JP4267670B2 - Manufacturing method of organic EL element - Google Patents
Manufacturing method of organic EL element Download PDFInfo
- Publication number
- JP4267670B2 JP4267670B2 JP2007134226A JP2007134226A JP4267670B2 JP 4267670 B2 JP4267670 B2 JP 4267670B2 JP 2007134226 A JP2007134226 A JP 2007134226A JP 2007134226 A JP2007134226 A JP 2007134226A JP 4267670 B2 JP4267670 B2 JP 4267670B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- compound
- organic
- layer
- light emitting
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 CC1C=C(C(C)=C(C=C(*)C=C2)C2=N2)C2=CC=C1 Chemical compound CC1C=C(C(C)=C(C=C(*)C=C2)C2=N2)C2=CC=C1 0.000 description 2
- YXFVVABEGXRONW-UHFFFAOYSA-N Cc1ccccc1 Chemical compound Cc1ccccc1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 2
- QQZVQGBXHFHFIC-UHFFFAOYSA-N Cc1c(cccc2)c2c(C)nn1 Chemical compound Cc1c(cccc2)c2c(C)nn1 QQZVQGBXHFHFIC-UHFFFAOYSA-N 0.000 description 1
- FKHNZQFCDGOQGV-UHFFFAOYSA-N Cc1nc2ccccc2nc1C Chemical compound Cc1nc2ccccc2nc1C FKHNZQFCDGOQGV-UHFFFAOYSA-N 0.000 description 1
Images


















Landscapes
- Electroluminescent Light Sources (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
本発明は、有機EL(電界発光)素子の製造方法に関し、詳しくは、有機化合物からなる積層構造薄膜に電界を印加して光を放出する素子の製造方法に関する。 The present invention relates to a method for manufacturing an organic EL (electroluminescence) device, and more particularly to a method for manufacturing a device that emits light by applying an electric field to a laminated thin film made of an organic compound.
有機EL素子は、蛍光性有機化合物を含む薄膜を、陰極と陽極とで挟んだ構成を有し、前記薄膜に電子および正孔を注入して再結合させることにより励起子(エキシトン)を生成させ、このエキシトンが失活する際の光の放出(蛍光・燐光)を利用して発光する素子である。 An organic EL element has a configuration in which a thin film containing a fluorescent organic compound is sandwiched between a cathode and an anode, and excitons (excitons) are generated by injecting electrons and holes into the thin film and recombining them. It is an element that emits light by utilizing light emission (fluorescence / phosphorescence) when this exciton is deactivated.
有機EL素子の特徴は、10V程度の低電圧で100〜10000cd/m2程度の高輝度の面発光が可能であり、また蛍光物質の種類を選択することにより青色から赤色までの発光が可能なことである。 The characteristics of the organic EL element are that it can emit surface light with a high luminance of about 100 to 10000 cd / m 2 at a low voltage of about 10 V, and can emit light from blue to red by selecting the type of fluorescent material. That is.
一方、有機EL素子の問題点は、発光寿命が短く、保存耐久性、信頼性が低いことであり、この原因としては、
(1)有機化合物の物理的変化
(結晶ドメインの成長などにより界面の不均一化が生じ、素子の電荷注入能の劣化・短絡・絶縁破壊の原因となる。特に分子量500以下の低分子化合物を用いると結晶粒の出現・成長が起こり、膜性が著しく低下する。また、ITO等の界面が荒れていても、顕著な結晶粒の出現・成長が起こり、発光効率の低下や、電流のリークを起こし、発光しなくなる。また、部分的非発光部であるダークスポットの原因にもなる。)
On the other hand, the problems of the organic EL element are that the light emission life is short, the storage durability and the reliability are low.
(1) Physical changes in organic compounds (non-uniformity of the interface occurs due to growth of crystal domains, etc., leading to deterioration of charge injection ability, short circuit, and dielectric breakdown of the device. Especially low molecular weight compounds with a molecular weight of 500 or less If used, the appearance and growth of crystal grains will occur and the film properties will be significantly reduced, and even if the interface of ITO etc. is rough, the appearance and growth of remarkable crystal grains will occur, resulting in a decrease in luminous efficiency and current leakage. (This also causes dark spots that are partially non-light emitting parts.)
(2)陰極の酸化・剥離
(電子の注入を容易にするために仕事関数の小さな金属としてNa・Mg・Alなどを用いてきたが、これらの金属は大気中の水分や酸素と反応したり、有機層と陰極の剥離が起こり、電荷注入ができなくなる。特に高分子化合物などを用い、スピンコートなどで成膜した場合、成膜時の残留溶媒や分解物が電極の酸化反応を促進し、電極の剥離が起こり部分的な非発光部を生じさせる。)
(2) Oxidation / peeling of the cathode (Na, Mg, Al, etc. have been used as metals with a small work function to facilitate the injection of electrons, but these metals react with moisture and oxygen in the atmosphere. In other words, when the organic layer and the cathode are peeled off, it becomes impossible to inject the charge, especially when a film is formed by spin coating using a polymer compound or the like, and the residual solvent and decomposition products during the film formation promote the oxidation reaction of the electrode. The electrode is peeled off to cause a partial non-light emitting portion.)
(3)発光効率が低く、発熱量が多いこと
(有機化合物中に電流を流すので、高い電界強度下に有機化合物を置かねばならず、発熱からは逃れられない。その熱のため、有機化合物の溶融・結晶化・熱分解などにより素子の劣化・破壊が起こる。)
(3) Low luminous efficiency and large calorific value (Because current flows in the organic compound, the organic compound must be placed under a high electric field strength and cannot escape from the heat generation. (Degradation / destruction of the device occurs due to melting, crystallization, thermal decomposition, etc.)
(4)有機化合物層の光化学的変化・電気化学的変化
などが挙げられる。
(4) Photochemical and electrochemical changes of the organic compound layer.
特に、青色発光素子に関しては、信頼性が高く安定な素子を提供する青色発光材料は少ない(例えば、非特許文献1)。一般に、青色発光材料は結晶性が高く、例えばアリールエテンは高い蛍光量子収率を持つにも関わらず、結晶性が高く、この化合物を発光材料に用いて、素子を作製しても高輝度・高効率で信頼性の高い素子を提供できなかった。また、今まで報告されたアリールエテン系化合物は、ウィッティッヒ ( Wittig ) 反応で合成されるため、エテンのすべてに、芳香環を導入することができず、化学的安定性が低いこと、分子量が小さく、コンフォメイションの安定構造が存在するため薄膜のアモルファス性が低く結晶化しやすいことが問題であった(例えば、特許文献1)。 In particular, with respect to blue light-emitting elements, there are few blue light-emitting materials that provide reliable and stable elements (for example, Non-Patent Document 1). In general, blue light-emitting materials have high crystallinity, for example, arylethene has high crystallinity despite high fluorescence quantum yield. Even if an element is manufactured using this compound as a light-emitting material, high luminance and A highly efficient and reliable device could not be provided. In addition, since the arylethene compounds reported so far are synthesized by the Wittig reaction, aromatic rings cannot be introduced into all ethene, resulting in low chemical stability and low molecular weight. The problem is that the amorphous structure of the thin film is low and it is easy to crystallize due to the presence of a stable structure of the conformation (for example, Patent Document 1).
また、特許文献2には、炭素数6〜20のアリール基の3個置換したトリアリールビニル基が炭素数6〜20のアリーレン基を介して連結されたジオレフィン化合物が開示されており、このジオレフィン化合物からなる発光層を有する有機EL素子が提案されている。このようなジオレフィン化合物は、ジアリールハロゲン化メタンをグリニャール化し、これとジベンゾイルアリールとを反応させて、さらに脱水することによって得ている。このため、副反応残留物が多く、EL素子に使用した場合蛍光を消光したりする欠点がある。
本発明の目的は、特に物理的変化や光化学的変化、電気化学的変化の少ない光・電子機能材料としてテトラアリールエテン誘導体を用い、信頼性および発光効率の高い種々の発光色を持った、特に青色の発光色を持った有機EL素子を実現することである。特に、分子量の大きな化合物を蒸着法で形成した有機薄膜を用い、素子の駆動時の駆動電圧上昇や輝度の低下、電流のリーク、部分的な非発光部の出現・成長を抑えた高信頼性の高輝度発光素子を実現することである。 The object of the present invention is to use a tetraarylethene derivative as an optical / electronic functional material that has little physical change, photochemical change, and electrochemical change, and has various emission colors with high reliability and luminous efficiency. An organic EL element having a blue emission color is realized. In particular, using an organic thin film formed by vapor deposition of a compound with a large molecular weight, high reliability with reduced drive voltage and reduced brightness, current leakage, and the appearance / growth of non-light emitting parts during device operation It is to realize a high-luminance light emitting element.
このような目的は、下記の本発明により達成される。
テトラアリールエテン誘導体を含有する有機化合物層を有する有機EL素子の製造方法であって、
芳香族残基三置換ハロゲン化エテンをグリニャール化し、ジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物とクロスカップリングするか、またはジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物をグリニャール化し、芳香族残基三置換ハロゲン化エテンとクロスカップリングするかによって、下記化1で表されるテトラアリールエテン誘導体を得る工程を備えることを特徴とする有機EL素子の製造方法。
Such an object is achieved by the present invention described below.
A method for producing an organic EL device having an organic compound layer containing a tetraarylethene derivative,
Grignard aromatic residue trisubstituted halogenated ethene and cross-couple with di, tri, tetra, penta or hexahalogenated aromatic compounds, or di, tri, tetra, penta or hexahalogenated aromatic compounds A method for producing an organic EL device comprising a step of obtaining a tetraarylethene derivative represented by the following

(作用)
本発明の有機EL素子は上記化1に示されるテトラアリールエテン誘導体を適宜選択して有機化合物層に用いるため、高輝度が安定して得られる。また、耐熱性・耐久性が高く、素子電流密度も100mAcm−2程度でも安定した駆動が可能である。
(Function)
In the organic EL device of the present invention, a tetraarylethene derivative represented by the above
上記のテトラアリールエテン誘導体の蒸着膜は安定なアモルファス状態なので、薄膜の膜物性が良好となりムラがなく均一な発光が可能である。また、大気下で、ほとんどの化合物が一年以上安定であり結晶化を起こさない。 Since the vapor-deposited film of the above-mentioned tetraarylethene derivative is in a stable amorphous state, the film physical properties are good and uniform light emission is possible without unevenness. Also, most compounds are stable for over a year in the atmosphere and do not cause crystallization.
また、本発明の有機EL素子は、低駆動電圧で効率よく発光する。 The organic EL device of the present invention emits light efficiently at a low driving voltage.
なお、本発明の有機EL素子の発光極大波長は、350〜700nm程度である。 In addition, the light emission maximum wavelength of the organic EL element of this invention is about 350-700 nm.
上記のテトラアリールエテン誘導体は、芳香族残基三置換ハロゲン化エテンをグリニャール化し、ジ〜ヘキサハロゲン化芳香族化合物とクロスカップリングするか、またはジ〜ヘキサハロゲン化芳香族化合物をグリニャール化し、芳香族残基三置換ハロゲン化エテンとクロスカップリングするかによって得ている。このため、高純度品が得られ、出発原料を適宜選択することによって、目的に応じ多種多様の反応生成物を得ることができる。 The above-mentioned tetraarylethene derivative is obtained by Grignard aromatic residue trisubstituted halogenated ethene and cross-coupling with di-hexahalogenated aromatic compound or Grignard di-hexahalogenated aromatic compound. It is obtained by cross-coupling with a group III trisubstituted halogenated ethene. For this reason, a high purity product is obtained, and various reaction products can be obtained according to the purpose by appropriately selecting the starting materials.
また、有機化合物層のうち発光層に上記化合物を用いる場合、テトラアリールエテン誘導体のうち、Lがアリーレン基であるものについては、アリーレン基の炭素数が21以上であるものが好ましい。アリーレン基の炭素数を21以上とすることで、薄膜状態での安定性が高くなり、結晶化がさらに抑制される。 Moreover, when using the said compound for a light emitting layer among organic compound layers, the thing whose carbon number of an arylene group is 21 or more is preferable about the thing whose L is an arylene group among tetraarylethene derivatives. By setting the number of carbon atoms of the arylene group to 21 or more, stability in a thin film state is increased, and crystallization is further suppressed.
なお、特開平6−100857号公報には炭素数6〜20のアリール基の3個置換したトリアリールビニル基が炭素数6〜20のアリーレン基を介して連結されたジオレフィン化合物が開示されており、このジオレフィン化合物からなる発光層を有する有機EL素子が提案されている。この場合のジオレフィン化合物は、本発明のものと一部重複するが、これらの化合物は、本発明と異なり、ジアリールハロゲン化メタンをグリニャール化し、これとジベンゾイルアリールとを反応させて、さらに脱水することによって得ている。 JP-A-6-1000085 discloses a diolefin compound in which a triarylvinyl group substituted with three aryl groups having 6 to 20 carbon atoms is linked via an arylene group having 6 to 20 carbon atoms. An organic EL device having a light emitting layer made of this diolefin compound has been proposed. In this case, the diolefin compound partially overlaps with that of the present invention, but these compounds are different from the present invention in that diaryl halogenated methane is Grignarded and reacted with dibenzoylaryl for further dehydration. Is gained by doing.
このように、連結基のアリーレン基の炭素数が6〜20であるものは、炭素数21以上のものに比べ、発光材料としたとき、薄膜状態での安定性にやや劣り、このため素子の寿命が短かくなりやすく、発光輝度が低下しやすくなる。 Thus, when the arylene group of the linking group has 6 to 20 carbon atoms, the stability in a thin film state is slightly inferior when the light emitting material is used compared to those having 21 or more carbon atoms. The lifetime tends to be short, and the light emission luminance tends to decrease.
また、上記の合成経路を経て得られるものは、本発明における合成経路を経て得られるものに比べ、不純物が多く薄膜安定性が低下し、素子としたとき寿命が短かくなり、発光輝度が低下する。 In addition, what is obtained through the synthesis route described above has more impurities and lowers the stability of the thin film than the product obtained through the synthesis route in the present invention, and the lifetime of the device is shortened, resulting in lower emission luminance. To do.
本発明に用いるテトラアリールエテン誘導体は、結晶性が低く、アモルファス状態の良好な膜を形成することができるので、有機EL素子用化合物、具体的には青色発光材料、電子注入輸送材料、あるいは正孔注入輸送材料として用いた場合、本発明の有機EL素子は連続発光光であり、輝度の低下が小さく、ダークスポットや電流リークの発生がない信頼性の高い素子となる。例えば発光層に用いた場合1万cd/m2以上の高輝度の青色発光が可能となる。 Since the tetraarylethene derivative used in the present invention has a low crystallinity and can form a film having a good amorphous state, it is a compound for an organic EL device, specifically, a blue light emitting material, an electron injecting and transporting material, or a positive film. When used as a hole injecting and transporting material, the organic EL device of the present invention is a continuous emission light, and is a highly reliable device with little decrease in luminance and no occurrence of dark spots or current leaks. For example, when used in the light emitting layer, blue light emission with high luminance of 10,000 cd / m 2 or more is possible.
以下、本発明の具体的構成について詳細に説明する。 Hereinafter, a specific configuration of the present invention will be described in detail.
本発明の有機EL素子(以下「EL素子」ともいう。)は、少なくとも1層の有機化合物層を有し、少なくとも1層の有機化合物層が化2で表されるテトラアリールエテン誘導体を含有する。本発明の有機EL素子の構成例を図1に示す。同図に示される有機EL素子1は、基板2上に、陽極3、正孔注入輸送層4、発光層5、電子注入輸送層6、陰極7を順次有する。
The organic EL device of the present invention (hereinafter also referred to as “EL device”) has at least one organic compound layer, and at least one organic compound layer contains a tetraarylethene derivative represented by Chemical Formula 2. . A configuration example of the organic EL element of the present invention is shown in FIG. The
発光層は、正孔および電子の注入機能、それらの輸送機能、正孔と電子の再結合により励起子を生成させる機能を有する。正孔注入輸送層は、陽極からの正孔の注入を容易にする機能、正孔を輸送する機能および電子の輸送を妨げる機能を有し、電子注入輸送層は、陰極からの電子の注入を容易にする機能、電子を輸送する機能および正孔の輸送を妨げる機能を有するものであり、これらの層は、発光層へ注入される正孔や電子を増大・閉じ込めさせ、再結合領域を最適化させ、発光効率を改善する。電子注入輸送層および正孔注入輸送層は、発光層に用いる化合物の電子注入、電子輸送、正孔注入、正孔輸送の各機能の高さを考慮し、必要に応じて設けられる。例えば、発光層に用いる化合物の正孔注入輸送機能または電子注入輸送機能が高い場合には、正孔注入輸送層または電子注入輸送層を設けずに、発光層が正孔注入輸送層または電子注入輸送層を兼ねる構成とすることができる。また、場合によっては正孔注入輸送層および電子注入輸送層のいずれも設けなくてよい。また、正孔注入輸送層および電子注入輸送層は、それぞれにおいて、注入機能をもつ層と輸送機能をもつ層とを別個に設けてもよい。 The light emitting layer has a hole and electron injection function, a transport function thereof, and a function of generating excitons by recombination of holes and electrons. The hole injecting and transporting layer has the function of facilitating the injection of holes from the anode, the function of transporting holes and the function of hindering the transport of electrons, and the electron injecting and transporting layer prevents the injection of electrons from the cathode. It has a function to facilitate, a function to transport electrons, and a function to prevent the transport of holes, and these layers increase and confine holes and electrons injected into the light-emitting layer and optimize the recombination region. To improve luminous efficiency. The electron injecting and transporting layer and the hole injecting and transporting layer are provided as necessary in consideration of the height of each function of electron injection, electron transport, hole injection and hole transport of the compound used for the light emitting layer. For example, when the hole injection / transport function or electron injection / transport function of the compound used in the light-emitting layer is high, the light-emitting layer is not provided with the hole injection / transport layer or the electron injection / transport layer, but the hole injection / transport layer or the electron injection It can be set as the structure which serves as a transport layer. In some cases, neither the hole injection transport layer nor the electron injection transport layer may be provided. Further, each of the hole injecting and transporting layer and the electron injecting and transporting layer may be provided with a layer having an injection function and a layer having a transport function.
化2で表されるテトラアリールエテン誘導体(以下「化2の化合物」ともいう。)は、その化合物の種類によって、正孔の注入ないし輸送機能をもつ正孔注入輸送性化合物として、電子の注入ないし輸送機能をもつ電子注入輸送性化合物として、また比較的ニュートラルな化合物は発光材料として適宜使用することができる。
Depending on the type of the compound, the tetraarylethene derivative represented by Chemical Formula 2 (hereinafter also referred to as “
また、組み合わせる発光層や電子注入輸送層や正孔注入輸送層のキャリア移動度やキャリア密度(イオン化ポテンシャル・電子親和力により決まる)を考慮しながら、膜厚をコントロールすることで、再結合領域・発光領域を自由に設計することが可能であり、発光色の設計や、両電極の干渉効果による発光輝度・発光スペクトルの制御や、発光の空間分布の制御を可能にできる。 In addition, the recombination region and light emission can be controlled by controlling the film thickness while considering the carrier mobility and carrier density (determined by the ionization potential and electron affinity) of the light-emitting layer, electron injection transport layer, and hole injection transport layer to be combined. The region can be designed freely, and it is possible to design the light emission color, control the light emission luminance / light emission spectrum by the interference effect of both electrodes, and control the spatial distribution of light emission.
本発明に用いられる化2のテトラアリールエテン誘導体について説明すると、化2において、Ar1、Ar2およびAr3は、各々芳香族残基を表し、これらは同一でも異なるものであってもよい。
The tetraarylethene derivative of
Ar1〜Ar3で表される芳香族残基としては、芳香族炭化水素基(アリール基)、芳香族複素環基が挙げられる。芳香族炭化水素基としては、単環もしくは多環の芳香族炭化水素基であってよく、縮合環や環集合も含まれる。芳香族炭化水素基は、総炭素数が6〜30のものが好ましく、置換基を有するものであってもよい。置換基を有する場合の置換基としては、アルキル基、アリール基、アルコキシ基、アリーロキシ基、アミノ基等が挙げられる。この置換基については後述する。芳香族炭化水素基としては、例えばフェニル基、アルキルフェニル基、アルコキシフェニル基、アリールフェニル基、アリーロキシフェニル基、アミノフェニル基、ビフェニル基、ナフチル基、アントリル基、ピレニル基、ペリレニル基などが挙げられる。 Examples of the aromatic residue represented by Ar 1 to Ar 3 include an aromatic hydrocarbon group (aryl group) and an aromatic heterocyclic group. The aromatic hydrocarbon group may be a monocyclic or polycyclic aromatic hydrocarbon group, and includes a condensed ring and a ring assembly. The aromatic hydrocarbon group preferably has a total carbon number of 6 to 30, and may have a substituent. In the case of having a substituent, examples of the substituent include an alkyl group, an aryl group, an alkoxy group, an aryloxy group, and an amino group. This substituent will be described later. Examples of the aromatic hydrocarbon group include a phenyl group, an alkylphenyl group, an alkoxyphenyl group, an arylphenyl group, an aryloxyphenyl group, an aminophenyl group, a biphenyl group, a naphthyl group, an anthryl group, a pyrenyl group, and a perylenyl group. It is done.
また、芳香族複素環基としては、ヘテロ原子としてO、N、Sを含むものが好ましく、5員環であっても6員環であってもよい。具体的には、チエニル基、フリル基、ピローリル基、ピリジル基などが挙げられる。 The aromatic heterocyclic group preferably includes O, N, and S as a hetero atom, and may be a 5-membered ring or a 6-membered ring. Specific examples include a thienyl group, a furyl group, a pyrrolyl group, and a pyridyl group.
Ar1〜Ar3で表される芳香族残基としては、特にフェニル基が好ましい。 As the aromatic residue represented by Ar 1 to Ar 3 , a phenyl group is particularly preferable.
nは2〜6の整数であり、特に2〜4の整数であることが好ましい。 n is an integer of 2 to 6, and preferably an integer of 2 to 4.
Lはn価の芳香族残基を表すが、特に芳香族炭化水素、芳香族複素環、芳香族エーテル(芳香族チオエーテルを含む。)または芳香族アミンから誘導される2〜6価、特に2〜4価の残基であることが好ましい。これらの芳香族残基は、さらに置換基を有するものであってもよい。 L represents an n-valent aromatic residue, and in particular, 2 to 6 valences derived from aromatic hydrocarbons, aromatic heterocycles, aromatic ethers (including aromatic thioethers) or aromatic amines, particularly 2 It is preferably a tetravalent residue. These aromatic residues may further have a substituent.
なお、このなかで、発光材料とするとき、Lは、オキシ基(−O−)、チオ基(−S−)、イミノ基(−NR0−:R0はアリール基)、複素環ジイル基、アルケニレン基およびアルキレン基のうちの1種以上が介在したアリーレン基、炭素数が21以上、好ましくは21〜100、さらに好ましくは24〜50のアリーレン基、芳香族炭化水素の3〜6価の残基または芳香族複素環、芳香族エーテルもしくは芳香族アミンの2〜6価の残基であるものが好ましい。 Of these, when a light emitting material is used, L is an oxy group (—O—), a thio group (—S—), an imino group (—NR 0 —: R 0 is an aryl group), a heterocyclic diyl group. , An arylene group in which one or more of an alkenylene group and an alkylene group are interposed, an arylene group having 21 or more carbon atoms, preferably 21 to 100, more preferably 24 to 50, and a trivalent to hexavalent aromatic hydrocarbon Those which are residues or divalent to hexavalent residues of aromatic heterocycles, aromatic ethers or aromatic amines are preferred.
化2のなかでも化3で示されるテトラアリールエテン誘導体が好ましい。
Of the
R1〜R3で表されるアルキル基としては、炭素数1〜10のものが好ましく、直鎖状であっても分岐を有するものであってもよく、さらには置換基を有するものであってもよく、例えばメチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、t−ブチル基等が挙げられる。 The alkyl group represented by R 1 to R 3 is preferably an alkyl group having 1 to 10 carbon atoms, which may be linear or branched, and further has a substituent. Examples thereof include a methyl group, an ethyl group, an n-propyl group, an i-propyl group, an n-butyl group, and a t-butyl group.
R1〜R3で表されるアリール基としては、炭素数6〜20のものが好ましく、置換基を有するものであってもよく、例えばフェニル基、o−トリル基、m−トリル基、p−トリル基、ナフチル基、アントリル基等が挙げられる。 The aryl group represented by R 1 to R 3 is preferably an aryl group having 6 to 20 carbon atoms and may have a substituent. For example, a phenyl group, an o-tolyl group, an m-tolyl group, p -A tolyl group, a naphthyl group, an anthryl group, etc. are mentioned.
R1〜R3で表されるアルコキシ基としては、アルコキシ基のアルキル基部分の炭素数が1〜6のものが好ましく、例えばメトキシ基、エトキシ基、t−ブトキシ基等が挙げられる。 As the alkoxy group represented by R 1 to R 3 , those having 1 to 6 carbon atoms in the alkyl group portion of the alkoxy group are preferable, and examples thereof include a methoxy group, an ethoxy group, and a t-butoxy group.
R1〜R3で表されるアリーロキシ基としては、フェノキシ基、4−メチルフェノキシ基、4−(t−ブチル)フェノキシ基等が挙げられる。 Examples of the aryloxy group represented by R 1 to R 3 include a phenoxy group, a 4-methylphenoxy group, and a 4- (t-butyl) phenoxy group.
R1〜R3で表されるアミノ基としては、置換基を有するものが好ましく、例えばジメチルアミノ基、ジエチルアミノ基、ジフェニルアミノ基、ビス(ビフェニル)アミノ基等が挙げられる。 The amino group represented by R 1 to R 3 preferably has a substituent, and examples thereof include a dimethylamino group, a diethylamino group, a diphenylamino group, and a bis (biphenyl) amino group.
s、tおよびuは、各々、0または1〜5の整数であり、s、t、uが2以上の整数であるとき、R1同士、R2同士、R3同士は、各々同一でも異なるものであってもよい。 s, t and u are each an integer of 0 or 1 to 5. When s, t and u are integers of 2 or more, R 1 s , R 2 s , and R 3 s are the same or different. It may be a thing.
化3において、s、tおよびuは、各々、0または1であることが好ましく、特に0であること、すなわち無置換のフェニル基であることが好ましい。
In
L1は、アリーレン基、アレーントリイル基、アレーンテトライル基、複素環ジイル基、複素環トリイル基、複素環テトライル基、トリアリールアミンもしくはその多量体のジイル基、トリアリールアミンもしくはその多量体のトリイル基、トリアリールアミンもしくはその多量体のテトライル基、アリール置換複素環ジイル基、アリール置換複素環トリイル基またはアリール置換複素環テトライル基を表す。これらはさらに置換されていてもよい。L1で表されるアリーレン基、アレーントリイル基、アレーンテトライル基は、オキシ基(−O−)、チオ基(−S−)、イミノ基(−NR0−:R0はフェニル基等のアリール基)、複素環ジイル基、アルケニル基およびアルキレン基のうちの1種以上が介在していてもよい。 L 1 is an arylene group, an arenetriyl group, an arenetetrayl group, a heterocyclic diyl group, a heterocyclic triyl group, a heterocyclic tetrayl group, a triarylamine or a multimeric diyl group, a triarylamine or a multimer thereof A triyl group, a triarylamine or a multimer thereof, a tetrayl group, an aryl-substituted heterocyclic diyl group, an aryl-substituted heterocyclic triyl group, or an aryl-substituted heterocyclic tetrayl group. These may be further substituted. The arylene group, arenetriyl group, and arenetetrayl group represented by L 1 are an oxy group (—O—), a thio group (—S—), an imino group (—NR 0 —: R 0 is a phenyl group, etc. Aryl group), a heterocyclic diyl group, an alkenyl group, and an alkylene group may be interposed.
このようなアリーレン基、アレーントリイル基、アレーンテトライル基は、総炭素数が6以上、さらには21以上、特に21〜100、さらに特には24〜50であることが好ましい。L1で表されるアリーレン基として、具体的にはフェニレン基、ビフェニレン基、ナフチレン基、ジフェニルエーテルジイル基、ジフェニルチオエーテルジイル基、ジフェニルメチルジイル基、ジフェニルオキサジアゾールジイル基、テルフェニレン基等が挙げられる。アレーントリイル基としては、ベンゼントリイル基、クアテルフェニルトリイル基等が挙げられる。アレーンテトライル基としては、テトラフェニルエテンテトライル基等が挙げられる。このような基にはフェニルエチリル基等が置換されていてもよい。 Such an arylene group, arenetriyl group, and arenetetrayl group preferably have a total carbon number of 6 or more, more preferably 21 or more, particularly 21 to 100, and more particularly 24 to 50. Specific examples of the arylene group represented by L 1 include a phenylene group, a biphenylene group, a naphthylene group, a diphenyletherdiyl group, a diphenylthioetherdiyl group, a diphenylmethyldiyl group, a diphenyloxadiazolediyl group, and a terphenylene group. It is done. Examples of the arenetriyl group include a benzenetriyl group and a quaterphenyltriyl group. Examples of the arenetetrayl group include a tetraphenylethenetetrayl group. Such a group may be substituted with a phenylethylyl group or the like.
L1で表される複素環ジイル基としては、チオフェンジイル基、フランジイル基、ピリジンジイル基、ビチオフェンジイル基、ビフランジイル基、ビピリジンジイル基、ピラジンジイル基、ピロールジイル基、ビピロールジイル基、キノリンジイル基、オキサジアゾールジイル基、キノキサリンジイル基、ジフェニルキノキサリンジイル基等が挙げられる。複素環トリイル基としてはイソキノリントリイル基等が挙げられ、複素環テトライル基としては、キノキサリンテトライル基等が挙げられる。これらの基は、さらにメトキシ基等の置換基を有していてもよい。 The heterocyclic diyl group represented by L 1 includes a thiophene diyl group, a furandiyl group, a pyridinediyl group, a bithiophenediyl group, a bifurandiyl group, a bipyridinediyl group, a pyrazinediyl group, a pyrrolediyl group, a bipyrrolediyl group, a quinolinediyl group, an oxaline group. Examples thereof include a diazole diyl group, a quinoxaline diyl group, and a diphenylquinoxaline diyl group. Examples of the heterocyclic triyl group include an isoquinoline triyl group, and examples of the heterocyclic tetrayl group include a quinoxaline tetrayl group. These groups may further have a substituent such as a methoxy group.
L1で表されるトリアリールアミンまたはその多量体のジイル基としては、トリフェニルアミンジイル基等が挙げられ、トリアリールアミンまたはその多量体のトリイル基としては、トリフェニルアミントリイル基等が挙げられる。またトリアリールアミンまたはその多量体のテトライル基としては、N,N’−テトラフェニル−4,4’−ジアミノ−1,1’−ビフェニルテトライル基等が挙げられる。なお、トリアリールアミンの多量体は通常2〜4量体程度のものである。 Examples of the triarylamine represented by L 1 or a diyl group of the multimer include a triphenylamine diyl group, and the triarylamine of the triarylamine or the multimer thereof includes a triphenylamine triyl group. Can be mentioned. Examples of the triylamine or multimer tetrayl group include N, N′-tetraphenyl-4,4′-diamino-1,1′-biphenyltetrayl group. In addition, the multimer of triarylamine is usually about 2 to 4 mer.
L1で表されるアリール置換複素環ジイル基としては、ジフェニルオキサジアゾールジイル基等が挙げられ、アリール置換複素環トリイル基としては、ジフェニルオキサジアゾールトリイル基、ジフェニルキノキサリントリイル基等が挙げられ、アリール置換複素環テトライル基としては、ジフェニルキノキサリンテトライル基等が挙げられる。 Examples of the aryl-substituted heterocyclic diyl group represented by L 1 include a diphenyloxadiazole diyl group, and examples of the aryl-substituted heterocyclic triyl group include a diphenyloxadiazole triyl group and a diphenylquinoxaline triyl group. Examples of the aryl-substituted heterocyclic tetrayl group include a diphenylquinoxaline tetrayl group.
L1の好適例を以下に示すが、本発明はこれらに限定されるものではない。 Preferred examples of L 1 are shown below, but the present invention is not limited thereto.
化3において、n1はL1の価数によるが、2〜4の整数であり、さらには2、3、特には2であることが好ましい。
In
なお、正孔注入輸送性化合物として用いるときのL1としては、複素環ジイル基、複素環トリイル基、複素環テトライル基、トリアリールアミン誘導体ジイル基、トリアリールアミン誘導体トリイル基、トリアリールアミン誘導体テトライル基またはイミノ基(−NR0−:R0はアリール基)が介在してもよいアリーレン基、アレーントリイル基もしくはアレーンテトライル基であることが好ましい。 In addition, as L 1 when used as a hole injecting and transporting compound, heterocyclic diyl group, heterocyclic triyl group, heterocyclic tetrayl group, triarylamine derivative diyl group, triarylamine derivative triyl group, triarylamine derivative It is preferably an arylene group, an arenetriyl group or an arenetetrayl group which may be intervened by a tetrayl group or an imino group (—NR 0 —: R 0 is an aryl group).
また、電子注入輸送性化合物として用いるときのL1としては、オキシ基(−O−)、チオ基(−S−)、複素環ジイル基およびアルキレン基のうちの1種以上が介在していてもよいアリーレン基、アレーントリイル基もしくはアレーンテトライル基、複素環ジイル基、複素環トリイル基、複素環テトライル基、アリール置換複素環ジイル基、アリール置換複素環トリイル基、またはアリール置換複素環テトライル基であることが好ましい。 In addition, as L 1 when used as an electron injecting and transporting compound, one or more of an oxy group (—O—), a thio group (—S—), a heterocyclic diyl group, and an alkylene group are present. Arylene group, arenetriyl group or arenetetrayl group, heterocyclic diyl group, heterocyclic triyl group, heterocyclic tetrayl group, aryl-substituted heterocyclic diyl group, aryl-substituted heterocyclic triyl group, or aryl-substituted heterocyclic tetrayl It is preferably a group.
また、発光材料として用いるときのL1としては、オキシ基(−O−)、チオ基(−S−)、イミノ基(−NR0−:R0はアリール基)、複素環ジイル基、アルケニレン基およびアルキレン基のうちの1種以上が介在したアリーレン基、炭素数が21以上、さらに好ましくは21〜100、特に好ましくは24〜50のアリーレン基、オキシ基(−O−)、チオ基(−S−)、イミノ基(−NR0:R0はアリール基)、複素環ジイル基、アルケニレン基およびアルキレン基のうちの1種以上が介在してもよいアレーントリイル基もしくはアレーンテトライル基、複素環ジイル基、複素環トリイル基、複素環テトライル基、トリアリールアミンもしくはその多量体のジイル基、トリアリールアミンもしくはその多量体のトリイル基、トリアリールアミンもしくはその多量体のテトライル基、アリール置換複素環ジイル基、アリール置換複素環トリイル基またはアリール置換複素環テトライル基であるものが好ましい。 In addition, as L 1 when used as a light emitting material, an oxy group (—O—), a thio group (—S—), an imino group (—NR 0 —: R 0 is an aryl group), a heterocyclic diyl group, an alkenylene An arylene group in which one or more of a group and an alkylene group are present, an arylene group having 21 or more carbon atoms, more preferably 21 to 100, and particularly preferably 24 to 50 carbon atoms, an oxy group (—O—), a thio group ( —S—), an imino group (—NR 0 : R 0 is an aryl group), a heterocyclic diyl group, an alkenylene group, and an alkylene group, an arenetriyl group or an arenetetrayl group that may be interposed. , Heterocyclic diyl group, heterocyclic triyl group, heterocyclic tetrayl group, triarylamine or multimeric diyl group, triarylamine or multimeric triyl group, Those which are a tetrayl group, an aryl-substituted heterocyclic diyl group, an aryl-substituted heterocyclic triyl group or an aryl-substituted heterocyclic tetrayl group of a rearylamine or a multimer thereof are preferable.
このようなテトラアリールエテン誘導体の好適例を以下に示すが、本発明はこれらに限定されるものではない。なお、化10は一般式であり、化11〜化18では化10の表示を用いて示している。R11〜R15、R21〜R25、R31〜R35については、すべて水素のときはHとし、いずれかが置換基のときは置換基のみを示すものとする。なお、併せて、化合物の属性を記す。正孔注入輸送性化合物のときはn、電子注入輸送性化合物のときはeとし、特に示さないものは弱い電子輸送性もしくはニュートラル(バイボール)とする。このなかの化合物のうち、青色発光材料とできるのは化合物No.1〜4、14、21、23〜26、32、42、43、47〜59等である。
Preferred examples of such tetraarylethene derivatives are shown below, but the present invention is not limited thereto. In addition,
本発明のテトラアリールエテン誘導体は、
(1)ハロゲン化トリフェニルエテン化合物等の芳香族残基三置換ハロゲン化エーテルをグリニャール化し、NiCl2(dppp)〔dppp:ジフェニルフォスフィノプロパン〕等のNi錯体などを用いて、ジハロゲン化アリール誘導体等のジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物とクロスカップリングする方法、
(2)ジハロゲン化アリール誘導体等のジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物をグリニャール化し、NiCl2(dppp)等のNi錯体などを用いてハロゲン化トリフェニルエテン誘導体等の芳香族残基三置換ハロゲン化エテンとクロスカップリングする方法、等を用いて合成できる。
The tetraarylethene derivative of the present invention is
(1) A dihalogenated aryl derivative using a Ni complex such as NiCl 2 (dppp) [dppp: diphenylphosphinopropane] by grinding an aromatic residue trisubstituted halogenated ether such as a halogenated triphenylethene compound. A method of cross-coupling with di-, tri-, tetra-, penta- or hexahalogenated aromatic compounds such as
(2) Di-, tri-, tetra-, penta- or hexa-halogenated aromatic compounds such as dihalogenated aryl derivatives are Grignarded, and aromatics such as halogenated triphenylethene derivatives using Ni complexes such as NiCl 2 (dppp) It can be synthesized using a method of cross-coupling with a residue trisubstituted halogenated ethene.
このようにして合成された化合物は、元素分析、質量分析、赤外吸収スペクトル、1H核磁気共鳴吸収(NMR)スペクトルなどによって同定することができる。 The compound thus synthesized can be identified by elemental analysis, mass spectrometry, infrared absorption spectrum, 1 H nuclear magnetic resonance absorption (NMR) spectrum, and the like.
本発明に用いる化2のテトラアリールエテン誘導体は、500〜2000程度の分子量をもち、200〜350℃の高融点を有し、80〜250℃のガラス転移温度(Tg)を示す。従って、通常の真空蒸着等により透明で室温以上でも安定なアモルファス状態の平滑で良好な膜を形成し、しかもその良好な膜の状態が長期間に渡って維持される。
The tetraarylethene derivative of
化2の化合物を発光層に用いる場合について説明する。発光層には化2の化合物のほか、他の蛍光性物質を用いてもよく、他の蛍光性物質としては、例えば、特開昭63−264692号公報に開示されているような化合物、例えば、キナクリドン、ルブレン、スチリル系色素やトリス(8−キノリノラト)アルミニウム等の金属錯体色素等の化合物から選択される少なくとも1種が挙げられる。このような蛍光性物質の含有量は、化2の化合物の5モル%以下とすることが好ましい。このような化合物を適宜選択して添加することにより、発光光を長波長側にシフトすることができる。
The case where the compound of
また、発光層には、一重項酸素クエンチャーが含有されていてもよい。このようなクエンチャーとしては、ニッケル錯体や、ルブレン、ジフェニルイソベンゾフラン、三級アミン等が挙げられる。このようなクエンチャーの含有量は、化2の化合物の10モル%以下とすることが好ましい。
The light emitting layer may contain a singlet oxygen quencher. Examples of such quenchers include nickel complexes, rubrene, diphenylisobenzofuran, and tertiary amines. The content of such a quencher is preferably 10 mol% or less of the compound of
化2の化合物を発光層に用いる場合、正孔注入輸送層および電子注入輸送層には、通常の有機EL素子に用いられている各種有機化合物、例えば、特開昭63−295695号公報、特開平2−191694号公報、特開平3−792号公報等に記載されている各種有機化合物を用いることができる。例えば、正孔注入輸送層には、芳香族三級アミン、ヒドラゾン誘導体、カルバゾール誘導体、トリアゾール誘導体、イミダゾール誘導体、アミノ基を有するオキサジアゾール誘導体等を用いることができ、また、電子注入輸送層には、アルミキノリノールなどの有機金属錯体誘導体、オキサジアゾール誘導体、ピリジン誘導体、ピリミジン誘導体、キノリン誘導体、キノキサリン誘導体、ジフェニルキノン誘導体、ペリレン誘導体、ニトロ置換フルオレン誘導体等を用いることができる。
When the compound of
正孔注入輸送層を正孔注入層と正孔輸送層とに分けて設層する場合は、正孔注入輸送層用の化合物のなかから好ましい組合せを選択して用いることができる。このとき、陽極(ITO等)側からイオン化ポテンシャルの小さい化合物の層の順に積層することが好ましい。また陽極表面には薄膜性の良好な化合物を用いることが好ましい。例えば、8個以上のオリゴチオフェンやポリチオフェンの蒸着膜を正孔注入兼ITO表面改質層として用い、テトラフェニルジアミノビフェニル誘導体(TPD)や化合物No.3を正孔輸送層として用いるのが好ましい。素子化する場合、蒸着を用いているので1〜10nm程度の薄い膜も、均一かつピンホールフリーとすることができるので、各種の化合物を用いても、発光色の色調変化や再吸収による効率の低下を防ぐことができる。 When the hole injecting and transporting layer is divided into a hole injecting layer and a hole transporting layer, a preferred combination can be selected from the compounds for the hole injecting and transporting layer. At this time, it is preferable to laminate in order of a compound layer having a small ionization potential from the anode (ITO or the like) side. Further, it is preferable to use a compound having a good thin film property on the anode surface. For example, a vapor deposition film of 8 or more oligothiophenes or polythiophenes is used as a hole injection and ITO surface modification layer, and tetraphenyldiaminobiphenyl derivative (TPD) or Compound No. 3 is preferably used as the hole transport layer. In the case of making an element, since vapor deposition is used, a thin film of about 1 to 10 nm can be made uniform and pinhole-free, so even if various compounds are used, the efficiency due to the color tone change and reabsorption of the emitted color Can be prevented.
電子注入輸送層を電子注入層と電子輸送層とに分けて設層する場合は、電子注入輸送層用の化合物のなかから好ましい組合せを選択して用いることができる。このとき、陰極側から電子親和力の値の大きい化合物の層の順に積層することが好ましい。このような積層順については電子注入輸送層を2層以上設けるときも同様である。 When the electron injecting and transporting layer is divided into an electron injecting layer and an electron transporting layer, a preferred combination can be selected from the compounds for the electron injecting and transporting layer. At this time, it is preferable to laminate in the order of the layer of the compound having a large electron affinity value from the cathode side. Such a stacking order is the same when two or more electron injecting and transporting layers are provided.
なお、本発明では、発光層を電子注入輸送性化合物と正孔注入輸送性化合物との混合層とすることも好ましい。そして、このような混合層に化2の化合物を含有させる。化2の化合物を蛍光性物質として含有させる場合、より具体的には、化2の化合物が電子注入輸送性化合物であるとき、他の正孔注入輸送性化合物をさらに添加することが好ましく、化2の化合物が正孔注入輸送性化合物であるときは、他の電子注入輸送性化合物をさらに添加することが好ましい。上記の混合層における電子注入輸送性化合物と正孔注入輸送性化合物との混合比は、重量比で、電子注入輸送性化合物:正孔注入輸送性化合物が60:40〜40:60であることが好ましく、特には50:50程度であることが好ましい。
In the present invention, the light emitting layer is preferably a mixed layer of an electron injecting and transporting compound and a hole injecting and transporting compound. And the compound of
この混合層に供する電子注入輸送性化合物は、上記の電子注入輸送層用の化合物のなかから、また正孔注入輸送性化合物は、上記の正孔注入輸送層用の化合物のなかから選択して用いることができる。また、場合によっては化2の化合物から選択して用いてもよく、化2の化合物同士で混合層を構成してもよい。さらに、混合層において、電子注入輸送性化合物、正孔注入輸送性化合物は各々1種のみ用いても2種以上を併用してもよい。また、混合層には発光強度を高めるために、化2の化合物や他の蛍光性物質をドープして用いてもよい。
The electron injecting and transporting compound to be provided to the mixed layer is selected from the above compounds for the electron injecting and transporting layer, and the hole injecting and transporting compound is selected from the above compounds for the hole injecting and transporting layer. Can be used. Moreover, depending on the case, you may select from the compound of
さらに、他の電子注入輸送性化合物および他の正孔注入輸送性化合物の混合層とし、このような混合層に化2の化合物をドープして用いてもよい。
Further, a mixed layer of another electron injecting and transporting compound and another hole injecting and transporting compound may be used, and such a mixed layer may be used by doping the compound of
このような混合層をEL素子に適用することによって、素子の安定性が向上する。 By applying such a mixed layer to an EL element, the stability of the element is improved.
次に化2の化合物を正孔注入輸送層に用いる場合について説明する。化2の化合物を正孔注入輸送層に用いる場合、発光層に用いる蛍光性物質は、化2の化合物より長波長の蛍光をもつものから選択すればよく、例えば、上記した、発光層において化2の化合物と併用される蛍光性物質の1種以上から適宜選択すればよい。なお、このような場合、発光層にも化2の化合物を用いることができる。また、化2の化合物は、正孔注入輸送層を兼ねた発光層にも用いることができる。
Next, the case where the compound of
さらに化2の化合物を電子注入輸送層に用いる場合について説明する。この場合、発光層に用いる蛍光性物質は、化2の化合物より長波長もしくは同程度の波長の蛍光をもつものを用いればよい。例えば、上記した発光層において化2の化合物と併用できる蛍光性物質のなかから選択して用いることができる。また、化2の化合物は、このような構成において、さらに発光層にも用いることができる。また、化2の化合物は電子注入輸送層を兼ねた発光層にも用いることができる。
Furthermore, the case where the compound of
なお、上記において、他の蛍光性物質を主に発光層に用いる場合、化2の化合物を蛍光性物質として10モル%以下添加して併用してもよい。
In addition, in the above, when other fluorescent substances are mainly used in the light emitting layer, the compound of
発光層の厚さ、正孔注入輸送層の厚さおよび電子注入輸送層の厚さは特に限定されず、形成方法によっても異なるが、通常、5〜1000nm程度、特に8〜200nmとすることが好ましい。 The thickness of the light emitting layer, the thickness of the hole injecting and transporting layer, and the thickness of the electron injecting and transporting layer are not particularly limited, and may vary depending on the forming method, but is usually about 5 to 1000 nm, and particularly 8 to 200 nm. preferable.
正孔注入輸送層の厚さおよび電子注入輸送層の厚さは、再結合・発光領域の設計によるが、発光層の厚さと同程度もしくは1/10〜10倍程度とすればよい。電子もしくは正孔の、各々の注入層と輸送層を分ける場合は、注入層は1nm以上、輸送層は10nm以上とするのが好ましい。このときの注入層、輸送層の厚さの上限は、通常、注入層で20nm程度、輸送層で100nm程度である。 The thickness of the hole injecting and transporting layer and the thickness of the electron injecting and transporting layer may be about the same as the thickness of the light emitting layer or about 1/10 to 10 times depending on the design of the recombination / light emitting region. When the injection layer and the transport layer for electrons or holes are separated, the injection layer is preferably 1 nm or more, and the transport layer is preferably 10 nm or more. The upper limit of the thickness of the injection layer and the transport layer at this time is usually about 20 nm for the injection layer and about 100 nm for the transport layer.
陰極には、仕事関数の小さい材料、例えば、Li、Na、Mg、Al、Ag、Inあるいはこれらの1種以上を含む合金を用いることが好ましい。また、陰極は結晶粒が細かいことが好ましく、特に、アモルファス状態であることが好ましい。陰極の厚さは10〜1000nm程度とすることが好ましい。 For the cathode, it is preferable to use a material having a low work function, such as Li, Na, Mg, Al, Ag, In, or an alloy containing one or more of these materials. Further, the cathode preferably has fine crystal grains, and particularly preferably in an amorphous state. The thickness of the cathode is preferably about 10 to 1000 nm.
EL素子を面発光させるためには、少なくとも一方の電極が透明ないし半透明である必要があり、上記したように陰極の材料には制限があるので、好ましくは発光光の透過率が80%以上となるように陽極の材料および厚さを決定することが好ましい。具体的には、例えば、ITO、SnO2、Ni、Au、Pt、Pd、ドーパントをドープしたポリピロールなどを陽極に用いることが好ましい。また、陽極の厚さは10〜500nm程度とすることが好ましい。また、素子の信頼性を向上するために駆動電圧が低いことが必要であるが、好ましいものとして10〜30Ω/□のITOが挙げられる。 In order to cause the EL element to emit light, at least one of the electrodes needs to be transparent or translucent, and as described above, the cathode material is limited, so that the transmittance of emitted light is preferably 80% or more. It is preferable to determine the material and thickness of the anode so that Specifically, for example, ITO, SnO 2 , Ni, Au, Pt, Pd, polypyrrole doped with a dopant, or the like is preferably used for the anode. The anode thickness is preferably about 10 to 500 nm. Moreover, in order to improve the reliability of an element, it is necessary for a drive voltage to be low, However As a preferable thing, 10-30 ohms / square ITO is mentioned.
基板材料に特に制限はないが、図示例では基板側から発光光を取り出すため、ガラスや樹脂等の透明ないし半透明材料を用いる。また、基板に色フィルター膜や誘電体反射膜を用いて発光色をコントロールしてもよい。 The substrate material is not particularly limited, but in the illustrated example, a transparent or translucent material such as glass or resin is used to extract emitted light from the substrate side. Further, the emission color may be controlled by using a color filter film or a dielectric reflection film on the substrate.
なお、基板に不透明な材料を用いる場合には、図1に示される積層順序を逆にしてもよい。 Note that when an opaque material is used for the substrate, the stacking order shown in FIG. 1 may be reversed.
次に、本発明の有機EL素子の製造方法を説明する。 Next, the manufacturing method of the organic EL element of this invention is demonstrated.
陰極および陽極は、蒸着法やスパッタ法等の気相成長法により形成することが好ましい。 The cathode and the anode are preferably formed by vapor deposition such as vapor deposition or sputtering.
正孔注入輸送層、発光層および電子注入輸送層の形成には、均質な薄膜が形成できることから真空蒸着法を用いることが好ましい。真空蒸着法を用いた場合、アモルファス状態または結晶粒径が0.1μm以下(通常0.005μm以上)の均質な薄膜が得られる。結晶粒径が0.1μmを超えていると、不均一な発光となり、素子の駆動電圧を高くしなければならなくなり、電荷の注入効率も著しく低下する。 For the formation of the hole injecting and transporting layer, the light emitting layer, and the electron injecting and transporting layer, it is preferable to use a vacuum deposition method because a homogeneous thin film can be formed. When the vacuum deposition method is used, a homogeneous thin film having an amorphous state or a crystal grain size of 0.1 μm or less (usually 0.005 μm or more) can be obtained. If the crystal grain size exceeds 0.1 μm, non-uniform light emission occurs, the drive voltage of the element must be increased, and the charge injection efficiency is also significantly reduced.
真空蒸着の条件は特に限定されないが、10−3Pa以下の真空度とし、蒸着速度は0.1〜1nm/sec程度とすることが好ましい。また、真空中で連続して各層を形成することが好ましい。真空中で連続して形成すれば、各層の界面に不純物が吸着することを防げるため、高特性が得られる。また、素子の駆動電圧を低くすることができる。 Although the conditions of vacuum deposition are not specifically limited, It is preferable to set it as the vacuum degree of 10 < -3 > Pa or less, and to set a vapor deposition rate to about 0.1-1 nm / sec. Moreover, it is preferable to form each layer continuously in a vacuum. If formed continuously in a vacuum, impurities can be prevented from adsorbing to the interface of each layer, so that high characteristics can be obtained. In addition, the driving voltage of the element can be lowered.
これら各層の形成に真空蒸着法を用いる場合において、1層に複数の化合物を含有させる場合、化合物を入れた各ボートを個別に温度制御して共蒸着することが好ましい。 In the case of using a vacuum deposition method for forming each of these layers, when a plurality of compounds are contained in one layer, it is preferable to co-deposit each boat containing the compounds by individually controlling the temperature.
本発明のEL素子は、通常、直流駆動型のEL素子として用いられるが、交流駆動またはパルス駆動することもできる。印加電圧は、通常、2〜20V程度とされる。 The EL element of the present invention is usually used as a direct current drive type EL element, but can be alternating current driven or pulse driven. The applied voltage is usually about 2 to 20V.
以下、本発明の具体的実施例を比較例とともに示し、本発明をさらに詳細に説明する。 Hereinafter, specific examples of the present invention will be shown together with comparative examples, and the present invention will be described in more detail.
<実施例1>
化合物No.1の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.488g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のテトラヒドロフラン(THF)溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.3gと4,4’−ジブロモビフェニル3.02g(9.4mmol)を加え、60℃で4時間還流した。この反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ジクロロメタンより再結晶し、3.0gの青色蛍光を示す白色固体を得た。
<Example 1>
Compound No. Synthesis of 1 To 0.488 g (20 mmol) of magnesium activated under argon in a Schlenk flask, 50 ml of a tetrahydrofuran (THF) solution of 6.70 g (20 mmol) of 2-bromo-1,1,2-triphenylethene was added dropwise. It became Grignard. To this reaction solution, 0.3 g of NiCl 2 (dppe) and 3.02 g (9.4 mmol) of 4,4′-dibromobiphenyl were added and refluxed at 60 ° C. for 4 hours. This reaction solution was poured into a 1N aqueous hydrochloric acid solution, extracted with toluene, washed with water, and dried over magnesium sulfate. After the solvent was distilled off, recrystallization from acetone / dichloromethane gave 3.0 g of a white solid exhibiting blue fluorescence.
この白色固体1.0gを昇華精製し、0.8gの純粋な固体を得た。 1.0 g of this white solid was purified by sublimation to obtain 0.8 g of a pure solid.
質量分析:m/e 662(M+)
元素分析: C H Br
計算値/% 94.22 5.78 0.0
測定値/% 94.31 5.54 0.0
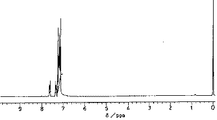
赤外吸収スペクトル:図2
1H−NMRスペクトル(270MHz):図3
示差走査熱量測定(DSC):融点304℃、ガラス転移温度110℃
イオン化ポテンシャル:5.90eV
Mass spectrometry: m / e 662 (M +)
Elemental analysis: C H Br
Calculated value /% 94.22 5.78 0.0
Measurement /% 94.31 5.54 0.0
Infrared absorption spectrum: Fig. 2
1 H-NMR spectrum (270 MHz): FIG.
Differential scanning calorimetry (DSC): melting point 304 ° C., glass transition temperature 110 ° C.
Ionization potential: 5.90 eV
<実施例2>
化合物No.2の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと1,4−ジブロモベンゼン2.35g(10mmol)を加え、60℃で4時間還流した。この反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ジクロロメタンより再結晶し、3.0gの青色蛍光を示す白色固体を得た。
<Example 2>
Compound No. Synthesis of 2 To 0.485 g (20 mmol) of magnesium activated under argon in a Schlenk flask, 50 ml of a THF solution of 6.70 g (20 mmol) of 2-bromo-1,1,2-triphenylethene was added dropwise to form a Grignard. . To this reaction solution, 0.4 g of NiCl 2 (dppe) and 2.35 g (10 mmol) of 1,4-dibromobenzene were added and refluxed at 60 ° C. for 4 hours. This reaction solution was poured into a 1N aqueous hydrochloric acid solution, extracted with toluene, washed with water, and dried over magnesium sulfate. After the solvent was distilled off, recrystallization from acetone / dichloromethane gave 3.0 g of a white solid exhibiting blue fluorescence.
この白色固体1.0gを昇華精製し、0.8gの純粋な固体を得た。 1.0 g of this white solid was purified by sublimation to obtain 0.8 g of a pure solid.
質量分析:m/e 586(M+)
元素分析: C H Br
計算値/% 94.16 5.84 0.0
測定値/% 94.15 5.53 0.0
赤外吸収スペクトル:図4
1H−NMRスペクトル:図5
示差走査熱量測定(DSC):融点250℃、ガラス転移温度83℃
イオン化ポテンシャル:5.95eV
Mass spectrometry: m / e 586 (M +)
Elemental analysis: C H Br
Calculated value /% 94.16 5.84 0.0
Measurement /% 94.15 5.53 0.0
Infrared absorption spectrum: Fig. 4
1 H-NMR spectrum: FIG.
Differential scanning calorimetry (DSC): melting point 250 ° C.,
Ionization potential: 5.95 eV
<実施例3>
化合物No.3の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.488g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.3gと4,4’,4”−トリブロモトリフェニルアミン3.00g(6.0mmol)を加え、60℃で4時間還流した。この反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ジクロロメタンにより再結晶後3.0gの青緑色蛍光を示す黄白色固体を得た。
<Example 3>
Compound No. Synthesis of 3 To 0.488 g (20 mmol) of magnesium activated under argon in a Schlenk flask, 50 ml of a THF solution of 6.70 g (20 mmol) of 2-bromo-1,1,2-triphenylethene was added dropwise to make a Grignard. . To this reaction solution, 0.3 g of NiCl 2 (dppe) and 3.00 g (6.0 mmol) of 4,4 ′, 4 ″ -tribromotriphenylamine were added and refluxed for 4 hours at 60 ° C. This reaction solution was 1N. The solution was poured into an aqueous hydrochloric acid solution, extracted with toluene, washed with water, dried over magnesium sulfate, evaporated, and recrystallized from acetone / dichloromethane to obtain 3.0 g of a yellowish white solid exhibiting blue-green fluorescence.
この白色固体1.0gを昇華精製し、0.8gの純粋な固体を得た。 1.0 g of this white solid was purified by sublimation to obtain 0.8 g of a pure solid.
質量分析:m/e 1007(M+)
元素分析: C H N Br
計算値/% 92.91 5.69 1.39 0.0
測定値/% 92.46 5.32 1.29 0.0
赤外吸収スペクトル:図6
1H−NMRスペクトル(270MHz):図7
示差走査熱量測定(DSC):融点300℃、ガラス転移温度129℃
イオン化ポテンシャル:5.45eV
Mass spectrometry: m / e 1007 (M +)
Elemental analysis: C H N Br
Calculated value /% 92.91 5.69 1.39 0.0
Measured value /% 92.46 5.32 1.29 0.0
Infrared absorption spectrum: FIG.
1 H-NMR spectrum (270 MHz): FIG.
Differential scanning calorimetry (DSC):
Ionization potential: 5.45 eV
<実施例4>
化合物No.7の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと1,3,5−トリブロモベンゼン2.07g(6.6mmol)を加え、60℃で4時間還流した。反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ヘキサンより再結晶後、トルエン/ヘキサンを展開溶媒としてシリカカラム精製し、1.0gの青色蛍光を示す白色固体を得た。
<Example 4>
Compound No. Synthesis of 7 To 0.485 g (20 mmol) of magnesium activated under argon in a Schlenk flask, 50 ml of a THF solution of 6.70 g (20 mmol) of 2-bromo-1,1,2-triphenylethene was added dropwise to form a Grignard. . To this reaction solution, 0.4 g of NiCl 2 (dppe) and 2.07 g (6.6 mmol) of 1,3,5-tribromobenzene were added and refluxed at 60 ° C. for 4 hours. The reaction solution was poured into 1N aqueous hydrochloric acid solution, extracted with toluene, washed with water, and dried over magnesium sulfate. After distilling off the solvent, the residue was recrystallized from acetone / hexane and purified by silica column using toluene / hexane as a developing solvent to obtain 1.0 g of a white solid exhibiting blue fluorescence.
この白色固体0.5gを昇華精製し、0.3gの純粋な固体を得た。 0.5 g of this white solid was purified by sublimation to obtain 0.3 g of a pure solid.
質量分析:m/e 840(M+)
赤外吸収スペクトル:図8
1H−NMRスペクトル(270MHz):図9
示差走査熱量測定(DSC):融点213℃、ガラス転移温度92℃
イオン化ポテンシャル:5.95eV
Mass spectrometry: m / e 840 (M +)
Infrared absorption spectrum: FIG.
1 H-NMR spectrum (270 MHz): FIG.
Differential scanning calorimetry (DSC): melting point 213 ° C.,
Ionization potential: 5.95 eV
<実施例5>
化合物No.8の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2,5−ジブロモチオフェン2.42g(10mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと1,2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)を加え、60℃で4時間還流した。反応溶液を1N塩酸水溶液に投入しトルエンで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトン/ヘキサンより再結晶後、1.0gの緑色蛍光を示す黄色固体を得た。
<Example 5>
Compound No. Synthesis of 8 To 0.485 g (20 mmol) of magnesium activated under argon in a Schlenk flask, 50 ml of a THF solution of 2.42 g (10 mmol) of 2,5-dibromothiophene was added dropwise to form a Grignard. To this reaction solution, 0.4 g of NiCl 2 (dppe) and 6.70 g (20 mmol) of 1,2-bromo-1,1,2-triphenylethene were added and refluxed at 60 ° C. for 4 hours. The reaction solution was poured into 1N aqueous hydrochloric acid solution, extracted with toluene, washed with water, and dried over magnesium sulfate. After distilling off the solvent, 1.0 g of a yellow solid showing green fluorescence was obtained after recrystallization from acetone / hexane.
この黄色固体1.0gを昇華精製し、0.8gの純粋な固体を得た。 1.0 g of this yellow solid was purified by sublimation to obtain 0.8 g of a pure solid.
質量分析:m/e 592(M+)
元素分析: C H S Br
計算値/% 89.15 5.44 5.41 0.0
測定値/% 89.05 5.32 5.05 0.0
イオン化ポテンシャル:5.40eV
Mass spectrometry: m / e 592 (M +)
Elemental analysis: C H S Br
Calculated value /% 89.15 5.44 5.41 0.0
Measured value /% 89.05 5.32 5.05 0.0
Ionization potential: 5.40 eV
なお、赤外吸収スペクトル、1H−NMRスペクトルの結果からも、上記化合物と同定した。 In addition, it identified with the said compound also from the result of the infrared absorption spectrum and the < 1 > H-NMR spectrum.
<実施例6>
化合物No.19の合成
シュレンクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.70g(20mmol)のTHF溶液50mlを滴下しグリニャール化した。この反応溶液にNiCl2(dppe)0.4gと2,3−ビス(4−ブロモフェニル)キノキサリン4.40g(20mmol)を加え、60℃で4時間還流した。反応溶液を0.1N塩酸水溶液に投入しクロロホルムで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトンより再結晶後、3.0gの緑色蛍光を示す黄色固体を得た。
<Example 6>
Compound No. Synthesis of 19 To 0.485 g (20 mmol) of magnesium activated under argon in a Schlenk flask, 50 ml of a THF solution of 6.70 g (20 mmol) of 2-bromo-1,1,2-triphenylethene was dropped to form a Grignard. . To this reaction solution, 0.4 g of NiCl 2 (dppe) and 4.40 g (20 mmol) of 2,3-bis (4-bromophenyl) quinoxaline were added and refluxed at 60 ° C. for 4 hours. The reaction solution was poured into a 0.1N hydrochloric acid aqueous solution, extracted with chloroform, washed with water, and dried over magnesium sulfate. After the solvent was distilled off, recrystallization from acetone gave 3.0 g of a yellow solid exhibiting green fluorescence.
この黄色固体1.0gを昇華精製し、0.8gの純粋な固体を得た。 1.0 g of this yellow solid was purified by sublimation to obtain 0.8 g of a pure solid.
質量分析:m/e 790(M+)
元素分析: C H N
計算値/% 91.10 5.35 3.54
測定値/% 91.03 5.28 3.40
赤外吸収スペクトル:図10
1H−NMRスペクトル:図11
イオン化ポテンシャル:5.98eV
Mass spectrometry: m / e 790 (M +)
Elemental analysis: C H N
Calculated value /% 91.10 5.35 3.54
Measurement /% 91.03 5.28 3.40
Infrared absorption spectrum: FIG.
1 H-NMR spectrum: FIG.
Ionization potential: 5.98 eV
<実施例7>
化合物No.4の合成
4,4’−ジブロモビフェニル3.02g(9.4mmol)のかわりに、ビス(p−ブロモフェニル)エーテル2.95g(9mmol)を用いるほかは実施例1と同様にして合成した。
<Example 7>
Compound No. Synthesis of 4 Synthesis was performed in the same manner as in Example 1 except that 2.95 g (9 mmol) of bis (p-bromophenyl) ether was used instead of 3.02 g (9.4 mmol) of 4,4′-dibromobiphenyl.
質量分析:m/e 678(M+)
赤外吸収スペクトル:図12
1H−NMRスペクトル:図13
示差走査熱量測定(DSC):融点254℃、ガラス転移温度90℃
なお、元素分析の測定値は計算値とよく一致した。
Mass spectrometry: m / e 678 (M +)
Infrared absorption spectrum: FIG.
1 H-NMR spectrum: FIG.
Differential scanning calorimetry (DSC): melting point 254 ° C.,
The measured values of elemental analysis agreed well with the calculated values.
<実施例8>
化合物No.33の合成
N,N’−(ビス(4−ブロモフェニル)−N,N’−ジフェニル−4,4’−ジアミノビフェニルの合成N,N’−ジフェニルベンジジン16.8g(50mmol)と4−ヨードブロモベンゼン42.4g(150mmol)と活性銅粉0.2gと炭酸カリウム20.7g(150mmol)を200mlナスフラスコに投入し、N2置換後200℃で24時間攪拌した。
<Example 8>
Compound No. Synthesis of 33 Synthesis of N, N ′-(bis (4-bromophenyl) -N, N′-diphenyl-4,4′-diaminobiphenyl 16.8 g (50 mmol) of N, N′-diphenylbenzidine and 4-iodo 42.4 g (150 mmol) of bromobenzene, 0.2 g of activated copper powder, and 20.7 g (150 mmol) of potassium carbonate were put into a 200 ml eggplant flask, and stirred for 24 hours at 200 ° C. after N 2 substitution.
トルエンを100ml加え、有機層を抽出し、水で3回洗浄した。トルエンとヘキサンの混合溶媒を抽出溶媒としてシリカカラムクロマト精製を3回行い、ヘキサン/ジクロロメタンより再結晶し、青色蛍光をもつ白色結晶15.2gを得た。 100 ml of toluene was added, the organic layer was extracted and washed 3 times with water. Silica column chromatography purification was performed 3 times using a mixed solvent of toluene and hexane as an extraction solvent, and recrystallization from hexane / dichloromethane gave 15.2 g of white crystals having blue fluorescence.
化合物No.33の合成シュレクフラスコにアルゴン下で活性化したマグネシウム0.485g(20mmol)に、2−ブロモ−1,1,2−トリフェニルエテン6.7g(20mmol)のTHF溶液50mlを滴下し、グリニャール化した。 Compound No. Into a synthetic Schlekk flask No. 33, 50 ml of THF solution of 6.7 g (20 mmol) of 2-bromo-1,1,2-triphenylethene was added dropwise to 0.485 g (20 mmol) of magnesium activated under argon to form Grignard. did.
この反応溶媒にNiCl2(dppe)0.4gとN,N’−ビス(4−ブロモフェニル)−N,N’−ジフェニル−4,4’−ジアミノビフェニル5.82g(9mmol)を加え、60〜70℃で4時間還流した。 To this reaction solvent, 0.4 g of NiCl 2 (dppe) and 5.82 g (9 mmol) of N, N′-bis (4-bromophenyl) -N, N′-diphenyl-4,4′-diaminobiphenyl were added. Refluxed at ˜70 ° C. for 4 hours.
反応溶液を10%塩酸水溶液に投入しトルエンとクロロホルムで抽出し、水洗後、硫酸マグネシウムで乾燥した。溶媒を留去後、アセトンで洗浄し、トルエンとヘキサンの混合溶媒を展開溶媒としてカラムクロマト精製し3.0gの緑黄色の蛍光をもつ黄白色固体を得た。 The reaction solution was poured into a 10% aqueous hydrochloric acid solution, extracted with toluene and chloroform, washed with water, and dried over magnesium sulfate. After the solvent was distilled off, the residue was washed with acetone and purified by column chromatography using a mixed solvent of toluene and hexane as a developing solvent to obtain 3.0 g of a yellowish white solid having greenish yellow fluorescence.
この黄色固体1.0gを昇華精製し、0.7gの純粋な固体を得た。 1.0 g of this yellow solid was purified by sublimation to obtain 0.7 g of a pure solid.
質量分析:m/e 996(M+)
赤外吸収スペクトル:図14
1H−NMRスペクトル:図15
示差走査熱量測定(DSC):融点333℃、ガラス転移温度132℃
イオン化ポテンシャル:5.38eV
なお、元素分析の測定値は計算値とよく一致した。
Mass spectrometry: m / e 996 (M +)
Infrared absorption spectrum: FIG.
1 H-NMR spectrum: FIG.
Differential scanning calorimetry (DSC): melting point 333 ° C., glass transition temperature 132 ° C.
Ionization potential: 5.38 eV
The measured values of elemental analysis agreed well with the calculated values.
<実施例9>
化合物No.42の合成
9,10−ビス(p−ブロモフェニル)アントラセンの合成
260mlフラスコにアルゴン下で、アントラキノン4.16g(20mmol)とトルエン100mlの中に4−ヨードブロモベンゼンとブチルリチウム(ヘキサン溶液)より合成した。4−ブロモフェニルリチウム6.52g(40mmol)のエーテル溶液を滴下した。室温で24時間攪拌後、水を100ml滴下した。沈澱物を濾過し、ジオール体を得た。
<Example 9>
Compound No. Synthesis of 42 Synthesis of 9,10-bis (p-bromophenyl) anthracene From 4-iodobromobenzene and butyllithium (hexane solution) in 4.16 g (20 mmol) of anthraquinone and 100 ml of toluene in a 260 ml flask under argon Synthesized. An ether solution of 6.52 g (40 mmol) of 4-bromophenyllithium was added dropwise. After stirring at room temperature for 24 hours, 100 ml of water was added dropwise. The precipitate was filtered to obtain a diol form.
このジオール体を酢酸100mlに溶解し、二塩化スズ2.0gの塩酸溶液を滴下し、100℃で1時間攪拌後トルエンで抽出し、水で5回洗浄した。硫酸マグネシウムで乾燥後、溶媒を留去してトルエンとヘキサンを抽出溶媒としてシリカカラムクロマトで精製した。 This diol was dissolved in 100 ml of acetic acid, a hydrochloric acid solution of 2.0 g of tin dichloride was added dropwise, stirred at 100 ° C. for 1 hour, extracted with toluene, and washed 5 times with water. After drying over magnesium sulfate, the solvent was distilled off and purified by silica column chromatography using toluene and hexane as extraction solvents.
その後トルエンで再結晶し、青色蛍光を示す白色結晶1gを得た。 Thereafter, it was recrystallized with toluene to obtain 1 g of white crystals exhibiting blue fluorescence.
化合物No.42の合成N,N’−ビス(p−ブロモフェニル)−N,N’−ジフェニル−4,4’−アミノビフェニル5.82g(9mmol)のかわりに、9,10−ビス(p−ブロモフェニル)アントラセン4.40g(9mmol)を用い、実施例8と同様に合成した。 Compound No. Synthesis of 42, instead of 5.82 g (9 mmol) of N, N′-bis (p-bromophenyl) -N, N′-diphenyl-4,4′-aminobiphenyl, 9,10-bis (p-bromophenyl) ) Synthesis was performed in the same manner as in Example 8 using 4.40 g (9 mmol) of anthracene.
質量分析:m/e 838(M+)
赤外吸収スペクトル:図16
1H−NMRスペクトル:図17
示差走査熱量測定(DSC):融点370℃、ガラス転移温度143℃
なお、元素分析の測定値は計算値とよく一致した。
Mass spectrometry: m / e 838 (M +)
Infrared absorption spectrum: FIG.
1 H-NMR spectrum: FIG.
Differential scanning calorimetry (DSC): melting point 370 ° C., glass transition temperature 143 ° C.
The measured values of elemental analysis agreed well with the calculated values.
<実施例10>
化合物No.65の合成
4,4’−ジブロモビフェニル3.02g(9.4mmol)のかわりに、2,6−ジクロロ−3メトキシアクリジン2.5g(9mmol)を用いるほかは実施例1と同様にして合成した。
<Example 10>
Compound No. Synthesis of 65 Synthesis was performed in the same manner as in Example 1 except that 2.5 g (9 mmol) of 2,6-dichloro-3methoxyacridine was used instead of 3.02 g (9.4 mmol) of 4,4′-dibromobiphenyl. .
質量分析:m/e 716(M+)
赤外吸収スペクトル:図18
1H−NMRスペクトル:図19
示差走査熱量測定(DSC):融点259.2℃、ガラス転移温度132.6℃
なお、元素分析の測定値は計算値とよく一致した。
Mass spectrometry: m / e 716 (M +)
Infrared absorption spectrum: FIG.
1 H-NMR spectrum: FIG.
Differential scanning calorimetry (DSC): melting point 259.2 ° C., glass transition temperature 132.6 ° C.
The measured values of elemental analysis agreed well with the calculated values.
化10〜化20に示される他の例示化合物も実施例1〜10に準じて合成した。これらの化合物は、元素分析、質量分析、赤外吸収スペクトル、1H−NMRスペクトルの結果から同定した。
Other exemplary compounds shown in
<実施例11>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Example 11>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、N,N’−ジフェニル−N,N’−m−トリル−4,4’−ジアミノ−1,1’−ビフェニル(TPD−1)を蒸着速度0.2nm/secで50nmの厚さに蒸着し、正孔注入輸送層とした。 Next, N, N′-diphenyl-N, N′-m-tolyl-4,4′-diamino-1,1′-biphenyl (TPD-1) was deposited to a thickness of 50 nm at a deposition rate of 0.2 nm / sec. It vapor-deposited and it was set as the positive hole injection transport layer.
次いで、実施例1の化合物No.1を50nmの厚さに蒸着し、発光層とした。 Subsequently, the compound No. 1 of Example 1 was used. 1 was deposited to a thickness of 50 nm to form a light emitting layer.
次いで、減圧状態を保ったまま、電子注入輸送層として、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで10nmの厚さに蒸着した。 Next, while maintaining the reduced pressure state, tris (8-quinolinolato) aluminum was deposited as an electron injecting and transporting layer to a thickness of 10 nm at a deposition rate of 0.2 nm / sec.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、19V、155mA/cm2で8500cd/m2の青色(発光極大波長λmax=485nm)の発光が確認され、この発光は乾燥窒素雰囲気中で200時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で10時間であった。 When a voltage was applied to the organic EL element and a current was passed, blue light of 8500 cd / m 2 (light emission maximum wavelength λmax = 485 nm) was confirmed at 19 V, 155 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for over 200 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 10 hours when driven at a constant current of 10 mA / cm 2 .
<実施例12>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Example 12>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、ポリ(チオフェン−2,5−ジイル)を10nmの厚さに蒸着し、正孔注入層とした。 Next, poly (thiophene-2,5-diyl) was deposited to a thickness of 10 nm to form a hole injection layer.
次いで、N,N’−ジフェニル−N,N’−m−トリル−4,4’−ジアミノ−1,1’−ビフェニル(TPD−1)を蒸着速度0.2nm/secで50nmの厚さに蒸着し、正孔輸送層とした。 Next, N, N′-diphenyl-N, N′-m-tolyl-4,4′-diamino-1,1′-biphenyl (TPD-1) was deposited to a thickness of 50 nm at a deposition rate of 0.2 nm / sec. It vapor-deposited and it was set as the positive hole transport layer.
次いで、実施例1の化合物No.1を50nmの厚さに蒸着し、発光層とした。 Subsequently, the compound No. 1 of Example 1 was used. 1 was deposited to a thickness of 50 nm to form a light emitting layer.
次いで、減圧状態を保ったまま、電子注入輸送層として、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで10nmの厚さに蒸着した。 Next, while maintaining the reduced pressure state, tris (8-quinolinolato) aluminum was deposited as an electron injecting and transporting layer to a thickness of 10 nm at a deposition rate of 0.2 nm / sec.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、16V、325mA/cm2で15000cd/m2の青色(発光極大波長λmax=485nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で100時間であった。 When a voltage was applied to the organic EL element and a current was applied, 15000 cd / m 2 of blue light (maximum light emission wavelength λmax = 485 nm) was confirmed at 16 V, 325 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 100 hours when driven at a constant current of 10 mA / cm 2 .
<実施例13>
実施例12と同様に素子を作製した。ただし、ホール輸送材料TPD−1の代わりに、N,N,N’,N’−テトラキス(3−ビフェニル)−4,4’−ジアミノ−1,1’−ビフェニル(TPD−2)を用いた。
<Example 13>
A device was produced in the same manner as in Example 12. However, N, N, N ′, N′-tetrakis (3-biphenyl) -4,4′-diamino-1,1′-biphenyl (TPD-2) was used instead of the hole transport material TPD-1. .
この有機EL素子に電圧を印加して電流を流したところ、18V、475mA/cm2で11500cd/m2の青色(発光極大波長λmax=485nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で200時間であった。 When a voltage was applied to the organic EL element and a current was passed, 11500 cd / m 2 of blue light (maximum light emission wavelength λmax = 485 nm) was confirmed at 18 V, 475 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 200 hours when driven at a constant current of 10 mA / cm 2 .
<実施例14>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Example 14>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、ポリ(チオフェン−2,5−ジイル)を10nmの厚さに蒸着し、正孔注入層とした。 Next, poly (thiophene-2,5-diyl) was deposited to a thickness of 10 nm to form a hole injection layer.
次いで、N,N’−ジフェニル−N,N’−m−トリル−4,4’−ジアミノ−1,1’−ビフェニル(TPD−1)を蒸着速度0.2nm/secで50nmの厚さに蒸着し、正孔輸送層とした。 Next, N, N′-diphenyl-N, N′-m-tolyl-4,4′-diamino-1,1′-biphenyl (TPD-1) was deposited to a thickness of 50 nm at a deposition rate of 0.2 nm / sec. It vapor-deposited and it was set as the positive hole transport layer.
次いで、実施例2の化合物No.2を50nmの厚さに蒸着し、発光層とした。 Subsequently, Compound No. 2 of Example 2 was used. 2 was deposited to a thickness of 50 nm to form a light emitting layer.
次いで、減圧状態を保ったまま、電子注入輸送層として、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで10nmの厚さに蒸着した。 Next, while maintaining the reduced pressure state, tris (8-quinolinolato) aluminum was deposited as an electron injecting and transporting layer to a thickness of 10 nm at a deposition rate of 0.2 nm / sec.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、15V、300mA/cm2で8000cd/m2の青色(発光極大波長λmax=485nm)の発光が確認され、この発光は乾燥窒素雰囲気中で100時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で20時間であった。 When a voltage was applied to this organic EL element and a current was passed, blue light emission of 8000 cd / m 2 (light emission maximum wavelength λmax = 485 nm) was confirmed at 15 V, 300 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 100 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 20 hours with a constant current drive of 10 mA / cm 2 .
<実施例15>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Example 15>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、ポリ(チオフェン−2,5−ジイル)を10nmの厚さに蒸着し、正孔注入層とした。 Next, poly (thiophene-2,5-diyl) was deposited to a thickness of 10 nm to form a hole injection layer.
次いで、N,N’−ジフェニル−N,N’−m−トリル−4,4’−ジアミノ−1,1’−ビフェニル(TPD−1)を蒸着速度0.2nm/secで50nmの厚さに蒸着し、正孔輸送層とした。 Next, N, N′-diphenyl-N, N′-m-tolyl-4,4′-diamino-1,1′-biphenyl (TPD-1) was deposited to a thickness of 50 nm at a deposition rate of 0.2 nm / sec. It vapor-deposited and it was set as the positive hole transport layer.
次いで、実施例3の化合物No.3を50nmの厚さに蒸着し、発光層とした。 Subsequently, compound No. 3 of Example 3 was used. 3 was deposited to a thickness of 50 nm to form a light emitting layer.
次いで、減圧状態を保ったまま、電子注入輸送層として、トリス(8−キノリノナト)アルミニウムを蒸着速度0.2nm/secで10nmの厚さに蒸着した。 Next, while maintaining the reduced pressure state, tris (8-quinolinonato) aluminum was deposited as an electron injecting and transporting layer to a thickness of 10 nm at a deposition rate of 0.2 nm / sec.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、14V、53mA/cm2で345cd/m2の青緑色(発光極大波長λmax=450nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で200時間であった。 When a voltage was applied to this organic EL element and a current was passed, 345 cd / m 2 of blue-green light (maximum light emission wavelength λmax = 450 nm) was confirmed at 14 V, 53 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 200 hours when driven at a constant current of 10 mA / cm 2 .
<実施例16>
実施例12において、発光層に化合物No.1を用いるかわりに、化合物No.42を用いるほかは同様にして有機EL素子を得た。
この有機EL素子に電圧を印加して電流を流したところ、15V、450mA/cm2で8020cd/m2の青色(発光極大波長λmax=470nm)の発光が確認され、この発光は乾燥窒素雰囲気中で3000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で300時間であった。
<Example 16>
In Example 12, the compound No. Instead of using
When a voltage was applied to this organic EL element and a current was passed, 8020 cd / m 2 of blue light (maximum light emission wavelength λmax = 470 nm) was confirmed at 15 V, 450 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for over 3000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 300 hours when driven at a constant current of 10 mA / cm 2 .
<実施例17>
実施例12において、発光層に化合物No.1を用いるかわりに、化合物No.49を用いるほかは同様にして有機EL素子を得た。
この有機EL素子に電圧を印加して電流を流したところ、14V、340mA/cm2で18000cd/m2の青色(発光極大波長λmax=480nm)の発光が確認され、この発光は乾燥窒素雰囲気中で5000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で500時間であった。
<Example 17>
In Example 12, the compound No. Instead of using
When a voltage was applied to the organic EL element and a current was passed, 18000 cd / m 2 of blue light (maximum light emission wavelength λmax = 480 nm) was confirmed at 14 V, 340 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for over 5000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 500 hours when driven at a constant current of 10 mA / cm 2 .
<実施例18>
実施例12において、発光層に化合物No.1を用いるかわりに、化合物No.53を用いるほかは同様にして有機EL素子を得た。
<Example 18>
In Example 12, the compound No. Instead of using
この有機EL素子に電圧を印加して電流を流したところ、16V、300mA/cm2で9040cd/m2の青色(発光極大波長λmax=470nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で200時間であった。 When a voltage was applied to the organic EL element and a current was passed, 9040 cd / m 2 of blue light (maximum light emission wavelength λmax = 470 nm) was confirmed at 16 V, 300 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 200 hours when driven at a constant current of 10 mA / cm 2 .
<実施例19>
実施例12において、発光層を形成した後、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで20nmの厚さに蒸着し、電子輸送層とした。次いで、テトラブチルジフェノキノンを10nmの厚さに蒸着し、電子注入層とした。その後、実施例12と同様にして有機EL素子を得た。
<Example 19>
In Example 12, after forming the light emitting layer, tris (8-quinolinolato) aluminum was evaporated to a thickness of 20 nm at a deposition rate of 0.2 nm / sec to form an electron transport layer. Next, tetrabutyldiphenoquinone was deposited to a thickness of 10 nm to form an electron injection layer. Thereafter, an organic EL device was obtained in the same manner as in Example 12.
この有機EL素子に電圧を印加して電流を流したところ、13V、105mA/cm2で4500cd/m2の青色(発光極大波長λmax=480nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で50時間であった。 When a voltage was applied to this organic EL element and a current was passed, 4500 cd / m 2 of blue light (maximum light emission wavelength λmax = 480 nm) was confirmed at 13 V, 105 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 50 hours with a constant current drive of 10 mA / cm 2 .
<実施例20>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Example 20>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、化合物No.7を蒸着速度0.2nm/secで50nmの厚さに蒸着し、正孔注入輸送層とした。 Subsequently, Compound No. 7 was deposited at a deposition rate of 0.2 nm / sec to a thickness of 50 nm to form a hole injecting and transporting layer.
次いで、トリス(8−キノリノラト)アルミニウムを50nmの厚さに蒸着し、電子注入輸送層を兼ねる発光層とした。 Next, tris (8-quinolinolato) aluminum was vapor-deposited to a thickness of 50 nm to form a light emitting layer that also served as an electron injection transport layer.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、11V、525mA/cm2で1759cd/m2の緑色(発光極大波長λmax=500nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は400時間であった。 When a voltage was applied to this organic EL element and a current was passed, 1759 cd / m 2 of green light emission (maximum light emission wavelength λmax = 500 nm) was confirmed at 11 V, 525 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of luminance was 400 hours.
<実施例21>
実施例20において、正孔注入輸送層に化合物No.7を用いるかわりに、化合物No.8を用いるほかは同様にして有機EL素子を得た。
<Example 21>
In Example 20, the compound No. Instead of using
この有機EL素子に電圧を印加して電流を流したところ、13V、300mA/cm2で4000cd/m2の緑色(発光極大波長λmax=520nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で50時間であった。 When a voltage was applied to this organic EL element and a current was passed, 4000 cd / m 2 of green light (maximum light emission wavelength λmax = 520 nm) was confirmed at 13 V, 300 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 50 hours with a constant current drive of 10 mA / cm 2 .
<実施例22>
実施例20において、正孔注入輸送層に化合物No.7を用いるかわりに、化合物No.33を用いるほかは同様にして有機EL素子を得た。
<Example 22>
In Example 20, the compound No. Instead of using
この有機EL素子に電圧を印加して電流を流したところ、12V、525mA/cm2で12000cd/m2の青緑色(発光極大波長λmax=503nm)の発光が確認され、この発光は乾燥窒素雰囲気中で4000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で500時間であった。 When a voltage was applied to this organic EL element and a current was passed, 12000 cd / m 2 of blue-green light (maximum light emission wavelength λmax = 503 nm) was confirmed at 12 V, 525 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for 4000 hours or more. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 500 hours when driven at a constant current of 10 mA / cm 2 .
<実施例23>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Example 23>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、ポリ(チオフェン−2,5−ジイル)を10nmの厚さに蒸着し、正孔注入層とした。 Next, poly (thiophene-2,5-diyl) was deposited to a thickness of 10 nm to form a hole injection layer.
次いで、N,N’−ジフェニル−N,N’−m−トリル−4,4’−ジアミノ−1,1’−ビフェニル(TPD−1)を蒸着速度0.2nm/secで50nmの厚さに蒸着し、正孔輸送層とした。 Next, N, N′-diphenyl-N, N′-m-tolyl-4,4′-diamino-1,1′-biphenyl (TPD-1) was deposited to a thickness of 50 nm at a deposition rate of 0.2 nm / sec. It vapor-deposited and it was set as the positive hole transport layer.
次いで、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secを50nmの厚さに蒸着し、発光層とした。 Next, tris (8-quinolinolato) aluminum was vapor-deposited at a deposition rate of 0.2 nm / sec to a thickness of 50 nm to form a light emitting layer.
次いで、減圧状態を保ったまま、電子注入輸送層として、実施例6の化合物No.19を蒸着速度0.2nm/secで10nmの厚さに蒸着した。 Next, while maintaining the reduced pressure state, as an electron injecting and transporting layer, Compound No. 19 was deposited to a thickness of 10 nm at a deposition rate of 0.2 nm / sec.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、14V、300mA/cm2で15000cd/m2の青緑色(発光極大波長λmax=495nm)の発光が確認され、この発光は乾燥窒素雰囲気中で500時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で200時間であった。 When a voltage was applied to the organic EL element and a current was passed, 15000 cd / m 2 of blue-green light (maximum light emission wavelength λmax = 495 nm) was confirmed at 14 V, 300 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for 500 hours or more. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 200 hours when driven at a constant current of 10 mA / cm 2 .
<実施例24>
実施例23において、電子注入輸送層に化合物No.19を用いるかわりに、化合物No.16を用いるほかは同様にして有機EL素子を得た。
<Example 24>
In Example 23, the compound No. Instead of using compound 19, compound no. An organic EL device was obtained in the same manner except that 16 was used.
この有機EL素子に電圧を印加して電流を流したところ、12V、450mA/cm2で18000cd/m2の緑色(発光極大波長λmax=500nm)の発光が確認され、この発光は乾燥窒素雰囲気中で1000時間以上安定していた。部分的非発光部の出現および成長は全くなかった。輝度の半減期は10mA/cm2の定電流駆動で400時間であった。 When a voltage was applied to this organic EL element and a current was passed, 18000 cd / m 2 of green light (maximum light emission wavelength λmax = 500 nm) was confirmed at 12 V, 450 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. And stable for over 1000 hours. There was no emergence or growth of partially non-light emitting parts. The half life of the luminance was 400 hours when driven at a constant current of 10 mA / cm 2 .
<実施例25>
実施例12と同様に正孔輸送層を形成した後、TPD−1と実施例1の化合物No.1を蒸着速度0.2nm/secで20nmの厚さに蒸着し、発光層とした。この場合、TPD−1と化合物No.1との比率は重量比で1:1とした。
<Example 25>
After forming the hole transport layer in the same manner as in Example 12, TPD-1 and the compound No. 1 in Example 1 were used. 1 was deposited to a thickness of 20 nm at a deposition rate of 0.2 nm / sec to form a light emitting layer. In this case, TPD-1 and Compound No. The ratio to 1 was 1: 1 by weight.
次いで、化合物No.1を用いるほかは、実施例12と同様に電子注入輸送層を形成し、さらに同様に陰極を形成して有機EL素子を得た。 Subsequently, Compound No. Except for using 1, an electron injecting and transporting layer was formed in the same manner as in Example 12, and a cathode was formed in the same manner to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、12V、250mA/cm2で12000cd/m2の青色(発光極大波長λmax=480nm)の発光が確認され、この発光は乾燥窒素雰囲気中で5000時間以上安定していた。部分的非発光部の出現および成長は全くなく、電流リークもみられなかった。輝度の半減期は10mA/cm2の定電流駆動で1000時間であった。 When a voltage was applied to the organic EL element and a current was applied, 12000 cd / m 2 of blue light (maximum light emission wavelength λmax = 480 nm) was confirmed at 12 V, 250 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. It was stable for over 5000 hours. There was no appearance and growth of a partial non-light emitting portion, and no current leakage was observed. The half life of the luminance was 1000 hours when driven at a constant current of 10 mA / cm 2 .
実施例11〜25において、化10〜化20に掲げた本発明の化合物の1種または2種以上を適宜選択して、上記実施例以外の組合せで、発光層、電子注入輸送層または正孔注入輸送層に用いたところ、有機EL素子の層構成等に応じて、上記実施例と同様の結果が得られた。
In Examples 11 to 25, one or more of the compounds of the present invention listed in
<比較例1>
厚さ100nmのITO透明電極(陽極)を有するガラス基板を、中性洗剤、アセトン、エタノールを用いて超音波洗浄し、煮沸エタノール中から引き上げて乾燥し、蒸着装置の基板ホルダーに固定して、1×10−4Paまで減圧した。
<Comparative Example 1>
A glass substrate having an ITO transparent electrode (anode) with a thickness of 100 nm is ultrasonically cleaned with a neutral detergent, acetone, and ethanol, and is lifted from boiling ethanol and dried, and fixed to a substrate holder of a vapor deposition apparatus. The pressure was reduced to 1 × 10 −4 Pa.
次いで、N,N’−ビス(−m−メチルフェニル)−N,N’−ジフェニル−1,1’−ビフェニル−4,4’−ジアミンを50nmの厚さに蒸着し、正孔注入輸送層とした。 Next, N, N′-bis (-m-methylphenyl) -N, N′-diphenyl-1,1′-biphenyl-4,4′-diamine was vapor-deposited to a thickness of 50 nm, and a hole injection transport layer was formed. It was.
次いで、減圧状態を保ったまま、1,3−ビス(5−(4−t−ブチルフェニル)−1,3,4−オキサジアゾ−2−イル)ベンゼン(OXD−7)を蒸着速度0.2nm/secで50nmの厚さに蒸着して、電子注入輸送・発光層とした。 Next, while maintaining the reduced pressure state, 1,3-bis (5- (4-t-butylphenyl) -1,3,4-oxadiazo-2-yl) benzene (OXD-7) was deposited at a deposition rate of 0.2 nm. The film was deposited to a thickness of 50 nm at / sec to form an electron injection transport / light emitting layer.
さらに、減圧状態を保ったまま、MgAg(重量比10:1)を蒸着速度0.2nm/secで200nmの厚さに蒸着して陰極とし、有機EL素子を得た。 Further, while maintaining the reduced pressure state, MgAg (weight ratio 10: 1) was vapor-deposited at a vapor deposition rate of 0.2 nm / sec to a thickness of 200 nm to form a cathode to obtain an organic EL device.
この有機EL素子に電圧を印加して電流を流したところ、14V、127mA/cm2で550cd/m2の青色(発光極大波長λmax=480nm)の発光が確認され、この発光は乾燥窒素雰囲気中で10時間にて、部分的非発光部の出現および成長が見られ、20時間にて絶縁破壊を起こした。輝度の半減期は20分であった。 When a voltage was applied to the organic EL element and a current was passed, 550 cd / m 2 of blue light (maximum light emission wavelength λmax = 480 nm) was confirmed at 14 V, 127 mA / cm 2 , and this light emission was in a dry nitrogen atmosphere. In 10 hours, the appearance and growth of a partial non-light emitting portion was observed, and dielectric breakdown occurred in 20 hours. The half life of the brightness was 20 minutes.
<比較例2>
C. Adachi et al., Appli. Phys. Lett., 56, 799(1990) に記載の1,1,4,4−テトラフェニル−1,3−ブタジエンを発光層に用いて、この文献と同様の構成の有機EL素子を組み立てた。すなわち、比較例1において、正孔注入輸送層を形成したのち、上記化合物を同様に50nmの厚さに蒸着して発光層とした。その後、トリス(8−キノリノラト)アルミニウムを蒸着速度0.2nm/secで10nmの厚さで蒸着して電子注入輸送層とし、比較例1と同様に陰極を形成し、有機EL素子を得た。
<Comparative example 2>
Similar to this document, using 1,1,4,4-tetraphenyl-1,3-butadiene described in C. Adachi et al., Appli. Phys. Lett., 56, 799 (1990) in the light emitting layer. An organic EL element having the structure was assembled. That is, in Comparative Example 1, after forming a hole injecting and transporting layer, the above compound was similarly deposited to a thickness of 50 nm to form a light emitting layer. Thereafter, tris (8-quinolinolato) aluminum was deposited at a deposition rate of 0.2 nm / sec to a thickness of 10 nm to form an electron injecting and transporting layer, and a cathode was formed in the same manner as in Comparative Example 1 to obtain an organic EL device.
この有機EL素子は、有機層が結晶化しており、両電極に電圧を印加したところ、ショートした状態であり、明確な発光は得られなかった。 In this organic EL element, the organic layer was crystallized, and when a voltage was applied to both electrodes, the organic EL element was in a short-circuited state, and clear light emission was not obtained.
<比較例3>
比較例2において、t−ブチルフェニルビフェニルオキサジアゾール(PBD)を電子注入輸送層とするほかは、同構成の有機EL素子を得た。
<Comparative Example 3>
In Comparative Example 2, an organic EL device having the same configuration was obtained except that t-butylphenylbiphenyloxadiazole (PBD) was used as the electron injecting and transporting layer.
この有機EL素子は、作製してから1時間後にはショートした状態となり、この状態で電圧を印加したところ青色の発光はみられたものの絶縁破壊した。 This organic EL element was short-circuited 1 hour after fabrication, and when a voltage was applied in this state, blue light emission was seen but dielectric breakdown occurred.
<比較例4>
特開平6−100857号公報の合成例1の方法に従い、ジフェニルクロロメタンをグリニャール化し、これと1,4−ジベンゾイルベンゼンとを反応させ、ギ酸存在下で脱水して、化合物No.2を得た。
<Comparative Example 4>
In accordance with the method of Synthesis Example 1 of JP-A-6-1000085, diphenylchloromethane is converted into Grignard, reacted with 1,4-dibenzoylbenzene, dehydrated in the presence of formic acid, and compound No. 1 is obtained. 2 was obtained.
このようにして得られた化合物No.2を発光層に用いるほかは実施例14と同様にして有機EL素子を得た。 Thus obtained compound No. An organic EL device was obtained in the same manner as in Example 14 except that 2 was used for the light emitting layer.
この有機EL素子について、実施例14と同様に特性を調べたところ、同じ駆動電流値によって半分程度の輝度しか得られなかった。 The characteristics of this organic EL element were examined in the same manner as in Example 14. As a result, only about half the luminance was obtained with the same drive current value.
なお、上記のEL素子を得るにあたり、化2の化合物および比較例に用いた化合物の蒸着膜の安定性を調べた。これらの一例を以下に示す。
In obtaining the above EL device, the stability of the vapor deposition film of the compound of
<実施例26>
実施例1で得られた化合物No.1をガラス基板上に、10−5Pa以下の減圧下で真空蒸着を行い、1000A厚の蒸着膜を形成した。
<Example 26>
Compound No. 1 obtained in Example 1 was obtained. 1 was vacuum-deposited on a glass substrate under a reduced pressure of 10 −5 Pa or less to form a 1000 A-thick deposited film.
この蒸着膜は初期においては透明なアモルファス状の膜であり、大気中に放置しても1ケ月程度は結晶化しなかった。 This deposited film was a transparent amorphous film in the initial stage, and did not crystallize for about one month even when left in the atmosphere.
<実施例27>
実施例2で得られた化合物No.2を用い、実施例26と同様に蒸着膜を形成し、実施例26と同様にして膜の安定性を調べたところ、10日程度は結晶化しなかった。
<Example 27>
Compound No. obtained in Example 2 2 was used to form a deposited film in the same manner as in Example 26, and the stability of the film was examined in the same manner as in Example 26. As a result, the film was not crystallized for about 10 days.
<比較例5>
特開平6−100857号公報の合成例1に従って得られた化合物No.2を用い、実施例26と同様に蒸着膜を形成し、実施例26と同様にして膜の安定性を調べたところ、初期においてはアモルファス状の膜であったが、1日後には結晶化した。
<Comparative Example 5>
Compound No. 1 obtained according to Synthesis Example 1 of JP-A-6-1000085. 2 was used to form a vapor-deposited film in the same manner as in Example 26, and the stability of the film was examined in the same manner as in Example 26. As a result, the film was initially amorphous but crystallized after one day. did.
<実施例28>
化合物(No.3、7、8、16、19、33、42、49、53)について、実施例26と同様にして各化合物の蒸着膜を形成し、同様に各膜の安定性を調べたところ、大気中に1年以上放置しても初期のアモルファス状態が維持されていることがわかった。
<Example 28>
For the compounds (Nos. 3, 7, 8, 16, 19, 33, 42, 49, 53), vapor-deposited films of the respective compounds were formed in the same manner as in Example 26, and the stability of each film was similarly examined. However, it was found that the initial amorphous state was maintained even after being left in the atmosphere for over a year.
1 有機EL素子
2 基板
3 陽極
4 正孔注入輸送層
5 発光層
6 電子注入輸送層
7 陰極
DESCRIPTION OF
Claims (1)
芳香族残基三置換ハロゲン化エテンをグリニャール化し、ジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物とクロスカップリングするか、またはジ、トリ、テトラ、ペンタもしくはヘキサハロゲン化芳香族化合物をグリニャール化し、芳香族残基三置換ハロゲン化エテンとクロスカップリングするかによって、下記化1で表されるテトラアリールエテン誘導体を得る工程を備えることを特徴とする有機EL素子の製造方法。
Grignard aromatic residue trisubstituted halogenated ethene and cross-couple with di, tri, tetra, penta or hexahalogenated aromatic compounds, or di, tri, tetra, penta or hexahalogenated aromatic compounds A method for producing an organic EL device comprising a step of obtaining a tetraarylethene derivative represented by the following chemical formula 1 depending on whether it is Grignarded and cross-coupled with an aromatic residue trisubstituted halogenated ethene.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007134226A JP4267670B2 (en) | 1994-04-28 | 2007-05-21 | Manufacturing method of organic EL element |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP11445694 | 1994-04-28 | ||
| JP2007134226A JP4267670B2 (en) | 1994-04-28 | 2007-05-21 | Manufacturing method of organic EL element |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004343976A Division JP3999781B2 (en) | 1994-04-28 | 2004-11-29 | Organic EL device |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007270153A JP2007270153A (en) | 2007-10-18 |
| JP4267670B2 true JP4267670B2 (en) | 2009-05-27 |
Family
ID=38673261
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007134226A Expired - Lifetime JP4267670B2 (en) | 1994-04-28 | 2007-05-21 | Manufacturing method of organic EL element |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4267670B2 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20210072209A (en) * | 2019-12-06 | 2021-06-17 | 삼성디스플레이 주식회사 | Organic electroluminescence device and amine compound for organic electroluminescence device |
-
2007
- 2007-05-21 JP JP2007134226A patent/JP4267670B2/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007270153A (en) | 2007-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3816969B2 (en) | Organic EL device | |
| JP5193074B2 (en) | Organic EL device | |
| JP3838816B2 (en) | Compound for organic EL device and organic EL device | |
| JP4623223B2 (en) | Organic EL device | |
| JP3712760B2 (en) | Organic EL device | |
| JP3828595B2 (en) | Organic EL device | |
| EP0681019B1 (en) | Phenylanthracene derivative and organic EL element | |
| US5142343A (en) | Organic electroluminescence device with oligomers | |
| JP3965063B2 (en) | Organic electroluminescence device | |
| JP3650200B2 (en) | Organic EL devices using quinoxaline compounds | |
| JP3642606B2 (en) | Organic EL device | |
| JP2003026616A (en) | Compound for organic el(electroluminescent) device and organic el device using it | |
| JP4364130B2 (en) | Organic EL device | |
| JP4588637B2 (en) | Conjugated molecules, electroluminescent devices, and electronic devices | |
| Mallesham et al. | Design and synthesis of novel anthracene derivatives as n-type emitters for electroluminescent devices: a combined experimental and DFT study | |
| JP3742176B2 (en) | Compound for organic EL device and organic EL device | |
| JP3993333B2 (en) | Organic EL device | |
| JP3999781B2 (en) | Organic EL device | |
| JP4320020B2 (en) | Organic EL device | |
| JP2016150920A (en) | Compound for organic electroluminescent element and organic electroluminescent element using the same | |
| JP4267670B2 (en) | Manufacturing method of organic EL element | |
| JP4190542B2 (en) | Phenylanthracene derivative | |
| JP4109292B2 (en) | Compounds for organic EL devices | |
| JP2000268963A (en) | Organic electroluminescent element | |
| JP4200152B2 (en) | Organic EL device |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070731 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090210 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090218 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120227 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120227 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R360 | Written notification for declining of transfer of rights |
Free format text: JAPANESE INTERMEDIATE CODE: R360 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R370 | Written measure of declining of transfer procedure |
Free format text: JAPANESE INTERMEDIATE CODE: R370 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R370 | Written measure of declining of transfer procedure |
Free format text: JAPANESE INTERMEDIATE CODE: R370 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130227 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140227 Year of fee payment: 5 |
|
| EXPY | Cancellation because of completion of term |