-ジオキサン(2mL)に溶解させ、窒素ガスの保護下でトリス(ジベンジリデンアセトン)ジパラ
ジウム(14.19mg、15.50μmol、0.1eq)、4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(8.97mg、15.50μmol、0.1eq)、炭酸セシウム(100.99mg、309.97μmol、2eq)、化合物5-3(60mg、154.98μmol、1eq)を加え、100℃で3時間撹拌した。反応溶液に水(10mL)を加えてクエンチングさせ、次に、酢酸エチル(10mL×3)で抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~95:5)で精製して化合物5-5を得た。
LCMS: m/z (ESI) = 581.20 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.40 -1.46 (m, 3 H), 2.38 (3 H), 2.53 - 2.65 (m, 4 H), 2.67 (s, 3 H), 2.83 -2.86 (m, 2 H), 3.23 - 3.37 (m, 6 H), 4.35 -4.39 (m, 2 H),
6.50 - 6.55 (m, 1 H), 7.12 -7.16 (m, 1 H), 8.24 -8.30 (m, 1 H), 8.30 (s, 1 H)。
化合物5-5(90mg、155.00μmol、1eq)をテトラヒドロフラン(2mL)に溶解させ、塩化アンモニウム(49.75mg、930.00μmol、6eq)を加え、窒素ガスの保護下で0℃で、1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(3.10mL、20eq)を加え、20℃で3時間撹拌した。反応系に水(10mL)を加えて反応をクエンチングさせ、次に、酢酸エチル(15mL×3)を加えて抽出し、分離し、有機相を合わせ、有機相を減圧濃縮して粗生成物を得、粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH 分取クロマトグラフ;カラム:C18 100×30mm×10μm;移動相A:
10mMの炭酸水素アンモニウム水溶液(0.05%のアンモニア水を含む)、移動相B:アセトニトリル;実行勾配:B%:30%~60%、8分間実行。)で分離・精製して化合物5を得た。
LCMS: m/z (ESI) = 552.20 [M+H]+。
1H NMR(400MHz、DMSO-d6)δ:8.77 (s, 1 H), 8.48 (s, 1 H), 8.02 - 8.06 (m, 1 H), 7.70
(s, 1 H), 7.47 (s, 2 H), 3.45 - 3.53 (m, 4 H), 3.10 - 3.19 (m, 2 H), 2.77 - 2.84 (m, 2 H), 2.61 (s, 3 H), 2.36 - 2.41 (m, 4 H), 2.20 (s, 3 H)。

炭酸カリウム(51.77g、374.58mmol、2eq)をジメチルスルホキシド(200mL)に溶解させ、次に、化合物6-1(21g、187.29mmol、1eq)を加え、20℃で10分間撹拌し、二硫化炭素(15.69g、206.02mmol、12.45mL、1.1eq)を加え、20℃で10分間撹拌し、次に、0℃でブロモ酢酸エチル(31.28g、187.29mmol、20.71mL、1eq)及びヨウ化メチル(26.58g、187.29mmol、11.66mL、1eq)の混合溶液を加え、温度を15~20℃に制御し、滴下完了後20℃で1時間撹拌した。反応溶液に水(500mL)及び飽和食塩水(300mL)を加え、酢酸エチル(500mL×3)で抽出し、分離し、有機相を合わせ、飽和食塩水(500mL)で洗浄し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して生成物を得た後、更にメチルtert-
ブチルエーテル(15mL)で1時間スラリー化させ、濾過し、ケーキを収集して化合物6-2を得た。
LCMS: m/z (ESI) =343.0 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.28 - 1.34 (m, 3 H), 1.34 - 1.42 (m, 3 H), 1.92 - 2.16 (m, 2 H), 2.48 - 2.68 (m, 2 H), 3.18 - 3.26 (m, 2 H), 3.86 (s, 2 H), 4.18 - 4.44 (m, 4 H)。
マイクロアルゴンガスの環境でラネーニッケル(5.20g、60.70mmol、4.00eq)及びエタノール(150mL)を加え、次に、化合物6-2(5.2g、15.19mmol、1eq)を加え、反応溶液を水素ガス(50psi)の条件で30℃で48時間反応させた。反応溶液を珪藻土を敷いた漏斗に通し、ケーキをエタノール(800mL)で濯ぎ、濾液を減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して生成物を得、生成物をメチルtert-ブチルエーテル(20mL)に溶解させた後、更に石油エーテル(30mL)を加え、1時間スラリー化させた後濾過し、ケーキを収集して化合物6-3を得た。
1H NMR (400 MHz, CDCl3) δ: 1.40 -1.50 (m, 3 H), 1.96 - 2.24 (m, 2 H), 2.41 - 2.75 (m, 2 H), 3.23 -3.26 (m, 2 H), 4.34 - 4.40 (m, 2 H), 8.29 (s, 1 H)。
化合物6-3(1g、4.46mmol、1eq)を無水エタノール(20mL)に溶解させ、水素化ホウ素ナトリウム(280mg、7.40mmol、1.66eq)を加え、20℃で2時間撹拌した。反応溶液に水(10mL)を加えた後、1Mの希塩酸水溶液でpHを6に調節した後、溶媒が減少しなくなるまで減圧濃縮し、粗生成物に水(10mL)及び飽和食塩水(10mL)を加え、酢酸エチル(40mL×2)で抽出し、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させた。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して化合物6-4を得た。
LCMS: m/z (ESI) =209.1 [M-17] +。
1H NMR (400 MHz, CDCl3) δ: 1.34 - 1.40 (m, 3 H), 1.54 - 1.83 (m, 3 H), 1.92 - 2.12 (m, 2 H), 3.03 - 3.09 (m, 2 H), 4.33 -4.38 (m, 2 H), 4.74 - 4.87 (m, 1 H), 7.53 (s, 1 H)。
化合物6-4(8g、35.35mmol、1eq)をジクロロメタン(110mL)に溶解させ、次に、塩化アセチル(11.10g、141.41mmol、10.09mL、4eq)及び4-ジメチルアミノピリジン(431.90mg、3.54mmol、
0.1eq)、ピリジン(13.98g、176.76mmol、14.27mL、5eq)を加え、20℃で2時間撹拌した。反応溶液を減圧濃縮して溶媒を原体積の1/3にさせた後、水(30mL)を加え、分離し、有機相を水(30mL)で洗浄して分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~85:15)で精製して化合物6-5を得た。
LCMS: m/z (ESI) =209.0 [M-59] +。
1H NMR (400 MHz, CDCl3) δ: 1.37 -1.40 (m, 3 H), 1.78 - 2.01 (m, 4 H), 2.08 (s, 3
H), 2.76 - 3.30 (m, 2 H), 4.33 -4.38 (m, 2 H), 5.93 -6.00 (m, 1 H), 7.52 (s, 1 H)。
化合物6-5(3.1g、11.55mmol、1eq)をN,N-ジメチルホルムアミド(31mL)に溶解させ、次に、N-ブロモスクシンイミド(6.37g、35.81mmol、3.1eq)を加え、50℃16時間撹拌した。反応溶液に酢酸エチル(50mL)及び飽和食塩水(50mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~95:5)で精製して化合物6-6を得た。
LCMS: m/z (ESI) =286.9,288.9 [M-59]+。
1H NMR (400 MHz, CDCl3) δ: 1.31 - 1.40 (m, 3 H), 1.71 - 1.89 (m, 3 H), 2.09 (s, 3 H), 2.19 (s, 1 H), 2.64 - 2.79 (m, 1 H), 3.29 - 3.49 (m, 1 H), 4.32 -4. 39 (m,
2 H), 5.86 - 6.20 (m, 1 H)。
化合物6-6(3.7g、10.66mmol、1eq)をエタノール(37mL)に溶解させ、次に、炭酸カリウム(1.47g、10.66mmol、1eq)を加え、40℃で16時間撹拌した後、60℃に昇温させて4時間撹拌した。反応溶液を濾過した後エタノール(500mL)でケーキを濯ぎ、濾液を収集し、減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して化合物6-7を得た。
LCMS: m/z (ESI) =286.9,288.8 [M-17] +。
1H NMR (400 MHz, CDCl3) δ: 1.36 -1.42 (m, 3 H), 1.77 - 1.80 (m, 4 H), 2.02 - 2.20 (m, 1 H), 2.61 - 2.81 (m, 1 H), 3.24 -
3.44 (m, 1 H), 4.32 -4.40 (m, 2 H), 4.88 -4.93 (m, 1 H)。
化合物6-7(1.76g、5.77mmol、1eq)をジクロロメタン(30mL
)に溶解させ、次に、クロロクロム酸ピリジニウム(3.73g、17.30mmol、3eq)及び酢酸ナトリウム(1.42g、17.30mmol、3eq)を加え、20℃で2時間撹拌した。反応溶液を珪藻土を敷いた漏斗に通して濾過し、酢酸エチル(50mL)及びジクロロメタン(50mL)で濯ぎ、次に、減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~80:20)で精製して化合物6-8を得た。
LCMS: m/z (ESI) =302.9,304.9 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.38 - 1.42 (m, 3 H), 2.10 -2.15 (m, 2 H), 2.59 - 2.65 (m, 2 H), 3.27 -3.30 (m, 2 H), 4.35 - 4.40 (m, 2 H)。
反応フラスコに化合物6-8(203mg、669.59μmol、1eq)、化合物6-9(332.79mg、1.34mmol、2eq)、2-ジシクロヘキシルホスフィノ-2,6-ジイソプロポキシ-1,1-ビフェニル(62.49mg、133.92μmol、0.2eq)、ビス(アセトニトリル)パラジウム(II)クロリド(17.37mg、66.96μmol、0.1eq)、炭酸セシウム(654.49mg、2.01mmol、3eq)を加え、次に、水(1mL)及びtert-ブタノール(1mL)の混合溶液を加え、窒素ガスで保護した後、100℃で16時間撹拌した。反応溶液に酢酸エチル(100mL)、飽和食塩水(50mL)及び水(50mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して化合物6-10を得た。
LCMS: m/z (ESI) =375.0 [M+Na] +。
1H NMR (400 MHz, CDCl3) δ: 1.38 -1.43 (m, 3 H), 1.54 - 1.57 (m, 6 H), 2.07 - 2.10
(m, 2 H), 2.47 - 2.62 (m, 2 H), 3.23 -3.30 (m, 2 H), 3.59 -3.62 (m, 6 H), 4.34 -4.37 (m, 2 H), 4.66 -4.70 (m, 1 H)。
化合物6-10(283mg、802.96μmol、1eq)をエタノール(10mL)に溶解させ、p-トルエンスルホン酸一水和物(158.48mg、833.15μmol、1.04eq)を加え、20℃で1時間撹拌した。反応溶液を減圧濃縮して溶媒を乾燥させて粗生成物を得、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~80:20)で精製して化合物6-11を得た。
LCMS: m/z (ESI) =268.9 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.38 - 1.41 (m, 3 H), 1.80 (s, 1 H), 2.01 - 2.17 (m, 2 H), 2.49 - 2.63 (m, 2 H), 3.24 -3.30 (m, 2 H), 3.51 -3.56 (m, 2 H), 3.95 -4.06
(m, 2 H), 4.34 - 4.38 (m, 2 H)。
化合物6-11(50mg、186.34μmol、1eq)を無水テトラヒドロフラン(7.5mL)に溶解させ、次に、tert-ブトキシビス(ジメチルアミノ)メタン(162.38mg、931.70μmol、192.39μL、5eq)を加え、80℃で12時間撹拌した。減圧濃縮して溶媒を乾燥させて化合物6-12を得た。
LCMS: m/z (ESI) =297.0 [M-26] +。
化合物6-12(60mg、185.53μmol、1eq)をN,N-ジメチルホルムアミド(1.2mL)に溶解させ、次に、化合物1-3(58.87mg、185.53μmol、1eq)を加え、110℃で12時間撹拌した。反応溶液に酢酸エチル(10mL)と水(5mL)及び飽和食塩水(5mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させた後、粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン:メタノール=100:0~90:10)で精製して化合物6-13を得た。
LCMS: m/z (ESI) =578.1 [M+H]+。
化合物6-13(8mg、13.85μmol、1eq)をテトラヒドロフラン(0.8mL)に溶解させ、0℃で窒素ガスで保護した後、塩化アンモニウム(30mg、560.84μmol、40.49eq)及び1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(567.85μL、41eq)を加え、20℃で1時間撹拌した。反応溶液にメタノール(5mL)を加えてクエンチングさせた後、減圧濃縮し溶媒を乾燥させ、次に、粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:Prep sunfire C18 100×30mm×10μm;移動相A:10mMの炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:35%~50%、8分間実行。)で分離・精製して化合物6を得た。
LCMS: m/z (ESI) =549.3 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 2.18 - 2.26
(m, 3 H), 2.29 - 2.38 (m, 4 H), 2.40 - 2.46 (m, 4 H), 2.68 - 2.81 (m, 4 H), 3.02 - 3.17 (m, 4 H), 4.55 - 4.70 (m, 1 H),
6.66 - 6.87 (m, 1 H), 7.11 - 7.27 (m, 2
H), 7.32 - 7.51 (m, 2 H), 8.27 - 8.37 (m, 1 H), 8.62 - 8.74 (m, 1 H)。

化合物7-1(4g、15.62mmol、1eq)をテトラヒドロフラン(40mL)に溶解させ、2-ジシクロヘキシルホスフィン-2-(N,N-ジメチルアミン)-ビフェニル(491.90mg、1.25mmol、0.08eq)、トリス(ジベンジリデンアセトン)ジパラジウム(1.14g、1.25mmol、0.08eq)を加え、窒素ガスで保護し、1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(37.50mL、2.4eq)及び1-Boc-ピペラジン(4.36g、23.44mmol、1.5eq)を加え、得られた反応溶液を窒素ガスで保護し、80℃で3時間撹拌して反応させた。反応系に水(60mL)を加えてクエンチングさせ、ジクロロメタン(50mL×3)で抽出し、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン:メタノール=100:0~98:2)で精製して化合物7-2を得た。
LCMS: m/z (ESI) = 362.10 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 7.00 - 7.06 (m, 1 H), 6.26 - 6.35 (m, 2 H), 3.78 - 3.86 (m, 2 H), 3.53 - 3.58 (m, 4 H), 3.02 - 3.14 (m, 4 H), 1.49 (s, 9 H)。
化合物7-2(2g、5.53mmol、1eq)をジメチルスルホキシド(60mL)溶液に溶解させ、亜硝酸ナトリウム(1.53g、22.14mmol、4eq)を加え、20℃で45%のヨウ化水素酸水溶液(3.78g、13.28mmol、2.22mL、2.4eq)を滴下し、次に、反応温度を35℃に昇温させて16時間撹拌した。
反応系に水(60mL)を加えて反応を希釈し、次に、酢酸エチル(50mL×3)を加えて抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~80:20)で精製して化合物7-3を得た。
LCMS: m/z (ESI) = 473.00 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 7.32 - 7.34 (m, 1 H), 7.11 - 7.16 (m, 1 H), 6.85 - 6.91 (m, 1 H), 3.55 - 3.61 (m, 4 H), 3.09 - 3.16 (m, 4 H), 1.49 (s, 9 H)。
化合物5-4(180.36mg、561.16μmol、1eq)を1,4-ジオキサン(8mL)溶液に溶解させ、在窒素ガスの保護下でトリス(ジベンジリデンアセトン)ジパラジウム(51.39mg、56.12μmol、0.1eq)、4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(32.47mg、56.12μmol、0.1eq)及び炭酸セシウム(365.67mg、1.12mmol、2eq)を加え、窒素ガスで3回置換し、化合物7-3(265mg、561.16μmol、1eq)を加え、得られた反応溶液を100℃で5時間撹拌した。反応系に水(20mL)を加えてクエンチングさせ、次に、酢酸エチル(20mL×3)を加えて抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~80:20)で精製して化合物7-4を得た。
LCMS: m/z (ESI) =666.20 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.31 (s, 1 H), 8.27 - 8.30 (m, 1 H), 7.09 - 7.19 (m, 1 H), 6.48 - 6.56 (m, 1 H), 5.28 - 5.35 (m, 1 H), 4.32 - 4.42 (m, 2 H), 3.53 - 3.63 (m, 4 H), 3.28 - 3.37 (m, 2 H), 3.12
- 3.24 (m, 4 H), 2.83 (t, J=7.2 Hz, 2 H), 2.66 (s, 3 H), 1.49 (s, 9 H), 1.38 - 1.43 (m, 3 H)。
化合物7-4(300mg、450.62μmol、1eq)を無水テトラヒドロフラン(10mL)に溶解させ、塩化アンモニウム(144.63mg、2.70mmol、6eq)を加え、窒素ガスの保護下で反応温度を0℃に冷却させ、1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(9.01mL、20eq)を加え、得られた反応溶液を20℃で3時間撹拌した。反応系に水(15mL)を加えてクエンチングさせ、酢酸エチル(20mL×3)で抽出し、分離し、有機相を合わせ、有機相を減圧濃縮し、乾燥させた後、粗生成物を薄層クロマトグラフィー(展開剤:ジクロロメタン/メタノール=10:1)で分離して化合物7-5を得た。
LCMS: m/z (ESI) = 637.20 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.25 - 8.31 (
m, 1 H), 8.14 - 8.24 (m, 1 H), 7.13 - 7.21 (m, 1 H), 6.52 - 6.63 (m, 1 H), 5.50 - 5.59 (m, 2 H), 3.54 - 3.65 (m, 4 H), 3.25 - 3.33 (m, 2 H), 3.13 - 3.25 (m, 4 H), 2.81 - 2.90 (m, 2 H), 2.64 (s, 3 H), 1.49 (s, 9 H)。
化合物7-5(100mg、157.06μmol、1eq)をジクロロメタン(5mL)溶液に溶解させ、トリフルオロ酢酸(3.85g、33.77mmol、2.50mL、214.99eq)を加え、20℃で2時間撹拌した。反応系を直接に減圧濃縮して乾燥させた後、粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge Prep OBD分取クロマトグラフ;カラム:C18 150×40mm×10μm;移動相A:10mMの炭酸水素アンモニウム水溶液(0.05%のアンモニア水)、移動相B:アセトニトリル;実行勾配:B%:20%~50%、8分間実行。)で分離・精製して化合物7を得た。
LCMS: m/z (ESI) = 537.10 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 8.46 (s, 1 H), 8.32 (s, 1 H), 7.47 - 7.51 (m, 1 H),
7.41 - 7.46 (m, 2 H), 7.13 - 7.20 (m, 1
H), 6.66 - 6.71 (m, 1 H), 3.08 - 3.15 (m, 2 H), 2.95 - 3.05 (m, 4 H), 2.79 - 2.84 (m, 4 H), 2.71 - 2.76 (m, 2 H), 2.54 (s, 3 H)。

化合物7(80mg、149.09μmol、1eq)を無水テトラヒドロフラン(5mL)及びジメチルスルホキシド(2.5mL)の混合溶液に溶解させ、トリエチルアミン(15.09mg、149.09μmol、20.75μL、1eq)でpHを7~8に調節し、更に、酢酸(35.81mg、596.36μmol、34.11μL、4eq)でpHを5~6に調節し、0℃で(tert-ブチルジメチルシリルオキシ)アセト
アルデヒド(64.97mg、372.72μmol、71.01μL、2.5eq)を加え、0.5時間撹拌し、0℃でトリアセトキシ水素化ホウ素ナトリウム(69.52mg、328.00μmol、2.2eq)を加え、得られた反応溶液を20℃で2時間撹拌した。反応系に水(3mL)を加え、更に、酢酸エチル(5mL×3)で抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して粗生成物を得た。粗生成物を薄層クロマトグラフィー(展開剤:ジクロロメタン/メタノール=10:1)で分離・精製して化合物8-1を得た。
LCMS: m/z (ESI) = 695.20 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.31 (s, 1 H), 8.20 - 8.27 (m, 1 H), 7.32 - 7.38 (m, 1 H), 7.12 - 7.20 (m, 1 H), 6.47 - 6.56 (m, 1 H), 5.65 - 5.75 (m, 2 H), 3.86 - 4.01 (m, 2 H), 3.37 - 3.44 (m, 2 H), 3.21
- 3.35 (m, 4 H), 2.81 - 2.99 (m, 6 H), 2.66 (s, 3 H), 1.24 - 1.29 (m, 2 H), 0.90 (s, 9 H), 0.09 (s, 6 H)。
化合物8-1(100mg、143.90μmol、1eq)を無水テトラヒドロフラン(0.5mL)溶液に溶解させ、1Mのフッ化テトラブチルアンモニウムテトラヒドロフラン溶液(287.81μL、2eq)を加え、20℃で16時間撹拌した。反応系に水(20mL)を加えて洗浄し、酢酸エチル(25mL×3)で抽出し、分離し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して粗生成物を得、分取HPLC(HPLC製造方法:Waters Xbridge Prep OBD分取クロマトグラフ;カラム:C18 150×40mm×10μm;移動相A:10mMの炭酸水素アンモニウム水溶液(0.05%のアンモニア水を含む)、移動相B:アセトニトリル;実行勾配:B%:20%~50%、8分間実行。)で分離・精製して化合物8を得た。
LCMS: m/z (ESI) = 581.20 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 8.44 - 8.48
(m, 1 H), 8.31 (s, 1 H), 7.49 - 7.52 (m, 1 H), 7.40 - 7.46 (m, 2 H), 7.13 - 7.19 (m, 1 H), 6.68 - 6.72 (m, 1 H), 4.39 -
4.45 (m, 1 H), 3.49 - 3.56 (m, 2 H), 3.29 (s, 3 H), 3.09 - 3.17 (m, 6 H), 2.71 - 2.77 (m, 2 H), 2.52 -2.60 (m, 4 H), 2.40 - 2.46 (m, 2 H)。

反応フラスコに化合物9-1(2.4g、11.06mmol、1eq)、ジクロロビス(トリフェニルホスフィン)パラジウム(II)(194.06mg、276.47μmol、0.025eq)、ヨウ化第一銅(105.31mg、552.94μmol、0.05eq)を加え、次に、ジエチルアミン(25mL)及び1-ジメチルアミノ-2-プロピン(1.15g、13.82mmol、1.47mL、1.25eq)を加え、窒素ガスで保護し、60℃で3時間撹拌した。反応完了後20℃に冷却させ、反応溶液を減圧濃縮して乾燥させた。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル:酢酸エチル=100:0~0:100)で精製して化合物9-2を得た。
LCMS: m/z (ESI)= 220.0 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 2.38 (s, 6 H), 2.84 (s, 3 H), 3.51 (s, 2 H), 8.24 -8.26 (m, 1 H), 8.69 -8.75 (m, 1 H)。
マイクロアルゴンガスの環境で、ラネーニッケル(1.4g、16.34mmol、2.56eq)、エタノール(50mL)及び化合物9-2(1.4g、6.39mmol、1eq)を加え、反応溶液を水素ガス(15psi)の条件下で25℃で4時間撹拌した。濾過し、濾液を減圧濃縮して溶媒を乾燥させた。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~95:5)で精製して化合物9-3を得た。
LCMS: m/z (ESI)= 194.1 [M+H]+。
ステップ3:化合物9-4の合成
化合物9-3を6Mの塩酸水溶液(517.36μL、6eq)に溶解させ、更に、アミノニトリル(174.00mg、4.14mmol、174.00μL、8eq)を加え、60℃で1時間撹拌した。反応溶液に水(5mL)及びジクロロメタン(5mL)を加え、分離し、水相に水酸化ナトリウム固体を加えて水相のpHを12を超えるように調節した後、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge Prep OBD分取クロマトグラフ;カラム:C18 150×40mm×10μm;移動相A:10mMの炭酸水素アンモ
ニウム水溶液(0.05%のアンモニア水を含む)、移動相B:アセトニトリル;実行勾配:B%:1%~15%、8分間実行。)で分離・精製して化合物9-4を得た。
LCMS: m/z (ESI) =236.3 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 1.56 - 1.63
(m, 2 H), 2.08 (s, 6 H), 2.10 -2.18 (m,
5 H), 2.40 -2.47 (m, 2 H), 5.08 (s, 4 H), 6.77 (s, 1 H), 7.82 (s, 1 H)。
化合物9-4(40mg、169.98μmol、1eq)をN,Nジメチルホルムアミド(1mL)に溶解させ、次に、化合物1-2(76.09mg、169.98μmol、1eq)を加え、110℃で12時間撹拌した。反応溶液に酢酸エチル(10mL)、飽和食塩水(5mL)及び水(5mL)を加え、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:C18 100×30mm×10μm;移動相A:0.04%の塩酸水溶液、移動相B:アセトニトリル;実行勾配:B%:30%~50%、8分間実行。)で分離・精製して化合物9-5を得た。
LCMS: m/z (ESI) =498.2 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 1.09 - 1.23
(m, 3 H), 1.92 - 2.07 (m, 2 H), 2.33 - 2.36 (m, 3 H), 2.54 - 2.57 (m, 3 H), 2.59 - 2.67 (m, 2 H), 2.68 - 2.72 (m, 6 H),
2.73 - 2.81 (m, 2 H), 2.95 - 3.02 (m, 2
H), 3.04 - 3.13 (m, 2 H), 4.02 - 4.17 (m, 2 H), 8.01 - 8.14 (m, 1 H), 8.24 - 8.36 (m, 1 H), 8.43 - 8.55 (m, 1 H)。
化合物9-5(24mg、48.22μmol、1eq)を1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(964.49μL、20eq)に溶解させ、次に、塩化アンモニウム(25.80mg、482.24μmol、10eq)を加え、窒素ガスで保護した後、20℃で16時間撹拌した。メタノール(5mL)を加えてクエンチングさせた後、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:C18 100×30mm×10μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:15%~45%、8分間実行。)で分離・精製して化合物9を得た。
LCMS: m/z (ESI) =469.1 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.76 - 1.84 (m, 2 H), 2.24 (s, 6 H), 2.33 - 2.37 (m, 2 H), 2.49 (s, 3 H), 2.55 (s, 3 H), 2.58
-2.63 (m, 2 H), 2.70 - 2.80 (m, 2 H), 3.15 -3.23 (m, 2 H), 5.49 (s, 2 H), 6.72
(s, 1 H), 8.00 (s, 1 H), 8.20 (s, 1 H), 8.29 (s, 1 H)。

化合物7(10mg、18.64μmol、1eq)をジクロロエタン(0.5mL)に溶解させた後、3-オキセタノン(1.48mg、20.50μmol、1.1eq)を加え、次に、テトラエトキシチタン(4.25mg、18.64μmol、3.86μL、1eq)を加え、得られた反応溶液を20℃で1時間撹拌し、次に、トリアセトキシ水素化ホウ素ナトリウム(4.34mg、20.50μmol、1.1eq)を加え、20℃で12時間撹拌した。反応系に水(5mL)を加え、大量の白色のフロックを析出させ、次に、酢酸エチル(5mL×5)を加えて抽出し、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して乾燥させて粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:Prep sunfire C18 100×30mm×10μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:35%~50%、8分間実行。)で分離・精製して化合物10を得た。
LCMS: m/z (ESI) = 593.20 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 8.29 (s, 1 H), 7.95 - 7.99 (m, 1 H), 7.16 - 7.21 (m, 1 H), 6.66 - 6.72 (m, 1 H), 4.70 - 4.75 (m, 2 H), 4.62 - 4.66 (m, 2 H), 3.53 - 3.59 (m, 1 H), 3.27 -3.30 (m, 4 H), 3.21 (t, J=7.20 Hz, 2 H), 2.80 - 2.85 (m, 2 H), 2.60 - 2.65 (m, 3 H), 2.49 - 2.55 (m,
4 H)。
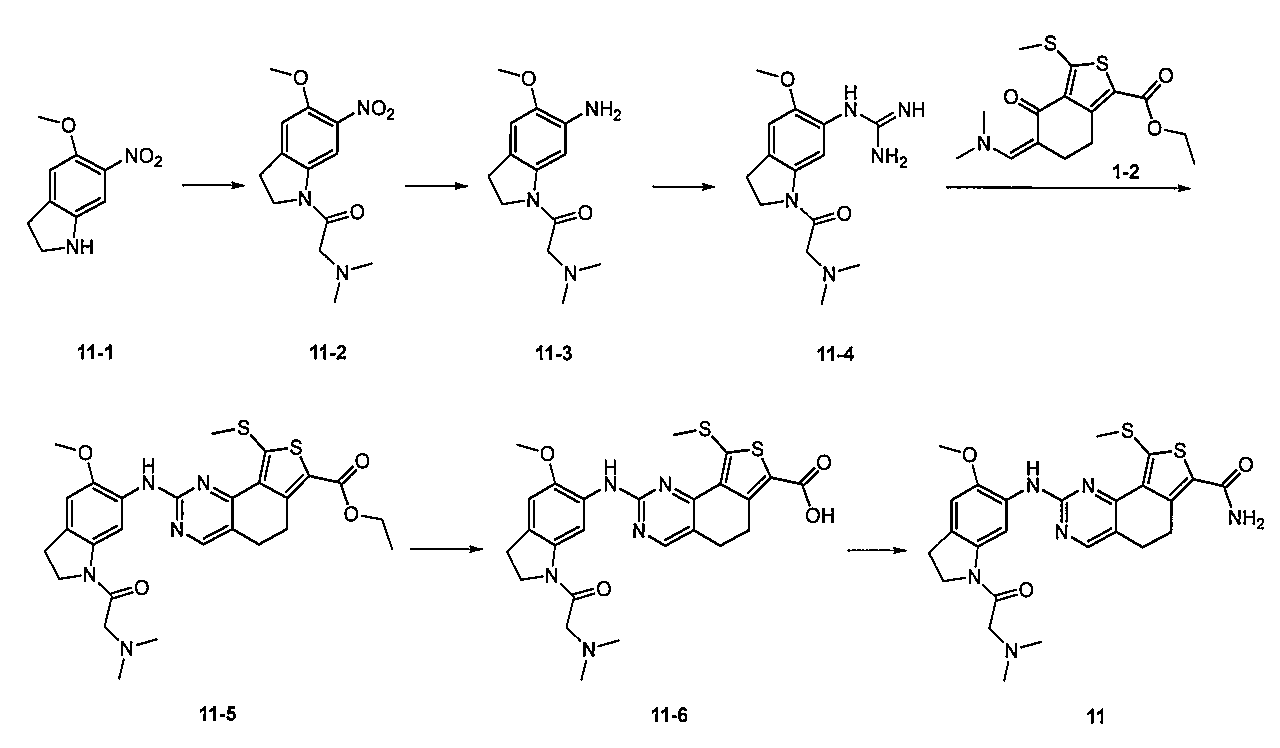
化合物11-1(2g、10.30mmol、1eq)及び炭酸カリウム(2.85g、20.60mmol、2eq)をテトラヒドロフラン(20mL)に溶解させ、反応混合物を窒素ガスの保護下で0℃に冷却させ、次に、臭化ブロモアセチル(3.12g、15.45mmol、1.34mL、1.5eq)を加え、0℃で10分間撹拌し、次に、0℃で40%のジメチルアミン水溶液(3.48g、30.90mmol、3.91mL、3eq)を加え、0℃で10分間撹拌した。反応溶液を氷水(200mL)にゆっくりと注いでクエンチングさせ、酢酸エチル(100mL×3)で抽出し、分離し、有機相を合わせ、有機相を飽和食塩水(100mL×3)で洗浄した後、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~95:5)で精製して化合物11-2を得た。
1H NMR (400 MHz, CDCl3) δ: 8.67 (s, 1 H), 6.92 (s, 1 H), 4.26 (t, J=8.60 Hz, 2 H), 3.92 (s, 3 H), 3.16 - 3.32 (m, 4 H), 2.41 (s, 6 H)。
マイクロアルゴンガスの環境で、10%の湿式パラジウム炭素(1g)を加え、次に、無水メタノール(2mL)、化合物11-2(1g、3.58mmol、1eq)を順次に加え、反応溶液を水素ガス(15psi)の条件下で、40℃で12時間撹拌した。反応溶液を珪藻土を敷いた漏斗を通し、ケーキをメタノール(50mL×2)で濯ぎ、濾液を収集し、濾液を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル+0.5%のアンモニア水=100:0~0:100)で精製して化合物11-3を得た。
1H NMR (400 MHz, DMSO-d6) δ: 7.55 (s, 1 H), 6.70 (s, 1 H), 4.63 (s, 2 H), 3.95 -
4.19 (m, 2 H), 3.65 - 3.71 (m, 3 H), 3.13 (s, 2 H), 2.87 - 3.04 (m, 2 H), 2.25 (s, 6 H)。
化合物11-3(200mg、802.22μmol、1eq)を6Mの塩酸水溶液(802.22μL、6eq)に溶解させ、次に、アミノニトリル(269.80mg、6.42mmol、269.80μL、8eq)を加え、100℃に昇温させて2時間撹拌し、次に、アミノニトリル(134.90mg、3.21mmol、134.90μL、4eq)を加え、反応溶液を100℃で12時間撹拌して反応させた。反応溶液に水(10mL)及びジクロロメタン(10mL)を加え、抽出し、分離して水相を収集し、水相を飽和水酸化ナトリウム水溶液でpHを11に調節した後、減圧濃縮して粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラフ;カラム:Waters Xbridge BEH C18 100×25mm×5μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:1%~25%、10分間実行。)で分離・精製して化合物11-4を得た。
1H NMR (400 MHz, DMSO-d6) δ: 7.61 (s, 1 H), 6.84 (s, 1 H), 5.50 (s, 3 H), 4.12 (t, J=8.40 Hz, 2 H), 3.67 (s, 3 H), 3.14 (s, 2 H), 3.05 (t, J=8.40 Hz, 2 H), 2.25
(s, 6 H)。
化合物1-2(67.02mg、205.94μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させた後、化合物11-4(60mg、205.94μmol、1eq)を加え、110℃で16時間撹拌して反応させた。反応溶液を水(30mL)に注いでクエンチングさせ、次に、酢酸エチル(15mL×3)を加えて抽出し、分離し、有機相を合わせ、有機相を飽和食塩水(15mL×3)で洗浄した後、無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して粗生成物を得た。粗生成物を薄層クロマトグラフィー(展開剤:ジクロロメタン/メタノール=10:1)で分離・精製して化合物11-5を得た。
1H NMR (400 MHz, CDCl3) δ: 9.25 (s, 1 H), 8.32 (s, 1 H), 6.76 (s, 1 H), 4.35 (q,
J=7.20 Hz, 2 H), 4.18 (t, J=8.20 Hz, 2 H), 3.82 - 3.95 (m, 3 H), 3.60 (s, 1 H),
3.25 - 3.36 (m, 2 H), 3.16 - 3.25 (m, 2
H), 3.09 - 3.16 (m, 1 H), 2.98 - 3.09 (m, 1 H), 2.73 - 2.89 (m, 2 H), 2.65 (s, 3 H), 2.39 (s, 6 H), 1.40 (t, J=7.20 Hz,
3 H)。
化合物11-5(50mg、90.30μmol、1eq)を無水テトラヒドロフラン
(4mL)及び無水エタノール(1mL)に溶解させ、次に、水酸化リチウム一水和物(18.95mg、451.51μmol、5eq)を水(1mL)に溶解させて反応溶液に加え、40℃で5時間撹拌した。反応溶液を1Mの希塩酸でpHを7に調節した後、冷凍乾燥させて化合物11-6を得た。
1H NMR (400 MHz, DMSO-d6) δ: 10.35 (s, 1
H), 8.82 (s, 1 H), 8.54 (s, 1 H), 8.34 (s, 1 H), 7.07 (s, 1 H), 4.45 (d, J=4.40
Hz, 2 H), 4.10 (t, J=8.20 Hz, 2 H), 3.80 (s, 3 H), 3.17 - 3.24 (m, 4 H), 2.88 (d, J=4.40 Hz, 6 H), 2.79 (t, J=7.00 Hz, 2 H), 2.56 (s, 3 H)。
化合物11-6(110mg、209.27μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させ、次に、N,N-ジイソプロピルエチルアミン(81.14mg、627.80μmol、109.35μL、3eq)、2-(7-アゾベンゾトリアゾール)-N,N,N,N-テトラメチルウロニウムヘキサフルオロホスフェート(103.44mg、272.05μmol、1.3eq)、炭酸水素アンモニウム(49.63mg、627.80μmol、51.70μL、3eq)を順次に加え、窒素ガスの保護下で20℃で2時間撹拌した。反応溶液を直接に濾過し、濾液を収集し、濾液を分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラフ;カラム:Phenomenex C18 80×40mm×3μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:30%~60%、8分間実行。)で分離・精製して化合物11を得た。
LCMS: m/z (ESI) = 525.30 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 8.67 (s, 1 H), 8.29 (s, 1 H), 7.84 (s, 1 H), 7.42 (s, 2 H), 6.96 (s, 1 H), 4.16 (t, J=8.20 Hz, 2 H), 3.79 (s, 3 H), 3.17 (s, 2 H), 3.11 (t, J=7.20 Hz, 4 H), 2.70 - 2.77 (m, 2 H), 2.53 (s, 3 H), 2.26 (s, 6 H)。

化合物6-3(200mg、891.76μmol、1eq)を無水テトラヒドロフラン(4mL)に溶解させ、次に、tert-ブトキシビス(ジメチルアミノ)メタン(466.25mg、2.68mmol、552.43μL、3eq)を加え、窒素ガスの保護下で80℃で2時間撹拌した。減圧濃縮して溶媒を乾燥させて化合物12-1を得た。
LCMS: m/z (ESI) =253.2 [M-26]+。
化合物12-1(100mg、357.97μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させ、次に、化合物1-3(113.59mg、357.97μmol、1eq)を加え、110℃で16時間撹拌した。反応溶液に酢酸エチル(20mL)と水(10mL)及び飽和食塩水(10mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~98:2)で精製して化合物12-2を得た。
LCMS: m/z (ESI) =534.3 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.42 (t, J=7.2 Hz, 3 H), 2.39 (s, 3 H), 2.62 (s, 4 H), 2.89 (t, J=7.2 Hz, 2 H), 3.26 - 3.43 (m, 6 H), 4.39 (q, J=7.2 Hz, 2 H), 6.50 -6.55 (m, 1 H), 7.15 -7.20 (m, 1 H), 7.35
(s, 1 H), 8.25 (s, 1 H), 8.37 (s, 1 H),
8.40 -8.46 (m, 1 H)。
化合物12-2(135mg、253.02μmol、1eq)を1Mの濃度のリチウムビス(トリメチルシリル)アミドテトラヒドロフラン溶液(2mL、7.90eq)に溶解させ、次に、塩化アンモニウム(54.14mg、1.01mmol、4eq)を加え、20℃で1時間撹拌した。反応溶液を直接に減圧濃縮して溶媒を乾燥させた後メタノール(2mL)を加え、継いて減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラ
フ;カラム:Phenomenex C18 80×40mm×3μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:25%~55%、8分間実行。)で分離・精製して化合物12を得た。
LCMS: m/z (ESI) =505.1 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 2.39 (s, 3 H), 2.57 - 2.69 (m, 4 H), 2.90 (t, J=7.15 Hz, 2 H), 3.26 - 3.38 (m, 6 H), 5.65 (d,
J=5.77 Hz, 2 H), 6.55 (dd, J=9.03, 3.01
Hz, 1 H), 7.16 (dd, J=8.91, 1.51 Hz, 1 H), 7.36 (s, 1 H), 8.18 (s, 1 H), 8.37 (s, 1 H), 8.41 (d, J=2.89 Hz, 1 H)。

化合物6-8(300mg、989.54μmol、1eq)をN,N-ジメチルホルムアミド(4.5mL)に溶解させ、次に、シアン化第一銅(265.88mg、2.97mmol、648.49μL、3eq)及びヨウ化カリウム(32.85mg、197.91μmol、0.2eq)を加え、窒素ガスの保護下で140℃で0.5時間撹拌した。反応溶液に酢酸エチル(20mL)及び水(20mL)を加え、濾過し、濾液を分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して化合物13-1を得た。
LCMS: m/z (ESI) =250.2 [M+1]+。
1H NMR (400 MHz, CDCl3) δ: 1.41 (t, J=7.2 Hz, 3 H), 2.15 (t, J=6.8 Hz, 2 H), 2.59 - 2.77 (m, 2 H), 3.27 (t, J=6.8 Hz, 2 H), 4.41 (q, J=7.2 Hz, 2 H)。
化合物13-1(145mg、581.66μmol、1eq)を無水テトラヒドロフラン(6mL)に溶解させ、次に、tert-ブトキシビス(ジメチルアミノ)メタン(304.12mg、1.74mmol、360.33μL、3eq)を加え、80℃で3時間撹拌した。反応溶液を直接に減圧濃縮して溶媒を乾燥させて化合物13-2を得た。
LCMS: m/z (ESI) =305.2 [M+H]+。
化合物13-2(177mg、581.54μmol、1eq)をDMF(4.5mL)に溶解させ、次に、化合物1-3(184.53mg、581.54μmol、1eq)を加え、110℃で16時間撹拌した。反応溶液に酢酸エチル(20mL)と水(10mL)及び飽和食塩水(10mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物13-3を得た。
LCMS: m/z (ESI) =559.3 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 1.43 (t, J=7.2 Hz, 3 H), 2.38 (s, 3 H), 2.62 (s, 4 H), 2.89 (t, J=7.2 Hz, 2 H), 3.17 - 3.44 (m, 6 H), 4.42 (q, J=6.8 Hz, 2 H), 6.51 -6.62 (m, 1 H), 7.10 -7.19 (m, 1 H), 7.39
(s, 1 H), 8.15 (d, J=2.8 Hz, 1 H), 8.46
(s, 1 H)。
化合物13-3(200mg、358.05μmol、1eq)を無水テトラヒドロフラン(5mL)に溶解させ、次に、水酸化リチウム一水和物(45.07mg、1.07mmol、3eq)及び水(1mL)を加え、40℃で1時間撹拌し、反応溶液を直接に濃縮して溶媒を乾燥させて化合物13-4を得た。
LCMS: m/z (ESI) =531.1 [M+1] +。
化合物13-4(200mg、376.99μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させ、次に、N,N-ジイソプロピルエチルアミン(146.17mg、1.13mmol、196.99μL、3eq)、2-(7-アゾベンゾトリアゾール)-N,N,N,N-テトラメチルウロニウムヘキサフルオロホスフェート(215.01mg、565.48μmol、1.5eq)、炭酸水素アンモニウム(89.41mg、1.13mmol、93.13μL、3eq)を順次に加え、20℃5時間撹拌した。反応溶液に酢酸エチル(10mL)と水(5mL)及び飽和食塩水(5mL)
を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させ濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラフ;カラム:Phenomenex C18 75×30mm×3μm;移動相A:0.1%の炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:25%~55%、8分間実行。)で分離・精製して化合物13を得た。
LCMS: m/z (ESI) =530.1 [M+1]+。
1H NMR (400 MHz, CDCl3) δ: 2.35 (s, 3 H), 2.52 - 2.70 (m, 4 H), 2.85 -2.92 (m, 2
H), 3.12 - 3.43 (m, 6 H), 5.81 (s, 2 H), 6.49 - 6.61 (m, 1 H), 7.10 - 7.20 (m, 1 H), 7.46 (s, 1 H), 8.05 - 8.19 (m, 1 H). 8.47 (s, 1 H)。

化合物14-1(2g、7.81mmol、1.1eq)を1,4-ジオキサン(30mL)に溶解させ、次に、1-メチル-3-オキソピペラジン(810.63mg、7.10mmol、1eq)を加え、窒素ガスで3回置換し、炭酸セシウム(4.63g、14.20mmol、2eq)を加え、更に窒素ガスで置換し、N,N-ジメチルエチレンジアミン(626.02mg、7.10mmol、775.73μL、1eq)を加え、最後に窒素ガスで置換し、ヨウ化第一銅(676.26mg、3.55mmol、0.5eq)を加え、120℃で16時間撹拌して反応させた。1-メチル-3-オキソピペラジン(810.63mg、7.10mmol、1eq)及びヨウ化第一銅(676.26mg、3.55mmol、0.5eq)を加え、120℃で24時間撹拌を続けた。反応系に水(50mL)を加え、分離し、水相を酢酸エチル(70mL×3)で抽出し、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮し粗生成
物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物14-2を得た。
LCMS: m/z (ESI) =290.0 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 2.41 (s, 3 H), 2.71 - 2.85 (m, 2 H), 3.23 - 3.32 (m, 2 H), 3.62 - 3.72 (t, J=5.20 Hz, 2 H), 3.94 ( s, 2 H), 6.58 - 6.66 (dd, J=8.8, 2.4 Hz, 1 H), 6.73 - 6.80 (d, J=2.4 Hz, 1
H), 7.10 - 7.15 (dd, J=8.8, 1.2 Hz, 1 H)。
化合物14-2(300mg、1.04mmol、1eq)に6Mの塩酸水溶液(1.20mL、6.94eq)を加え、次に、アミノニトリル(348.82mg、8.30mmol、348.82μL、8eq)を加え、60℃で16時間撹拌した。反応系に水(2mL)を加えて希釈し、酢酸エチル(5mL×2)で抽出し、分離した。水相を1Mの水酸化カリウム水溶液でpH=13に調節し、次に、更に酢酸エチル(5mL×3)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させて濾過し、濾液を減圧濃縮して化合物14-3を得た。
LCMS: m/z (ESI) =332.0 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 2.26 (s, 3 H), 2.65 - 2.73 (m, 2 H), 3.08 (s, 2 H),
3.60 - 3.67 (t, J=5.20 Hz, 2 H), 5.27 -
5.34 (m, 2 H), 5.36 - 5.45 (m, 2 H), 6.81 - 6.89 (m, 2 H), 7.14 - 7.20 (m, 1 H)。
化合物14-3(196.47mg、603.70μmol、1eq)のN,N-ジメチルホルムアミド(2mL)に化合物1-2(200mg、603.70μmol、1eq)を加え、110℃で16時間撹拌して反応させた。反応系に水(5mL)を加えて洗浄し、次に、酢酸エチル(5mL×3)を加えて抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物をTLCプレート(展開剤:ジクロロメタン/メタノール=10:1)で分離して化合物14-4を得た。
LCMS: m/z (ESI) =594.1 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 1.26 - 1.33
(m, 3 H), 2.28 (s, 3 H), 2.59 (s, 3 H),
2.60 - 2.70 (m, 2 H), 2.76 - 2.82 (m, 2
H), 3.11 (s, 2 H), 3.22 ( t, J=7.2 Hz, 2 H), 3.63 - 3.69 (t, J=5.2 Hz, 2 H), 4.18 - 4.28 (q, J=7.2 Hz, 2 H), 7.07 - 7.14 (d, J=8.4 Hz, 1 H), 7.35 - 7.42 (d, J=8.4 Hz, 1 H), 8.12 - 8.20 (m, 1 H), 8.39
(s, 1 H), 8.82 (s, 1 H)。
化合物14-4(80mg、134.76μmol、1eq)を無水テトラヒドロフラン(1.6mL)溶液に溶解させ、無水エタノール(0.4mL)、水(0.4mL)を加え、次に、水酸化リチウム一水和物(28.28mg、673.81μmol、5eq)を加え、窒素ガスで3回パージングし、添加完了後、温度を45℃に上昇させて5時間撹拌した。1Mの塩酸水溶液で反応系のpHを7に調節し、次に、反応系の有機溶媒を水ポンプでスピン乾燥させ、残りの水溶液を更に凍結乾燥させて粗生成物を得た。粗生成物にメタノール(5mL)を加え、5分間超音波処理し、濾過し、ケーキを水ポンプで乾燥させて化合物14-5を得た。
LCMS: m/z (ESI) =566.0 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 2.55 (s, 3 H), 2.67 (s, 3 H), 2.75 - 2.80 (m, 4 H),
3.14 (s, 2 H), 3.20 ( t, J=7.2 Hz, 2 H), 3.77 - 3.85 (m, 2 H), 7.08 - 7.15 (m, 1 H), 7.40 - 7.48 (m, 1 H), 8.05 - 8.13 (m, 1 H), 8.37 (s, 1 H)。
化合物14-5(30mg、53.04μmol、1eq)を無水テトラヒドロフラン(0.5mL)に溶解させ、0℃で塩化オキサリル(53.86mg、424.34μmol、37.14μL、8eq)を加え、窒素ガスで3回置換し、次に、N,N-ジメチルホルムアミド(387.71μg、5.30μmol、4.08e-1μL、0.1eq)を加え、0℃で0.5時間撹拌した。20℃で反応系を25%のアンモニア水(5mL)に注ぎ、0.5時間撹拌した。反応系に酢酸エチル(15mL×4)を加えて抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge
BEH分取クロマトグラフ;カラム:Phenomenex C18 100×30mm×10μm;移動相A:10mMの炭酸水素アンモニウム的水溶液、移動相B:アセトニトリル;実行勾配B%:20%~55%、10分)で分離・精製して化合物14を得た。
LCMS: m/z (ESI) =565.1 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 2.29 (s, 3 H), 2.56 (s, 3 H), 2.69 - 2.79 (m, 4 H),
3.07 - 3.16 (m, 4 H), 3.63 - 3.70 (t, J=5.2 Hz, 2H), 7.06 - 7.12 (m, 1 H), 7.36
- 7.42 (m, 1 H), 7.42 - 7.49 (m, 2 H), 8.19 - 8.24 (d , J=2.4 Hz, 1 H), 8.38 (s, 1 H), 8.75 (s, 1 H)。

化合物15-1(1g、2.73mmol、1eq)、化合物15-2(540.37mg、2.73mmol、1eq)、4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(157.70mg、272.55μmol、0.1eq)、トリス(ジベンジリデンアセトン)ジパラジウム(249.58mg、272.55μmol、0.1eq)、ナトリウムtert-ブトキシド(785.80mg、8.18mmol、3eq)を無水トルエン(20mL)に溶解させ、反応混合物を窒素ガスの保護下で60℃で16時間撹拌した。反応完了後、反応溶液を減圧濃縮して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~90:10)で精製して化合物15-3を得た。
LCMS: m/z (ESI) = 381.0, 383.0 [M-55]+。
1H NMR (400 MHz, CDCl3) δ: 7.18 - 7.22 (m, 1 H), 6.92 (d, J=3.0 Hz, 1 H), 6.62 -
6.66 (m, 1 H), 4.31 ( d, J=5.0 Hz, 2 H), 3.67 - 4.04 (m, 2 H), 3.27 (d, J=10.4 Hz, 2 H), 2.68 - 2.72 (m, 1 H), 1.48 (d,
J=8.7 Hz, 1 H), 1.39 (s, 9 H)。
化合物5-4(240mg、746.69μmol、1eq)、化合物15-3(391.79mg、896.03μmol、1.2eq)、酢酸パラジウム(8.38mg、37.33μmol、0.05eq)、4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(21.60mg、37.33μmol、0.05eq)、炭酸セシウム(729.86mg、2.24mmol、3eq)を1,4-ジオキサン(12mL)に溶解させ、窒素ガスの環境下で、110℃で40時間撹拌した。反応溶液に酢酸エチル(10mL)、水(5mL)及び飽和食塩水(5mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させた後濾過し、濾液を減圧濃縮して粗生成物を得、化合物15
-4を得た。
LCMS: m/z (ESI) = 678.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.30 (s, 1 H), 7.92 (s, 1 H), 7.30 - 7.41 (m, 1 H), 7.15 - 7.19 (m, 1 H), 6.32 -6.44 (m, 1 H), 4.19 - 4.46 (m, 4 H), 3.71 - 4.14 (m, 2 H), 3.21 - 3.47 (m, 4 H), 2.83 (t, J=7.2 Hz, 2 H), 2.65 (s, 4 H), 1.49 (d, J=8.4 Hz, 1 H), 1.36 - 1.43 (m, 12 H)。
化合物15-4(150mg、221.32μmol、1eq)をジクロロメタン(5mL)に溶解させ、次に、トリフルオロ酢酸(770.00mg、6.75mmol、0.5mL、30.51eq)を加え、20℃で16時間撹拌した。減圧濃縮して溶媒を乾燥させた。粗生成物に水(10mL)及び酢酸エチル(10mL)を加え、次に、飽和炭酸水素ナトリウム水溶液でpHを8以上に調節した後、分離し、有機相を無水硫酸ナトリウムで乾燥させて濾過し、減圧濃縮して溶媒を乾燥させて化合物15-5を得た。
LCMS: m/z (ESI) = 578.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.31 (s, 1 H), 7.93 (d, J=3.0 Hz, 1 H), 7.26 (s, 1 H), 7.18 - 7.22 (m, 1 H), 6.33 -6.35 (m, 1
H), 5.31 (s, 1 H), 4.36 -4.40 (m, 2 H),
3.92 (d, J=5.8 Hz, 2 H), 3.51 - 3.73 (m, 4 H), 3.33 (t, J=7.2 Hz, 2 H), 2.83 (t, J=7.2 Hz, 3 H), 2.64 (s, 3 H), 1.60 -1.65 (m, 1 H), 1.40 (t, J=7.2 Hz, 3 H)。
化合物15-5(120mg、207.74μmol、1eq)を無水テトラヒドロフラン(6.5mL)に溶解させ、37%の純度のホルムアルデヒド水溶液(238.46mg、2.94mmol、218.77μL、14.14eq)を加え、次に、酢酸(229.45mg、830.97μmol、218.52μL、4eq)を加え、25℃で15分間撹拌し、次に、ナトリウムトリアセトキシボロヒドリド(176.12mg、830.97μmol、4eq)を加え、窒素ガスの保護下で25℃で、45分間撹拌した。反応溶液に酢酸エチル(10mL)と水(5mL)及び飽和炭酸水素ナトリウム水溶液(5mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させた後、濾過して粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物15-6を得た。
LCMS: m/z (ESI) = 592.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.32 (s, 1 H), 8.03 (d, J=2.9 Hz, 1 H), 7.30 (s, 1 H), 7.20 -7.22 (m, 1 H), 6.34 -6.40 (m, 1 H), 4.36 - 4.40 (m, 2 H), 4.04 (d, J=4.1
Hz, 2 H), 3.54 - 3.74 (m, 4 H), 3.33 (t
, J=7.2 Hz, 2 H), 2.84 (t, J=7.2 Hz, 2 H), 2.64 (s, 3 H), 2.34 (s, 3 H), 1.76 (d, J=9.2 Hz, 1 H), 1.40 (t, J=7.2 Hz, 3 H)。
化合物15-6(90mg、152.11μmol、1eq)及び塩化アンモニウム(48.82mg、912.68μmol、6eq)を1Mのリチウムビス(トリメチルシリル)アミドのn-ヘキサン溶液(1.52mL、10eq)に溶解させ、窒素ガスの環境下で、20℃で2時間撹拌した。反応溶液にメタノール(3mL)を加えてクエンチングさせた後、減圧濃縮して溶媒を乾燥させて、粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:Phenomenex C18 100×30mm×10μm;移動相A:10mMの炭酸水素アンモニウム的水溶液、移動相B:アセトニトリル;実行勾配:アセトニトリル%:15%~85%、8分)で分離・精製して化合物15を得た。
LCMS: m/z (ESI) = 563.2 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 8.30 (s, 1 H), 7.63 (d, J=2.8 Hz, 1 H), 7.21 (d, J=9.0 Hz, 1 H), 6.50 -6.55 (m, 1 H), 3.60 - 3.80 (m, 4 H), 3.44 - 3.57 (m, 2 H), 3.23 (t, J=7.2 Hz, 2 H), 2.84 (t, J=7.2 Hz,
2 H), 2.50 - 2.72 (m, 4 H), 2.19 (s, 3 H), 1.70 (d, J=8.4 Hz, 1 H)。

反応フラスコに化合物6-8(400mg、1.32mmol、1eq)、シクロプロピルボロン酸(147.33mg、1.72mmol、1.3eq)、リン酸カリウム(1.01g、4.75mmol、3.6eq)、酢酸パラジウム(29.62mg、131.94μmol、0.1eq)、トリシクロヘキシルホスフィン(111.00mg、395.82μmol、128.32μL、0.3eq)を加え、次に、無水トルエン(
12mL)及び水(0.6mL)を加え、窒素ガスで3回パージングし、80℃で16時間撹拌した。反応完了後、反応溶液を減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物16-1を得た。
LCMS: m/z (ESI) = 265.1 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 4.30 -4.38 (m, 2 H), 3.41 (s, 1 H), 3.21 (t, J=6.2 Hz, 2 H), 2.50 - 2.65 (m, 2 H), 2.05 (t, J=6.3 Hz, 2 H), 1.37 (t, J=7.2 Hz, 3 H), 1.25 - 1.32 (m, 2 H), 0.77 - 0.91 (m, 2 H)。
化合物16-1(225mg、851.18μmol、1eq)を無水テトラヒドロフラン(4.5mL)に溶解させ、次に、tert-ブトキシビス(ジメチルアミノ)メタン(445.04mg、2.55mmol、527.30μL、3eq)を加え、80℃で20時間撹拌した。反応完了後、減圧濃縮して溶媒を乾燥させて粗生成物化合物16-2を得、精製せず、直接次の反応に使用した。
LCMS: m/z (ESI) = 293.1 [M-26]+。
化合物16-2(270mg、845.29μmol、1eq)をN,N-ジメチルホルムアミド(3.5mL)に溶解させ、次に、化合物1-3(268.22mg、845.29μmol、1eq)を加え、110℃で20時間撹拌した。反応完了後、反応溶液に酢酸エチル(50mL)及び水(50mL)を加え、分離し、有機相を無水硫酸ナトリウムで乾燥させた後、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物16-3を得た。
LCMS: m/z (ESI) = 574.3 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.35 (s, 1 H), 8.09 (d, J=2.8 Hz, 1 H), 7.11 - 7.16 (m, 2 H), 6.52 - 6.54 (m, 1 H), 4.34 (d, J=7.2 Hz, 2 H), 3.55 (s, 1 H), 3.28 -3.30 (m, 6 H), 2.79 -2.82 (m, 2 H), 2.57 - 2.73 (m, 4 H), 2.42 (s, 3 H), 1.38 (t, J=7.2 Hz, 3 H), 1.17 - 1.25 (m, 2 H), 0.82 - 0.90 (m, 2 H)。
化合物16-3(120mg、209.19μmol、1eq)及び塩化アンモニウム(67.14mg、1.26mmol、6eq)を1Mのリチウムビス(トリメチルシリル)アミドのn-ヘキサン溶液(2.09mL、10eq)に溶解させ、次に、窒素ガスの環境下で20℃で1時間撹拌した。反応完了後、反応溶液にメタノール(3mL)を加えてクエンチングさせ、次に、減圧濃縮して溶媒を乾燥させた。粗生成物を分取HPLC
(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:Phenomenex C18 100×30mm×10μm;移動相A:10mMの炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配B%:15%~85%、8分)で分離・精製して化合物16を得た。
LCMS: m/z (ESI) = 545.3 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 8.32 (s, 1 H), 7.60 (m, 1 H), 7.19 -7.23 (m, 1 H), 6.76 - 6.72 (m, 1 H), 3.39 -3.48 (m, 1 H),
3.25 (s, 4 H), 3.17 (t, J=6.8 Hz, 2 H),
2.80 (t, J=6.8 Hz, 2 H), 2.67 (s, 4 H),
2.40 (s, 3 H), 1.04 - 1.10 (m, 2 H), 0.75 - 0.79 (m, 2 H)。

-75℃で、窒素ガスの保護下で、化合物17-1(1.25g、8.65mmol、1eq)の無水テトラヒドロフラン溶液(5mL)を0.1Mのリチウム2,2,6,6-テトラメチルピペリジンのテトラヒドロフラン溶液(172.94mL、2eq)に加え、0.5時間撹拌し、次に、-75℃でヨード(2.59g、10.20mmol、2.06mL、1.18eq)を加え、3時間撹拌した。反応系に飽和塩化アンモニウム水溶液(150mL)を加えて洗浄し、分離し、酢酸エチル(100mL×3)で抽出し、有機相を合わせ、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物
を得た。シリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~95:5)で分離・精製して化合物17-2を得た。
LCMS: m/z (ESI) =271.0 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 4.05 (s, 3 H), 8.44 (s, 1 H)。
化合物17-2(600mg、2.22mmol、1eq)のジメチルスルホキシド(12mL)にp-メトキシベンジルアミン(912.99mg、6.66mmol、861.31μL、3eq)、フッ化カリウム(386.66mg、6.66mmol、155.91μL、3eq)を加え、120℃で3時間撹拌した。反応系に水(10mL)を加え、酢酸エチル(10mL×3)で抽出し、分離し、有機相を合わせて減圧濃縮した。シリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~60:40)で精製して化合物17-3を得た。
LCMS: m/z (ESI) =279.9 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 3.71 (s, 3 H), 4.00 (s, 3 H), 4.28 - 4.38 (d, J=6.4
Hz 2 H), 6.45 (s, 1 H), 6.85 - 6.92 (d,
J=8.8 Hz, 2 H), 7.21 - 7.29 (d, J=8.8 Hz, 2 H), 7.50 - 7.58 (m, 1 H)。
化合物17-3(250mg、893.75μmol、1eq)を1,4-ジオキサン(2mL)に溶解させ、2-ジシクロヘキシルホスフィノ-2,6-ジイソプロポキシ-1,1-ビフェニル(208.53mg、446.87μmol、0.5eq)、(2-ジシクロヘキシルホスフィノ-2,6-ジイソプロポキシ-1,1-ビフェニル)[2-(2-アミノ-1,1-ビフェニル)]パラジウム(II)(373.75mg、446.87μmol、0.5eq)を加え、窒素ガスで3回置換し、更に、ナトリウムtert-ブトキシド(171.78mg、1.79mmol、2eq)、N-メチルピペラジン(179.04mg、1.79mmol、198.27μL、2eq)を加え、110℃で4時間撹拌した。反応系に水(10mL)を加えて洗浄し、次に、酢酸エチル(10mL×4)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。シリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物17-4を得た。
LCMS: m/z (ESI) =344.2 [M+H] +。
1H NMR (400 MHz, CDCl3) δ: 2.36 (s, 3 H), 2.49 - 2.63 (m, 4 H), 3.47 - 3.50 (m, 4 H), 3.82 (s, 3 H), 4.08 (s, 3 H), 4.23
- 4.30 (d, J=5.2 Hz, 2 H), 5.93 (s, 1 H), 6.89 - 6.93 (d, J=8.8 Hz, 2 H), 7.23 - 7.26 (d, J=8.8 Hz, 2 H)。
化合物17-4(190mg、553.25μmol、1eq)にトリフルオロ酢酸(
6mL)を加え、50℃で16時間撹拌した。反応系を直接に減圧濃縮して粗生成物を得た。反応系に水(5mL)を加え、次に、ジクロロメタン(5mL×3)で洗浄し、水相を1Mの水酸化ナトリウム水溶液でpHを14に調節し、ジクロロメタン(20mL×6)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して化合物17-5を得た。
LCMS: m/z (ESI) =224.0 [M+H] +。
1H NMR (400 MHz, CD3OD) δ: 2.35 (s, 3 H), 2.52 - 2.62 (m, 4 H), 3.40 - 3.44 (m, 4 H), 3.99 (s, 3 H), 6.30 (s, 1 H)。
化合物5-4(200mg、622.24μmol、1eq)のエチレングリコールジメチルエーテル(20mL)にヨウ化セシウム(96.88mg、684.47μmol、59.43μL、1.1eq)、ヨード(86.86mg、342.23μmol、68.94μL、0.55eq)、ヨウ化第一銅(37.92mg、199.12μmol、0.32eq)、亜硝酸イソアミル(116.63mg、995.59μmol、134.06μL、1.6eq)を順次に加え、70℃で18時間撹拌した。反応系にアンモニア水(10mL)を加えて洗浄し、次に、飽和チオ硫酸ナトリウム水溶液(20mL)で洗浄し、有機相を合わせ、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。シリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物17-6を得た。
LCMS: m/z (ESI) =432.9 [M+H] +。
化合物17-5(50mg、223.94μmol、1.5eq)を1,4-ジオキサン(2mL)に溶解させ、化合物17-6(64.54mg、149.29μmol、1eq)、4,5-ビス(ジフェニルホスフィノ-9,9-ジメチルキサンテン(3.46mg、5.97μmol、0.04eq)、炭酸セシウム(145.93mg、447.88μmol、3eq)を加え、窒素ガスで3回置換した後、酢酸パラジウム(670.35μg、2.99μmol、0.02eq)を加え、窒素ガスで3回置換し、110℃で2時間撹拌した。反応系に水(5mL)を加え、更に酢酸エチル(10mL×3)を加えて抽出し、分離し、有機相を直接に減圧濃縮して粗生成物を得た。TLCプレート(展開剤:ジクロロメタン/メタノール=10:1)で精製して化合物17-7を得た。
LCMS: m/z (ESI) =528.1 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 1.29 - 1.32
(m, 3 H), 2.31 - 2.35 (m, 4 H), 2.41 (s, 3 H), 2.65 - 2.69 (m, 4 H), 2.71 (s, 3
H), 2.82 - 2.87 (m, 2 H), 3.24 - 3.27 (m, 2 H), 4.02 (s, 3 H), 4.27 - 4.31 (m, 2 H), 8.04 - 8.13 (m, 1 H), 8.18 (s, 1 H), 8.56 (s, 1 H)。
化合物17-7(35mg、66.33μmol、1eq)の無水テトラヒドロフラン
(3.5mL)溶液に塩化アンモニウム(21.29mg、397.98μmol、6eq)を加え、次に、1Mのリチウムビス(トリメチルシリル)アミドのテトラヒドロフラン溶液(663.30μL、10eq)を加え、20℃で2時間撹拌して反応させた。反応系に飽和塩化アンモニウム水溶液(10mL)を加え、酢酸エチル(10mL×3)で抽出し、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Waters Xbridge BEH分取クロマトグラフ;カラム:C18 100×30mm×10μm;移動相A:10mMの炭酸水素アンモニウムの水溶液、移動相B:アセトニトリル;実行勾配B%:10%~40%、8分)で分離・精製して化合物17を得た。
LCMS: m/z (ESI) =499.2 [M+H] +。
1H NMR (400 MHz, DMSO-d6) δ: 2.22 (s, 3 H), 2.41 - 2.45 (m, 4 H), 2.66 (s, 3 H),
2.78 - 2.85 (m, 2 H), 3.10 - 3.17 (m, 2
H), 3.41 - 3.45 (m, 4 H), 4.02 (s, 3 H), 7.49 (s, 2 H), 8.03 (s, 1 H), 8.20 (s,
1 H), 8.53 (s, 1 H)。

0℃で窒素ガスの保護下で、シクロプロパノール(192.08mg、3.31mmol、2eq)の無水テトラヒドロフラン溶液(8mL)に60%の純度の水素化ナトリウム(132.29mg、3.31mmol、2eq)を加え、0℃で0.5時間撹拌して反応させ、化合物2-1(500mg、1.65mmol、1eq)を加え、0℃で1時間反応を続けた。反応完了後、水(5mL)を加え、酢酸エチル(10mL×2)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、薄層クロマトグラフィー(展開剤:石油エーテル/酢酸エチル=100:0~
60:40)で分離して化合物18-1を得た。
LCMS: m/z (ESI) =280.9 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 0.90 - 0.97 (m, 2 H), 1.06 - 1.14 (m, 2 H), 1.33 - 1.42 (t, J=7.2 Hz, 3 H), 1.96 - 2.06 (m, 2
H), 2.44 - 2.53 (t, J=6.0 Hz, 2 H), 3.16 - 3.23 (t, J=7.2 Hz, 2 H), 4.05 - 4.12
(m, 1 H), 4.28 - 4.39 (q, J=7.2 Hz, 2 H)。
化合物18-1を無水トルエン(10mL)に溶解させ、tert-ブトキシビス(ジメチルアミノ)メタン(1.99g、11.41mmol、2.36mL、20eq)を加え、90℃で2時間撹拌した。反応完了後、反応系に水(10mL)を加え、酢酸エチル(10mL×3)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して化合物18-2を得た。
LCMS: m/z (ESI) =308.9 [M-26]+。
化合物18-2(150mg、447.20μmol、1eq)をN,Nジメチルホルムアミド(2mL)に溶解させ、化合物1-3(141.90mg、447.20μmol、1eq)を加え、110℃で4時間撹拌して反応させた。反応完了後、反応系に水(5mL)を加え、酢酸エチル(10mL×3)で抽出し、分離し、有機相を合わせ、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、カラムクロマトグラフィー(勾配溶出剤:ジクロロメタン/メタノール=100:0~90:10)で分離・精製して化合物18-3を得た。
LCMS: m/z (ESI) =590.1 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 0.88 - 0.97 (m, 2 H), 1.05 - 1.14 (m, 2 H), 1.38 - 1.42 (t, J=7.2 Hz, 3 H), 2.41 (s, 3 H), 2.57 - 2.71 (m, 4 H), 2.78 - 2.88 (t, J=7.2 Hz, 2 H), 3.26 - 3.36 (m, 6 H), 4.14 -
4.17 (m, 1 H), 4.31 - 4.39 (m, 2 H), 6.46 - 6.52 (m, 1 H), 7.11 - 7.17 (m, 1 H), 7.30 (s, 1 H), 8.28 (s, 1 H), 8.31 - 8.34 (d, J =2.8 Hz 1 H)。
化合物18-3(100mg、169.60μmol、1eq)を無水テトラヒドロフラン(4mL)、メタノール(1mL)及び水(1mL)の混合溶媒に溶解させ、水酸化リチウム一水和物(35.58mg、847.99μmol、5eq)を加え、45℃で3時間撹拌した。反応完了後、反応系を減圧濃縮して有機溶媒を除去し、更に、2NのHCl水溶液でpHを7に調節し、直接に減圧濃縮して化合物18-4を得た。
LCMS: m/z (ESI) =562.1 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 0.71 - 0.81
(m, 4 H), 2.21 (s, 3 H), 2.44 - 2.46 (m, 4 H), 2.57 - 2.63 (m, 2 H), 3.09 - 3.15 (m, 4 H), 3.17 - 3.22 (m, 2 H), 3.98 -
4.07 (m, 1 H), 6.57 - 6.68 (m, 1 H), 7.10 - 7.22 (m, 1 H), 7.79 - 7.86 (d, J=2.8 Hz, 1 H), 7.97 (s, 1 H), 8.22 (s, 1 H)。
化合物18-4(171.77mg、1.35mmol、118.46μL、8eq)を無水テトラヒドロフラン(3mL)に溶解させ、0℃で塩化オキサリル(171.77mg、1.35mmol、118.46μL、8eq)を加え、窒素ガスで3回置換し、N,Nジメチルホルムアミド(1.24mg、16.92μmol、1.30μL、0.1eq)を加え、0℃で0.5時間撹拌し、25%の純度のアンモニア水(9mL)を加え、20℃で0.5時間撹拌した。反応完了後、反応系に酢酸エチル(10mL×3)を加え、抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、分取HPLC(HPLC製造方法:Waters Xbridge
BEH分取クロマトグラフ;カラム:C18 75×30mm×3μm;移動相A:10mMの炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;勾配B%:30%~70%、8分)で分離・精製して化合物18を得た。
LCMS: m/z (ESI) =561.2 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 0.79 - 0.85
(m, 4 H), 2.21 (s, 3 H), 2.43 - 2.47 (m, 4 H), 2.66 - 2.70 (t, J=7.2 Hz, 2 H), 3.04 - 3.09 (t, J=7.2 Hz, 2 H), 3.11 - 3.15 (m, 4 H), 4.11 - 4.18 (m, 1 H), 6.65
- 6.71 (m, 1 H), 7.13 - 7.19 (d, J=8.8 Hz, 1 H), 7.35 (s, 2 H), 7.65 - 7.70 (d,
J=2.8 Hz, 1 H), 8.20 (s, 1 H), 8.29 (s,
1 H)。

化合物6-3(1.2g、5.35mmol、1eq)、(2-ブロモエチニル)トリイソプロピルシラン(1.47g、5.62mmol、1.05eq)、酢酸銀(893.06mg、5.35mmol、273.95μL、1eq)、酢酸パラジウム(120.13mg、535.06μmol、0.1eq)をアセトニトリル(45mL)に溶解させ、80℃で72時間撹拌した。反応完了後、20℃に冷却させ、反応溶液を減圧濃縮して乾燥させて粗生成物を得、シリカゲルカラムクロマトグラフィー(勾配溶出:石油エーテル/酢酸エチル=100:0~98:2)で精製して化合物19-1を得た。
LCMS: m/z (ESI) = 405.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 4.30 - 4.39 (m, 2 H), 3.18 - 3.25 (m, 2 H), 2.51 - 2.65 (m, 2 H), 1.97 - 2.14 (m, 2 H), 1.32 - 1.42 (m, 3 H), 1.17 (s, 21 H)。
化合物19-1(155mg、383.06μmol、1eq)を無水テトラヒドロフラン(2.5mL)に溶解させ、次に、tert-ブトキシビス(ジメチルアミノ)メタン(211.00mg、1.21mmol、0.25mL、3.16eq)を加え、窒素ガスでパージングした後、80℃で2時間撹拌した。反応完了後、20℃に冷却させ、減圧濃縮して溶媒を乾燥させて粗生成物化合物19-2を得、直接に次の反応に使用した。
LCMS: m/z (ESI) = 433.2 [M-26]+。
ステップ3:化合物19-3の合成
化合物19-2(170mg、369.79μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させ、次に、化合物1-3(117.34mg、369.79μmol、1eq)を加え、110℃で20時間撹拌した。反応完了後、反応溶液に水(10mL)及び飽和食塩水(10mL)を加え、酢酸エチル(20mL)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得、シリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~98:2)で精製して化合物19-3を得た。
LCMS: m/z (ESI) = 714.3 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.38 (s, 1 H), 8.20 (m, 1 H), 7.19 (s, 1 H), 7.14 (m,
1 H), 6.52 (m, 1 H), 4.37 (m, 2 H), 3.19 - 3.40 (m, 6 H), 2.81 ( 2 H), 2.53 - 2.70 (m, 4 H), 2.39 (s, 3 H), 1.40 (t, J=7.2 Hz, 3 H), 1.09 - 1.19 (m, 21 H)。
化合物19-3(60mg、84.04μmol、1eq)及び塩化アンモニウム(26.97mg、504.25μmol、6eq)を1Mのリチウムビス(トリメチルシリル)アミドのn-ヘキサン溶液(2.09mL、10eq)に溶解させ、窒素ガスの環境下で20℃で1時間撹拌し、反応溶液にメタノール(3mL)を加えて反応をクエンチングさせ、減圧濃縮して溶媒を乾燥させて粗生成物化合物19-4を得た。
LCMS: m/z (ESI) = 685.3 [M+H]+。
化合物19-4(55mg、80.31μmol、1eq)及び1Mのテトラブチルアンモニウムフルオリドのテトラヒドロフラン溶液(1.2mL、14.94eq)を無水テトラヒドロフラン(2mL)に溶解させ、20℃で20時間撹拌した。反応溶液に飽和食塩水(5mL)及び水(5mL)を加え、更に、酢酸エチル(10mL)を加え、溶液を濾過した後分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して溶媒を乾燥させて粗生成物を得、分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラフ;カラム:C18 75×30mm×3μm;移動相A:H2O(0.2%のギ酸を含む)、移動相B:アセトニトリル;実行勾配:B%:1%~40%、8分間実行。)で分離・精製して化合物19的ギ酸塩を得た。
LCMS: m/z (ESI) = 529.2 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 9.01 - 9.13 (m, 1 H), 8.41 - 8.55 (m, 1 H), 7.98 - 8.09 (m, 1 H), 7.69 - 7.78 (m, 1 H), 7.08 - 7.24 (m, 1 H), 6.87 - 6.98 (m, 1 H), 6.66 - 6.82 (m, 1 H), 4.53 - 4.64 (m, 1 H), 3.39 - 3.46 (m, 2 H), 3.28 - 3.32 (m,
4 H), 3.05 - 3.12 (m, 2 H), 2.93 - 3.02
(m, 4 H), 2.54 - 2.72 (m, 3 H)。

化合物6-8(200mg、659.69μmol、1eq)、2-(トリブチルスタニル)フラン(235.59mg、659.69μmol、208.48μL、1eq)、1,1-ビス(ジフェニルホスフィノ)フェロセンジクロロパラジウム(48.27mg、65.97μmol、0.1eq)をN,N-ジメチルホルムアミド(5mL)に溶解させ、120℃で16時間撹拌した。反応溶液に水(10mL)及び飽和食塩水(10mL)を加え、酢酸エチル(20mL)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して溶媒を乾燥させて粗生成物を得、シリカゲルカラムクロマトグラフィー(勾配溶出剤:石油エーテル/酢酸エチル=100:0~70:30)で精製して化合物20-1を得た。
LCMS: m/z (ESI) = 291.0 [M+H]+。
化合物20-1(145mg、499.43μmol、1eq)を無水テトラヒドロフラン(5mL)に溶解させ、次に、tert-ブトキシビス(ジメチルアミノ)メタン(6.12g、35.11mmol、7.25mL、70.30eq)を加え、窒素ガスの環境下で、80℃で16時間撹拌した。反応完了後、減圧濃縮して溶媒を乾燥させて化合物20-2を得た。
LCMS: m/z (ESI) = 319.0 [M-26]+。
化合物20-2(170mg、492.17μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させ、化合物1-3(156.17mg、492.17μmol、1eq)を加え、110℃で20時間撹拌した。反応完了後、20℃に冷却させ、反応溶液に水(10mL)及び飽和食塩水(10mL)を加え、酢酸エチル(20mL)で抽出し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮し
て乾燥させて粗生成物を得、シリカゲルカラムクロマトグラフィー(勾配溶出:ジクロロメタン/メタノール=100:0~98:2)で精製して化合物20-3を得た。
LCMS: m/z (ESI) = 600.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.41 (s, 1 H), 8.11 - 8.13 (m, 1 H), 7.40 - 7.57 (m, 2 H), 7.23 (s, 1 H), 7.13 -7.16 (m, 1 H), 6.49 -6.52 (m, 1 H), 6.40 - 6.45 (m, 1
H), 4.39 - 4.45 (m, 2 H), 3.33 -3.36 (m, 2 H), 3.04 - 3.16 (m, 4 H), 2.82 - 2.86 (m, 2 H), 2.44 - 2.59 (m, 4 H), 2.34 (s, 3 H), 1.42 (t, J=7.2 Hz, 3 H)。
化合物20-3(130mg、216.80μmol、1eq)及び塩化アンモニウム(69.58mg、1.30mmol、6eq)を1Mのビス(トリメチルシリル)アミノのn-ヘキサン溶液(1.73mL、8eq)に溶解させ、窒素ガスの環境下で、20℃で2時間撹拌した。反応溶液にメタノール(5mL)を加えた後、減圧濃縮して溶媒を乾燥させて粗生成物を得、分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラフ;カラム:C18 80×40mm×3μm;移動相A:10mMの炭酸水素アンモニウムの水溶液、移動相B:アセトニトリル;実行勾配:B%:25%~55%、8分間実行。)で分離・精製して化合物20を得た。
LCMS: m/z (ESI) = 571.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.41 (s, 1 H), 8.11 (s, 1 H), 7.45 (s, 2 H), 7.24 (s,
1 H), 7.13 -7.16 (m, 1 H), 6.50 -6.55 (m, 1 H), 6.40 (s, 1 H), 5.66 (s, 2 H), 3.30 -3.36 (m, 2 H), 3.01 - 3.20 (m, 4 H), 2.82 - 2.90 (m, 2 H), 2.54 -2.60 (m, 4
H), 2.36 (s, 3 H)。

化合物5-4(50mg、155.56μmol、1eq)を無水ジクロロメタン(2mL)に加え、0℃に冷却させ、次に、85%の純度のm-クロロ過安息香酸(63.17mg、311.12μmol、2eq)を加え、25℃に昇温させて12時間撹拌した。反応完了後、反応溶液を飽和炭酸水素ナトリウム水溶液(4mL)にゆっくりと注ぎ、ジクロロメタン(4mL×2)で抽出し、有機相を合わせ、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、シリカゲルカラムクロマトグラフィー(勾配溶出剤:ジクロロメタン/メタノール=100:0~90:10)で精製して化合物21-1を得た。
LCMS: m/z (ESI) = 354.1 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.29 (s, 1H),
5.05 (s, 2H), 4.40 (q, J = 7.2 Hz, 2H),
3.76 (s, 3H), 3.33 (t, J = 7.2 Hz, 2H),
2.80 (t, J = 7.2 Hz, 2H), 1.41 (t, J = 7.2 Hz, 3H)。
化合物21-1(50mg、141.48μmol、1eq)及び化合物7-3(66.81mg、141.48μmol、1eq)を1,4-ジオキサン(2mL)に加え、トリス(ジベンジリデンアセトン)ジパラジウム(12.96mg、14.15μmol、0.1eq)、4,5-ビス(ジフェニルホスフィノ-9,9-ジメチルキサンテン(
8.19mg、14.15μmol、0.1eq)、炭酸セシウム(92.19mg、282.95μmol、2eq)を加え、100℃に上昇させ、12時間撹拌した。反応完了後、反応溶液を水(3mL)にゆっくりと注ぎ、クエンチングさせ、酢酸エチル(3mL×3)で抽出し、有機相を合わせ、飽和食塩水(3mL×3)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、薄層クロマトグラフィー(展開剤:石油エーテル/酢酸エチル=100:100)で分離して化合物21-2を得た。
LCMS: m/z (ESI) = 698.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.45 (s, 1H),
7.96 - 8.04 (m, 1H), 7.14 - 7.20 (m, 1H), 7.07 - 7.13 (m, 1H), 6.56 - 6.65 (m, 1H), 4.34 - 4.56 (m, 2H), 3.58 - 3.66 (m, 4H), 3.53 - 3.57 (m, 3H), 3.32 - 3.39 (m, 2H), 3.17 - 3.24 (m, 4H), 2.82 - 2.90 (m, 2H), 1.47 - 1.52 (m, 9H), 1.38 - 1.45 (m, 3H)。
化合物21-2(80mg、114.66μmol、1eq)を無水ジクロロメタン(2mL)に加え、トリフルオロ酢酸(273.78mg、2.40mmol、177.78μL、20.94eq)を加え、25℃で2時間撹拌し、反応溶液を減圧濃縮して化合物21-3のトリフルオロ酢酸塩を得、粗生成物を直接に次のステップに使用した。
LCMS: m/z (ESI) = 598.1 [M+H]+。
化合物21-3のトリフルオロ酢酸塩(75mg、105.39μmol、1eq)を無水テトラヒドロフラン(1mL)に加え、トリエチルアミン(10.66mg、105.39μmol、14.67μL、1eq)、37%の純度のホルムアルデヒド水溶液(34.21mg、421.55μmol、31.38μL、4eq)及び氷酢酸(25.32mg、421.55μmol、24.11μL、4eq)を順次に加え、25℃で0.5時間撹拌し、次に、トリアセトキシ水素化ホウ素ナトリウム(89.34mg、421.55μmol、4eq)を加え、25℃で12時間撹拌し、反応溶液に飽和炭酸水素ナトリウム水溶液(2mL)を加え、酢酸エチル(2mL×2)で抽出し、分離し、乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、薄層クロマトグラフィー(展開剤:ジクロロメタン/メタノール=100:10)で分離・精製して化合物21-4を得た。
LCMS: m/z (ESI) = 612.2 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 8.41 - 8.46 (m, 1H), 7.91 - 7.95 (m, 1H), 7.09 - 7.18
(m, 2H), 6.55 - 6.63 (m, 1H), 4.34 - 4.44 (m, 2H), 3.50 - 3.56 (m, 3H), 3.30 - 3.36 (m, 2H), 3.23 - 3.30 (m, 4H), 2.78 - 2.89 (m, 2H), 2.59 - 2.66 (m, 4H), 2.35 - 2.41 (m, 3H), 1.37 - 1.43 (m, 3H)。
化合物21-4(60mg、98.09μmol、1eq)を無水テトラヒドロフラン(1mL)及び水(1mL)に加え、次に、水酸化リチウム一水和物(12.35mg、294.28μmol、3eq)を加え、25℃で12時間撹拌し、反応完了後、直接に濃縮し、2Nの塩酸でpH=6~7に調節し、次に、ジクロロメタン(3×3mL)で抽出し、分離し、有機相を合わせ、無水硫酸ナトリウムで乾燥させ、濾過し、濃縮し、化合物21-5を得た。
LCMS: m/z (ESI) = 584.2 [M+H]+。
化合物21-5(50mg、85.67μmol、1eq)を無水N,N-ジメチルホルムアミド(0.5mL)に加え、炭酸水素アンモニウム(8.80mg、111.38μmol、9.17μL、1.3eq)、2-(7-アゾベンゾトリアゾール)-N,N,N,N-テトラメチルウロニウムヘキサフルオロホスフェート(65.15mg、171.35μmol、2eq)及びN,N-ジイソプロピルエチルアミン(33.22mg、257.02μmol、44.77μL、3eq)を加え、25℃で12時間撹拌し、反応完了後濾過し、濾液を減圧濃縮して粗生成物を得、分取HPLC(HPLC製造方法:Waters 2767/QDa分取クロマトグラフ;カラム:C18 80×30mm×3μm;移動相A:10mMの炭酸水素アンモニウム水溶液、移動相B:アセトニトリル;実行勾配:B%:25%~55%、8分間実行。)で分離・精製して化合物21を得た。
LCMS: m/z (ESI) = 583.2 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 8.75 - 8.84
(m, 1H), 8.44 - 8.50 (m, 1H), 7.77 - 7.99 (m, 2H), 7.18 - 7.24 (m, 1H), 7.12 - 7.16 (m, 1H), 6.82 - 6.87 (m, 1H), 3.30 - 3.33 (m, 3H), 3.13 - 3.21 (m, 4H), 3.04 - 3.11 (m, 2H), 2.71 - 2.78 (m, 2H), 2.41 - 2.45 (m, 4H), 2.19 - 2.24 (m, 3H)。

エチレングリコールモノメチルエーテル(151.00mg、1.98mmol、156.47μL、2eq)をテトラヒドロフラン溶液(5mL)に溶解させ、0℃に冷却させ、60%の純度の水素化ナトリウム(79.37mg、1.98mmol、2eq)を加え、0℃で0.5時間反応させ、化合物2-1(300mg、992.18μmol、1eq)を加え、0℃で0.5時間反応を続け、反応完了後、水(20mL)を加え、酢酸エチル(10mL×3)で抽出し、飽和食塩水(20mL)で洗浄し、分離し、有機相を無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、カラムクロマトグラフィー(勾配溶出剤:石油エーテル/酢酸エチル=100:0~50:50)で分離・精製して化合物22-1を得た。
LCMS: m/z (ESI) =298.9 [M+H]+。
1H NMR (400 MHz, CDCl3) δ: 1.38 (t, J=7.09 Hz, 3 H), 1.98 - 2.08 (m, 2 H), 2.42 - 2.58 (m, 2 H), 3.19 - 3.23 (m, 2 H), 3.46 - 3.55 (m, 3 H), 3.81 - 3.95 (m, 2 H), 4.33 (q, J=7.09 Hz, 2 H), 4.38 - 4.42
(m, 2 H)。
化合物22-1(100mg、335.17μmol、1eq)をトルエン(3mL)に溶解させ、tert-ブトキシビス(ジメチルアミノ)メタン(350.49mg、2.01mmol、415.27μL、6eq)を加え、90℃で12時間撹拌した。反応完了後、飽和塩化アンモニウム水溶液(5mL)を加え、酢酸エチル(3mL×3)で抽
出し、分離し、有機相を飽和塩化ナトリウム水溶液(5mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して化合物22-2を得た。
LCMS: m/z (ESI) =327.0 [M-26]+。
化合物22-2(200mg、565.88μmol、1eq)及び化合物1-3(179.56mg、565.88μmol、1eq)をN,Nジメチルホルムアミド(2mL)に溶解させ、110℃で12時間撹拌し反応させた。反応完了後、水(5mL)を加えて反応をクエンチングさせ、酢酸エチル(5mL×3)で抽出し、有機相を合わせ、有機相を飽和塩化ナトリウム溶液(5mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得、分取HPLC(HPLC製造方法:Waters Xbridge Prep OBD分取クロマトグラフ;カラム:C18 150×40mm×10μm;移動相A:10mMの炭酸水素アンモニウム水溶液(0.05%のアンモニア水を含む)、移動相B:アセトニトリル;勾配B%:50%~80%、8分)で分離・精製して化合物22-3を得た。
LCMS: m/z (ESI) =608.3 [M+H]+。
1H NMR (400 MHz, CD3OD) δ: 1.38 (t, J=7.13 Hz, 3 H), 2.37 (s, 3 H), 2.59 - 2.68 (m, 4 H), 2.80 - 2.82 (m, 2 H), 3.24 - 3.31 (m, 9 H), 3.75 - 3.80 (m, 2 H), 4.33
(q, J=7.13 Hz, 2 H), 4.41 - 4.50 (m, 2 H), 6.60 - 6.70 (m J=9.07, 1 H), 7.15 - 7.23 (m, 1 H), 8.20 - 8.24 (m, 1 H), 8.32 (s, 1 H)。
化合物22-3(40mg、65.83μmol、1eq)をテトラヒドロフラン(1mL)、メタノール(0.25mL)及び水(0.25mL)の混合溶媒に溶解させ、水酸化リチウム一水和物(13.81mg、329.14μmol、5eq)を加え、40℃で12時間撹拌し、反応完了後、減圧濃縮して溶媒の大部分を除去し、2Nの塩酸でpHを6~7に調節し、酢酸エチル(2mL×3)で抽出し、有機相を合わせ、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して化合物22-4を得た。
LCMS: m/z (ESI) =580.1 [M+H]+。
化合物22-4(35mg、60.39μmol、1eq)をN,Nジメチルホルムアミド(2mL)に溶解させ、次に、N,N-ジイソプロピルエチルアミン(78.05mg、603.88μmol、105.18μL、10eq)、2-(7-アゾベンゾトリアゾール)-N,N,N,N-テトラメチルウロニウムヘキサフルオロホスフェート(137.77mg、362.33μmol、6eq)、炭酸水素アンモニウム(47.74mg、603.88μmol、49.73μL、10eq)を順次に加え、20℃で12時間撹拌した。反応完了後、飽和塩化アンモニウム水溶液(2mL)を加え、酢酸エチル(2mL×3)で抽出し、有機相を合わせ、飽和食塩水(3mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して乾燥させて粗生成物を得、分取HPL
C(HPLC製造方法:Phenomenex 分取クロマトグラフ;カラム:C18 75×30mm×3μm;移動相A:10mMの炭酸水素アンモニウム水溶液(0.05%のアンモニア水を含む)、移動相B:アセトニトリル;実行勾配:B%:30%~55%、8分間実行。)で分離・精製して化合物22を得た。
LCMS: m/z (ESI) =579.1 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 2.25 (s, 3 H), 2.71 (s, 2 H), 3.00 - 3.24 (m, 9 H),
3.35 (s, 4 H), 3.59 (s, 2 H), 4.26 (s, 2 H), 6.60 - 6.70 (m, 1 H), 7.15 - 7.20 (m, 1 H), 7.38 (s, 2 H), 7.80 (s, 1 H), 8.21 (s, 1 H), 8.34 (s, 1 H)。

化合物1-4(14g、24.15mmol、1eq)を水(140mL)、テトラヒドロフラン(70mL)及びメタノール(70mL)の混合溶液に加え、水酸化リチウム一水和物(3.04g、72.46mmol、3eq)を加え、25℃で12時間撹拌した。反応完了後、反応溶液を原体積の1/2まで減圧濃縮し、2-メチルテトラヒドロフラン(30mL)を加え、6Nの塩酸でpHを6~7に調節し、20℃で2時間撹拌した後濾過し、乾燥させて化合物23-1を得た。
LCMS: m/z (ESI) = 552.1 [M+H]+。
化合物23-1(50mg、90.64μmol、1eq)をN,N-ジメチルホルムアミド(2mL)に溶解させ、2-(7-アゾベンゾトリアゾール)-N,N,N,N-テトラメチルウロニウムヘキサフルオロホスフェート(68.93mg、181.29μmol、2eq)、N,N-ジイソプロピルエチルアミン(35.14mg、271.93μmol、47.36μL、3eq)を加え、20℃で0.5時間撹拌し、次に、O-(テトラヒドロ-2H-ピラン)-2-ヒドロキシルアミン(21.24mg、181.29μmol、2eq)を加え、20℃で1時間撹拌を続け、反応完了後、反応系に水(3mL)を加え、濾過し、得られたケーキを乾燥させて化合物23-2を得た。
LCMS: m/z (ESI) =651.0 [M+H]+。
化合物23-2(50mg、76.84μmol、1eq)を無水メタノール(5mL)に溶解させ、p-トルエンスルホン酸(35.71mg、207.40μmol、2.70eq)を加え、20℃で1時間撹拌して反応させた。反応完了後、反応系を減圧濃縮し、水(5mL)を加え、酢酸エチル(5mL×3)で抽出し、分離し、有機相を合わせて無水硫酸ナトリウムで乾燥させ、濾過し、濾液を減圧濃縮して粗生成物を得た。粗生成物を分取HPLC(HPLC製造方法:Phenomenex Luna分取クロマトグラフ;カラム:C18 75×30mm×3μm;移動相A:水(0.05%のギ酸を含む)、移動相B:アセトニトリル;実行勾配:B%:20%~60%、8分間実行。)で分離・精製して化合物23のギ酸塩を得た。
LCMS: m/z (ESI) = 567.1 [M+H]+。
1H NMR (400 MHz, DMSO-d6) δ: 10.53 - 10.85 (m, 1 H), 8.93 - 9.36 (m, 1 H), 8.47 (s, 1 H), 8.32 (s, 1 H), 8.16 (s, 1 H), 7.50 (d, J=2.8 Hz, 1 H), 7.17 (dd, J=8.8, 1.6 Hz, 1 H), 6.71 (dd, J=9.2, 2.8 Hz,
1 H), 3.12 - 3.18 (m, 4 H), 3.07 - 3.10
(m, 2 H), 2.73 (t, J=7.2 Hz, 2 H), 2.53
(s, 3 H), 2.43 - 2.46 (m, 4 H), 2.22 (s, 3 H)。
試験例1:in vitro PLK1キナーゼ活性評価
33P同位体標識キナーゼ活性試験(Reaction Biology Corp)を使用して、IC50値を測定し、それによりヒトPLK1プロテインキナーゼに対する試験化合物の阻害能力を評価した。

33P同位体標識キナーゼ活性試験(Reaction Biology Corp)を使用して、IC50値を測定し、それによりヒトPLKファミリープロテインキナーゼPLK2/PLK3/PLK4に対する試験化合物の阻害能力を評価した。
紙(Whatman#3698-915)にスポットした。0.75%のリン酸溶液で濾紙を繰り返して洗浄した後、濾紙上に残った放射性リン酸化基質のレベルを測定した。%キナーゼ活性=キナーゼ活性試験化合物/キナーゼ活性ブランク群(DMSO)×100%であり、Prism4ソフトウェア(GraphPad)を用いたカーブフィッティングによりIC50値を得、実験結果は表2に示された通りである。

実験材料:
McCoy’s 5A培地、ペニシリン/ストレプトマイシン抗生物質はVicenteから購入し、ウシ胎児血清はBioseraから購入した。3D CellTiter-Glo(細胞生存率の化学発光検出試薬)試薬はPromegaから購入した。HCT116細胞株は、Nanjing Cobioer Biosciences Co.、Ltd.から購入した。Envisionマルチラベルアナライザー(PerkinElmer)。
HCT116細胞を超低吸着96ウェルUプレートに播種し、各ウェルに80μLの細胞懸濁液とし、その中に1000個のHCT116細胞が含まれるようにした。細胞プレートを二酸化炭素インキュベーターで一晩培養した。
試験化合物をピペットで9個の濃度に5倍希釈し、即ち、2μMから5.12nMに希釈し、同じ条件で2つのウェルを設置した。78μLの培地をミドルプレートに加え、更に対応する位置に従って、2μL/ウェルの勾配希釈化合物をミドルプレートに移し、均一に混合した後20μL/ウェルを細胞プレートに移した。細胞プレートに移された化合物濃度の範囲は、10μM~0.0256nMであった。細胞プレートを二酸化炭素インキュベーターに入れ、5日間培養した。別の細胞プレートを用意し、薬物添加当日の最大値(下式のMax値)として信号値を読み取り、データ解析に参与させた。
当該細胞プレートの各ウェルに100μLの細胞生存率化学発光検出試薬を加え、室温で10分間培養して、発光信号を安定させた。マルチラベルアナライザーでデータを読み取った。
式(Sample-Min)/(Max-Min)×100%を使用して元のデータを阻害率に変換すると、IC50値は、4つのパラメーターを使用したカーブフィッティングによって求めることができた(GraphPad Prismのlog(inhibitor)vs.response--Variable slopeモードで求めることができる)。表3は、HCT116細胞増殖に対する本発明の化合物の阻害活性を提供している。

試験目的:
本実験の目的は、静脈内注射及び経口投与後のCD-1オスマウスの血漿中の試験化合物の薬物動態を研究するためである。
静脈内注射群:適量の試験化合物を秤量し、20%のSBE-b-CDの水溶液で溶解させ、6Mの塩酸でpHを4~5に調節し、2分間ボルテックスして透明な溶液を得、その後、5Nの水酸化ナトリウムでpHを約7に調節し、1分間ボルテックスして1.5mg/mLの透明な溶液を得、使用のために0.22μmの微多孔膜で濾過した。6~10週齢のCD-1オスマウスを選択し、試験化合物溶液を静脈内注射した。試料収集時間は:0.083、0.25、0.5、1、2、4、6、8、24時間であった。
各時点で頸静脈から約50uLの全血を収集し、HPLC-タンデム質量分析(LC-MS/MS)に用いられる血漿を調製して、濃度を測定した。最後の時点でPK試料を収集した後、すべての動物をCO2麻酔で安楽死させた。WinNonlinTM Version 6.3(Pharsight、Mountain View、CA)薬物動態学ソフトの非コンパートメントモデルを使用して血漿濃度を処理し、線形対数ラダー法によって薬物動態パラメーターを計算し、実験結果は、下記の表4に示される通りである。

化合物はCD-1マウスの薬物動態研究において低い薬物クリアランスを示し、経口投与した後、快速にピークに達し、比較的に高い経口吸收バイオアベイラビリティを示した。
1.実験目的
本実験では、ヒト結腸癌HCT116皮下移植腫瘍BALB/cヌードマウスモデルを使用して、試験化合物のin vivo抗腫瘍効果を評価する。
2.1モデルの構築
HCT116細胞の培養:McCoy’s 5A培地に10%のウシ胎児血清を加え、37℃の5%のCO2細胞インキュベータで培養した。細胞の飽和度が80%~90%であり、細胞の数が要件に達した時、細胞を収集してカウントし、BALB/cヌードマウス(オス、6~7週齢)の皮下に接種した。
腫瘍がある程度成長した時点で、腫瘍体積が大きすぎる動物、小さすぎる動物又は腫瘍の形が不規則な動物を排除し、腫瘍体積が103.12~174.35mm3の範囲の動物を選択し、腫瘍体積に応じて、無作為群分け法により動物を6つの群に分け、各群に6匹のマウスにし、平均腫瘍体積は約147.12mm3であり、実験の群分け及び投与方法は下記の表5に示される通りである。動物の健康と死亡を毎日モニタリングし、定期的に腫瘍の成長及び例えば、行為活動、食物と水の摂取量、体重の変化(週に2回体重を測定)、腫瘍の大きさ(週に2回腫瘍体積を測定)、身体的兆候又はその他の異常の行動などの動物の日常行動に対する薬物治療の影響を検出した。

PO:経口投与
QD:1日1回、5日間投与し、2日間休薬。
腫瘍体積(tumor volume、TV)の計算式は:1/2×a×b2であり、ここで、a、bはそれぞれ腫瘍の長さと幅を表す。腫瘍阻害率TGI(%)の計算式は:TGI(%)=[(1-(特定の処理群の投与終了時の平均腫瘍体積-当該処理群の投与開始時の平均腫瘍体積)/(溶媒対照群の治療終了時の平均腫瘍体積-溶媒対照群の治療開始時の平均腫瘍体積)]×100%である。
本研究では、実験データはいずれもMean±SEMで表した。統計分析は、試験終了時のRTVデータに基づいてIBM SPSS Statisticsソフトを使用して実行された。2つの群間の比較にはT testを使用し、3つの群又はそれ以上の群間の比較にはone-way ANOVAを使用して分析し、分散が均一である場合(F値に有意差がない場合)、Tukey’s法で分析し、分散が均一ではない場合(F値に有意差がある場合)、Games-Howell法で試験した。p<0.05は、有意差があると見なされた。
本実験では、ヒト結腸癌HCT-116細胞皮下移植腫瘍BALB/cヌードマウスモデルにおける試験化合物1、化合物2、化合物8の薬効を評価した。本試験では、投与期間を通じてすべての実験群で動物の死亡が発生せず、マウスは良好な耐薬性を示した。実験結果は、表6及び図1、図2に示される通りである。

Claims (13)
- 式(P)で表される化合物又はその薬学的に許容される塩。

T1は、CR1及びNから選択され、
T2は、CH及びNから選択され、
R1は、Hから選択され、
R2は、H、CN、C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ、S(=O)2C1-3アルキル、C2-3アルキニル、C3-5シクロアルキル、-O-C3-5シクロアルキル及び5員ヘテロアリールから選択され、前記C1-3アルキル、C1-3アルコキシ、C1-3アルキルチオ、S(=O)2C1-3アルキル、C2-3アルキニル、C3-5シクロアルキル、-O-C3-5シクロアルキル及び5員ヘテロアリールは、任意選択で1、2又は3つのRbにより置換され、
R3は、C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルから選択され、前記C1-4アルキル、ピペラジニル及び7~9員ヘテロシクロアルキルは、任意選択で1、2又は3つのRcにより置換され、
R4は、C1-3アルキル及びC1-3アルコキシから選択され、前記C1-3アルキル及びC1-3アルコキシは、任意選択で1、2又は3つのRdにより置換され、
R5は、H及びOHから選択され、
或いは、R1及びR3と連結された原子と環を形成し、構造フラグメント
から選択され、
各Rbは、それぞれ独立してF、Cl、Br、I、OH及びOCH3から選択され、
各Rcは、それぞれ独立して=O、C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルから選択され、前記C1-3アルキル、C1-4アルキルアミノ及びヘテロシクロブチルは、任意選択で1、2又は3つのRにより置換され、
各Rdは、それぞれ独立してF、Cl、Br及びIから選択され、
各Rは、それぞれ独立してF、Cl、Br、I及びOHから選択され、
前記「ヘテロシクロブチル」及び「7~9員ヘテロシクロアルキル」のヘテロ原子は、N、O及びSから選択される。) - R2は、H、CN、SCH3、SCH2CH3、SCH(CH3)2、S(=O)2CH3、OCH3、OCH2CH3、OCH(CH3)2、CH3、CH2CH3、CH(CH3)2、シクロプロピル、
から選択され、前記SCH3、SCH2CH3、SCH(CH3)2、S(=O)2CH3、OCH3、OCH2CH3、OCH(CH3)2、CH3、CH2CH3、CH(CH3)2、シクロプロピル、
は、任意選択で1、2又は3つのRbにより置換される、請求項1に記載の化合物又はその薬学的に許容される塩。
- R2は、H、CN、SCH3、SCH2CH2OH、S(=O)2CH3、OCH2CH3、OCH2CH2OH、OCH2CH2OCH3、CH3、CH2CH2OH、CH2OCH3、シクロプロピル、
から選択される、請求項2に記載の化合物又はその薬学的に許容される塩。
- Rcは、=O、CH3、CH2CH3、N(CH3)2及び
から選択され、前記CH3、CH2CH3及びN(CH3)2は、任意選択で1、2又は3つのRにより置換される、請求項1に記載の化合物又はその薬学的に許容される塩。
- Rcは、=O、CH3、CH2CH2OH、N(CH3)2及び
から選択される、請求項4に記載の化合物又はその薬学的に許容される塩。
- R3は、CH2CH2CH3、
から選択され、前記CH2CH2CH3、
は、任意選択で1、2又は3つのRcにより置換される、請求項1に記載の化合物又はその薬学的に許容される塩。
- R3は、
から選択される、請求項1、4~6のいずれか1項に記載の化合物又はその薬学的に許容される塩。
- R4は、CH3、OCH3、OCHF2及びOCF3から選択される、請求項1に記載の化合物又はその薬学的に許容される塩。
- 下記の式から選択される、請求項1~8のいずれか1項に記載の化合物又はその薬学的に許容される塩。

- 下記の式から選択される、化合物又はその薬学的に許容される塩。


- 固形腫瘍を治療するための医薬の製造における、請求項1~10のいずれか一項に記載の化合物又はその薬学的に許容される塩の使用。
- 選択性PLK1阻害剤に関連する固形腫瘍を治療する薬物の製造における、請求項1~10のいずれか一項に記載の化合物又はその薬学的に許容される塩の使用。
- 前記固形腫瘍は結直腸癌を指す、請求項11又は12に記載の使用。
Applications Claiming Priority (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110172946 | 2021-02-08 | ||
| CN202110172946.6 | 2021-02-08 | ||
| CN202110655630.2 | 2021-06-11 | ||
| CN202110655630 | 2021-06-11 | ||
| CN202110864387.5 | 2021-07-29 | ||
| CN202110864387 | 2021-07-29 | ||
| CN202111137961 | 2021-09-27 | ||
| CN202111137961.3 | 2021-09-27 | ||
| CN202111473661 | 2021-11-29 | ||
| CN202111473661.2 | 2021-11-29 | ||
| PCT/CN2022/074110 WO2022166725A1 (zh) | 2021-02-08 | 2022-01-26 | 5,6-二氢噻吩并[3,4-h]喹唑啉类化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2024505714A JP2024505714A (ja) | 2024-02-07 |
| JP7545622B2 true JP7545622B2 (ja) | 2024-09-05 |
Family
ID=82740912
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023547663A Active JP7545622B2 (ja) | 2021-02-08 | 2022-01-26 | 5,6-ジドヒロチエノ[3,4-h]キナゾリン系化合物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20240140962A1 (ja) |
| EP (1) | EP4289852A1 (ja) |
| JP (1) | JP7545622B2 (ja) |
| CN (1) | CN116761806A (ja) |
| WO (1) | WO2022166725A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118382626A (zh) * | 2021-12-10 | 2024-07-23 | 山东绿叶制药有限公司 | 蛋白激酶抑制剂及其制备方法和应用 |
| WO2024032558A1 (zh) * | 2022-08-08 | 2024-02-15 | 南京明德新药研发有限公司 | 一种5,6-二氢噻吩并[3,4-h]喹唑啉类化合物的盐型、晶型及其制备方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007502851A (ja) | 2003-05-22 | 2007-02-15 | ファルマシア・イタリア・エス・ピー・エー | ピラゾロ−キナゾリン誘導体、その製造方法、およびキナーゼ阻害剤としてのその使用 |
| JP2007509848A (ja) | 2003-10-14 | 2007-04-19 | バーテックス ファーマシューティカルズ インコーポレイテッド | プロテインキナーゼのインヒビターとして有用な組成物 |
| CN101484457A (zh) | 2006-04-12 | 2009-07-15 | 弗特克斯药品有限公司 | 作为用于治疗增殖病症的蛋白激酶PLK1抑制剂的4,5-二氢-[1,2,4]三唑并[4,3-f]蝶啶 |
| WO2009112324A1 (de) | 2008-03-11 | 2009-09-17 | Vita Zahnfabrik H. Rauter Gmbh & Co.Kg | Dental-sinterofen sowie verfahren zum sintern keramischer dental-elemente |
| CN101563351A (zh) | 2006-12-21 | 2009-10-21 | 内尔维阿诺医学科学有限公司 | 取代的吡唑并-喹唑啉衍生物、它们的制备方法和它们作为激酶抑制剂的用途 |
| JP2010536760A (ja) | 2007-08-15 | 2010-12-02 | バーテックス ファーマシューティカルズ インコーポレイテッド | 増殖性疾患の処置のための、ヒトタンパク質キナーゼplk1ないしplk4の阻害剤としての4−(9−(3,3−ジフルオロシクロペンチル)−5,7,7−トリメチル−6−オキソ−6,7,8,9−テトラヒドロ−5h−ピリミド[4,5−b[1,4]ジアゼパン−2−イルアミノ]−3−メトキシベンザミド誘導体 |
| JP2013533276A (ja) | 2010-07-30 | 2013-08-22 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | タンパク質キナーゼ活性の調整剤としてのイソオキサゾロ−キナゾリン |
| JP2016515537A (ja) | 2013-03-15 | 2016-05-30 | ダナ ファーバー キャンサー インスティテュート,インコーポレイテッド | ピリミド−ジアゼピノン化合物および障害の治療方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ584760A (en) * | 2007-09-25 | 2012-03-30 | Takeda Pharmaceutical | Polo-like kinase inhibitors |
| EP2100894A1 (en) * | 2008-03-12 | 2009-09-16 | 4Sc Ag | Pyridopyrimidines used as Plk1 (polo-like kinase) inhibitors |
| US8735386B2 (en) * | 2010-07-23 | 2014-05-27 | Boehringer Ingelheim International Gmbh | Aminopyrazoloquinazolines |
| CN112771049B (zh) * | 2018-09-27 | 2024-01-26 | 贝达药业股份有限公司 | Fgfr4抑制剂及其应用 |
-
2022
- 2022-01-26 JP JP2023547663A patent/JP7545622B2/ja active Active
- 2022-01-26 US US18/276,226 patent/US20240140962A1/en active Pending
- 2022-01-26 EP EP22749004.2A patent/EP4289852A1/en active Pending
- 2022-01-26 WO PCT/CN2022/074110 patent/WO2022166725A1/zh active Application Filing
- 2022-01-26 CN CN202280011925.9A patent/CN116761806A/zh active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007502851A (ja) | 2003-05-22 | 2007-02-15 | ファルマシア・イタリア・エス・ピー・エー | ピラゾロ−キナゾリン誘導体、その製造方法、およびキナーゼ阻害剤としてのその使用 |
| JP2007509848A (ja) | 2003-10-14 | 2007-04-19 | バーテックス ファーマシューティカルズ インコーポレイテッド | プロテインキナーゼのインヒビターとして有用な組成物 |
| CN101484457A (zh) | 2006-04-12 | 2009-07-15 | 弗特克斯药品有限公司 | 作为用于治疗增殖病症的蛋白激酶PLK1抑制剂的4,5-二氢-[1,2,4]三唑并[4,3-f]蝶啶 |
| CN101563351A (zh) | 2006-12-21 | 2009-10-21 | 内尔维阿诺医学科学有限公司 | 取代的吡唑并-喹唑啉衍生物、它们的制备方法和它们作为激酶抑制剂的用途 |
| JP2010536760A (ja) | 2007-08-15 | 2010-12-02 | バーテックス ファーマシューティカルズ インコーポレイテッド | 増殖性疾患の処置のための、ヒトタンパク質キナーゼplk1ないしplk4の阻害剤としての4−(9−(3,3−ジフルオロシクロペンチル)−5,7,7−トリメチル−6−オキソ−6,7,8,9−テトラヒドロ−5h−ピリミド[4,5−b[1,4]ジアゼパン−2−イルアミノ]−3−メトキシベンザミド誘導体 |
| WO2009112324A1 (de) | 2008-03-11 | 2009-09-17 | Vita Zahnfabrik H. Rauter Gmbh & Co.Kg | Dental-sinterofen sowie verfahren zum sintern keramischer dental-elemente |
| JP2013533276A (ja) | 2010-07-30 | 2013-08-22 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | タンパク質キナーゼ活性の調整剤としてのイソオキサゾロ−キナゾリン |
| JP2016515537A (ja) | 2013-03-15 | 2016-05-30 | ダナ ファーバー キャンサー インスティテュート,インコーポレイテッド | ピリミド−ジアゼピノン化合物および障害の治療方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4289852A1 (en) | 2023-12-13 |
| WO2022166725A1 (zh) | 2022-08-11 |
| US20240140962A1 (en) | 2024-05-02 |
| CN116761806A (zh) | 2023-09-15 |
| JP2024505714A (ja) | 2024-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111484477B (zh) | 一种苯并吡啶酮杂环化合物及其用途 | |
| US20230118795A1 (en) | Aryl or heteroaryl pyridone or pyrimidine derivative, preparation method and use thereof | |
| JP2022549171A (ja) | 縮合ピリドン化合物、ならびにその調製方法および使用 | |
| JP7333342B2 (ja) | 2,3-ジヒドロ-1h-ピロリジン-7-ホルムアミド誘導体およびその使用 | |
| CN112745335B (zh) | 一种三并杂环化合物及其用途 | |
| EP4129996A1 (en) | Novel aminopyrimidine egfr inhibitor | |
| JP7545622B2 (ja) | 5,6-ジドヒロチエノ[3,4-h]キナゾリン系化合物 | |
| CN113024544B (zh) | 一种含氰基并杂环化合物及其用途 | |
| CN112851663B (zh) | 一种并杂环化合物及其用途 | |
| JP2021514997A (ja) | ピラゾロピリミジン誘導体及びその使用 | |
| RU2771314C1 (ru) | Производные хинолинпирролидин-2-она и их применение | |
| CN112707905B (zh) | 一种三并杂环化合物及其制备方法和用途 | |
| JP2023501110A (ja) | Cdk2/4/6三重阻害剤としてのアミノピリミジン化合物 | |
| WO2021129817A1 (zh) | 具有果糖激酶(khk)抑制作用的嘧啶类化合物 | |
| CN115942937A (zh) | 嘧啶并环类化合物 | |
| CA3200649A1 (en) | Novel camptothecin derivative, composition containing same, and use thereof | |
| CN114591315A (zh) | 一种组织蛋白酶c小分子抑制剂 | |
| CN113825757B (zh) | 取代的稠合双环类衍生物、其制备方法及其在医药上的应用 | |
| JP2022506802A (ja) | 大環状チロシンキナーゼ阻害剤及びその用途 | |
| US7465724B2 (en) | Bis-pyrrolo[2,1-c][1,4]benzodiazepine-anthraquinone conjugates and a process for the preparation thereof | |
| JP2023522863A (ja) | Egfr阻害剤としての三環式化合物 | |
| CN114685520B (zh) | 三并环化合物及其药物组合物和应用 | |
| CN113045569B (zh) | 用作ret激酶抑制剂的化合物及其应用 | |
| AU2015360133A1 (en) | Nitromidazole compound, preparation method therefor and use thereof in drug manufacturing | |
| JP7275444B2 (ja) | チエノ[2,3-c]ピリダジン-4(1H)-オン系誘導体及びその使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230814 |
|
| TRDD | Decision of grant or rejection written | ||
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20240627 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20240703 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20240730 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240730 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20240730 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7545622 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |