CN107531695B - Jak抑制剂 - Google Patents
Jak抑制剂 Download PDFInfo
- Publication number
- CN107531695B CN107531695B CN201680021913.9A CN201680021913A CN107531695B CN 107531695 B CN107531695 B CN 107531695B CN 201680021913 A CN201680021913 A CN 201680021913A CN 107531695 B CN107531695 B CN 107531695B
- Authority
- CN
- China
- Prior art keywords
- reaction
- pyrazol
- cyanomethyl
- esi
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940122245 Janus kinase inhibitor Drugs 0.000 title abstract description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 74
- 150000003839 salts Chemical class 0.000 claims abstract description 47
- -1 1,3, 4-triazolyl Chemical group 0.000 claims description 207
- 229910052757 nitrogen Inorganic materials 0.000 claims description 47
- 125000005842 heteroatom Chemical group 0.000 claims description 32
- 125000003118 aryl group Chemical group 0.000 claims description 27
- 229910052799 carbon Inorganic materials 0.000 claims description 14
- 229910052717 sulfur Inorganic materials 0.000 claims description 14
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 11
- 229910052736 halogen Inorganic materials 0.000 claims description 10
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 8
- 150000002367 halogens Chemical class 0.000 claims description 8
- 125000003282 alkyl amino group Chemical group 0.000 claims description 7
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- 229910052794 bromium Inorganic materials 0.000 claims description 5
- 229910052801 chlorine Inorganic materials 0.000 claims description 5
- 229910052740 iodine Inorganic materials 0.000 claims description 5
- 125000006716 (C1-C6) heteroalkyl group Chemical group 0.000 claims description 4
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 4
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 4
- XZMCDFZZKTWFGF-UHFFFAOYSA-N Cyanamide Chemical compound NC#N XZMCDFZZKTWFGF-UHFFFAOYSA-N 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 4
- 125000002883 imidazolyl group Chemical group 0.000 claims description 3
- 125000002971 oxazolyl group Chemical group 0.000 claims description 3
- 125000000335 thiazolyl group Chemical group 0.000 claims description 3
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 217
- 238000006243 chemical reaction Methods 0.000 description 176
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 162
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 144
- 239000000243 solution Substances 0.000 description 105
- 238000002360 preparation method Methods 0.000 description 95
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 74
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 73
- 239000000203 mixture Substances 0.000 description 70
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 63
- 239000007787 solid Substances 0.000 description 63
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 59
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 58
- 238000005160 1H NMR spectroscopy Methods 0.000 description 56
- 229910001868 water Inorganic materials 0.000 description 54
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 53
- AIMMVWOEOZMVMS-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1 AIMMVWOEOZMVMS-UHFFFAOYSA-N 0.000 description 50
- 235000019439 ethyl acetate Nutrition 0.000 description 49
- 150000003852 triazoles Chemical class 0.000 description 46
- 239000000047 product Substances 0.000 description 40
- 239000012074 organic phase Substances 0.000 description 39
- 239000011541 reaction mixture Substances 0.000 description 39
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 38
- 239000000543 intermediate Substances 0.000 description 38
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 36
- 238000004364 calculation method Methods 0.000 description 35
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 35
- 238000005259 measurement Methods 0.000 description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 32
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 31
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 31
- 238000004809 thin layer chromatography Methods 0.000 description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 30
- 125000001424 substituent group Chemical group 0.000 description 28
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical group OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 27
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 26
- 238000001514 detection method Methods 0.000 description 25
- 239000000706 filtrate Substances 0.000 description 24
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 24
- 150000003254 radicals Chemical group 0.000 description 21
- 239000012043 crude product Substances 0.000 description 19
- 238000002953 preparative HPLC Methods 0.000 description 19
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- 239000002253 acid Substances 0.000 description 17
- 125000000217 alkyl group Chemical group 0.000 description 17
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 17
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 17
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 17
- 125000004429 atom Chemical group 0.000 description 16
- 238000000746 purification Methods 0.000 description 16
- 238000012746 preparative thin layer chromatography Methods 0.000 description 15
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 15
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 14
- 239000002585 base Substances 0.000 description 14
- 125000005605 benzo group Chemical group 0.000 description 14
- 238000001035 drying Methods 0.000 description 14
- 239000003208 petroleum Substances 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 13
- 125000001072 heteroaryl group Chemical group 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- 238000010898 silica gel chromatography Methods 0.000 description 13
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- 238000000034 method Methods 0.000 description 12
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 11
- 125000000623 heterocyclic group Chemical group 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 11
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 11
- QHOCXLIPZASCFO-UHFFFAOYSA-N 2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-c]pyrimidin-5-yl)pyrazol-1-yl]-1-cyclopropylsulfonylazetidin-3-yl]acetonitrile Chemical compound NC1=NN2C(=NC=CC2=N1)C=1C=NN(C=1)C1(CN(C1)S(=O)(=O)C1CC1)CC#N QHOCXLIPZASCFO-UHFFFAOYSA-N 0.000 description 10
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 10
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 239000007858 starting material Substances 0.000 description 10
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 10
- 239000000460 chlorine Substances 0.000 description 9
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- 230000035772 mutation Effects 0.000 description 9
- 125000004433 nitrogen atom Chemical group N* 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 8
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 8
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 8
- 239000013543 active substance Substances 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 229910052760 oxygen Inorganic materials 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- QUERMGFVJPRMJL-UHFFFAOYSA-N tert-butyl azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC1 QUERMGFVJPRMJL-UHFFFAOYSA-N 0.000 description 8
- GRGCWBWNLSTIEN-UHFFFAOYSA-N trifluoromethanesulfonyl chloride Chemical compound FC(F)(F)S(Cl)(=O)=O GRGCWBWNLSTIEN-UHFFFAOYSA-N 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- 125000000753 cycloalkyl group Chemical group 0.000 description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- PLRCVBKYFLWAAT-UHFFFAOYSA-N 3,3-difluorocyclobutane-1-carboxylic acid Chemical compound OC(=O)C1CC(F)(F)C1 PLRCVBKYFLWAAT-UHFFFAOYSA-N 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 6
- RZOUMGQHOPTVHG-UHFFFAOYSA-N ethyl 6-(bromomethyl)-5-ethyl-2-(trifluoromethyl)pyrimidine-4-carboxylate Chemical compound CCOC(=O)c1nc(nc(CBr)c1CC)C(F)(F)F RZOUMGQHOPTVHG-UHFFFAOYSA-N 0.000 description 6
- 125000000524 functional group Chemical group 0.000 description 6
- 125000004404 heteroalkyl group Chemical group 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- AWQVKAURKXXOCG-UHFFFAOYSA-N n-cyclopropylformamide Chemical compound O=CNC1CC1 AWQVKAURKXXOCG-UHFFFAOYSA-N 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 125000004434 sulfur atom Chemical group 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- ZYWSXGRMDPBISP-UHFFFAOYSA-N 1-nitro-2-phenylmethoxybenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1OCC1=CC=CC=C1 ZYWSXGRMDPBISP-UHFFFAOYSA-N 0.000 description 5
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 5
- 208000017733 acquired polycythemia vera Diseases 0.000 description 5
- 125000002252 acyl group Chemical group 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- AVFGMPNUNSNLGU-UHFFFAOYSA-N ethyl 6-[(dimethylamino)methyl]-5-ethyl-2-(trifluoromethyl)pyrimidine-4-carboxylate Chemical compound CCOC(=O)c1nc(nc(CN(C)C)c1CC)C(F)(F)F AVFGMPNUNSNLGU-UHFFFAOYSA-N 0.000 description 5
- 125000001183 hydrocarbyl group Chemical group 0.000 description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000012046 mixed solvent Substances 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 125000004430 oxygen atom Chemical group O* 0.000 description 5
- 208000037244 polycythemia vera Diseases 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- UHGULLIUJBCTEF-UHFFFAOYSA-N 2-aminobenzothiazole Chemical compound C1=CC=C2SC(N)=NC2=C1 UHGULLIUJBCTEF-UHFFFAOYSA-N 0.000 description 4
- AMUFZBGBFWPJAY-UHFFFAOYSA-N 6-[(dimethylamino)methyl]-2-(trifluoromethyl)pyrimidine-4-carboxylic acid Chemical compound CN(C)CC1=CC(C(O)=O)=NC(C(F)(F)F)=N1 AMUFZBGBFWPJAY-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 4
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 4
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 4
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 4
- 108010024121 Janus Kinases Proteins 0.000 description 4
- 102000015617 Janus Kinases Human genes 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 108091000080 Phosphotransferase Proteins 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- KVVMXWRFYAGASO-UHFFFAOYSA-N azetidine-1-carboxylic acid Chemical compound OC(=O)N1CCC1 KVVMXWRFYAGASO-UHFFFAOYSA-N 0.000 description 4
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 210000000845 cartilage Anatomy 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- MVEAAGBEUOMFRX-UHFFFAOYSA-N ethyl acetate;hydrochloride Chemical compound Cl.CCOC(C)=O MVEAAGBEUOMFRX-UHFFFAOYSA-N 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- HBNYJWAFDZLWRS-UHFFFAOYSA-N ethyl isothiocyanate Chemical compound CCN=C=S HBNYJWAFDZLWRS-UHFFFAOYSA-N 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 229940093915 gynecological organic acid Drugs 0.000 description 4
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 235000005985 organic acids Nutrition 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 102000020233 phosphotransferase Human genes 0.000 description 4
- 208000003476 primary myelofibrosis Diseases 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 239000002994 raw material Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 230000019491 signal transduction Effects 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- QRZNHJHKTCPFAO-UHFFFAOYSA-N tert-butyl 3-(cyanomethyl)azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(CC#N)C1 QRZNHJHKTCPFAO-UHFFFAOYSA-N 0.000 description 4
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- XBRVSIPVHYWULW-UHFFFAOYSA-N (3-bromophenyl)thiourea Chemical compound NC(=S)NC1=CC=CC(Br)=C1 XBRVSIPVHYWULW-UHFFFAOYSA-N 0.000 description 3
- IJAMXMAVDDCHHD-UHFFFAOYSA-N 1-bromo-3-nitro-2-phenylmethoxybenzene Chemical compound [O-][N+](=O)C1=CC=CC(Br)=C1OCC1=CC=CC=C1 IJAMXMAVDDCHHD-UHFFFAOYSA-N 0.000 description 3
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- KGCHVYYLIWLLFD-UHFFFAOYSA-N 3,3-difluorocyclobutane-1-carbonyl chloride Chemical compound FC1(F)CC(C(Cl)=O)C1 KGCHVYYLIWLLFD-UHFFFAOYSA-N 0.000 description 3
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 102000042838 JAK family Human genes 0.000 description 3
- 108091082332 JAK family Proteins 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- 208000014767 Myeloproliferative disease Diseases 0.000 description 3
- AXLLMCGIXUZGRB-UHFFFAOYSA-N N-[8-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-2,2,2-trifluoroacetamide Chemical compound C(#N)CC1(CN(C1)S(=O)(=O)CC)N1N=CC(=C1)C=1C=2N(C=CC=1)N=C(N=2)NC(C(F)(F)F)=O AXLLMCGIXUZGRB-UHFFFAOYSA-N 0.000 description 3
- DKIGFJIQIMSXPC-UHFFFAOYSA-N N-[8-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(C(=CC=C2)C=2C=NN(C=2)C2(CN(C2)S(=O)(=O)CC)CC#N)=N1 DKIGFJIQIMSXPC-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 102100034196 Thrombopoietin receptor Human genes 0.000 description 3
- 239000004012 Tofacitinib Substances 0.000 description 3
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- OPGPYIKNRCXNQY-UHFFFAOYSA-N benzyl 3-oxocyclobutane-1-carboxylate Chemical compound C1C(=O)CC1C(=O)OCC1=CC=CC=C1 OPGPYIKNRCXNQY-UHFFFAOYSA-N 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical group CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000006911 enzymatic reaction Methods 0.000 description 3
- GCKWGKCORGRVAT-UHFFFAOYSA-N ethyl n-carbamothioylcarbamate Chemical compound CCOC(=O)NC(N)=S GCKWGKCORGRVAT-UHFFFAOYSA-N 0.000 description 3
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- UEDBWPMPTRCSAB-UHFFFAOYSA-N tert-butyl 4-(4-aminopyrimidin-2-yl)pyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=C(C=N1)C1=NC=CC(N)=N1 UEDBWPMPTRCSAB-UHFFFAOYSA-N 0.000 description 3
- 229960001350 tofacitinib Drugs 0.000 description 3
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- 230000007306 turnover Effects 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 2
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 2
- HQUIOHSYUKWGOM-UHFFFAOYSA-N 2-(1-ethylsulfonylazetidin-3-ylidene)acetonitrile Chemical compound CCS(=O)(=O)N1CC(=CC#N)C1 HQUIOHSYUKWGOM-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- DHYHYLGCQVVLOQ-UHFFFAOYSA-N 3-bromoaniline Chemical compound NC1=CC=CC(Br)=C1 DHYHYLGCQVVLOQ-UHFFFAOYSA-N 0.000 description 2
- APWNCDGAOZSDNR-UHFFFAOYSA-N 4-(3-nitro-2-phenylmethoxyphenyl)-1H-pyrazole Chemical compound C(C1=CC=CC=C1)OC1=C(C=CC=C1[N+](=O)[O-])C=1C=NNC=1 APWNCDGAOZSDNR-UHFFFAOYSA-N 0.000 description 2
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 206010010356 Congenital anomaly Diseases 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102000003951 Erythropoietin Human genes 0.000 description 2
- 108090000394 Erythropoietin Proteins 0.000 description 2
- 108010075944 Erythropoietin Receptors Proteins 0.000 description 2
- 102100036509 Erythropoietin receptor Human genes 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 2
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 101150009057 JAK2 gene Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 2
- DAZXNSBGQYEODS-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)-1-(trifluoromethylsulfonyl)azetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-c]pyrimidin-2-yl]-3,3-difluorocyclobutane-1-carboxamide Chemical compound C=1N=C(N2C(=NC(=N2)NC(=O)C2CC(F)(F)C2)C=1)C=1C=NN(C=1)C1(CC#N)CN(C1)S(=O)(=O)C(F)(F)F DAZXNSBGQYEODS-UHFFFAOYSA-N 0.000 description 2
- APASPURLTSWCMY-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)-1-(trifluoromethylsulfonyl)azetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-c]pyrimidin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(=NC=CC2=N1)C=1C=NN(C=1)C1(CN(C1)S(=O)(=O)C(F)(F)F)CC#N APASPURLTSWCMY-UHFFFAOYSA-N 0.000 description 2
- IZOOJMJUQKUBJL-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)-1-cyclopropylsulfonylazetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-c]pyrimidin-2-yl]cyclopropanecarboxamide Chemical compound C(#N)CC1(CN(C1)S(=O)(=O)C1CC1)N1N=CC(=C1)C1=NC=CC=2N1N=C(N=2)NC(=O)C1CC1 IZOOJMJUQKUBJL-UHFFFAOYSA-N 0.000 description 2
- RGGPJGQLSGRLMR-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(C=CC=C2C=2C=NN(C=2)C2(CN(C2)S(=O)(=O)CC)CC#N)=N1 RGGPJGQLSGRLMR-UHFFFAOYSA-N 0.000 description 2
- KUXAHNPBDXEPTH-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-c]pyrimidin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(=NC=CC2=N1)C=1C=NN(C=1)C1(CN(C1)S(=O)(=O)CC)CC#N KUXAHNPBDXEPTH-UHFFFAOYSA-N 0.000 description 2
- LUDFAJPSAAFHAG-UHFFFAOYSA-N N-[8-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-a]pyrazin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(C(=NC=C2)C=2C=NN(C=2)C2(CN(C2)S(=O)(=O)CC)CC#N)=N1 LUDFAJPSAAFHAG-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 102000036693 Thrombopoietin Human genes 0.000 description 2
- 108010041111 Thrombopoietin Proteins 0.000 description 2
- 206010052779 Transplant rejections Diseases 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 125000006242 amine protecting group Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- RRSMHVQRSRIXGF-UHFFFAOYSA-N benzyl 3,3-difluorocyclobutane-1-carboxylate Chemical compound C1C(F)(F)CC1C(=O)OCC1=CC=CC=C1 RRSMHVQRSRIXGF-UHFFFAOYSA-N 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- DGJMPUGMZIKDRO-UHFFFAOYSA-N cyanoacetamide Chemical compound NC(=O)CC#N DGJMPUGMZIKDRO-UHFFFAOYSA-N 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000006735 deficit Effects 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- JYQLKKNUGGVARY-UHFFFAOYSA-N difluoromethanesulfonyl chloride Chemical compound FC(F)S(Cl)(=O)=O JYQLKKNUGGVARY-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 229940105423 erythropoietin Drugs 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 230000036244 malformation Effects 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 2
- NCZHGZFMOSPBNL-UHFFFAOYSA-N n-(4-ethylphenyl)-2-[5-(1,3,7-trimethyl-2,6-dioxopurin-8-yl)sulfanyl-1,2,4-triazol-1-yl]acetamide Chemical compound C1=CC(CC)=CC=C1NC(=O)CN1C(SC=2N(C=3C(=O)N(C)C(=O)N(C)C=3N=2)C)=NC=N1 NCZHGZFMOSPBNL-UHFFFAOYSA-N 0.000 description 2
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 125000004287 oxazol-2-yl group Chemical group [H]C1=C([H])N=C(*)O1 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- DMEUDSHBHKHLPD-UHFFFAOYSA-N pyrazin-2-amine Chemical compound NC1=CN=C=C[N]1 DMEUDSHBHKHLPD-UHFFFAOYSA-N 0.000 description 2
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 238000009987 spinning Methods 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 125000005309 thioalkoxy group Chemical group 0.000 description 2
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 238000005292 vacuum distillation Methods 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- LKCKVKCBUPTMHR-UHFFFAOYSA-N (6-bromopyridin-2-yl)carbamothioyl-ethylcarbamic acid Chemical compound C(C)N(C(O)=O)C(NC1=NC(=CC=C1)Br)=S LKCKVKCBUPTMHR-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000000196 1,4-pentadienyl group Chemical group [H]C([*])=C([H])C([H])([H])C([H])=C([H])[H] 0.000 description 1
- DNUTZBZXLPWRJG-UHFFFAOYSA-N 1-Piperidine carboxylic acid Chemical compound OC(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- HCRPLJAPJBCTMH-UHFFFAOYSA-N 2-(1-cyclopropylsulfonylazetidin-3-ylidene)acetonitrile Chemical compound C1(CC1)S(=O)(=O)N1CC(C1)=CC#N HCRPLJAPJBCTMH-UHFFFAOYSA-N 0.000 description 1
- KLFRDMGSQKVORW-UHFFFAOYSA-N 2-(1-ethylsulfonylazetidin-3-yl)acetonitrile Chemical compound C(#N)CC1CN(C1)S(=O)(=O)CC KLFRDMGSQKVORW-UHFFFAOYSA-N 0.000 description 1
- AIOFIJZHNMZOMX-UHFFFAOYSA-N 2-[3-[4-(1H-1,2,4-triazol-5-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile Chemical compound C(#N)CC1(CNC1)N1N=CC(=C1)C1=NNC=N1 AIOFIJZHNMZOMX-UHFFFAOYSA-N 0.000 description 1
- MKWFBLFEVFFTEE-UHFFFAOYSA-N 2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-a]pyridin-8-yl)pyrazol-1-yl]-1-cyclopropylsulfonylazetidin-3-yl]acetonitrile Chemical compound NC1=NN2C(C(=CC=C2)C=2C=NN(C=2)C2(CN(C2)S(=O)(=O)C2CC2)CC#N)=N1 MKWFBLFEVFFTEE-UHFFFAOYSA-N 0.000 description 1
- LHCFKGGJKXCMRY-UHFFFAOYSA-N 2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-a]pyridin-8-yl)pyrazol-1-yl]-1-ethylsulfonylazetidin-3-yl]acetonitrile Chemical compound NC1=NN2C(C(=CC=C2)C=2C=NN(C=2)C2(CN(C2)S(=O)(=O)CC)CC#N)=N1 LHCFKGGJKXCMRY-UHFFFAOYSA-N 0.000 description 1
- FSOYLIKIBWZTIR-UHFFFAOYSA-N 2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-c]pyrimidin-5-yl)pyrazol-1-yl]-1-ethylsulfonylazetidin-3-yl]acetonitrile Chemical compound NC1=NN2C(=NC=CC2=N1)C=1C=NN(C=1)C1(CN(C1)S(=O)(=O)CC)CC#N FSOYLIKIBWZTIR-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- VEJSIOPQKQXJAT-UHFFFAOYSA-N 2-bromo-6-nitrophenol Chemical compound OC1=C(Br)C=CC=C1[N+]([O-])=O VEJSIOPQKQXJAT-UHFFFAOYSA-N 0.000 description 1
- LPBDZVNGCNTELM-UHFFFAOYSA-N 2-chloropyrimidin-4-amine Chemical compound NC1=CC=NC(Cl)=N1 LPBDZVNGCNTELM-UHFFFAOYSA-N 0.000 description 1
- GEQZTCMVWVDEDF-UHFFFAOYSA-N 2-cyanoacetyl chloride Chemical compound ClC(=O)CC#N GEQZTCMVWVDEDF-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- MTEZLAATISORQK-UHFFFAOYSA-N 2-methoxyacetamide Chemical compound COCC(N)=O MTEZLAATISORQK-UHFFFAOYSA-N 0.000 description 1
- JJKWHOSQTYYFAE-UHFFFAOYSA-N 2-methoxyacetyl chloride Chemical compound COCC(Cl)=O JJKWHOSQTYYFAE-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- AEVSSZHXGJAPIE-UHFFFAOYSA-N 3-chloropyrazin-2-amine Chemical compound NC1=NC=CN=C1Cl AEVSSZHXGJAPIE-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- ZSHGVMYLGGANKU-UHFFFAOYSA-N 3-hydroxycyclobutane-1-carboxylic acid Chemical compound OC1CC(C(O)=O)C1 ZSHGVMYLGGANKU-UHFFFAOYSA-N 0.000 description 1
- IENOFRJPUPTEMI-UHFFFAOYSA-N 3-oxocyclobutane-1-carboxylic acid Chemical compound OC(=O)C1CC(=O)C1 IENOFRJPUPTEMI-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- NUKYPUAOHBNCPY-UHFFFAOYSA-N 4-aminopyridine Chemical compound NC1=CC=NC=C1 NUKYPUAOHBNCPY-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- GNJSVBWXALJBTF-UHFFFAOYSA-N 5-(1H-pyrazol-4-yl)-[1,2,4]triazolo[1,5-c]pyrimidin-2-amine Chemical compound N1N=CC(=C1)C1=NC=CC=2N1N=C(N=2)N GNJSVBWXALJBTF-UHFFFAOYSA-N 0.000 description 1
- TVGFHUIJNZRKFW-UHFFFAOYSA-N 5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-amine Chemical compound C1=CC=C(Br)N2N=C(N)N=C21 TVGFHUIJNZRKFW-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- BKLJUYPLUWUEOQ-UHFFFAOYSA-N 6-bromopyridin-2-amine Chemical compound NC1=CC=CC(Br)=N1 BKLJUYPLUWUEOQ-UHFFFAOYSA-N 0.000 description 1
- YHKASBDRJLTLHH-UHFFFAOYSA-N 7-bromo-1,3-benzothiazol-2-amine Chemical compound C1=CC(Br)=C2SC(N)=NC2=C1 YHKASBDRJLTLHH-UHFFFAOYSA-N 0.000 description 1
- AKEJQBRNKKIWIQ-UHFFFAOYSA-N 8-chloro-[1,2,4]triazolo[1,5-a]pyrazin-2-amine Chemical compound ClC1=NC=CN2N=C(N)N=C21 AKEJQBRNKKIWIQ-UHFFFAOYSA-N 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- AXICIBPYBONRSP-UHFFFAOYSA-N Anofinic acid Chemical compound C1=C(C(O)=O)C=C2C=CC(C)(C)OC2=C1 AXICIBPYBONRSP-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 229910014263 BrF3 Inorganic materials 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- KVDOLHZUQZCFRM-UHFFFAOYSA-N C(P(OCC)(O)OCC)C#N Chemical compound C(P(OCC)(O)OCC)C#N KVDOLHZUQZCFRM-UHFFFAOYSA-N 0.000 description 1
- FPDHMYIOWOJSLE-UHFFFAOYSA-N CCS(=O)(=O)N1CC(C1)C2=CC=CC=C2C#N Chemical compound CCS(=O)(=O)N1CC(C1)C2=CC=CC=C2C#N FPDHMYIOWOJSLE-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- GHOKWGTUZJEAQD-UHFFFAOYSA-N Chick antidermatitis factor Natural products OCC(C)(C)C(O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108010005939 Ciliary Neurotrophic Factor Proteins 0.000 description 1
- 102100031614 Ciliary neurotrophic factor Human genes 0.000 description 1
- YPZMPEPLWKRVLD-PJEQPVAWSA-N D-Glycero-D-gulo-Heptose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C=O YPZMPEPLWKRVLD-PJEQPVAWSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102100031939 Erythropoietin Human genes 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 241000282414 Homo sapiens Species 0.000 description 1
- 101000617830 Homo sapiens Sterol O-acyltransferase 1 Proteins 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical group NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000000646 Interleukin-3 Human genes 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 101150069380 JAK3 gene Proteins 0.000 description 1
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 1
- 206010072206 Janus kinase 2 mutation Diseases 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102100032352 Leukemia inhibitory factor Human genes 0.000 description 1
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- DOHFULAMGIIDHL-UHFFFAOYSA-N N-(8-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-2,2,2-trifluoroacetamide Chemical compound BrC=1C=2N(C=CC=1)N=C(N=2)NC(C(F)(F)F)=O DOHFULAMGIIDHL-UHFFFAOYSA-N 0.000 description 1
- JFJSKASPXVFXEO-UHFFFAOYSA-N N-[5-(1H-pyrazol-4-yl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide Chemical compound C1CC1C(=O)NC(=NN12)N=C1C=CC=C2C=1C=NNC=1 JFJSKASPXVFXEO-UHFFFAOYSA-N 0.000 description 1
- VSJHXEIAMYZDAJ-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)-1-(trifluoromethylsulfonyl)azetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(C=CC=C2C=2C=NN(C=2)C2(CN(C2)S(=O)(=O)C(F)(F)F)CC#N)=N1 VSJHXEIAMYZDAJ-UHFFFAOYSA-N 0.000 description 1
- WPYUYWQUYMQXHL-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)azetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(C=CC=C2C=2C=NN(C=2)C2(CNC2)CC#N)=N1 WPYUYWQUYMQXHL-UHFFFAOYSA-N 0.000 description 1
- BCZPOOSGSHMFOX-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)azetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-c]pyrimidin-2-yl]-3,3-difluorocyclobutane-1-carboxamide Chemical compound C(#N)CC1(CNC1)N1N=CC(=C1)C1=NC=CC=2N1N=C(N=2)NC(=O)C1CC(C1)(F)F BCZPOOSGSHMFOX-UHFFFAOYSA-N 0.000 description 1
- FBZCHBARQFORIA-UHFFFAOYSA-N N-[5-[1-[3-(cyanomethyl)azetidin-3-yl]pyrazol-4-yl]-[1,2,4]triazolo[1,5-c]pyrimidin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(=NC=CC2=N1)C=1C=NN(C=1)C1(CNC1)CC#N FBZCHBARQFORIA-UHFFFAOYSA-N 0.000 description 1
- QYEQEIMTBYDQFI-UHFFFAOYSA-N N-[8-(1H-pyrazol-4-yl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide Chemical compound C1(CC1)C(=O)NC1=NN2C(C(=CC=C2)C=2C=NNC=2)=N1 QYEQEIMTBYDQFI-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 108090000630 Oncostatin M Proteins 0.000 description 1
- 102100031942 Oncostatin-M Human genes 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- 101710113246 Pectinesterase 3 Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 108010072819 STAT Transcription Factors Proteins 0.000 description 1
- 102000007078 STAT Transcription Factors Human genes 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 102100021993 Sterol O-acyltransferase 1 Human genes 0.000 description 1
- 101000697584 Streptomyces lavendulae Streptothricin acetyltransferase Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102100034195 Thrombopoietin Human genes 0.000 description 1
- 108010070774 Thrombopoietin Receptors Proteins 0.000 description 1
- 102000000887 Transcription factor STAT Human genes 0.000 description 1
- 108050007918 Transcription factor STAT Proteins 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- BBAWTPDTGRXPDG-UHFFFAOYSA-N [1,3]thiazolo[4,5-b]pyridine Chemical compound C1=CC=C2SC=NC2=N1 BBAWTPDTGRXPDG-UHFFFAOYSA-N 0.000 description 1
- PNDPGZBMCMUPRI-XXSWNUTMSA-N [125I][125I] Chemical compound [125I][125I] PNDPGZBMCMUPRI-XXSWNUTMSA-N 0.000 description 1
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical group 0.000 description 1
- 125000005101 aryl methoxy carbonyl group Chemical group 0.000 description 1
- 125000005002 aryl methyl group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000004566 azetidin-1-yl group Chemical group N1(CCC1)* 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- QARVLSVVCXYDNA-UHFFFAOYSA-N bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001735 carboxylic acids Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- VLOHNGJRIFIHMO-UHFFFAOYSA-N chloromethane;hydrate Chemical compound O.ClC VLOHNGJRIFIHMO-UHFFFAOYSA-N 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical compound C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- 125000006255 cyclopropyl carbonyl group Chemical group [H]C1([H])C([H])([H])C1([H])C(*)=O 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- FRYHCSODNHYDPU-UHFFFAOYSA-N ethanesulfonyl chloride Chemical compound CCS(Cl)(=O)=O FRYHCSODNHYDPU-UHFFFAOYSA-N 0.000 description 1
- BXHVHUIFLOJUNW-UHFFFAOYSA-N ethyl 4-cyano-5-[[2-[(4-ethyl-5-pyridin-4-yl-1,2,4-triazol-3-yl)sulfanyl]acetyl]amino]-3-methylthiophene-2-carboxylate Chemical compound CC1=C(C(=O)OCC)SC(NC(=O)CSC=2N(C(C=3C=CN=CC=3)=NN=2)CC)=C1C#N BXHVHUIFLOJUNW-UHFFFAOYSA-N 0.000 description 1
- VDWKYJAKPKFAHJ-UHFFFAOYSA-N ethyl 6-(bromomethyl)-2-(trifluoromethyl)pyrimidine-4-carboxylate Chemical compound CCOC(=O)C1=CC(CBr)=NC(C(F)(F)F)=N1 VDWKYJAKPKFAHJ-UHFFFAOYSA-N 0.000 description 1
- FBSFWDOFMQDRDV-UHFFFAOYSA-N ethyl 6-methyl-2-(trifluoromethyl)pyrimidine-4-carboxylate Chemical compound CCOC(=O)C1=CC(C)=NC(C(F)(F)F)=N1 FBSFWDOFMQDRDV-UHFFFAOYSA-N 0.000 description 1
- BDTDECDAHYOJRO-UHFFFAOYSA-N ethyl n-(sulfanylidenemethylidene)carbamate Chemical compound CCOC(=O)N=C=S BDTDECDAHYOJRO-UHFFFAOYSA-N 0.000 description 1
- JTAYHXCQRWVPKL-UHFFFAOYSA-N ethyl n-[(3-chloropyrazin-2-yl)carbamothioyl]carbamate Chemical compound CCOC(=O)NC(=S)NC1=NC=CN=C1Cl JTAYHXCQRWVPKL-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000008303 genetic mechanism Effects 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 208000035474 group of disease Diseases 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 102000049918 human JAK1 Human genes 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940076264 interleukin-3 Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical group CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- RMIODHQZRUFFFF-UHFFFAOYSA-N methoxyacetic acid Chemical compound COCC(O)=O RMIODHQZRUFFFF-UHFFFAOYSA-N 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- XONPDZSGENTBNJ-UHFFFAOYSA-N molecular hydrogen;sodium Chemical compound [Na].[H][H] XONPDZSGENTBNJ-UHFFFAOYSA-N 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- DXASQZJWWGZNSF-UHFFFAOYSA-N n,n-dimethylmethanamine;sulfur trioxide Chemical group CN(C)C.O=S(=O)=O DXASQZJWWGZNSF-UHFFFAOYSA-N 0.000 description 1
- XLBCMNCPMMPNHH-UHFFFAOYSA-N n-(5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide Chemical compound N=1N2C(Br)=CC=CC2=NC=1NC(=O)C1CC1 XLBCMNCPMMPNHH-UHFFFAOYSA-N 0.000 description 1
- MRLMSFNIIPBOGC-UHFFFAOYSA-N n-(8-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide Chemical compound N1=C2C(Br)=CC=CN2N=C1NC(=O)C1CC1 MRLMSFNIIPBOGC-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- QNNHQVPFZIFNFK-UHFFFAOYSA-N oxazolo[4,5-b]pyridine Chemical compound C1=CC=C2OC=NC2=N1 QNNHQVPFZIFNFK-UHFFFAOYSA-N 0.000 description 1
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229940055726 pantothenic acid Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 229960004838 phosphoric acid Drugs 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- ZNNZYHKDIALBAK-UHFFFAOYSA-M potassium thiocyanate Chemical compound [K+].[S-]C#N ZNNZYHKDIALBAK-UHFFFAOYSA-M 0.000 description 1
- 229940116357 potassium thiocyanate Drugs 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 238000004537 pulping Methods 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- GQIXFHWAAHPMSO-UHFFFAOYSA-N pyrimidin-2-amine Chemical compound NC1=NC=C=C[N]1 GQIXFHWAAHPMSO-UHFFFAOYSA-N 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 238000009666 routine test Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 150000003413 spiro compounds Chemical class 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- 229950000244 sulfanilic acid Drugs 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 125000006253 t-butylcarbonyl group Chemical group [H]C([H])([H])C(C(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000001648 tannin Substances 0.000 description 1
- 235000018553 tannin Nutrition 0.000 description 1
- 229920001864 tannin Polymers 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- DONSYFVBIUXSCL-UHFFFAOYSA-N tert-butyl 3-(cyanomethyl)-3-[4-(3-nitro-2-phenylmethoxyphenyl)pyrazol-1-yl]azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(CC#N)(C1)N1C=C(C=N1)C1=C(OCC2=CC=CC=C2)C(=CC=C1)[N+]([O-])=O DONSYFVBIUXSCL-UHFFFAOYSA-N 0.000 description 1
- YHPRTDGQZFXVQJ-UHFFFAOYSA-N tert-butyl 3-(cyanomethyl)-3-[4-[2-(cyclopropanecarbonylamino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]pyrazol-1-yl]azetidine-1-carboxylate Chemical compound C(#N)CC1(CN(C1)C(=O)OC(C)(C)C)N1N=CC(=C1)C1=CC=CC=2N1N=C(N=2)NC(=O)C1CC1 YHPRTDGQZFXVQJ-UHFFFAOYSA-N 0.000 description 1
- BESFCRTTXQYNBW-UHFFFAOYSA-N tert-butyl 3-(cyanomethylidene)azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(=CC#N)C1 BESFCRTTXQYNBW-UHFFFAOYSA-N 0.000 description 1
- KMQWAOHEIKYYIJ-UHFFFAOYSA-N tert-butyl 3-[4-(2-amino-[1,2,4]triazolo[1,5-c]pyrimidin-5-yl)pyrazol-1-yl]-3-(cyanomethyl)azetidine-1-carboxylate Chemical compound NC1=NN2C(=NC=CC2=N1)C=1C=NN(C=1)C1(CN(C1)C(=O)OC(C)(C)C)CC#N KMQWAOHEIKYYIJ-UHFFFAOYSA-N 0.000 description 1
- IPISOFJLWYBCAV-UHFFFAOYSA-N tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole-1-carboxylate Chemical compound C1=NN(C(=O)OC(C)(C)C)C=C1B1OC(C)(C)C(C)(C)O1 IPISOFJLWYBCAV-UHFFFAOYSA-N 0.000 description 1
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 1
- SCSLUABEVMLYEA-UHFFFAOYSA-N tert-butyl pentanoate Chemical compound CCCCC(=O)OC(C)(C)C SCSLUABEVMLYEA-UHFFFAOYSA-N 0.000 description 1
- BSJFXAFLSWDUPK-UHFFFAOYSA-N tert-butyl pyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC=N1 BSJFXAFLSWDUPK-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000011345 viscous material Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/428—Thiazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本发明公开了一系列JAK抑制剂,具体涉及式(Ⅰ)化合物或其药学上可接受的盐。
Description
技术领域
本发明涉及一系列JAK抑制剂,具体涉及式(Ⅰ)化合物或其药学上可接受的盐。
背景技术
JAK属于参与炎症、自身免疫疾病、增殖性疾病、移植排斥、涉及软骨更新(turnover)受损的疾病、先天软骨畸形和/或与IL6分泌过多相关的疾病的酪氨酸激酶家族。本发明还提供所述化合物、含有所述化合物的药物组合物的生产方法和通过施用本发明化合物预防和/或治疗炎症、自身免疫疾病、增殖性疾病、移植排斥、涉及软骨更新受损的疾病、先天软骨畸形和/或与IL6分泌过多相关的疾病的方法。
Janus激酶(JAK)是转导细胞因子信号从膜受体到STAT转录因子的细胞质酪氨酸激酶。现有技术已经描述了四种JAK家族成员:JAK1、JAK2、JAK3和TYK2。当细胞因子与其受体结合时,JAK家族成员自磷酸化和/或彼此转磷酸化,随后STATs磷酸化,然后迁移至细胞核内以调节转录。JAK-STAT细胞内信号转导适用于干扰素、大多数白细胞介素以及多种细胞因子和内分泌因子,例如EPO、TPO、GH、OSM、LIF、CNTF、GM-CSF和PRL(Vainchenker W.等人(2008))。
遗传学模型和小分子JAK抑制剂的组合研究揭示了几种JAKs的治疗潜能。JAK2基因突变研究是近年来血液病研究的突破性进展之一。现有技术公开了骨髓增殖性疾病(myeloproliferative diseases,MPD),包括真性红细胞增多症(polycythemia vela,PV)、原发性血小板增多症(essential thrombocythemia,ET)和特发性骨髓纤维化(idiopathicmyelofibrosis,IMF)是一组造血干细胞病变引发的恶性疾病,2005年研究人员在本组疾病中发现存在JAK2点突变(JAK2V617F),才使MPD的诊治进入了一个新纪元。JAK2V617是在第14外显子v617位点发生的点突变,缬氨酸(valine,V)被苯丙氨酸(phenylalanine,F)取代。在JAK2的结构中,JHl为激酶域;而Val617位于与JHl相邻的JH2,后者为假激酶域,与JHl结合并抑制其激活。V617F突变使JH2失去了对JHl激酶活性的抑制作用,导致了JAK2的持续活化,由此造成细胞的增殖活性增强[Kilpivaara 0,LevineRL.JAK2and MPL mutations in myeloprolifer-ative neoplasms:discoveryandscience.Leukemia.2008;22(10):1813-7]。JAK2V617F突变在真性红细胞增多症、原发性血小板增多症和特发性骨髓纤维化患者中存在很高的发生率。通过等位基因特异性多聚酶链反应检测到JAK2 V617F突变在真性红细胞增多症患者中的发生率为90%;原发性血小板增多症和特发性骨髓纤维化患者为50%-60%[Baxter EJ,Scott LM,Campbell PJ,etal.Lancet.2005;365(9464):1054-61]。在未发现JAK2突变缺乏V617F突变患者的这些疾病的分子基础尚不清楚。2007年,有研究在JAK2V617F突变阴性的MPD患者中发现了外显子12的突变,该突变同样也可造成JH2丧失对JH激酶活性的抑制,这为JAK2 V617F阴性的骨髓增殖性疾病患者提供了分子标志物和遗传机制[Scott LM,Tong W,Levine RL,et al.JAK2exon 12mutations in polycythemia vera and idiopathic erythrocytosis.NEngl JMed 2007;356:459-68·]。[0004]正常生理情况下,JAK2介导促红细胞生成素(EP0)、促血小板生成素(TP0)、粒-巨噬细胞集落刺激因子、白细胞介素_3和生长因子在内的多种细胞因子的信号转导,并调节和促进细胞增殖。JAK2基因在造血调节中发挥重要作用,其下游的STAT家族是一种能与DNA结合的蛋白家族,与JAK磷酸化信号通路偶联(JAK-STAT信号通路),发挥转录调控作用。JAK-STAT能把细胞外信号与基因表达调控直接联系起来,启动响应基因的转录和表达,完成细胞因子受体如促红细胞生成素受体(EP0R)和促血小板生成素受体(MPL/TP0R)介导的信号转导过程,产生细胞增殖效应。
Tofacitinib是一种pan jak抑制剂,非高特异性JAK2抑制剂,其结构式如下所示:

发明内容
本发明的目的在于提供式(Ⅰ)化合物或其药学上可接受的盐,

其中,
R1选自H、或选自任选被1、2、3或4个R取代的:C1-6烷基、C1-6杂烷基、C3-7环烷基、3~7元杂环烷基、5~6元芳基、5~6元杂芳基、
L1、L2分别独立地选自单键、-S(=O)2-、-S(=O)-、-C(=O)-、-NHC(=O)-;
R2选自:H、或选自任选被1、2、3或4个R取代的:NH2、C1-6烷基、C1-6杂烷基、C3-7环烷基、3~7元杂环烷基、5~6元芳基、5~6元杂芳基;
环A选自5~6元杂芳基;
X分别独立地选自N、C;
T选自N或C(R);
R选自H、卤素、NH2、CN、OH、或选自任选被1、2、3或4个R’取代的:C1-3烷基、C1-3杂烷基、C3-6环烷基、3~6元杂环烷基、5~6元芳基、5~6元杂芳基;
R’选自卤素、OH、CN、NH2;
所述“杂”代表杂原子或杂原子团,分别独立地选自O、S、N、C(=O)、S(=O)或S(=O)2;
杂原子或杂原子团的数目分别独立地选自0、1、2、3或4。
本发明的一个方案中,上述R分别独立地选自H、卤素、OH、NH2、CN,或选自任选被1、2、3或4个R’取代的C1-3烷基、C1-3烷氧基、C1-3烷胺基。
本发明的一个方案中,上述R选自H、F、Cl、Br、I、OH、CN、NH2、Me、Et、N(CH3)2、NH(CH3)。
本发明的一个方案中,上述R1选自H,或任选被1、2、3或4个R取代的C1-3烷基、C3-6环烷基、C3-6杂环烷基、C1-3烷基-O-C1-3烷基-、C1-3烷基-S-C1-3烷基-、C1-3烷基-NH-C1-3烷基-、C1-6烷氧基、C1-6烷氨基、
本发明的一个方案中,上述R1选自H,或选自任选被1、2、3或4个R取代的Me、

本发明的一个方案中,上述R1选自:H、Me、

本发明的一个方案中,上述R1-L1-选自H,或选自任选被R取代的:

本发明的一个方案中,上述R1-L1-选自:H、

本发明的一个方案中,上述R2选自H、NH2,或任选被1、2、3或4个R取代的C1-3烷基、C3-6环烷基、3~6元杂环烷基、C1-3烷基-O-C1-3烷基-、C1-3烷基-S-C1-3烷基-、C1-3烷基-NH-C1-3烷基-、C1-6烷氧基、C1-6烷氨基;
本发明的一个方案中,上述R2选自H、NH2,或选自任选被R取代的:Me、

本发明的一个方案中,上述R2选自:H、NH2、

本发明的一个方案中,上述R2-L2-选自H,或任选被R’取代的:NH2、

本发明的一个方案中,上述R2-L2-选自:H、NH2、

本发明的一个方案中,上述环A选自:1,3,4-三氮唑基、咪唑基、恶唑基、噻唑基。
本发明的一个方案中,上述环A选自:
本发明的一个方案中,上述结构单元 选自:
选自: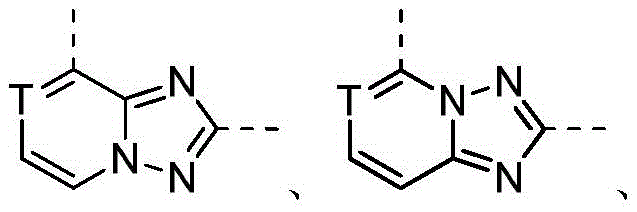

本发明的一个方案中,上述结构但单元 选自:
选自:

本发明的化合物,其选自:



定义和说明
除非另有说明,本文所用的下列术语和短语旨在具有下列含义。一个特定的术语或短语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文中出现商品名时,意在指代其对应的商品或其活性成分。
C1-6选自C1、C2、C3、C4、C5和C6;C3-7选自C3、C4、C5、C6和C7;3~7元选自3元、4元、5元、6元和7元。
这里所采用的术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定取代基的化合物与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物的中性形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机氨或镁盐或类似的盐。当本发明的化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物的中性形式接触的方式获得酸加成盐。药学上可接受的酸加成盐的实例包括无机酸盐,所述无机酸包括例如盐酸、氢溴酸、硝酸、碳酸,碳酸氢根,磷酸、磷酸一氢根、磷酸二氢根、硫酸、硫酸氢根、氢碘酸、亚磷酸等;以及有机酸盐,所述有机酸包括如乙酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸和甲磺酸等类似的酸;还包括氨基酸(如精氨酸等)的盐,以及如葡糖醛酸等有机酸的盐(参见Berge et al.,"PharmaceuticalSalts",Journal of Pharmaceutical Science 66:1-19(1977))。本发明的某些特定的化合物含有碱性和酸性的官能团,从而可以被转换成任一碱或酸加成盐。
优选地,以常规方式使盐与碱或酸接触,再分离母体化合物,由此再生化合物的中性形式。化合物的母体形式与其各种盐的形式的不同之处在于某些物理性质,例如在极性溶剂中的溶解度不同。
本文所用的“药学上可接受的盐”属于本发明化合物的衍生物,其中,通过与酸成盐或与碱成盐的方式修饰所述母体化合物。药学上可接受的盐的实例包括但不限于:碱基比如胺的无机酸或有机酸盐、酸根比如羧酸的碱金属或有机盐等等。药学上可接受的盐包括常规的无毒性的盐或母体化合物的季铵盐,例如无毒的无机酸或有机酸所形成的盐。常规的无毒性的盐包括但不限于那些衍生自无机酸和有机酸的盐,所述的无机酸或有机酸选自2-乙酰氧基苯甲酸、2-羟基乙磺酸、乙酸、抗坏血酸、苯磺酸、苯甲酸、碳酸氢根、碳酸、柠檬酸、依地酸、乙烷二磺酸、乙烷磺酸、富马酸、葡庚糖、葡糖酸、谷氨酸、乙醇酸、氢溴酸、盐酸、氢碘酸盐、羟基、羟萘、羟乙磺酸、乳酸、乳糖、十二烷基磺酸、马来酸、苹果酸、扁桃酸、甲烷磺酸、硝酸、草酸、双羟萘酸、泛酸、苯乙酸、磷酸、多聚半乳糖醛、丙酸、水杨酸、硬脂酸、亚乙酸、琥珀酸、氨基磺酸、对氨基苯磺酸、硫酸、单宁、酒石酸和对甲苯磺酸。
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的这些化合物与化学计量的适当的碱或酸反应来制备。一般地,优选醚、乙酸乙酯、乙醇、异丙醇或乙腈等非水介质。
除了盐的形式,本发明所提供的化合物还存在前药形式。本文所描述的化合物的前药容易地在生理条件下发生化学变化从而转化成本发明的化合物。此外,前体药物可以在体内环境中通过化学或生化方法被转换到本发明的化合物。
本发明的某些化合物可以以非溶剂化形式或者溶剂化形式存在,包括水合物形式。一般而言,溶剂化形式与非溶剂化的形式相当,都包含在本发明的范围之内。
本发明的某些化合物可以具有不对称碳原子(光学中心)或双键。外消旋体、非对映异构体、几何异构体和单个的异构体都包括在本发明的范围之内。
本文中消旋体、ambiscalemic and scalemic或者对映体纯的化合物的图示法来自Maehr,J.Chem.Ed.1985,62:114-120。1985年,62:114-120。除非另有说明,用楔形键和虚线键表示一个立体中心的绝对构型。当本文所述化合物含有烯属双键或其它几何不对称中心,除非另有规定,它们包括E、Z几何异构体。同样地,所有的互变异构形式均包括在本发明的范围之内。
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对对映体、(R)-和(S)-对映体、非对映异构体、(D)-异构体、(L)-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。烷基等取代基中可存在另外的不对称碳原子。所有这些异构体以及它们的混合物,均包括在本发明的范围之内。
可以通过的手性合成或手性试剂或者其他常规技术制备光学活性的(R)-和(S)-异构体以及D和L异构体。如果想得到本发明某化合物的一种对映体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对映体混合物分离,并且辅助基团裂开以提供纯的所需对映异构体。或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的拆分方法进行非对映异构体拆分,然后回收得到纯的对映体。此外,对映异构体和非对映异构体的分离通常是通过使用色谱法完成的,所述色谱法采用手性固定相,并任选地与化学衍生法相结合(例如由胺生成氨基甲酸盐)。
本发明的化合物可以在一个或多个构成该化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素标记化合物,比如氚(3H),碘-125(125I)或C-14(14C)。本发明的化合物的所有同位素组成的变换,无论放射性与否,都包括在本发明的范围之内。
术语“药学上可接受的载体”是指能够递送本发明有效量活性物质、不干扰活性物质的生物活性并且对宿主或者患者无毒副作用的任何制剂或载体介质代表性的载体包括水、油、蔬菜和矿物质、膏基、洗剂基质、软膏基质等。这些基质包括悬浮剂、增粘剂、透皮促进剂等。它们的制剂为化妆品领域或局部药物领域的技术人员所周知。关于载体的其他信息,可以参考Remington:The Science and Practice of Pharmacy,21st Ed.,Lippincott,Williams&Wilkins(2005),该文献的内容通过引用的方式并入本文。
术语“赋形剂”通常是指配制有效的药物组合物所需要载体、稀释剂和/或介质。
针对药物或药理学活性剂而言,术语“有效量”或“治疗有效量”是指无毒的但能达到预期效果的药物或药剂的足够用量。对于本发明中的口服剂型,组合物中一种活性物质的“有效量”是指与该组合物中另一种活性物质联用时为了达到预期效果所需要的用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
术语“活性成分”、“治疗剂”,“活性物质”或“活性剂”是指一种化学实体,它可以有效地治疗目标紊乱、疾病或病症。
术语“被取代的”是指特定原子上的任意一个或多个氢原子被取代基取代,只要特定原子的价态是正常的并且取代后的化合物是稳定的。当取代基为酮基(即=O)时,意味着两个氢原子被取代。酮取代不会发生在芳香基上。术语“任选被取代的”是指可以被取代,也可以不被取代,除非另有规定,取代基的种类和数目在化学上可以实现的基础上可以是任意的。
当任何变量(例如R)在化合物的组成或结构中出现一次以上时,其在每一种情况下的定义都是独立的。因此,例如,如果一个基团被0-2个R所取代,则所述基团可以任选地至多被两个R所取代,并且每种情况下的R都有独立的选项。此外,取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。
当其中一个变量选自单键时,表示其连接的两个基团直接相连,比如A-L-Z中L代表单键时表示该结构实际上是A-Z。
当一个取代基的键可以交叉连接到一个环上的两个原子时,这种取代基可以与这个环上的任意原子相键合。当所列举的取代基中没有指明其通过哪一个原子连接到化学结构通式中包括但未具体提及的化合物时,这种取代基可以通过其任何原子相键合。取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。例如,结构单元 表示其可在环己基或者环基二烯上的任意一个位置发生取代。
表示其可在环己基或者环基二烯上的任意一个位置发生取代。
烷基和杂烷基原子团的取代基一般被称为“烷基取代基”,它们可以选自但不限于下列基团中的一个或多个:-R’、-OR’、=O、=NR’、=N-OR’、-NR’R”、-SR’、卤素、-SiR’R”R”’、OC(O)R’、-C(O)R’、-CO2R’、-CONR’R”、-OC(O)NR’R”、-NR”C(O)R’、NR’C(O)NR”R”’、-NR”C(O)2R’、-NR””’-C(NR’R”R’”)=NR””、NR””C(NR’R”)=NR’”、-S(O)R’、-S(O)2R’、-S(O)2NR’R”、NR”SO2R’、-CN、–NO2、-N3、-CH(Ph)2和氟代(C1-C4)烷基,取代基的数目为0~(2m’+1),其中m’是这类原子团中碳原子的总数。R'、R”、R”'、R””和R””’各自独立地优选氢、被取代或未被取代的杂烷基、被取代或未被取代的芳基(例如被1~3个卤素取代芳基)、被取代或未被取代的烷基、烷氧基、硫代烷氧基基团或芳烷基。当本发明的化合物包括一个以上的R基团时,例如,每一个R基团是独立地加以选择的,如同当存在一个以上的R'、R”、R”'、R””和R””’基团时的每个这些基团。当R'和R”附着于同一个氮原子时,它们可与该氮原子结合形成5-,6-或7-元环。例如,-NR'R“意在包括但不仅限于1-吡咯烷基和4-吗啉基。根据上述关于取代基的讨论中,本领域技术人员可以理解,术语“烷基”意在包括碳原子键合于非氢基团所构成的基团,如卤代烷基(例如-CF3、-CH2CF3)和酰基(例如-C(O)CH3、-C(O)CF3、-C(O)CH2OCH3等)。
与烷基原子团所述取代基相似,芳基和杂芳基取代基一般统称为“芳基取代基”,选自例如-R’、-OR’、-NR’R”、-SR’、-卤素,-SiR’R”R”’、OC(O)R’、-C(O)R’、-CO2R’、-CONR’R”、-OC(O)NR’R”、-NR”C(O)R’、NR’C(O)NR”R”’、-NR”C(O)2R’、-NR””’-C(NR’R”R’”)=NR””、NR””C(NR’R”)=NR’”、-S(O)R’、-S(O)2R’、-S(O)2NR’R”、NR”SO2R’、-CN、–NO2、-N3、-CH(Ph)2、氟(C1-C4)烷氧基和氟(C1-C4)烷基等,取代基的数量为0到芳香环上开放化合价的总数之间;其中R’、R”、R”’、R””和R””’独立地优选自氢、被取代或未被取代的烷基、被取代或未被取代的杂烷基、被取代或未被取代的芳基和被取代或未被取代的杂芳基。当本发明的化合物包括一个以上的R基团时,例如,每个R基团是独立地加以选择的,如同当存在一个以上R’、R”、R”’、R””和R””’基团时的每个这些基团。
除非另有规定,芳基或杂芳基环的相邻原子上的两个取代基可以任选地被通式为–T-C(O)-(CRR’)q-U-的取代基所取代,其中T和U独立地选自-NR-、-O-、CRR'-或单键,q是0到3的整数。作为替代选择,芳基或杂芳基环的相邻原子上的两个取代基可以任选地被通式为–A(CH2)rB-的取代基所取代,其中A和B独立的选自–CRR’-、-O-、-NR-、-S-、-S(O)-、S(O)2-、-S(O)2NR’-或单键,r是1~4的整数。任选地,由此形成的新环上的一个单键可以替换为双键。作为替代选择,芳基或杂芳基环的相邻原子上的两个取代基可以任选地被通式为–(CRR’)s-X-(CR”R”’)d-取代基所取代,其中s和d分别独立的选自0~3的整数,X是–O-、-NR’、-S-、-S(O)-、-S(O)2-或–S(O)2NR’-。取代基R、R’、R”和R”’分别独立地优选自氢和被取代或未被取代的(C1-C6)烷基。
除非另有规定,术语“卤代素”或“卤素”本身或作为另一取代基的一部分表示氟、氯、溴或碘原子。此外,术语“卤代烷基”意在包括单卤代烷基和多卤代烷基。例如,术语“卤代(C1-C4)烷基”意在包括但不仅限于三氟甲基、2,2,2-三氟乙基、4-氯丁基和3-溴丙基等等。
卤代烷基的实例包括但不仅限于:三氟甲基、三氯甲基、五氟乙基,和五氯乙基。“烷氧基”代表通过氧桥连接的具有特定数目碳原子的上述烷基。C1-6烷氧基包括C1、C2、C3、C4、C5和C6的烷氧基。烷氧基的例子包括但不限于:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基和S-戊氧基。“环烷基”包括饱和环基,如环丙基、环丁基或环戊基。3-7环烷基包括C3、C4、C5、C6和C7环烷基。“链烯基”包括直链或支链构型的烃链,其中该链上任何的稳定位点上存在一个或多个碳-碳双键,例如乙烯基和丙烯基。
术语“卤”或“卤素”是指氟、氯、溴和碘。
除非另有规定,术语“杂”表示杂原子或杂原子团(即含有杂原子的原子团),包括碳(C)和氢(H)以外的原子以及含有这些杂原子的原子团,例如包括氧(O)、氮(N)、硫(S)、硅(Si)、锗(Ge)、铝(Al)、硼(B)、-O-、-S-、=O、=S、-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)、-S(=O)2-,以及任选被被取代的-C(=O)N(H)-、-N(H)-、-C(=NH)-、-S(=O)2N(H)-或-S(=O)N(H)-。
除非另有规定,“环”表示被取代或未被取代的环烷基、杂环烷基、环烯基、杂环烯基、环炔基、杂环炔基、芳基或杂芳基。所谓的环包括单环、联环、螺环、并环或桥环。环上原子的数目通常被定义为环的元数,例如,“5~7元环”是指环绕排列5~7个原子。除非另有规定,该环任选地包含1~3个杂原子。因此,“5~7元环”包括例如苯基、吡啶和哌啶基;另一方面,术语“5~7元杂环烷基环”包括吡啶基和哌啶基,但不包括苯基。术语“环”还包括含有至少一个环的环系,其中的每一个“环”均独立地符合上述定义。
除非另有规定,术语“杂环”或“杂环基”意指稳定的含杂原子或杂原子团的单环、双环或三环,它们可以是饱和的、部分不饱和的或不饱和的(芳族的),它们包含碳原子和1、2、3或4个独立地选自N、O和S的环杂原子,其中上述任意杂环可以稠合到一个苯环上形成双环。氮和硫杂原子可任选被氧化(即NO和S(O)p)。氮原子可以是被取代的或未取代的(即N或NR,其中R是H或本文已经定义过的其他取代基)。该杂环可以附着到任何杂原子或碳原子的侧基上从而形成稳定的结构。如果产生的化合物是稳定的,本文所述的杂环可以发生碳位或氮位上的取代。杂环中的氮原子任选地被季铵化。一个优选方案是,当杂环中S及O原子的总数超过1时,这些杂原子彼此不相邻。另一个优选方案是,杂环中S及O原子的总数不超过1。如本文所用,术语“芳族杂环基团”或“杂芳基”意指稳定的5、6、7元单环或双环或7、8、9或10元双环杂环基的芳香环,它包含碳原子和1、2、3或4个独立地选自N、O和S的环杂原子。氮原子可以是被取代的或未取代的(即N或NR,其中R是H或本文已经定义过的其他取代基)。氮和硫杂原子可任选被氧化(即NO和S(O)p)。值得注意的是,芳香杂环上S和O原子的总数不超过1。桥环也包含在杂环的定义中。当一个或多个原子(即C、O、N或S)连接两个不相邻的碳原子或氮原子时形成桥环。优选的桥环包括但不限于:一个碳原子、两个碳原子、一个氮原子、两个氮原子和一个碳-氮基。值得注意的是,一个桥总是将单环转换成三环。桥环中,环上的取代基也可以出现在桥上。
杂环化合物的实例包括但不限于:吖啶基、吖辛因基、苯并咪唑基、苯并呋喃基、苯并巯基呋喃基、苯并巯基苯基、苯并恶唑基、苯并恶唑啉基、苯并噻唑基、苯并三唑基、苯并四唑基、苯并异恶唑基、苯并异噻唑基、苯并咪唑啉基、咔唑基、4aH-咔唑基、咔啉基、苯并二氢吡喃基、色烯、噌啉基、十氢喹啉基、2H,6H-1,5,2-二噻嗪基、二氢呋喃并[2,3-b]四氢呋喃基、呋喃基、呋咱基、咪唑烷基、咪唑啉基、咪唑基、1H-吲唑基、吲哚烯基、二氢吲哚基、中氮茚基、吲哚基、3H-吲哚基、isatino基、异苯并呋喃基、吡喃、异吲哚基、异二氢吲哚基、异吲哚基、吲哚基、异喹啉基、异噻唑基、异恶唑基、亚甲二氧基苯基、吗啉基、萘啶基,八氢异喹啉基、恶二唑基、1,2,3-恶二唑基、1,2,4-恶二唑基、1,2,5-恶二唑基、1,3,4-恶二唑基、恶唑烷基、恶唑基、异恶唑基、羟吲哚基、嘧啶基、菲啶基、菲咯啉基、吩嗪、吩噻嗪、苯并黄嘌呤基、酚恶嗪基、酞嗪基、哌嗪基、哌啶基、哌啶酮基、4-哌啶酮基、胡椒基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑烷基、吡唑啉基、吡唑基、哒嗪基、吡啶并恶唑、吡啶并咪唑、吡啶并噻唑、吡啶基、嘧啶基、吡咯烷基、吡咯啉基、2H-吡咯基、吡咯基、吡唑基、喹唑啉基、喹啉基、4H-喹嗪基、喹喔啉基、奎宁环基、四氢呋喃基、四氢异喹啉基、四氢喹啉基、四唑基,6H-1,2,5-噻二嗪基、1,2,3-噻二唑基、1,2,4-噻二唑基、1,2,5-噻二唑基、1,3,4-噻二唑基、噻蒽基、噻唑基、异噻唑基噻吩基、噻吩基、噻吩并恶唑基、噻吩并噻唑基、噻吩并咪唑基、噻吩基、三嗪基、1,2,3-三唑基、1,2,4-三唑基、1,2,5-三唑基、1,3,4-三唑基和呫吨基。还包括稠环和螺环化合物。
除非另有规定,术语“烃基”或者其下位概念(比如烷基、烯基、炔基、苯基等等)本身或者作为另一取代基的一部分表示直链的、支链的或环状的烃原子团或其组合,可以是完全饱和的、单元或多元不饱和的,可以是单取代、二取代或多取代的,可以是一价(如甲基)、二价(如亚甲基)或者多价(如次甲基),可以包括二价或多价原子团,具有指定数量的碳原子(如C1-C10表示1至10个碳)。“烃基”包括但不限于脂肪烃基和芳香烃基,所述脂肪烃基包括链状和环状,具体包括但不限于烷基、烯基、炔基,所述芳香烃基包括但不限于6-12元的芳香烃基,例如苯、萘等。在一些实施例中,术语“烃基”表示直链的或支链的原子团或它们的组合,可以是完全饱和的、单元或多元不饱和的,可以包括二价和多价原子团。饱和烃原子团的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、仲丁基、异丁基、环己基、(环己基)甲基、环丙基甲基,以及正戊基、正己基、正庚基、正辛基等原子团的同系物或异构体。不饱和烷基具有一个或多个双键或三键,其实例包括但不限于乙烯基、2-丙烯基、丁烯基、巴豆基、2-异戊烯基、2-(丁二烯基)、2,4-戊二烯基、3-(1,4-戊二烯基)、乙炔基、1-和3-丙炔基,3-丁炔基,以及更高级的同系物和异构体。
除非另有规定,术语“杂烃基”或者其下位概念(比如杂烷基、杂烯基、杂炔基、杂芳基等等)本身或者与另一术语联合表示稳定的直链的、支链的或环状的烃原子团或其组合,有一定数目的碳原子和至少一个杂原子组成。在一些实施例中,术语“杂烷基”本身或者与另一术语联合表示稳定的直链的、支链的烃原子团或其组合物,有一定数目的碳原子和至少一个杂原子组成。在一个典型实施例中,杂原子选自B、O、N和S,其中氮和硫原子任选地被氧化,氮杂原子任选地被季铵化。杂原子或杂原子团可以位于杂烃基的任何内部位置(包括该烃基附着于分子其余部分的位置)。实例包括但不限于-CH2-CH2-O-CH3、-CH2-CH2-NH-CH3、-CH2-CH2-N(CH3)-CH3、-CH2-S-CH2-CH3、-CH2-CH2、-S(O)-CH3、-CH2-CH2-S(O)2-CH3、-CH=CH-O-CH3、-CH2-CH=N-OCH3和–CH=CH-N(CH3)-CH3。至多两个杂原子可以是连续的,例如-CH2-NH-OCH3。
除非另有规定,术语“烷氧基”、“烷氨基”和“烷硫基”(或硫代烷氧基)属于惯用表达,是指分别通过一个氧原子、氨基或硫原子连接到分子的其余部分的那些烷基基团。
除非另有规定,术语“环烃基”、“杂环烃基”或者其下位概念(比如芳基、杂芳基、环烷基、杂环烷基、环烯基、杂环烯基、环炔基、杂环炔基等等)本身或与其他术语联合分别表示环化的“烃基”、“杂烃基”。此外,就杂烃基或杂环烃基(比如杂烷基、杂环烷基)而言,杂原子可以占据该杂环附着于分子其余部分的位置。环烷基的实例包括但不限于环戊基、环己基、1-环己烯基、3-环己烯基、环庚基等。杂环基的非限制性实例包括1-(1,2,5,6-四氢吡啶基)、1-哌啶基、2-哌啶基,3-哌啶基、4-吗啉基、3-吗啉基、四氢呋喃-2-基、四氢呋喃吲哚-3-基、四氢噻吩-2-基、四氢噻吩-3-基,1-哌嗪基和2-哌嗪基。
除非另有规定,术语“芳基”表示多不饱和的芳族烃取代基,可以是单取代、二取代或多取代的,可以是一价、二价或者多价,它可以是单环或多环(比如1至3个环;其中至少一个环是芳族的),它们稠合在一起或共价连接。术语“杂芳基”是指含有一至四个杂原子的芳基(或环)。在一个示范性实例中,杂原子选自B、N、O和S,其中氮和硫原子任选地被氧化,氮原子任选地被季铵化。杂芳基可通过杂原子连接到分子的其余部分。芳基或杂芳基的非限制性实施例包括苯基、1-萘基、2-萘基、4-联苯基、1-吡咯基、2-吡咯基、3-吡咯基、3-吡唑基、2-咪唑基、4-咪唑基、吡嗪基、2-恶唑基、4-恶唑基、2-苯基-4-恶唑基、5-恶唑基、3-异恶唑基、4-异恶唑基、5-异恶唑基、2-噻唑基、4-噻唑基、5-噻唑基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-嘧啶基、4-嘧啶基、5-苯并噻唑基、嘌呤基、2-苯并咪唑基、5-吲哚基、1-异喹啉基、5-异喹啉基、2-喹喔啉基、5-喹喔啉基、3-喹啉基和6-喹啉基。
除非另有规定,芳基在与其他术语联合使用时(例如芳氧基、芳硫基、芳烷基)包括如上定义的芳基和杂芳基环。因此,术语“芳烷基”意在包括芳基附着于烷基的那些原子团(例如苄基、苯乙基、吡啶基甲基等),包括其中碳原子(如亚甲基)已经被例如氧原子代替的那些烷基,例如苯氧基甲基、2-吡啶氧甲基3-(1-萘氧基)丙基等。
术语“离去基团”是指可以被另一种官能团或原子通过取代反应(例如亲和取代反应)所取代的官能团或原子。例如,代表性的离去基团包括三氟甲磺酸酯;氯、溴、碘;磺酸酯基,如甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯、对甲苯磺酸酯等;酰氧基,如乙酰氧基、三氟乙酰氧基等等。
术语“保护基”包括但不限于“氨基保护基”、“羟基保护基”或“巯基保护基”。术语“氨基保护基”是指适合用于阻止氨基氮位上副反应的保护基团。代表性的氨基保护基包括但不限于:甲酰基;酰基,例如链烷酰基(如乙酰基、三氯乙酰基或三氟乙酰基);烷氧基羰基,如叔丁氧基羰基(Boc);芳基甲氧羰基,如苄氧羰基(Cbz)和9-芴甲氧羰基(Fmoc);芳基甲基,如
苄基(Bn)、三苯甲基(Tr)、1,1-二-(4'-甲氧基苯基)甲基;甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。术语“羟基保护基”是指适合用于阻止羟基副反应的保护基。代表性羟基保护基包括但不限于:烷基,如甲基、乙基和叔丁基;酰基,例如链烷酰基(如乙酰基);芳基甲基,如苄基(Bn),对甲氧基苄基(PMB)、9-芴基甲基(Fm)和二苯基甲基(二苯甲基,DPM);甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。
本发明所使用的溶剂可经市售获得。本发明采用下述缩略词:aq代表水;HATU代表O-(7-氮杂苯并三唑-1-基)-N,N,N',N'-四甲基脲六氟磷酸盐;EDC代表N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺盐酸盐;eq代表当量、等量;CDI代表羰基二咪唑;DCM代表二氯甲烷;PE代表石油醚;DIAD代表偶氮二羧酸二异丙酯;DMF代表N,N-二甲基甲酰胺;DMSO代表二甲亚砜;EtOAc代表乙酸乙酯;EtOH代表乙醇;MeOH代表甲醇;CBz代表苄氧羰基,是一种胺保护基团;BOC代表叔丁基羰基是一种胺保护基团;HOAc代表乙酸;NaCNBH3代表氰基硼氢化钠;r.t.代表室温;O/N代表过夜;THF代表四氢呋喃;Boc2O代表二-叔丁基二碳酸酯;TFA代表三氟乙酸;DIPEA代表二异丙基乙基胺;TsOH代表对甲苯磺酸;NFSI代表N-氟-N-(苯磺酰基)苯磺酰胺;NCS代表1-氯吡咯烷-2,5-二酮;n-Bu4NF代表氟化四丁基铵;iPrOH代表2-丙醇;mp代表熔点;LDA代表二异丙基胺基锂;MsCl代表甲基磺酰氯;THF代表四氢呋喃;Pd(dppf)Cl2代表[1,1'-双(二苯基磷)二茂铁]二氯化钯;DBU代表1.8-二氮杂二环十一烷-7-烯;TFA代表三氟乙酸;EtOAc或EA代表乙酸乙酯;EDCI代表N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺盐酸盐;DMAP代表4-二甲氨基吡啶;DIEA代表二异丙基胺;MTBE代表甲基叔丁基醚;BnBr代表苄溴;DAST代表二乙胺基三氟化硫。
化合物经手工或者 软件命名,市售化合物采用供应商目录名称。
软件命名,市售化合物采用供应商目录名称。
技术效果:
本发明化合物针对JAK2的选择性优于Tofacitinib。
具体实施方式
中间体化合物的制备
中间体1~5的制备

步骤1:制备4-(4-氨基嘧啶-2-基)吡唑-1-羧酸叔丁酯(2)
将2-氯-4-氨基嘧啶(3.0g,23.2mmol)、4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)吡唑-1-羧酸叔丁酯(8.2g,27.8mmol)、碳酸钾(9.6g,69.5mmol)溶于二氧六环(30mL)和水(5mL)的混合溶剂中。稍后加入Pd(dppf)Cl2(1.7g,2.3mmol),抽真空并用氮气置换。氮气保护下,将反应液置于80℃油浴中搅拌2小时,TLC跟踪检测反应完全。反应液冷却后,硅藻土过滤,乙酸乙酯(100mL)和四氢呋喃(100mL)洗涤滤饼。滤液经无水硫酸钠干燥,过滤,减压浓缩。将残余物用硅胶色谱柱法纯化(石油醚/乙酸乙酯=2/1~1/1洗脱),得到浅黄色油状物4-(4-氨基嘧啶-2-基)吡唑-1-羧酸叔丁酯(4.50g,产率为59.49%)。1H NMR(400MHz,DMSO-d6)δ=8.50(s,1H),8.17(s,1H),8.07(d,J=5.8Hz,1H),6.92(br.s.,2H),6.30(d,J=5.8Hz,1H),1.58(s,9H).MS(ESI)计算值C12H15N5O2[M+H]+262,测定值262.
步骤2:制备4-(4-(3-(乙氧羰基)硫脲)嘧啶-2-基)-1H-吡唑-1-甲酸叔丁酯(3)
向溶有4-(4-氨基嘧啶-2-基)吡唑-1-羧酸叔丁酯(4.0g,15.3mmol)的四氢呋喃(40mL)和二氯甲烷(40mL)的溶液中加入乙氧羰基异硫氰酸酯(4g,30.6mmol)。将反应液加热到70℃搅拌16小时。TLC显示反应完全后。反应液减压浓缩,经硅胶色谱柱法纯化(石油醚:乙酸乙酯=10:1~2:1洗脱),得到浅黄色油状物4-(4-(3-(乙氧羰基)硫脲)嘧啶-2-基)-1H-吡唑-1-甲酸叔丁酯(4.00g,产率为63.25%)。1H NMR(400MHz,DMSO-d6)δ=12.49(br.s.,1H),12.16(br.s.,1H),8.78(d,J=5.8Hz,1H),8.72(s,1H),8.32(s,1H),8.22(br.s.,1H),4.27(q,J=7.1Hz,2H),1.62(s,9H),1.30(t,J=7.2Hz,3H).MS(ESI)计算值C16H20N6O4S[M+H]+393,测定值393.
步骤3:制备5-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-c]嘧啶-2-胺(中间体1)
向溶有盐酸羟胺(3.5g,50.9mmol)的甲醇(50mL)和乙醇(50mL)溶液中加入DIEA(4.0g,30.6mmol)。生成的浑浊液在26℃下搅拌1小时后,加入4-(4-(3-(乙氧羰基)硫脲)嘧啶-2-基)-1H-吡唑-1-甲酸叔丁酯(4.0g,10.2mmol),然后将反应液加热到90℃回流3小时,TLC显示反应完全。反应液减压浓缩,加入水(20mL),过滤收集生成的沉淀,真空干燥得到白色固体5-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-c]嘧啶-2-胺(1.7g,产率为82.9%)。1HNMR(400MHz,DMSO-d6)δ=13.53(br.s.,1H),8.87(br.s.,1H),8.47(br.s.,1H),8.10(d,J=6.0Hz,1H),7.23(d,J=6.0Hz,1H),6.50(s,2H).MS(ESI)计算值C8H7N7[M+H]+202,测定值202.
步骤4:制备3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-3-(氰基甲基)氮杂环丁烷-1-羧酸叔丁酯(中间体2)
向微溶有中间体1(500mg,2.5mmol)的乙腈(10mL)的悬浊液中加入3-(氰基甲基烯基)氮杂环丁烷-1-羧酸叔丁酯(600mg,3.1mmol)和DBU(756mg,4.97mmol)。反应液在26℃下搅拌16小时。TLC显示反应完全。反应液减压浓缩,经硅胶色谱柱法纯化(石油醚:乙酸乙酯=1:1~1:3洗脱),得到白色固体3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-3-(氰基甲基)氮杂环丁烷-1-羧酸叔丁酯(800mg,产率为81.3%)。1H NMR(400MHz,CDCl3)δ=9.03(s,1H),8.64(s,1H),8.17(d,J=6.0Hz,1H),7.24(d,J=6.0Hz,1H),4.83(s,2H),4.57(d,J=9.8Hz,2H),4.33(d,J=9.8Hz,2H),3.36(s,2H),1.49(s,9H).MS(ESI)计算值C18H21N9O2[M+H]+396,测定值396.
步骤5:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]环丁胺-3-基]乙腈(中间体3)
往中间体2(500mg,1.3mmol)的DCM(10mL)溶液中于15℃下加入TFA(4mL)并在此温度下搅拌反应3小时。反应完成后,减压浓缩干反应液得棕色固体2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]环丁胺-3-基]乙腈(515mg,产率为99.9%,TFA盐)。MS(ESI)计算值C13H13N9[M+H]+296,测定值296.
步骤6:制备3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基]吡唑-1-基]氮杂环丁烷-1-羧酸叔丁酯(中间体4)
向微溶有3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-3-(氰基甲基)氮杂环丁烷-1-羧酸叔丁酯(400mg,1.0mmol)的乙腈(8mL)的浑浊液中加入环丙基甲酰氯(317mg,3.0mmol)和三乙胺(307mg,3.0mmol)。反应液在26℃下搅拌16小时。TLC显示反应完全,LC-MS显示全部生成为二取代产物。反应液减压浓缩后,加入甲胺的乙醇溶液(27%~32%,3mL),26℃搅拌0.5小时。LC-MS显示全部生成为一取代目标产物。反应液减压浓缩,经硅胶色谱柱法纯化(石油醚:乙酸乙酯=1:1~1:3洗脱),得到白色固体3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基]吡唑-1-基]氮杂环丁烷-1-羧酸叔丁酯(420mg,产率为89.7%)。1H NMR(400MHz,CDCl3)δ=9.31(s,1H),9.11(s,1H),8.67(s,1H),8.28(d,J=6.0Hz,1H),7.42(d,J=6.0Hz,1H),4.58(d,J=9.5Hz,2H),4.34(d,J=9.5Hz,2H),3.37(s,2H),1.50(s,9H),1.31-1.22(m,3H),1.03(qd,J=3.7,7.4Hz,2H).MS(ESI)计算值C22H25N9O3[M+H]+464,测定值464.
步骤7:制备N-[5-[1-[3-(氰基甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(中间体5)
向溶解有3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基]吡唑-1-基]氮杂环丁烷-1-羧酸叔丁酯(220mg,474.7umol)的二氯甲烷(8mL)溶液中加入三氟乙酸(2mL)。反应液在26℃下搅拌2小时。TLC显示反应完全。反应液减压浓缩,得到浅黄色固体N-[5-[1-[3-(氰基甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(280mg,粗品直接用于下一步反应)。MS(ESI)计算值C17H17N9O[M+H]+364,测定值364.
中间体6~8的制备

步骤1:制备乙基-N-[(6-溴-2-吡啶基)硫代氨基甲酰基]氨基甲酸(2)
往溶有6-溴吡啶-2-胺(30g,173.4mmol)的二氯甲烷(400mL)中慢慢滴加异硫氰酸乙酯(25.0g,190.7mmol),加完后25℃反应16个小时。TLC监测显示反应完全后,反应液减压蒸馏,得到的残余物用200mL石油醚搅拌洗涤30分钟,过滤,收集滤饼干燥得到淡红色固体乙基-N-[(6-溴-2-吡啶基)硫代氨基甲酰基]氨基甲酸(51g,产率为96.7%).1H NMR(400MHz,DMSO-d6)δ=12.17(s,1H),11.66(br.s.,1H),8.65(d,J=7.54Hz,1H),7.82(t,J=7.92Hz,1H),7.49(d,J=7.78Hz,1H),4.22(q,J=7.18Hz,2H),1.25(t,J=7.16Hz,3H).MS(ESI)计算值C9H10BrN3O2S[M+H]+304,测定值304.
步骤2:制备5-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(3)
将盐酸羟胺(35.2g,503.1mmol)、二异丙基乙胺(54.1g,419.3mmol)溶于乙醇(500mL)和甲醇(500mL)的混合溶剂中25℃搅拌1小时后,加入乙基-N-[(6-溴-2-吡啶基)硫代氨基甲酰基]氨基甲酸(51.0g,167.7mmol)经氮气换气3次,加热到80℃反应3小时,冷却。TLC监测显示反应完全后,反应液减压蒸馏,得到的残留物用(500mL)水搅拌洗涤10分钟,过滤,收集滤饼干燥得到白色固体5-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(32g,产率为85.1%).1H NMR(400MHz,DMSO-d6)7.30-7.39(m,1H),7.20(dd,J=6.78,1.76Hz,1H),6.27(s,2H).MS(ESI)计算值C6H5Br N4[M+H]+215,测定值215.
步骤3:制备N-(5-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙甲酰胺(4)
在0℃下,往溶有5-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(15.00g,70.41mmol和三乙胺(21.4g,211.2mmol)的乙腈(150mL)中慢慢滴加环丙基甲酰氯(8.8g,84.5mmol),加完后混合液升到室温反应16个小时。TLC监测显示原料反应完全后,反应液减压蒸馏,得到的残留物溶解于甲胺醇(150mL)溶液中,加热到80℃反应1小时,冷却,减压蒸馏再次得到残留物溶于水(100mL)和乙酸乙酯(200mL)的混合液中,分层萃取,合并有机相并用无水硫酸钠干燥,过滤,滤液减压蒸馏得到的粗产品用硅胶色谱柱法纯化(乙酸乙酯/石油醚=0~70%洗脱),得到淡黄色固体N-(5-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙甲酰胺(7.2g,产率为56.64%).1H NMR(400MHz,DMSO-d6)δ=11.20(br.s.,1H),7.68-7.73(m,1H),7.52-7.58(m,1H),7.46-7.51(m,1H),1.96-2.09(m,1H),0.82(d,J=6.28Hz,4H).MS(ESI)计算值C10H9Br N4O[M+H]+282测定值282.
步骤4:制备N-[5-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺(中间体6)
在氮气的氛围下,向溶有N-(5-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺(3.0g,10.67mmol)、4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-1H-吡唑(2.4g,12.9mmol)和碳酸钾(3.7g,26.7mmol)的二氧六环(30mL)和(5mL)水的混合液中加入Pd(dppf)Cl2(260mg),得到的混合液加热到110℃反应3小时。冷却至室温,TLC监测显示原料反应完全后,反应液过滤,滤液用水(150mL)洗涤,乙酸乙酯(150mL×3)萃取,合并有机相并用无水硫酸钠干燥,过滤,滤液减压蒸馏得到的粗产品用硅胶色谱柱法纯化(乙酸乙酯/石油醚=50~100%洗脱),得到灰色固体N-[5-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺(2.1g,产率为62.4%).1H NMR(400MHz,DMSO-d6)δ=13.37(br.s.,1H),11.15(br.s.,1H),8.96(s,1H),8.53(s,1H),7.57-7.72(m,2H),7.51(d,J=8.28Hz,1H),2.06(br.s.,1H),0.78-0.91(m,4H).MS(ESI)计算值C13H12N6O[M+H]+269,测定值269.
步骤5:制备3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三氮唑并[1,5-a]吡啶-5-基]吡唑-1-基]氮杂环丁烷-1-羧酸叔丁酯(中间体7)
向溶有N-[5-(1H-吡唑-4-基-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺(200mg,745.5umol)、3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁酯(144.8mg,745.5umol)的乙腈(5mL)液中加入DBU(340.49mg,2.3mmol),室温反应16小时,LCMS监测显示原料反应完全后,反应液减压蒸馏,得到的残留物溶于水(20mL)和乙酸乙酯(20mL)的混合液中,分层萃取,合并有机相并用无水硫酸钠干燥,过滤,滤液减压蒸馏得到的粗产品用制备型TLC法(纯乙酸乙酯)纯化,得到淡黄色固体3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三氮唑并[1,5-a]吡啶-5-基]吡唑-1-基]氮杂环丁烷-1-羧酸叔丁酯(170mg,产率为44.4%).MS(ESI)计算值C23H26N8O3[M+H]+463,测定值463.
步骤6:制备N-[5-[1-[3-(氰基甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺(中间体8)(WX00)
向溶有3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三氮唑并[1,5-a]吡啶-5-基]吡唑-1-基]氮杂环丁烷-1-羧酸叔丁酯(150mg,324.3umol)的5mL二氯甲烷(5mL)溶液中加入三氟乙酸(1mL)室温反应2小时。LCMS监测显示反应完全后,反应液减压蒸馏得到黄色油状N-[5-[1-[3-(氰基甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺(100mg)的粗产品直接用于下一步。1H NMR(400MHz,DMSO-d6)δ=11.16(br.s.,1H),9.18(s,1H),8.71(s,1H),7.75-7.63(m,2H),7.61-7.50(m,1H),4.00(d,J=9.0Hz,2H),3.71(d,J=9.0Hz,2H),3.57(s,2H),2.13(br.s.,1H),0.95-0.81(m,4H).MS(ESI)计算值C18H18N8O[M+H]+363,测定值363.
中间体9的制备

步骤1:制备3-(氰甲基)氮杂环丁烯-1-碳酸叔丁酯(1)
冰浴冷却下,向钠氢(1.2g,30.7mmol)的四氢呋喃(50mL)溶液中滴加氰甲基亚磷酸二乙酯(5.7g,32.1mmol)的四氢呋喃(50mL)溶液。滴加完成后,混合物于25℃搅拌1hr后再冷却至0℃。然后于一小时内滴加3-氮杂环丁酮-1-碳酸叔丁酯(5.0g,29.2mmol)的四氢呋喃(50mL)溶液。混合物于25℃搅拌反应16小时。反应完成后,反应液用水(80mL)淬灭后用乙酸乙酯(80mL×3)萃取,合并有机相用饱和食盐水洗涤,无水硫酸钠干燥后,旋干得到黄色固体粗产物3-(氰甲基)氮杂环丁烯-1-碳酸叔丁酯(5.2g,产率为78.0%),粗产物可直接用于下一步反应,无须进行进一步纯化。1H NMR(400MHz,CDCl3)δ=5.38(t,J=2.5Hz,1H),4.73-4.68(m,2H),4.61(td,J=2.4,4.3Hz,2H),1.45(s,9H).MS(ESI)计算值C10H14N2O2[M+H]+195,测定值195.
步骤2:制备2-(氮杂环丁-3-基烯)甲基氰(2)
将3-(氰甲基)氮杂环丁烯-1-碳酸叔丁酯(5.2g,26.8mmol)用少量乙酸乙酯(5mL)浸润,搅拌均匀后于0℃加入盐酸乙酸乙酯(150mL),0℃下搅拌一小时。TLC(石油醚:乙酸乙酯=5:1)检测反应完成。反应所得黄色悬浊液过滤,固体用少量冷的乙酸乙酯(5mL×2)洗涤,真空下干燥得到白色固体2-(氮杂环丁-3-基烯)甲基氰(2.8g,产率为80.0%)。1H NMR(400MHz,D2O)δ=5.69-5.65(m,1H),4.95(d,J=2.5Hz,2H),4.88(br.s.,2H).MS(ESI)计算值C5H6N2[M+H]+95测定值95.
步骤3:制备2-(1-(乙基磺酰基)氮杂环丁-3-基烯)甲基氰(中间体9)
氮气保护下,于0℃下向2-(氮杂环丁-3-基烯)甲基氰(2.8g,21.4mmol)和DIPEA(8.3g,64.3mmol)的二氯甲烷(30mL)溶液中滴加乙基磺酰氯(4.1g,32.1mmol),滴加时保持温度在2℃以下。反应混合物于25℃搅拌反应16小时。TLC(石油醚:乙酸乙酯=1:1)检测反应完成。反应液用水淬灭后,用二氯甲烷(30mL×2)萃取。合并有机相用饱和食盐水(20mL×2)洗涤,无水硫酸钠干燥,过滤旋干。残留物用柱层析法(二氯甲烷:乙酸乙酯=3/1)纯化得到浅黄色固体2-(1-(乙基磺酰基)氮杂环丁-3-基烯)甲基氰(1.4g,产率为33.0%)。1H NMR(400MHz,CDCl3)δ=5.50-5.41(m,1H),4.79(d,J=3.0Hz,2H),4.71(d,J=2.5Hz,2H),3.06(q,J=7.4Hz,2H),1.40(t,J=7.4Hz,3H).MS(ESI)计算值C7H10N2O2S[M+H]+187测定值187.
步骤3’:制备2-(1-环丙基磺酰基氮杂环丁烷-3-烯基)乙腈(中间体10)
运用制备中间体9的同样方法制备中间体10.2-(1-环丙基磺酰基氮杂环丁烷-3-烯基)乙腈(1.5g),浅黄色固体,MS(ESI)计算值C7H10N2O2S[M+H]+199,测定值199.
实施例1

步骤1:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-乙基磺酰基氮杂环丁烷-3-基]乙腈(1)
向微溶有中间体1(150mg,745.6umol)乙腈(4mL)和DMF(2mL)的悬浊液中加入中间体9(208mg,1.1mmol)和DBU(227mg,1.5mmol)。该反应液在26℃下搅拌16小时。LC-MS显示反应完全。过滤收集析出的固体,并用冷乙腈(5mL)洗涤,减压干燥得到白色固体2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-乙基磺酰基氮杂环丁烷-3-基]乙腈(200mg,产率为69.2%)。1H NMR(400MHz,DMSO-d6)δ=9.16(s,1H),8.69(s,1H),8.16(d,J=6.3Hz,1H),7.31(d,J=6.0Hz,1H),6.56(s,2H),4.53(d,J=9.0Hz,2H),4.28(d,J=9.0Hz,2H),3.70(s,2H),3.25(q,J=7.4Hz,2H),1.25(t,J=7.4Hz,3H).MS(ESI)计算值C15H17N9O2S[M+H]+388,测定值388.
步骤2:制备N-[5-[1-[3-(氰基甲基)-1-乙磺酰基氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(WX01)
向微溶有2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-乙基磺酰基氮杂环丁烷-3-基]乙腈(100mg,258.1umol)的乙腈(2mL)和四氢呋喃(1mL)的悬浊液中加入环丙基甲酰氯(80.9mg,774.4umol)和三乙胺(78mg,774.4umol)。反应液在26℃下搅拌16小时,TLC显示反应完全,LC-MS显示全部生成为二取代产物。反应液减压浓缩后,加入甲胺的乙醇溶液(27%~32%,3mL),并在26℃下搅拌反应0.5小时后。LC-MS显示全部生成为一取代产物。反应液减压浓缩,并通过制备型HPLC(碱性条件)进行纯化,得到N-[5-[1-[3-(氰基甲基)-1-乙磺酰基氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(25mg,产率为21.1%)。1H NMR(400MHz,DMSO-d6)δ=11.43(br.s.,1H),9.25(s,1H),8.82(s,1H),8.32(d,J=6.0Hz,1H),7.60(d,J=6.3Hz,1H),4.50(d,J=9.0Hz,2H),4.28(d,J=9.0Hz,2H),3.70(s,2H),3.24(q,J=7.3Hz,2H),2.18-2.02(m,1H),1.23(t,J=7.3Hz,3H),0.95-0.80(m,4H).MS(ESI)计算值C19H21N9O3S[M+H]+456,测定值456.
实施例2

步骤1:制备N-[5-[1-[3-(氰基甲基)-1-(三氟甲基磺酰基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(WX02)
向微溶有中间体5(100mg,209.5umol)的二氯甲烷(3mL)浑浊液中加入三氟甲磺酰氯(53mg,314.2umol)和三乙胺(106mg,1.1mmol)。反应液在26℃下搅拌16小时,LC-MS显示反应完全。反应液减压浓缩,并通过制备型HPLC(碱性条件)进行纯化,得到N-[5-[1-[3-(氰基甲基)-1-(三氟甲基磺酰基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(25mg,产率为24.1%)。1H NMR(400MHz,DMSO-d6)δ=11.45(br.s.,1H),9.27(s,1H),8.90(s,1H),8.35(d,J=6.0Hz,1H),7.63(d,J=6.3Hz,1H),4.90(d,J=9.0Hz,2H),4.72(d,J=9.0Hz,2H),3.85(s,2H),1.30-1.23(m,1H),0.97-0.87(m,4H).MS(ESI)计算值C18H16F3N9O3S[M+H]+496,测定值496.
实施例3

步骤1:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-环丙基磺酰基-氮杂环丁烷-3-基]乙腈(2)
向溶解有中间体1(150mg,745.6umol)的乙腈(4mL)悬浊液中加入中间体10(192mg,969.2umol)和DBU(227mg,1.5mmol)。反应液在26℃下搅拌16小时,TLC显示反应完全。过滤收集析出的固体,并用冷乙腈(5mL)洗涤,减压干燥得到白色固体2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-环丙基磺酰基-氮杂环丁烷-3-基]乙腈(200mg,产率为67.2%)。1H NMR(400MHz,DMSO-d6)δ=9.18(s,1H),8.70(s,1H),8.15(d,J=6.3Hz,1H),7.30(d,J=6.0Hz,1H),6.56(s,2H),4.59(d,J=9.3Hz,2H),4.33(d,J=9.3Hz,2H),3.70(s,2H),2.90-2.82(m,1H),1.09-1.03(m,2H),1.03-0.96(m,2H).MS(ESI)计算值C16H17N9O2S[M+H]+400,测定值400.
步骤2:制备N-[5-[1-[3-(氰基甲基)-1-环丙基磺酰基-氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(WX03)
向微溶有2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-环丙基磺酰基-氮杂环丁烷-3-基]乙腈(80mg,200.3umol)的乙腈(2mL)悬浑浊液中加入环丙基甲酰氯(63mg,600.9umol)和三乙胺(61mg,600.9umol)。反应液在26℃下搅拌16小时,80℃下再搅拌3小时,LC-MS显示为一取代产物和二取代产物的混合物。反应液减压浓缩后,加入甲胺的乙醇溶液(27%~32%,3mL),26℃搅拌0.5小时,LC-MS显示全部生成为一取代产物。反应液减压浓缩,并通过制备型HPLC(碱性条件)进行纯化,得到N-[5-[1-[3-(氰基甲基)-1-环丙基磺酰基-氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丙烷甲酰胺(60mg,产率为64.1%)。1H NMR(400MHz,DMSO-d6)δ=11.43(s,1H),9.27(s,1H),8.81(s,1H),8.32(d,J=6.0Hz,1H),7.60(d,J=6.0Hz,1H),4.57(d,J=9.3Hz,2H),4.32(d,J=9.3Hz,2H),3.70(s,2H),2.92-2.79(m,1H),2.07(d,J=13.6Hz,1H),1.07-0.96(m,4H),0.91-0.83(m,4H).MS(ESI)计算值C20H21N9O3S[M+H]+468,测定值468.
实施例4

步骤1:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-甲磺酰基-环丁胺-3-基]乙腈(WX04)
往溶有中间体3(1.0g,2.4mmol,TFA盐)和三乙胺(617mg,6.1mmol)的DCM(50mL)溶液中在15℃下滴加MsCl(307mg,2.7mmol),滴加完成后,反应混合物在15℃下搅拌2小时。反应完成后,将反应物浓缩干,所得固体经制备HPLC(碱性方法)分离纯化得2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-三氟甲磺酰基-环丁胺-3-基]乙腈(800mg,产率为87.8%)。1H-NMR(400MHz,DMSO-d6)δ=9.17(s,1H),8.70(s,1H),8.15(d,J=6.0Hz,1H),7.30(d,J=6.0Hz,1H),6.56(brs,2H),4.55(d,J=9.2Hz,2H),4.31(d,J=9.2Hz,2H),3.69(s,2H),3.14(s,3H).MS(ESI)计算值C14H12F3N9O2S[M+H]+428,测定值428.
实施例5

步骤1:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑[1,5-c]嘧啶-5-基)吡唑-1-基]-1-(三氟甲磺酰基)环丁胺-3-基]乙腈(WX05)
往溶有中间体3(515mg,1.7mmol)和TEA(264mg,2.6mmol)的DCM(10mL)溶液中,于15℃、氮气保护下滴加三氟甲磺酰氯(323mg,1.9mmol),滴加完成后将反应混合物在15℃下搅拌3小时。反应完全后,浓缩干燥,所得固体用水(20mL)打浆、过滤,滤饼干燥后得白色固体2-[3-[4-(2-氨基-[1,2,4]三氮唑[1,5-c]嘧啶-5-基)吡唑-1-基]-1-(三氟甲磺酰基)环丁胺-3-基]乙腈(700mg,产率为94.1%)。1H-NMR(400MHz,DMSO-d6)δ=9.18(s,1H),8.74(s,1H),8.15(d,J=6.0Hz,1H),7.30(d,J=6.0Hz,1H),6.55(brs,2H),4.91(d,J=9.2Hz,2H),4.70(d,J=9.2Hz,2H),3.82(s,2H).MS(ESI)计算值C14H12F3N9O2S[M+H]+428,测定值428.
步骤2:制备N-[5-[1-[3-(腈甲基)-1-(三氟甲磺酰基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丁胺-3-甲酰胺(WX06)
往N-叔丁基甲酸环丁胺-3-甲酸(198mg,982.8umol)和DMF(100uL)的DCM(10mL)溶液中在氮气保护下,于0℃滴加草酰氯(156mg,1.23mmol)的DCM(1mL)溶液滴加完成后,将反应混合物在0℃下搅拌2小时。然后将反应混合物在15℃减压浓缩干,所得液体溶解在DCM(2mL)中,将其在氮气保护0℃下通过注射器滴加到2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基)吡唑-1-基]-1-三氟甲磺酰基-环丁胺-3-基]乙腈(350mg,818.9umol)的DCM(10mL)溶液中。滴加完成后,反应混合物在0℃下搅拌2小时。将反应液直接用制备薄层色谱(DCM/MeOH=10/1)纯化得Boc保护的产物(10mg)。将该产物溶解在DCM(2mL)中,在15℃下,往该溶液中加入TFA(2mL),所得混合物在15℃搅拌1小时。反应完成后,浓缩干燥,所得固体经制备HPLC(碱性方法)分离纯化得N-[5-[1-[3-(腈甲基)-1-(三氟甲磺酰基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-c]嘧啶-2-基]环丁胺-3-甲酰胺(2mg,产率为0.42%)。MS(ESI)计算值C18H17F3N10O3S[M+H]+511,测定值511.
实施例6

步骤1:制备叔丁基-3-(腈基甲基)-3-[4-[2-[(3-羟基环丁烷甲酰基)氨基]-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基]吡唑-1-基]环丁烷-1-甲酸酯(1)
将中间体2(300mg,758.7umol),3-羟基环丁烷甲酸(106mg,910.4umol)和EDCI(218mg,1.1mmol)的混合物加入到吡啶(10mL)中,然后将所得混合物在氮气保护下加热回流16小时。浓缩干后。剩余固体经制备薄层色谱(DCM/MeOH=10/1)纯化得白色固体叔丁基-3-(腈基甲基)-3-[4-[2-[(3-羟基环丁烷甲酰基)氨基]-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基]吡唑-1-基]环丁烷-1-甲酸酯(36mg,产率为9.61%)。MS(ESI)计算值C23H27N9O4[M+H]+494,测定值494.
步骤2:制备N-[5-[1-[3-(腈基甲基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3-羟基-环丁烷甲酰胺(2)
往叔丁基-3-(腈基甲基)-3-[4-[2-[(3-羟基环丁烷甲酰基)氨基]-[1,2,4]三氮唑并[1,5-c]嘧啶-5-基]吡唑-1-基]环丁烷-1-甲酸酯(36mg,72.9umol)的二氯甲烷(2.00mL)溶液中、于15℃下滴加TFA(1mL),所得混合物在15℃下搅拌30min。LCMS显示反应完全。反应混合物在30℃下浓缩干得黄色固体N-[5-[1-[3-(腈基甲基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3-羟基-环丁烷甲酰胺(37mg,产率为99.9%,TFA盐)。MS(ESI)计算值C18H19N9O2[M+H]+394,测定值394.
步骤3:制备N-[5-[1-[3-(腈甲基)-1-(三氟甲磺酰基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3-羟基-环丁烷甲酰胺(WX07)
往N-[5-[1-[3-(腈基甲基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3-羟基-环丁烷甲酰胺(15mg,29.6umol)和三乙胺(9mg,88.7umol)的DCM(5.00mL)溶液中,并于20℃、N2保护下滴加三氟甲磺酰氯(7mg,44.34umol)的DCM(1mL)溶液。加完后,将该混合物在20℃下搅拌1小时。反应完成后,浓缩反应液至干。所得固体用制备HPLC(0.1%NH4OH为添加物)分离纯化得N-[5-[1-[3-(腈甲基)-1-(三氟甲磺酰基)环丁胺-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3-羟基-环丁烷甲酰胺(8mg,产率为51.50%)。1H-NMR(400MHz,MeOD-d4)δ=9.29(s,1H),8.73(s,1H),8.32(d,J=6.4Hz,1H),7.49(d,J=6.0Hz,1H),5.00(d,J=9.6Hz,2H),4.70(d,J=9.2Hz,2H),4.05-4.13(m,1H),3.70(s,2H),2.90(brs,1H),2.50-2.66(m,2H),2.20-2.35(m,2H).MS(ESI)计算值C19H18F3N9O4S[M+H]+526,测定值526.
实施例7

步骤1:制备N-(5-(1-(3-(氰基甲基)-1-(三氟甲基磺酰基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺
在0℃下,往溶有中间体8(100mg,275.9umol)和三乙胺(84mg,827.9umol)的二氯甲烷(5mL)中慢慢滴加三氟甲基磺酰氯(56mg,331.4umol),加完后混合液升到室温反应16个小时。LCMS监测显示反应完全后,反应液用水(20mL)稀释,二氯甲烷(20mL×3)萃取,合并有机相并用无水硫酸钠干燥,过滤,滤液减压蒸馏得到的残留物用制备薄层色谱法(乙酸乙酯)纯化,得到白色固体N-(5-(1-(3-(氰基甲基)-1-(三氟甲基磺酰基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺(WX09,45mg,产率为31.33%).1H NMR(400MHz,METHANOL-d4)δ=9.21(s,1H),8.59(s,1H),7.71-7.77(m,1H),7.60(dd,J=14.44,8.16Hz,2H),5.00(d,J=9.04Hz,2H),4.70(d,J=9.04Hz,2H),3.68(s,2H),1.28-1.39(m,1H),1.11(quin,J=3.84Hz,2H)0.97-1.04(m,2H).MS(ESI)计算值C19H17N8O3F3S[M+H]+495,测定值495.
WX08的制备:用类似于WX09(步骤1)的制备方法制备。得到N-(5-(1-(3-(氰基甲基)-1-(环丙基磺酰基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺(WX08)。1H NMR(400MHz,METHANOL-d4)δ=9.23(s,1H),8.58(s,1H),7.71-7.76(m,1H),7.58-7.65(m,2H),4.70(d,J=9.28Hz,2H),4.39(d,J=9.04Hz,2H),3.64(s,2H),2.72(dt,J=12.74,6.31Hz,1H),1.78(d,J=7.04Hz,1H),1.08-1.14(m,6H),1.00(dd,J=7.28,3.26Hz,2H).MS(ESI)计算值C21H22N8O3S[M+H]+467测定值467.
实施例8
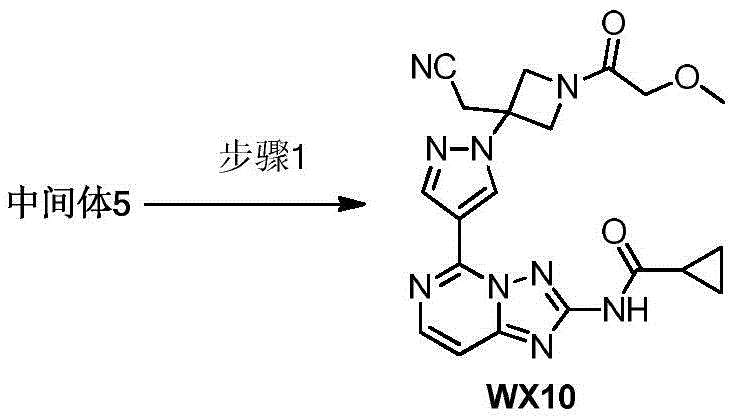
步骤1:制备N-[5-[1-[3-(氰甲基)-1-(2-甲氧基乙酰基)氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺
将2-甲氧基乙酸(11mg,128.4umol)溶于混合溶剂DCM/DMF(6mL,5:1),依次加入HOBt(35mg,256.9umol)和EDCI(49mg,256.9umol)。得到的混合物搅拌反应1小时。加入中间体5(70mg,192.6umol)和DIEA(50mg,385.3umol),15℃搅拌反应12小时。LC-MS显示原料消耗完全,目标产物生成。反应液减压浓缩除去DCM和DMF。制备HPLC(碱性)分离得到N-[5-[1-[3-(氰甲基)-1-(2-甲氧基乙酰基)氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺(30mg,产率为53.7%)。1H NMR(400MHz,DMSO-d6)δ=9.26(s,1H),8.80(s,1H),8.34(d,J=6.0Hz,1H),7.61(d,J=6.3Hz,1H),4.81(d,J=10.0Hz,1H),4.61(d,J=10.0Hz,1H),4.46(d,J=10.5Hz,1H),4.33(d,J=10.3Hz,1H),4.01(s,2H),3.72(s,2H),3.32(s,3H),2.12(br.s.,1H),0.98-0.84(m,4H).MS(ESI)计算值C20H21N9O3[M+H]+436,测定值436.
实施例9

步骤1:制备N-[5-[1-[3-(氰甲基)-1-(环丙甲酰基)氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺
将中间体5(79mg,216.3umol)溶解于二氯甲烷(3mL),加入DIEA(84mg,648.9umol)。然后5分钟内用针筒将环丙酰氯(27mg,259.6umol)加入,15℃反应液搅拌反应3小时。LC-MS显示原料消耗完全,目标产物生成。反应液减压浓缩除去DCM和DMF,制备型HPLC(碱性)分离得到N-[5-[1-[3-(氰甲基)-1-(环丙甲酰基)氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺(50mg,53.6%)。1H NMR(400MHz,DMSO-d6)δ=9.25(s,1H),8.83-8.78(m,1H),8.32(dd,J=3.8,6.0Hz,1H),7.60-7.52(m,1H),4.88(d,J=9.3Hz,1H),4.68(d,J=9.5Hz,1H),4.44(d,J=10.5Hz,1H),4.29(d,J=10.3Hz,1H),3.72(d,J=5.5Hz,2H),3.13(br.s.,1H),1.69-1.55(m,1H),0.98-0.85(m,4H),0.77(br.s.,4H)。MS(ESI)calcd for C21H21N9O2[M+H]+432,测定值432.
实施例10

步骤1:制备N-[5-[1-[3-(氰甲基)-1-甲磺酰基-氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺(WX12)
将中间体5(100mg,275.2umol)混悬于二氯甲烷(8mL)中,依次加入DIEA(107mg,825.6umol)和MsCl(140mg,1.2mmol)。反应液在15℃下搅拌反应2小时。LC-MS显示原料消耗完全,并监测到目标产物。反应液减压浓缩除去DCM,制备HPLC(碱性)分离得到N-[5-[1-[3-(氰甲基)-1-甲磺酰基-氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺(39mg,产率为31.1%)。1H NMR(400MHz,DMSO-d6)δ==9.28(s,1H),8.86(s,1H),8.35(d,J=6.3Hz,1H),7.63(d,J=6.3Hz,1H),6.08(br.s.,1H),4.55(d,J=9.5Hz,2H),4.33(d,J=9.3Hz,2H),3.72(s,2H),3.16(s,3H),2.10(d,J=14.8Hz,1H),1.01-0.79(m,4H).MS(ESI)计算值C18H19N9O3S[M+H]+442,测定值442.
实施例11

步骤1:制备N-[5-[1-[3-(氰甲基)-1-(二氟甲磺酰基)-氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺(WX13)
将中间体5(100mg,275.2umol)混悬于二氯甲烷(8mL)中,依次加入DIEA(178mg,1.4mmol)和二氟甲磺酰氯(62mg,412.8umol)。反应液在15℃搅拌反应12小时。LC-MS显示原料消耗完全,并监测到目标产物。反应液减压浓缩除去DCM,残余物用DMF和MeOH稀释成溶液(5ml),制备HPLC(碱性)分离得到N-[5-[1-[3-(氰甲基)-1-(二氟甲磺酰基)-氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]嘧啶2-]环丙基甲酰胺(8mg,6.1%)。1H NMR(400MHz,DMSO-d6)δ=11.46(br.s.,1H),9.27(s,1H),8.87(s,1H),8.34(d,J=6.3Hz,1H),7.63(d,J=6.0Hz,1H),7.42-7.06(m,1H),4.79(d,J=9.0Hz,2H),4.67-4.49(m,2H),3.78(s,2H),2.26-1.96(m,1H),0.97-0.84(m,4H).MS(ESI)计算值C18H17F2N9O3S[M+H]+478,测定值478.
实施例12

步骤1:制备N-[5-[1-[3-(氰甲基)1-(二氟甲磺酰基)氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]吡啶-2-]环丙基甲酰胺(WX14)
将中间体8(300mg,629.7umol,三氟乙酸盐)混悬于DCM(4mL)中,依次加入DMAP(8mg,63umol),DIEA(407mg,3.2mmol)和二氟甲磺酰氯(142mg,944.6umol)。混合物在15℃搅拌反应12小时。LC-MS显示原料消耗完全,并监测到目标产物。反应液减压浓缩除去DCM,用DMF和MeOH稀释成溶液(5ml),用制备型HPLC(碱性)分离,得到N-[5-[1-[3-(氰甲基)1-(二氟甲磺酰基)氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-c]吡啶-2-]环丙基甲酰胺(8mg,产率为2.7%)。1H NMR(400MHz,METHANOL-d4)δ=9.99(s,1H),9.57-9.44(m,1H),8.52-8.44(m,1H),8.39(d,J=7.3Hz,1H),8.33(d,J=8.3Hz,1H),8.01-7.68(m,1H),5.57(d,J=8.8Hz,2H),5.36(d,J=9.0Hz,2H),4.47(s,2H),1.81-1.61(m,4H).MS(ESI)计算值C19H18F2N8O3S[M+H]+477,测定值477.
实施例13

步骤1:制备N-[5-[1-[3-(氰甲基)-1-甲磺酰基-氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-a]嘧啶2-]环丙基甲酰胺
将中间体8(150mg,314.9umol,三氟乙酸盐)混悬于二氯甲烷(2mL),依次加入DIEA(203mg,1.6mmol),DMAP(11mg,94.5umol)和MsCl(180mg,1.6mmol)。反应液在15℃搅拌反应12小时。LC-MS显示原料消耗完全,并监测到目标产物。反应液减压浓缩除去DCM,用MeOH稀释成溶液(5ml)。制备HPLC(碱性)分离得到N-[5-[1-[3-(氰甲基)-1-甲磺酰基-氮杂环丁烷-3-]吡唑-4-]-[1,2,4]三氮唑[1,5-a]嘧啶2-]环丙基甲酰胺(10mg,7.15%)。1H NMR(400MHz,DMSO-d6)δ=11.17(br.s.,1H),9.27(s,1H),8.79(s,1H),7.83-7.68(m,1H),7.62(dd,J=7.8,17.6Hz,2H),4.51(d,J=9.0Hz,2H),4.34(d,J=9.0Hz,2H),3.69(s,2H),3.16(s,3H),2.25-2.00(m,1H),0.95-0.80(m,4H).MS(ESI)计算值C19H20N8O3S[M+H]+441,测定值441.
实施例14

步骤1:制备N-(5-(1-(3-(氰甲基)-1-(乙基磺酰基)氮杂环丁-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-a]吡啶-2-基)环丙甲酰胺(WX16)
氮气保护下,向中间体6(1.5g,5.6mmol)和中间体9(1.5g,7.8mmol)的乙腈(15mL)溶液中滴加DBU(1.7g,11.2mmol)。反应混合物于25℃搅拌反应16小时。TLC(石油醚:乙酸乙酯=0:1)检测反应完成。于0℃下将反应液倒入甲醇(200mL)中,立即析出大量固体,搅拌10min后过滤,所得固体用少量甲醇(5mL)洗涤后,真空下干燥得到产物N-(5-(1-(3-(氰甲基)-1-(乙基磺酰基)氮杂环丁-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-a]吡啶-2-基)环丙甲酰胺(1.60g,产率为60%)。1H NMR(400MHz,DMSO-d6)δ=9.25(s,1H),8.79(s,1H),7.76-7.70(m,1H),7.64(d,J=7.0Hz,1H),7.60(d,J=8.5Hz,1H),4.49(d,J=9.0Hz,2H),4.32(d,J=9.0Hz,2H),3.68(s,2H),1.26(t,J=7.3Hz,3H),0.91-0.85(m,4H).MS(ESI)计算值C20H22N8O3S[M+H]+455,测定值455.
实施例15

步骤1:制备3-[4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶-1-]-3-(氰基乙基)氮杂环丁烷-1-甲酸叔丁酯
5-(1H-吡唑-4-)-[1,2,4]三氮唑[1,5-a]吡啶-2-胺(700mg,3.5mmol)和3-(氰甲基)氮杂环丁烷-1-甲酸叔丁酯(747mg,3.9mmol)溶于乙腈(20.00mL),加入DBU(1.6g,10.5mmol),混合物在40℃反应3小时。LC-MS显示原料反应完全,并检测到目标产物。混合物倒入水中(30mL),搅拌30分钟。水相用乙酸乙酯(20mL×3)萃取。合并有机层,饱和食盐水(20mL×2)洗涤,无水Na2SO4干燥,过滤,减压浓缩得到棕色固体3-[4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶-1-]-3-(氰基乙基)氮杂环丁烷-1-甲酸叔丁酯(1.33g,粗品)。MS(ESI)计算值C19H22N8O2[M+H]+395,测定值395.
步骤2:制备3-(氰甲基-3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]氮杂环丁烷-1-甲酸叔丁酯
3-[4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶-1-]-3-(氰基乙基)氮杂环丁烷-1-甲酸叔丁酯(150mg,380.3umol)溶于四氢呋喃(2mL),加入t-BuONO(59mg,570.5umol),15℃搅拌反应3小时。LC-MS显示原料反应完全,并检测到目标产物。反应液减压浓缩,用DCM(4ml)稀释,制备TLC分离(DCM:MeOH=10:1),得到3-(氰甲基-3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]氮杂环丁烷-1-甲酸叔丁酯(80mg,产率为55.4%)。MS(ESI)计算值C19H21N7O2[M+H]+380,测定值380.
步骤3:制备2-[3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]氮杂环丁烷-3-]乙腈
3-(氰甲基-3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]氮杂环丁烷-1-甲酸叔丁酯(80mg,210.9umol)混悬于DCM(1.5mL),加入三氟乙酸(857mg,7.5mmol),15℃搅拌反应3小时。LC-MS显示反应完全并检测到目标产物MS。反应液减压浓缩溶剂和剩余的三氟乙酸,得到的棕色粘稠物2-[3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]氮杂环丁烷-3-]乙腈(129mg,粗品)。MS(ESI)计算值C14H13N7[M+H]+280,测定值280.
步骤4:制备2-[3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]-1-(三氟甲磺酰基)氮杂环丁烷-3-]乙腈2-[3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]氮杂环丁烷-3-]乙腈(60mg,214.8umol)溶于DCM(2mL),加入DMAP(13mg,107.4umol)和Et3N(109mg,1.1mmol),然后在15℃滴加三氟甲磺酰氯(47mg,279.3umol)。反应物搅拌反应在15℃反应4小时。LC-MS显示原料反应完全并检测到目标产物MS。反应液减压浓缩溶剂。制备型HPLC分离(碱性)得到2-[3-[4-([1,2,4]三氮唑[1,5-a]吡啶-5-)吡唑-1-]-1-(三氟甲磺酰基)氮杂环丁烷-3-]乙腈(25mg,产率为28.3%)。1H NMR(400MHz,DMSO-d6)δ=9.28(br.s.,1H),8.73(d,J=17.8Hz,2H),7.82(br.s.,3H),5.22-4.50(m,4H),3.86(br.s.,2H).MS(ESI)计算值C15H12F3N7O2S[M+H]+412,测定值412.
实施例16

步骤1:制备2-(3-(4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶)-1H-吡唑)-1-(三氟甲磺酰基)环丁烷)乙腈(WX18)
将2-(3-(4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶)-1H-吡唑)环丁烷-3)乙腈(200mg,489.8umol)溶解在DCM(10mL)中,然后加入TEA(198mg,2mmol)。所得到的混合物冷却到0℃,将三氟甲磺酰氯(107mg,636umol)缓慢的滴加到该溶液中,滴加完毕将该反应回到室温,在室温下反应12小时。LC-MS显示反应完全后。溶剂减压旋干,将残余物用DMF溶解,进一步通过制备型HPLC(HCl)纯化、冻干。得到制备2-(3-(4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶)-1H-吡唑)-1-(三氟甲磺酰基)环丁烷)乙腈。1H-NMR(400MHz,MeOD-d4)δ=9.14(s,1H),8.56(s,1H),7.58(t,J=7.8,1H),7.41(d,J=7.5Hz,1H),7.30(d,J=8.8Hz,1H),4.98(d,J=9.3Hz,2H),4.68(d,J=9.0Hz,2H),3.67(s,2H).MS(ESI)计算值C15H13F3N8O2S[M+H]+427,测定值427.
制备WX19:运用同WX18(步骤1)的制备方法制备WX19。制备型HPLC(HCl)纯化、冻干。得到2-(3-(4-(2-氨基-[1,2,4]三氮唑[1,5-a]吡啶)-1H-吡唑)-1-(甲磺酰基)环丁烷)乙腈。1H-NMR(400MHz,MeOD-d4)δ=9.13(s,1H),8.53(s,1H),7.57(m,1H),7.41(d,J=6.8Hz,1H),7.30(d,J=8.5Hz,1H),4.64(d,J=9.3Hz,2H),4.35(d,J=9.3Hz,2H),3.62(s,2H),3.08(s,3H).MS(ESI)计算值C15H16N8O2S[M+H]+373,测定值373.
实施例17

步骤1:制备N-(8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺
向溶有8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(1.0g,4.7mmol),三乙胺(1.4g,14.1mmol)的乙腈(15.0mL)中,滴加环丙基甲酰氯(1.5g,14.1mmol)。然后将该混合物在26℃下搅拌反应12小时。LC-MS显示反应完全,减压蒸馏除去乙腈,残余物加入H2O(5mL),水层用DCM(15mL x 3)萃取两次。合并有机相并用饱和食盐水(15mL)洗涤,无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过硅胶柱色谱法纯化(DCM/MeOH=20/1)得到黄色固体(700mg,产率为47.8%)。MS(ESI)计算值C10H9N4OBr[M+H]+282,测定值282.
步骤2:制备N-[8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺
向溶有N-(8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺(700mg,2.5mmol),4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-1H-吡唑(579mg,3.0mmol)的二氧六环(25mL)和水(6mL)混合溶剂中,分别加入碳酸钾(1.0g,7.5mmol)和Pd(dppf)Cl2(182mg,249umol)。体系抽真空并充入氮气保护。然后将该混合物加热回流1小时。LC-MS显示反应完全后,减压蒸馏除去溶剂,将残余物溶解于DCM(50mL)和水(10mL)中。分出有机层,水层用DCM(2x 50mL)萃取两次。合并有机相,饱和食盐水(10mL)洗涤,并用无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过硅胶柱色谱法纯化(EA/PE=3/1至1/1)得到黄色固体(300mg,产率为40.4%)。MS(ESI)计算值C13H12N6O[M+H]+269,测定值269.
步骤3:制备化合物N-(8-(1-(3-(氰甲基)-1-(乙磺酰基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-a]吡啶-2-基)环丙烷甲酰胺(WX20)
向溶有N-[8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-基]环丙烷甲酰胺(100mg,372.8umol),2-(1-乙基磺酰基氮杂环丁烷-3-基)乙腈(83mg,447.3umol)的乙腈(15mL)中,滴加DBU(68mg,447.3umol)。形成的混合物在26℃下搅拌反应12小时。TLC显示反应完全后,减压蒸馏除去溶剂,将残余物溶解于DCM(15mL)和水(10mL)中。分出有机层,水层用DCM(15mL x 2)萃取两次。合并有机相,饱和食盐水(10mL)洗涤,并用无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过制备型HPLC纯化(碱性方法)得到(WX20)(65mg,产率为37.98%):1H NMR(400MHz,METHANOL-d4)δ=8.72(s,1H),8.37(d,J=6.8Hz,1H),8.28(s,1H),7.71(d,J=7.3Hz,1H),6.98(t,J=7.0Hz,1H),4.63(d,J=9.0Hz,2H),4.28(d,J=9.0Hz,2H),3.58(s,2H),3.20(q,J=7.3Hz,2H),1.44-1.31(m,3H),1.07(quin,J=3.8Hz,2H),0.96(qd,J=3.7,7.3Hz,2H).MS(ESI)计算值C20H22N8O3S[M+H]+455,测定值455.
制备WX21:运用同WX21(步骤3)的制备方法制备WX21。1H NMR(400MHz,CDCl3)δ=8.93-8.82(m,1H),8.67(s,1H),8.42(d,J=6.3Hz,1H),8.15(s,1H),7.63(d,J=7.3Hz,1H),6.96(t,J=7.2Hz,1H),4.62(d,J=9.3Hz,2H),4.25(d,J=9.3Hz,2H),3.42(s,2H),2.54-2.42(m,1H),1.87(br.s.,1H),1.25-1.17(m,4H),1.13-1.06(m,2H),0.94(dd,J=3.0,7.5Hz,2H).MS(ESI)计算值C21H22N8O3S[M+H]+467,测定值467.
实施例18

步骤1:制备N-(8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)-2,2,2-三氟-乙酰胺
向溶有8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(1.0g,4.7mmol),三乙胺(1.4g,14.1mmol)的二氯甲烷(25.00mL)溶液中滴加三氟乙酸(3.0g,14.1mmol)。形成的反应液在26℃下搅拌反应12小时。LC-MS显示反应完全后,减压蒸馏除去溶剂,将残余物溶解于DCM(50mL)和饱和水(10mL)中。分出有机层,水层用DCM(50mL×2)萃取两次。合并有机相,饱和食盐水(10mL)洗涤,并用无水硫酸钠干燥,过滤、滤液减压蒸馏除去,得到白色粗产品N-(8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)-2,2,2-三氟-乙酰胺(1.1g),未经纯化,直接使用。MS(ESI)计算值C8H4N4OBrF3[M+H]+310,测定值310.步骤2:制备8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺
向溶有N-(8-溴-[1,2,4]三氮唑并[1,5-a]吡啶-2-基)-2,2,2-三氟-乙酰胺(1.1g,3.6mmol),4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-1H-吡唑(1.0g,5.3mmol)的二氧六环(25mL)和水(6mL)中,分别加入碳酸钾(492mg,3.6mmol)和Pd(dppf)Cl2(260mg,356umol)。体系抽真空并充入氮气保护。然后将该混合物加热回流1小时。LC-MS显示反应完全后,过滤、滤液用水(10mL)洗涤,然后再用EA(30mL x 3)萃取,合并有机相,饱和食盐水(10mL)洗涤,并用无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过硅胶柱色谱法纯化(DCM/MeOH=DCM至20/1)得到黄色固体8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(430mg,产率为57.3%)。MS(ESI)计算值C9H8N6[M+H]+201,测定值201.
步骤3:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-a]吡啶-8-基)吡唑-1-基]-1-乙基磺酰基-氮杂环丁烷-3-基]乙腈
向溶有8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(50mg,249.7umol),2-(1-乙基磺酰基氮杂环丁烷-3-亚基)乙腈(56mg,299.7umol)的乙腈(8mL)中,滴加DBU(46mg,299.7umol)。该反应液在26℃下搅拌反应12小时。LC-MS显示反应完全后,减压蒸馏除去乙腈,残余物加水(10mL),然后再用EA(10mLx3)萃取,合并有机相,饱和食盐水(10mL)洗涤,并用无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过硅胶柱色谱法纯化(DCM/MeOH=20/1)得到白色固体2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-a]吡啶-8-基)吡唑-1-基]-1-乙基磺酰基-氮杂环丁烷-3-基]乙腈(50mg,产率为49.22%)。MS(ESI)计算值C16H18N8SO2[M+H]+387,测定值387。
步骤4:制备N-(8-(1-(3-(氰甲基)-1-(乙磺酰基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-a]吡啶-2-基)-2,2,2-三氟乙酰胺(WX22)
向溶有2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-a]吡啶-8-基)吡唑-1-基]-1-乙基磺酰基-氮杂环丁烷-3-基]乙腈(50mg,129.4umol),三乙胺(39.28mg,388.2umol)的二氯甲烷(5mL)溶液中,滴加三氟乙酸酐(81.5mg,388.2umol)。该反应液在26℃下搅拌反应12小时。TLC显示反应完全后,用加入H2O(5mL),分出有机层,水层用DCM(15mL x 3)萃取两次。合并有机相,饱和食盐水(10mL)洗涤,并用饱和食盐水(15mL)洗涤,无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过制备薄层色谱(DMC:MeOH=20:1)纯化得到(WX22)(29mg,46.5%)。1H NMR(400MHz,METHANOL-d4)δ=8.93-8.89(m,1H),8.63-8.59(m,1H),8.48(s,1H),8.02(dd,J=1.0,7.3Hz,1H),7.28-7.21(m,1H),4.68-4.58(m,4H),4.32(s,2H),3.60(s,2H),3.19(q,J=7.4Hz,2H),1.38(t,J=7.3Hz,3H).MS(ESI)计算值C18H17N8SO3F3[M+H]+483,测定值483.
实施例19

步骤1:制备2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-a]吡啶-8-基)吡唑-1-基]-1-环丙基磺酰基-氮杂环丁烷-3-基]乙腈
向溶有8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡啶-2-胺(250mg,1.3mmol),2-(1-环丙磺酰基氮杂环丁烷-3-亚基)乙腈(297mg,1.5mmol)的乙腈(25.00mL)中,滴加DBU(228mg,1.5mmol)。该反应液在26℃下搅拌反应12小时。LC-MS显示反应完全后,加入H2O(5mL),分出有机层,水层用DCM(2x 15mL)萃取两次。合并有机相,并用饱和食盐水(15mL)洗涤,无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过硅胶柱色谱法纯化(DCM/MeOH=20/1)得到黄色固体2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-a]吡啶-8-基)吡唑-1-基]-1-环丙基磺酰基-氮杂环丁烷-3-基]乙腈(250mg,产率为45.2%)。MS(ESI)计算值C17H18N8SO2[M+H]+399,测定值399。
步骤2:制备化合物WX23
向溶有2-[3-[4-(2-氨基-[1,2,4]三氮唑并[1,5-a]吡啶-8-基)吡唑-1-基]-1-环丙基磺酰基-氮杂环丁烷-3-基]乙腈(50mg,125.5umol),三乙胺(38mg,376.5umol)的DCM(5mL)溶液中,滴加三氟乙酸酐(79mg,376.5umol)。该反应液在26℃下搅拌反应12小时。LC-MS显示反应完全后,加入H2O(5mL),分出有机层,水层用DCM(2x 15mL)萃取两次。合并有机相,并用饱和食盐水(15mL)洗涤,无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过制备型HPLC(碱性,0-60)纯化得到(WX23)(17mg,产率为27.4%)。1H NMR 1H NMR(400MHz,CDCl3)δ=9.09-9.04(m,1H),8.74(s,1H),8.55-8.52(m,1H),8.19(s,1H),7.79-7.74(m,1H),7.12(t,J=7.0Hz,1H),4.64(d,J=9.3Hz,2H),4.27(d,J=9.3Hz,2H),3.44(s,2H),2.50-2.43(m,1H),1.25-1.21(m,2H),1.15-1.08(m,2H).MS(ESI)计算值C19H17N8SO3[M+H]+495,测定值495.
实施例20

步骤1:制备3-(氰甲基)-3-(4-(2-(2-甲氧基乙酰胺)-[1,2,4]三氮唑[1,5-c]吡啶-5-基)-1H-吡唑-1-基)氮杂环
丁烷-1-羧酸叔丁酯(1)
向溶解有中间体2(0.1g,0.25mmol)和三乙胺(0.15mL,1.2mmol)的DMF(10.00mL)中加入2-甲氧基乙酰氯(65mg,0.5mmol,)。将所得混合物在60℃下搅拌反应16小时,直至LC-MS显示反应完全。将混合物倒入10ml水中,乙酸乙酯(10ml×3)萃取,合并有机相,并用饱和食盐水(20mL)洗涤,无水硫酸钠干燥,浓缩后得到粗品(120mg),直接用于下一反应。MS(ESI)计算值C21H25N9O4[M+H]+468,测定值468.
步骤2:制备N-(5-(1-(3-(氰甲基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)-2-甲氧基乙酰胺(2)
将3-(氰甲基)-3-(4-(2-(2-甲氧基乙酰胺)-[1,2,4]三氮唑[1,5-c]吡啶-5-基)-1H-吡唑-1-基)氮杂环丁烷-1-羧酸叔丁酯(100mg,0.2mmol)溶于二氯甲烷(5mL)中,然后加入TFA(5ml)。所得混合物在10℃下搅拌反应1小时后,LC-MS显示反应完全,将溶剂浓缩后直接得到100mg粗品,直接用于下一反应。MS(ESI)计算值C16H17N9O2[M+H]+482,测定值482.
步骤3:制备N-(5-(1-(3-(氰甲基)-1-((三氟甲基)磺酰)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)-2-甲氧基乙酰胺(WX24)
将N-(5-(1-(3-(氰甲基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)-2-甲氧基乙酰胺(50mg,0.14mmol)溶于二氯甲烷(5mL)中,先后加入三乙胺(42mg,0.4mmol)和三氟甲磺酰氯(47mg,0.28mmol),所得混合物在10℃下搅拌反应1小时后,LC-MS显示反应完全,将溶剂浓缩后直接得到粗品(50mg),粗品通过制备HPLC(碱性)分离,得到白色固体N-(5-(1-(3-(氰甲基)-1-((三氟甲基)磺酰)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)-2-甲氧基乙酰胺(WX24,10mg)。1H NMR(400MHz,DMSO-d6)9.30(s,1H),8.89(s,1H),8.36(d,J=6.27Hz,1H),7.66(d,J=6.02Hz,1H),4.74(s,2H),3.86(s,2H),3.40(s,4H)。MS(ESI)计算值C17H16F3N9O4S[M+H]+495,测定值495.
制备WX25:运用同WX24(步骤3)的制备方法制备N-(5-(1-(3-(氰甲基)-1-((甲基磺酰)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)-2-甲氧基乙酰胺(WX25)。1H NMR(400MHz,CDCl3)δ=9.29-9.32(m,1H),8.67-8.71(m,1H),8.28-8.33(m,1H),7.44-7.48(m,1H),4.60-4.68(m,1H),4.30-4.36(m,2H),4.13-4.19(m,1H),3.58(s,2H),3.41-3.48(m,1H),3.04(s,3H),1.45(s,3H).MS(ESI)计算值C17H19N9O4S[M+H]+446,测定值446.
实施例21

步骤1:制备3-(4-(2-(2-氰基乙酰胺)-[1,2,4]三氮唑[1,5-c]吡啶-5-基)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁烷-1-羧酸叔丁酯(1)
向溶解有中间体2(0.1g,0.3mmol)和三乙胺(0.17ml,1.3mmol)的DMF(10mL)中加入2-氰基乙酰氯(131mg,1.3mmol,)。将所得混合物在60℃下搅拌反应2小时,直至LC-MS显示反应完全。将混合物倒入水(10mL)中,乙酸乙酯(10mL×3)萃取,合并有机相,无水硫酸钠干燥,浓缩后得到3-(4-(2-(2-氰基乙酰胺)-[1,2,4]三氮唑[1,5-c]吡啶-5-基)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁烷-1-羧酸叔丁酯(100mg,粗品),直接用于下一反应。MS(ESI)计算值C21H22N10O3[M+H]+463,测定值463.
步骤2:制备2-氰基-N-(5-(1-(3-(氰甲基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)乙酰胺(2)
将3-(4-(2-(2-氰基乙酰胺)-[1,2,4]三氮唑[1,5-c]吡啶-5-基)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁烷-1-羧酸叔丁酯(1)(100mg,0.2mmol)溶于二氯甲烷(5mL)中,然后加入TFA(5mL)。所得混合物在10℃下搅拌反应1小时后,LC-MS显示反应完全,将溶剂浓缩后直接得到2-氰基-N-(5-(1-(3-(氰甲基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)乙酰胺(2)(100mg,粗品),直接用于下一反应。MS(ESI)计算值C16H14N10O[M+H]+463,测定值463.
步骤3:制备2-氰基-N-(5-(1-(3-(氰甲基)-1-(甲基磺酰胺)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)乙酰胺(WX26)
将2-氰基-N-(5-(1-(3-(氰甲基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)乙酰胺(50mg,0.14mmol)溶于二氯甲烷(5mL),先后加入三乙胺(42mg,0.4mmol),甲磺酰氯(47mg,0.28mmol),所得混合物在10℃下搅拌反应1小时后,LC-MS显示反应完全,将溶剂浓缩后直接得到50mg粗品,粗品通过制备HPLC(碱性)分离,得到2-氰基-N-(5-(1-(3-(氰甲基)-1-(甲基磺酰胺)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三氮唑[1,5-c]吡啶-2-基)乙酰胺(WX26 10mg)。1H NMR(400MHz,DMSO-d6)9.27(s,1H),8.83(s,1H),8.25-8.52(m,1H),7.67(d,J=6.27Hz,1H),4.56(d,J=9.29Hz,4H),4.34(d,J=9.29Hz,4H),3.73(s,3H)。MS(ESI)计算值C17H16N10O3S[M+H]+441,测定值441.
实施例22

步骤1:制备1-(3-溴苯基)硫脲(2)
室温下,向3-溴苯胺(30.0g,174mmol)的稀盐酸溶液(1M,50mL)中加入硫氰酸钾(20.0g,205.8mmol)。混合物于100℃搅拌反应12小时。TLC(PE:EA=1:1)检测3-溴苯胺还有部分剩余(约20%)。反应液冷却至0℃后用氨水碱化至pH=10。所得紫色乳浊液继续搅拌半小时,然后用乙酸乙酯萃取(200mL×4)。合并有机相用饱和食盐水(30mL)洗涤,无水硫酸钠干燥,过滤旋干得到浓稠的紫色悬浊液。稍冷却加入二氯甲烷(50mL),冰浴下冷却至0℃。不溶的淡紫色固体抽滤后,用少量二氯甲烷(10mL×2)洗涤,真空干燥得到产物1-(3-溴苯基)硫脲(25g,收率为55.8%)。1H NMR(400MHz,CDCl3)7.46(s,1H),7.39-7.44(m,1H),7.28-7.32(m,1H),7.25(s,1H).MS(ESI)计算值C7H7BrN2S[M+H]+230,测定值230.
步骤2:制备7-溴苯并[d]噻唑-2-胺(3)
0℃下,向(3-溴苯基)硫脲(5.0g,21.6mmol)的乙酸(50mL)溶液中滴加液溴(4.7g,29.2mmol)的氯仿溶液(5mL)。混合物于85℃搅拌反应3小时。TLC(PE:EA=1:1)检测显示原料完全消耗并且拿到两个新点。反应液趁热过滤,不溶的固体用少量二氯甲烷(10mL×2)洗涤并干燥得到黄色固体产物(4.2g,收率50%)。滤液旋干得黄色悬浊液,残余物用二氯甲烷(20mL)打浆,不溶物抽滤,用少量DCM(5mL×2)洗涤后,真空干燥即得到黄色固体产物(1.2g,收率13%)。分别取一小份产物(P1和P2)溶于水(1mL)中,用氨水碱化至pH=10,再用乙酸乙酯(0.2mL)萃取。两份萃取液分别用氮气吹干后即可直接用于NMR检测。核磁显示P1中主要是5-溴代异构体副产物,而P2中主要是所需的7-溴代产物。所得粗产物可直接用于下一步反应,无须进一步纯化。1H NMR(400MHz,DMSO-d6)7.13-7.16(m,1H),7.16-7.22(m,2H),7.32(dd,J=7.28,1.51Hz,1H),7.48(d,J=1.76Hz,1H),7.62(d,J=8.53Hz,1H),7.71(br.s.,1H),7.73(s,2H).MS(ESI)计算值C7H5BrN2S[M+H]+228,测定值228.
步骤3:制备N-(7-溴苯并[d]噻唑-2-基)环丙甲酰胺(4)
氮气保护下,于0℃下向7-溴苯并[d]噻唑-2-胺(1.2g,3.9mmol,HBr盐)和三乙胺(1.6g,15.5mmol)的乙腈(50mL)溶液中滴加环丙甲酰氯(1.2g,11.6mmol)。混合物于30℃搅拌反应12小时。TLC(PE:EA=1:1)检测显示主点为单取代所需产物。反应液用水(60mL)淬灭,乙酸乙酯(30mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤旋干。残余物用柱层析法(PE:EA=5:1)纯化得到黄色固体N-(7-溴苯并[d]噻唑-2-基)环丙甲酰胺(580mg,收率40%)。1H NMR(400MHz,DMSO-d6)δ=12.85(s,1H),7.75(d,J=8.03Hz,1H),7.52(d,J=7.78Hz,1H),7.38-7.43(m,1H),1.98-2.05(m,1H),0.95-1.02(m,4H),.MS(ESI)计算值C11H9BrN2OS[M+H]+296,测定值296.
步骤4:制备N-(7-(1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺(5)
氮气保护下,向N-(7-溴苯并[d]噻唑-2-基)环丙甲酰胺(480mg,1.6mmol)和4-频哪醇硼酸酯-1H-吡唑(317mg,1.6mmol)的二氧六环(15mL)溶液中加入Pd(dppf)Cl2(119mg,162umol),K2CO3(672mg,4.9mmol)和H2O(2.5mL)。混合物于90℃搅拌反应12小时。TLC(PE:EA=1:1)检测显示到新点。LC-MS检测到目标产物。反应液用水(100mL)稀释后,乙酸乙酯(30mL×3)萃取。合并有机相用饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤旋干。残余物用柱层析法(PE:EA=1:1)纯化得到黄色固体产物N-(7-(1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺(80mg,收率15.63%)。1H NMR(400MHz,DMSO-d6)δ=13.22(br.s.,1H),12.70(s,1H),8.23(s,1H),7.99(s,1H),7.64(d,J=8.03Hz,1H),7.52-7.55(m,1H),7.45-7.50(m,1H),2.03(t,J=4.52Hz,1H),0.95-1.00(m,4H).MS(ESI)计算值C14H12N4OS[M+H]+285,测定值285.
步骤5:制备7-(1H-吡唑-4-基)苯并[d]噻唑-2-胺(6)
向N-(7-(1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺(120mg,422.1umol)的甲醇(3mL)溶液中逐滴加入NaOH(240mg,6mmol)水溶液(1mL)。混合物于80℃搅拌反应12小时。LCMS检测显示反应完成。反应液用水稀释(50mL),用1M HCl中和至pH=7,乙酸乙酯萃取(15mL×4)。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物当作粗产品7-(1H-吡唑-4-基)苯并[d]噻唑-2-胺(100mg,收率87.7%),直接用于下一步反应,无须进一步纯化。MS(ESI)计算值C10H8N4S[M+H]+216,测定值216.8.
步骤6:制备3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁基-1-碳酸叔丁酯(7)
氮气保护下,向7-(1H-吡唑-4-基)苯并[d]噻唑-2-胺(100mg,462.4umol)和3-(氰甲基烯)氮杂环丁基-1-碳酸叔丁酯(90mg,463.4umol)的乙腈(3mL)溶液中滴加DBU(140.8mg,924.8umol)。混合物于30℃搅拌反应12小时。LC-MS检测显示反应完成。反应液用水(50mL)稀释,乙酸乙酯(20mL×3)萃取。合并有机相经饱和食盐水(30mL)洗涤,无水硫酸钠干燥,抽滤旋干。残余物作为产物3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁基-1-碳酸叔丁酯(190mg,收率80%)的粗产物直接用于下一步反应,无须进一步纯化。MS(ESI)计算值C20H22N6O2S[M+H]+410,测定值411.
步骤7:制备2-(3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)氮杂环丁-3-基)甲基氰(8)
3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁基-1-碳酸叔丁酯(180mg,438.5umol)和盐酸乙酸乙酯溶液(30mL)的混合物于25℃下搅拌反应1小时。LCMS检测反应完成。反应液直接旋干。所得黄色固体作为产物2-(3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)氮杂环丁-3-基)甲基氰(150mg,收率78.9%,HCl盐)的粗品,直接用于下一步反应,无须进一步纯化处理。MS(ESI)计算值C15H14N6S[M+H]+310,测定值310.
步骤8:制备2-(3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(WX27)
氮气保护下,于0℃向2-(3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)氮杂环丁-3-基)甲基氰(150mg,345.9umol,HCl盐)以及Et3N(140mg,1.4mmol)的二氯甲烷(3mL)溶液中逐滴加入MsCl(80mg,698.9umol)。混合物于0℃搅拌反应1小时。LC-MS检测显示反应完成。反应液用水(50mL)淬灭,二氯甲烷萃取(15mL×3)。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物用制备级薄层色谱(DCM:MeOH=10:1)纯化得到黄色固体产物2-(3-(4-(2-氨基苯并[d]噻唑-7-基)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(30mg,收率20.1%)。1H NMR(400MHz,DMSO-d6)δ=8.55(s,1H),8.07(s,1H),7.56(s,2H),7.28-7.31(m,2H),7.25-7.28(m,1H),4.54(d,J=9.29Hz,2H),4.27(d,J=9.29Hz,2H),3.66(s,2H),3.14(s,3H).MS(ESI)计算值C16H16N6O2S2[M+H]+389,测定值389.
实施例23

步骤1:制备3-(氰甲基)-3-(4-(2-(环丙甲酰胺)苯并[d]噻唑-7-基)-1H-吡唑-1-基)氮杂环丁基-1-碳酸叔丁酯
氮气保护下,向N-[7-(1H-吡唑-4-基)-1,3-苯并噻唑-2-基]环丙甲酰胺(100mg,351.7umol)和3-(氰甲基烯)氮杂环丁基-1-碳酸叔丁酯(100mg,513.5umol)的乙腈(3mL)溶液中滴加DBU(107mg,703.4umol)。混合物于25℃下搅拌反应12小时。TLC(PE:EA=1:1)检测显示反应完全。反应液经水(50mL)稀释,乙酸乙酯(20mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物用制备级TLC(PE:EA=1:1)分离纯化得到黄色固体产物3-(氰甲基)-3-(4-(2-(环丙甲酰胺)苯并[d]噻唑-7-基)-1H-吡唑-1-基)氮杂环丁基-1-碳酸叔丁酯(80mg,收率47.5%)。1H NMR(400MHz,CDCl3)δ=11.30(br.s.,1H),8.05(d,J=12.05Hz,2H),7.75(d,J=7.78Hz,1H),7.48-7.53(m,1H),7.43-7.47(m,1H),4.58(d,J=9.79Hz,2H),4.32(d,J=9.54Hz,2H),3.31(s,2H),1.74(dq,J=7.97,3.95Hz,1H),1.51(s,9H),1.27-1.31(m,2H),1.01-1.09(m,2H).MS(ESI)计算值C24H26N6O3S[M+H]+479,测定值479.
步骤2:制备N-(7-(1-(3-(氰甲基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺
3-(氰甲基)-3-(4-(2-(环丙甲酰胺)苯并[d]噻唑-7-基)-1H-吡唑-1-基)氮杂环丁基-1-碳酸叔丁酯(80mg,167.2umol)和盐酸乙酸乙酯(30mL)的混合物于25℃搅拌反应1小时。LCMS检测反应完成。反应液直接旋干。残余物溶于水(50mL)中,用饱和NaHCO3水溶液调至弱碱性(pH>7)后乙酸乙酯(15mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。所得黄色固体作为产物N-(7-(1-(3-(氰甲基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺(60mg,收率85.4%,纯度90%)的粗品,可直接用于下一步反应,无须进一步纯化处理。MS(ESI)计算值C19H18N6OS[M+H]+378,测定值378.
步骤3:制备N-(7-(1-(3-(氰甲基)-1-(甲磺酰基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺
氮气保护下,于向N-(7-(1-(3-(氰甲基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺(60mg,158.54umol)和三乙胺(50mg,494.6umol)的二氯甲烷溶液(5mL)中滴加甲烷磺酰氯(50mg,436.5umol)。混合物于0℃下搅拌0.5小时并且在25℃下继续搅拌1小时。LC-MS及TLC检测都显示反应完成。反应液用水(50mL)淬灭,乙酸乙酯(15mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物用制备级TLC(PE:EA=1:1)分离纯化得到白色固体产物N-(7-(1-(3-(氰甲基)-1-(甲磺酰基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]噻唑-2-基)环丙甲酰胺(45mg,收率62%)。1H NMR(400MHz,CDCl3)δ=12.26(br.s.,1H),8.07(s,2H),7.74(d,J=7.78Hz,1H),7.46-7.52(m,1H),7.39-7.45(m,1H),4.63(d,J=9.03Hz,2H),4.30(d,J=9.03Hz,2H),3.41(s,2H),3.04(s,3H),1.72-1.80(m,1H),1.26(d,J=3.26Hz,2H),1.01(dd,J=7.40,2.89Hz,2H).MS(ESI)计算值C20H20N6O3S2[M+H]+457,测定值457.
实施例24

步骤1:制备2-(苄氧基)-1-溴-3-硝基苯
向2-溴-6-硝基苯酚(5.2g,23.9mmol)以及K2CO3(3.6g,26.3mmol)的乙腈(100mL)溶液中加入苄溴(4.3g,25.3mmol)。混合物于100℃搅拌反应3小时。TLC(PE:EA=10:1)检测显示反应完成。反应液抽滤,固体用乙酸乙酯(10mL×3)淋洗。滤液旋干后,残余物溶解于乙酸乙酯(100mL)中,经水(20mL)和饱和食盐水(20mL)洗涤。有机相用无水硫酸钠干燥,抽滤后旋干。所得黄色固体作为产物2-(苄氧基)-1-溴-3-硝基苯(7.50g,收率91.51%)的粗品,可直接用于下一步反应,无须进一步纯化处理。1HNMR(400MHz,CDCl3)7.84(d,J=8.03Hz,1H),7.79(d,J=8.03Hz,1H),7.56(d,J=6.53Hz,2H),7.37-7.45(m,3H),7.16(t,J=8.28Hz,1H),5.21(s,2H).MS(ESI)计算值C13H10BrNO3[M+H]+308,测定值308.
步骤2:制备4-(2-(苄氧基)-3-硝基苯)-1H-吡唑
氮气保护下,向2-(苄氧基)-1-溴-3-硝基苯(1.0g,3.3mmol)和4-频哪醇硼酸酯吡唑-1-碳酸叔丁酯(1.0g,3.4mmol)的二氧六环(30mL)溶液中加入Pd(dppf)Cl2(250mg,341.7umol),碳酸钾(1.4g,9.8mmol)以及水(5mL)。混合物于100℃搅拌反应12小时。TLC(PE:EA=1:1)以及LC-MS检测都显示反应完成。反应液用水(20mL)稀释,乙酸乙酯(20mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物用柱层析法(PE:EA=1:1)分离纯化得到黄色油状产物4-(2-(苄氧基)-3-硝基苯)-1H-吡唑(900mg,收率89.1%)。1H NMR(400MHz,CDCl3)δ=8.03(s,2H),7.70-7.78(m,2H),7.33-7.39(m,5H),7.28-7.32(m,1H),4.87(s,2H).MS(ESI)计算值C16H13N3O3[M+H]+296,测定值296.
步骤3:制备3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁-1-碳酸叔丁酯
氮气保护,于0℃下向4-(2-(苄氧基)-3-硝基苯)-1H-吡唑(900mg,3.1mmol)以及3-(氰甲烯基)氮杂环丁-1-碳酸叔丁酯(900mg,4.6mmol)的乙腈(20mL)溶液中滴加DBU(928mg,6.1mmol)。混合物于25℃搅拌反应3小时。TLC(PE:EA=1:1)检测显示反应完成。反应液经水(60mL)淬灭,乙酸乙酯(20mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物用柱层析法(PE:EA=1:1)分离纯化得到黄色油状产物3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁-1-碳酸叔丁酯(1.00g,收率60.3%)。1H NMR(400MHz,CDCl3)δ=7.96(d,J=9.03Hz,2H),7.71-7.76(m,2H),7.32-7.40(m,5H),7.27-7.31(m,1H),4.90(s,2H),4.24(d,J=9.54Hz,2H),4.12(d,J=9.03Hz,2H),3.16(s,2H),1.48(s,9H).MS(ESI)计算值C26H27N5O5[M+H]+490,测定值490.
步骤4:制备2-(3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)氮杂环丁-3-基)甲基氰
3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)-3-(氰甲基)氮杂环丁-1-碳酸叔丁酯(1.0g,2.04mmol)和盐酸乙酸乙酯(50mL)的混合物于25℃搅拌反应1小时。LC-MS检测显示反应完成。反应液直接旋干后,残余物溶于水中(50mL),用饱和NaHCO3溶液调至中性pH=7后,乙酸乙酯萃取(15mL×3)。合并有机相经饱和食盐水(2mL)洗涤,无水硫酸钠干燥,抽滤后旋干。所得橙色油状物作为产物2-(3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)氮杂环丁-3-基)甲基氰(900mg,收率90.6%)的粗品,可直接用于下一步反应,无须进一步纯化处理。MS(ESI)计算值C21H19N5O3[M+H]+390,测定值390.
步骤5:制备2-(3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰
氮气保护,于0℃下向2-(3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)氮杂环丁-3-基)甲基氰(900mg,2.3mmol)和三乙胺(730mg,7.2mmol)的二氯甲烷(10mL)溶液中滴加甲烷磺酰氯(529mg,4.6mmol)。混合物于0℃搅拌反应1小时。LC-MS检测显示反应完成。反应液用水(50mL)淬灭后,乙酸乙酯(20mL×3)萃取。合并有机相经饱和食盐水(5mL)洗涤,无水硫酸钠干燥,抽滤后旋干。残余物用柱层析法(PE:EA=1:1(0.5%Et3N))分离纯化得到黄色油状产物2-(3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(450mg,收率37.50%)。1H NMR(400MHz,CDCl3)δ=7.98(d,J=12.55Hz,2H),7.71-7.78(m,2H),7.34-7.41(m,5H),7.30(t,J=8.03Hz,1H),4.88-4.93(m,2H),4.33(d,J=9.03Hz,2H),4.09(d,J=9.54Hz,2H),3.21(s,2H),2.95(s,3H).MS(ESI)计算值C22H21N5O5S[M+H]+468,测定值468.
步骤6:制备2-(3-(4-(3-氨基-2-羟基苯)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰
氢气氛围下,向2-(3-(4-(2-(苄氧基)-3-硝基苯)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(400mg,855.6umol)的乙酸乙酯(10mL)溶液中加入Pd/C(200mg,1.9mmol)。混合物于25℃搅拌反应1.5小时。LC-MS检测显示反应完成。反应液抽滤去除Pd/C后低温旋蒸稍浓缩。所得的淡黄色产物2-(3-(4-(3-氨基-2-羟基苯)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(300mg,收率80.75%),可直接用于下一步反应,无须进一步纯化处理。MS(ESI)计算值C15H17N5O3S[M+H]+348,测定值348.步骤7:制备2-(3-(4-(2-氨基苯并[d]恶唑-7-基)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(WX29)
氮气保护下,向2-(3-(4-(3-氨基-2-羟基苯)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(300mg,863.58umol)的乙酸乙酯(5mL)溶液中加入溴氰(100mg,944.1umol)。混合物于50℃搅拌反应12小时。LC-MS及TLC(DCM:MeOH=10:1)检测显示反应完成。反应液直接旋干后,溶解于DCM:MeOH=10:1(3mL),经制备级TLC(DCM:MeOH=10:1)分离纯化得到黄色固体产物2-(3-(4-(2-氨基苯并[d]恶唑-7-基)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(200mg,收率56.0%)。1H NMR(400MHz,CDCl3)δ=8.26(br.s.,1H),8.10(s,1H),7.26-7.28(m,2H),7.24(d,J=7.28Hz,1H),4.65(d,J=9.03Hz,2H),4.28(d,J=9.29Hz,2H),3.45(s,2H),3.04(s,3H).MS(ESI)计算值C16H16N6O3S[M+H]+373,测定值373.
步骤8:制备N-(7-(1-(3-(氰甲基)-1-(甲磺酰基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]恶唑-2-基)环丙甲酰胺(WX30)
氮气保护下,向2-(3-(4-(2-氨基苯并[d]恶唑-7-基)-1H-吡唑-1-基)-1-(甲磺酰基)氮杂环丁-3-基)甲基氰(100mg,268.5umol)及三乙胺(54mg,537.1umol)的乙腈(3mL)溶液中滴加环丙甲酰氯(30mg,287.3umol)。混合物于30℃下搅拌反应12小时。LCMS检测显示反应收率较低。反应液直接旋干,残余物经制备级TLC(DCM:MeOH=10:1)分离纯化得到黄色固体产物N-(7-(1-(3-(氰甲基)-1-(甲磺酰基)氮杂环丁-3-基)-1H-吡唑-4-基)苯并[d]恶唑-2-基)环丙甲酰胺(10mg,收率8.45%)。1H NMR(400MHz,DMSO-d6)δ=12.05(br.s.,1H),8.77(s,1H),8.34(s,1H),7.60(d,J=7.53Hz,1H),7.44(d,J=7.53Hz,1H),7.31-7.38(m,1H),4.52(d,J=9.29Hz,2H),4.30(d,J=9.03Hz,2H),3.66(s,2H),3.15(s,3H),2.14(br.s.,1H),0.96(d,J=5.52Hz,4H).MS(ESI)计算值C20H20N6O4S[M+H]+441,测定值441.
实施例25

步骤1:制备N-[(3-氯吡嗪-2-基)硫代氨基甲酰基]氨基甲酸乙酯
向溶解有3-氯吡嗪-2-胺(8.7g,67.2mmol)的THF(100mL)中。加入异硫氰酸乙酯(9.7g,73.9mmol)。将所得混合物在27℃下搅拌反应48小时,直至LC-MS显示反应完全。将混合物溶剂THF减压旋干,得粗产物N-[(3-氯吡嗪-2-基)硫代氨基甲酰基]氨基甲酸乙酯用甲基叔丁基醚,洗涤、干燥。无需进一步纯化(纯度足够)直接用于下一步反应。MS(ESI)计算值C8H9ClN4O2S[M+H]+261,测定值261.
步骤2:制备8-氯-[1,2,4]三氮唑并[1,5-a]吡嗪-2-胺
将盐酸羟胺(20.0g,287.7mmol)悬浮于100mL的乙醇和甲醇(1:1)的混合物中,然后加入DIEA(22.3g,172.6mmol)。所得混合物在27℃下搅拌反应1小时后,将N-[(3-氯吡嗪-2-基)硫代氨基甲酰基]氨基甲酸乙酯(15.0g,57.54mmol)加入到该反应体系中并缓慢回流(70℃)3小时。LC-MS显示反应完全后,将反应物冷却至室温,过滤,用水和MTBE洗涤,然后经真空干燥(60℃),得到黄色固体8-氯-[1,2,4]三氮唑并[1,5-a]吡嗪-2-胺(6.50g,产率为64.62%)。MS(ESI)计算值C5H4ClN5[M+H]+170,测定值170.
步骤3:制备N-(8-氯-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基)环丙烷甲酰胺
5℃下,向溶解有8-氯-[1,2,4]三氮唑并[1,5-a]吡嗪-2-胺(2.0g,11.8mmol)的无水CH3CN(30mL)中,加入Et3N(3g,29.5mmol),接着加入环丙烷甲酰氯(3.1g,29.5mmol)。加完后,将反应混合物升至28℃中并搅拌直至LC-MS显示所有的原料被消耗。如果需要的话,进一步加入Et3N(7.1mmol)和环丙烷甲酰氯(7.1mmol),以保证反应完全。溶剂减压旋干得到残余物,残余物用Et2O(50mL)打浆,固体通过过滤收集,用H2O(2×50mL),丙酮(50mL)和Et2O(50mL)洗涤,然后真空干燥,得到所需的黄色固体N-(8-氯-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基)环丙烷甲酰胺(1.2g)。MS(ESI)计算值C9H8ClN5O[M+H]+237,测定值237.
步骤4:制备N-[8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基]环丙烷甲酰胺
向溶解有N-(8-氯-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基)环丙烷甲酰胺(100mg,420.8umol),4-(4,45,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-1H-吡唑(122.5mg,631.2umol)和K2CO3(175mg,1.3mmol)的混合溶剂H2O(1mL)/二恶烷(4mL)中加入Pd(dppf)Cl2.CH2Cl2(34mg,42.08umol)。抽真空充入氮气后,将混合物在100℃下搅拌反应3小时。LC-MS显示反应完全后,冷却至室温,将混合物通过硅藻土床过滤,硅藻土用用DCM(30mL)洗涤。分离有机层,水层用DCM(3×50mL)萃取。合并有机相,饱和盐水洗涤,无水Na2SO4干燥。溶剂减压旋干,将粗产物通过制备薄层色谱纯化(DCM/MeoH=10:1),得到浅黄色固体N-[8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基]环丙烷甲酰胺(70mg,产率为8.1%)。MS((ESI))计算值C12H11N7O[M+H]+270,测定值270.
步骤5:制备N-[8-[1-[3-(氰基甲基)-1-乙磺酰基氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基]环丙烷甲酰胺(WX31)
向溶解有N-[8-(1H-吡唑-4-基)-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基]环丙烷甲酰胺(120mg,445.7umol),2-(1-乙磺酰基氮杂环丁烷-3-亚基)乙腈(125mg,668.5umol)的DMF(10mL)溶液中,加入DBU(136mg,891umol)。所得该混合物在40℃搅拌反应16小时。LC-MS显示反应完全后。溶剂减压旋干,将残余物溶解于EtOAc(50mL)中。此溶液依次用1N HCl(10mL)和盐水(20mL)洗涤,有机相用无水Na2SO4干燥。溶剂减压旋干,将残余物用制备薄层色谱纯化(PE/EA=1:4),得到粗化合物,将其进一步通过制备型HPLC(HCl)纯化、冻干。得到白色固体N-[8-[1-[3-(氰基甲基)-1-乙磺酰基氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑并[1,5-a]吡嗪-2-基]环丙烷甲酰胺(40mg)。1H-NMR(400MHz,MeOD-d4)δ=8.07(d,J=8Hz,2H),7.86(dd,J=7.2,13.2Hz,2H),7.67(d,J=8.4Hz,2H),7.44-7.42(m,1H),4.19(s,2H),3.44(d,J=4.8Hz,4H),3.26(d,J=4.8Hz,4H),2.95(m,1H),0.87(m,2H),0.74(m,2H).MS(ESI)计算值C19H21N9O3S[M+H]+456,测定值456.
实施例26

步骤1:制备3-氧代环丁烷基甲酸苄酯
将溶有3-氧代环丁烷基羧酸(3.0g,26.3mmol),溴化苄(6.7g,39.4mmol)和碳酸钾(7.3g,52.6mmol)的丙酮(30mL)溶液,加热回流10小时。TLC显示反应完全后,反应液减压浓缩除去溶剂,加入水(20mL),乙酸乙酯(150mL×2)萃取。合并的有机相用饱和食盐水(50mL)洗涤,无水硫酸钠干燥,过滤并真空浓缩。将残余物用硅胶色谱柱(石油醚:乙酸乙酯=10:1)纯化,得到3-氧代环丁烷基甲酸苄酯(2.5g,产率为41.9%)为无色液体。MS(ESI)计算值C12H12O3[M+H]+205测定值205。
步骤2:制备3,3-二氟环丁烷基甲酸苄酯
在-60℃下,向溶有3-氧代环丁烷基甲酸苄酯(1.0g,4.9mmol)的二氯甲烷(35mL)溶液中,逐滴加入DAST(1.6g,9.8mmol),滴加完后,反应液慢慢升温至15℃并搅拌10小时。TLC显示反应完全后,将反应液冷却至0℃,加入饱和碳酸氢钠溶液(10mL)淬灭反应,水相用二氯甲烷(30mL×2)萃取。合并的有机相用饱和食盐水(10mL)洗涤,无水硫酸钠干燥,过滤并真空浓缩。将残余物用硅胶色谱柱(石油醚:乙酸乙酯=20:1~10:1)纯化,得到3,3-二氟环丁烷基甲酸苄酯(450mg,产率为36.54%)为无色油状。MS(ESI)计算值C12H12F2O2[M+H]+227测定值227。
步骤3:制备3,3-二氟环丁烷基羧酸
向溶有3,3-二氟环丁烷基甲酸苄酯(450mg,2.0mmol)的乙醇(10mL)溶液中,加入Pd/C(10%,40mg),反应液在氢气(15Psi)氛围中,常温搅拌反应10小时。TLC显示反应完全后,将固体滤去,滤液真空浓缩得到3,3-二氟环丁烷基羧酸(250mg,产率为83.1%)为白色固体。MS(ESI)计算值C5H6F2O2[M+H]+137测定值137。
步骤4:制备3,3-二氟环丁烷基甲酰氯
在0℃下,向溶有3,3-二氟环丁烷基羧酸(230mg,1.69mmol)和DMF(13mg,169.0umol)的二氯甲烷(5mL)溶液中,逐滴加入草酰氯(322mg,2.5mmol),滴加完后,反应液常温搅拌3小时。TLC显示反应完全后,反应液真空浓缩得到3,3-二氟环丁烷基甲酰氯(300mg,粗品)为黄色固体,该产物无需纯化,直接用于下一步骤。MS(ESI)计算值C5H5ClF2O[M+H]+155,测定值155。
步骤5:制备叔丁基3-(氰甲基)-3-[4-[2-[(3,3-二氟环丁烷基羧酸)氨基]-[1,2,4]三氮唑[1,5-c]嘧啶-5-基吡唑-1-基]氮杂环丁烷-1-羧酸
在0℃下,向溶有叔丁基3-[4-(2-氨基-[1,2,4]三氮唑[1,5-c]嘧啶-5-基)吡唑-1-基]-3-(氰甲基)氮杂环丁烷-1-羧酸(395mg,1.0mmol),DMAP(13mg,110umol)and吡啶(396mg,5mmol)的二氯甲烷(8mL)溶液,加入3,3-二氟环丁烷基甲酰氯(294mg,1.9mmol),将混合物加热至40℃并搅拌10小时。LCMS显示反应完成。将反应液倾入水(5mL)中,水相用二氯甲烷萃取(15mL×2)。将合并的有机相用饱和食盐水(10mL)洗涤,用无水硫酸钠干燥,过滤和真空浓缩。残余物通过制备型TLC纯化(乙酸乙酯:石油醚=1:0)得到叔丁基3-(氰甲基)-3-[4-[2-[(3,3-二氟环丁烷基羧酸)氨基]-[1,2,4]三氮唑[1,5-c]嘧啶-5-基吡唑-1-基]氮杂环丁烷-1-羧酸(80mg,产率为12.00%)为白色固体。MS(ESI)calcd.C23H25F2N9O3[M+H]+514测定值514。
步骤6:制备N-[5-[1-[3-(氰甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3,3-二氟-环丁烷基甲酰胺
向溶有叔丁基3-(氰甲基)-3-[4-[2-[(3,3-二氟环丁烷基羧酸)氨基]-[1,2,4]三氮唑[1,5-c]嘧啶-5-基吡唑-1-基]氮杂环丁烷-1-羧酸(80mg,155.8umol)的二氯甲烷(5mL)溶液中,滴加入三氟乙酸(765mg,6.7mmol),反应液常温搅拌2小时。LCMS显示反应完成。反应液真空浓缩得到N-[5-[1-[3-(氰甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3,3-二氟-环丁烷基甲酰胺(100mg,粗品,TFA盐)为褐色固体,该产物无需纯化,直接用于下一步骤。MS(ESI)计算值C18H17F2N9O[M+H]+414测定值414。
步骤7:制备N-[5-[1-[3-(氰甲基)-1-(三氟甲磺酰基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3,3-二氟-环丁烷基甲酰胺(WX32)
向溶有N-[5-[1-[3-(氰甲基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3,3-二氟-环丁烷基甲酰胺(100mg,189.61umol,TFA盐)的二氯甲烷(8mL)溶液中,加入三氟甲基磺酰氯(38mg,227.5mmol),后再加入三乙胺(96mg,948.1umol),反应液常温搅拌10小时。LCMS显示反应完成。反应液真空浓缩,残余物通过制备型HPLC纯化得到N-[5-[1-[3-(氰甲基)-1-(三氟甲磺酰基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三氮唑[1,5-c]嘧啶-2-基]-3,3-二氟-环丁烷基甲酰胺(WX32)(3mg,产率为2.9%)为白色固体。MS(ESI)计算值C19H16F5N9O3S[M+H]+546测定值546。
实施例27

步骤1:制备叔丁基4-[3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]吡丁烷-1-基]氮杂环丁烷-1-基]哌啶-1-甲酸叔丁酯(1)
向溶有中间体8(250mg,689.9umol),4-氧代哌啶羧酸叔丁酯(137mg,689.9umol)和NaBH(OAc)3(292mg,1.4mmol)的THF(3mL)中加入DIEA(446mg,3.5mmol)。体系抽真空并充入氮气保护。然后将该混合物在26℃先搅拌反应12小时。LC-MS显示反应完全后,过滤、滤液用水(3ml)洗,然后再用EA(5ml×3)萃取,合并有机相并,再用饱和食盐水(3mL)洗,无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过硅胶柱色谱法纯化(DCM/MeOH=20/1)得到黄色固体(143.00mg,产率为37.99%)。MS ESI计算值C28H35N9O3[M+H]+546,测定值546。
步骤2:制备N-[5-[1-[3-(氰基甲基)-1-(4-哌啶基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三唑并[1,5-]吡啶-2-基]环丙烷甲酰胺(2)
向溶有叔丁基4-[3-(氰基甲基)-3-[4-[2-(环丙烷羰基氨基)-[1,2,4]三唑并[1,5-a]吡啶-5-基]吡丁烷-1-基]氮杂环丁烷-1-基]哌啶-1-甲酸叔丁酯(143mg,262.1umol)的二氯甲烷(5mL)中滴加三氟乙酸(2mL)。然后将该反应在26℃下搅拌反应3小时。LC-MS显示反应完全后,减压蒸馏除去溶剂,得到黄色固体(56mg,粗产品),未经纯化,直接用于下一步反应。MS ESI计算值C23H27N9O[M+H]+446,测定值446。
步骤3:制备乙基6-(溴甲基)-2-(三氟甲基)嘧啶-4-羧酸乙酯(3)
向溶有6-甲基-2-(三氟甲基)嘧啶-4-羧酸乙酯(2.0g,8.5mmol)的乙酸(12mL)中滴加Br2(1.4g,8.5mmol)。该反应液在80℃下搅拌反应30分钟。LC-MS显示反应结束后,减压蒸馏除去溶剂。残余物通过制备分离(PE/EA=3/1)得到黄色油状液体(610mg,产率为12.3%)。MS ESI计算值C9H8BrF3N2O2[M+H]+313,测定值313。
步骤4:制备乙基6-[(二甲基氨基)甲基]-2-(三氟甲基)嘧啶-4-羧酸乙酯(4)
向溶有乙基6-(溴甲基)-2-(三氟甲基)嘧啶-4-羧酸乙酯(610mg,2.0mmol)和N-二甲基甲胺(318mg,3.9mmol)的二氯甲烷(20mL)中滴加三乙胺(592mg,5.9mmol).该反应液在26℃下搅拌0.5小时。LC-MS显示反应完全后,滤液用水(20mL)洗,然后再用EA(20mL×3)萃取,合并有机相,用饱和食盐水(20mL)洗涤,无水硫酸钠干燥,过滤、滤液减压蒸馏除去。残余物通过制备分离纯化(PE/EA=3/1)得到黄色液体(310mg,产率为51.6%)。MS ESI计算值C11H14F3N3O2[M+H]+278,测定值278。
步骤5:制备6-[(二甲基氨基)甲基]-2-(三氟甲基)嘧啶-4-羧酸(5)
向溶有乙基6-[(二甲基氨基)甲基]-2-(三氟甲基)嘧啶-4-羧酸乙酯(310mg,1.1mmol)的四氢呋喃(8mL)和水(2mL)加入氢氧化锂(54mg,2.3mmol)。该反应液在26℃下搅拌反应0.5小时。LC-MS显示反应完全后,减压蒸馏除去溶剂得到黄色油状(314mg)液体,未经纯化直接使用。MS ESI计算值C9H10F3N3O2[M+H]+250,测定值250。
步骤6:制备N-(5-(1-(3-(氰基甲基)-1-(1-(6-((二甲基氨基)甲基)-2-(三氟甲基)嘧啶-4-羰基)哌啶-4-基)氮杂环丁烷-3-基)-1H-吡唑-4-基)-[1,2,4]三唑并[1,5-a]吡啶-2-基)环丙烷甲酰胺(WX33)
向溶有N-[5-[1-[3-(氰基甲基)-1-(4-哌啶基)氮杂环丁烷-3-基]吡唑-4-基]-[1,2,4]三唑并[1,5-]吡啶-2-基]环丙烷甲酰胺(480mg,107.7umol),化合物2(27mg,107.7umol),EDCI(52mg,269.4umol)和HOBt(36mg,269.4umol)的DMF(3mL)溶液中加入TEA(55mg,538.7umol)。该反应液在26℃下搅拌反应12小时。LC-MS显示反应完全后,将反应液溶于EA(10mL)和水(10mL)中。分出有机层,水层用EA(2×15mL)萃取两次。合并有机相,经饱和食盐水(5ml)洗并用无水硫酸钠干燥,过滤、滤液减压蒸馏除去。经制备分离得到白色固体(WX33)(5mg,产率为6.9%)。1H NMR(400MHz,METHANOL-d4)δ=9.18(s,1H),8.54(s,1H),7.94(s,1H),7.75-7.69(m,1H),7.57(ddd,J=1.1,8.1,18.9Hz,2H),3.87-3.73(m,6H),3.53(s,2H),3.43-3.35(m,1H),3.30-3.21(m,1H),2.74-2.66(m,1H),2.37(s,6H),2.06-1.93(m,2H),1.50(d,J=10.3Hz,1H),1.38-1.28(m,4H),1.11-1.05(m,2H),1.01-0.95(m,2H)。MS ESI计算值C33H35F3N12O2[M+H]+677,测定值677。
Jak1,2,Jak3激酶体外活性测试
实验材料
重组人源JAK1、JAK2、JAK3蛋白酶均购自Life technology。LANCE UltraULightTM-JAK-1(Tyr1023)peptide和LANCE Eu-W1024Anti-phosphotyrosine(PT66)均购自PerkinElmer。使用多联酶标仪Envision(PerkinElmer)读板。
实验方法
将测试化合物进行3倍浓度梯度稀释,终浓度为10uM到0.17nM 11个浓度,每个浓度两个复孔;DMSO在检测反应中的含量为1%。
JAK1酶反应:
2nM JAK1蛋白激酶,50nM LANCE Ultra ULightTM-JAK-1(Tyr1023)peptide,38uMATP,50mM HEPES(pH 7.5),10mM MgCl2,1mM EGTA,2mM DTT,0.01%BRIJ-35。检测板为White Proxiplate 384-Plus plate(PerkinElmer),室温反应90分钟,反应体系为10ul。
JAK2酶反应:
0.02nM JAK2蛋白激酶,50nM LANCE Ultra ULightTM-JAK-1(Tyr1023)peptide,12uM ATP,50mM HEPES(pH 7.5),10mM MgCl2,1mM EGTA,2mM DTT,0.01%BRIJ-35。检测板为White Proxiplate 384-Plus plate(PerkinElmer),室温反应60分钟,反应体系为10ul。
JAK3酶反应:
0.05nM JAK3蛋白激酶,50nM LANCE Ultra ULightTM-JAK-1(Tyr1023)peptide,4uM ATP,50mM HEPES(pH 7.5),10mM MgCl2,1mM EGTA,2mM DTT,0.01%BRIJ-35。检测板为White Proxiplate 384-Plus plate(PerkinElmer),室温反应90分钟,反应体系为10ul。
反应检测:
加10ul检测试剂至反应板中,其中LANCE Eu-W1024Anti-phosphotyrosine(PT66)终浓度为2nM,EDTA终浓度为10mM,室温孵育60分钟,Envision仪器读板。
数据分析
通过下列公式将读数转化成抑制率(%)=(Min-Ratio)/(Max-Min)×100%。4参数曲线拟合(Model 205 in XLFIT5,iDBS)测得IC50数据,具体见表1。
表1


A≤10nM;10<B≤100nM;100<C≤1000nM;1≤F1≤5;5<F2≤10;10<F3≤25;25<F4≤100
结论:本发明化合物针对JAK2的选择性优于Tofacitinib。
Claims (18)
1.式(Ⅰ)化合物或其药学上可接受的盐,

其中,
R1选自H、或选自任选被1、2、3或4个R取代的:C1-6烷基、C1-6杂烷基、C3-7环烷基、3~7元杂环烷基、6元芳基、5~6元杂芳基、
L1、L2分别独立地选自单键、-S(=O)2-、-S(=O)-、-C(=O)-、-NHC(=O)-;
R2选自:H、或选自任选被1、2、3或4个R取代的:NH2、C1-6烷基、C1-6杂烷基、C3-7环烷基、3~7元杂环烷基、6元芳基、5~6元杂芳基;
环A选自5~6元杂芳基;
X分别独立地选自N、C;
T选自N或C(R);
R选自H、卤素、NH2、CN、OH、或选自任选被1、2、3或4个R’取代的:C1-3烷基、C1-3杂烷基、C3-6环烷基、3~6元杂环烷基、6元芳基、5~6元杂芳基;
R’选自卤素、OH、CN、NH2;
所述“杂”代表杂原子或杂原子团,分别独立地选自O、S、N、C(=O)、S(=O)或S(=O)2;
杂原子或杂原子团的数目分别独立地选自0、1、2、3或4。
2.根据权利要求1所述化合物或其药学上可接受的盐,其中,R分别独立地选自H、卤素、OH、NH2、CN,或选自任选被1、2、3或4个R’取代的C1-3烷基、C1-3烷氧基、C1-3烷胺基。
3.根据权利要求1所述化合物或其药学上可接受的盐,其中,R选自H、F、Cl、Br、I、OH、CN、NH2、Me、Et、N(CH3)2、NH(CH3)。
4.根据权利要求1所述化合物或其药学上可接受的盐,其中,R1选自H,或任选被1、2、3或4个R取代的C1-3烷基、C3-6环烷基、C3-6杂环烷基、C1-3烷基-O-C1-3烷基-、C1-3烷基-S-C1-3烷基-、C1-3烷基-NH-C1-3烷基-、C1-6烷氧基、C1-6烷氨基、
5.根据权利要求1所述化合物或其药学上可接受的盐,其中,R1选自H,或选自任选被1、2、3或4个R取代的Me、
6.根据权利要求1所述化合物或其药学上可接受的盐,其中,R1选自:H、Me、

7.根据权利要求1所述化合物或其药学上可接受的盐,其中,R1-L1-选自H,或选自任选被R取代的:

8.根据权利要求1所述化合物或其药学上可接受的盐,其中,R1-L1-选自:H、

9.根据权利要求1所述化合物或其药学上可接受的盐,其中,R2选自H、NH2,或任选被1、2、3或4个R取代的C1-3烷基、C3-6环烷基、3~6元杂环烷基、C1-3烷基-O-C1-3烷基-、C1-3烷基-S-C1-3烷基-、C1-3烷基-NH-C1-3烷基-、C1-6烷氧基、C1-6烷氨基。
10.根据权利要求1所述化合物或其药学上可接受的盐,其中,R2选自H、NH2,或选自任选被R取代的:Me、
11.根据权利要求1所述化合物或其药学上可接受的盐,其中,R2选自:H、NH2、

12.根据权利要求1所述化合物或其药学上可接受的盐,其中,R2-L2-选自H,或任选被R’取代的:NH2、
13.根据权利要求1所述化合物或其药学上可接受的盐,其中,R2-L2-选自:H、NH2、

14.根据权利要求1所述化合物或其药学上可接受的盐,其中,环A选自:1,3,4-三氮唑基、咪唑基、噁唑基、噻唑基。
15.根据权利要求1所述化合物或其药学上可接受的盐,其中,环A选自:

16.根据权利要求1或14所述化合物或其药学上可接受的盐,其中,结构单元 选自:
选自:
17.根据权利要求1或14所述化合物或其药学上可接受的盐,其中,结构单元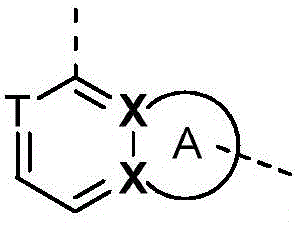 选自:
选自:
18.根据权利要求1所述化合物,其选自:


Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510213187 | 2015-04-29 | ||
| CN2015102131878 | 2015-04-29 | ||
| PCT/CN2016/080208 WO2016173484A1 (zh) | 2015-04-29 | 2016-04-26 | Jak抑制剂 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107531695A CN107531695A (zh) | 2018-01-02 |
| CN107531695B true CN107531695B (zh) | 2020-03-27 |
Family
ID=57198164
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680021913.9A Active CN107531695B (zh) | 2015-04-29 | 2016-04-26 | Jak抑制剂 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US10174036B2 (zh) |
| EP (1) | EP3290418B1 (zh) |
| JP (1) | JP6600365B2 (zh) |
| KR (1) | KR102006684B1 (zh) |
| CN (1) | CN107531695B (zh) |
| AU (1) | AU2016254385B2 (zh) |
| CA (1) | CA2982493C (zh) |
| DK (1) | DK3290418T3 (zh) |
| EA (1) | EA036058B1 (zh) |
| ES (1) | ES2734048T3 (zh) |
| HU (1) | HUE044240T2 (zh) |
| MX (1) | MX370933B (zh) |
| PL (1) | PL3290418T3 (zh) |
| PT (1) | PT3290418T (zh) |
| TR (1) | TR201909694T4 (zh) |
| TW (1) | TWI694998B (zh) |
| UA (1) | UA119701C2 (zh) |
| WO (1) | WO2016173484A1 (zh) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT3419978T (pt) | 2016-02-24 | 2020-06-01 | Pfizer | Derivados de pirazolo[1,5-a]pirazin-4-ilo como inibidores de jak |
| KR102492378B1 (ko) | 2016-07-26 | 2023-01-27 | 톈진 롱보진 파마시우티컬 씨오., 엘티디. | 선택적 jak 저해제로서 화합물, 및 이의 염 및 치료학적 용도 |
| WO2019034973A1 (en) * | 2017-08-14 | 2019-02-21 | Pfizer Inc. | PYRAZOLO [1,5-A] PYRAZIN-4-YL AND RELATED DERIVATIVES |
| KR20190043437A (ko) | 2017-10-18 | 2019-04-26 | 씨제이헬스케어 주식회사 | 단백질 키나제 억제제로서의 헤테로고리 화합물 |
| US12065441B2 (en) | 2018-06-06 | 2024-08-20 | Gengle Therapeutics, Inc. | Pyrazolopyrimidine derivative, use thereof and pharmaceutical composition |
| JP2022503932A (ja) * | 2018-09-27 | 2022-01-12 | フォチョン・ファーマシューティカルズ・リミテッド | Retキナーゼ阻害剤としての置換イミダゾ[1,2-a]ピリジン及び[1,2,4]トリアゾロ[1,5-a]ピリジン化合物 |
| CN111320624B (zh) * | 2018-12-14 | 2023-05-12 | 中国医药研究开发中心有限公司 | 三唑并吡啶类和咪唑并吡啶类化合物及其制备方法和医药用途 |
| AU2020242287A1 (en) | 2019-03-21 | 2021-09-02 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A Dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021023207A1 (zh) * | 2019-08-06 | 2021-02-11 | 江苏柯菲平医药股份有限公司 | Jak激酶抑制剂及其用途 |
| CN114761006A (zh) | 2019-11-08 | 2022-07-15 | Inserm(法国国家健康医学研究院) | 对激酶抑制剂产生耐药性的癌症的治疗方法 |
| CN111039963B (zh) * | 2019-12-31 | 2021-03-19 | 卓和药业集团有限公司 | Wxfl10203614水溶性类似物及其合成方法 |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| TW202246233A (zh) | 2021-02-19 | 2022-12-01 | 英商蘇多生物科學有限公司 | Tyk2抑制劑及其用途 |
| WO2022175752A1 (en) | 2021-02-19 | 2022-08-25 | Sudo Biosciences Limited | Tyk2 inhibitors and uses thereof |
| AR125588A1 (es) | 2021-03-04 | 2023-08-02 | Lilly Co Eli | Compuestos inhibidores de fgfr3 |
| CN114539247B (zh) * | 2022-01-19 | 2023-03-21 | 暨南大学 | 一种[1,2,4]三唑并[1,5-a]吡啶类化合物及其制备方法和应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011130146A1 (en) * | 2010-04-14 | 2011-10-20 | Array Biopharma Inc. | 5, 7-substituted-imidazo [1, 2-c] pyrimidines as inhibitors of jak kinases |
| WO2012177606A1 (en) * | 2011-06-20 | 2012-12-27 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as jak inhibitors |
| WO2013055645A1 (en) * | 2011-10-12 | 2013-04-18 | Array Biopharma Inc. | 5,7-substituted-imidazo[1,2-c]pyrimidines |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7160890B2 (en) | 1999-12-02 | 2007-01-09 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| JP4579497B2 (ja) | 2000-12-01 | 2010-11-10 | オーエスアイ・ファーマスーティカルズ・インコーポレーテッド | アデノシンa1、a2a、およびa3受容体に特異的な化合物、ならびにそれらの使用 |
| US7301023B2 (en) | 2001-05-31 | 2007-11-27 | Pfizer Inc. | Chiral salt resolution |
| EA019504B1 (ru) * | 2005-12-13 | 2014-04-30 | Инсайт Корпорейшн | ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПИРРОЛО[2,3-b]ПИРИДИНЫ И ПИРРОЛО[2,3-b]ПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ |
| GB0719803D0 (en) | 2007-10-10 | 2007-11-21 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| JO3041B1 (ar) * | 2008-07-25 | 2016-09-05 | Galapagos Nv | مركبات جديدة مفيدة لمعالجة الأمراض التنكسية والالتهابية |
| CN102131812B (zh) | 2008-08-20 | 2014-04-09 | 硕腾有限责任公司 | 吡咯并[2,3-d]嘧啶化合物 |
| US8551542B1 (en) | 2012-09-20 | 2013-10-08 | Basic Research L.L.C. | Methods and compositions for increasing growth hormones |
| EP2994454B1 (en) | 2013-12-09 | 2019-03-06 | Unichem Laboratories Limited | An improved process for the preparation of (3r,4r)-(1-benzyl-4-methylpiperidin-3-yl)-methylamine |
-
2016
- 2016-04-26 MX MX2017013798A patent/MX370933B/es active IP Right Grant
- 2016-04-26 CN CN201680021913.9A patent/CN107531695B/zh active Active
- 2016-04-26 TR TR2019/09694T patent/TR201909694T4/tr unknown
- 2016-04-26 AU AU2016254385A patent/AU2016254385B2/en active Active
- 2016-04-26 PT PT16785915T patent/PT3290418T/pt unknown
- 2016-04-26 ES ES16785915T patent/ES2734048T3/es active Active
- 2016-04-26 DK DK16785915.6T patent/DK3290418T3/da active
- 2016-04-26 EP EP16785915.6A patent/EP3290418B1/en active Active
- 2016-04-26 EA EA201792021A patent/EA036058B1/ru unknown
- 2016-04-26 JP JP2017556667A patent/JP6600365B2/ja active Active
- 2016-04-26 HU HUE16785915 patent/HUE044240T2/hu unknown
- 2016-04-26 UA UAA201709833A patent/UA119701C2/uk unknown
- 2016-04-26 KR KR1020177034258A patent/KR102006684B1/ko active IP Right Grant
- 2016-04-26 CA CA2982493A patent/CA2982493C/en active Active
- 2016-04-26 PL PL16785915T patent/PL3290418T3/pl unknown
- 2016-04-26 WO PCT/CN2016/080208 patent/WO2016173484A1/zh active Application Filing
- 2016-04-26 US US15/570,499 patent/US10174036B2/en active Active
- 2016-04-29 TW TW105113453A patent/TWI694998B/zh active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011130146A1 (en) * | 2010-04-14 | 2011-10-20 | Array Biopharma Inc. | 5, 7-substituted-imidazo [1, 2-c] pyrimidines as inhibitors of jak kinases |
| WO2012177606A1 (en) * | 2011-06-20 | 2012-12-27 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as jak inhibitors |
| WO2013055645A1 (en) * | 2011-10-12 | 2013-04-18 | Array Biopharma Inc. | 5,7-substituted-imidazo[1,2-c]pyrimidines |
Also Published As
| Publication number | Publication date |
|---|---|
| EA036058B1 (ru) | 2020-09-21 |
| CA2982493C (en) | 2020-01-07 |
| EP3290418A4 (en) | 2018-10-17 |
| KR20170140370A (ko) | 2017-12-20 |
| UA119701C2 (uk) | 2019-07-25 |
| EA201792021A1 (ru) | 2018-07-31 |
| EP3290418A1 (en) | 2018-03-07 |
| HUE044240T2 (hu) | 2019-10-28 |
| WO2016173484A1 (zh) | 2016-11-03 |
| US20180179209A1 (en) | 2018-06-28 |
| JP6600365B2 (ja) | 2019-10-30 |
| CA2982493A1 (en) | 2016-11-03 |
| MX370933B (es) | 2020-01-09 |
| CN107531695A (zh) | 2018-01-02 |
| KR102006684B1 (ko) | 2019-08-02 |
| TW201704231A (zh) | 2017-02-01 |
| TR201909694T4 (tr) | 2019-07-22 |
| DK3290418T3 (da) | 2019-07-01 |
| AU2016254385B2 (en) | 2018-05-10 |
| US10174036B2 (en) | 2019-01-08 |
| PL3290418T3 (pl) | 2019-11-29 |
| JP2018514551A (ja) | 2018-06-07 |
| MX2017013798A (es) | 2018-03-21 |
| AU2016254385A1 (en) | 2017-11-02 |
| PT3290418T (pt) | 2019-07-16 |
| EP3290418B1 (en) | 2019-05-15 |
| ES2734048T3 (es) | 2019-12-04 |
| TWI694998B (zh) | 2020-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107531695B (zh) | Jak抑制剂 | |
| US10829501B2 (en) | Spiroheptane salicylamides and related compounds as inhibitors of ROCK | |
| CN107531711B (zh) | Janus激酶抑制剂 | |
| TWI690531B (zh) | Jak抑制劑 | |
| JP6726677B2 (ja) | 抗がん剤としての置換2−h−ピラゾール誘導体 | |
| CN105461711B (zh) | 作为PI3K抑制剂的吡啶并[1,2-a]嘧啶酮类似物 | |
| CN106470992B (zh) | 作为pi3k抑制剂的吡啶并[1,2-a]嘧啶酮类似物 | |
| WO2018108167A1 (zh) | Cdk4/6抑制剂 | |
| JP6511692B2 (ja) | ヒドロキシプリン類化合物及びその応用 | |
| KR102412045B1 (ko) | 혈액응고인자 Xa 억제제로서의 히드라지드 화합물 | |
| JP6900406B2 (ja) | Akt阻害剤としてのジヒドロピラゾロアゼピン系化合物 | |
| IL266659A (en) | fgfr4 inhibitor and method for its preparation and use | |
| CN107614501B (zh) | 羟基嘌呤类化合物及其应用 | |
| US20240132464A1 (en) | Heterocyclic compounds as e3 ligase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |