CN102391250B - 一种达比加群酯化合物、制备方法及其药物组合物 - Google Patents
一种达比加群酯化合物、制备方法及其药物组合物 Download PDFInfo
- Publication number
- CN102391250B CN102391250B CN 201110249228 CN201110249228A CN102391250B CN 102391250 B CN102391250 B CN 102391250B CN 201110249228 CN201110249228 CN 201110249228 CN 201110249228 A CN201110249228 A CN 201110249228A CN 102391250 B CN102391250 B CN 102391250B
- Authority
- CN
- China
- Prior art keywords
- dabigatran
- derivative
- pharmaceutical composition
- dabigatran etcxilate
- hour
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
一种达比加群酯化合物、制备方法及其药物组合物,所述达比加群酯化合物为3-[(2-{[4-(己氧羰基氨基-亚氨基-甲基)-苯基氨基]-甲基}-1-甲基-1H-苯并咪唑-5-羰基)-吡啶-2-基-氨基]-丙酸乙酯的甲磺酸盐一水合物,本发明还涉及该化合物的制备方法以及以该化合物为活性物质的药物组合物。本发明化合物与达比加群酯甲磺酸盐相比,具有更好的稳定性,更适于制备各种形式的药物制剂及贮存、使用。
Description
技术领域
本发明涉及一种新的达比加群酯化合物,具体讲是涉及3-[(2-{[4-(己氧羰基氨基-亚氨基-甲基)-苯基氨基]-甲基}-1-甲基-1H-苯并咪唑-5-羰基)-吡啶-2-基-氨基]-丙酸乙酯即达比加群酯的甲磺酸盐水合物、及其制备方法和含其药物组合物,属医药技术领域。
背景技术
心房纤颤,简称房颤,是临床上最常见的心律失常。调查显示,我国房颤的总患病率约0.6%,且呈现随年龄增长而上升的趋势,80岁以上年龄组达7.5%,显著高于其他年龄组。所有房颤病人中瓣膜型、非瓣膜型及孤立性房颤所占比例分别为12.9%,65.2%和21.9%,非瓣膜型显著高于其他两种类型。心源性卒中是房颤的主要并发症之一,约20%的患者会由于房颤引发脑卒中,而且房颤导致的卒中往往更为严重,伴有增高的死亡风险(20%)和致残风险(60%)。房颤病人脑卒中(主要是缺血性脑卒冲)发病率明显高于非房颤人群。由于社会人口老龄化的原因以及目前心血管疾病人群生存率的提高,使得近年来房颤发生率急剧上升,目前仅我国房颤患者已达1000万人,所以房颤现已引起社会广泛关注。
目前房颤的治疗方法中,导管消融术虽然已获成功,但存在费用昂贵、仅少数医院能够开展的问题,外科手术成功率较高,但创伤也相对较大,因此人们试图寻找药物和损伤性小的非手术方法治疗房颤。
因为心源性脑卒中是房颤患者最主要的并发症之一,而血栓栓塞并发症是其致死致残的主要原因,所以房颤的抗凝药物治疗是非常必要的。目前华法林是唯一获得FDA批准,用来预防手术后VTE及心房颤动的口服抗血栓药物,但其存在治疗窗较窄、剂量个体差异大、干扰因素多、需频繁监测凝血指标(INR)等缺陷,因此安全、有效的口服抗凝剂成为此领域的研发热点。
达比加群(dabigatran)最早公开于WO 98/37075中,后由德国Boehringer Ingelheim(勃林格殷格翰)公司开发,是一种新型口服抗凝血药,属于非肽类凝血酶抑制剂。该药于2008年4月首先在德国和英国上市,2010年10月又被FDA批准用于减少非瓣膜性房颤患者发生中风及全身性栓塞风险。
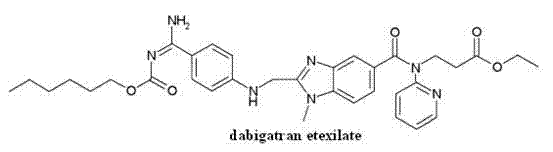
(Ⅰ)。
该化合物为达比加群的前药—达比加群酯,化学名称为3-[(2-{[4-(己氧羰基氨基-亚氨基-甲基)-苯基氨基]-甲基}-1-甲基-1H-苯并咪唑-5-羰基)-吡啶-2-基-氨基]-丙酸乙酯,在体内转化为有活性的达比加群,直接抑制凝血酶发挥抗凝血效应。其具有口服生物利用度高、强效、无需特殊用药监测、药物相互作用少等优点。目前市售的达比加群制剂为达比加群酯甲磺酸盐,商品名为Pradaxa。
研究开发具有良好制剂特性的达比加群新技术以提高并拓展其应用对于医药领域而言无疑是自主创新的新路径。
发明内容
本发明的目的在于提供一种具有良好稳定性的3-[(2-{[4-(己氧羰基氨基-亚氨基-甲基)-苯基氨基]-甲基}-1-甲基-1H-苯并咪唑-5-羰基)-吡啶-2-基-氨基]-丙酸乙酯即达比加群酯药用盐化合物,更具体的讲,所述盐为甲磺酸盐,所述化合物为一水合物,其具有式(Ⅰ)结构:

CH4SO3·H2O。
本发明所述的达比加群酯化合物分子式为C34H41N7O5·CH4SO3·H2O,分子量为741。
本发明的达比加群酯化合物在常温下非常稳定,并且与现有的达比加群酯甲磺酸盐无水物分别在高温60℃、高湿92.5%、光照4500Lx条件下相比,具有更稳定的特性,从而更有利于药物制剂的生产和贮存。
本发明的另一目的是提供上述达比加群酯化合物的制备方法,该方法包括如下步骤:
a.制备达比加群酯甲磺酸盐:称取达比加群酯,溶解于有机溶剂中,边搅拌便加入含甲磺酸的同种有机溶剂溶液,继续搅拌1小时使充分反应,冷却,再搅拌40分钟,然后过滤分离,滤饼用有机溶剂洗涤,在循环空气干燥器中55℃下干燥3h,得达比加群酯甲磺酸盐;
b.制备目标化合物:取步骤a所得达比加群酯甲磺酸盐,溶于60℃热水中,逐步冷却并搅拌,析出结晶,过滤分离出该结晶,再在一定条件下干燥,得本发明所述达比加群酯化合物。
上述制备化合物的方法,所述步骤a中有机溶剂选自乙酸乙酯、丙酮或异丙醇。
上述制备化合物的方法,所述步骤b中逐步冷却并搅拌为首先冷却至15~20℃搅拌1小时,再冷却至5~10℃搅拌1小时,最后冷却至-5-0℃,搅拌10小时。
上述制备化合物的方法,所述步骤b中干燥的条件为温度20-40℃、相对湿度50-70%、干燥4-10小时;其中温度优选30-35℃,相对湿度优选60-65%,干燥时间优选6-8小时。
本发明所述制备达比加群酯化合物的方法,具有有机溶剂使用种类少,无污染,易操作等特点,适合于规模化应用。
本发明再一个目的是提供含有上述达比加群酯化合物的药物组合物。
本发明所述的达比加群酯化合物可与一种或多种药学上可接受的载体或赋形剂制成药物组合物,也可与其他药用活性成分制成药物组合物。药物组合物最小单元中含有本发明所述达比加群酯化合物的量以达比加群酯计为10~200mg,优选 50~110mg。
上述药物组合物,可以是任何临床上可接受的剂型形式,包括口服及肠胃外给药形式的各种剂型。用于口服时,可以是片剂、胶囊、软胶囊、口服液、糖浆、颗粒、滴丸、口崩片、缓释片、缓释胶囊、控释片、控释胶囊;用于肠胃外给药途径时,可以是水针、冻干粉针、无菌粉针、输液。本发明药物组合物优选口服固体制剂,包括含有本发明达比加群酯化合物的片剂或胶囊剂等。
上述药物组合物,所述药学上可接受的载体或赋形剂可选自适用于口服制剂的药用赋形剂,包括填充剂、粘合剂、润滑剂、崩解剂、助溶剂、表面活性剂、吸附载体等。
上述药物组合物,所述药学上可接受的载体或赋形剂可选自适用于注射剂的药用赋形剂,包括溶剂、抗氧剂、助溶剂、吸附剂、渗透压调节剂、PH调节剂。
药物组合物最小单元是指一片,一颗胶囊,一袋颗粒或一支注射剂等。
本发明的达比加群酯化合物与上市的达比加群酯甲磺酸盐活性形式都是达比加群,故适用于使用达比加群酯甲磺酸的所有疾病场合。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
实施例1:达比加群酯甲磺酸盐的制备:
将6.278g(0.01mol)达比加群酯(如WO 98/37075中所述方法),溶于400ml乙酸乙酯中,室温下边搅拌边加入溶有甲磺酸0.961g(0.01mol)的乙酸乙酯溶液40ml,加完后继续搅拌60分钟,然后置冰浴中再搅拌40分钟,过滤,滤饼用乙酸乙酯80ml洗涤,在循环空气干燥器中55℃下干燥3h,得6.85g达比加群酯甲磺酸盐无水物,收率94.6%。熔点:178-179℃。
元素分析:
| 元素分析 | 实际值% | 理论值% |
| C | 58.02 | 58.09 |
| H | 6.32 | 6.22 |
| N | 13.60 | 13.55 |
| O | 17.24 | 17.27 |
| S | 4.47 | 4.43 |
。 [0027] 实施例2:本发明所述达比加群酯化合物的制备:
取实施例1中达比加群酯甲磺酸盐2g,加入100ml60℃的热水溶解,冷至15~20℃搅拌1小时,再冷至5~10℃搅拌1小时,最后冷却至-5-0℃,搅拌10小时,析出结晶,过滤,将滤饼于35℃、65%相对湿度条件下干燥6小时,得到本发明所述达比加群酯化合物1.68g,收率82%。
元素分析:
| 元素分析 | 实际值% | 理论值% |
| C | 56.56 | 56.68 |
| H | 6.38 | 6.34 |
| N | 13.25 | 13.22 |
| O | 19.46 | 19.43 |
| S | 4.27 | 4.32 |
用卡尔-费休氏法测得本发明所述达比加群酯化合物中的水分为2.2%(理论:2.4%);热重分析结果表明为一水合物的特征。
实施例3:本发明所述达比加群酯化合物的制备:
取实施例1中达比加群酯甲磺酸盐2g,加入100ml 60℃的热水溶解,冷至15~20℃搅拌1小时,再冷至5~10℃搅拌1小时,最后冷却至-5-0℃,搅拌10小时,析出结晶,过滤,将滤饼于30℃、60%相对湿度条件下干燥8小时,得到本发明所述达比加群酯化合物1.63g,收率80%。
元素分析:
| 元素分析 | 实际值% | 理论值% |
| C | 56.61 | 56.68 |
| H | 6.29 | 6.34 |
| N | 13.28 | 13.22 |
| O | 19.51 | 19.43 |
| S | 4.26 | 4.32 |
用卡尔-费休氏法测得本发明所述达比加群酯化合物中的水分为2.5%(理论:2.4%);热重分析结果表明为一水合物的特征。
实施例4:本发明所述达比加群酯化合物片剂(75mg)的制备:
配方:达比加群酯甲磺酸盐一水合物 75g(以达比加群酯计):
微晶纤维素 70g;
可压性淀粉 80g;
羧甲基淀粉钠 15g;
2%羟丙基甲基纤维素乙醇液 适量;
硬脂酸镁 2g;
滑石粉 1g;
制成 1000片。
工艺:
1、原、辅料分别粉碎过80目筛备用;
2、取2%HPMC加浓度为30~95%药用乙醇制成5~10%的溶液,即得;
3、取达比加群酯甲磺酸盐一水合物、微晶纤维素、可压性淀粉、羧甲基淀粉钠混合均匀,加入2%HPMC乙醇溶液制软材,16目筛制粒,60℃干燥;
4、16目筛整粒,加入硬脂酸镁、滑石粉混合10分钟,使均匀,压片即得。
实施例5:本发明所述达比加群酯化合物胶囊(110mg)的制备:
配方:达比加群酯甲磺酸盐一水合物 110g(以达比加群酯含量计);
微晶纤维素 50g;
可压性淀粉 70g;
羧甲基淀粉钠 15g;
2%羟丙基甲基纤维素乙醇液 适量;
硬脂酸镁 2g;
制成 1000粒。
工艺:
1、原、辅料分别粉碎过80目筛备用;
2、取2%HPMC加浓度为30~95%药用乙醇制成5~10%的溶液,即得;
3、取达比加群酯甲磺酸盐一水合物、微晶纤维素、可压性淀粉、羧甲基淀粉钠混合均匀,加入2%HPMC乙醇溶液制软材,16目筛制粒,60℃干燥;
4、16目筛整粒,加入硬脂酸镁混合10分钟,使均匀,灌装胶囊即得。
实施例6:本发明所述达比加群酯化合物胶囊(150mg)的制备:
配方:达比加群酯甲磺酸盐一水合物 150g(以达比加群酯含量计);
微晶纤维素 40g;
可压性淀粉 40g;
羧甲基淀粉钠 15g;
2%羟丙基甲基纤维素 适量;
硬脂酸镁 2g;
制成 1000粒。
工艺:
1、原、辅料分别粉碎过80目筛备用;
2、取2%HPMC加浓度为30~95%药用乙醇制成5~10%的溶液,即得;
3、取达比加群酯甲磺酸盐一水合物、微晶纤维素、可压性淀粉、羧甲基淀粉钠混合均匀,加入2%HPMC乙醇溶液制软材,16目筛制粒,60℃干燥;
4、16目筛整粒,加入硬脂酸镁混合10分钟,使均匀,灌装胶囊即得。
实施例7:本发明所述达比加群酯化合物与达比加群酯甲磺酸盐在高温条件下的稳定性对比试验:
取上述两种化合物置扁形称量瓶中,摊成≤5mm厚的薄层,分别置密封洁净容器中,在60℃条件下放置5天;分别于第0天和第5天取样,检测,结果如下:
在60℃高温条件放置变化情况
| 时间 | 0天:有关物质 | 5天:有关物质 | 有关物质变化 |
| 达比加群酯甲磺酸盐 | 0.45% | 1.49% | 1.04% |
| 达比加群酯甲磺酸盐一水合物 | 0.43% | 1.10% | 0.67% |
由上表可见,在高温60℃条件下放置5天后,达比加群酯甲磺酸盐的有关物质升高1.04%,对高温比较稳定;本发明所述达比加群酯化合物的有关物质变化不大,对高温的稳定性很好,明显优于达比加群酯甲磺酸盐。
实施例8:本发明所述达比加群酯化合物与达比加群酯甲磺酸盐在高湿条件下的稳定性对比试验。
取上述两种化合物置扁形称量瓶中,摊成≤5mm厚的薄层,置恒湿密闭容器中,于92.5%相对湿度条件下放置5天;分别于第0天和第5天取样,检测,结果如下:
在92.5%相对湿度高湿条件下放置变化情况
| 时间 | 0天:有关物质 | 5天:有关物质 | 有关物质变化 |
| 达比加群酯甲磺酸盐 | 0.45% | 5.23% | 4.78% |
| 达比加群酯甲磺酸盐一水合物 | 0.43% | 1.75% | 1.32% |
由上表可见,在高湿92.5%相对湿度条件下放置5天后,达比加群酯甲磺酸盐的有关物质升高4.78%,对高湿极不稳定;本发明所述达比加群酯化合物有关物质有一定升高,对高湿较稳定,优于达比加群酯甲磺酸盐。
实施例9:本发明所述达比加群酯化合物与达比加群酯甲磺酸盐在强光照射条件下的稳定性对比试验。
取上述两种化合物置扁形称量瓶中,摊成≤5mm厚的薄层,置光照箱,于照度5000Lx条件下放置5天,分别于第0天和第5天取样,检测,结果如下:
在光照5000Lx条件下放置变化情况
| 时间 | 0天:有关物质 | 5天:有关物质 | 有关物质变化 |
| 达比加群酯甲磺酸盐 | 0.45% | 2.60% | 2.15% |
| 达比加群酯甲磺酸盐一水合物 | 0.43% | 0.85% | 0.42% |
由上表可见,在光照5000Lx条件下放置5天后,达比加群酯甲磺酸盐的有关物质升高2.15%,对光照的稳定性较差;本发明所述达比加群酯化合物的有关物质变化不大,对光照的稳定性很好,显著优于达比加群酯甲磺酸盐。
Claims (10)
1.一种达比加群酯衍生物,其特征在于,所述达比加群酯为3-[(2-{[4-(己氧羰基氨基-亚氨基-甲基)-苯基氨基]-甲基}-1-甲基-1H-苯并咪唑-5-羰基)-吡啶-2-基-氨基]-丙酸乙酯,所述衍生物具有式(1)结构:
CH4SO3·H2O。
2.一种制备如权利要求1所述衍生物的方法,其特征在于,它按如下步骤进行:
a.制备达比加群酯甲磺酸盐:称取达比加群酯,溶解于有机溶剂中,边搅拌边加入含甲磺酸的同种有机溶剂溶液,继续搅拌1小时使充分反应,冷却,再搅拌40分钟,然后过滤分离,滤饼用有机溶剂洗涤,在循环空气干燥器中55℃下干燥3h,得达比加群酯甲磺酸盐;
b.制备目标衍生物:取步骤a所得达比加群酯甲磺酸盐,溶于60℃热水中,逐步冷却并搅拌,析出结晶,过滤分离出该结晶,再在一定条件下干燥,得权利要求1所述的达比加群酯衍生物。
3.根据权利要求2所述制备衍生物的方法,其特征在于,所述步骤a中有机溶剂选自乙酸乙酯、丙酮或异丙醇。
4.根据权利要求3所述制备衍生物的方法,其特征在于,所述步骤b中的逐步冷却并搅拌是指首先冷却至15~20℃搅拌1小时,再冷却至5~10℃搅拌1小时,最后冷却至-5-0℃,搅拌10小时。
5.根据权利要求4所述制备衍生物的方法,其特征在于,所述步骤b中干燥的条件为温度20-40℃、相对湿度50-70%、干燥4-10小时。
6.根据权利要求5所述制备衍生物的方法,其特征在于,所述步骤b中干燥的温度为30-35℃,相对湿度为60-65%,干燥时间为6-8小时。
7.一种药物组合物,其特征在于,它以权利要求1所述的达比加群酯衍生物作为活性成分。
8.根据权利要求7所述的药物组合物,其特征在于,所述药物组合物最小单元中含有达比加群酯衍生物的量以达比加群酯计为10~200mg;所述药物组合物最小单元是指一片,一颗胶囊,一袋颗粒或一支注射剂。
9.根据权利要求7所述的药物组合物,其特征在于,所述药物组合物是任何临床上可接受的药物剂型。
10.根据权利要求9所述的药物组合物,其特征在于,所述剂型为口服固体制剂。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201110249228 CN102391250B (zh) | 2011-08-29 | 2011-08-29 | 一种达比加群酯化合物、制备方法及其药物组合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201110249228 CN102391250B (zh) | 2011-08-29 | 2011-08-29 | 一种达比加群酯化合物、制备方法及其药物组合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102391250A CN102391250A (zh) | 2012-03-28 |
| CN102391250B true CN102391250B (zh) | 2013-06-19 |
Family
ID=45858656
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 201110249228 Active CN102391250B (zh) | 2011-08-29 | 2011-08-29 | 一种达比加群酯化合物、制备方法及其药物组合物 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102391250B (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103420980A (zh) * | 2012-05-22 | 2013-12-04 | 北京美倍他药物研究有限公司 | 达比加群衍生物 |
| CN103420985B (zh) * | 2012-05-24 | 2015-09-23 | 天津药物研究院 | 作为前药的达比加群酯衍生物及其制备方法和用途 |
| CN103420982B (zh) * | 2012-05-24 | 2015-07-08 | 天津药物研究院 | 达比加群酯衍生物及其制备方法和用途 |
| CN103420994B (zh) * | 2012-05-24 | 2016-04-06 | 天津药物研究院 | 作为前药的达比加群酯衍生物及其制备方法和用途 |
| CN103420983B (zh) * | 2012-05-24 | 2015-07-08 | 天津药物研究院 | 达比加群酯衍生物及其制备方法和用途 |
| CN103420984B (zh) * | 2012-05-24 | 2015-07-08 | 天津药物研究院 | 作为前药的达比加群酯衍生物及其制备方法和用途 |
| CN103539779B (zh) * | 2012-07-13 | 2016-12-21 | 四川海思科制药有限公司 | 一种达比加群酯的羟基取代苯磺酸盐及其制备方法和用途 |
| CN104892574A (zh) * | 2014-03-04 | 2015-09-09 | 浙江海正药业股份有限公司 | 达比加群酯甲磺酸盐的晶型及其制备方法和用途 |
| CN104974137A (zh) * | 2014-04-04 | 2015-10-14 | 江苏天士力帝益药业有限公司 | 达比加群酯甲磺酸盐新晶型及其制备方法 |
| CN103951654B (zh) * | 2014-05-13 | 2016-08-24 | 南京生命能科技开发有限公司 | 甲磺酸达比加群酯的晶体v及其制备方法 |
| CN103965164A (zh) * | 2014-05-13 | 2014-08-06 | 南京生命能科技开发有限公司 | 甲磺酸达比加群酯的晶体ⅵ及其制备方法 |
| CN105572275B (zh) * | 2014-10-08 | 2017-09-29 | 华仁药业股份有限公司 | 一种甲磺酸达比加群酯含量的检测方法 |
| CN106916141A (zh) * | 2017-04-06 | 2017-07-04 | 南京生命能科技开发有限公司 | 一种甲磺酸达比加群酯的制备方法 |
| JP2019014712A (ja) * | 2017-07-03 | 2019-01-31 | エルメッド エーザイ株式会社 | 安定なダビガトラン製剤 |
| CN108570035A (zh) * | 2018-07-01 | 2018-09-25 | 李万强 | 一种甲磺酸达比加群酯的提纯方法 |
| CN111334537A (zh) * | 2020-04-01 | 2020-06-26 | 中山万汉制药有限公司 | 酶催化的达比加群酯中间体的合成方法 |
| CN113307792A (zh) * | 2021-05-21 | 2021-08-27 | 杭州国瑞生物科技有限公司 | 一种达比加群酯的精制方法及其特定降解杂质的控制方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE540943T1 (de) * | 2002-03-07 | 2012-01-15 | Boehringer Ingelheim Pharma | 3-ä(2-ää4-(hexyloxycarbonylamino-imino-methyl)- phenylaminoümethylü-1-methyl-1h-benzimidazol-5- carbonyl)-pyridin-2-yl-aminoü-propionsäure- ethylester methansulfonat |
| DE10337697A1 (de) * | 2003-08-16 | 2005-03-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Tablette enthaltend 3-[(2-{[4-(Hexyloxycarbonylamino-imino-methyl)-phenyl-amino]-methyl}-1-methyl-1H-benzimidazol-5-carbonyl)-pyridin-2-yl-amino]-propionsäure-ethylester oder dessen Salze |
| WO2010020602A1 (en) * | 2008-08-19 | 2010-02-25 | Boehringer Ingelheim International Gmbh | Dabigatran for percutaneous interventional cardiac catheterisation |
-
2011
- 2011-08-29 CN CN 201110249228 patent/CN102391250B/zh active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN102391250A (zh) | 2012-03-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102391250B (zh) | 一种达比加群酯化合物、制备方法及其药物组合物 | |
| CN102351853B (zh) | 一种阿齐沙坦酯化合物、制备方法及其药物组合物 | |
| CN111825547B (zh) | 一种芳基丙酸类化合物的盐及其制药用途 | |
| CN108530382A (zh) | 一种非布索坦川芎嗪共晶体及其制备方法和用途 | |
| CN102349902B (zh) | 一种含有左旋氨氯地平和坎地沙坦酯的药物组合物及其制备方法 | |
| CN103664881A (zh) | 结晶变体形态b的达比加群酯及其制备方法和用途 | |
| CN102285970B (zh) | 一种埃索美拉唑化合物、制备方法及其药物组合物 | |
| CN102321007B (zh) | 一种奥拉西坦化合物、制备方法及其药物组合物 | |
| CN102304088B (zh) | 一种伊伐布雷定化合物、制备方法及其药物组合物 | |
| CN112194624A (zh) | 一种异喹啉类化合物的晶型及其制备方法 | |
| TW201311240A (zh) | 非晶形哌啶基化合物之生物可利用組合物 | |
| CN105367551A (zh) | 达比加群酯乙醇酸盐及其制备方法和应用 | |
| CN105440017B (zh) | 达比加群酯香草酸盐及其制备方法和应用 | |
| CN107868009B (zh) | 一种酒石酸美托洛尔晶体及含该晶体的药物组合物及其制备方法 | |
| CN102351881B (zh) | 一种稳定的盐酸左氧氟沙星化合物 | |
| CN103570679A (zh) | 达比加群酯葡萄糖酸盐及其制备方法和应用 | |
| CN102093234B (zh) | 一种二元酯酸的氨丁三醇盐化合物及其制备方法和药物应用 | |
| CN105348261A (zh) | 达比加群酯丙酮酸盐及其制备方法和应用 | |
| CN102295619B (zh) | 一种非布索坦化合物、制备方法及其药物组合物 | |
| CN104546899A (zh) | 一种含有奥美拉唑的口服固体药物组合物 | |
| CN104829467A (zh) | 盐酸氨溴索二水化合物 | |
| WO2015106636A1 (zh) | 去甲伊伐布雷定盐及其制备方法和应用 | |
| CN105726495B (zh) | 一种注射用短效苯并二氮杂卓盐药物组合物及其制备方法 | |
| CN103509004A (zh) | 达比加群酯咖啡酸盐及其制备方法和应用 | |
| CN103570680A (zh) | 达比加群酯果糖酸盐及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20201204 Address after: No.88, Yangzi Road, economic and Technological Development Zone, Shijiazhuang City, Hebei Province Patentee after: CSPC PHARMACEUTICAL GROUP OUYI PHARMA Co.,Ltd. Patentee after: SHIJIAZHUAN PHARMA GROUP NBP PHARMACEUTICAL Co.,Ltd. Address before: 050051 No. 276 West Zhongshan Road, Hebei, Shijiazhuang Patentee before: CSPC PHARMACEUTICAL GROUP OUYI PHARMA Co.,Ltd. |
|
| TR01 | Transfer of patent right |