CN114380800B - Pyridine-pyrimidine amine-benzimidazole derivative, and preparation method and application thereof - Google Patents
Pyridine-pyrimidine amine-benzimidazole derivative, and preparation method and application thereof Download PDFInfo
- Publication number
- CN114380800B CN114380800B CN202210096582.2A CN202210096582A CN114380800B CN 114380800 B CN114380800 B CN 114380800B CN 202210096582 A CN202210096582 A CN 202210096582A CN 114380800 B CN114380800 B CN 114380800B
- Authority
- CN
- China
- Prior art keywords
- compound
- cancer
- derivative
- mmol
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 7
- OIMWEHOYHJJPJD-UHFFFAOYSA-N pyridine;pyrimidine Chemical compound C1=CC=NC=C1.C1=CN=CN=C1 OIMWEHOYHJJPJD-UHFFFAOYSA-N 0.000 title abstract description 7
- 150000001875 compounds Chemical class 0.000 claims abstract description 101
- 239000003814 drug Substances 0.000 claims abstract description 15
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 13
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 claims abstract description 11
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 claims abstract description 11
- 229940043355 kinase inhibitor Drugs 0.000 claims abstract description 10
- 239000003757 phosphotransferase inhibitor Substances 0.000 claims abstract description 10
- 150000003839 salts Chemical class 0.000 claims abstract description 9
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 7
- 208000003174 Brain Neoplasms Diseases 0.000 claims abstract description 7
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 claims abstract 3
- 102100026804 Cyclin-dependent kinase 6 Human genes 0.000 claims abstract 3
- 239000001257 hydrogen Substances 0.000 claims description 34
- 229910052739 hydrogen Inorganic materials 0.000 claims description 34
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- 239000000463 material Substances 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 206010006187 Breast cancer Diseases 0.000 claims description 5
- 208000026310 Breast neoplasm Diseases 0.000 claims description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 5
- 201000005202 lung cancer Diseases 0.000 claims description 5
- 208000020816 lung neoplasm Diseases 0.000 claims description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 4
- 208000008770 Multiple Hamartoma Syndrome Diseases 0.000 claims description 4
- 206010038389 Renal cancer Diseases 0.000 claims description 4
- 208000029742 colonic neoplasm Diseases 0.000 claims description 4
- 201000010982 kidney cancer Diseases 0.000 claims description 4
- 201000007815 Bannayan-Riley-Ruvalcaba syndrome Diseases 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims description 2
- 206010005969 Bone giant cell tumour Diseases 0.000 claims description 2
- 201000002847 Cowden syndrome Diseases 0.000 claims description 2
- 206010014967 Ependymoma Diseases 0.000 claims description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 2
- 208000005726 Inflammatory Breast Neoplasms Diseases 0.000 claims description 2
- 206010021980 Inflammatory carcinoma of the breast Diseases 0.000 claims description 2
- 208000022010 Lhermitte-Duclos disease Diseases 0.000 claims description 2
- 206010025323 Lymphomas Diseases 0.000 claims description 2
- 208000000172 Medulloblastoma Diseases 0.000 claims description 2
- 206010033128 Ovarian cancer Diseases 0.000 claims description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 2
- 206010060862 Prostate cancer Diseases 0.000 claims description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 206010039491 Sarcoma Diseases 0.000 claims description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 2
- 208000008383 Wilms tumor Diseases 0.000 claims description 2
- 239000004480 active ingredient Substances 0.000 claims description 2
- 201000011143 bone giant cell tumor Diseases 0.000 claims description 2
- 201000010099 disease Diseases 0.000 claims description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 2
- 206010017758 gastric cancer Diseases 0.000 claims description 2
- 208000005017 glioblastoma Diseases 0.000 claims description 2
- 201000010536 head and neck cancer Diseases 0.000 claims description 2
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 2
- 201000004653 inflammatory breast carcinoma Diseases 0.000 claims description 2
- 208000032839 leukemia Diseases 0.000 claims description 2
- 201000007270 liver cancer Diseases 0.000 claims description 2
- 208000014018 liver neoplasm Diseases 0.000 claims description 2
- 206010025135 lupus erythematosus Diseases 0.000 claims description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 2
- 201000001441 melanoma Diseases 0.000 claims description 2
- 201000008968 osteosarcoma Diseases 0.000 claims description 2
- 201000002528 pancreatic cancer Diseases 0.000 claims description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 2
- 239000000825 pharmaceutical preparation Substances 0.000 claims description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 2
- 201000011549 stomach cancer Diseases 0.000 claims description 2
- 201000002510 thyroid cancer Diseases 0.000 claims description 2
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 2
- 230000000694 effects Effects 0.000 abstract description 15
- 230000005764 inhibitory process Effects 0.000 abstract description 15
- 230000002401 inhibitory effect Effects 0.000 abstract description 14
- 210000004881 tumor cell Anatomy 0.000 abstract description 9
- 229940079593 drug Drugs 0.000 abstract description 7
- 238000001727 in vivo Methods 0.000 abstract description 5
- 230000004614 tumor growth Effects 0.000 abstract description 5
- 230000008499 blood brain barrier function Effects 0.000 abstract description 4
- 210000001218 blood-brain barrier Anatomy 0.000 abstract description 4
- 239000000651 prodrug Substances 0.000 abstract description 4
- 229940002612 prodrug Drugs 0.000 abstract description 4
- 239000012453 solvate Substances 0.000 abstract description 4
- 238000006243 chemical reaction Methods 0.000 description 56
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 42
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 39
- 239000002994 raw material Substances 0.000 description 33
- 150000002431 hydrogen Chemical class 0.000 description 28
- 239000007787 solid Substances 0.000 description 28
- 229910052736 halogen Inorganic materials 0.000 description 27
- 150000002367 halogens Chemical class 0.000 description 27
- 229910052757 nitrogen Inorganic materials 0.000 description 24
- 210000004027 cell Anatomy 0.000 description 21
- 238000004896 high resolution mass spectrometry Methods 0.000 description 20
- 238000001308 synthesis method Methods 0.000 description 20
- -1 hydroxy, carboxyl Chemical group 0.000 description 19
- 125000000217 alkyl group Chemical group 0.000 description 16
- 238000012360 testing method Methods 0.000 description 16
- 239000000203 mixture Substances 0.000 description 15
- 125000003545 alkoxy group Chemical group 0.000 description 14
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 14
- 239000000243 solution Substances 0.000 description 14
- 238000002474 experimental method Methods 0.000 description 13
- 238000000746 purification Methods 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- SFHYNDMGZXWXBU-LIMNOBDPSA-N 6-amino-2-[[(e)-(3-formylphenyl)methylideneamino]carbamoylamino]-1,3-dioxobenzo[de]isoquinoline-5,8-disulfonic acid Chemical compound O=C1C(C2=3)=CC(S(O)(=O)=O)=CC=3C(N)=C(S(O)(=O)=O)C=C2C(=O)N1NC(=O)N\N=C\C1=CC=CC(C=O)=C1 SFHYNDMGZXWXBU-LIMNOBDPSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 12
- 101000715943 Caenorhabditis elegans Cyclin-dependent kinase 4 homolog Proteins 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- 238000000034 method Methods 0.000 description 10
- 238000012544 monitoring process Methods 0.000 description 10
- KOFLVDBWRHFSAB-UHFFFAOYSA-N 1,2,4,5-tetrahydro-1-(phenylmethyl)-5,9b(1',2')-benzeno-9bh-benz(g)indol-3(3ah)-one Chemical compound C1C(C=2C3=CC=CC=2)C2=CC=CC=C2C23C1C(=O)CN2CC1=CC=CC=C1 KOFLVDBWRHFSAB-UHFFFAOYSA-N 0.000 description 9
- 241000699670 Mus sp. Species 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 238000005406 washing Methods 0.000 description 9
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 8
- 102000013701 Cyclin-Dependent Kinase 4 Human genes 0.000 description 8
- 102000013698 Cyclin-Dependent Kinase 6 Human genes 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 239000012295 chemical reaction liquid Substances 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 230000035755 proliferation Effects 0.000 description 8
- HUXJXNSHCKHFIL-UHFFFAOYSA-N 1-(2-bromoethoxy)-2-methoxyethane Chemical compound COCCOCCBr HUXJXNSHCKHFIL-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- 239000005457 ice water Substances 0.000 description 7
- 239000012046 mixed solvent Substances 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- LCCCTXULXHJDLA-UHFFFAOYSA-N 1-[2-(2-bromoethoxy)ethoxy]-2-methoxyethane Chemical compound COCCOCCOCCBr LCCCTXULXHJDLA-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 description 6
- 230000004663 cell proliferation Effects 0.000 description 6
- 239000012091 fetal bovine serum Substances 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- LJXQPZWIHJMPQQ-UHFFFAOYSA-N pyrimidin-2-amine Chemical compound NC1=NC=CC=N1 LJXQPZWIHJMPQQ-UHFFFAOYSA-N 0.000 description 6
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 210000005013 brain tissue Anatomy 0.000 description 5
- 201000011510 cancer Diseases 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 5
- UOXJNGFFPMOZDM-UHFFFAOYSA-N 2-[di(propan-2-yl)amino]ethylsulfanyl-methylphosphinic acid Chemical compound CC(C)N(C(C)C)CCSP(C)(O)=O UOXJNGFFPMOZDM-UHFFFAOYSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 108091000080 Phosphotransferase Proteins 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 239000012065 filter cake Substances 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 230000005917 in vivo anti-tumor Effects 0.000 description 4
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 102000020233 phosphotransferase Human genes 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 102000016736 Cyclin Human genes 0.000 description 3
- 108050006400 Cyclin Proteins 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 229950001573 abemaciclib Drugs 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 230000022131 cell cycle Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical compound C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 239000011550 stock solution Substances 0.000 description 3
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 3
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 2
- HTFNVAVTYILUCF-UHFFFAOYSA-N 2-[2-ethoxy-4-[4-(4-methylpiperazin-1-yl)piperidine-1-carbonyl]anilino]-5-methyl-11-methylsulfonylpyrimido[4,5-b][1,4]benzodiazepin-6-one Chemical compound CCOc1cc(ccc1Nc1ncc2N(C)C(=O)c3ccccc3N(c2n1)S(C)(=O)=O)C(=O)N1CCC(CC1)N1CCN(C)CC1 HTFNVAVTYILUCF-UHFFFAOYSA-N 0.000 description 2
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 2
- 238000011725 BALB/c mouse Methods 0.000 description 2
- 102000003910 Cyclin D Human genes 0.000 description 2
- 108090000259 Cyclin D Proteins 0.000 description 2
- 206010012735 Diarrhoea Diseases 0.000 description 2
- 208000010201 Exanthema Diseases 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 108010019160 Pancreatin Proteins 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 201000005884 exanthem Diseases 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 229940055695 pancreatin Drugs 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 206010037844 rash Diseases 0.000 description 2
- 238000007789 sealing Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 238000009987 spinning Methods 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- ZDFBKZUDCQQKAC-UHFFFAOYSA-N 1-bromo-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(Br)C=C1 ZDFBKZUDCQQKAC-UHFFFAOYSA-N 0.000 description 1
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 1
- HUUFTVUBFFESEN-UHFFFAOYSA-N 2-bromo-5-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Br)N=C1 HUUFTVUBFFESEN-UHFFFAOYSA-N 0.000 description 1
- IVAQXQBQSNCSDU-UHFFFAOYSA-N 4-(2-methoxyethyl)piperazine-1-carbaldehyde Chemical compound COCCN1CCN(C=O)CC1 IVAQXQBQSNCSDU-UHFFFAOYSA-N 0.000 description 1
- KEBLLGGDPFLQMX-UHFFFAOYSA-N 4-[2-(2-methoxyethoxy)ethyl]piperazine-1-carbaldehyde Chemical compound COCCOCCN1CCN(CC1)C=O KEBLLGGDPFLQMX-UHFFFAOYSA-N 0.000 description 1
- ATXXLNCPVSUCNK-UHFFFAOYSA-N 5-bromo-2-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Br)C=N1 ATXXLNCPVSUCNK-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 108091007914 CDKs Proteins 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 1
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 1
- 102000001388 E2F Transcription Factors Human genes 0.000 description 1
- 108010093502 E2F Transcription Factors Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 230000010190 G1 phase Effects 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 1
- 206010067482 No adverse event Diseases 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 241000233805 Phoenix Species 0.000 description 1
- 108050002653 Retinoblastoma protein Proteins 0.000 description 1
- 230000018199 S phase Effects 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 1
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 1
- 241000934136 Verruca Species 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940058303 antinematodal benzimidazole derivative Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000012864 cross contamination Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 239000003684 drug solvent Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethyl mercaptane Natural products CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- ZFGMDIBRIDKWMY-PASTXAENSA-N heparin Chemical compound CC(O)=N[C@@H]1[C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O[C@@H]1O[C@@H]1[C@@H](C(O)=O)O[C@@H](O[C@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@@H](O[C@@H]3[C@@H](OC(O)[C@H](OS(O)(=O)=O)[C@H]3O)C(O)=O)O[C@@H]2O)CS(O)(=O)=O)[C@H](O)[C@H]1O ZFGMDIBRIDKWMY-PASTXAENSA-N 0.000 description 1
- 229960001008 heparin sodium Drugs 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 208000013403 hyperactivity Diseases 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- 239000011565 manganese chloride Substances 0.000 description 1
- 235000002867 manganese chloride Nutrition 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000012353 t test Methods 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 125000003698 tetramethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The invention provides a pyridine-pyrimidine amine-benzimidazole derivative, a preparation method and application thereof, and belongs to the field of medicines. The derivative is a compound shown in a formula I, or a salt thereof, or a stereoisomer thereof, or a solvate thereof, or a hydrate thereof, or a prodrug thereof. The derivative has good inhibitory activity on CDK4 and CDK6 kinases, and can be used for preparing CDK4 and CDK6 kinase inhibitors; meanwhile, the derivative has remarkable inhibition activity on various tumor cells, can inhibit the growth of tumors in vivo, and has excellent effect; the derivative has small administration dosage and high safety; in addition, the derivative can pass through the blood brain barrier, and solves one of the biggest problems in brain tumor treatment. The derivative can be used for preparing medicines for preventing and/or treating tumors and autoimmune diseases, and has good application prospects.
Description
Technical Field
The invention belongs to the field of medicines, and particularly relates to a pyridine-pyrimidine amine-benzimidazole derivative, and a preparation method and application thereof.
Background
Malignant tumors have high mortality and the incidence rate of the malignant tumors increases year by year, and the malignant tumors exceed cardiovascular and cerebrovascular diseases and become the first killer for human health. CDK4/6, cyclin dependent kinases 4 and 6, are key conditional proteins of the human cell division proliferation cycle, and are common downstream targets of a plurality of growth promoting signal pathways such as ER, PI3K/mTOR and the like.
CDK4/6 regulates the cell cycle through the Cyclin D-CDK4/6-RB pathway. During cell proliferation, CDK4/6 forms a complex with Cyclin D (Cyclin D) and phosphorylates retinoblastoma protein (Rb). Rb, once phosphorylated, releases the tightly bound transcription factor E2F. E2F activation further pushes the cell cycle from G1 phase to S phase, into the cell proliferation cycle.
It was found that there is a deregulation of the pathway Cyclin D-CDK4/6-RB in almost all malignant tumors. This pathway is continuously activated, which manifests as CDK4/6 hyperactivity, ultimately leading to uncontrolled cell growth and proliferation and spread of cancer cells. And inhibit CDK4/6, make tumor cell unable to form Cyclin D-CK4/6 complex, block cell cycle in growing phase, thus achieve the goal of inhibiting tumor proliferation. Thus, targeted inhibition of CDK4/6 with novel small molecule inhibitors is a potential therapeutic approach.
Disclosure of Invention
The invention aims to provide pyridine-pyrimidine amine-benzimidazole derivatives, and a preparation method and application thereof.
The present invention provides a compound of formula I, or a salt thereof, or a stereoisomer thereof, or a solvate thereof, or a hydrate thereof, or a prodrug thereof:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 an integer selected from 0 to 5;
the dotted line is a bond or none;
X 1 、Y 1 independently selected from N or CR 3 ;
X 2 、Y 2 Independently selected from N or CR 4 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 3 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 4 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxy, amino, nitro.
Further, the method comprises the steps of,
n 1 an integer selected from 1 to 3;
n 2 selected from 0 or 1;
the dotted line is a bond or none;
X 1 、Y 1 independently selected from N or CR 3 ;
X 2 、Y 2 Independently selected from N or CR 4 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 4 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen.
Further, the compound is represented by formula II:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 an integer selected from 0 to 5;
X 1 、Y 1 independently selected from N or CR 3 ;
X 2 、Y 2 Independently selected from N or CR 4 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 3 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 4 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
preferably, the method comprises the steps of,
n 1 an integer selected from 1 to 3;
n 2 selected from 0 or 1;
X 1 、Y 1 independently selected from N or CR 3 ;
X 2 、Y 2 Independently selected from N or CR 4 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 4 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen.
Further, the compound is represented by formula III:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 an integer selected from 0 to 5;
X 1 、Y 1 independently selected from N or CR 3 ;
X 2 、Y 2 Independently selected from N or CR 4 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 3 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 4 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
preferably, the method comprises the steps of,
n 1 an integer selected from 1 to 3;
n 2 selected from 0 or 1;
X 1 、Y 1 independently selected from N or CR 3 ;
X 2 、Y 2 Independently selected from N or CR 4 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 4 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen.
Further, the compound is represented by formula IV:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 an integer selected from 0 to 5;
the dotted line is a bond or none;
X 1 、Y 1 independently selected from N or CR 3 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
each R 3 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
preferably, the method comprises the steps of,
n 1 an integer selected from 1 to 3;
n 2 selected from 0 or 1;
the dotted line is a bond or none;
X 1 、Y 1 independently selected from N or CR 3 ;
R 1 、R 2 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen.
Further, the compound is represented by formula V:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 an integer selected from 0 to 5;
the dotted line is a bond or none;
X 1 、Y 1 independently selected from N or CR 3 ;
Each R 3 Are independently selected from hydrogen, C 1 ~C 8 Alkyl, C 1 ~C 8 Alkoxy, halogen, hydroxy, carboxyl, amino, nitro;
preferably, the method comprises the steps of,
n 1 an integer selected from 1 to 3;
n 2 selected from 0 or 1;
the dotted line is a bond or none;
X 1 、Y 1 independently selected from N or CR 3 ;
Each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen.
Further, the compound is represented by formula Va:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 selected from 0 or 1;
X 1 、Y 1 independently selected from N or CR 3 ;
Each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen;
alternatively, the compound is of formula Vb:
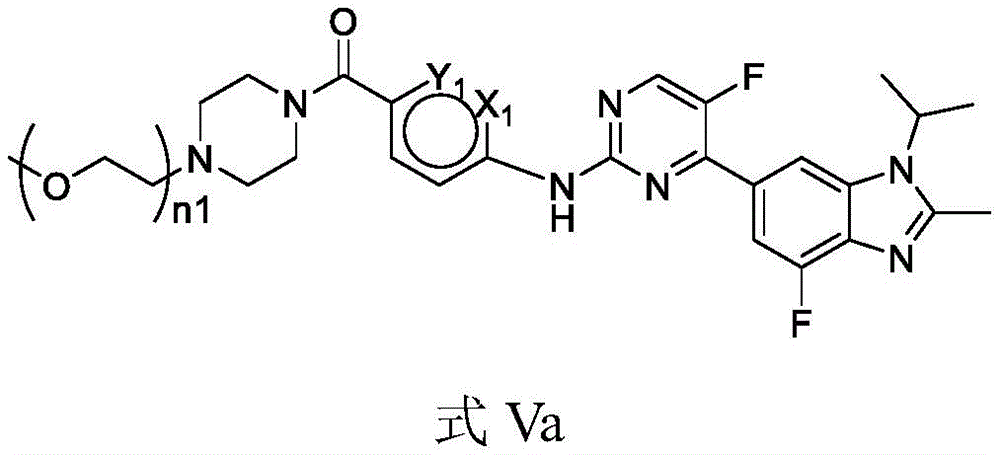
wherein,,
n 1 an integer selected from 1 to 3;
X 1 、Y 1 independently selected from N or CR 3 ;
Each R 3 Are independently selected from hydrogen, C 1 ~C 3 Alkyl, C 1 ~C 3 Alkoxy, halogen.
Further, the compound is one of the following compounds:



the invention also provides the use of a compound as hereinbefore described, or a salt thereof, or a stereoisomer thereof, or a solvate thereof, or a hydrate thereof, or a prodrug thereof, in the preparation of a CDK4 and/or CDK6 kinase inhibitor.
Further, the kinase inhibitor is a medicament for preventing and/or treating tumors;
preferably, the tumor is brain cancer, glioblastoma, leukemia, lymphoma, bannayan-Zonana syndrome, coden disease, lhermitte-Duclos disease, breast cancer, inflammatory breast cancer, wilms 'tumor, ewing's sarcoma, rhabdomyosarcoma, ependymoma, medulloblastoma, colon cancer, stomach cancer, bladder cancer, head and neck cancer, kidney cancer, lung cancer, liver cancer, melanoma, kidney cancer, ovarian cancer, pancreatic cancer, prostate cancer, sarcoma, osteosarcoma, bone giant cell tumor or thyroid cancer.
Further, the kinase inhibitor is a medicament for preventing and/or treating autoimmune diseases;
preferably, the autoimmune disease is psoriasis or lupus erythematosus.
The invention also provides a medicine which is a pharmaceutical preparation prepared by taking the compound, or a salt thereof, or a stereoisomer thereof, or a solvate thereof, or a hydrate thereof, or a prodrug thereof as an active ingredient and adding pharmaceutically acceptable auxiliary materials or auxiliary ingredients.
The compounds and derivatives provided in the present invention are named according to IUPAC (international union of pure and applied chemistry) or CAS (chemical abstract service, columbus, OH) naming system.
In the present invention, the minimum and maximum values of the carbon atom content in the hydrocarbon group are represented by prefixes, for example, C a~b Alkyl indicates any alkyl group containing from "a" to "b" carbon atoms. Thus, for example, C 1 ~C 8 Alkyl refers to straight or branched alkyl groups containing 1 to 8 carbon atoms; c (C) 1 ~C 8 Alkoxy refers to an alkoxy group containing 1 to 8 carbon atoms.
In the present invention, halogen is fluorine, chlorine, bromine or iodine.
In the formula I of the invention, n 1 When 1, the structure is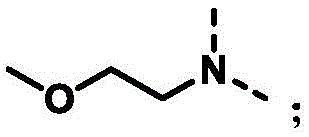 n 1 When 2, the structure is +.>
n 1 When 2, the structure is +.> n 1 In case of 3, the structure is +.>
n 1 In case of 3, the structure is +.>
In the formula I, the dotted line is zero, and the structure is When the dotted line is a bond, the structure is +>
When the dotted line is a bond, the structure is +>
n 2 0, 1, 2, 3, 4 or 5.
The invention provides a pyridine-pyrimidine amine-benzimidazole derivative which has good inhibitory activity on CDK4 and CDK6 kinase and can be used for preparing CDK4 and CDK6 kinase inhibitors; meanwhile, the derivative has remarkable inhibition activity on various tumor cells, can inhibit the growth of tumors in vivo, and has excellent effect; the derivative has small administration dosage and high safety; in addition, the derivative can pass through the blood brain barrier, and solves one of the biggest problems in brain tumor treatment. The derivative can be used for preparing medicines for preventing and/or treating tumors and autoimmune diseases, and has good application prospects.
It should be apparent that, in light of the foregoing, various modifications, substitutions and alterations can be made herein without departing from the spirit and scope of the invention as defined by the appended claims.
The above-described aspects of the present invention will be described in further detail below with reference to specific embodiments in the form of examples. It should not be understood that the scope of the above subject matter of the present invention is limited to the following examples only. All techniques implemented based on the above description of the invention are within the scope of the invention.
Drawings
FIG. 1 shows the tumor growth of NOD-Bablc mice in each group.
Figure 2 shows the distribution of compound A2 in blood and brain tissue at various time points.
Detailed Description
Unless otherwise indicated, the materials and equipment used in the embodiments of the present invention are all known products and are obtained by purchasing commercially available products.
Example 1, compound A1 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2-methoxyethyl) piperazin-1-yl) methyl) pyridin-2-yl) pyrimidin-2-amine

The synthetic route for compound A1 is as follows:

the first step:
raw material 1 (1 g,4.61 mmol) was dissolved in anhydrous DMF (15 mL), anhydrous potassium carbonate (862mg, 6.24 mmol) was added, and stirred at room temperature for 5min. Subsequently, raw material 2 (1.03 g,5.532 mmol) was slowly added and the reaction was stirred at 55℃for 2h. TLC monitors the reaction to be complete, the reaction liquid is cooled to room temperature, a proper amount of ice water is added under the stirring condition, a large amount of white solid is precipitated in the reaction system, the reaction system is filtered, a filter cake is washed with water for 3 times, and intermediate 3 is obtained after drying, the white solid is 1.321g, and the yield is 89%.
And a second step of:
intermediate 3 (1.2 g,3.72 mmol) was dissolved in methanol (25 mL), 10% palladium on carbon (390 mg,0.372 mmol) was added, the mixture was evacuated, replaced three times with hydrogen, and the mixture was stirred at room temperature under a hydrogen atmosphere for 2 hours. TLC monitoring the reaction completion, the reaction solution was filtered with celite, washed 3 times with a mixed solvent of dichloromethane/methanol (10/1, v/v), and the filtrate was concentrated to dryness under reduced pressure to give crude product intermediate 4. Without further purification, it was used directly in the next reaction.
And a third step of:
toward intermediate 4 (3.72 mmol), starting material 5 (1.282 g,3.72 mmol), pd 2 (dba) 3 Toluene (20 mL) was added to a mixture of (3411 mg,0.372 mmol), X-phos (178 mg,0.372 mmol) and cesium carbonate (1.82 g,5.58 mmol), the mixture was evacuated and replaced 3 times with nitrogen, and the reaction mixture was stirred under nitrogen at 100℃for 5 hours. TLC monitoring reaction completion, cooling the reaction liquid to room temperature, filtering with diatomite, washing 3 times with dichloromethane/methanol (10/1, v/v) mixed solvent, concentrating the filtrate under reduced pressure, separating by silica gel column chromatography to obtain intermediate 6, 1.1g of off-white solid, and the yield is 51%.
Fourth step:
intermediate 6 (1 g,1.73 mmol) was dissolved in dichloromethane (15 mL) and trifluoroacetic acid (4 mL) was slowly added dropwise with stirring and reacted at room temperature for 2h. TLC monitoring reaction completion, the reaction solution was concentrated to dryness under reduced pressure, dissolved again with a small amount of dichloromethane, dried by spin, and operated repeatedly 3 times to obtain crude product intermediate 7. Without further purification, it was used directly in the next reaction.
Fifth step:
intermediate 7 (0.173 mmol) was dissolved in anhydrous DMF (4 mL), cesium carbonate (113 mg, 0.348 mmol) was added, and after stirring at room temperature for 5min, starting material 8 (25. Mu.L, 0.2595 mmol) was slowly added and the temperature was raised to 60℃for 3h. TLC monitoring reaction completion, cooling the reaction solution to room temperature, adding appropriate amount of ice water, extracting with ethyl acetate three times, mixing organic phases, washing with water, washing with saturated saline, drying with anhydrous sodium sulfate, filtering, concentrating the filtrate under reduced pressure to dryness, further drying with siliconThe target compound A1 is separated by gel column chromatography, the white solid is 68mg, and the yield is 73%. HR-MS (ESI-TOF) m/z calcd for C 28 H 34 F 2 N 8 O[M+H] + 537.2902,found 537.2900.
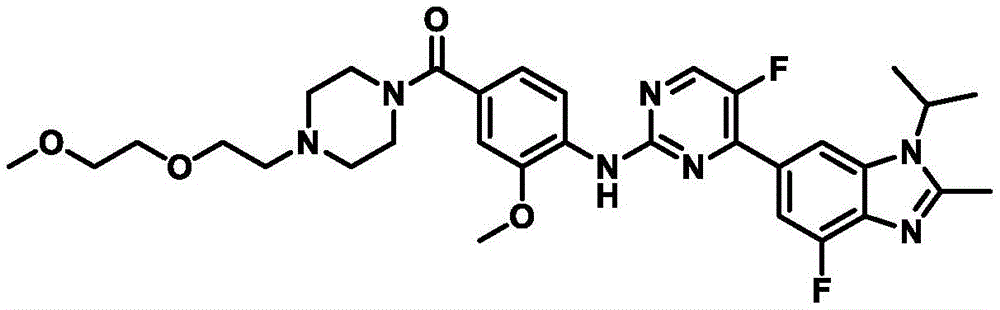
EXAMPLE 2 Compound A2 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) methyl) pyridin-2-yl) pyrimidin-2-amine

The intermediate 7 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A2 is obtained by referring to a fifth step of the synthesis method of the compound A1, and the yield is 78 percent. HR-MS (ESI-TOF) m/z calcd for C 30 H 38 F 2 N 8 O 2 [M+H] + 581.3164,found 581.3163.
EXAMPLE 3 Compound A3 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2- (2-methoxyethoxy) ethoxy) ethyl) piperazin-1-yl) methyl) pyridin-2-amine

The intermediate 7 and diethylene glycol-2-bromoethyl methyl ether are used as raw materials, and the compound A3 is obtained by referring to a fifth step of compound A1 synthesis method, and the yield is 69%. HR-MS (ESI-TOF) m/z calcd for C 32 H 42 F 2 N 8 O 3 [M+H] + 625.3426,found 625.3425.
Example 4 Compound A4 (5- ((5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) pyrimidin-2-yl) amino) pyridin-2-yl) (4- (2-methoxyethyl) piperazin-1-yl) methanone

The synthetic route for compound A4 is as follows:

the first step:
raw material 9 (2 g,11.9 mmol) was dissolved in anhydrous THF (30 mL), DIEA (2.95 mL,17.85 mmol) and HATU (4.98 g,13.09 mmol) were added sequentially, and after stirring at room temperature for 10min, raw material 2 (3.32 g,17.85 mmol) was slowly added and the reaction was continued under stirring at room temperature for 4h. TLC was used to monitor completion of the reaction, a proper amount of water was added, extraction was performed three times with ethyl acetate, the organic phases were combined, washed with water, then with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated to dryness under reduced pressure, followed by separation by silica gel column chromatography to give intermediate 10 as a white solid 2.092g in 52% yield.
And a second step of:
the second step synthesis of reference compound A1 gives crude intermediate 11 starting from intermediate 10. Without further purification, it was used directly in the next reaction.
And a third step of:
the intermediate 12 is obtained as a yellow solid with a yield of 82% by referring to the third step of the synthesis method of the compound A1 by taking the intermediate 11 and the raw material 5 as raw materials.
Fourth step:
the fourth step of the synthesis method, which uses intermediate 12 as a raw material, refers to compound A1, gives a crude product intermediate 13. Without further purification, it was used directly in the next reaction.
Fifth step:
the intermediate 13 and the raw material 8 are used as raw materials, and the compound A4 is obtained by referring to a fifth step of synthesis method of the compound A1, and is yellow solid with the yield of 77%. HR-MS (ESI-TOF) m/z calcd for C 28 H 32 F 2 N 8 O 2 [M+H] + 551.2694,found 551.2692.
Example 5 Compound A5 (5- ((5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) pyrimidin-2-yl) amino) pyridin-2-yl) (4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) methanone

The intermediate 13 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A5 is obtained by referring to a fifth step of the synthesis method of the compound A1, and the yield is 66 percent. HR-MS (ESI-TOF) m/z calcd for C 30 H 36 F 2 N 8 O 3 [M+H] + 595.2956,found 595.2952.
Example 6 Compound A6 (5- ((5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) pyrimidin-2-yl) amino) pyridin-2-yl) (4- (2- (2-methoxyethoxy) ethoxy) piperazin-1-yl) methanone

The intermediate 13 and diethylene glycol-2-bromoethyl methyl ether are used as raw materials, and the compound A6 is obtained by referring to a fifth step of compound A1 synthesis method, and the yield is 74 percent. HR-MS (ESI-TOF) m/z calcd for C 32 H 40 F 2 N 8 O 4 [M+H] + 639.3219,found 639.3218.
EXAMPLE 7 Compound A7 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (6- ((4- (2-methoxyethyl) piperazin-1-yl) methyl) pyridin-3-yl) pyrimidin-2-amine

The synthetic route for compound A7 is as follows:

the first step:
intermediate 11 (918 mg,3.0 mmol) was dissolved in anhydrous THF (20 mL) and borane/dimethyl sulfide was slowly added dropwiseComplex BH 3 /S(CH 3 ) 2 (2M in THF) (6 mL) and then the reaction was warmed to 60℃and stirred for 3h. TLC monitored completion of the reaction, the reaction mixture was cooled to room temperature, piperidine (2.5 mL) was added and stirring continued at room temperature for 30min. The reaction mixture was concentrated to dryness under reduced pressure, and intermediate 14 was isolated by silica gel column chromatography as a white solid (362 mg) in 41% yield.
And a second step of:
the intermediate 15, a white solid, was obtained in a yield of 76% by the third step of the synthesis method with reference to compound A1, starting from intermediate 14 and starting material 5.
And a third step of:
the fourth step of the synthesis, with reference to compound A1, gives the crude intermediate 16 starting from intermediate 15. Without further purification, it was used directly in the next reaction.
Fourth step:
the intermediate 16 and the raw material 8 are used as raw materials, and the compound A7 is obtained by referring to a fifth step of the synthesis method of the compound A1, and is white solid with the yield of 63%. HR-MS (ESI-TOF) m/z calcd for C 28 H 34 F 2 N 8 O[M+H] + 537.2902,found 537.2902.
Example 8, compound A8 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (6- ((4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) methyl) pyridin-3-yl) pyrimidin-2-amine

The intermediate 16 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A8 is obtained by referring to a fifth step of the synthesis method of the compound A1, and the yield is 70 percent. HR-MS (ESI-TOF) m/z calcd for C 30 H 38 F 2 N 8 O 2 [M+H] + 581.3164,found 581.3161.
Example 9, compound A9 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (6- ((4- (2- (2-methoxyethoxy) ethoxy) ethyl) piperazin-1-yl) methyl) pyridin-3-yl) pyrimidin-2-amine

The intermediate 16 and diethylene glycol-2-bromoethyl methyl ether are used as raw materials, and the compound A9 is obtained by referring to a fifth step of compound A1 synthesis method, so that the yield is 59%. HR-MS (ESI-TOF) m/z calcd for C 32 H 42 F 2 N 8 O 3 [M+H] + 625.3426,found 625.3431.
Example 10 Compound A10 (4- ((5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) pyrimidin-2-yl) amino) -3-methoxyphenyl) (4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) methanone
The synthetic route for compound a10 is as follows:

the first step:
the intermediate 18, a white solid, was obtained in 69% yield by the first step synthesis of reference compound A4 starting from starting material 17.
And a second step of:
the second step synthesis of reference compound A1 gives crude intermediate 19 starting from intermediate 18. Without further purification, it was used directly in the next reaction.
And a third step of:
the intermediate 20 is obtained as a yellow solid in 77% yield by referring to the third step of the synthesis method of the compound A1 by taking the intermediate 19 and the raw material 5 as raw materials.
Fourth step:
the fourth step of the synthesis, starting from intermediate 20, is to provide crude intermediate 21, with reference to compound A1. Without further purification, it was used directly in the next reaction.
Fifth step:
the intermediate 21 and the raw material 8 are used as raw materials, and the compound A10 is obtained by referring to a fifth step of synthesis method of the compound A1, and is yellow solid with the yield of 65%. HR-MS (ESI-TOF) m/z calcd for C 32 H 39 F 2 N 7 O 4 [M+Na] + 646.2930,found 646.2950.
Example 11 Compound A11 (4- ((5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) pyrimidin-2-yl) amino) -3-methoxyphenyl) (4- (2- (2-methoxyethoxy) ethoxy) piperazin-1-yl) methanone

The intermediate 21 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A11 is obtained by referring to a fifth step of the synthesis method of the compound A1, and is yellow solid with the yield of 71%. HR-MS (ESI-TOF) m/z calcd for C 34 H 43 F 2 N 7 O 5 [M+Na] + 690.3192,found 690.3185.
EXAMPLE 12 Compound A12 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2-methoxyethyl) piperazin-1-yl) pyridin-2-yl) pyrimidin-2-amine

The synthetic route for compound a12 is as follows:

the first step:
5-bromo-2-nitropyridine (1 g,4.93 mmol) was dissolved in anhydrous DMF (15 mL), anhydrous potassium carbonate (921 mg,6.67 mmol) was added, and stirred at room temperature for 5min. Subsequently, raw material 2 (1.03 g,5.532 mmol) was slowly added and the reaction was stirred at 55℃for 2h. TLC monitors the reaction completely, cool the reaction liquid to room temperature, add a proper amount of ice water under stirring, precipitate a large amount of white solid in the reaction system, filter, wash the filter cake with water for 3 times, dry to get intermediate 22, 1.211g of white solid, yield 89%.
And a second step of:
intermediate 22 (1.2 g,3.89 mmol) was dissolved in methanol (25 mL), 10% palladium on carbon (390 mg,0.372 mmol) was added, the mixture was evacuated, replaced three times with hydrogen, and the mixture was stirred at room temperature under a hydrogen atmosphere for 2 hours. TLC showed that the reaction was complete, the reaction solution was filtered through celite, washed 3 times with a mixed solvent of dichloromethane/methanol (10/1, v/v), and the filtrate was concentrated to dryness under reduced pressure to give crude product intermediate 23. Without further purification, it was used directly in the next reaction.
And a third step of:
toward intermediate 23 (3.72 mmol), starting material 5 (1.282 g,3.72 mmol), pd 2 (dba) 3 Toluene (20 mL) was added to a mixture of (3411 mg,0.372 mmol), X-phos (178 mg,0.372 mmol) and cesium carbonate (1.82 g,5.58 mmol), the mixture was evacuated and replaced 3 times with nitrogen, and the reaction mixture was stirred under nitrogen at 100℃for 5 hours. TLC monitoring reaction completion, cooling the reaction liquid to room temperature, filtering with diatomite, washing 3 times with dichloromethane/methanol (10/1, v/v) mixed solvent, concentrating the filtrate under reduced pressure, separating by silica gel column chromatography to obtain intermediate 24, 1.05g of off-white solid, yield 52%.
Fourth step:
intermediate 24 (1 g,1.73 mmol) was dissolved in dichloromethane (15 mL) and trifluoroacetic acid (4 mL) was slowly added dropwise with stirring and reacted at room temperature for 2h. TLC monitored the reaction was complete, the reaction was concentrated to dryness under reduced pressure, dissolved again with a small amount of dichloromethane, dried by spinning, and operated repeatedly 3 times to afford crude intermediate 25. Without further purification, it was used directly in the next reaction.
Fifth step:
intermediate 25 (0.172 mmol) was dissolved in anhydrous DMF (4 mL), cesium carbonate (112 mg,0.345 mmol) was added, and after stirring at room temperature for 5min, starting material 8 (25. Mu.L, 0.2595 mmol) was slowly added and the temperature was raised to 60℃for 3h. TLC monitoring reaction is complete, the reaction solution is cooled to room temperature, a proper amount of ice water is added, extraction is carried out three times by ethyl acetate, the organic phases are combined,washing with water, washing with saturated saline, drying with anhydrous sodium sulfate, filtering, concentrating the filtrate under reduced pressure to dryness, and further separating by silica gel column chromatography to obtain the target compound A12, 70mg of white solid, and yield 74%. HR-MS (ESI-TOF) m/z calcd for C 27 H 32 F 2 N 8 O[M+H] + 522.2667,found 522.2667.
EXAMPLE 13 Compound A13 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) pyridin-2-yl) pyrimidin-2-amine

The intermediate 25 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A13 is obtained by referring to a fifth step of the synthesis method of the compound A12, and the yield is 79 percent. HR-MS (ESI-TOF) m/z calcd for C 29 H 36 F 2 N 8 O 2 [M+H] + 566.2929,found 566.2929.
EXAMPLE 14 Compound A14 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2- (2-methoxyethoxy) ethoxy) ethyl) piperazin-1-yl) pyridin-2-yl) pyrimidin-2-amine

The intermediate 25 and diethylene glycol-2-bromoethyl methyl ether are used as raw materials, and the compound A14 is obtained by referring to a fifth step of compound A12 synthesis method, and the yield is 70%. HR-MS (ESI-TOF) m/z calcd for C 31 H 40 F 2 N 8 O 3 [M+H] + 610.3191,found 610.3191.
Example 15 Compound A15 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (6- ((4- (2-methoxyethyl) piperazin-1-yl) pyridin-3-yl) pyrimidin-2-amine

The synthetic route for compound a15 is as follows:

the first step:
2-bromo-5-nitropyridine (1 g,4.93 mmol) was dissolved in anhydrous DMF (15 mL), anhydrous potassium carbonate (922 mg,6.70 mmol) was added, and stirred at room temperature for 5min. Subsequently, raw material 2 (1.02 g,5.530 mmol) was slowly added and the reaction stirred at 55℃for 2h. The TLC monitoring reaction is complete, the reaction liquid is cooled to room temperature, a proper amount of ice water is added under the stirring condition, a large amount of white solid is precipitated in the reaction system, the reaction system is filtered, the filter cake is washed with water for 3 times, and intermediate 26 is obtained after drying, the white solid is 1.210g, and the yield is 89%.
And a second step of:
intermediate 26 (1.19 g,3.88 mmol) was dissolved in methanol (25 mL), 10% palladium on carbon (399mg, 0.371 mmol) was added, the mixture was evacuated, replaced with hydrogen gas three times, and the mixture was stirred under a hydrogen atmosphere at room temperature for 2 hours. TLC showed that the reaction was complete, the reaction solution was filtered through celite, washed 3 times with a mixed solvent of dichloromethane/methanol (10/1, v/v), and the filtrate was concentrated to dryness under reduced pressure to give crude intermediate 27. Without further purification, it was used directly in the next reaction.
And a third step of:
the intermediate 28, a yellow solid, was obtained in 78% yield from intermediate 27 and starting material 5 by the third step synthesis of compound a 12.
Fourth step:
starting from intermediate 28, the fourth step synthesis of reference compound a12 affords crude intermediate 29. Without further purification, it was used directly in the next reaction.
Fifth step:
the intermediate 29 and the raw material 8 are used as raw materials, and the compound A15 is obtained by referring to a fifth step of the synthesis method of the compound A12, and is white solid with the yield of 63%. HR-MS (ESI-TOF) m/z calcd for C 27 H 32 F 2 N 8 O[M+H] + 522.2667,found 522.2667.
EXAMPLE 16 Compound A16 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (6- ((4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) pyridin-3-yl) pyrimidin-2-amine

The intermediate 29 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A16 is obtained by referring to a fifth synthesis method of the compound A12, and the yield is 70 percent. HR-MS (ESI-TOF) m/z calcd for C 29 H 36 F 2 N 8 O 2 [M+H] + 566.2929,found 566.2929.
EXAMPLE 17 Compound A17 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (6- ((4- (2- (2-methoxyethoxy) ethoxy) ethyl) piperazin-1-yl) pyridin-3-yl) pyrimidin-2-amine

The intermediate 29 and diethylene glycol-2-bromoethyl methyl ether are used as raw materials, and the compound A17 is obtained as a pale yellow solid by referring to a fifth step of the compound A1, and the yield is 59%. HR-MS (ESI-TOF) m/z calcd for C 31 H 40 F 2 N 8 O 3 [M+H] + 610.3191,found 610.3191.
EXAMPLE 18 Compound A18 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2-methoxyethyl) piperazin-1-yl) phenyl-2-yl) pyrimidin-2-amine

The synthetic route for compound a18 is as follows:

the first step:
1-bromo-4-nitrobenzene (1 g,4.61 mmol) was dissolved in anhydrous DMF (15 mL), anhydrous potassium carbonate (860 mg,6.22 mmol) was added, and stirred at ambient temperature for 5min. Subsequently, raw material 2 (1.01 g,5.530 mmol) was slowly added and the reaction was stirred at 55℃for 2h. The TLC monitoring reaction is complete, the reaction liquid is cooled to room temperature, a proper amount of ice water is added under the stirring condition, a large amount of white solid is precipitated in the reaction system, the reaction system is filtered, the filter cake is washed with water for 3 times, and intermediate 30, white solid 1.221g and yield 86% are obtained by drying.
And a second step of:
intermediate 30 (1.2 g,3.71 mmol) was dissolved in methanol (25 mL), 10% palladium on carbon (399mg, 0.371 mmol) was added, the mixture was evacuated, replaced with hydrogen three times, and the mixture was stirred under a hydrogen atmosphere at room temperature for 2 hours. TLC showed that the reaction was complete, the reaction solution was filtered through celite, washed 3 times with a mixed solvent of dichloromethane/methanol (10/1, v/v), and the filtrate was concentrated to dryness under reduced pressure to give crude intermediate 31. Without further purification, the reaction mixture was used in the next reaction.
And a third step of:
intermediate 31 (3.72 mmol), starting material 5 (1.281g, 3.71 mmol), pd 2 (dba) 3 Toluene (20 mL) was added to a mixture of (340 mg,0.3712 mmol), X-phos (177 mg,0.371 mmol) and cesium carbonate (1.81 g,5.57 mmol), the mixture was evacuated and replaced 3 times with nitrogen, and the reaction mixture was stirred under nitrogen at 100℃for 5 hours. TLC monitoring reaction completion, cooling the reaction liquid to room temperature, filtering with diatomite, washing 3 times with dichloromethane/methanol (10/1, v/v) mixed solvent, concentrating the filtrate under reduced pressure, separating by silica gel column chromatography to obtain intermediate 32, white solid 1.05g, yield 50%.
Fourth step:
intermediate 32 (1.0 g,1.73 mmol) was dissolved in dichloromethane (15 mL) and trifluoroacetic acid (4 mL) was slowly added dropwise with stirring and reacted at room temperature for 2h. TLC monitored the reaction was complete, the reaction was concentrated to dryness under reduced pressure, dissolved again with a small amount of dichloromethane, dried by spinning, and operated repeatedly 3 times to afford crude intermediate 33. Without further purification, it was used directly in the next reaction.
Fifth step:
intermediate 33 (0.173 mmol) was dissolved in anhydrous DMF (4 mL), cesium carbonate (113 mg, 0.348 mmol) was added, and after stirring at room temperature for 5min, starting material 8 (25. Mu.L, 0.2595 mmol) was slowly added and the temperature was raised to 60℃for 3h. TLC monitoring reaction completion, cooling the reaction liquid to room temperature, adding a proper amount of ice water, extracting with ethyl acetate three times, combining organic phases, washing with water, washing with saturated saline, drying with anhydrous sodium sulfate, filtering, concentrating the filtrate under reduced pressure to dryness, further separating the target compound A18 by silica gel column chromatography, and obtaining a white solid with a yield of 73%. HR-MS (ESI-TOF) m/z calcd for C 28 H 33 F 2 N 7 O[M+H] + 522.2715,found 522.2715.
EXAMPLE 19 Compound A19 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2- (2-methoxyethoxy) ethyl) piperazin-1-yl) phenyl-2-yl) pyrimidin-2-amine

The intermediate 33 and 1-bromo-2- (2-methoxyethoxy) ethane are used as raw materials, and the compound A2 is obtained by referring to a fifth synthesis method of the compound A1, and is white solid with the yield of 78%. HR-MS (ESI-TOF) m/z calcd for C 30 H 37 F 2 N 7 O 2 [M+H] + 565.2977,found 565.2977.
EXAMPLE 20 Compound A20 5-fluoro-4- (4-fluoro-1-isopropyl-2-methyl-1H-benzo [ d ] imidazol-6-yl) -N- (5- (4- (2- (2-methoxyethoxy) ethoxy) ethyl) piperazin-1-yl) phenyl-2-amine

Intermediate 33 and diethylene glycol-2-bromoethyl methyl ether are used as raw materials, and the compound A20 is obtained by referring to a fifth step of compound A1 synthesis method, and the yield is 68%. HR-MS (ESI-TO)F)m/z:calcd for C 32 H 41 F 2 N 7 O 3 [M+H] + 609.3239,found 609.3239.
The beneficial effects of the invention are demonstrated by the following in vitro tests.
Test example 1, CDK4, CDK6 kinase inhibitory Activity assay
The aim of this experiment was to detect the inhibitory activity of the compounds of the invention against in vitro kinases by means of isotope labelling. In this experiment, CDK4 and CDK6 kinases were tested for inhibition of in vitro activity, respectively. Abemaciclib is a positive control. IC for kinase inhibitory Activity of test Compounds 50 (half inhibition concentration). IC (integrated circuit) 50 Values can be obtained by calculation of the inhibition rate of the kinase activity by the test compound at a range of different concentrations.
1. Experimental materials
20mM 3- (N-morpholino) propanesulfonic acid (MOPS); 1mM ethylenediamine tetraacetic acid (EDTA); 0.01% Brij 35 (Brij-35); 5% Glycerol (Glycerol); 0.1% mercaptoethanol (mercptoethanol); 1mg/ml Bovine Serum Albumin (BSA); l0mM manganese dichloride solution (MnC 1 2 ) The method comprises the steps of carrying out a first treatment on the surface of the Stop buffer (3% phosphate buffer); washing buffer (75 mM phosphate solution); methanol (methanol); filtermat a film; CDK4/6 kinase, test compounds.
2. Experimental method
Adding the kinase to be detected and the corresponding substrates and the compounds to be detected or DMSO with different concentrations into a reaction buffer solution (8 mM propanesulfonate, 0.2mM EDTA,10mM magnesium acetate and Km concentration gamma-33P-ATP solution), and incubating for 40min at room temperature; adding 3% phosphate solution to terminate the reaction; pipette 10. Mu.L of the reaction mixture drop onto P30 filter paper; the filter paper was washed 3 times with 75mM phosphate solution; cleaning the filter paper with methanol for 1 time; after the filter paper was air-dried, scintillation fluid was added and the phosphorylated substrate was measured by scintillation counting. Inhibition% = (1-compound treatment group count/blank group count) ×100%; half maximal Inhibitory Concentration (IC) 50 ) Fitting the inhibition ratios corresponding to the concentrations.
3. Experimental results
The test method tests theCompounds of the invention are directed to the inhibitory activity of CDK4/6 kinase, respectively. Table 1 shows the IC of the test compounds for CDK4/6 kinase inhibitory activity 50 Values.
Experimental results show that the tested compound has strong inhibitory activity on CDK4/6 kinase. Can be used for preparing CDK4 kinase and CDK6 kinase inhibitors.
TABLE 1 IC of test compounds for CDK4/6 kinase inhibitory Activity 50 Value of

*A:IC 50 <50nM;B:50nM≤IC 50 <100nM;C:100nM≤IC 50 <150nM;D:IC 50 >150nM
Test example 2 in vitro tumor cell proliferation inhibition experiments of Compounds
The aim of the experiment is to detect the proliferation inhibition activity of the compound on in vitro tumor cells, and the adopted method is MTT (tetramethyl azoazole salt) colorimetric method.
1. Experimental materials
1.1 major reagents
Breast cancer cell lines MCF-7, MDA-MB-231, MDA-MB-436 and MDA-MB-468, colorectal cancer cell line SW620, lung cancer cell A549 were all purchased from American standard biological Collection (American Type Culture Collection, ATCC), RPMI 1640 medium, fetal Bovine Serum (FBS) were all purchased from GIBICO company, USA; penicillin and streptomycin were purchased from Dalianbao biosome; cultured cells were purchased from Corning company using plates, 96-well plates, etc.; centrifuge tubes of various sizes were purchased from BD company; MTT reagent was purchased from the Japan same-kernel chemical institute (Donjindo). The tested compounds are synthesized by the inventor, 10mM stock solution is prepared by using 100% DMSO in vitro experiments, the stock solution is stored in a refrigerator at the temperature of minus 20 ℃ for standby, and the stock solution is diluted to the required concentration in the test.
1.2 cell lines and cultures
The breast cancer cell lines MCF-7, MDA-MB-231, MDA-MB-436 and MDA-MB-468, colorectal cancer cell line SW620 and lung cancer cell line A549 used in the experimentConventional RPMI 1640 complete medium containing 10% Fetal Bovine Serum (FBS), 100IU/mL penicillin, 100. Mu.g/mL streptomycin was cultured at 37℃in 5% CO 2 Is cultured in an incubator of (a).
2. Experimental method
The cell concentration is regulated to be 1 to 2 multiplied by 10 by using the complete cell culture solution 4 Each/ml of cell suspension (HCC 827 cell concentration 6X 10) 4 H1975 cells 4X 10 per ml 3 At/ml), 200 μl of cell suspension per well was inoculated in 96-well plates and incubated overnight. The next day, the supernatant was aspirated and the cells were then individually treated with gradient concentrations of the test compound. Simultaneously setting a negative control group without medicine and an equal volume solvent control group, wherein the concentration of DMSO is 0.1%, 3 compound holes are arranged in each dosage group, and the concentration of CO is 5% at 37 DEG C 2 Culturing under the condition. After 72 hours, 20. Mu.L of MTT reagent at a concentration of 5mg/ml was added to each well, and after further incubation for 2 to 4 hours, the supernatant was discarded, 150. Mu.L of DMSO was added to each well, and the mixture was mixed by shaking for 15 minutes, and the absorbance (A) value (A value proportional to the number of living cells) was measured by an enzyme-labeled instrument (lambda=570 nm), and the average value was obtained. Relative cell proliferation inhibition ratio= (control group a 570-experimental group a 570)/control group a570×100%. Experiments were repeated at least 3 times. Experimental data are expressed by mean, data statistics are tested by t-test, P<A difference of 0.05 is statistically significant. IC is used for inhibiting cell proliferation by the following compounds 50 And (3) representing.
3. Experimental results
By the method, proliferation inhibition activity tests are carried out on breast cancer cell strains MCF-7, MDA-MB-231, MDA-MB-436 and MDA-MB-468, colorectal cancer cell strain SW620 and lung cancer cell A549. Table 2 shows the proliferation inhibitory activity (IC) 50 ). The results show that most of the compounds of the examples show remarkable proliferation inhibition activity on the tested tumor cell lines, wherein the proliferation inhibition activity of the compounds A8 and A9 on various tumor cell lines is in a low micromolar level, and the compounds have certain therapeutic advantages compared with the positive compound Abemaciclib.
TABLE 2 proliferation of Compounds against tumor cell lines (MTT method)

Note that: ND: non-determined.
A:IC 50 <0.1μM;B:0.1μM≤IC 50 <1μM;C:IC 50 ≥1μM.
Test example 3 in vivo antitumor test of Compound A2
The aim of this experiment was to detect the in vivo antitumor effect of the compounds of the invention. Experimental BALB/c mice were tested for anti-tumor activity in vivo in a subcutaneous tumor model. The cell line used was human colon cancer cell line COLO205.
1. Experimental materials
Fetal bovine serum, culture medium, pancreatin, etc. were purchased from Gibco BRL company (Invitrogen Corporation, USA), culture medium was purchased from ATCC (American Type Culture Collection), human colon cancer cell line COLO205 was purchased from ATCC company, USA, and NOD-Bablc mice were purchased from the beijing verruca animal experiment center.
2. Experimental method
NOD-Bablc mice were used for 6-8 weeks at about 1X 10 7 The concentration of each cell per 0.1 ml/COLO 205 was inoculated into the subcutaneous posterior rib of the mice, and after the tumor was grown to a certain volume, the mice were randomly grouped and started to be orally administrated by gastric lavage and tail vein administration.
And (3) observing the indexes: the tumor length and diameter of the mice body weight meter and the neck were measured every 3 days and the tumor volume was calculated (length diameter x short diameter 2 x 0.52), and the presence or absence of diarrhea, cramps, rash, significant weight loss and other reactions were observed.
3. Experimental results
The experimental results of the tumor growth of the various groups are shown in FIG. 1. The experimental results show that the tested compound A2 has obvious in-vivo growth inhibition effect on COLO205. No adverse reactions such as weight loss, rash, diarrhea and the like were found in the mice during the administration, indicating that the toxicity of the test compound A2 was very low in the administration dosage range.
Test example 4 blood brain tissue distribution experiment of Compound A2
The aim of this experiment was to detect the in vivo antitumor effect of the compounds of the invention. In-situ model of BALB/c mice brain tumor is used for experiment, and the in-vivo anti-tumor activity of the compound A2 is tested. The cell strain used was human brain astrocyte U87-LUC.
1. Experimental materials
Fetal bovine serum, culture medium, pancreatin, etc. were purchased from Gibco BRL company (Invitrogen Corporation, USA), culture medium was purchased from ATCC (American Type Culture Collection), NOD-Bablc mice were purchased from the Beijing Warcang animal laboratory center.
2. Experimental method
SPF grade ICR mice were used, weighed prior to dosing, and dosing amounts were calculated from body weight. Administration is by intravenous injection.
Plasma sample treatment: blood was collected by cardiac puncture, each sample was collected at about 0.2mL, heparin sodium was anticoagulated, placed on ice after collection, and plasma was centrifuged within 1 hour (centrifugation conditions: 6800g,6 minutes, 2-8deg.C). The plasma samples were stored in a-80 ℃ freezer prior to analysis.
Tissue sample treatment: after animals were euthanized, brain tissue was collected, rinsed with normal saline to avoid cross contamination, blotted dry with filter paper, and then placed in labeled tubes/self-sealing bags (one tissue per tube/self-sealing bag), and samples were temporarily placed on ice before storage in an ultra-low temperature refrigerator.
Experimental grouping: drug solvent control (12.5% EL+12.5% EtOH+75% Water)
50mg/kg q.d of compound A2-M;
100mg/kg q.d of compound A2-H;
compound Abemaciclib 50mg/kg q.d;
(each group of drugs was dissolved in 12.5% EL+12.5% EtOH+75% water)
Analysis of results: pharmacokinetic parameters were calculated using Phoenix WinNonlin7.0 from blood concentration data at different time points, providing parameters such as AUC0-T, AUC 0-infinity, MRT 0-infinity, cmax, tmax, and T1/2, as well as their mean and standard deviation.
3. Experimental results
The distribution of compound A2 in blood and brain tissue at various time points is shown in figure 2. The experimental results show that the concentration of the drug in brain tissue is higher than that in plasma of the tested compound A2 at different time points. The compound A2 can penetrate through the blood brain barrier, directly enter the brain to play a role, and can be used for developing medicaments for treating brain tumors.
In summary, the present invention provides a pyridine-pyrimidine amine-benzimidazole derivative, which has good inhibitory activity on CDK4 and CDK6 kinases, and can be used to prepare CDK4 and CDK6 kinase inhibitors; meanwhile, the derivative has remarkable inhibition activity on various tumor cells, can inhibit the growth of tumors in vivo, and has excellent effect; the derivative has small administration dosage and high safety; in addition, the derivative can pass through the blood brain barrier, and solves one of the biggest problems in brain tumor treatment. The derivative can be used for preparing medicines for preventing and/or treating tumors and autoimmune diseases, and has good application prospects.
Claims (8)
1. A compound, or salt thereof, characterized by: the compound is represented by formula Va:

wherein,,
n 1 an integer selected from 1 to 3;
n 2 selected from 1;
X 1 selected from CR 3 ;
Y 1 Selected from N;
R 3 selected from hydrogen.
2. The compound, or salt thereof, according to claim 1, wherein: the compound is one of the following compounds:

3. use of a compound according to claim 1 or 2, or a salt thereof, in the preparation of a CDK4 and/or CDK6 kinase inhibitor.
4. Use according to claim 3, characterized in that: the kinase inhibitor is a medicament for preventing and/or treating tumors.
5. Use according to claim 4, characterized in that: the tumor is brain cancer, glioblastoma, leukemia, lymphoma, bannayan-Zonana syndrome, coden disease, lhermitte-Duclos disease, breast cancer, inflammatory breast cancer, wilms 'tumor, ewing's sarcoma, rhabdomyosarcoma, ependymoma, medulloblastoma, colon cancer, gastric cancer, bladder cancer, head and neck cancer, renal cancer, lung cancer, liver cancer, melanoma, renal cancer, ovarian cancer, pancreatic cancer, prostate cancer, sarcoma, osteosarcoma, bone giant cell tumor or thyroid cancer.
6. Use according to claim 3, characterized in that: the kinase inhibitor is a medicament for preventing and/or treating autoimmune diseases.
7. Use according to claim 6, characterized in that: the autoimmune disease is psoriasis or lupus erythematosus.
8. A medicament, characterized in that: a pharmaceutical preparation prepared by adding pharmaceutically acceptable auxiliary materials or auxiliary components into the compound or the salt thereof as an active ingredient in the invention as claimed in claim 1 or 2.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210096582.2A CN114380800B (en) | 2022-01-26 | 2022-01-26 | Pyridine-pyrimidine amine-benzimidazole derivative, and preparation method and application thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210096582.2A CN114380800B (en) | 2022-01-26 | 2022-01-26 | Pyridine-pyrimidine amine-benzimidazole derivative, and preparation method and application thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN114380800A CN114380800A (en) | 2022-04-22 |
| CN114380800B true CN114380800B (en) | 2023-05-19 |
Family
ID=81204696
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210096582.2A Active CN114380800B (en) | 2022-01-26 | 2022-01-26 | Pyridine-pyrimidine amine-benzimidazole derivative, and preparation method and application thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114380800B (en) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016015604A1 (en) * | 2014-07-26 | 2016-02-04 | Sunshine Lake Pharma Co., Ltd. | Compounds as cdk small-molecule inhibitors and uses thereof |
| CN113880816A (en) * | 2020-07-01 | 2022-01-04 | 杭州百诚医药科技股份有限公司 | Piperazine-containing aminopyrimidine derivative and application thereof |
| CN113880809B (en) * | 2020-07-03 | 2022-10-18 | 盛世泰科生物医药技术(苏州)有限公司 | Pyrimidine derivative and preparation method and application thereof |
-
2022
- 2022-01-26 CN CN202210096582.2A patent/CN114380800B/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN114380800A (en) | 2022-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021129820A1 (en) | Spiro ring-containing quinazoline compound | |
| CN110563703B (en) | Compound for inducing PARP-1 degradation based on CRBN ligand, preparation method and application | |
| CA2692922A1 (en) | Azaindole-indole coupled derivatives, preparation methods and uses thereof | |
| KR20220038453A (en) | Crystalline Forms of CD73 Inhibitors | |
| CN105732615A (en) | CDK kinase inhibitor | |
| CN105461720B (en) | Morpholine class tyrosine kinase inhibitor | |
| WO2021023272A1 (en) | Crystalline form of atr inhibitor and use thereof | |
| CN114380800B (en) | Pyridine-pyrimidine amine-benzimidazole derivative, and preparation method and application thereof | |
| CN109369620B (en) | Pyridine compound, preparation method thereof and application thereof in resisting gastric cancer | |
| CN111484495B (en) | Preparation method and application of derivative containing dihydropteridine diketone framework | |
| CN118076614A (en) | Polymorphic forms of EGFR inhibitors | |
| CN114315837A (en) | Crystalline form of ERK inhibitor and preparation method thereof | |
| CN115867552A (en) | Imidazolopyrimidine derivative, preparation method thereof and application thereof in medicines | |
| CN115109049B (en) | Triazine compound containing aryl urea structure and application thereof | |
| CN117355528B (en) | Salt type pyrrolotriazine compound, crystal form and preparation method thereof | |
| CN116239594B (en) | 6- (imidazo [1,2-a ] pyridin-6-yl) quinazoline derivatives and uses thereof | |
| CN112300235B (en) | Benzimidazole derivative BI321 and preparation method and application thereof | |
| CN111875606B (en) | Purine compound obtained based on virtual docking and preparation method and application thereof | |
| CN112375112B (en) | Benzimidazole derivative BI361 and preparation method and application thereof | |
| CN111171064B (en) | Azafluoroborodipyrrole compound, preparation method thereof and application thereof as Fyn kinase inhibitor | |
| WO2020224585A1 (en) | Salt and crystal form of mtorc1/2 dual kinase activity inhibitor and preparation method therefor | |
| CN108047231B (en) | Hydrochloride of [1,2,4] triazino [6,1-a ] isoindole compound and application thereof | |
| CN111171017B (en) | Pyrimidine-based derivatives, their preparation and use | |
| WO2020221358A1 (en) | Crystal form of wee1 inhibitor compound and use thereof | |
| CN118108710A (en) | Indole iodized salt-pyridine bishemicyanine compound and preparation method and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |