CN101426764A - 具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性的二烷基苯基化合物 - Google Patents
具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性的二烷基苯基化合物 Download PDFInfo
- Publication number
- CN101426764A CN101426764A CNA2007800140984A CN200780014098A CN101426764A CN 101426764 A CN101426764 A CN 101426764A CN A2007800140984 A CNA2007800140984 A CN A2007800140984A CN 200780014098 A CN200780014098 A CN 200780014098A CN 101426764 A CN101426764 A CN 101426764A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- methyl
- ethyl
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
本发明涉及式(I)化合物,其中R1和R2均如说明书中所定义,或其医药上可接受的盐或溶剂合物或立体异构体。本发明也涉及包含这些化合物的医药组合物和组合,制备这些化合物的方法和中间体,和使用这些化合物(例如)治疗诸如慢性阻塞性肺病和哮喘等肺病的方法。
Description
技术领域
本发明涉及新颖二烷基苯基化合物,其具有β2肾上腺素能激动剂和毒蕈碱受体拮抗剂活性。本发明也涉及包含这些化合物的医药组合物,制备这些化合物的方法和中间体,和使用这些化合物治疗(例如)肺病的方法。
背景技术
肺病,例如哮喘和慢性阻塞性肺病(COPD),一般是用支气管扩张药来治疗。用于治疗肺病的一类支气管扩张药是由β2肾上腺素能受体(肾上腺素受体)激动剂组成,例如沙丁胺醇(albuterol)、福莫特罗(formoterol)和沙美特罗(salmeterol)。这些化合物一般是通过吸入来投与。另一类支气管扩张药是由毒蕈碱受体拮抗剂(抗胆碱能化合物)组成,例如异丙托铵(ipratropium)和噻托铵(tiotropium)。这些化合物通常也是通过吸入来投与。
业内已知含有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂的组合的医药组合物可用于治疗肺病。例如,2002年8月13日颁布的美国专利第6,433,027号揭示药剂组合物,其含有诸如噻托溴铵(tiotropiumbromide)等毒蕈碱受体拮抗剂和诸如福莫特罗反丁烯二酸盐等β2肾上腺素能受体激动剂。
此外,具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性两者的化合物是业内已知的。例如,2006年11月28日颁布的美国专利第7,141,671号揭示具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性两者的联苯基化合物。人们高度期望同时具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性的化合物,因为这些化合物可通过两种独立作用模式提供支气管扩张同时具有单一分子药物代谢动力学。
在治疗肺病时,特别有效的是提供在通过吸入投与时具有长作用持续时间(即至少约24小时的持续时间)的治疗药剂,以使患者只需每天一次或以更低频率投与所述治疗药剂。并非所有先前文献中所揭示的双作用化合物都具有这种期望性质。
因此,业内仍需要具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性两者的新颖化合物,并且在通过吸入投与患者时,其具有长作用持续时间。
发明内容
本发明提供具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性两者的新颖二烷基苯基化合物。已发现本发明化合物其它在通过吸入投与哺乳动物时尤其具有长作用持续时间,即至少约24小时的持续时间。因此,预期本发明化合物可有利地用作治疗肺病的治疗药剂。
因此,在本发明组合物的一方面中,本发明涉及式I化合物:

其中
R1为甲基或乙基;R2为甲基或乙基;或其医药上可接受的盐或溶剂合物或立体异构体。
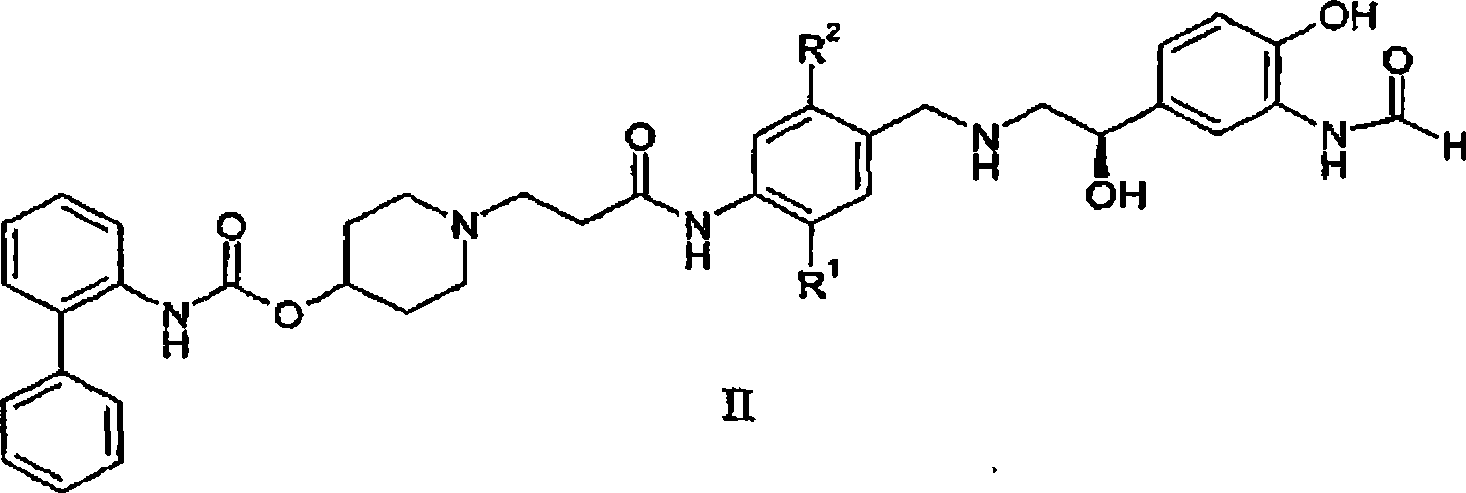
在本发明的特定方面中,式I化合物是具有式II的化合物:

其中R1和R2如本文中所定义(包括任何特定或优选实施例);或其医药上可接受的盐或溶剂合物。
在本发明组合物的另一方面中,本发明涉及医药组合物,其包含医药上可接受的载剂和式I化合物。
如果需要,本发明化合物可与其它治疗药剂组合投与,例如类固醇消炎药。因此,在本发明组合物的另一方面中,本发明涉及医药组合物,其包含(a)式I化合物;和(b)第二治疗药剂。在本发明组合物的另一方面中,本发明涉及医药组合物,其包含(a)式I化合物;(b)第二治疗药剂;和(c)医药上可接受的载剂。
于本发明组合物的另一方面中,本发明涉及治疗药剂的组合,所述组合包含(a)式I化合物;和(b)第二治疗药剂。在本发明组合物的另一方面中,本发明涉及医药组合物的组合,所述组合包含(a)第一医药组合物,其包含式I化合物和医药上可接受的第一载剂;和(b)第二医药组合物,其包含第二治疗药剂和医药上可接受的第二载剂。本发明也涉及含有这些医药组合物的试剂盒。
本发明化合物同时具有β2肾上腺素能受体激动剂活性和毒蕈碱受体拮抗剂活性。因此,预期式I化合物可用作治疗肺病的治疗药剂,例如哮喘和慢性阻塞性肺病。
因此,在本发明方法的一方面中,本发明涉及治疗肺病的方法,所述方法包含对需要治疗的患者投与治疗有效量的式I化合物。本发明也涉及治疗慢性阻塞性肺病或哮喘的方法,所述方法包含对患者投与治疗有效量的式I化合物。此外,在本发明方法的另一方面中,本发明涉及在哺乳动物中产生支气管扩张的方法,所述方法包含对哺乳动物投与支气管扩张产生量的式I化合物。本发明也涉及在哺乳动物中拮抗毒蕈碱受体并激动β2肾上腺素能受体的方法,所述方法包含对所述哺乳动物投与式I化合物。
由于本发明化合物同时具有β2肾上腺素能受体激动剂活性和毒蕈碱受体拮抗剂活性,因此这些化合物也可用作研究工具。因此,在本发明方法的另一方面中,本发明涉及使用式I化合物作为研究工具的方法,所述方法包含使用式I化合物进行生物学分析。
本发明化合物也可用于评估新颖化学化合物。因此,在本发明方法的另一方面中,本发明涉及在生物学分析中评估测试化合物的方法,所述方法包含:(a)用测试化合物进行生物学分析以提供第一分析值;(b)用式I化合物进行生物学分析以提供第二分析值;其中步骤(a)是在步骤(b)之前或之后进行或同时进行两个步骤;以及(c)比较来自步骤(a)的第一分析值与来自步骤(b)的第二分析值。
本发明也涉及可用于制备式I化合物的方法和新颖中间体。因此,在本发明方法的另一方面中,本发明涉及制备式I化合物的方法,所述方法包含使式6化合物(如本文所定义)去保护以提供式I化合物。
在本发明方法的另一方面中,本发明涉及制备式I化合物的方法,所述方法包含:(a)在还原剂存在下使式4化合物与式5化合物反应以提供式6化合物;和(b)使式6化合物去保护以提供式I化合物;其中化合物4、5和6如本文所定义。
在本发明具体实施例中,式I化合物是通过使式6化合物去保护来制备,其中羟基保护基为甲硅烷基。因此,在本发明方法的另一方面中,本发明涉及制备式I化合物的方法,所述方法包含使式6a化合物去保护:

其中Ra、Rb及Rc独立选自C1-4烷基、苯基、-C1-4烷基-(苯基),或R1a、R1b和R1c之一为-O-(C1-4烷基);以提供式I化合物。
在其它实施例中,本文所述方法另外包含形成式I化合物医药上可接受的盐的步骤。在其它实施例中,本发明涉及本文所述其它方法;以及通过任何本文所述方法制成的产物。
在特定实施例中,本发明涉及式III化合物:

或其盐或立体异构体,其中Y1是选自-CHO、-CN、-CH2OH、-CH(OR3a)OR3b、-C(O)OH、-C(O)OR3c、溴和碘,其中R3a和R3b独立选自C1-6烷基,或R3a和R3b接合形成C2-6亚烷基,R3c是选自C1-6烷基;并且R1和R2如本文所定义(包括任何特定或优选实施例),在式I化合物制备中所述化合物可用作中间体。在式III的特定实施例中,R1和R2为甲基。在式III的另一特定实施例中,Y1为-CHO。在式III的另一特定实施例中,R1和R2为甲基并且Y1为-CHO。
本发明也涉及式I化合物的治疗用途。此外,本发明涉及式I化合物在制造治疗肺病的药剂中的用途;以及式I化合物作为研究工具的用途。本文中揭示本发明的其它方面和实施例。
附图说明
具体实施方式
在本发明组合物的一方面中,本发明涉及新颖式I化合物。式I化合物含有一或多个手性中心,并且除非另外说明,否则本发明因此涉及外消旋混合物;纯立体异构体(即对映异构体或非对映异构体);富含立体异构体的混合物等。当本文中显示或指称特定立体异构体时,熟习此项技术者应理解,除非另外说明,否则较少量其它立体异构体可存在于本发明组合物中,条件是组合物整体的利用性不会因为这些其它异构体的存在而被消除。
具体来说,在以下部分化学式中,式I化合物在由符号*所指示的碳原子处含有手性中心:
在本发明特定实施例中,通过符号*标识的碳原子具有(R)构型。在此实施例中,式I化合物在由符号*标识的碳原子处具有(R)构型,或在此碳原子处富含具有(R)构型的立体异构体型。
式I化合物也含有数个碱性基团(例如氨基),并且式I化合物因此可以游离碱型或以各种盐型存在。所有这些形式都包含在本发明范围内。此外,式I化合物或其盐的溶剂合物也包含在本发明范围内。
因此,熟习此项技术者应了解,除非另外说明,否则本文提及化合物时(例如提及式I化合物或化合物6时)包括提及所述化合物的盐和立体异构体和溶剂合物。
此外,除非所用上下文明确表示其它含义,否则本文所用单数形式「一」和「所述」包括相应的复数形式。
本文中所用命名本发明化合物及其中间体的命名法一般已经可使用市售奥托诺姆(AutoNom)软件(MDL,圣莱昂纳多,加利福尼亚)来导出。通常式I化合物可命名为联苯-2-基氨基甲酸的哌啶-4-基酯衍生物。
代表性实施例
下列取代基和数值意欲提供本发明各方面和实施例的代表性实例。这些代表性数值意欲进一步定义和说明这些方面和实施例,并且不意欲排除其它实施例或限制本发明范围。就这一点来说,除非明确说明,否则在描述特定数值或取代基是优选数值或取代基时并不意味着以任何方式将其它数值或取代基自本发明排除。
在一实施例中,R1为甲基并且R2为甲基。
在另一实施例中,R1为乙基并且R2为乙基。
在另一实施例中,R1为甲基并且R2为乙基。
在另一实施例中,R1为乙基并且R2为甲基。
因此,在本发明组合物的一方面中,本发明涉及选自以下的式I化合物:
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIa);
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二乙基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIb);
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2-甲基-5-乙基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIc);
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2-乙基-5-甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IId);
或其医药上可接受的盐。
定义
当描述本发明化合物、组合物、方法和过程时,除非另外说明,否则下述术语具有下述意义。
术语「烷基」是指可能为直链或具支链的单价饱和烃基。除非另有定义,否则这些烷基通常含有1至10个碳原子。代表性烷基包括(例如)甲基、乙基、正-丙基、异丙基、正-丁基、第二-丁基、异丁基、第三-丁基、正-戊基、正-己基、正-庚基、正-辛基、正-壬基、正-癸基等。
当欲表示本文所用特定术语的特定碳原子数时,碳原子数显示在术语前。例如,术语「C1-3烷基」意指具有1至3个碳原子的烷基,其中碳原子呈任何化学上可接受的构型。
术语「亚烷基」是指可能为直链或具支链的二价饱和烃基。除非另有定义,否则这些亚烷基通常含有1至10个碳原子。代表性亚烷基包括(例如)亚甲基、乙烷-1,2-二基(「亚乙基」)、丙烷-1,2-二基、丙烷-1,3-二基、丁烷-1,4-二基、戊烷-1,5-二基等。
术语「氨基保护基」意指适合在氨基处防止不期望反应的保护基。代表性氨基保护基包括(但不限于)第三-丁氧羰基(BOC)、三苯甲基(Tr)、苯甲氧基羰基(Cbz)、9-茀基甲氧基羰基(Fmoc)、苯甲基、甲酰基、三甲基甲硅烷基(TMS)、第三-丁基二甲基甲硅烷基(TBS)等。
术语「羧基保护基」意指适合在羧基处防止不期望反应的保护基。代表性羧基保护基包括(但不限于)酯类,例如甲基、乙基、第三-丁基、苯甲基(Bn)、对-甲氧基苯甲基(PMB)、9-茀基甲基(Fm)、三甲基甲硅烷基(TMS)、第三-丁基二甲基甲硅烷基(TBS)、二苯甲基(二苯甲基,DPM)等。
除非另外说明,否则本文所用术语「本发明化合物」或「式I化合物」或「式II化合物」意指所指定化合物或其医药上可接受的盐或溶剂合物或立体异构体。
术语「卤素」意指氟、氯、溴和碘。
术语「羟基保护基」意指适合在羟基处防止不期望反应的保护基。代表性羟基保护基包括(但不限于)甲硅烷基,包括三(C1-6烷基)甲硅烷基,例如三甲基甲硅烷基(TMS)、三乙基甲硅烷基(TES)、第三-丁基二甲基甲硅烷基(TBS)等;酯类(酰基),包括C1-6烷酰基,例如甲酰基、乙酰基等;芳甲基,例如苯甲基(Bn)、对-甲氧基苯甲基(PMB)、9-茀基甲基(Fm)、二苯甲基(二苯甲基,DPM)等。此外,两个羟基也可被保护成亚烷基,例如通过(例如)与诸如丙酮等酮反应而形成的亚丙-2-基。
术语「离去基团」意指在取代反应(例如亲核取代反应)中,可被另一官能团或原子置换的官能团或原子。例如,代表性离去基团包括(但不限于)氯、溴和碘基团;磺酸酯基,例如甲磺酸酯、甲苯磺酸酯、溴苯磺酸酯、硝基苯磺酸酯等;以及酰氧基,例如乙酰氧基、三氟乙酰氧基等。
在用于表示粒子时,术语「质量中位直径」或「MMD」意指其中一半质量的粒子为直径大于所述直径的粒子而另一半为直径小于所述直径的粒子时的直径。
除非另外说明,否则术语「微粉化」或「呈微粉化型」意指粒子中至少约90%粒子的直径小于约10μm。
本文所用术语「或其医药上可接受的盐或溶剂合物或立体异构体」意欲包括盐、溶剂合物和立体异构体的所有排列,例如式I化合物立体异构体的医药上可接受的盐的溶剂合物。
术语「医药上可接受的盐」意指可接受用于投与患者(例如哺乳动物)的盐(例如对给定剂量方案具有可接受哺乳动物安全性的盐)。代表性医药上可接受的盐包括乙酸、抗坏血酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、乙磺酸、乙二磺酸、反丁烯二酸、龙胆酸、葡萄糖酸、葡萄糖醛酸、谷氨酸、马尿酸、氢溴酸、盐酸、羟乙磺酸、乳酸、乳糖醛酸、顺丁烯二酸、苹果酸、苯乙醇酸、甲磺酸、粘酸、萘磺酸、萘-1,5-二磺酸、萘-2,6-二磺酸、烟碱酸、硝酸、乳清酸、双羟萘酸、泛酸、磷酸、琥珀酸、硫酸、酒石酸、对-甲苯磺酸和爱克辛那弗酸(xinafoic acid)等的盐。
术语「其经保护衍生物」意指特定化合物的衍生物,其中化合物的一或多个官能团经保护或封闭基团保护或封闭以免经历不期望的反应。可被保护的官能团包括(例如)羧酸基、氨基、羟基、硫醇基、羰基等。对这些官能团的适当保护基是熟习此项技术者熟知的,其例示于T.W.格林(T.W.Greene)和G.M.伍兹(G.M.Wuts),有机合成中的保护基(Protecting Groups in Organic Synthesis),第三版,威雷(Wiley),纽约,1999及其中所引述参考资料的教示中。
术语「其盐」意指当酸的氢被阳离子替代时所形成的化合物,所述阳离子例如金属阳离子或有机阳离子等。在本发明中,阳离子通常包含式I化合物的质子化型,意即其中一或多个氨基已被酸质子化。所述盐优选为医药上可接受的盐,但不欲供投与患者用的中间化合物的盐并不需要如此。
术语「溶剂合物」意指由一或多个溶质分子(即式I化合物或其医药上可接受的盐)和一或多个溶剂分子形成的复合物或聚集体。这些溶剂合物通常为结晶固体,具有实质上固定摩尔比的溶质和溶剂。代表性溶剂包括(例如)水、甲醇、乙醇、异丙醇、乙酸等。当溶剂为水时,所形成溶剂合物为水合物。
术语「治疗有效量」意指在投与需要治疗的患者时足以达成治疗的量。
本文所用术语「治疗」意指在患者(例如哺乳动物(特别是人类))中治疗疾病或医疗病况(例如COPD或哮喘),其包括:
(a)预防疾病或医疗病况发生,意即患者的预防性治疗;
(b)改善疾病或医疗病况,意即在患者中消除疾病或医疗病况或使其消退;
(c)抑制疾病或医疗病况,意即在患者中减慢或遏制疾病或医疗病况的发展;或
(d)在患者中减轻疾病或医疗病况的症状。
本文所用所有其它术语都意欲具有熟习其所属领域技术者所知的其一般意义。
一般合成程序
本发明化合物可使用下述一般方法和程序或通过使用熟习此项技术者易于取得或已知的其它信息自易于取得的起始物质来制备。虽然下述程序可阐释本发明的特定实施例或方面,但熟习此项技术者可明了,可使用相同或类似方法或通过使用熟习此项技术者已知的其它方法、试剂和起始物质来制备本发明的其它实施例或方面。也应明了,除非另外说明,否则尽管给定了典型或优选方法条件(即反应温度、时间、反应物的摩尔比、溶剂、压力等),但也可使用其它方法条件。虽然最适宜反应条件通常根据各种反应参数(例如所使用的反应物、溶剂和数量)而改变,但熟习此项技术者使用例行最佳化程序可容易地确定适宜反应条件。
此外,正如熟习此项技术者所明了,可能需要或期望习用保护基来防止某些官能团发生不期望反应。特定官能团的适当保护基以及用于这些官能团的保护和去保护的适当条件和试剂的选择是业内所熟知的。如果需要,可使用本文所述程序中所示者以外的保护基。
在一实施例中,如方案I中所示合成式I化合物:
方案I

其中P1为羟基保护基;并且R3a和R3b独立选自C1-6烷基,或R3a和R3b接合形成C2-6亚烷基。
如方案I中所示,化合物1可与约0.95至约1.05摩尔当量的化合物2反应以提供化合物3。此迈克尔(Michael)反应通常是在约0℃至约75℃、通常约40℃至约45℃范围的温度下进行约8至约24小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如二氯甲烷或二氯甲烷与甲醇或乙醇的混合物等。在反应完成后,通常使用习用程序来分离产物,例如萃取、再结晶、色谱等。或者,含有化合物3的反应混合物可直接用于下一合成步骤中。
然后使化合物3与含水酸反应以水解缩醛基并提供醛化合物4。在此反应中可采用任何适当酸,包括(例如)盐酸、硫酸、甲磺酸、对-甲苯磺酸等。水解反应通常是在约0℃至约30℃、通常约20℃至约25℃范围的温度下进行约1至约6小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲醇、乙醇、异丙醇、二氯甲烷/乙醇、乙腈等。在反应完成后,通常使用习用程序来分离产物,例如萃取、再结晶、色谱等。
然后使化合物4与约0.95至约1.5摩尔当量的化合物5在还原剂存在下反应来获得化合物6。此反应中可使用任何适当还原剂,包括(例如)金属氢化物试剂(例如硼氢化钠、三乙酰氧基硼氢化钠、氰基硼氢化钠等)或氢及金属催化剂(例如钯/碳等)。此还原性烷基化反应通常是在约-20℃至约30℃、通常约0℃至约5℃范围的温度下进行约1至约6小时或直至反应基本完成。一般来说,此反应是在适当稀释剂和质子性溶剂中进行,例如二氯乙烷和甲醇等。在反应完成后,通常使用习用程序来分离产物,例如萃取、再结晶、色谱等。
然后使化合物6去保护以提供式I化合物。用于使化合物6去保护的特定条件可取决于所采用保护基。例如,当P1为甲硅烷基保护基(即如本文所定义的式6a化合物)时,此去保护反应通常是通过使化合物6a与氟离子源接触来进行,P1是例如第三-丁基二甲基甲硅烷基、第三-丁基二苯基甲硅烷基、二苯甲基甲硅烷基、二-第三-丁基甲基甲硅烷基、第三-丁氧基二苯基甲硅烷基等。在特定实施例中,氟离子源为三乙胺三氢氟酸盐。其它适当氟离子源包括氟化四丁基铵、氟化钾-18-冠-6、氟化氢、氟化氢吡啶等。此反应通常是在约0℃至约50℃、通常约10℃至约25℃范围的温度下进行约24至约72小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如二氯乙烷等。在反应完成后,通常使用习用程序来分离产物,例如萃取、再结晶、色谱等。
化合物1是业内已知的或可使用已知程序自市售可起始物质和试剂来制备。参阅(例如)美国专利申请公开案第2004/0167167A1号和R.奈托(R.Naito)等人,化学药物公报(Chem.Pharm.Bull.),46(8)1286-1294(1998)。例如,可如方案II中所示制备化合物1:
方案II

如方案II中所示,使2-异氰酸联苯基酯(7)与N-保护的4-羟基哌啶8反应(其中P2为氨基保护基诸如苯甲基等)以提供氨基甲酸酯中间体9。此反应通常是在约20℃至约100℃、通常约60℃至约80℃范围的温度下进行约6至约24小时或直至反应基本完成。如果需要,则此反应可在适当稀释剂中进行,例如二氯甲烷、甲苯等。或者,可在不存在稀释剂的情况下进行此反应。在反应完成后,通常使用习用程序来分离产物9,例如萃取、再结晶、色谱等。或者,含有化合物9的反应混合物可直接用于下一合成步骤中。
然后使用习用程序自化合物9移除氨基保护基P2而获得化合物1。例如,当P2为苯甲基时,可在催化剂(例如钯催化剂)存在下使用氢或甲酸铵使化合物9去保护。代表性催化剂包括(例如)钯/碳、氢氧化钯/碳等。此反应通常是在约20℃至约50℃范围的温度下(通常约40℃)进行约6至约24小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲醇、乙醇、异丙醇等。在反应完成后,通常使用习用程序来分离化合物1,例如萃取、再结晶、色谱等。
式2化合物是业内已知的或可使用已知程序自市售起始物质和试剂制备。例如,可根据方案III中所示来制备式2化合物:
方案III

如方案III中所示,首先在氨基处保护式10苯胺化合物(其中X1为溴或碘)以提供式11化合物,其中P3a为氨基保护基并且P3b为氢或氨基保护基。可使用任何适当氨基保护基,例如苯甲基、4-甲氧基苯甲基、三氟乙酰基等。例如,可使式10化合物与约2或更多摩尔当量、优选地约2.5至约3.0摩尔当量的苯甲基卤化物(例如苯甲基氯化物、溴化物或碘化物)反应而获得化合物11,其中P3a和P3b都是苯甲基。此反应通常是在约0℃至约50℃范围的温度下(通常为约30℃)进行约18至约24小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲醇、乙醇、异丙醇等。通常,此反应也可在适当碱存在下进行,例如碳酸钾、碳酸钠等。在反应完成后,通常使用习用程序来分离化合物11,例如萃取、再结晶、色谱等。
可用于此反应中的代表性式10化合物包括2,5-二甲基-4-碘苯胺、2,5-二乙基-4-碘苯胺、2-乙基-4-碘-5-甲基苯胺、5-乙基-4-碘-2-甲基苯胺、4-溴-2,5-二甲基苯胺、4-溴-2,5-二乙基苯胺、4-溴-2-乙基-5-甲基苯胺、4-溴-5-乙基-2-甲基苯胺等。这些化合物是市售可得(例如得自波谱(Spectra)集团有限公司,米尔伯里(Millbury),OH)或可使用习用程序制自市售起始物质和试剂。
然后使化合物11与约1至约2摩尔当量的烷基锂试剂(例如正-丁基锂或第三-丁基锂)接触以形成相应阴离子,其中X1基团已被交换成锂。此反应通常是在约-70℃至约0℃范围的温度下(通常为约-20℃)进行约0.25至约1小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲苯、二甲苯、四氢呋喃等。
不分离所形成锂阴离子,而是使其与摩尔过量的N,N-二甲基甲酰胺就地反应以提供化合物12。一般来说,使用约2至约4摩尔当量的N,N-二甲基甲酰胺。此反应通常是在约-70℃至约0℃、通常约-20℃至约0℃范围的温度下进行约0.5至约2小时或直至反应基本完成。在反应完成后,通常使用习用程序来分离化合物12,例如萃取、再结晶、色谱等。
然后通过在酸性催化剂存在下使化合物12与醇或二醇反应将化合物12的醛基保护成缩醛。任何适当醇或二醇都可用于此反应中。例如,代表性醇类和二醇类包括甲醇、乙醇、正丙醇、乙二醇、丙二醇等。一般来说,此反应中采用摩尔过量的醇或二醇,优选为约2至约4摩尔当量。
此反应中可使用任何适当酸催化剂以促进缩醛的形成。代表性酸催化剂包括(例如)对甲苯磺酸、苯磺酸、盐酸等。
此反应通常是在约50℃至约100℃、通常约60℃至约80℃范围的温度下进行约12至约24小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲苯、二甲苯等。通常,此反应的实施方式使得可去除所产生水,例如通过共沸蒸馏或通过使用分子筛来实施。在反应完成后,通常使用习用程序来分离化合物13,例如萃取、再结晶、色谱等。或者,含有化合物13的反应混合物可直接用于下一合成步骤中。
在形成缩醛后,使用标准试剂和条件使化合物13的氨基去保护以形成化合物14。例如,如果P3a和P3b为苯甲基,则可使用氢和催化剂(例如钯催化剂)来使化合物13去保护。代表性催化剂包括(例如)钯/碳、氢氧化钯/碳等。此反应通常是在约20℃至约50℃、通常约25℃至约30℃范围的温度下进行约4至约12小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲醇、乙醇、乙醇/乙酸乙酯混合物等。在反应完成后,通常使用习用程序来分离化合物14,例如萃取、再结晶、色谱等。
然后使化合物14与丙烯酰基卤化物15(其中X2为氯、溴或碘)反应以形成化合物2。此反应通常是在约-20℃至约25℃、通常约0℃至约5℃范围的温度下进行约0.5至约6小时或直至反应基本完成。一般来说,此反应是在适当稀释剂(例如二氯甲烷等)中于适当碱(例如二异丙基乙胺、三乙胺等)存在下进行。一般来说,此反应中使用约1至1.2摩尔当量的丙烯酰基卤化物和约1至约2摩尔当量的碱。在反应完成后,通常使用习用程序来分离化合物2,例如萃取、再结晶、色谱等。
式5化合物是业内已知的或可使用已知程序制自市售起始物质和试剂。例如,可如方案IV中所示来制备式5化合物:
方案IV

如方案IV中所示,可自式16化合物制备式5化合物,其中P4为羟基保护基,例如苯甲基,并且X3为离去基团,例如氯、溴或碘。式16化合物是业内已知的。例如,2001年7月31日颁布的美国专利第6,268,533 B1号;和R.海特(R.Hett)等人,有机过程研究和发展1998,2,96-99;描述自2-溴-4′-苯甲氧基-3′-硝基苯乙酮开始制备N-[2-苯甲氧基-5-((R)-2-溴-1-羟乙基)苯基]甲酰胺(即化合物16的(R)对映异构体,其中P4为苯甲基并且X3为溴),所述起始物质的合成描述于K.莫里斯(K.Murase)等人,化学药物公报,25(6)1368-1377(1977)中。如果需要,可利用非手性性还原剂(例如硼烷二甲硫醚复合物)还原2-溴-4′-苯甲氧基-3′-硝基苯乙酮来制备化合物16的外消旋形式。
使用习用程序和试剂保护化合物16的羟基以提供化合物17,其中P1为羟基保护基。在特定实施例中,羟基保护基为甲硅烷基保护基,例如二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、第三-丁基二甲基甲硅烷基、第三-丁基二苯基甲硅烷基、二苯甲基甲硅烷基等。例如,可在约1.1至约1.3摩尔当量的咪唑存在下使化合物16与约0.95至约1.2摩尔当量的氯化第三-丁基二甲基硅烷反应以提供化合物17,其中P1为第三-丁基二甲基甲硅烷基。此反应通常是在约0℃至约50℃范围的温度下(通常在室温下)进行约24至约48小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如N,N-二甲基甲酰胺等。在反应完成后,通常使用习用程序来分离化合物17,例如萃取、色谱等。例如,N-{2-苯甲氧基-5-[(R)-2-溴-1-(第三-丁基二甲基硅烷氧基)乙基]苯基}甲酰胺的合成描述于2004年12月9日公开的美国专利公开案第2004/0248985A1号的实例2中。
然后使化合物17与苄胺(即Bn1-NH2)反应或得化合物18,其中Bn1是在苯甲基苯环上具有1至3个独立选自C1-4烷基或C1-4烷氧基的取代基的未经取代苯甲基或经取代苯甲基。代表性苄胺类包括苄胺、3,4-二甲氧基苄胺、4-甲氧基苄胺、4-甲基苄胺等。此反应通常是通过使化合物17与约2至约4摩尔当量的苄胺在约40℃至约100℃、通常约80℃至约90℃范围的温度下接触约5至约24小时或直至反应基本完成来实施。一般来说,此反应是在适当稀释剂中进行,例如N-甲基-2-吡咯烷酮(NMP)等。在反应完成后,通常使用习用程序来分离化合物18,例如萃取、色谱、再结晶等。
然后使用习用程序和试剂来移除苯甲基Bn1和P4以获得化合物5。在一实施例中,Bn1和P4两者都是在相同反应混合物中移除的苯甲基。通常,此反应是通过在催化剂(例如钯催化剂)存在下使化合物5与氢接触来进行。代表性催化剂包括氢氧化钯/碳、钯/碳等。一般来说,此脱苯甲基化反应是在酸存在下进行,例如乙酸、甲酸等。此反应通常是在约10℃至约50℃范围的温度下(通常在室温下)进行约6至约24小时或直至反应基本完成。一般来说,此反应是在适当稀释剂中进行,例如甲醇、乙醇等。在反应完成后,通常使用习用程序来分离化合物5,例如萃取、色谱等。在特定实施例中,以乙酸盐型分离化合物5。
熟习此项技术者可明了,也可通过其它合成程序制备式I化合物。例如,实施合成步骤的具体顺序可以改变,或可采用不同中间体。例如,可使用习用程序和试剂来还原式III化合物:

(其中Y1为-CN、-C(O)OH或-C(O)OR3c)以提供醛4(即其中Y1为-CHO)。此外,可将这些化合物还原成醇,即其中Y1为-CH2OH,并且之后可使用标准程序和试剂将醇氧化以提供醛4(即其中Y1为-CHO)。
如果需要,则可通过使式I化合物的游离碱型与医药上可接受的酸接触来制备式I化合物的医药上可接受的盐。
关于制备本发明代表性化合物或其中间体的具体反应条件及其它程序的其它细节描述于下文所述实例中。
医药组合物、组合和调配物
本发明化合物通常是以医药组合物或调配物的形式投与患者。这些医药组合物可通过任何可接受投与途径投与患者,包括(但不限于)吸入、经口、经鼻、局部(包括经皮)和不经肠投与模式。应明了,适合特定投与模式的本发明化合物的任何形式(即游离碱、医药上可接受的盐、溶剂合物等)都可用于本文所论述医药组合物中。
因此,在本发明组合物的一方面中,本发明涉及医药组合物,其包含医药上可接受的载剂或赋形剂和式I化合物。如果需要,这些医药组合物可视情况含有其它治疗性和/或调配性药剂。
本发明医药组合物通常含有治疗有效量的式I化合物。但是,熟习此项技术者应明了,医药组合物可含有大于治疗有效量(即总体组合物)或低于治疗有效量(即经设计用于多次投与以达成治疗有效量的个别单位剂量)。
通常,医药组合物可含有约0.01重量%至约95重量%的治疗药剂;包括约0.01重量%至约30重量%;例如约0.01重量%至约10重量%的治疗药剂。
任何习用载剂或赋形剂都可使用于本发明医药组合物中。特定载剂或赋形剂、或载剂或赋形剂的组合的选择可取决于治疗特定患者投与模式或医疗病况或疾病状态的类型。就这一点来说,熟习医药领域技术者精通用于特定投与模式的适当医药组合物的制备。
在本发明医药组合物中使用市售载剂或赋形剂。例如,这些物质可购自西格玛(Sigma)(圣刘易斯(St.Louis),MO)。例如,习用调配技术是熟习此项技术者熟知的,其例示于雷鸣登(Remington):药物科学与实践(The Science and Practice of Pharmacy),第20版,利平科特威廉姆斯与怀特公司(Lippincott Williams & White),巴尔的摩(Baltimore),马里兰(Maryland)(2000);和H.C.安塞尔(H.C.Ansel)等人,医药剂型与药物递送系统(Pharmaceutical Dosage Forms and Drug Delivery Systems),第7版,利平科特威廉姆斯与怀特公司,巴尔的摩,马里兰(1999)的教示中。可用作医药上可接受载剂的物质的代表性实例包括(但不限于)以下物质:(1)糖类,例如乳糖、葡萄糖和蔗糖;(2)淀粉,例如玉米淀粉和马铃薯淀粉;(3)纤维素及其衍生物,例如羧甲基纤维素钠、乙基纤维素和纤维素乙酸酯;(4)粉末状黄蓍胶;(5)麦芽;(6)明胶;(7)滑石;(8)赋形剂,例如可可脂和栓剂蜡;(9)油类,例如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;(10)二醇类,例如丙二醇;(11)多元醇类,例如甘油、山梨醇、甘露醇和聚乙二醇;(12)酯类,例如油酸乙酯和月桂酸乙酯;(13)琼脂;(14)缓冲液,例如氢氧化镁和氢氧化铝;(15)海藻酸;(16)无热原水;(17)等渗盐水;(18)林格氏(Ringer′s)溶液;(19)乙醇;(20)磷酸盐缓冲溶液;(21)压缩推进剂气体,例如氯氟烃类和氢氟烃类;以及(22)医药组合物中所用其它无毒性可相容物质。
本发明医药组合物通常是通过将本发明化合物与医药上可接受的载剂和任何可选成份充分和密切混合或掺合来制备。如果需要或期望,之后可使用习用程序和设备将所得均匀掺合混合物制成片剂、胶囊、丸剂等或装填至药罐、药筒、分配器等之中。
在一实施例中,本发明医药组合物适用于吸入投与。适用于吸入投与的医药组合物通常可呈气溶胶或粉末型。这些组合物一般是使用习知递送装置来投与,例如雾化罐吸入器、定量式吸入器(MDI)、干粉吸入器(DPI)或类似递送装置。
在本发明具体实施例中,包含治疗药剂的医药组合物是使用雾化罐吸入器通过吸入来投与。这些雾化罐装置通常产生高速度空气流,其使包含治疗药剂的医药组合物以雾气型被喷雾输送至患者的呼吸道中。因此,当经调配用于雾化罐吸入器时,通常使治疗药剂溶于适当载剂中以形成溶液。或者,治疗药剂可被微粉化并与适当载剂合并形成微粉化粒子的悬浮液。适用于吸入投与治疗药剂的雾化罐装置是熟习此项技术者熟知的,或这些装置是市售装置。例如,代表性雾化罐装置或产品包括肺部输药微雾吸入器(Respimat Softmist Inhalaler)(勃林格殷格翰(Boehringer Ingelheim));AERx肺递送系统(阿拉迪金(Aradigm)公司);PARILC Plus可重复使用雾化罐(Pari GmbH)等。
用于雾化罐吸入器中的代表性医药组合物包含等渗水溶液,其包含约0.05μg/mL至约10mg/mL的式I化合物。在一实施例中,此一溶液具有约4至约6的pH。
在本发明的另一具体实施例中,使用干粉吸入器通过吸入来投与包含治疗药剂的医药组合物。这些干粉吸入器通常以自由流动性粉末型投与治疗药剂,所述自由流动粉末在吸气期间分散于患者的气流中。为达成自由流动性粉末,通常将治疗药剂与适当赋形剂一起调配,所述赋形剂例如乳糖、淀粉、甘露醇、右旋糖、聚乳酸(PLA)、聚交酯-共-乙交酯(PLGA)或其组合。通常,治疗药剂经微粉化并与适当载剂合并以形成适用于吸入的掺合物。因此,于本发明一实施例中,式I化合物是呈微粉化型。
用于干粉吸入器的代表性医药组合物包含干磨乳糖和式I化合物的微粉化粒子。
此—干粉调配物可(例如)通过将乳糖与治疗药剂合并然后干掺合所述组份来制造。或者,如果需要,可在不使用赋形剂的情况下调配治疗药剂。然后,通常将医药组合物装填至干粉分配器中,或装填至吸入药筒或胶囊中以供与干粉递送装置一起使用。
适用于吸入投与治疗药剂的干粉吸入器递送装置是熟习此项技术者熟知的或这些装置是市售装置。例如,代表性干粉吸入器递送装置或产品包括伊欧雷斯(Aeolizer)(诺华(Novartis));埃尔麦克斯(Airmax)(IVAX);单击式吸入器(ClickHaler)(伊诺威达生物医药公司(Innovata Biomed));碟式吸入器(Diskhaler)(葛兰素史克(GlaxoSmithKline));迪斯科斯/阿库吸入器(Diskus/Accuhaler)(葛兰素史克);易吸干粉定量吸入器(Easyhaler)(芬恩制药公司(Orion Pharma));伊克利普斯(Eclipse)(安万特(Aventis));福鲁开普(FlowCaps)(好利安(Hovione));便利型吸入器(Handihaler)(勃林格殷格翰);帕维诺(Pulvinal)(凯西(Chiesi));旋转吸入器(Rotahaler)(葛兰素史克);斯基吸入器/斯而特吸入器(SkyeHaler/Certihaler)(斯基制药(SkyePharma));旋蝶形吸入器(Twisthaler)(先灵葆雅(Schering-Plough));涡轮吸入器(Turbuhaler)(阿斯利康(AstraZeneca));储库型吸入器(Ultrahaler)(安万特)等。
在本发明另一具体实施例中,包含治疗药剂的医药组合物是使用定量式吸入器通过吸入来投与。这些定量式吸入器通常使用压缩推进剂气体来排放经测量数量的治疗药剂。因此,使用定量式吸入器投与的医药组合物通常包含存于液化推进剂中的治疗药剂溶液或悬浮液。可采用任何适当液化推进剂,包括氯氟烃类(例如CCl3F)和氢氟烷类(HFA)(例如1,1,1,2-四氟乙烷(HFA134a)和1,1,1,2,3,3,3-七氟-正-丙烷(HFA227))。由于顾虑氯氟烃类会影响臭氧层,因此一般含有HFA的调配物是优选的。HFA调配物的其它可选组份包括共溶剂(例如乙醇或戊烷)和表面活性剂(例如山梨醇三油酸酯、油酸、卵磷脂和甘油)。
用于定量式吸入器的代表性医药组合物包含约0.01重量%至约5重量%的式I化合物;约0重量%至约20重量%的乙醇;和约0重量%至约5重量%的表面活性剂;其余部分为HFA推进剂。
这些组合物通常是通过将经冷冻或加压的氢氟烷添加至含有治疗药剂、乙醇(如果存在)和表面活性剂(如果存在)的适当容器中来制备。为制备悬浮液,将治疗药剂微粉化然后将其与推进剂合并。然后,将调配物装填至气溶胶罐中,所述罐形成定量式吸入器装置的一部分。
适用于吸入投与治疗药剂的定量式吸入器装置是熟习此项技术者熟知的,或这些装置是市售装置。例如,代表性定量式吸入器装置或产品包括氟尼松气雾(AeroBid)吸入器系统(森林制药公司(ForestPharmaceuticals));爱全乐(Atrovent)吸入气溶胶(勃林格殷格翰);弗洛温特(Flovent)(葛兰素史克);吡布特罗(Maxair)吸入器(3M);舒喘灵(Proventil)吸入器(先灵);施立稳(Serevent)吸入气溶胶(葛兰素史克)等。
在另一实施例中,本发明医药组合物适用于口服投与。口服投与的适当医药组合物可呈胶囊、片剂、丸剂、锭剂、扁囊剂、糖衣锭、粉末、颗粒型;或作为存于水性或非水性液体中的溶液或悬浮液;或作为水包油或油包水液体乳液;或作为酏剂或糖浆;及其类似物;其各自含有预定量的本发明化合物作为活性成份。
当欲以固体剂型(即作为胶囊、片剂、丸剂等)口服投与时,本发明医药组合物通常可包含作为活性成份的本发明化合物和一或多种医药上可接受的载剂,例如柠檬酸钠或磷酸二钙。视情况或替代地,这些固体剂型也可包含:(1)填充剂或增量剂,例如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和/或硅酸;(2)粘合剂,例如羧甲基纤维素、藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和/或阿拉伯胶;(3)保湿剂,例如甘油;(4)崩解剂,例如琼脂、碳酸钙、马铃薯或木薯淀粉、海藻酸、某些硅酸盐和/或碳酸钠;(5)溶液阻滞剂,例如石蜡;(6)吸收促进剂,例如季铵化合物;(7)润湿剂,例如鲸蜡醇和/或单硬脂酸甘油酯;(8)吸收剂,例如高岭土(kaolin)和/或膨润土(bentonite clay);(9)润滑剂,例如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠和/或其混合物;(10)着色剂;以及(11)缓冲液。
离型剂、润湿剂、涂布剂,增甜、矫味和芳香剂,防腐剂和抗氧化剂也可存在于本发明医药组合物中。医药上可接受抗氧化剂的实例包括:(1)水溶性抗氧化剂,例如抗坏血酸、半胱氨酸盐酸盐、硫酸氢钠、偏亚硫酸钠、亚硫酸钠等;(2)油溶性抗氧化剂,例如抗坏血酸棕榈酸酯、丁羟茴醚(BHA)、丁羟甲苯(BHT)、卵磷脂、没食子酸丙酯、α-生育酚等;以及(3)金属螯合剂,例如柠檬酸、乙二胺四乙酸(EDTA)、山梨醇、酒石酸、磷酸等。用于片剂、胶囊、丸剂等的涂布剂包括用于肠溶包衣者,例如纤维素乙酸酞酸酯(CAP)、聚酞酸乙酸乙烯酯(PVAP)、羟丙基甲基纤维素酞酸酯、甲基丙烯酸、甲基丙烯酸酯共聚物、乙酸均苯三甲酸纤维素(CAT)、羧甲基乙基纤维素(CMEC)、羟丙基甲基纤维素乙酸琥珀酸酯(HPMCAS)等。
如果需要,也可以不同比例使用(例如)羟丙基甲基纤维素、或其它聚合体基质、脂质体和/或微球体来调配本发明医药组合物以提供活性成份的缓慢或受控释放。
此外,本发明医药组合物可视情况含有遮光剂并且可经调配以使其只在或优先在胃肠道的某一部分中(视情况)以延迟方式释放活性成份。可用包埋组合物的实例包括聚合物质和蜡。如果适当,活性成份也可与一或多种上述赋形剂一起呈微囊封型。
用于口服投与的适当液体剂型包括(例如)医药上可接受的乳液、微乳液、溶液、悬浮液、糖浆和酏剂。这些液体剂型通常包含活性成份和惰性稀释剂,例如水或其它溶剂、增溶剂和乳化剂,例如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苄醇、苯甲酸苄基酯、丙二醇、1,3-丁二醇、油类(尤其棉籽油、花生油、玉米油、胚油、橄榄油、蓖麻油及芝麻油)、甘油、四氢呋喃醇、聚乙二醇和山梨醇的脂肪酸酯、及其混合物。除了活性成份以外,悬浮液可含有悬浮剂,例如乙氧基化异硬脂醇、聚氧乙烯山梨醇和山梨醇酯、微晶性纤维素、偏氢氧化铝、膨润土、琼脂和黄蓍胶、及其混合物。
当欲用于口服投与时,可以单位剂型包装本发明医药组合物。术语「单位剂型」意指适合患者服药的物理离散单位,即各单位含有经计算可单独或与一或多个其它单位组合产生期望治疗效果的预定量活性药剂。例如,这些单位剂型可为胶囊、片剂、丸剂等。
也可使用已知经皮递送系统和赋形剂以经皮方式投与本发明化合物。例如,可将本发明化合物与渗透增强剂(例如丙二醇、聚乙二醇单月桂酸酯、氮杂环烷-2-酮等)互混并将其纳入贴剂或类似递送系统中。如果需要可将其它赋形剂(包括胶凝剂、乳化剂和缓冲液)用于这些经皮组合物中。
此外,本发明化合物可以不经肠方式投与,即以静脉内方式、皮下方式或肌内方式投与。对于不经肠投与,通常使式I化合物溶于不经肠投与可接受的载剂中,例如无菌水、盐水、植物油等。例如,静脉内组合物通常包含式I化合物的无菌水溶液,其中所述溶液的pH在约4至约7范围内。
如果需要,本发明化合物可与一或多种其它治疗药剂组合投与。在此实施例中,以物理方式将本发明化合物与另一治疗药剂混合来形成含有两种药剂的组合物;或各药剂存在于分开的不同组合物中,并将其同时或相继投与患者。
例如,可使用习用程序和设备将式I化合物与第二治疗药剂组合以形成包含式I化合物和第二治疗药剂的组合物。此外,可将治疗药剂与医药上可接受的载剂组合以形成包含式I化合物、第二治疗药剂和医药上可接受载剂的医药组合物。在此实施例中,通常将组合物的组份混合或掺合以产生物理混合物。然后,使用本文所述任何途径以治疗有效量投与所述物理混合物。
或者,在投与患者前,可使治疗药剂保持分开和不同。在此实施例中,在投与前并未以物理方式将治疗药剂混合在一起,而是将其作为分开的组合物同时或相继投与。例如,可使用吸入递送装置以吸入方式同时或相继投与式I化合物与另一治疗药剂,所述装置对各治疗药剂采用分开地隔室(例如泡壳包装)。或者,可使用分开递送装置投与组合,即每种治疗药剂都各自使用一种递送装置。此外,可通过不同投与途径递送治疗药剂,即一种通过吸入投与而另一种通过口服投与。
可与本发明化合物相容的任何治疗药剂都可与这些化合物组合使用。在特定实施例中,第二治疗药剂是可通过吸入有效投与者。例如,可与本发明化合物一起使用的治疗药剂的代表性类型包括(但不限于)消炎药,例如类固醇消炎药(包括皮质类固醇和糖皮质激素)、非类固醇消炎药(NSAID)和PDE4抑制剂;支气管扩张药,例如PDE3抑制剂、腺苷2b调制剂和β2肾上腺素能受体激动剂;抗感染药,例如革兰氏(Gram)阳性抗生素、革兰氏阴性抗生素和抗病毒药;抗组胺药;蛋白酶抑制剂;传入阻断剂,例如D2激动剂和神经激肽调节剂;和毒蕈碱受体拮抗剂(抗胆碱能剂)。这些治疗药剂的许多实例是业内熟知的。与本发明化合物组合投与的其它治疗药剂的适当剂量通常在约0.05μg/天至约500mg/天的范围内。
在本发明的特定实施例中,式I化合物是与类固醇消炎药组合投与。可与本发明化合物组合使用的类固醇消炎药的代表性实例包括(但不限于)二丙酸倍氯米松(beclomethasone dipropionate);布地奈德(budesonide);丙酸布替可特(butixocortpropionate);20R-16α,17α-[亚丁基双(氧基)]-6α,9α-二氟-11β-羟基-17β-(甲硫基)雄甾-4-烯-3-酮(RPR-106541);环索奈德(ciclesonide);地塞米松(dexamethasone);6α,9α-二氟-17α-[(2-呋喃基羰基)氧基]-11β-羟基-16α-甲基-3-氧代雄甾-1,4-二烯-17β-硫代甲酸S-氟甲酯;6α,9α-二氟-11β-羟基-16α-甲基-17α-[(4-甲基-1,3-噻唑-5-羰基)氧基]-3-氧代雄甾-1,4-二烯-17β-硫代甲酸S-氟甲酯;6α,9α-二氟-11β-羟基-16α-甲基-3-氧-17α-丙酰氧基雄甾-1,4-二烯-17β-硫代甲酸(S)-(2-氧代四氢呋喃-3S-基)酯;氟尼缩松(flunisolide);丙酸氟替卡松(fluticasone propionate);甲基泼尼松龙(methyl prednisolone);糠酸莫米松(mometasone furoate);泼尼松龙(prednisolone);泼尼松(prednisone);罗氟奈德(rofleponide);ST-126;曲安奈德(triamcinolone acetonide)等,或其医药上可接受的盐。这些类固醇消炎药是市售的或可使用习用程序和试剂来制备。例如,类固醇消炎药的制备和用途描述于2004年6月15日颁布的美国专利第6,750,210 B2号;2004年7月6日颁布的美国专利第6,759,398 B2号;2003年3月25日颁布的美国专利第6,537,983号;2002年2月14日公开的美国专利申请公开案第2002/0019378 A1号;及其中所引用参考资料。
在使用时,类固醇消炎药通常是以在与本发明化合物共投与时可产生治疗上有利效应的量来投与。通常,类固醇消炎药可以足以提供每剂量约0.05μg至约500μg的量来投与。
下述实例阐释本发明代表性医药组合物:
实例A
干粉组合物
使经微粉化的本发明化合物(100mg)与经研磨乳糖(25g)(例如其中约85%以下粒子的MMD为约60μm至约90μm并且15%以上粒子的MMD低于15μm的乳糖)掺合。然后,将此掺合混合物装填至可剥离泡壳包装的单独泡壳中,其量足以提供每剂量约10μg至约500μg本发明化合物。使用干粉吸入器投与气泡的内含物。
实例B
干粉组合物
使微粉化本发明化合物(1g)与经研磨乳糖(200g)掺合以形成化合物与经研磨乳糖的重量比为1:200的总体组合物。将掺合组合物包装至干粉吸入装置中,其每剂量能够递送约10μg至约500μg本发明化合物。
实例C
干粉组合物
使微粉化本发明化合物(100mg)和微粉化类固醇消炎药(500mg)与经研磨乳糖(30g)掺合。然后,将此掺合混合物装填至可剥离泡壳包装的单独泡壳中,其量足以提供每剂量约10μg至约500μg的本发明化合物。使用干粉吸入器投与泡壳内含物。
实例D
定量式吸入器组合物
使经微粉化本发明化合物(10g)分散于通过使卵磷脂(0.2g)溶解在脱矿质水(200mL)中所制成的溶液内。将所形成的悬浮液喷雾干燥然后微粉化以形成包含平均直径小于约1.5μm的粒子的经微粉化组合物。然后将经微粉化组合物装填至含有加压1,1,1,2-四氟乙烷的定量式吸入器药筒中,当通过定量式吸入器投与时装填量足以提供每剂量约10μg至约500μg的本发明化合物。
实例E
雾化罐组合物
使本发明化合物(25mg)溶于柠檬酸盐缓冲(pH 5)等渗盐水(125mL)中。将混合物搅拌并实施超声处理直至化合物溶解。检查溶液的pH值并且如果需要就通过慢慢添加1N氢氧化钠水溶液调整至pH5。使用雾化罐装置投与溶液,其可提供每剂量约10μg至约500μg的本发明化合物。
实例F
硬明胶胶囊
将本发明化合物(50g)、经喷雾干燥乳糖(440g)和硬脂酸镁(10g)充分地掺合。将所形成组合物装填至以口服方式投与的硬明胶胶囊(每胶囊500mg组合物)中。
实例G
口服悬浮液
将下列成份充分混合以形成口服投与用的悬浮液:

所形成悬浮液含有100mg活性成份/10mL悬浮液。悬浮液是以口服方式投与。
实例H
可注射组合物
将本发明化合物(0.2g)与0.4M乙酸钠缓冲溶液(2.0mL)掺合。根据需要使用0.5N盐酸水溶液或0.5N氢氧化钠水溶液将所形成溶液的pH值调整至pH 4,然后添加足够注射用水以提供20mL的总体积。然后将混合物经过无菌滤器(0.22微米)过滤以提供适合通过注射投与的无菌溶液。
效用
本发明化合物同时具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性,因此预期这些化合物可用作治疗药剂来治疗由β2肾上腺素能受体或毒蕈碱受体所介导的医疗病况,即通过以β2肾上腺素能受体激动剂或毒蕈碱受体拮抗剂治疗可改善的医疗病况。这些医疗病况是熟习此项技术者熟知的,其例示于如埃格林(Eglen)等人,毒蕈碱受体亚型:医药学与治疗功效(Muscarinic Receptor Subtypes:Pharmacology andTherapuetic Potential),DN&P 10(8),462-469(1997);艾美林(Emilien)等人,β-肾上腺素能受体激动剂和拮抗剂的当前治疗应用与功效(Current Therapeutic Uses andPotential of beta-Adrenoceptor Agonists and Antagonists),欧洲临床医药杂志(European J.Clinical Pharm.),53(6),389-404(1998);和其中所引用参考文献的教示中。这些医疗病况包括(例如)肺病或与可逆气道阻塞相关的疾病,例如慢性阻塞性肺病(例如慢性和喘息性支气管炎和气肿)、哮喘、肺纤维化等。其它病况包括早产、抑郁、充血性心力衰竭、皮肤病(例如炎症性、过敏性、干癣性和增生性皮肤病),其中期望降低胃液素酸度的病况(例如消化性溃疡和胃溃疡),以及肌肉萎缩疾病。
因此,在一实施例中,本发明涉及治疗肺病的方法,所述方法包含对需要治疗的患者投与治疗有效量的式I化合物。当用于治疗肺病时,本发明化合物通常是通过吸入以每天多次剂量、每天单次剂量或每周单次剂量投与。一般来说,治疗肺病的剂量是在约10μg/天至约500μg/天范围内。
在本发明方法的一方面中,本发明涉及治疗慢性阻塞性肺病或哮喘的方法,所述方法包含对患者投与治疗有效量的式I化合物。一般来说,治疗COPD或哮喘的剂量是在约10μg/天至约500μg/天范围内。熟习此项技术者可理解术语「COPD」包括多种呼吸病况,包括慢性阻塞支气管炎和气肿,其例示于伯纳斯(Barnes),慢性阻塞性肺病(Chronic Obstructive Pulmonary Disease),新英格兰医学期刊(N.Engl.J.Med.),2000:343:269-78,及其中所引用参考文献的教示中。
当通过吸入投与时,本发明化合物通常具有产生支气管扩张的效应。因此,在本发明方法的另一方面中,本发明涉及在哺乳动物中产生支气管扩张的方法,所述方法包含对哺乳动物投与支气管扩张产生量的式I化合物。一般来说,产生支气管扩张的剂量是在约10μg/天至约500μg/天范围内。
当用作治疗药剂时,本发明化合物视情况与其它治疗药剂组合投与。具体来说,通过将本发明化合物与类固醇消炎药组合投与,仅使用两种治疗药剂即可达成三联疗法(即β2肾上腺素能受体激动剂活性、毒蕈碱受体拮抗剂活性和消炎活性)。由于含有两种治疗药剂的医药组合物(和组合)通常比含有三种治疗药剂的组合物更易于调配和/或投与,因此这些二组份组合物与含有三种治疗药剂的组合物相比具有显着优势。因此,在特定实施例中,本发明医药组合物、组合和方法另外包含类固醇消炎药。
由于本发明化合物同时具有β2肾上腺素能激动剂活性和毒蕈碱受体拮抗剂活性,因此这些化合物也可用作研究工具来探查或研究具有β2肾上腺素能受体或毒蕈碱受体的生物系统或试样。此外,这些化合物可用于筛选分析以发现(例如)同时具有β2肾上腺素能激动剂活性和毒蕈碱受体拮抗剂活性的新化合物。这些生物系统或试样可包含β2肾上腺素能受体和/或毒蕈碱受体。具有β2肾上腺素能和/或毒蕈碱受体的任何适当生物系统或试样都可用于这些可在活体外或活体内进行的研究中。适合这些研究的代表性生物系统或试样包括(但不限于)细胞、细胞提取物、质膜、组织试样、哺乳动物(例如小鼠、大鼠、天竺鼠、兔子、狗、猪等)等。
当作为研究工具使用时,通常使包含β2肾上腺素能受体和/或毒蕈碱受体的生物系统或试样与β2肾上腺素能受体激动量或毒蕈碱受体拮抗量的本发明化合物接触。然后在哺乳动物中使用习用程序和设备测定或测量化合物对生物系统或试样产生的效应,例如测量放射性配体结合分析中的结合或功能性分析中的配体介导改变,或在支气管保护分析中测定由化合物所产生的支气管保护量。在功能性分析中代表性配体介导改变包括胞内环腺苷酸(cAMP)中的配体介导改变;腺苷基环化酶(其合成cAMP)活性中的配体介导改变;在通过[35S]GTP S对GDP的受体催化交换将鸟苷5′-O-(硫基)三磷酸盐([35S]GTP S)纳入经分离膜中的配体介导改变;胞内游离钙离子中的配体介导改变(例如用得自分子装置(Molecular Devices)公司的荧光连结成像板读取器或 来测量)等。在本文所列功能性分析或类似性质的分析中,预期本发明化合物可激动β2肾上腺素能受体或使其活化并且拮抗毒蕈碱受体或降低其活化。本发明化合物通常可以约0.1毫微摩尔至约100毫微摩尔范围的浓度用于这些研究中。
来测量)等。在本文所列功能性分析或类似性质的分析中,预期本发明化合物可激动β2肾上腺素能受体或使其活化并且拮抗毒蕈碱受体或降低其活化。本发明化合物通常可以约0.1毫微摩尔至约100毫微摩尔范围的浓度用于这些研究中。
此外,本发明化合物可用作评估其它化学化合物的研究工具。在本发明此方面中,式I化合物在分析中用作标准品以使得可比较使用测试化合物与式I化合物所获得的结果。例如,比较测试化合物或一组测试化合物的β2肾上腺素能受体和/或毒蕈碱受体结合数据(如通过(例如)活体外放射性配体置换分析所测定)与式I化合物的β2肾上腺素能受体和/或毒蕈碱受体结合数据以鉴定具有期望结合的那些测试化合物,即结合程度大致等于或优于式I化合物的测试化合物(如果存在)。或者,例如可在哺乳动物的支气管保护分析中测定测试化合物和式I化合物的支气管保护效应,并比较此数据以鉴定可提供大致相当或更佳支气管保护效应的测试化合物。作为个别实施例,本发明此方面包括(i)比较数据的产生(使用适当分析)和(ii)测试数据的分析以鉴定目标测试化合物。
可使用熟习此项技术者所习知的各种活体外和活体内分析来证实本发明化合物的性质和效用。例如,代表性分析更详细地描述于下述实例中。
实例
提供下述制备和实例来说明本发明具体实施例和方面。但是,除非明确地指出,否则具体实施例和方面的说明并不意欲以任何方式限制本发明范围。
除非另外说明,否则下述实例中使用的所有试剂、起始物质和溶剂都是购自市场供应商(例如奥德里奇(Aldrich)、弗路卡(Fluka)、西格玛等)并且都是未经进一步纯化就使用。
在下述实例中,HPLC分析通常是使用由安捷伦(Agilent)提供、具有Zorbax Bonus型RP 2.1 x 50毫米管柱(C14管柱)并且粒径为3.5微米的安捷伦(帕洛阿尔托(PaloAlto),CA))系列1100仪器来进行。通过214nm下的UV吸光率来实施检测。流动相「A」为2%乙腈、97.9%水和0.1%三氟乙酸(v/v/v);并且流动相「B」为89.9%乙腈、10%水和0.1%三氟乙酸(v/v/v)。HPLC(10-70)数据是用10%至70%流动相B梯度以0.5mL/分钟的流速经6分钟获得;HPLC(5-35)数据是用5%至35%流动相B梯度以0.5mL/分钟的流速经5分钟获得;且HPLC(10-90)数据是用10%至90%流动相B梯度以0.5mL/分钟的流速经5分钟获得。
液相色谱质谱(LCMS)数据通常是以应用生物系统(福斯特市(Foster City),CA)型API-150EX仪器来获得。LCMS10-90数据是以10%至90%流动相B梯度经5分钟获得。
小规模纯化通常是使用得自应用生物系统(Applied Biosystems)的API150EX制备工作站系统来进行。流动相「A」为含有0.05%三氟乙酸(v/v)的水;并且流动相「B」为含有0.05%三氟乙酸(v/v)的乙腈。对于小型试样(约3至50mg的回收样品尺寸)通常使用以下条件:20mL/min流速;15min梯度和粒径为5微米的20mm x 50mm普锐斯(Prism)RP管柱(塞默飞世尔-拱石公司(Thermo Hypersil-Keystone),白露枫丹(Bellefonte),PA)。对于较大试样(即粗试样大于约100mg)通常使用以下条件:60mL/min流速;30min梯度和粒径为10微米的41.4mm x 250mm麦克若陶伯(Microsorb)BDS管柱(瓦瑞安(Varian),帕洛阿尔托,CA)。
实例1
联苯-2-基氨基甲酸哌啶-4-基酯
在70℃下将2-异氰酸联苯基酯(97.5g,521mmol)和4-羟基-1-苯甲基哌啶(105g,549mmol)(两者都可购自奥德里奇,密尔沃基(Milwaukee),WI)一起加热12小时,在此期间通过LCMS监测联苯-2-基氨基甲酸1-苯甲基哌啶-4-基酯的形成。然后,使反应混合物冷却至50℃并添加乙醇(1L),之后慢慢添加6M盐酸(191mL)。然后,使反应混合物冷却至环境温度并添加甲酸铵(98.5g,1.56mol),并且使氮气激烈鼓泡经过溶液20min。然后添加钯(10重量%(以干重计)存于活性碳上)(20g)。在40℃下将反应混合物加热12小时,然后经硅藻土垫过滤。然后在减压下移除溶剂,并将1M盐酸(40mL)添加至粗制残留物中。然后添加氢氧化钠(10N)以调整pH至12。用乙酸乙酯(2 x 150mL)萃取水层并干燥(硫酸镁),之后在减压下移除溶剂而获得联苯-2-基氨基甲酸哌啶-4-基酯(155g,100%)。HPLC(10-70)Rt=2.52;MS m/z:[M+H+]计算值C18H20N2O2297.15;实验值297.3。
实例2
3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酸
向联苯-2-基氨基甲酸哌啶-4-基酯(50g,67.6mmol)存于二氯甲烷(500mL)中的溶液中添加丙烯酸(15.05mL,100mmol)。在50℃及回流下将所形成混合物加热18小时,然后移除溶剂。添加甲醇(600mL)并在75℃下将此混合物加热2小时,之后冷却至室温以形成浓稠浆液。通过过滤收集固体,用甲醇(50mL)洗涤并风干而获得呈白色粉末形式的3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酸(61g,96%纯度)。
实例3
N-{5-[(R)-2-氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-羟苯基}-甲酰胺乙酸盐
步骤A-N-{5-[(R)-2-苄氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-苯甲氧基苯基}甲酰胺
向500mL三颈圆底烧瓶中添加N-{2-苯甲氧基-5-[(R)-2-溴-1-(第三-丁基二甲基硅烷氧基)乙基]苯基}甲酰胺(100g,215mmol)和N-甲基-2-吡咯烷酮(300mL)。添加苄胺(69.4mL,648mol)并用氮冲洗反应混合物。然后将反应混合物加热至90℃并搅拌约8小时。然后使反应混合物冷却至室温并添加水(1.5L)和乙酸乙酯(1.5L)。分离各层并用水(500mL)、水与饱和盐水的1:1混合物(总计500mL)洗涤有机层,之后再次用水(500mL)洗涤。然后经无水硫酸镁干燥有机层,过滤并在减压下浓缩以提供呈橙褐色浓稠油形式的粗制N-{5-[(R)-2-苄氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-苯甲氧基苯基}甲酰胺(100g,90%产率,75-80%纯度)。
步骤B-N-{5-[(R)-2-氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-羟苯基}-甲酰胺乙酸盐
使粗制N-{5-[(R)-2-苄氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-苯甲氧基苯基}甲酰胺(100g,194mmol)溶于甲醇(1L)和乙酸(25mL,291mmol)中。用干燥氮吹扫所形成混合物,然后添加氢氧化钯/碳(20g,20重量%,约50%水)。使氢鼓泡经过反应混合物,同时在室温下搅拌约10小时。然后用干燥氮吹扫混合物,并经硅藻土过滤混合物。在回转式蒸发器上浓缩滤液并将乙酸乙酯(600mL)添加至残留物中。将此混合物搅拌约2小时,此时已形成浓稠黄色浆液。过滤浆液并使沉淀物风干以提供呈黄白色固体形式的N-{5-[(R)-2-氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-羟苯基}甲酰胺乙酸盐(48g,98%纯度)。LCMS(10-70)Rt=3.62;[M+H+]实验值311.3。
实例4
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
步骤A-2,5-二甲基-4-硝基苯甲酸甲酯
在0℃和干燥氮下向2,5-二甲基-4-硝基苯甲酸(480mg,2.4mmol)存于无水甲醇(8.2mL)中的经搅拌溶液中添加亚硫酰氯(0.538mL,7.38mmol)。使所形成混合物升温至室温并搅拌约7小时。另外添加亚硫酰氯(0.300mL)并在室温下持续搅拌过夜。于减压下移除溶剂并使残留物溶于乙酸乙酯中。用饱和碳酸氢钠水溶液洗涤此溶液,经无水硫酸钠干燥并在减压下浓缩以提供呈淡黄色固体形式的2,5-二甲基-4-硝基苯甲酸甲酯(578mg)。HPLC(10-70)Rt=4.61;1H NMR(300MHz,CDCl3)δ 2.57(3H,s),2.61(3H,s),3.94(3H,s),7.82(1H,s),7.87(1H,s)。
步骤B-4-氨基-2,5-二甲基苯甲酸甲酯
在0℃下向2,5-二甲基-4-硝基苯甲酸甲酯(523mg,2.5mmol)存于甲醇和水的9:1混合物(总计25mL)中的经搅拌溶液中添加氯化铵(401mg,7.5mmol)。逐份添加锌(1.63g,25mmol)并在室温下将所形成混合物搅拌过夜。然后经硅藻土过滤反应混合物并用甲醇洗涤硅藻土垫。使滤液在减压下浓缩并且使所得残留物溶于乙酸乙酯中。用饱和碳酸氢钠水溶液洗涤此溶液,以无水硫酸钠干燥并在减压下浓缩以提供呈黄色油形式的4-氨基-2,5-二甲基苯甲酸甲酯(450mg)。1H NMR(300MHz,CDCl3)δ 2.14(3H,s),2.53(3H,s),3.83(3H,s),3.85(2H,br s),6.48(1H,s),7.72(1H,s)。
步骤C-4-{3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酰基氨基}-2,5-二甲基苯甲酸甲酯
向3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酸(670mg,1.82mmol)和4-氨基-2,5-二甲基苯甲酸甲酯(390mg,2.18mmol)存于二氯甲烷(3.6mL)和二异丙基乙胺(0.413mL)中的经搅拌溶液中添加六氟磷酸O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲阳离子(O-(7-azabenzotriazol-l-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate)(HATU)(829mg,2.18mmol)。在室温下将所形成混合物搅拌过夜。然后用饱和碳酸氢钠水溶液洗涤混合物,以无水硫酸钠干燥,过滤并在减压下浓缩。通过硅胶色谱纯化残留物,其中用含有3%至5%甲醇的二氯甲烷洗脱以提供4-{3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酰基氨基}-2,5-二甲基苯甲酸甲酯(568mg,59%产率)。LCMS(10-70)Rt=4.55;[M+H+]实验值530.4。
步骤D-联苯-2-基氨基甲酸1-[2-(4-羟甲基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
在0℃下向1M氢化锂铝存于THF中的经搅拌溶液(1.52mL,1.52mmol)中添加4-{3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酰基氨基}-2,5-二甲基苯甲酸甲酯(400mg,0.76mmol)。在0℃下将所形成混合物搅拌30分钟,然后添加1M氢氧化钠水溶液(5mL)与水(5mL)的1:1混合物,并持续搅拌2小时。添加二氯甲烷并分离有机层,以硫酸钠干燥并在减压下移除溶剂。通过硅胶色谱纯化残留物,其中用含有5%甲醇的二氯甲烷洗脱以提供联苯-2-基氨基甲酸1-[2-(4-羟甲基-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯。LCMS(10-70)Rt=3.94;[M+H+]实验值502.5。
步骤E-联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
在0℃下向联苯-2-基氨基甲酸1-[2-(4-羟甲基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(151mg,0.3mmol)存于二氯甲烷(3mL)中的溶液中添加二甲亚砜(128μL,1.8mmol)和二异丙基乙胺(157μL,0.9mmol)。15分钟后,添加三氧化硫吡啶复合物(143mg,0.9mmol),并在0℃下持续搅拌1小时。添加水以终止反应,并分离各层。以无水硫酸钠干燥有机层,过滤并在减压下移除溶剂而获得联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(150mg,100%产率),其不经进一步纯化就可使用。[M+H+]实验值500.4。
步骤F-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
在室温下将联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(150mg,0.30mmol)和N-{5-[(R)-2-氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-羟苯基}甲酰胺(112mg,0.36mmol)存于二氯甲烷与甲醇的1:1混合物(总计3.0mL)中的溶液搅拌30分钟。添加三乙酰氧基硼氢化钠(191mg,0.9mmol)并在室温下将所形成混合物搅拌过夜。添加乙酸以终止反应,并在减压下浓缩混合物而获得联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯,其不经进一步纯化就可使用。LCMS(10-70)Rt=4.55;[M+H+]实验值794.6。
步骤G-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
向联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯(238mg,0.30mmol)存于二氯甲烷(3.0mL)中的悬浮液中添加三乙胺三氢氟酸盐(147μL,0.90mmol)。在室温下将此混合物搅拌过夜,然后在减压下浓缩混合物。然后通过制备型RP-HPLC(梯度:存于含0.05% TFA的水中的2-50%乙腈)纯化残余物。收集适当流份并将其合并并且冻干而获得呈二(三氟乙酸盐)型的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(50mg,97%纯度)。LPLC(2-90)Rt=2.76;[M+H+]实验值680.8。
实例5
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
步骤A-二苯甲基-(4-碘-2,5-二甲基苯基)胺
向装有顶置式搅拌器、温度控制和加料漏斗的2升圆底烧瓶中添加4-碘-2,5-二甲基苯胺(100.0g,0.405mol)(得自波谱集团有限公司,米尔伯里,OH)。添加乙醇(1L)和固体碳酸钾(160g,1.159mol),然后以一份添加纯溴化苄(140mL,1.179mol)。在30℃下将所形成混合物搅拌约18小时,此时HPLC显示大于98%的转化率。然后使混合物冷却至室温并添加己烷(1L)。将此混合物搅拌15分钟,然后经滤纸过滤以移除固体,并用己烷(200mL)洗涤滤饼。使用回转式蒸发器使滤液的体积降低至约500mL并添加浓盐酸(30mL)。然后,将使用回转式蒸发器移除残留溶剂。向所得残留物中添加己烷(500mL),并将此混合物搅拌约30分钟,此时自由流动性浆液已形成。过滤浆液并用己烷(200mL)洗涤滤饼并干燥以提供呈绿色固体形式的二苯甲基-(4-碘-2,5-二甲基苯基)胺盐酸盐(115g,62%产率,97.5%纯度)。
将二苯甲基-(4-碘-2,5-二甲基苯基)胺盐酸盐转移至3L烧瓶中,并添加甲苯(1L)和1M氢氧化钠水溶液(1L)。将所形成混合物搅拌1小时,然后分离各层。用稀盐水(500mL)洗涤有机层并通过回转式蒸发器移除溶剂以提供呈半固体浓稠油形式的二苯甲基-(4-碘-2,5-二甲基苯基)氨(80g)。(或者,在此步骤中可使用二氯甲烷来替代甲苯)。1H NMR(300MHz,DMSO-d6)δ 2.05(3H,s),2.19(3H,s),3.90(4H,s),6.91(1H,s),7.05-7.20(10H,m),7.42(1H,s);MS[M+H+]实验值428。
步骤B-4-二苯甲基氨基-2,5-二甲基苯甲醛盐酸盐
向装有顶置式搅拌器、温度控制和加料漏斗的1升3颈圆底烧瓶中添加二苯甲基-(4-碘-2,5-二甲基苯基)胺(15g,35mmol)。添加甲苯(300mL),并将所形成混合物搅拌约15分钟。用干燥氮吹扫反应烧瓶,并冷却至约-20℃,并且通过加料漏斗逐滴添加存于己烷中的1.6M正-丁基锂(33mL,53mmol)。在添加期间,使反应混合物的内部温度保持低于-10℃。当添加完成时,在约-15℃下将所形成混合物搅拌15分钟。然后逐滴添加N,N-二甲基甲酰胺(10mL,129mmol),同时保持内部反应温度低于0℃。然后在-20℃至0℃下将所形成混合物搅拌约1小时。然后经5分钟添加1M盐酸水溶液(200mL),并将所形成混合物搅拌15分钟。然后分离各层并用稀盐水(100mL)洗涤有机层。然后以无水硫酸钠干燥有机层,将其过滤并在减压下移除溶剂以提供呈浓稠油形式的4-二苯甲基氨基-2,5-二甲基苯甲醛盐酸盐(11.5g,90%产率,95%纯度),其在静置后固化。产物含有约3-5%脱碘副产物。1H NMR(300MHz,CDCl3)δ 2.42(3H,s),2.50(3H,s),4.25(4H,s),6.82(1H,s),7.10-7.30(10H,m),7.62(1H,s),10.15(1H,s);MS[M+H+]实验值330.3。
步骤C-4-[1,3]二氧戊环-2-基-2,5-二甲基苯胺
向500mL圆底烧瓶中添加4-二苯甲基氨基-2,5-二甲基苯甲醛盐酸盐(11.5g,31.4mmol)和甲苯(150mL),并搅拌所形成混合物直至盐完全溶解。然后,用干燥氮将反应烧瓶吹扫5分钟。添加乙二醇(5.25mL,94.2mmol)和对-甲苯磺酸(760mg,6.2mmol),并在60℃至80℃下将所形成混合物加热约20小时。然后在40℃下于回转式蒸发器上慢慢移除溶剂(经约40分钟)。将甲苯(100mL)添加至残留物中,并在40℃下于回转式蒸发器上再次慢慢地移除溶剂。使用另一等份甲苯(100mL)重复此过程,并使混合物蒸发至干燥。将乙酸乙酯(150mL)和饱和碳酸氢钠水溶液(100mL)添加至残留物中并分离各层。用盐水(50mL)洗涤有机层,然后以无水硫酸钠干燥,过滤并在减压下浓缩以提供粗制二苯甲基-(4-[1,3]二氧戊环-2-基-2,5-二甲基-苯基)胺(11.4g)。
使粗制二苯甲基-(4-[1,3]二氧戊环-2-基-2,5-二甲基-苯基)胺溶于乙醇与水的2:1混合物(总计150mL)中,并用干燥氮将所形成混合物吹扫5分钟。添加钯/碳(2.3g,10wt%,含有约50%水)和固态碳酸氢钠(1.0g),并在约1atm的氢气和25℃至30℃下使所形成混合物氢化约8小时。然后,经硅藻土过滤混合物,并在回转式蒸发器上浓缩滤液以提供呈浓稠油形式的粗制4-[1,3]二氧戊环-2-基-2,5-二甲基苯胺(5.6g,92%产率)。1H NMR(300MHz,DMSO-d6)δ 2.05(3H,s),2.22(3H,s),3.7-3.9(4H,m),3.95(4H,s),5.59(1H,s),6.72(1H,s),7.0-7.25(11H,m)。
步骤D-N-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基)丙烯酰胺
向500mL圆底烧瓶中添加粗制4-[1,3]二氧戊环-2-基-2,5-二甲基苯胺(5.6g,29mmol)、二氯甲烷(100mL)和二异丙基乙胺(7.6mL,43.5mmol)。在室温下搅拌所形成混合物直至成份溶解,然后使混合物冷却至0℃。然后经5分钟逐滴添加丙烯酰氯(2.35mL,29mmol)。在0℃至5℃下将反应混合物搅拌1小时,之后添加水(50mL)并持续搅拌约30分钟,此时微细固体已形成。过滤混合物以收集固体。然后分离滤液各层,并在减压下使有机层浓缩至干燥。将二氯甲烷(50mL)添加至残留物中并将此混合物搅拌至形成自由流动性浆液。过滤浆液(使用收集上文微细固体所用的相同漏斗),并用二氯甲烷(10mL)洗涤滤饼并干燥以提供呈白色至灰白色固体形式的N-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基)丙烯酰胺(3.1g,97%纯度)。
然后,使得自上文的滤液蒸发至干燥,并将甲醇(10mL)添加至残留物中。将此混合物搅拌15分钟之后通过过滤收集沉淀物,用甲醇(5mL)洗涤并干燥而获得第二批N-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基)丙烯酰胺(0.8g,95%纯度)。1H NMR(300MHz,CD3OD)δ 2.10(3H,s),2.23(3H,s),3.85-4.10(4H,m),5.60-6.40(3H,m),5.59(1H,s),7.18(1H,s),7.23(1H,s)。
步骤E-联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯盐酸盐
向50mL圆底烧瓶中添加联苯-2-基氨基甲酸哌啶-4-基酯(1.2g,4.04mmol)和N-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基)丙烯酰胺(1.0g,4.04mmol)。添加乙醇(10mL)和二氯甲烷(10mL)以形成浆液。在45℃至50℃下将反应混合物加热约18小时,然后冷却至室温。添加1M盐酸水溶液(10mL),并将所形成混合物剧烈搅拌约3小时。添加二氯甲烷(10mL),并将所形成混合物搅拌约5分钟。然后分离各层,并以无水硫酸钠干燥有机层,过滤并在回转式蒸发器上浓缩以提供粗制联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯盐酸盐(1.9g)。1H NMR(300MHz,DMSO-d6)δ 1.2-1.4(2H,m),1.58-1.75(2H,m),2.0-2.17(2H,m),2.19(3H,s),2.38(3H,s),2.41-2.50(4H,m),2.5-2.75(2H,m),4.31-4.42(1H,m),7.10-7.35(9H,m),7.55(1H,s),7.75(1H,s),8.59(1H,s),9.82(1H,s),9.98(1H,s);MS[M+H+]实验值500.2。
步骤F-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯
向2L三颈圆底烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯盐酸盐(38g,70mmol)和N-{5-[(R)-2-氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-羟苯基}甲酰胺乙酸盐(33.6g,91mmol)。添加二氯甲烷(500mL)和甲醇(500mL),并在室温和干燥氮下将所形成混合物搅拌约3小时。然后,使反应混合物冷却至0℃至5℃,并经10分钟逐份添加固体三乙酰氧基硼氢化钠(44.5g,381mmol)。使反应混合物经约2小时自0℃慢慢升温至室温,然后冷却至0℃。添加饱和碳酸氢钠水溶液(500mL)和二氯甲烷(500mL)。将此混合物充分搅拌之后分离各层。用盐水(500mL)洗涤有机层,以无水硫酸钠将其干燥,过滤并在减压下浓缩以获得呈黄色固体形式的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟基苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯(55g,86%纯度)。
使粗产物(30g)溶于含有2%甲醇的二氯甲烷(总计150mL)中,并将其装填至已经用含有2%甲醇和0.5%氢氧化铵的二氯甲烷填装并平衡的硅胶管柱(300g)上。使用以下洗脱液自管柱洗脱产物:含有2%甲醇和0.5%氢氧化铵的二氯甲烷(1L);含有4%甲醇和0.5%氢氧化铵的二氯甲烷(1L);和含有5%甲醇和0.5%氢氧化铵的二氯甲烷(约3L)。收集流份(200mL)并合并那些纯度大于90%的流份,并且在减压下将其浓缩以提供呈黄色固体形式的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯(21.6g,96.5%纯度)。MS[M+H+]实验值794.6。
步骤G-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯氢氟酸盐
向1L圆底烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(21.5g,27.1mmol)和二氯甲烷(200mL)。在室温下搅拌所形成混合物直至成份溶解,然后添加三乙胺三氢氟酸盐(8.85mL,54.2mmol),并在25℃下将所形成混合物搅拌约48小时。在回转式蒸发器上移除溶剂以提供浓稠糊剂。将二氯甲烷(100mL)和乙酸乙酯(200mL)添加至糊剂中,并将所形成混合物搅拌30分钟。在干燥氮下慢慢过滤所形成的浆液,并用二氯甲烷与乙酸乙酯的1:2混合物(总计100mL)洗涤滤饼,在氮气下将其干燥2小时,之后在真空下干燥过夜以提供呈氢氟酸盐型的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(25g,96.9%纯度),其为硬质粘土状固体。MS[M+H+]实验值680.8。
步骤H-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯
在6英时反相管柱(麦克若陶伯固相)上,使用存于水(含有1%三氟乙酸)中的乙腈的10%至50%混合物作为流动相,以三等量批次纯化联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯氢氟酸盐(25g)。合并纯度大于99%的流份,然后用一体积水稀释。使所形成混合物冷却至0℃并添加固态碳酸氢钠直至混合物的pH为约7.5至8.0。在约5分钟内,形成成白色浆液。将浆液搅拌30分钟后过滤。用水(500mL)洗涤滤饼,将其风干约4小时,之后在真空中干燥过夜以提供呈半结晶游离碱型的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯(12g,99+%纯度)。
实例6
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
步骤A-联苯-2-基氨基甲酸1-[2-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
向500mL圆底烧瓶中添加联苯-2-基氨基甲酸哌啶-4-基酯(17.0g,58mmol)和N-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基)丙烯酰胺(13.1g,52.9mmol)。添加乙醇(150mL)和二氯甲烷(150mL)以形成浆液。在50℃至55℃下将反应混合物加热约24小时,然后冷却至室温。在回转式蒸发器上移除大部分溶剂而获得浓稠浆液。添加乙醇(试剂级)以形成约200mL的总体积,并将所形成混合物加热至80℃,然后慢慢冷却至室温。过滤所形成的浓稠白色浆液,用乙醇(20mL)洗涤并在真空中干燥以提供呈白色固体形式的联苯-2-基氨基甲酸1-[2-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(23.8g,约98%纯度)。
步骤B-联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯
向500mL圆底烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-[1,3]二氧戊环-2-基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(15g,27.6mmol)和乙腈(150mL)以形成浆液。添加2M盐酸水溶液(75mL)并在30℃下将所形成混合物搅拌1小时。然后使混合物冷却至室温,并添加乙酸乙酯(150mL)。添加2M氢氧化钠水溶液(75mL),检查pH值之后另外添加2M氢氧化钠直至溶液的pH到达9至10范围内。分离各层并用稀盐水(75mL;1:1盐水/水)洗涤有机层,以无水硫酸钠干燥,并在回转式蒸发器上移除溶剂而获得联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯(12.5g,约98%纯度)。如果需要,可通过以下步骤来提高此中间体的纯度:用乙醇(3体积乙醇)形成浆液,将浆液加热至80℃,之后慢慢冷却至室温,并通过过滤来分离。
步骤C-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙基亚氨基]甲基}-2,5-二甲基-苯基氨甲酰基)乙基]哌啶-4-基酯
向250mL圆底烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(7.1g,14.2mmol)和N-{5-[(R)-2-氨基-1-(第三-丁基二甲基硅烷氧基)乙基]-2-羟苯基}甲酰胺乙酸盐(5.8g,15.6mmol)。添加甲醇(100mL)以形成浆液,并在45℃至50℃及氮气下将此混合物搅拌1小时。然后使混合物冷却至室温并添加甲苯(50mL),并且在35℃至45℃范围的温度下于回转式蒸发器上移除溶剂。将甲苯(50mL)添加至残留物中,并移除溶剂以提供呈黄-橙色固体形式的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙基亚氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(12g)。
步骤D-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯
向氢化作用烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙基亚氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(4.6g)和2-甲基四氢呋喃(50mL)。搅拌所形成混合物直至固体溶解(约5分钟),然后用氮吹扫混合物。添加铂/碳(920mg,5wt.%,活性碳载体)并在50psi下使混合物氢化(帕尔(Parr)振荡器)6小时。然后经硅藻土过滤混合物,并用2-甲基四氢呋喃(10mL)洗涤硅藻土。向滤液中添加经硫丙基改质的硅胶(溶液重量的20%,硅环(Silicycle)),并在25℃至30℃下将此混合物搅拌3小时。然后,经硅藻土过滤混合物并浓缩以移除溶剂。使残余物溶于甲醇(5mL/克残余物)中之后将所得溶液缓慢添加至经剧烈搅拌的饱和碳酸氢钠水溶液与水的1:1混合物中(40mL/克残余物)。将所得灰白色浆液搅拌20分钟后过滤。用水(20体积)洗涤滤饼,将其风干3小时,之后在真空中于室温下干燥过夜以提供联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(80%回收率,约96%纯度)。
步骤E-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯L-酒石酸盐
向200mL圆底烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(第三-丁基二甲基硅烷氧基)-2-(3-甲酰氨基-4-羟苯基)乙氨基]-甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(3.8g,4.8mmol)和2-甲基四氢呋喃(40mL)。在室温下搅拌所形成混合物直至成份溶解(约15分钟),然后添加三乙胺三氢氟酸盐(0.94mL,5.76mmol),并在25℃下将所形成混合物搅拌约24小时。向此混合物中添加饱和碳酸氢钠水溶液与水的1:1混合物(40mL)和2-甲基四氢呋喃,并搅拌所形成混合物直至固体溶解(溶液pH为约8)。分离各层,并用盐水(30mL)洗涤有机层,以无水硫酸钠干燥,过滤并在回转式蒸发器上浓缩。使残留物溶于2-甲基四氢呋喃(50mL)中,并添加固体L-酒石酸(650mg)。在25℃至30℃下将所形成混合物搅拌18小时,然后经滤纸过滤。用2-甲基四氢呋喃(10mL)、异丙醇(10mL)洗涤滤饼,并将其立即置于真空下以提供联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯L-酒石酸盐(3.7g,>97%纯度)。
步骤F-联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯
向250mL圆底烧瓶中添加联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯L-酒石酸盐(3.5g)和甲醇(35mL),并将所形成混合物搅拌15分钟。经5分钟添加饱和碳酸氢钠水溶液与水的1:1混合物(70mL),并持续搅拌2小时。过滤所形成的灰白色浆液并用水(20mL)洗涤滤饼,风干2小时之后在真空中干燥过夜以提供呈半结晶游离碱型的联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯(2.3g)。
在环境温度下,使用JEOL ECX-400NMR波谱仪获得联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯试样(22.2mg,存于约0.75mL DMSO-d6中)的1H和13C NMR波谱:
1H NMR(400MHz,DMSO-d6),主要异构体δ 9.64(br,1H),9.54(br s,1H),9.43(s,1H),8.67(s,1H),8.26(s,1H),8.03(d,J=1.9,1H),7.25-7.45(m,9H),~7.3(nd,1H),7.07(s,1H),6.88(dd,J=8.2,1.9,1H),6.79(d,J=8.2,1H),5.15(br,1H),4.53(dd,J=7.3,4.7,1H),4.47(m,1H),~3.65和~3.60(AB对,2H),2.68(br m,2H),~2.59(nd,4H),2.44(br t,J=6.5,2H),2.20(s,3H),~2.17(br m,2H),2.14(s,3H),1.73(br,2H),1.44(br q,J=~9.0,2H)。
1H NMR(400MHz,DMSO-d6),次要异构体δ 9.64(br,1H),9.43(s,1H),9.26(br d,J=~7.0,1H),8.67(s,1H),8.50(br d,J=~7.0,1H),7.25-7.45(m,9H),~7.3(nd,1H),7.07(s,1H),~7.07(nd,1H),6.95(dd,J=8.3,1.8,1H),6.83(d,J=8.3,1H),5.15(br,1H),4.47(m,1H),~3.65和~3.60(AB对,2H),2.68(br m,2H),~2.59(nd,2H),2.44(br t,J=6.5,2H),2.20(s,3H),~2.17(br m,2H),2.14(s,3H),1.73(br,2H),1.44(br q,J=~9.0,2H)。
13C NMR(100MHz,DMSO-d6),主要异构体δ 170.0,159.9,153.9,145.5,139.3,137.6,135.2,135.0,134.8,133.4,133.4,130.2,130.2,128.6,128.2,127.8,127.4,127.2,127.0,126.1,125.7,125.6,121.7,118.6,114.5,71.4,70.0,57.4,53.9,50.3,50.1,33.7,30.7,18.2,17.5。
13C NMR(100MHz,DMSO-d6),次要异构体δ 170.0,163.4,153.9,147.8,139.3,137,6,135.7,135.2,135.0,134.8,133.4,133.4,130.2,130.2,128.6,128.2,127.8,127.4,127.2,127.0,126.1,125.7,123.0,119.6,115.6,71.0,70.0,57.3,53.9,50.3,50.1,33.7,30.7,18.2,17.5。
1H和13C NMR波谱显示主要异构体(约82mol%)和次要异构体(约18mol%)的存在,据信其都是得自环绕-NH-C(O)H键的受阻旋转的旋转异构体。据信在主要异构体中苯基与羰基氧在同侧,而在次要异构体中在对侧。
实例7
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯的型II的种晶
使半结晶型联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯(500mg)溶于甲醇(50mL)中,并添加水直至达成浊点。在25℃下将所形成混合物搅拌3小时,并通过过滤来分离所形成结晶物质以提供结晶联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(420mg)。据测定,此结晶游离碱的差示扫描量热法(DSC)的图形在吸热流中于约142℃至约150℃处显示峰值;并且粉末x-射线衍射(PXRD)图尤其在2θ值为约20.7±0.3、21.6±0.3、22.5±0.3和23.2±0.3处具有显着衍射峰。此结晶游离碱型称为型II。关于此化合物的型II和其它结晶游离碱型的其它信息揭示于和本案在同一日期提出申请的共同让渡的美国专利申请案第____号(代理档案号P-222-US1)和在2006年4月25日提出申请的美国临时申请案第60/794,709号中;其揭示内容是全文以引用方式并入本文中。
实例8
联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯的型II的结晶
向装有顶置式搅拌器、温度控制和加料漏斗的3L三颈圆底烧瓶中添加半结晶型联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(14g)和甲醇(1.4L)。以一份添加水(500mL),然后另外慢慢添加水(200mL)直至达成浊点。添加联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯的型II的种晶(50mg),并在25℃下将所形成混合物搅拌3小时,此时已形成自由流动性浆液。经15分钟添加水(300mL),并在25℃下将所形成混合物搅拌过夜。然后过滤混合物并用水(100mL)洗涤滤饼,风干约2小时之后在真空和室温下干燥48小时以提供结晶联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(12.5g,99.6%纯度)。此结晶盐经测定为型II。
实例9
联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]-哌啶-4-基酯
步骤A-4-碘-2,5-二甲基苯胺
向2,5-二甲基苯胺(20g,165mmol)存于二氯甲烷与甲醇的1:1混合物(400mL)中的溶液中添加碳酸氢钠(20.8g,250mmol)和四甲基二氯碘酸(I)铵(44.7g,165mmol)。在室温下将所形成混合物搅拌1小时,然后添加水(500mL)。移除有机层,并用5%硫代硫酸钠水溶液(500mL)和盐水(500mL)洗涤。然后以无水硫酸镁干燥有机层,过滤并在真空下浓缩以提供4-碘-2,5-二甲基苯胺(39.6g,98%产率)。不经进一步纯化就使用此产物。
步骤B-N-(4-碘-2,5-二甲基苯基)丙烯酰胺
向4-碘-2,5-二甲基苯胺(37.2g,151mmol)存于二氯甲烷(500mL)中的溶液中添加碳酸氢钠(25.4g,302mmol)。使所形成混合物冷却至0℃,并经25分钟慢慢添加丙烯酰氯(12.3mL,151mmol)。在室温下将所形成混合物搅拌过夜然后过滤。使滤液的体积降低至约100mL并形成沉淀物。过滤沉淀物,将其干燥并用水(1L)洗涤,之后再一次干燥而获得N-(4-碘-2,5-二甲基苯基)丙烯酰胺(42.98g,95%纯度,90%产率)。不经进一步纯化就使用此产物。
步骤C-联苯-2-基氨基甲酸1-[2-(4-碘-2,5-二甲基苯基氨甲酰基)-乙基]哌啶-4-基酯
向N-(4-碘-2,5-二甲基苯基)丙烯酰胺(32.2g,107mmol)存于N,N-二甲基甲酰胺与异丙醇的6:1 v/v混合物(700mL)中的溶液中添加联苯-2-基氨基甲酸哌啶-4-基酯(36.3g,123mmol)。在50℃下将所形成混合物加热24小时,之后在80℃下加热24小时。然后,使反应混合物冷却至室温并在真空下浓缩。使残留物溶于二氯甲烷(1L)中,并用1N盐酸水溶液(500mL)、水(500mL)、盐水(500mL)和饱和碳酸氢钠水溶液(500mL)洗涤此溶液。然后以无水硫酸镁干燥有机层并过滤。添加乙醇(400mL),并在真空下使所形成混合物浓缩至体积为约400mL,此时沉淀物已形成。过滤沉淀物并干燥而获得联苯-2-基氨基甲酸1-[2-(4-碘-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(59.6g,84%纯度,79%产率)。m/z:[M+H+]对C29H32IN3O3的计算值598.49;实验值598.5。
步骤D-4-{3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酰基氨基}-2,5-二甲基苯甲酸甲酯
向联苯-2-基氨基甲酸1-[2-(4-碘-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(56g,94mmol)存于N,N-二甲基甲酰胺与甲醇的5:1 v/v混合物(600mL)中的溶液中添加二异丙基乙胺(49mL,281mmol)、1,3-双(二苯基膦基)丙烷(3.9g,9.4mmol)和乙酸钯(II)(2.1g,9.4mmol)。用一氧化碳吹扫所形成混合物,然后在70℃至80℃及一氧化碳气氛(气囊压力)下搅拌过夜。在真空下浓缩反应混合物,并使残留物溶于二氯甲烷(500mL)中。用1N盐酸水溶液(500mL)、水(500mL)洗涤此混合物,之后用盐水(500mL)洗涤。然后以无水硫酸镁干燥有机层,将其过滤之后在真空下浓缩。将残留物与乙醇混合(约5:1 v/w乙醇对残留物),且加热混合物直至所有固体物质溶解。使此溶液慢慢冷却至室温,并通过过滤来分离所形成沉淀物而获得4-{3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酰基氨基}-2,5-二甲基苯甲酸甲酯(47.3g,97%纯度,92%产率)。m/z:[M+H+]对C31H35N3O5的计算值530.63;实验值530.4.
步骤E-联苯-2-基氨基甲酸1-[2-(4-羟甲基-2,5-二甲基-苯基氨甲酰基)乙基]哌啶-4-基酯
使4-{3-[4-(联苯-2-基氨甲酰基氧基)哌啶-1-基]丙酰基氨基}-2,5-二甲基苯甲酸甲酯(49.8g,93.9mmol)存于四氢呋喃(200mL)中的溶液冷却至0℃,并逐份(10 x 1.07g)添加氢化锂铝(10.7g,281.7mmol)。将所形成混合物搅拌3小时,然后添加水(10.7mL),之后添加1N氢氧化钠水溶液(10.7mL)和其它水(32.1mL)。将此混合物搅拌过夜之后过滤。在真空下浓缩有机层,并将残留物与乙酸乙酯混合(约5:1 v/w乙酸乙酯对残留物)。加热此混合物直至所有固体物质溶解,然后使溶液冷却至室温。过滤所得沉淀物并干燥而获得联苯-2-基氨基甲酸1-[2-(4-羟甲基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(24.6g,95%纯度,47.5%产率)。不经进一步纯化就使用此物质。m/z:[M+H+]对C30H35N3O4的计算值502.62;实验值502.5。
步骤F-联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯
向联苯-2-基氨基甲酸1-[2-(4-羟甲基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(5.0g,10mmol)存于二氯甲烷(200mL)中的溶液中添加二异丙基乙胺(8.7mL,50mmol)和二甲亚砜(5.6mL,100mmol)。使所形成混合物冷却至0℃,并添加三氧化硫吡啶复合物(8.0g,50mmol)。在0℃下将反应混合物搅拌1小时,然后添加水(300mL)。移除有机层,并用1N盐酸水溶液(300mL)和盐水(300mL)洗涤。然后以无水硫酸镁干燥有机层并过滤。不经进一步纯化就使用所得含有联苯-2-基氨基甲酸1-[2-(4-甲酰基-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯的溶液。m/z:[M+H+]对C30H33N3O4的计算值500.60;实验值500.4。
实例10
自表达人类M1、M2、M3和M4毒蕈碱受体的细胞实施细胞培养和膜制备
使稳定表达经克隆人类hM1、hM2、hM3和hM4毒蕈碱受体亚型的CHO细胞系分别在补加10% FBS和250μg/mL遗传霉素的哈莫斯(Hams)F-12培养基中生长至几乎汇合。使细胞在5% CO2下于37℃培养器中生长,并用存于dPBS中的2mM EDTA提取出来。通过在650xg下离心5分钟来收集细胞,并将细胞沉淀在-80℃下冷冻储存或立即制备膜供使用。对于膜的制备,使细胞沉淀再悬浮于溶胞缓冲液中,并用保利通(Polytron)PT-2100组织破碎器匀浆(凯恩迈特格(Kinematica)AG;20秒x 2次破裂)。在4℃下以40,000xg将粗制膜离心15分钟。然后用再悬浮缓冲液使膜沉淀再悬浮,并用保利通组织破碎器再次匀浆。膜悬浮液的蛋白质浓度是通过劳瑞(Lowry)等人,1951,生物化学期刊(Journal of Biochemistry),193,265中所述方法来测定。在-80℃下等份冷冻储存所有膜或将其立即使用。所制成hM5受体膜的等份是直接购自铂金埃尔默(PerkinElmer)公司(韦尔兹利(Wellesley),MA),并储存在-80℃下直至使用。
实例11
对毒蕈碱受体的放射性配体结合分析
对经克隆毒蕈碱受体的放射性配体结合分析是在96-孔微滴定板中以100μL总分析体积进行。使稳定表达hM1、hM2、hM3、hM4或hM5毒蕈碱亚型的CHO细胞膜在分析缓冲液中稀释至以下具体目标蛋白浓度(μg/孔):10μg hM1、10-15μg hM2、10-20μg hM3、10-20μg hM4、和10-12μg hM5以获得相似信号(cpm)。在分析板添加前,使用保利通组织破碎器对膜实施短暂匀浆(10秒)。测定放射性配体KD值的饱和结合研究是以0.001nM至20nM范围的浓度使用L-[N-甲基-3H]氯化甲基东莨菪碱([3H]-NMS)(TRK666,84.0Ci/mmol,安法玛西亚生物制药公司(Amersham PharmaciaBiotech),白金汉郡(Buckinghamshire),英格兰)来进行。用于测定测试化合物Ki值的置换分析是以1nM使用[3H]-NMS和十一种不同测试化合物浓度来进行。首先使测试化合物以400μM的浓度溶于稀释缓冲液中,然后以稀释缓冲液连续性稀释5x至10pM至100μM的最终浓度。向分析板中添加的顺序和体积如下所述:25μL放射性配体、25μL经稀释测试化合物、和50μL膜。在37℃下将分析板培养60分钟。通过经已用1% BSA预处理的GF/B玻璃纤维滤板(铂金埃尔默公司)快速过滤来终止结合反应。用洗涤缓冲液(10mM HEPES)将滤板冲洗三次以移除未结合的放射活性。然后使板风干,并将50μL微森特(Microscint)-20液态闪烁流体(铂金埃尔默公司)添加至各孔中。然后在铂金埃尔默拓普康(TopCount)液体闪烁计数器(铂金埃尔默公司)上对板实施计数。结合数据是通过非线性回归分析使用格拉夫派得普锐斯(GraphPad Prism)软件包(格拉夫派得(GraphPad)软件公司,圣地亚哥,CA)以单一位点竞争模式来分析。测试化合物的Ki值是自所观测到放射性配体的IC50值和KD值使用程-普鲁萨福(Cheng-Prusoff)方程(程Y(Cheng Y);普鲁萨福WH.(Prusoff WH.)(1973),生化医药(Biochemical Pharmacology),22(23):3099-108)来计算。使Ki值转化成pKi值以测定几何平均和95%置信区间。然后,将这些汇总统计值转化回Ki值以用于数据报告。
在此分析中,较低Ki值表示测试化合物对所测试受体具有较高结合亲和力。发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)对M1、M2、M3、M4和M5毒蕈碱受体亚型的Ki值低于10nM。
实例12
自表达人类β1、β2或β3肾上腺素能受体的细胞实施细胞培养和膜制备
在500μg/mL遗传霉素存在下,使稳定表达克隆人类β1和β2肾上腺素能受体的人类胚肾(HEK-293)细胞系或稳定表达克隆人类β3肾上腺素能受体的中国仓鼠卵巢(CHO)细胞系在具有10% FBS的DMEM或哈莫斯F-12培养基中生长至几乎汇合。用存于PBS中的2mM EDTA将细胞单层提取出来。通过在1,000rpm下离心使细胞沉淀,并将细胞沉淀在-80℃下冷冻储存或立即制备膜供使用。对于表达β1和β2受体的膜的制备,使细胞沉淀再悬浮于溶胞缓冲液(10mM HEPES/HCl,10mM EDTA,在4℃下pH 7.4)中,并使用紧配杜恩斯(Dounce)玻璃匀浆器(30次冲程)在冰上匀浆。对于对蛋白酶更敏感的表达β3受体的膜,在溶胞缓冲液(10mM Tris/HCl,pH 7.4)中匀浆细胞沉淀,其中每50mL缓冲液补加有一片「完全蛋白酶抑制剂混合片剂和2mM EDTA」(罗氏分子生化公司(RocheMolecular Biochemicals),印第安纳波利斯,IN)。使匀浆在20,000xg下离心,并通过上述再悬浮和离心用溶胞缓冲液将所形成沉淀洗涤一次。然后使最终沉淀再悬浮于冰冷结合分析缓冲液(75mM Tris/HCl,pH 7.4,12.5mM MgCl2,1mM EDTA)中。膜悬浮液的蛋白质浓度是通过劳瑞等人,1951,生物化学期刊,193,265;和布莱德伏特(Bradford),分析生物化学(AnalyticalBiochemistry),1976,72,248-54中所述方法来测定。将全部膜以数等份冷冻储存在-80℃下或立即使用。
实例13
对人类β1、β2和β3肾上腺素能受体的放射性配体结合分析
在96-孔微滴定板中以100μL总分析体积使用存于分析缓冲液(75mM Tris/HCl,pH 7.4,在25℃下,12.5mM MgCl2,1mM EDTA,0.2% BSA)中的含有人类β1、β2或β3肾上腺素能受体的10-15μg膜蛋白质来进行结合分析。用于测定放射性配体的Kd值的饱和结合研究是以介于0.01nM与20nM之间的10或11种不同浓度针对β1和β2受体使用[3H]-二氢烯丙洛尔(dihydroalprenolol)(NET-720,100Ci/mmol,铂金埃尔默生命科学公司,波士顿,MA)以及使用[125I]-(-)-碘氰吲哚洛尔(iodocyanopindolol)(NEX-189,220Ci/mmol,铂金埃尔默生命科学公司,波士顿,MA)来进行。用于测定测试化合物Ki值的置换分析是针对介于10pM至10μM之间的10或11种不同浓度的测试化合物使用1nM的[3H]-二氢烯丙洛尔和0.5nM的[125I]-(-)-碘氰吲哚洛尔来进行。非特异性结合是在10μM普萘洛尔(propranolol)存在下测定。在37℃下将分析物培养1小时,然后通过经已在0.3%聚乙烯亚胺中预浸泡的GF/B(针对β1和β2受体)或GF/C玻璃纤维滤板(帕克(Packard)生物科技公司,梅里登,CT)(针对β3受体)快速过滤来终止结合反应。用过滤缓冲液(75mM Tris/HCl,在4℃下pH 7.4,12.5mMMgCl2,1mM EDTA)将滤板洗涤三次以移除未结合放射活性。然后使板干燥并添加50μL微森特-20液态闪烁流体(帕克生物科技公司,梅里登,CT),并且在帕克拓普康液体闪烁计数器(帕克生物科技公司,梅里登,CT)上对板实施计数。结合数据是通过非线性回归分析以格拉夫派得普锐斯软件包(格拉夫派得软件公司,圣地亚哥,CA)使用单一位点竞争的3-参数模式来分析。将曲线最低值固定为在10μM普萘洛尔存在下测定的非特异性结合的数值。测试化合物的Ki值是使用程-普鲁萨福方程(程Y和普鲁萨福WH.,生化药理学,1973,22,23,3099-108)自所观测到放射性配体的IC50值和Kd值来计算。
在此分析中,较低Ki值表示测试化合物对所测试的受体具有较高结合亲和力。发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)对β2肾上腺素能受体的Ki值低于10nM,并且对β1和β3肾上腺素能受体的Ki值大于1000nM。
实例14
对毒蕈碱受体亚型的拮抗作用的功能性分析
分析A-cAMP积累的激动剂介导抑制的阻断
在此分析中,测试化合物作为hM2受体拮抗剂的功能性功效是通过测量测试化合物在表达hM2受体的CHO-K1细胞中阻断福司柯林(forskolin)介导cAMP积累的氧代震颤素抑制的能力来测定。cAMP分析是根据制造商说明书使用具有125I-cAMP(NENSMP004B,铂金埃尔默生命科学公司,波士顿,MA)的闪光板腺苷酸环化酶活化分析系统在放射性免疫分析格式下进行。用dPBS将细胞冲洗一次并用胰蛋白酶-EDTA溶液(0.05%胰蛋白酶/0.53mM EDTA)将细胞提取出来,如上文细胞培养和膜制备部分中所述。通过在650xg下离心五分钟将已分离细胞在50mL dPBS中洗涤两次。然后使细胞沉淀再悬浮于10mL dPBS中,并用Coulter Z1双粒子计数器(贝克曼库尔特(Beckman Coulter),富勒顿,CA)对细胞实施计数。将细胞在650xg下再离心五分钟,并以1.6 x 106至2.8 x 106个细胞/mL的分析浓度再悬浮于刺激缓冲液中。
首先以400μM的浓度使测试化合物溶于稀释缓冲液(补加有1mg/mL BSA(0.1%)的dPBS)中,然后用稀释缓冲液连续性稀释至介于100μM至0.1nM之间的最终摩尔浓度。以类似方式稀释氧代震颤素。
为测量腺苷酸环化酶活性的氧代震颤素抑制,将25μL福司柯林(以25μM最后浓度稀释于dPBS中)、25μL经稀释氧代震颤素和50μL细胞添加至激动剂分析孔中。为测量测试化合物阻断氧代震颤素抑制的腺苷酸环化酶活性的能力,将25μL福司柯林和氧代震颤素(分别以25μM和5μM的最后浓度稀释于dPBS中)、25μL经稀释测试化合物和50μL细胞添加至其余分析孔中。
在37℃下将反应物培养10分钟,并通过添加100μL冰冷检测缓冲液终止反应。将板密封,在室温下培养过夜,并在隔天早上在铂金埃尔默拓普康液体闪烁计数器(铂金埃尔默公司,韦尔兹利,MA)上计数。所产生cAMP量(pmol/孔)是根据制造商的使用者手册基于所观测到试样和cAMP标准品的计数来计算。数据是通过非线性回归分析以格拉夫派得普锐斯软件包(格拉夫派得软件公司,圣地亚哥,CA)使用非线性回归单一位点竞争方程来分析。使用程-普鲁萨福方程来计算Kobs,其中分别使用氧代震颤素浓度-反应曲线的EC50和氧代震颤素分析浓度来作为KD和[L]。
在此分析中,较低Kobs值表示测试化合物对所测试的受体具有较高功能性活性。发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)在表达hM2受体的CHO-K1细胞中阻断福司柯林介导cAMP积累的氧代震颤素抑制的Kobs值低于约10nM。
分析B-激动剂介导[35S]GTPγS结合的阻断
在此功能性分析中,测试化合物作为hM2受体拮抗剂的功能性功效是通过测量测试化合物在表达hM2受体的CHO-K1细胞中阻断氧代震颤素刺激的[35S]GTPγS结合的能力来测定。
在使用时,使冷冻膜解冻,然后以5-10μg蛋白质/孔的最终目标组织浓度使其稀释于分析缓冲液中。使用保利通PT-2100组织破碎器将膜短暂匀浆,然后添加至分析板。
在每个实验中测定激动剂氧代震颤素刺激[35S]GTPγS结合的EC90值(90%最大反应的有效浓度)。
为测定测试化合物抑制氧代震颤素刺激[35S]GTPγS结合的能力,将以下物质添加至96孔板的各孔中:25μL具有[35S]GTPγS(0.4nM)的分析缓冲液、25μL氧代震颤素(EC90)和GDP(3μM)、25μL经稀释测试化合物和25μL表达hM2受体的CHO细胞膜。然后在37℃下将分析板培养60分钟。用经1% BSA预处理并且使用铂金埃尔默96-孔采集器的GF/B滤器过滤分析板。用冰冷洗涤缓冲液将板冲洗3x3秒,然后风干或真空干燥。将微森特-20闪烁液体(50μL)添加至各孔中,并将各板密封并在拓普康(铂金埃尔默)上计数放射性活性。数据是通过非线性回归分析以格拉夫派得普锐斯软件包(格拉夫派得软件公司,圣地亚哥,CA)使用非线性回归单一位点竞争方程来分析。使用程-普鲁萨福方程来计算Kobs,其中分别使用测试化合物的浓度-反应曲线的IC50值和分析中氧代震颤素的浓度作为KD和[L](配体浓度)。
在此分析中,较低Kobs值表示测试化合物对所测试的受体具有较高功能性活性。发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)在表达hM2受体的CHO-K1细胞中对阻断氧代震颤素刺激的[35S]GTPγS结合的Kobs值低于约10nM。
分析C-通过FLIPR分析激动剂介导钙释放的阻断
在此功能性分析中,测试化合物作为hM1、hM3和cM5受体拮抗剂的功能性功效是通过测量测试化合物抑制激动剂介导细胞内钙增加的能力来测定。
在实施分析的前一晚,将稳定表达受体的CHO细胞接种于96-孔FLIPR板中。使用细胞洗涤器(Cellwash)(MTX雷勃系统(Labsystems)公司)以FLIPR缓冲液(10mMHEPES,pH 7.4,2mM氯化钙,2.5mM羧苯磺胺(probenecid),存于不含钙和镁的Hank氏缓冲盐溶液(HBSS)中)将所接种细胞洗涤两次以移除生长培养基。在洗涤后,各孔含有50μL FLIPR缓冲液。然后,在37℃和5%二氧化碳下将细胞与50μL/孔4μMFLUO-4AM(制成2X溶液)一起培养40分钟。在染料培养期之后,用FLIPR缓冲液将细胞洗涤两次,在各孔中留下50μL的最终体积。
测定氧代震颤素对细胞内Ca2+释放的剂量依赖性刺激以使得可在EC90浓度下针对氧代震颤素刺激来测量测试化合物。首先,将细胞与化合物稀缓冲液一起培养20分钟,然后添加氧代震颤素。根据下文FLIPR测量和数据简化部分中所详述方法,结合公式ECF=((F/100-F)^1/H)*EC50来获得氧代震颤素的EC90值。在刺激板中制备3 xECF浓度的氧代震颤素以将EC90浓度的氧代震颤素添加至测试分析板的各孔中。
用于FLIPR的参数是:0.4秒的暴露时间、0.5瓦特的激光强度、488nm的攻击波长、和550nm的发射波长。在添加氧代震颤素前,通过测量10秒内的荧光变化来确定基线。在氧代震颤素刺激之后,FLIPR在1.5分钟内每0.5至1秒连续测量荧光变化以捕获最大荧光变化。
荧光变化表示为各孔的最大荧光减去基线荧光。原始数据是通过非线性回归以格拉夫派得普锐斯(格拉夫派得软件公司,圣地亚哥,CA)使用S形剂量-反应的内建模式针对测试化合物浓度的对数来分析。拮抗剂Kobs值是根据程-普鲁萨福方程(程和普鲁萨福,1973)通过普锐斯来测定,其中使用氧代震颤素EC50值作为KD并用氧代震颤素EC90作为配体浓度。
在此分析中,较低Kobs值表示测试化合物对所测试的受体具有较高功能性活性。发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)在稳定表达hM1、hM3和cM5受体的CHO细胞中对阻断激动剂介导的钙释放的Kobs值低于约10nM。
实例15
在异源性表达人类β1、β2或β3肾上腺素能受体的HEK-293和CHO细胞系中实施全细胞cAMP闪光板分析
cAMP分析是根据制造商说明书使用具有[125I]-cAMP(NEN SMP004,铂金埃尔默生命科学公司,波士顿,MA)的闪光板腺苷酸环化酶活化分析系统以放射性免疫分析格式来进行。为测定β1和β2受体激动剂功效(EC50),使稳定表达克隆人类β1和β2受体的HEK-293细胞系在补加有10% FBS和遗传霉素(500μg/mL)的DMEM中生长至几乎长满。为测定β3受体激动剂功效(EC50),使稳定表达克隆人类或β3肾上腺素能受体的CHO-K1细胞系在补加有10% FBS和遗传霉素(250μg/mL)的哈莫斯F-12培养基中生长至几乎长满。用PBS冲洗细胞并在含有2mM EDTA的dPBS(达尔贝科(Dulbecco)氏磷酸盐缓冲的盐水,不含CaCl2和MgCl2)或胰蛋白酶-EDTA溶液(0.05%胰蛋白酶/0.53mM EDTA)中使细胞分离。在Coulter细胞计数器中对细胞实施计数后,通过以1,000rpm离心来使细胞沉淀,并使细胞在预升温至室温并含有IBMX的刺激缓冲液中(铂金埃尔默试剂盒)再悬浮至1.6 x 106至2.8 x 106个细胞/mL的浓度。在此分析中,每孔使用约40,000至80,000个细胞。在贝克曼贝尔麦克-2000中,将测试化合物(10mM,存于DMSO中)稀释至含有0.1% BSA的PBS中,并以100μM至1pM范围内的11种不同浓度测试。在37℃下将反应物培养10分钟,并通过添加100μL含有125I-cAMP(NEN SMP004,铂金埃尔默生命科学公司,波士顿,MA)的冷检测缓冲液来使反应停止。所产生cAMP的量(pmol/孔)是根据制造商的使用者手册基于所观察到试样和cAMP标准品的计数来计算。数据是通过非线性回归分析以格拉夫派得普锐斯软件包(格拉夫派得软件公司,圣地亚哥,CA)使用S形方程进行分析。使用程-普鲁萨福方程(程Y和普鲁萨福WH.,生化药理学,1973,22,23,3099-108)来计算EC50值。
在此分析中,较低EC50值表示测试化合物对测试受体具有较高功能性活性。发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)对β2肾上腺素能受体的EC50值低于约10nM;对β1肾上腺素能受体的EC50值为约30nM;并且对β3肾上腺素能受体的EC50值大于700nM。
实例16
以内源性表达人类β2肾上腺素能受体的肺上皮细胞系实施全细胞cAMP闪光板分析
在此分析中,测试化合物的激动剂功效和内在活性是使用表达内源含量的β2肾上腺素能受体的细胞系来测定。在无血清完全培养基(含有肾上腺素和视黄酸的LHC-9培养基,本源国际(Biosource International),卡马里奥,CA)中使得自人类肺上皮细胞系(BEAS-2B)(ATCCC RL-9609,美国模式培养物保藏所(American Type CultureCollection),马纳萨斯,VA)的细胞生长至75-90%汇合(翟纽尔瑞(January)B等人,英国药理学期刊(British Journal of Pharmacology),1998,123,4,701-11)。在分析前一天,将培养基转换成LHC-8(无肾上腺素或视黄酸,本源国际,卡马里奥,CA)。cAMP分析是根据制造商说明书使用具有[125I]-cAMP的闪光板腺苷酸环化酶活化分析系统(NEN SMP004,铂金埃尔默生命科学公司,波士顿,MA)以放射性免疫分析格式来进行。
在分析当天,用PBS冲洗细胞,通过用存于PBS中的5mM EDTA磨擦将细胞提取出来并实施计数。通过以1,000rpm离心使细胞沉淀,并以600,000个细胞/mL的最终浓度使其再悬浮于预热至37℃的刺激缓冲液中。在此分析中,细胞是在100,000至120,000个细胞/孔的最终浓度下使用。在贝克曼贝尔麦克(Biomek)-2000中,将测试化合物连续性稀释至分析缓冲液(75mM Tris/HCl,在25℃下pH 7.4,12.5mM MgCl2,1mM EDTA,0.2% BSA)中。在分析中,以10μM至10pM范围内的11种不同浓度来测试测试化合物。在37℃下将反应物培养10分钟,并通过添加100μL冰冷检测缓冲液来终止反应。将板密封,在4℃下培养过夜并在隔天早上在拓普康闪烁计数器(帕克生物科技公司,梅里登,CT)中计数。根据制造商的使用者手册中所述,基于所观察到试样和cAMP标准品的计数来计算每mL反应物所产生的cAMP量。数据是通过非线性回归分析以格拉夫派得普锐斯软件包(格拉夫派得软件公司,圣地亚哥,CA)使用S形剂量-反应的4-参数模式来分析。
在此分析中,较低EC50值表示测试化合物对受体测试具有较高功能性活性。发现与完整β2激动剂异丙基肾上腺素(1.0)相比,联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯(化合物IIa)的EC50值低于10nM,并且内在活性值大于0.3。
实例17
用于测定支气管保护性效能和持续时间的爱因托芬(Einthoven)分析
在此分析中,使用天竺鼠来测定测试化合物的支气管保护性效能和持续时间。此分析是得自爱因托芬(1892)欧洲生理学杂志(Pfugers Arch.)51:367-445;和默罕默德(Mohammed)等人(2000),肺脏药理学与治疗学(Pulm Pharmacol Ther.),13(6):287-92中所述程序。在此分析中,使用换气压力的变化作为气道阻力的替代量度。在用测试化合物预处理后,在普萘洛尔存在下使用对静脉内乙酰甲胆碱的支气管收缩剂量-反应曲线来测定毒蕈碱拮抗剂功效。同样地,使用组胺来测定β2激动剂支气管保护性功效。在不存在普萘洛尔的情况下使用乙酰甲胆碱来测定组合支气管保护性功效。
使用体重在250与400g之间的雄性邓肯-哈特利(Duncan-Hartley)天竺鼠(哈兰(Harlan),印第安纳波利斯,IN)来进行分析。在全身暴露投药室(R+S模具,圣卡洛斯,CA)中使用5mL投药溶液经10分钟通过吸入(IH)投用测试化合物或媒剂(即无菌水)。使动物暴露于自LC星式(Star)雾化罐组(22F51型,PARI呼吸设备公司(PARIRespiratory Equipment,Inc.),中洛锡安(Midlothian),VA)产生并且以22psi的压力通过生物掺合物(Bioblend)(5% CO2、21% O2、与74% N2的混合物)喷出的气溶胶中。在吸入投药后,在不同时间点评估肺功能。
在分析开始前七十五分钟时,通过肌内(IM)注射氯胺酮(43.7mg/kg)/甲苯噻嗪(3.5mg/kg)/乙酰丙嗪(1.05mg/kg)的混合物来使天竺鼠麻醉。根据需要投与此混合物的补充剂量(首次剂量的50%)。分离颈静脉与颈动脉并用填充盐水的聚乙烯导管(分别为微瑞那散(renathane)和PE-50,贝克曼迪金逊(Beckton Dickinson),斯巴克斯(Sparks),MD)进行插管。将颈动脉连接至压力传感器以允许测量血压,并且将颈静脉插管用于乙酰甲胆碱或组胺的IV注射。然后将气管分割开来,并用14G针头(第NE-014号,小部件(Small Parts),迈阿密湖,FL)实施插管。一旦完成插管,使用呼吸器(683型,哈佛(Harvard)装置公司,MA)对天竺鼠实施换气,其中每搏量设定为1mL/100g体重但不超过2.5mL体积,并且速率设定100搏/分钟。换气压力(VP)是在气管插管中使用连接至贝尔帕克(Biopac)(TSD 137C)预放大器的贝尔帕克传感器来测量。使用加热垫将体温保持在37℃。在开始收集数据之前,以腹膜腔内方式(IP)投与戊巴比妥(pentobarbital)(25mg/kg)以抑制自主呼吸并获得稳定基线。在贝尔帕克窗口(Windows)数据收集界面上记录VP变化。经至少5分钟收集基线值,在此之后以非累积方式使用2-倍增加剂量的支气管收缩剂(乙酰甲胆碱或组胺)IV攻击天竺鼠。当使用乙酰甲胆碱作为支气管收缩剂时,用普萘洛尔(5mg/kg,IV)将动物预处理以隔离测试化合物的抗毒蕈碱效应。在构建对乙酰甲胆碱或组胺的剂量反应曲线前30分钟投与普萘洛尔。使用艾克诺类(Acknowledge)数据收集软件(圣巴巴拉(Santa Barbara),CA)记录VP的变化。在研究完成后,使动物安乐死。
以水的cm数测量VP的变化。VP的变化(cm H2O)=峰值压力(支气管收缩剂攻击后)-峰值基线压力。使用格拉夫派得普锐斯的窗口3.00版(格拉夫派得软件,圣地亚哥,加利福尼亚)将对乙酰甲胆碱或组胺的剂量反应曲线拟合至四参数对数方程。使用下列方程:
Y=Min+(Max-Min)/(1+10((log ID50-X)′希尔斜率e))
其中X为剂量的对数,Y为反应。Y始于Min并且以S形形状渐近地到达Max。
在每种测试化合物剂量下使用以下方程来计算对次极大剂量乙酰甲胆碱或组胺的支气管收缩反应的抑制百分比:反应的%抑制=100-((峰值压力(在支气管收缩剂攻击后,经处理)-峰值基线压力(经处理)*100%/(峰值压力(在支气管收缩剂攻击后,水)-峰值基线压力(水)x 100)。使用来自格拉夫派得软件的四参数对数方程拟合抑制曲线。如果适宜,同样估计ID50(产生支气管收缩剂反应的50%抑制所需剂量)和Emax(最高抑制)。
使用吸入测试化合物后不同时间点的支气管保护强度来估计药效半衰期(PDT1/2)。使用非线性回归拟合以一阶指数衰减方程(格拉夫派得普锐斯,4.00版)来测定PD T1/2:Y=Span*exp(-K*X)+稳定期;始于Span+稳定期并衰减至速率常数为K的稳定期。PD T1/2=0.69/K。稳定期降低至0。
在投药后1.5小时,发现联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基氨甲酰基)乙基]哌啶-4-基酯(IIa)对乙酰甲胆碱所诱导的支气管收缩和组胺所诱导的支气管收缩两者的ID50低于约50μg/mL。
此外,当以单一次极大剂量(100μg/mL)投与时,此化合物在最长约72小时内产生显着支气管保护。在此分析中,沙美特罗(3μg/mL)(β2肾上腺素能受体激动剂)在6至14小时内表现显着支气管保护;并且噻托铵(10μg/mL)(毒蕈碱受体拮抗剂)在大于72小时内表现显着支气管保护。
实例18
用于测定支气管保护性效能和持续时间的体积描记器天竺鼠分析
在此分析中,使用天竺鼠分析来测定测试化合物的支气管保护性效能和持续时间。
通过笼子卡片分别标识体重在250与350g之间雄性天竺鼠组(每组6只)(邓肯-哈特利(HsdPoc:DH)哈兰,麦迪逊,WI)。在整个研究中,允许动物无限制地获取食物和水。在全身暴露投药室(R&S模具,圣卡洛斯,CA)中经10分钟通过吸入投与测试化合物。投药室的安排方式使气溶胶可从中央歧管同时递送至6个独室中。使天竺鼠暴露于测试化合物或媒剂(WFI)的气溶胶中。使用LC星式雾化罐组(22F51型,PARI呼吸设备公司,中洛锡安,VA)自水溶液产生气溶胶并在22psi的压力下通过气体混合物(CO2=5%,O2=21%,并且N2=74%)喷出气溶胶。在此操作压力下,经过雾化罐的气流是每分钟大约3L。通过正压将所产生气溶胶喷至室中。在气溶胶化溶液递送期间未使用稀释空气。在10分钟雾化期间,大约1.8mL溶液被雾化。通过比较经填充雾化罐在雾化之前和之后的重量以重量分析方式测量此数值。
通过吸入投与的测试化合物的支气管保护作用是在投药后1.5、24、48和72小时使用全身体积描记法来评估。在肺评估开始前四十五分钟,通过肌内注射氯胺酮(43.75mg/kg)、甲苯噻嗪(3.50mg/kg)和乙酰丙嗪(1.05mg/kg)使各天竺鼠麻醉。将手术位置刮毛并用70%醇清理,并在颈部腹面实施2至3cm中线切开术。分离颈静脉并用填充盐水的聚乙烯导管(PE-50,Becton Dickinson,Sparks,MD)插管以允许静脉内输注存于盐水中的乙酰胆碱或组胺。然后,将气管分割开来,并用14G特富龙(teflon)管(第NE-014号,小部件,迈阿密湖,FL)实施插管。如果需要,通过另外实施肌内注射麻醉混合物来保持麻醉。监测麻醉的深度并在动物反应夹缩其爪或呼吸速率大于每分钟100次呼吸时进行调整。
一旦完成插管,就将动物放置在体积描记器(第PLY3114号,布克思科(Buxco)电子公司,沙郎(Sharon),CT)中,并插入食管压力插管(PE-160,贝克曼迪金逊,斯巴克斯,MD)以测量肺驱动压力。将特富龙导气管连接至体积描记器的开孔以允许天竺鼠呼吸来自室外的屋内空气。然后将室密封。使用加热灯来保持体温并以4mL空气使用10mL校准注射器(第5520系列,汉斯鲁道夫(Hans Rudolph),堪萨斯市,MO)使天竺鼠的肺膨大三倍,以确保下气道未曾陷缩并且动物不发生换气过度。
在测定出顺应性的基线值在0.3-0.9mL/cm H2O范围内并且阻力的基线值在0.1-0.199cm H2O/mL/秒范围内后,开始肺评估。使用布克思科肺测量电脑程序来收集和衍生肺数值。在程序启动的同时开始实施实验方案和数据收集。通过布克思科压力传感器来测量在体积描记期间伴随每次呼吸发生的体积随时间的改变。通过将此信号随时间积分,计算每次呼吸的流量测量值。使用赛西姆(Sensym)压力传感器(TRD4100)收集的此信号和肺驱动压力变化,是通过布克思科(MAX2270)前置放大器连接至数据收集界面(SFT3400和SFT3813)。所有其它肺参数都是衍生自者两种输入。
收集5分钟内的基线值,在此之后用乙酰胆碱或组胺来攻击天竺鼠。当评估测试化合物的毒蕈碱拮抗剂作用时,在用乙酰胆碱攻击前15分钟投与心得安(propanolol)(5mg/kg,iv)(西格玛-奥德里奇,圣刘易斯,MO)。在反应开始时以以下剂量和预定时间经1分钟自注射泵(sp210iw,世界精密仪器(World Precision Instruments)公司,萨拉索塔,FL)静脉内注入乙酰胆碱(西格玛-奥德里奇,圣刘易斯,MO)(0.1mg/mL):在5分钟时以1.9μg/分钟注入,在10分钟时以3.8μg/分钟注入,在15分钟时以7.5μg/分钟注入,在20分钟时以15.0μg/分钟注入,在25分钟时以30μg/分钟注入,以及在30分钟时以60μg/分钟注入。或者,在未经心得安预处理的乙酰胆碱攻击模型中评价测试化合物的支气管保护效应。
在评估测试化合物的β2肾上腺素能受体激动剂效应时,在反应开始时以以下剂量和预定时间经1分钟自注射泵静脉内注入组胺(25μg/mL)(西格玛-奥德里奇,圣刘易斯,MO):在5分钟时以0.5μg/分钟注入,在10分钟时以0.9μg/分钟注入,在15分钟时以1.9μg/分钟注入,在20分钟时以3.8μg/分钟注入,在25分钟时以7.5μg/分钟注入,以及在30分钟时以15μg/分钟注入。如果在每次乙酰胆碱或组胺剂量后3分钟阻力或顺应性未返回基线值,则用4mL空气自10mL校准注射器使天竺鼠的肺膨大3倍。所记录的肺参数包括呼吸频率(每分钟呼吸数)、顺应性(mL/cm H2O)和肺阻力(cm H2O/mL/秒)。一旦在此方案中于第35分钟时完成肺功能测量,就将天竺鼠移出体积描记器并通过二氧化碳窒息使其安乐死。
以两种方式之一来评估数据:
(a)自压力变化与流量变化的比例来计算肺阻力(RL)(cm H2O/mL/秒)。计算媒剂和测试化合物的对乙酰胆碱(60μg/min,IH)的RL反应。在每个预处理时间计算经媒剂处理动物中的平均乙酰胆碱反应,并将其用于在相应预处理时间和每个测试化合物剂量下计算乙酰胆碱反应的抑制百分比。使用格拉夫派得普锐斯的窗口3.00版(格拉夫派得软件,圣地亚哥,加利福尼亚)将‘RL’的抑制剂量-反应曲线与四参数对数方程拟合,以估计支气管保护性ID50(将乙酰胆碱(60μg/min)支气管收缩反应抑制50%所需剂量)。使用下列方程:

其中X为剂量对数,Y为反应(乙酰胆碱所诱导RL增加的抑制百分比)。Y始于Min并且以S形形状渐近地到达Max。
(b)数量PD2被定义为使基线肺阻力加倍所需乙酰胆碱或组胺的量,其使用得自一系列乙酰胆碱或组胺攻击的流量和压力的肺阻力值使用以下方程(衍生自美国胸科学会(American Thoracic Society),乙酰甲胆碱和实践攻击测试导引-1999.国际呼吸医学杂志(Am J Respir Crit Care Med.),2000;161:309-329中所述用于计算PC20值的方程)来计算:

其中:
C1=在C2前的乙酰胆碱或组胺的浓度
C2=导致肺阻力(RL)增加至少2倍的乙酰胆碱或组胺的浓度
R0=基线RL值
R1=C1后的RL值
R2=C2后的RL值
数据的统计分析是使用二尾式司徒登氏(Student’s)t-测试来进行。P值<0.05被认为有意义。
在2004年8月24日公开的美国专利公开案第2004/0167167A1中所述化合物50是在此分析中测试。化合物50的化学结构如下:
此化合物缺少存于本发明化合物苯环上的烷基。在此分析中,对于3μg/mL至300μg/mL的剂量,在投药后24小时化合物50未显示显着支气管保护。在24小时化合物50的PD2x值与媒剂(水)组类似。
在此分析中,沙美特罗(100μg/mL)(β2肾上腺素能受体激动剂)在至少24小时内表现显着支气管保护;并且噻托铵(10μg/mL)(毒蕈碱受体拮抗剂)在至少24小时内表现显着支气管保护。
虽然已参考其特定方面或实施例来描述本发明,但熟习此项技术者应理解,在不偏离本发明的真实精神和范围的情况下可进行各种改变或可取代等效内容。此外,根据适用专利法和条例所允许的程度,本文中所引用的所有出版物、专利和专利申请案都是全文以引用方式并入本文中,其并入程度如同每个文件均单独地以引用方式并入本文中一般。
Claims (27)
1、一种式I化合物:
其中
R1为甲基或乙基;
R2为甲基或乙基;
或其医药上可接受的盐或溶剂合物或立体异构体。
2、如权利要求1所述的化合物,其具有式II:
其中
R1为甲基或乙基;
R2为甲基或乙基;
或其医药上可接受的盐。
3、如权利要求1所述的化合物,其具有式IIa:

4、如权利要求1所述的化合物,其中所述化合物是式IIa化合物的医药上可接受的盐:
5、一种医药组合物,其包含医药上可接受的载剂和如权利要求1至4中任一权利要求所述的化合物。
6、一种医药组合物,其包含:
(a)如权利要求1至4中任一权利要求所述的化合物;
(b)类固醇消炎药;和
(c)医药上可接受的载剂。
7、一种治疗药剂的组合,其包含:
(a)如权利要求1至4中任一权利要求所述的化合物;和
(b)类固醇消炎药。
8、一种试剂盒,其包含:
(a)第一医药组合物,其包含如权利要求1至4中任一权利要求所述的化合物和医药上可接受的第一载剂;和
(b)第二医药组合物,其包含类固醇消炎药和医药上可接受的第二载剂;
其中所述第一和第二医药组合物是分开的医药组合物。
9、一种治疗肺病的方法,所述方法包含向需要治疗的患者投与治疗有效量的如权利要求1至4中任一权利要求所述的化合物。
10、一种治疗慢性阻塞性肺病或哮喘的方法,所述方法包含向患者投与治疗有效量的如权利要求1至4中任一权利要求所述的化合物。
11、一种在哺乳动物中产生支气管扩张的方法,所述方法包含向哺乳动物投与支气管扩张产生量的如权利要求1至4中任一权利要求所述的化合物。
12、一种在哺乳动物中拮抗毒蕈碱受体并激动β2肾上腺素能受体的方法,所述方法包含向所述哺乳动物投与如权利要求1至4中任一权利要求所述的化合物。
13、一种使用如权利要求1至4中任一权利要求所述的化合物作为研究工具的方法,所述方法包含使用如权利要求1至4中任一权利要求所述的化合物进行生物学分析。
14、一种在生物学分析中评估测试化合物的方法,所述方法包含:
(a)使用测试化合物进行生物学分析以提供第一分析值;
(b)使用如权利要求1至4中任一权利要求所述的化合物进行所述生物学分析以提供第二分析值;其中步骤(a)是在步骤(b)之前、之后或同时进行;和
(c)比较得自步骤(a)的第一分析值与得自步骤(b)的第二分析值。
15、如权利要求14所述的方法,其中所述生物学分析为毒蕈碱受体结合分析或β2肾上腺素能受体结合分析。
16、如权利要求14所述的方法,其中所述生物学分析是哺乳动物中的支气管保护分析。
17、一种制备如权利要求1所述的化合物的方法,所述方法包含使式6化合物去保护:

其中P1为羟基保护基;以提供式I化合物。
18、一种制备如权利要求1所述的化合物的方法,所述方法包含使式6a化合物去保护:

其中Ra、Rb和Rc独立选自C1-4烷基、苯基、-C1-4烷基-(苯基),或R1a、R1b和R1c之一为-O-(C1-4烷基);以提供式I化合物。
19、一种制备如权利要求1所述化合物的方法,所述方法包含:
(a)使式4化合物:

与式5化合物:

其中P1为羟基保护基;于还原剂存在下反应以提供式6化合物:

和
(b)使所述式6化合物去保护以提供式I化合物。
20、一种制备如权利要求1所述的化合物的医药上可接受的盐的方法,所述方法包含使呈游离碱型的式I化合物与医药上可接受的酸接触。
21、一种制备如权利要求1至4中任一权利要求所述的化合物的中间体,其中所述中间体为式III化合物:

其中
Y1选自-CHO、-CN、-CH2OH、-CH(OR3a)OR3b、-C(O)OH、-C(O)OR3c、溴和碘,其中R3a和R3b独立选自C1-6烷基,或R3a和R3b连接形成C2-6亚烷基,R3c选自C1-6烷基;
R1为甲基或乙基;
R2为甲基或乙基;
或其盐或立体异构体。
22、如权利要求21所述的化合物,其中R1和R2为甲基。
23、如权利要求21所述的化合物,其中Y1为-CHO。
24、如权利要求21所述的化合物,其中Y1为-CHO;并且R1和R2为甲基。
25、如权利要求4所述的化合物,其中所述化合物为联苯-2-基氨基甲酸1-[2-(4-{[(R)-2-(3-甲酰氨基-4-羟苯基)-2-羟乙基氨基]甲基}-2,5-二甲基苯基-氨甲酰基)乙基]哌啶-4-基酯L-酒石酸盐。
26、如权利要求1至4中任一权利要求所述的化合物,其用于治疗中。
27、一种如权利要求1至4中任一权利要求所述的化合物的用途,其用于制造供治疗肺病的药物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79470206P | 2006-04-25 | 2006-04-25 | |
| US60/794,702 | 2006-04-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101426764A true CN101426764A (zh) | 2009-05-06 |
Family
ID=38577470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2007800140984A Pending CN101426764A (zh) | 2006-04-25 | 2007-04-24 | 具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性的二烷基苯基化合物 |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US7524965B2 (zh) |
| EP (1) | EP2010489B1 (zh) |
| JP (1) | JP2009541209A (zh) |
| KR (1) | KR20090005389A (zh) |
| CN (1) | CN101426764A (zh) |
| AR (1) | AR060647A1 (zh) |
| AU (1) | AU2007243481A1 (zh) |
| BR (1) | BRPI0710767A2 (zh) |
| CA (1) | CA2650530A1 (zh) |
| CO (1) | CO6140054A2 (zh) |
| ES (1) | ES2391584T3 (zh) |
| IL (1) | IL194334A0 (zh) |
| MA (1) | MA30556B1 (zh) |
| MX (1) | MX2008013555A (zh) |
| NO (1) | NO20084603L (zh) |
| PE (1) | PE20080094A1 (zh) |
| RU (1) | RU2008146383A (zh) |
| TW (1) | TW200811105A (zh) |
| WO (1) | WO2007127196A2 (zh) |
| ZA (1) | ZA200808817B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114409556A (zh) * | 2022-01-28 | 2022-04-29 | 兰州康鹏威耳化工有限公司 | 一种3,4-二取代-2-氨基苯甲醛的制备方法 |
| CN117924216A (zh) * | 2024-01-12 | 2024-04-26 | 王叔和生物医药(武汉)有限公司 | 一种1-哌啶丙酸的合成方法 |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6693202B1 (en) * | 1999-02-16 | 2004-02-17 | Theravance, Inc. | Muscarinic receptor antagonists |
| PE20040950A1 (es) | 2003-02-14 | 2005-01-01 | Theravance Inc | DERIVADOS DE BIFENILO COMO AGONISTAS DE LOS RECEPTORES ADRENERGICOS ß2 Y COMO ANTAGONISTAS DE LOS RECEPTORES MUSCARINICOS |
| TWI341836B (en) | 2004-03-11 | 2011-05-11 | Theravance Inc | Biphenyl compounds useful as muscarinic receptor antagonists |
| WO2006023457A1 (en) * | 2004-08-16 | 2006-03-02 | Theravance, Inc. | COMPOUNDS HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| TW200811104A (en) * | 2006-04-25 | 2008-03-01 | Theravance Inc | Crystalline forms of a dimethylphenyl compound |
| TW200811105A (en) * | 2006-04-25 | 2008-03-01 | Theravance Inc | Dialkylphenyl compounds having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| CN102083839B (zh) * | 2008-02-06 | 2014-03-26 | 阿斯利康(瑞典)有限公司 | 化合物 |
| LT2599778T (lt) | 2009-04-23 | 2017-08-25 | Theravance Respiratory Company, Llc | Diamido junginiai, pasižymintys muskarininiam receptoriui antagonistiniu ir beta 2 adrenerginiam receptoriui agonistiniu aktyvumu |
| US20110020423A1 (en) | 2009-07-22 | 2011-01-27 | Puretech Ventures | Methods and compositions for treatment of disorders ameliorated by muscarinic receptor activation |
| US10265311B2 (en) | 2009-07-22 | 2019-04-23 | PureTech Health LLC | Methods and compositions for treatment of disorders ameliorated by muscarinic receptor activation |
| WO2011081937A1 (en) | 2009-12-15 | 2011-07-07 | Gilead Sciences, Inc. | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy |
| GB201009801D0 (en) | 2010-06-11 | 2010-07-21 | Astrazeneca Ab | Compounds 950 |
| CA3180743A1 (en) | 2018-09-28 | 2020-04-02 | Karuna Therapeutics, Inc. | Composition comprising xanomeline and trospium for treating disorders ameliorated by muscarinic receptor activation |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU7545894A (en) | 1993-09-02 | 1995-03-22 | Yamanouchi Pharmaceutical Co., Ltd. | Carbamate derivative and medicine containing the same |
| EP0747355A4 (en) | 1994-02-10 | 1997-04-09 | Yamanouchi Pharma Co Ltd | NEW CARBAMATES AND MEDICINAL PRODUCTS CONTAINING THEM |
| US6040344A (en) | 1996-11-11 | 2000-03-21 | Sepracor Inc. | Formoterol process |
| US6541669B1 (en) * | 1998-06-08 | 2003-04-01 | Theravance, Inc. | β2-adrenergic receptor agonists |
| US6693202B1 (en) | 1999-02-16 | 2004-02-17 | Theravance, Inc. | Muscarinic receptor antagonists |
| DE19921693A1 (de) | 1999-05-12 | 2000-11-16 | Boehringer Ingelheim Pharma | Neuartige Arzneimittelkompositionen auf der Basis von anticholinergisch wirksamen Verbindungen und ß-Mimetika |
| UA73965C2 (en) | 1999-12-08 | 2005-10-17 | Theravance Inc | b2 ADRENERGIC RECEPTOR ANTAGONISTS |
| WO2002076933A1 (en) | 2001-03-22 | 2002-10-03 | Glaxo Group Limited | Formailide derivatives as beta2-adrenoreceptor agonists |
| US20030018019A1 (en) | 2001-06-23 | 2003-01-23 | Boehringer Ingelheim Pharma Kg | Pharmaceutical compositions based on anticholinergics, corticosteroids and betamimetics |
| TWI249515B (en) | 2001-11-13 | 2006-02-21 | Theravance Inc | Aryl aniline beta2 adrenergic receptor agonists |
| PE20040950A1 (es) * | 2003-02-14 | 2005-01-01 | Theravance Inc | DERIVADOS DE BIFENILO COMO AGONISTAS DE LOS RECEPTORES ADRENERGICOS ß2 Y COMO ANTAGONISTAS DE LOS RECEPTORES MUSCARINICOS |
| CA2543858C (en) | 2003-11-21 | 2014-04-15 | Theravance, Inc. | Compounds having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| US7456199B2 (en) | 2004-03-11 | 2008-11-25 | Theravance, Inc. | Biphenyl compounds useful as muscarinic receptor antagonists |
| WO2006023457A1 (en) | 2004-08-16 | 2006-03-02 | Theravance, Inc. | COMPOUNDS HAVING β2 ADRENERGIC RECEPTOR AGONIST AND MUSCARINIC RECEPTOR ANTAGONIST ACTIVITY |
| EP1833822A2 (en) | 2004-08-16 | 2007-09-19 | Theravance, Inc. | Compounds having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
| TWI374883B (en) | 2004-08-16 | 2012-10-21 | Theravance Inc | Crystalline form of a biphenyl compound |
| TW200811104A (en) * | 2006-04-25 | 2008-03-01 | Theravance Inc | Crystalline forms of a dimethylphenyl compound |
| TW200811105A (en) | 2006-04-25 | 2008-03-01 | Theravance Inc | Dialkylphenyl compounds having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity |
-
2007
- 2007-04-23 TW TW096114175A patent/TW200811105A/zh unknown
- 2007-04-24 JP JP2009507757A patent/JP2009541209A/ja active Pending
- 2007-04-24 MX MX2008013555A patent/MX2008013555A/es active IP Right Grant
- 2007-04-24 PE PE2007000509A patent/PE20080094A1/es not_active Application Discontinuation
- 2007-04-24 AR ARP070101777A patent/AR060647A1/es unknown
- 2007-04-24 CA CA002650530A patent/CA2650530A1/en not_active Abandoned
- 2007-04-24 CN CNA2007800140984A patent/CN101426764A/zh active Pending
- 2007-04-24 BR BRPI0710767-6A patent/BRPI0710767A2/pt not_active IP Right Cessation
- 2007-04-24 AU AU2007243481A patent/AU2007243481A1/en not_active Abandoned
- 2007-04-24 RU RU2008146383/04A patent/RU2008146383A/ru not_active Application Discontinuation
- 2007-04-24 ES ES07776093T patent/ES2391584T3/es active Active
- 2007-04-24 EP EP07776093A patent/EP2010489B1/en active Active
- 2007-04-24 WO PCT/US2007/009925 patent/WO2007127196A2/en active Application Filing
- 2007-04-24 KR KR1020087028589A patent/KR20090005389A/ko not_active Application Discontinuation
- 2007-04-24 US US11/789,300 patent/US7524965B2/en active Active
-
2008
- 2008-09-25 IL IL194334A patent/IL194334A0/en unknown
- 2008-10-15 ZA ZA200808817A patent/ZA200808817B/xx unknown
- 2008-10-29 CO CO08115666A patent/CO6140054A2/es unknown
- 2008-10-30 NO NO20084603A patent/NO20084603L/no not_active Application Discontinuation
- 2008-11-14 MA MA31382A patent/MA30556B1/fr unknown
-
2009
- 2009-03-18 US US12/406,283 patent/US7687521B2/en active Active
-
2010
- 2010-02-05 US US12/701,108 patent/US8134006B2/en not_active Expired - Fee Related
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114409556A (zh) * | 2022-01-28 | 2022-04-29 | 兰州康鹏威耳化工有限公司 | 一种3,4-二取代-2-氨基苯甲醛的制备方法 |
| CN114409556B (zh) * | 2022-01-28 | 2024-10-18 | 兰州康鹏威耳化工有限公司 | 一种3,4-二取代-2-氨基苯甲醛的制备方法 |
| CN117924216A (zh) * | 2024-01-12 | 2024-04-26 | 王叔和生物医药(武汉)有限公司 | 一种1-哌啶丙酸的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2008146383A (ru) | 2010-05-27 |
| PE20080094A1 (es) | 2008-02-17 |
| WO2007127196A3 (en) | 2007-12-21 |
| US8134006B2 (en) | 2012-03-13 |
| ZA200808817B (en) | 2010-03-31 |
| CA2650530A1 (en) | 2007-11-08 |
| ES2391584T3 (es) | 2012-11-28 |
| TW200811105A (en) | 2008-03-01 |
| US20100137603A1 (en) | 2010-06-03 |
| US7687521B2 (en) | 2010-03-30 |
| EP2010489A2 (en) | 2009-01-07 |
| MX2008013555A (es) | 2008-11-04 |
| MA30556B1 (fr) | 2009-07-01 |
| US20090176833A1 (en) | 2009-07-09 |
| BRPI0710767A2 (pt) | 2011-06-07 |
| IL194334A0 (en) | 2009-08-03 |
| JP2009541209A (ja) | 2009-11-26 |
| CO6140054A2 (es) | 2010-03-19 |
| EP2010489B1 (en) | 2012-08-22 |
| US7524965B2 (en) | 2009-04-28 |
| AR060647A1 (es) | 2008-07-02 |
| WO2007127196A2 (en) | 2007-11-08 |
| AU2007243481A1 (en) | 2007-11-08 |
| KR20090005389A (ko) | 2009-01-13 |
| US20070249675A1 (en) | 2007-10-25 |
| NO20084603L (no) | 2008-11-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101426764A (zh) | 具有β2肾上腺素能受体激动剂和毒蕈碱受体拮抗剂活性的二烷基苯基化合物 | |
| CN101426765A (zh) | 二甲基苯基化合物的晶形 | |
| KR101270183B1 (ko) | 비페닐 화합물의 결정형 | |
| KR101223991B1 (ko) | 베타2 아드레날린 수용체 작용제 및 무스카린 수용체길항제 활성을 갖는 비페닐 유도체 | |
| CN1930125B (zh) | 适用作毒蕈碱性受体拮抗剂的联苯基化合物 | |
| CN103936716B (zh) | 具蕈毒碱受体拮抗剂和β2肾上腺素受体激动剂活性的二酰胺化合物 | |
| CN101006077B (zh) | 晶体形式的联苯化合物 | |
| JP2008510014A (ja) | β2アドレナリン作用性レセプターアゴニスト活性およびムスカリン性レセプターアンタゴニスト活性を有する化合物 | |
| US20060205949A1 (en) | Crystalline forms of a biphenyl compound | |
| US8697724B2 (en) | Crystalline oxalate salts of a diamide compound | |
| CN101239969A (zh) | 联苯衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20090506 |