CN113636973A - 一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 - Google Patents
一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 Download PDFInfo
- Publication number
- CN113636973A CN113636973A CN202111047083.6A CN202111047083A CN113636973A CN 113636973 A CN113636973 A CN 113636973A CN 202111047083 A CN202111047083 A CN 202111047083A CN 113636973 A CN113636973 A CN 113636973A
- Authority
- CN
- China
- Prior art keywords
- acid
- piperazine
- water
- formula
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- RMULRXHUNOVPEI-UHFFFAOYSA-N tert-butyl 4-(6-aminopyridin-3-yl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=CC=C(N)N=C1 RMULRXHUNOVPEI-UHFFFAOYSA-N 0.000 title claims abstract description 24
- 238000002360 preparation method Methods 0.000 title claims abstract description 13
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims abstract description 46
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 45
- 239000002253 acid Substances 0.000 claims abstract description 35
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 claims abstract description 22
- 239000003960 organic solvent Substances 0.000 claims abstract description 20
- 239000011230 binding agent Substances 0.000 claims abstract description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 13
- 238000000034 method Methods 0.000 claims abstract description 12
- 239000003054 catalyst Substances 0.000 claims abstract description 11
- 238000009776 industrial production Methods 0.000 claims abstract description 7
- 239000012046 mixed solvent Substances 0.000 claims abstract description 7
- 238000009903 catalytic hydrogenation reaction Methods 0.000 claims abstract description 6
- 238000006243 chemical reaction Methods 0.000 claims description 31
- 150000001875 compounds Chemical class 0.000 claims description 27
- 239000012535 impurity Substances 0.000 claims description 27
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 24
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 24
- 239000000047 product Substances 0.000 claims description 21
- 239000007787 solid Substances 0.000 claims description 20
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 18
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 15
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 14
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 12
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 12
- 238000001914 filtration Methods 0.000 claims description 12
- 238000005406 washing Methods 0.000 claims description 11
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 8
- ATXXLNCPVSUCNK-UHFFFAOYSA-N 5-bromo-2-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Br)C=N1 ATXXLNCPVSUCNK-UHFFFAOYSA-N 0.000 claims description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 8
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 claims description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- 239000001632 sodium acetate Substances 0.000 claims description 8
- 235000017281 sodium acetate Nutrition 0.000 claims description 8
- 239000002904 solvent Substances 0.000 claims description 8
- 238000010533 azeotropic distillation Methods 0.000 claims description 7
- 238000001035 drying Methods 0.000 claims description 7
- 239000000203 mixture Substances 0.000 claims description 7
- 150000003839 salts Chemical class 0.000 claims description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 6
- DSVGQVZAZSZEEX-UHFFFAOYSA-N [C].[Pt] Chemical compound [C].[Pt] DSVGQVZAZSZEEX-UHFFFAOYSA-N 0.000 claims description 6
- 235000011054 acetic acid Nutrition 0.000 claims description 6
- 239000003513 alkali Substances 0.000 claims description 6
- 239000007788 liquid Substances 0.000 claims description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 6
- 239000012295 chemical reaction liquid Substances 0.000 claims description 5
- 238000000926 separation method Methods 0.000 claims description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 4
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 4
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 claims description 4
- 239000012043 crude product Substances 0.000 claims description 4
- 238000002425 crystallisation Methods 0.000 claims description 4
- 230000008025 crystallization Effects 0.000 claims description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 4
- 239000000706 filtrate Substances 0.000 claims description 4
- 238000010438 heat treatment Methods 0.000 claims description 4
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 claims description 4
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 claims description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 4
- 239000012044 organic layer Substances 0.000 claims description 4
- 239000002798 polar solvent Substances 0.000 claims description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 3
- 238000005119 centrifugation Methods 0.000 claims description 3
- 239000010410 layer Substances 0.000 claims description 3
- 239000000463 material Substances 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- 230000001502 supplementing effect Effects 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 2
- 239000007868 Raney catalyst Substances 0.000 claims description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 claims description 2
- 229910000564 Raney nickel Inorganic materials 0.000 claims description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 claims description 2
- 239000007864 aqueous solution Substances 0.000 claims description 2
- 235000015165 citric acid Nutrition 0.000 claims description 2
- 235000019253 formic acid Nutrition 0.000 claims description 2
- 235000006408 oxalic acid Nutrition 0.000 claims description 2
- 235000011056 potassium acetate Nutrition 0.000 claims description 2
- 235000019260 propionic acid Nutrition 0.000 claims description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 claims description 2
- 238000011084 recovery Methods 0.000 claims description 2
- 235000017550 sodium carbonate Nutrition 0.000 claims description 2
- 239000000243 solution Substances 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims 7
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims 2
- 229960001701 chloroform Drugs 0.000 claims 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 claims 1
- 235000017557 sodium bicarbonate Nutrition 0.000 claims 1
- -1 6-aminopyridine-3-yl Chemical group 0.000 abstract description 13
- UBCDLQPOKISIDX-UHFFFAOYSA-N 1-(6-nitropyridin-3-yl)piperazine Chemical compound C1=NC([N+](=O)[O-])=CC=C1N1CCNCC1 UBCDLQPOKISIDX-UHFFFAOYSA-N 0.000 abstract description 9
- SUWKOEMQNOBJEQ-UHFFFAOYSA-N tert-butyl 4-(6-nitropyridin-3-yl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=CC=C([N+]([O-])=O)N=C1 SUWKOEMQNOBJEQ-UHFFFAOYSA-N 0.000 abstract description 7
- 238000010534 nucleophilic substitution reaction Methods 0.000 abstract description 3
- 239000002994 raw material Substances 0.000 abstract description 3
- 238000007670 refining Methods 0.000 abstract description 3
- 238000003912 environmental pollution Methods 0.000 abstract description 2
- 238000004821 distillation Methods 0.000 description 6
- 230000007613 environmental effect Effects 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 230000009286 beneficial effect Effects 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 5
- 229960004390 palbociclib Drugs 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 4
- 108091007914 CDKs Proteins 0.000 description 3
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 3
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- YXVDXGVAUQKDPH-UHFFFAOYSA-N 1-(6-nitropyridin-3-yl)piperazine;hydrochloride Chemical compound Cl.C1=NC([N+](=O)[O-])=CC=C1N1CCNCC1 YXVDXGVAUQKDPH-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 238000007256 debromination reaction Methods 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 238000012805 post-processing Methods 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 239000008213 purified water Substances 0.000 description 2
- 238000004064 recycling Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 239000002699 waste material Substances 0.000 description 2
- YUBHMOQVHOODEI-UHFFFAOYSA-N 5-chloro-2-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Cl)C=N1 YUBHMOQVHOODEI-UHFFFAOYSA-N 0.000 description 1
- 241000272875 Ardeidae Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 241000244269 Peucedanum Species 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000032798 delamination Effects 0.000 description 1
- YROXEBCFDJQGOH-UHFFFAOYSA-N ditert-butyl piperazine-1,4-dicarboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(C(=O)OC(C)(C)C)CC1 YROXEBCFDJQGOH-UHFFFAOYSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000007674 genetic toxicity Effects 0.000 description 1
- 231100000025 genetic toxicology Toxicity 0.000 description 1
- 231100000024 genotoxic Toxicity 0.000 description 1
- 230000001738 genotoxic effect Effects 0.000 description 1
- 231100000734 genotoxic potential Toxicity 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000005748 halopyridines Chemical class 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/74—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明公开了一种4‑(6‑氨基吡啶‑3‑基)哌嗪‑1‑羧酸叔丁酯的工业化制备方法,该方法采用5‑溴‑2‑硝基吡啶与哌嗪为起始原料,在醇类有机溶剂和水的混合溶剂中,以酸为催化剂通过亲核取代反应制备得到高纯度1‑(6‑硝基吡啶‑3‑基)哌嗪,再与Boc酸酐在有机溶剂和水的存在下,以弱碱为缚酸剂,制备得到高纯度4‑(6‑硝基吡啶‑3‑基)哌嗪‑1‑羧酸叔丁酯,再通过催化氢化,精制脱色,得到高纯度、浅色泽的4‑(6‑氨基吡啶‑3‑基)哌嗪‑1‑羧酸叔丁酯。该方法操作简便、对环境污染少、收率高、成本低、产品质量好,更适合于工业化生产。
Description
技术领域
本发明属于化学合成技术领域,具体涉及一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法。
背景技术
帕布昔利布(Palbociclib),化学名为2-[(4-哌啶基)苄基]-6-乙酰基-8-环戊基-5-甲基吡啶并[2,3-d]嘧啶-7(8H)-酮,由辉瑞公司研制开发,2015年2月在美国率先批准上市,是一种周期蛋白-依赖激酶(CDK)4和6的抑制剂,主要通过调节细胞周期、抑制(CDK)4和6活性来阻止细胞由G1期到S期进而抑制DNA的合成,临床主要用于治疗晚期乳腺癌患者。
4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯(式(1))是合成帕布昔利布的关键中间体,CAS号为571188-59-5,结构式为:

文献报道的4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的合成方法主要有以下两种:
方法一:Imaeda,Yasuhiro等人(Bioorganic&Medicinal Chemistry,16(6),3125-3140;2008)采用5-溴-2-硝基吡啶与1-Boc-哌嗪在N-甲基吡咯烷酮中反应,得到4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯,再在乙醇中用钯碳催化氢化还原硝基得到目标产物,反应路线如下:

该合成方法的路线短,收率尚可,但因制备1-Boc-哌嗪(哌嗪与Boc酸酐反应)的收率低,价格较高的Boc酸酐利用率低,三废多,不利于环保要求,造成1-Boc-哌嗪不仅价格高昂,而且含有大量的双-Boc-哌嗪,纯度低,致使上述反应成本高。
方法二:JV卡列尼等人在中国专利CN105384741B中报道了5-氯-2-硝基吡啶首先与哌嗪在正丁醇中通过亲核取代反应得到1-(6-硝基吡啶-3-基)哌嗪盐酸盐,再在四氢呋喃中,以碳酸钾做缚酸剂,与Boc酸酐反应,再通过催化还原,得到4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工艺路线,反应路线如下:
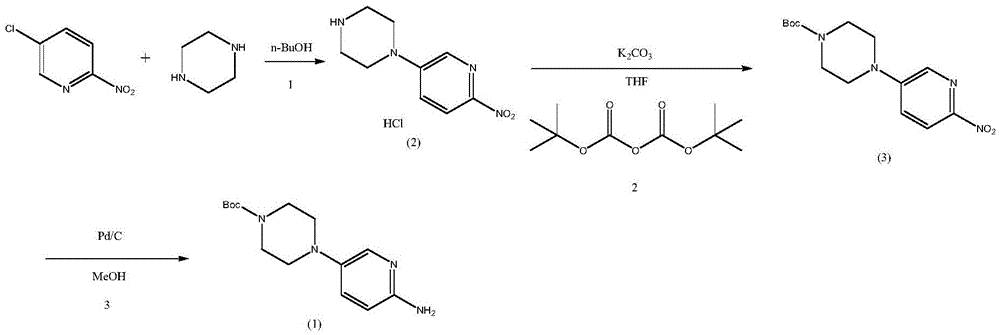
该路线制备1-(6-硝基吡啶-3-基)哌嗪盐酸盐的步骤,文献虽然给出了82.3%的收率和熔点范围(mp>230℃),但在碱性条件下,哌嗪两端的氮容易与双分子卤代吡啶发生潜在副反应,生成双分子缩合副产物(Tetrahedron Letters 39(1998)617-620)。
在实际制备过程中,我们观测到2个哌嗪双取代杂质,杂质式(4)和杂质式(5),其反应路线如下:

虽然杂质式(4)和杂质式(5),均不与Boc哌嗪反应,但其在正丁醇和水中的溶解度均较小,虽然专利CN105384741B中通过步骤2的过滤,杂质式(4)和杂质式(5)均大部分去除,但因其在四氢呋喃中有一定的溶解度,带入4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯中,通过加氢还原,得到其衍生物杂质式(6)和杂质式(7):

杂质式(6)和杂质式(7)在4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯中均难以去除,从而无法得到高纯度的产品。
我们在研究钯碳催化还原硝基的过程中,发现微量的杂质式(5),在钯碳还原过程中因发生脱溴反应,生成微量的氢溴酸,进而促使4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的Boc基团水解,生成一系列杂质,其中包含具有潜在基因毒性的杂质,进而影响产品的质量。

另外,文献报道的4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的色泽,均为棕黄色固体,未见白色至类白色固体的报道。但作为以医药原料药及中间体为主的企业,客户给我们提出了浅黄色固体的要求。
作为重磅炸弹级新药,帕布昔利布的用量很大,其关键中间体4-(6-氨基-3-吡啶基)哌嗪-1-羧酸叔丁酯的用量也会逐年增多。随着医药化工企业对环境保护意识的逐年增强,同时为了满足追求高品质产品客户的需求,开发一种低成本、高收率、高纯度、低毒性、环境友好的4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的合成方法是当前需要解决的问题。
发明内容
针对现有技术的不足,本发明提供了一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法,该方法采用5-溴-2-硝基吡啶与哌嗪为起始原料,在正丁醇和水的混合溶剂中,以酸为催化剂通过亲核取代反应制备得到高纯度1-(6-硝基吡啶-3-基)哌嗪,再与Boc酸酐在有机溶剂和水的存在下,以弱碱为缚酸剂,制备得到高纯度4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯,再通过催化氢化,精制脱色,得到高纯度、浅色泽的4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯。该方法操作简便、对环境污染少、收率高、成本低、产品质量好,更适合于工业化生产。
本发明的技术方案是:一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法,其特征是,包括以下步骤:
S1:在醇类有机溶剂与水的混合溶剂中,以酸为催化剂,5-溴-2-硝基吡啶和哌嗪反应生成化合物式(2)化合物的酸式盐,反应结束后,加水并通过共沸蒸馏回收溶剂,得到含有式(2)化合物酸式盐的水溶液,通过过滤除去不溶杂质,用碱调pH10以上,离心,得到高纯度的式(2)化合物湿品;
S2:式(2)化合物湿品在有机溶剂与水的混合溶剂中,在缚酸剂的存在下,与Boc酸酐反应,反应液后处理得到式(3)化合物;
S3:式(3)化合物在极性溶剂中,在催化剂和缚酸剂的存在下,催化氢化,后处理生成式(1)化合物粗品;
S4:将式(1)化合物粗品加入水中,再加入酸以使固体溶解,活性炭脱色,用氢氧化钠溶液调pH碱性,经过析晶、离心、洗涤、烘干,得到高纯度(≥99.8%),颜色为白色至浅黄色的4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯。
合成路线如下:

其中,
步骤S1的酸为盐酸、氢溴酸、磷酸、硫酸、醋酸、柠檬酸等的一种或两种混合,优选氢溴酸,所使用酸与哌嗪的摩尔比为0.5~1.5:1,进一步优选为1:1。通过哌嗪成单酸盐的方式,保护哌嗪的氨基的一端,防止双哌嗪取代产物的生成。
步骤S1的醇类有机溶剂为正丁醇、正戊醇、异戊醇、叔丁醇、仲丁醇、1-丙醇中的一种,优选正丁醇;其与水的比例为10:0.3~5(V/V),优选10:0.5~2,进一步优选为10:1。
步骤S1中的5-溴-2-硝基吡啶、哌嗪的摩尔比为1:1.2~2,优选1:1.2。哌嗪用量增多,虽有利于哌嗪的单取代产物生成,减少哌嗪双取代产物,但从环保角度考虑,在不影响反应结果的前提下,尽量减少哌嗪的用量,有利于三废处理和环保要求。
步骤S1中的反应温度为50~100℃,优选60~65℃。
步骤S1中的共沸回收溶剂,通过分批次补充水的方式,共沸蒸馏得到溶剂与水的混合物,通过分液分出有机溶剂和水,有机溶剂直接用于下一批反应,水用于下一批次物料共沸蒸馏,实现溶剂的零排放。
步骤S1中通过过滤除去式(4)及式(5)双聚杂质,再用碱调至碱性,得到高纯度的式(2)化合物。

反应过程产生的哌嗪双取代杂质式(4)和式(5)两个杂质,在酸性条件下,略溶解于有机溶剂,不溶于水。采用共沸除有机溶剂的方式,将反应液替换成水,中间体式(2)酸式盐溶解于水中,少量的哌嗪双取代杂质直接通过过滤去除,再采用式(2)化合物在强碱中溶解度较小的特点,将反应液调pH值至10以上,析出中间体式(2),通过离心得到中间体式(2)湿品,不用干燥,可直接进行下一步反应。
步骤S2中的缚酸剂为碳酸钠、碳酸钾、三乙胺、二异丙基乙胺、醋酸钠、氢氧化钠、氢氧化钾中的一种,优选碳酸钠,式(2)化合物与缚酸剂、Boc酸酐的摩尔比为1:1.2~1.8:1.0~1.5。
步骤S2中的有机溶剂为二氯甲烷、三氯甲烷、甲苯、四氢呋喃、二氧六环中的一种,优选甲苯和二氯甲烷。
步骤S2中的后处理为:升温至固体溶解后,静置分液,分出水层,有机层洗涤,减压浓缩,重结晶。
步骤S3中的催化剂为钯碳、铂碳、雷尼镍中的一种,优选铂碳,式(3)化合物与催化剂的质量比为1:0.01~1:0.05,优选1:0.03。
步骤S3中的缚酸剂为醋酸钠、醋酸钾、三乙胺、氨水、碳酸钠、碳酸氢钠中的一种,优选醋酸钠。式(3)化合物与缚酸剂的摩尔比为1:0.01~0.1,优选1:0.01~0.05,进一步优选为1:0.02。硝基的催化氢化还原,微量式(5)杂质的脱溴反应,生成的溴化氢溶解于氢化还原产生的水,导致反应液具有一定的酸性,导致Boc集团发生微量降解,导致生成多个式(1)化合物中的微量杂质,其中包含具有遗传毒性警示结构的杂质。采用增加少量缚酸剂,中和产生的氢溴酸的方式,以阻止Boc基团的降解,有利于提高产品的纯度和质量。
步骤S3中的极性溶剂为甲醇、乙醇、异丙醇、正丁醇中的一种,优选甲醇。甲醇更有利于回收处理,经济成本也更低,同时,甲醇对缚酸剂醋酸钠也有一定的溶解性。
步骤S3中的反应温度为10~60℃,优选10~30℃。
步骤S3的后处理为:过滤,滤液减压浓缩至干,正庚烷重结晶。
步骤S4的酸为醋酸、甲酸、柠檬酸、草酸、丙酸中的一种,优选醋酸。
本发明的技术特点和优益效果:
1、本发明利用5-溴-2-硝基吡啶与哌嗪,以正丁醇等有机溶剂与水为混合溶剂,以氢溴酸等为催化剂,制备得到1-(6-硝基吡啶-3-基)哌嗪的酸式盐,通过共沸蒸馏,使溶剂实现回收利用,通过过滤去除产生的微量哌嗪双取代,再通过调碱,得到高纯度的1-(6-硝基吡啶-3-基)哌嗪(式(2))。该步骤反应条件温和,后处理简洁,产品无需烘干,质量好,收率高,直接按100%投料。有机溶剂可重复利用,废水为无机盐类,易于处理,更适合工业化生产。
2、本发明直接采用1-(6-硝基吡啶-3-基)哌嗪湿品,在两相体系中与Boc酸酐反应,通过分液,洗涤去除无机物,减压蒸馏,再结晶得到高纯度的4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯(式(3)),蒸出的有机溶剂可以直接回收套用。
3、本发明通过加入醋酸钠等缚酸剂的方式,采用更廉价的铂碳等做催化剂进行硝基的氢化还原,制备4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯(式(1))。缚酸剂的加入阻止了Boc基团的降解,阻碍了潜在的基因毒性杂质的产生,提高了产品的安全性,进一步的提高了帕布昔利布的安全性。
4、本发明创造性的通过4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯在弱酸性溶解的特点,采用活性炭脱色的方式,去除影响色泽的高分子集团,通过调碱得到高纯度、浅色泽的4-(6-氨基-3-吡啶基)哌嗪-1-羧酸叔丁酯。
综合以上,本发明为4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的合成,提供了一条高质量、低成本、对环境友好、高收率(≥85%)、高纯度(纯度可达99.9%)的适合工业化生产的制备方法。
具体实施方式
下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。
实施例1:1-(6-硝基吡啶-3-基)哌嗪(式(2)化合物)的实验室制备
2000ml反应瓶中,加入5-溴-2-硝基吡啶101.5g(0.5mol),无水哌嗪51.6g(0.6mol),正丁醇1000ml,水100ml及氢溴酸(48%)100g,控制60~65℃搅拌反应24h。反应毕,加入300ml水,减压蒸馏,再通过阶段补充的方式,补充1200ml水,蒸馏至馏出液变清。过滤除去不溶固体,母液用30%氢氧化钠调pH12,析出亮黄色固体。过滤,收集固体得1-(6-硝基-3-吡啶基)哌嗪湿品,以100%计算收率,直接投下一步反应,HPLC纯度≥99.5%。
实施例2:4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯(式(3)化合物)的实验室制备
2000ml反应瓶中,加入上步制备的1-(6-硝基吡啶-3-基)哌嗪湿品全部,甲苯1000ml,水500ml,碳酸钠79.5g(0.75mol),控制反应温度10~30℃,缓慢加入Boc酸酐131g(0.6mol),加毕继续反应1h,TLC检测至原料消失。反应毕,升温至95℃,固体溶解后,静置分液,分出水层,有机层加入500ml水,保持90~95℃,洗涤一次,减压蒸除约500ml甲苯,降温至0~10℃搅拌析晶,过滤,烘干得亮黄色固体142g,两步反应收率92.2%,纯度99.8%(HPLC)。
实施例3:1-(6-硝基吡啶-3-基)哌嗪(式(2)化合物)的工业化制备
2000L搪玻璃反应釜中,依次投入正丁醇1000L,水100kg,氢溴酸(48%)100kg,5-溴-2-硝基吡啶101.5kg,无水哌嗪51.6kg,保温60~65℃搅拌反应24h。反应毕,加入300kg纯化水,减压蒸馏,再补充1200kg纯化水,观察馏出液不再浑浊后,停止蒸馏。采用囊式过滤器压滤,50kg水洗涤,滤液用30%氢氧化钠调pH值至12,降温至0-10℃,离心,得亮黄色固体135kg(湿重),HPLC纯度99.7%,按100%计直接投下一步反应。
实施例4:4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯(式(3)化合物)的工业化制备
2000L不锈钢反应釜中,依次投入二氯甲烷1000L,水500kg,碳酸钠80kg及上步制备的1-(6-硝基吡啶-3-基)哌嗪湿品135kg,降温至10℃,缓慢加入131kg Boc酸酐,加毕继续反应1h,TLC控制反应终点。反应毕,静置分层,有机层用500kg水洗涤一次,常压蒸馏回收二氯甲烷,减压蒸馏残余溶剂。加入500L甲苯,升温至固体溶解,降温至0~10℃搅拌析晶,离心,烘干得145kg亮黄色固体,两步收率94.2%,纯度99.9%(HPLC)。
实施例5:4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯(式(1)化合物)的工业化制备
2000L高压釜中,投入1000L无水甲醇,145kg 4-(6-硝基吡啶-3-基)哌嗪-1-羧酸叔丁酯,醋酸钠0.77kg,通氮气置换3次,将5kg铂碳分散于100L甲醇中,打入高压釜。氮气置换三次,通氢反应3h。压滤,100L甲醇洗涤,收集铂碳回收套用,滤液减压浓缩至干,加入1000L正庚烷,降温至室温,搅拌析晶,离心,正庚烷洗涤,烘干得褐色固体126kg,收率96.3%,纯度99.7%。
3000L反应釜中,加入2000kg水,126kg 4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯粗品,30kg醋酸,室温搅拌至固体溶解,加入3kg活性炭(日本白鹭),继续搅拌1h,过滤,100kg水洗涤。母液用10%氢氧化钠调pH值至10,降温至0~10℃析晶2h,离心,水洗,烘干得类白色固体122kg,精制收率96.8%,纯度99.9%(HPLC)。
Claims (9)
1.一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法,其特征是,包括以下步骤:
S1:在醇类有机溶剂与水的混合溶剂中,以酸为催化剂,5-溴-2-硝基吡啶和哌嗪反应生成化合物式(2)化合物的酸式盐,反应结束后,加水并通过共沸蒸馏回收溶剂,得到含有式(2)化合物酸式盐的水溶液,通过过滤除去不溶杂质,用碱调pH10以上,离心,得到高纯度的式(2)化合物湿品;
S2:式(2)化合物湿品在有机溶剂与水的混合溶剂中,在缚酸剂的存在下,与Boc酸酐反应,反应液后处理得到式(3)化合物;
S3:式(3)化合物在极性溶剂中,在催化剂和缚酸剂的存在下,催化氢化,后处理生成4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的粗品;
S4:将粗品加入水中,再加入酸以使固体溶解,活性炭脱色,用碱溶液调pH碱性,经过析晶、离心、洗涤、烘干,得到4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯纯品;
所述式(2)化合物、式(3)化合物的结构式如下:

2.如权利要求1所述的工业化制备方法,其特征是,步骤S1中,
所述酸为盐酸、氢溴酸、磷酸、硫酸、醋酸、柠檬酸等的一种或两种的混合;
所述醇类有机溶剂为正丁醇、正戊醇、异戊醇、叔丁醇、仲丁醇、1-丙醇中的一种;其与水的体积比为10:0.3~5。
3.如权利要求1所述的工业化制备方法,其特征是,步骤S1中,
所述反应温度为50~100℃;
所述共沸回收溶剂,通过分批次补充水的方式,共沸蒸馏得到溶剂与水的混合物,通过分液分出有机溶剂和水,有机溶剂直接用于下一批反应,水用于下一批次物料共沸蒸馏。
4.如权利要求1所述的工业化制备方法,其特征是,步骤S2中,
所述缚酸剂为碳酸钠、碳酸钾、三乙胺、二异丙基乙胺、醋酸钠、氢氧化钠、氢氧化钾中的一种;
所述有机溶剂为二氯甲烷、三氯甲烷、甲苯、四氢呋喃、二氧六环中的一种。
5.如权利要求1所述的工业化制备方法,其特征是,步骤S2中的后处理为:升温至固体溶解后,静置分液,分出水层,有机层洗涤,减压浓缩,重结晶。
6.如权利要求1所述的工业化制备方法,其特征是,步骤S3中,
所述催化剂为钯碳、铂碳、雷尼镍中的一种;
所述缚酸剂为醋酸钠、醋酸钾、三乙胺、氨水、碳酸钠、碳酸氢钠中的一种。
7.如权利要求1所述的工业化制备方法,其特征是,步骤S3中,
所述极性溶剂为甲醇、乙醇、异丙醇、正丁醇中的一种;
反应温度为10~60℃。
8.如权利要求1所述的工业化制备方法,其特征是,步骤S3的后处理为:过滤,滤液减压浓缩至干,正庚烷重结晶。
9.如权利要求1-8中任一项所述的工业化制备方法,其特征是,步骤S4的酸为醋酸、甲酸、柠檬酸、草酸、丙酸中的一种。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111047083.6A CN113636973B (zh) | 2021-09-07 | 2021-09-07 | 一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111047083.6A CN113636973B (zh) | 2021-09-07 | 2021-09-07 | 一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN113636973A true CN113636973A (zh) | 2021-11-12 |
| CN113636973B CN113636973B (zh) | 2023-04-07 |
Family
ID=78425280
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111047083.6A Active CN113636973B (zh) | 2021-09-07 | 2021-09-07 | 一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN113636973B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112724078A (zh) * | 2021-01-13 | 2021-04-30 | 山东邹平大展新材料有限公司 | 一种哌柏西利中间体杂质的去除方法 |
| CN114805194A (zh) * | 2022-06-29 | 2022-07-29 | 南京威凯尔生物医药科技有限公司 | 一种2-硝基吡啶衍生物的连续加氢方法及其应用 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101671297A (zh) * | 2008-09-11 | 2010-03-17 | 上海药明康德新药开发有限公司 | 4-(3-碘-2-吡啶基)哌嗪类化合物的合成方法 |
| CN103168039A (zh) * | 2010-03-11 | 2013-06-19 | 吉利德康涅狄格公司 | 咪唑并吡啶类syk抑制剂 |
| CN103201275A (zh) * | 2010-11-10 | 2013-07-10 | 诺华有限公司 | 7-环戊基-2-(5-哌嗪-1-基-吡啶-2-基氨基)-7h-吡咯并[2,3-d]嘧啶-6-羧酸二甲酰胺的盐及其制备方法 |
| WO2017045648A1 (zh) * | 2015-09-18 | 2017-03-23 | 正大天晴药业集团股份有限公司 | 一种氘代化合物的制备方法 |
| WO2017211245A1 (zh) * | 2016-06-06 | 2017-12-14 | 深圳市塔吉瑞生物医药有限公司 | 一种取代的吡咯并嘧啶化合物及其应用 |
| CN109369517A (zh) * | 2018-10-24 | 2019-02-22 | 郑传花 | 一种治疗乳腺癌药物用中间体的制备方法 |
| CN110036012A (zh) * | 2016-11-28 | 2019-07-19 | 帝人制药株式会社 | 吡啶并[3,4-d]嘧啶衍生物及其药学上可接受的盐 |
-
2021
- 2021-09-07 CN CN202111047083.6A patent/CN113636973B/zh active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101671297A (zh) * | 2008-09-11 | 2010-03-17 | 上海药明康德新药开发有限公司 | 4-(3-碘-2-吡啶基)哌嗪类化合物的合成方法 |
| CN103168039A (zh) * | 2010-03-11 | 2013-06-19 | 吉利德康涅狄格公司 | 咪唑并吡啶类syk抑制剂 |
| CN103201275A (zh) * | 2010-11-10 | 2013-07-10 | 诺华有限公司 | 7-环戊基-2-(5-哌嗪-1-基-吡啶-2-基氨基)-7h-吡咯并[2,3-d]嘧啶-6-羧酸二甲酰胺的盐及其制备方法 |
| CN105384741A (zh) * | 2010-11-10 | 2016-03-09 | 诺华有限公司 | 一种细胞周期调节蛋白依赖性激酶抑制剂的盐及其制备方法 |
| WO2017045648A1 (zh) * | 2015-09-18 | 2017-03-23 | 正大天晴药业集团股份有限公司 | 一种氘代化合物的制备方法 |
| WO2017211245A1 (zh) * | 2016-06-06 | 2017-12-14 | 深圳市塔吉瑞生物医药有限公司 | 一种取代的吡咯并嘧啶化合物及其应用 |
| CN108290899A (zh) * | 2016-06-06 | 2018-07-17 | 深圳市塔吉瑞生物医药有限公司 | 一种取代的吡咯并嘧啶化合物及其应用 |
| CN110036012A (zh) * | 2016-11-28 | 2019-07-19 | 帝人制药株式会社 | 吡啶并[3,4-d]嘧啶衍生物及其药学上可接受的盐 |
| CN109369517A (zh) * | 2018-10-24 | 2019-02-22 | 郑传花 | 一种治疗乳腺癌药物用中间体的制备方法 |
Non-Patent Citations (1)
| Title |
|---|
| SOLOMON TADESSE等: "Highly Potent,Selective,and Orally Bioavailable 4-Thiazol-N-(pyridine-2-yl)pyrimidin-2-amine Cyclin-Dependent Kinases 4 and 6 Inhibitors as Anticancer Drug Candidates:Design,Synthesis,and Evaluation", 《J.MED.CHEM.》 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112724078A (zh) * | 2021-01-13 | 2021-04-30 | 山东邹平大展新材料有限公司 | 一种哌柏西利中间体杂质的去除方法 |
| CN114805194A (zh) * | 2022-06-29 | 2022-07-29 | 南京威凯尔生物医药科技有限公司 | 一种2-硝基吡啶衍生物的连续加氢方法及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN113636973B (zh) | 2023-04-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107793418B (zh) | 一种枸橼酸托法替布的工业化生产方法 | |
| CN113636973B (zh) | 一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 | |
| CN111470983B (zh) | 一种盐酸溴己新的制备方法 | |
| CN112062726B (zh) | 一种2-氨基-4,6-二氯-5-甲酰胺基嘧啶的制备方法 | |
| CN105440034A (zh) | 一种利格列汀及其中间体的制备方法 | |
| CN105153169B (zh) | 一种盐酸依匹斯汀的合成方法 | |
| CN106674084A (zh) | 一种2‑异丙基氧基‑5‑甲基‑4‑(哌啶‑4‑基)苯胺二盐酸盐的制备方法 | |
| CN113278021B (zh) | 1,7-二氮杂螺[3.5]壬烷-7-甲酸叔丁酯及其草酸盐的制备方法 | |
| CN115960059A (zh) | 一种高产率高纯度合成呋塞米杂质d的方法 | |
| KR101485418B1 (ko) | 고순도 미르타자핀의 제조방법 | |
| CN110483388B (zh) | 一种烟酸衍生物的制备方法 | |
| JP6947354B2 (ja) | リナグリプチンの製造法 | |
| CN111233864B (zh) | 一种工业化生产多索茶碱的方法 | |
| CN116768762B (zh) | 一种美索巴莫降解衍生物的制备方法 | |
| CN112176011B (zh) | 一种酶催化制备盐酸伐昔洛韦的方法 | |
| CN112552299B (zh) | 一种治疗ii型糖尿病利格列汀的制备方法 | |
| CN112300070B (zh) | 一种米力农的纯化方法 | |
| CN111606929B (zh) | 德高替尼的制备方法 | |
| CN111574540B (zh) | 一种德高替尼的制备方法 | |
| CN108623608B (zh) | 扎布沙星中间体的制备方法 | |
| CN114560862A (zh) | 一种吡咯并[1,2-a]喹喔啉-4(5h)-酮及其衍生物的合成方法 | |
| CN118791491A (zh) | 一种Istradefylline的制备方法 | |
| CN118126029A (zh) | 一种吡贝地尔的制备方法 | |
| CN109748885B (zh) | 一种色瑞替尼中间体及色瑞替尼的制备方法 | |
| CN118125995A (zh) | 一种1-(2-甲氧基苯基)哌嗪盐酸盐的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| CB02 | Change of applicant information |
Address after: 251400 No. 12, Taixing East Street, Jibei Economic Development Zone, Jiyang District, Jinan City, Shandong Province Applicant after: Shandong Baoyuan Pharmaceutical Co.,Ltd. Address before: Strong in Jiyang County of Ji'nan City, 251400 North Street, Shandong Province Economic Development Zone Applicant before: SHANDONG BOYUAN PHARMACEUTICAL Co.,Ltd. |
|
| CB02 | Change of applicant information | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |