ES2754248T3 - Inhibidores de proteína cinasa C y métodos para su uso - Google Patents
Inhibidores de proteína cinasa C y métodos para su uso Download PDFInfo
- Publication number
- ES2754248T3 ES2754248T3 ES15750138T ES15750138T ES2754248T3 ES 2754248 T3 ES2754248 T3 ES 2754248T3 ES 15750138 T ES15750138 T ES 15750138T ES 15750138 T ES15750138 T ES 15750138T ES 2754248 T3 ES2754248 T3 ES 2754248T3
- Authority
- ES
- Spain
- Prior art keywords
- amino
- pyridin
- pyrazin
- carboxamide
- trifluoromethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 Cc1ccnc(-c(nc2C(Nc(nccc3)c3N(CC[C@]3*)C[C@@]3F)=O)cnc2N)c1OC(F)(F)F Chemical compound Cc1ccnc(-c(nc2C(Nc(nccc3)c3N(CC[C@]3*)C[C@@]3F)=O)cnc2N)c1OC(F)(F)F 0.000 description 4
- JQNAONNYNBBDBJ-UHFFFAOYSA-N CC(C)(C)OC(NC(CO)(CC1)CCN1c(cccn1)c1[N+]([O-])=O)=O Chemical compound CC(C)(C)OC(NC(CO)(CC1)CCN1c(cccn1)c1[N+]([O-])=O)=O JQNAONNYNBBDBJ-UHFFFAOYSA-N 0.000 description 1
- PYGPOWSSBHTNHQ-UHFFFAOYSA-N CCOC(CC(CCN(C1)c(cccn2)c2[N+]([O-])=O)(C1F)NC(OC(C)(C)C)=O)=O Chemical compound CCOC(CC(CCN(C1)c(cccn2)c2[N+]([O-])=O)(C1F)NC(OC(C)(C)C)=O)=O PYGPOWSSBHTNHQ-UHFFFAOYSA-N 0.000 description 1
- KAIILUUOYYOIHZ-UHFFFAOYSA-N Cc1c(-c(nc2C(O)=O)cnc2N)nccc1 Chemical compound Cc1c(-c(nc2C(O)=O)cnc2N)nccc1 KAIILUUOYYOIHZ-UHFFFAOYSA-N 0.000 description 1
- ZIHFJEMRYPMALO-UHFFFAOYSA-N Nc1ncc(-c(nccc2)c2F)nc1C(Nc1ncccc1N(CC1)CCC1NI)=O Chemical compound Nc1ncc(-c(nccc2)c2F)nc1C(Nc1ncccc1N(CC1)CCC1NI)=O ZIHFJEMRYPMALO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Transplantation (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Un compuesto de fórmula II**Fórmula** o una sal farmacéuticamente aceptable del mismo, en donde: X es N o CR; R, R2, R3 y R4 son cada uno independientemente H, 2H, halo, hidroxilo (-OH), alcoxi C1-3, haloalquilo C1-3 o alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con hidroxilo, halo, alcoxi C1-3 o haloalcoxi C1-3; R5 es independientemente H, 2H, CH3, CH2F, CHF2, CF3, CH2OH, alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con F, OH, alcoxi C1-3 o haloalcoxi C1-3; cada uno de R5a y R5b es independientemente H, 2H, alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con F, OH o alcoxi C1-3, o R5a y R5b se unen para formar un grupo puente de metileno o etileno; cada uno de R5c y R5d es independientemente H, 2H, F, -OH, alcoxi C1-3, alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con F, OH o alcoxi C1-3 o R5c y R5d se unen para formar un grupo puente de metileno, etileno o -CH2-O-; cada uno de R6, R7 y R8 se selecciona independientemente entre H, 2H, halo, alquilo C1-3, haloalquilo C1-3, alcoxi C1-3, haloalcoxi C1-3, cicloalquilo C3-7 y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho alquilo C1-3 opcionalmente sustituido con F, OH, alcoxi C1-3 o haloalcoxi C1-3; o en donde R6 y R8 opcionalmente forman un anillo carbocíclico o anillo heterobicíclico con el anillo heteroarilo, dicho anillo carbocíclico o anillo heterobicíclico opcionalmente sustituido con de 1 a 3 grupos seleccionados entre: 2H, halo, haloalquilo C1-3, alcoxi C1-3, haloalcoxi C1-3, cicloalquilo C3-7 y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S.
Description
DESCRIPCIÓN
Inhibidores de proteína cinasa C y métodos para su uso
Campo de la invención
La presente invención se refiere a nuevos compuestos y sus tautómeros y estereoisómeros y sales farmacéuticamente aceptables de los mismos, composiciones de los nuevos compuestos junto con transportadores farmacéuticamente aceptables y a estos compuestos, ya sea solos o en combinación con al menos un agente terapéutico adicional, para su uso en la profilaxis o tratamiento del cáncer.
Antecedentes
El melanoma uveal es el tumor maligno intraocular primario más común en adultos. Se describen ciertos inhibidores de proteína cinasa en las Publicaciones Internacionales n.° WO 02/38561 y WO 2008/106692. Se ha demostrado que otro inhibidor de proteína cinasa C (PKC), sotrastaurina, tiene actividad contra ciertos isotipos de PKC y tan solo recientemente se ha demostrado que inhibe de manera selectiva el crecimiento de células de melanoma uveal que portan mutaciones GNAQ actuando de manera selectiva sobre las vías de PKC/ERK1/2 y PKC/NF-kB (véase X. Wu, et al en Mol. Cancer Ther., Vol. 11, páginas 1905-1914, 2012). Se encuentra en desarrollo un ensayo clínico que estudia el uso de sotrastaurina para tratar a pacientes que tienen melanoma uveal. Sin embargo, sigue habiendo una necesidad no satisfecha para proporcionar inhibidores de PKC de última generación para tratar el melanoma uveal que tengan eficacia mejorada a menores cantidades de dosis para lograr la regresión tumoral, una potencia mejorada, actividad hERG, absorción, tolerancia gastrointestinal y selectividad de cinasa.
El linfoma difuso de linfocitos B grandes (DLBCL) representa el subtipo más común de melanoma maligno y es heterogéneo con respecto a su morfología, biología y presentación clínica. El inhibidor de PKC, sotrastaurina (AEB071), ha demostrado inhibir de manera selectiva el crecimiento de células de DLBCL mutantes para CD79 (véase T. Naylor, et al en Cancer Res., Vol. 71(7), 2643-2653, 2011). Además, el estudio sugirió que la sotrastaurina muestra una sinergia significativa cuando se combina con el inhibidor de mTOR, everolimus (Afinitor™). Se encuentra en desarrollo un ensayo clínico que estudia el uso de sotrastaurina para tratar a pacientes que tienen DLBCL que porten la mutación de CD79. Sin embargo, sigue habiendo una necesidad no satisfecha para proporcionar inhibidores de PKC de última generación para tratar el DLBCL que tengan eficacia mejorada a menores cantidades de dosis para lograr la regresión tumoral, una potencia mejorada, perfil PK, absorción, tolerancia gastrointestinal y selectividad de cinasa.
Sumario
La presente invención proporciona un compuesto de fórmula (II):

o una sal farmacéuticamente aceptable del mismo,
en donde:
X es N o CR;
R, R2, R3 y R4 son cada uno independientemente H, 2H, halo, hidroxilo (-OH), alcoxi C1-3, haloalquilo C1-3 o alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con hidroxilo, halo, alcoxi C1-3 o haloalcoxi C1-3;
R5 es independientemente H, 2H, CH3, CH2F, CHF2, CF3, CH2OH, alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con F, OH, alcoxi C1-3 o haloalcoxi C1-3;
cada uno de R5a y R5b es independientemente H, 2H, alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con
F, OH o alcoxi C1-3, o R5a y R5b se unen para formar un grupo puente de metileno o etileno;
cada uno de R5c y R5d es independientemente H, 2H, F, -OH, alcoxi C1-3, alquilo C1-3, dicho alquilo C1-3
opcionalmente sustituido con F, OH o alcoxi C1-3, o R5c y R5d se unen para formar un grupo puente de metileno,
etileno o -CH2-O-;
cada uno de R6, R7 y R8 se selecciona independientemente entre H, 2H, halo, alquilo C1-3, haloalquilo C1-3, alcoxi
C1-3, haloalcoxi C1-3, cicloalquilo C3-7 y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados
entre N, O y S, dicho alquilo C1-3 opcionalmente sustituido con F, OH, alcoxi C1-3 o haloalcoxi C1-3; o
en donde R6 y R8 opcionalmente forman un anillo carbocíclico o anillo heterobicíclico parcialmente saturado con el
anillo heteroarilo, dicho anillo carbocíclico o anillo heterobicíclico opcionalmente sustituido con de 1 a 3 grupos
seleccionados entre: 2H, halo, haloalquilo C1-3, alcoxi C1-3, haloalcoxi C1-3, cicloalquilo C3-7 y miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S.
En una realización preferida,
X es N o CR;
R, R2, R3 y R4 son cada uno independientemente H, 2H, halo, hidroxi (-OH), alcoxi C1-3, haloalquilo C1-3 o alquilo C1-3,
dicho alquilo C1-3 opcionalmente sustituido con de uno a dos de hidroxi, halo, alcoxi C1-3 o haloalcoxi C1-3;
R5 es -H, 2H, CH3, CH2F, CHF2, CF3, CH2OH, alquilo C1-3, CH2-O-alquilo C1-3 o CH2-O-haloalquilo C1-3, dicho alquilo
C1-3 opcionalmente sustituido con F, OH, alcoxi C1-3 o haloalcoxi C1-3;
cada uno de R5a y R5b es independientemente H, 2H, alquilo C1-3, dicho alquilo C1-3 opcionalmente sustituido con F,
OH o alcoxi C1-3, o R5a y R5b se unen para formar un grupo puente de metileno o etileno;
cada uno de R5c y R5d es independientemente H, 2H, F, -OH, alquilo C1-3 o alcoxi C1-3, dicho alquilo C1-3 opcionalmente
sustituido con F, OH o alcoxi C1-3, o R5c y R5d se unen para formar un grupo puente de metileno, etileno o -CH2-O-; y
cada uno de R6, R7 y R8 se selecciona independientemente entre H, 2H, halo, alquilo C1-3, haloalquilo C1-3, alcoxi C1-3,
haloalcoxi C1-3, cicloalquilo C3-7 y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre
N, O y S, dicho alquilo C1-3 opcionalmente sustituido con F, OH, alcoxi C1-3 o haloalcoxi C1-3; o
en donde R6 y R8 opcionalmente forman un anillo carbocíclico o anillo heterobicíclico parcialmente saturado con el
anillo heteroarilo, dicho anillo carbocíclico o anillo heterobicíclico opcionalmente sustituido con de 1 a 3 grupos
seleccionados entre: 2H, halo, haloalquilo C1-3, alcoxi C1-3, haloalcoxi C1-3, cicloalquilo C3-7 y heterociclilo de 4-7
miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S.
En otros aspectos, la presente invención proporciona una composición farmacéutica que comprende: un compuesto
de fórmula (II) o una sal farmacéuticamente aceptable del mismo y al menos un transportador o excipiente
farmacéuticamente aceptable.
Los compuestos de la invención son útiles en el tratamiento de trastornos relacionados con la proteína cinasa C,
específicamente trastornos relacionados con isoformas de proteína cinasa C alfa y/o theta (PKCa/0).
Por lo tanto, la presente invención proporciona un compuesto de fórmula (II) o una sal farmacéuticamente aceptable
del mismo para su uso en el tratamiento de cánceres, incluyendo, por ejemplo, melanoma, melanoma maligno uveal,
linfoma, linfoma difuso de linfocitos B grandes (DLBCL) y cánceres resistentes a ibrutinib.
En otro aspecto la presente invención proporciona un compuesto de fórmula (II) o una sal farmacéuticamente aceptable
del mismo para su uso en el tratamiento de trastornos relacionados con el sistema inmunológico seleccionados entre
enfermedades autoinmunes, reacciones alérgicas y rechazo de trasplante de tejidos.
Breve descripción de los dibujos
La figura 1 resume que el ejemplo 2 reduce la proliferación en un 92,1 de xenoinjertos de melanoma uveal de una
manera dependiente de la dosis, en comparación con sotrastaurina.
La figura 2 resume que el ejemplo 9 reduce la proliferación en un 92,1 de xenoinjertos de melanoma uveal de una
manera dependiente de la dosis, en comparación con sotrastaurina.
La figura 3 representa la reducción en el volumen tumoral con el paso del tiempo después de la administración del
ejemplo 10 y el ejemplo 9, en comparación con el vehículo.
Descripción detallada
La expresión "alquilo" se refiere a grupos alquilo que no contienen heteroátomos. Así, la expresión incluye grupos
alquilo de cadena lineal tales como metilo, etilo, propilo, butilo, pentilo, hexilo, heptilo, octilo, nonilo, decilo, undecilo,
dodecilo y similares. La expresión también incluye isómeros de cadena ramificada de grupos alquilo de cadena lineal,
que incluyen, pero sin limitación, los siguientes que se proporcionan a modo de ejemplo: -CH(CH3)2, -CH(CH3)(CH2CH3), -CH(CH2CH3)2, -C(CH3)3, -C(CH2CH3)3, -CH2CH(CH3)2, -CH2 CH(CH3)(CH2CH3), -CH2CH(CH2CH3)2, -CH2C(CH3)3, -CH2C(CH2CH3)3, -CH(CH3)-CH(CH3)(CH2CH3), -CH2CH2CH(CH3)2, -CH2CH2CH(CH3)(CH2CH3), -CH2CH2CH(CH2CH3)2, -CH2CH2C(CH3)3, -CH2CH2C(CH2CH3)3, -CH(CH3)CH2-CH(CH3)2,
-CH(CH3)CH(CH3)CH(CH3)2, -CH(CH2CH3)CH(CH3)CH(CH3)(CH2CH3) y otros. La expresión también incluye grupos
alquilo cíclicos tales como ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y ciclooctilo y dichos anillos
sustituidos con grupos alquilo de cadena lineal y ramificada como se han definido anteriormente. Por lo tanto, el término
"grupo alquilo C1-12" incluye grupos alquilo primarios, grupos alquilo secundarios y grupos alquilo terciarios. Los grupos alquilo incluyen grupos alquilo de cadena lineal y ramificada y grupos alquilo cíclicos que tienen de 1 a 12 átomos de carbono.
Tal como se usa en el presente documento, "alquilo C1-6" incluye grupos alquilo de cadena lineal o ramificada tanto sustituidos como sin sustituir que tienen de 1 a 6 átomos de carbono. Los grupos alquilo C1-6 representativos incluyen, por ejemplo, metilo, etilo, propilo, isopropilo, n-butilo, ferc-butilo, neopentilo, trifluorometilo, pentafluoroetilo y similares. Los grupos alquilo C1-6 pueden estar sustituidos, tal como con grupos halo, hidroxi, amino, nitro y/o ciano y similares. Los grupos haloalquilo C1-3 e hidroxialquilo C1-3 incluyen clorometilo, triclorometilo, trifluorometilo, fluorometilo, fluoroetilo, cloroetilo, hidroximetilo, hidroxietilo y similares. Otros restos alquilo C1-3 sustituidos adecuados incluyen, por ejemplo, aralquilo, aminoalquilo, aminoaralquilo, carbonilaminoalquilo, alquilcarbonilaminoalquilo, arilcarbonilaminoalquilo, aralquilcarbonilaminoalquilo, aminoalcoxialquilo y arilaminoalquilo.
Tal como se usa en el presente documento, "alcoxi C1-6" como se usa en el presente documento se refiere al radical RO-, en donde R es alquilo C1-6. Los ejemplos representativos de grupos alcoxi C1-6 incluyen metoxi, etoxi, t-butoxi, trifluorometoxi y similares.
Tal como se usa en el presente documento, el término "halógeno" o "halo" se refiere a grupos cloro, bromo, fluoro y yodo. "Haloalquilo" se refiere a un radical alquilo C1-3 sustituido con uno o más átomos de halógeno. El término "haloalcoxi" se refiere a un radical alcoxi C1-3 sustituido con uno o más átomos de halógeno. Hidroxi se refiere al grupo -OH.
En el presente documento "amino" se refiere al grupo -NH2. En el presente documento la expresión "alquilamino C1-3" se refiere al grupo -NRR' en donde cada uno de R y R' se selecciona independientemente entre hidrógeno o un alquilo C1-3. En el presente documento el término "arilamino" se refiere al grupo -NRR' en donde R es arilo C6-10, incluyendo fenilo, y R' es hidrógeno, un alquilo C1-3 o arilo C6-10, incluyendo fenilo. En el presente documento el término "aralquilamino" se refiere al grupo -NRR' en donde R es un aralquilo y R' es hidrógeno, un alquilo C1-3, un arilo, incluyendo fenilo, o un aralquilo.
El término "alcoxialquilo" se refiere al grupo -alk1-O-alk2 en donde alk i es alquilo C1-3 y alk2 es alquilo C1-3. El término "ariloxialquilo" se refiere al grupo -alquil C1-3-O-arilo, en donde arilo es arilo C6-10, incluyendo fenilo. El término "aralcoxialquilo" se refiere al grupo -alquilenil-O-aralquilo, en donde aralquilo es un aralquilo inferior.
En el presente documento el término "aminocarbonilo" se refiere al grupo -C(O)-NH2. En el presente documento "aminocarbonilo sustituido" se refiere al grupo -CO-NHR- o -C(O)-NRR' en donde R es alquilo C1-3 o arilo C6-10 y R' es hidrógeno, alquilo C1-3 o arilo C6-10. En algunas realizaciones, R y R', junto con el átomo N unido a ellos pueden tomarse juntos parar formar un grupo "heterocicloalquilcarbonilo". El término "carboxiamido" también se refiere al grupo -CONH2. En el presente documento la expresión "carboxiamida sustituida" se refiere al grupo -CO-NHR- o -CO-NRR' en donde R es alquilo C1-3, arilo C6-10 o y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3 y R' es hidrógeno, alquilo C1-3, arilo C6-10 o y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3. En el presente documento el término "arilaminocarbonilo" se refiere al grupo -C(O)-NRR' en donde R es un arilo y R' es hidrógeno, alquilo C1-3 o arilo. En el presente documento el término "aralquilaminocarbonilo" se refiere al grupo -C(O)-NRR' en donde R es aralquilo y R' es hidrógeno, alquilo C1-3, arilo, fenilo o aralquilo.
En el presente documento el término "aminosulfonilo" se refiere al grupo -SO2-NH2. En el presente documento "aminosulfonilo sustituido" se refiere al grupo -SO2-NHR- o SO2-NRR' en donde R es alquilo C1-3 o arilo C6-10 y R' es hidrógeno o un alquilo C1-3 o arilo C6-10. El término "sulfonamido" se refiere al grupo -SONH2. En el presente documento el término "sulfonamida sustituida" se refiere al grupo -SO-NHR- o -SO-NRR' en donde R es alquilo C1-3, arilo C6-10 o y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3 y R' es hidrógeno o un alquilo C1-3, arilo C6-10 o y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3. En el presente documento el término "aralquilaminosulfonilarilo" se refiere al grupo -aril-S(O)2-NH-aralquilo.
El término "carbonilo" se refiere al grupo divalente -C(O)-. "Carboxi" se refiere a -C(=O)-OH. "Alcoxicarbonilo" se refiere a éster -C(=O)-OR en donde R es alquilo C1-3. "Cicloalquiloxicarbonilo" se refiere a -C(=O)-OR en donde R es cicloalquilo. El término "ariloxicarbonilo" se refiere a -C(=O)-OR en donde R es arilo. El término "heterocicliloxicarbonilo" se refiere a -C(=O)-OR en donde R es heterociclilo.
En el presente documento el término "aralcoxicarbonilo" se refiere al grupo -(C=O)-O-aralquilo, en donde el aralquilo es aralquilo C1-3.
En el presente documento el término "sulfonilo" se refiere al grupo -SO2-. En el presente documento el término "sulfanilo" se refiere al grupo -S-. "Alquilsulfonilo" se refiere a un sulfonilo sustituido de estructura -SO2R- en el que R es alquilo C1-3. "Alquilsulfanilo" se refiere a un sulfanilo sustituido de estructura -SR- en el que R es alquilo C1-3. Por lo tanto, los grupos alquilsulfonilo y alquilsulfanilo inferior habituales empleados en compuestos de la presente invención incluyen, por ejemplo, metilsulfonilo y metilsulfanilo (es decir, en donde R es metilo), etilsulfonilo y etilsulfanilo (es decir, en donde R es etilo), propilsulfonilo y propilsulfanilo (es decir, en donde R es propilo) y similares. En el presente documento el término "arilsulfonilo" se refiere al grupo -SO2-arilo. En el presente documento el término "aralquilsulfonilo" se refiere al grupo -SO2-aralquilo, en el que el aralquilo es aralquilo C1-3. En el presente documento el término "sulfonamido" se refiere a -SO2NH2.
Como alternativa, el término "amido" se refiere a -C(=O)NH2 y "carbonilamino" se refiere al grupo divalente -NH-(C=O)-en el que el átomo de hidrógeno del nitrógeno de la amida del grupo carbonilamino puede estar sustituido con un alquilo C1-3, arilo C6-10, aralquilo o y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con uno o dos sustituyentes cada uno independientemente seleccionado entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3. Dichos grupos incluyen restos tales como ésteres de carbamato (-NH-C(O)-O-R) y amidas -NH-C(O)-R, en donde R es un alquilo C1-3, cicloalquilo C3-8 o arilo C6-10 de cadena lineal o ramificada, incluyendo fenilo, aralquilo o y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3.
El término "cicloalquilo C3-8" se refiere a un sustituyente alquilo C3-8 mono o policíclico, heterocíclico o carbocíclico. Los sustituyentes cicloalquilo habituales tienen de 3 a 8 átomos de cadena principal (es decir, anillo) en los que cada átomo de cadena principal es un carbono o un heteroátomo. En el presente documento el término "heterocicloalquilo" se refiere a sustituyentes cicloalquilo que tienen de 1 a 5 y más habitualmente de 1 a 4 heteroátomos en la estructura del anillo. Los heteroátomos adecuados empleados en los compuestos de la presente invención son nitrógeno, oxígeno y azufre. Los restos heterocicloalquilo representativos incluyen, por ejemplo, morfolino, piperazinilo, piperidinilo y similares. Los grupos carbocicloalquilo son grupos cicloalquilo en los que todos los átomos del anillo son carbono. Cuando se usa junto con sustituyentes cicloalquilo, en el presente documento el término "policíclico" se refiere a estructuras cíclicas de alquilo condensadas y no condensadas. El término "carbobicíclico o carbobiciclilo" se refiere a un anillo carbocíclico saturado o parcialmente insaturado condensado a otro anillo carbocíclico, anillo arilo, anillo heterocíclico o anillo heteroarilo. El grupo cicloalquilo está sustituido o sin sustituir.
El término "heterociclo sustituido" o "grupo heterocíclico" o "heterociclilo", tal como se usa en el presente documento, se refiere a cualquier anillo de 3 o 4 miembros que contiene un heteroátomo seleccionado entre nitrógeno, oxígeno o azufre o un anillo de 5, 6 o 7 miembros que contiene de uno a tres heteroátomos seleccionados entre el grupo que consiste en nitrógeno, oxígeno o azufre; en donde el anillo de 5 miembros tiene 0-1 dobles enlaces y los anillos de 6 y 7 miembros tienen 0-1 dobles enlaces o anillos condensados que tienen 0-2 dobles enlaces; en donde el átomo de nitrógeno y azufre opcionalmente puede oxidarse; en donde los heteroátomos de nitrógeno y azufre opcionalmente pueden cuaternizarse e incluye cualquier grupo bicíclico en el que cualquiera de los anillos heterocíclicos anteriores está condensado a un anillo de benceno o a otro anillo heterocíclico de 5 o 6 miembros definido de manera independiente anteriormente y se denomina anillo heterobicíclico o grupo heterobicíclico. El grupo heterociclilo está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3.
Por lo tanto, el término "heterociclo" incluye anillos en los que el nitrógeno es el heteroátomo así como anillos parcial y completamente saturados. Los heterociclos ejemplares incluyen, pero sin limitación, por ejemplo: piperidinilo, piperazinilo, 1,2-oxazinano, 2-oxopiperazinilo, 2-oxopiperidinilo, N-metil piperazinilo y morfolinilo, cada uno opcionalmente sustituido.
Los restos heterocíclicos pueden estar sin sustituir o monosustituidos o disustituidos con diversos sustituyentes seleccionados de manera independiente entre hidroxi, halo, oxo (C=O), alquilimino (RN=, en donde R es un grupo alquilo C1-3 o alcoxi C1-3), amino, alquilamino C1-3, dialquilamino C1-3, acilaminoalquilo, alcoxi C1-3, alquilo C1-3, cicloalquilo o haloalquilo C1-3.
Los grupos heterocíclicos (heterociclilo) pueden estar unidos en diversas posiciones como será evidente para los expertos en las técnicas de la química orgánica y medicinal junto con la divulgación en el presente documento. Los ejemplos representativos de los grupos heterociclilo, heterobiciclilo y heterociclilo sustituido usados de acuerdo con la invención se enumeran a continuación:

El término "arilo C6-10" se refiere a grupos aromáticos monocíclicos y poM cíclicos opcionalmente sustituidos que tienen de 6 a 10 o de 3 a 14 carbonos o heteroátomos de cadena principal y que incluyen tanto grupos arilo carbocíclicos como grupos arilo heterocíclicos. Los grupos arilo carbocíclicos son grupos arilo C6-10 en los que todos los átomos del anillo en el anillo aromático son carbono. Los restos de arilo C6-10 ejemplares empleados como sustituyentes en los compuestos de la presente invención incluyen fenilo, naftilo, isonaftilo y similares.
"Aralquilo" se refiere a un grupo alquilo C1-3 o alquilo C1-6 sustituido con un grupo arilo C6-10. Típicamente, los grupos aralquilo empleados en los compuestos de la presente invención tienen de 1 a 6 átomos de carbono incorporados dentro de la porción alquilo del grupo aralquilo. Los grupos aralquilo adecuados empleados en los compuestos de la presente invención incluyen, por ejemplo, bencilo, picolilo y similares.
El término "heteroarilo" se refiere a sistema de anillos carbocíclico de 5-10 miembros, incluyendo sistemas de anillos condensados, que tienen de 1 a 4 heteroátomos seleccionados cada uno de ellos independientemente entre el grupo que consiste en: O, N y S. Dicho heteroarilo puede estar opcionalmente sustituido con uno o dos sustituyentes. En el presente documento el término "heteroarilo" también se refiere a grupos arilo C6-10 que tienen de 1 a 4 heteroátomos como átomos del anillo en un anillo aromático siendo el resto de los átomos del anillo átomos de carbono. Los sustituyentes de ejemplo incluyen, pero sin limitación: halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3, haloalcoxi C1-3, cicloalquilo C3-7 y heterociclilo de 4-7 miembros que tiene 1 o 2 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3. Los grupos heteroarilo representativos incluyen, por ejemplo, los que se muestran a continuación. Los heteroarilos representativos incluyen, por ejemplo, imidazolilo, piridinilo (también denominado aspiridilo), pirazinilo, azetidinilo, tiazolilo, triazolilo, benzoimidazolilo, benzotiazolilo, tiazolilo, tiazolidinilo, isotiazolilo, isotiazolidinilo, indolilo, quinolinilo, isoquinolinilo, azetidinilo, N-metilazetidinilo, pirimidinilo, piridazinilo, oxazolilo, oxazolidinilo, isoxazolilo, isoazolidinilo, benzoimidazolilo, benzotiazolilo, benzoxazolilo, furilo, tienilo, triazolilo, benzotienilo diazapinilo, pirrilo, pirrolinilo, pirrolidinilo, pirazolilo, pirazolinilo, pirazolidinilo, imidazoilo, imidazolinilo, imidazolidinilo y benzoxazolilo. El heteroarilo está sin sustituir o sustituido con de 1 a 3 sustituyentes seleccionado cada uno de manera independiente entre el grupo que consiste en: H, 2H, halo, alquinilo C2-3, alquenilo C2-3, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3, haloalcoxi C1-3, cicloalquilo C3-7, CONH2, CONHalquilo C1-3, CONHarilo C6-10, SO2NH2, SO2NHalquilo C1-3, SO2NHarilo C6-10 y heterociclilo de 4-7 miembros que tienen de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con uno o dos sustituyentes cada uno independientemente seleccionado entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3.
Los grupos heteroarilo pueden estar sustituidos adicionalmente y pueden estar unidos en diversas posiciones como será evidente para los expertos en las técnicas de la química orgánica y medicinal junto con la divulgación en el
presente documento. Los ejemplos representativos de grupos heteroarilo y heteroarilo sustituido usados de acuerdo con la invención se enumeran a continuación:



y
"Opcionalmente sustituido" o "sustituido" se refiere a la sustitución de uno o más átomos de hidrógeno con un radical monovalente o divalente. Los grupos de sustitución adecuados incluyen, por ejemplo, H, 2H, halo, alquinilo C2-3, alquenilo C2-3, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3, haloalcoxi C1-3, cicloalquilo C3-7, CONH2, CONHalquilo C1-3, CONHarilo C6-10, SO2NH2, SO2NHalquilo C1-3, SO2NHarilo C6-10 y heterociclilo de 4-7 miembros que tienen de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho heterociclilo opcionalmente sustituido con uno o dos sustituyentes cada uno independientemente seleccionado entre el grupo que consiste en: H, 2H, halo, CN, alquilo C1-3, alcoxi C1-3, haloalquilo C1-3 y haloalcoxi C1-3; y similares.
El grupo de sustitución puede a su vez estar sustituido. El grupo sustituido en el grupo de sustitución puede ser carboxilo, halo; nitro, amino, ciano, hidroxi, alquilo C1-3, alcoxi C1-3, aminocarbonilo, -SR, tioamido, -SO3H, -SO2R o cicloalquilo C3-8, en donde R es habitualmente hidrógeno, hidroxilo o alquilo C1-3.
Cuando el sustituyente sustituido incluye un grupo de cadena lineal, la sustitución puede tener lugar tanto en la cadena (por ejemplo, 2-hidroxipropilo, 2-aminobutilo y similares) o en el extremo de la cadena (por ejemplo, 2-hidroxietilo, 3-cianopropilo y similares). Los sustituyentes sustituidos pueden ser disposiciones de cadena lineal, ramificada o cíclicas de carbonos o heteroátomos unidos covalentemente.
El término "2H" se refiere a un isótopo pesado de hidrógeno que también se denomina deuterio (D). Se entiende que las definiciones anteriores no pretenden incluir patrones de sustitución no permitidos (por ejemplo, metilo sustituido con cinco grupos flúor o un átomo de halógeno sustituido con otro átomo de halógeno). Dichos patrones de sustitución no permitidos son bien conocidos por el experto en la técnica.
Los compuestos de la invención, o sus tautómeros, así como las sales farmacéuticamente aceptables, pueden comprender átomos de carbono sustituidos de manera asimétrica. Dichos átomos de carbono sustituidos de manera asimétrica pueden dar como resultado que los compuestos de la invención existan como enantiómeros, diastereómeros y otras formas estereoisoméricas que pueden definirse, en términos de estereoquímica absoluta, tales como en formas (R) o (S). Como resultado, todos esos posibles isómeros, estereoisómeros individuales en sus formas ópticamente puras, mezclas de los mismos, mezclas racémicas (o "racematos"), mezclas de diastereómeros, así como diastereómeros puros de los compuestos de la invención están incluidos dentro de la presente invención. Los términos
configuración "S" y "R", tal como se usa en el presente documento, son como se definen por la IUPAC 1974 RECOMMENDATIONS FOR SECTION E, FUNDAMENTAL STEREOCHEMISTRY, Pure Appl. Chem. 45:13-30 (1976). Los términos a y p se emplean para las posiciones en el anillo de los compuestos cíclicos. El lado a del plano de referencia es el lado en el que se encuentra el sustituyente preferido en la posición numérica más baja. A los sustituyentes que se encuentran en el lado opuesto del plano de referencia se les asigna un descriptor p. Cabe destacar que este uso difiere del de los estereoparentes cíclicos, en los que "a" significa "por debajo del plano" y denota la configuración absoluta. Los términos configuración a y p, tal como se usa en el presente documento, como se define por el CHEMICAL ABSTRACTS INDEX GUIDE-APPENDIX IV (1987) párrafo 203.
En otra realización, un compuesto o un estereoisómero, tautómero o una sal farmacéuticamente aceptable del mismos se selecciona entre: 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3- amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4- metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3- amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-ciclopropil-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-cloropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-((1R,5S,8s)-8-amino-3-azabiciclo[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metoxipiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-((1S,5R,8S)-8-amino-6-oxa-3-azabiciclo[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(5,6,7,8-tetrahidroquinazolin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(7H-pirrolo[2,3-d]pirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-(dimetilamino)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-((1R,5S,8s)-8-amino-3-azabiciclo[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-(2-metoxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4- metilpiperidin-1-il)piridin-2-il)-6-(6-fluoroquinazolin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(5-fluoro-7H-pirrolo[2,3-d]pirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3,6-bis(trifluorometil)piridin-2-il)pirazin-2-carboxamida, (±) 3-amino-N-(3-((cis)-4-amino-3-fluoropiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1- il)piridin-2-il)-6-(2-morfolinopirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2- il)-6-(2-(3,6-dihidro-2H-piran-4-il)-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-morfolinoquinazolin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(7-fluoroisoquinolin-1-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-morfolinopiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(7-cloroisoquinolin-1-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-(azetidin-1-il)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-((3S,4R)-4-amino-3-fluoropiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-(dimetilamino)-3- (trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-(4,4-difluoropiperidin-1-il)-5-fluoropirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(5-fluoro-2-morfolinopirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-morfolinoquinazolin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 4-(5-amino-6-((3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)carbamoil)pirazin-2-il)-5-fluoropirimidin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-amino-5-cloropirimidin-4-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3morfolinoisoquinolin-1-il)pirazin-2-carboxamida, 3-ammo-N-(3-(4-ammo-4-iTietNpiperidin-1-N)piridin-2-N)-6-(4-doro-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidm-1-N)piridm-2-N)-6-(6-iTiorfoNn-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida, 3-ammo-N-(3-(4-ammopiperidin-1-N)-6-iTietNpindin-2-N)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-3-iTietoxipiperidin-1-N)pindin-2-N)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-iTietNpiperidin-1-N)pmdm-2-N)-6-(3-fluoro-4-metilpiridin-2-il)pirazin-2-carboxamida, 3-ammo-N-(3-(4-amino-4-iTietNpipendin-1-N)piridin-2-N)-6-(4-etoxi-3-fluoropiridin-2-il)pirazin-2-carboxamida, 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(hidroximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida y 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(metoximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida.
En otra realización, un compuesto o un estereoisómero, tautómero o sal farmacéuticamente aceptable del mismo, se selecciona entre:





La invención proporciona compuestos, sus tautómeros, estereoisómeros o sales farmacéuticamente aceptables del mismo o ésteres que tienen restos potenciadores de la solubilidad de la fórmula (II), tal como se ha definido anteriormente:

En una realización separada, se proporcionan compuestos, sus tautómeros, estereoisómeros o sales
farmacéuticamente aceptables de los mismos de la fórmula (II),
en donde:
X es CR;
cada uno de R2, R3 y R4 es H;
R5 es independientemente H, CH3, CH2F, CHF2, CF3, CH2OH, CH2-O-alquilo C1-3
cada uno de R5a y R5b es H o R5a y R5b se unen para formar un grupo puente de metileno o etileno;
cada uno de R5c y R5d es independientemente H, F, alquilo C1-3 o alcoxi C1-3 o R5c y R5d se unen para formar un grupo puente de metileno, etileno o -CH2-O-; y
cada uno de R6 y R7 se selecciona independientemente entre H, halo, haloalquilo C1-3, haloalcoxi C1-3, cicloalquilo C3-7, morfolino, piperinilo y piperazinilo.
En otra realización, un compuesto o una sal farmacéuticamente aceptable del mismo se selecciona entre:
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-cloropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metoxipiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(2-metoxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
(±)3-amino-N-(3-((cis)-4-amino-3-fluoropiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-((3S,4R)-4-amino-3-fluoropiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-cloro-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-3-metoxipiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metilpiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(hidroximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida; y
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(metoximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida.
En otra realización, un compuesto o una sal farmacéuticamente aceptable del mismo se selecciona entre:
3-amino-N-(3-(4-amino-4-(etoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(etoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-((difluorometoxi)metil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(5-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-3-fluoro-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-3-fluoro-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-((3S,4R)-4-amino-3-fluoro-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; y
3-amino-N-(3-(4-amino-3-fluoro-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida.
En otra realización, un compuesto o una sal farmacéuticamente aceptable del mismo se selecciona entre:
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3 morfolinoisoquinolin-1-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(5,6,7,8-tetrahidroquinazolin-4-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(7H-pirrolo[2,3-d]pirimidin-4-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-fluoroquinazolin-4-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(5-fluoro-7H-pirrolo[2,3-d]pirimidin-4-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-morfolinoquinazolin-4-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(7-fluoroisoquinolin-1-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(7-cloroisoquinolin-1-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-morfolinoquinazolin-4-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida; y
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida.
En otra realización un compuesto o una sal farmacéuticamente aceptable del mismo se selecciona entre:
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida; y 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida.
Tal como se usa en el presente documento, el término "sales farmacéuticamente aceptables" se refiere las sales de ácidos o metales alcalinotérreos no tóxicas de los compuestos de la invención. Estas sales se pueden preparar in situ durante el aislamiento final y la purificación de los compuestos de la invención o por separado haciendo reaccionar las funciones de base o ácido con un ácido o una base orgánica o inorgánica adecuada, respectivamente. Las sales representativas incluyen, pero sin limitación, las siguientes: acetato, adipato, alginato, citrato, aspartato, benzoato, bencenosulfonato, bisulfato, butirato, alcanforato, alcanforsulfonato, digluconato, ciclopentanopropionato, dodecilsulfato, etanosulfonato, glucoheptanoato, glicerofosfato, hemisulfato, heptanoato, hexanoato, fumarato, clorhidrato, bromhidrato, yodhidrato, 2-hidroxietanosulfonato, lactato, maleato, metanosulfonato, nicotinato, 2-naftalenosulfonato, oxalato, pamoato, pectinato, persulfato, 3-fenilproionato, picrato, pivalato, propionato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato y undecanoato. Además, los grupos que contienen nitrógeno básico pueden cuaternizarse con agentes tales como haluros de alquilo inferior, tales como cloruro, bromuros y yoduros de metilo, etilo, propilo y butilo; dialquil sulfatos como dimetil, dietil, dibutil y diamil sulfatos, haluros de cadena larga tales como cloruros, bromuros y yoduros de decilo, laurilo, miristilo y estearilo, haluros de aralquilo como bromuros de bencilo y fenetilo y otros. De este modo se obtienen productos solubles o dispersables en agua o aceite.
Los ejemplos de ácidos que se pueden emplear para formar sales de adición de ácido farmacéuticamente aceptables incluyen ácidos inorgánicos tales como ácido clorhídrico, ácido sulfúrico y ácido fosfórico y ácidos orgánicos tales como ácido oxálico, ácido maleico, ácido metanosulfónico, ácido succínico y ácido cítrico. Pueden prepararse sales de adición básicas in situ durante el aislamiento y purificación final de los compuestos de fórmula (I) o por separado haciendo reaccionar restos de ácido carboxílico con una base adecuada tal como el hidróxido, el carbonato o el bicarbonato de un catión metálico farmacéuticamente aceptable o con amoniaco o una amina orgánica primaria, secundaria o terciaria. Las sales farmacéuticamente aceptables incluyen, pero no se limitan a, cationes basados en los metales alcalinos y alcalinotérreos, tales como sales de sodio, litio, potasio, calcio, magnesio, aluminio y similares, así como cationes no tóxicos de amonio, amonio cuaternario y amina, incluyendo, pero sin limitación amonio, tetrametilamonio, tetraetilamonio, metilamina, dimetilamina, trimetilamina, trietilamina, etilamina y similares. Otras aminas orgánicas representativas útiles para la formación de sales de adición de base incluyen dietilamina, etilendiamina, etanolamina, dietanolamina, piperazina y similares.
Tal como se usa en el presente documento, la expresión "éster farmacéuticamente aceptable" se refiere a ésteres, que se hidrolizan in vivo e incluyen aquellos que se rompen con facilidad en el cuerpo humano para dejar el compuesto precursor o una sal del mismo. Los grupos éster adecuados incluyen, por ejemplo, los obtenidos a partir de ácidos carboxílicos alifáticos farmacéuticamente aceptables, en particular ácidos alcanoicos, alquenoicos, cicloalcanoicos y alcanodioicos, en los que cada resto alquilo o alquenilo ventajosamente no tiene más de 6 átomos de carbono. Ejemplos de ésteres particulares incluyen formiatos, acetatos, propionatos, butiratos, acrilatos y etilsuccinatos.
El término "profármacos farmacéuticamente aceptables", tal como se usa en el presente documento, se refiere a aquellos profármacos que son, dentro del alcance del buen criterio médico, adecuados para su uso en contacto con los tejidos de seres humanos y animales inferiores sin excesiva toxicidad, irritación, respuesta alérgica y similares, de acuerdo con una relación beneficio/riesgo razonable y eficaces para su uso previsto, así como las formas zwiteriónicas, cuando sea posible, de los compuestos de la invención. El término "profármaco" se refiere a compuestos que se transforman rápidamente in vivo para producir el compuesto precursor de la fórmula anterior, por ejemplo mediante hidrólisis en sangre. Se proporciona un análisis minucioso en T. Higuchi y V. Stella, Pro-drugs as Novel Delivery Systems, vol. 14 de la A.C.S. Symposium Series y en Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association y Pergamon Press, 1987.
Será evidente para los expertos en la técnica que los compuestos de la invención o sus tautómeros, profármacos y estereoisómeros, así como las sales farmacéuticamente aceptables, ésteres y profármacos de cualquiera de ellos, pueden procesarse in vivo a través de metabolismo en un cuerpo o célula humano o animal para producir metabolitos. El término "metabolito", tal como se usa en el presente documento, se refiere a la fórmula de cualquier derivado producido en un sujeto tras la administración de un compuesto precursor. Los derivados pueden producirse a partir del compuesto precursor mediante diversas transformaciones bioquímicas en el sujeto, tales como, por ejemplo, oxidación, reducción, hidrólisis o conjugación e incluyen, por ejemplo, óxidos y derivados desmetilados. Los metabolitos se pueden identificar usando técnicas rutinarias conocidas en la materia. Véase, por ejemplo, Bertolini, G. et al., J. Med. Chem. 40:2011-2016 (1997); Shan, D. et al., J. Pharm. Sci. 86(7):765-767; Bagshawe K., Drug Dev. Res. 34:220-230 (1995); Bodor, N., Advances in Drug Res. 13:224-331 (1984); Bundgaard, H., Design of Prodrugs (Elsevier Press 1985); y Larsen, I. K., Design and Application of Prodrugs, Drug Design and Development (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991).
Los compuestos de la invención son útiles en el tratamiento de trastornos relacionados con la proteína cinasa C en un sujeto humano o animal con necesidad reconocida de dicho tratamiento. El tratamiento puede comprender administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para inhibir la actividad de p Kc en el sujeto. Los inhibidores de PKC útiles en la práctica de la presente invención pueden inhibir varias isoformas de la PKC, en particular, pueden inhibir selectivamente isoformas específicas de PKC (por ejemplo, inhibidores selectivos de PKC o inhibidores de PKC selectivos de isozima). Los inhibidores de PKC son capaces de inhibir de manera selectiva a isoformas de PKC que se seleccionan entre las isoformas clásicas de PKC (a, p1, p2 , y) e isoformas novedosas de PKC (8, £, n, 0) o isoformas atípicos (Z, i), más preferentemente, seleccionadas entre las isoformas de PKC a, p (isoformas p1 y p2) y 0. Los inhibidores de PKC preferidos son capaces de inhibir selectivamente a las isoformas de PKC a y 0. Los inhibidores de PKC adecuados incluyen derivados de maleimida, tales como los compuestos descritos en las Patentes de los Estados Unidos n.° 5.545.636; 5.668.152; 5.672.681; 5.698.578; 5.710.145; 6.645.970; 7.220.774; 7.235.555; la Publicación de los Estados Unidos n.° 2008/0318975; las Patentes Europeas n.° 0776895 B1; 0817627 B1; 1449529 B1; 1337527 B1; y las publicaciones PCT n.° WO03/082859; y W O07/006.533. Como se usa en el presente documento, la expresión "inhibidor de PKC" se refiere a un inhibidor de proteína cinasa C que puede ser general (de múltiples subtipos) o selectivo para una o más isozimas PKC. El término PKC se refiere generalmente a la familia completa de isoformas: isoformas convencionales; alfa, beta y gamma, nuevas isoformas; delta, épsilon, eta y teta e isoformas atípicas; zeta y iota. La expresión "inhibidor selectivo de PKC" se refiere a un inhibidor de PKC que posee una selectividad de al menos aproximadamente 20 veces por una o más isoformas de PKC en comparación con las otras isoformas de PKC. Preferentemente, esta selectividad es de al menos aproximadamente 100 veces, más preferentemente, de al menos aproximadamente 500 veces, lo más preferentemente, de al menos aproximadamente 1.000 o al menos aproximadamente 2.000 veces. La expresión "inhibidor selectivo de PKC alfa/teta", "inhibidor selectivo de PKC a/0" se refiere a un inhibidor de proteína cinasa C que es más selectivo por la isoforma PKC alfa y/o teta de PKC que las otras isoformas divulgadas de PKC. Por ejemplo, PKC alfa o PKC alfa y teta, frente a las otras isoformas de PKC nombradas de al menos aproximadamente 20 veces (preferentemente al menos aproximadamente 100, más preferentemente, al menos aproximadamente 500, lo más preferentemente, de al menos aproximadamente 1.000 o al menos aproximadamente 2.000 veces).
La regulación diferencial de G S K 3p mediante isotipos de proteína cinasa C se describió por Goode et al. en la publicación, J. Biol. Chem., Vol. 267, págs. 16878-16882 (1992). Más recientemente, se ha descrito que la regulación dual de G S K 3a/p por un isotipo alfa de proteína cinasa C promueve la activación de trombina mediada por integrina anb/p3 y la secreción de gránulos en plaquetas por Moore, et al en la publicación J. Biol. Chem., Vol. 288, págs. 3918 3928 (2013).
Los compuestos de la invención son útiles en el tratamiento de trastornos relacionados con proteína cinasa, específicamente, trastornos relacionados con proteína cinasa C alfa, teta (PKCa/0) en un sujeto humano o animal con necesidad reconocida de dicho tratamiento. El tratamiento puede comprender administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para tratar el cáncer o el crecimiento tumoral asociado con la PKCa /0 en el sujeto.
En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar trastornos relacionados con la inmunidad, incluyendo, pero sin limitación, enfermedades autoinmunitarias, reacciones alérgicas y rechazo de trasplante de tejidos, en un sujeto humano o animal con necesidad reconocida de
dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto. En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar tumores sólidos malignos en un sujeto humano o animal con necesidad reconocida de dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto. Además de una función potencial en el tratamiento contra el cáncer y enfermedades mieloproliferativas, los compuestos de la invención podrían ser útiles para controlar la expansión de células inmunitarias en otras patologías, tales como enfermedades autoinmunitarias, reacciones alérgicas y síndromes de rechazo de trasplante de órganos. Se proporcionan pruebas que respaldan que el inhibidor selectivo de PKC inventado de la presente invención podría ser eficaz en el tratamiento de trastornos relacionados con la inmunidad por la reciente divulgación de que la sotrastaurina representa una nueva clase de agente inmunosupresor que afecta a la activación temprana de linfocitos T (Evenou et al., "The Journal of Pharmacology and Experimental Therapeutics", Vol. 330 págs. 792-801,2009).
En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar el cáncer, tumores en un sujeto humano o animal con necesidad reconocida de dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto. En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar tumores sólidos malignos en un sujeto humano o animal con necesidad reconocida de dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto.
En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar el melanoma uveal, incluyendo melanoma uveal que porta las mutaciones GNAQ o GNA11 en un sujeto humano o animal con necesidad reconocida de dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto.
En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar el linfoma, incluyendo el linfoma difuso de linfocitos B grandes (DLBCL), en un sujeto humano o animal con necesidad reconocida de dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto.
En otros aspectos, la presente invención proporciona un compuesto de la invención para su uso en un método para tratar cánceres resistentes a ibrutinib en un sujeto humano o animal con necesidad reconocida de dicho tratamiento, que comprende administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral en el sujeto. La PKC se encuentra inmediatamente aguas debajo de la tirosina cinasa de Bruton con respecto a los linfomas de linfocitos B y los cánceres hematológicos y aporta pruebas de que los inhibidores de PKC inventados podrían ser eficaces para tratar cánceres y enfermedades resistentes a ibrutinib. Woyach, et al han descrito e identificado ciertas mutaciones específicas que pueden mediar la resistencia al ibrutinib en la publicación, J. New England Medicine, DOI: 10.1056/NEJMoa1400029, 2014.
Los compuestos de la invención son útiles en el tratamiento de trastornos relacionados con proteína cinasa, específicamente, trastornos relacionados con proteína cinasa C (PKCa/0) en un sujeto humano o animal con necesidad reconocida de dicho tratamiento. El tratamiento puede comprender administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento tumoral asociado con el trastorno relacionado con PKCa/0 en el sujeto. La expresión "una cantidad terapéuticamente eficaz" de un inhibidor de PKC se refiere a una cantidad del inhibidor de PKC que provocará una respuesta biológica o médica en un sujeto, por ejemplo, reducción o inhibición de una enzima o la actividad de una proteína y/o mejorar los síntomas, aliviar afecciones, frenar o retrasar la progresión de la enfermedad o prevenir una enfermedad, etc. En una realización no limitante, la expresión "una cantidad terapéuticamente eficaz" se refiere a la cantidad de inhibidor de PKC, que cuando se administra a un sujeto, es eficaz para, al menos parcialmente, (1) aliviar, inhibir, prevenir y/o mejorar una afección o un trastorno o una enfermedad mediado por o asociado con la actividad de PKC, tal como, por ejemplo, linfoma de linfocitos B que tiene señalización activa crónica del receptor de linfocitos B (por ejemplo, linfoma difuso de linfocitos B grandes mutante para CD79) o melanoma uveal que porta las mutaciones GNAQ o GNA11; y/o es eficaz para, al menos parcialmente (2) reducir el tamaño (volumen tumoral) o inhibir el crecimiento adicional de tumores (sólidos o líquidos). En otra realización no limitante, la expresión "una cantidad terapéuticamente eficaz" se refiere a la cantidad del compuesto de la presente invención que, cuando se administra a un sujeto, célula o tejido o a un material biológico no celular o a un medio, es eficaz para, al menos parcialmente, reducir o inhibir el crecimiento de un linfoma de linfocitos B que tiene una señalización activa crónica del receptor de linfocitos B (preferentemente, un linfoma difuso de linfocitos B grandes mutante para CD79) o melanoma uveal que porta las mutaciones GNAQ o GNA11.
Como se usa en el presente documento, el término "sujeto" se refiere a un animal. Normalmente, el animal es un mamífero. Un sujeto también se refiere a, por ejemplo, primates (por ejemplo, seres humanos, de sexo masculino o femenino), vacas, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, peces, aves y similares. En ciertas realizaciones, el sujeto es un primate. En otras realizaciones más, el sujeto es un ser humano. Como se usa en el presente documento, el término "inhibir", "inhibición" o "inhibe" se refiere a la reducción o supresión de una afección, síntoma o trastorno o enfermedad o a una disminución significativa en la actividad inicial de una actividad o proceso
biológico. Como se usa en el presente documento, el término "tratar", "tratamiento" o "tratar" de cualquier enfermedad o trastorno, se refiere (i) a mejorar la enfermedad o trastorno (es decir, ralentizar o detener o reducir el desarrollo de la enfermedad o al menos uno de los síntomas clínicos de la misma; (ii) aliviar o mejorar al menos un parámetro físico, incluyendo aquellos que pueden no ser discernibles por el paciente; (iii) modular la enfermedad o el trastorno, ya sea físicamente, (por ejemplo, estabilización de un síntoma discernible), fisiológicamente, (por ejemplo, estabilización de un parámetro físico) o ambos; o (iv) prevenir o retrasar el inicio o el desarrollo o la progresión de la enfermedad o trastorno. En general, el término "tratar" o "tratamiento" describe la gestión y cuidado de un paciente con el fin de combatir la enfermedad, la afección o el trastorno, e incluye la administración de un inhibidor de PKC para prevenir la aparición de los síntomas o complicaciones, aliviar los síntomas o complicaciones o eliminar la enfermedad, afección o trastorno.
Los compuestos de la invención son útiles en el tratamiento de trastornos relacionados con PKC, incluyendo los cánceres divulgados en el presente documento, en un sujeto humano o animal que necesite dicho tratamiento. El tratamiento puede comprender administrar a dicho sujeto una cantidad de un compuesto de la invención eficaz para reducir o prevenir el crecimiento del tumor en el sujeto, en combinación con al menos un agente adicional para el tratamiento del cáncer. En la presente invención se contempla una serie de agentes anticáncer adecuados para su uso como agentes terapéuticos de combinación. De hecho, la presente invención contempla, pero sin limitación, la administración de numerosos agentes anticáncer, tales como: agentes que inducen la apoptosis; polinucleótidos (por ejemplo, ribozimas); polipéptidos (por ejemplo, enzimas); fármacos; miméticos biológicos; alcaloides; agentes alquilantes; antibióticos antitumorales; antimetabolitos; hormonas; compuestos de platino; anticuerpos monoclonales conjugados con fármacos anticancerosos, toxinas y/o radionúclidos; modificadores de la respuesta biológica (por ejemplo, interferones [por ejemplo, IFN-a, etc.] e interleucinas [por ejemplo, IL-2, etc.], etc.); agentes de inmunoterapia adoptiva; factores de crecimiento hematopoyéticos; agentes que inducen la diferenciación de células tumorales (por ejemplo, ácido todo trans retinoico, etc.); reactivos de terapia génica; reactivos y nucleótidos de terapia antisentido; vacunas tumorales; inhibidores de la angiogénesis y similares. Los expertos en la materia conocen numerosos ejemplos distintos de compuestos quimioterapéuticos y terapias anticáncer adecuadas para su coadministración con los compuestos divulgados de la invención.
En realizaciones preferidas, los agentes anticáncer para su uso en combinación con los compuestos de la presente invención comprenden agentes que inducen o estimulan la apoptosis. Los agentes que inducen la apoptosis incluyen, pero sin limitación, radiación (por ejemplo, W); inhibidores de cinasa (por ejemplo, inhibidor de cinasa de receptor de factor de crecimiento epidérmico [EGFR], inhibidor de cinasa de receptor de factor de crecimiento vascular [VGFR], inhibidor de cinasa de receptor de factor de crecimiento de fibroblastos [FGFR], inhibidor de cinasa I de receptor de factor de crecimiento derivado de plaquetas [PGFR] e inhibidores de Bcr-Abl cinasa, tales como STI-571, Gleevec y Glivec]); moléculas antisentido; anticuerpos [por ejemplo, Herceptin y Rituxan]; anti-estrógenos [por ejemplo, raloxifeno y tamoxifeno]; anti-andrógenos [por ejemplo, flutamida, bicalutamida, finasterida, aminoglutetamida, ketoconazol y corticosteroides]; inhibidores de ciclooxigenasa 2 (COX-2) [por ejemplo, Celecoxib, meloxicam, NS-398 y fármacos antiinflamatorios no esteroideos (AINES)]; y fármacos quimioterapéuticos para el cáncer [por ejemplo, irinotecán (Camptosar), CPT-11, fludarabina (Fludara), dacarbazina (DTIC), dexametasona, mitoxantrona, Mylotarg, VP-16, cisplatino, 5-FU, doxorrubicina, taxotere o taxol]; moléculas de señalización celular; ceramidas y citocinas; y estaurosporina y similares.
En otros aspectos más, la presente invención proporciona composiciones terapéuticas que comprenden al menos un compuesto de la invención, en combinación con un vehículo farmacéuticamente aceptable y opcionalmente, con uno o más agentes adicionales para el tratamiento del cáncer, como se emplean comúnmente en la terapia para el cáncer.
El término "cáncer" se refiere a enfermedades cancerosas que pueden tratarse de manera beneficiosa mediante la inhibición de PKC, incluyendo, por ejemplo, cánceres sólidos, tales como carcinomas (por ejemplo, de los pulmones, páncreas, tiroides, ovario, vejiga, mama, próstata o colon), melanomas, trastornos mieloides (por ejemplo, melanoma uveal, leucemia mieloide, mieloma múltiple y eritroleucemia), adenomas (por ejemplo, adenoma velloso del colon) y sarcomas (por ejemplo, osteosarcoma).
"Inhibidor de PKC" se usa en el presente documento para hacer referencia a un compuesto que muestra una CI50 con respecto a la actividad de PKCa/0 de menos de aproximadamente 100 nM, medida en los ensayos descritos en el presente documento. En algunas realizaciones, un inhibidor de PKC tiene una CI50 con respecto a la actividad de PKCa/0 de menos de aproximadamente 50 nM, medida en los ensayos descritos más adelante en el presente documento. En otras realizaciones más, un inhibidor de PKC tiene una CI50 con respecto a la actividad de PKCa/0 de menos de aproximadamente 10 nM, medida en los ensayos descritos más adelante en el presente documento.
Los compuestos de la invención son útiles para inhibir al menos una isoforma de PKC en un sujeto o para tratar una afección biológica mediada por la isoforma de PKC, incluyendo la vía de señalización de la isoforma de PKC, en un sujeto. La inhibición o el tratamiento puede comprender la etapa de administrar una composición terapéutica que comprende al menos un compuesto de la invención eficaz para inhibir a la isoforma de PKC (PKCa, PKC0) o la vía de señalización de la isoforma de PKC en el sujeto. Las composiciones terapéuticas son útiles para tratar a pacientes que necesitan dichos inhibidores (por ejemplo, aquellos que padecen un cáncer mediado por la señalización anormal de PKC).
Los compuestos de la invención son útiles para inhibir al menos una serina/treonina cinasa seleccionada entre PKCa o PKC0 en un sujeto o para tratar una afección biológica mediada por al menos una de PKCa o PKC0. La inhibición o el tratamiento puede comprender la etapa de administrar una composición terapéutica que comprende al menos un compuesto de la invención eficaz para inhibir la cinasa en el sujeto. Los compuestos terapéuticos son útiles para tratar a pacientes que necesitan dichos inhibidores (por ejemplo, aquellos que padecen cáncer mediado por la señalización anormal del receptor de PKC).
Los compuestos de la invención son útiles para inhibir la actividad de PKCa o PKC0 en un sujeto o para tratar una afección biológica mediada por al menos uno de PKCa o PKC0 en un sujeto humano o animal que necesita dicho tratamiento. La inhibición o el tratamiento puede comprender la etapa de administrar al sujeto al menos un compuesto de la invención en una cantidad eficaz para inhibir la cinasa en el sujeto. Los compuestos terapéuticos son útiles para tratar a pacientes que necesitan dichos inhibidores (por ejemplo, aquellos que padecen un cáncer mediado por una señalización anormal de un receptor de serina/treonina cinasa).
En otros aspectos, la presente divulgación se refiere a los procesos para preparar los compuestos de la invención y a los intermedios sintéticos útiles en dichos procesos, tal como se describe en detalle a continuación.
Métodos sintéticos
Los compuestos de la invención pueden obtenerse mediante procedimientos conocidos por los expertos en la técnica (Métodos 1-6). Por ejemplo, como se muestra en el esquema 1 (método 1), puede prepararse ácido 3-amino-6-sustituido-pirazin-2-carboxílico a partir de su correspondiente 3-amino-6-sustituido-pirazin-2-carboxilato de metilo partiendo de 3-amino-6-bromopirazin-2-carboxilato de metilo. Un compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido (por ejemplo, (1-(2-aminopiridin-3-il)piperidin-4-il)carbamato de tere-butilo), se prepara después partiendo de 3-fluoro-2-nitropiridina. El 3-amino-6-substituido-pirazin-2-carboxilato de metilo se hace reaccionar después con un compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido, después se desprotege para producir un compuesto 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida.
Esquema 1

ntermedio 1 ntermedio 2 ntermedio 3 ntermedio 4

Grupo Grupo
protector protector Intermedio 6 Intermedio 7

Grupo rupo protector protector Intermedio 7 Intermedio 8

Compuesto de formula I
Como alternativa, los compuestos 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida, pueden prepararse en una etapa sintética menos, tal como se muestra en el método 2 (Esquema 2). Por ejemplo, tal como se muestra en el esquema 2, puede prepararse ácido 3-amino-6-sustituido-pirazin-2-carboxílico a partir de su correspondiente 3-amino-6-sustituido-pirazin-2-carboxilato de metilo partiendo de 3-amino-6-bromopirazin-2-carboxilato de metilo. Un compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido (por ejemplo, (1-(2-aminopiridin-3-il)piperidin-4-il)carbamato de ferc-butilo), se prepara después partiendo de 3-fluoro-2-nitropiridina. El 3-amino-6-sustituido-pirazin-2-carboxilato de metilo se hace reaccionar después con un compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido, después se desprotege para producir un compuesto 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida.
Esquema 2

Interm edio 1 Interm edio 3 In term edio 4

Grupo Grupo protector protector ntermedio 6 ntermedio 7

Grupo Grupo protector protector
Intermedio 7 In term edio 8

Compuesto de formula I
En un modo alternativo, se pueden obtener compuestos 3-amino-N-(3-(4-amino-4-(metoximetN)piperidin-1-N)piridin-2-il)-6-substituido-pirazin-2-carboxamida tal como se muestra en el método 3 (Esquema 3). Por ejemplo, partiendo de 3 fluoro-2-nitropiridina, se prepara un compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido. El compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido se hace reaccionar después con ácido 3-amino-6-bromopirazin-2-carboxílico o un ácido protegido (por ejemplo, 3-amino-6-bromopirazin-2-carboxilato de metilo) para producir una 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-bromo-pirazin-2-carboxamida. La 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-bromo-pirazin-2-carboxamida se convierte en dos etapas en el compuesto 3
amino-N-(3-(4-aminopiperidin-1-il)piridin-2-N)-6-sustituido-pirazin-2-carboxamida.
Esquema 3

rupo v rupo
protector protector
Intermedio 6 Intermedio 7

Grupo rupo
protector protector
Intermedio 7 Intermedio 10

Grupo rupo
protector protector
Intermedio 11 ntermedio 8

Compuesto de fórmula I
En un modo alternativo, se pueden obtener compuestos 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-N)piridin-2-il)-6-substituido-pirazin-2-carboxamida tal como se muestra en el método 4 (Esquema 4). Por ejemplo, partiendo de 3-fluoro-2-nitropiridina, se prepara un compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido. El compuesto (1-(2-aminopiridin-3-il)piperidin-4-il) protegido se hace reaccionar después con ácido 3-amino-6-bromopirazin-2-carboxílico o un ácido protegido (por ejemplo, 3-amino-6-bromopirazin-2-carboxilato de metilo) para producir una 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-bromo-pirazin-2-carboxamida. La 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-bromo-pirazin-2-carboxamida se convierte en menos etapas en el compuesto
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida.
Esquema 4

Grupo Grupo protector protector ntermedio 6 ntermedio 7

Grupo v Grupo protector protector Intermedio 7 Intermedio 7a

Grupo
ntermedio 8 protector Compuesto de fórmula I
En un modo alternativo, se pueden obtener compuestos 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-substituido-pirazin-2-carboxamida tal como se muestra en el método 5 (Esquema 5). Por ejemplo, puede prepararse ácido 3-amino-6-sustituido-pirazin-2-carboxílico a partir de su correspondiente 3-amino-6-sustituidopirazin-2-carboxilato de metilo partiendo de 3-amino-6-bromopirazin-2-carboxilato de metilo. El ácido 3-amino-6-sustituido-pirazin-2-carboxílico se hace reaccionar después con 2-amino-3-yodo-piridina para producir el intermedio 12, que se convierte después en compuesto (1-(2-aminopiridin-3-il)piperidin-4-il), después se desprotege para producir un compuesto 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida.

En otro modo alternativo más, pueden prepararse compuestos 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida en una etapa sintética menos tal como se muestra en el método 6 (Esquema 6). Por ejemplo, puede prepararse un ácido 3-amino-6-halo-sustituido-pirazin-2-carboxílico a partir de su correspondiente 3-amino-6-sustituido-pirazin-2-carboxilato de metilo partiendo de 3-amino-6-bromopirazin-2-carboxilato de metilo en una etapa menos. El ácido 3-amino-6-sustituido-pirazin-2-carboxílico se hace reaccionar después con 2-amino-3-yodo-piridina para producir el intermedio 12, que se convierte después en compuesto (1-(2-aminopiridin-3-il)piperidin-4-il), después se desprotege para producir un compuesto 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-sustituido-pirazin-2-carboxamida.
Esquema 6

Intermedio 1 Intermedio 3 ntermedio 4

ntermedio 12 Grupo
protector
Intermedio 8

Compuesto de formula I
Los compuestos de la invención son útiles in vitro o in vivo para inhibir el crecimiento de las células cancerosas. Los compuestos pueden usarse en solitario o en composiciones junto con un vehículo o excipiente farmacéuticamente aceptable. Los vehículos o excipientes farmacéuticamente aceptables adecuados incluyen, por ejemplo, agentes de procesamiento y modificadores y potenciadores de liberación de fármaco, tales como, por ejemplo, fosfato de calcio, estearato de magnesio, talco, monosacáridos, disacáridos, almidón, gelatina, celulosa, metilcelulosa, carboximetilcelulosa sódica, dextrosa, hidroxipropil-P-ciclodextrina, polivinilpirrolidona, ceras de bajo punto de fusión, resinas de intercambio iónico y similares, así como combinaciones de dos cualesquiera o más de los mismos. Otros excipientes farmacéuticamente aceptables adecuados se describen en "Remington's Pharmaceutical Sciences", Mack Pub. Co., Nueva Jersey (1991).
Las cantidades eficaces de los compuestos de la invención generalmente incluyen cualquier cantidad suficiente para inhibir de forma detectable la actividad de la isoforma de PKC divulgada mediante cualquiera de los ensayos descritos en el presente documento, mediante otros ensayos de actividad de PKC conocidos por los expertos habituales en la técnica o detectando una inhibición o alivio de los síntomas de cáncer.
La cantidad de principio activo que puede combinarse con los materiales de transporte para producir una forma de dosificación única variará dependiendo del hospedador tratado y del modo de administración particular. Se entenderá, sin embargo, que el nivel de dosis específico para cualquier paciente particular dependerá de una diversidad de factores que incluyen la actividad del compuesto específico empleado, la edad, peso corporal, estado de salud general, sexo, dieta, tiempo de administración, vía de administración, velocidad de excreción, la combinación de fármacos y la gravedad de la enfermedad particular que se someta a terapia. La cantidad terapéuticamente eficaz para una situación dada se puede determinar con facilidad mediante experimentación rutinaria y está dentro de la habilidad y el criterio del médico.
Para los fines de la presente invención, una dosis terapéuticamente eficaz será generalmente una dosis diaria total administrada a un hospedador en dosis individuales o divididas puede estar en cantidades, por ejemplo, de desde 0,001 hasta 1000 mg/kg de peso corporal diarios y más preferentemente desde 1,0 hasta 30 mg/kg de peso corporal diarios. Las composiciones de unidades de dosificación pueden contener dichas cantidades de submúltiplos de los mismos para componer la dosis diaria.
Los compuestos de la presente invención se pueden administrar por vía oral, por vía parenteral, por vía sublingual, por aerosolización o pulverización por inhalación, por vía rectal o por vía tópica en formulaciones de unidad de dosificación que contienen transportadores, adyuvantes y vehículos farmacéuticamente aceptables, no tóxicos, convencionales, según se desee. La administración tópica también puede implicar el uso de administración transdérmica tal como parches transdérmicos o dispositivos de ionoforesis. El término parenteral, tal como se usa en el presente documento, incluye inyecciones subcutánea, intravenosa, intramuscular, inyección intraesternal o técnicas de infusión.
Pueden formularse preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas inyectables estériles, de acuerdo con la técnica conocida, usando agentes dispersantes o humectantes y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente no tóxico parenteralmente aceptable, por ejemplo, en forma de una solución en 1,3-propanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse están agua, solución de Ringer y solución isotónica de cloruro de sodio. Además, aceites fijos estériles, se usan convencionalmente en forma de un disolvente o un medio de suspensión. Para este fin puede emplearse cualquier aceite suave no volátil, incluyendo mono o diglicéridos sintéticos. Además, los ácidos grasos tales como ácido oleico encuentran uso en la preparación de inyectables.
Los supositorios para administración rectal del fármaco pueden prepararse mezclando el fármaco con un excipiente no irritante adecuado tal como manteca de cacao y polietilenglicoles, que son sólidos a temperatura normal pero líquidos a la temperatura rectal y por lo tanto se derretirán en el recto y liberarán el fármaco.
Las formas farmacéuticas sólidas para administración oral pueden incluir cápsulas, comprimidos, píldoras, polvos y gránulos. En dichas formas farmacéuticas sólidas, el compuesto activo puede mezclarse con al menos un diluyente inerte tal como sacarosa, lactosa o almidón. Dichas formas farmacéuticas también pueden comprender, como es práctica normal, sustancias adicionales distintas de los diluyentes inertes, por ejemplo, agentes lubricantes tales como estearato de magnesio. En el caso de cápsulas, comprimidos y píldoras, las formas farmacéuticas también pueden comprender agentes tamponantes. Los comprimidos y píldoras pueden prepararse adicionalmente con recubrimientos entéricos.
Las formas de dosificación líquidas para administración oral pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados habitualmente en la técnica, tales como agua. Dichas composiciones también pueden comprender adyuvantes, tales como agentes humectantes, agentes emulsionantes y de suspensión, ciclodextrinas y agentes edulcorantes, saporíferos y perfumantes.
Los compuestos de la presente invención también pueden administrarse en forma de liposomas. Tal como se conoce en la técnica, los liposomas se obtienen generalmente a partir de fosfolípidos u otras sustancias lipídicas. Los liposomas se forman mediante cristales líquidos mono o multilamelares hidratados que se dispersan en un medio acuoso. Puede usarse cualquier lípido no tóxico, fisiológicamente aceptable y metabolizable capaz de formar liposomas. Las presentes composiciones en forma de liposoma pueden contener, además de un compuesto de la presente invención, estabilizantes, conservantes, excipientes y similares. Los lípidos preferidos son los fosfolípidos y las fosfatidilcolinas (lecitinas), tanto naturales como sintéticos. Se conocen en la técnica métodos para formar liposomas. Véase, por ejemplo, Prescott, Ed., Methods in Cell Biology, volumen XIV, Academic Press, Nueva York, N.W., pág. 33 y siguientes (1976).
Si bien los compuestos de la invención pueden administrarse como el único agente farmacéutico activo, también pueden usarse en combinación con uno o más de otros agentes usados en el tratamiento del cáncer. Los compuestos de la presente invención también son útiles en combinación con agentes terapéuticos y agentes antineoplásicos conocidos y las combinaciones de los compuestos divulgados actualmente con otros agentes antineoplásicos o quimioterapéuticos están dentro del alcance de la invención. Pueden encontrarse ejemplos de dichos agentes en Cancer Principles and Practice of Oncology, V. T. Devita y S. Hellman (editores), 6a edición (15 de febrero de 2001), Lippincott Williams y Wilkins Publishers. Un experto habitual en la técnica sería capaz de discernir qué combinaciones de agentes serían útiles basándose en las características particulares de los fármacos y del cáncer en cuestión. Dichos agentes antineoplásicos incluyen, pero no se limitan a, los siguientes: moduladores de los receptores de estrógenos, moduladores de los receptores de andrógenos, moduladores de receptores de retinoides, agentes citotóxicos/citostáticos, agentes antiproliferativos, inhibidores de la prenil-proteína transferasa, inhibidores de la HMG-CoA reductasa y otros inhibidores de la angiogénesis, inhibidores de la señalización de la proliferación y la supervivencia celular, agentes inductores de apoptosis y agentes que interfieren con los puntos de control del ciclo celular. Los compuestos de la invención también son útiles cuando se administran junto con radioterapia.
Por lo tanto, en una realización de la invención, los compuestos de la invención también se usan junto con agentes antineoplásicos conocidos que incluyen, por ejemplo, moduladores de los receptores de estrógenos, moduladores de los receptores de andrógenos, moduladores de receptores de retinoides, agentes citotóxicos, agentes antiproliferativos, inhibidores de la prenil-proteína transferasa, inhibidores de HMG-CoA reductasa, inhibidores de la proteasa de VIH, inhibidores de la transcriptasa inversa y otros inhibidores de la angiogénesis.
Los moduladores de los receptores de estrógenos son compuestos que interfieren con o inhiben la unión del estrógeno al receptor, independientemente del mecanismo. Los ejemplos de los moduladores de los receptores de estrógenos incluyen, pero no se limitan a, tamoxifeno, raloxifeno, idoxifeno, LY353381, LY117081, toremifeno, fulvestrant, 4-[7-(2,2-dimetil-1-oxopropoxi-4-metil-2-[4-[2-(1-piperidinil)etoxi]fenil]-2H-1-benzopiran-3-il]-fenil-2,2-dimetilpropanoato, 4,4'-dihidroxibenzofenona-2,4-dinitrofenil-hidrazona y SH646.
Los moduladores de los receptores de andrógenos son compuestos que interfieren con o inhiben la unión de los andrógenos a un receptor de andrógenos. Los ejemplos representativos de moduladores de los receptores de andrógenos incluyen finasterida y otros inhibidores de la 5a-reductasa, nilutamida, flutamida, bicalutamida, liarozol y acetato de abiraterona. Los moduladores de los receptores retinoides son compuestos que interfieren o inhiben la unión de los retinoides a un receptor de retinoides. Los ejemplos de moduladores de los receptores de retinoides incluyen bexaroteno, tretinoína, ácido 13-cis-retinoico, ácido 9-cis-retinoico, a-difluorometilornitina, LX23-7553, trans-N-(4'-hidroxifenil) retinamida y N4-carboxifenilo retinamida.
Los agentes citotóxicos y/o citostáticos son compuestos que provocan la muerte celular o inhiben la proliferación celular principalmente interfiriendo directamente con el funcionamiento de la célula o inhibiendo o interfiriendo con la mitosis celular, incluyendo agentes de alquilación, factores de necrosis tumoral, intercaladores, compuestos activables por hipoxia, agentes inhibidores de los microtúbulos/estabilizadores de los microtúbulos, inhibidores de las quinesinas mitóticas, inhibidores de las quinasas implicadas en la progresión mitótica, antimetabolitos; modificadores de la respuesta biológica; agentes terapéuticos hormonales/antihormonales, factores de crecimiento hematopoyético, agentes terapéuticos dirigidos contra anticuerpos monoclonales, inhibidores de la topoisomerasa, inhibidores de proteasoma e inhibidores de ubiquitina ligasa. Los ejemplos de agentes citotóxicos incluyen, pero no se limitan a, sertenef, caquectina, ifosfamida, tasonermina, lonidamina, carboplatino, altretamina, prednimustina, dibromodulcitol, ranimustina, fotemustina, nedaplatino, oxaliplatino, temozolomida, heptaplatino, estramustina, tosilato de improsulfano, trofosfamida, nimustina, cloruro de dibrospidio, pumitepa, lobaplatino, satraplatino, profiromicina, cisplatino, irofulveno, dexifosfamida, c/s-aminadicloro(2-metil-piridina)platino, bencilguanina, glufosfamida, GPX100, (trans, trans, trans)-bis-mu-(hexan-1,6-diamin)-mu-[diamin-platino (II)] tetracloruro de bis[diamina(cloro)platino (Il)], diarizidinilespermina, trióxido de arsénico, 1-(11-dodecilamino-10-hidroxiundecil)-3,7-dimetilxantina, zorrubicina, idarrubicina, daunorrubicina, bisantreno, mitoxantrona, pirarrubicina, pinafida, valrubicina, amrubicina, antineoplaston, 3'-desamino-3'-morfolin-13-desoxo-10-hidroxicarminomicina, anamicina, galarrubicina, elinafida, MEN10755 y 4-demetoxi-3-desamino-3-aziridinil-4-metilsulfonil-daunorrubicina (véase el documento WO 00/50032). Un ejemplo representativo de un compuesto activable por hipoxia es tirapazamina. Los inhibidores del proteasoma incluyen, pero no se limitan a, lactacistina y bortezomib. Los ejemplos de agentes inhibidores de microtúbulos/agentes estabilizantes de microtúbulos incluyen paclitaxel, sulfato de vindesina, 3',4'-didehidro-4'-desoxi-8'-norvincaleucoblastina, docetaxol, rizoxina, dolastatina, isetionato de mivobulina, auristatina, cemadotina, RPR109881, BMS184476, vinflunina, criptoficina, 2,3,4,5,6-pentafluoro-N-(3-fluoro-4-metoxifenil)benceno sulfonamida, anhidrovinblastina, N,N-dimetil-L-valil-L-valil-N-metil-L-valil-L-prolil-L-prolin-t-butilamida, TDX258, los epotilones (véanse por ejemplo las patentes de Estados Unidos n.° 6.284.781 y 6.288.237) y BMS188797. Los ejemplos representativos de inhibidores de la topoisomerasa incluyen topotecán, hicaptamina, irinotecán, rubitecán, 6-etoxipropionil-3',4'-O-exo-bencilidenchartreusina, 9-metoxi-N,N-dimetil-5-nitropirazolo[3,4,5-kl]acridin-2-(6H)propanamina, 1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3',4':b,7]-indolizino[1,2b]quinolin-10,13(9H,15H)diona, lurtotecán, 7-[2-(N-isopropilamino)etil]-(20S)camptotecina, BNP1350, BNPI1100, BN80915, BN80942, fosfato de etopósido, tenipósido, sobuzoxano, 2'-dimetilamino-2'-desoxi-etopósido, GL331, N-[2-(dimetilamino)etil]-9-hidroxi-5,6-dimetil-6H-pirido[4,3-b]carbazol-1-carboxamida, asulacrina, (5a, 5aB, 8aa, 9b)-9-[2-[N-[2-(dimetilamino)etil]-N-metilamino]etil]-5-[4-hidroOxi-3,5-dimetoxifenil]-5,5a,6,8,8a,9-hexahidrofuro-(3',4':6,7)nafto(2,3-d)-1,3-dioxol-6-ona, 2,3-(metilenodioxi)-5-metil-7-hidroxi-8-metoxibenzo[c]-fenantridinio, 6,9-bis[(2-aminoetil)amino]benzo[g]isoquinolin-5,10-diona, 5-(3-aminopropilamino)-7,10-dihidroxi-2-(2-hidroxietilaminometil)-6H-pirazolo[4,5,1'-de]acridin-6-ona, N -[l-[2(dietilamino)etilamino]-7-metoxi-9-oxo-9H-tioxanten-4-ilmetil]formamida, N-(2-(dimetilamino)etil)acridin-4-carboxamida, 6-[[2-(dimetilamino)etil]amino]-3-hidroxi-7H-indeno[2,1-c]quinolin-7-ona y dimesna. Se describen ejemplos de inhibidores de las quinesinas mitóticas, tales como la quinesina mitótica humana KSP, en las publicaciones PCT WO 01/30768 y WO 01/98278, WO 03/050.064 (19 de junio de 2003), WO 03/050.122 (19 de junio de 2003), WO 03/049.527 (19 de junio de 2003), WO 03/049.679 (19 de junio de 2003), WO 03/049.678 (19 de junio de 2003) y WO 03/39460 (15 de mayo de 2003) y solicitudes PCT pendientes. n.° US03/06403 (presentada el 4 de marzo de 2003), US03/15861 (presentada el 19 de marzo de 2003), US03/15810 (presentada el 19 de marzo de 2003), US03/18482 (presentada el 12 de junio de 2003) y US03/18694 (presentada el 12 de junio de 2003). En una realización, los inhibidores de las quinesinas mitóticas incluyen, pero no se limitan a inhibidores de KSP, inhibidores de MKLP1, inhibidores de CENP-E, inhibidores de MCAK, inhibidores de Kif14, inhibidores de Mfosf1 e inhibidores de Rab6-KIFL.
Los inhibidores de las cinasas implicadas en la progresión mitótica incluyen, pero no se limitan a, inhibidores de la
quinasa aurora, inhibidores de las cinasas de tipo Polo (PLK) (por ejemplo, inhibidores de PLK-1), inhibidores de bub-1 e inhibidores de bub-R1. Los agentes antiproliferativos incluyen oligonucleótidos de ARN y ADN antisentido tales como G3139, ODN698, RVASKRAS, GEM231 e INX3001 y antimetabolitos tales como enocitabina, carmofur, tegafur, pentostatina, doxifluridina, trimetrexato, fludarabina, capecitabina, galocitabina, ocfosfato de citarabina, fosteabina de sodio hidratada, raltitrexed, paltitrexid, emitefur, tiazofurina, decitabina, nolatrexed, pemetrexed, nelzarabina, 2'-desoxi-2'-metilidencitidina, 2'-fluorometilen-2'-desoxicitidina, N-[5-(2,3-dihidro-benzofuril)sulfonil]-N'-(3,4-diclorofenil}urea, N6-[4-desoxi-4-[N2-[2(E),4(E)-tetradecadienoil]glicilamino]-L-glicero-B-L-manoheptopiranosil]adenina, aplidina, ecteinascidina, troxacitabina, ácido 4-[2-amino-4-oxo-4,6,7,8-tetrahidro-3H-pirimidino[5,4-b][1,4]tiazin-6-il-(S)-etil]-2,5-tienoil-L-glutámico, aminopterina, 5-fluorouracilo, alanosina, éster del ácido 11-acetil-8-(carbamoiloximetil)-4-formil-6-metoxi-14-oxa-1,1-diazatetraciclo(7.4,1.0.0)-tetradeca-2,4,6-trien-9-il acético, swainsonina, lometrexol, dexrazoxano, metioninasa, 2'-ciano-2'-desoxi-N4-palmitoil-1-B-D-arabinofuranosil citosina y 3-aminopiridin-2-carboxaldehído tiosemicarbazona. Ejemplos de agentes terapéuticos dirigidos contra anticuerpos monoclonales incluyen aquellos agentes terapéuticos que tienen agentes citotóxicos o radioisótopos unidos a una célula cancerosa específica o a un anticuerpo monoclonal específico de una célula diana. Los ejemplos incluyen, por ejemplo, Bexxar. Los inhibidores de la HMG-CoA reductasa son inhibidores de la 3-hidroxi-3-metilglutaril-CoA reductasa. Los compuestos que tienen actividad inhibidora para la HMG-CoA reductasa pueden identificarse fácilmente usando ensayos bien conocidos en la técnica tales como los descritos o citados en las patentes de Estados Unidos n.° 4.231.938 y WO 84/02131. Los ejemplos de inhibidores de la HMG-CoA reductasa que se pueden usar incluyen, pero no se limitan a, lovastatina (MEVACOR®; véanse las patentes de Estados Unidos n.° 4.231.938, 4.294.926 y 4.319.039), simvastatina (ZOCOR®; véanse las patentes de Estados Unidos n.° 4.444.784, 4.820.850 y 4.916.239), pravastatina (PRAVACHOL®.; véanse las patentes de Estados Unidos n.° 4.346.227, 4.537.859, 4.410.629, 5.030.447 y 5.180.589), fluvastatina (LESCOL®; véanse las patentes de Estados Unidos n.° 5.354.772, 4.911.165, 4.929.437, 5.189.164, 5.118.853, 5.290.946 y 5.356.896) y atorvastatina (LIPITOR®; véanse las patentes de Estados Unidos n.° 5.273.995, 4.681.893, 5.489.691 y 5.342.952). Las fórmulas estructurales de estos inhibidores y de inhibidores de la HMG-CoA reductasa adicionales que se pueden usar en los presentes métodos se describen en la página 87 de M. Yalpani, "Cholesterol Lowering Drugs", Chemistry & Industry, págs. 85-89 (5 de febrero de 1996) y patentes de Estados Unidos n.° 4.782.084 y 4.885.314. En una realización, el inhibidor de la HMG-CoA reductasa se selecciona entre lovastatina y simvastatina.
Los inhibidores de la prenil-proteína transferasa son compuestos que inhiben cualquiera o cualquier combinación de las enzimas prenil-proteína transferasas, incluyendo farnesil-proteína transferasa (FPTasa), geranilgeranil-proteína transferasa de tipo I (GGPTasa-I) y geranilgeranil-proteína transferasa de tipo II (GGPTasa-II, denominada también Rab GGPTasa). Los ejemplos de compuestos la prenil-proteína transferasa incluyen (±)-6-[amino(4-clorofenil)(1-metil-1H-imidazol-5-il)metil]-4-(3-clorofenil)-1-metil-2(1H)quinolinona, (-)-6-[amino(4-clorofenil)(1-metil-1H-imidazol-5-il)metil]-4-(3-clorofenil)-1-metil-2(1H)-quinolinona, (+)-6-[amino(4-clorofenil)(1-metil-1H-imidazol-5-il)metil]-4-(3-clorofenil)-1-metil-2(1H)-quinolinona, 5(S)-n-butil-1-(2,3-dimetilfenil)-4-[1-(4-cianobencil)-5-imidazolilmetil-2-piperazinona, (S)-1-(3-clorofenil)-4-[1-(4-cianobencil)-5-imidazolilmetil]-5-[2-(etanosulfonil)metil)-2-piperazinona, 5(S)-n-butil-1-(2-metilfenil)-4-[1-(4-cianobencil)-5-imidazolilmetil]-2-piperazinona, 1-(3-clorofenil)-4-[1-(4-cianobencil)-2-metil-5-imidazolilmetil]-2-piperazinona, 1-(2,2-difeniletil)-3-[N-(1-(4-cianobencil)-1H-imidazol-5-iletil)carbamoil]piperidina, 4-{-[4-hidroximetil-4-(4-cloropiridin-2-ilmetil)-piperidin-1-ilmetil]-2-metilimidazol-1-ilmetil}benzonitrilo, 4-{-5-[4-hidroximetil-4-(3-clorobencil)-piperidin-1-ilmetil]-2-metilimidazol-1-il-metil}benzonitrilo, 4-{3-[4-(2-oxo-2H-piridin-1-il)bencil]-3H-imidazol-4-ilmetil}-benzonitrilo, 4-{3-[4-(5-cloro-2-oxo-2H-[1,2']bipiridin-5'-ilmetil]-3H-imidazol-4-il-metil}benzonitrilo, 4-{3-[4-(2-oxo-2H-[1,2']bipiridin-5'-ilmetil]-3H-imidazol4-il-metil}benzonitrilo, 4-[3-(2-oxo-1-fenil-1,2-dihidropiridin-4-ilmetil)-3H-imidazol-4-ilmetil}benzonitrilo, 18,19-dihidro-19-oxo-5H,17H-6,10:12,16-dimeten-1 H-imidazo[4,3-c][1,11,4]dioxaazaciclo-nonadecin-9-carbonitrilo, (±)-19,20-dihidro-19-oxo-5H-18,21-etan-12,14-eten-6,10-meten-22H-benzo[d]imidazo[4,3-k][1,6,9,12]oxatriaza-ciclooctadecin-9-carbonitrilo, 19,20-dihidro-19-oxo-5H,17H-18,21-etan-6,10:12,16-dimeten-22H-imidazo[3,4-h][1,8,11,14]oxatriazacicloeicosin-9-carbonitrilo y (.+-.)-19,20-dihidro-3-metil-19-oxo-5H-18,21-etan-12,14-eten-6,10-meten-22H-benzo[d]imidazo[4,3-k][1,6,9,12]oxa-triazaciclooctadecin-9-carbonitrilo. Se pueden encontrar otros ejemplos de inhibidores de la prenilproteína transferasa en las siguientes publicaciones y patentes: documento WO 96/30343, documento WO 97/18813, documento WO 97/21701, documento WO 97/23478, documento WO 97/38665, documento WO 98/28980, documento WO 98/29119, documento WO 95/32987, patente de los Estados Unidos n.° 5.420.245, patente de los Estados Unidos n.° 5.523.430, patente de los Estados Unidos n.° 5.532.359, patente de los Estados Unidos n.° 5.510.510, patente de los Estados Unidos n.° 5.589.485, patente de los Estados Unidos n.° 5.602.098, publicación de patente europea 0618 221, publicación de patente europea 0675 112, publicación de patente europea 0604 181, publicación de patente europea 0 696 593, documento W o 94/19357, documento WO 95/08542, documento WO 95/11917, documento WO 95/12612, documento WO 95/12572, documento WO 95/10514, patente de los Estados Unidos n.° 5.661.152, documento WO 95/10515, documento WO 95/10516, documento WO 95/24612, documento WO 95/34535, documento WO 95/25086, documento WO 96/05529, documento WO 96/06138, documento WO 96/06193, documento WO 96/16443, documento WO 96/21701, documento WO 96/21456, documento WO 96/22278, documento WO 96/24611, documento WO 96/24612, documento WO 96/05168, documento WO 96/05169, documento WO 96/00736, patente de los Estados Unidos n.° 5.571.792, documento WO 96/17861, documento WO 96/33159, documento WO 96/34850, documento WO 96/34851, documento WO 96/30017, documento WO 96/30018, documento WO 96/30362, documento WO 96/30363, documento WO 96/31111, documento WO 96/31477, documento WO 96/31478, documento WO 96/31501, documento WO 97/00252, documento WO 97/03047, documento WO 97/03050, documento WO 97/04785, documento WO 97/02920, documento WO 97/17070, documento WO 97/23478, documento WO 97/26246, documento
WO 97/30053, documento WO 97/44350, documento WO 98/02436 y patente de los Estados Unidos n.° 5.532.359. Para un ejemplo del papel de un inhibidor de la prenil-proteína transferasa sobres la angiogénesis, véase European J. of Cancer 35(9):1394-1401 (1999).
Inhibidores de la angiogénesis se refiere a compuestos que inhiben la formación de nuevos vasos sanguíneos, independientemente del mecanismo. Ejemplos de inhibidores de la angiogénesis incluyen, pero no se limitan a, inhibidores de tirosina quinasa, tales como los inhibidores de los receptores de la tirosina quinasa Flt-1 (VEGFR1) y Flk-1/KDR (VEGFR2), inhibidores de los factores de crecimiento derivados de epidermis, derivados de fibroblastos o derivados de plaquetas, inhibidores de la MMP (metaloproteasa de la matriz), bloqueadores de la integrina, interferón alfa, interleucina-12, polisulfato de pentosán, inhibidores de la ciclooxigenasa, incluyendo antiinflamatorios no esteroideos (AINE) tales como aspirina e ibuprofeno, así como inhibidores selectivos de la ciclooxigenasa-2 tales como celecoxib y rofecoxib (PNAS 89:7384 (1992); JNCI 69:475 (1982); Arch. Ophthalmol. 108:573 (1990); Anat. Rec., (238):68 (1994); FEBS Letters 372:83 (1995); Clin, Orthop. 313:76 (1995); J. Mol. Endocrinol. 16:107 (1996); Jpn. J. Pharmacol. 75:105 (1997); Cancer Res. 57:1625 (1997); Cell 93:705 (1998); Intl. J. Mol. Med. 2:715 (1998); J. Biol. Chem. 274:9116 (1999)), antiinflamatorios esteroideos (tales como corticosteroides, mineralocorticoides, dexametasona, prednisona, prednisolona, metilpred, betametasona), carboximidotriazol, combretastatina A4, escualamina, 6-O-cloroacetil-carbonol)-fumagilol, talidomida, angiostatina, troponina-1, antagonistas de la angiotensina II (véase Fernández et al., J. Lab. Clin. Med. 105:141-145 (1985)) y anticuerpos contra VEGF (véase, Nature Biotechnology, 17:963-968 (octubre de 1999); Kim et al., Nature, 362:841-844 (1993); documento WO 00/44777 y documento WO 00/61186). Otros agentes terapéuticos que modulan o inhiben la angiogénesis y también pueden usarse en combinación con los compuestos de la presente invención incluyen agentes que modulan o inhiben los sistemas de coagulación y fibrinolisis (véase una revisión en Clin. Chem. La. Med. 38:679-692 (2000)). Ejemplos de dichos agentes que modulan o inhiben las rutas de coagulación y fibrinolisis incluyen, pero no se limitan a, heparina (véase, Thromb. Haemost. 80:10-23 (1998)), heparinas de bajo peso molecular e inhibidores de carboxipeptidasa U (también conocidos como inhibidores del inhibidor de fibrinolisis activable por trombina activa [TAFIa]) (véase Trombosis Res. 101:329-354 (2001)). Se han descrito inhibidores de TAFIa en la publicación PCT WO 03/013.526 y n.° de serie de Estados Unidos n.° 60/349.925 (presentada el 18 de enero de 2002). La invención también incluye combinaciones de los compuestos de la invención con AINEs que son inhibidores selectivos de COX-2 (generalmente definidos como aquellos que poseen una especificidad para inhibir COX-2 sobre COX-1 de al menos 100 veces, como se midió por la relación de la CI50 para c OX-2 sobre la CI50 para COX-1 evaluadas mediante ensayos celulares o microsomales). Dichos compuestos incluyen, pero sin limitación, los desvelados en la patente de Estados Unidos n.° 5.474.995, emitida el 12 de diciembre de 1995, patente de los Estados Unidos n.° 5.861.419, emitida el 19 de enero de 1999, patente de los Estados Unidos n.° 6.001.843, emitida el 14 de diciembre de 1999, patente de los Estados Unidos n.° 6.020.343, emitida el 1 de febrero de 2000, patente de los Estados Unidos n.° 5.409.944, emitida el 25 de abril de 1995, patente de los Estados Unidos n.° 5.436.265, emitida el 25 de julio de 1995, patente de los Estados Unidos n.° 5.536.752, emitida el 16 de julio de 1996, patente de los Estados Unidos n.° 5.550.142, emitida el 27 de agosto de 1996, patente de los Estados Unidos n.° 5.604.260, emitida el 18 de febrero de 1997, patente de los Estados Unidos n.° 5.698.584, emitida el 16 de diciembre de 1997, patente de los Estados Unidos n.° 5.710.140, emitida el 20 de enero de 1998, documento WO 94/15932, publicada el 21 de julio de 1994, patente de los Estados Unidos n.° 5.344.991, emitida el 6 de junio de 1994, patente de los Estados Unidos n.° 5.134.142, emitida el 28 de julio de 1992, patente de los Estados Unidos n.° 5.380.738, emitida el 10 de enero de 1995, patente de los Estados Unidos n.° 5.393.790, emitida el 20 de febrero de 1995, patente de los Estados Unidos n.° 5.466.823, emitida el 14 de noviembre de 1995, patente de los Estados Unidos n.° 5.633.272, emitida el 27 de mayo de 1997 y la patente de Estados Unidos n.° 5.932.598, emitida el 3 de agosto de 1999. Inhibidores representativos de COX-2 que son útiles en los métodos de la presente invención incluyen 3-fenil-4-(4-(metilsulfonil)fenil)-2-(5H)-furanona; y 5-cloro-3-(4-metilsulfonil)fenil-2-(2-metil-5-piridinil)piridina. Los compuestos que se describen como inhibidores específicos de COX-2 y son por lo tanto útiles en la presente invención, y los métodos de síntesis de los mismos, pueden encontrarse en las patentes siguientes, solicitudes pendientes y publicaciones: documento WO 94/15932, publicada el 21 de julio de 1994, patente de los Estados Unidos n.° 5.344.991, emitida el 6 de junio de 1994, patente de los Estados Unidos n.° 5.134.142, emitida el 28 de julio de 1992, patente de los Estados Unidos n.° 5.380.738, emitida el 10 de enero de 1995, patente de los Estados Unidos n.° 5.393.790, emitida el 20 de febrero de 1995, patente de los Estados Unidos n.° 5.466.823, emitida el 14 de noviembre de 1995, patente de los Estados Unidos n.° 5.633.272, emitida el 27 de mayo de 1997, patente de los Estados Unidos n.° 5.932.598, emitida el 3 de agosto de 1999, patente de los Estados Unidos n.° 5.474.995, emitida el 12 de diciembre de 1995, patente de los Estados Unidos n.° 5.861.419, emitida el 19 de enero de 1999, patente de los Estados Unidos n.° 6.001.843, emitida el 14 de diciembre de 1999, patente de los Estados Unidos n.° 6.020.343, emitida el 1 de febrero de 2000, patente de los Estados Unidos n.° 5.409.944, emitida el 25 de abril de 1995, patente de los Estados Unidos n.° 5.436.265, emitida el 25 de julio de 1995, patente de los Estados Unidos n.° 5.536.752, emitida el 16 de julio de 1996, patente de los Estados Unidos n.° 5.550.142, emitida el 27 de agosto de 1996, patente de los Estados Unidos n.° 5.604.260, emitida el 18 de febrero de 1997, patente de los Estados Unidos n.° 5.698.584, emitida el 16 de diciembre de 1997 y la patente de Estados Unidos n.° 5.710.140, emitida el 20 de enero de 1998. Otros ejemplos de inhibidores de la angiogénesis incluyen, pero no se limitan a, endostatina, ucraína, ranpirnasa, IM862, 5-metoxi-4-[2-metil-3-(3-metil-2-butenil)oxiranil]-1-oxaespiro[2,5]oct-6-il(cloroacetil)carbamato, acetildinanalina, 5-amino-1-[[3,5-dicloro-4-(4-clorobenzoil)fenil]metil]-1H-1,2,3-triazol-4-carboxamida, CM101, escualamina, combretastatina, RPI4610, NX31838, fosfato de manopentanosa sulfatado, 7,7-(carbonil-bis[imino-N-metil-4,2-pirrolocarbonilimino[N-metil-4,2-pirrol]-carbonilimino]-bis-(1,3-naftalen disulfonato) y 3-[(2,4-dimetilpirrol-5-il)metilen]-2-indolinona (SU5416).
Los agentes que interfieren con los puntos de control del ciclo celular son compuestos que inhiben a las proteínas cinasas que transducen señales de punto de control del ciclo celular, sensibilizando de este modo a la célula cancerosa frente a agentes que dañan al ADN. Dichos agentes incluyen inhibidores de las cinasas ATR, ATM, Chk1 y Chk2 e inhibidores de cdk y cdc cinasa y se ilustran específicamente por 7-hidroxiestaurosporina, flavopiridol, CYC202 (Cyclacel) y BMS-387032.
Los inhibidores de la proliferación celular y la vía de señalización de supervivencia son agentes farmacéuticos que inhiben a receptores de la superficie celular y cascadas de transducción de señales aguas abajo de dichos receptores de la superficie. Dichos agentes incluyen inhibidores de EG FR (por ejemplo gefitinib y erlotinib), inhibidores de ERB-2 (por ejemplo, trastuzumab), inhibidores de IGFR, inhibidores de receptores de citocinas, inhibidores de MET, inhibidores de PI3K (por ejemplo, LY294002), serina/treonina quinasas (incluyendo, pero sin limitación, inhibidores de Akt tales como los descritos en los documentos WO 02/083064, WO 02/083139, WO 02/083140 y WO 02/083138), inhibidores de Raf cinasa (por ejemplo, BAY-43-9006), inhibidores de MEK (por ejemplo, CI-1040 y PD-098059) e inhibidores de mTOR (por ejemplo, rapamicina, everolimus y Wyeth CCI-779). Dichos agentes incluyen compuestos inhibidores de molécula pequeña y antagonistas de anticuerpos.
Los agentes que inducen la apoptosis incluyen activadores de miembros de la familia de receptor de TNF (incluyendo los receptores TRAIL).
En ciertas realizaciones de la invención preferidas en el presente documento, los agentes representativos útiles en combinación con los compuestos de la invención para su uso en el tratamiento del cáncer incluyen, por ejemplo, irinotecán, topotecán, gemcitabina, 5-fluorouracilo, leucovorina, carboplatino, cisplatino, taxanos, tezacitabina, ciclofosfamida, alcaloides de la vinca, imatinib (Gleevec™), nilotinib (Tasigna™), everolimus (Afinitor™), antraciclinas, rituximab, trastuzumab, así como otros agentes quimioterapéuticos para el cáncer.
Los anteriores compuestos a emplear en combinación con los compuestos de la invención pueden usarse en cantidades terapéuticas tal como se indica en el Physicians’ Desk Reference (PDR) 47a edición (1993) o dichas cantidades terapéuticamente útiles tal como serían conocidas por un experto habitual en la técnica.
Los compuestos de la invención y los otros agentes antineoplásicos pueden administrarse a las dosis clínicas máximas recomendadas o a dosis menores. Los niveles de dosificación de los compuestos activos en las composiciones de la invención pueden variarse para obtener una respuesta terapéutica deseada dependiendo de la vía de administración, la gravedad de la enfermedad y la respuesta del paciente. La combinación puede administrarse en forma de composiciones separadas o como una forma farmacéutica que contiene ambos agentes. Cuando se administra en forma de una combinación, los agentes terapéuticos pueden formularse como composiciones separadas, que se administran al mismo tiempo o en momentos diferentes, o los agentes terapéuticos pueden administrarse como una única composición.
Los antiestrógenos, tales como el tamoxifeno, inhiben el crecimiento del cáncer de mama mediante la inducción de la detención del ciclo celular, efectuando la acción del inhibidor del ciclo celular p27Kip. Recientemente, se ha demostrado que la activación de la vía de Ras-Raf-MAP cinasa altera el estado de fosforilación de p27Kip, de tal forma que se atenúa su actividad inhibidora en la detención del ciclo celular, contribuyendo de este modo a la resistencia a los antiestrógenos (Donovan et al., J. Biol. Chem. 276:40888, 2001). Como se informa por Donovan et al., la inhibición de la señalización de MAPK mediante el tratamiento con el inhibidor de MEK cambió el estado de fosforilación de p27 en líneas celulares de cáncer de mama resistentes a hormonas y al hacer esto, se restauró la sensibilidad a las hormonas. Por consiguiente, en un aspecto, los compuestos de la invención pueden usarse en el tratamiento de cánceres dependientes de hormonas, tales como los cánceres de mama y próstata, para revertir la resistencia a las hormonas comúnmente observada en estos cánceres con agentes anticáncer convencionales.
En los cánceres hematológicos, tales como la leucemia mielógena crónica (CML), la traslocación cromosómica es responsable de la tirosina cinasa BCR-AB1 activada de manera constitutiva. Los pacientes afectados son sensibles a Gleevec, un inhibidor de tirosina cinasa de molécula pequeña, como resultado de la inhibición de la actividad de Abl cinasa. Sin embargo, muchos pacientes con enfermedad en estadio avanzado responden inicialmente a Gleevec, pero posteriormente tienen una recidiva debido a mutaciones que confieren resistencia en el dominio de cinasa de Ab1. Los estudios in vitro han demostrado que BCR-Av1 emplea la vía de Raf cinasa para provocar sus efectos. Además, la inhibición de más de una cinasa en la misma vía proporciona protección adicional contra mutaciones que confieren resistencia. Por consiguiente, en otro aspecto de la invención, los compuestos de la invención se usan en combinación con al menos un agente adicional, tal como Gleevec™ o Tasigna™ en el tratamiento de cánceres hematológicos, tales como la leucemia mielógena crónica (CML), para revertir o prevenir la resistencia al al menos un agente adicional. Los compuestos de la invención son útiles para inhibir al menos una serina/treonina cinasa en la vía de señalización de Jak/Stat en un sujeto o para tratar una afección biológica mediada por una vía de señalización de PKC en un sujeto. La inhibición o el tratamiento puede comprender administrar una composición terapéutica que comprende al menos un compuesto de la invención eficaz para inhibir la actividad de la al menos una serina/treonina cinasa en la vía de señalización de PKC en el sujeto.
Las composiciones terapéuticas de acuerdo con este aspecto de la invención son útiles para tratar a pacientes que necesitan dichos inhibidores (por ejemplo, aquellos que padecen cáncer mediado por señalización anormal de PKC). Los tipos de cáncer mediados por la señalización anormal de PKC incluyen, por ejemplo, melanoma, melanoma uveal, linfoma, linfoma difuso de linfocitos B grandes (DLBCL) y cánceres resistentes a ibrutinib, cáncer papilar, cáncer de tiroides, cáncer de ovario, cáncer de colon, cáncer de páncreas, cáncer de pulmón no microcítico (CPNM), cánceres hematológicos, leucemia mielógena crónica (CML), leucemia linfoblástica aguda (ALL) y leucemia mieloide aguda. Los compuestos de la invención son útiles para inhibir a PKCa, PKC0 y GSKp en un sujeto humano o animal. La inhibición puede comprender administrar una cantidad eficaz de un compuesto o una sal farmacéuticamente aceptable del mismo, de cualquiera de las realizaciones de compuestos de la invención a un sujeto que lo necesite.
La presente invención se comprenderá más fácilmente haciendo referencia a los ejemplos siguientes, que se proporcionan a modo de ilustración y no pretenden ser limitantes de la presente invención.
Las cadenas laterales representativas para su uso en los compuestos de los ejemplos siguientes generalmente pueden prepararse de acuerdo con los procedimientos siguientes:
Ejemplos
En referencia a los ejemplos siguientes, los compuestos de las realizaciones preferidas se sintetizaron usando los métodos descritos en el presente documento u otros métodos, que se conocen en la técnica. Los ejemplos que detallan compuestos que no son de fórmula (II) se incluyen con fines de referencia.
Los compuestos y/o intermedios se caracterizaron mediante cromatografía líquida de alto rendimiento (HPLC) usando un sistema de cromatografía Waters Millenium con un módulo de separación 2695 (Milford, MA). Las columnas analíticas eran Phenomenex Luna C 18 -5 p de fase inversa, 4,6 x 50 mm, de Alltech (Deerfield, IL). Se usó una elución en gradiente (velocidad de 2,5 ml/min), normalmente partiendo con acetonitrilo al 5 % /agua al 95 % y progresando a acetonitrilo al 100 % durante un periodo de 10 minutos. Todos los disolventes contenían ácido trifluoroacético al 0,1 % (TFA). Los compuestos se detectaron por absorción de luz ultravioleta (UV) a 220 o 254 nm. Los disolventes de la HPLC eran de Burdick y Jackson (Muskegan, MI) o Fisher Scientific (Pittsburgh, PA).
En algunos casos, la pureza se evaluó por cromatografía de capa fina (TLC) usando placas de gel de sílice con respaldo de vidrio o plástico, tales como, por ejemplo, láminas flexibles Baker-Flex Silica Gel 1B2-F. Los resultados de la TLC se detectaron visualmente con facilidad bajo luz ultravioleta o empleando vapor de yodo y otras técnicas de tinción distintas bien conocidas.
Los compuestos y/o intermedios se caracterizaron por LCMS. Las condiciones generales son las siguientes.
Los espectros de masas de baja y alta resolución se obtuvieron en sistemas LC/MS usando métodos de ionización por electronebulización de una gama de instrumentos de las siguientes configuraciones: Baja resolución - Sistema Agilent 1100 HPLC-UV equipado con espectrómetro de masas Waters ZQ y detectores Schimadzu ELSD; Baja resolución - Sistema Waters AcQuity UPLC-UV equipado con espectrómetro de masas Waters SQ y detectores Thermo CAD; Alta resolución - Sistema Waters AcQuity UPLC-UV equipado con un espectrómetro de masas Waters LCT Premier. [M+H]+ se refiere al ion molecular protonado de la especie química.
Métodos de instrumentos analíticos
Métodos de MS de baja resolución
Agilent 1100 HPLC-UV con espectrómetro de masas Waters ZQ
Método ácido: Columna: Sunfire C18, 3x30 mm, 3,5 pm, temperatura 40 °C, volumen de inyección 2 pl; Disolvente A: TFA al 0,05 % en agua; Disolvente B: acetonitrilo; Gradiente: 5-95 % .
Método básico: Columna: Xbridge C18, 3x30 mm, 3,5 pm, temperatura 40 °C, volumen de inyección 2 pl; Disolvente A: NH4OH 5 mM en agua; Disolvente B: acetonitrilo; Gradiente: 5-95 % .
Método de MS de baja resolución
Waters AcQuity equipado con espectrómetro de masas Waters SQ
Método ácido: Columna: Acquity UPLC BEH C18, 2,1x50 mm, 1,7 pm, temperatura 50 °C, volumen de inyección 1,5 pl; Disolvente A: TFA al 0,05 % en agua; Disolvente B: acetonitrilo; Gradiente: 2-98 % en 1,7 min.
Método neutro: Columna: Acquity BEH C 18 1,7 pm 2,1x50 mm - 50 °C; Disolvente A: agua Amm Ace 3,75 mM
CAN al 2 % ; Disolvente B: ACN Amm Ace 3,75 mM agua al 5 % ; Gradiente: del 2 al 98 % de B durante 1,7 min -velocidad 1 ml/min.
Método de HRMS
Waters AcQuity UPLC-UV equipado con espectrómetro de masas Waters LCT Premier
Método ácido: Columna: ACQUITY UPLC BEH C18, 130 A, 1,7 um, 2,1 mm X 50 mm - temp: 50 °C; Disolvente A: AGUA ácido fórmico al 0,1 % ; Disolvente B: Acetonitrilo ácido fórmico al 0,1 % ; Gradiente: 2-98 % de disolvente B en 7,5 min; Velocidad de escaneo: 0,2 s, sobre el intervalo de 120-1100 Daltons.
Método básico: Columna: ACQUITY UPLC BEH C18, 130 A, 1,7 um, 2,1 mm X 50 mm - temp: 50 °C; Disolvente A: agua NH4OH 5 mM; Disolvente B: NH4OH 5 mM en acetonitrilo; Gradiente 2-98 % en 7,5 min; Velocidad de escaneo: 0,2 s, sobre el intervalo de 120 -1100 Daltons.
El análisis por resonancia magnética nuclear (RMN) se realizó en un espectrómetro de RMN Bruker de 400 MHz usando ICON-RMN, con el programa de control TopSpin. Los espectros se midieron a 298 K, a menos que se indique otra cosa y se referenciaron en relación con el desplazamiento químico del disolvente.
La pureza de algunos de los compuestos se evalúa por análisis elemental (Desert Analytics, Tucson, AZ).
Los puntos de fusión se determinan en un aparato Mel-Temp de Laboratory Devices (Holliston, MA).
Las separaciones preparativas se llevaron a cabo usando
un sistema de HPLc Waters 2545 equipado con detección de espectrómetro de masas Waters PDA 2998 y/o Waters 3100.
UV desencadenado por ácido: agua/acetonitrilo con un modificador de TFA al 0,1 % , caudal 75 ml/min, volumen de inyección 1,5 ml; Columna: Waters Sunfire 30 mm ID x 50 mm, partícula de 5 pm.
UV desencadenado por base: agua/acetonitrilo con NH4OH 5 mM, caudal 75 ml/min, volumen de inyección 1,5 ml; Columna: Waters X-Bridge 30 mm ID x 50 mm, partícula de 5 pm.
Métodos:
Todos los métodos ejecutan un gradiente enfocado desde el % inicial de acetonitrilo hasta el % final de acetonitrilo durante 3,5 minutos con una parada inicial de 10 segundos. Después del gradiente, todos los métodos pasan a acetonitrilo al 95 % durante 30 segundos y se mantienen ahí durante 1,5 minutos antes de volver a las condiciones iniciales. Las condiciones iniciales y finales para cada gradientes son como siguen:
Método 0: acetonitrilo al 5 -12 %
Método 1: acetonitrilo al 7,5-20 %
Método 2: acetonitrilo al 10-30 %
Método 3: acetonitrilo al 15-40 %
Método 4: acetonitrilo al 25-50 %
Método 5: acetonitrilo al 35-60 %
Método 6: acetonitrilo al 45-70 %
Método 7: acetonitrilo al 55-80 %
Método 8: acetonitrilo al 65-95 %
Debería entenderse que los compuestos orgánicos de acuerdo con las realizaciones preferidas pueden mostrar el fenómeno de tautomerismo. Como las estructuras químicas incluidas en esta memoria descriptiva solo pueden representar una de las posibles formas tautoméricas, debería entenderse que las realizaciones preferidas abarcan cualquier forma tautomérica de la estructura dibujada.
Se entiende que la invención no se limita a las realizaciones mostradas en el presente documento para ilustración, pero abarca todas las formas de las mismas que entran dentro del alcance de la anterior divulgación.
En los ejemplos siguientes así como en toda la aplicación, las abreviaturas siguientes tienen los siguientes significados. Si no se definen, los términos tienen sus significados habitualmente aceptados.
ABREVIATURAS


MS Espectro de masas
HRMS Espectros de masas de alta resolución n-BuLi n-Butillitio
DBAD diisobutilazadicarboxilato
TFA ácido trifluoroacético
h hora
g gramo
l litro
equiv equivalente
min minuto
mmol milimoles
NaHCO3 bicarbonato sódico
n2 nitrógeno
MTBE metil tertbutiléter
ml mililitro
SiO 2 gel de sílice
NaH hidruro sódico
TLC cromatografía de capa fina
KMnO4 permanganato de potasio
NH4Cl cloruro de amonio
HPLC cromatografía líquida de alto rendimiento AMRI Albany Molecular Research Inc NH4OH hidróxido de amonio
DIAD diisopropilazadicarboxilato
HCl ácido clorhídrico
DCE dicloroetano
NH3 amoniaco
HCOOH ácido fórmico
Boc carboxilato de ferc-butilo
IPA isopropanol
mg miligramo
Ejemplo 1: 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

1) Síntesis de ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico

Etapa 1. Síntesis de 3-amino-6-(trimetilestannil)pirazin-2-carboxilato de metilo

En un matraz de fondo redondo de dos bocas de 500 ml equipado con un agitador magnético y un condensador de reflujo, se suspendieron 3-amino-6-bromopirazin-2-carboxilato de metilo (25 g, 108 mmol) y Pd(PPh3)4 (6,23 g, 5,39 mmol) en 1,2-dimetoxietano (200 ml) a ta (temperatura ambiente) en atmósfera de argón. La mezcla se desgasificó y se lavó abundantemente con argón (dos veces) y se añadió hexametilditina (29,0 ml, 140 mmol) mediante una jeringa a través de un septo de goma, la mezcla se desgasificó y se lavó abundantemente con argón otra vez y se calentó a 90 °C durante 2 h. La mezcla se enfrió a ta y se concentró al vacío. Se añadieron agua (400 ml) y acetato de etilo (200 ml), se agitó durante 20 minutos y se filtró a través de celite. El filtrado se transfirió a un embudo de separación, las fases se separaron y la fase orgánica se lavó con salmuera, se secó sobre sulfato sódico y el disolvente se eliminó a presión reducida. El residuo se filtró sobre gel de sílice con heptano, después un disolvente mezcla de (acetato de etilo 1:9 heptano) y finalmente una mezcla de disolventes (acetato de etilo 2:8 heptano) que dio 3-amino-6-(trimetilestannil)pirazin-2-carboxilato de metilo (20,92 g) de un sólido de color amarillo. LC-MS (Método básico): tiempo de ret. = 1,07 min, M+H = 317,8.
Etapa 2. Síntesis de 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxilato de metilo

En un matraz de fondo redondo de 500 ml equipado con un agitador magnético y entrada de argón, se disolvieron 3-amino-6-(trimetilestannil)pirazin-2-carboxilato de metilo (20,92 g, 55,0 mmol), 2-bromo-3-(trifluorometil)piridina (14,38 g, 60,5 mmol), Pd2(dba)3 (5,54 g, 6,05 mmol) y P(o-Tol)3 (3,79 g, 12,09 mmol) en DMF (100 ml) a ta, seguido de la adición de NEt3 (10,72 ml, 77 mmol). La mezcla de reacción se calentó a 110 °C en atmósfera de argón durante 1 h. Después de enfriar a ta, la mezcla de reacción se filtró a través de celite, se lavó con acetato de etilo y se concentró a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice usando acetato de etilo heptano que dio 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxilato de metilo (7,8 g) en forma de un sólido de color amarillo. LC-M S (Método básico): tiempo de ret. = 0,93 min, M+H = 299,0.
Etapa 3. Síntesis de ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico

En un matraz de fondo redondo de 250 ml equipado con un agitador magnético, se disolvió 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxMato de metilo (7,8 g, 23,80 mmol) en dioxano (100 ml) a ta. Se disolvió LiOH monohidrato (2,008 g, 47,6 mmol) en agua (25 ml) y se añadió a ta y se agitó durante 1 h. La suspensión se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 100 ml), la capa orgánica se extrajo de nuevo con agua (2 x 50 ml). La capa acuosa se ajustó a pH 3 con HCl conc. y se extrajo con acetato de etilo (3x 100 ml), las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se concentraron lo que dio ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico (6,89 g) en forma de un polvo de color amarillo. LC-MS (Método básico) M+H = 284,9. RMN 1H (DMSO-d6): 8 (ppm)= 13,04 (s ancho, 0,84 H), 8,92 (d, 1H, J = 4,9 Hz), 8,71 (s, 1H), 8,33 (d, 1H, J = 8,1 Hz), 7,70-7,67 (m, 3H).
2) Síntesis de 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1. (1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo
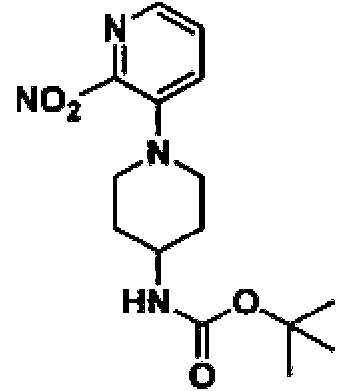
A un matraz de fondo redondo de 100 ml equipado con un agitador magnético y una entrada de nitrógeno, se le añadió THF (20 ml), 3-fluoro-2-nitropiridina (1,524 g, 10,73 mmol), piperidin-4-ilcarbamato de ferc-butilo (2,256 gramos, 11,26 mmol), N-etil-N-isopropilpropan-2-amina (3,47 g, 26,8 mmol). La mezcla se calentó a 70 °C durante 24 horas. La mezcla se enfrió y se concentró hasta un residuo espeso. El residuo se purificó por cromatografía sobre gel de sílice usando acetato de etilo-heptano lo que dio (1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (3,24 g, 98 % de rendimiento). LC-M S (Método básico): tiempo de ret. = 1,30 min, M+H = 323,3
Etapa 2. (1-(2-aminopiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

A un matraz de fondo redondo de 250 ml equipado con una barra de agitación magnética purgado con nitrógeno se le añadió (1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (3,2 gramos, 9,93 mmol), THF (75 ml) y Pd/C (1,1 g, Pd al 10 % sobre carbón húmedo). La mezcla resultante se agitó en atmósfera de hidrógeno hasta que se consumió todo el (1-(2-aminopiridin-3-il)piperidin-4-il)carbamato de ferc-butilo. Después la reacción se purgó con nitrógeno, se
añadió sulfato de magnesio y se agitó. Después, la mezcla se filtró a través de un lecho de celite. La almohadilla del filtro se lavó con exceso de DMC. El filtrado se concentró hasta un residuo espeso que se solidificó al vacío. El sólido se secó hasta un peso constante y se usó directamente (2,9 gramos, 99,9 % de rendimiento).
LC-MS (Método básico): tiempo de ret. = 1,04 min, M+H = 293.
Etapa 3. Síntesis de (1 -(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo.
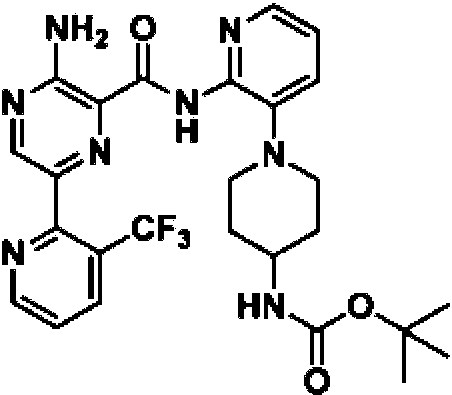
A un matraz de 25 ml se le añadió ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico (332 mg, 1,168 mmol), DMF (4 ml), hexafluorofosfato de 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio (V) (454 mg, 1,194 mmol) y N-etil-N-isopropilpropan-2-amina (0,8 ml, 4,58 mmol). La mezcla se dejó en agitación durante aproximadamente 5 minutos seguido de la adición de (1-(2-aminopiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (311 mg, 1,064 mmol). La mezcla resultante se dejó en agitación durante una noche. El residuo se inactivó con solución sat. de NaCl (150 ml) y se extrajo con EtOAc (2 x 250 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron hasta un sólido oscuro que se purificó mediante cromatografía sobre gel de sílice usando un acetato de etilo-heptano. LC-M S (Método básico): tiempo de ret. = 1,22 min, M+H = 559.
Etapa 4. Síntesis de 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

A un matraz de 100 ml se le añadió un agitador magnético, (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo y diclorometano (10 ml). La mezcla se agitó hasta que todos los sólidos se disolvieron y después se enfrió en un baño de agua enfriada con hielo en atmósfera de nitrógeno. A esta mezcla se le añadió ácido trifluoroacético (10 ml). El baño de hielo se retiró y la mezcla se agitó durante 3 horas a TA. Después, la mezcla se concentró y después el residuo se co-evaporó con tolueno (20 ml). Después el residuo resultante se mezcló con salmuera (20 ml), se saturó con NaHCO3 y se extrajo con diclorometano (3 x 50 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron hasta un sólido. El sólido se disolvió en diclorometano y se precipitó con heptano. El sólido se filtró y se secó al vacío hasta un peso constante lo que dio 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (223 mg) con un rendimiento del 95 % . LC-MS (Método básico): tiempo de ret. = 1,1 min, M+H= 459 RMN 1H (400 MHz, cloroformo-d) 810,74 (s, 1H), 8,97 - 8,84 (m, 1H), 8,77 (s, 1H), 8,30 (dd, J = 5,2, 1,9 Hz, 1H), 8,20 (dd, J = 8,0, 1,6 Hz, 1H), 7,53 (dd, J = 8,1, 4,8 Hz, 1H), 7,42 (dd, J = 7,9, 1,7 Hz, 1H), 7,06 (dd, J = 7,9, 4,9 Hz, 1H), 3 ,11 (dd, J = 11 ,1 , 4,9 Hz, 2H), 2,87 - 2,49 (m, 3H), 1,45 - 1,14 (m, 3H).
Ejemplo 2: 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida
La 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al ejemplo 1 (Método 1) en donde se usó ácido 3-amino-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxílico en lugar de ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico lo que dio 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-M)pirazin-2-carboxamida (1,21 g) con un rendimiento del 77 % . LC-MS (Método ácido): tiempo de ret. = 1,12 min, M+H = 475,2
RMN 1H (400 MHz, metanol-d4) 88,86 (s, 1H), 8,77 (dd, J = 4,7, 1,4 Hz, 1H), 8,14 (dd, J = 5,0, 1,6 Hz, 1H), 8,05 (dp, J = 8,5, 1,4 Hz, 1H), 7,72 - 7,57 (m, 2H), 7,20 (dd, J = 7,9, 4,9 Hz, 1H), 3,29 (s, 3H), 3,10 (dt, J = 12,7, 4,0 Hz, 2H), 2,82 - 2,71 (m, 2H), 2,69 - 2,53 (m, 1H), 1,93 - 1,69 (m, 2H), 1,37 (dtd, J = 13,9, 10,5, 3,6 Hz, 2H).
Ejemplo 3: 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de (4-(hidroximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

A un matraz con forma de pera de 25 ml se le añadió 3-fluoro-2-nitropiridina, (0,441 g, 3,1 mmol), (4-(hidroximetil)piperidin-4-il)carbamato de ferc-butilo (0,65 g, 2,82 mmol), N-etil-N-isopropilpropan-2-amina, (0,839 g, 6,49 mmol) y tetrahidrofurano (10 ml) y un agitador magnético. La mezcla se agitó en atmósfera de nitrógeno y se calentó a 70 °C durante 3 días. Después, la mezcla se enfrió y se concentró hasta un residuo espeso y se cromatografió directamente sobre gel de sílice (acetato de etiloheptano) lo que dio (4-(hidroximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (0,668 g, 1,858 mmol) con un rendimiento del 65,8 % .
l C-M S (Método básico): tiempo de ret. = 1,13 min, M+H = 353,5.
Etapa 2: Síntesis de (4-(metoximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

A un matraz con forma de pera de 25 ml se le añadió tolueno (8 ml), dioxano (4 ml), (4-(hidroximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (0,546 g, 1,549 mmol), sulfato de dimetilo (0,293 g, 2,324 mmol), hidróxido sódico (0,124 g, 1,549 mmol) y N,N,N-trimetil-1, cloruro de fenilmetanaminio (0,288 g, 1,549 mmol). La mezcla resultante se agitó durante 18 horas. La reacción se diluyó con 40 ml de acetato de etilo y se agitó, seguido de la adición de una pequeña cantidad (cucharada) de MgSO4. La mezcla se agitó durante aproximadamente 5 min, se filtró y se concentró. El residuo resultante se cromatografió sobre gel de sílice (gradiente de acetato de etilo-heptano al 10 100 % ) lo que dio (4-(metoximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (0,556 g, 1,487 mmol) con un rendimiento del 96 % .
LC-MS (Método básico): tiempo de ret. = 1,44 min, M+H = 367,4.
Etapa 3: Síntesis de 1-(2-aminopiridin-3-il)-4-(metoximetil) piperidin-4-il)carbamato de ferc-butilo

A un matraz de fondo redondo de 100 ml se le añadió (4-(metoximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (0,697 g, 1,902 mmol), acetato de etilo (20 ml) y paladio al 10 % sobre carbono húmedo (aproximadamente 0,7 g). El matraz se purgó con hidrógeno y se agitó en atmósfera de hidrógeno durante 16 h. A la mezcla resultante se le añadió después MgSO4 (5 gramos) y se agitó. Después, la mezcla se filtró a través de un lecho de MgSO4 debajo de un cono de nitrógeno. El filtrado se concentró a sequedad lo que dio (1-(2-aminopiridin-3-il)-4-(metoximetil)piperidin-4-il)carbamato de ferc-butilo (0,454 g, 1,322 mmol) con un rendimiento del 98 % .
LC-M S (Método básico): tiempo de ret. = 0,88 min, M+H = 337,5.
Etapa 4: Síntesis de (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-(metoximetil)piperidin-4-il)carbamato de ferc-butilo

A un matraz de 25 ml se le añadió ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico (0,188 g, 0,66 mmoles), DMF (2 ml), hexafluorofosfato de 2-(1H-benzo[d][1,2,3]triazol-1-il)-1,1,3,3-tetrametilisouronio (HBTU), (0,25 g, 0,66 mmol) y N-etil-N-isopropilpropan-2-amina (0,18 ml, 0,99 mmol). La mezcla se dejó en agitación durante aproximadamente 60 minutos seguido de la adición de (1-(2-aminopiridin-3-il)-4-(metoximetil)piperidin-4-il)carbamato de ferc-butilo (0,111 g, 0,33 mmol). La mezcla resultante se dejó en agitación durante 18 horas y después se concentró hasta un residuo espeso. El residuo se cromatografió directamente sobre gel de sílice usando acetato de etilo y heptano lo que dio (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-(metoximetil)piperidin-4-il}carbamato de ferc-butilo (0,432 g).
LC-M S (Método básico): tiempo de ret. = 1,12 min, M+H = 603,4.
Etapa 5: Síntesis de 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il) piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida
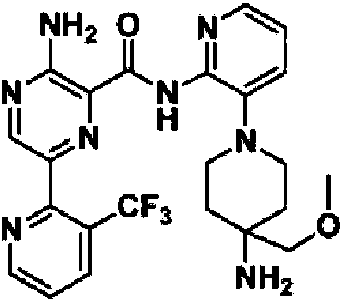
A un matraz de 100 ml se le añadió un agitador magnético, (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo (0,199 g, 0,33 mmol) y diclorometano (10 ml). La mezcla se agitó hasta que todos los sólidos se disolvieron y después se enfrió en un baño de agua enfriada con hielo en atmósfera de nitrógeno. A esta mezcla se le añadió ácido trifluoroacético (25 ml). El baño de hielo se retiró y la mezcla se agitó durante 3 horas a temperatura ambiente. Después, la mezcla se concentró y el residuo se co-evaporó después con tolueno (30 ml) 3 veces hasta un residuo espeso. El residuo se purificó por HPLC de fase inversa (método 3) lo que dio 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (0,142 g, 0,277 mmol) con un rendimiento del 84 % .
LC-M S (Método básico): tiempo de ret. = 1,04 min, M+H = 503,2. RMN 1H (400 MHz, DMSO-cfe) 810,56 (s, 1H), 9,00 (dd, J = 4,8, 1,5 Hz, 1H), 8,45 (dd, J = 8,1, 1,4 Hz, 1H), 8,11 (dd, J = 4,8, 1,6 Hz, 3H), 7,78 (dd, J = 8,0, 4,8 Hz, 1H), 7,55 (dd, J = 8,1, 1,8 Hz, 1H), 7,16 (dd, J = 7,9, 4,8 Hz, 1H), 3,13 (s, 3H), 2,92 (dt, J = 11,0, 7,1 Hz, 2H), 2,68 (dt, J = 11,2, 3,4 Hz, 2H), 1,38 - 1,00 (m, 9H), 0,94 - 0,77 (m, 1H).
Ejemplo 4: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3 morfolinoisoquinolin-1-il)pirazin-2-carboxamida
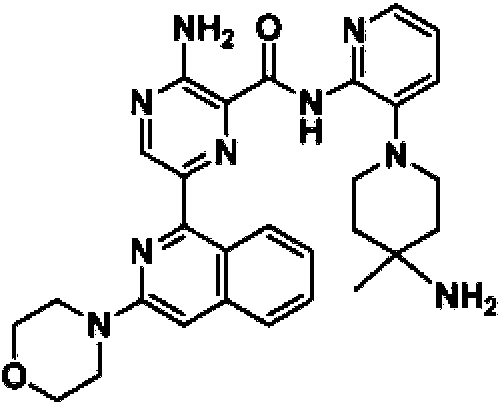
Síntesis de ácido 3-amino-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxílico

Etapa 1. Síntesis de 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo

En un matraz de fondo redondo de 500 ml equipado con un agitador magnético, una solución de 3-amino-6-bromopirazin-2-carboxilato de metilo (10 g, 43,1 mmol), bis(pinacolato)diboro (13,68 g, 53,9 mmol), KOAc (7,61 g, 78 mmol) y PdCb(dppf) (79 mg, 0,108 mmol) en dioxano (200 ml) se desgasificó y se lavó abundantemente con nitrógeno (dos veces) y después se calentó a 80 °C durante 3 h. La mezcla de reacción se enfrió a 25 °C y se añadieron 30 ml de DCM y se filtró a través de celite. Al filtrado se le añadió 60 ml de heptano. La suspensión se concentró a 1/2 volumen y se filtró. El sólido se lavó con heptano (3 X 20 ml) y se secó al vacío lo que dio 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo (12,85 g, 46,0 mmol).
LC-M S (método ácido): : tiempo de ret. = 1,04 min, M+H = 198,1
Etapa 2. Síntesis de 3-amino-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxilato de metilo

En un matraz de fondo redondo de 15 ml equipado con un agitador magnético, una solución de 4-(1-cloroisoquinolin-3-il)morfolina (673 mg, 2,412 mmol), 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo (673 mg, 2,412 mmol), K3 PO4 (3,02 ml, 3,02 mmol, 1 M) y PdCb(dppf) (118 mg, 0 , 1 6 1 mmol) en dioxano (12 ml) se desgasificó y se lavó abundantemente con nitrógeno (dos veces). La mezcla se calentó a 85 °C durante 3 h. La reacción se enfrió a ta y se añadió agua (100 ml) y se extrajo con EtOAc (3 x 50 ml), se secó sobre Na2SO4 y se concentró. El producto en bruto se purificó por columna de HPLC ácida (Método 4) lo que dio 3-amino-6-(3morfolinoisoquinolin-1-il)pirazin-2-carboxilato de metilo (460 mg, 1,259 mmol) con un rendimiento del 62 % .
LC-MS (método ácido): : tiempo de ret. = 1,39 min, M+H = 366,4.
Etapa 3. Síntesis de ácido 3-amino-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxílico

En un matraz de fondo redondo de 100 ml equipado con un agitador magnético, a una solución de 3-amino-6-(3morfolinoisoquinolin-1-il)pirazin-2-carboxilato de metilo (460 mg, 1,259 mmol) en THF (4 ml) y MeOH (4,00 ml) se le añadió LiOH.H2 O (3,15 ml, 6,29 mmol) en agua (4 ml) y se agitó a 25 °C durante 2 h. La mezcla de reacción se concentró. Después se añadieron 5 ml de agua y se acidificó con HCl 0,5 N a pH 5. La mezcla de reacción se filtró y se lavó con agua (3 X 20 ml) y se secó, lo que proporcionó ácido 3-amino-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxílico (377 mg, 1,073 mmol) con un rendimiento del 85 % . LC-MS (método ácido): : tiempo de ret. = 1,02 min, M+H = 352,4.
Etapa 4. Síntesis de 3-amino-N-(3-(4-metil-4-pivalamidopiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1 -il)pirazin-2-carboxamida

En un matraz de fondo redondo de 100 ml equipado con un agitador magnético, a una solución de ácido 3-amino-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxílico en DMF (3 ml) se le añadió DIEA (0,149 ml, 0,854 mmol) y HATU (156 mg, 0,410 mmol), seguido de (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (105 mg, 0,342 mmol) y se agitó a 25 °C durante 60 h. A la mezcla de reacción se le añadió 80 ml de agua y se extrajo con EtOAc (3 X 40 ml), se secó sobre Na2SO4 y se concentró. El producto en bruto se purificó por HPLC básica (Método 3) lo que dio 3-amino-N-(3-(4-metil-4-pivalamidopiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida (108 mg, 0,169 mmol) con un rendimiento del 49 % .
LC-MS (método ácido): : tiempo de ret. = 2,30 min, M+H = 640,7.
Etapa 5. Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida

En un matraz de fondo redondo de 100 ml equipado con un agitador magnético, se enfrió TFA (0,650 ml, 8,44 mmol) a -20 °C, se añadió una solución de 3-amino-N-(3-(4-metil-4-pivalamidopiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida (108 mg, 0,169 mmol) en DCM (0,18 ml) lentamente. La mezcla de reacción se agitó a 25 °C durante 45 min. La mezcla de reacción se concentró. El producto en bruto se purificó por HPLC básica (método 3) lo que dio 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-morfolinoisoquinolin-1-il)pirazin-2-carboxamida (33 mg, 0,058 mmol) con un rendimiento del 34 % . LC-M S (método ácido): : tiempo de ret. = 1,22 min, M+H = 540,6. RMN 1H (METANOL-d4) d: 8,89 (s, 1H), 8,47 (d, J = 8,5 Hz, 1H), 8,11 (dd, J = 4,9, 1,6 Hz, 1H), 7,76 (d, J = 8,5 Hz, 1H), 7,47-7,64 (m, 2H), 7,28 (ddd, J = 8,5, 7,0, 1,1 Hz, 1H), 7,14 (dd, J = 7,8, 5,0 Hz, 1H), 7,03 (s, 1H), 3,82-3,91 (m, 4H), 3,56-3,65 (m, 4H), 2,69-2,80 (m, 2H), 2,56-2,68 (m, 2H), 0,79-1,02 (m, 4H), 0,49 (s a, 3H).
Ejemplo 5: 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Síntesis de (1-(2-aminopiridin-3-il)-4-(((íercbutildimetMsilM)oxi)metM)piperidin-4-M)carbamato de ferc-butilo

Etapa 1. Síntesis de (4-(hidroximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

En un matraz de fondo redondo de 25 ml se combinó 3-fluoro-2-nitropiridina (0,441 g, 3,1 mmol), (4-(hidroximetil)piperidin-4-il)carbamato de ferc-butilo (0,65 g, 2,82 mmol), N-etil-N-isopropilpropan-2-amina (1,2 ml) y THF (10 ml). La mezcla se calentó a 70 °C durante 48 h. La mezcla se enfrió y se concentró hasta un residuo espeso. El residuo se purificó por cromatografía sobre gel de sílice usando acetato de etilo y heptano lo que dio (4-(hidroximetil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (0,668 g) con un rendimiento del 98 % .
LC-MS (Método básico): tiempo de ret. = 1,13 min, M+H = 353,5.
Etapa 2. Síntesis de (4-(((ferc-butildimetilsilil)oxi)metil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo.

En un matraz de fondo redondo de 50 ml se combinó (4-(hidroximetil)-1-(2-nitropiridin-3-il)piperidin-4-il}carbamato de ferc-butilo (1,08 g, 3,06 mmol), DMF (10 ml) e imidazol (0,459 g, 6,74 mmol) seguido de ferc-butilclorodimetilsilano (0,554 g, 3,68 mmol). La mezcla se dejó en agitación hasta que todo el alcohol se convirtió en el silil éter. Después, la mezcla se concentró y se cromatografió sobre gel de sílice usando acetato de etilo y heptano lo que dio (4-(((ferc
butildimetilsilil)oxi)metil)-1-(2-nitropiridin-3-M)piperidin-4-il)carbamato de ferc-butilo (1,178 g) con un rendimiento del 99 % .
LC-MS (Método básico): tiempo de ret. = 1,84 min, M+H = 467,3
Etapa 3. Síntesis de (1-(2-aminopiridin-3-il)-4-(((fercbutildimetilsilil)oxi)metil)piperidin-4-il)carbamato de ferc-butilo

Comenzando con (4-(((ferc-butildimetilsilil)oxi)metil)-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (1,178 g), el compuesto nitro se trató con hidrógeno y paladio como se describe en ejemplo 1 lo que dio (1-(2-aminopiridin-3-il)-4-(((fercbutildimetilsilil)oxi)metil)piperidin-4-il)carbamato de ferc-butilo (0,954 g) con un rendimiento del 85 % .
LC-MS (Método básico): tiempo de ret. = 1,78 min, M+H = 437,3
Etapa 4. Síntesis de (1-(2-(3-amino-6-(3-(trifluorometN)pindin-2-N)pirazin-2-carboxamido)piridin-3-N)-4-(((fercbutildimetilsilil)oxi)metil)piperidin-4-il)carbamato de ferc-butilo

Comenzando con (1-(2-aminopiridin-3-il)-4-(((fercbutildimetilsilil)oxi)metil)piperidin-4-il)carbamato de ferc-butilo (0,2 g, 0,459 mmol), se preparó (1-(2-(3-amino-6-(3-(tnfluorometN)piridin-2-N)pirazin-2-carboxamido)pindin-3-N)-4-(((tercbutildimetilsilil)oxi)metil)piperidin-4-il)carbamato de ferc-butilo como se describe en el ejemplo 1 y se usó directamente.
Etapa 5. Síntesis de 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

El (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-(((fercbutildimetilsilil)oxi)metil)piperidin-4-il)carbamato de ferc-butilo de la etapa 4 se trató con ácido trifluoroacético de un modo como el descrito en el ejemplo 1 lo que dio 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (0,108 g, 0,217 mmol) con un rendimiento del 21 % . LC-M S (Método básico): tiempo de ret. = 0,92 min, M+H = 489,2. RMN 1H (400 MHz, DMSO-de) 8 10,55 (s, 1H), 8,97 (dd, J = 4,8, 1,5 Hz, 1H), 8,77 (s, 1H), 8,39 (dd, J = 8,2, 1,5 Hz, 1H), 8,26 - 7,65 (m, 4H), 7,56 (dd, J = 8,2, 1,9 Hz, 1H), 7,16 (dd, J = 7,9, 4,7 Hz, 1H), 4,34 (s, 1H), 3,23 - 3,07 (m, 3H), 2,91 (td, J = 10,6, 4,2 Hz, 2H), 2,76 - 2,38 (m, 13H), 1,69 - 1,43 (m, 3H).
Ejemplo 6 : 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-M)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (0,0865 g) con un rendimiento del 85 % .
LC-MS (método ácido): tiempo de ret. = 1,24 min, M+H = 505,2. RMN 1H (DMSO-d6) 8: 10,75 (s, 1H), 8,92 (s, 1H), 8,77 (dd, J = 4,6, 1,3 Hz, 1H), 8,11 (dd, J = 4,8, 1,6 Hz, 1H), 8,08 (s a, 1H), 8,06 (dt, J = 8,4, 1,4 Hz, 1H), 7,65 (dd, J = 8,4, 4,6 Hz, 1 H), 7,60 (dd, J = 8,0, 1,7 Hz, 1H), 7 ,17 (dd, J = 7,8, 4,8 Hz, 1H), 4,48 (s, 1H), 2,97 (td, J = 11,3 , 3,0 Hz, 2H), 2,75 (dc, J = 7,6, 3,9 Hz, 4H), 1,77 (s, 2H), 1,47 - 1,16 (m, 4H).
Ejemplo 7: 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Síntesis de (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo

Etapa 1. Síntesis de (1-(2-(3-amino-6-bromopirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo

En un matraz de fondo redondo de 100 ml equipado con un agitador magnético, una solución de ácido 3-amino-6-bromopirazin-2-carboxílico (1,044 g, 4,79 mmol), (1-(2-aminopiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (1,4 g, 4,79 mmol), DIPEA (2,091 ml, 11,97 mmol) y HATU (2,185 g, 5,75 mmol) en DMF (15 ml) se agitó a 25 °C durante 15 h. La mezcla de reacción se inactivó con 30 ml de agua y se extrajo con EtOAc (3 X 20 ml). El lavado con acetato de
etilo se secó sobre Na2SO4 y se concentró. El producto en bruto se purificó por cromatografía sobre gel de sílice usando acetato de etilo y heptano lo que dio 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-bromopirazin-2-carboxamida (1,76 g, 3,57 mmol) con un rendimiento del 74 % . LC-MS (método ácido): : tiempo de ret. = 1 ,17 min, M+H = 492,3.
Etapa 2. Síntesis de (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo

En un matraz de fondo redondo de 15 cerrado herméticamente equipado con un agitador magnético, una solución de 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-bromopirazin-2-carboxamida (220 mg, 0,447 mmol), bis(pinacolato)diboro (142 mg, 0,559 mmol), KOAc (79 mg, 0,804 mmol) y PdCl2(dppf) (16,35 mg, 0,022 mmol) en dioxano (2,5 ml) se desgasificó y se lavó abundantemente con nitrógeno (dos veces) y se calentó a 80 °C durante 3 h. La mezcla de reacción se enfrió a ta, se diluyó con 30 ml de DCM y se filtró a través de celite. El filtrado se diluyó después con 60 ml de heptano y después se concentró hasta 1/2 del volumen. La mezcla se filtró, el sólido se lavó con heptano (3 X 20 ml) y se secó al vacío lo que dio (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo (173 mg, 0,321 mmol) con un rendimiento del 71 % . LC-M S (método ácido): : tiempo de ret. = 0,91 min, M+H = 458,4.
Etapa 3. Síntesis de (1-(2-(3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo

En un matraz de fondo redondo de 15 ml equipado con un agitador magnético, una solución de (1-(2-(3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo (316 mg, 0,497 mmol), 4-(6-cloro-5-(trifluorometil)piridin-2-il)morfolina (190 mg, 0,710 mmol), K3 PO4 (1 M) (0,923 ml, 0,923 mmol) y PdCb(dppf) (41,6 mg, 0,057 mmol) en dioxano (6 ml) se desgasificó y se lavó abundantemente con nitrógeno (dos veces). Después de agitarse a 80 °C durante 2 h., la reacción se enfrió a ta. La mezcla de reacción se añadió a agua (100 ml) y se extrajo con EtOAc (3 x 50 ml), después se secó sobre Na2SO4 y se concentró. El producto en bruto se purificó por HPLC (método ácido 3) lo que dio (1-(2-(3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de ferc-butilo (62 mg, 0,096 mmol) 13 % de rendimiento. l C-M S (método ácido): tiempo de ret. = 1,18 min, M+H= 645,7.
Etapa 4. Síntesis de 3-amino-N-(3 -(4-aminopiperidin-1-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida
En un matraz de fondo redondo de 100 ml equipado con un agitador magnético, se enfrió TFA (0,370 ml, 4,81 mmol) a -20 °C, se añadió una solución de 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (62 mg, 0,096 mmol) en DCM (2 ml) y se agitó a 25 °C durante 45 min. La mezcla de reacción se concentró. El producto en bruto se purificó por HPLC (método básico 3) lo que dio 3-amino-N-(3-(4-aminopiperidin-1-M)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (26,3 mg, 0,048 mmol) con un rendimiento del 50 % .
LC-MS (método ácido): tiempo de ret. = 1,03 min, M+H = 545,6 (M+H). RMN 1H (METANOL-d4) d: 8,89 (s, 1H), 8,76 (s, 1H), 8,12 (dd, J = 4,9, 1,4 Hz, 1H), 7,63 (dd, J = 8,0, 1,5 Hz, 1H), 7,20 (dd, J = 7,9, 4,9 Hz, 1H), 3,89-4,02 (m, 4H), 3,78 (t, J = 4,9 Hz, 4H), 3,07 (d, J = 12,0 Hz, 2H), 2,52-2,79 (m, 3H), 1,79 (d, J = 10,8 Hz, 2H), 1,18 -1,44 (m, 2H).
Ejemplo 8: 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamida (Ejemplo de referencia)

Etapa 1: Síntesis de (1-(2-(3-amino-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de tere-butilo

A un matraz de fondo redondo de 25 ml se le añadió ácido (5-amino-6-((3-(4-((tere-butoxicarbonil)amino)piperidin-1-il)piridin-2-il)carbamoil)pirazin-2-il)borónico (0,36 g, 0,787 mmol), 4-(4-clorotiazol-2-il)morfolina (0,161 g, 0,787 mmol), dicloruro de Pd (dppe) (0,085 g, 0,116 mmol), fosfato potásico 1 M (1 ml) y un agitador magnético. La mezcla resultante se desgasificó con nitrógeno y después se colocó en un baño de aceite precalentado a 80 °C y se calentó durante 2 horas. La reacción se retiró del calor, se enfrió y después se vertió en 100 ml de diclorometano. Se añadió sulfato de magnesio para secar la reacción, seguido de filtración y concentración hasta un residuo espeso. El residuo se cromatografió sobre gel de sílice usando acetato de etilo y heptano lo que dio (1-(2-(3-amino-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de tere-butilo (0,187 g, 0,289 mmol) con un rendimiento del 37 % .
LC-MS (método básico): tiempo de ret. = 1,30 min, M+H = 582,5.
Etapa 2: Síntesis de 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamida

A un matraz de 100 ml se le añadió un agitador magnético, (1-(2-(3-amino-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamido)piridin-3-il)piperidin-4-il)carbamato de tere-butilo (0,113 g, 0,194 mmol) y diclorometano (5 ml). La mezcla se agitó hasta que todos los sólidos se disolvieron y después se enfrió en un baño de agua enfriada con hielo en
atmósfera de nitrógeno. A esta mezcla se le añadió ácido trifluoroacético (15 ml). El baño de hielo se retiró y la mezcla se agitó durante 3 horas a temperatura ambiente. Después, la mezcla se concentró y el residuo se co-evaporó después con tolueno (30 ml) 3 veces hasta un residuo espeso. El residuo se purificó por el método 3, lo que dio 3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(2-morfolinotiazol-4-il)pirazin-2-carboxamida (0,026 g, 0,052 mmol) con un rendimiento del 27 % .
LC-MS (Método básico): tiempo de ret. = 1,16 min, M+H = 482,6. RMN 1H (400 MHz, cloroformo-d) 8 10,97 (s, 2H), 8,85 (s, 2H), 8,23 (d, J = 4,8 Hz, 2H), 7,36 (d, J = 8,0 Hz, 3H), 7,22 (d, J = 18,0 Hz, 4H), 7,03 - 6,95 (m, 3H), 3,83 -3,76 (m, 10H), 3,62 (s, 0H), 3,53 - 3,45 (m, 10H), 3,08 (d, J = 11,5 Hz, 5H), 2,74 (s, 2H), 2,66 (t, J = 11,8 Hz, 6H), 1,89 (d, J = 12,7 Hz, 5H), 1,54 (d, J = 11,9 Hz, 6H), 1,46 (s, 8H), 1,18 (s, 5H).
Ejemplo 9: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de (4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

A una solución de 3-fluoro-2-nitropiridina (11,2 g, 81 mmol) en dioxano (200 ml) se le añadió (4-metilpiperidin-4-il)carbamato de ferc-butilo (26 g, 121 mmol). se añadió base de Huenig (28,3 ml, 162 mmol) y la mezcla se calentó a 85 °C durante 18 h. La reacción se enfrió a TA y se concentró para dar un sólido de color pardo. Los sólidos se lavaron con 200 ml de 4:1 de heptano:EtOAc. La suspensión se concentró hasta la mitad de su volumen y se filtró para recoger (26,2 g, 78 mmol, 96 % ) un sólido de color pardo. LC-M S (Método ácido): tiempo de ret. = 1,46 min, M+H = 337,4 Etapa 2: Síntesis de (4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

A una solución de (4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (11,6 g, 37,2 mmol) en acetato de etilo (200 ml). Se le añadió Pd al 10 % -C (3,48 g) y se agitó en un globo de presión de H2 a TA durante 4 h. Se añadió una pequeña cantidad de MgSO4 a la reacción y después la mezcla de reacción se filtró a través de un lecho de celite, después se lavó con acetato de etilo (100 ml) y el filtrado se concentró para proporcionar un sólido de color pardo (8,54 g, 27,9 mmol, 85 % ). LC-MS (Método ácido): tiempo de ret. = 0,91 min, M+H = 307,4.
Etapa 3: Síntesis de (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo

A una solución de acido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazm-2-carboxmco en dimetil formamida (125 ml) se le añadió hexafluorofosfato de ((1H-benzo[d][1,2,3]triazol-1-il)oxi)tris(dimetilamino) fosfonio (V) (1,8 g, 4,24 mmol) y 4-metilmorfolina (1 ml, 9,79 mmol). La reacción se agitó a TA durante 40 minutos. Se añadió (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo en dimetilformamida (25 ml) y la reacción se agitó durante 16 h a TA. La mezcla de reacción se diluyó con EtOAc y se lavó con NaHCO3 (ac.) (3 x 200 ml) y salmuera (1x 200 ml). La fase orgánica se secó con Na2SO4, se filtró y se concentró. El producto en bruto se recogió en acetonitrilo (30 ml) y la mezcla se dejó en reposo a TA durante un periodo de tiempo. Se recogió un sólido de color amarillo por filtración (1,39 g, 74 % ). L C -m S (Método ácido): tiempo de ret. = 1,13 min, M+H = 573,3.
Etapa 4: Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Una solución de (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (1,39 g, 2,06 mmol) en diclorometano (10 ml) se enfrió a 0 °C. Se añadió ácido 2,2,2-trifluoroacético (2,4 ml, 31 mmol) gota a gota a la solución. La mezcla se dejó calentar a 22 °C y se agitó durante 4 h. La mezcla de reacción se concentró para eliminar el DCM y el exceso de TFA. Se produjo un aceite de color rojo, que se recogió en 100 ml de CHCb/IPA 3:1 y se añadió NaHCO3 ac. saturado para neutralizar la solución. Después, la mezcla se agitó a 22 °C durante 16 h. La mezcla se transfirió a un embudo de decantación y las capas acuosas se lavaron con CHCb/IPA 3:1 (3X 100 ml). Las fases orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron para proporcionar un sólido de color amarillo. El producto en bruto se recristalizó en acetonitrilo. Se recogió un sólido de color amarillo por filtración (0,82 g, 83 % ). LC-MS (Método ácido): tiempo de ret. = 0,75 min, M+H = 473,2. RMN 1H (400 MHz, metanol-d4) 88,92 (dd, J = 5,1, 1,4 Hz, 1H), 8,68 (s, 1H), 8,47 - 8,27 (m, 1H), 8,12 (dd, J = 4,9, 1,6 Hz, 1H), 7,83 - 7,50 (m, 2H), 7,18 (dd, J = 7,9, 4,9 Hz, 1H), 3,02 - 2,65 (m, 4H), 1,54 - 1,24 (m, 4H), 0,74 (s, 3H).
Ejemplo 10: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

Se preparó la 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida de un modo como el descrito para el ejemplo 1, método 1 (1,41 g) con un rendimiento del 77 % . LC-MS (método ácido): tiempo de ret. = 1,0 min, M+H = 489,1 RMN 1H (400 MHz, metanol-d4) 88,81 (s, 1H), 8,73 (dd, J = 4,7, 1,3 Hz, 1H), 8,13 (dd, J = 4,9, 1,6 Hz, 1H), 8,01 (dp, J = 8,4, 1,4 Hz, 1H), 7,75 - 7,54 (m, 2H), 7,19 (dd, J = 7,9, 4,9 Hz, 1H), 3,04 - 2,74 (m, 4H), 1,67 - 1,35 (m, 4H), 0,82 (s, 3H).
Ejemplo 11: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamida

Etapa 1: Síntesis de 4-(4-doro-5-(trifluorometil)pirimidin-2-il)morfolina

A un matraz de fondo redondo se le añadió morfolina (0,897 g, 10,3 mmol) y una solución de dicloroetano-tercbutanol (1:1, 30 ml) agitado en atmósfera de nitrógeno y enfriado en un baño de agua enfriada con hielo. A esta mezcla se le añadió cloruro de cinc (5,45 g, 40 mmol) en una porción y se agitó durante 30 minutos seguido de la adición de 2,4-dicloro-5-(trifluorometil)pirimidina (2,17 g, 10 mmol). La solución resultante se agitó a temperatura de baño de agua enfriada en hielo seguido de la adición rápida gota a gota de N-etil-N-isopropilpropan-2-amina. La reacción se mantuvo en agitación a temperatura de baño de agua enfriada en hielo durante 2 horas y después se dejó calentar a temperatura ambiente y se agitó durante 18 horas más. La reacción se vertió en 200 ml de DCM, se agitó y se filtró. El filtrado se concentró y se cromatografió sobre gel de sílice usando acetato de etilo heptano lo que proporcionó 4-(4-cloro-5-(trifluorometil)pirimidin-2-il)morfolina (2,1 g, 7,61 mmol) con un rendimiento del 76 % . LC-M S (método básico): tiempo de ret. = 1,40 min, M+H = 268,4.
Etapa 2: Síntesis de (1-(2-(3-amino-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de terc-butilo

A un recipiente de 10 ml con tapón de rosca se le añadió ácido 3-amino-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxílico (0,104 g, 0,281 mmol), que se preparó de modo análogo al ejemplo 4, DMF (2 ml), N-etil-N-isopropilpropan-2-amina (0,12 ml, 0,689 mmol) y hexafluorofosfato de 2-(1H-benzo[d][1,2,3]triazol-1-il)-1,1,3,3-tetrametilisouronio (V) (0,128 g, 0,337 mmol). La mezcla se agitó durante 1 hora. A la mezcla resultante se le añadió (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de terc-butilo (0,095 g, 0,309 mmol) y se agitó durante 20 horas. La reacción se concentró y se obtuvo (1-(2-(3-amino-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de terc-butilo con un rendimiento del 14 % y se usó directamente. LC-M S (método básico): tiempo de ret. = 1,46 min, M+H = 659,4.
Etapa 3: Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamida

A un matraz de 25 ml se le añadió (1-(2-(3-amino-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (0,185 g, 0,281 mmol) y diclorometano (10 ml) que después se agitó y se enfrió en un baño de agua enfriada con hielo en atmósfera de nitrógeno. A esta mezcla resultante se le añadió ácido trifluoroacético (20 ml) y se agitó y después se dejó calentar a temperatura ambiente. La mezcla se agitó durante dos horas y media. Después, la mezcla se concentró y después se coevaporó con tolueno (30 ml). Esta coevaporación se realizó tres veces. La mezcla se cromatografió después por el método de HPLC 4. Después el sólido resultante se trituró con agua caliente, se dejó enfriar. El material sólido obtenido se filtró y se secó hasta un peso constante lo que dio 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(2-morfolin-5-(trifluorometil)pirimidin-4-il)pirazin-2-carboxamida (0,0104 g, 0,281 mmol) con un rendimiento del 14 % . LC-MS (método básico): tiempo de ret. = 1,15 min, M+H = 559,4. RMN 1H (400 Mh z , cloroformo-d) 810,55 (s, 1H), 8,84 (s, 1H), 8,69 (s, 1 H), 8,29 (dd, J = 4,9, 1,6 Hz, 1H), 7,45 (dd, J = 8,0, 1,7 Hz, 1H), 7,32 (s, 0H), 7,10 (dd, J = 7,8, 4,8 Hz, 1H), 5,64 (d, J = 81,5 Hz, 1H), 5,32 (s, 1H), 3,97 (t, J = 4,8 Hz, 4H), 3,81 (t, J = 4,7 Hz, 4H), 3,06 - 2,75 (m, 4H), 1,61 (ddd, J = 13,5, 9,3, 4,0 Hz, 2H), 1,42 (dt, J = 13,3, 3,9 Hz, 2H), 1,26 (s, 4H), 0,97 (s, 3H), 0,94 - 0,76 (m, 1H).
Ejemplo 12: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamida

Etapa 1: (1-(2-(3-amino-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo
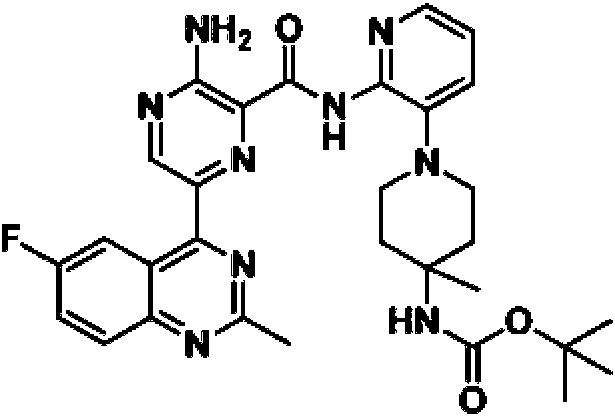
La mezcla de (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (293 mg, 0,529 mmol), (4-cloro-5-fluoro-7H-pirrolo[2,3-d]pirimidina (125 mg, 0,637 mmol) y K3 PO4 (1591 pl, 1,591 mmol, solución acuosa 1 M) en dioxano (2 ml) se desgasificó con corriente de N2 durante 10 min. Después se añadió PdCb(dppf) (19,41 mg, 0,027 mmol). La mezcla de reacción se desgasificó durante 5 min y después se calentó a 80 °C en atmósfera de N2 durante 5 h. La mezcla de reacción se enfrió a TA y se filtró a través de un lecho de Celite y se lavó con DCM. Al filtrado se le añadió agua y DCM. La fase acuosa se extrajo otra vez con DCM 2x. Las fases de DCM combinadas se concentraron al vacío. El residuo se disolvió en MeOH y varias gotas de agua, después se filtró. La solución resultante se separó después con HPLC prep. (columna C-18, ACN al 25-50 % /H 2 O con TFA al 0,1 % ). Las fracciones deseadas se combinaron y después se añadió más solución acuosa de DCM y Na2CO32 M para hacer a fase acuosa de pH 8. La fase ac. se extrajo con DCM 2x. Las fases combinadas de DCM se evaporaron para proporcionar 141 mg (69 % de rendimiento) como (1-(2-(3-amino-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo. LC/MS: m/z M+H= 588,6
Etapa 2: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2
carboxamida

A una solución de TFA (336 pl, 4,36 mmol) en DCM (1 ml) a 0 °C se le añadió (1-(2-(3-amino-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (128 mg, 0,218 mmol) en DCM (2 ml). La mezcla de reacción se concentró al vacío. El residuo se diluyó con DCM y agua y después se añadió Na2CO32 N para alcanzar un pH 12 de la fase acuosa. La fase acuosa básica se extrajo mediante DCM 3x. Las fases orgánicas combinadas se concentraron. El residuo se disolvió en MeOH/MeCN y después se separaron por HPLC prep. (columna C-18, 10-30 % de ACN/H2 O con TFA al 0,1 %). Las fracciones deseadas se combinaron y se añadió Na2CO3 2 N para lograr pH 11. La solución básica resultante se extrajo mediante DCM 3x. Las fases de DCM combinadas se concentraron al vacío, lo que proporcionó 74 mg (70 % de rendimiento) de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-fluoro-2-metilquinazolin-4-il)pirazin-2-carboxamida. LC/MS: m/z M+H = 488,2 RMN 1H (METANOL-d4) 8: 9,15 (s, 1H), 8,52 (dd, J = 9,7, 2,9 Hz, 1H), 8,17 (dd, J = 5,0, 1,5 Hz, 1H), 8,10 (dd, J = 9,3, 5,3 Hz, 1H), 7,87 (ddd, J = 9,2, 8,2, 2,9 Hz, 1H), 7,68 (dd, J = 7,9, 1,6 Hz, 1H), 7,21 (dd, J = 7,9, 4,9 Hz, 1H), 3,37 (s, 2H), 2,93 (s, 3H), 2,80-2,90 (m, 2H), 2,68-2,78 (m, 2H), 0,98-1,07 (m, 2H), 0,87-0,97 (m, 2H), 0,42 (s, 3H) Ejemplo 13: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

1) Síntesis de ácido 3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico

Etapa 1. Síntesis de 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo

Una mezcla de 3-amino-6-bromopirazin-2-carboxilato de metilo (8,8 g, 38 mmol), 4,4,4',4',5,5,5',5'-octametil-2,2'bi(1,3,2-dioxaborolano) (9,6 g, 38 mmol) y acetato potásico (11 g, 110 mmol) en dioxano (200 ml) se desgasificó y después se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (N) (1,4 g, 1,9 mmol). La mezcla resultante se agitó y se calentó a 80 °C en atmósfera de nitrógeno durante 15 h. Se añadió más 4,4,4',4',5,5,5',5'-octametil-2,2'-bi(1,3,2-dioxaborolano) (1,9 g, 7,6 mmol) y la mezcla de reacción se calentó durante otras 3 h. La mezcla de reacción se enfrió a temperatura ambiente y la mezcla se diluyó con diclorometano y se filtró a través de un lecho de tierra de diatomeas. El filtrado se concentró a presión reducida y se purificó por cromatografía sobre gel de sílice con un gradiente de 0 % -10 % de metanol en diclorometano para proporcionar el producto deseado en forma de un sólido de color pardo (9,5 g, 90 % de rendimiento). LC-M S (método ácido) tiempo de ret. = 0,42 min, M+H = 198,1 (LC-MS método ácido).
Etapa 2. Síntesis de 2-bromo-4-metoxi-3-(trifluorometil)piridina

Una mezcla de 2-bromo-4-cloro-3-(trifluorometil)piridina (200 mg, 0,768 mmol) e hidróxido sódico acuoso (6 N, 0,640 ml) en metanol (7 ml) se agitó a temperatura ambiente durante 2 h. La mezcla se extrajo con acetato de etilo (3 x 10 ml) y las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío para obtener 165 mg, 84 % de rendimiento) de un polvo de color blanco. LC-M S (método ácido): tiempo de ret. = 1 ,11 min, M+H = 257,9.
Etapa 3. Síntesis de 3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxilato de metilo

Una solución de 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo (180 mg, 0,645 mmol), 2-bromo-4-metoxi-3-(trifluorometil)piridina (165 mg, 0,645 mmol) y fosfato potásico 1 M (0,838 ml, 0,838 mmol) en tetrahidrofurano (4 ml) se desgasificó al vacío durante 10 min y después se añadió cloro(2-diciclohexilfosfino-2',4',6'-triisopropil-1,1'-bifenil)[2-(2'-amino-1,1'-bifen-il)paladio (II) (25 mg, 0,032 mmol). La mezcla se calentó a 50 °C en atmósfera de nitrógeno durante 3 h. La reacción se enfrió a temperatura ambiente y se diluyó con acetato de etilo. A la capa acuosa se le añadió cloruro de amonio sat. y se extrajo de nuevo con más acetato de etilo. La solución orgánica combinada se purificó por cromatografía sobre gel de sílice con un gradiente de 0 % -100 % de acetato de etilo en heptano para proporcionar el producto deseado en forma de un sólido de color blanco (14 mg, 7 % de rendimiento). LC-MS (método ácido): tiempo de ret. = 0,92 min, M+H= 329,3.
Etapa 4. Síntesis de ácido 3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico

Una mezcla de 3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxilato de metilo (14 mg, 0,043 mmol) e hidróxido sódico acuoso (6 N, 0,071 ml, 0,43 mmol) en metanol (3 ml) se agitó a temperatura ambiente durante 5 h. Después, se calentó a 60 °C durante 1 h. Se ajustó el pH a 5 usando HCl conc. La solución se concentró al vacío para obtener un polvo de color blanco. Este se usó en la etapa siguiente sin más purificación. LC-MS (método ácido): tiempo de ret. = 0,81 min, M+H = 315,0.
2) Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida
Etapa 1. Síntesis de (4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

Una mezcla de 3-fluoro-2-nitropiridina (550 mg, 3,87 mmol), (4-metilpiperidin-4-il)carbamato de ferc-butilo (871 mg, 4,06 mmol) y trietilamina (1,61 ml, 11,6 mmol) en dioxano (10 ml) se calentó a 100 °C durante 6 h. La mezcla se enfrió y se concentró hasta un residuo espeso. El residuo se diluyó con agua y se extrajo con diclorometano (3 x 20 ml). La capa orgánica combinada se purificó por cromatografía sobre gel de sílice con un gradiente de 0 % -100 % de acetato de etilo en heptano para proporcionar el producto deseado en forma de un sólido de color amarillo (1,3 g, 100 % de rendimiento). LC-MS (Método básico): tiempo de ret. = 1,28 min, M+H = 337,2.
Etapa 2. Síntesis de (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo

A una solución de (4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (550 mg, 1,64 mmol) en etanol (10 ml) se le añadió Pd/C (17 mg, Pd al 10 % sobre carbón húmedo). La mezcla resultante se agitó en atmósfera de hidrógeno hasta que se consumió todo el (4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo. Después la reacción se purgó con nitrógeno. Después, la mezcla se filtró a través de un lecho de celite. La almohadilla del filtro se lavó con exceso de DMC. El filtrado se concentró hasta un residuo espeso que solidificó al vacío. El sólido se secó hasta peso constante y se usó directamente (500 mg, 100 % de rendimiento). LC-MS (Método básico): tiempo de ret. = 0,78 min, M+H = 308,3.
Etapa 3. Síntesis de (1-(2-(3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo.

Una mezcla de ácido 3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico (14 mg, 0,043 mmol), (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (13 mg, 0,043 mmol), N-etil-N-isopropilpropan-2-amina (0,019 ml, 0,11 mmol) y hexafluorofosfato de (1-ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilamino-morfolinocarbenio (COMU) (44 mg, 0,10 mmol) en dimetilformamida (0,4 ml) se agitó en atmósfera de nitrógeno a temperatura ambiente durante 60 h. El residuo se concentró hasta un sólido de color oscuro que se purificó por HPLC (Sunfire 30x50 mm 5 |jm columna ACN/H2O con TFA al 0,1 % 75 ml/min, inyección de ,5 ml) para proporcionar un sólido de color amarillo (25 mg, 81 % de rendimiento). LC-M S (Método básico): tiempo de ret. = 1,18 min, M+H = 603,2.
Etapa 4. Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Una mezcla de (1-(2-(3-amino-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (25 mg, 0,041 mmol) y ácido trifluoroacético (0,32 ml, 4,2 mmol) en diclorometano (2 ml) se agitó a temperatura ambiente durante 12 h. El residuo resultante se concentró hasta una goma de color oscuro que se purificó por HPLC (X-Bridge 30x50 mm 5 um columna ACN/H 2 O con NH4OH 5 mM 75 ml/min., en inyección de 5 ml) para proporcionar un sólido de color amarillo (5 mg, 23 % de rendimiento). LC-M S (Método básico): tiempo de ret. = 0,79 min, M+H = 502,9. RMN 1H (400 MHz, METANOL-d4) 8 ppm 8,70 (d, J = 5,77 Hz, 1 H) 8,53 (s, 1 H) 8,11 (dd, J = 5,02, 1,51 Hz, 1 H) 7,67 (dd, J = 7,91, 1,63 Hz, 1H) 7,40 (d, J = 5,77 Hz, 1 H) 7,16 (dd, J = 7,91, 4,89 Hz, 1 H) 4,08 (s, 3 H) 2,85 - 3,00 (m, 2 H) 2,72 - 2,85 (m, 2 H) 1,28 - 1,50 (m, 4 H) 0,81 (s, 3 H).
Ejemplo 14: 3-ammo-N-(3-(4-ammo-4-metMpipendm-1-M)pmdm-2-N)-6-(6-(3,3-difluoroazetidm-1-N)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1. Síntesis de (1-(2-(3-amino-6-bromopirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de fercbutilo.

En un matraz de fondo redondo de 100 ml equipado con un agitador magnético HBTU (2,23 g, 5,87 mmol), ácido 3-amino-6-bromopirazin-2-carboxílico (1,17 g, 5,39 mmol) y N-etil-N-isopropilpropan-2-amina (1,28 ml, 7,34 mmol) se dejó en agitación en DMF (15 ml) durante 15 minutos después de lo cual se añadió (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (1,5 g, 4,9 mmol) en una porción. La reacción se dejó en agitación durante 16 h. La reacción se vertió entonces en DMF y se extrajo tres veces con 50 ml de acetato de etilo. Los extractos orgánicos se combinaron y se secaron con salmuera seguida de sulfato sódico anhidro. El residuo se purificó por gel de sílice usando cromatografía con gradiente de etanol - acetato de etilo al 0 -10 % lo que proporcionó (1-(2-(3-amino-6-bromopirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo puro (1,86 g, 67,5 % de rendimiento) hasta que se evaporaron las fracciones que contenían el producto deseado. LC-M S (método ácido): tiempo de ret. = 1,17 min, M+H = 508,3.
E ta p a 2. S ín te s is d e (1 -(2 -(3 -a m in o -6 -(4 ,4 ,5 ,5 - te t ra m e ti l-1 ,3 ,2 -d io x a b o ro la n -2 - i l) p ira z in -2 -c a rb o x a m id o )p ir id in -3 - i l) -4 -m e t i lp ip e r id in -4 - i l)c a rb a m a to d e fe rc -b u tilo .

En un matraz de fondo redondo de 100 ml equipado con un agitador magnético, (1-(2-(3-amino-6-bromopirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (1,9 g, 3,7 mmol), bis(pinacolato)diboro (1 g, 4 mmol) y acetato potásico (0,54 g, 5,5 mmol) se suspendieron en dioxano (15 ml). La suspensión formada se burbujeó con gas nitrógeno durante 20 minutos para eliminar el oxígeno disuelto. Después se añadió 1,1 '-Bis(difenilfosfino)ferroceno]dicloropaladio (II) (0,13 g, 0,18 mmol) y la reacción se calentó a 90 °C en un baño de aceite. Después de 3 h los volátiles se eliminaron y el residuo se suspendió en acetato de etilo que se lavó con agua para eliminar el exceso de acetato potásico. El residuo se disolvió en diclorometano y se trituró con heptano hasta que se formaron sólidos que se filtraron y se lavaron con más porciones de heptano para producir (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (1,32 g, 46 % de rendimiento) en forma de un polvo de color pardo de pureza suficiente para más transformaciones. LC-MS (método ácido): tiempo de ret. = 0,91 min, M+H = 554,4.
Etapa 3. Síntesis de 2-cloro-6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridina.

En un matraz de fondo redondo de 100 ml se disolvió 2,6-dicloro-3-(trifluorometil)piridina (1,25 g, 5,79 mmol) en DMF (30 ml) junto con 3,3-difluoroazetidina (0,75 g, 5,79 mmol) y N-etil-N-isopropilpropan-2-amina (1,5 ml, 8,7 mmol). Después de 16 h la reacción se enfrió, se vertió en agua y se extrajo tres veces con 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron con salmuera y sulfato sódico anhidro. Los volátiles se eliminaron y el residuo se purificó por cromatografía sobre gel de sílice usando gradiente de 0 - 60 % de acetato de etilo en heptano para producir 2-cloro-6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridina pura (935 mg, 53 % de rendimiento). LC-MS (método ácido): tiempo de ret. = 1,56 min, M+H = 273,3.
Etapa 4. Síntesis de (1-(2-(3-amino-6-(6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo.

En un matraz de fondo redondo de 10 ml se combinó (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (159 mg, 0,23 mmol) con 2-cloro-6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridina (50 mg, 0,183 mmol) y se disolvió en THF junto con fosfato tripotásico acuoso 1 M (0,3 ml, 0,3 mmol). La suspensión se desgasificó por evacuación y purgado con nitrógeno tres veces, después se agitó en atmósfera de nitrógeno durante 15 minutos. Se añadió 1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) y después la mezcla de reacción se calentó con un baño de aceite precalentado a 50 °C durante 16 h. Los volátiles de la reacción se eliminaron y el residuo se disolvió en DCM y se filtró a través de celite. Después se eliminaron los volátiles y el residuo se purificó por cromatografía sobre gel de sílice usando gradiente de 0-60 % de acetato de etilo en heptano para obtener el (1-(2-(3-amino-6-(6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo puro deseado (70 mg, 54 % de rendimiento). LC-MS (método ácido): tiempo de ret. = 1,27 min, M+H = 664,7.
Etapa 5. Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(6-(3,3-difluoroazetidin-1 -il)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida.

En un matraz de fondo redondo de 10 ml se disolvió (1-(2-(3-amino-6-(6-(3,3-difluoroazetidin-1-N)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo en 1,5 ml de diclorometano y se trató con ácido 2,2,2-trifluoroacético (0,2 ml, 2,2 mmol). Después de 16 horas la reacción se trató con una solución saturada de bicarbonato sódico hasta que se alcanzó pH 9. Se formó un precipitado sólido y lentamente se volvió a disolver. La capa orgánica se separó después se secó con salmuera, sulfato sódico anhidro y después se evaporó. El residuo se purificó por cromatografía sobre gel de sílice usando gradiente de 0 - 10 % de etanol en etilacetato para producir la 3-amino-N-(3-(4-amino-4-metNpiperidin-1-N)piridin-2-N)-6-(6-(3,3-difluoroazetidin-1-il)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida pura (49 mg, 78 % de rendimiento). LC-M S (método ácido): tiempo de ret. = 0,88 min, M+H = 564,2. RMN 1H (400 MHz, metanol-d4) 88,64 (s, 1H), 8,11 (dd, J = 5,0, 1,5 Hz, 1H), 8,03 (d, J = 8,8 Hz, 1H), 7,65 (dd, J = 7,9, 1,6 Hz, 1H), 7 ,17 (dd, J = 7,9, 4,9 Hz, 1H), 6,72 (d, J = 8,6 Hz, 1H), 4,51 (t, J = 12,0 Hz, 4H), 2,99 - 2,74 (m, 4H), 1,52 - 1,33 (m, 4H), 0,81 (s, 3H).
Ejemplo 15: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-ciclopropil-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1. Síntesis de 2-cloro-6-ciclopropil-3-(trifluorometil)piridina.

A un matraz de fondo redondo de 100 ml se le añadió tolueno (20 ml), 2,6-dicloro-3-(trifluorometil)piridina (3 g, 13,8 mmol), ácido ciclopropilborónico (1,3 g, 15,3 mmol), triciclohexil fosfina (0,39 g, 1,4 mmol), fosfato potásico tribásico (3,2 g, 15,3 mmol) y agua (1 ml). La suspensión se desgasificó por evacuación y purgado con nitrógeno tres veces, después se agitó en atmósfera de nitrógeno durante 15 minutos. Se añadió diacetoxipaladio (0,16 g, 0,7 mmol) y después se calentó la mezcla de reacción con un baño de aceite precalentado a 100 °C durante 16 horas. Después de este tiempo, se eliminaron los volátiles de la reacción, el residuo se disolvió en DCM, se filtró y después se purificó por cromatografía sobre gel de sílice usando gradiente de 0 - 60 % de acetato de etilo en heptano para obtener el producto puro deseado 2-cloro-6-ciclopropil-3-(trifluorometil)piridina (2 g, 59 % de rendimiento). LC-M S (método ácido): tiempo de ret. = 1,61 min, M+H = 222,2
E ta p a 2. S ín te s is d e (1 -(2 -(3 -a m in o -6 -(6 -c ic lo p ro p il-3 -( t r i f lu o ro m e t i l)p ir id in -2 - il) p ira z in -2 -c a rb o x a m id o )p ir id in -3 - i l) -4 -m e t i lp ip e r id in -4 - i l) c a rb a m a to d e fe rc -b u t ilo

A un matraz de fondo redondo de 10 ml equipado con un agitador magnético y purgado con nitrógeno se le añadió 2-doro-6-cidopropil-3-(trifluorometil)piridina (50 mg, 0,23 mmol), dioxano (2 ml), (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (156 mg, 0,23 mmol) y fosfato potásico tribásico acuoso 1 M (0,29 ml, 0,29 mmol). La mezcla resultante se agitó en atmósfera de nitrógeno en un baño de aceite a 90 °C hasta que se consumió toda la 2-cloro-6-ciclopropil-3-(trifluorometil)piridina (16 h). Después, la mezcla se enfrió y se filtró a través de un lecho de celite. La almohadilla del filtro se lavó con exceso de cloruro de metileno. Los filtrados combinados se concentraron hasta un residuo espeso y se purificaron por cromatografía sobre gel de sílice usando gradiente de 0 - 75 % de acetato de etilo en heptano para producir (50 mg, 34 % de rendimiento). LC-M S (método ácido): tiempo de ret. = 1,31 min, M+H = 613,2.
Etapa 3. Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-ciclopropil-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida.

A un matraz de 10 ml se le añadió (1-(2-(3-amino-6-(6-ciclopropil-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo, diclorometano (2 ml) y ácido 2,2,2-trifluoroacético (0,1 ml, 1,2 mmol). Después de 16 h la reacción se trató con exceso de una solución acuosa saturada de bicarbonato sódico hasta que se alcanzó pH 9. La capa orgánica se separó, después se secó con salmuera y sulfato sódico anhidro. El compuesto impuro final se purificó por cromatografía sobre gel de sílice usando gradiente de 0 - 10 % de etanol en acetato de etilo para producir 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-ciclopropil-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida pura (41 mg, 93 % de rendimiento). LC-MS (método ácido): tiempo de ret. = 1,22 min, M+H = 513,2. RMN 1H (400 MHz, metanol-d4) 88,63 (s, 1H), 8,14 - 8,09 (m, 2H), 7,64 (dd, J = 8,0, 1,6 Hz, 1H), 7,49 (d, J = 8,2 Hz, 1H), 7 ,17 (dd, J = 7,9, 4,9 Hz, 1H), 2,97 - 2,73 (m, 4H), 2,26 (ddd, J = 7,7, 4,5, 2,5 Hz, 1H), 1,45 - 1,31 (m, 4H), 1,17 - 1,12 (m, 4H), 0,75 (s, 3H).
Ejemplo 16: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

E ta p a 1. S ín te s is d e (1 -(2 -(3 -a m in o -6 -(6 -m e to x i-3 -( t r i f lu o ro m e t i l)p ir id in -2 - i l)p ira z in -2 -c a rb o x a m id o )p ir id in -3 - i l) -4 -m e t i lp ip e r id in -4 - i l)c a rb a m a to d e fe rc -b u t ilo

En un recipiente de 40 ml equipado con un agitador magnético, 2-doro-6-metoxi-3-(trifluorometil)piridina (J. Heterociclic Chem., 28, 971 (1991)) (40 mg, 0,190 mmol), (1-(2-(3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (100 mg, 0,181 mmol), PdCb(dppf) (6,61 mg, 0,09 mmol) y fosfato potásico (1 M, 0,271 ml) se suspendieron en 1,4-dioxano (2 ml), se desgasificó con N2 durante 10 min. y se calentó a 90 °C durante 4 h. La mezcla se enfrió a ta y se añadió EtOAc (20 ml), se filtró a través de Celite lavando con EtOAc 920 ml), se concentró al vacío. El sólido se purificó por cromatografía líquida de alta presión de fase inversa usando un método con un gradiente de 35-60 % de ACN 3,5 min a través de una columna X -Bridge 30x50 mm 5 um ACN/H 2 O con NH4OH 5 mM 75 ml/min, inyección de 5 ml 3 veces lo que dio 50 mg (sólido de color amarillo). LC-M S (método ácido): tiempo de ret. = 1,17 min, M+H = 603,7
Etapa 2. Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

En un recipiente de 40 ml equipado con un agitador magnético, se añadió ácido 2,2,2-trifluoroacético (64 ml, 0,830 mmol) a una solución de (1-(2-(3-amino-6-(6-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (50 mg, 0,083 mmol) en diclorometano (5 ml) y se dejó en agitación durante 30 min. a temperatura ambiente. La reacción se concentró al vacío y se neutralizó para proporcionar 40 mg del compuesto del título. LC-M S (método ácido): tiempo de ret. = 0,83 min, M+H = 503,5 RMN 1H (400 MHz, metanold4) 88,77 (s, 1H), 8,24 - 8,10 (m, 2H), 7,67 (dd, J = 8,0, 1,6 Hz, 1H), 7,21 (dd, J = 7,9, 4,9 Hz, 1H), 7,03 (d, J = 8,8 Hz, 1H), 4,08 (s, 3H), 3,37 (s, 1H), 2,94 (ddd, J = 12,6, 9,9, 3,1 Hz, 2H), 2,84 (dt, J = 12,0, 4,6 Hz, 2H), 1,51 (ddd, J = 13,6, 9,8, 4,1 Hz, 2H), 1,39 (dt, J = 13,3, 3,8 Hz, 2H), 0,82 (s, 3H).
Ejemplo 17: 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1. Síntesis de (4-etil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo.

A un recipiente de microondas equipado con un agitador magnético se le añadió 3-fluoro-2-nitropiridina (205 mg, 1,44 mmol), piperidin-4-ilcarbamato de ferc-butilo (329 mg, 1,443 mmol), N-etil-N-isopropilpropan-2-amina (559 mg, 4,33 mmol) en etanol (10 ml). La mezcla se calentó en un horno de microondas Biotage a 100 °C durante 30 minutos. La mezcla se enfrió y se concentró hasta un residuo espeso. El residuo se purificó por cromatografía sobre gel de sílice (usando metanol/diclorometano como eluyente) para dar (4-etil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (337 mg, 67 % de rendimiento). LC-M S (Método básico): tiempo de ret. = 1,45 min, M+H = 351,0 RMN 1H (400 MHz, DMSO-de) 88,04 (dd, J = 4,4, 1,3 Hz, 1H), 7,89 (dd, J = 8,3, 1,4 Hz, 1H), 7,63 (dd, J = 8,3, 4,4 Hz, 1H), 6,52 (s, 1H), 3,02 - 2,91 (m, 4H), 2,18 - 2,06 (m, 2H), 1,62 (c, J = 7,4 Hz, 2H), 1,43 - 1,31 (m, 11H), 0,75 (t, J = 7,4 Hz, 3H).
Etapa 2. Síntesis de (1-(2-aminopiridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo.

A un matraz de fondo redondo equipado con un agitador magnético y purgado con nitrógeno se le añadió (4-etil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo (337 mg, 0,96 mmol), etanol (10 ml) y Pd/C (41 mg, Pd al 10 % sobre carbón húmedo). La mezcla resultante se agitó en atmósfera de hidrógeno hasta que se consumió todo el (4-etil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo. La reacción se filtró a través de un lecho de celite, después se aclaró con metanol y acetato de etilo. El filtrado se concentró al vacío, después el residuo obtenido se disolvió en diclorometano y se filtró a través de un lecho pequeño de sulfato de magnesio. El filtrado se concentró para obtener (1-(2-aminopiridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo, en forma de un sólido de color blanco (268 mg, 87 % de rendimiento). LC-M S (Método básico): tiempo de ret. = 1,32 min, M+H = 321,1.
Etapa 3. Síntesis de (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo.

A un recipiente de centelleo de 20 ml equipado con un agitador magnético se le añadió ácido 3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico (61 mg, 0,22 mmol), hexafluorofosfato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametilaminio (120 mg, 0,32 mmol), N-etil-N-isopropilpropan-2-amina (73 mg, 0,56 mmol) en DMF (2 ml). La mezcla se dejó en agitación durante 5 minutos a ta seguido de la adición de (1-(2-aminopiridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo (60 mg, 0,187 mmol). La mezcla resultante se dejó en agitación durante una noche a ta, tras lo cual se filtró. El filtrado se concentró, después el residuo se purificó por HPLC de fase inversa (35-60 % de ACN 3.5 min de gradiente, X-Bridge 30x50 mm 5 pm columna ACN/H 2 O con 5 mm de NH4OH, 75 ml/min, 4 inyecciones a 1.5 ml/inyección) para obtener (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo (81 mg, 66 % de rendimiento). LC-M S (Método básico): tiempo de ret. = 1,42 min, M+H = 587,0.
Etapa 4. Síntesis de 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida.
A un recipiente de centelleo de 20 ml equipado con una barra de agitación magnética se le añadió (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo (81 mg, 0,14 mmol) y 1,4-dioxano (1 ml). Después se añadió una solución de HCl 4 N/1,4-dioxano (0,69 ml, 2,76 mmol) gota a gota. La mezcla resultante se agitó a ta durante 3 horas, punto en el cual se consumió todo el (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo. Se añadió acetonitrilo a mezcla de reacción, después el sólido se filtró y se aclaró con más acetonitrilo. El sólido obtenido se secó sobre un liofilizador para obtener 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (57 mg, 77 % de rendimiento), en forma de su sal clorhidrato. LC-M S (método ácido): tiempo de ret. = 0,83 min, M+H = 487,2. RMN 1H (400 MHz, DMSO-d6) 810,59 (s, 1H), 8,98 (dd, J = 4,9, 1,5 Hz, 1H), 8,81 (s, 1H), 8,44 (dd, J = 8,1, 1,5 Hz, 1H), 8,20 (dd, J = 5,1, 1,5 Hz, 1H), 8,08 (s, 5H), 7,81 - 7,71 (m, 2H), 7,33 (dd, J = 7,9, 5,1 Hz, 1 H), 3,17 - 3,07 (m, 2H), 2,93 - 2,84 (m, 2H), 1,68 - 1,58 (m, 2H), 1,35 - 1,26 (m, 2H), 1,06 (c, J = 7,5 Hz, 2H), 0,53 (t, J = 7,5 Hz, 3H).
Ejemplo 18: 3-ammo-N-(3-(4-ammo-4-metMpiperidm-1-M)piridm-2-M)-6-(3-cloropiridm-2-M)pirazm-2-carboxamida

Etapa 1. Síntesis de 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo.

En un matraz de fondo redondo equipado con un agitador magnético y entrada de nitrógeno se cargó 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo (906 mg, 3,25 mmol), 2-bromo-3-cloropiridina (500 mg, 2,6 mmol), PdCb(dppf) DCM (149 mg, 0,18 mmol), fosfato potásico (3,6 ml, 3,6 mmol, 1 M ac.) y THF (10 ml). La reacción se desgasificó con nitrógeno durante 5 minutos, después se calentó la mezcla a 55 °C en atmósfera de nitrógeno durante 16 h. Después de enfriar a ta, la mezcla de reacción se diluyó con acetato de etilo, después se dejó que se separaran las capas. La capa orgánica se secó con sulfato sódico, se secó y se concentró. El residuo obtenido se purificó por cromatografía sobre gel de sílice (usando gradiente de acetato de etilo/heptano 30-70 % como eluyente) para obtener 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo (191 mg). Debido a la baja pureza del producto, este por lo tanto se volvió a purificar por HPLC de fase inversa (15-40 % de ACN 3,5 min. de gradiente, X-Bridge 30x50 mm 5 pm columna ACN/H 2 O con 5 mm de NH4OH, 75 ml/min, 2 inyecciones a 1,5 ml/inyección) para obtener 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo (91 mg, 13 % de rendimiento). LC-MS (Método básico): tiempo de ret. = 0,92 min, M+H = 265,0.
Etapa 2. Síntesis de 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo.

En un matraz de fondo redondo equipado con un agitador magnético, se disolvió 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo (91 mg, 0,34 mMoles) en metanol (4 ml) a ta. Se añadió hidróxido de litio (0,52 ml, 1,03 mmol, 2 N ac.) a ta y se agitó durante 16 h. Los volátiles se eliminaron al vacío, después el residuo acuoso se acidificó con HCl ac. 2 N hasta que se obtuvo pH 2.
El precipitado obtenido se filtró y se aclaró con agua, después se secó sobre un liofilizador para proporcionar 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo (77 mg, 89 % de rendimiento), en forma de un sólido de color
amarillo claro. LC-M S (método ácido): tiempo de ret. = 0,68 min, M+H = 251,4.
Etapa 3. Síntesis de (1-(2-(3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo.

A un recipiente de centelleo de 20 ml equipado con una barra de agitación magnética se le añadió 3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxilato de metilo (38 mg, 0,15 mmol), hexafluorofosfato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametilaminio (84 mg, 0,22 mmol), N-etil-N-isopropilpropan-2-amina (51 mg, 0,39 mmol) en DMF (1,5 ml). La mezcla se dejó en agitación durante 5 minutos a ta seguido de la adición de (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (40 mg, 0,13 mmol). La mezcla resultante se dejó en agitación durante una noche a ta, tras lo cual se filtró. El filtrado se concentró, después el residuo se purificó por HPLC de fase inversa (35-60 % de ACN 3.5 min de gradiente, X-Bridge 30x50 mm 5 pm columna ACN/H2 O con 5 ml de NH4OH, 75 ml/min, 3 inyecciones a 1.5 ml/inyección) para obtener (1-(2-(3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (46 mg, 65 % de rendimiento), LC-MS (Método básico): tiempo de ret. = 1,35 min, M+H = 538,9.
Etapa 4. Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-cloropiridin-2-il)pirazin-2-carboxamida.

A un recipiente de centelleo de 20 equipado con una barra de agitación magnética se le añadió (1-(2-(3-amino-6-(3-cloropiridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (46 mg, 0,085 mmol) y diclorometano (1 ml). Después se añadió ácido trifluoroacético (146 mg, 1,28 mmol) gota a gota. La mezcla resultante se agitó a ta durante 16 horas, punto en el cual se consumió todo el (1-(2-(3-amino-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-etilpiperidin-4-il)carbamato de ferc-butilo. La mezcla de reacción se concentró, después se añadieron diclorometano y solución acuosa 2 N de carbonato sódico al residuo obtenido en dos fases. Las fases orgánica y acuosa (capas) se separaron. La capa orgánica se lavó una vez con más solución acuosa 2 N de carbonato sódico, después se secó con sulfato sódico, se filtró y se concentró. La mezcla en bruto se purificó por HPLC de fase inversa (25-50 % de ACN 3,5 min de gradiente, X-Bridge 30x50 mm 5 pm columna ACN/H 2 O con 5 mm de NH4OH, 75 ml/min, 2 inyecciones a 1,5 ml/inyección) para obtener 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-cloropiridin-2-il)pirazin-2-carboxamida (25 mg, 67 % de rendimiento), LC-MS (método ácido): tiempo de ret. = 0,75 min, M+H = 439,0. RMN 1H (400 MHz, DMSO-de) 810,82 (s, 1H), 8,77 (s, 1H), 8,67 (dd, J = 4,7, 1,5 Hz, 1H), 8,14 - 8,06 (m, 2H), 7,59 (dd, J = 7,9, 1,7 Hz, 1H), 7,52 (dd, J = 8,2, 4,6 Hz, 1H), 7,15 (dd, J = 7,9, 4,8 Hz, 1H), 2,97 - 2,88 (m, 2H), 2,77 - 2,67 (m, 2H), 1,46 - 1,22 (m, 6H), 0,71 (s, 3H).
Ejemplo 19: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida

Síntesis de ácido 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxílico

Etapa 1. Síntesis de 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxilato de metilo

Un recipiente de microondas equipado con una barra de agitación se cargó con 3-amino-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirazin-2-carboxilato de metilo (515 mg, ~80 % , 1,847 mmol), PdCte(dppf)-DCM (81 mg, 0,099 mmol), 2-bromo-3-fluoropiridina (250 mg, 1,421 mmol), carbonato de cesio (741 mg, 2,273 mmol) y dioxano (24 ml). La mezcla se desgasificó durante 5 minutos, después la reacción se calentó en un reactor de microondas a 110 °C durante 45 minutos. Después de enfriarse a ta, la mezcla de reacción se filtró a través de celite, se lavó con EtOAc (35 ml) y se concentró a presión reducida. El residuo se diluyó después con MeOH (25 ml), lo que condujo a la precipitación de un sólido de color pardo. La filtración de este sólido dio 250 mg de 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxilato de metilo. LC-MS (método 3, básico): tiempo de ret. = 0,84 min, M+H = 249,0.
Etapa 2. Síntesis de ácido 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxílico

En un recipiente de 40 ml se disolvió parcialmente 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxilato de metilo (950 mg, 3,83 mmol) en metanol (5 ml). A esta mezcla se le añadió LiOH (860 mg, 11,48 mmol) en agua (0,5 ml) y la mezcla se agitó durante 3 h a temperatura ambiente. Durante la reacción precipitó un sólido y se filtró lo que dio 1 , 13 g de ácido 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxílico. LC-M S (método ácido): tiempo de ret. = 0,70 min, M+H = 235,2.
Etapa 3: Síntesis de (1-(2-(3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo

A una solución de ácido 3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxílico (500 mg, 2,135 mmol) en DCM/DMA (2:1, 4 ml/2 ml) se le añadió (1-(2-aminopiridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (654 mg, 2,135 mmol), HBTU (1619 mg, 4,27 mmol) y DIPEA (1,492 ml, 8,54 mmol). La reacción se agitó a ta durante 16 h, después se interrumpió con agua. La reacción se diluyó después con DCM (25 ml) y se lavó con agua (15 ml) y salmuera (15 ml). Después la capa orgánica se separó, se secó sobre MgSO4 y se evaporó para dar un sólido de color parduzco que se purificó después por HPLC básica, lo que proporcionó 424,5 mg de (1-(2-(3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo. LC-M S (Método básico): tiempo de ret. = 1,32 min, M+H = 523,3.
4) Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida

En un recipiente de 40 ml se disolvió (1-(2-(3-amino-6-(3-fluoropiridin-2-il)pirazin-2-carboxamido)piridin-3-il)-4-metilpiperidin-4-il)carbamato de ferc-butilo (424,5 mg, 0,812 mmol) en DCM (2,5 ml). A esta mezcla se le añadió HCl/dioxano (2031 pl, 8,12 mmol) lentamente. Durante la adición se formó un sólido de color naranja. La reacción se dejó en agitación durante 18 h antes de filtrar el precipitado. El sólido obtenido se diluyó después con NaHCO3 (10 ml) y se extrajo con DCM (2x15 ml). Las capas orgánicas se combinaron, se secaron sobre MgSO4 y se evaporaron para dar 170 mg de 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida. LC-M S (Método básico): tiempo de ret. = 0,98 min, M+H = 423,3. RMN 1H (400 MHz, DMSO-cfó) 810,81 (s, 1H), 8,94 (s, 1H), 8,58 (dd, J = 4,3, 1,8 Hz, 1H), 8,11 (dd, J = 4,8, 1,6 Hz, 2H), 7,90 (ddd, J = 11,3 , 8,4, 1,3 Hz, 2H), 7,65 - 7,52 (m, 2H), 7,16 (dd, J = 7,8, 4,8 Hz, 1H), 3,57 (s, 1H), 2,96 (td, J = 11,0, 2,7 Hz, 2H), 2,74 (dt, J = 11,6, 4,2 Hz, 2H), 1,50 (ddd, J = 13,7, 10,3, 3,8 Hz, 2H), 1,39 - 1,26 (m, 4H), 0,79 (s, 3H).
Ejemplo 20: 3-ammo-N-(3-((1R,5S,8s)-8-ammo-3-azabiddo[3.2,1]octan-3-M)pmdm-2-N)-6-(6-morfoMn-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de ((1R,5S,8s)-3-(2-nitropiridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo

A un matraz con forma de pera de 25 ml se le añadió 3-fluoro-2-nitropiridina, (0,56 g, 3,94 mmol), (1R,5S,8s)-3-azabiciclo[3.2.1]octan-8-ilcarbamato de ferc-butilo (0,849 g, 3,75 mmol), N-etil-N-isopropilpropan-2-amina, (1,11 g, 8,59 mmol) y tetrahidrofurano (14 ml) y un agitador magnético. La mezcla se agitó en atmósfera de nitrógeno y se calentó a 70 °C durante 1 día. Después, la mezcla se enfrió y se concentró hasta un residuo espeso y se cromatografió directamente sobre gel de sílice (acetato de etilo-heptano) lo que dio ((1R,5S,8s)-3-(2-nitropiridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo (0,993 g, 2,71 mmol) con un rendimiento del 72 % . LC-M S (Método básico): tiempo de ret. = 1,41 min, M+H = 349,6.
Etapa 2: Síntesis de ((1R,5S,8s)-3-(2-aminopiridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo

A un matraz de fondo redondo de 100 ml se le añadió ((1R,5S,8s)-3-(2-nitropiridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo (0,99 g, 2,84 mmol), acetato de etilo (50 ml) y paladio al 10 % sobre carbono húmedo (1 g). El matraz se purgó con hidrógeno y se agitó en atmósfera de hidrógeno durante 16 h. A la mezcla resultante se le añadió después MgSO4 (5 gramos) y se agitó. Después, la mezcla se filtró a través de un lecho de MgSO4 debajo de un cono de nitrógeno. El filtrado se concentró a sequedad, lo que dio ((1R,5S,8s)-3-(2-aminopiridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo (2,52 g, 1,322 mmol) con un rendimiento del 98 % . LC-MS (Método básico): tiempo de ret. = 1,00 min, M+H = 319,5.
Etapa 3: Síntesis de ((1R,5S,8s)-3-(2-(3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo

A un recipiente de 10 ml con tapón de rosca se le añadió ácido 3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxílico (0,257 g, 0,696 mmol) que se preparó de modo análogo al ejemplo 1 (Método 1), DMF (2,5 ml), N-etil-N-isopropilpropan-2-amina (0,334 g, 0.2,58 mmol) y hexafluorofosfato de 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio (V) (0,421 g, 1,107 mmol). La mezcla se dejó en agitación durante 15 minutos. A la mezcla resultante se le añadió ((1R,5S,8s)-3-(2-aminopiridin-3-il)-3-azabiciclo[3.2,1]octan-8-il)carbamato de ferc-butilo (0,235 g, 0,738 mmol) y se dejó en agitación durante 18 horas. La reacción se concentró y se purificó por cromatografía sobre gel de sílice usando acetato de etilo y heptano lo que dio ((1R,5S,8s)-3-(2-(3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo (0,494 g, 0,556 mmol) con un rendimiento del 80 % . LC-M S (Método básico): tiempo de ret. = 1,28 min, M+H = 670,8. Etapa 4: Síntesis de 3-amino-N-(3-((1R,5S,8s)-8-amino-3-azabiciclo[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

A un matraz de 100 ml se le añadió un agitador magnético, ((1R,5S,8s)-3-(2-(3-amino-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamido)piridin-3-il)-3-azabiciclo[3.2.1]octan-8-il)carbamato de ferc-butilo (0,61 g, 0,923 mmol) y diclorometano (10 ml). La mezcla se agitó hasta que todos los sólidos se disolvieron y después se enfrió en un baño de agua enfriada con hielo en atmósfera de nitrógeno. A esta mezcla se le añadió ácido trifluoroacético (25 ml). El baño de hielo se retiró y la mezcla se agitó durante 2,5 horas a temperatura ambiente. Después, la mezcla se concentró y el residuo se co-evaporó después con tolueno (30 ml) 3 veces hasta un residuo espeso. El residuo se purificó por un método de HPLC de fase inversa lo que dio 3-amino-N-(3-((1R,5S,8s)-8-amino3-azabicido[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida (0,084 g, 0,277 mmol) con un rendimiento del 15 % . LC-M S (Método básico): tiempo de ret. = 1,12 min, M+H = 570,6. RMN 1H (400 MHz, cloroformo-d) 8 10,48 (s, 3H), 8,56 (s, 3H), 8,22 (d, J = 4,8 Hz, 3H), 7,77 (d, J = 9,0 Hz, 3H), 7,37 (d, J = 7,9 Hz, 3H), 6,99 (dd, J = 8,0, 4,8 Hz, 3H), 6,58 (d, J = 9,0 Hz, 3H), 3,90 - 3,67 (m, 12H), 3,59 (t, J = 4,8 Hz, 12H), 3,06 - 2,74 (m, 9H), 2,66 (d, J = 10,7 Hz, 6H), 1,83 (s, 6H), 1,20 (d, J = 12,5 Hz, 16H).
Ejemplo 21: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metoxipiridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metoxipiridin-2-il)pirazin-2-carboxamida se preparó de un modo como el descrito para el ejemplo 1, método 1 (0,185 g) con un rendimiento del 56 % . LC-MS (método ácido): tiempo de ret. = 1,11 min, M+H = 453,2
RMN 1H (400 MHz, DMSO-d6) 810,83 (s, 1H), 8,91 (s, 1H), 8,41 (d, J = 5,5 Hz, 1H), 8,11 (dd, J = 4,8, 1,6 Hz, 1H), 7,62 (dd, J = 7,9, 1,7 Hz, 1H), 7,33 (t, J = 5,8 Hz, 1H), 7,16 (dd, J = 7,8, 4,8 Hz, 1H), 3,97 (s, 3H), 2,95 (dd, J = 12,2, 9,5 Hz, 2H), 2,82 - 2,66 (m, 2H), 1,53 (ddd, J = 13,7, 10,4, 3,9 Hz, 2H), 1,44 - 1,13 (m, 4H), 0,82 (s, 3H).
Ejemplo 22: 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (0,01 g) con un rendimiento del 58 % . LC-MS (método básico): tiempo de ret. = 2 ,11 min, M+H = 452,2.
RMN 1H (400 MHz, metanol-d4) 88,91 (d, J = 1,2 Hz, 1H), 8,57 (dt, J = 4,6, 1,5 Hz, 1H), 8,13 (dd, J = 5,0, 1,6 Hz, 1H), 7,83 (ddd, J = 10,9, 8,4, 1,3 Hz, 1H), 7,70 (dd, J = 7,9, 1,6 Hz, 1H), 7,57 (ddd, J = 8,3, 4,6, 3,8 Hz, 1H), 7,20 (dd, J = 7,9, 4,9 Hz, 1H), 3,48 (c, J = 7,0 Hz, 7H), 3,23 (s, 19H), 3,06 - 2,84 (m, 6H), 1,66 (ddd, J = 14,8, 11,1,4 ,3 Hz, 2H), 1,52 (dt, J = 13,1, 3,0 Hz, 2H), 1,18 (t, J = 7,0 Hz, 10H).
Ejemplo 23: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-ciano-3-fluoropiridin-2-il)pirazin-2-carboxamida (Ejemplo de referencia)

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(6-ciano-3-fluoropiridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 7, método 2 (0,016 g) con un rendimiento del 78 % . LC-MS (método ácido): tiempo de ret. = 0,72 min, M+H= 448,3 RMN 1H (400 MHz, DMSO-de) 8 ppm 0,79 (s, 3 H) 1,29 - 1,38 (m, 2 H) 1,39 - 1,52 (m, 2 H) 2,73 (d, J = 11,80 Hz, 2 H) 2,96 (t, J = 9,79 Hz, 2 H) 3,17 (d, J = 3,51 Hz, 3 H) 4,12 (d, J = 4,27 Hz, 1 H) 7 ,17 (dd, J = 7,78, 4,77 Hz, 1 H) 7,62 (dd, J = 7,78, 1,51 Hz, 1 H) 8,11 (dd, J = 4,77, 1,51 Hz, 1 H) 8,18 - 8,29 (m, 2 H) 8,93 (s, 1 H) 10,70 (s, 1 H).
Ejemplo 24: 3-ammo-N-(3-(4-ammo-4-metNpiperídm-1-N)piridm-2-N)-6-(3-cianopiridm-2-N)pirazm-2-carboxamida (Ejemplo de referencia)

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-cianopiridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (0,109 g) con un rendimiento del 69 % . LC-M S (Método básico): tiempo de ret. = 1,92 min, M+H= 429,2.
RMN 1H (400 MHz, DMSO-de) 810,56 (s, 1H), 9,18 (s, 1H), 8,95 (dd, J = 4,8, 1,7 Hz, 1H), 8,52 (dd, J = 8,0, 1,7 Hz, 1H), 8,29 (s, 1H), 8,26 - 8,15 (m, 5H), 7,87 - 7,60 (m, 3H), 7,35 (dd, J = 7,9, 5,0 Hz, 1H), 3,20 - 3,02 (m, 3H), 2,97 -2,79 (m, 3H), 2,01 - 1,73 (m, 3H), 1,64 (ddd, J = 13,5, 9,2, 4,1 Hz, 2H), 1,36 (s, 1H), 1,05 (s, 3H).
Ejemplo 25: 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (3,15 g) con un rendimiento del 83 % . LC-MS (Método básico): tiempo de ret. = 1,02 min, M+H= 503,2.
RMN 1H (400 MHz, DMSO-d6) 810,80 (s, 1H), 8,84 (s, 1H), 8,76 (dd, J = 4,7, 1,3 Hz, 1H), 8,16 (s a, 1H), 8,10 (dd, J = 4,8, 1,6 Hz, 1H), 8,07 (dt, J = 8,4, 1,4 Hz, 1H), 7,96 (s a, 1H), 7,65 (dd, J = 8,4, 4,6 Hz, 1H), 7,59 (dd, J = 7,9, 1,6 Hz, 1H), 7,16 (dd, J = 7,8, 4,8 Hz, 1H), 2,95 (td, J = 11,5 , 2,9 Hz, 2H), 2,75 - 2,68 (m, 2H), 1,35 - 1,18 (m, 4H), 1,07 (s, 2H), 0,81 (c, J = 7,4 Hz, 2H), 0,56 (t, J = 7,4 Hz, 3H).
Ejemplo 26: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-ciano-4-metoxipiridin-2-il)pirazin-2-carboxamida (Ejemplo de referencia)

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-ciano-4-metoxipiridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 7, método 2 (0,018 g) con un rendimiento del 49 % . LC-MS (método ácido): tiempo de ret. = 0,69 min, M+H= 460,2.
RMN 1H (400 MHz, METANOL-d4) 8 ppm 0,92 (s, 4 H) 1,56 (d, J = 13,05 Hz, 2 H) 1,65 - 1,80 (m, 2 H) 2,90 - 2,98 (m, 2 H) 2,98 - 3,07 (m, 2 H) 4,13 (s, 3 H) 7,24 (dd, J = 8,03, 5,02 Hz, 1 H) 7,31 (d, J = 6,02 Hz, 1 H) 7,71 (dd, J = 7,91, 1,38 Hz, 1 H) 8,14 (dd, J = 4,89, 1,38 Hz, 1 H) 8,74 (d, J = 6,02 Hz, 1 H) 9,11 (s, 1 H).
Ejemplo 27: 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (0,018 g) con un rendimiento del 49 % . LC-MS (método ácido): tiempo de ret. = 0,79 min, M+H = 5 l 9,2.
RMN 1H (400 MHz, DMSO-d6) 810,78 (s, 1H), 8,89 (s, 1H), 8,79 (dd, J = 4,6, 1,3 Hz, 1H), 8,17 (s, 1H), 8,14 - 8,06 (m, 2H), 7,98 (s, 1H), 7,69 (dd, J = 8,3, 4,6 Hz, 1H), 7,59 (dd, J = 8,0, 1,7 Hz, 1H), 7,16 (dd, J = 7,9, 4,8 Hz, 1H), 3,09 (s, 3H), 2,97 (d, J = 11,6, 2,7 Hz, 2H), 2,73 (dt, J = 11,4, 3,7 Hz, 2H), 2,61 (s, 2H), 1,37 (dt, J = 11,9, 6,5 Hz, 3H), 1,31 - 1,21 (m, 2H).
Ejemplo 28: 3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-M)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (0,039 g) con un rendimiento del 38 % . LC-MS (Método básico): tiempo de ret. = 0,96 min, M+H= 503,2. RMN 1H (400 MHz, DMSO-d6) 810,61 (s, 1H), 8,95 (dd, J = 4,9, 1,5 Hz, 1H), 8,76 (s, 1H), 8,40 (dd, J = 8,1, 1,5 Hz, 1H), 8,22 - 8,06 (m, 2H), 7,97 (s a, 1H), 7,71 (dd, J = 8,0, 4,8 Hz, 1H), 7,56 (dd, J = 7,9, 1,7 Hz, 1H), 7,16 (dd, J = 7,9, 4,8 Hz, 1H), 4,77 (s, 1H), 3,29 (t, J = 6,6 Hz, 2H), 2,94 - 2,83 (m, 2H), 2,71 - 2,63 (m, 2H), 1,39 (s, 2H), 1,27 - 1,13 (m, 4H), 0,92 (t, J = 6,6 Hz, 2H).
Ejemplo 29: 3-amino-N-(3-((1S,5R,8S)-8-amino-6-oxa-3-azabiciclo[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-((1S,5R,8S)-8-amino-6-oxa-3-azabiciclo[3.2.1]octan-3-il)piridin-2-il)-6-(6-morfolin-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1 (0,069 g) con un rendimiento del 14 % . LC-M S (método básico): tiempo de ret. = 1,69 min, M+H= 572,6. RMN 1H (400 MHz, cloroformo-d) 810,35 (s, 1H), 8,64 (s, 1H), 8,22 (d, J = 4,8 Hz, 1H), 7,80 (d, J = 8,9 Hz, 1H), 7,44 (d, J = 7,9 Hz, 1H), 7,00 (dd, J = 7,9, 4,8 Hz, 1H), 6,59 (d, J = 9,0 Hz, 1H), 3,99 (d, J = 8,3 Hz, 1H), 3,87 (d, J = 4,2 Hz, 1H), 3,85 - 3,67 (m, 4H), 3,69 - 3,54 (m, 4H), 3,29 (dd, J = 8,2, 4,8 Hz, 1H), 3,21 - 2,93 (m, 3H), 2,81 (dd, J = 27,2, 11,4 Hz, 2H). Ejemplos 30-85
Los ejemplos 30-85, preparados mediante los métodos sintéticos (métodos 1-6) divulgados anteriormente, se resumen en la tabla 1.
Tabla 1

(c o n t in u a c ió n )

(c o n t in u a c ió n )

(c o n t in u a c ió n )

(c o n t in u a c ió n )

(c o n t in u a c ió n )

(c o n t in u a c ió n )

(c o n t in u a c ió n )

Ejemplo 79: 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de trifluorometanosulfonato de 6-metil-2-nitropiridin-3-ilo

A una solución de 3-mdroxi-6-metil-2-nitropmdina (1,0 g, 6,5 mmol) en DCM (24 ml) a 0 °C en atmosfera de N2 se le añadió trietilamina (1,35 ml, 9,73 mmol) y seguido de anhídrido tríflico (1,32 ml, 7,79 mmol). La mezcla se agitó durante 1 hora a 0 °C y después se inactivó con agua. La capa orgánica se separó, se lavó con agua y se secó sobre MgSO4. Después de la filtración y la concentración a presión reducida, la mezcla en bruto se purificó por cromatografía ultrarrápida sobre columna de gel de sílice eluyendo con 0-70 % de EtOAc/Heptano para proporcionar el producto deseado en forma de un aceite de color amarillo (1,8 g, 98 % de rendimiento). LC-MS (Método ácido): tiempo de ret. = 1,21 min, M+H = 287,0. RMN 1H (400 MHz, CDCla) ó ppm 7,81 (d, 2 H) 7,59 (d, 1 H) 2,70 (s, 3 H).
Etapa 2. Síntesis de (1-(6-metil-2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

Una mezcla de trifluorometanosulfonato de 6-metil-2-nitropiridin-3-ilo (0,70 g, 2,45 mmol), piperidin-4-ilcarbamato de ferc-butilo (1,23 g, 6,11 mmol) y trietilamina (0,85 ml, 6,11 mmol) en acetonitrilo (20 ml) se calentó a reflujo durante 8 h. La reacción se enfrió a temperatura ambiente y se concentró a presión reducida. La mezcla en bruto se inactivó con agua y se extrajo con DCM. La capa orgánica en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (0-100 % de EtOAc/Heptano) para proporcionar rendimiento cuantitativo del producto deseado en forma de un sólido de color amarillo. LC-M S (Método ácido): tiempo de ret. = 1,27 min, M+H = 337,2.
Síntesis de 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (94 mg) con un rendimiento del 16 % . LC-MS (método ácido): tiempo de ret. = 0,66 min, M+H = 489,3. RMN 1H (400 MHz, metanol-d4) ó 8,85 (s, 1H), 8,73 (dd, J = 4,7, 1,4 Hz, 1 H), 8,00 (dt, J = 8,4, 1,4 Hz, 1H), 7,61 (dd, J = 8,4, 4,7 Hz, 1H), 7,53 (d, J = 8,1 Hz, 1H), 7,05 (d, J = 8,0 Hz, 1H), 3,05 (dt, J = 12,4, 3,6 Hz, 2H), 2,76 - 2,60 (m, 3H), 2,50 (s, 3H), 1,85 - 1,75 (m, 2H), 1,42 - 1,29 (m, 2H). Ejemplo 80: 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida se preparó de un modo similar al descrito para el ejemplo 1, método 1 (105 mg) con un rendimiento del 18 % . LC-MS (método ácido): tiempo de ret. = 0,64 min, M+H = 473,3. RMN 1H (400 MHz, metanol-d4) 88,92 (dd, J = 4,8, 1,5 Hz, 1H), 8,74 (s, 1H), 8,37 (dd, J = 8,1, 1,5 Hz, 1H), 7,83 - 7,62 (m, 1H), 7,49 (d, J = 8,0 Hz, 1H), 7,03 (d, J = 8,0 Hz, 1H), 3,31 (p, J = 1,6 Hz, 1 H), 2,97 (dt, J = 12,4, 3,6 Hz, 2H), 2,64 (td, J = 11,5 , 2,4 Hz, 2H), 2,55 (s a, 1H), 2,49 (s, 3H), 1,70 (dc, J = 15,0, 3,1 Hz, 2H), 1,37 - 0,98 (m, 2H).
Ejemplo 81: 3-amino-N-(3-(4-amino-3-metoxipiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de 7-oxa-3-azabiciclo[4.1.0]heptan-3-carboxilato de bencilo

A una solución de 5,6-dihidropiridin-1(2H)-carboxilato de bencilo (3,8 g, 17 mmol) en DCM (60 ml) a -15 °C se le añadió ácido 3-clorobenzoperoxoico (4,7 g, 70 % puro, 19 mmol). La mezcla se agitó a temperatura ambiente durante 12 h y después se lavó con solución de bicarbonato sódico. La capa orgánica se secó sobre sulfato sódico, se filtró y se concentró a presión reducida. La mezcla en bruto se purificó por cromatografía ultrarrápida sobre gel de sílice (30 % de EtOAc/Heptano) para proporcionar el producto deseado en forma de un aceite incoloro. LC-M S (Método ácido): tiempo de ret. = 1,08 min, M+H = 234,3.
Etapa 2. Síntesis de 4-(bis(2,4-dimetoxibencil)amino)-3-hidroxipiperidin-1-carboxilato de bencilo

Una suspensión de bromuro de litio seco (5,2 g, 60 mmol) en acetonitrilo (15 ml) se agitó a 60 °C hasta que se formó una solución transparente. Se añadió 7-oxa-3-azabicido[4.1.0]heptan-3-carboxilato de bencilo (8 g, 34 mmol) en acetonitrilo (35 ml), seguido de bis(2,4-dimetoxibencil)amina (12 g, 38 mmol) y acetonitrilo (50 ml). La mezcla se agitó a 60 °C durante 46 h. La reacción se enfrió a temperatura ambiente y se concentró a presión reducida. La mezcla en bruto se inactivó con agua y se extrajo con DCM. La capa acuosa se extrajo adicionalmente con DCM dos veces. La capa orgánica combinada se purificó por cromatografía ultrarrápida sobre gel de sílice (0-100 % de EtOAc/Heptano) para dar dos productos con igual M+H 551,0. El producto deseado salió primero en forma de un aceite incoloro. LC-MS (Método ácido): tiempo de ret. = 1,18 min, M+H = 551,0.
Etapa 3. Síntesis de 4-(bis(2,4-dimetoxibencil)amino)-3-metoxipiperidin-1-carboxilato de bencilo

A una solución de 4-(bis(2,4-dimetoxibencil)amino)-3-hidroxipiperidin-1-carboxilato de bencilo (5,0 g, 9,1 mmol) en THF (100 ml) se le añadió hidruro sódico (0,55 g, 60 % puro, 14 mmol) a 0 °C en atmósfera de nitrógeno. Se añadió yodometano (2,1 ml, 33 mmol). La mezcla se agitó a temperatura ambiente durante 12 h. La mezcla en bruto se inactivó con cloruro de amonio acuoso saturado y se extrajo con EtOAc. La capa acuosa se extrajo adicionalmente con EtOAc dos veces. La capa orgánica combinada se lavó con salmuera y se purificó por cromatografía ultrarrápida sobre gel de sílice (0-50 % de EtOAc/Heptano) para dar el producto deseado. l C-M S (Método ácido): tiempo de ret. = 1,14 min, M+H = 565,0.
Etapa 4. Síntesis de N,N-bis(2,4-dimetoxibencil)-3-metoxipiperidin-4-amina
Una mezcla de 4-(bis(2,4-dimetoxibencil)amino)-3-metoxipiperidin-1-carboxilato de bencilo (4,8 g, 8,4 mmol), paladio sobre carbono (0,45 g, 10 % puro, 0,42 mmol) en etanol (100 ml) se agitó en un globo de hidrógeno durante 2,5 h. Se filtró y se concentró para dar el producto deseado en forma de un aceite incoloro. Este se usó en la etapa siguiente sin más purificación. LC-MS (Método ácido): tiempo de ret. = 0,68 min, M+H = 431,3.
Síntesis de 3-amino-N-(3-(4-amino-3-metoxipiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida
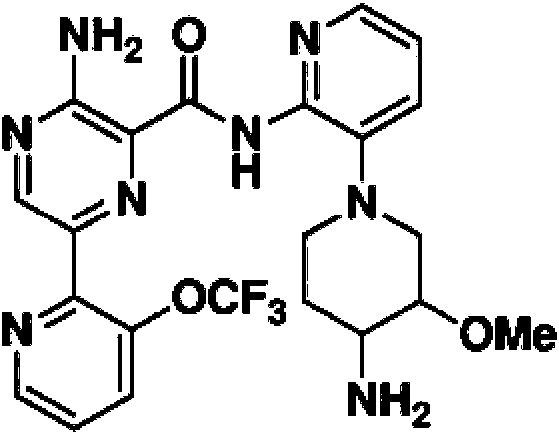
La 3-amino-N-(3-(4-amino-3-metoxipiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida se preparó de un modo como el descrito para el ejemplo 1, método 1 (43 mg) con un rendimiento del 75 % . LC-MS (método ácido): tiempo de ret. = 0,71 min, M+H = 505,3. RMN 1H (400 MHz, cloroformo-d) 8 10,96 (s, 1H), 8,88 (s, 1H), 8,82 (dd, J = 4,5, 1,4 Hz, 1H), 8,34 (dd, J = 4,9, 1,6 Hz, 1H), 7,81 (dt, J = 8,3, 1,4 Hz, 1H), 7,52 - 7,41 (m, 2H), 7,09 (dd, J = 7,9, 4,8 Hz, 1H), 3,51 (s, 1H), 3,41 (ddd, J = 11,0, 4,5, 2,1 Hz, 1H), 3,22 (s, 3H), 3,14 - 2,94 (m, 2H), 2,70 (td, J = 12,0, 2,5 Hz, 1H), 2,56 (ddd, J = 11,6, 9,1, 4,6 Hz, 1H), 2,41 (t, J = 10,5 Hz, 1H), 1,90 - 1,79 (m, 1H).
Ejemplo 82: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metilpiridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metilpiridin-2-il)pirazin-2-carboxamida se preparó de un modo como el descrito para el ejemplo 1, método 1 (8,6 mg) con un rendimiento del 7,5 % . LC-MS (método ácido): tiempo de ret. = 1,04 min, M+H = 437,1 RMN 1H (400 MHz, metanol-d4) 88,83 (d, J = 1,3 Hz, 1H), 8,39 (d, J = 4,8 Hz, 1H), 8,12 (dd, J = 5,0, 1,6 Hz, 1H), 7,68 (dd, J = 7,9, 1,6 Hz, 1H), 7,40 (td, J = 5,2, 0,9 Hz, 1H), 7,18 (dd, J = 7,9, 4,9 Hz, 1H), 3,10 - 2,77 (m, 4H), 2,45 (d, J = 1,9 Hz, 3H), 1,81 - 1,40 (m, 4H), 0,87 (s, 3H).
Ejemplo 83: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-fluoropiridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de 2-bromo-4-etoxi-3-fluoropiridina

Una mezcla de 2-bromo-3-fluoropiridin-4-ol (0,20 g, 1,0 mmol), yodoetano (0,22 ml, 2,1 mmol) y carbonato potásico (0,29 g, 2,1 mmol) en acetona (7 ml) se calentó a reflujo durante 4 h. La mezcla de reacción se concentró a presión reducida y se purificó por cromatografía ultrarrápida sobre columna de gel de sílice eluyendo con 0-100 % de EtOAc/Heptano para proporcionar el producto deseado en forma de un sólido de color blanco (0,19 g, 83 % de rendimiento). LC-MS (Método ácido): tiempo de ret. = 1,07 min, M+H = 221,6.
Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-fluoropiridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-fluoropiridin-2-il)pirazin-2-carboxamida se preparó de un modo como el descrito para el ejemplo 1, método 1 (57 mg) con un rendimiento del 91 % . LC-MS (método ácido): tiempo de ret. = 1,06 min, M+H = 467,2 RMN 1H (400 MHz, metanol-d4) 88,86 (d, J = 0,8 Hz, 1H), 8,35 (d, J = 5,5 Hz, 1H), 8,12 (dd, J = 4,9, 1,6 Hz, 1H), 7,70 (dd, J = 7,9, 1,6 Hz, 1H), 7,30 - 7,14 (m, 2H), 4,28 (c, J = 7,0 Hz, 2H), 3,05 - 2,83 (m, 4H), 1,73 (ddd, J = 13,2, 9,2, 3,9 Hz, 2H), 1,64 - 1,53 (m, 2H), 1,51 (t, J = 7,0 Hz, 3H), 0,98 (s, 3H).
Ejemplo 84: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(hidroximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de (2-cloro-3-(trifluorometil)piridin-4-il)metanol

A una solución de 2-cloro-3-(triflurometil)isonicotinaldehído (0,97 g, 4,6 mmol) en MeOH (10 ml) a 0 °C se le añadió borohidruro sódico (0,23 g, 6,0 mmol). La mezcla se agitó a temperatura ambiente durante 20 minutos. Después se concentró y se repartió entre DCM y agua. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró para dar un sólido de color amarillo. (0,91 g, 93 % de rendimiento). LC-M S (Método ácido): tiempo de ret. = 0,90 min, M+H = 212,0.
Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1 -il)piridin-2-il)-6-(4-(hidroximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(hidroximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida se preparó de un modo como el descrito para el ejemplo 7, método 3 (36 mg) con un rendimiento del 94%. LC-MS (método ácido): tiempo de ret. = 0,62 min, M+H = 503,1. RMN 1H (400 MHz, DMSO-d6) 810,64 (s, 1H), 8,89 (d, J = 5,1 Hz, 1H), 8,68 (s, 1H), 8,10 (dd, J = 4,8, 1,6 Hz, 1H), 7,90 (d, J = 5,1 Hz, 1H), 7,57 (dd, J = 7,9, 1,7 Hz, 1H), 7,14 (dd, J = 7,9, 4,8 Hz, 1H), 4,79 (s, 2H), 4,10-4,04 (a, 1H), 2,88 (ddd, J = 12,4, 7,5, 5,2 Hz, 2H), 2,64 (dt, J = 11,4, 4,5 Hz, 2H), 1,23 - 1,11 (m, 4H), 0,65 (s, 3H).
Ejemplo 85: 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(metoximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

Etapa 1: Síntesis de 2-cloro-4-(metoximetil)-3-(trifluorometil)piridina

A una solución de 2-cloro-3-(triflurometil)piridin-4-il)metanol (0,15 mg, 0,69 mmol) en THF (5 ml) se le anadió terc-butóxido de potasio (0,10 g, 0,89 mmol). La mezcla se agitó a temperatura ambiente durante 10 minutos. Después se le anadió yoduro de metilo (0,22 ml, 3,4 mmol) y se agitó a temperatura ambiente durante 17 h. Se anadió más terc-butóxido de potasio (0,10 g, 0,89 mmol) y se agitó a temperatura ambiente durante otras 12 h. Después, la mezcla se filtró y se lavó con EtOAc. La capa orgánica combinada se purificó por cromatografía ultrarrápida sobre columna de gel de sílice eluyendo con 0-30 % de EtOAc/Heptano para proporcionar el producto deseado en forma de un aceite incoloro (25 mg, 16 % de rendimiento). LC-M S (Método ácido): tiempo de ret. = 1,16 min, M+H = 226,0.
Síntesis de 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(metoximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida

La 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(metoximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida se preparó de un modo como el descrito para el ejemplo 7, método 3 (7,5 mg) con un rendimiento del 13 % . LC-M S (método ácido): tiempo de ret. = 1,11 min, M+H = 517,1. RMN 1H (400 MHz, metanol-d4) 88,84 (d, J =
,1 Hz, 1H), 8,61 (s, 1H), 8,11 (dd, J = 5,0, 1,6 Hz, 1H), 7,88 (dd, J = 5,0, 1,1 Hz, 1H), 7,66 (dd, J = 7,9, 1,7 Hz, 1H), ,16 (dd, J = 7,9, 4,9 Hz, 1H), 4,80 - 4,74 (m, 2H), 3,56 (s, 3H), 3,03 - 2,69 (m, 4H), 1,46 - 1,25 (m, 4H), 0,76 (s, 3H).

continuación

Los siguientes compuestos puede prepararse de acuerdo con los métodos sintéticos (Métodos 1-6) descritos en el presente documento.

continuación

continuación

continuación

c o n t in u a c ió n

c o n t in u a c ió n

(c o n t in u a c ió n )

Ejemplo 110: Síntesis de (3-fluoro-4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de tere-butilo (Intermedio)

Etapa 1. Síntesis de 3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-ona

A un matraz de fondo redondo de 100 ml equipado con un agitador magnético y una entrada de nitrógeno, se le añadió dioxano (10 ml), DMF ( 6 ml), 3-fluoropiperidin-4-ona (550 mg, 3,58 mmol), 3-fluoro-2-nitropiridina (509 mg, 3,58 mmol) y diisopropiletilamina (1,876 ml, 10,74 mmol). La solución se agitó a 70 °C en un baño de aceite durante 4 horas. La mezcla se repartió después entre acetato de etilo ( 2 0 ml) y agua ( 2 0 ml) y los extractos orgánicos se lavaron con agua (10 ml) dos veces y salmuera (10 ml) y después se secaron sobre Na2 SÜ 4. Después de la evaporación del disolvente, se recogió un aceite de color ámbar como producto en bruto, 3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-ona. Se llevó este producto en bruto a la etapa siguientes directamente, sin más purificación.
LC-M S (Método ácido): tiempo de ret. = 0,63 min, M+H = 240,1
Etapa 2. Síntesis de 2-(3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-ilideno)acetato de (Z)-etilo

A un matraz de fondo redondo de 100 ml equipado con un agitador magnético y una entrada de nitrógeno, se le añadió NaH (169 mg, 4,21 mmol) y THF (10 ml). La mezcla se enfrió a 0 °C y se añadió 2-(dimetoxifosforil)acetato de etilo (827 mg, 4,21 mmol) gota a gota. La reacción se agitó a temperatura ambiente durante 20 minutos, después se añadió una solución de 3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-ona (840 mg, 3,51 mmol) en THF (5 ml). La mezcla se agitó a temperatura ambiente durante 3 horas y se inactivó mediante la adición de agua (25 ml) a 0 °C. La mezcla acuosa se extrajo con acetato de etilo (2 x 50 ml). La capa orgánica se lavó con salmuera, se secó sobre Na2 SÜ 4 , se filtró y se concentró. El material en bruto se purificó por cromatografía sobre gel de sílice usando acetato de etilo-heptano (Rf ~ 0,5 al 50 % de acetato de etilo en heptano). Se obtuvo 2-(3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-ilideno)acetato de (Z)-etilo en forma de un sólido de color amarillo claro.
LC-M S (Método ácido): tiempo de ret. = 1,44 min, M+H = 310,2
Etapa 3. Síntesis de 2-(4-amino-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acetato de etilo

Se disolvió 2-(3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-ilideno)acetato de (Z)-etilo (4,02 g, 13 mmol) en 10 ml de NH3 7 N en MeOH en un recipiente a presión. El recipiente se selló y se calentó a 80 °C durante 12 horas. Los disolventes
se evaporaron y después se aplicó cromatografía sobre gel de sílice (Rf ~ 0,4 a 5 % d e MeOH (0,5 % de NH4OH) en DCM) para recoger 2-(4-amino-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acetato de etilo con >80 % de pureza. LC-M S (Método ácido): tiempo de ret. = 0,68 min, M+H = 327,3
Etapa 4. Síntesis de 2-(4-((ferc-butoxicarbonil)amino)-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acetato de etilo

A un matraz de 100 ml se le añadió un agitador magnético y 2-(4-amino-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acetato de etilo (3 g, 9,19 mmol) y BOC anhídrido (2,006 g, 9,19 mmol) en THF (Volumen: 33 ml, proporción: 1,000), agua (volumen: 33,0 ml, proporción: 1,000) y THF (volumen: 8,0 ml, proporción: 1,000). Se añadió DIPEA (1,606 ml, 9,19 mmol). La mezcla se calentó a 85 °C durante 4 horas. La mezcla se enfrió a temperatura ambiente y se observó un precipitado de color amarillo. La mezcla se filtró y el sólido se aclaró con agua. El sólido se purificó por cromatografía sobre gel de sílice (Rf ~ 0,7 a 70 % de acetato de etilo en heptano) para recoger 2-(4-((ferc-butoxicarbonil)amino)-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acetato de etilo.
LC-MS (Método ácido): tiempo de ret. = 1,51 min, M+H = 427,2
Etapa 5. Síntesis de ácido 2-(4-((ferc-butoxicarbonil)amino)-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acético

A un matraz de 100 ml se le añadió un agitador magnético y 2-(4-((ferc-butoxicarbonil)amino)-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acetato de etilo (2,4 g, 5,82 mmol) en MeOH ( 6 ml) y THF (3 ml). Se añadió una solución acuosa 3 M de NaOH (9,7 ml) y la reacción se calentó a 55 °C durante 2 horas. Se diluyó la mezcla de reacción con agua (10 ml) y se lavó con Et2 O (20 ml). La capa acuosa se acidificó con HCl 1 N lentamente a pH~ 6 y después se extrajo con acetato de etilo (2 x 50 ml); las capas de acetato de etilo combinadas se secaron sobre Na2 SO 4 , se filtraron y se concentraron. Se purificó por cromatografía sobre gel de sílice (Rf ~ 0,3 a 70 % de acetato de etilo en heptano) para recoger ácido 2-(4-((ferc-butoxicarbonil)amino)-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acético en forma de un sólido de color amarillo. RMN 1H (400 MHz, cloroformo-d) 8 8,15 (d, J = 4,3 Hz, 1H), 7,67 (d, J = 8,1 Hz, 1H), 7,51 (dd, J = 8,2, 4,4 Hz, 1H), 5,02 (s, 1H), 4,92 (s, 1H), 3,36 (dd, J = 26,9, 16,7 Hz, 2H), 3,10 (dt, J = 22,4, 11 ,5 Hz, 2H), 2,91 (d, J = 17,1 Hz, 2H), 1,89 (s, 1H), 1,46 (s, 9H), 0,87 (s, 1H). LC-M S (Método ácido): tiempo de ret. = 1,05 min, M-56+H = 343,1
Etapa 6. Síntesis de (3-fluoro-4-metil-1-(2-nitropiridin-3-il)piperidin-4-il)carbamato de ferc-butilo

A la mezcla de ácido 2-(4-((terc-butoxicarbonil)amino)-3-fluoro-1-(2-nitropiridin-3-il)piperidin-4-il)acético (1 g, 2,51 mmol) y HOTT (1,118 g, 3,01 mmol) en acetonitrilo (15 ml), se le añadió trietilamina (1,399 ml, 10,04 mmol) en THF (5 ml), se aisló el recipiente de reacción de la luz mediante una lámina de aluminio. Se añadió DMAP (0,031 g, 0,251 mmol) y se mantuvo la reacción en agitación a temperatura ambiente durante 2 horas. A la mezcla de reacción se le añadió terc-dodecil mercaptano (2,363 ml, 10,04 mmol) en acetonitrilo (5 ml) y la reacción se llevó a temperatura de reflujo durante 18 h. La mezcla de reacción se concentró y se diluyó con agua (50 ml) y se extrajo con acetato de etilo. Se recogió la capa de acetato de etilo y se secó sobre Na2SO4; se filtró y se evaporó. Se purificó por cromatografía sobre gel de sílice con DCM/MeOH (0 - 7 % ) para recoger el producto deseado en forma de un sólido de color amarillo con >50 % de pureza.
LC-M S (Método básico): tiempo de ret. = 1,38 min, M+H = 355,1
Este intermedio se usó después como se describe en el método 3.
Actividad biológica
Actividad de inhibición de PIM cinasa
Para una comparación entre ciertos inhibidores de PKC de la presente solicitud e inhibidores de PIM cinasa estructuralmente comparables, se midió la actividad de PIM2 usando un ensayo de cinasa Caliper in vitro. Las etapas de manejo de líquidos y de incubación se efectuaron en un dispositivo Innovadyne Nanodrop Express equipado con un brazo robótico (Thermo CatX, Caliper Twister II) y una incubadora (Liconic STX40, Thermo Cytomat 2C450). Las placas de ensayo de microtitulación de 384 pocillos se prepararon mediante la adición de 50 nl por pocillo de solución de compuesto en DMSO al 90 % . Las reacciones de cinasa se iniciaron mediante la adición por etapas de 4,5 pl por pocillo de solución de péptido/ATP (H EPES 50 mM, pH 7,5, DTT 1 mM, Tween20 al 0,02 % , BSA al 0,02 % , DMs O al 0,6 % , beta-glicerofosfato 10 mM y ortovanadato de sodio 10 pM, MgCb 1 mM, ATP 25 pM y péptido S62 pM) y 4,5 pl por pocillo de solución enzimática (HEPES 50 mM, pH 7,5, DTT 1 mM, Tween20 al 0,02 % , BSA al 0,02 % , DMs O al 0,6 % , beta-glicerofosfato 10 mM y ortovanadato de sodio 10 pM, MgCb 1 mM y enzima PIM20,6 nM).
Las reacciones de cinasa se incubaron a 30 °C durante 60 minutos y posteriormente se terminaron mediante la adición de 16 pl por pocillo de solución de parada (H EPES 100 mM a pH 7,5, DMSO al 5 % , reactivo de recubrimiento Caliper al 0,1 % , EDTA 10 mM y Brij35 al 0,015 % ). Las placas con reacciones de cinasa terminadas se transfirieron a las estaciones de trabajo Caliper LC3000 para su lectura. Los péptidos fosforilados y no fosforilados se separaron usando la tecnología de cambio de movilidad de microfluidos de Caliper. Brevemente, se aplicaron a la microplaca muestras de reacciones de cinasa terminadas. Los analitos se transportan por la microplaca mediante un flujo de tampón constante y se monitoriza la migración del péptido de sustrato mediante la señal de fluorescencia de su marcador. Se separan el péptido S6 fosforilado (producto) y el péptido S6 no fosforilado (sustrato) en un campo eléctrico por medio de su relación carga/masa. Las actividades cinasa se calcularon a partir de las cantidades de péptido fosforilado formado. Los valores de CI50 se determinaron a partir de los valores de porcentaje de inhibición a diferentes concentraciones de compuesto mediante análisis de regresión no lineal.
Los compuestos de los ejemplos anteriores se ensayaron mediante el ensayo de Pim2 cinasa y se observó que mostraban valores de CI50 como se muestran en la tabla 4. La CI50, la concentración inhibidora semimáxima, representa la concentración de un compuesto de ensayo de la invención que es necesaria para una inhibición del 50 % de su diana in vitro.
Ensayo de GSKbeta
Los tipos de ensayo de G SK-3 usados para evaluar la selectividad/potencial fuera de la diana de los compuestos de la invención con respecto a la inhibición de PKC a/0 incluyen los siguientes: Tipo 1: El péptido específico para GSK-3 usado en este ensayo se obtuvo del sitio de fosforilación de la glucógeno sintasa y su secuencia es: YR R A A VPPSPSLSR H SSPH Q (S)ED EEE. (S) está prefosforilado, como en la glucógeno sintasa in vivo y los tres sitios consenso para la fosforilación específica de G SK-3 están subrayados. El tampón usado para producir el péptido de glucógeno sintasa y [y-33P] ATP consistió en MOPS 25 mM, EDTA 0,2 mM, acetato de magnesio 10 mM, Tween-20 al 0,01 % y mercaptoetanol 7,5 mM a pH 7,00. Los compuestos se disolvieron en dimetilsulfóxido (DMSO) a una
concentración final de 100 mM. Se prepararon varias concentraciones en DMSO y se mezclaron con la solución de sustrato (péptido GSK-3) (a una concentración final de 20 j M) descrita en la sección anterior, junto con G SK -3a y GSK-3p (concentración final de 0,5 jM/ml de enzima). Las reacciones se iniciaron con la adición de [y-33P] ATP (500 cpm/pmol) añadido a una mezcla de ATP (concentración final de 10 j M). Después de 30 minutos a temperatura ambiente, la reacción se terminó mediante la adición de 10 j l de H3 PO 4/O.OP / 0 Tween-20 (2,5 % ). Se añadió un volumen (10 jl) de la mezcla a papel de fosfocelulosa P-30 (Wallac & Berthold, EG&G Instruments Ltd, Milton Keynes). El papel se lavó cuatro veces en H3 PO4 (0,5 % ), 2 minutos para cada lavado, se secó al aire y el fosfato radiactivo incorporado en el péptido de glucógeno sintasa sintético, que se une al papel de fosfocelulosa P-30, se contó en un contador de centelleo Wallac Microbeta. Análisis de los datos: Se calcularon los valores de CI50 para cada inhibidor ajustando una curva logística de cuatro parámetros al modelo: cpm=inferior+(superior-inferior) /(1 (concentración CI50) pendiente).
Tipo 2: Este protocolo se basa en la capacidad de la cinasa para fosforilar un péptido biotinilado, cuya secuencia se obtiene del sitio de fosforilación de la glucógeno sintasa y su secuencia es: Biot-KYR R AA VPPSPSLSR H SSPH Q (S)ED EEE. (S) está una serina prefosforilada, como en la glucógeno sintasa in vivo y los tres sitios consenso para la fosforilación específica de G SK-3 están subrayados. Posteriormente, se captura el péptido biotinilado fosforilado sobre perlas de SpA recubiertas con estreptavidina (Amersham Technology), donde se amplifica la señal del 33P a través del centelleante contenido en las perlas. La cinasa se ensayó a una concentración final de 10 nM en tampón MOPS 25 mM, pH 7,0 que contenía Tween-20 al 0,01 % , 2-mercaptoetanol 7,5 mM, acetato de magnesio 10 mM y [y-33P]-ATP 10 jM . Después de 60 minutos de incubación a temperatura ambiente, se detuvo la reacción mediante la adición de solución de EDTA 50 mM que contenía las perlas de SPA recubiertas de estreptavidina para dar una cantidad final de 0,5 mg de perlas por pocillo de ensayo en un formato de placa de microtitulación de 384 pocillos. Se generaron soluciones madre 10 mM de los compuestos de la invención en DMSO al 100 % como primera etapa en el proceso de exploración. La segunda etapa implica la creación de placas de respuesta a la dosis, donde se diluyen estos compuestos a lo largo de la placa, donde las concentraciones finales bajas y altas han de ser 0,008 y 10 jM en el ensayo de cinasa. La tercera etapa implica la creación de las placas de ensayo. Esto se logra transfiriendo los compuestos de cuatro placas de 96 pocillos de respuesta a la dosis a una placa de ensayo de 384 pocillos en el sistema Robocon Robolab. La cuarta etapa es llevar a cabo el ensayo como se ha descrito y contar las placas resultantes en el dispositivo T riluc (contador de centelleo líquido y de luminiscencia Wallac 1450 MicroBeta). La etapa final es la adquisición y el análisis de datos, donde se generan los valores de CI50 para cada compuesto por duplicado, ajustando una curva logística de cuatro parámetros al modelo: cpm = inferior (superior-inferior) / (1 (concentración/ C I50) pendiente) de un modo discontinuo. Los compuestos más potentes contra PKC de la presente invención muestran valores de CI50 para GSKbeta en el intervalo de entre 100 a 100.000 nM.
Actividad de inhibición de PKCa/9 in vitro
Se ensayó la actividad de los compuestos de fórmula I sobre diferentes isoformas diferentes de PKC, de acuerdo con un método publicado (D. Geiges et al. Biochem. Pharmacol. 1997;53:865-875). El ensayo se lleva a cabo en una placa de microtitulación de polipropileno de 96 pocillos (Costar 3794) que se había siliconizado previamente con Sigmacote (Sigma SL-2). La mezcla de reacción (50 jl) contiene 10 j l de la isozima PKC relevante junto con 25 j l del compuesto inhibidor de PKC y 15 j l de una solución de mezcla que contiene 200 jg/m l de sulfato de protamina, Mg(NO3)210 mM, ATP 10 jM (Boehringer 519987) y 3750 Bq de 33P-ATP (Hartmann Analytic SFC 301, 110TBq/mmol) en tampón de Tris 20 mM a pH 7,4 BSA al 0,1 % . La incubación se llevó a cabo durante 15 minutos a 32 °C en una incubadora con agitación de placas de microtitulación (Biolabo Scientific Instruments). La reacción se detuvo añadiendo 10 j l de Na2 EDTA 0,5 M, pH 7,4. Se pipetearon 50 j l de mezcla sobre un papel de fosfocelulosa prehumedecido (Whatmann 3698-915) con presión suave. El ATP no incorporado se elimina por lavado con 100 j l de H2 O bidestilada. El papel se lava dos veces en H3 PO4 al 0,5 % durante 15 minutos, seguido de 5 minutos en EtOH. Posteriormente, se seca el papel y se coloca en un dispositivo Ominifilter (Packard 6005219) y se cubre con 10 jl/pocillo de Microscint-O (Packard 6013611) antes de contar en un contador de radiactividad T opcount (Packard). La medición de la C I50 se lleva a cabo de manera rutinaria incubando una dilución seriada de inhibidor a concentraciones en el intervalo entre 1-1000 jM de acuerdo con los procedimientos descritos anteriormente. Los valores de CI50 se calculan a partir de la gráfica mediante ajuste de curvas sigmoidales.
2. Ensayo de proteína cinasa C a
Se usa PKCa humana recombinante en las condiciones de ensayo descritas anteriormente. En este ensayo, los compuestos de fórmula I inhiben a PKC a con una CI50 < 1 jM . Los compuestos de los ejemplos 2, 9, 75 y 76 inhiben a PKCa en este ensayo con una CI50 <10 nM.
3. Ensayo de proteína cinasa C 9
Se obtuvo PKC9 humana recombinante de Oxford Biomedical Research y se usa en las condiciones de ensayo descritas en la sección A.1 anterior: los compuestos de fórmula I inhiben a PKC a con una CI50 < 1 jM . Los compuestos de los ejemplos 2, 9, 75 y 76 inhiben a PKC9 en este ensayo con una CI50 <10 nM.
Ensayos celulares
Para evaluar la capacidad de los compuestos de la invención para inhibir la actividad de PKC en ensayos celulares, se evaluó la capacidad de los compuestos para inhibir de manera selectiva la proliferación de células de melanoma uveal 92.1 y células de linfoma de linfocitos B TMD8, en relación con células SK-MEL-28. Las células de melanoma uveal 92.1 son dependientes de la expresión de una forma mutante de subunidad alfa de proteína G, GNAQ, que señaliza a través de PKC para permitir el crecimiento y la proliferación. Las células TMD8 son dependientes de la expresión de una forma mutante de CD79, que señaliza a través de PKC para permitir el crecimiento y la proliferación. Las células SK-M EL-28 son dependientes de la expresión de una forma mutante de B-Raf que no señaliza a través de PKC para permitir el crecimiento y la señalización. Por lo tanto, se espera que los inhibidores de PKC tengan actividad antiproliferativa contra células 92.1 y/o TMD8, pero no contra células SK-MEL-28. Se obtuvieron células 92.1 (mutantes para GNAQ) de Martine Jager (Leiden University Medical Center, 2300 RC Leiden, Países Bajos). Las células SK-M EL-28 pueden obtenerse de la Colección Americana de Cultivos Tipo (ATCC). Las células se mantuvieron en medio RPMI 1640 (Lonza) y FBS al 10 % (Lonza).
Ensayo de proliferación
Para cada línea celular, puede ajustarse la densidad celular a 40000 células/ml y se añaden 50ul (2000 células) por pocillo de una placa de ensayo de 384 pocillos. Los compuestos de ensayo se resuspenden en DMSO a una concentración de 10 mM. Se llevó a cabo una dilución seriada de factor tres de cada compuesto con DMSO en placas de 384 pocillos usando el dispensador de líquidos Janus (PerkinElmer). Se transfirieron 50 nl de cada dilución de compuesto a la placa de ensayo que contenía células hasta alcanzar concentraciones de ensayo finales de 10 j M, 3,33 j M, 1,11 j M, 0,37 j M, 0,12 j M, 0,041 j M, 0,014 j M, 0,0046 j M, 0,0015 j M, 0,00051 j M.
Las células pueden incubarse a 37 grados centígrados en un ambiente humidificado con dióxido de carbono al 5 % durante 72 horas. Se preparó ATPlite (Perkin Elmer) de acuerdo con las instrucciones del fabricante y se añadieron 25 j l a cada pocillo de la placa de ensayo. Las placas se incuban durante 10 minutos y se detecta la luminiscencia en un lector de placas multimodo EnVision (Perkin Elmer). El grado de luminiscencia se correlaciona con el número de células viables en cada pocillo. Se calculó el efecto de cada inhibidor y pueden generarse los valores de CI50.
Los valores de CI50 de la isoforma alfa y teta de PKC para los inhibidores de PKC (ejemplos 1-29) se resumen en la tabla 2. Los datos presentados en el presente documento representan la media de al menos dos replicados.
T l 2 D I inhi i i n PK l i n r l m l 1-2

continuación

Los valores de CI50 de la isoforma alfa y teta de PKC para los inhibidores de PKC (ejemplos 30-123) se resumen en la tabla 3.
T l D I inhi i i n PK l i n r l m l -11

continuación

continuación

continuación

La tabla 4 presenta datos comparativos de inhibición de PKCa/0, así como otros datos de actividad de cinasa para el ejemplo 1, un inhibidor de PKC de la presente solicitud y diversos inhibidores de PIM cinasa.
Tabla 4. Comparación de datos de CI50 de cinasas seleccionadas entre un inhibidor de PIM cinasa y diversos inhibidores de PKC

continuación

continuación

La tabla 4 presenta una comparación directa de los datos de actividad de cinasa para varios inhibidores de PIM cinasa divulgados en el documento WO2008/160692 y el ejemplo 1, un inhibidor de PKC ilustrativo divulgado en la presente solicitud. Estos datos revelan que el ejemplo 1 es inesperadamente selectivo con respecto a GSK3p fuera de la diana e inesperadamente potente para suprimir la proliferación de células 92.1 (melanoma uveal), en comparación con los inhibidores de PIM ejemplares. Esta selectividad aumentada es probablemente el resultado de la porción bisagra de piridin-3-ilo acoplada al resto de piperidin-4-ilo, que está presente en el ejemplo 1. La porción bisagra de piridin-3-ilo se encuentra en los todos los compuestos inhibidores de PKC de la presente solicitud. Ninguno de los inhibidores de PIM cinasa ejemplares en la tabla 4 posee la porción bisagra de piridin-3-ilo y como se muestra en la tabla 4, los inhibidores de PIM cinasa son relativamente no selectivos. Por consiguiente, los inhibidores de PIM cinasa estructuralmente similares divulgados en el documento WO2008/160692 no tienen o no se espera que tengan la selectividad de los inhibidores de PKC divulgados en el presente documento. Además, los datos en la tabla 4 demuestran que el ejemplo 1 tiene una actividad contra PIM2 escasa o nula, lo que diferencia adicionalmente a los compuestos inhibidores de PKC de la presente solicitud de los inhibidores de PIM cinasa.
Los datos de CI50 de cinasa en la diana y fuera de la diana para los inhibidores de PKC ejemplares adicionales de la invención se resumen en la tabla 5.
T l D I inhi i i n in r inhi i r PK l r n li i

Modelos de eficacia in vivo - estudios de xenoinjerto de melanoma uveal 92.1 de inhibidores de PKC seleccionados
Se implantó a ratones células de melanoma uveal 92.1 mutantes para GNAQ para evaluar la eficacia in vivo de los inhibidores de PKC. Se inyectó a cada ratón por vía subcutánea (región axilar) 5x106 células mezcladas en 50 pl de Matrigel y 50 pl de PBS. Se monitorizó el crecimiento tumoral hasta que los tumores alcanzaron un volumen de 150 250 mm3. El tamaño tumoral, en mm3, se calculó a partir de: Volumen tumoral = (a2 x l)/2 donde a = ancho y l = longitud, en mm, del tumor. Cuando los tumores alcanzaron el tamaño requerido, se administraron los compuestos de ensayo con las dosis y pautas requeridas en un volumen de dosis de 10 ml/kg. Se pesó a los animales dos veces a la semana y se ajustaron consecuentemente los volúmenes de dosificación. El volumen tumoral se midió dos veces a la semana usando mediciones con un calibre y se calcularon los volúmenes tumorales como longitud x ancho2/2. Se generaron
datos in vivo para ciertos inhibidores de PKC potentes y selectivos (ejemplos 2, 9) y se compararon con AEB071. Los inhibidores de PKC inventados lograron la regresión tumoral, en comparación con la estasis tumoral para AEB071 a dosis inferiores que las de AEB071, que solo lograron la estasis en el modelo.
El ejemplo 2 reduce la proliferación celular en xenoinjertos de melanoma uveal 92.1 de una manera dependiente de la dosis, en comparación con sotrastaurina (figura 1). Además, el ejemplo 2 muestra una reducción significativa en la dosis para lograr eficacia mejorada (regresión) frente a sotrastaurina (estasis). Basándose en los datos presentados en el presente documento, los compuestos divulgados en la presente solicitud o inducen o se espera que induzcan regresión tumoral en un modelo de melanoma uveal que porta las mutaciones GNAQ, así como que se logre una eficacia mejorada (regresión) frente a sotrastaurina (estasis), como se muestra en la figura 1. Por ejemplo, como se observa en la figura 3, el ejemplo 9 y el ejemplo 10 también muestran eficacia mejorada (regresión) en un modelo de xenoinjerto de melanoma uveal 92.1 en comparación con vehículo. Por consiguiente, se espera que los compuestos divulgados en el presente documento induzcan de manera selectiva la regresión tumoral in vivo.
Una comparación de los datos farmacocinéticos de ratón y rata in vivo para el ejemplo 2 y sotrastaurina (tablas 6 y 7) demuestra que el ejemplo 2 tiene una PK mejorada frente a sotrastaurina.
Tabla 6. Com aración de datos farmacocinéticos de ratón C57BL/6 in vivo

Tabla 7. Com aración de datos farmacocinéticos en ratas Wistar-Han no canuladas in vivo

Los compuestos de la presente invención representan una nueva clase mejorada de inhibidores selectivos de molécula pequeña de PKC con actividad y selectividad antitumoral in vivo, en comparación con sotrastaurina. Además, los inhibidores de PKC de la presente solicitud muestran por lo general una potencia, perfil PK, absorción, tolerancia gastrointestinal y selectividad de cinasa mejorados, en comparación con inhibidores de PKC conocidos.
Claims (15)
1. Un compuesto de fórmula II

o una sal farmacéuticamente aceptable del mismo, en donde:
X es N o CR;
R, R2, R3 y R4 son cada uno independientemente H, 2H, halo, hidroxilo (-OH), alcoxi C 1 - 3 , haloalquilo C 1 -3 o alquilo C 1 - 3 , dicho alquilo C 1 -3 opcionalmente sustituido con hidroxilo, halo, alcoxi C 1 -3 o haloalcoxi C 1 - 3 ;
R5 es independientemente H, 2H, CH 3 , CH 2 F, C H F 2 , C F 3 , CH 2 OH, alquilo C 1 - 3 , dicho alquilo C 1 -3 opcionalmente sustituido con F, OH, alcoxi C 1 -3 o haloalcoxi C 1 - 3 ;
cada uno de R5a y R5b es independientemente H, 2H, alquilo C 1 - 3 , dicho alquilo C 1 -3 opcionalmente sustituido con F, OH o alcoxi C 1 - 3 , o R5a y R5b se unen para formar un grupo puente de metileno o etileno;
cada uno de R5c y R5d es independientemente H, 2H, F, -OH, alcoxi C 1 - 3 , alquilo C 1 - 3 , dicho alquilo C 1 -3 opcionalmente sustituido con F, OH o alcoxi C 1 -3 o R5c y R5d se unen para formar un grupo puente de metileno, etileno o -C H 2 -O-; cada uno de R6, R7 y R8 se selecciona independientemente entre H, 2H, halo, alquilo C 1 - 3 , haloalquilo C 1 - 3 , alcoxi C 1 - 3 , haloalcoxi C 1 - 3 , cicloalquilo C 3 -7 y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S, dicho alquilo C 1 -3 opcionalmente sustituido con F, OH, alcoxi C 1 -3 o haloalcoxi C1-3; o
en donde R6 y R8 opcionalmente forman un anillo carbocíclico o anillo heterobicíclico con el anillo heteroarilo, dicho anillo carbocíclico o anillo heterobicíclico opcionalmente sustituido con de 1 a 3 grupos seleccionados entre: 2H, halo, haloalquilo C 1 - 3 , alcoxi C 1 - 3 , haloalcoxi C 1 - 3 , cicloalquilo C 3 -7 y heterociclilo de 4-7 miembros que tiene de 1 a 3 heteroátomos seleccionados entre N, O y S.
2. Un compuesto de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde X es N y R2, R3 y R4 son cada uno independientemente H o halo.
3. Un compuesto de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde X es CR; y R2, R3, R4 y R8 son cada uno independientemente H o halo.
4. Un compuesto de acuerdo con la reivindicación 3 o una sal farmacéuticamente aceptable del mismo, en donde cada uno de R6 y R7 se selecciona independientemente entre H, halo, haloalquilo C 1 - 3 , haloalcoxi C 1 - 3 , cicloalquilo C 3 - 7 , morfolino, piperidinilo y piperazinilo.
5. Un compuesto de acuerdo con la reivindicación 4 o una sal farmacéuticamente aceptable del mismo, en donde R5 es -H, 2H o CH 3 ; y cada uno de R5a y R5b es H y cada uno de R5c y R5d es H.
6. Un compuesto de acuerdo con la reivindicación 1 que se selecciona entre:
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-doropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metoxipiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(2-metoxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
(+)3-amino-N-(3-((cis)-4-amino-3-fluoropiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-((3S,4R)-4-amino-3-fluoropiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-etilpiperidin-1-il)piridin-2-il)-6-(3-fluoropiridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-cloro-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-aminopiperidin-1-il)-6-metilpiridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-3-metoxipiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-fluoro-4-metilpiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-etoxi-3-fluoropiridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(hidroximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida y
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(metoximetil)-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida o una sal farmacéuticamente aceptable de los mismos.
7. Un compuesto de acuerdo con la reivindicación 1 que se selecciona entre:
3-amino-N-(3-(4-amino-4-(etoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-(etoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-((difluorometoxi)metil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(5-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-3-fluoro-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-3-fluoro-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-((3S,4R)-4-amino-3-fluoro-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida y
3-amino-N-(3-(4-amino-3-fluoro-4-(2-hidroxietil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
o una sal farmacéuticamente aceptable de los mismos.
8. Un compuesto de acuerdo con la reivindicación 1 que se selecciona entre:
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(metoximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-(hidroximetil)piperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida; 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida y 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(4-metoxi-3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida
o una sal farmacéuticamente aceptable de los mismos.
9. Un compuesto de acuerdo con la reivindicación 8 que se selecciona entre:
3-amino-N-(3-(4-aminopiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida;
3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida y 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometoxi)piridin-2-il)pirazin-2-carboxamida o una sal farmacéuticamente aceptable de los mismos.
10. Un compuesto de acuerdo con la reivindicación 8 que es 3-amino-N-(3-(4-amino-4-metilpiperidin-1-il)piridin-2-il)-6-(3-(trifluorometil)piridin-2-il)pirazin-2-carboxamida o una sal farmacéuticamente aceptable de la misma.
11. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-10 o una sal farmacéuticamente aceptable del mismo y un vehículo o excipiente farmacéuticamente aceptable.
12. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 - 10 para su uso en el tratamiento de cánceres.
13. Un compuesto o una sal farmacéuticamente aceptable del mismo para su uso de acuerdo con la reivindicación 12 en donde el cáncer se selecciona entre melanoma, melanoma maligno uveal, linfoma, linfoma difuso de linfocitos B grandes y cánceres resistentes a ibrutinib.
14. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 - 10 para su uso en el tratamiento de trastornos relacionados con el sistema inmunitario seleccionados entre enfermedades autoinmunes, reacciones alérgicas y rechazo de trasplante de tejidos.
15. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 - 10 junto con otro agente antineoplásico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462033679P | 2014-08-06 | 2014-08-06 | |
| PCT/IB2015/055951 WO2016020864A1 (en) | 2014-08-06 | 2015-08-05 | Protein kinase c inhibitors and methods of their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2754248T3 true ES2754248T3 (es) | 2020-04-16 |
Family
ID=53836145
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15750138T Active ES2754248T3 (es) | 2014-08-06 | 2015-08-05 | Inhibidores de proteína cinasa C y métodos para su uso |
Country Status (42)
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JO3589B1 (ar) | 2014-08-06 | 2020-07-05 | Novartis Ag | مثبطات كيناز البروتين c وطرق استخداماتها |
| BR112018000496A2 (pt) * | 2015-08-14 | 2018-09-11 | Novartis Ag | combinações farmacêuticas e seu uso |
| JP2019524700A (ja) | 2016-07-14 | 2019-09-05 | ミングサイト ファーマシューティカルズ,インク. | 癌の処置 |
| WO2019053595A1 (en) * | 2017-09-12 | 2019-03-21 | Novartis Ag | INHIBITORS OF PROTEIN KINASE C FOR THE TREATMENT OF CHOROIDAL MELLANOMA |
| BR112020018094A2 (pt) | 2018-03-08 | 2020-12-22 | Incyte Corporation | Compostos de aminopirazina diol como inibidores de pi3k-¿ |
| CN110496223B (zh) * | 2018-05-17 | 2021-09-10 | 复旦大学附属肿瘤医院 | 一种治疗非霍奇金氏淋巴瘤的药物组合物 |
| US11046658B2 (en) | 2018-07-02 | 2021-06-29 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| WO2020092924A1 (en) * | 2018-11-02 | 2020-05-07 | Board Of Regents, The University Of Texas System | Combination therapy for the treatment of egfr tyrosine kinase inhibitor resistant cancer |
| EP3908282A1 (en) * | 2019-01-07 | 2021-11-17 | Ideaya Biosciences, Inc. | Treatment of cancer having gnaq or gna11 genetic mutations with protein kinase c inhibitors |
| US11505542B2 (en) * | 2019-01-16 | 2022-11-22 | Shanghai Yingli Pharmaceutical Co., Ltd | Preparation method for morpholinquinazoline compound and intermediates thereof |
| WO2020198469A1 (en) * | 2019-03-27 | 2020-10-01 | Ideaya Biosciences Inc. | Method for treating epidermal growth factor receptor-driven cancers with protein kinase c inhibitors in combination with an egfr-tyrosine kinase inhibitor |
| CA3161953A1 (en) * | 2019-11-18 | 2021-05-27 | Ideaya Biosciences, Inc. | Dosing regimens for a protein kinase c inhibitor |
| JP2023527792A (ja) | 2020-05-27 | 2023-06-30 | アキシャル セラピューティクス,インク. | Tlr2調節剤化合物、医薬組成物、及びそれらの使用 |
| PL243663B1 (pl) * | 2020-06-29 | 2023-09-25 | Univ Medyczny Im Piastow Slaskich We Wroclawiu | Zastosowanie wanikozydów |
| MX2023002757A (es) * | 2020-09-08 | 2023-07-25 | Ideaya Biosciences Inc | Combinacion farmaceutica y tratamiento antitumoral. |
| WO2024125543A1 (zh) * | 2022-12-16 | 2024-06-20 | 苏州科睿思制药有限公司 | 达洛色替的晶型及其制备方法和用途 |
Family Cites Families (170)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US15677A (en) * | 1856-09-02 | Improved cutting device for harvesters | ||
| US4319039A (en) | 1979-06-15 | 1982-03-09 | Merck & Co., Inc. | Preparation of ammonium salt of hypocholesteremic fermentation product |
| US4231938A (en) | 1979-06-15 | 1980-11-04 | Merck & Co., Inc. | Hypocholesteremic fermentation products and process of preparation |
| US4294926A (en) | 1979-06-15 | 1981-10-13 | Merck & Co., Inc. | Hypocholesteremic fermentation products and process of preparation |
| US4444784A (en) | 1980-08-05 | 1984-04-24 | Merck & Co., Inc. | Antihypercholesterolemic compounds |
| DK149080C (da) | 1980-06-06 | 1986-07-28 | Sankyo Co | Fremgangsmaade til fremstilling af derivater af ml-236b-carboxylsyre |
| JPS5889191A (ja) | 1981-11-20 | 1983-05-27 | Sankyo Co Ltd | 3−ヒドロキシ−ml−236b誘導体の製造法 |
| US5354772A (en) | 1982-11-22 | 1994-10-11 | Sandoz Pharm. Corp. | Indole analogs of mevalonolactone and derivatives thereof |
| AU570021B2 (en) | 1982-11-22 | 1988-03-03 | Novartis Ag | Analogs of mevalolactone |
| US4911165A (en) | 1983-01-12 | 1990-03-27 | Ethicon, Inc. | Pliabilized polypropylene surgical filaments |
| US4681893A (en) | 1986-05-30 | 1987-07-21 | Warner-Lambert Company | Trans-6-[2-(3- or 4-carboxamido-substituted pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis |
| US4782084A (en) | 1987-06-29 | 1988-11-01 | Merck & Co., Inc. | HMG-COA reductase inhibitors |
| US4885314A (en) | 1987-06-29 | 1989-12-05 | Merck & Co., Inc. | Novel HMG-CoA reductase inhibitors |
| US4820850A (en) | 1987-07-10 | 1989-04-11 | Merck & Co., Inc. | Process for α-C-alkylation of the 8-acyl group on mevinolin and analogs thereof |
| US5180589A (en) | 1988-03-31 | 1993-01-19 | E. R. Squibb & Sons, Inc. | Pravastatin pharmaceuatical compositions having good stability |
| US5030447A (en) | 1988-03-31 | 1991-07-09 | E. R. Squibb & Sons, Inc. | Pharmaceutical compositions having good stability |
| US4916239A (en) | 1988-07-19 | 1990-04-10 | Merck & Co., Inc. | Process for the lactonization of mevinic acids and analogs thereof |
| US5118853A (en) | 1988-10-13 | 1992-06-02 | Sandoz Ltd. | Processes for the synthesis of 3-disubstituted aminoacroleins |
| US5290946A (en) | 1988-10-13 | 1994-03-01 | Sandoz Ltd. | Processes for the synthesis of 3-(substituted indolyl-2-yl)propenaldehydes |
| US4929437A (en) | 1989-02-02 | 1990-05-29 | Merck & Co., Inc. | Coenzyme Q10 with HMG-CoA reductase inhibitors |
| US5189164A (en) | 1989-05-22 | 1993-02-23 | Sandoz Ltd. | Processes for the synthesis of syn-(E)-3,5-dihydroxy-7-substituted hept-6-enoic and heptanoic acids and derivatives and intermediates thereof |
| FI94339C (fi) | 1989-07-21 | 1995-08-25 | Warner Lambert Co | Menetelmä farmaseuttisesti käyttökelpoisen /R-(R*,R*)/-2-(4-fluorifenyyli)- , -dihydroksi-5-(1-metyylietyyli)-3-fenyyli-4-/(fenyyliamino)karbonyyli/-1H-pyrroli-1-heptaanihapon ja sen farmaseuttisesti hyväksyttävien suolojen valmistamiseksi |
| PH27357A (en) | 1989-09-22 | 1993-06-21 | Fujisawa Pharmaceutical Co | Pyrazole derivatives and pharmaceutical compositions comprising the same |
| US5420245A (en) | 1990-04-18 | 1995-05-30 | Board Of Regents, The University Of Texas | Tetrapeptide-based inhibitors of farnesyl transferase |
| US5475085A (en) | 1991-02-07 | 1995-12-12 | Molecumetics, Ltd. | Conformationally restricted mimetics of beta turns and beta bulges and peptides containing the same |
| HU217629B (hu) | 1991-12-12 | 2000-03-28 | Novartis Ag. | Eljárás fluvasztatint tartalmazó stabilizált gyógyszerkészítmények előállítására |
| US5604260A (en) | 1992-12-11 | 1997-02-18 | Merck Frosst Canada Inc. | 5-methanesulfonamido-1-indanones as an inhibitor of cyclooxygenase-2 |
| EP0604181A1 (en) | 1992-12-21 | 1994-06-29 | Eli Lilly And Company | Antitumor compositions and method of treatment |
| CA2152792C (en) | 1993-01-15 | 2000-02-15 | Stephen R. Bertenshaw | Novel 3,4-diaryl thiophenes and analogs thereof having use as antiinflammatory agents |
| WO1994019357A1 (en) | 1993-02-23 | 1994-09-01 | Merrell Dow Pharmaceuticals Inc. | Farnesyl:protein transferase inhibitors as anticancer agents |
| US5298627A (en) | 1993-03-03 | 1994-03-29 | Warner-Lambert Company | Process for trans-6-[2-(substituted-pyrrol-1-yl)alkyl]pyran-2-one inhibitors of cholesterol synthesis |
| US5409944A (en) | 1993-03-12 | 1995-04-25 | Merck Frosst Canada, Inc. | Alkanesulfonamido-1-indanone derivatives as inhibitors of cyclooxygenase |
| CA2118985A1 (en) | 1993-04-02 | 1994-10-03 | Dinesh V. Patel | Heterocyclic inhibitors of farnesyl protein transferase |
| US5843941A (en) | 1993-05-14 | 1998-12-01 | Genentech, Inc. | Ras farnesyl transferase inhibitors |
| US5602098A (en) | 1993-05-18 | 1997-02-11 | University Of Pittsburgh | Inhibition of farnesyltransferase |
| US5380738A (en) | 1993-05-21 | 1995-01-10 | Monsanto Company | 2-substituted oxazoles further substituted by 4-fluorophenyl and 4-methylsulfonylphenyl as antiinflammatory agents |
| GB9602877D0 (en) | 1996-02-13 | 1996-04-10 | Merck Frosst Canada Inc | 3,4-Diaryl-2-hydroxy-2,5- dihydrofurans as prodrugs to cox-2 inhibitors |
| US5474995A (en) | 1993-06-24 | 1995-12-12 | Merck Frosst Canada, Inc. | Phenyl heterocycles as cox-2 inhibitors |
| US5436265A (en) | 1993-11-12 | 1995-07-25 | Merck Frosst Canada, Inc. | 1-aroyl-3-indolyl alkanoic acids and derivatives thereof useful as anti-inflammatory agents |
| US5728830A (en) | 1993-09-22 | 1998-03-17 | Kyowa Hakko Kogyo Co., Ltd. | Farnesyltransferase inhibitor |
| US5719148A (en) | 1993-10-15 | 1998-02-17 | Schering Corporation | Tricyclic amide and urea compounds useful for inhibition of g-protein function and for treatment of proliferative diseases |
| US5661152A (en) | 1993-10-15 | 1997-08-26 | Schering Corporation | Tricyclic sulfonamide compounds useful for inhibition of G-protein function and for treatment of proliferative diseases |
| AU699043B2 (en) | 1993-10-15 | 1998-11-19 | Schering Corporation | Tricyclic carbamate compounds useful for inhibition of G-protein function and for treatment of proliferative diseases |
| ES2164717T3 (es) | 1993-10-15 | 2002-03-01 | Schering Corp | Compuestos triciclicos de sulfonamida utiles para inhibir la funcion de la proteina-g y para el tratamiento de enfermedades proliferativas. |
| US5721236A (en) | 1993-10-15 | 1998-02-24 | Schering Corporation | Tricyclic carbamate compounds useful for inhibition of G-protein function and for treatment of proliferative diseases |
| IL111235A (en) | 1993-10-15 | 2001-03-19 | Schering Plough Corp | Medicinal preparations for inhibiting protein G activity and for the treatment of malignant diseases, containing tricyclic compounds, some such new compounds and a process for the preparation of some of them |
| KR100378615B1 (ko) | 1993-10-25 | 2003-10-10 | 파크데이비스앤드캄파니 | 단백질:파르네실트랜스퍼라제의치환된테트라및펜타펩티드억제제 |
| US5344991A (en) | 1993-10-29 | 1994-09-06 | G.D. Searle & Co. | 1,2 diarylcyclopentenyl compounds for the treatment of inflammation |
| US5783593A (en) | 1993-11-04 | 1998-07-21 | Abbott Laboratories | Inhibitors of squalene synthetase and protein farnesyltransferase |
| EP0677039B1 (en) | 1993-11-04 | 1999-03-10 | Abbott Laboratories | Cyclobutane derivatives as inhibitors of squalene synthetase and protein farnesyltransferase |
| WO1995012612A1 (en) | 1993-11-05 | 1995-05-11 | Warner-Lambert Company | Substituted di- and tripeptide inhibitors of protein:farnesyl transferase |
| US5466823A (en) | 1993-11-30 | 1995-11-14 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides |
| PE49195A1 (es) | 1993-12-07 | 1996-01-06 | Lilly Co Eli | Inhibidor de proteinquinasa |
| US5484799A (en) | 1993-12-09 | 1996-01-16 | Abbott Laboratories | Antifungal dorrigocin derivatives |
| US5545636A (en) | 1993-12-23 | 1996-08-13 | Eli Lilly And Company | Protein kinase C inhibitors |
| EP1449529B1 (en) | 1993-12-23 | 2010-01-27 | Eli Lilly And Company | Protein Kinase C Inhibitors |
| US5393790A (en) | 1994-02-10 | 1995-02-28 | G.D. Searle & Co. | Substituted spiro compounds for the treatment of inflammation |
| WO1995024612A1 (de) | 1994-03-07 | 1995-09-14 | International Business Machines Corporation | Verfahren und vorrichtung zur schnellen interpolation von zwischenwerten aus periodischen phasenverschobenen signalen und zur erkennung von defekten in einem drehkörper |
| US5840918A (en) | 1994-03-15 | 1998-11-24 | Eisai Co., Ltd. | Isoprenyl transferase inhibitors |
| HUT72440A (en) | 1994-03-31 | 1996-04-29 | Bristol Myers Squibb Co | Imidazole-containing inhibitors of farnesyl protein transferase and pharmaceutical compositions containing them |
| US5523430A (en) | 1994-04-14 | 1996-06-04 | Bristol-Myers Squibb Company | Protein farnesyl transferase inhibitors |
| US5510510A (en) | 1994-05-10 | 1996-04-23 | Bristol-Meyers Squibb Company | Inhibitors of farnesyl protein transferase |
| US5563255A (en) | 1994-05-31 | 1996-10-08 | Isis Pharmaceuticals, Inc. | Antisense oligonucleotide modulation of raf gene expression |
| TW365602B (en) | 1994-06-10 | 1999-08-01 | Rhone Poulenc Rorer Sa | New farnesyl transferase inhibitors, their preparation and the pharmaceutical compositions which contain them |
| US5571792A (en) | 1994-06-30 | 1996-11-05 | Warner-Lambert Company | Histidine and homohistidine derivatives as inhibitors of protein farnesyltransferase |
| WO1996005529A1 (en) | 1994-08-09 | 1996-02-22 | Micron Optics, Inc. | Temperature compensated fiber fabry-perot filters |
| EP0776884B1 (en) | 1994-08-11 | 2000-01-05 | Banyu Pharmaceutical Co., Ltd. | Substituted amide derivative |
| CA2155448A1 (en) | 1994-08-11 | 1996-02-12 | Katerina Leftheris | Inhibitors of farnesyl protein transferase |
| AU3192495A (en) | 1994-08-12 | 1996-03-07 | Banyu Pharmaceutical Co., Ltd. | N,n-disubstituted amic acid derivative |
| DE4429506B4 (de) | 1994-08-19 | 2007-09-13 | Degussa Gmbh | Verfahren zur Extraktion natürlicher Carotinoid-Farbstoffe |
| DE4429653C2 (de) | 1994-08-20 | 1997-04-03 | Anton Dr More | Konverter und Verfahren zum Frischen von Metallschmelzen insbesondere von Roheisen zu Stahl |
| DE69507284T2 (de) | 1994-11-22 | 1999-07-01 | Philips Electronics N.V., Eindhoven | Halbleiter mit einem träger auf dem ein substrat mit einem halbleiter-element mittels einer klebeschicht und ein leiterbahn-muster befestigt sind |
| AU3971295A (en) | 1994-12-09 | 1996-06-26 | Warner-Lambert Company | Substituted tetra- and pentapeptide inhibitors of protein:farnesyl transferase |
| CN1176652A (zh) | 1995-01-09 | 1998-03-18 | 马格拉国际有限公司 | 胶乳表面的耐磨图象印刷 |
| JP3929069B2 (ja) | 1995-01-12 | 2007-06-13 | ユニバーシティ オブ ピッツバーグ | プレニルトランスフェラーゼの阻害剤 |
| FR2729390A1 (fr) | 1995-01-18 | 1996-07-19 | Rhone Poulenc Rorer Sa | Nouveaux inhibiteurs de farnesyl transferase, leur preparation et les compositions pharmaceutiques qui les contiennent |
| FR2730491B1 (fr) | 1995-02-09 | 1997-03-14 | Rhone Poulenc Rorer Sa | Nouveaux inhibiteurs de farnesyl transferase, leur preparation et les compositions pharmaceutiques qui les contiennent |
| FR2730492B1 (fr) | 1995-02-09 | 1997-03-14 | Rhone Poulenc Rorer Sa | Nouveaux inhibiteurs de farnesyl transferase, leur preparation et les compositions pharmaceutiques qui les contiennent |
| US5633272A (en) | 1995-02-13 | 1997-05-27 | Talley; John J. | Substituted isoxazoles for the treatment of inflammation |
| US5684013A (en) | 1995-03-24 | 1997-11-04 | Schering Corporation | Tricyclic compounds useful for inhibition of g-protein function and for treatment of proliferative diseases |
| US5700806A (en) | 1995-03-24 | 1997-12-23 | Schering Corporation | Tricyclic amide and urea compounds useful for inhibition of G-protein function and for treatment of proliferative diseases |
| IL117580A0 (en) | 1995-03-29 | 1996-07-23 | Merck & Co Inc | Inhibitors of farnesyl-protein transferase and pharmaceutical compositions containing them |
| US5891872A (en) | 1995-04-07 | 1999-04-06 | Schering Corporation | Tricyclic compounds |
| US5712280A (en) | 1995-04-07 | 1998-01-27 | Schering Corporation | Tricyclic compounds useful for inhibition of G-protein function and for treatment of proliferative diseases |
| ES2194986T3 (es) | 1995-04-07 | 2003-12-01 | Schering Corp | Compuestos de carbonil-piperazinilo y piperidinilo que inhiben la farnesil-protein-transferasa. |
| IL117798A (en) | 1995-04-07 | 2001-11-25 | Schering Plough Corp | Tricyclic compounds useful for inhibiting the function of protein - G and for the treatment of malignant diseases, and pharmaceutical preparations containing them |
| US5831115A (en) | 1995-04-21 | 1998-11-03 | Abbott Laboratories | Inhibitors of squalene synthase and protein farnesyltransferase |
| IL118101A0 (en) | 1995-05-03 | 1996-09-12 | Abbott Lab | Inhibitors of farnesyltransferase |
| US5919780A (en) | 1995-06-16 | 1999-07-06 | Warner Lambert Company | Tricyclic inhibitors of protein farnesyltransferase |
| FR2736641B1 (fr) | 1995-07-10 | 1997-08-22 | Rhone Poulenc Rorer Sa | Nouveaux inhibiteurs de farnesyl transferase, leur preparation et les compositions pharmaceutiques qui les contiennent |
| AT402617B (de) | 1995-07-11 | 1997-07-25 | Datacon Schweitzer & Zeindl Gm | Anlage zum automatisierten, hermetischen anlage zum automatisierten, hermetischen verschliessen von gehäusen verschliessen von gehäusen |
| FR2736638B1 (fr) | 1995-07-12 | 1997-08-22 | Rhone Poulenc Rorer Sa | Nouveaux inhibiteurs de farnesyl transferase, leur preparation et les compositions pharmaceutiques qui les contiennent |
| CH690163A5 (fr) | 1995-07-28 | 2000-05-31 | Symphar Sa | Dérivés gem-diphosphonates substitués utiles en tant qu'agents anti-cancers. |
| US6020343A (en) | 1995-10-13 | 2000-02-01 | Merck Frosst Canada, Inc. | (Methylsulfonyl)phenyl-2-(5H)-furanones as COX-2 inhibitors |
| CA2235986C (en) | 1995-11-06 | 2006-09-12 | University Of Pittsburgh | Inhibitors of protein isoprenyl transferases |
| PT1186606E (pt) | 1995-11-17 | 2004-08-31 | Biotechnolog Forschung Mbh Gbf | Derivados do epotilone sua preparacao e utilizacao |
| ATE172199T1 (de) | 1995-11-20 | 1998-10-15 | Lilly Co Eli | Proteinkinase-c-inhibitor |
| CA2238081A1 (en) | 1995-11-22 | 1997-05-29 | S. Jane Desolms | Inhibitors of farnesyl-protein transferase |
| EE03484B1 (et) | 1995-12-08 | 2001-08-15 | Janssen Pharmaceutica N.V. | Farnesüülproteiintransferaasi inhibeerivad (5-imidasolüül)metüül-2-kinolinooni derivaadid ja nende kasutamine |
| EP1019392B1 (en) | 1995-12-22 | 2005-11-09 | Schering Corporation | Tricyclic amides useful for inhibition of g-protein function and for treatment of proliferative diseases |
| WO1997026246A1 (en) | 1996-01-16 | 1997-07-24 | Warner-Lambert Company | Substituted histidine inhibitors of protein farnesyltransferase |
| US6673927B2 (en) | 1996-02-16 | 2004-01-06 | Societe De Conseils De Recherches Et D'applications Scientifiques, S.A.S. | Farnesyl transferase inhibitors |
| WO1997038665A2 (en) | 1996-04-03 | 1997-10-23 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| ES2194195T3 (es) | 1996-04-12 | 2003-11-16 | Searle & Co | N-((4-(5-metil-3-fenililxazol-4-il)fenil)sulfonilpropanamida y su sal sodica como profarmacos de inhibnidores de cox-2. |
| JP2000511527A (ja) | 1996-05-22 | 2000-09-05 | ワーナー―ランバート・コンパニー | タンパク質ファルネシルトランスフェラーゼの阻止 |
| CA2260216A1 (en) | 1996-07-15 | 1998-01-22 | Bristol-Myers Squibb Company | Thiadioxobenzodiazepine inhibitors of farnesyl protein transferase |
| US5861419A (en) | 1996-07-18 | 1999-01-19 | Merck Frosst Canad, Inc. | Substituted pyridines as selective cyclooxygenase-2 inhibitors |
| EP0977563B1 (en) | 1996-12-03 | 2005-10-12 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| EP1003374A1 (en) | 1996-12-30 | 2000-05-31 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| JP2001507699A (ja) | 1996-12-30 | 2001-06-12 | メルク エンド カンパニー インコーポレーテッド | ファルネシル蛋白トランスフェラーゼ阻害薬 |
| EP1151002A4 (en) | 1999-01-29 | 2002-05-02 | Imclone Systems Inc | KDR-SPECIFIC ANTIBODIES AND USES THEREOF |
| GB9904387D0 (en) | 1999-02-25 | 1999-04-21 | Pharmacia & Upjohn Spa | Antitumour synergistic composition |
| AU4972900A (en) | 1999-04-08 | 2000-11-14 | Arch Development Corporation | Use of anti-vegf antibody to enhance radiation in cancer therapy |
| US6545004B1 (en) | 1999-10-27 | 2003-04-08 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
| PL203998B1 (pl) | 1999-10-27 | 2009-11-30 | Cytokinetics Inc | Nowy związek chinazolinowy, zastosowanie związku chinazolinowego, sposób jego otrzymywania i kompozycja farmaceutyczna zawierająca związek chinazolinowy |
| AU2001231143A1 (en) | 2000-01-27 | 2001-08-07 | Cytovia, Inc. | Substituted nicotinamides and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US6737957B1 (en) * | 2000-02-16 | 2004-05-18 | Verance Corporation | Remote control signaling using audio watermarks |
| ATE445613T1 (de) | 2000-11-07 | 2009-10-15 | Novartis Pharma Gmbh | Indolylmaleimidderivative als proteinkinase-c- inhibitoren |
| US6645970B2 (en) | 2000-11-07 | 2003-11-11 | Novartis Ag | Indolylmaleimide derivatives |
| WO2002083139A1 (en) | 2001-04-10 | 2002-10-24 | Merck & Co., Inc. | Inhibitors of akt activity |
| ATE400274T1 (de) | 2001-04-10 | 2008-07-15 | Merck & Co Inc | Hemmstoffe der akt aktivität |
| US6958334B2 (en) | 2001-04-10 | 2005-10-25 | Merck & Co., Inc. | Inhibitors of Akt activity |
| WO2002083064A2 (en) | 2001-04-10 | 2002-10-24 | Merck & Co., Inc. | A method of treating cancer |
| SE0102439D0 (sv) | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| SE0102438D0 (sv) | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| WO2003013526A1 (en) | 2001-08-08 | 2003-02-20 | Merck & Co. Inc. | Anticoagulant compounds |
| AU2002363429B2 (en) | 2001-11-07 | 2008-05-08 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| EP1463733B1 (en) | 2001-12-06 | 2007-09-05 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| DE60234278D1 (de) | 2001-12-06 | 2009-12-17 | Merck & Co Inc | Mitotische kinesin-hemmer |
| JP4391825B2 (ja) | 2001-12-06 | 2009-12-24 | メルク エンド カムパニー インコーポレーテッド | 有糸分裂キネシン阻害剤 |
| JP4464136B2 (ja) | 2001-12-06 | 2010-05-19 | メルク・シャープ・エンド・ドーム・コーポレイション | 有糸分裂キネシン阻害薬 |
| AU2002364128B2 (en) | 2001-12-06 | 2008-03-06 | Merck Sharp & Dohme Corp. | Mitotic kinesin inhibitors |
| TW200918046A (en) | 2002-04-03 | 2009-05-01 | Novartis Ag | Indolylmaleimide derivatives |
| SE0203754D0 (sv) | 2002-12-17 | 2002-12-17 | Astrazeneca Ab | New compounds |
| CA2536964A1 (en) | 2003-04-25 | 2004-11-11 | Ortho-Mcneil Pharmaceutical, Inc. | C-fms kinase inhibitors |
| WO2005079802A1 (en) | 2004-02-12 | 2005-09-01 | Merck & Co., Inc. | Bipyridyl amides as modulators of metabotropic glutamate receptor-5 |
| DE102004039789A1 (de) | 2004-08-16 | 2006-03-02 | Sanofi-Aventis Deutschland Gmbh | Arylsubstituierte polycyclische Amine, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| CA2584485C (en) | 2004-10-20 | 2013-12-31 | Resverlogix Corp. | Stilbenes and chalcones for the prevention and treatment of cardiovascular diseases |
| EP1809620B1 (en) | 2004-11-04 | 2010-12-29 | Addex Pharma SA | Novel tetrazole derivatives as positive allosteric modulators of metabotropic glutamate receptors |
| US7781438B2 (en) | 2005-07-11 | 2010-08-24 | Novartis Ag | Indolylmaleimide derivatives |
| TW200815426A (en) | 2006-06-28 | 2008-04-01 | Astrazeneca Ab | New pyridine analogues II 333 |
| BRPI0807107A2 (pt) * | 2007-02-08 | 2014-05-06 | Univ California | Mutações gnaq no melanoma |
| MX2009009304A (es) * | 2007-03-01 | 2009-11-18 | Novartis Ag | Inhibidores de cinasa pim y metodos para su uso. |
| BRPI0814441A2 (pt) | 2007-07-19 | 2015-07-14 | Schering Corp | Compostos de amida heterocíclica como inibidores de proteína cinase |
| PE20091309A1 (es) | 2007-12-21 | 2009-09-30 | Astrazeneca Ab | Derivados de ciclohexilo como inhibidores de acetil coenzima a carboxilasa |
| US8168794B2 (en) | 2008-03-03 | 2012-05-01 | Novartis Ag | Pim kinase inhibitors and methods of their use |
| CN101971057B (zh) | 2008-03-18 | 2013-03-27 | 诺瓦提斯公司 | 眼用透镜涂覆方法 |
| CN104311480A (zh) * | 2008-09-02 | 2015-01-28 | 诺华股份有限公司 | 作为激酶抑制剂的吡啶甲酰胺衍生物 |
| WO2010046780A2 (en) | 2008-10-22 | 2010-04-29 | Institut Pasteur Korea | Anti viral compounds |
| US8703962B2 (en) | 2008-10-24 | 2014-04-22 | Purdue Pharma L.P. | Monocyclic compounds and their use as TRPV1 ligands |
| KR101958632B1 (ko) | 2008-12-19 | 2019-03-15 | 버텍스 파마슈티칼스 인코포레이티드 | Atr 키나제의 억제제로서 유용한 피라진 유도체 |
| CN102245604A (zh) | 2008-12-23 | 2011-11-16 | 雅培制药有限公司 | 抗病毒化合物 |
| JP2012529512A (ja) * | 2009-06-08 | 2012-11-22 | アブラクシス バイオサイエンス リミテッド ライアビリティー カンパニー | トリアジン誘導体類及びそれらの治療応用 |
| US8415381B2 (en) | 2009-07-30 | 2013-04-09 | Novartis Ag | Heteroaryl compounds and their uses |
| AU2010291199A1 (en) * | 2009-09-04 | 2012-03-08 | Novartis Ag | Pyrazinylpyridines useful for the treatment of proliferative diseases |
| JP5802677B2 (ja) * | 2009-12-01 | 2015-10-28 | ライジェル ファーマシューティカルズ, インコーポレイテッド | プロテインキナーゼc阻害剤およびその使用 |
| RU2012146083A (ru) * | 2010-03-30 | 2014-05-10 | Новартис Аг | Ингибиторы ркс для лечения в-клеточной лимфомы с хроническими активным сигнальным каскадом в-клеточного рецептора |
| US20130109682A1 (en) | 2010-07-06 | 2013-05-02 | Novartis Ag | Cyclic ether compounds useful as kinase inhibitors |
| ES2553610T3 (es) | 2010-12-14 | 2015-12-10 | Electrophoretics Limited | Inhibidores de la caseína cinasa 1 delta (CK1delta) |
| WO2012101064A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyrimidine biaryl compounds as protein kinase inhibitors |
| UY33930A (es) * | 2011-03-04 | 2012-10-31 | Novartis Ag | Inhibidores novedosos de quinasas |
| CN103429572A (zh) | 2011-03-04 | 2013-12-04 | 诺瓦提斯公司 | 作为激酶抑制剂的四取代的环己基化合物 |
| WO2012129562A2 (en) | 2011-03-24 | 2012-09-27 | The Scripps Research Institute | Compounds and methods for inducing chondrogenesis |
| CN104109161B (zh) | 2011-07-27 | 2016-08-17 | 阿斯利康(瑞典)有限公司 | 2-(2,4,5-取代苯胺)嘧啶衍生物作为egfr调谐子用于治疗癌症 |
| JP6204462B2 (ja) * | 2012-05-21 | 2017-09-27 | ノバルティス アーゲー | キナーゼ阻害剤としての新規環置換n−ピリジニルアミド |
| CN103570625A (zh) | 2012-07-19 | 2014-02-12 | 南京英派药业有限公司 | N-(3-杂芳基芳基)-4-芳基芳基甲酰胺和类似物作为Hedgehog通路抑制剂及其应用 |
| US20150336960A1 (en) | 2012-12-19 | 2015-11-26 | Novartis Ag | Aryl-substituted fused bicyclic pyridazine compounds |
| WO2014181287A1 (en) | 2013-05-09 | 2014-11-13 | Piramal Enterprises Limited | Heterocyclyl compounds and uses thereof |
| JO3589B1 (ar) | 2014-08-06 | 2020-07-05 | Novartis Ag | مثبطات كيناز البروتين c وطرق استخداماتها |
| WO2019053595A1 (en) * | 2017-09-12 | 2019-03-21 | Novartis Ag | INHIBITORS OF PROTEIN KINASE C FOR THE TREATMENT OF CHOROIDAL MELLANOMA |
-
2015
- 2015-08-04 JO JOP/2015/0187A patent/JO3589B1/ar active
- 2015-08-05 AR ARP150102513A patent/AR101456A1/es unknown
- 2015-08-05 MX MX2017001640A patent/MX2017001640A/es active IP Right Grant
- 2015-08-05 KR KR1020177005674A patent/KR102518945B1/ko active IP Right Grant
- 2015-08-05 RS RS20191472A patent/RS59692B1/sr unknown
- 2015-08-05 JP JP2017506392A patent/JP6644765B2/ja active Active
- 2015-08-05 AU AU2015298364A patent/AU2015298364B2/en active Active
- 2015-08-05 EA EA201790323A patent/EA033361B1/ru not_active IP Right Cessation
- 2015-08-05 EP EP15750138.8A patent/EP3177608B1/en active Active
- 2015-08-05 ES ES15750138T patent/ES2754248T3/es active Active
- 2015-08-05 MY MYPI2017700303A patent/MY195488A/en unknown
- 2015-08-05 PT PT157501388T patent/PT3177608T/pt unknown
- 2015-08-05 UY UY0001036258A patent/UY36258A/es active IP Right Grant
- 2015-08-05 DK DK15750138T patent/DK3177608T3/da active
- 2015-08-05 LT LTEP15750138.8T patent/LT3177608T/lt unknown
- 2015-08-05 BR BR112017002263-0A patent/BR112017002263B1/pt active IP Right Grant
- 2015-08-05 KR KR1020237011340A patent/KR102605181B1/ko active IP Right Grant
- 2015-08-05 WO PCT/IB2015/055951 patent/WO2016020864A1/en active Application Filing
- 2015-08-05 TN TN2017000032A patent/TN2017000032A1/en unknown
- 2015-08-05 US US14/818,778 patent/US9452998B2/en active Active
- 2015-08-05 SI SI201530945T patent/SI3177608T1/sl unknown
- 2015-08-05 HU HUE15750138A patent/HUE047862T2/hu unknown
- 2015-08-05 TW TW104125461A patent/TWI680968B/zh active
- 2015-08-05 CN CN202110433787.0A patent/CN113214246A/zh active Pending
- 2015-08-05 CA CA2956996A patent/CA2956996C/en active Active
- 2015-08-05 SG SG11201700664UA patent/SG11201700664UA/en unknown
- 2015-08-05 CN CN201580053955.6A patent/CN106795151B/zh active Active
- 2015-08-05 PE PE2017000177A patent/PE20170241A1/es unknown
- 2015-08-05 PL PL15750138T patent/PL3177608T3/pl unknown
- 2015-08-05 EP EP19160124.4A patent/EP3514151A1/en active Pending
- 2015-08-05 CU CU2017000010A patent/CU24465B1/es unknown
- 2015-08-05 AP AP2017009729A patent/AP2017009729A0/en unknown
- 2015-08-28 CR CR20170036A patent/CR20170036A/es unknown
-
2016
- 2016-08-16 US US15/237,880 patent/US9845309B2/en active Active
-
2017
- 2017-01-26 IL IL250318A patent/IL250318B/en active IP Right Grant
- 2017-02-01 ZA ZA2017/00786A patent/ZA201700786B/en unknown
- 2017-02-03 DO DO2017000035A patent/DOP2017000035A/es unknown
- 2017-02-03 CL CL2017000290A patent/CL2017000290A1/es unknown
- 2017-02-03 CO CONC2017/0001049A patent/CO2017001049A2/es unknown
- 2017-02-03 PH PH12017500215A patent/PH12017500215A1/en unknown
- 2017-02-06 NI NI201700012A patent/NI201700012A/es unknown
- 2017-02-06 SV SV2017005373A patent/SV2017005373A/es unknown
- 2017-02-06 GT GT201700025A patent/GT201700025A/es unknown
- 2017-03-06 EC ECIEPI201713903A patent/ECSP17013903A/es unknown
- 2017-12-04 US US15/830,769 patent/US20180179181A1/en not_active Abandoned
-
2019
- 2019-03-15 US US16/354,416 patent/US10508101B2/en active Active
- 2019-10-28 US US16/666,108 patent/US11059804B2/en active Active
- 2019-11-21 CY CY20191101222T patent/CY1122407T1/el unknown
- 2019-12-11 HR HRP20192224TT patent/HRP20192224T1/hr unknown
-
2021
- 2021-03-12 US US17/200,509 patent/US11505541B2/en active Active
-
2023
- 2023-10-09 US US18/483,436 patent/US20240308979A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2754248T3 (es) | Inhibidores de proteína cinasa C y métodos para su uso | |
| KR101514853B1 (ko) | Pim 키나제 억제제 및 이들의 사용 방법 | |
| EP2152700B1 (en) | Csf-1r inhibitors, compositions, and methods of use | |
| TWI574962B (zh) | 作爲pi3激酶調節劑的芳雜環化合物及其使用方法和用途 | |
| JP2016516710A (ja) | ピリジン系cdk9キナーゼ阻害薬 | |
| JP2018536681A (ja) | インフルエンザウイルス複製の阻害剤、その適用方法および使用 | |
| EP3116506A1 (en) | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors | |
| EP3876928B1 (en) | Inhibitors of histone deacetylase useful for the treatment or prevention of hiv infection | |
| CN104016979B (zh) | 取代的环化合物及其使用方法和用途 |