CN102079737B - Method for preparing apigenin - Google Patents
Method for preparing apigenin Download PDFInfo
- Publication number
- CN102079737B CN102079737B CN201010616467A CN201010616467A CN102079737B CN 102079737 B CN102079737 B CN 102079737B CN 201010616467 A CN201010616467 A CN 201010616467A CN 201010616467 A CN201010616467 A CN 201010616467A CN 102079737 B CN102079737 B CN 102079737B
- Authority
- CN
- China
- Prior art keywords
- solid
- solution
- adds
- apigenin
- weighing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- XADJWCRESPGUTB-UHFFFAOYSA-N apigenin Natural products C1=CC(O)=CC=C1C1=CC(=O)C2=CC(O)=C(O)C=C2O1 XADJWCRESPGUTB-UHFFFAOYSA-N 0.000 title claims abstract description 26
- KZNIFHPLKGYRTM-UHFFFAOYSA-N apigenin Chemical compound C1=CC(O)=CC=C1C1=CC(=O)C2=C(O)C=C(O)C=C2O1 KZNIFHPLKGYRTM-UHFFFAOYSA-N 0.000 title claims abstract description 26
- 229940117893 apigenin Drugs 0.000 title claims abstract description 26
- 235000008714 apigenin Nutrition 0.000 title claims abstract description 26
- 238000000034 method Methods 0.000 title claims abstract description 13
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 claims abstract description 12
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 claims abstract description 11
- 239000003513 alkali Substances 0.000 claims abstract description 8
- 239000002994 raw material Substances 0.000 claims abstract description 7
- FBUBVLUPUDBFME-UHFFFAOYSA-N xanthoxylline Natural products COC1=CC(O)=C(C(C)=O)C(OC)=C1 FBUBVLUPUDBFME-UHFFFAOYSA-N 0.000 claims abstract description 4
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 claims description 32
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 28
- 238000005303 weighing Methods 0.000 claims description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 17
- 235000010265 sodium sulphite Nutrition 0.000 claims description 16
- 150000001875 compounds Chemical class 0.000 claims description 15
- 238000003756 stirring Methods 0.000 claims description 15
- 238000000967 suction filtration Methods 0.000 claims description 15
- 239000007788 liquid Substances 0.000 claims description 14
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 12
- 235000014493 Crataegus Nutrition 0.000 claims description 11
- 241001092040 Crataegus Species 0.000 claims description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 10
- 239000007787 solid Substances 0.000 claims description 9
- 238000006243 chemical reaction Methods 0.000 claims description 8
- 238000001914 filtration Methods 0.000 claims description 8
- 238000001816 cooling Methods 0.000 claims description 7
- 238000001035 drying Methods 0.000 claims description 7
- 239000012065 filter cake Substances 0.000 claims description 7
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical class [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 claims description 6
- 239000012295 chemical reaction liquid Substances 0.000 claims description 5
- 230000001143 conditioned effect Effects 0.000 claims description 5
- 239000012153 distilled water Substances 0.000 claims description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 claims description 5
- 238000002156 mixing Methods 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 3
- 229940071870 hydroiodic acid Drugs 0.000 claims description 3
- 239000000376 reactant Substances 0.000 claims description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims 1
- 229910052740 iodine Inorganic materials 0.000 claims 1
- 239000011630 iodine Substances 0.000 claims 1
- 238000003786 synthesis reaction Methods 0.000 abstract description 5
- 230000002378 acidificating effect Effects 0.000 abstract description 3
- 239000003054 catalyst Substances 0.000 abstract description 3
- 238000004519 manufacturing process Methods 0.000 abstract description 2
- 238000000746 purification Methods 0.000 abstract description 2
- 238000000926 separation method Methods 0.000 abstract description 2
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 abstract 1
- 238000006555 catalytic reaction Methods 0.000 abstract 1
- 238000006482 condensation reaction Methods 0.000 abstract 1
- 238000010438 heat treatment Methods 0.000 abstract 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 abstract 1
- 238000007363 ring formation reaction Methods 0.000 abstract 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 7
- 239000002904 solvent Substances 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 4
- 239000013065 commercial product Substances 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 240000007087 Apium graveolens Species 0.000 description 2
- 235000015849 Apium graveolens Dulce Group Nutrition 0.000 description 2
- 235000010591 Appio Nutrition 0.000 description 2
- 102100033270 Cyclin-dependent kinase inhibitor 1 Human genes 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 102000003945 NF-kappa B Human genes 0.000 description 2
- 108010057466 NF-kappa B Proteins 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000000452 restraining effect Effects 0.000 description 2
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102000012199 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 1
- 108050002772 E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000944380 Homo sapiens Cyclin-dependent kinase inhibitor 1 Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 235000010627 Phaseolus vulgaris Nutrition 0.000 description 1
- 244000046052 Phaseolus vulgaris Species 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000002785 anti-thrombosis Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- FXNFHKRTJBSTCS-UHFFFAOYSA-N baicalein Chemical compound C=1C(=O)C=2C(O)=C(O)C(O)=CC=2OC=1C1=CC=CC=C1 FXNFHKRTJBSTCS-UHFFFAOYSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000025084 cell cycle arrest Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 229930003935 flavonoid Natural products 0.000 description 1
- -1 flavonoid compound Chemical class 0.000 description 1
- 235000017173 flavonoids Nutrition 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000003075 phytoestrogen Substances 0.000 description 1
- 229960004839 potassium iodide Drugs 0.000 description 1
- 235000007715 potassium iodide Nutrition 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention provides a method for preparing apigenin, which comprises the following steps: performing the condensation reaction of the raw material of 2,4-dimethoxy-6-hydroxyacetophenone with p-methoxybenzaldehyde with the catalysis of an alkali to obtain a compound II; performing the cyclization reaction with the presence of a catalyst to obtain a compound III; heating to remove the methoxy group at an oxygen-isolated and acidic condition so as to obtain apigenin. The invention prepares apigenin by chemical synthesis, and has the advantages of simple process, easy processing for product separation and purification, high yield, mass production potential, abundant raw material sources, and low cost.
Description
Technical field
The present invention relates to a kind of method for preparing apigenin, belong to the synthetic field of medicine.
Background technology
Apigenin, systematic naming method are 4 ˊ, 5,7-trihydroxyflavone, and its structural formula is suc as formula shown in the I.Apigenin is naturally occurring a kind of flavonoid compound, and the title of " phytoestrogen " is arranged, and extensively is present in multiple fruit, vegetables, beans and the tealeaves, and wherein content is the highest in the celery.Have multiple pharmacological effect, like: antitumor, anti-inflammatory, arteriosclerosis, antithrombotic, anxiety, effect such as antibiotic, antiviral, anti-oxidant, at present clinical be mainly used in antitumor.
Ⅰ
The anticancer scope of apigenin is wider, and in vitro tests proves that it all has restraining effect to kinds of tumor cells, as: breast cancer cell, stomach cancer cell, prostate cancer cell, liver cancer cell, ovarian cancer cell etc. all have restraining effect.
Apigenin can activate the pl4ARF-Mdm2-p53 approach through the expression of downward modulation Mdm2; Also can be through activating the p53/waf1 approach; Increase the stability of p53; P53 albumen in the tumour cell, P21/WAF1 protein level are raise, induce the cell retardance of multiple cancerous cell line, with cell-cycle arrest at G
2/ M the phase, and through p53 approach inducing apoptosis of tumour cell.Nuclear Factor-Kappa B (NF-κ B), B cell lymphoma/white blood disease-2 (Bcl-2), Bax have also participated in the effect of apigenin inducing apoptosis of tumour cell.
Because the content of apigenin in crude drug is very low, all adopt the method for from plant, directly extracting to prepare apigenin usually.Prepare apigenin through chemical synthesis and can improve apigenin production efficiency, and can improve the quality of medicine greatly, therefore also received domestic and international pharmacy worker's attention.At present, the method report for preparing apigenin with chemical synthesis in a large number is few, and with 2,4-dimethoxy-6-hydroxy acetophenone is that raw material synthetic celery element does not appear in the newspapers yet.
Summary of the invention
Prepare apigenin for solving chemical synthesis, the present invention provides a kind of abundant raw material, productive rate height, technology simply to prepare the method for apigenin, and the present invention realizes through following technical scheme:
A kind of method for preparing apigenin, following each step of process:
(1) take by weighing 2,4-dimethoxy-6-hydroxy acetophenone is by 2; The mol ratio of 4-dimethoxy-6-hydroxy acetophenone and aubepine is that 1 ︰ 1 ~ 1.2 takes by weighing aubepine; Be that 1 ︰, 10 adding solvents dissolve by solid-to-liquid ratio again, add alkali again, making the concentration of alkali in solution is 10 ~ 25%; Stir 20 ~ 40h down at 20 ~ 40 ℃, and then get compound ii after handling;

Ⅱ
(2) getting compound ii, is that 1 ︰ 1 ~ 10 adds solvent by solid-to-liquid ratio, adds the catalyzer of raw materials quality 0.5 ~ 3%, is heated to 80 ~ 150 ℃, stirs 3 ~ 30h, handles obtaining the compound III after the cooling again;

Ⅲ
(3) take by weighing the compound III, by solid solid than or solid-to-liquid ratio be that 1 ︰ 5 ~ 20 adds an acidic catalysts, after mixing, under nitrogen protection, be heated to 100 ~ 180 ℃, reacted 6 ~ 10 hours, be cooled to room temperature then, obtain apigenin after the processing again, i.e. chemical compounds I.
Alkali in the said step (1) is Pottasium Hydroxide or sodium hydroxide.
Solvent in the said step (1) is methyl alcohol or ethanol.
Processing again in the said step (1) is to pass through with concentrated hydrochloric acid solution conditioned reaction liquid pH=3 ~ 4, and with the reaction solution suction filtration, filter cake is used distilled water wash, and filter cake carries out drying again.
Catalyzer in the said step (2) is iodine, iodized salt.
Solvent in the said step (2) is a DMSO 99.8MIN..
Handling again in the said step (2) is through taking by weighing the sodium sulfite anhy 96 with quality such as catalyzer; Add 5 ~ 10 times water of solvent volume, be made into sodium sulfite solution, cooling back solution in the step (2) is added in the sodium sulfite solution; Stir 0.5 ~ 1h; Hold over night, behind the suction filtration with filtration cakes torrefaction
An acidic catalyst in the said step (3) is solid pyridine hydrochloride, hydrobromic acid solution or hydroiodic acid HI acetum.
The mass concentration of the hydrobromic acid solution in the said step (3) is 48%.
The mass concentration of the hydroiodic acid HI acetum in the said step (3) is 56%.
Handling again in the said step (3) is that the volume ratio through by reactant and water is that 1 ︰ 10 ~ 20 adds in the entry, hold over night, suction filtration after drying.
Said reagent is commercial product, and wherein methyl alcohol, ethanol, methyl-sulphoxide are solution, and concentration is the concentration of conventional commercial product; Aubepine, Pottasium Hydroxide, sodium hydroxide, iodine, iodized salt, pyridine hydrochloride, sodium sulfite anhy 96 are solid, are the commercial product of routine.
The present invention utilizes chemical synthesis to prepare apigenin, and its advantage is:
1, technology is simple, and the product separation and purification treatment is easy;
2, productive rate is high; Reaction mole total recovery reaches more than 50%, possesses the potentiality of scale prodn.
3, raw material sources are abundant, and cost is low.
Embodiment
Below in conjunction with embodiment the present invention is done and to further describe.
Embodiment 1
(1) take by weighing 19.6g2,4-dimethoxy-6-hydroxy acetophenone is by 2; The mol ratio of 4-dimethoxy-6-hydroxy acetophenone and aubepine is that 1 ︰ 1.2 takes by weighing the 16.3g aubepine, is that 1 ︰, 10 adding 356mL methyl alcohol dissolve by solid-to-liquid ratio again, adds sodium hydroxide again; Making the concentration of alkali in solution is 15%; Stir 40h down at 20 ℃, use concentrated hydrochloric acid solution conditioned reaction liquid pH=3 then, the reaction solution suction filtration; Filter cake is used distilled water wash, gets compound ii behind the filtration cakes torrefaction; Yield 90%; M.p.:112 ~ 116;
1HNMR (CDCl
3) characterize: δ 3.82 (s, 3H), 3.85 (s, 3H), 3.92 (s, 3H), 5.97 (d, J=1.9Hz, 1H), 6.11 (d, J=1.9Hz, 1H), 6.93 (d, J=7.5Hz, 2H), 7.56 (d, J=7.5 Hz, 2H), 12.78 (s, 1H);
(2) getting the 15.7g compound ii, is that 1 ︰ 1 adds the 15.7mL DMSO 99.8MIN. by solid-to-liquid ratio, adds 0.0785g iodine, is heated to 150 ℃, stirs 3h, cooling; Take by weighing the sodium sulfite anhy 96 of 7.85g, add the water of 78.5mL, be made into sodium sulfite solution, will cool off back solution and add in the sodium sulfite solution, stir 1h, hold over night with filtration cakes torrefaction, obtains the compound III behind the suction filtration; Yield 84%; M.p.:158 ~ 160;
1HNMR (CDCl
3) characterize: δ 3.99 (s, 3H), 4.03 (s, 3H), 4.07 (s, 3H), 6.49 (d, J=2.2Hz, 1H); 6.68 (d, J=2.2Hz, 1H); 6.81 (s, 1H); 7.13 (d, J=7.5Hz, 2H); 7.95 (d, J=7.5Hz, 2H);
(3) take by weighing 3.1g compound III, by solid solid than being 1 ︰, 5 adding 15.5g pyridine hydrochlorides, after mixing; Under nitrogen protection, be heated to 150 ℃, reacted 6 hours, be cooled to room temperature then; Volume ratio by reactant and water is that 1 ︰ 15 adds in the entry, hold over night, suction filtration after drying; Obtain apigenin, i.e. chemical compounds I; Yield 82%; M.p.:345 ~ 350;
1HNMR (DMSO) characterizes: δ 6.18 (s, 1H), 6.47 (s, 1H), 6.78 (s, 1H), 6.91 (d, J=7.8Hz, 2H), 7.91 (d, J=7.8Hz, 2H), 10.36 (s, 1H), 10.85 (s, 1H), 12.95 (s, 1H).
Embodiment 2
(1) take by weighing 19.6g2,4-dimethoxy-6-hydroxy acetophenone is by 2; The mol ratio of 4-dimethoxy-6-hydroxy acetophenone and aubepine is that 1 ︰ 1 takes by weighing the 13.6g aubepine, is that 1 ︰, 10 adding 332mL ethanol dissolve by solid-to-liquid ratio again, adds Pottasium Hydroxide again; Making the concentration of alkali in solution is 10%, stirs 20h down at 40 ℃, uses concentrated hydrochloric acid solution conditioned reaction liquid pH=4 then; With the reaction solution suction filtration, filter cake is used distilled water wash, gets compound ii behind the filtration cakes torrefaction; Yield 90%; M.p.:112 ~ 116;
1HNMR (CDCl
3) characterize: δ 3.82 (s, 3H), 3.85 (s, 3H), 3.92 (s, 3H), 5.97 (d, J=1.9Hz, 1H), 6.11 (d, J=1.9Hz, 1H), 6.93 (d, J=7.5Hz, 2H), 7.56 (d, J=7.5 Hz, 2H), 12.78 (s, 1H);
(2) getting the 15.7g compound ii, is that 1 ︰ 10 adds the 157mL DMSO 99.8MIN. by solid-to-liquid ratio, adds the 0.157g potassiumiodide, is heated to 80 ℃, stirs 30h, cooling; Take by weighing the sodium sulfite anhy 96 of 15.7g, add the water of 125.6mL, be made into sodium sulfite solution, will cool off back solution and add in the sodium sulfite solution, stir 0.5h, hold over night with filtration cakes torrefaction, obtains the compound III behind the suction filtration; Yield 80%; M.p.:158 ~ 160;
1HNMR (CDCl
3) characterize: δ 3.99 (s, 3H), 4.03 (s, 3H), 4.07 (s, 3H), 6.49 (d, J=2.2Hz, 1H); 6.68 (d, J=2.2Hz, 1H); 6.81 (s, 1H); 7.13 (d, J=7.5Hz, 2H); 7.95 (d, J=7.5Hz, 2H);
(3) taking by weighing 3.1g compound III, is that 1 ︰, 10 adding 31mL mass concentrations are 56% Hydrogen bromide acetum by solid-to-liquid ratio, after mixing; Under nitrogen protection, be heated to 100 ℃, reacted 10 hours, be cooled to room temperature then; Volume ratio by reaction solution and water is that 1 ︰ 10 adds in the 310mL water hold over night, suction filtration after drying; Obtain apigenin, i.e. chemical compounds I; Yield 70%; M.p.:345 ~ 350;
1HNMR (DMSO) characterizes: δ 6.18 (s, 1H), 6.47 (s, 1H), 6.78 (s, 1H), 6.91 (d, J=7.8Hz, 2H), 7.91 (d, J=7.8Hz, 2H), 10.36 (s, 1H), 10.85 (s, 1H), 12.95 (s, 1H).
Embodiment 3
(1) take by weighing 19.6g2,4-dimethoxy-6-hydroxy acetophenone is by 2; The mol ratio of 4-dimethoxy-6-hydroxy acetophenone and aubepine is that 1 ︰ 1 takes by weighing the 13.6g aubepine, is that 1 ︰, 10 adding 332mL ethanol dissolve by solid-to-liquid ratio again, adds Pottasium Hydroxide again; Making the concentration of alkali in solution is 25%, stirs 30h down at 30 ℃, uses concentrated hydrochloric acid solution conditioned reaction liquid pH=3 then; With the reaction solution suction filtration, filter cake is used distilled water wash, gets compound ii behind the filtration cakes torrefaction; Yield 88%; M.p.:112 ~ 116;
1HNMR (CDCl
3) characterize: δ 3.82 (s, 3H), 3.85 (s, 3H), 3.92 (s, 3H), 5.97 (d, J=1.9Hz, 1H), 6.11 (d, J=1.9Hz, 1H), 6.93 (d, J=7.5Hz, 2H), 7.56 (d, J=7.5 Hz, 2H), 12.78 (s, 1H);
(2) getting the 15.7g compound ii, is that 1 ︰ 5 adds the 78.5mL DMSO 99.8MIN. by solid-to-liquid ratio, adds the 0.471g Soiodin, is heated to 100 ℃, stirs 20h, cooling; Take by weighing the sodium sulfite anhy 96 of 15.7g, add the water of 785mL, be made into sodium sulfite solution, will cool off back solution and add in the sodium sulfite solution, stir 0.5h, hold over night with filtration cakes torrefaction, obtains the compound III behind the suction filtration; Yield 83%; M.p.:158 ~ 160;
1HNMR (CDCl
3) characterize: δ 3.99 (s, 3H), 4.03 (s, 3H), 4.07 (s, 3H), 6.49 (d, J=2.2Hz, 1H); 6.68 (d, J=2.2Hz, 1H); 6.81 (s, 1H); 7.13 (d, J=7.5Hz, 2H); 7.95 (d, J=7.5Hz, 2H);
(3) taking by weighing 3.1g compound III, is that 1 ︰, 20 adding 62mL mass concentrations are 48% hydrobromic acid solution by solid-to-liquid ratio, after mixing; Under nitrogen protection, be heated to 180 ℃, reacted 8 hours, be cooled to room temperature then; Volume ratio by reaction solution and water is that 1 ︰ 20 adds in the 1240mL water hold over night, suction filtration after drying; Obtain apigenin, i.e. chemical compounds I; Yield 75%; M.p.:345 ~ 350;
1HNMR (DMSO) characterizes: δ 6.18 (s, 1H), 6.47 (s, 1H), 6.78 (s, 1H), 6.91 (d, J=7.8Hz, 2H), 7.91 (d, J=7.8Hz, 2H), 10.36 (s, 1H), 10.85 (s, 1H), 12.95 (s, 1H).
The above only is a preferred implementation of the present invention; Should be pointed out that for those skilled in the art, under the prerequisite that does not break away from the principle of the invention; Can also make some improvement and retouching, these improvement and retouching also should be regarded as protection scope of the present invention.
Claims (1)
1. method for preparing apigenin is characterized in that through following each step:
(1) take by weighing 2,4-dimethoxy-6-hydroxy acetophenone is by 2; The mol ratio of 4-dimethoxy-6-hydroxy acetophenone and aubepine is that 1 ︰ 1 ~ 1.2 takes by weighing aubepine, is that 1 ︰ 10 adds methyl alcohol or ethanol dissolves by solid-to-liquid ratio again, adds Pottasium Hydroxide or sodium hydroxide again; Making the concentration of alkali in solution is 10 ~ 25%; Stir 20 ~ 40h down at 20 ~ 40 ℃, process is with concentrated hydrochloric acid solution conditioned reaction liquid pH=3 ~ 4, with the reaction solution suction filtration then; Filter cake is used distilled water wash, and filter cake carries out getting compound ii after the drying again;
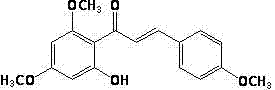
Ⅱ
(2) getting compound ii, is that 1 ︰ 1 ~ 10 adds DMSO 99.8MIN. by solid-to-liquid ratio, adds the catalyzer iodine or the iodized salt of raw materials quality 0.5 ~ 3%; Be heated to 80 ~ 150 ℃, stir 3 ~ 30h, after the cooling again through taking by weighing the sodium sulfite anhy 96 with quality such as catalyzer; Add 5 ~ 10 times water of DMSO 99.8MIN. volume, be made into sodium sulfite solution, cooling back solution in the step (2) is added in the sodium sulfite solution; Stir 0.5 ~ 1h, hold over night obtains the compound III with filtration cakes torrefaction behind the suction filtration;
Ⅲ
(3) take by weighing the compound III, by solid solid than or solid-to-liquid ratio be that 1 ︰, 5 ~ 20 adding solid pyridine hydrochlorides, mass concentration are that 48% hydrobromic acid solution or mass concentration are 56% hydroiodic acid HI acetum, after mixing; Under nitrogen protection, be heated to 100 ~ 180 ℃, reacted 6 ~ 10 hours, be cooled to room temperature then; Be that 1 ︰ 10 ~ 20 adds in the entry through volume ratio again by reactant and water; Hold over night obtains apigenin behind the suction filtration after drying, i.e. chemical compounds I.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010616467A CN102079737B (en) | 2010-12-31 | 2010-12-31 | Method for preparing apigenin |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010616467A CN102079737B (en) | 2010-12-31 | 2010-12-31 | Method for preparing apigenin |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102079737A CN102079737A (en) | 2011-06-01 |
| CN102079737B true CN102079737B (en) | 2012-09-05 |
Family
ID=44085958
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201010616467A Expired - Fee Related CN102079737B (en) | 2010-12-31 | 2010-12-31 | Method for preparing apigenin |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102079737B (en) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104341380A (en) * | 2013-08-02 | 2015-02-11 | 江苏柯菲平医药股份有限公司 | Preparation method of novel flavonoid compounds |
| CN104031016A (en) * | 2014-06-24 | 2014-09-10 | 陕西嘉禾植物化工有限责任公司 | Synthetic method of apigenin |
| CN105348245A (en) * | 2015-12-01 | 2016-02-24 | 陕西嘉禾生物科技股份有限公司 | Eriodictyol synthesis method |
| CN111303106B (en) * | 2020-03-23 | 2023-06-02 | 武汉轻工大学 | Preparation method of 5, 6-diacetoxy-7-hydroxy flavone |
| CN113557234B (en) * | 2020-06-19 | 2023-12-01 | 邦泰生物工程(深圳)有限公司 | Semi-synthesis method of apigenin |
| CN113956226A (en) * | 2021-11-19 | 2022-01-21 | 南京科技职业学院 | Method for synthesizing carvachin |
| CN116041302A (en) * | 2022-12-02 | 2023-05-02 | 泉州海创医药科技有限公司 | Method for preparing flavonoid compound containing hydroxyl substituent |
-
2010
- 2010-12-31 CN CN201010616467A patent/CN102079737B/en not_active Expired - Fee Related
Non-Patent Citations (1)
| Title |
|---|
| Farooq, M. O.等.Die Anthoxanthinglykoside von Apium petroselinum und eine neue Synthses des Apigenins.《Archiv der Pharmazie》.1959,第292卷(第12期),792-796. * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102079737A (en) | 2011-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102079737B (en) | Method for preparing apigenin | |
| CN104086379B (en) | The synthetic method of the clean intermediate of Da Gelie | |
| CN111675710B (en) | Preparation method of duloxetine | |
| CN104045669A (en) | Separation method suitable for chemical synthesis of salidroside for industrial production | |
| CN102532130A (en) | Method for full chemical synthesis of fibrauretin anti-bacterial anti-inflammatory medicine | |
| CN114044777B (en) | Preparation method of tricitabinib phosphate | |
| CN107417603B (en) | Preparation method of crizotinib intermediate | |
| CN103880623A (en) | 4,2',4'-triethoxy-5' substituted chalcone derivatives and preparation method and application thereof | |
| CN103012268B (en) | Novel preparation method for ivabradine | |
| CN102180914A (en) | Preparation method of 2-deoxidizing-D-glucose | |
| CN100391943C (en) | Prepn. of 3-position substituted indole derivative | |
| CN100376555C (en) | Process for preparing diindolylmethane derivatives | |
| CN106238098B (en) | A kind of preparation method and its catalyst for preparing of 1,2,4,5- tetra- substituted ramification of imidazole | |
| CN103351291B (en) | It is a kind of that natural phlorizin is semi-synthetic prepares Phloretin technique | |
| CN104961787B (en) | Synthetic method of cordycepin | |
| CN102040529A (en) | Method for synthesizing synephrine hydrochloride | |
| CN116986986A (en) | Synthesis method of 3-oxo-1-cyclobutanecarboxylic acid intermediate | |
| CN110256325B (en) | Process method for synthesizing 3,3' -diindolylmethane | |
| CN102766108A (en) | Method for preparing benzoxazole C2 position ammoniated derivatives | |
| CN103554005A (en) | Novel simple synthesis method of L-5-hydroxytryptophan | |
| CN104109182A (en) | Preparation method of gemcitabine hydrochloride | |
| CN113004248A (en) | Method for synthesizing carbazole compound by catalyzing hydrocarbon amination reaction with cobalt | |
| CN102070510A (en) | 3-hydroxyindole derivatives and synthesis method and use thereof | |
| CN101792451A (en) | Full synthesis method of 4'',5''-dihydroxyl-5-methoxyl-[6'',6''-dimethyl pyran (2'',3'':7,8)] Hirtellanine A | |
| CN112830876A (en) | Synthesis method of octadecyl gallate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20120905 Termination date: 20141231 |
|
| EXPY | Termination of patent right or utility model |