CN101735238A - 抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用 - Google Patents
抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用 Download PDFInfo
- Publication number
- CN101735238A CN101735238A CN201010010170A CN201010010170A CN101735238A CN 101735238 A CN101735238 A CN 101735238A CN 201010010170 A CN201010010170 A CN 201010010170A CN 201010010170 A CN201010010170 A CN 201010010170A CN 101735238 A CN101735238 A CN 101735238A
- Authority
- CN
- China
- Prior art keywords
- hydroxyl
- compound
- expression
- derivative
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images




Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及一种抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用。抗肿瘤药物羟地吗啉和其衍生物,具有通式[I]所示的结构。式中,R1表示羟基、羰基、酰胺、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基;R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基;R3表示羟基、羰基或酰胺。本发明得到的抗肿瘤药物羟地吗啉和其衍生物,具有更优良的抗肿瘤活性和安全性,可在治疗肝癌、肺癌、乳腺癌、白血病、结肠癌、卵巢癌胃癌及神经癌等肿瘤中的应用,因而治疗窗宽,所以在医药领域中作为抗肿瘤剂是非常有应用价值的。
Description
技术领域
本发明属于医药领域,尤其涉及抑制肿瘤细胞生长、发挥抗肿瘤效果的羟地吗啉和其衍生物及制备方法及用途。
背景技术
在冯威健等进行的中药抗癌筛选中,瑞香狼毒(Stellera chamaejasme L.)的甲醇提取物显示了较强的抗癌活性(中华肿瘤杂志1995年1月第17卷第1期)。瑞香狼毒可用于治疗肿瘤性疾病,临床报道其粗提物对治疗肺癌和肝癌等有效。冯威健等以小鼠白血病为活性指标,对瑞香狼毒进行了分离提取,得到了一种二萜活性成份,其中尼地吗琳具有较强的抗癌作用,其结构为:

尼地吗琳对常规使用的L-1210腹水型及实体瘤Lewis肺癌、B-16和结肠癌26显示较强的抗癌活性,尼地吗琳对体外培养的人白血病和胃癌具有明显的细胞生长抑制作用和克隆形成抑制作用,对小鼠体内体外抗癌活性均较强,表明其抗癌机制是通过对癌细胞的直接作用而发挥的。
但尼地吗琳的腹腔给药剂量超过0.05mg/kg时即出现毒性反应,说明其活性剂量与毒性剂量比较接近,治疗窗非常窄,临床应用安全性较差,对其它肿瘤细胞的抑制作用不明显。
发明概述
为解决以上问题,本发明的目的是提供比尼地吗啉具有更优良的抗肿瘤活性和安全性,及治疗窗宽的抗肿瘤药物羟地吗啉和其衍生物。
本发明的另一目的是提供抗肿瘤药物羟地吗啉和其衍生物的制备方法及其应用。
本发明中,为改变尼地吗啉的上述弱点,我们对其进行结构修饰,合成多种羟地吗啉等衍生物,对其抗肿瘤活性进行研究,结果发现用下述通式[I]和[II]表示的化合物是具有极其优良的抗肿瘤活性、稳定性和安全性的新化合物。
本发明涉及用通式[I]表示的化合物及其药学上可允许的盐。

式中,
R1表示羟基、羰基、酰胺、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基。
R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基。
R3表示羟基、羰基或酰胺。
所述的低级烷基是指C1~C6的烷基。
优选的:R1为羟基,R2为C2的不饱和烃基,R3为羟基的化合物1及其药学上可允许的盐。其结构式如(1)所示:

优选的:R1为羟基,R2为四氮唑,R3为羟基的化合物6(即,羟地吗啉)及其药学上可允许的盐。其结构式如(6)所示:

本发明涉及用通式[II]表示的化合物及其药学上可允许的盐。

式中,
R1表示羟基、羰基、酰胺、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基。
R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基。
R3表示羟基、羰基或酰胺。
所述的低级烷基是指C1~C6的烷基。
优选的:R1为羟基,R2为四氮唑,R3为羟基的化合物7及其药学上可允许的盐。其结构式如(7)所示:

对于本发明通式[I]和[II]表示的化合物及中间体,制备方法如下:
图1和图2中,
R1表示羟基、羰基、酰胺、低级烷基、苄氧甲基、芳烷基和C2-5的不饱和烃基。
R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基和C2-5的不饱和烃基。
R3表示羟基、羰基或酰胺。
(一)通式[I]表示的化合物的制备方法如图1所示,
尼地吗啉在甲醇和三乙胺的作用下,直接水解获得多羟基化合物;乙酸酐对羟基进行保护,15位的双键经加成,获得16位为卤代烃的卤代化合物;经取代可获得16位为低级烷基、苄氧甲基、芳烷基和C2-5的不饱和烃基,或16位卤代化合物经与氰化钠反应获得氰甲基,再与叠氮化钠反应获得16位为四氮唑基团;得到具有通式[III]表示的中间体。

通式[III]表示的中间体,经脱保护获得R1表示羟基,R3表示羟基的本发明通式[I]表示的化合物。其脱保护以二丁基氧化锡为催化剂,因而保持6、7位环氧结构不该变。
通式[III]表示的中间体,优选的,
R2为C2的不饱和烃基,其结构式如(2)所示;
或R2为卤代烃,优选为溴甲基,其结构式如(3)所示;
或R2为氰甲基,其结构式如(4)所示;
或R2为四氮唑,其结构式如(5)所示。
制备方法还可以是:
尼地吗啉通过乙酸酐对4、5、20和28位的羟基进行保护后,再进行水解,获得3、18位为羟基,对3、18位的羟基再进行修饰,3位可获得羰基、酰胺、低级烷基、苄氧甲基、芳烷基和C2-5的不饱和烃基,18位可获得羰基或酰胺,得通式[IV]表示的化合物。
通式[IV]表示的化合物,15位的双键经加成,获得16位为卤代烃的卤代化合物,经取代可获得16位为低级烷基、苄氧甲基、芳烷基和C2-5的不饱和烃基;或16位卤代化合物经与氰化钠反应获得氰甲基,再与叠氮化钠反应获得16位为四氮唑基团,得通式[V]表示的化合物。
通式[V]表示的化合物经脱保护获得本发明通式[I]表示的化合物。其脱保护以二丁基氧化锡为催化剂,因而保持6、7位环氧结构不该变。
(二)通式[II]表示的化合物的制备方法如图2所示
通式[V]表示的化合物经脱保护获得本发明通式[II]表示的化合物。其脱保护以醋酸锌为催化剂,因而获得6、7位双键。
本发明的有益效果是:本发明得到的抗肿瘤药物羟地吗啉和其衍生物,具有更优良的抗肿瘤活性和安全性,可在治疗肝癌、肺癌、乳腺癌、白血病、结肠癌、卵巢癌胃癌及神经癌等肿瘤中的应用,抗癌谱广,治疗窗宽,所以在医药领域中作为抗肿瘤剂是非常有应用价值的。
附图说明
图1是本发明通式[I]表示的化合物及其药学上可允许的盐的合成机理图;
图2是本发明通式[II]表示的化合物及其药学上可允许的盐的合成机理图;
图3是本发明化合物1和化合物6的合成工艺路线图;
图4是本发明化合物7的合成工艺路线图;
具体实施方式
以下举出实施例,进一步具体地说明本发明,但本发明不受这些实施例的限制。
用图3表示化合物1和化合物6的制备工艺。
实施例1 化合物1及其制备方法,化合物1的结构式如(1)所示
取一个干燥的50ml的茄型瓶,加入磁力搅拌棒、尼地吗啉1.548g(2mmol)、加甲醇∶三乙胺∶水(v∶v∶v)=5∶1∶1共21ml溶液溶解,将反应体系置于集热式恒温加热磁力搅拌器上剧烈搅拌,密闭反应体系,在干燥的氮气保护下,70℃下回流20小时后停止反应,将反应体系于50℃下减压浓缩蒸干得到棕黄色初产物,采用硅胶柱层析分离得到化合物1,共0.46g,收率40.6%,mp 150~152℃。
元素分析实测值:C,63.54;H,8.19。
分子式(C30H46O10)计算值:C,63.58;H:8.18。
质谱MS(EI,70ev)m/z(%):567(M+1)。
红外IR(KBr)cm-1:3432,3298,2986,1652,1383,739。
核磁共振氢谱1H-NMR(CDCl3)δ:5.23(s,1H),4.88(s,1H),4.44(d,J=3.0Hz,1H),3.99-3.80(m,1H),3.92(d,J=10.0Hz,1H),3.87-3.68(d,J=7.4Hz,1H),3.85(t,J=10.0Hz,1H),3.78(d,1H),3.72(d,J=10.0Hz,1H),3.67(t,J=10.0Hz,1H),3.40(s,1H),3.12(d,J=2.9Hz,1H),2.99(d,J=12.5Hz,1H),2.74(m,1H),2.56(dd,J=12.5Hz,1H),2.32(d,J=14.7Hz,1H),2.26(m,1H),2.11-1.65(m,18H),2.00(d,J=7.5Hz,1H),1.89(s,3H),1.74(m,1H),1.24(d,J=7.6Hz,3H),1.06(d,J=7.6Hz,3H).
核磁共振氢谱检测氢数为46,核磁共振碳谱检测碳数为30,结构分析与目标产物一致。
实施例2 化合物6(即,羟地吗啉)及其制备方法
(一)中间体1的制备,中间体1的结构式如下:

取一个干燥的50ml的茄型瓶,加入磁力搅拌棒、0.452g(0.8mmo1)化合物1后,加入四氢呋喃溶剂10ml,乙酸酐1ml,吡啶1ml,将反应体系置于集热式恒温加热磁力搅拌器上室温下密闭剧烈搅拌8小时后停止反应。45℃下减压旋出反应瓶中四氢呋喃,加入无水乙醚20ml,室温下剧烈搅拌有大量白色固体析出,过滤干燥后得到中间体1,共0.62g,收率95.1%,mp 156~158℃。
元素分析实测值:C,61.56;H,7.16。
分子式(C42H58O16)计算值:C,61.60;H,7.14。
质谱MS(EI,70ev)m/z(%):819(M+1)。
红外IR(KBr)cm-1:3308,3029,1742,1648,1350,649。
核磁共振氢谱检测氢数为58,核磁共振碳谱检测碳数为42,结构分析与目标产物一致。
(二)中间体2及其制备方法,中间体2的结构式如下:

取一个干燥的50ml的茄型瓶,加入磁力搅拌棒、0.41克(0.5mmol)中间体1,加30%溴化氢乙酸溶液15ml,30%双氧水溶液2ml,将反应体系密闭,置于集热式恒温加热磁力搅拌器上50℃剧烈搅拌2小时后停止反应,反应液在50℃下减压浓缩至油状棕黄色初产物,加入乙酸乙酯30ml溶解,并用饱和碳酸氢钠水溶液50毫升*3洗涤三次,分出有机相用无水硫酸镁干燥后浓缩,柱层析硅胶分离得到中间体2,共0.30克,收率67.3%。
元素分析实测值:C,56.11;H,6.64;Br,8.79。
分子式(C42H59BrO16)计算值:C,56.06;H,6.61;Br,8.88。
质谱MS(EI,70ev)m/z(%):899(M+1)。
红外IR(KBr)cm-1:3352,2988,1745,1432,832。
核磁共振氢谱检测氢数为59,核磁共振碳谱检测碳数为42,结构分析与目标产物一致。
(三)中间体3及其制备方法,中间体3的结构式如下:

取一个干燥的50ml的茄型瓶,加入磁力搅拌棒、0.27g(0.3mmol)中间体2,加入氰化钠0.18g(0.36mmol),加DMSO溶剂5ml溶液溶解,密闭反应体系,在干燥的氮气保护下,将反应体系置于集热式恒温加热磁力搅拌器上剧烈搅拌,25℃下反应12小时后停止反应,将反应液加入20ml乙酸乙酯,25ml水,充分震荡分层后用分液漏斗分出有机相,并分别用25ml、25ml、10ml去离子水洗有机相,分出有机相并用无水硫酸镁干燥后,滤液在50℃下减压浓缩,经柱层析硅胶分离得到中间体3,共0.19g,收率72.8%,mp 162~166℃。
元素分析实测值:C,61.24;H,7.11;N,1.52。
分子式(C43H59NO16)计算值:C,61.05;H,7.03;N,1.66。
质谱MS(EI,70ev)m/z(%):846(M+1)。
红外IR(KBr)cm-1:3024,2928,2231,1754,1324,1250,895。
核磁共振氢谱检测氢数为59,核磁共振碳谱检测碳数为43,结构分析与目标产物一致。
(四)中间体4及其制备方法,中间体4的结构式如下:

取一个干燥的50ml的茄型瓶,加入磁力搅拌棒、0.17克(0.2mmol)中间体3、叠氮化钠0.14g(0.21mmol)、氯化锌0.27g(0.2mmol),加入甲苯溶剂20ml后,密闭反应体系,在干燥的氮气保护下,滴加30%盐酸水溶液1毫升,将反应体系置于集热式恒温加热磁力搅拌器上剧烈搅拌,110℃下反应8小时后停止反应,将反应液在50℃下减压浓缩,经柱层析硅胶分离得到中间体4,共0.16g,收率87.6%,mp 192~194℃。
元素分析实测值:C,58.11;H,6.88;N,6.23。
分子式(C43H60N4O16)计算值:C,58.10;H,6.80;N,6.30。
质谱MS(EI,70ev)m/z(%):889(M+1)。
红外IR(KBr):cm-13411,3024,2984,1752,1668,1338,1223,766。
核磁共振氢谱检测氢数为60,核磁共振碳谱检测碳数为43,结构分析与目标产物一致。
(五)化合物6及其制备方法,化合物6的结构式如下:
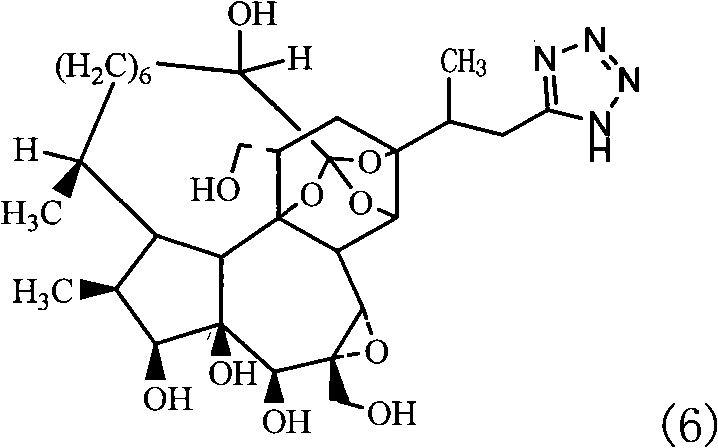
取一个干燥的25ml的茄型瓶,加入磁力搅拌棒、0.14g(1.6mmol)中间体4、二丁基氧化锡0.08g,加入无水甲醇10ml,密闭反应体系,在干燥的氮气保护下,将反应体系置于集热式恒温加热磁力搅拌器上剧烈搅拌,70℃下回流8小时后停止反应,将反应液在50℃下减压浓缩,经柱层析硅胶分离得到化合物6,共0.04g,收率36.5%,mp 189~191℃。
元素分析实测值:C,58.52;H,7.66;N,8.66。
分子式(C31H48N4O10)计算值:C,58.48;H,7.60;N,8.80。
质谱MS(EI,70ev)m/z(%):637(M+1)。
红外IR(KBr)cm-1:3432,3335,3266,2998,1659,1374,1148,856。
核磁共振氢谱1H-NMR(CDCl3)δ:15.1(s,1H),4.59(d,J=3.0Hz,1H),4.02-3.86(m,1H),3.90(d,J=11Hz,1H),3.85(d,J=7.4Hz,1H),3.81(t,J=11Hz,1H),3.76(d,1H),3.70(d,J=10.0Hz,1H),3.65(t,J=10.0Hz,1H),3.35(s,1H),3.22(d,J=3.0Hz,1H),3.03(d,J=12.6Hz,1H),2.74-2.36(m,4H),2.66(dd,J=12.6Hz,1H),2.42(d,J=15Hz,1H),2.33(m,1H),2.05-1.56(m,18H),2.01(d,J=7.4Hz,1H),1.68(s,3H),1.64(m,1H),1.35(d,J=7.5Hz,3H),1.16(d,J=7.5Hz,3H)。
核磁共振氢谱检测氢数为48,核磁共振碳谱检测碳数为31,结构分析与目标产物一致。
当然,以上(一)至(四)反应得到的各中间体,即中间体1、中间体2和中间体3都可以按第(五)步揭示的中间体4的脱保护方法,以二丁基氧化锡为催化剂,进行脱保护,得到具有相应结构的通式[I]表示的化合物:由中间体1得到,R1为羟基,R2为C2的不饱和烃基,R3为羟基的化合物;由中间体2得到,R1为羟基,R2为溴甲基,R3为羟基的化合物;由中间体3得到,R1为羟基,R2为氰甲基,R3为羟基的化合物。这对于本领域的技术人员来说是显而易见。
实施例3 化合物7及其制备方法
用图4表示化合物7的制备工艺,化合物7的结构式如(7)所示,
取一个干燥的25ml的茄型瓶,加入磁力搅拌棒、0.14g(0.16mmol)中间体4、醋酸锌0.12g,加入无水甲醇15ml,密闭反应体系,在干燥的氮气保护下,将反应体系置于集热式恒温加热磁力搅拌器上剧烈搅拌,70℃下回流8小时后停止反应,将反应液在50℃下减压浓缩,经柱层析硅胶分离得到化合物7共0.03g,收率25.8%,,mp 176~179℃。
元素分析实测值:C,60.06;H,7.81;N,8.95。
分子式(C31H48N4O9)计算值:C,59.98;H,7.79;N,9.03。
质谱MS(EI,70ev)m/z(%):621(M+1)。
红外IR(KBr)cm-1:3458,3297,3011,1668,1642,1274,956。
核磁共振氢谱1H-NMR(CDCl3)δ:14.9(s,1H),5.76(m,1H),4.49(d,J=3.0Hz,1H),3.98-3.77(m,1H),3.89(d,J=10.2Hz,1H),3.84(d,J=7.5Hz,1H),3.80(t,J=10.0Hz,1H),3.71(d,J=10.0Hz,1H),3.64(t,J=10.0Hz,1H),3.50(d,1H),3.28(d,J=3.0Hz,1H),3.08(d,J=12.6Hz,1H),2.74-2.36(m,4H),2.66(d,J=15.0Hz,1H),2.60(dd,J=12.5Hz,1H),2.30-1.66(m,19H),2.11(d,J=7.5Hz,1H),1.76(m,1H),1.60(s,3H),1.33(d,J=7.6Hz,3H),1.06(d,J=7.6Hz,3H)。
核磁共振氢谱检测氢数为48,核磁共振碳谱检测碳数为31,结构分析与目标产物一致。
实施例4 化合物6和化合物7的药效学试验
以实施例2和实施例3提供的化合物6和化合物7为受试样品,表示了如以下药效学试验所示的优良抗肿瘤作用。
(1)对于各种癌细胞的抑制生长活性(GI50)测定方法:
肿瘤细胞经胰蛋白酶消化后,分散成单个细胞,并使其悬浮在含青霉素(25U/ml)和链霉素(25μg/ml)的RPMI1640培养基中。将细胞接种于96孔培养板(CorningIncorporated),在37℃,含5%CO2的空气,相对湿度100%条件下培养24小时后,弃去培养液,加入含一系列浓度受试样品的培养液,每一浓度设平行孔,培养24小时后,弃去含受试样品的培养液,加入常规培养液培养48小时后,弃去培养液,再代之以含噻唑蓝(MTT,美国Sigma公司产品)培养液,MTT终浓度为0.5g/L,继续温育4小时后加二甲基亚砜溶解液,1小时后紫色结晶完全溶解,在SK601型酶标仪(日本国Seikagaku公司产品)检测570nm/630nm的光密度(OD)。按下式计算受试样品对肿瘤细的半数生长抑制率:
(T-T0)/(C-T0)×100%
注:C表示对照组细胞的OD值
T表示加受试样品组细胞的OD值
T0表示加受试样品时对照平板细胞的OD值
受试样品对于各种癌细胞的抑制作用,结果见表1。
表1 化合物6和化合物7对肿瘤细胞的抑制作用

(2)对于肝癌SMMC-7721和肺癌A549的抑制作用
取生长旺盛期的瘤组织剪切成1.5mm3左右,在无菌条件下,接种于裸小鼠右侧腋窝皮下。裸小鼠移植瘤用游标卡尺测量移植瘤直径,待肿瘤生长至100~300mm3后将动物随机分组。使用测量瘤径的方法,动态观察被试物抗肿瘤的效应。肿瘤直径的测量次数为每周2次,每次测量时同时称鼠重。实验组每周静脉给药3次,阳性对照组每周静脉给药3次,阴性对照组同时给等量生理盐水。肿瘤体积(tumor volume,TV)的计算公式为:
TV=1/2×a×b2
其中a、b分别表示长宽。根据测量的结果计算出相对肿瘤体积(relative tumorvolume,RTV),计算公式为:RTV=Vt/V0。其中V0为分笼给药时(即d0)测量所得肿瘤体积,Vt为每一次测量时的肿瘤体积。抗肿瘤活性的评价指标为相对肿瘤增殖率T/C(%),计算公式如下:
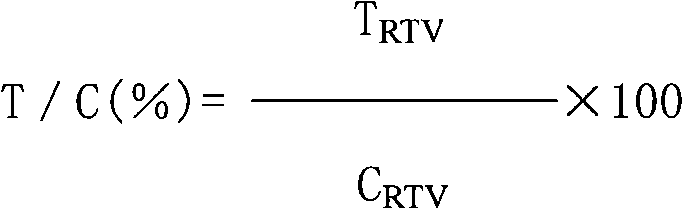
TRTV:实验组RTV;CRTV:阴性对照组RTV。
受试样品对人肝癌SMMC-7721和人肺癌A549的抑制作用,结果见表2、3。
表2 化合物6和化合物7对异种移植于裸鼠人肝癌SMMC-7721的抑制作用

本发明提供的化合物6和化合物7,与对照化合物环磷酰胺相比较,显示了如上述药理试验结果所示的更优良对肿瘤的抑制作用。
实施例5 化合物6和化合物7的安全性试验(急性毒性)
化合物6和化合物7通过小鼠尾静脉给药结果显示对呼吸系统有一定的毒性,大剂量动物在给药后1~2小时出现呼吸困难、俯卧、活动减少等临床症状,死亡发生在给药后5~10小时内,对死亡小鼠进行解剖,肉眼观察小鼠胸腔内存有一定量的积液,肺脏有大面积出血点,其余脏器未见异常,肺脏病理组织学检查显示肺脏有出血,大量炎性细胞浸润,肺泡周围有实变。对存活小鼠连续观察14天,摄食、饮水、一般状态及活动情况均未见异常,观察期结束后处死小鼠,肉眼观察肺脏及主要器官均未见异常。
化合物6对呼吸系统有一定的毒性,给药后造成小鼠呼吸衰竭死亡,其LD50为20.66mg/kg,95%的可信限为16.32mg/kg~24.51mg/kg,毒性靶器官主要为肺脏。
化合物7对呼吸系统有一定的毒性,给药后造成小鼠呼吸衰竭死亡,其LD50为22.58mg/kg,95%的可信限为17.19mg/kg~25.32mg/kg,毒性靶器官主要为肺脏。
如上述药理试验结果表明,本发明化合物显示了优良的抗肿瘤作用,作为抗肿瘤剂,对于预防、治疗疾病,特别是处置癌是有用的。将本发明的化合物用于这样的用途时,可制成含有本发明化合物的有效量和药学容许的载体或赋形剂的制剂。
作为抗肿瘤剂使用本发明化合物的给药形态,可选择各种形态,例如可举出片剂、胶囊剂、粉剂、颗粒剂或液剂等的经口制剂、或例如溶液或悬浮液等的杀菌了的液状非经口制剂、注射剂、栓剂、软膏剂等。
固体的制剂可直接以片剂、胶囊剂、颗粒剂或粉末的形态进行制造,但也可使用适当的添加剂制造。作为这样的添加剂,例如可举出乳糖或葡萄糖等的糖类,例如玉米、小麦或米等的淀粉类,例如硬脂酸等的脂肪酸、例如偏硅酸铝酸镁或无水磷酸钙等的无机盐、例如聚乙烯吡咯烷酮或聚亚烷基二醇等的合成高分子,例如硬酯酸钙或硬质酸镁等的脂肪酸盐、例如十八烷醇或苄醇等的醇类,例如甲基纤维素、羧甲基纤维素、乙基纤维素或羟丙基甲基纤维素等的合成纤维素衍生物,其他,明胶、滑石、植物油、阿拉伯树胶等通常可使用的添加物。
这些片剂、胶囊剂、颗粒剂和粉末等的固形制剂,通常可含有0.1~99%(w/w),优选的是0.1~50%(w/w)的有效成分。
液状制剂,可在水、醇类或例如大豆油、花生油、芝麻油等植物油的液状制剂中,使用通常所用的适当添加物、以悬浮液、糖浆剂、注射剂、点滴剂等形态制造。
特别是作为以非经口的肌肉注射、静脉注射或皮下注射的形式给药时的适当溶剂,例如可举出注射用蒸馏水、生理食盐水、葡萄糖水溶液、乙醇、聚乙二醇、静脉注射用液体(例如柠檬酸和柠檬酸钠等的水溶液)或电解质溶液(点滴静脉注射和静脉注射用)等,或这些的混合溶液。
除了这些预先溶解的注射剂之外,也可作成加有粉末或适当的添加剂的在使用时溶解的形态。这些注射液通常可含有0.1~20%(w/w)、优选的是0.5~5%(w/w)的有效成分。
另外,经口给药用的悬浮剂、糖浆剂等的剂型,通常可含0.5~10%(w/w)的有效成分。
本发明化合物优选的给药量,可根据使用的化合物的种类、配合的组合物种类、适用频度和应该治疗的特定部位、病情的轻重、患者的年龄、医生的诊断、肿瘤的种类等而变化,但作为大致目标,例如每天每1个成人的给药量,在经口给药时,可在0.01~200mg范围内,另外,在非经口给药时,在静脉注射时,优选的是每天在0.01~50mg范围内。另外,给药次数,根据给药方法和症状而不同,但1天是1~3次。另外,也可使用隔日给药、隔二日给药等间歇给药等给药方法。
Claims (10)
1.用通式[I]表示的化合物及其药学上可允许的盐:

式中,
R1表示羟基、羰基、酰胺、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基;
R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基;
R3表示羟基、羰基或酰胺。
2.用通式[II]表示的化合物及其药学上可允许的盐:

式中,
R1表示羟基、羰基、酰胺、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基;
R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基;
R3表示羟基、羰基或酰胺。
3.如权利要求1所述的化合物及其药学上可允许的盐,其特征在于:R1为羟基,R2为C2的不饱和烃基,R3为羟基,其结构式如(1)所示,

4.如权利要求1所述的化合物及其药学上可允许的盐,其特征在于:R1为羟基,R2为四氮唑,R3为羟基,其结构式如(6)所示,

5.一种合成权利要求1或2所述的化合物及其药学上可允许的盐的中间体,其特征在于:具有通式[III]的结构,

式中,
R2表示四氮唑、卤原子、卤代烃、氰基、氰甲基、低级烷基、苄氧甲基、芳烷基或C2-5的不饱和烃基。
6.如权利要求5所述的中间体,其特征在于:R2为C2的不饱和烃基、卤代烃、氰甲基或四氮唑。
7.如权利要求6所述的中间体,其特征在于所述的卤代烃为溴甲基。
8.如权利要求2所述的化合物及其药学上可允许的盐,其特征在于:R1为羟基,R2为四氮唑,R3为羟基,其结构式如(7)所示,
9.权利要求1、2、3、4或8所述的化合物及其药学上可允许的盐作为有效成分在抗肿瘤药物上的应用。
10.权利要求5、6或7所述的中间体在制备抗肿瘤药物羟地吗啉和其衍生物上的应用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010010170XA CN101735238B (zh) | 2010-01-21 | 2010-01-21 | 抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010010170XA CN101735238B (zh) | 2010-01-21 | 2010-01-21 | 抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101735238A true CN101735238A (zh) | 2010-06-16 |
| CN101735238B CN101735238B (zh) | 2011-11-09 |
Family
ID=42459315
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201010010170XA Expired - Fee Related CN101735238B (zh) | 2010-01-21 | 2010-01-21 | 抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101735238B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101766599B (zh) * | 2010-02-09 | 2012-07-25 | 辽宁大学 | 含有羟地吗啉或羟地吗啉药用复合物的药物组合物制剂 |
| CN115160337A (zh) * | 2022-08-19 | 2022-10-11 | 沈阳药科大学 | 1α-烷基瑞香烷型二萜类化合物及其制备方法和应用 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2409992A (en) * | 1991-08-08 | 1993-03-02 | Tsumura & Co. | Carcinostatic compound and production thereof |
-
2010
- 2010-01-21 CN CN201010010170XA patent/CN101735238B/zh not_active Expired - Fee Related
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101766599B (zh) * | 2010-02-09 | 2012-07-25 | 辽宁大学 | 含有羟地吗啉或羟地吗啉药用复合物的药物组合物制剂 |
| CN115160337A (zh) * | 2022-08-19 | 2022-10-11 | 沈阳药科大学 | 1α-烷基瑞香烷型二萜类化合物及其制备方法和应用 |
| CN115160337B (zh) * | 2022-08-19 | 2023-11-10 | 沈阳药科大学 | 1α-烷基瑞香烷型二萜类化合物及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101735238B (zh) | 2011-11-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102295635B (zh) | 抗肿瘤药物四氢化萘酰胺类化合物及其药学上可接受的盐及制备方法和应用 | |
| CN106905313B (zh) | 一氧化氮供体型原小檗碱类衍生物及其制备方法和用途 | |
| CN103044395B (zh) | 含有地氯雷他定结构的氨基酸类衍生物、其制备方法和用途 | |
| CN101402667B (zh) | 糖基化修饰的一氧化氮供体型齐墩果酸类化合物、其制备方法及用途 | |
| CN110343033A (zh) | 厚朴酚系列衍生物及其制备方法和用途 | |
| CN101735238B (zh) | 抗肿瘤药物羟地吗啉和其衍生物及制备方法和应用 | |
| CN101519423B (zh) | 白桦酸类似物、其制备方法和用途 | |
| CN108484632A (zh) | 青蒿素-苯胺基喹唑啉类衍生物及其制备方法和应用 | |
| CN107141284B (zh) | 黄连碱类衍生物、其制备方法、药物组合物及抗肿瘤用途 | |
| CN108752404B (zh) | 一种三氮唑糖修饰的小檗碱盐衍生物及其制备方法和用途 | |
| CN101974016A (zh) | 酰胺类化合物及其制备方法和用途 | |
| CN102786458B (zh) | 吡咯甲酰胺衍生物、其制备方法和用途 | |
| CN109232703A (zh) | 含16-(1′-芳香基-1′,2′,3′-三氮唑)亚甲基-雄甾-17-酮衍生物 | |
| CN104804047A (zh) | 新型含氮氧自由基的二茂铁衍生物的制备方法及其用途 | |
| CN104292211A (zh) | 地氯雷他定类一氧化氮供体及其制备方法和用途 | |
| CN110078770B (zh) | 一种具有喹啉酮四价铂结构的化合物、制备方法及其在制备抗肿瘤药物中的应用 | |
| CN109369634B (zh) | 具有抗肿瘤活性的2-甲氧基烟酰胺衍生物制备方法及用途 | |
| CN104672191B (zh) | 胡枝子酚e1类化合物及制备方法和应用 | |
| CN101891795A (zh) | 具有抗肿瘤活性的熊果酸二乙醇胺类衍生物及其制备方法 | |
| CN102633855B (zh) | 齐墩果酸-尿嘧啶核苷缀合物及其制备方法和应用 | |
| CN102702116B (zh) | 4-(3-氯-4-甲氧基苯胺基)-6-(3-胺基苯基)喹唑啉类化合物或其药学上可接受的盐和制备方法与应用 | |
| CN104961794B (zh) | 一种丹参酮iia衍生物及其制备和应用 | |
| CN101967163A (zh) | 对癌细胞有选择性的铂(ⅱ)抗癌配合物 | |
| CN102417514B (zh) | 吡啶衍生物、其制备方法和用途 | |
| CN102924387B (zh) | 4-(3-氯-4-甲氧基苯胺基)-6-(3,4-取代苯基)喹唑啉及盐和制法与应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20111109 Termination date: 20150121 |
|
| EXPY | Termination of patent right or utility model |