CN101274934A - 具有hm74a受体活性的药物 - Google Patents
具有hm74a受体活性的药物 Download PDFInfo
- Publication number
- CN101274934A CN101274934A CNA2008100992548A CN200810099254A CN101274934A CN 101274934 A CN101274934 A CN 101274934A CN A2008100992548 A CNA2008100992548 A CN A2008100992548A CN 200810099254 A CN200810099254 A CN 200810099254A CN 101274934 A CN101274934 A CN 101274934A
- Authority
- CN
- China
- Prior art keywords
- purine
- dihydro
- chloro
- diketone
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明提供了具有式(II)的为黄嘌呤衍生物的治疗活性化合物,制备所述衍生物的方法,含有所述活性化合物的药物制剂以及所述化合物在治疗、特别是在治疗由HM74A受体激活不足所引起的疾病或者受益于激活该受体的疾病中的用途,其中R1选自:氢以及可任选被一个或多个选自CN和CF3中的基团取代的C1-4烷基,R2选自:未被取代的C2-10烷基、被一个或多个选自氟和CN中的基团取代的C1-10烷基、C5链烯基、直链C4链烯基、以及被环烷基取代的C1-4烷基,和R3选自卤素和CN。
Description
本申请是申请日为2005年2月10日、申请号为200580011433.6、发明名称为“具有HM74A受体活性的药物”的发明专利申请的分案申请。
本发明涉及为黄嘌呤衍生物的治疗活性化合物,制备所述衍生物的方法,含有所述活性化合物的药物制剂以及所述化合物在治疗、特别是在治疗由HM74A受体激活不足(under-activation)所引起的疾病或者受益于激活HM74A受体的疾病中的用途。
血脂障碍是用于描述个体脂蛋白异常的统称。在临床上,用于治疗患有血脂障碍以及由此引发的心血管疾病高危患者的主要化合物类型是斯特汀(statins)、贝特类(fibrates)、胆汁酸结合树脂以及烟酸。烟酸(Niacin,维生素B)在临床上用于治疗患有不同形式的血脂障碍的患者已超过40余年。烟酸的主要作用模式是通过抑制对激素敏感的甘油三酯脂肪酶(HSL),使得血浆非酯化脂肪酸(NEFA)减少,进一步降低了肝脏脂肪代谢,从而减少LDL和VLDL(低密度脂蛋白和极低密度脂蛋白)输出。据信VLDL水平降低可以降低胆固醇脂转移蛋白(CETP)活性,导致HDL(高密度脂蛋白)水平升高,这可能是观察到心血管受益的原因。因此,烟酸对脂蛋白产生了非常令人满意的改变;降低了VLDL和LDL水平而同时升高了HDL水平。另外还证实烟酸具有调节疾病的优势,可以减轻动脉粥样硬化病变的恶化并促进其复原,以及在多个试验中还减少了引发心血管事件的次数。
采用烟酸治疗所观察到的HSL抑制作用是通过细胞内环磷腺苷(cAMP)减少来调节,而细胞内环磷腺苷(cAMP)减少是由G-蛋白介导的腺苷酸环化酶抑制作用引起的。最近,已经鉴定G-蛋白偶联的受体HM74和HM74A是烟酸的受体(PCT专利申请WO02/84298;Wise等人.J Biol Chem.,2003,278(11),9869-9874)。人HM74A的DNA序列可在Genbank中找到;登录号AY148884。其它两篇论文支持了上述发现(Tunaru等人.Nature Medicine,2003,9(3),352-255和Soga等人.Biochem Biophys Res Commun.,2003,303(1)364-369),但是其中所用术语有细微差异。在Tunaru的论文中,所称人HM74实际上是HM74A,而在Soga的论文中,HM74b就是HM74A。转染表达HM74A和/或HM74的细胞在曝露于烟酸之后,获得了引发Gi G-蛋白介导响应的能力。在缺乏HM74A(m-PUMA-G)同系物的小鼠中,烟酸不能降低血浆NEFA水平。
现有技术中已经合成并公开了某些黄嘌呤衍生物。例如,EP0389282公开了用作脑血管障碍的潜在调节剂的黄嘌呤衍生物。Jacobson等人在J.Med.Chem.,1993,36,2639-2644中证实大量的黄嘌呤衍生物均为腺苷受体拮抗剂。
目前我们发现一类黄嘌呤衍生物,它是烟酸受体HM74A的选择性激动剂,因而其在治疗、预防和抑制由该受体激活不足所引起的疾病或者受益于激活该受体的疾病中是有益的。
发明概述
本发明提供了具有治疗活性的黄嘌呤衍生物以及所述衍生物在治疗、特别是在治疗由HM74A受体激活不足所引起的疾病或者受益于激活该受体的疾病中的用途,所述疾病特别是脂类代谢疾病包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍(mixed dyslipidaemia),心力衰竭,血胆脂醇过多症(hypercholesteraemia),心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症。本发明化合物同样还可以用作冠状动脉疾病,血栓症,心绞痛,慢性肾衰竭,周围血管疾病和中风,以及与II型糖尿病、I型糖尿病、胰岛素耐受性、高脂血症、神经性厌食症、肥胖症有关的心血管适应症的治疗剂。本发明化合物还可用于治疗如下面所述的各种炎性疾病或病症。
本文所述的各种中间体、制剂、方法以及步骤构成了本发明另一方面。
发明详述
根据本发明一方面,提供了式(I)化合物及其生理学功能的衍生物,

其中
R1选自:氢以及可任选被一个或多个选自CN和CF3中的基团取代的C1-4烷基;
R2选自:未被取代的C3-10烷基、被一个或多个选自氟和CN中的基团取代的C1-10烷基、C5链烯基、直链C4链烯基、以及被环烷基取代的C1-4烷基;
和R3选自卤素和CN;
条件是:
(i)当R3表示Cl,且R1表示乙基时,R2不是丙基;
(ii)当R3表示Br,且R1表示丙基时,R2不是丙基;
(iii)当R3表示Cl或Br,且R1表示丁基时,R2不是丁基;以及
(iv)当R1表示C1-4烷基、CH2CN或(CH2)3CF3时,R2不是支链烷基。
本发明化合物可用于治疗由HM74A受体激活不足所引起的疾病或者受益于激活该受体的疾病,特别是脂类代谢疾病包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍,心力衰竭,血胆脂醇过多症,心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症。本发明化合物同样还可以用作冠状动脉疾病,血栓症,心绞痛,慢性肾衰竭,周围血管疾病和中风,以及与II型糖尿病、I型糖尿病、胰岛素耐受性、高脂血症、神经性厌食症、肥胖症有关的心血管适应症的治疗剂。本发明化合物同样也可以用作HM74A的激动剂或部分激动剂(HM74A调节剂)。
在具体实施方案中,R1选自:氢、C1-4烷基、CH2CN和(CH2)3CF3;在更具体的实施方案中,R1选自:氢和甲基。
在部分实施方案中,R2选自:未被取代的C3-10烷基、被一个或多个CN取代基取代的C1-6烷基、被一个或多个氟取代基取代的C1-10烷基、C5链烯基、直链C4链烯基、以及被环烷基取代的C1-4烷基。具体地,R2选自:未被取代的C3-10烷基;(CH2)1-5CN;被一个或多个氟取代基取代的C2-5烷基;C5链烯基;以及被环烷基取代的C1-4烷基。更具体地,R2选自未被取代的C4-6正烷基,例如戊基;(CH2)1-3CN,例如(CH2)CN或(CH2)3CN;具有一个或多个氟取代基的C3-4烷基,特别是其中末端碳完全用氟饱和的C3-4烷基,例如(CH2)2-3CF3;以及C5链烯基,特别是其中仅有一个双键的C5链烯基,例如其中双键位于第四个和第五个碳之间的C5链烯基(末端链烯基)。
在具体实施方案中,R3表示卤素。更具体地,R3选自:氯和溴。最具体地,R3表示氯。
应该理解的是,本发明包括这些具体实施方案的任何组合,并且覆盖上述具体取代基的所有组合。
具体的本发明化合物包括:
(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)乙腈,
3-丁基-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-溴-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(3,3,3-三氟丙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-1-丙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
3-丁基-8-氯-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮,
(3-丁基-8-氯-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈,
8-氯-3-(2-环丙基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮,
4-(8-氯-1-甲基-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈,
8-氯-1-乙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
1-甲基-2,6-二氧代-3-戊基-2,3,6,7-四氢-1H-嘌呤-8-腈,
8-氯-3-丙基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(3-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮,
3-丁基-1-甲基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈,
8-氯-3-(4-戊烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-己基-3,7-二氢-1H-嘌呤-2,6-二酮,
4-(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈,
8-氯-3-己基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮,
3-丁基-8-氯-1-乙基-3,7-二氢-1H-嘌呤-2,6-二酮,
[8-氯-3-(2-环丙基乙基)-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-1-基]乙腈,
(8-氯-2,6-二氧代-3-丙基-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈,
8-氯-1-(4,4,4-三氟丁基)-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
2,2′-(8-氯-2,6-二氧代-6,7-二氢-1H-嘌呤-1,3(2H)-二基)二乙腈,
8-氯-1-甲基-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(2-环己基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
1,3-二丁基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈,
1,3-二丁基-8-碘-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(4-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(6-甲基庚基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-辛基-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-癸基-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(环己基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮,
(+/-)-8-氯-3-(3-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(2-环戊基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(环丙基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮,
(+/-)-8-氯-3-(2-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮,
(+/-)-8-氯-3-(2-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(环丁基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(环戊基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(3-环丙基丙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(2-环丁基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(4-氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(3-氟丙基)-3,7-二氢-1H-嘌呤-2,6-二酮,
8-氯-3-(5-氟戊基)-3,7-二氢-1H-嘌呤-2,6-二酮,
4-(8-氯-1-甲基-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈,
3-(3-丁烯-1-基)-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮,
6-(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)-2,2-二甲基己腈,
8-氯-3-(6-氟己基)-3,7-二氢-1H-嘌呤-2,6-二酮。
本说明书和权利要求书中通篇使用的措辞“含有”和“包括”及其各种变型是指包含在内的意思。也就是说,在上下文允许的情形下,这些措辞意味着可以含有其它没有明确指明的元素和个体。
本文所使用的术语“卤素”或“卤”是指氟、氯、溴和碘。
本文所使用的术语“烷基”(当被用作基团或基团中的一部分使用时)是指直链或支链烃链,除非另有限定,是指含有指定数目的碳原子的直链或支链烃链。例如,C3-C10烷基是指含有至少3个、至多10个碳原子的直链或支链烃链。本文所使用的烷基实例包括但不限于甲基(Me)、乙基(Et)、正丙基和异丙基。术语“正烷基”具体是指直链烃链。
本文所使用的术语“环烷基”是指含有3-6个碳原子、不含杂原子或共轭双键的烃环。本文所使用的环烷基的实例包括但不限于环丙基和环己基。
本文所使用的术语“链烯基”是指含有指定数目的碳原子、一个或多个双键的直链或支链烃链。
在本文中,当某基团被称作被另一基团“取代”或者具有“一个或多个取代基”时,除非明确指出了所述取代基的具体位置,应该理解所述取代基可以存在于该基团的任意位置上。
本文所使用的术语“生理学功能的衍生物”是指本发明化合物的任意可药用衍生物例如其酰胺,包括式(I)化合物的任意可药用盐、以及式(I)化合物的任意可药用溶剂化物,向哺乳动物例如人施用之后,它们能够提供(直接或间接地)式(I)化合物或其活性代谢物或残余物。本领域普通技术人员应该理解的是,可以在式(I)化合物的任意功能基团上对其进行修饰得到其生理学功能的衍生物,并且式(I)化合物可以在不止一个位置上进行上述修饰。
在本文中,与可包含在向患者施用的药物制剂中的成分(活性成分或赋形剂)关联使用的术语“可药用”,是指在与该药物制剂中其它任意成分配伍方面可以接受、并且对其接受者无毒的成分。
本文所使用的术语“溶剂化物”是指由溶质(在本发明中为式(I)化合物、或其盐或其生理学功能的衍生物)与溶剂形成的具有可变化学计量的络合物。满足本发明目的的这类溶剂不会干扰所述溶质的生理活性。所用溶剂可以是可药用溶剂。适宜的可药用溶剂的实例包括水、乙醇和乙酸。可使用溶剂的实例是水,在该情形中,所述溶剂化物可称作被讨论溶质的水合物。
对于药学应用应该理解,上述“盐或溶剂化物”可以为可药用盐或溶剂化物。当然,其它的盐或溶剂化物可用于例如式(I)化合物或者其可药用盐或溶剂化物的制备中。
可药用盐包括Berge,Bighley和Monkhouse,J.Pharm.Sci.,1977,66,1-19中所述的盐。适宜的可药用盐包括由加入碱金属例如碱金属氢氧化物形成的碱金属盐。适宜的碱金属盐的实例是钠盐或钾盐。其它适宜的可药用盐包括碱土金属盐,例如钙盐或镁盐、铵盐;或与有机碱例如乙醇胺、三乙醇胺、乙二胺、三乙胺、胆碱和葡甲胺形成的盐;或者与氨基酸例如精氨酸、赖氨酸和组氨酸形成的盐。
式(I)化合物在治疗和缓解诸多脂类代谢疾病症状中具有潜在的治疗效果,所述疾病包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍,心力衰竭,血胆脂醇过多症,心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症。本发明化合物同样还可用作冠状动脉疾病、血栓症、心绞痛、慢性肾衰竭、周围血管疾病和中风的治疗剂。
此外,据信HM74和HM74A受体与炎症有关。炎症代表了对于创伤的一系列血管、细胞和神经响应。可以将炎症描述为炎症细胞例如单核细胞、嗜中性粒细胞和粒性白细胞向组织内的移动。这通常伴随着内皮屏障作用降低和组织内水肿。与疾病相关的炎症通常被称作慢性炎症,甚至可以持续终生。这类慢性炎症可以通过疾病症状表现出来。因此,抗炎治疗的目的在于减轻慢性炎症,使得愈合和组织修复的生理步骤得以进行。
本发明化合物所适用的炎性疾病或病症的实例包括关节炎性疾病或病症,特别是关节炎(例如类风湿性关节炎、骨关节炎、假关节失灵(prostheticjoint failure))、或者胃肠道炎症(例如溃疡性结肠炎、克罗恩氏病、以及其它炎性肠病和胃肠疾病、由感染引起的胃炎和粘膜炎症、由非甾体消炎药物引起的肠病)、肺部发炎(例如成人呼吸窘迫综合征、哮喘、囊性纤维化病、或慢性阻塞性肺疾患)、心脏发炎(例如心肌炎)、神经组织炎症(例如多发性硬化)、胰腺炎症(例如与糖尿病及其并发症相关的炎症、肾脏发炎(例如肾小球肾炎)、皮肤发炎(例如皮炎、银屑病、湿疹、荨麻疹、烧伤)、眼部发炎(例如青光眼)、以及移植器官(例如排异反应)和多器官疾病(例如系统性红斑狼疮、脓毒症)炎症、和病毒或细菌感染的炎性后遗症、以及与动脉粥样硬化相关的炎症和接下来出现在例如大脑或缺血性心脏病中的含氧量低的或缺血性(ischaemic)创伤(包括或不包括再灌注)。
具体地说,本发明化合物可用于治疗和预防炎症、糖尿病和心血管疾病或病症,包括动脉粥样硬化、动脉硬化、高甘油三酯血症、和混合血脂障碍。
烟酸具有明显的副作用,这可能是归于其按照高水平(每日的克数量(gram quantities))给药的缘故。最常见的副作用是强烈的皮肤发红。在本发明的某些实施方案中,本发明化合物具有相对于烟酸而言更低的副作用。已经鉴定HM74A是烟酸的高亲和力受体,而HM74是低亲和力受体。本发明化合物可用作具有选择性的HM74A激动剂或部分激动剂;在这样的情形下,本发明化合物对HM74A显示出比对HM74更高的亲和力。
式(I)化合物活化HM74A的效能可以采用下述酶和体外全细胞测定加以证实:
体外测试
为了进行瞬时转染,将HEK293T细胞(稳定表达SV40大T-抗原的HEK293细胞)保持在含有10%胎儿小牛血清和2mM谷氨酰胺的DMEM中。细胞接种在90mm培养皿中,在转染前生长至60-80%融合(18-24小时)。将人HM74A(GenBankTM登录号AY148884)亚克隆在哺乳动物表达载体(pcDNA3;Invitrogen)中,使用Lipofectamine试剂进行转染。为了进行转染,将9μg DNA与30μl Lipofectamine混和在0.6ml Opti-MEM(LifeTechnologies Inc.)中,在室温下培养30分钟,然后加入1.6ml Opti-MEM。将细胞曝露于Lipofectamine/DNA混合物下5小时,然后加入6ml 20%(v/v)胎儿小牛血清的DMEM溶液。细胞在转染后收获(harvested)48小时。通过以50ngml-1向介质中补充16小时,完成百日咳毒素治疗。全部瞬时转染研究涉及受体与Gi/oG蛋白Go1α的共转染。
为了生成稳定的细胞系,上述方法被用于转染接种在六孔皿中、且生长至30%融合的CHO-K1细胞。将这些细胞保持在含有10%胎儿小牛血清和2mM谷氨酰胺的DMEM F-12HAM介质中。转染48小时后,将介质用400μg/ml遗传霉素(G418,Gibco)补充,进行抗菌素耐受性细胞选择。在加入烟酸之后,稳定表达HM74A的克隆CHO-K1细胞系通过[35S]-GTPγS结合测量加以确认。
P2膜制备-由收获(harvest)后冷冻于-80℃下的细胞团(cell paste)制备得到含有P2颗粒部分的质膜。全部步骤在4℃下进行。将细胞小球(pellet)再次悬浮于1ml的10mM Tris-HCl和0.1mM EDTA,pH 7.5(缓冲液A)中,使用Ultra Turrax匀化20秒钟,接着通过(5次)25-号针。将细胞裂解物(lysates)在微量离心机中、在1,000g下离心10分钟,使胞核和未破损细胞形成小球,P2颗粒部分通过在16,000g下微量离心30分钟进行回收。将P2颗粒部分再次悬浮于缓冲液A中,储存在-80℃下直到被需要时。
[35S]-GTPγS结合-根据先前所述的方法(Wieland,T.和Jakobs,K.H.(1994)Methods Enzymol.237,3-13)在室温下的384-孔中进行测定。简单地说,制备标准或测试化合物的稀释品,然后以体积为10μl加入至384-孔板中。将膜(HM74A或HM74)稀释在测试缓冲液(20mM HEPES,100mM NaCl,10mMMgCl2,pH7.4)中,用皂苷(60μg/ml)、Leadseeker WGA珠(Amersham;250μg/孔)和10μM GDP补充,使得加入至各孔中的20μl体积含有5μg膜。将[35S]-GTPγS(1170Ci/mmol,Amersham)稀释(1∶1500)在测试缓冲液中,向每个孔中加入20μl。加入放射性配体之后,将板密封,脉冲旋转(pulse spun),并在室温下孵育4小时。在孵育期结束时,在Leadseeker机器(VIEWLUX PLUS;Perkin-Elmer)上读板,测定特异性结合水平。
体内试验
在研究前已禁食至少12天的雄性Spague-Dawley大鼠(200-250g)中测试HM74A激动剂。化合物通过静脉内(5ml/kg)或经口强饲法(oral gavage)(10ml/kg)给药。在给药前以及给药后的三个时间点(从给药后15分钟至8小时的时间段)采集血样(0.3ml尾部静脉血)。将每个血样转移至肝素试管(BectonDickinson Microtainer,PST LH)中,离心(10,000g,5分钟)得到血浆样本。使用商购得到的试剂盒(Randox)测定该血浆样本的非酯化脂肪酸(NEFA)水平。采用对血浆NEFA水平的抑制(相对于给药前的水平而言)表示HM74A激动剂活性。
为了确定HM74A化合物是否显示出与烟酸相关的发红响应,将其给药于麻醉后的豚鼠。雄性Dunkin Hartley豚鼠(300-800g)在用含有盐酸氯胺酮(Vetalar,40mg/kg i.m.)、赛拉嗪(Rompun,8mg/kg i.m.)和戊巴比妥钠(Sagatal,30mg/kg i.p.)的混合物麻醉之前,禁食12小时。麻醉后,进行气管造口术,将动物用室内空气机械通风(10-12mL/kg,60次呼吸/分钟)。向颈静脉和颈动脉插入导管以静脉内给药测试化合物,然后分别收集血液。在左耳顶部3-5mm处放置红外温度传感器(Extech Instruments)。在给药测试化合物之前5分钟至给药测试化合物后40分钟内,每分钟记录一次温度测量结果。在Psion计算机上自动收集数据,然后在Excel表格程序中转换进行数据分析。在给药化合物之前以及之后的频繁时间点上,通过颈动脉插管采集血样(0.3ml),并转移至含有肝素锂(lithium heparin)的Microtainer(BD)试管中。样本在血液滚筒(roller)上充分混和,然后储存在冰上,之后在1200g下离心5分钟。
烟酸(10mg/kg i.v.)引起相当于10.42+1.44(曲线下面积;随机单元;n=6)的耳朵温度的平均(+s.e.m.)升高。通过对照,实施例30的化合物(10mg/kg i.v.)产生相当于1.52+0.39(曲线下面积;随机单元;n=6)的耳朵温度的平均(+s.e.m.)升高,减少85%。
合成得到根据式(I)的化合物(参见下面的合成实施例),并在上述的一个或多个所述试验中进行测试。所有的实施例化合物均具有4.9(+/-0.3对数单位)或更高的pEC50以及30%或更高的效力。下面例举了部分具体化合物。
一般纯化和分析方法:
质谱(MS)是在使用电喷射阳极电离作用[(ES+ve得到MH+和M(NH4)+分子离子]或者电喷射阴极电离作用[(ES-ve得到(M-H)-分子离子]模式的FisonsVG Platform质谱仪上记录的。
1H NMR谱使用Bmker DPX 400MHz光谱仪记录,使用四甲基甲硅烷作为外标。
BiotageTM色谱法是指使用由Dyax Corporation销售的装置(Flash 40i或Flash 150i)和预先用KPSil装填的柱进行纯化。
质量定向自动化制备(Mass directed autoprep)是指这样一种方法:其中物质通过高效液相色谱法、在HPLCABZ+5μm柱(5cm x 10mm i.d.)上使用0.1%HCO2H的水溶液以及95%MeCN、5%水(0.5%HCO2H)进行纯化,采用下述梯度洗脱条件:0-1.0分钟5%B,1.0-8.0分钟5→30%B,8.0-8.9分钟30%B,8.9-9.0分钟30→95%B,9.0-9.9分钟95%B,9.9-10分钟95→0%B,流速为8ml分钟-1(体系2)。Gilson 202-部分收集器在检测感兴趣的质量峰时由VG Platform质谱仪启动。
制备性h.p.l.c.是指这样一种方法:其中物质通过高效液相色谱法、在HPLCABZ+5μm柱(10cm x 21.2mm i.d.)上使用0.1%HCO2H的水溶液(A)和MeCN(0.5%HCO2H)(B)进行纯化,采用具有下述梯度体系表达为“x至y”的一般梯度洗脱条件:0-1.45分钟x%B,1.45-20分钟x→y%B,20-24分钟y→95%B,24-30分钟95%B,32-34分钟95→x%B,流速为8ml分钟-1。Gilson233部分收集器由UV(254nm)启动。
SPE(固相萃取)是指采用由International Sorbent Technology Ltd销售的柱。
Strata Phenyl SPE是指使用由Phenomenex销售的柱。将化合物装填在预先经MeCN调节(conditioned)、然后用5%MeCN的水溶液平衡的柱上。将化合物用0.1%HCO2H的水溶液和MeCN(0.5%HCO2H),按照适宜的梯度在Combiflash Optix 10上洗脱。
如上所示,发现式(I)化合物可用于人药或兽药,特别是用作HM74A激活剂用于控制血脂障碍和高脂蛋白血症。
因此,本发明另一方面提供了式(I)化合物或其生理学功能的衍生物用于人药或兽药,特别是用于治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍,心力衰竭,血胆脂醇过多症,心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症。本发明化合物同样被提供用于治疗冠状动脉疾病、血栓症、心绞痛、慢性肾衰竭、周围血管疾病和中风。
本发明另一方面提供了式(I)化合物或其生理学功能的衍生物用于制备治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍,心力衰竭,血胆脂醇过多症,心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症的药物。本发明化合物同样被提供用于治疗冠状动脉疾病、血栓症、心绞痛、慢性肾衰竭、周围血管疾病和中风。
应该理解的是,本文中的治疗延伸包括预防、防止症状的复发和抑制以及对已确诊病症的治疗。
根据本发明另一方面,提供了式(II)化合物及其生理学功能的衍生物,

其中:
R1选自:氢以及可任选被一个或多个选自CN和CF3中的基团取代的C1-4烷基;
R2选自:未被取代的C2-10烷基、被一个或多个选自氟和CN中的基团取代的C1-10烷基、C5链烯基、直链C4链烯基、以及被环烷基取代的C1-4烷基;
和R3选自卤素和CN;
在制备用于治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症的药物中的用途。具体地,所述用途为提供式(II)化合物在制备用于治疗糖尿病血脂障碍或混合血脂障碍,心力衰竭,血胆脂醇过多症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症,冠状动脉疾病,血栓症,心绞痛,慢性肾衰竭,中风以及心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症的药物中的用途。
在本发明一实施方案中,提供了式(II)化合物用于治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症。具体地,所述用途为提供式(II)在制备用于治疗糖尿病血脂障碍或混合血脂障碍,心力衰竭,血胆脂醇过多症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症,冠状动脉疾病,血栓症,心绞痛,慢性肾衰竭,中风以及心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症的药物中的用途。
在具体实施方案中,R1选自:氢、C1-4烷基、CH2CN和(CH2)3CF3。在更具体的实施方案中,R1选自:氢和甲基。
在某些实施方案中,R2选自:未被取代的C3-10烷基、被一个或多个选自氟和CN中的基团取代的C1-10烷基、C5链烯基、直链C4链烯基、以及被环烷基取代的C1-4烷基。具体地,R2选自:未被取代的C3-10烷基、具有一个或多个CN取代基的C1-6烷基、具有一个或多个氟取代基的C1-10烷基、C5链烯基、直链C4链烯基、以及被环烷基取代的C1-4烷基。更具体地,R2选自:未被取代的C3-10烷基;(CH2)1-5CN;具有一个或多个氟取代基的C2-5烷基;C5链烯基;以及被环烷基取代的C1-4烷基。最具体地,R2选自未被取代的C4-6正烷基,例如戊基;(CH2)1-3CN,例如(CH2)CN或(CH2)3CN;具有一个或多个氟取代基的C3-4烷基,特别是其中末端碳完全用氟饱和的C3-4烷基,例如(CH2)2-3CF3;以及C5链烯基,特别是其中仅有一个双键的C5链烯基,例如其中双键位于第四个和第五个碳之间的C5链烯基(末端链烯基)。
在具体实施方案中,R3表示卤素。更具体地,R3选自氯和溴。最具体地,R3表示氯。
用于治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症、或者制备用于治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症的药物的具体化合物包括:
(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)乙腈、
3-丁基-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-溴-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(3,3,3-三氟丙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-1-丙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
3-丁基-8-氯-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮、
(3-丁基-8-氯-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈、
8-氯-3-(2-环丙基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮、
4-(8-氯-1-甲基-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈、
8-氯-1-乙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
1-甲基-2,6-二氧代-3-戊基-2,3,6,7-四氢-1H-嘌呤-8-腈、
8-氯-3-丙基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(3-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮、
3-丁基-1-甲基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈、
8-氯-3-(4-戊烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-己基-3,7-二氢-1H-嘌呤-2,6-二酮、
4-(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈、
8-氯-3-己基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮、
3-丁基-8-氯-1-乙基-3,7-二氢-1H-嘌呤-2,6-二酮、
[8-氯-3-(2-环丙基乙基)-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-1-基]乙腈、
(8-氯-2,6-二氧代-3-丙基-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈、
8-氯-1-(4,4,4-三氟丁基)-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
2,2′-(8-氯-2,6-二氧代-6,7-二氢-1H-嘌呤-1,3(2H)-二基)二乙腈、
8-氯-1-甲基-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(2-环己基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
1,3-二丁基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈、
1,3-二丁基-8-碘-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(4-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(6-甲基庚基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-辛基-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-癸基-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(环己基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮、
(+/-)-8-氯-3-(3-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(2-环戊基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(环丙基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮、
(+/-)-8-氯-3-(2-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮、
(+/-)-8-氯-3-(2-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(环丁基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(环戊基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(3-环丙基丙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(2-环丁基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(4-氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(3-氟丙基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-(5-氟戊基)-3,7-二氢-1H-嘌呤-2,6-二酮、
4-(8-氯-1-甲基-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈、
3-(3-丁烯-1-基)-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮、
6-(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)-2,2-二甲基己腈、
8-氯-3-(6-氟己基)-3,7-二氢-1H-嘌呤-2,6-二酮、
8-氯-3-乙基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮。
应该理解的是,本发明该方面包括这些具体实施方案的任何组合,并且覆盖针对式(II)化合物的上述具体取代基的所有组合。
此外,本发明提供了式(I)化合物或其生理学功能的衍生物在制备用于治疗关节炎性疾病或病症,特别是关节炎(例如类风湿性关节炎、骨关节炎、假关节失灵)、或者胃肠道炎症(例如溃疡性结肠炎、克罗恩氏病、以及其它炎性肠病和胃肠疾病、由感染引起的胃炎和粘膜炎症、由非甾体消炎药物引起的肠病)、肺部发炎(例如成人呼吸窘迫综合征、哮喘、囊性纤维化病、或慢性阻塞性肺疾患)、心脏发炎(例如心肌炎)、神经组织炎症(例如多发性硬化)、胰腺炎症(例如与糖尿病及其并发症相关的炎症、肾脏发炎(例如肾小球肾炎)、皮肤发炎(例如皮炎、银屑病、湿疹、荨麻疹、烧伤)、眼部发炎(例如青光眼)、以及移植器官(例如排异反应)和多器官疾病(例如系统性红斑狼疮、脓毒症)炎症、和病毒或细菌感染的炎性后遗症、以及与动脉粥样硬化相关的炎症和接下来出现在例如大脑或缺血性心脏病中的含氧量低的或缺血性创伤(包括或不包括再灌注)的药物中的用途。
在另一或替代方面,提供了治疗患有由HM74A受体激活不足所引起的病症或者受益于激活HM74A受体的病症的人或动物的方法,所述方法包括向所述人或动物对象给药有效量的式(I)化合物或其生理学上可接受的盐或溶剂化物。
同样应该理解的是,本发明该方面包括这些具体实施方案的任何组合,并且覆盖针对式(I)化合物的上述具体取代基的所有组合。
更具体地,本发明提供了治疗脂类代谢障碍包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍,心力衰竭,血胆脂醇过多症,心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症的方法,所述方法包括向所述人或动物对象给药有效量的式(I)化合物或其生理学上可接受的盐或溶剂化物。同样,这些化合物还可用于治疗冠状动脉疾病、血栓症、心绞痛、慢性肾衰竭、周围血管疾病和中风的方法中,所述方法包括向所述人或动物对象给药有效量的式(I)化合物。
获得理想生物效应所需的HM74A调节剂的量当然取决于各种因素,例如给药模式和接受者具体的临床病症。一般来说,日剂量可以为0.1mg-1g/kg,通常为0.1-100mg/kg。静脉内给药剂量可以例如为0.01mg-0.1g/kg,通常为0.01mg-10mg/kg,其可以方便地通过0.1μg-1mg/分钟输注给药。适合上述目的的输注液体可以含有例如0.01μg-0.1mg/毫升。单位剂量中可以含有例如0.01μg-1g HM74A调节剂。因此,注射安瓿中可以含有例如0.01μg-0.1g,口服单位剂量制剂例如片剂或胶囊剂中可以含有例如0.1mg-1g。当本发明化合物以上述剂量范围给药时,未发现/预期到毒性效应。
本发明化合物本身可用于治疗由HM74A受体激活不足所引起的疾病或者受益于激活该受体的疾病,其实例是将本发明化合物与可接受载体以药物制剂的形式存在。当然,所述载体在能与制剂中其它成分配伍方面必须是可接受的,同时对接受者必须无毒。所述载体可以是固体或液体、或者两者兼有,可以将HM74A调节剂制备成单位剂量制剂形式,例如片剂,其中含有0.05重量%-95重量%的HM74A调节剂。
所述制剂包括那些适合口服、直肠、局部、口腔(例如舌下)和肠胃外(例如皮下、肌内、皮内或静脉内)给药的制剂。
根据本发明还提供了制备这类药物组合物的方法,所述方法包括将各种成分进行混和。
适合口服给药的制剂可以以个别的单位形式存在,例如各自含有预定量的HM74A调节剂的胶囊剂、扁囊剂、锭剂或片剂;粉剂或颗粒剂形式;在水或非水液体中的溶液剂或混悬剂形式;或者水包油或油包水乳剂形式。一般来说,通过将活性HM74A调节剂与液体或细分的固体载体、或者两者兼有进行均匀且致密的混和,然后如果需要的话,使产品成型,这样可以制备得到上述制剂。例如,片剂可以通过将HM74A调节剂的粉末或颗粒与任选的一种或多种辅助成分压制或成型而制备得到。压制片剂可以通过在适宜的机器中将自由流动(free-flowing)形式的本发明化合物例如任选与(一种或多种)粘合剂、润滑剂、惰性稀释剂和/或表面活性剂和/或分散剂混合的粉末或颗粒进行压制而制备得到。模制(Moulded)片剂可以通过在适宜的机器中将用惰性液体稀释剂润湿的粉状化合物进行模制而制备得到。
口服给药的片剂和胶囊剂中可以含有常规赋形剂例如:粘合剂,如糖浆、阿拉伯胶、明胶、山梨糖醇、西黄蓍胶、淀粉粘液或聚乙烯吡咯烷酮;填充剂,如乳糖、微晶纤维素、糖、玉米淀粉、磷酸钙或山梨糖醇;润滑剂,如硬脂酸镁、硬脂酸、滑石、聚乙二醇或二氧化硅;崩解剂,如马铃薯淀粉、交联羧甲基纤维素钠或淀粉羟乙酸钠;或者润湿剂,如十二烷基硫酸钠。片剂可以按照本领域熟知的方法包衣。口服液体制剂可以是例如水性或油性混悬剂、溶液剂、乳剂、糖浆剂或酏剂形式,或者也可以是在使用前与水或其它适宜载体重构(constitution)的干燥产品形式。这种液体制剂可以含有常规添加剂例如:助悬剂,如山梨糖醇糖浆、甲基纤维素、葡萄糖/食用糖糖浆、明胶、羟甲基纤维素、羧甲基纤维素、硬脂酸铝凝胶或者氢化食用脂;乳化剂,如卵磷脂、脱水山梨糖醇单油酸酯或阿拉伯胶;非水载体(其中可以含有食用油),如杏仁油、分馏椰子油(fractionated coconut oil)、油性酯、丙二醇或乙醇;或者防腐剂,如对羟基苯甲酸甲酯或丙酯或者山梨酸。如果适合的话,制剂中还可以含有缓冲盐、调味剂、着色剂和/或甜味剂(例如甘露醇)。
适合口腔(舌下)给药的制剂包括在调味基质(通常为蔗糖和阿拉伯胶或西黄蓍胶)中含有HM74A调节剂的锭剂、以及在惰性基质例如明胶和甘油或者蔗糖和阿拉伯胶中含有HM74A调节剂的软锭剂。
适合肠胃外给药的本发明制剂通常包括HM74A调节剂的无菌含水制剂,所述制剂可与预期接受者体内的血液等渗。这些制剂可以通过静脉内给药,当然还可以通过皮下、肌内、或皮内注射实现。这类制剂通常可以通过将HM74A调节剂与水进行混和,然后将所得到的溶液灭菌并使其与血液等渗而制备得到。根据本发明的可注射组合物通常含有0.1-5%w/w HM74A调节剂。
因此,可以将含有本发明化合物的适合肠胃外给药的本发明制剂配制成通过快速浓注(bolus injection)或连续输注进行肠胃外给药的形式,并且可以以单位剂量形式存在,例如安瓿、小瓶(vials)、小体积输液或者预装注射器形式,或者以具有添加的防腐剂的多剂量容器形式存在。所述组合物可以采取例如在水或非水载体(vehicles)中的溶液剂、混悬剂、或乳剂形式,并且可以含有各种配制制剂例如抗氧化剂、缓冲剂、抗微生物剂和/或毒性调节剂。或者,所述活性成分可以为在使用之前与适宜载体例如无菌无热原的水重构的粉末形式。所述干燥固体形式可以通过将无菌粉末无菌填入单独的无菌容器中制备得到,或者通过将无菌溶液无菌填入各容器中,然后再冻干而制备得到。
适合直肠给药的制剂可以以单位剂量栓剂形式存在。所述单位剂量栓剂可以通过将HM74A调节剂与一种或多种常规固体载体例如可可豆脂或甘油酯混和,然后再使所得到的混合物成型而制备得到。
适合局部应用至皮肤的制剂可以采取软膏剂、乳剂、洗剂、糊剂、凝胶剂、喷雾剂、气雾剂、或油剂形式。可使用的载体包括凡士林、羊毛脂、聚乙二醇、醇类、以及其中的两种或多种的混合物。所述HM74A调节剂通常以占组合物0.1-15%w/w、例如0.5-2%w/w的浓度存在。
本文所使用的局部给药包括吹入和吸入给药。用于局部给药的各种类型的制剂实例包括软膏剂、乳剂、洗剂、粉剂、阴道栓剂、喷雾剂、气雾剂、胶囊剂、用于吸入器或吹入器中的药筒或者滴剂(例如滴眼剂或滴鼻剂)。
软膏剂和乳剂可以通过例如使用水性或油性基质,同时加入适宜的增稠剂和/或胶凝剂和/或溶剂配制得到。因此,这类基质可以例如包括水和/或油例如液体凡士林或者植物油例如花生油或蓖麻油或者溶剂例如聚乙二醇。可使用的增稠剂包括软石蜡、硬脂酸铝、十八醇十六醇混合物、聚乙二醇、微晶蜡和蜂蜡。
洗剂可以使用水性或油性基质配制得到,并且一般来说,还可以含有一种或多种乳化剂、稳定剂、分散剂、助悬剂或增稠剂。
用于外敷的粉剂可以借助于适宜的粉末基质例如滑石、乳糖或淀粉形成。滴剂可以使用水性或非水性基质配制得到,并且还可以含有一种或多种分散剂、增溶剂或助悬剂。
喷雾组合物可以被配制成例如水溶液剂或混悬剂形式或者由加压包装中借助适宜的推进剂递送的气雾剂,其中推进剂例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、1,1,1,2,3,3,3-七氟丙烷、1,1,1,2-四氟乙烷、二氧化碳或其它适宜的气体。
可以将用于吸入器或吹入器中的胶囊剂和药筒(cartridges)(例如明胶)配制成包含本发明化合物与适宜的粉末基质例如乳糖或淀粉的粉末的混合物。
根据本发明的药物组合物还可以与其它的治疗剂组合(combination)使用,例如与其它类型的血脂障碍药物(例如斯特汀、贝特类(fibrates)、胆汁酸结合树脂或烟酸)组合使用。
本发明化合物可以与一种或多种其它治疗剂组合使用,例如与其它类型的血脂障碍药物组合使用,如3-羟基-3-甲基戊二酰辅酶A还原酶抑制剂(斯特汀)或贝特类或胆汁酸结合树脂或者烟酸。因此本发明另一方面提供了所述组合(combination)在治疗由HM74A受体激活不足所引起的疾病或者受益于激活该受体的疾病中的用途,以及式(I)或(II)化合物或其可药用盐、溶剂化物或生理学功能的衍生物在制备用于联合治疗(combination therapy)脂类代谢障碍包括血脂障碍或高脂蛋白血症例如糖尿病血脂障碍和混合血脂障碍,心力衰竭,血胆脂醇过多症,心血管疾病包括动脉粥样硬化、动脉硬化、和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症或肥胖症的药物中的用途。
当本发明化合物与其它治疗剂组合使用时,这些化合物可以通过任何常规途径依次或者同时给药。
前面所称的组合可以方便地通过使用药物制剂的形式来实现,因此含有上述组合以及任选的可药用载体或赋形剂的药物制剂构成了本发明另一方面。这类组合中的单独组分可以以单独或合并的药物制剂形式依次或同时给药。
当被组合在同一制剂中时,应该理解这两种组分必须是稳定的,并且能够彼此配伍以及与制剂中的其它组分同样能够配伍,并且还可以配制用于给药。当分别配制时,它们可以以任意的常规制剂形式(通常是本领域针对这类化合物已知的方式)提供。
当与对抗相同疾病的第二治疗剂组合时,每种组分的剂量可以与化合物单独使用的剂量不同。适宜的剂量可以方便地由本领域技术人员加以确定。
因此,本发明另一方面提供了含有式(I)或(II)化合物或其生理学上可接受的盐或溶剂化物与其它治疗活性剂的组合。
前面所称组合可以方便地通过使用药物制剂的形式来实现,因此含有上述组合与其可药用载体的药物制剂构成了本发明另一方面。
本发明化合物具有有益的作用持续时间。
本发明化合物及其盐和溶剂化物可以按照下文中所述的方法制备,这构成了本发明的另一方面。
方法A:
根据本发明的制备其中R1是H或者与R2相同、和R3是Cl的式(I)或式(II)化合物的方法,所述方法包括:

i)使用烯丙基溴烷基化鸟嘌呤
ii)使用亚硝酸钠重氮化,接着再水解形成黄嘌呤
iii)氯化
iv)在N3上烷基化和/或在N1和N3上二烷基化
v)钯催化除去烯丙基
方法B:
根据本发明的制备其中R3是CN的式(I)或式(II)化合物的方法,所述方法包括方法A中的步骤(i)和(ii),接着:

iii)在N3上烷基化
iv)在N1上烷基化
v)通过用LiHMDS锂化和DMF猝灭,在C8上形成醛
vi)将醛转化为腈
vii)钯催化除去烯丙基
方法C:
根据本发明的制备其中R3是Cl或Br的式(I)或式(II)化合物的方法,所述方法包括方法B中的步骤(i)-(iv),接着:

i)在C8上用NCS或NBS卤化
ii)钯催化除去烯丙基
方法D:
根据本发明的制备其中R3是CN的式(I)或式(II)化合物的方法,所述方法包括方法B中的步骤(i)-(iv),接着:

v)形成酯
vi)甲基酯的水解
vii)将酸转化为酰胺
viii)将酰胺转化为腈
ix)钯催化除去烯丙基
方法E:
根据本发明的制备其中R3是Cl的式(I)或式(II)化合物的方法,所述方法包括:

i)在N3上烷基化
ii)在N1上烷基化
iii)脱苄基反应
iv)在C8上氯化
方法F:
根据本发明的制备其中R1与R2不同、且R3是Cl的式(I)或式(II)化合物的方法,所述方法包括方法A中的步骤(i)-(iv),接着:

v)在N1上烷基化
vi)钯催化除去烯丙基
方法G:
根据本发明的制备其中R1与R2不同、和R3是Cl的式(I)或式(II)化合物的方法,所述方法包括方法F中的步骤(i)-(v)(其中方法F中的R2具体为SEM或MEM),接着:

vi)裂解MEM或SEM保护基团
vii)在N3上烷基化,接着钯催化除去烯丙基
方法H:
根据本发明的制备其中R3是Cl、Br、I或F的式(I)或式(II)化合物的方法,所述方法包括方法B中的步骤(i)-(iv),接着:

v)钯催化除去烯丙基
vi)在C8上使用NCS、NBS或NIS卤化
方法I:
根据本发明的制备其中R1是H或烷基、R2是烷基、和R3是Cl的式(I)或式(II)化合物的方法,所述方法包括:

i)形成嘧啶二酮
ii)亚硝基化
iii)使用Na2S2O4或者类似的还原剂还原
iv)形成黄嘌呤
v)在N1上烷基化(任选)
vi)在C8上使用NCS卤化
如果适合或者需要的话,作为任意一种上述合成方法中的最后步骤,可以将所得到的式(I)或式(II)化合物转化为生理学上可接受的盐形式,或者反之亦然,将一种盐形式转化为另一种生理学上可接受的盐形式。
缩写
THF 四氢呋喃
Ac 乙酰基
DCM 二氯甲烷
DMEM Dulbecco改进的Eagle培养基
HEPES 4-(2-羟基乙基)哌嗪-1-乙磺酸
DMSO 二甲亚砜
NBS N-溴代琥珀酰亚胺
NCS N-氯代琥珀酰亚胺
NIS N-碘代琥珀酰亚胺
DMF 二甲基甲酰胺
LiHMDS 六甲基二甲硅烷基氨基化锂
DBAD 偶氮二羧酸二苄基酯
DIPEA 二异丙基乙基胺
PyBOP 苯并三唑-1-基氧基三吡咯烷基磷
鎓(tripyrrolidinophosphonium)六氟磷酸盐
MEM 甲氧基乙氧基甲基
SEM 2-(三甲基甲硅烷基)乙氧基甲基
TFA 三氟乙酸
RT 室温
△ 加热
利用下述非限制性实施例对本发明进行示例性说明:
合成实施例
实施例1:8-氯-3-(4-戊烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮
a)2-氨基-7-(2-丙烯-1-基)-1,7-二氢-6H-嘌呤-6-酮

在室温、氮气氛下,将含有鸟苷(20g,0.071mol)、烯丙基溴(14.7ml,0.169mol)和无水DMSO(100ml)的混合物搅拌18小时。以一批加入浓HCl(50ml,37%),将混合物搅拌45分钟后,倾入MeOH(600ml)中。该甲醇溶液用2M NaOH(aq)溶液中和,通过过滤收集所得到的白色沉淀。该白色固体在50℃下真空干燥18小时,得到标题化合物(16g粗产物,119%)。m/z 192.2[MH+]。
b)7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮
将2-氨基-7-(2-丙烯-1-基)-1,7-二氢-6H-嘌呤-6-酮(40g,0.209mol)在AcOH(900ml)和水(100ml)中的混合物在55℃下加热。逐滴加入亚硝酸钠(57.74g,0.837mol)的水(100ml)溶液。注意有毒气体。加料完毕后(大约25分钟),反应混合物冷却至环境温度,然后浓缩至其初始体积的大约1/3。加入水(500ml),所得到的沉淀通过过滤收集。残余物用水洗涤,然后在50℃下用P2O5真空干燥2小时,得到标题化合物(17.20g)。含水部分浓缩后,加入水(100ml)。所得到的固体再次过滤并干燥。这次得到更多的标题化合物(2.31g)。合并产物(19.52g,49%)。m/z 193.2[MH+]。
c)8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮)(10.52g,54.7mmol)的无水DMF(60ml)溶液中加入NCS(8.04g,60.2mmol)。反应混合物在氮气氛、20℃下搅拌6小时。反应混合物真空浓缩得到琥珀色油状物。加入MeOH,放置18小时。所得到的残余物过滤并真空干燥,得到标题化合物(7.69g,62%)。m/z 227.2[MH+]。
d)8-氯-3-(4-戊烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.10g,0.44mmol)溶解于含有碳酸钠(0.12g,0.49mmol)和5-溴戊烯(0.07g,0.49mmol)的DMF(1.5ml)中,混合物搅拌18小时。烷基化完毕后,加入吗啉(0.5ml)和四(三苯基膦)钯(0)(0.08g,0.07mmol),继续搅拌3.5小时。反应用乙酸乙酯(10ml)稀释,依次用2N盐酸(2x5ml)和盐水(3x5ml)洗涤,分离有机相,干燥(MgSO4)并浓缩。将粗产物悬浮于甲醇(2ml)中,然后在氨基丙基SPE(5g)上纯化,先用甲醇洗脱,再用5%乙酸的甲醇溶液洗脱,浓缩后分离得到为白色固体的标题化合物(0.039g,35%)。NMR;(400MHz,d6-DMSO)1.75(m,2H),2.05(m,2H),3.85(t,2H,J=7Hz),4.95(m,1H),5.05(m,1H),5.8(m,1H),11.1(br s,1H),一个可交换质子未观察到δH 13;m/z 255[MH+]。
实施例2:8-氯-3-己基-3,7-二氢-1H-嘌呤-2,6-二酮

使用己基碘化物,按照类似于实施例1的方式制备,得到标题化合物。NMR;δH(400MHz,d6-DMSO)0.85(t,3H,J=7Hz),1.25(br s,6H),1.6(m,2H),3.85(t,2H,J=8Hz),11.2(br.s,1H),一个可交换质子未观察到δH 13;m/z 271[MH+]。
实施例3和4:(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)乙腈和 2,2′-(8-氯-2,6-二氧代-6,7-二氢-1H-嘌呤-1,3(2H)-二基)二乙腈
a)[8-氯-2,6-二氧代-7-(2-丙烯-1-基)-1,2,6,7-四氢-3H-嘌呤-3-基]乙腈和2,2′-[8-氯-2,6-二氧代-7-(2-丙烯-1-基)-6,7-二氢-1H-嘌呤-1,3(2H)-二基]二乙腈

将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.445g,2.0mmol)的DMF(8ml)溶液用碳酸钠(0.18g,1.7mmol)和溴代乙腈(0.1ml,1.4mmol)处理。搅拌后的混合物在70℃下加热3小时,然后冷却至50℃,进一步用溴代乙腈(0.06ml,0.8mmol)处理。混合物在50℃下继续保持2小时,然后冷却至环境温度并蒸发至干。残余物用1M盐酸水溶液(20ml)处理,并用乙酸乙酯(2x50ml)萃取。合并有机部分,用硫酸镁干燥,过滤并蒸发。残余物溶解于二氯甲烷(2ml)中,20分钟后,滤出生成的沉淀固体(未反应的8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮),进一步用二氯甲烷洗涤。滤液真空浓缩后,经快速色谱法处理,使用梯度洗脱为1∶3至4∶1的乙酸乙酯/环己烷作为洗脱剂。得到两个标题化合物:
[8-氯-2,6-二氧代-7-(2-丙烯-1-基)-1,2,6,7-四氢-3H-嘌呤-3-基]乙腈
白色固体(0.084g,16%);m/z 266[MH+]。
2,2′-[8-氯-2,6-二氧代-7-(2-丙烯-1-基)-6,7-二氢-1H-嘌呤-1,3(2H)-二基]二乙腈
白色固体(0.195g,32%);m/z 305[MH+]。
b)(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)乙腈
将[8-氯-2,6-二氧代-7-(2-丙烯-1-基)-1,2,6,7-四氢-3H-嘌呤-3-基]乙腈(0.084g,0.32mmol)的THF(5ml)溶液通过连续向反应混合物施加真空和氮气压除去气体。溶液依次用吗啉(0.3ml,3.4mmol)和四(三苯基膦)钯(0)(0.03g,0.03mmol)处理。2小时后,混合物用2M盐酸水溶液(3ml)和氯仿(5ml)处理。混合物分离后,蒸发有机相。产物由残余物中使用质量定向(mass-directed)HPLC纯化,得到为白色固体的标题化合物(0.018g,25%)。NMR δH(400MHz,d6-DMSO)4.95(s,2H),11.49(s,1H),14.63(br.s,1H);m/z 226[MH+]。
c)2,2′-(8-氯-2,6-二氧代-6,7-二氢-1H-嘌呤-1,3(2H)-二基)二乙腈

使用合成(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)乙腈所描述的条件,由2,2′-[8-氯-2,6-二氧代-7-(2-丙烯-1-基)-6,7-二氢-1H-嘌呤-1,3(2H)-二基]二乙腈制备得到标题化合物。得到为白色固体的标题化合物0.06g(4%);NMR δH(400MHz,d6-DMSO)4.88(s,2H),5.06(s,2H),NH未观察到δH 14;m/z 282[MNH4 +]。
实施例5:8-氯-3-(3,3,3-三氟丙基)-3,7-二氢-1H-嘌呤-2,6-二酮

使用3-溴-1,1,1-三氟丙烷作为烷基化试剂,按照类似于实施例3的方式制备,得到标题化合物。
NMR δH(400MHz,d6-DMSO)2.64-2.76(m,2H),4.12(t,2H,J=7Hz),11.30(s,1H),14.46(br.s,1H);m/z 283[MH+]。
实施例6:8-氯-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

使用2-溴-1,1,1-三氟乙烷作为烷基化试剂和碳酸氢钠作为碱,按照类似于实施例3的方式制备,得到标题化合物。
δH(400MHz,d4-MeOD)4.68(q,2H,J=8.5Hz);m/z 267.1[M-H]-。
实施例7和8:8-氯-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮和8- 氯-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮

a)8-氯-7-(2-丙烯-1-基)-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮和8-氯-7-(2-丙烯-1-基)-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.5g,6.64mmol)、碳酸钠(844mg,7.9mmol)和4-溴-1,1,1-三氟丁烷(1.39g,7.3mmol)在二甲基甲酰胺(25ml,干燥)中搅拌7天。反应混合物在乙酸乙酯和水之间分配。有机相分离后用盐酸(2N)、盐水洗涤,干燥(MgSO4)并蒸发至干。粗产物用乙醚研磨,固体通过过滤收集,得到为白色固体的8-氯-7-(2-丙烯-1-基)-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.23g,57%)。m/z 337[MH+]。
浓缩后的滤液在二氧化硅、SPE柱(20g)上色谱处理。用环己烷∶乙酸乙酯(10∶1至2∶1)洗脱,得到为浆状物的8-氯-7-(2-丙烯-1-基)-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮(480mg,16%)。m/z 447[MH+]。
b)8-氯-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮
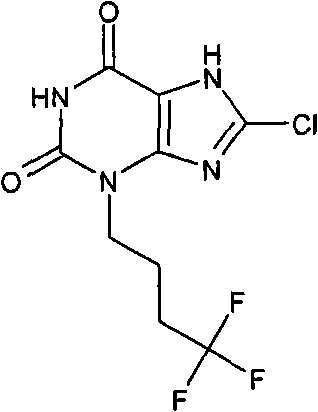
将8-氯-7-(2-丙烯-1-基)-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮(84mg,0.25mmol)和吗啉(220μl,2.5mmol)用氮气在四氢呋喃(3ml)中除气,然后加入四(三苯基膦)钯(0)(29mg,0.025mmol),反应在室温下搅拌过夜。通过过滤收集白色沉淀,用四氢呋喃和乙醚洗涤,得到标题化合物的吗啉盐(59mg)。将其用2N HCl和甲醇处理,溶剂蒸发至干,然后重新溶解于DMSO/MeOH中,通过制备性HPLC纯化,使用10-40%梯度洗脱,得到标题化合物(11mg,14.9%)。NMR δH(400MHz,d4-MeOD)1.92-2.03(m,2H),2.19-2.33(m,2H),4.06(t,2H,J=7Hz);m/z 297[MH+]。
c)8-氯-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮
将8-氯-7-(2-丙烯-1-基)-1,3-双(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮(478mg,1.1mmol)和吗啉(937μl,11mmol)用氮气在四氢呋喃(10ml)中除气,然后加入四(三苯基膦)钯(0)(123mg,0.11mmol),反应在室温下搅拌过夜。反应混合物在二氯甲烷和盐酸2N之间分配。有机相分离后,浓缩得到粗产物。将其通过氨基丙基SPE(5g)纯化,然后由乙腈重结晶,得到标题化合物(75.5mg,16.9%)。NMR.δH(400MHz,CDCl3)1.96-2.13(m,4H),2.15-2.29(m,4H),4.15-4.23(m,4H),12.94(br.s,1H);m/z 407[MH+]。
实施例9:8-氯-3-(2-环丙基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

a)8-氯-3-(2-环丙基乙基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.5g,6.64mmol)、碳酸钠(844mg,7.9mmol)和甲磺酸2-环丙基乙酯(1.19g,7.3mmol)在80℃下的二甲基甲酰胺(25ml,无水)中搅拌2天。反应混合物在乙酸乙酯和水之间分配。有机相分离后,用盐酸(2N)、盐水洗涤,干燥(MgSO4)并蒸发至干。粗产物用乙醚研磨,通过过滤收集固体,得到为白色固体的标题化合物(0.96g,49%)。m/z 295[MH+]。
b)8-氯-3-(2-环丙基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-3-(2-环丙基乙基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(74mg,0.25mmol)和吗啉(220μl,2.5mmol)用氮气在四氢呋喃(3ml)中除气,然后加入四(三苯基膦)钯(0)(29mg,0.025mmol),反应在室温下搅拌过夜。通过过滤收集白色沉淀,用四氢呋喃和乙醚洗涤,得到标题化合物的吗啉盐(52mg)。将其用2N HCl和甲醇处理,溶剂蒸发至干,然后重新溶解于DMSO/MeOH中,通过制备性HPLC纯化,使用10-40%梯度洗脱,得到标题化合物(22mg,34.6%)。NMR δH(400MHz,d4-MeOD)0.00-0.05(m,2H),0.37-0.43(m,2H),0.67-0.77(m,1H),1.61(q,2H,J=7Hz),4.06-4.11(m,2H);m/z 255[MH+]。
实施例10:3-丁基-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮

a)3-丁基-8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向3-丁基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(3.34g,13.4mmol)的无水DMF(19ml)溶液中加入NCS(1.97g,14.8mmol),然后在室温和氮气氛下搅拌22小时。混合物真空浓缩,得到黄色固体,过滤后用甲醇洗涤。滤液浓缩后重复上述步骤。滤液最后一次洗涤后,通过SPE(Si,20g)柱纯化,用1∶1的EtOAc∶环己烷洗脱。合并的固体真空干燥后,得到标题化合物(2.42g,64%);m/z 283.3[MH+]。
b)3-丁基-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮

将3-丁基-8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.35mmol)在无水THF(4ml)和无水DMSO(0.4ml)中的溶液用Pd(PPh3)4(61mg,0.053mmol)处理。混合物在轻度真空下除气,加入吗啉(308μL,3.5mmol),在室温和氮气氛下搅拌4小时。黄色溶液在2M HCl(aq)和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩。残余物溶于(taken up)MeOH中,通过氨基丙基SPE(5g),用MeOH洗脱,再用5%AcOH/MeOH洗脱。各部分产物合并后真空浓缩,得到为灰白色固体的标题化合物(30mg,35%)。NMR;δH(400MHz,d6-DMSO)0.89(t,3H,J=7.5Hz),1.23-1.34(m,2H),1.55-1.65(m,2H),3.85(t,2H,J=7Hz),11.17(s,1H),14.37(br.s,1H);m/z 243.3[MH+]。
实施例11:8-氯-3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮

将3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮(J.Med.Chem,1993,36(10),1380-6)(0.3g,1.5mmol)和N-氯代琥珀酰亚胺(0.21g,1.5mmol)溶解于DMF(5ml)中,溶液搅拌5小时。溶液浓缩后,固体残余物用甲醇洗涤,过滤得到为白色固体的产物(0.148g,42%)。NMR;δH(400MHz,d6-DMSO)0.85(t,3H,J=7Hz),1.65(m,2H),3.8(t,2H,J=7Hz),11.2(s,1H),一个可交换质子未观察到δH 13;m/z 229[MH+]。
实施例12:8-氯-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮
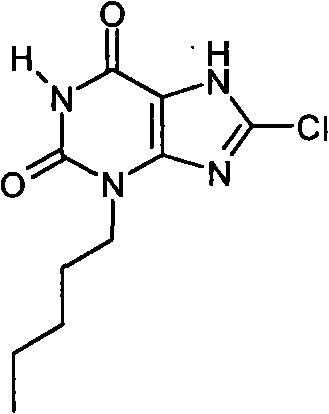
a)8-氯-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.44mmol)的无水DMF(3ml)溶液中加入碳酸钠(0.051g,0.484mmol)。在室温下搅拌10分钟后,加入戊基碘化物(0.063ml,0.484mmol),在氮气氛、室温下继续搅拌18小时。反应混合物用水(25ml)稀释,用EtOAc(2x25ml)萃取。干燥合并的有机萃取物(MgSO4)、过滤并蒸发。通过SPE(Si,5g)纯化,用4∶1EtOAc/环己烷洗脱,得到为白色固体的标题化合物(96mg,74%);m/z 297.2[MH+]。
b)8-氯-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮

将含有四(三苯基膦)钯(0)(56mg,0.049mmol)的烧瓶用氮气冲洗,然后加入8-氯-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(96mg,0.323mmol)的无水THF(1.5ml)溶液,接着加入DMSO(0.1ml)和吗啉(0.28ml,0.049mmol)。所得到的混合物在室温、氮气氛下搅拌72小时。反应混合物溶解于EtOAc(25ml)中,用2M HCl水溶液(25ml)洗涤。干燥有机萃取物(MgSO4)、过滤并减压蒸发。通过装填氨基丙基SPE(2g)纯化,用甲醇洗涤,然后产物用5%乙酸的甲醇溶液洗脱。含有产物的部分蒸发后,得到为白色固体的标题化合物(27mg,33%)。NMR;δH(400MHz,d6-DMSO)0.85(t,3H,J=7Hz),1.20-1.34(m,4H),1.57-1.67(m,2H),3.84(t,2H,J=7Hz),11.19(s,1H),14.38(br.s,1H);m/z 257.2[MH+]。
实施例13:8-氯-3-(3-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮

a)8-氯-3-(3-甲基丁基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.5g,6.6mmol)的DMF(40ml)溶液用碳酸钠(0.9g,8.5mmol)和1-溴-3-甲基丁烷(1.04g,6.9mmol)处理。搅拌后的混合物在50℃下加热18小时,然后冷却并蒸发至干。残余物用水(60ml)处理,用乙酸乙酯(3x80ml)萃取。有机部分合并后,用硫酸镁干燥,过滤并蒸发。残余物用二乙醚和环己烷的混合物研磨,得到为白色固体的产物,将其滤出后干燥。得到为白色固体的标题化合物m/z 297[MH+]。
b)8-氯-3-(3-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-3-(3-甲基丁基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.074g,0.25mmol)的THF(2ml)溶液用吗啉(0.035ml,4.0mmol)处理,混合物通过向反应器中反复施加真空和氮气来除气。混合物随后用四(三苯基膦)钯(0)(0.03g,0.026mmol)在除气的THF(0.5ml)中的溶液处理。2小时后,混合物用2M盐酸水溶液(2ml)和二乙醚(3ml)处理。滤出沉淀析出的产物,用二乙醚洗涤并干燥。得到为白色固体的标题化合物(0.036g,56%)。NMR δH(400MHz,d6-DMSO);0.91(d,6H,J=6.3Hz),1.47-1.62(m,3H),3.87(t,2H,J=7.5Hz),11.19(br.s,1H),14.38(br.s,1H);m/z 257,259[MH+]。
实施例14:4-(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈

使用4-溴丁腈作为烷基化试剂,按照实施例13制备。
NMR δH(400MHz,d6-DMSO);1.89-2.00(m,2H),2.55(t,2H,J=7.0Hz),3.95(t,2H,J=6.5Hz),11.25(br.s,1H),14.40(br.s,1H);m/z 254[MH+]。
实施例15:8-氯-3-(2-环己基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.442mmol)与碳酸钠(52mg,0.486mmol)在无水DMF(3ml)中搅拌30分钟。加入环己基乙基溴化物(93mg,0.486mmol),混合物在37-40℃、氮气氛下搅拌65小时,接着在90℃下加热18小时。冷却后,溶液通过抽空和引入氮气数次而除气,加入四(三苯基膦)钯(0)(76mg,0.066mmol)和吗啉(0.385ml,4.42mmol),然后混合物搅拌18小时。继续加入一定量的四(三苯基膦)钯(0)(50mg,0.043mmol)和吗啉(0.2ml)后,继续搅拌1小时。加入乙酸乙酯和2M HCl水溶液(各为大约10ml),有机层分离后,用盐水洗涤并蒸发。残余物溶解于THF中,装填在5g氨基丙基SPE柱上。柱用THF洗涤,再用MeOH洗涤,酸性产物用AcOH的MeOH溶液(由5%升至10%)洗脱。所得到的产物进一步通过自动制备性(autoprep)HPLC纯化,得到标题化合物,5.5mg,3%。
NMR δH(400MHz,d6-DMSO)0.80-0.95(m,2H),1.05-1.35(m,4H),1.45-1.55(m,2H),1.55-1.70(m,3H),1.70-1.80(m,2H),3.86(t,2H,J=8Hz),11.07(s,1H),一个可交换质子未观察到。m/z 297(MH+)。
实施例16:3-丁基-1-甲基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈

a)3-丁基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将搅拌后的7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(10g,52mmol)的无水DMF(100ml)溶液用K2CO3(7.91g,57.2mmol)处理,10分钟后加入BuI(6.51ml,57.2mmol)。反应2天后,反应混合物在2M HCl(aq)和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并真空浓缩,得到灰白色固体。将其用热环己烷洗涤并真空干燥,得到标题化合物(8.87g,68%);m/z249.3[MH+]。
b)3-丁基-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将搅拌后的3-丁基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.0g,4.03mmol)的无水DMF(10ml)溶液用Na2CO3(470mg,4.43mmol)处理,再用甲基碘化物(275μl,4.43mmol)处理。混合物在35℃下加热17小时。加入K2CO3(500mg,3.6mmol)和甲基碘化物(275μl,4.43mmol),然后在50℃下继续搅拌18小时。反应混合物放冷后,在2M HCl(aq)和EtOAc之间分配。有机层分离后,水层用EtOAc萃取不止一次。合并的萃取物用盐水洗涤,干燥(MgSO4)并浓缩得到黄色/褐色油状物(1.24g)。产物通过二氧化硅SPE(10g)纯化,用EtOAc/环己烷混合物洗脱。产物部分合并后,浓缩得到为浅黄色固体的标题化合物(1.11g,定量);m/z 263.3[MH+]。
c)3-丁基-1-甲基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-甲醛

将装填有3-丁基-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(300mg,1.14mmol)和无水THF(6ml)的预干燥烧瓶在氮气氛下冷却至-75℃,然后用LiHMDS(1.37ml的1.0M THF溶液)处理。所得到的溶液在1.5小时内温热至-60℃,然后加入无水DMF(177μl,2.29mmol)。溶液在3小时内温热至-10℃,然后将其用饱和NH4Cl(aq)溶液猝灭。混合物在1M HCl(aq)和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩得到褐色油状物(350mg)。产物通过SPE(Si,10g)纯化,用EtOAc/环己烷混合物洗脱,得到为白色固体的标题化合物(131mg,39%);NMR;δH(400MHz,d6-DMSO)0.91(t,3H,J=7.5Hz),1.28-1.39(m,2H),1.63-1.73(m,2H),3.25(s,3H),4.02(t,2H,J=7.5Hz),5.03(dd,1H,J=17和1Hz),5.17(dd,1H,J=10和1Hz),5.31(app.d,2H,J=5.5Hz),5.98-6.09(m,1H),9.88(s,1H)。
d)3-丁基-1-甲基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-腈
将3-丁基-1-甲基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-甲醛的无水吡啶(5ml)溶液用羟胺盐酸盐(63mg,0.91mmol)处理,同时在50℃下加热1小时。混合物放冷后,浓缩,用乙酸酐(5ml)处理,然后在100℃下加热2.5小时,在125℃下加热45分钟。混合物再次放冷后,在水和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩,得到为黄色残余物的标题化合物(230mg粗产物,114%);m/z 288.3[MH+]。
e)3-丁基-1-甲基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈

将3-丁基-1-甲基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-腈(230mg,0.80mmol)在无水THF(5ml)和无水DMSO(0.5ml)中的溶液用Pd(PPh3)4(185mg,0.16mmol)处理。混合物在轻度真空下除气,加入吗啉(698μL),在室温、氮气氛下放置搅拌2小时。黄色溶液在2M HCl(aq)和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩。残余物溶于MeOH中,通过氨基丙基SPE(5g),依次用MeOH、5%AcOH、10%、20%和30%AcOH/MeOH混合物洗脱。产物部分合并后浓缩得到浅黄色固体(116mg)。将其用MeOH洗涤,为白色固体的标题化合物通过过滤收集,然后真空干燥(55mg,28%)。NMR;δH(400MHz,d6-DMSO)0.90(t,3H,J=7.5Hz),1.25-1.35(m,2H),1.59-1.68(m,2H),3.24(s,3H),3.96(t,2H,J=7Hz),NH未观察到δH 15;m/z 248.2[MH+]。
实施例17:1-甲基-2,6-二氧代-3-戊基-2,3,6,7-四氢-1H-嘌呤-8-腈

a)3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.61g,3.2mmol)、碳酸钠(0.60g,5.7mmol)和戊基碘化物(0.64g,3.2mmol)在50℃的DMF(5ml)中搅拌18小时。溶液冷却后,在乙酸乙酯和盐水之间分配,有机层分离后干燥(MgSO4)并浓缩。在二氧化硅上色谱处理(用二氯甲烷至5∶1二氯甲烷/乙酸乙酯梯度洗脱),得到为浅黄色固体的标题化合物(0.47g,56%)。m/z 263[MH+]。
b)1-甲基-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.20g,0.76mmol)、碳酸钾(0.4g,2.9mmol)和甲基碘化物(0.5ml,4.9mmol)在50℃的DMF(5ml)中搅拌加热3小时。溶液放冷后在乙酸乙酯和盐水之间分配。有机层分离后,干燥(MgSO4)并浓缩,得到标题化合物(0.21g,100%)。m/z 277[MH+]。
c)1-甲基-2,6-二氧代-3-戊基-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-甲醛

在-78℃下,向1-甲基-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.05g,3.6mmol)的THF(15ml)溶液中在10分钟内加入LiHMDS(4ml,1M己烷溶液,4mmol),溶液搅拌0.5小时。加入DMF(0.5ml),溶液在-78℃下继续搅拌0.5小时,然后用冷却浴在2小时内温热至环境温度。反应用2N盐酸(3ml)猝灭,然后在乙酸乙酯和盐水之间分配。有机层分离后干燥并浓缩。粗产物在二氧化硅上色谱处理(用二氯甲烷至5∶1二氯甲烷/乙酸乙酯梯度洗脱),得到为白色固体的标题化合物(0.35g,30%)。m/z 305[MH+]。
d)1-甲基-2,6-二氧代-3-戊基-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-腈

将1-甲基-2,6-二氧代-3-戊基-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-甲醛(0.18g,0.6mmol)和羟胺盐酸盐(0.053g,0.76mmol)在50℃的吡啶(5ml)中加热1小时,然后冷却至环境温度。加入乙酸酐(0.08g,0.78mmol)后,溶液搅拌18小时。溶液浓缩得到乙酸酯,将其溶解于乙酸酐(3ml)中,同时加热至130℃,持续3小时,冷却后浓缩得到粗产物。在二氧化硅上色谱处理(用二氯甲烷洗脱),得到为透明油状物的标题化合物(0.17g,95%)。m/z 302[MH+]。
e)1-甲基-2,6-二氧代-3-戊基-2,3,6,7-四氢-1H-嘌呤-8-腈

将1-甲基-2,6-二氧代-3-戊基-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-腈(0.17g,0.56mmol)和吗啉(0.6ml,6.7mmol)溶解于含有DMSO(0.5ml)的THF(5ml)中。将含有该溶液的烧瓶置于真空下,用氮气替换其中的空气(x3)。加入四(三苯基膦)钯(0)(0.13g,0.11mmol),溶液搅拌2.5小时。溶液在乙酸乙酯(20ml)和2N盐酸(10ml)之间分配,有机层分离后,用盐水(3x10ml)洗涤。然后有机层用2N氢氧化钠溶液(2x10ml)洗涤,水层用2N盐酸酸化,用乙酸乙酯(2x10ml)萃取。有机层分离后,干燥(MgSO4)并浓缩得到标题化合物(0.026g,18%)。NMR;δH(400MHz,CDCl3)0.92(t,3H,J=7Hz),1.32-1.43(m,4H),1.79(m,2H),3.54(s,3H),4.15(t,2H,J=7.5Hz),14.35(b r.s,1H);m/z 262[MH+]。
实施例18:8-氯-3-己基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮
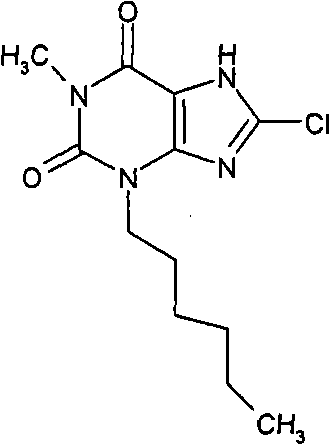
a)8-氯-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(6g,26.5mmol)的无水DMF(30ml)溶液中加入碳酸钠(3.09g,29.15mmol)。在室温下搅拌10分钟后,加入甲氧乙氧基甲基氯化物(3.03ml,26.5mmol),继续在氮气氛和室温下搅拌66小时。反应混合物真空浓缩后,残余物溶解于EtOAc(100ml)中,用盐水(100ml)洗涤,含水萃取物用DCM(100ml)萃取,干燥有机萃取物(MgSO4)并合并后真空浓缩。残余物用EtOAc研磨,滤出固体。滤液浓缩得到浅褐色油状物,将其吸附在二氧化硅上,通过SPE(Si,50g)纯化,用梯度为1∶1的EtOAc/环己烷-EtOAc洗脱,得到为白色固体的标题化合物(2g,24%),m/z315.2[MH+]。
b)8-氯-1-甲基-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(2g,6.37mmol)的无水DMF(15ml)溶液中加入碳酸钠(0.743g,7mmol)。在室温下搅拌10分钟后,加入甲基碘化物(0.44ml,7mmol),在氮气氛和室温下继续搅拌18小时。反应混合物真空浓缩后,残余物溶解于EtOAc(100ml)中,用盐水(100ml)洗涤。干燥有机萃取物(MgSO4)、过滤并蒸发得到为棕褐色油状物的标题化合物(85%纯度)(2.98g,定量),m/z 329.2[MH+]。
c)8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-1-甲基-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(2.9g,6.37mmol)的二噁烷(20ml)和水(20ml)溶液中加入5M HCl(20ml)。所得到的混合物在100℃和氮气氛下加热18小时。反应混合物随后真空浓缩,残余物溶解于EtOAc(100ml)中,用水洗涤。干燥有机萃取物(MgSO4)、过滤并蒸发。通过SPE(Si,20g)纯化,用2∶3 EtOAc/环己烷洗脱,得到为白色固体的标题化合物(1.04g,68%)。m/z 241.1[MH+]。
或者,8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮也可以采用SEM保护制备得到。
a)8-氯-7-(2-丙烯-1-基)-3-({[2-(三甲基甲硅烷基)乙基]氧基}甲基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(5g,22.1mmol)的DMF(80ml)溶液中加入2-2-(三甲基甲硅烷基)乙氧基甲基氯化物(4.3ml,24.2mmol)和碳酸钠(2.6g,24.2mmol)。在室温下搅拌过夜后,继续加入2-2-(三甲基甲硅烷基)乙氧基甲基氯化物(4.3ml,24.2mmol)和碳酸钠(1.3g,12.1mmol),继续搅拌2小时。反应混合物随后在5%LiCl水溶液和乙酸乙酯之间分配。有机萃取物分离后,用盐水洗涤,干燥(MgSO4)并浓缩。通过使用二氧化硅柱的BiotageTM色谱法纯化,用1∶4-1∶2乙酸乙酯/环己烷洗脱,得到标题化合物(3.14g,40%);m/z 374.2[MNH4 +]。
b)8-氯-1-甲基-7-(2-丙烯-1-基)-3-({[2-(三甲基甲硅烷基)乙基]氧基}甲基)-3,7-二氢-1H-嘌呤-2,6-二酮
向8-氯-7-(2-丙烯-1-基)-3-({[2-(三甲基甲硅烷基)乙基]氧基}甲基)-3,7-二氢-1H-嘌呤-2,6-二酮(3.14g,8.82mmol)的DMF(50ml)溶液中加入甲基碘化物(0.659ml,10.58mmol)和碳酸铯(3.45g,10.58mmol),反应混合物在室温下搅拌过夜。反应混合物在水和乙酸乙酯之间分配。有机萃取物分离后,用盐水洗涤,干燥(MgSO4)并浓缩,得到标题化合物2.99g(92%);m/z 388[MNH4 +]。
c)8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-1-甲基-7-(2-丙烯-1-基)-3-({[2-(三甲基甲硅烷基)乙基]氧基}甲基)-3,7-二氢-1H-嘌呤-2,6-二酮(2.99g,8.08mmol)的DCM(20ml)溶液中加入TFA(10ml),反应在室温下搅拌2.5小时。反应混合物随后浓缩,残余物再用DCM处理,同时蒸发不止一次。通过SPE(Si)纯化,用1∶9-4∶1乙酸乙酯/环己烷洗脱,得到不纯产物(1.31g),将其溶解于甲醇(20ml)中,用饱和碳酸钾水溶液(20ml)处理。搅拌过夜后,混合物在含有2M HCl(1ml)的水和乙酸乙酯之间分配。有机萃取物分离后,用盐水洗涤,干燥(MgSO4)并浓缩得到标题化合物0.87g(45%);m/z 241.1[MH+]。
d)8-氯-3-己基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.42mmol)的无水DMF(3ml)溶液中加入碳酸钠(58mg,0.54mmol),搅拌10分钟后,加入己基碘化物(0.08ml,0.54mmol),反应混合物在室温和氮气氛下搅拌90小时。然后加入Pd(PPh3)4(73mg,0.063mmol),反应器抽空后用氮气冲洗(x3),加入吗啉(0.37ml,4.3mmol),在室温和氮气氛下继续搅拌4小时。反应混合物用EtOAc(25ml)稀释,用2M HCl水溶液(25ml)洗涤。干燥有机萃取物(MgSO4)、过滤并蒸发。通过将化合物装填入氨基丙基SPE(5g)纯化,用MeOH洗涤后,产物用5%AcOH/MeOH洗脱,得到为白色固体的标题化合物(65mg,54%)。NMR;δH(400MHz,d6-DMSO)0.85(t,3H,J=7Hz),1.23-1.33(m,6H),1.58-1.68(m,2H),3.22(s,3H),3.91(t,2H,J=7.5Hz),14.46(br.s,1H);m/z 285.3[MH+]。
实施例19:8-氯-1-甲基-3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮
按照类似于实施例18的方式制备,但是使用丙基碘化物在N3上烷基化。NMR δH(400MHz,d6-DMSO)0.87(t,3H,J=7.5Hz),1.61-1.73(m,2H),3.22(s,3H),3.89(t,2H,J=7.5Hz),14.45(br.s,1H),m/z 243[MH+]。
实施例20:1,3-二丁基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈

a)1,3-二丁基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

将1,3-二-N-丁基黄嘌呤(10g,38mmol)的无水DMF(80ml)溶液用K2CO3(5.2g,38mmol)处理,再用烯丙基溴(3.6ml,42mmol)处理。混合物在55℃和氮气氛下加热18小时。冷却至室温后,混合物在水和EtOAc之间分配。加入几毫升2M HCl(aq)帮助分离。有机层分离后,水层用EtOAc萃取不止一次。合并的萃取物用盐水洗涤,干燥(MgSO4)并浓缩,得到为灰白色固体的标题化合物(12.23g,106%)。m/z 305.3[MH+]。
b)1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-羧酸甲酯

将1,3-二丁基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(3.0g,9.9mmol)的无水THF(30ml)溶液冷却至-50℃,并用LiHMDS(18ml的1.0M THF溶液,17.8mmol)处理。在-50℃下1小时后,加入氯甲酸甲酯(1.9ml,24.6mmol),混合物在2小时内温热至-30℃,然后用饱和NH4Cl(aq)溶液猝灭。混合物在EtOAc和1M HCl(aq)之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩,得到暗橙色油状物(4.07g)。将该油状物溶于(taken up)15%EtOAc/环己烷中,通过Si BiotageTM色谱柱。产物部分合并后,浓缩得到为黄色固体的标题化合物(1.35g,38%)。m/z 363.2[MH+]。
c)1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-羧酸

将搅拌后的1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-羧酸甲酯(1.30g,3.6mmol)的MeOH(15ml)溶液用LiOH(215mg)和水(1.5ml)处理。在室温下处理3小时后,混合物用水稀释,用2M HCl(aq)调节pH至大约pH 5。加入EtOAc后分离,用盐水洗涤,干燥(MgSO4)并浓缩,得到为黄色固体的标题化合物85%纯度(1.2g,88%)。m/z.349.2[MH+]。
d)1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-甲酰胺

将搅拌后的1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-羧酸(1.0g,2.9mmol)的无水DMF(10ml)溶液依次用DIPEA(1.1ml)、PyBOP、和2M NH3(3.6ml)处理。2小时后,将产物混合物在2M HCl(aq)和EtOAc之间分配。有机层分离后,用饱和NaHCO3(aq)溶液、盐水洗涤,干燥(MgSO4)并浓缩,得到橙色油状物(大约2g)。产物通过BiotageTM色谱法纯化,用5%→40%EtOAc/环己烷混合物洗脱。适宜部分合并后浓缩得到酰胺90%纯度(790mg,78%)。m/z.392.3[M+甲酸-H]-。
e)1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-腈(carbonitrile)

将1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-甲酰胺(300mg)的无水DMF(7ml)溶液在0℃下逐滴用POCl3(237μL)处理。除去冰浴,2小时后混合物在水和Et2O之间分配。水层再次用Et2O萃取,合并的萃取物分离后,用水(x2)、盐水洗涤,然后干燥(MgSO4)并浓缩,得到黄色油状物(312mg)。将该油状物溶于(taken up)环己烷中,通过SPE(Si,10g)纯化,用EtOAc/环己烷混合物洗脱。产物部分浓缩后,得到为无色油状物的标题化合物(150mg,53%);m/z.330.3[MH+]。
f)1,3-二丁基-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-8-腈
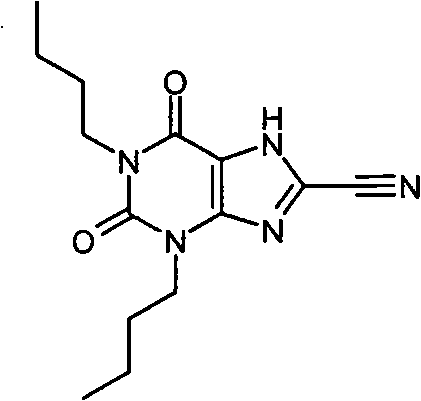
将1,3-二丁基-2,6-二氧代-7-(2-丙烯-1-基)-2,3,6,7-四氢-1H-嘌呤-8-腈(140mg,0.43mmol)在无水THF(4ml)和无水DMSO(0.4ml)中的溶液用Pd(PPh3)4(74mg,0.064mmol)处理。混合物在轻度真空下除气,加入吗啉(371μL)。在室温和氮气氛下放置搅拌4小时。黄色溶液在2M HCl(aq)和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩。将残余物溶于(taken up)MeOH中,通过氨基丙基SPE(5g),依次用MeOH、5%→50%AcOH/MeOH洗脱。洗脱的产物浓缩后含有少许杂质,用环己烷洗净后得到为灰白色固体的标题化合物(30mg,24%)。NMR δH(400MHz,d6-DMSO)0.89(app.td,6H,J=7和3Hz),1.25-1.35(m,4H),12.48-1.55(m,2H),1.58-1.69(m,2H),3.87(t,2H,J=7Hz),3.95(t,2H,J=7Hz),NH未观察到δH 15;m/z 290.3[MH+]。
实施例21:1,3-二丁基-8-碘-3,7-二氢-1H-嘌呤-2,6-二酮

将搅拌后的1,3-二-N-丁基黄嘌呤(100mg,3.39mmol)的无水DMF(3ml)溶液用NIS(94mg,3.75mmol)处理,然后在室温和氮气氛下放置搅拌23小时。混合物在饱和Na2SO3(aq)溶液和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并真空浓缩。产物经通过SPE(Si,5g)柱纯化,用EtOAc/环己烷混合物洗脱。产物部分浓缩得到为白色固体的标题化合物(75mg,51%);NMR;δH(400MHz,d6-DMSO)(app.td,6H,J=7.5和4Hz),1.21-1.34(m,4H),1.45-1.54(m,2H),1.56-1.66(m,2H),3.84(t,2H,J=7.5Hz),3.93(t,2H,J=7.5Hz),14.10(s,1H);m/z 391.3[MH+]。
实施例22:(3-丁基-8-氯-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈
向3-丁基-8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(200mg,0.707mmol)和Cs2CO3(254mg,0.778mmol)的无水DMF(5ml)溶液中加入氯乙腈(0.054ml,0.85mmol)。混合物在50℃下加热18小时,然后冷却至室温,在轻度真空下除气,随后引入氮气。重复上述步骤两次。加入Pd(PPh3)4(82mg,0.071mmol),混合物除气不止一次,然后加入吗啉(0.617ml,7.07mmol),混合物在室温下放置搅拌3小时。混合物在2M HCl(aq)和EtOAc之间分配。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩。将残余物溶于(takenup)MeOH中,通过氨基丙基SPE(5g),用MeOH洗脱,再用5-10%AcOH/MeOH洗脱。产物部分浓缩得到标题化合物52mg(26%);NMR;δH(400MHz,d6-DMSO)0.90(t,3H,J=7.5Hz),1.26-1.37(m,2H),1.60-1.69(m,2H),3.94(t,2H,J=7.5Hz),4.87(s,2H),14.72(br s,1H);m/z 299.2[MNH4 +]。
实施例23:(8-氯-2,6-二氧代-3-丙基-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈

a)8-氯-7-(2-丙烯-1-基)-3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮
将8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.5g,6.6mmol)、1-碘丙烷(1.2g,6.9mmol)和碳酸钠(0.9g,8.5mmol)在DMF(40ml)中的混合物在50℃下加热18小时。反应混合物真空浓缩后,残余物用水(60ml)处理,用乙酸乙酯(3x80ml)萃取。干燥合并的有机萃取物(MgSO4)、过滤并蒸发。残余物用乙醚/环己烷研磨,固体滤出后干燥得到标题化合物(0.82g,46%);m/z269.1[MH+]。
b)(8-氯-2,6-二氧代-3-丙基-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈

将8-氯-7-(2-丙烯-1-基)-3-丙基-3,7-二氢-1H-嘌呤-2,6-二酮(0.067g,0.25mmol)的DMF(2ml)溶液用碳酸铯(0.082g,0.25mmol)和溴代乙腈(0.044g,0.37mmol)处理。混合物在80℃下加热4小时,然后冷却至环境温度。真空除去DMF,残余物用THF(2ml)处理。溶剂通过连续向反应混合物施加真空和氮气来除气。混合物随后用吗啉(0.035ml,0.4mmol)和四(三苯基膦)钯(0)(0.03g,0.026mmol)处理。2小时后,混合物用2M盐酸水溶液(2ml)处理,产物用氯仿(3x5ml)萃取。有机部分合并后蒸发。残余物通过质量定向HPLC纯化,得到为白色固体的标题化合物(0.022g,33%)。NMR;δH(400MHz,d6-DMSO),0.88(t,3H,J=7.5Hz),1.63-1.74(m,2H),3.91(t,2H,J=7.5Hz),4.87(s,2H),NH未观察到δH 14;m/z 268[MH+]。
实施例24:[8-氯-3-(2-环丙基乙基)-2,6-二氧代-2,3,6,7-四氢-1H-嘌呤-1- 基]乙腈

使用8-氯-3-(2-环丙基乙基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮,按照(8-氯-2,6-二氧代-3-丙基-2,3,6,7-四氢-1H-嘌呤-1-基)乙腈(实施例23)制备。
NMR δH(400MHz,d6-DMSO)-0.06-0.00(m,2H),0.31-0.39(m,2H),0.64-0.74(m,1H),1.57(q,2H,J=7Hz),4.04(t,2H,J=7Hz),4.87(s,2H),14.68(br.s,1H);m/z 294[MH+]。
实施例25:8-氯-1-乙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

a)8-氯-7-(2-丙烯-1-基)-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(1.5g,6.62mmol)的无水DMF(50ml)溶液中加入碳酸氢钠(0.98g,9.25mmol),再加入1,1,1-三氟-2-碘乙烷(1.20g,5.72mmol),混合物在50℃和氮气氛下搅拌加热6小时。溶液在10小时内冷却至环境温度,然后在120℃下加热48小时。加入另外的1,1,1-三氟-2-碘乙烷(0.43g,2.05mmol),混合物加热至120℃,继续持续3小时。减压除去溶剂后,残余物用DCM研磨,然后过滤。
使用8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(3.80g,16.8mmol)、碳酸氢钠(2.45g,23.1mmol)和1,1,1-三氟-2-碘乙烷(4.05g,19.3mmol)在无水DMF(125ml)中重复上述反应。混合物在120℃下加热16小时,减压除去溶剂后,残余物用DCM研磨,然后过滤。
合并前两次所得到的DCM滤液,减压浓缩后,使用BiotageTM色谱法纯化(依次用环己烷/乙酸乙酯1∶1、7∶3洗脱),得到为白色固体的标题化合物(1.6g,23%)。m/z 309[MH+]。
b)8-氯-1-乙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-7-(2-丙烯-1-基)-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.070g,0.23mmol)的无水DMF(2ml)溶液中加入碳酸铯(0.085g,0.26mmol),再加入1-碘乙烷(0.061g,0.39mmol)。混合物在80℃下加热5小时,然后在环境温度和氮气氛下搅拌16小时。使用真空离心机减压除去溶剂,残余物溶解于无水THF(2.5ml)中。向混合物中加入四(三苯基膦)钯(0)(0.030g,0.026mmol)和吗啉(0.040g,0.45mmol),反应混合物用氮气除气,然后在环境温度下搅拌72小时。混合物在氯仿和2N HCl水溶液之间分配,水层重新萃取。有机萃取物合并后,在氮气流下蒸发,然后使用氨基丙基SPE纯化(用乙酸∶甲醇∶DCM,1∶2∶2洗脱),得到为白色固体的标题化合物>95%纯度(0.041g,60%)。NMR δH(400MHz,d4-MeOD)1.20(t,3H,J=7Hz),4.03(q,2H,J=7Hz),4.73(q,2H,J=8.5Hz),m/z 297[MH+]。
实施例26:8-氯-1-丙基-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤-2,6-二酮
使用丙基碘化物在N1上烷基化,按照类似于实施例25的方式制备。
NMR δH(400MHz,CDCl3)0.99(t,3H,J=7.5Hz),1.68-1.79(m,2H),4.07(t,2H,J=7.5Hz),4.77(q,2H,J=8.5Hz),NH未观察到δH 13;m/z 311[MH+]。
实施例27:8-氯-1-(4,4,4-三氟丁基)-3-(2,2,2-三氟乙基)-3,7-二氢-1H-嘌呤 -2,6-二酮

使用4-溴-1,1,1-三氟丁烷在N1上烷基化,按照类似于实施例25的方式制备。
NMR;δH(400MHz,d4-MeOD)1.83-1.95(m,2H),2.14-2.32(m,2H),4.06(t,2H,J=7Hz),4.74(q,2H,J=8.5Hz),m/z 377[M-H]-。
实施例28:8-溴-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮
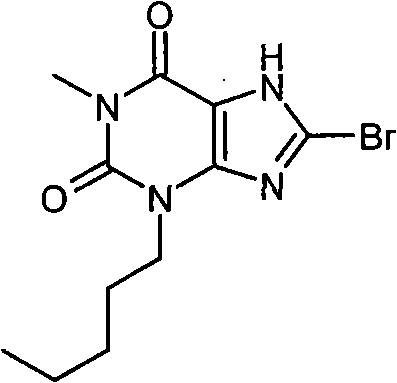
a)1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮

将1-甲基-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.45g,1.63mmol)、苯基硅烷(0.25ml,2.03mmol)和四(三苯基膦)钯(0)(0.35g,0.3mmol)溶解于含有乙酸(6ml)的DCM(10ml)中。通过抽空烧瓶,然后填入氮气(x3),用氮气置换出烧瓶中的空气,然后将反应混合物加热至45℃,持续4小时。溶液冷却后,用DCM稀释,然后用水和饱和碳酸氢钠溶液洗涤。有机层分离后,干燥并浓缩得到粗产物。通过SPE(二氧化硅)纯化,用乙醚洗脱得到产物0.06g,16%。m/z 237[MH+]。
b)8-溴-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮

将1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮(0.06g,0.25mmol)溶解于DMF(2ml)中,加入N-溴代琥珀酰亚胺(0.045g,0.25mmol)。混合物搅拌18小时后,浓缩,粗产物通过氨基丙基SPE(5g)洗脱纯化,先用甲醇再用5%乙酸/甲醇洗脱产物。产物进一步通过质量定向自动化制备(mass directed autoprep)纯化,得到为白色固体的标题化合物(0.01g,12%)。NMR δH(400MHz,d6-DMSO)0.86(t,3H,J=7Hz),1.21-1.35(m,4H),1.59-1.68(m,2H),3.22(s,3H),3.91(t,2H,J=7.5Hz),14.39(br.s,1H);m/z 315,317[MH+]。
实施例29:8-氯-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮
a)8-氯-1-甲基-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮
向8-氯-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(3.9g,13.3mmol)的DMF(35ml)溶液中加入碳酸铯,混合物搅拌10分钟,然后加入碘甲烷(0.91ml,14.6mmol),混合物搅拌18小时。反应在乙酸乙酯和2N HCl溶液之间分配,有机层分离后,干燥(MgSO4)并浓缩。在二氧化硅SPE上色谱处理,用环己烷/乙酸乙酯(5%-20%)洗脱,得到为油状物的产物2.78g,68%。m/z 311[MH+]。
b)8-氯-1-甲基-3-戊基-3,7-二氢-1H-嘌呤-2,6-二酮

将四(三苯基膦)钯(1.0,0.90mmol)置于抽空后再充满氮气(x3)的烧瓶中。加入8-氯-1-甲基-3-戊基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(2.78g,8.96mmol)在50ml THF中的溶液,烧瓶抽空不止一次,然后引入氮气。加入DMSO(4.5ml)和吗啉(7.8ml,89.6mmol),溶液搅拌5小时。溶液在乙酸乙酯和2N HCl溶液之间分配,有机部分用盐水洗涤,干燥(MgSO4)并浓缩。粗产物通过氨基丙基SPE纯化,先用甲醇再用含有0-15%乙酸的甲醇洗脱,得到为白色固体的标题化合物1.12g,46%。NMR δH(400MHz,d6-DMSO)0.86(t,3H,J=7Hz),1.21-1.35(m,4H),1.59-1.68(m,2H),3.22(s,3H),3.91(t,2H,J=7.5Hz),NH not observed;m/z 271[MH+]。
实施例30:3-丁基-8-氯-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮

使用3-丁基-8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮作为原料,按照类似于实施例29的方式制备。
NMR δH(400MHz,d6-DMSO)0.88(t,3H,J=7Hz),1.25-1.35(m,2H),1.6-1.66(m,2H),3.22(s,3H),3.91(t,2H,J=7.5Hz),14.46(br s,1H);m/z 257[MH+]。
实施例31:4-(8-氯-1-甲基-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)丁腈
向8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(70mg,0.292mmol)和Na2CO3(37mg,0.35mmol)在DMF(3ml)中的混合物中加入4-溴丁腈(0.035ml,0.35mmol)。混合物在室温下搅拌过夜,然后在轻度真空下除气并引入氮气。然后依次加入Pd(PPh3)4(50mg,0.044mmol)和吗啉(0.254ml,2.92mmol)。在室温下搅拌2小时后,进一步加入现制的Pd(PPh3)4(50mg,0.044mmol),继续搅拌过夜。反应混合物在乙酸乙酯(20ml)和水(20ml)之间分配,同时加入少量2M HCl以帮助分离。有机层分离后,用盐水洗涤,干燥(MgSO4)并浓缩。残余物溶于(taken up)MeOH中,通过氨基-丙基SPE(5g),依次用MeOH、3-5%AcOH/MeOH洗脱。产物部分浓缩得到标题化合物39.7mg(51%);NMR;δH(400MHz,d6-DMSO)1.91-2.00(m,2H),2.55(t,2H,J=7Hz),3.22(s,3H),4.03(t,2H,J=7Hz),14.49(br.s,1H);m/z 268.1[MH+]。
实施例32:8-氯-1-甲基-3-(4,4,4-三氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮 
将8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.048g,0.2mmol)的THF(1ml)溶液用碳酸铯(0.78g,0.24mmol)和4-溴-1,1,1-三氟丁烷(0.044g,0.25mmol)处理。混合物在环境温度下搅拌1小时,然后在50℃下加热4小时,随后冷却。混合物通过交替向混合物施加真空和氮气压来除气,然后用吗啉(0.17ml,2mmol)和四(三苯基膦)钯(0)(0.023g,0.02mmol)处理,2小时后,混合物小心地用2M盐酸水溶液(2ml)处理,产物用氯仿(2x4ml)萃取。合并的有机层蒸发后,产物通过反相质量定向HPLC纯化,得到标题化合物6.2mg(10%);NMR;δH(400MHz,d6-DMSO);1.84-1.92(m,2H),2.28-2.35(m,2H),3.22(s,3H),3.99-4.03(m,2H)14.31(br.s,1H);m/z 311.2[MH+]。
实施例33:3-丁基-8-氯-1-乙基-3,7-二氢-1H-嘌呤-2,6-二酮

a)3-丁基-7-(苯基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮

在40℃下,将7-苄基-3,7-二氢-1H-嘌呤-2,6-二酮(17.14g,70.8mmol)[Synthetic Communications,20(16),2459-2467,1990]和碳酸钾(11.43g,82.8mmol)悬浮于DMF(400ml)中。搅拌30分钟后,加入丁基碘化物(8.76mL,77.0mmol),混合物在40℃下搅拌过夜。加入50%乙酸水溶液(60ml),溶液减压浓缩。残余物悬浮于水(500ml)中,产物用氯仿萃取。有机层合并后浓缩,产物用快速色谱法分离出来,用1%甲醇的二氯甲烷溶液洗脱,得到产物(9.49g,45%);1H NMR(400MHz;CDCl3)δ:0.95(3H,t),1.34-1.41(2H,m),1.70-1.78(2H,m),4.05(2H,t),5.46(2H,s),7.31-7.40(5H,m),7.56(1H,s),8.21(1H,br.s);m/z 299[MH+]。
b)3-丁基-1-乙基-7-(苯基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮

将3-丁基-7-(苯基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.429g,1.24mmol)和碳酸钾(0.256g,1.85mmol)悬浮于DMF(8ml)中,加入碘乙烷(0.113mL,1.42mmol)。反应混合物在环境温度下搅拌过夜。反应混合物蒸发至干后,残余物在水和乙酸乙酯之间分配。有机层用水洗涤,再用盐水洗涤,经无水硫酸钠干燥,减压浓缩得到标题化合物;1H NMR(400MHz;CDCl3)δ:0.96(3H,t),1.25(3H,t),1.36-1.45(2H,m),1.72-1.76(2H,m),4.05-4.13(4H,m),5.50(2H,s),7.32-7.40(5H,m),7.52(1H,s);m/z 327[MH+]。
c)3-丁基-1-乙基-3,7-二氢-1H-嘌呤-2,6-二酮
将3-丁基-1-乙基-7-(苯基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮(0.353g,1.08mmol)溶解于乙酸(30ml)中,加入20%氢氧化钯-碳(0.238g),混合物在氢气氛(50psi)下振摇过夜。催化剂通过Celite 过滤除去,用乙酸洗涤。滤液减压浓缩得到标题化合物(0.227g,89%);1H NMR(400MHz;CDCl3)δ:0.97(3H,t),1.28(3H,t),1.38-1.47(2H,m),1.74-1.82(2H,m),4.12-4.17(4H,m),7.80(1H,s);m/z 237[MH+]。
过滤除去,用乙酸洗涤。滤液减压浓缩得到标题化合物(0.227g,89%);1H NMR(400MHz;CDCl3)δ:0.97(3H,t),1.28(3H,t),1.38-1.47(2H,m),1.74-1.82(2H,m),4.12-4.17(4H,m),7.80(1H,s);m/z 237[MH+]。
d)3-丁基-8-氯-1-乙基-3,7-二氢-1H-嘌呤-2,6-二酮

将3-丁基-1-乙基-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.42mmol)和NCS(56mg,0.42mmol)悬浮于MeCN(5ml)中,在120℃下微波辐射加热。反应混合物减压浓缩后,通过使用HPLC分离标题化合物。[用于纯化的HPLC条件:23分钟运行时间。溶剂:0.1%TFA的MeCN溶液和0.1%TFA的水溶液。MeCN在15分钟内由5%线性地升至95%。保持在95%持续2分钟。然后在1分钟内线性地降至5%,在5%平衡5分钟后进行下一次注射。];1HNMR(400MHz;CDCl3)δ:0.97(3H,t),1.31(3H,t),1.38-1.45(2H,m),1.72-1.80(2H,m),4.09-4.20(4H,m),13.40(1H,br.s);m/z 271[MH+]。
实施例34:8-氯-3-(4-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮
由1-溴-4-甲基戊烷(81mg)出发
由MeOH重结晶
收率34.8mg(29%),NMR;(400MHz,d6-DMSO)δH 0.83(d,6H,J=8Hz),1.12-1.22(m,2H),1.55(七重峰,1H,J=8Hz),1.58-1.68(m,2H),3.83(t,2H,J=7.5Hz),11.20(s,1H);m/z 271[MH+]。
实施例35:6-(8-氯-2,6-二氧代-1,2,6,7-四氢-3H-嘌呤-3-基)-2,2-二甲基己 腈

由6-溴-2,2-二甲基己腈(100mg)出发
由MeOH重结晶
收率48.5mg(35%);NMR;(400MHz,d6-DMSO)δH 1.27(s,6H),1.35-1.44(m,2H),1.54-1.59(m,2H),1.63-1.72(m,2H),3.88(t,2H,J=7Hz),11.24(s,1H);m/z 310[MH+]。
实施例36:8-氯-3-(6-甲基庚基)-3,7-二氢-1H-嘌呤-2,6-二酮

由1-溴-6-甲基庚烷(95mg)出发
由MeOH重结晶
收率36mg(27%),NMR;(400MHz,d6-DMSO)δH 0.83(d,6H,J=7.5Hz),1.10-1.17(m,2H),1.20-1.34(m,4H),1.48(七重峰,1H,J=7.5Hz),1.58-1.68(m,2H),3.84(t,2H,J=8Hz),11.22(s,1H);m/z 299[MH+]。
实施例37:8-氯-3-辛基-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.44mmol)与碳酸钠(52mg,0.49mmol)在无水DMF(3ml)中搅拌20分钟,然后加入1-碘辛烷(118mg,0.49mmol),混合物在氮气氛和40℃下搅拌65小时。冷却至室温后,混合物通过抽空容器和重新填入氮气数次来彻底除气。加入四(三苯基膦)钯(0)(102mg,0.09mmol),混合物再次除气,然后加入吗啉(0.385ml,4.4mmol),继续搅拌6.5小时。加入2M HCl和EtOAc,过滤两相体系。产物主要包含在过滤后的固体中,将其由THF-乙腈重结晶,再由MeOH重结晶,过滤后得到纯的标题化合物。
收率48mg(36%);NMR;(400MHz,d6-DMSO)δH 0.84(t,3H,J=7Hz),1.18-1.30(m,10H),1.57-1.66(m,2H),3.84(t,2H,J=7.5Hz),11.22(s,1H);m/z299[MH+]。
实施例38:8-氯-3-癸基-3,7-二氢-1H-嘌呤-2,6-二酮
由1-溴癸烷(108mg)出发,按照实施例37的方法制备。通过由MeOH重结晶,再质量定向自动化制备进行进一步的纯化。
收率2mg(1.4%);NMR;(400MHz,d4-甲醇)δH 0.89(t,3H,J=7Hz),1.26-1.38(m,14H),1.68-1.76(m,2H),3.97(t,2H,J=7.5Hz);m/z 327[MH+]。
实施例39:8-氯-3-(环己基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮

由(溴甲基)环己烷(87mg)出发,除了在80℃下额外加热18小时外,按照类似于实施例37的方式制备。
由MeOH重结晶。
收率31mg(25%);NMR;(400MHz,d6-DMSO)δH 0.90-1.02(m,2H),1.08-1.20(m,3H),1.53-1.69(m,5H),1.77-1.87(m,1H),3.70(d,2H,J=7.5Hz),11.21(s,1H);m/z 283[MH+]。
实施例40-46的一般方法:
向8-氯-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.442mmol)的无水THF(3ml)溶液中加入醇(0.442mmol)。混合物在0℃下搅拌,同时加入偶氮二羧酸二苄基酯(280mg,94%纯度,0.88mmol)的无水THF(2ml)溶液,接着在5分钟内逐滴加入三苯基膦(232mg,0.88mmol)的无水THF溶液。在0℃下持续30分钟后,在室温下继续搅拌18小时。混合物通过抽空和用氮气再填充反应器数次来除气,然后加入四(三苯基膦)钯(0)(102mg,0.088mmol),再加入吗啉(0.385ml,4.42mmol),继续搅拌4.5小时。加入EtOAc和2M HCl,混合物过滤后除去黄色沉淀析出的固体。分离滤液,有机相浓缩后重新溶解于含有THF和MeOH的混合物中。将上述溶液通过氨基丙基SPE,依次用THF-MeOH(1∶1)、MeOH以及5%AcOH的DCM-MeOH(1∶1)溶液洗脱。所得到的产物部分浓缩后,由MeOH重结晶得到纯标题化合物。
实施例40:(+/-)-8-氯-3-(3-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮

由(+/-)-3-甲基-1-戊醇45mg出发
收率20.2mg(17%);NMR;(400MHz,d6-DMSO)δH 0.83(t,3H,J=7.5Hz),0.90(d,3H,J=6.5Hz),1.12-1.21(m,1H),1.30-1.48(m,3H),1.58-1.68(m,1H),3.87(t,2H,J=7.5Hz),11.21(s,1H);m/z 271[MH+]。
实施例41:8-氯-3-(2-环戊基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮
由2-环戊基乙醇50mg出发
收率24.6mg(20%);NMR;(400MHz,d6-DMSO)δH 1.04-1.15(m,2H),1.40-1.67(m,6H),1.70-1.82(m,3H),3.86(t,2H,J=7.5Hz),11.22(s,1H);m/z283[MH+]。
实施例42:8-氯-3-(环丙基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮
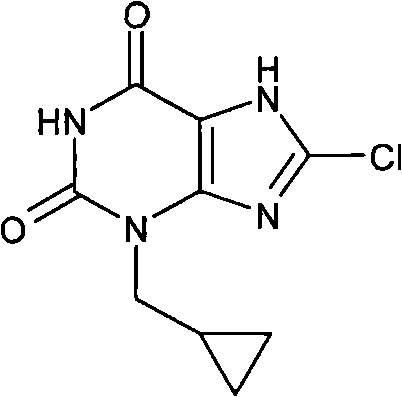
由环丙基甲醇32mg出发
收率22.3mg(21%);NMR;(400MHz,d6-DMSO)δH 0.34-0.40(m,2H),0.40-0.48(m,2H),1.17-1.27(m,1H),3.74(d,2H,J=7.5Hz),11.23(s,1H);m/z241[MH+]。
实施例43:(+/-)-8-氯-3-(2-甲基丁基)-3,7-二氢-1H-嘌呤-2,6-二酮

由(+/-)-2-甲基-1-丁醇39mg出发
收率12mg(9.5%);NMR;(400MHz,d6-DMSO)δH 0.81(d,3H,J=7Hz),0.86(t,3H,J=7.5Hz),1.06-1.17(m,1H),1.30-1.41(m,1H),1.90-2.00(m,1H),3.68(dd,1H,J=13.5和8Hz),3.75(dd,1H,J=13.5和7.5Hz),11.22(s,1H);m/z257[MH+]。
实施例44:(+/-)-8-氯-3-(2-甲基戊基)-3,7-二氢-1H-嘌呤-2,6-二酮

由(+/-)-2-甲基-1-戊醇45mg出发
收率22.4mg(19%);NMR;(400MHz,d6-DMSO)δH 0.81(d,3H,J=7Hz),0.84(t,3H,J=7.5Hz),1.05-1.16(m,1H),1.16-1.43(m,3H),1.98-2.09(m,1H),3.67(dd,1H,J=13.5和8Hz),3.74(dd,1H,J=13.5和7Hz),11.22(s,1H);m/z271[MH+]。
实施例45:8-氯-3-(环丁基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮

由环丁基甲醇38mg出发
收率30.5mg(27%);NMR;(400MHz,d6-DMSO)δH 1.73-1.85(m,4H),1.86-1.97(m,2H),2.66-2.79(m,1H),3.90(d,2H,J=7.5Hz),11.22(s,1H);m/z255[MH+]。
实施例46:8-氯-3-(环戊基甲基)-3,7-二氢-1H-嘌呤-2,6-二酮

由环戊基甲醇44mg出发
收率15mg(13%);NMR;(400MHz,d6-DMSO)δH 1.20-1.32(m,2H),1.42-1.54(m,2H),1.54-1.66(m,4H),2.32-2.45(m,1H),3.79(d,2H,J=8Hz),11.22(s,1H);m/z 269[MH+]。
实施例47:8-氯-3-(3-环丙基丙基)-3,7-二氢-1H-嘌呤-2,6-二酮

由3-环丙基-1-丙醇(P.J.Wagner,J.Amer.Chem.Soc.,1981,103,3837-3841)(44mg)出发
收率27.7mg(23%);NMR;(400MHz,d6-DMSO)δH-0.03-+0.03(m,2H),0.34-0.40(m,2H),0.65-0.75(m,1H),1.15-1.23(m,2H),1.66-1.76(m,2H),3.87(t,2H,J=7Hz),11.15(s,1H);m/z 269[MH+]。
实施例48:8-氯-3-(2-环丁基乙基)-3,7-二氢-1H-嘌呤-2,6-二酮

由2-环丁基乙醇(P.Vergnon,Eur.J.Med.Chem.,1975,10,65-71)(44mg)出发
收率21.5mg(18%);NMR;(400MHz,d6-DMSO)δH 1.53-1.64(m,2H),1.68-1.85(m,4H),1.93-2.03(m,2H),2.19-2.30(m,1H),3.78(t,2H,J=7Hz),11.20(s,1H);m/z 269[MH+]。
实施例49:8-氯-3-(4-氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮

a)8-氯-3-(4-氟丁基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

在装备有搅拌器的1.5ml微波瓶中,向8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(200mg,0.88mmol,1eq)的无水DMSO(1ml)溶液中加入碳酸氢钠(113mg,1.07mmol,1.2eq),再加入1-溴-4-氟丁烷(114μl,165mg,1.06mmol,1.2eq)。小瓶密封后,用微波搅拌加热,保持温度为120℃,持续25分钟,最大功率输出为300W。所得到的暗褐色溶液用甲醇(1ml)稀释,通过质量定向自动化制备性HPLC纯化,得到为白色固体的标题化合物(159mg,60%)。m/z 301.3[MH+]。
8-氯-3-(4-氟丁基)-3,7-二氢-1H-嘌呤-2,6-二酮
向8-氯-3-(4-氟丁基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.33mmol,1eq)的无水DCM(2ml)悬浮液中加入四(三苯基膦)钯(0)(38mg,0.033mmol,10%bw),再加入乙酸(115μl,121mg,2.01mmol,6eq)和苯基硅烷(410μl,360mg,3.33mmol,10eq)。所得到的浅黄色溶液在环境温度下搅拌16小时,得到暗紫色溶液。在氮气流下除去溶剂后,残余物加热溶解于DMSO/甲醇溶液(3ml,2∶1)中。使凝胶状混合物冷却至环境温度,过滤后通过质量定向自动化制备性HPLC纯化,得到为白色固体的标题化合物(35mg,43%)。m/z 261.2[MH+]NMR(400MHz,MeOD),δH 4.45(2H,dt,J=47和6Hz),4.03(2H,t,J=7Hz),1.90-1.65(4H,m)。
下述化合物按照类似的方式制备,同时适当地通过制备性或质量定向自动化制备性HPLC纯化:
实施例50:8-氯-3-(3-氟丙基)-3,7-二氢-1H-嘌呤-2,6-二酮

NMR(400MHz,MeOD),δH 4.51(2H,dt,J=47和6Hz),4.11(2H,t,J=7Hz),2.18-2.03(2H,m)。m/z 247[MH+]。
实施例51:8-氯-3-(5-氟戊基)-3,7-二氢-1H-嘌呤-2,6-二酮

NMR(400MHz,MeOD),δH 4.41(2H,dt,J=48和6Hz),3.99(2H,t,J=8Hz),1.84-1.63(4H,m),1.52-1.40(2H,m)。m/z 273.29[MH-]。
实施例52:3-(3-丁烯-1-基)-8-氯-3,7-二氢-1H-嘌呤-2,6-二酮

将8-氯-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.44mmol)与碳酸钠(52mg,0.49mmol)在无水DMF(3ml)中搅拌45分钟,然后加入4-溴-1-丁烯(66mg,0.49mmol),混合物在氮气氛和40℃下搅拌65小时。冷却至室温后,混合物通过抽空容器再填充氮气数次来彻底除气。加入四(三苯基膦)钯(0)(102mg,0.09mmol),混合物再次除气,然后加入吗啉(0.385ml,4.4mmol),继续搅拌6.5小时。加入2M HCl和EtOAc,过滤两相体系除去沉淀析出的黄色固体。滤液的有机相分离后蒸发。将残余物温热溶解于THF-MeOH(1∶1)中,装填在氨基丙基SPE(5g)上,依次用THF-MeOH(1∶1)、MeOH和5%AcOH的MeOH-DCM(1∶1)溶液洗脱。产物部分进一步通过质量定向自动化制备纯化,得到标题化合物。
收率27.5mg(26%),NMR;(400MHz,d6-DMSO)δH 2.40(dt,2H,J=7和6Hz),3.93(t,2H,J=7Hz),4.97-5.07(m,2H),5.74-5.85(m,1H),11.22(s,1H);m/z 241[MH+]。
实施例53:8-氯-3-(6-氟己基)-3,7-二氢-1H-嘌呤-2,6-二酮
NMR(400MHz,MeOD),δH 4.40(2H,dt,48和6Hz),3.98(2H,t,8Hz),1.80-1.60(4H,m),1.52-1.35(4H,m)。m/z 287[MH-]。
实施例54:8-氯-3-乙基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮
a)8-氯-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮
向8-氯-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(6g,26.5mmol)的无水DMF(30ml)溶液中加入碳酸钠(3.09g,29.15mmol)。在室温下搅拌10分钟后,加入甲氧基乙氧基甲基氯化物(3.03ml,26.5mmol),继续在氮气氛和室温下搅拌66小时。反应混合物真空浓缩后,将残余物溶解于EtOAc(100ml)中,用盐水(100ml)洗涤,将含水萃取物用DCM(100ml)萃取,干燥有机萃取物(MgSO4)、合并后真空浓缩。残余物用EtOAc研磨后,滤出固体。滤液浓缩得到浅褐色油状物,将其吸附在二氧化硅上,通过SPE(Si,50g)纯化,用梯度1∶1 EtOAc/环己烷-EtOAc洗脱,得到为白色固体的标题化合物(2g,24%),m/z 315.2[MH+]。
b)8-氯-1-甲基-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(2g,6.37mmol)的无水DMF(15ml)溶液中加入碳酸钠(0.743g,7mmol)。在室温下搅拌10分钟后,加入甲基碘化物(0.44ml,7mmol),继续在氮气氛和室温下搅拌18小时。反应混合物真空浓缩后,残余物溶解于EtOAc(100ml)中,用盐水(100ml)洗涤。干燥有机萃取物(MgSO4)、过滤并蒸发得到为黄褐色油状物的标题化合物(85%纯度)(2.98g,定量),m/z 329.2[MH+]。
c)8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-1-甲基-3-({[2-(甲氧基)乙基]氧基}甲基)-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(2.9g,6.37mmol)在二噁烷(20ml)和水(20ml)的溶液中加入5M HCl水溶液(20ml)。所得到的混合物在100℃和氮气氛下加热18小时。反应混合物随后真空浓缩,残余物溶解于EtOAc(100ml)中,用水洗涤。干燥有机萃取物(MgSO4)、过滤并蒸发。通过SPE(Si,20g)纯化,用2∶3 EtOAc/环己烷洗脱,得到为白色固体的标题化合物(1.04g,68%)。m/z 241.1[MH+]。
d)8-氯-3-乙基-1-甲基-3,7-二氢-1H-嘌呤-2,6-二酮

向8-氯-1-甲基-7-(2-丙烯-1-基)-3,7-二氢-1H-嘌呤-2,6-二酮(100mg,0.42mmol)的无水DMF(3ml)溶液中加入碳酸钠(58mg,0.54mmol),搅拌10分钟后,加入乙基碘化物(0.043ml,0.54mmol),反应混合物在室温和氮气氛下搅拌90小时。随后加入Pd(PPh3)4(73mg,0.063mmol),反应器抽空后用氮气冲洗(x3),加入吗啉(0.37ml,4.3mmol)并在室温和氮气氛下继续搅拌4小时。反应混合物用EtOAc(25ml)稀释,用2M HCl水溶液(25ml)洗涤。干燥有机萃取物(MgSO4)、过滤并蒸发。通过将化合物装填在氨基丙基SPE(5g)上纯化,用MeOH洗涤后,产物用5%AcOH/MeOH洗脱,得到为白色固体的标题化合物(67mg,70%)。NMR;dH(400MHz,d6-DMSO)1.20(t,3H,J=7Hz),3.22(s,3H),3.97(q,2H,J=7Hz),14.46(1H,br s);m/z 227.2[M-H]-。
在此将本说明书中所引用的全部出版物,包括但并不限于各种专利和专利申请引入作为参考,就好像每篇独立出版物中的全部内容被具体和独立引入作为参考一样。
Claims (13)
1.式(I)化合物或其药学可接受的盐或溶剂化物,

其中
R1选自:氢和甲基;
R2表示被C3-6环烷基取代的C1-4烷基;
和R3表示氯。
2.根据权利要求1的化合物,其用于人或兽药。
3.根据权利要求1的化合物,其用于治疗脂类代谢障碍。
4.根据权利要求1的化合物,其用于治疗糖尿病血脂障碍,混合血脂障碍,心力衰竭,血胆脂醇过多症,动脉粥样硬化,动脉硬化,和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症,冠状动脉疾病,血栓症,心绞痛,慢性肾衰竭,周围血管疾病或者中风。
5.根据权利要求1的化合物,其用于治疗血脂障碍,高脂蛋白血症,血胆脂醇过多症或高甘油三酯血症。
6.根据权利要求1的化合物在制备用于治疗人或动物对象的药物中的用途,所述的人或动物对象患有由HM74A受体激活不足所引起的疾病或者受益于激活该受体的疾病。
7.根据权利要求1的化合物在制备用于治疗脂类代谢障碍的药物中的用途。
8.根据权利要求1的化合物在制备用于治疗糖尿病血脂障碍,混合血脂障碍,心力衰竭,血胆脂醇过多症,动脉粥样硬化,动脉硬化,和高甘油三酯血症,II型糖尿病,I型糖尿病,胰岛素耐受性,高脂血症,神经性厌食症,肥胖症,冠状动脉疾病,血栓症,心绞痛,慢性肾衰竭或者中风的药物中的用途。
9.根据权利要求1的化合物在制备用于治疗血脂障碍,高脂蛋白血症,血胆脂醇过多症或高甘油三酯血症的药物中的用途。
10.药物制剂,其含有根据权利要求1的化合物和一种或多种生理学可接受的稀释剂、赋形剂或载体。
11.组合,用于一起或分别,顺序或同时给药单独或合并形式的药物制剂,所述组合包含根据权利要求1的化合物以及其它治疗活性药物。
12.药物制剂,包含
(i)根据权利要求1的化合物;
(ii)一种或多种选自斯特汀、贝特类、胆汁酸结合树脂和烟酸中的活性成分;以及
(iii)一种或多种生理学上可接受的稀释剂、赋形剂或载体。
13.制备根据权利要求1的化合物的方法,该方法包括:
(i)在N7保护的黄嘌呤的N1或N3上烷基化,或者在N1和N3上二烷基化;
(ii)在C8上氯化;以及
(iii)脱保护基;
可以以任何顺序,条件是在烷基化之后进行脱保护基。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0403282A GB0403282D0 (en) | 2004-02-14 | 2004-02-14 | Chemical compounds |
| GB0403282.7 | 2004-02-14 | ||
| GB0423562.8 | 2004-10-22 | ||
| GB0428375.0 | 2004-12-24 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2005800114336A Division CN101103030B (zh) | 2004-02-14 | 2005-02-10 | 具有hm74a受体活性的药物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101274934A true CN101274934A (zh) | 2008-10-01 |
Family
ID=32011901
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2008100992548A Pending CN101274934A (zh) | 2004-02-14 | 2005-02-10 | 具有hm74a受体活性的药物 |
Country Status (3)
| Country | Link |
|---|---|
| CN (1) | CN101274934A (zh) |
| GB (1) | GB0403282D0 (zh) |
| ZA (1) | ZA200605785B (zh) |
-
2004
- 2004-02-14 GB GB0403282A patent/GB0403282D0/en not_active Ceased
-
2005
- 2005-02-10 CN CNA2008100992548A patent/CN101274934A/zh active Pending
-
2006
- 2006-07-13 ZA ZA200605785A patent/ZA200605785B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| GB0403282D0 (en) | 2004-03-17 |
| ZA200605785B (en) | 2008-02-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101103030B (zh) | 具有hm74a受体活性的药物 | |
| CA2584904A1 (en) | Xanthine derivatives with hm74a receptor activity | |
| US20080200468A1 (en) | 2-Substituted 5-Membered Heteroaryl Carboxylates as Hm74a Receptor Agonists | |
| US20080221108A1 (en) | Anthranilic Acid Derivatives As Hm74A Receptor Agonists | |
| EP1670749A1 (en) | 2-substituted benzoic acid derivatives as hm74a receptor agonists | |
| KR20080034993A (ko) | 선택적 hm74a 작동제로서의 크산틴 유도체 | |
| JP2009504593A (ja) | 選択的hm74aアゴニストとしてのキサンチン誘導体 | |
| EP1689699A2 (en) | Anthranilic acid derivatives and their use as activators of the hm74a receptor | |
| CN110092779B (zh) | 一种取代的苯基化合物及其应用 | |
| CN101274934A (zh) | 具有hm74a受体活性的药物 | |
| RU2395511C2 (ru) | Новые соединения | |
| US20090209561A1 (en) | Xanthine Derivatives with HM74A Receptor Activity | |
| CN101087790A (zh) | 具有hm74a受体活性的黄嘌呤衍生物 | |
| MXPA06009269A (en) | Novel compounds | |
| ES2354723T3 (es) | Derivados de xantina como agonistas selectivos de hm74a. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Open date: 20081001 |