JP4267670B2 - 有機el素子の製造方法 - Google Patents
有機el素子の製造方法 Download PDFInfo
- Publication number
- JP4267670B2 JP4267670B2 JP2007134226A JP2007134226A JP4267670B2 JP 4267670 B2 JP4267670 B2 JP 4267670B2 JP 2007134226 A JP2007134226 A JP 2007134226A JP 2007134226 A JP2007134226 A JP 2007134226A JP 4267670 B2 JP4267670 B2 JP 4267670B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- compound
- organic
- layer
- light emitting
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 CC1C=C(C(C)=C(C=C(*)C=C2)C2=N2)C2=CC=C1 Chemical compound CC1C=C(C(C)=C(C=C(*)C=C2)C2=N2)C2=CC=C1 0.000 description 2
- YXFVVABEGXRONW-UHFFFAOYSA-N Cc1ccccc1 Chemical compound Cc1ccccc1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 2
- QQZVQGBXHFHFIC-UHFFFAOYSA-N Cc1c(cccc2)c2c(C)nn1 Chemical compound Cc1c(cccc2)c2c(C)nn1 QQZVQGBXHFHFIC-UHFFFAOYSA-N 0.000 description 1
- FKHNZQFCDGOQGV-UHFFFAOYSA-N Cc1nc2ccccc2nc1C Chemical compound Cc1nc2ccccc2nc1C FKHNZQFCDGOQGV-UHFFFAOYSA-N 0.000 description 1
Images


















Landscapes
- Electroluminescent Light Sources (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
(1)有機化合物の物理的変化
(結晶ドメインの成長などにより界面の不均一化が生じ、素子の電荷注入能の劣化・短絡・絶縁破壊の原因となる。特に分子量500以下の低分子化合物を用いると結晶粒の出現・成長が起こり、膜性が著しく低下する。また、ITO等の界面が荒れていても、顕著な結晶粒の出現・成長が起こり、発光効率の低下や、電流のリークを起こし、発光しなくなる。また、部分的非発光部であるダークスポットの原因にもなる。)
(電子の注入を容易にするために仕事関数の小さな金属としてNa・Mg・Alなどを用いてきたが、これらの金属は大気中の水分や酸素と反応したり、有機層と陰極の剥離が起こり、電荷注入ができなくなる。特に高分子化合物などを用い、スピンコートなどで成膜した場合、成膜時の残留溶媒や分解物が電極の酸化反応を促進し、電極の剥離が起こり部分的な非発光部を生じさせる。)
(有機化合物中に電流を流すので、高い電界強度下に有機化合物を置かねばならず、発熱からは逃れられない。その熱のため、有機化合物の溶融・結晶化・熱分解などにより素子の劣化・破壊が起こる。)
などが挙げられる。
C.Adachi,et al., Appli.Phys.Lett,.56,799(1990)
テトラアリールエテン誘導体を含有する有機化合物層を有する有機EL素子の製造方法であって、
芳香族残基三置換ハロゲン化エテンをグリニャール化し、ジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物とクロスカップリングするか、またはジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物をグリニャール化し、芳香族残基三置換ハロゲン化エテンとクロスカップリングするかによって、下記化1で表されるテトラアリールエテン誘導体を得る工程を備えることを特徴とする有機EL素子の製造方法。

本発明の有機EL素子は上記化1に示されるテトラアリールエテン誘導体を適宜選択して有機化合物層に用いるため、高輝度が安定して得られる。また、耐熱性・耐久性が高く、素子電流密度も100mAcm−2程度でも安定した駆動が可能である。
(1)ハロゲン化トリフェニルエテン化合物等の芳香族残基三置換ハロゲン化エーテルをグリニャール化し、NiCl2(dppp)〔dppp:ジフェニルフォスフィノプロパン〕等のNi錯体などを用いて、ジハロゲン化アリール誘導体等のジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物とクロスカップリングする方法、
(2)ジハロゲン化アリール誘導体等のジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物をグリニャール化し、NiCl2(dppp)等のNi錯体などを用いてハロゲン化トリフェニルエテン誘導体等の芳香族残基三置換ハロゲン化エテンとクロスカップリングする方法、等を用いて合成できる。
化合物No.1の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.488g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のテトラヒドロフラン(THF)溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.3gと4,4’−ジブロモビフェニル3.02g(9.4mmol)を加え、60℃で4時間還流した。この反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ジクロロメタンより再結晶し、3.0gの青色蛍光を示す白色固体を得た。
元素分析: C H Br
計算値/% 94.22 5.78 0.0
測定値/% 94.31 5.54 0.0
赤外吸収スペクトル:図2
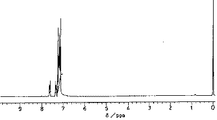
1H−NMRスペクトル(270MHz):図3
示差走査熱量測定(DSC):融点304℃、ガラス転移温度110℃
イオン化ポテンシャル:5.90eV
化合物No.2の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと1,4−ジブロモベンゼン2.35g(10mmol)を加え、60℃で4時間還流した。この反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ジクロロメタンより再結晶し、3.0gの青色蛍光を示す白色固体を得た。
元素分析: C H Br
計算値/% 94.16 5.84 0.0
測定値/% 94.15 5.53 0.0
赤外吸収スペクトル:図4
1H−NMRスペクトル:図5
示差走査熱量測定(DSC):融点250℃、ガラス転移温度83℃
イオン化ポテンシャル:5.95eV
化合物No.3の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.488g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.3gと4,4’,4”−トリブロモトリフェニルアミン3.00g(6.0mmol)を加え、60℃で4時間還流した。この反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ジクロロメタンにより再結晶後3.0gの青緑色蛍光を示す黄白色固体を得た。
元素分析: C H N Br
計算値/% 92.91 5.69 1.39 0.0
測定値/% 92.46 5.32 1.29 0.0
赤外吸収スペクトル:図6
1H−NMRスペクトル(270MHz):図7
示差走査熱量測定(DSC):融点300℃、ガラス転移温度129℃
イオン化ポテンシャル:5.45eV
化合物No.7の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと1,3,5−トリブロモベンゼン2.07g(6.6mmol)を加え、60℃で4時間還流した。反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ヘキサンより再結晶後、トルエン/ヘキサンを展開溶媒としてシリカカラム精製し、1.0gの青色蛍光を示す白色固体を得た。
赤外吸収スペクトル:図8
1H−NMRスペクトル(270MHz):図9
示差走査熱量測定(DSC):融点213℃、ガラス転移温度92℃
イオン化ポテンシャル:5.95eV
化合物No.8の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2,5−ジブロモチオフェン2.42g(10mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと1,2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)を加え、60℃で4時間還流した。反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ヘキサンより再結晶後、1.0gの緑色蛍光を示す黄色固体を得た。
元素分析: C H S Br
計算値/% 89.15 5.44 5.41 0.0
測定値/% 89.05 5.32 5.05 0.0
イオン化ポテンシャル:5.40eV
化合物No.19の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと2,3−ビス(4−ブロモフェニル)キノキサリン4.40g(20mmol)を加え、60℃で4時間還流した。反応溶液を0.1N塩酸水溶液に投入しクロロホルムで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトンより再結晶後、3.0gの緑色蛍光を示す黄色固体を得た。
元素分析: C H N
計算値/% 91.10 5.35 3.54
測定値/% 91.03 5.28 3.40
赤外吸収スペクトル:図10
1H−NMRスペクトル:図11
イオン化ポテンシャル:5.98eV
化合物No.4の合成
4,4’−ジブロモビフェニル3.02g(9.4mmol)のかわりに、ビス(p−ブロモフェニル)エーテル2.95g(9mmol)を用いるほかは実施例1と同様にして合成した。
赤外吸収スペクトル:図12
1H−NMRスペクトル:図13
示差走査熱量測定(DSC):融点254℃、ガラス転移温度90℃
なお、元素分析の測定値は計算値とよく一致した。
化合物No.33の合成
N,N’−(ビス(4−ブロモフェニル)−N,N’−ジフェニル−4,4’−ジアミノビフェニルの合成N,N’−ジフェニルベンジジン16.8g(50mmol)と4−ヨードブロモベンゼン42.4g(150mmol)と活性銅粉0.2gと炭酸カリウム20.7g(150mmol)を200mlナスフラスコに投入し、N2置換後200℃で24時間攪拌した。
赤外吸収スペクトル:図14
1H−NMRスペクトル:図15
示差走査熱量測定(DSC):融点333℃、ガラス転移温度132℃
イオン化ポテンシャル:5.38eV
なお、元素分析の測定値は計算値とよく一致した。
化合物No.42の合成
9,10−ビス(p−ブロモフェニル)アントラセンの合成
260mlフラスコにアルゴン下で、アントラキノン4.16g(20mmol)とトルエン100mlの中に4−ヨードブロモベンゼンとブチルリチウム(ヘキサン溶液)より合成した。4−ブロモフェニルリチウム6.52g(40mmol)のエーテル溶液を滴下した。室温で24時間攪拌後、水を100ml滴下した。沈澱物を濾過し、ジオール体を得た。
赤外吸収スペクトル:図16
1H−NMRスペクトル:図17
示差走査熱量測定(DSC):融点370℃、ガラス転移温度143℃
なお、元素分析の測定値は計算値とよく一致した。
化合物No.65の合成
4,4’−ジブロモビフェニル3.02g(9.4mmol)のかわりに、2,6−ジクロロ−3メトキシアクリジン2.5g(9mmol)を用いるほかは実施例1と同様にして合成した。
赤外吸収スペクトル:図18
1H−NMRスペクトル:図19
示差走査熱量測定(DSC):融点259.2℃、ガラス転移温度132.6℃
なお、元素分析の測定値は計算値とよく一致した。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
実施例12と同様に素子を作製した。ただし、ホール輸送材料TPD−1の代わりに、N,N,N’,N’−テトラキス(3−ビフェニル)−4,4’−ジアミノ−1,1’−ビフェニル(TPD−2)を用いた。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
実施例12において、発光層に化合物No.1を用いるかわりに、化合物No.42を用いるほかは同様にして有機EL素子を得た。
この有機EL素子に電圧を印加して電流を流したところ、15V、450mA/cm2で8020cd/m2の青色(発光極大波長λmax=470nm)の発光が確認され、この発光は乾燥窒素雰囲気中で3000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で300時間であった。
実施例12において、発光層に化合物No.1を用いるかわりに、化合物No.49を用いるほかは同様にして有機EL素子を得た。
この有機EL素子に電圧を印加して電流を流したところ、14V、340mA/cm2で18000cd/m2の青色(発光極大波長λmax=480nm)の発光が確認され、この発光は乾燥窒素雰囲気中で5000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で500時間であった。
実施例12において、発光層に化合物No.1を用いるかわりに、化合物No.53を用いるほかは同様にして有機EL素子を得た。
実施例12において、発光層を形成した後、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで20nmの厚さに蒸着し、電子輸送層とした。次いで、テトラブチルジフェノキノンを10nmの厚さに蒸着し、電子注入層とした。その後、実施例12と同様にして有機EL素子を得た。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
実施例20において、正孔注入輸送層に化合物No.7を用いるかわりに、化合物No.8を用いるほかは同様にして有機EL素子を得た。
実施例20において、正孔注入輸送層に化合物No.7を用いるかわりに、化合物No.33を用いるほかは同様にして有機EL素子を得た。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
実施例23において、電子注入輸送層に化合物No.19を用いるかわりに、化合物No.16を用いるほかは同様にして有機EL素子を得た。
実施例12と同様に正孔輸送層を形成した後、TPD−1と実施例1の化合物No.1を蒸着速度0.2nm/secで20nmの厚さに蒸着し、発光層とした。この場合、TPD−1と化合物No.1との比率は重量比で1:1とした。
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
C. Adachi et al., Appli. Phys. Lett., 56, 799(1990) に記載の1,1,4,4−テトラフェニル−1,3−ブタジエンを発光層に用いて、この文献と同様の構成の有機EL素子を組み立てた。すなわち、比較例1において、正孔注入輸送層を形成したのち、上記化合物を同様に50nmの厚さに蒸着して発光層とした。その後、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで10nmの厚さで蒸着して電子注入輸送層とし、比較例1と同様に陰極を形成し、有機EL素子を得た。
比較例2において、t−ブチルフェニルビフェニルオキサジアゾール(PBD)を電子注入輸送層とするほかは、同構成の有機EL素子を得た。
特開平6−100857号公報の合成例1の方法に従い、ジフェニルクロロメタンをグリニャール化し、これと1,4−ジベンゾイルベンゼンとを反応させ、ギ酸存在下で脱水して、化合物No.2を得た。
実施例1で得られた化合物No.1をガラス基板上に、10−5Pa以下の減圧下で真空蒸着を行い、1000A厚の蒸着膜を形成した。
実施例2で得られた化合物No.2を用い、実施例26と同様に蒸着膜を形成し、実施例26と同様にして膜の安定性を調べたところ、10日程度は結晶化しなかった。
特開平6−100857号公報の合成例1に従って得られた化合物No.2を用い、実施例26と同様に蒸着膜を形成し、実施例26と同様にして膜の安定性を調べたところ、初期においてはアモルファス状の膜であったが、1日後には結晶化した。
化合物(No.3、7、8、16、19、33、42、49、53)について、実施例26と同様にして各化合物の蒸着膜を形成し、同様に各膜の安定性を調べたところ、大気中に1年以上放置しても初期のアモルファス状態が維持されていることがわかった。
2 基板
3 陽極
4 正孔注入輸送層
5 発光層
6 電子注入輸送層
7 陰極
Claims (1)
- テトラアリールエテン誘導体を含有する有機化合物層を有する有機EL素子の製造方法であって、
芳香族残基三置換ハロゲン化エテンをグリニャール化し、ジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物とクロスカップリングするか、またはジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物をグリニャール化し、芳香族残基三置換ハロゲン化エテンとクロスカップリングするかによって、下記化1で表されるテトラアリールエテン誘導体を得る工程を備えることを特徴とする有機EL素子の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007134226A JP4267670B2 (ja) | 1994-04-28 | 2007-05-21 | 有機el素子の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP11445694 | 1994-04-28 | ||
| JP2007134226A JP4267670B2 (ja) | 1994-04-28 | 2007-05-21 | 有機el素子の製造方法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004343976A Division JP3999781B2 (ja) | 1994-04-28 | 2004-11-29 | 有機el素子 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007270153A JP2007270153A (ja) | 2007-10-18 |
| JP4267670B2 true JP4267670B2 (ja) | 2009-05-27 |
Family
ID=38673261
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007134226A Expired - Lifetime JP4267670B2 (ja) | 1994-04-28 | 2007-05-21 | 有機el素子の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4267670B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20210072209A (ko) * | 2019-12-06 | 2021-06-17 | 삼성디스플레이 주식회사 | 유기 전계 발광 소자 및 유기 전계 발광 소자용 아민 화합물 |
-
2007
- 2007-05-21 JP JP2007134226A patent/JP4267670B2/ja not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007270153A (ja) | 2007-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3816969B2 (ja) | 有機el素子 | |
| JP5193074B2 (ja) | 有機el素子 | |
| JP3838816B2 (ja) | 有機el素子用化合物および有機el素子 | |
| JP4623223B2 (ja) | 有機el素子 | |
| JP3712760B2 (ja) | 有機el素子 | |
| JP3828595B2 (ja) | 有機el素子 | |
| EP0681019B1 (en) | Phenylanthracene derivative and organic EL element | |
| US5142343A (en) | Organic electroluminescence device with oligomers | |
| JP3965063B2 (ja) | 有機エレクトロルミネッセンス素子 | |
| JP3650200B2 (ja) | キノキサリン系化合物を用いた有機el用素子 | |
| JP3642606B2 (ja) | 有機el素子 | |
| JP2003026616A (ja) | 有機el素子用化合物、有機el素子 | |
| JP4364130B2 (ja) | 有機el素子 | |
| JP4588637B2 (ja) | 共役分子、電界発光素子、及び電子機器 | |
| Mallesham et al. | Design and synthesis of novel anthracene derivatives as n-type emitters for electroluminescent devices: a combined experimental and DFT study | |
| JP3742176B2 (ja) | 有機el素子用化合物および有機el素子 | |
| JP3993333B2 (ja) | 有機el素子 | |
| JP3999781B2 (ja) | 有機el素子 | |
| JP4320020B2 (ja) | 有機el素子 | |
| JP2016150920A (ja) | 有機電界発光素子用化合物およびこれを用いた有機電界発光素子 | |
| JP4267670B2 (ja) | 有機el素子の製造方法 | |
| JP4190542B2 (ja) | フェニルアントラセン誘導体 | |
| JP4109292B2 (ja) | 有機el素子用化合物 | |
| JP2000268963A (ja) | 有機el素子 | |
| JP4200152B2 (ja) | 有機el素子 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070731 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090210 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090218 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120227 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120227 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R360 | Written notification for declining of transfer of rights |
Free format text: JAPANESE INTERMEDIATE CODE: R360 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R370 | Written measure of declining of transfer procedure |
Free format text: JAPANESE INTERMEDIATE CODE: R370 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R370 | Written measure of declining of transfer procedure |
Free format text: JAPANESE INTERMEDIATE CODE: R370 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140227 Year of fee payment: 5 |
|
| EXPY | Cancellation because of completion of term |