CN113087667B - 一种咪唑啉酮衍生物的合成方法 - Google Patents
一种咪唑啉酮衍生物的合成方法 Download PDFInfo
- Publication number
- CN113087667B CN113087667B CN202110314112.4A CN202110314112A CN113087667B CN 113087667 B CN113087667 B CN 113087667B CN 202110314112 A CN202110314112 A CN 202110314112A CN 113087667 B CN113087667 B CN 113087667B
- Authority
- CN
- China
- Prior art keywords
- compound
- derivative
- imidazolinone
- solvent
- synthesized
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000008624 imidazolidinones Chemical class 0.000 title claims description 6
- 238000001308 synthesis method Methods 0.000 title abstract description 7
- CAAMSDWKXXPUJR-UHFFFAOYSA-N 3,5-dihydro-4H-imidazol-4-one Chemical class O=C1CNC=N1 CAAMSDWKXXPUJR-UHFFFAOYSA-N 0.000 claims abstract description 34
- 239000003054 catalyst Substances 0.000 claims abstract description 25
- 229910017052 cobalt Inorganic materials 0.000 claims abstract description 25
- 239000010941 cobalt Substances 0.000 claims abstract description 25
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims abstract description 25
- -1 amidine compound Chemical class 0.000 claims abstract description 22
- 238000010189 synthetic method Methods 0.000 claims abstract description 13
- 238000006243 chemical reaction Methods 0.000 claims abstract description 11
- 239000000126 substance Substances 0.000 claims abstract description 11
- 150000005846 sugar alcohols Polymers 0.000 claims abstract description 11
- 150000001875 compounds Chemical class 0.000 claims abstract description 10
- 239000002904 solvent Substances 0.000 claims description 26
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 20
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 16
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 239000001257 hydrogen Substances 0.000 claims description 13
- 238000000034 method Methods 0.000 claims description 13
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 12
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 9
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 claims description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 7
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 6
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 6
- 229920005862 polyol Polymers 0.000 claims description 5
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 claims description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 4
- 125000003118 aryl group Chemical group 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 4
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 3
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 claims description 3
- 239000004408 titanium dioxide Substances 0.000 claims description 3
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- WCVRQHFDJLLWFE-UHFFFAOYSA-N pentane-1,2-diol Chemical compound CCCC(O)CO WCVRQHFDJLLWFE-UHFFFAOYSA-N 0.000 claims description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- 230000035484 reaction time Effects 0.000 claims description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 2
- 239000011787 zinc oxide Substances 0.000 claims description 2
- AMWRITDGCCNYAT-UHFFFAOYSA-L hydroxy(oxo)manganese;manganese Chemical compound [Mn].O[Mn]=O.O[Mn]=O AMWRITDGCCNYAT-UHFFFAOYSA-L 0.000 claims 2
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 claims 1
- 230000015572 biosynthetic process Effects 0.000 claims 1
- 230000008901 benefit Effects 0.000 abstract description 4
- 229910052751 metal Inorganic materials 0.000 abstract description 4
- 239000002184 metal Substances 0.000 abstract description 4
- 239000002994 raw material Substances 0.000 abstract description 4
- 239000002028 Biomass Substances 0.000 abstract description 2
- 125000000524 functional group Chemical group 0.000 abstract description 2
- 239000007800 oxidant agent Substances 0.000 abstract description 2
- 230000001590 oxidative effect Effects 0.000 abstract description 2
- 239000000758 substrate Substances 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 57
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 41
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 24
- 239000003208 petroleum Substances 0.000 description 19
- 238000004809 thin layer chromatography Methods 0.000 description 17
- 239000000203 mixture Substances 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- 238000003756 stirring Methods 0.000 description 11
- 230000005526 G1 to G0 transition Effects 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical group O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 239000003480 eluent Substances 0.000 description 9
- 239000012046 mixed solvent Substances 0.000 description 9
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- 238000012512 characterization method Methods 0.000 description 8
- 238000002390 rotary evaporation Methods 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- PFURGBBHAOXLIO-UHFFFAOYSA-N cyclohexane-1,2-diol Chemical compound OC1CCCCC1O PFURGBBHAOXLIO-UHFFFAOYSA-N 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- 238000001228 spectrum Methods 0.000 description 5
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- LZCZIHQBSCVGRD-UHFFFAOYSA-N benzenecarboximidamide;hydron;chloride Chemical compound [Cl-].NC(=[NH2+])C1=CC=CC=C1 LZCZIHQBSCVGRD-UHFFFAOYSA-N 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 125000006539 C12 alkyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- OWBTYPJTUOEWEK-UHFFFAOYSA-N butane-2,3-diol Chemical compound CC(O)C(C)O OWBTYPJTUOEWEK-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229910001867 inorganic solvent Inorganic materials 0.000 description 2
- 239000003049 inorganic solvent Substances 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 229910052748 manganese Inorganic materials 0.000 description 2
- 239000011572 manganese Substances 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 239000011943 nanocatalyst Substances 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- RXAOGVQDNBYURA-UHFFFAOYSA-N (4-chlorobenzenecarboximidoyl)azanium;chloride Chemical compound Cl.NC(=N)C1=CC=C(Cl)C=C1 RXAOGVQDNBYURA-UHFFFAOYSA-N 0.000 description 1
- HBMWQILXZYMGKB-UHFFFAOYSA-N 1-(8-fluoro-1,3,4,5-tetrahydropyrido[4,3-b]indol-2-yl)ethanone Chemical compound N1C2=CC=C(F)C=C2C2=C1CCN(C(=O)C)C2 HBMWQILXZYMGKB-UHFFFAOYSA-N 0.000 description 1
- MFGOFGRYDNHJTA-UHFFFAOYSA-N 2-amino-1-(2-fluorophenyl)ethanol Chemical compound NCC(O)C1=CC=CC=C1F MFGOFGRYDNHJTA-UHFFFAOYSA-N 0.000 description 1
- CFCNTIFLYGKEIO-UHFFFAOYSA-N 2-isocyanoacetic acid Chemical compound OC(=O)C[N+]#[C-] CFCNTIFLYGKEIO-UHFFFAOYSA-N 0.000 description 1
- JQDATBKJKUWNGA-UHFFFAOYSA-N 4-fluorobenzenecarboximidamide;hydrochloride Chemical compound Cl.NC(=N)C1=CC=C(F)C=C1 JQDATBKJKUWNGA-UHFFFAOYSA-N 0.000 description 1
- AJOSDIDPIBJFAI-UHFFFAOYSA-N 4-methoxybenzenecarboximidamide;hydrochloride Chemical compound Cl.COC1=CC=C(C(N)=N)C=C1 AJOSDIDPIBJFAI-UHFFFAOYSA-N 0.000 description 1
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VRJJZMJUNZRSLS-UHFFFAOYSA-N Cl.N1=CC(=CC=C1)C1=CC=CC=C1C(=N)N Chemical compound Cl.N1=CC(=CC=C1)C1=CC=CC=C1C(=N)N VRJJZMJUNZRSLS-UHFFFAOYSA-N 0.000 description 1
- XZMCDFZZKTWFGF-UHFFFAOYSA-N Cyanamide Chemical compound NC#N XZMCDFZZKTWFGF-UHFFFAOYSA-N 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 238000012271 agricultural production Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Inorganic materials [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 1
- 238000001354 calcination Methods 0.000 description 1
- 150000005323 carbonate salts Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- GVPFVAHMJGGAJG-UHFFFAOYSA-L cobalt dichloride Chemical compound [Cl-].[Cl-].[Co+2] GVPFVAHMJGGAJG-UHFFFAOYSA-L 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 150000001470 diamides Chemical class 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000002143 encouraging effect Effects 0.000 description 1
- 238000003912 environmental pollution Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 125000005059 halophenyl group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 239000004009 herbicide Substances 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 125000006303 iodophenyl group Chemical group 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- SQQMAOCOWKFBNP-UHFFFAOYSA-L manganese(II) sulfate Chemical compound [Mn+2].[O-]S([O-])(=O)=O SQQMAOCOWKFBNP-UHFFFAOYSA-L 0.000 description 1
- 229910000357 manganese(II) sulfate Inorganic materials 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000006452 multicomponent reaction Methods 0.000 description 1
- KERBAAIBDHEFDD-UHFFFAOYSA-N n-ethylformamide Chemical compound CCNC=O KERBAAIBDHEFDD-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- XLMFDCKSFJWJTP-UHFFFAOYSA-N pentane-2,3-diol Chemical compound CCC(O)C(C)O XLMFDCKSFJWJTP-UHFFFAOYSA-N 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/70—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description








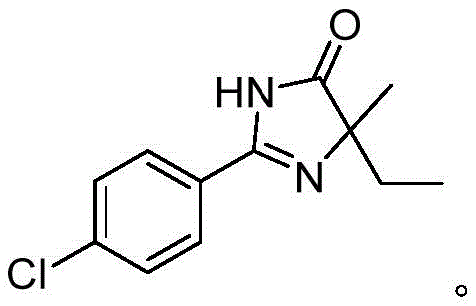
Claims (7)
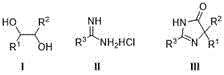
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110314112.4A CN113087667B (zh) | 2021-03-24 | 2021-03-24 | 一种咪唑啉酮衍生物的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110314112.4A CN113087667B (zh) | 2021-03-24 | 2021-03-24 | 一种咪唑啉酮衍生物的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN113087667A CN113087667A (zh) | 2021-07-09 |
| CN113087667B true CN113087667B (zh) | 2022-06-24 |
Family
ID=76669735
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110314112.4A Active CN113087667B (zh) | 2021-03-24 | 2021-03-24 | 一种咪唑啉酮衍生物的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN113087667B (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114737210B (zh) * | 2022-04-26 | 2023-04-18 | 南京工业大学 | 一种利用电化学微通道反应装置连续制备1,3-茚二酮螺咪唑啉类化合物的方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101273018A (zh) * | 2005-09-26 | 2008-09-24 | 惠氏公司 | 用于β分泌酶(BACE)抑制剂的氨基-5-[4-(二氟甲氧基)苯基]-5-苯基咪唑酮化合物 |
| JP2011231022A (ja) * | 2010-04-23 | 2011-11-17 | Ajinomoto Co Inc | イミダゾロン誘導体 |
| CN103214420A (zh) * | 2013-04-26 | 2013-07-24 | 台州职业技术学院 | 一种2-丁基-4-氯-5-甲酰基咪唑的制备方法 |
| CN104177298A (zh) * | 2014-09-18 | 2014-12-03 | 湘潭大学 | 4,4-二取代-4,5-二氢-1h–咪唑-5-酮、衍生物及其合成方法 |
| CN108976170A (zh) * | 2018-08-14 | 2018-12-11 | 华南理工大学 | 一种5-三氟甲基-4h-咪唑啉-4-酮衍生物及合成方法 |
-
2021
- 2021-03-24 CN CN202110314112.4A patent/CN113087667B/zh active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101273018A (zh) * | 2005-09-26 | 2008-09-24 | 惠氏公司 | 用于β分泌酶(BACE)抑制剂的氨基-5-[4-(二氟甲氧基)苯基]-5-苯基咪唑酮化合物 |
| JP2011231022A (ja) * | 2010-04-23 | 2011-11-17 | Ajinomoto Co Inc | イミダゾロン誘導体 |
| CN103214420A (zh) * | 2013-04-26 | 2013-07-24 | 台州职业技术学院 | 一种2-丁基-4-氯-5-甲酰基咪唑的制备方法 |
| CN104177298A (zh) * | 2014-09-18 | 2014-12-03 | 湘潭大学 | 4,4-二取代-4,5-二氢-1h–咪唑-5-酮、衍生物及其合成方法 |
| CN108976170A (zh) * | 2018-08-14 | 2018-12-11 | 华南理工大学 | 一种5-三氟甲基-4h-咪唑啉-4-酮衍生物及合成方法 |
Non-Patent Citations (3)
| Title |
|---|
| Efficient 4,5-dihydro-1H-imidazol-5-one formation from amidines and ketones under transition-metal free conditions;Yanjun Xie et al.;《Green Chem.》;20140926;第17卷;第209-213页 * |
| OMS-2 nanorod-supported cobalt catalyst for aerobic dehydrocyclization of vicinal diols and amidines: Access to functionalized imidazolones;Feng Xie et al.;《Journal of Catalysis》;20210504;第398卷;第192-197页 * |
| Synthesis and evaluation of 5,5-diphenylimidazolones as potent human neuropeptide Y5 receptor antagonists;Kevin W. Gillman et al.;《Bioorganic & Medicinal Chemistry》;20060511;第14卷;第5517-5526页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN113087667A (zh) | 2021-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111205279B (zh) | 一种多取代苯并二氢呋喃并杂环类化合物及其制备方法和应用 | |
| CN101723771B (zh) | 功能离子液体催化制备β-氨基酮、酯、腈和酰胺衍生物的方法 | |
| CN101798279B (zh) | 铁催化的吡咯及吡咯并环类化合物的制备方法 | |
| CN107674044B (zh) | 一种利用二氧化碳、胺和芳基重氮乙酸酯合成氨基甲酸酯的方法 | |
| CN113087667B (zh) | 一种咪唑啉酮衍生物的合成方法 | |
| CN101146812A (zh) | 光学活性铵盐化合物、其制造中间体和制造方法 | |
| CN115233243A (zh) | 一种电催化下2,4,5-三取代噁唑衍生物的制备方法 | |
| CN108863890B (zh) | 一种4-吡咯啉-2-酮衍生物及其制备方法 | |
| CN103242371A (zh) | 联芳基吡啶环钯氮杂环卡宾化合物及其制备方法和用途 | |
| CN112174842A (zh) | 一种制备(s)-3-氨基-2-苄基丙酸的方法 | |
| CN111333543A (zh) | 一种利匹韦林中间体的合成方法 | |
| JP4157766B2 (ja) | 置換イミダゾピリジン化合物の製造方法 | |
| CN115160211B (zh) | 一种异吲哚啉酮类化合物的绿色合成方法 | |
| CN111233745B (zh) | (e)1-(9-烷基-咔唑-3-)-丙烯酸及其制备方法 | |
| Ma et al. | N-Heterocyclic carbene-catalyzed (NHC) three-component domino reactions: highly stereoselective synthesis of functionalized acyclic ε-ketoesters | |
| CN111393437B (zh) | 三取代吲嗪类化合物及其制备方法 | |
| CN110577529A (zh) | N-(杂)芳基-7-氮杂吲哚的α-酮类化合物及制备方法 | |
| CN103214394B (zh) | 一种炔基亚胺衍生物 | |
| CN113004236A (zh) | 一种合成3-亚甲基苯并呋喃-1(3h)-酮类化合物的方法 | |
| CN111732552A (zh) | 一种钯催化合成1,3-噁唑-2-硫酮的方法 | |
| CN110804007B (zh) | 一种多取代吡咯衍生物及其制备方法 | |
| CN101643450B (zh) | 一种多取代2,3-二氢-4(1h)-嘧啶硫酮的合成方法 | |
| CN109096139A (zh) | 一种α-羰基酰胺衍生物的制备方法 | |
| CN114797968B (zh) | 一种负载型磷酸催化剂及其制备方法和用途 | |
| CN110590641B (zh) | 一种3-羟基异吲哚-1-酮系列化合物的绿色制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20240426 Address after: 1003, Building A, Zhiyun Industrial Park, No. 13 Huaxing Road, Tongsheng Community, Dalang Street, Longhua District, Shenzhen City, Guangdong Province, 518000 Patentee after: Shenzhen Wanzhida Enterprise Management Co.,Ltd. Country or region after: China Address before: No.22, Dongcheng village, Pengjiang district, Jiangmen City, Guangdong Province Patentee before: WUYI University Country or region before: China |
|
| TR01 | Transfer of patent right | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20240611 Address after: 233000 south of Kaiyuan Avenue, east of Jinli Road, mohekou Industrial Park, Huaishang District, Bengbu City, Anhui Province Patentee after: ANHUI KANGMU INTERNATIONAL FERTILIZER CO.,LTD. Country or region after: China Address before: 1003, Building A, Zhiyun Industrial Park, No. 13 Huaxing Road, Tongsheng Community, Dalang Street, Longhua District, Shenzhen City, Guangdong Province, 518000 Patentee before: Shenzhen Wanzhida Enterprise Management Co.,Ltd. Country or region before: China |
|
| TR01 | Transfer of patent right |