CN109071567B - 抗流感小分子化合物及其制备方法和用途 - Google Patents
抗流感小分子化合物及其制备方法和用途 Download PDFInfo
- Publication number
- CN109071567B CN109071567B CN201780013432.8A CN201780013432A CN109071567B CN 109071567 B CN109071567 B CN 109071567B CN 201780013432 A CN201780013432 A CN 201780013432A CN 109071567 B CN109071567 B CN 109071567B
- Authority
- CN
- China
- Prior art keywords
- substituted
- unsubstituted
- membered
- unsaturated
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
本发明属于化学医药领域,具体涉及抗流感小分子化合物及其制备方法和用途。本发明要解决的技术问题是目前临床上能运用的抗流感药物数量和种类都较少,而且这些抗流感药物存在耐药性、治疗窗窄、毒副作用大、临床疗效差等问题。发明解决上述技术问题的方案是提供一种抗流感小分子化合物,其结构如式I所示。本发明还提供了上述抗流感小分子化合物的制备方法和用途。本发明提供的化合物可能克服目前临床使用的神经氨酸酶抑制剂和M2离子通道抑制剂等药物的耐药性、疗效差、治疗窗窄等问题,具有较大的开发价值。
Description
技术领域
本发明属于化学医药领域,具体涉及抗流感小分子化合物及其制备方法和用途。
背景技术
流行性感冒(简称流感,Flu)是由流感病毒(Influenza virus)引起的呼吸系统疾病。根据病毒核蛋白和基质蛋白的抗原决定簇的差异,可将流感病毒分为甲(A)、乙(B)、丙(C)三种类型,其中A型流感最为常见,致病性强,易发生大范围流行,严重威胁人类生命与健康。禽流感病毒(Avian influenza virus)属于A型流感病毒,正常情况下仅在禽类等动物之间传播。但是有些变异株直接由禽类等动物传染给人类,给人类带来严重健康威胁,例如高致病性禽流感H5N1、H7N9等,其致死率可达30%。历史上曾发生过多次严重的流感大流行,造成了上百万人死亡,给人类社会带来严重损失。
预防和治疗流感,通常采用疫苗接种和抗流感药物治疗两种方法。疫苗接种是目前预防流感的一种有效措施。成年人接种后可以达到较好预防效果,但是婴幼儿、老年人等免疫力较低者接种后效果并不理想。而且流感病毒不断地变异,旧疫苗难以对抗新病毒。化学药物是治疗流感的另一种重要手段,但是迄今为止,上市抗流感化学药物数量少,应用较多的是M2离子通道抑制剂、神经氨酸酶(NA)抑制剂以及核苷类抗病毒药物。
目前,上市的M2离子通道抑制剂仅有金刚烷胺(Amantadine,1966年上市)、金刚乙胺(Rimantadine,1987年上市),但仅对A型流感病毒有效,且金刚烷胺具有较大中枢神经系统不良反应,近年来涌现了较多耐药病毒株,耐药性成为了该类药物面临的重大问题。神经氨酸酶抑制剂(NAI)是一类强效的抗流感药物,除了FDA批准上市的奥司他韦(Oseltamivir,达菲,1999年上市)和扎拉米韦(Zanamivir,1999年上市),还有在局部地区上市的帕拉米韦(Peramivir,日本、韩国、中国上市)和拉尼那米韦(Laninamivir,日本上市)。尽管这类药物在体外显示了较好的抗流感效果,但来自国际循证医学协作组(Cochrane Collaboration)的报告指出,在临床上,目前并无证据支持奥司他韦具有防止流感病毒传播、或降低患者住院率和并发症风险的作用。另外,患者在感染流感病毒48h后,服用奥司他韦并不能起到抗流感效果。而且NAI也出现了严重的耐药情况。核苷类代表药物是利巴韦林(Ribavirin)和法匹拉韦(Favipiravir,T-705)。利巴韦林是一种广谱抗病毒上市药物,对DNA和RNA病毒均有抑制效果,作用机制尚不明确,在体内能引起溶血性贫血、心肺方面的毒性反应,所以临床应用受限。法匹拉韦主要抑制RNA病毒的复制,与利巴韦林相比具有活性较高、细胞毒性较小的优点,目前仅在日本上市。另外,盐酸阿比朵尔(arbidolhydrochloride)是1993年在俄罗斯上市抗流感病毒药物。它的作用机制尚不明确,可能是抑制HA,通过阻止流感病毒外壳与宿主细胞细胞膜的接触、黏附和融合,抑制病毒与细胞浆膜的融合及病毒与内吞囊泡之间的膜融合而达到抗病毒作用。
近年来,流感RNA聚合酶(RdRp)受到了广泛关注。RdRp是由PA、PB1和PB2三个亚基组成的异源三聚体,在流感病毒基因组转录和复制过程中发挥重要作用。流感病毒RNA的转录具有特殊的“夺帽”机制,在此过程中,PB2亚基负责识别和结合宿主前体mRNA的“帽子结构”,然后PA亚基剪切宿主mRNA得到引物,启动转录过程。抑制“夺帽”可以阻断转录过程,达到抑制流感病毒增殖的效果。因此,PB2被认为是很有前途的抗流感药物靶标,已引起了制药公司和学术研究机构的高度重视。
发明内容
本发明要解决的技术问题是目前临床上能运用的抗流感药物数量和种类都较少,而且这些抗流感药物存在耐药性、治疗窗窄、毒副作用大、临床疗效差等问题。
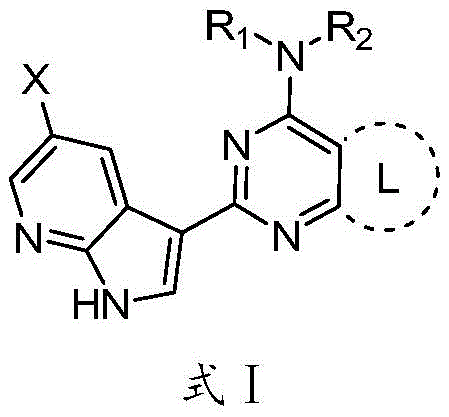
本发明解决上述技术问题的方案是提供一种抗流感小分子化合物,其结构如式I所示:

其中,X为-H、卤素、氰基、-CF3、C1~C4烷氧基、C1~C4烷基、氨基或C1~C3氨酰基;
L为饱和或不饱和的4~10元杂环烷基、饱和或不饱和的C4~C10环烷基;所述饱和或不饱和的4~10元杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3;
R1和R2组合成取代或未取代的5~8元饱和杂环烷基,所述的杂原子为N,杂原子个数为1~2个;所述取代5~8元饱和杂环烷基的取代基为-H、-NH2、卤素、-CF3、氰基、C1~C4烷基、 a、b为0~4;
a、b为0~4;
或者R1为-H或C1~C10烷基;R2为 n为0~4;
n为0~4;
R4、R5独立的为-H或C1~C10烷基;或者R4和R5组合形成环,所述的环为C5~C10环烷基、C5~C10的桥环烷基、C5~C10的稠环烷基、5~10元饱和杂环烷基、5~10元桥环杂环烷基或5~10元螺环杂环烷基;所述5~10元饱和杂环烷基、5~10元桥环杂环烷基、5~10元螺环杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3;R3为 R9为C1~C5烷基,R10为氢或C1~C5烷基;
R9为C1~C5烷基,R10为氢或C1~C5烷基;
R6、R7独立的为C1~C4烷基、取代或未取代的C5~C10芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C10芳基或苄基的取代基为C1~C6烷基、C1~C6烷氧基、卤素、-CF3或氰基;a、b为0~4;
或-CONH2;所述取代C5~C10芳基或苄基的取代基为C1~C6烷基、C1~C6烷氧基、卤素、-CF3或氰基;a、b为0~4;
R8为取代或未取代的C5~C10环烷基、取代或未取代的5~10元饱和或不饱和杂环、取代或未取代的C5~C10不饱和环或取代或未取代的C5~C15的桥环烷基;所述5~10元饱和或不饱和杂环的杂原子为N、O、S,杂原子个数为1~3个;所述取代C5~C10环烷基的取代基为-H、 卤素、C1~C4烷基或-COOH;所述取代C5~C10不饱和环的取代基为-H、-OH、-SO2NH2、-CF3、卤素、C1~C4烷基或-COOH;所述取代5~1O元饱和或不饱和杂环的取代基为卤素取代的苄基、卤素、苯基、苄基、-OH、-CF3、C1~C4烷基、C1~C4羰基、-COOH或-NH2;所述取代C5~C15的桥环烷基的取代基为-H、-OH、-COOH、C1~C4烷基或-NH2;a为0~4。
卤素、C1~C4烷基或-COOH;所述取代C5~C10不饱和环的取代基为-H、-OH、-SO2NH2、-CF3、卤素、C1~C4烷基或-COOH;所述取代5~1O元饱和或不饱和杂环的取代基为卤素取代的苄基、卤素、苯基、苄基、-OH、-CF3、C1~C4烷基、C1~C4羰基、-COOH或-NH2;所述取代C5~C15的桥环烷基的取代基为-H、-OH、-COOH、C1~C4烷基或-NH2;a为0~4。
作为本发明优选的方案,当R1为H,R2为 R3为
R3为 时,其结构如式II所示:
时,其结构如式II所示:

其中,X为卤素或C1~C4烷基;L为饱和或不饱和的5~8元杂环烷基、饱和或不饱和的C5~C8环烷基;所述5~8元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;R4、R5独立地为-H或C1~C8烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基、C5~C8的稠环烷基、5~8元饱和杂环烷基、5~8元桥环杂环烷基或5~10元螺环杂环烷基;所述5~8元饱和杂环烷基、5~8元桥环杂环烷基、5~10元螺环杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3。
优选的,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C8环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基、C5~C8的稠环烷基、5~8元饱和杂环烷基、5~8元桥环杂环烷基或5~10元螺环杂环烷基;所述5~8元饱和杂环烷基、5~8元桥环杂环烷基、5~10元螺环杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3。
再进一步优选的,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基、C5~C8的稠环烷基或5~8元饱和杂环烷基;所述5~8元饱和杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3。
更进一步优选的,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基、C5~C8的稠环烷基或5~8元饱和杂环烷基;所述5~8元饱和杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3。
优选的,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基、C5~C8的稠环烷基或5~8元饱和杂环烷基;所述5~8元饱和杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~2。
进一步优选的,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基或C5~C8的稠环烷基。
更进一步优选的,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基或C5~C8的桥环烷基。
最优的,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1个;R4、R5独立地为-H或C1~C4烷基;或R1和R2组合形成环,所述的环为C5~C6环烷基或C5~C8的桥环烷基。
上述抗流感小分子化合物中,当R1为H,R2为 时,其结构如式III所示:
时,其结构如式III所示:

其中,X为卤素或C1~C4烷基;L为饱和或不饱和的5~8元杂环烷基、饱和或不饱和的C5~C8环烷基;所述5~8元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;R6、R7独立的为C1~C4烷基、取代或未取代的C5~C8芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C8芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~3。
或-CONH2;所述取代C5~C8芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~3。
优选的,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C8环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;R6、R7独立的为C1~C4烷基、取代或未取代的C5~C6芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C6芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~2。
或-CONH2;所述取代C5~C6芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~2。
再进一步优选的,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;R6、R7独立的为C1~C4烷基、取代或未取代的苯基、取代或未取代的苄基、 或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基或卤素;a、b为0~2。
或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基或卤素;a、b为0~2。
更进一步优选的,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;R6、R7独立的为C1~C4烷基、取代或未取代的苯基、取代或未取代的苄基、 或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基、-F、-Cl或-Br;a、b为0~2。
或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基、-F、-Cl或-Br;a、b为0~2。
最优的,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1个;R6、R7独立的为C1~C4烷基、取代或未取代的苯基、取代或未取代的苄基、 或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基、-F、-Cl或-Br;a、b为0~2。
或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基、-F、-Cl或-Br;a、b为0~2。
上述抗流感小分子化合物,当R1为H,R2为 时,其结构如式IV所示:
时,其结构如式IV所示:

其中,n为0~4,X为卤素或C1~C4烷基;L为饱和或不饱和的5~8元杂环烷基、饱和或不饱和的C5~C8环烷基;所述5~8元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;
R8为取代或未取代的C5~C8环烷基、取代或未取代的5~8元芳杂基、取代或未取代的5~8元饱和杂环烷基、取代或未取代的5~8元不饱和杂环烷基、取代或未取代的C5~C10芳基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~8元饱和或不饱和杂环烷基、5~8元芳杂基的杂原子为N、O、S,杂原子个数为1~3个;所述取代C5~C8环烷基的取代基为-H或 所述取代5~8元芳杂基、5~8元饱和杂环烷基、5~8元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C4羰基;所述取代C5~C10芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、卤素、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH,a为0~3。
所述取代5~8元芳杂基、5~8元饱和杂环烷基、5~8元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C4羰基;所述取代C5~C10芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、卤素、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH,a为0~3。
优选的,n为0~4,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C8环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~3个;
R8为取代或未取代的C5~C6环烷基、取代或未取代的5~6元芳杂基、取代或未取代的5~6元饱和杂环烷基、取代或未取代的5~6元不饱和杂环烷基、取代或未取代的C5~C8芳基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~6元饱和或不饱和杂环烷基、5~6元芳杂基的杂原子为N、O、S,杂原子个数为1~2个;所述取代C5~C6环烷基的取代基为-H或 所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C8芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH;a为0~3。
所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C8芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH;a为0~3。
更优选的,n为0~3,X为卤素;L为饱和或不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;
R8为取代或未取代的C5~C6环烷基、取代或未取代的5~6元芳杂基、取代或未取代的5~6元饱和杂环烷基、取代或未取代的5~6元不饱和杂环烷基、取代或未取代的C5~C6芳基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~6元饱和或不饱和杂环烷基、5~6元芳杂基的杂原子为N、O、S,杂原子个数为1~2个;所述取代C5~C6环烷基的取代基为-H或 所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C6芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH;a为0~3。
所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C6芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH;a为0~3。
进一步优选的,n为0~3,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1~2个;
R8为取代或未取代的C5~C6环烷基、取代或未取代的5~6元芳杂基、取代或未取代的5~6元饱和杂环烷基、取代或未取代的5~6元不饱和杂环烷基、取代或未取代的苯基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~6元饱和或不饱和杂环烷基、5~6元芳杂基的杂原子为N、O,杂原子个数为1~2个;所述取代C5~C6环烷基的取代基为-H或 所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为氟取代的苄基、-F、-Cl、-Br、苯基、苄基、-CF3或C1~C2羰基;所述取代苯基的取代基为-F、-Cl、-Br、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH;a为0~2。
所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为氟取代的苄基、-F、-Cl、-Br、苯基、苄基、-CF3或C1~C2羰基;所述取代苯基的取代基为-F、-Cl、-Br、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-H、-OH或-COOH;a为0~2。
最优的,n为0~3,X为卤素;L为不饱和的5~6元杂环烷基、饱和或不饱和的C5~C6环烷基;所述5~6元杂环烷基的杂原子为N、O或S,所述的杂原子为1个;
R8为

上述抗流感小分子化合物的具体结构式为:


本发明还提供了上述抗流感小分子化合物的制备方法,其合成路线为:

其中,X为-H、卤素、氰基、-CF3、C1~C4烷氧基、C1~C4烷基、氨基或C1~C3氨酰基;
L为饱和或不饱和的4~10元杂环烷基、饱和或不饱和的C4~C10环烷基;所述饱和或不饱和的4~10元杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3;
R1和R2组合成取代或未取代的5~8元饱和杂环烷基,所述的杂原子为N,杂原子个数为1~2个;所述取代5~8元饱和杂环烷基的取代基为-H、-NH2、卤素、-CF3、氰基、C1~C4烷基、 a、b为0~4;
a、b为0~4;
或者R1为-H或C1~C10烷基;R2为 n为0~4;
n为0~4;
R4、R5独立的为-H或C1~C10烷基;或者R4和R5组合形成环,所述的环为C5~C10环烷基、C5~C10的桥环烷基、C5~C10的稠环烷基、5~10元饱和杂环烷基、5~10元桥环杂环烷基或5~10元螺环杂环烷基;所述5~10元饱和杂环烷基、5~10元桥环杂环烷基、5~10元螺环杂环烷基的杂原子为N、O或S,所述杂原子的个数为1~3;R3为 R9为C1~C5烷基,R10为氢或C1~C5烷基;
R9为C1~C5烷基,R10为氢或C1~C5烷基;
R6、R7独立的为C1~C4烷基、取代或未取代的C5~C10芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C10芳基或苄基的取代基为C1~C6烷基、C1~C6烷氧基、卤素、-CF3或氰基;a、b为0~4;
或-CONH2;所述取代C5~C10芳基或苄基的取代基为C1~C6烷基、C1~C6烷氧基、卤素、-CF3或氰基;a、b为0~4;
R8为取代或未取代的C5~C10环烷基、取代或未取代的5~10元饱和或不饱和杂环、取代或未取代的C5~C10不饱和环或取代或未取代的C5~C15的桥环烷基;所述5~10元饱和或不饱和杂环的杂原子为N、O、S,杂原子个数为1~3个;所述取代C5~C10环烷基的取代基为-H、 卤素、C1~C4烷基或-COOH;所述取代C5~C10不饱和环的取代基为-H、-OH、-SO2NH2、-CF3、卤素、C1~C4烷基或-COOH;所述取代5~10元饱和或不饱和杂环的取代基为卤素取代的苄基、卤素、苯基、苄基、-OH、-CF3、C1~C4烷基、C1~C4羰基、-COOH或-NH2;所述取代C5~C15的桥环烷基的取代基为-H、-OH、-COOH、C1~C4烷基或-NH2;a为0~4。
卤素、C1~C4烷基或-COOH;所述取代C5~C10不饱和环的取代基为-H、-OH、-SO2NH2、-CF3、卤素、C1~C4烷基或-COOH;所述取代5~10元饱和或不饱和杂环的取代基为卤素取代的苄基、卤素、苯基、苄基、-OH、-CF3、C1~C4烷基、C1~C4羰基、-COOH或-NH2;所述取代C5~C15的桥环烷基的取代基为-H、-OH、-COOH、C1~C4烷基或-NH2;a为0~4。
上述抗流感小分子化合物制备方法的操作步骤包括:
a、原料1与卤代试剂进行卤代反应制备得到中间体1;所述的卤代试剂为NIS(N-碘代丁二酰亚胺)、NBS(N-溴代丁二酰亚胺)、Br2、I2、ICl、IBr等中的任意一种;所述原料1与卤代试剂的摩尔比为1∶1.2~2;所述反应的温度为0℃~80℃;所述反应的时间为0.5h~8h。
b、中间体1在碱存在下与酰氯反应制备得到中间体2;所述的碱为NaH、KOH、NaOH、K2CO3、Na2CO3、Cs2CO3等中的任意一种;所述的酰氯为TsCl(对甲苯磺酰氯)、苯磺酰氯、甲磺酰氯等中的任意一种;所述中间体1与碱、酰氯的摩尔比为1∶2~4∶1~1.5;所述反应的温度为0℃~50℃;所述反应的时间为0.5h~4h。
c、中间体2在过渡金属催化下,与硼酸酯试剂在碱存在下反应制备得到中间体3;所述的过渡金属为Pd4(PPh3)4、PdAc2、Pd2(dba)3、Pd(PPh3)2Cl2、Pd(PPh3)2Cl2DCM等中的任意一种;所述的硼酸酯试剂为联硼酸频那醇酯等;所述的碱为KOAc、K2CO3、Na2CO3、Cs2CO3等中的任意一种;所述中间体2与过渡金属、硼酸酯试剂、碱的摩尔比为1∶0.05~0.2∶1.5~2∶2~4;所述反应的温度为60℃~90℃;所述反应的时间为0.5h~24h。
d、原料2或中间体4与原料3在碱存在下反应制备得到中间体5;所述的碱为DIEA(N,N-二异丙基乙胺)、TEA(三乙胺)、KOAc、K2CO3、Na2CO3、Cs2CO3等中的任意一种;所述中间体4与原料3、碱的摩尔比为1∶1~1.2∶2~4;所述反应的温度为室温~90℃;所述反应的时间为5h~48h。若中间体5含有异构体,则采用制备SFC或制备HPLC进行手性拆分。
其中中间体4的制备为:若原料2中R2包含-COOH,需经酸或二氯亚砜(SOCl2)催化成酯反应制备得到中间体4;所述的酸为浓硫酸等;所述原料2与酸或SOCl2的摩尔比为1∶5~15;所述反应的温度为0℃~90℃;所述反应的时间为4h~24h。
e、中间体5与中间体3在过渡金属催化下、碱存在下发生偶联反应,制备得到中间体6;所述的过渡金属为Pd4(PPh3)4、PdAc2、Pd2(dba)3、Pd(PPh3)2Cl2、Pd(PPh3)2Cl2DCM等中的任意一种;所述的碱为Cs2CO3、KOAc、K2CO3、Na2CO3、KOH、NaOH等中的任意一种;所述中间体5与中间体3、过渡金属、碱的摩尔比为1∶1~1.2∶0.05~0.2∶2~4;所述反应的温度为室温~90℃;所述反应的时间为5h~48h。
f、中间体6在碱存在下,0~50℃反应脱去保护基R和上述中间体4引入的酯保护,可制备得到化合物7;所述的碱为KOH、NaOH、甲醇钠、乙醇钠等中的任意一种;所述中间体6与碱的摩尔比为1∶4~10;所述碱性反应的时间为5h~24h。
本发明上述抗流感小分子化合物包括其互变异构体、立体异构体及其所有比例的混合物,还包括其同位素取代的化合物。
本发明还提供了上述抗流感小分子化合物药学上可接受的盐。
本发明所用的术语“药学上可接受的”是指在在合理的医学判断范围,能适于用来与人类和其他哺乳动物的组织接触,而没有不当毒性、刺激、过敏反应等,其在对受者给药时能直接或间接地提供本发明的化合物或化合物的前药。
本发明还提供了上述抗流感小分子化合物药学上可接受的水合物。术语“水合物”表示进一步通过非共价分子间作用力结合化学计量或非化学计量的水的化合物。
本发明还提供了上述抗流感小分子化合物药学上可接受的多晶型物。术语“多晶型物”表示化合物或其复合物的固体结晶形式,其可以通过物理方法,例如X.射线粉末衍射图或红外光谱进行表征。
本发明还提供了上述抗流感小分子化合物药学上可接受的药物组合物,这种药物组合物是由式I所示的抗流感小分子化合物及其盐或水合物添加药学上可以接受的辅助性成分制备而成的。
上述药物组合物可以为液体形式或固体形式。其中,所述的液体形式可以为水溶液形式。所述的固体形式可以为粉末、颗粒、片剂或冻干粉形式。该药物组合物还含有注射用水、盐水溶液、葡萄糖水溶液、注射/输注用盐水、注射/输注用葡萄糖、格林氏溶液或含有乳酸盐的格林氏溶液。
式I所示的抗流感小分子化合物及其盐、水合物或药物组合物在制备抗流感药物中的用途。
本发明还提供了上述式I所示的抗流感小分子化合物及其盐、水合物或药物组合物在制备口服或静脉注射制剂中的用途。所述的口服或静脉注射制剂至少包含一种式I所示的抗流感小分子化合物及其盐、水合物或药物组合物以及任意的赋形剂和/或佐剂。
本发明的有益效果在于:首先,本系列化合物在体外对流感病毒具有很强的抑制效果,在抗流感活性测试中,大部分化合物对流感病毒H1N1(A/PR/8/34)的EC50(半数有效浓度)达到了纳摩尔级水平,活性最优的化合物7z对流感病毒H1N1感染导致的小鼠死亡和肺部炎症有显著保护和抑制效果作用。其次,本系列化合物毒性较小,大部分化合物对MDCK细胞(马丁狗肾细胞)的CC50(半数毒性浓度)在10uM以上,化合物选择性较好。再次,本系列化合物针对的是靶标是流感RNA聚合酶(RdRP)的PB2亚基(碱性蛋白2),该亚基是近年来的开发抗流感药物的热点靶标,但是目前尚无上市药物。针对该靶标的抑制剂,可能克服目前临床使用的神经氨酸酶抑制剂和M2离子通道抑制剂等药物的耐药性、疗效差、治疗窗窄等问题,具有较大的开发价值。
附图说明
图1化合物7j的ITC测试结果:得到7j与PB2318-483蛋白的Kd值为0.87~1.6μM,ΔH为-14530±782,8cal/mol,ΔS为-21.5cal/mol/deg。
具体实施方式
实施例1:3-碘-5-氯-1H-吡咯并[2,3-b]吡啶(中间体1a)的制备

向500mL圆底烧瓶中加入5-氯-1H-吡咯并[2,3-b]吡啶(5g,32.77mmol),用150mL丙酮溶解完全,分批加入NIS(8.86g,39.4mmol),常温搅拌,10min后有白色固体析出,30min后反应完毕,减压蒸馏除去溶剂,加入1M的硫代硫酸钠水溶液20mL,常温搅拌,滤出固体,用水洗涤3次,干燥后重结晶得到白色固体,产率91%。
1H NMR(400MHz,DMSO-d6)12.35(s,1H),8.30(d,J=2.0Hz,1H),7.94(d,J=1.6Hz,1H),7.84(d,J=2.0Hz,1H)ppm;ESI-MS m/z:278.6[M+H]+。
按照中间体1a类似的制备方法,以5-X-1H-吡咯并[2,3-b]吡啶为原料,以NIS或NBS等为卤代试剂,以DMF、THF、丙酮等为溶剂,常温、低温或加热反应,可得到中间体1b-e。
表1中间体1b-e的结构、1H NMR和ESI-MS

实施例2:3-碘-5-氯-1-对甲苯磺酰基-吡咯并[2,3-b]吡啶(中间体2a)的制备

向250mL圆底烧瓶中加入3-碘-5-氯-1H-吡咯并[2,3-b]吡啶(8.35g,30mmol),溶于50mL无水四氢呋喃,冰浴,搅拌,缓慢分批加入NaH(1.44g,60mmol),有气泡产生。15min后,缓慢滴加TsCl(6.86g,36mmol)四氢呋喃溶液20mL,30min后滴加完毕,移至室温反应30min。反应完毕后,减压蒸馏除去溶剂,得到固体粗品,加入40mL二氯甲烷溶解粗品,用水萃取3次,分离出DCM层,用无水硫酸镁干燥,过滤,蒸干滤液即得到产品,浅黄色固体,产率99%;ESI-MS m/z:432.9[M+H]+。
按照中间体2a类似的制备方法,3-卤-5-X-1H-吡咯并[2,3-b]吡啶为原料,与对甲苯磺酰氯、苯磺酰氯等反应,可得
表2中间体2b-g的结构、1H NMR和ESI-MS


实施例3:3-硼酸频哪醇酯-5-氯-1-对甲苯磺酰基-吡咯并[2,3-b]吡啶(中间体3a)的制备

向50mL双颈瓶中加入3-碘-5-氯-1-对甲苯磺酰基-吡咯并[2,3-b]吡啶(10.7g,25mmol)、醋酸钾(4.9g,50mmol)、PdCl2(dppf)DCM(2.04g,2.5mmol)以及联硼酸频哪醇酯(7.62g,30mmol),溶于20mL的1,4-二氧六环中,抽真空,氮气置换3次,80℃反应过夜。反应完毕后,减压蒸馏除去溶剂,重新加入DCM溶解,用弗洛里硅土过滤,滤液蒸干后,用正己烷和甲基叔丁基醚重结晶可得到产品3a,黄棕色固体,产率52%。
1H NMR(400MHz,CDCl3):8.36(d,J=1.6Hz,1H),8.17(d,J=1.6Hz,2H),8.09(m,2H),7.30(m,2H),2.39(s,3H),1.37(s,12H)ppm;ESI-MS m/z:433.1[M+H]+。
按照中间体3a类似的制备方法,可以得到中间体3b-e。
表3中间体3b-e的结构、1H NMR和ESI-MS

实施例4:(S)-3-氨基丁酸甲酯盐酸盐(中间体4a)的合成

将(S)-3-氨基丁酸(500mg,4.85mmol)分散于20mL甲醇中,冰浴,缓慢滴加3mL二氯亚砜,固体逐渐消失,反应液变澄清。滴加完毕后,移至80℃反应过夜。反应完全后,旋干溶剂,得到无色油状物即为产品,产率约100%。
1H NMR(400MHz,CDCl3)δ8.48(br s,2H),7.73(br s,1H),,3.84~3.70(m,1H),3.75(s,3H),2.92(dd,J=17.2,7.2Hz,1H),2.77(dd,J=17.2,4.8Hz,1H),1.53(d,J=6.4Hz,3H)ppm;ESI-MS m/z:118.0[M+H]+。
按照中间体4a类似的制备方法,可以得到中间体4b-j。
表4中间体4b-j的结构、1H NMR和ESI-MS


实施例5:(1S,2S)-2-氨基环己甲酸甲酯(中间体4k)的合成

将(1S,2S)-2-氨基环己甲酸(500mg,3.49mmol)溶于15mL甲醇,冰浴下缓慢加入2mL浓硫酸,滴加完毕后,升温至80℃回流过夜。反应完毕后,蒸干溶剂,加入10mL冰水,用稀氢氧化钠溶液调节pH至8~9,用DCM萃取水层3次,合并有机层,用无水硫酸镁干燥,过滤,蒸干滤液即得到产品,无色油状物,产率96%。
1H NMR(400MHz,DMSO-d6)δ3.59(s,3H),2.63(td,J=10.8,4.0Hz,1H),2.01(ddd,J=12.0,10.0,3.6Hz,1H),1.84~1.71(m,2H),1.71~1.57(m,2H),1.51(br s,2H),1.39~0.96(m,4H)ppm;ESI-MS m/z:158.3[M+H]+。
按照中间体4k类似的制备方法,可得到中间体4l。
表5中间体4l的结构、1H NMR和ESI-MS

实施例6:(S)-3-((2-氯呋喃并[3,2-d]嘧啶-4-基)氨基)丁酸甲酯(中间体5a)的制备

将2,4-二氯呋喃并[3,2-d]嘧啶(250mg,1.32mmol)、DIEA(0.87mL,5.28mmol)和中间体4a(203mg,1.32mmol)用20mL甲醇溶解完全,80℃回流过夜,反应完毕后,减压蒸馏除去溶剂,拌样,柱层析,以PE∶EA=3∶1洗脱得到产品,白色固体,产率60%。
1H NMR(400MHz,CDCl3)δ7.73(d,J=2.0Hz,1H),6.78(d,J=2.0Hz,1H),5.95(d,J=2.8Hz,1H),4.79(ddt,J=9.2,6.8,5.2Hz,1H),3.72(s,3H),2.70(qd,J=16.0,5.2Hz,2H),1.40(d,J=6.8Hz,3H)ppm;ESI-MS m/z:270.1[M+H]+。
按照中间体5a类似的制备方法,以含不同取代基的胺类化合物与氯代嘧啶类衍生物为原料,以DIEA、碳酸钠、碳酸铯等有机碱或无机碱作碱,乙醇、乙腈、四氢呋喃等作溶剂,常温或加热反应,得到中间体5b-5z、5aa-af,表征数据如下:
表6中间体5b-5z、5aa-af的结构、1H NMR和ESI-MS






实施例7:(R)-3-((2-氯-5-甲苯磺酰-5H-吡咯并[3,2-d]嘧啶-4-基)氨基)-4,4-二甲基戊酸甲酯(中间体5ag)的制备

第一步,制备2,4-二氯-5-对甲苯磺酰-5H-吡咯并[3,2-d]嘧啶:
将2,4-二氯吡咯并[3,2-d]嘧啶(500mg,2.67mmol)溶于15mL无水THF,冰浴,缓慢分批加入NaH(64mg,5.34mmol)。15min后,缓慢滴入TsCl(510mg,2.67mmol)的THF溶液10mL,30min后滴加完毕,室温反应1h。减压蒸馏除去溶剂,残留物用DCM溶解,用水萃取三次,DCM层用无水硫酸镁干燥,过滤除去硫酸镁,蒸干滤液,即得到中间体2,4-二氯-5-对甲苯磺酰-5H-吡咯并[3,2-d]嘧啶,白色固体,产率98%。
1H NMR(400MHz,DMSO-d6)δ8.69(d,J=4.0Hz,1H),7.92(d,J=8.4Hz,2H),7.49(d,J=8.0Hz,2H),7.15(d,J=4.0Hz,1H),2.40(s,3H)ppm。
第二步,制备(R)-3-((2-氯-5-甲苯磺酰-5H-吡咯并[3,2-d]嘧啶-4-基)氨基)-4,4-二甲基戊酸甲酯:
将上一步得到的中间体(342mg,1.0mmol)用20mL的MeCN溶解,加入三乙胺(202mg,2mmol)和中间体4d(235mg,1.2mmol),80℃回流过夜。反应完毕后,减压蒸干溶剂,残留物拌样过柱,以PE∶EA=4∶1洗脱得到产物5ag,白色固体,产率43%。
1H NMR(400MHz,DMSO-d6)δ8.12(d,J=4.0Hz,1H),7.71(d,J=8.4Hz,2H),7.65(d,J=8.0Hz,1H),7.44(d,J=8.0Hz,2H),6.82(d,J=4.0Hz,1H),4.63(td,J=10.0,3.2Hz,1H),3.47(s,3H),2.84(dd,J=14.8,3.2Hz,1H),2.42(dd,J=14.8,10.0Hz,1H),2.36(s,3H),0.98(s,9H)ppm。ESI-MS m/z:465.1[M+H]+。
实施例8:(R)-3-((2-氯-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)氨基)-4,4-二甲基戊酸甲酯(中间体5ah)的制备

第一步,制备2-脲基环戊-1-烯-1-羧酸甲酯:
将甲基-2-氧代环戊烷-1-羧酸(5g,35.17mmol)用25mL甲醇溶解,加入尿素(4.225g,70.3mmol)以及2mL浓盐酸,80℃回流,10min后有白色固体析出,回流过夜。反应完全后,过滤得到中间体2-脲基环戊-1-烯-1-羧酸甲酯,产率90%。1H NMR(400MHz,DMSO-d6)δ9.41(s,1H),6.75(s,2H),3.64(s,3H),3.02(t,J=7.6Hz,2H),2.38(t,J=7.6Hz,2H),1.82-1.70(m,2H)ppm。
第二步,制备2,4-二氯-6,7-二氢-5H-环戊二烯并[d]嘧啶:
将上一步得到的中间体用10mL三氯氧磷溶解,80℃回流,反应液黄棕色。5h反应完全后,减压蒸馏除去溶剂,得到棕色油状物,加入10mL冰水,立即有棕色固体析出,过滤,干燥即得中间体2,4-二氯-6,7-二氢-5H-环戊二烯并[d]嘧啶,黄棕色固体,产率54%。
1H NMR(400MHz,DMSO-d6)δ3.04(t,J=7.6Hz,2H),2.93(t,J=7.6Hz,2H),2.20-2.07(m,2H)ppm;ESI-MS m/z:188.9[M+H]+。
第三步,制备(R)-3-((2-氯噻吩并[3,2-d]嘧啶-4-基)氨基)-4,4-二甲基戊酸甲酯:
将上一步得到的中间体(500mg,2.64mmol)用20mL乙腈溶解,加入DIEA(0.8ml,5.28mmol)和中间体4d(442mg,2.77mmol),升温至80℃反应。24h后,反应完毕,拌样柱层析,以PE∶EA=4∶1洗脱得到中间体5ah,白色固体,产率31%。
1H NMR(400MHz,DMSO-d6)δ6.88(d,J=9.2Hz,1H),4.50(t,J=8.8Hz,1H),3.49(s,3H),2.76-2.65(m,3H),2.65-2.57(dd,J=11.2,3.2Hz,3H),2.10-1.92(m,2H),0.88(s,9H)ppm;ESI-MS m/z:312.1[M+H]+。
实施例9:中间体5ai-ao的合成

按照实施例6所述的合成方法,可得到5ai-ao的消旋体,再用制备级超临界流体色谱进行手性拆分,可得到中间体5ai-ao,表征数据如下:
表7中间体5ai-ao的结构、1H NMR和ESI-MS



实施例10:(S)-3-((5-氯-1-对甲苯磺酰基-1H-吡咯并[2,3-d]吡啶-3-基)呋喃并[3,2-d]嘧啶-4-基)氨基)-丁酸甲酯(中间体6a)的合成

将(R)-3-((2-氯呋喃并[3,2-d]嘧啶-4-基)氨基)-丁酸甲酯5a(160mg,0.593mmol)、中间体3a(308mg,0.7119mmol)、碳酸铯(390mg,1.2mmol)和PdCl2(dppf)DCM(49mg,0.06mmol)用10mL的1,4-二氧六环溶解,加入2mL水,抽真空,氮气置换三次,置于80℃反应,6h后反应完毕。减压蒸馏除去溶剂,残留物用DCM溶解,拌样,柱层析,以PE∶EA=4∶1洗脱得到中间体6a,白色固体,产率85%。
1H NMR(400MHz,DMSO-d6)8.94(d,J=2.0Hz,1H),8.49(s,1H),8.48(d,J=2.4Hz,1H),8.28(d,J=2.4Hz,1H),8.07(d,J=8.4Hz,2H),7.99(d,J=9.2Hz,1H),7.45(d,J=8.4Hz,2H),7.08(d,J=2.0Hz,1H),4.83(dt,J=14.0,7.2Hz,1H),3.55(s,3H),2.81(dd,J=15.2,7.2Hz,1H),2.63(dd,J=15.2,6.6Hz,1H),2.36(s,3H),1.35(d,J=6.6Hz,3H)ppm;ESI-MS m/z:540.1[M+H]+。
按照中间体6a类似的制备方法,可以得到中间体6b-6z、6aa-6az和6ba-6bb,表征数据如下:
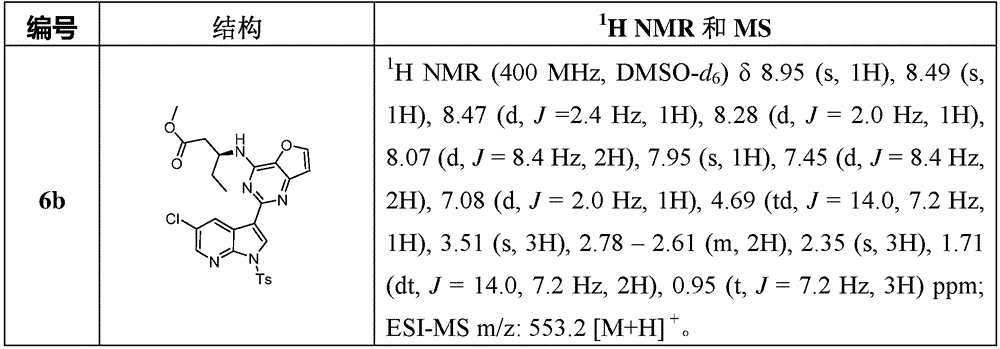
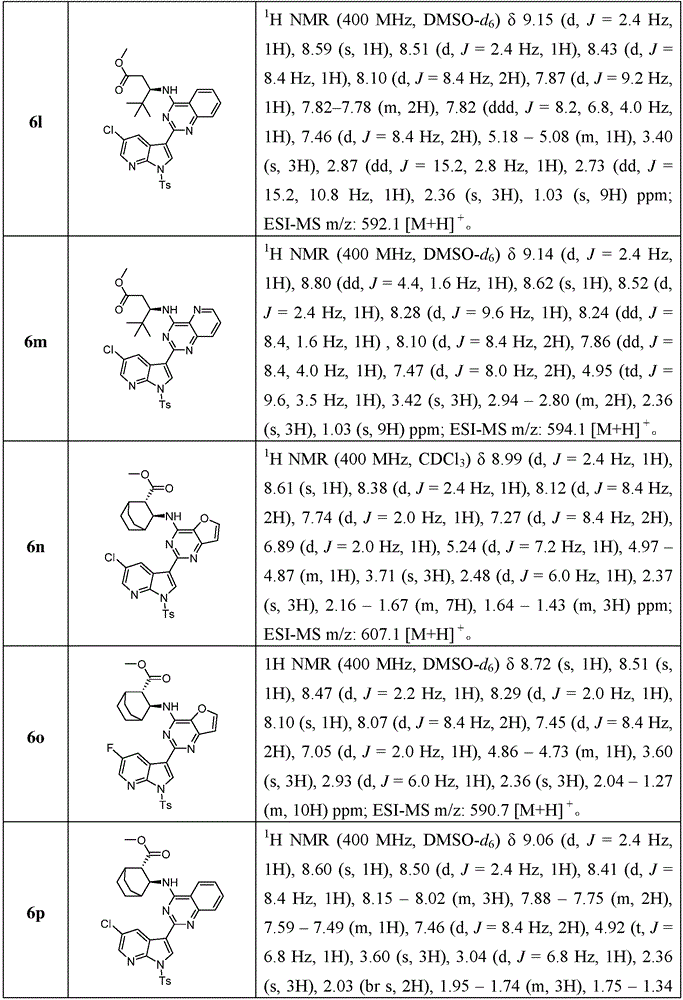
表8中间体6b-6z、6aa-6az和6ba-6bb的结构、1H NMR和ESI-MS











实施例11:(S)-3-((2-(5-氯-1H-吡咯并[2,3-b]吡啶-3-基)呋喃并[3,2-d]嘧啶-4-基)氨基)丁酸(化合物7a)的制备

将得到的中间体6a(80mg,0.15mmol)用MeOH∶THF=1∶1溶解完全,加入3mL饱和氢氧化钠水溶液,50℃搅拌过夜。反应完全后,减压蒸馏除去溶剂,加入5mL水,EA萃取一次,弃去有机层。水层用盐酸调pH至7,有大量白色固体析出,过滤,用水洗涤所得固体3次,干燥,重结晶即得产物7a,近白色固体,产率81%。
1H NMR(400MHz,DMSO-d6)δ15.04(br s,1H),12.94(s,1H),9.45(br s,1H),8.94(s,1H),8.78(s,1H),8.49(s,1H),8.40(d,J=2.0Hz,1H),7.16(d,J=1.6Hz,1H),5.01~4.81(m,1H),2.82(dd,J=16.0,7.2Hz,1H),2.72~2.56(m,1H),1.41(d,J=6.6Hz,3H)ppm;ESI-MS m/z:370.6[M-H]-。
化合物7b-z、7aa-7az及7ba-bb用上述类似方法可得到,其表征数据如表9所示:
表9化合物7b-z、7aa-7az及7ba-bb的结构、1H NMR和ESI-MS











实施例12:化合物的抗流感活性测试
实验采用测定流感病毒神经氨酸酶(NA)活性来检测病毒的复制水平。
实验材料:MUNANA(2′-(4-Methylumbellifery)-α-D-N-acetylneuraminic acid,2′-(4-甲基伞型酮)-α-D-N-乙酰神经氨酸)、MDCK细胞(马丁狗肾细胞)、流感病毒H1N1(A/PR/8/34)、利巴韦林(Ribavirin)均来源于武汉威立得生物医药有限公司。VX-787((2S,3S)-3-((5-氟-2-(5-氟-1H-吡咯[2,3-b]吡啶-3-基)嘧啶-4-基)氨基)二环[2.2.2]辛烷-2-羧酸)来源于四川大学生物治疗国家重点实验室。

实验原理:流感病毒NA是一种具有酶活性的表面糖蛋白,在病毒感染末期,能水解HA-SA糖苷键,切断病毒与宿主细胞的联系,对流感病毒的释放与传播有重要作用。MUNANA是流感病毒NA的特异性荧光底物,在NA作用下产生的催化产物在激发光照射下,可以产生荧光,荧光强度的变化,可以灵敏反映NA活性与数量,间接反映流感病毒增殖情况。
方法步骤:MDCK细胞(马丁狗肾细胞)接种于96孔细胞培养板中,37℃培养过夜后备用。MDCK细胞用PBS洗两遍后同时加入药物(本发化合物)及流感病毒H1N1(A/PR/8/34),药物设置八个浓度,两个复孔。加药完毕后,置于37℃细胞培养箱培养24h,显微镜下观察细胞病变(CPE),并取培养液上清进行神经氨酸酶活力检测。实验设置空白对照孔(正常MDCK细胞),病毒对照孔(病毒感染后未加药物),阳性药物对照孔(感染后加利巴韦林或者VX-787)。
抑制率(%)=100-(样品孔-空白对照)/(病毒对照-空白对照)*100%
最后用Graphpad Prism软件拟合得出半数有效浓度(EC50)。
实施例13:药物对MDCK细胞的毒性
采用了 (Invitrogen)试剂盒检测药物对细胞的毒性作用。
(Invitrogen)试剂盒检测药物对细胞的毒性作用。
实验原理: 是一种氧化还原指示剂,能根据代谢活性产生吸光度变化和荧光信号。
是一种氧化还原指示剂,能根据代谢活性产生吸光度变化和荧光信号。 易溶于水,其氧化形式进入细胞后经线粒体酶还原产生可测量的荧光及颜色变化,用于细胞活性和细胞增殖的定量分析以及体外细胞毒性研究。这种测定是基于具有代谢活性的细胞将试剂转换成荧光和比色指示剂的能力,受损和无活性细胞具有较低的天然代谢活性,对应的信号较低。因此荧光信号强弱,可以反映细胞活性的高低。
易溶于水,其氧化形式进入细胞后经线粒体酶还原产生可测量的荧光及颜色变化,用于细胞活性和细胞增殖的定量分析以及体外细胞毒性研究。这种测定是基于具有代谢活性的细胞将试剂转换成荧光和比色指示剂的能力,受损和无活性细胞具有较低的天然代谢活性,对应的信号较低。因此荧光信号强弱,可以反映细胞活性的高低。
方法步骤:MDCK细胞接种于96孔细胞培养板中,细胞贴壁后加入含药培养基。药物终浓度为100μM、33.33μM、11.11μM、3.70μM、1.23μM、0.41μM、0.14μM、0.046μM共八个浓度,两个复孔。加药培养后,光镜下观察药物引起的细胞病变效应(CPE),加入 37℃孵育2h,荧光检测
37℃孵育2h,荧光检测 的还原情况,激发光570nm,发射光595nm。
的还原情况,激发光570nm,发射光595nm。
细胞活性(%)=(样品孔-空白对照)/(细胞对照-空白对照)*100%
最后用Graphpad Prism软件拟合得出细胞半数抑制浓度(CC50)。
表10为测试化合物对流感病毒H1N1(A/PR/8/34)的抑制活性以及细胞毒性,其中,A表示<100nM,B表示100nM~500nM,C表示500nM~1μM,D表示1μM~10μM,E表示>10μM。
表10化合物对流感病毒H1N1(A/PR/8/34)的抑制活性以及细胞毒性



由上表可知,该系列化合物对流感病毒H1N1(A/PR/8/34)具有很好的抑制活性。EC50均明显优于阳性对照利巴韦林。大部分化合物与VX-787相比,EC50、CC50处于相同数量级水平。
实施例14:测定PB2318-483蛋白与小分子之间的Kd值
实验目的:检测化合物7j与PB2318-483蛋白的结合能力。选择等温滴定量热仪(ITC)测定小分子和蛋白的Kd值。
实验材料:PB2318-483蛋白(流感RNA聚合酶PB2亚基的318-483肽段,即“帽子”结合域)来源于四川大学生物治疗国家重点实验室。
实验步骤:
1)利用过分子筛的磷酸盐缓冲液和DMSO(二甲基亚砜),配制不同浓度的PB2318-483蛋白溶液和小分子溶液。蛋白溶液的终浓度分别为5μM、10μM、20μM、50μM,DMSO含量为1%。再根据蛋白和小分子浓度为1∶10的比例配制小分子溶液。
2)取3S0μL蛋白溶液和80μL小分子溶液,13000rpm离心2min。将蛋白溶液缓慢的加到ITC的样品池内,防止产生气泡;将小分子溶液加入到滴定针内,开始滴定。设置滴定条件为25℃;第一滴0.5μL,2s,其余每滴2μL,4s,共20滴;间隔150s。滴定完毕后,分析滴定结果。
实验结果:图1为化合物7j的ITC测试结果,得到7j与PB2318-483蛋白的Kd值为0.87~1.6μM,ΔH(焓变)为-14530±782.8cal/mol,ΔS(熵变)为-21.5cal/mol/deg,其中ΔH反映小分子与蛋白的特异性结合能力,ΔS反映溶剂效应以及化合物构象转变等。由数据可知化合物7j能特异性结合PB2318-483蛋白,Kd值处于微摩尔级水平。
实施例15:化合物的药代动力学研究
实验目的:单次给予SD大鼠受试药物,于不同时间点采集血样,LC-MS/MS测定给予受试物后大鼠血浆中受试物的浓度并计算相关参数。
实验材料:SPF级SD大鼠(雄性,6周,体重160-180g)来源于上海西普尔-必凯实验动物有限公司。受试药物最终配置浓度为1mg/mL,溶媒为5%DMSO+10%(乙醇∶蓖麻油=1∶1)+85%saline。
实验步骤:
1)所有动物给药前禁食10-14小时,SD大鼠单次静脉注射和口服给予受试药物,给药后4小时恢复给食。
2)样品采集与处理:经颈静脉穿刺采血,每个样品采集约0.20mL,肝素钠抗凝,采血时间点如下:静脉给药组采血时间:给药后2min,5min,15min,30min,1h,2h,3h,4h,6h,8h,12h,24h,36h,48h,72h。口服给药组采血时间:给药后5min,10min,15min,30min,1h,2h,3h,4h,6h,8h,12h,24h,6h,48h,72h。血液样本采集后置于冰上,离心分离血浆(离心条件:8000转/分钟,6分钟,2-8℃,收集的血浆分析前存放于-80℃。
3)药物代谢动力学分析:根据药物的血药浓度数据,使用药代动力学计算软件WinNonlin5.2非房室模型分别计算供试品的药代动力学参数。
实验结果:以7z为例,其药代动力学参数如表11所示。
表11 SD大鼠单次静脉注射或口服7z的主要药代动力学参数

实施例16:化合物对甲型流感病毒A/FM/1/47(H1N1)感染小鼠的保护作用
实验材料:甲型流感病毒鼠肺适应株A/FM/1/47(H1N1),接种鸡胚,收集尿囊液保存。ICR小鼠,体重18~22g,来源于扬州大学比较医学中心,许可证号:SCXK(苏)2012-0004。给药期间自由进食、饮水,每天12小时光照,12小时黑暗,温度22±2℃,湿度55-70%。
实验方法:适应性饲养3天后,开始进行实验。除未感染对照组以外,其它各组小鼠用乙醚轻度麻醉,鼻腔内接种用生理盐水稀释的相当于8×LD50的流感病毒A/FM/1/47(H1N1)的鸡胚尿囊液50μL/只,阳性对照奥司他韦组小鼠于感染后2h首次灌胃给药,给药剂量为100mg/kg,供试给药组于病毒感染24h后以40mg/kg首次灌胃给药,以后每日1次,病毒对照组及未感染对照组同法口服生理盐水,每日1次,给药体积为0.1mL/10g体重。共7天。第5天,记录体重损失百分比。自感染之日起,连续14d,记录小鼠每日体重、死亡数、死亡时间,计算死亡保护率。
死亡保护率(%)=病毒对照组死亡率(%)-实验组死亡率(%)。
实施例17:药物对流感病毒H1N1感染导致的小鼠肺部炎症的缓解作用
实验方法:适应性饲养3天后,开始进行实验。除未感染对照组以外,其它各组小鼠用乙醚轻度麻醉,鼻腔内接种用生理盐水稀释的相当于8×LD50的流感病毒A/FM/1/47(H1N1)的鸡胚尿囊液50μL/只,阳性对照奥司他韦组小鼠于感染后2h首次灌胃给药,给药剂量为100mg/kg,供试给药组于病毒感染24h后以40mg/kg首次灌胃给药,以后每日1次,病毒对照组及未感染对照组同法口服生理盐水,每日1次,给药体积为0.1mL/10g体重。共5天。第6天每组取3只小鼠称重,摘除眼球放血致死,取出全肺,称重,计算肺指数及肺指数抑制率。
肺指数=小鼠肺重/小鼠体重×100

表12化合物7z对流感病毒感染小鼠体重、生存率及肺部炎症的影响

实验结果:以7z为例,口服给药40mg/kg/d,小鼠存活率达到了90%;在第五天,病毒对照组小鼠平均重量损失30%,化合物7z治疗组平均重量损失10%;小鼠的肺指数为1.16,与病毒对照组(2.09)相比有明显降低,肺指数抑制率达到了60.99%。因此化合物7z对流感病毒引起的死亡及肺部炎症有明显的保护和抑制作用,且效果优于奥司他韦对照组。
Claims (17)
1.抗流感小分子化合物,其结构如式Ⅰ所示:
其中,X为-H、卤素、氰基、-CF3、C1~C4烷氧基或C1~C4烷基;
L为不饱和的4~10元杂环烷基;所述不饱和的4~10元杂环烷基的杂原子为O或S,所述杂原子的个数为1;
R1和R2组合成取代或未取代的5~8元饱和杂环烷基,所述取代或未取代的5~8元饱和杂环烷基的杂原子为N,杂原子个数为1个;所述取代5~8元饱和杂环烷基的取代基为 a、b为0~4;
a、b为0~4;
或者R1为-H或C1~C10烷基;R2为 n为0~4;R4、R5独立的为-H或C1~C10烷基;或者R4和R5组合形成环,所述的环为C5~C10环烷基、C5~C10的桥环烷基或C5~C10的稠环烷基;R3为
n为0~4;R4、R5独立的为-H或C1~C10烷基;或者R4和R5组合形成环,所述的环为C5~C10环烷基、C5~C10的桥环烷基或C5~C10的稠环烷基;R3为 R9为C1~C5烷基;
R9为C1~C5烷基;
R6、R7独立的为C1~C4烷基、取代或未取代的C5~C10芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C10芳基或苄基的取代基为C1~C6烷基、C1~C6烷氧基、卤素、-CF3或氰基;a、b为0~4;
或-CONH2;所述取代C5~C10芳基或苄基的取代基为C1~C6烷基、C1~C6烷氧基、卤素、-CF3或氰基;a、b为0~4;
R8为取代或未取代的C5~C10环烷基、取代或未取代的5~10元饱和或不饱和杂环、取代或未取代的C5~C10不饱和环或取代或未取代的C5~C15的桥环烷基;所述5~10元饱和或不饱和杂环的杂原子为N、O、S,杂原子个数为1~3个;所述取代C5~C10环烷基的取代基为 卤素、C1~C4烷基或-COOH;所述取代C5~C10不饱和环的取代基为-OH、-SO2NH2、-CF3、卤素、C1~C4烷基或-COOH;所述取代5~10元饱和或不饱和杂环的取代基为卤素取代的苄基、卤素、苯基、苄基、-OH、-CF3、C1~C4烷基、C1~C4羰基、-COOH或-NH2;所述取代C5~C15的桥环烷基的取代基为-OH、-COOH、C1~C4烷基或-NH2;a为0~4。
卤素、C1~C4烷基或-COOH;所述取代C5~C10不饱和环的取代基为-OH、-SO2NH2、-CF3、卤素、C1~C4烷基或-COOH;所述取代5~10元饱和或不饱和杂环的取代基为卤素取代的苄基、卤素、苯基、苄基、-OH、-CF3、C1~C4烷基、C1~C4羰基、-COOH或-NH2;所述取代C5~C15的桥环烷基的取代基为-OH、-COOH、C1~C4烷基或-NH2;a为0~4。
2.根据权利要求1所述的抗流感小分子化合物,当R1为H,R2为 R3为
R3为 时,其结构如式Ⅱ所示:
时,其结构如式Ⅱ所示:

其中,X为卤素或C1~C4烷基;L为不饱和的5~8元杂环烷基;所述5~8元杂环烷基的杂原子为O或S,所述的杂原子为1个;
R4、R5独立地为-H或C1~C8烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基或C5~C8的稠环烷基。
3.根据权利要求2所述的抗流感小分子化合物,其特征在于:
X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;R4、R5独立地为-H或C1~C4烷基;或R4和R5组合形成环,所述的环为C5~C8环烷基、C5~C8的桥环烷基或C5~C8的稠环烷基。
4.根据权利要求1所述的抗流感小分子化合物,其特征在于:当R1为H,R2为 时,其结构如式Ⅲ所示:
时,其结构如式Ⅲ所示:

其中,X为卤素或C1~C4烷基;L为不饱和的5~8元杂环烷基;所述5~8元杂环烷基的杂原子为O或S,所述的杂原子为1个;R6、R7独立的为C1~C4烷基、取代或未取代的C5~C8芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C8芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~3。
或-CONH2;所述取代C5~C8芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~3。
5.根据权利要求4所述的抗流感小分子化合物,其特征在于:
X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;R6、R7独立的为C1~C4烷基、取代或未取代的C5~C6芳基、取代或未取代的苄基、 或-CONH2;所述取代C5~C6芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~2。
或-CONH2;所述取代C5~C6芳基或苄基的取代基为C1~C4烷基、卤素、-CF3或氰基;a、b为0~2。
6.根据权利要求5所述的抗流感小分子化合物,其特征在于:
X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;R6、R7独立的为C1~C4烷基、取代或未取代的苯基、取代或未取代的苄基、 或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基或卤素;a、b为0~2。
或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基或卤素;a、b为0~2。
7.根据权利要求5所述的抗流感小分子化合物,其特征在于:
X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;R6、R7独立的为C1~C4烷基、取代或未取代的苯基、取代或未取代的苄基、 或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基、-F、-Cl或-Br;a、b为0~2。
或-CONH2;所述取代苯基或苄基的取代基为C1~C4烷基、-F、-Cl或-Br;a、b为0~2。
8.根据权利要求1所述的抗流感小分子化合物,其特征在于:当R1为H,R2为 时,其结构如式Ⅳ所示:
时,其结构如式Ⅳ所示:

其中,n为0~4,X为卤素或C1~C4烷基;L为不饱和的5~8元杂环烷基;所述5~8元杂环烷基的杂原子为O或S,所述的杂原子为1个;
R8为取代或未取代的C5~C8环烷基、取代或未取代的5~8元芳杂基、取代或未取代的5~8元饱和杂环烷基、取代或未取代的5~8元不饱和杂环烷基、取代或未取代的C5~C10芳基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~8元饱和或不饱和杂环烷基、5~8元芳杂基的杂原子为N、O、S,杂原子个数为1~3个;所述取代C5~C8环烷基的取代基为 所述取代5~8元芳杂基、5~8元饱和杂环烷基、5~8元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C4羰基;所述取代C5~C10芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、卤素、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH,a为0~3。
所述取代5~8元芳杂基、5~8元饱和杂环烷基、5~8元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C4羰基;所述取代C5~C10芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、卤素、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH,a为0~3。
9.根据权利要求8所述的抗流感小分子化合物,其特征在于:
n为0~4,X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;
R8为取代或未取代的C5~C6环烷基、取代或未取代的5~6元芳杂基、取代或未取代的5~6元饱和杂环烷基、取代或未取代的5~6元不饱和杂环烷基、取代或未取代的C5~C8芳基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~6元饱和或不饱和杂环烷基、5~6元芳杂基的杂原子为N、O、S,杂原子个数为1~2个;所述取代C5~C6环烷基的取代基为 所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C8芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH;a为0~3。
所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C8芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH;a为0~3。
10.根据权利要求9所述的抗流感小分子化合物,其特征在于:
n为0~3,X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;
R8为取代或未取代的C5~C6环烷基、取代或未取代的5~6元芳杂基、取代或未取代的5~6元饱和杂环烷基、取代或未取代的5~6元不饱和杂环烷基、取代或未取代的C5~C6芳基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~6元饱和或不饱和杂环烷基、5~6元芳杂基的杂原子为N、O、S,杂原子个数为1~2个;所述取代C5~C6环烷基的取代基为 所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C6芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH;a为0~3。
所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为卤素取代的苄基、卤素、苯基、苄基、-CF3或C1~C3羰基;所述取代C5~C6芳基的取代基为卤素、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH;a为0~3。
11.根据权利要求9所述的抗流感小分子化合物,其特征在于:
X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;
R8为取代或未取代的C5~C6环烷基、取代或未取代的5~6元芳杂基、取代或未取代的5~6元饱和杂环烷基、取代或未取代的5~6元不饱和杂环烷基、取代或未取代的苯基、取代或未取代的C5~C10不饱和环烷基或取代或未取代的C5~C12的桥环烷基;所述5~6元饱和或不饱和杂环烷基、5~6元芳杂基的杂原子为N、O,杂原子个数为1~2个;所述取代C5~C6环烷基的取代基为 所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为氟取代的苄基、-F、-Cl、-Br、苯基、苄基、-CF3或C1~C2羰基;所述取代苯基的取代基为-F、-Cl、-Br、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH;a为0~2。
所述取代5~6元芳杂基、5~6元饱和杂环烷基、5~6元不饱和杂环烷基的取代基为氟取代的苄基、-F、-Cl、-Br、苯基、苄基、-CF3或C1~C2羰基;所述取代苯基的取代基为-F、-Cl、-Br、-OH、-SO2NH2、-CF3或-COOH;所述取代C5~C10不饱和环烷基的取代基为-OH、-CF3或-COOH;所述取代C5~C12的桥环烷基的取代基为-OH或-COOH;a为0~2。
12.根据权利要求9所述的抗流感小分子化合物,其特征在于:
n为0~3,X为卤素;L为不饱和的5~6元杂环烷基;所述5~6元杂环烷基的杂原子为O或S,所述的杂原子为1个;
R8为

13.抗流感小分子化合物,其特征在于:其结构式为:


14.权利要求1~13任一项所述抗流感小分子化合物药学上可接受的盐。
15.药物组合物,这种药物组合物是由权利要求1~13任一项所述抗流感小分子化合物或权利要求14所述的盐添加药学上可以接受的辅助性成分制备而成的。
16.权利要求1~13任一项所述抗流感小分子化合物、权利要求14所述的盐或权利要求15所述的药物组合物在制备抗流感药物中的用途。
17.权利要求1~13任一项所述抗流感小分子化合物、权利要求14所述的盐或权利要求15所述的药物组合物在制备口服或静脉注射制剂中的用途。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610334744 | 2016-05-19 | ||
| CN2016103347446 | 2016-05-19 | ||
| PCT/CN2017/084301 WO2017198122A1 (zh) | 2016-05-19 | 2017-05-15 | 抗流感小分子化合物及其制备方法和用途 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109071567A CN109071567A (zh) | 2018-12-21 |
| CN109071567B true CN109071567B (zh) | 2021-03-23 |
Family
ID=60326421
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201780013432.8A Active CN109071567B (zh) | 2016-05-19 | 2017-05-15 | 抗流感小分子化合物及其制备方法和用途 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN109071567B (zh) |
| WO (1) | WO2017198122A1 (zh) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10501444B2 (en) | 2016-08-16 | 2019-12-10 | Sunshine Lake Pharma Co., Ltd. | Inhibitors of influenza virus replication, application methods and uses thereof |
| CN109641868B (zh) | 2016-08-30 | 2021-12-03 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其使用方法和用途 |
| CN108218873B (zh) | 2016-12-15 | 2020-07-07 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其用途 |
| CN108276401B (zh) | 2017-01-05 | 2020-12-22 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其用途 |
| CN110446711B (zh) | 2017-03-02 | 2022-02-15 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其用途 |
| WO2018200425A1 (en) | 2017-04-24 | 2018-11-01 | Cocrystal Pharma, Inc. | Pyrrolopyrimidine derivatives useful as inhibitors of influenza virus replication |
| SG11202109036WA (en) * | 2019-03-01 | 2021-09-29 | Revolution Medicines Inc | Bicyclic heteroaryl compounds and uses thereof |
| EP4355732A1 (en) * | 2021-06-15 | 2024-04-24 | Johnson Matthey Public Limited Company | Process for preparing pexidartinib |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005095400A1 (en) * | 2004-03-30 | 2005-10-13 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 (en) * | 2006-01-17 | 2007-07-26 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| WO2010148197A1 (en) * | 2009-06-17 | 2010-12-23 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| CN106854205A (zh) * | 2015-12-09 | 2017-06-16 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其使用方法和用途 |
-
2017
- 2017-05-15 CN CN201780013432.8A patent/CN109071567B/zh active Active
- 2017-05-15 WO PCT/CN2017/084301 patent/WO2017198122A1/zh active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005095400A1 (en) * | 2004-03-30 | 2005-10-13 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 (en) * | 2006-01-17 | 2007-07-26 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| WO2010148197A1 (en) * | 2009-06-17 | 2010-12-23 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| CN106854205A (zh) * | 2015-12-09 | 2017-06-16 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其使用方法和用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017198122A1 (zh) | 2017-11-23 |
| CN109071567A (zh) | 2018-12-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN109071567B (zh) | 抗流感小分子化合物及其制备方法和用途 | |
| JP6134338B2 (ja) | B型肝炎ウイルス共有結合閉環状dna形成の阻害剤およびそれらの使用方法 | |
| US11786533B2 (en) | Use of EZH2 inhibitors for treating cancer | |
| CN100432059C (zh) | 可用作抗痛觉过敏剂的喹唑啉酮衍生物 | |
| CN105566276B (zh) | 作为dpp-4抑制剂的苯并六元环衍生物及其应用 | |
| KR20190056380A (ko) | 항인플루엔자 바이러스 피리미딘 유도체 | |
| JP2009500451A (ja) | ピラノピリジン化合物 | |
| CN108473477A (zh) | 用于在流感病毒感染中使用的芳基取代的嘧啶 | |
| CN111253391A (zh) | 一种含氘氮杂环二酮化合物用于治疗流感 | |
| WO2022107745A1 (ja) | Covid-19の治療剤又は予防剤 | |
| WO2018223030A1 (en) | Use of ezh2 inhibitors for treating cancer | |
| CN112771048B (zh) | 流感病毒复制抑制剂及其中间体和用途 | |
| CN102633796B (zh) | 一种苦参酸类衍生物的制备方法 | |
| CN112724156B (zh) | 一种多环吡啶酮衍生物和药物组合物及其应用 | |
| CN109369623B (zh) | 一种取代1,2,3三氮唑类二芳基嘧啶衍生物及其制备方法与应用 | |
| CN102731296A (zh) | 迷迭香酸衍生物及其制备方法与在制备抗结核药物中的应用 | |
| CN113563319A (zh) | 具有磷酸二酯酶4b抑制活性的吲唑杂环类化合物 | |
| TW202146413A (zh) | 含吡啶酮稠環類衍生物抑制劑、其製備方法和應用 | |
| CN114621204B (zh) | 一种含有嘧啶二酮酰基多取代哌嗪类衍生物及其制备方法与应用 | |
| SK28298A3 (en) | Carboxylic acid derivatives, their preparation and their use | |
| JP2022516922A (ja) | フッ素含有置換ベンゾチオフェン化合物ならびにその医薬組成物および応用 | |
| CN115381823B (zh) | 一种周期蛋白依赖性激酶9抑制剂的用途 | |
| WO2024061048A1 (zh) | 一类石蒜碱衍生物、其药物组合物及其在制备抗病毒药物中的用途 | |
| EP2867213A1 (en) | Dihydropyrimidin-2(1h)-ones and dihydropyrimidin-2(1h)-thiones as inhibitors of sodium iodide symporter | |
| CN114890963B (zh) | 苯亚甲基噻唑烷二酮类衍生物及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |